



EPIDERMOLISIS BULOSA
Achmad Fitrah Khalid, S.KedPembimbing Dr. Sarah Diba, SPKK
Bagian/Departemen Ilmu Kesehatan Kulit dan Kelamin Fakultas Kedokteran Universitas Sriwijaya/Rumah Sakit Dr. Mohammad Hoesin
Palembang
Pendahuluan
Epidermolisis bulosa (EB) adalah kelompok penyakit genetik langka yang ditandai
dengan terbentuknya lepuh akibat trauma ringan.1 Klasifikasi kelompok penyakit ini sangat
sulit mengingat banyak sekali subtipe yang telah ditemukan. Pearson berhasil
mengkategorikan EB menjadi tiga kelompok utama, yaitu EB simpleks, junctional EB, dan
dystrophic EB dengan subtipe masing-masing.2
Seluruh tipe dan subtipe EB jarang ditemukan. Angka kejadian EB tercatat kisaran 19
per satu juta angka kelahiran. Penelitian yang dilakukan National EB Registry (NEBR)
mencatat bahwa selama periode 16 tahun (1986 – 2002), sebanyak 3.300 penderita EB
berhasil di identfikasi.3 Sebuah penelitian yang dilakukan di negara-negara benua eropa
menunjukkan tidak ditemukan kecenderungan jenis kelamin, ras, etnik, dan geografis
terhadap angka kejadian EB.5
Gejala penderita EB pada umumnya adalah mudah terbentuk lepuh pada permukaan
kulit ataupun jaringan mukosa yang biasanya disebabkan kerapuhan kulit dan trauma.
Epidermolisis bulosa berasal dari defek perlekatan basal keratinosit terhadap dermis. Defek
ini bisa berasal dari membran plasma atau ekstraselular dermal-epidermal basement
membrane zone (BMZ). Defek pada perlekatan BMZ inilah yang berujung pada perbedaan
tipe EB.4
Referat ini membahas tentang ultrastruktur dermal epidermal junction, klasifikasi,
etiopatogensis, gejala klinis dan diagnosis EB diturunkan yang bertujuan untuk menambah
khasanah pengetahuan mengenai EB.
Klasifikasi
EB dibagi menjadi dua yaitu EB diturunkan (inherited EB) dan EB didapat (acquired EB). EB
diturunkan ditandai dengan kulit rapuh sebagai hasil mutasi paling tidak sepuluh gen yang
mengkode protein struktural yang berada pada epidermis, dermo-epidermal junction, atau
dermis pars papilare. Letak mutasi protein struktural ini menentukan lokasi ultrastruktural
dimana lepuh dan akan memberikan ciri khas pada tipe dan subtipe EB (Gambar 1).5 EB
1

diturunkan menjadi beberapa tipe, yaitu epidermis bulosa simpleks, junctional EB, dan
distrofik EB. Berikut akan dibahas masing-masing tipe EB diturunkan.
