Review The malonyl CoA axis as a potential target for treating ischaemic heart disease John R. Ussher and Gary D. Lopaschuk* Cardiovascular Research Group, Department of Pediatrics, University of Alberta, Edmonton, Canada Received 23 January 2008; revised 11 May 2008; accepted 16 May 2008; online publish-ahead-of-print 22 May 2008 Time for primary review: 20 days Cardiovascular disease is the leading cause of death and disability for people living in western societies, with ischaemic heart disease accounting for the majority of this health burden. The primary treatment for ischaemic heart disease consists of either improving blood and oxygen supply to the heart or redu- cing the heart’s oxygen demand. Unfortunately, despite recent advances with these approaches, ischae- mic heart disease still remains a major health problem. Therefore, the development of new treatment strategies is still required. One exciting new approach is to optimize cardiac energy metabolism, par- ticularly by decreasing the use of fatty acids as a fuel and by increasing the use of glucose as a fuel. This approach is beneficial in the setting of ischaemic heart disease, as it allows the heart to produce energy more efficiently and it reduces the degree of acidosis associated with ischaemia/reper- fusion. Malonyl CoA is a potent endogenous inhibitor of cardiac fatty acid oxidation, secondary to inhi- biting carnitine palmitoyl transferase-I, the rate-limiting enzyme in the mitochondrial uptake of fatty acids. Malonyl CoA is synthesized in the heart by acetyl CoA carboxylase, which in turn is phosphorylated and inhibited by 5 0 AMP-activated protein kinase. The degradation of myocardial malonyl CoA occurs via malonyl CoA decarboxylase (MCD). Previous studies have shown that inhibiting MCD will significantly increase cardiac malonyl CoA levels. This is associated with an increase in glucose oxidation, a decrease in acidosis, and an improvement in cardiac function and efficiency during and following ischaemia. Hence, the malonyl CoA axis represents an exciting new target for the treatment of ischaemic heart disease. KEYWORDS Ischaemic heart disease; Malonyl CoA; Malonyl CoA decarboxylase; Acetyl CoA carboxylase; AMP-activated protein kinase; Fatty acid oxidation 1. Introduction Cardiovascular disease (CVD) is a major health problem worldwide, and it is predicted to be the number one killer by 2010. 1 The underlying cause for the majority living with CVD is a diminished oxygen supply to the cardiac muscle, termed ‘ischaemic heart disease’. Fortunately, epidemiological studies and randomized clinical trials have provided compelling evidence that ischaemic heart disease is largely manageable. 2 Current treatment regimens, which consist of either percutaneous or surgical techniques to improve myocardial blood supply, or pharmacotherapy to limit myocardial oxygen demand, have greatly aided in the overall prognosis of ischaemic heart disease patients. Yet, there are still patients who prove to be ineligible or refractory to conventional treat- ment, and percutaneous or surgical revascularization is associated with a distinct set of risks. Therefore, new approaches to treat such patients are necessary. One such exciting new therapy is the optimization of cardiac energy metabolism. In the setting of ischaemic heart disease, the general premise for the optimization of cardiac energy metabolism is to either stimulate the oxidation of glucose or inhibit the oxidation of fatty acids for energy production. 3 As the oxidation of one molecule of glucose consumes less oxygen than that of a fatty acid, this allows the heart to produce energy more efficiently. Furthermore, stimulating glucose oxidation either directly, or secondarily due to an inhibition of fatty acid oxidation, results in improved coupling between glycolysis and glucose oxidation, which decreases proton production and alleviates myocardial acidosis, improving cardiac efficiency. 4,5 There are numerous ways to inhibit cardiac fatty acid oxi- dation, some of which include 2 the inhibition of fatty acid transport into the cardiac myocyte, 3 the inhibition of fatty acid uptake into the mitochondria, 4 and the inhibition of the enzymatic machinery of the b-oxidative pathway itself. Although there are existing agents that target all three of these approaches, this review will focus on the inhibition of mitochondrial fatty acid uptake approach, 3 and in particular, the use of agents that increase levels of * Corresponding author: 423 Heritage Medical Research Center, University of Alberta, Edmonton, Canada T6G 2S2. Tel: þ1 780 492 2170; fax: þ1 780 492 9753. E-mail address: [email protected] Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2008. For permissions please email: [email protected]. Cardiovascular Research (2008) 79, 259–268 doi:10.1093/cvr/cvn130 Downloaded from https://academic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Review
The malonyl CoA axis as a potential target for treatingischaemic heart disease
John R. Ussher and Gary D. Lopaschuk*
Cardiovascular Research Group, Department of Pediatrics, University of Alberta, Edmonton, Canada
Received 23 January 2008; revised 11 May 2008; accepted 16 May 2008; online publish-ahead-of-print 22 May 2008
Time for primary review: 20 days
Cardiovascular disease is the leading cause of death and disability for people living in western societies,with ischaemic heart disease accounting for the majority of this health burden. The primary treatmentfor ischaemic heart disease consists of either improving blood and oxygen supply to the heart or redu-cing the heart’s oxygen demand. Unfortunately, despite recent advances with these approaches, ischae-mic heart disease still remains a major health problem. Therefore, the development of new treatmentstrategies is still required. One exciting new approach is to optimize cardiac energy metabolism, par-ticularly by decreasing the use of fatty acids as a fuel and by increasing the use of glucose as a fuel.This approach is beneficial in the setting of ischaemic heart disease, as it allows the heart toproduce energy more efficiently and it reduces the degree of acidosis associated with ischaemia/reper-fusion. Malonyl CoA is a potent endogenous inhibitor of cardiac fatty acid oxidation, secondary to inhi-biting carnitine palmitoyl transferase-I, the rate-limiting enzyme in the mitochondrial uptake of fattyacids. Malonyl CoA is synthesized in the heart by acetyl CoA carboxylase, which in turn is phosphorylatedand inhibited by 50AMP-activated protein kinase. The degradation of myocardial malonyl CoA occurs viamalonyl CoA decarboxylase (MCD). Previous studies have shown that inhibiting MCD will significantlyincrease cardiac malonyl CoA levels. This is associated with an increase in glucose oxidation, a decreasein acidosis, and an improvement in cardiac function and efficiency during and following ischaemia.Hence, the malonyl CoA axis represents an exciting new target for the treatment of ischaemic heartdisease.
KEYWORDSIschaemic heart disease;
Malonyl CoA;
Malonyl CoA decarboxylase;
Acetyl CoA carboxylase;
AMP-activated protein kinase;
Fatty acid oxidation
1. Introduction
Cardiovascular disease (CVD) is a major health problemworldwide, and it is predicted to be the number one killerby 2010.1 The underlying cause for the majority living withCVD is a diminished oxygen supply to the cardiac muscle,termed ‘ischaemic heart disease’.
Fortunately, epidemiological studies and randomizedclinical trials have provided compelling evidence thatischaemic heart disease is largely manageable.2 Currenttreatment regimens, which consist of either percutaneousor surgical techniques to improve myocardial blood supply,or pharmacotherapy to limit myocardial oxygen demand,have greatly aided in the overall prognosis of ischaemicheart disease patients. Yet, there are still patients whoprove to be ineligible or refractory to conventional treat-ment, and percutaneous or surgical revascularization isassociated with a distinct set of risks. Therefore, newapproaches to treat such patients are necessary. One such
exciting new therapy is the optimization of cardiac energymetabolism.
In the setting of ischaemic heart disease, the generalpremise for the optimization of cardiac energy metabolismis to either stimulate the oxidation of glucose or inhibitthe oxidation of fatty acids for energy production.3 As theoxidation of one molecule of glucose consumes less oxygenthan that of a fatty acid, this allows the heart to produceenergy more efficiently. Furthermore, stimulating glucoseoxidation either directly, or secondarily due to an inhibitionof fatty acid oxidation, results in improved couplingbetween glycolysis and glucose oxidation, which decreasesproton production and alleviates myocardial acidosis,improving cardiac efficiency.4,5
There are numerous ways to inhibit cardiac fatty acid oxi-dation, some of which include2 the inhibition of fatty acidtransport into the cardiac myocyte,3 the inhibition of fattyacid uptake into the mitochondria,4 and the inhibition ofthe enzymatic machinery of the b-oxidative pathwayitself. Although there are existing agents that target allthree of these approaches, this review will focus on theinhibition of mitochondrial fatty acid uptake approach,3
and in particular, the use of agents that increase levels of
* Corresponding author: 423 Heritage Medical Research Center, Universityof Alberta, Edmonton, Canada T6G 2S2. Tel: þ1 780 492 2170; fax: þ1 780492 9753.
E-mail address: [email protected]
Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2008.For permissions please email: [email protected].
Cardiovascular Research (2008) 79, 259–268doi:10.1093/cvr/cvn130
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

malonyl CoA, a potent endogenous inhibitor of carnitinepalmitoyltransferase I (CPT-I), the rate-limiting enzyme inthe mitochondrial uptake of fatty acids.6 We will reviewthe literature on the regulation of malonyl CoA via bothits synthesis and degradation, how the malonyl CoA axishas been manipulated in animal models of ischaemia/reperfusion (focusing on the most recent studies involvingtransgenic mice to manipulate malonyl CoA), and thepotential for this axis to be manipulated in humans.
2. Cardiac energy metabolism
In the normal healthy heart, almost all (.95%) ATP gener-ated in the heart comes from mitochondrial oxidative phos-phorylation, with the remainder derived from glycolysis.7
Despite producing more ATP than carbohydrates, fattyacids are not as oxygen-efficient, requiring �10% moreoxygen to produce an equivalent amount of ATP.8 This is ofparticular importance when oxygen becomes a limitingfactor for oxidative metabolism, as seen with ischaemicheart disease. In addition, fatty acids directly inhibit theoxidation of carbohydrates through a phenomenon termedthe ‘Randle cycle’.9 This uncouples glycolysis from glucoseoxidation, which results in an increased proton productionthat reduces cardiac efficiency during reperfusion followingischaemia10 (see 11,12 for review).
As will be discussed, an emerging approach to optimizingcardiac energy metabolism is to keep levels of malonyl CoAhigh in the heart. This inhibits the mitochondrial uptake offatty acids, leading to a subsequent inhibition of fatty acidb-oxidation and secondary increase in glucose oxidation,thereby making oxygen utilization and cardiac energy pro-duction more efficient, while preventing the production ofprotons and development of acidosis.10,11
3. Cardiac carbohydrate metabolism
The metabolism of glucose can be separated into two majorcomponents, glycolysis and glucose oxidation (see 3,8 forreview). Glycolysis results in the production of pyruvateand accounts for ,10% of the total ATP produced by theaerobic heart.13 If glycolysis is coupled to glucose oxidation,the pyruvate generated from glycolysis is converted toacetyl-CoA (which is subsequently oxidized in the TCAcycle) by the enzymatic action of the multi-enzymecomplex, pyruvate dehydrogenase (PDH).
The PDH complex is under tight regulation by an upstreamkinase, PDH kinase, which phosphorylates and inhibits itsactivity.9 This PDH kinase is positively regulated by acetylCoA and NADH. As mitochondrial acetyl CoA/CoA andNADH/NAD ratios are increased by elevated rates of fattyacid oxidation, it leads to a potent inhibition of PDH andglucose oxidation. This phenomenon was first describedby Randle et al.9 in the 1960s and has been termed the‘Randle cycle’.
4. Cardiac fatty acid metabolism
Fatty acids enter the cardiac myocyte by either passive dif-fusion or protein-mediated transport across the sarco-lemma14 (see 3,8 for review). Once transported across thesarcolemma, fatty acids are subsequently activated byesterification to fatty acyl CoA by fatty acyl CoA synthetase.
This acyl CoA can either be esterified to intracellular lipidsor converted to long-chain fatty acyl carnitine by CPT-I.7
Fatty acid b-oxidation occurs predominantly in the mito-chondria and to a smaller extent in the peroxisomes.15 Formitochondrial fatty acid b-oxidation to begin, the cyto-plasmic long-chain fatty acyl CoA must first be transportedinto the mitochondrial matrix through a carnitine-dependent transport system.8,16 This carnitine transportsystem involves three enzymes: CPT-I, carnitine acyltranslo-case, and CPT-II. Of these three enzymes, CPT-I is rate limit-ing in regard to mitochondrial fatty acid uptake and issubject to potent inhibition via malonyl CoA, the compoundwhose regulation will be the major focus of this review.6
Once in the mitochondria, b-oxidation repeatedly cleavesoff two carbon acetyl CoA units from fatty acyl CoA, gener-ating NADH and reduced flavine adenine dinucleotide in theprocess. The b-oxidation process involves four enzymaticallycatalysed reactions, with the last step regenerating acyl CoAfor another round of b-oxidation and releasing acetyl CoAfor the citric acid cycle. As mentioned earlier, oxidation offatty acids increases the acetyl CoA/CoA ratio, which inhi-bits PDH and glucose oxidation.
5. Regulation of malonyl CoA
As mentioned previously, malonyl CoA is a potent endogen-ous inhibitor of CPT-I, the rate-limiting enzyme in the mito-chondrial uptake of fatty acids. Thus, malonyl CoAdecreases the uptake of fatty acids into the mitochondria,thereby reducing mitochondrial fatty acid b-oxidation.Because the turnover of malonyl CoA is quite rapid, witha half life of �1.25 min,17 both the production and thedegradation of malonyl CoA control its levels, and thereforeof fatty acid oxidation rates.
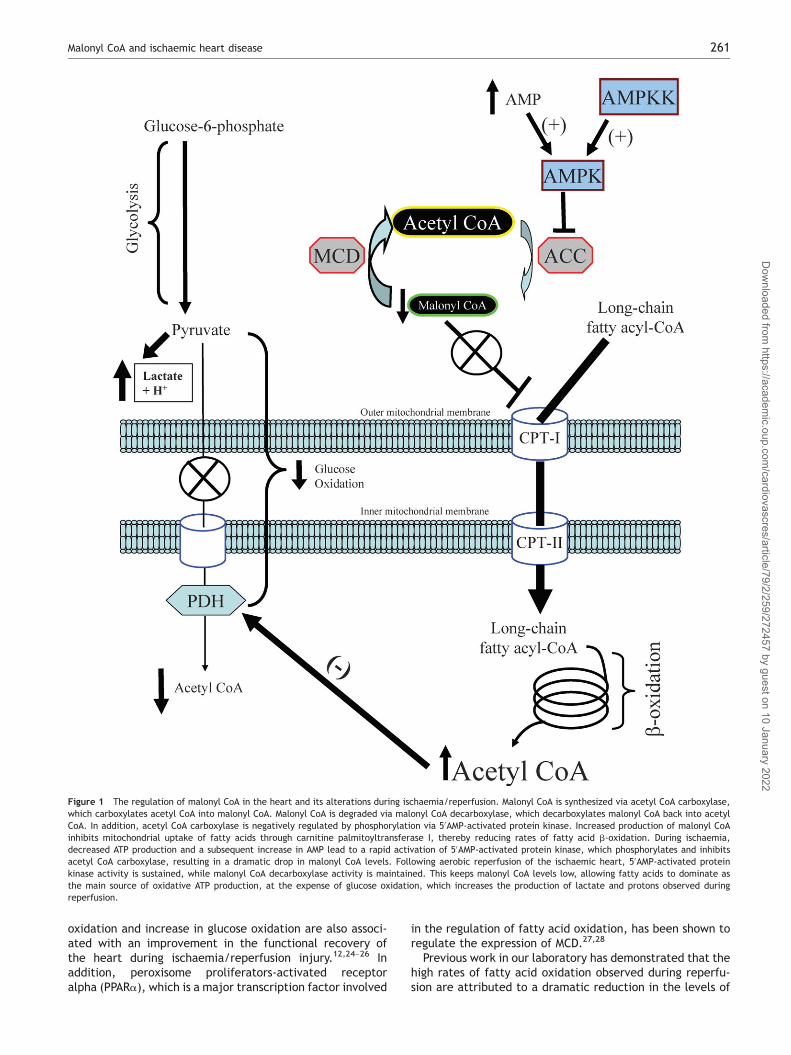
The production of malonyl CoA is primarily attributed tothe enzymatic activity of acetyl CoA carboxylase (ACC),which catalyses the carboxylation of acetyl CoA to malonylCoA (Figure 1).18,19 There are two isoforms of ACC in theheart, a and b, with a predominance of ACCb.19 This leadsto the suggestion that the malonyl CoA produced by thisisoform is more involved in the regulation of fatty acid oxi-dation, as opposed to the high abundance of ACCa in theliver, where the malonyl CoA produced by this isoform ismore involved in the regulation of fatty acid synthesis.Studies from our laboratory have confirmed the key role ofACCb in regulating cardiac fatty acid oxidation.20 The regu-lation of ACC is under phosphorylation/dephosphorylationcontrol, with 50AMP-activated protein kinase (AMPK) havinga major role in its regulation in the heart (Figure 1).21,22
As will be discussed in the following section, this AMPK regu-lation of ACC becomes very important during times of energystarvation in the heart, as seen in ischaemia/reperfusion.