Gambar 1. Komponen dermal-epidermal basal membrane zone (BMZ).4
1. Epidermolisis Bulosa Simpleks
Epidermolisis bulosa simpleks (EBS) merupakan kelainan kulit ditandai dengan terbentuk
celah intraepidermal dan seringkali berhubungan dengan mutasi gen keratin.4 Sebagian
besar subtipe EBS bersifat autosomal dominan. Mutasi gen keratin 5 (K5) atau keratin 14
(K14) yang berada pada lapisan basal epidermis diduga berperan dalam terjadinya EBS.5
Tipe EBS yang paling sering ditemukan adalah Dowling-Meara EBS (Herpetiformis EBS),
EBS generalisata (Koebner EBS), dan EBS lokalisata (Weber-Cockayne EBS). Tipe EBS
yang jarang ditemukan, yaitu EBS Ogna, EBS dengan distrofi otot, EBS dengan
pigmentasi. EBS biasanya tidak diikuti dengan retardasi pertumbuhan atau anemia.4
Patologi EBS
EBS merupakan suatu kelainan genetik disebabkan oleh mutasi gen pengkode keratin 5
(K5) dan keratin 14 (K14) yang banyak terdapat pada lapisan basal epidermis. Mutasi ini
menyebabkan terpisahnya kulit pada jaringan midbasal (Gambar 2). Hemidesmosom dan
struktur BMZ lainnya dalam keadaan normal jika dilihat menggunakan mikroskop
elektron. Mayoritas mutasi gen keratin pada EBS diakibatkan oleh abnormalitas dari
pembentukan multinumerik keratin filamen.4 Mutasi ini dapat menyebabkan kerapuhan
dari sel basal dan pembentukan lepuh. Mutasi keratin 14 yang terjadi secara heterozigot
memberikan gambaran lepuh yang ditemukan pada tangan dan kaki, namun mutasi
homozigot memiliki predileksi lepuh yang lebih luas dan parah.1
2

Gambar 2. Gambaran ultrastruktural BMZ pada EB simpleks.5
a. Dowling-Meara EBS (Herpetiformis EBS)
Subtipe ini seringkali muncul pada kelahiran, ditandai dengan lesi generalisata dan
dianggap sebagai subtipe EBS yang paling berat. EBS Dowling-Meara berbeda
dengan EBS generalisata karena keterlibatan mukosa mulut dengan gambaran erosi.
Subtipe ini memberikan gambaran lepuh yang herpetiformis. Lepuh biasa muncul
pada regio trunkus dan ekstrimitas proksimal yang sembuh tanpa meninggalkan skar.