Until recently, it was much less clear as to what enzymesmight be responsible for the degradation of malonyl CoA.One enzyme that has emerged as being important in control-ling cardiac malonyl CoA degradation is malonyl CoA decar-boxylase (MCD), whose catalytic activity is responsible forthe decarboxylation of malonyl CoA back into acetyl CoA(Figure 1).23 Studies in both rat and mouse have demon-strated that MCD is indeed involved in regulating cardiacmalonyl CoA levels, and that inhibition of MCD can limitrates of fatty acid oxidation, leading to a secondary increasein glucose oxidation (Figure 2). This decrease in fatty acid
J.R. Ussher and G.D. Lopaschuk260
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022
oxidation and increase in glucose oxidation are also associ-ated with an improvement in the functional recovery ofthe heart during ischaemia/reperfusion injury.12,24–26 Inaddition, peroxisome proliferators-activated receptoralpha (PPARa), which is a major transcription factor involved
in the regulation of fatty acid oxidation, has been shown toregulate the expression of MCD.27,28
Previous work in our laboratory has demonstrated that thehigh rates of fatty acid oxidation observed during reperfu-sion are attributed to a dramatic reduction in the levels of
Figure 1 The regulation of malonyl CoA in the heart and its alterations during ischaemia/reperfusion. Malonyl CoA is synthesized via acetyl CoA carboxylase,which carboxylates acetyl CoA into malonyl CoA. Malonyl CoA is degraded via malonyl CoA decarboxylase, which decarboxylates malonyl CoA back into acetylCoA. In addition, acetyl CoA carboxylase is negatively regulated by phosphorylation via 50AMP-activated protein kinase. Increased production of malonyl CoAinhibits mitochondrial uptake of fatty acids through carnitine palmitoyltransferase I, thereby reducing rates of fatty acid b-oxidation. During ischaemia,decreased ATP production and a subsequent increase in AMP lead to a rapid activation of 50AMP-activated protein kinase, which phosphorylates and inhibitsacetyl CoA carboxylase, resulting in a dramatic drop in malonyl CoA levels. Following aerobic reperfusion of the ischaemic heart, 50AMP-activated proteinkinase activity is sustained, while malonyl CoA decarboxylase activity is maintained. This keeps malonyl CoA levels low, allowing fatty acids to dominate asthe main source of oxidative ATP production, at the expense of glucose oxidation, which increases the production of lactate and protons observed duringreperfusion.
Malonyl CoA and ischaemic heart disease 261
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022
malonyl CoA, as opposed to direct alterations in the charac-teristics of CPT-I.21 In addition, we have shown that thisreduction in malonyl CoA levels is associated with a rapidactivation of AMPK during ischaemia, which persists intoreperfusion, causing the phosphorylation-induced inacti-vation of ACC (Figure 1).21 It has also been suggested that
MCD is a direct target of AMPK, but our laboratory hasbeen unable to reproduce that data.29 In summary,decreased ACC activity via AMPK phosphorylation, coupledwith a maintained MCD activity, is a key factor responsiblefor the rapid decrease in cardiac malonyl CoA levelsobserved during ischaemia/reperfusion.
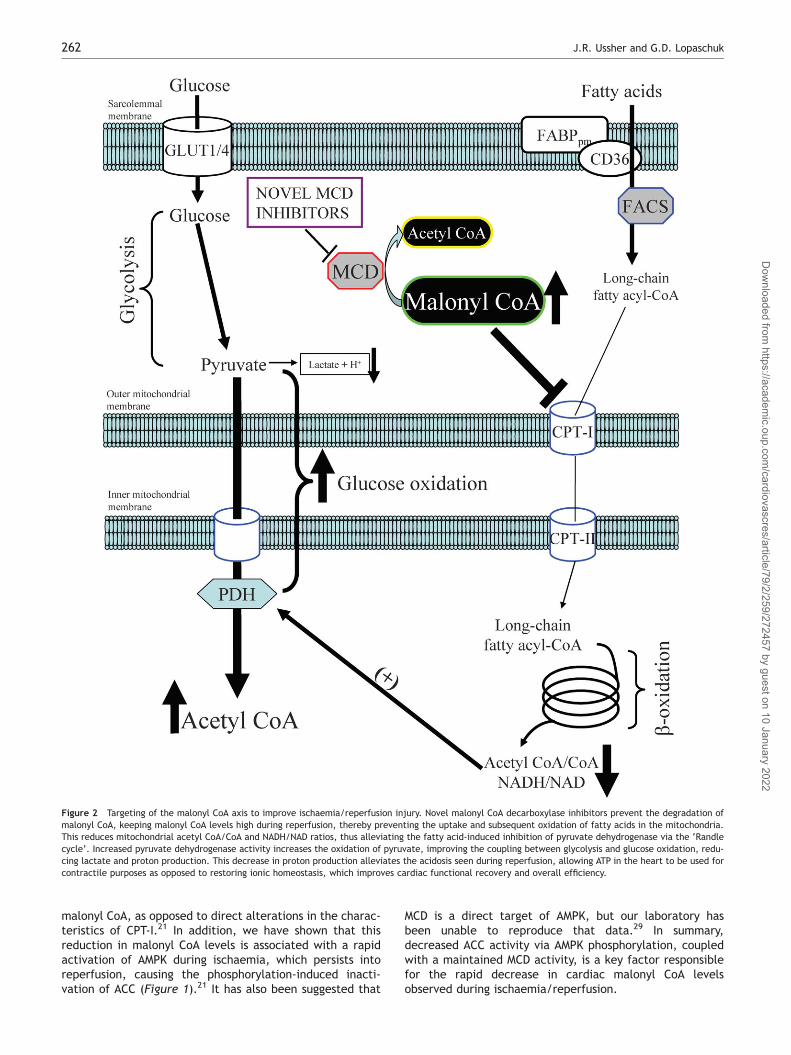
Figure 2 Targeting of the malonyl CoA axis to improve ischaemia/reperfusion injury. Novel malonyl CoA decarboxylase inhibitors prevent the degradation ofmalonyl CoA, keeping malonyl CoA levels high during reperfusion, thereby preventing the uptake and subsequent oxidation of fatty acids in the mitochondria.This reduces mitochondrial acetyl CoA/CoA and NADH/NAD ratios, thus alleviating the fatty acid-induced inhibition of pyruvate dehydrogenase via the ‘Randlecycle’. Increased pyruvate dehydrogenase activity increases the oxidation of pyruvate, improving the coupling between glycolysis and glucose oxidation, redu-cing lactate and proton production. This decrease in proton production alleviates the acidosis seen during reperfusion, allowing ATP in the heart to be used forcontractile purposes as opposed to restoring ionic homeostasis, which improves cardiac functional recovery and overall efficiency.
J.R. Ussher and G.D. Lopaschuk262
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

Cardiac malonyl CoA levels can also increase secondary toan increase in glucose oxidation. Previous work demon-strated that the acetyl CoA produced by PDH was moreaccessible to carnitine acetyltransferase than that producedfrom fatty acid b-oxidation.30 Thus, we hypothesized andshowed that increased glucose oxidation could increasecytosolic acetyl CoA supply for ACCb, increasing malonylCoA levels that then inhibit fatty acid b-oxidation.20
It is of interest to note that although malonyl CoA is apotent inhibitor of CPT-I, the majority of malonyl CoA inthe heart is inaccessible to CPT-I. The cytosolic concen-tration of malonyl CoA in the heart is �5 mM, whereas theIC50 of CPT-I inhibition via malonyl CoA is between 50 and100 nM.20,31 Therefore, if all the malonyl CoA in the heartwere accessible to CPT-I, fatty acid oxidation would alwaysbe completely inhibited, which of course is not the case.The reason why the majority of malonyl CoA is not accessi-ble to CPT-I is not known, but may have to do with compart-mentation of malonyl CoA. Recent evidence supporting thismay come from the observation that a large proportion ofMCD exists in the peroxisomes.32 In addition, it has beensuggested that the majority of malonyl CoA produced inthe heart originates from the peroxisomes.33
6. Cardiac energy metabolism duringischaemia/reperfusion
Cardiac energy metabolism is drastically altered duringischaemia, starting with an acceleration of glycolysis, inan attempt to provide an anaerobic source of ATP to makeup for the reduction in oxidative ATP production. Also ofimportance is that fatty acids dominate as the substratefor what residual oxidative metabolism remains in theoxygen-deprived ischaemic heart,34 at the expense ofglucose oxidation. This results in an uncoupling between gly-colysis and glucose oxidation, contributing to the acidosisobserved in the ischaemic heart, which reduces cardiacefficiency.4,10
During reperfusion of the ischaemic heart, glycolytic ratesremain elevated, while fatty acids dominate as a source foroxidative energy production.10 These high rates of fatty acidmetabolism can account for .90% of energy production inthe reperfused heart, and this results in a drastic inhibitionof glucose oxidation. Thus, similar to ischaemia, reperfusionof the ischaemic heart is accompanied by an increased pro-duction of protons that lowers cardiac efficiency. A majorfocus of this review has been the emphasis that high ratesof fatty acid oxidation can cause myocardial acidosis byuncoupling glucose oxidation from glycolysis. Althoughother studies have suggested that acidosis may protect theheart during reperfusion by reducing contractile force35–38
to preserve the viability of hibernating myocardium, orkeeping the mitochondrial permeability transition pore ina closed state,39,40 such matters are beyond the scope ofthis review.
The reason for the excessive use of fatty acids as a sourceof fuel during and following ischaemia is primarily the resultof two factors: (1) plasma levels of fatty acids increase dra-matically during and following ischaemia,41–44 and (2) sub-cellular changes occur in the heart itself, resulting in adecreased control over fatty acid oxidation.21,45 In particu-lar, the levels of malonyl CoA decrease in the heart due to
the rapid activation of AMPK during ischaemia, which per-sists into reperfusion, resulting in the phosphorylationinduced inhibition of ACC.11,21,23,45 This leads to the accel-erated mitochondrial uptake of fatty acids and subsequentoxidation.
7. Targeting the malonyl CoA axis to treatcardiac ischaemia
There are a number of ways to manipulate malonyl CoA inthe heart, which include manipulation of the enzymesinvolved in regulating its production, i.e. AMPK, ACC, andMCD. The following sections will examine each of thelatter three enzymes individually as potential treatmentsfor ischaemia/reperfusion in more detail.
7.1 Targeting 50AMP-activated protein kinaseto treat ischaemia/reperfusion
Since its initial identification in 1988 by Sim and Hardie,46
AMPK has become a protein, with wide interest amongmany investigators, due to its ability to increase energymetabolism in times of stress. In regard to the stress ofischaemia/reperfusion injury, many groups have postulatedthat AMPK activation would be beneficial in this scenariovia increasing glucose uptake to provide an anaerobicsource of ATP for the energy-starved heart.47–49 However,our group previously showed that AMPK activity candecrease cardiac malonyl CoA and increase fatty acid oxi-dation in the ischaemic heart.21 We therefore hypothesizedthat inhibition of AMPK would be beneficial for ischaemia/reperfusion injury by increasing malonyl CoA levels andreducing fatty acid oxidation rates, thereby alleviatingmyocardial acidosis. Supporting the former proposal,isolated working heart studies in a transgenic mouse modelof a dominant negative (DN) AMPKa2 with nearly a completeloss of myocardial AMPK activity have been shown to recoverworse during reperfusion following a low-flow ischaemia.50
Hearts from the transgenic DN-AMPKa2 mice were unableto increase GLUT4 translocation and glucose uptakeduring the low-flow ischaemia or reperfusion periods, andthey had accelerated rates of apoptosis as indicated byincreased caspase 3 cleavage and TUNEL staining.However, DN-AMPKa2 transgenic mice had significantcontractile dysfunction compared with wild-type mice inthe normal setting, which may explain why they did notrecover as well during ischaemia/reperfusion. Furthermore,there were no differences in myocardial metabolismbetween the control and DN-AMPKa2 transgenic mice,which suggests that there may have been no differences inmalonyl CoA levels from the hearts of these animals.
Another study investigating the beneficial effects ofadiponectin during ischaemia/reperfusion injury noted areduced phosphorylation of AMPK at its threonine 172residue (which is necessary for AMPK activation) in anadiponectin-deficient mouse model 48 h after a 30 minocclusion of the left anterior descending (LAD) coronaryartery.51 The authors demonstrated that inhibition of AMPKprevented the anti-apoptotic effects of adiponectin oncardiac myocytes and fibroblasts subjected to hypoxia/reox-ygenation. Nonetheless, the anti-apoptotic effects of AMPKwere performed only in culture, and the beneficial effectsof adiponectin during reperfusion following LAD occlusion
Malonyl CoA and ischaemic heart disease 263
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

could also be explained via cyclooxygenase II-dependenteffects.
More recently, a study in an AMPKa2 subunit-deficientmouse model reported that although AMPKa2 deficiencyaccelerated the appearance of contracture during ischae-mia, there was no effect on reperfusion recovery of thesehearts, suggesting that inhibition of AMPK is not detrimentalin the heart as an earlier study had reported.52 Moreover,the perfusions in this study were glucose-only perfusions,and thus high rates of fatty acid oxidation would not be aproblem in ischaemia/reperfusion. It is possible that thebeneficial effect of reducing the extremely high rates offatty acid oxidation during reperfusion may have beenmasked because of this.
The regulation of AMPK by its upstream kinases, the AMPKkinases (AMPKKs), likely plays an important role in activatingAMPK during ischaemia/reperfusion. Previous work in ourlaboratory has shown that although AMPK is activated inthe rat heart during reperfusion following ischaemia, myo-cardial levels of AMP quickly return to normal. Thus,alternative mechanisms of AMPK activation likely existoutside of changes in AMP levels. One possibility is AMPKK,which phosphorylates and activates AMPK. IdentifiedAMPKKs include the tumour-suppressor LKB1, calmodulin-dependent protein kinase kinase b (CamKKb), and morerecently, transforming growth factor-activatedkinase-1.22,53,54 Our group55 and others47 have shown thatAMPKK is rapidly activated in the ischaemic heart. To date,the only AMPKK that has been extensively studied in theheart is LKB1. We have shown that AMPKK activationduring ischemia is not due to LKB1 activation.55 In contrast,a previous study investigating the role of LKB1 on AMPK acti-vation in the heart during ischaemia and anoxia reportedthat LKB1 was responsible for phosphorylating and activatingthe AMPKa2 subunit at its threonine 172 residue, but not theAMPKa1 subunit.56 Although hearts from these mice hadenlarged atria and reduced weights, echocardiographicassessment revealed no cardiac dysfunction. However,these studies did not examine functional differencesduring reperfusion, and perfusions were again performedwith glucose as the sole substrate. Further studies are there-fore required to determine what role LKB1 and the otherAMPKKs have on AMPK during ischaemia/reperfusion,malonyl CoA levels, and functional recovery.