Subtipe ini seringkali melibatkan kuku berupa distrofi atau pertumbuhan kuku
dengan hiperkeratosis sub ungual.4
b. EBS Generalisata (Koebner EBS)
Subtipe ini ditemukan 1 per 500.000 kelahiran. Subtipe ini menunjukan onset lepuh
generalisata saat lahir, ditandai dengan pertumbuhan vesikel, bula, dan milia pada
sendi tangan, siku, lutut, kaki, dan tempat yang sering terkena trauma berulang.1 Lesi
yang sudah sembuh seringkali memberikan gambaran hiperpigmentasi atau
hipopigmentasi paska inflamasi. Atrofi dan milia dapat ditemukan pada EBS
generalisata, namun tidak sebanyak pada Dowling-Meara EBS. Hiperkeratosis dan
erosi pada palmoplantar sering kali ditemukan.
c. EBS Lokalisasta (Weber-Cockayne EBS)
EBS Lokalisata merupakan subtipe EBS yang paling ringan dan paling sering
ditemukan pada masa infantil dan anak-anak. Banyak kasus EBS lokalisata yang
tidak terdiagnosa karena gejala yang ringan dan sering tidak terdeteksi. Hiperhidrosis
3

pada telapak tangan dan kaki merupakan gejala yang sering ditemukan.4 Lepuh
seringkali ditemukan pada kaki dan tangan, hanya 10% pasien yang memiliki lepuh
pada tempat lain, seperti pinggang dan leher. Lepuh biasanya menjadi semakin buruk
pada cuaca panas.1 Perubahan pigmentasi paska inflamasi sering ditemukan pada tipe
ini, namun milia dan skar tidak pernah ditemukan.4
Gambar 3. Gambaran lepuh pada Dowling-Meara EBS (A); lepuh generalisata pada EBS generalisata (B); dan lepuh akibat trauma berulang pada EBS lokalisata (C).4
2. Junctional EB (JEB)
Seluruh tipe JEB merupakan penyakit keturunan autosomal resesif yang menyebabkan
disfungsi pada anchoring filaments yang terdapat pada lamina lucida.5 Penyakit keturunan
ini memiliki gambaran klinis yang bervariasi tergantung dari defek secara molekuler.4 JEB
terbagi menjadi dua tipe yaitu tipe Herlitz dan non-Herlitz.2 Pada JEB-Herlitz terjadi
mutasi heterozigot yang menyebabkan terminasi kodon yang prematur pada gen yang
mengkode sub-unit protein laminin 332, sebuah protein penyokong lamina lucida pada
dermo-epidermal junction.5,6