Another way to inhibit AMPK is via insulin administration.A number of studies have examined the role of glucose–insulin–potassium (GIK) for the treatment of acute myocar-dial infarction.57–59 We initially hypothesized that insulinwould benefit the aerobically reperfused ischaemic heart.This would arise from the inhibition of myocardial AMPKduring ischaemia and subsequent reperfusion, thereby redu-cing rates of fatty acid oxidation and increasing glucose oxi-dation rates, alleviating myocardial acidosis and improvingfunctional recovery. Unfortunately, the majority of studiesexamining the beneficial effects of insulin on functionalrecovery of the heart during reperfusion have performedtheir perfusions with glucose as the sole substrate. Arecent study in our laboratory has demonstrated that fattyacids in the perfusate interfere with insulin’s ability toinhibit AMPK, and although insulin is still able to reducefatty acid oxidation, a greater stimulation of glycolysisthan glucose oxidation actually increases proton productionand worsens functional recovery during reperfusion.60
Therefore, it is unlikely that any beneficial effects ofinsulin during reperfusion involve an inhibition of AMPK,and it is possible that our previous results can explain thelack of mortality benefit with GIK for patients undergoingan acute myocardial infarction during the recent CREATE-ECLA trial.61 In fact, a recent study suggests that GIK mayactually increase mortality in the early post-AMI period.62
Owing to the limitations of the first two studies, and theresults of the aforementioned third study, we believe thatthere is insufficient evidence to state that AMPK activationis beneficial during ischaemia/reperfusion injury. What isneeded to reconcile these discrepancies is more studiesdone using in vivo ischaemia/reperfusion models investi-gating the effect of AMPK on myocardial function, as wellas actual measurements of myocardial malonyl CoA inthese systems.
7.2 Targeting acetyl CoA carboxylase to treatischaemia/reperfusion
In regard to ACC, there is very little literature investigatingits effects on ischaemia/reperfusion, as the majority of lit-erature on ACC modulation has focused on inhibiting ACCin the liver and adipose tissue. It has been postulated thatinhibiting ACC in these tissues to decrease malonyl CoAwould decrease fatty acid biosynthesis.63–65 Unfortunately,no studies have investigated the effects of ACC inhibitionon cardiac function. There are no selective pharmacologicalinhibitors of ACC available, and they would have to bespecific to the b-isoform, which predominates in the heartand is more tightly linked to the regulation of fatty acid oxi-dation.20 Nonetheless, mice deficient for ACCb are availableand have been characterized and show significant increasesin fatty acid oxidation.66 It would be of interest to conductischaemia/reperfusion studies in these mice, as it is possiblethat hearts from these mice would have a depressed recov-ery of cardiac function during ischaemia/reperfusion due tohigh rates of fatty acid oxidation and subsequent acidosis.
7.3 Targeting malonyl CoA decarboxylaseto treat ischaemia/reperfusion
Recent studies in our laboratory have shown that MCD is amajor regulator of fatty acid oxidation rates in the heart,and that inhibition of this enzyme is a viable target for thetreatment of ischaemia/reperfusion injury.25,26 First, usingnovel inhibitors of MCD, we have shown that inhibition ofMCD in an isolated working rat heart perfusion system signifi-cantly increases malonyl CoA levels.26 This was associatedwith a significant decrease in fatty acid oxidation ratesand a subsequent increase in glucose oxidation rates.These metabolic effects induced via inhibition of MCDcaused a significant reduction in proton production duringboth the aerobic setting and during low-flow ischaemia.Furthermore, inhibition of MCD resulted in a significantimprovement in cardiac functional recovery of aerobicallyreperfused ischaemic rat hearts. Another study from ourlaboratory and Stanley’s demonstrated in an in vivo pigmodel of demand-induced ischaemia that MCD inhibitiononce again significantly increased malonyl CoA levelsand glucose oxidation rates.24,26 Moreover, this wasaccompanied by a significant reduction in myocardiallactate production and a complete restoration of leftventricular work. Last, a third study from our laboratory
J.R. Ussher and G.D. Lopaschuk264
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

investigated the effects of chronic MCD inhibition in awhole-body MCD-deficient mouse model.25 Although heartsfrom these animals had a significant increase in malonylCoA levels, isolated aerobic working heart perfusionsdemonstrated no differences in glucose and fatty acidmetabolism compared with control hearts. This may havebeen attributed to the significant upregulation in themRNA of a number of different PPARa target transcriptssuch as CD36 and CPT-I. Nonetheless, when hearts fromthe MCD-deficient mice were subjected to the stress ofischaemia/reperfusion injury, a significant improvement inthe recovery of cardiac power and function was observed.This enhanced recovery was associated with a significantincrease in glucose oxidation rates, whereby glucoseoxidation became the major source of ATP production inthe heart. Thus, hearts from the MCD-deficient micehad improved coupling between glycolysis and glucoseoxidation, reduced proton production, and less myocardialacidosis.
As mentioned earlier, although the inhibition of fatty acidoxidation in the heart improves function in the setting ofischaemic heart disease, others have postulated that theinhibition of fatty acid oxidation in peripheral tissues, suchas the muscle and liver, will exacerbate insulin resistanceand type 2 diabetes.67 This is an important point to consider,a significant number of patients with ischaemic heartdisease are also obese/type 2 diabetic. From a clinicalstandpoint, oral delivery of MCD inhibitors for the prophy-lactic treatment of ischaemic heart disease would be practi-cal. However, this would affect the peripheral tissues suchas the muscle, and could cause insulin resistance orworsen the diabetic state. A recent collaboration by our lab-oratory and that of Muoio and colleagues68 has shown thatthis is not the case. Obesity and insulin resistance inducedby high-fat diet are actually accompanied by increasedrates of incomplete fatty acid oxidation as opposed toimpaired fatty acid oxidation.68 In fact, we demonstratedthat the MCD-deficient mouse was protected fromobesity-induced insulin resistance, which was associatedwith decreased rates of incomplete fatty acid oxidation.Furthermore, potential toxic intramuscular fatty acidmetabolites that have been postulated to cause insulinresistance due to impaired fatty acid oxidation, such aslong-chain acyl CoAs, actually trended to increase in theMCD KO mice, suggesting that they are not direct mediatorsof insulin resistance.
As mentioned earlier, MCD expression is transcriptionallyregulated by PPARa.27,28 Thus, manipulating PPARa offersanother approach to regulating malonyl CoA levels andrates of fatty acid oxidation. Indeed, previous work in ourlaboratory has shown that hearts from mice deficient forPPARa have reduced expression of MCD and increasedlevels of malonyl CoA.69 These animals subsequently havelower rates of fatty acid oxidation and increased rates ofglucose oxidation. Moreover, PPARa-deficient mice have animproved recovery of cardiac power during reperfusion fol-lowing a global no-flow ischaemia, whereas mice overex-pressing PPARa have a worse recovery of cardiac powerunder identical conditions.70 These results are contrary toreports utilizing acute systemic activation of PPARa withligands that caused beneficial effects against ischaemia/reperfusion injury.71,72 The benefit seen with PPARa inhi-bition could be due to the optimization of cardiac energy
metabolism during reperfusion, whereas the benefit seenwith PPARa activation could be the result of its anti-inflammatory properties.73–75 Because PPARa has anti-inflammatory properties and regulates the expression ofmany genes involved in fatty acid uptake and oxidationbesides MCD,73,76,77 inhibiting MCD to decrease fatty acidoxidation in the heart may be a more plausible approachthan inhibiting PPARa.
8. The potential for malonyl CoA axismanipulation in humans
Currently, there are a number of agents used clinically thatoptimize cardiac energy metabolism. This includes, trimeta-zidine, a 3-ketoacyl CoA thiolase inhibitor. The Cochranecollaboration meta-analysis recently showed trimetazidineto be an effective therapy for stable angina compared withplacebo (�40% reduction in the mean number of anginaattacks per week), alone or combined with conventional anti-anginal agents.78 In addition, perhexiline, a CPT-1 inhibitor,has been shown to have clinical utility in refractory anginapectoris,79 aortic stenosis,80 and chronic heart failure,81
where it improves symptomatic status, left ventricular ejec-tion fraction, and quality of life. Direct stimulation of glucoseoxidation with dichloroacetate, a PDH kinase inhibitor,improves left ventricular stroke volume and cardiac efficiencyin the setting of heart failure.82
To date, there are no agents clinically available for thetreatment of ischaemia/reperfusion injury that target themalonyl CoA axis to optimize cardiac energy metabolism.On the basis of the positive human trials with trimetazidine,a direct fatty acid b-oxidation inhibitor, and the positiveresults seen with both acute and chronic MCD inhibition inanimals, manipulating the malonyl CoA axis in humansappears plausible and could yield positive effects for thetreatment of ischaemia/reperfusion injury. In regard toMCD inhibition, two potential setbacks are that CPT-1 inhi-bition has been shown in past studies to cause hypertrophy,and that MCD deficiency in humans is associated with cardi-omyopathy.83 A number of studies in the early 1980sreported that chronic treatment in dogs and rats with theCPT-1 inhibitor, oxfenicine, results in the development ofcardiac hypertrophy.84,85 However, more recent studieshave actually shown that the CPT-1 inhibitors, oxfenicineand etomoxir, can actually delay adverse left ventricularmodelling in a dog model of pacing-induced heart failure86
and rat model of aortic constriction.87 Some of the reporteddiscrepancy may lie in the fact that the earlier studies didnot address whether physiological or pathological hypertro-phy was occurring. Furthermore, these inhibitors of CPT-1were irreversible, whereas manipulating malonyl CoA viaMCD would be an indirect inhibition of CPT-1. In addition,MCD-deficient mice have normal cardiac function and showno signs of cardiac hypertrophy.26 To address the cardiomyo-pathy issue, only 18 patients with MCD deficiency have beenreported in the literature,88 and of these 18, only five devel-oped a cardiomyopathy.83,88–92 Of further interest, in thosepatients who completely lack MCD, no cardiomyopathieswere observed, and those patients deficient in MCD thatdeveloped cardiomyopathies often possessed mutations inthe gene that result in subcellular mistargeting and notthe complete absence of MCD protein.92 Therefore,
Malonyl CoA and ischaemic heart disease 265
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

inhibition of MCD in the heart appears to be safe, and futurestudies are warranted to explore the role of MCD inhibitionas a therapy for ischaemic heart disease. As of yet, thereare no studies looking at regulating the malonyl CoA axisvia ACC in the heart to warrant manipulation of thisenzyme in human studies of ischaemia/reperfusion. More-over, animal studies of AMPK manipulation have yet to inves-tigate the effects of AMPK on malonyl CoA levels in theheart, and results of AMPK’s effects on cardiac functionare mixed. Thus, further work in animal models is necessarybefore AMPK manipulation is used as a therapeutic target inhumans for ischaemia/reperfusion injury.
9. Summary
The optimization of cardiac energy metabolism representsan exciting new therapeutic approach for the treatment ofischaemia/reperfusion injury. One approach to optimizecardiac energy metabolism is to regulate the levels ofmalonyl CoA, a potent endogenous inhibitor of cardiacfatty acid oxidation, secondary to its inhibition of CPT-1,the rate-limiting enzyme in the mitochondrial uptake offatty acids. A number of studies have recently been pub-lished showing that the inhibition of MCD increases cardiacmalonyl CoA levels. These studies reported improved func-tional recovery of the heart during ischaemia/reperfusioninjury, which was attributed to increased glucose oxidationand decreased proton production. Thus, targeting themalonyl CoA axis in the heart represents a potential excitingnew therapy for the treatment of ischaemic heart disease.
Conflict of interest: J.R.U. is a trainee of the Alberta HeritageFoundation for Medical Research and Tomorrow’s Research Cardio-vascular Health Professionals (TORCH). G.D.L. is a medical scientistof the Alberta Heritage Foundation for Medical Research. J.R.U. hasno conflicts to declare. G.D.L. is the President and CEO of MetabolicModulators Research Ltd, a company which has interests in develop-ing metabolic drugs to treat heart disease. This includes agents thatmodify the malonyl CoA axis.
Funding
Supported by a grant from the Canadian Institutes for HealthResearch Grant to G.D.L.
References1. NHLBI Morbidity and Mortality Chart-book. Bethesda, MD: National
Heart, Lung, and Blood Institute; 2004. http://www.nhlbi.nih.gov/resources/docs/cht-book.htm (23 August 2007).
2. Cooper R, Cutler J, Desvigne-Nickens P, Fortmann SP, Friedman L,Havlik R et al. Trends and disparities in coronary heart disease, stroke,and other cardiovascular diseases in the United States: findings of thenational conference on cardiovascular disease prevention. Circulation2000;102:3137–3147.
3. Ussher JR, Lopaschuk GD. Clinical implications of energetic problems incardiovascular disease. Heart Metab 2006;32:9–17.
4. Liu Q, Docherty JC, Rendell JC, Clanachan AS, Lopaschuk GD. High levelsof fatty acids delay the recovery of intracellular pH and cardiac efficiencyin post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol2002;39:718–725.
5. Folmes CD, Clanachan AS, Lopaschuk GD. Fatty acid oxidation inhibitorsin the management of chronic complications of atherosclerosis. CurrAtheroscler Rep 2005;7:63–70.
6. Folmes CD, Lopaschuk GD. Role of malonyl-CoA in heart disease and thehypothalamic control of obesity. Cardiovasc Res 2007;73:278–287.
7. Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO. Regulation offatty acid oxidation in the mammalian heart in health and disease.Biochim Biophys Acta 1994;1213:263–276.
8. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolismin the normal and failing heart. Physiol Rev 2005;85:1093–1129.
9. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acidcycle. Its role in insulin sensitivity and the metabolic disturbances of dia-betes mellitus. Lancet 1963;1:785–789.
10. Liu B, Clanachan AS, Schulz R, Lopaschuk GD. Cardiac efficiency isimproved after ischemia by altering both the source and fate ofprotons. Circ Res 1996;79:940–948.
11. Dyck JR, Lopaschuk GD. Malonyl CoA control of fatty acid oxidation in theischemic heart. J Mol Cell Cardiol 2002;34:1099–1109.
12. Cuthbert KD, Dyck JR. Malonyl-CoA decarboxylase is a major regulator ofmyocardial fatty acid oxidation. Curr Hypertens Rep 2005;7:407–411.
13. Neely JM, Morgan HE. Relationship between carbohydrate metabolismand energy balance of heart muscle. Annu Rev Physiol 1974;36:413–459.
14. van der Vusse GJ, van Bilsen M, Glatz JF. Cardiac fatty acid uptake andtransport in health and disease. Cardiovasc Res 2000;45:279–293.
15. Kunau WH, Dommes V, Schulz H. Beta-oxidation of fatty acids in mito-chondria, peroxisomes, and bacteria: a century of continued progress.Prog Lipid Res 1995;34:267–342.
16. Schulz H. Regulation of fatty acid oxidation in heart. J Nutr 1994;124:165–171.
17. Reszko AE, Kasumov T, Comte B, Pierce BA, David F, Bederman IR et al.Assay of the concentration and 13C-isotopic enrichment of malonyl-coenzyme A by gas chromatography-mass spectrometry. Anal Biochem2001;298:69–75.
18. Bianchi A, Evans JL, Iverson AJ, Nordlund AC, Watts TD, Witters LA.Identification of an isozymic form of acetyl-CoA carboxylase. J BiolChem 1990;265:1502–1509.
19. Thampy KG. Formation of malonyl coenzyme A in rat heart. Identificationand purification of an isozyme of A carboxylase from rat heart. J BiolChem 1989;264:17631–17634.