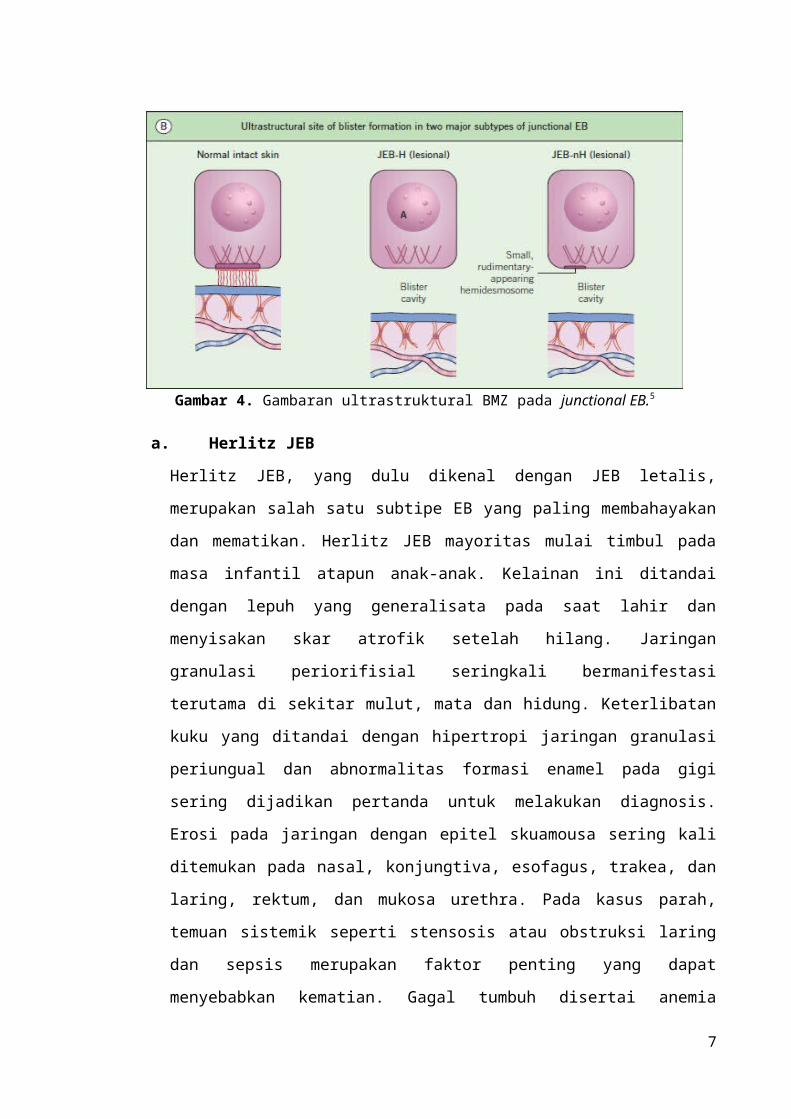
Patologi JEB
JEB merupakan suatu kelainan genetik autosomal resesif yang ditandai dengan mutasi
pada gen pengkode α3, β3, dan γ2 pada sub-unit laminin 332. Kehilangan salah satu dari
tiga rantai tersebut akan memberikan klinis lepuh. Pada penderita JEB Herlitz, ditemukan
terminasi kodon secara prematur yang menyebabkan laminin 332 tidak berfungsi secara
keseluruhan. Sementara itu, pada penderita Non-Herlitz JEB, ditemukan kehilangan parsial
terhadap fungsi dari laminin 332 (Gambar 4).4
4
A CB

Gambar 4. Gambaran ultrastruktural BMZ pada junctional EB.5
a. Herlitz JEB
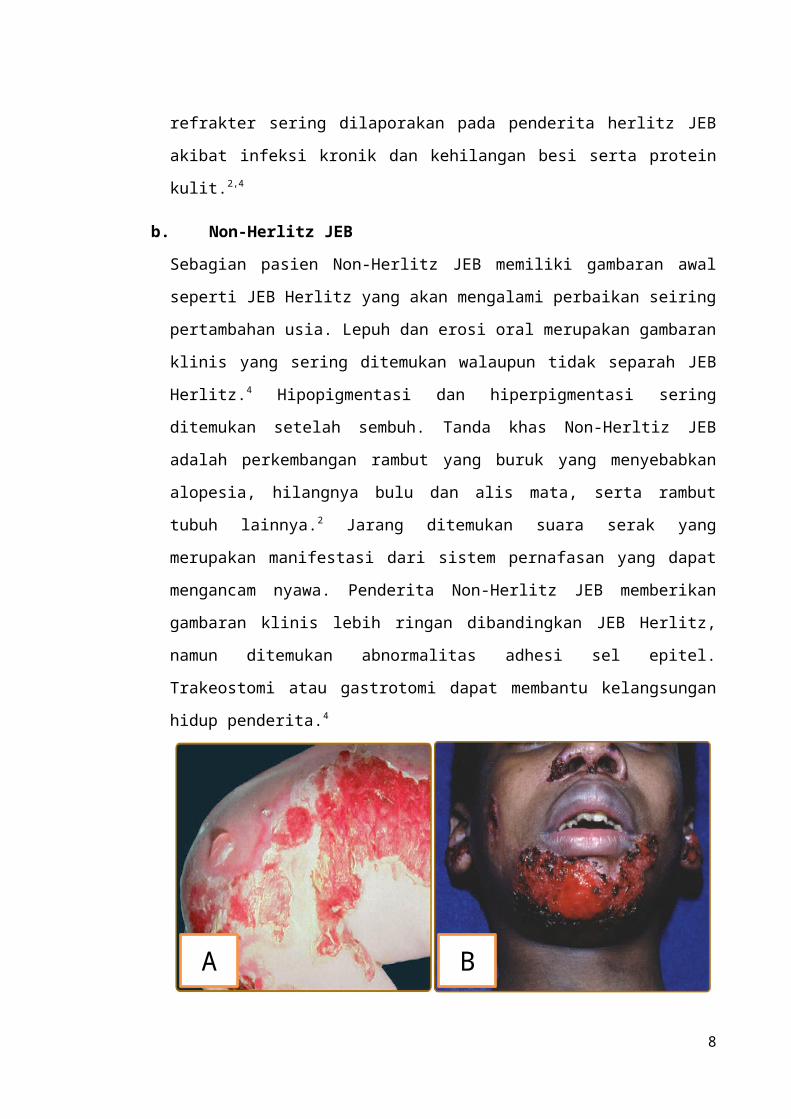
Herlitz JEB, yang dulu dikenal dengan JEB letalis, merupakan salah satu subtipe EB
yang paling membahayakan dan mematikan. Herlitz JEB mayoritas mulai timbul
pada masa infantil atapun anak-anak. Kelainan ini ditandai dengan lepuh yang
generalisata pada saat lahir dan menyisakan skar atrofik setelah hilang. Jaringan
granulasi periorifisial seringkali bermanifestasi terutama di sekitar mulut, mata dan
hidung. Keterlibatan kuku yang ditandai dengan hipertropi jaringan granulasi
periungual dan abnormalitas formasi enamel pada gigi sering dijadikan pertanda
untuk melakukan diagnosis. Erosi pada jaringan dengan epitel skuamousa sering kali
ditemukan pada nasal, konjungtiva, esofagus, trakea, dan laring, rektum, dan mukosa
urethra. Pada kasus parah, temuan sistemik seperti stensosis atau obstruksi laring dan
sepsis merupakan faktor penting yang dapat menyebabkan kematian. Gagal tumbuh
disertai anemia refrakter sering dilaporakan pada penderita herlitz JEB akibat infeksi
kronik dan kehilangan besi serta protein kulit.2,4
b. Non-Herlitz JEB
Sebagian pasien Non-Herlitz JEB memiliki gambaran awal seperti JEB Herlitz yang
akan mengalami perbaikan seiring pertambahan usia. Lepuh dan erosi oral
merupakan gambaran klinis yang sering ditemukan walaupun tidak separah JEB
Herlitz.4 Hipopigmentasi dan hiperpigmentasi sering ditemukan setelah sembuh.