20. Saddik M, Gamble J, Witters LA, Lopaschuk GD. Acetyl-CoA carboxylaseregulation of fatty acid oxidation in the heart. J Biol Chem 1993;268:25836–25845.
21. Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acidoxidation during reperfusion of ischemic hearts are associated with adecrease in malonyl-CoA levels due to an increase in 50-AMP-activatedprotein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem 1995;270:17513–17520.
22. Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and path-ology: enemy or ally? J Physiol 2006;574:95–112.
23. Dyck JR, Barr AJ, Barr RL, Kolattukudy PE, Lopaschuk GD. Characteriz-ation of cardiac malonyl-CoA decarboxylase and its putative role in reg-ulating fatty acid oxidation. Am J Physiol 1998;275:H2122–H2129.
24. Stanley WC, Morgan EE, Huang H, McElfresh TA, Sterk JP, Okere IC et al.Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation andreduces lactate production during demand-induced ischemia. Am JPhysiol Heart Circ Physiol 2005;289:H2304–H2309.
25. Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe Met al. Absence of malonyl coenzyme A decarboxylase in mice increasescardiac glucose oxidation and protects the heart from ischemic injury.Circulation 2006;114:1721–1728.
26. Dyck JR, Cheng JF, Stanley WC, Barr R, Chandler MP, Brown S et al.Malonyl coenzyme a decarboxylase inhibition protects the ischemicheart by inhibiting fatty acid oxidation and stimulating glucose oxidation.Circ Res 2004;94:e78–e84.
27. Lee GY, Kim NH, Zhao ZS, Cha BS, Kim YS. Peroxisomal-proliferator-activated receptor alpha activates transcription of the rat hepaticmalonyl-CoA decarboxylase gene: a key regulation of malonyl-CoAlevel. Biochem J 2004;378:983–990.
28. Young ME, Goodwin GW, Ying J, Guthrie P, Wilson CR, Laws FA et al. Regu-lation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fattyacids. Am J Physiol Endocrinol Metab 2001;280:E471–E479.
29. Saha AK, Schwarsin AJ, Roduit R, Masse F, Kaushik V, Tornheim K et al.Activation of malonyl-CoA decarboxylase in rat skeletal muscle bycontraction and the AMP-activated protein kinase activator5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside. J Biol Chem2000;275:24279–24283.
30. Lysiak W, Toth PP, Suelter CH, Bieber LL. Quantitation of the efflux ofacylcarnitines from rat heart, brain, and liver mitochondria. J BiolChem 1986;261:13698–13703.
J.R. Ussher and G.D. Lopaschuk266
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

31. McGarry JD, Mills SE, Long CS, Foster DW. Observations on the affinity forcarnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase Iin animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem J 1983;214:21–28.
32. Sacksteder KA, Morrell JC, Wanders RJ, Matalon R, Gould SJ. MCDencodes peroxisomal and cytoplasmic forms of malonyl-CoA decarboxy-lase and is mutated in malonyl-CoA decarboxylase deficiency. J BiolChem 1999;274:24461–24468.
33. Reszko AE, Kasumov T, David F, Jobbins KA, Thomas KR, Hoppel CL et al.Peroxisomal fatty acid oxidation is a substantial source of the acetylmoiety of malonyl-CoA in rat heart. J Biol Chem 2004;279:19574–19579.
34. Whitmer JT, Idell-Wenger JA, Rovetto MJ, Neely JR. Control of fatty acidmetabolism in ischemic and hypoxic hearts. J Biol Chem 1978;253:4305–4309.
35. Lorkovic H. Influence of changes in pH on the mechanical activity ofcardiac muscle. Circ Res 1966;19:711–720.
36. Pannier JL, Leusen I. Contraction characteristics of papillary muscleduring changes in acid–base composition of the bathing-fluid. Arch IntPhysiol Biochim 1968;76:624–634.
37. Poole-Wilson PA, Langer GA. Effect of pH on ionic exchange and functionin rat and rabbit myocardium. Am J Physiol 1975;229:570–581.
38. Solaro RJ, Lee JA, Kentish JC, Allen DG. Effects of acidosis on ventricularmuscle from adult and neonatal rats. Circ Res 1988;63:779–787.
39. Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability tran-sition pore opening during myocardial reperfusion—a target for cardio-protection. Cardiovasc Res 2004;61:372–385.
40. Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Yet al.The mitochondrial permeability transition in cell death: a common mech-anism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998;1366:177–196.
41. Wisneski JA, Gertz EW, Neese RA, Mayr M. Myocardial metabolism of freefatty acids. Studies with 14C-labeled substrates in humans. J Clin Invest1987;79:359–366.
42. Lopaschuk GD, Collins-Nakai R, Olley PM, Montague TJ, McNeil G, Gayle Met al. Plasma fatty acid levels in infants and adults after myocardialischemia. Am Heart J 1994;128:61–67.
43. Kurien VA, Oliver MF. Free fatty acids during acute myocardial infarction.Prog Cardiovasc Dis 1971;13:361–373.
44. Mueller HS, Ayres SM. Metabolic response of the heart in acute myocar-dial infarction in man. Am J Cardiol 1978;42:363–371.
45. Kudo N, Gillespie JG, Kung L, Witters LA, Schulz R, Clanachan AS et al.Characterization of 50AMP-activated protein kinase activity in the heartand its role in inhibiting acetyl-CoA carboxylase during reperfusion fol-lowing ischemia. Biochim Biophys Acta 1996;1301:67–75.
46. Sim AT, Hardie DG. The low activity of acetyl-CoA carboxylase in basaland glucagon-stimulated hepatocytes is due to phosphorylation by theAMP-activated protein kinase and not cyclic AMP-dependent proteinkinase. FEBS Lett 1988;233:294–298.
47. Baron SJ, Li J, Russell RR III, Neumann D, Miller EJ, Tuerk R et al. Dualmechanisms regulating AMPK kinase action in the ischemic heart. CircRes 2005;96:337–345.
48. Beauloye C, Marsin AS, Bertrand L, Krause U, Hardie DG,Vanoverschelde JL et al. Insulin antagonizes AMP-activated proteinkinase activation by ischemia or anoxia in rat hearts, without affectingtotal adenine nucleotides. FEBS Lett 2001;505:348–352.
49. Russell R III. Stress signaling in the heart by AMP-activated protein kinase.Curr Hypertens Rep 2006;8:446–450.
50. Russell RR III, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M et al.AMP-activated protein kinase mediates ischemic glucose uptake and pre-vents postischemic cardiac dysfunction, apoptosis, and injury. J ClinInvest 2004;114:495–503.
51. Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K et al. Adi-ponectin protects against myocardial ischemia–reperfusion injurythrough AMPK- and COX-2-dependent mechanisms. Nat Med 2005;11:1096–1103.
52. Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur ACet al. Role of the alpha2-isoform of AMP-activated protein kinase in themetabolic response of the heart to no-flow ischemia. Am J PhysiolHeart Circ Physiol 2006;291:H2875–H2883.
53. Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1protein kinase in yeast and phosphorylates AMP-activated proteinkinase in vitro. J Biol Chem 2006;281:25336–25343.
54. Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M et al. A pivotal rolefor endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activatedprotein kinase energy-sensor pathway. Proc Natl Acad Sci USA 2006;103:17378–17383.
55. Altarejos JY, Taniguchi M, Clanachan AS, Lopaschuk GD. Myocardial ische-mia differentially regulates LKB1 and an alternate 50-AMP-activatedprotein kinase kinase. J Biol Chem 2005;280:183–190.
56. Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur AC, Dutta A, Prescott ARet al. Deficiency of LKB1 in heart prevents ischemia-mediated activationof AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab 2006;290:E780–E788.
57. Fath-Ordoubadi F, Beatt KJ. Glucose–insulin–potassium therapy for treat-ment of acute myocardial infarction: an overview of randomizedplacebo-controlled trials. Circulation 1997;96:1152–1156.
58. Timmer JR, van der Horst IC, Ottervanger JP, De Luca G, van’t Hof AW,Bilo HJ et al. Glucose–insulin–potassium infusion as adjunctive therapyin myocardial infarction: current evidence and potential mechanisms.Ital Heart J 2004;5:727–731.