Tanda khas Non-Herltiz JEB adalah perkembangan rambut yang buruk yang
menyebabkan alopesia, hilangnya bulu dan alis mata, serta rambut tubuh lainnya.2
Jarang ditemukan suara serak yang merupakan manifestasi dari sistem pernafasan
5

yang dapat mengancam nyawa. Penderita Non-Herlitz JEB memberikan gambaran
klinis lebih ringan dibandingkan JEB Herlitz, namun ditemukan abnormalitas adhesi
sel epitel. Trakeostomi atau gastrotomi dapat membantu kelangsungan hidup
penderita.4
Gambar 5. Lepuh generalisata pada anak dengan herlitz JEB (A); Erosi periorifisial dan hipertrofik jaringan granulasi pada non-herlitz JEB (B).5
3. Distrofik EB (DEB)
Distrofik EB (DEB) merupakan penyakit keturunan yang bersifat autosomal dominan
(DDEB) atau resesif (RDEB). Kelainan ini ditandai dengan kerapuhan kulit, lepuh, skar,
perubahan kuku dan pembentukan milia.2 Salah satu alasan penting untuk membedakan
dari kedua subtipe ini adalah peningkatan angka kejadian karsinoma skuamousa invasif
yang sering dikaitkan dengan bentuk resesif.4
Patologi DEB
Distrofik EB telah dibuktikan berkaitan dengan mutasi pada gen pengkode kolagen VII
(COL7A1). Abnomralitas pada anchoring fibrils juga berperan pada penderita DEB yang
akan menyebabkan lepuh distrofik (Gambar 4). Pada analisa mikroskopis, terkadang
ditemukan retensi sitoplasma kolagen VII di keratinosit.4
6
A B
Gambar 6. Gambaran ultrastruktural BMZ pada distrofik EB.5
a. Dominan Distrofik EB Lokalisata (DDEB Lokalisata)
Kelainan ini biasanya mulai bermanifestasi pada masa anak-anak, namun tidak
jarang muncul pada kelahiran. Keluhan yang sering disampaikan penderita adalah
lepuh yang terlokalisir terutama pada daerah yang sering mengalami trauma berulang
seperti lutut, sakrum, dan permukaan tangan. Pada area tersebut didapatkan skar
hipertrofik. Milia sering ditemukan pada proses penyembuhan. Distrofi kuku dan
pelepasan kuku disertai skar atrofik pada jari sering ditemukan. Abnormalitas pada
kuku hanya ditemukan pada DDEB.4
b.Dominan Distrofik EB Generalisata (DDEB Generalisata)
DDEB generalisata, sering disebut subtipe Pasini, memberikan gambaran lepuh yang
lebih parah dan luas dibandingkan dengan tipe lokalisata pada saat kelahiran. Pada
proses penyembuhan sering ditemukan milia. Papul yang tampak pada badan
menjadi ciri khas DDEB generalisata. Lepuh akan lebih terlokalisir di ekstrimitas
seiring bertambahnya usia. Beberapa penderita menunjukkan klinis berupa distrofi
bahkan hilangnya kuku. Erosi pada sekitar mulut dan defek pada enamel gigi dapat
terjadi namun tidak ekstensif.4
c. Resesif Distrofik EB (RDEB)
Resesif Distrofik EB (RDEB) sangat bervariasi dalam derajat keparahannya. Sama
seperti DDEB lokalisata, RDEB lokalisata dapat terjadi pada daerah yang mengalami
trauma berulang. Skar dan milia biasanya terbentuk pada masa penyembuhan.4
7
Secara garis besar, RDEB memiliki dua subtipe, yaitu subtipe mitis (mild)