59. Yusuf S, Mehta SR, Diaz R, Paolasso E, Pais P, Xavier D et al. Challenges inthe conduct of large simple trials of important generic questions inresource-poor settings: the CREATE and ECLA trial program evaluatingGIK (glucose, insulin and potassium) and low-molecular-weight heparinin acute myocardial infarction. Am Heart J 2004;148:1068–1078.
60. Folmes CD, Clanachan AS, Lopaschuk GD. Fatty acids attenuate insulinregulation of 50-AMP-activated protein kinase and insulin cardioprotec-tion after ischemia. Circ Res 2006;99:61–68.
61. Mehta SR, Yusuf S, Diaz R, Zhu J, Pais P, Xavier D et al. Effect of glucose–insulin–potassium infusion on mortality in patients with acuteST-segment elevation myocardial infarction: the CREATE-ECLA random-ized controlled trial. JAMA 2005;293:437–446.
62. Diaz R, Goyal A, Mehta SR, Afzal R, Xavier D, Pais P et al. Glucose–insulin–potassium therapy in patients with ST-segment elevation myocar-dial infarction. JAMA 2007;298:2399–2405.
63. Choi CS, Savage DB, Abu-Elheiga L, Liu ZX, Kim S, Kulkarni A et al. Con-tinuous fat oxidation in acetyl-CoA carboxylase 2 knockout miceincreases total energy expenditure, reduces fat mass, and improvesinsulin sensitivity. Proc Natl Acad Sci USA 2007;104:16480–16485.
64. Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE et al. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglycerideaccumulation without affecting glucose homeostasis. Proc Natl Acad SciUSA 2006;103:8552–8557.
65. Oh W, Abu-Elheiga L, Kordari P, Gu Z, Shaikenov T, Chirala SS et al.Glucose and fat metabolism in adipose tissue of acetyl-CoA carboxylase2 knockout mice. Proc Natl Acad Sci USA 2005;102:1384–1389.
66. Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fattyacid oxidation and reduced fat storage in mice lacking acetyl-CoA car-boxylase 2. Science 2001;291:2613–2616.
67. Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J et al. Mitochondrialdysfunction due to long-chain acyl-CoA dehydrogenase deficiencycauses hepatic steatosis and hepatic insulin resistance. Proc Natl AcadSci USA 2007;104:17075–17080.
68. Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O et al.Mitochondrial overload and incomplete fatty acid oxidation contribute toskeletal muscle insulin resistance. Cell Metab 2008;7:45–56.
69. Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD et al. Arole for peroxisome proliferator-activated receptor alpha (PPARalpha) inthe control of cardiac malonyl-CoA levels: reduced fatty acid oxidationrates and increased glucose oxidation rates in the hearts of micelacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem2002;277:4098–4103.
70. Sambandam N, Morabito D, Wagg C, Finck BN, Kelly DP, Lopaschuk GD.Chronic activation of PPARalpha is detrimental to cardiac recoveryafter ischemia. Am J Physiol Heart Circ Physiol 2006;290:H87–H95.
71. Khandoudi N, Delerive P, Berrebi-Bertrand I, Buckingham RE, Staels B,Bril A. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma, inhibits the Jun NH(2)-terminal kinase/activating protein 1pathway and protects the heart from ischemia/reperfusion injury. Dia-betes 2002;51:1507–1514.
72. Yue TL, Bao W, Jucker BM, Gu JL, Romanic AM, Brown PJ et al. Activationof peroxisome proliferator-activated receptor-alpha protects the heartfrom ischemia/reperfusion injury. Circulation 2003;108:2393–2399.
73. Han SH, Quon MJ, Koh KK. Beneficial vascular and metabolic effects ofperoxisome proliferator-activated receptor-alpha activators. Hyperten-sion 2005;46:1086–1092.
74. Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M et al. Tar-geted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponec-tin binding and metabolic actions. Nat Med 2007;13:332–339.
Malonyl CoA and ischaemic heart disease 267
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022

75. Marx N, Duez H, Fruchart JC, Staels B. Peroxisome proliferator-activatedreceptors and atherogenesis: regulators of gene expression in vascularcells. Circ Res 2004;94:1168–1178.
76. Jay MA, Ren J. Peroxisome proliferator-activated receptor (PPAR) inmetabolic syndrome and type 2 diabetes mellitus. Curr Diabetes Rev2007;3:33–39.
77. Sugden MC, Holness MJ. Potential role of peroxisome proliferator-activated receptor-alpha in the modulation of glucose-stimulatedinsulin secretion. Diabetes 2004;53(Suppl. 1):S71–S81.
78. Ciapponi A, Pizarro R, Harrison J. Trimetazidine for stable angina.Cochrane Database Syst Rev 2005; CD003614.
79. Cole PL, Beamer AD, McGowan N, Cantillon CO, Benfell K, Kelly RA et al.Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circu-lation 1990;81:1260–1270.
80. Unger SA, Robinson MA, Horowitz JD. Perhexiline improves symptomaticstatus in elderly patients with severe aortic stenosis. Aust N Z J Med1997;27:24–28.
81. Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P,Williams L et al. Metabolic modulation with perhexiline in chronicheart failure: a randomized, controlled trial of short-term use of anovel treatment. Circulation 2005;112:3280–3288.
82. Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW,Pepine CJ. Myocardial metabolic and hemodynamic effects of dichloroa-cetate in coronary artery disease. Am J Cardiol 1988;61:65–70.
83. Yano S, Sweetman L, Thorburn DR, Mofidi S, Williams JC. A new case ofmalonyl coenzyme A decarboxylase deficiency presenting with cardio-myopathy. Eur J Pediatr 1997;156:382–383.
84. Greaves P, Martin J, Michel MC, Mompon P. Cardiac hypertrophy in the dogand rat induced by oxfenicine, an agent which modifies muscle metab-olism. Arch Toxicol Suppl 1984;7:488–493.
85. Higgins AJ, Faccini JM, Greaves P. Coronary hyperemia and cardiac hyper-trophy following inhibition of fatty acid oxidation. Evidence of a regulat-ory role for cytosolic phosphorylation potential. Adv Myocardiol 1985;6:329–338.
86. Lionetti V, Linke A, Chandler MP, Young ME, Penn MS, Gupte S et al. Car-nitine palmitoyl transferase-I inhibition prevents ventricular remodelingand delays decompensation in pacing-induced heart failure. CardiovascRes 2005;66:454–461.
87. Turcani M, Rupp H. Modification of left ventricular hypertrophy by chronicetomixir treatment. Br J Pharmacol 1999;126:501–507.
88. de Wit MC, de Coo IF, Verbeek E, Schot R, Schoonderwoerd GC, Duran Met al. Brain abnormalities in a case of malonyl-CoA decarboxylasedeficiency. Mol Genet Metab 2006;87:102–106.
89. Ficicioglu C, Chrisant MR, Payan I, Chace DH. Cardiomyopathy and hypo-tonia in a 5-month-old infant with malonyl-coa decarboxylase deficiency:potential for preclinical diagnosis with expanded newborn screening.Pediatr Cardiol 2005;26:881–883.
90. Gao J, Waber L, Bennett MJ, Gibson KM, Cohen JC. Cloning and muta-tional analysis of human malonyl-coenzyme A decarboxylase. J LipidRes 1999;40:178–182.
91. Matalon R, Michaels K, Kaul R, Whitman V, Rodriguez-Novo J, Goodman Set al. Malonic aciduria and cardiomyopathy. J Inherit Metab Dis 1993;16:571–573.
92. Wightman PJ, Santer R, Ribes A, Dougherty F, McGill N, Thorburn DR et al.MLYCD mutation analysis: evidence for protein mistargeting as a cause ofMLYCD deficiency. Hum Mutat 2003;22:288–300.
J.R. Ussher and G.D. Lopaschuk268
Dow
nloaded from https://academ
ic.oup.com/cardiovascres/article/79/2/259/272457 by guest on 10 January 2022
Related Documents