dan severe (Hallopeau-Siemens). Pada tipe mitis, lepuh hanya terbatas pada tangan,
kaki, lutut, siku, dan juga memiliki komplikasi yang tidak luas. Sedangkan pada tipe
severe ditemukan lepuh jaringan mukosa dan kutan yang generalisata. Gambaran
deformitas mitten-like merupakan ciri khas pada jari-jari penderita RDEB tipe severe
yang muncul pada usia 25 tahun (Gambar 7).1
Orofaring dapat terlibat baik pada tipe dominan ataupun resesif dengan
gambaran erosi generalisata yang akan menjadi skar dan membatasi pergerakan lidah
dan gangguan membuka mulut. Trakeostomi terkadang dilakukan pada penderita
yang mengalami keterlibatan pada trakea ataupun laring. Erosi pada esofagus sering
berujung pada striktur. Seluruh gangguan tersebut membuat banyak penderita RDEB
mengalami malnutrisi, gangguan pertumbuhan, dan anemia defisiensi besi.4
Pada penderita severe RDEB, 50 -80% dilaporkan mengalami karsinoma dan
dilaporkan meninggal akibat metastasis. Karsinoma yang diakibatkan oleh RDEB
dipisahkan dari karsinoma sel skuama lainnya karena sifatnya yang agresif dengan
kemungkinan metastasis yang tinggi.4
Gambar 7. Lepuh generalisata pada severe RDEB (A); Gambaran deformitas
mitten-like pada RDEB (B).2
Diagnosis
Langkah pertama mendiagnosis EB dimulai dari anamnesis dan pemeriksaan fisik. Pada
anamnesis, riwayat onset lepuh muncul dan keberadaan lepuh pada keluarga dapat membantu
diagnosis. Riwayat gangguan sistem gastro intestinal, respirasi, mata, gigi, tulang dan
genitourinaria merupakan evaluasi penting dalam perjalanan penyakit. Pada pemeriksaan
8
A B

fisik, pemeriksaan status dermatologikus saja tidak cukup, perlu dilakukan evaluasi secara
menyeluruh baik pada jaringan mukosa, rambut, kuku, dan gigi. Pada pemeriksaan
laboratorium, evaluasi terhadap anemia dan kadar albumin untuk mengukur tingkat malnutrisi
perlu dilakukan.4
Biopsi kulit merupakan langkah penting dalam mendiagnosis. Analisa histologis dapat
menyingkirkan penyebab lain dari lepuh walaupun tidak dapat mendiagnosis EB. Perlu
dilakukan transmission electron microscopy (TEM) atau indirect immnofluorescent
microscopy (IDIF) untuk melihat pemisahan pada BMZ.4
Kesimpulan
Epidermolisis bulosa (EB) merupakan suatu kelainan genetik pada kulit yang ditandai
timbulnya bula akibat trauma ringan. EB dapat bersifat autosomal resesif ataupun autosomal
dominan tergantung pada subtipe. EB dibedakan menjadi EB diturunkan dan EB didapat. EB
diturunkan dibagi menjadi tiga tipe berdasarkan letak defek pada BMZ, yaitu EB simpleks
(EBS), junctional EB (JEB), dan distrofik EB (DEB) yang masing-masing memiliki subtipe.
Diagnosa dini EB sangat penting untuk mencegah progresifitas dan komplikasi. Anamnesis
dan pemeriksaan yang baik dapat mendiagnosis EB, walaupun pemeriksaan mikroskop
elektron (TEM) merupakan gold standard untuk mendiagnosa jenis EB.
9






























