Characterization of synthetic routes to ‘Bromo-DragonFLY’ and benzodifuranyl isopropylamine homologues utilizing ketone intermediates. Part 1: Synthesis of ketone precursors Richard E. O’Connor and John J. Keating* Bromo-DragonFLY (BDF) and many of its analogues are misused as recreational drugs due to their potency as psychoactive sub- stances. To date, none of the published routes to these designer amphetamines have exploited a ketone intermediate. It is well known that benzyl methyl ketone (BMK) can be employed as a precursor in the synthesis of amphetamine. Similarly, it is reason- able to assume that ketone precursors may potentially be utilized in the clandestine synthesis of BDF and its homologues. This paper describes the multifaceted synthesis of novel precursor ketones structurally related to BDF, namely benzodifuranyl propanone 16, its tetrahydrobenzodifuranyl homologue 8, and their brominated analogues 12 and 20. Their characterization by Fourier transform infrared spectroscopy (FTIR), proton nuclear magnetic resonance spectroscopy ( 1 H-NMR), carbon nuclear magnetic resonance spectroscopy ( 13 C-NMR), high performance liquid chromatography (HPLC), gas chromatography (GC) and mass spectrometry (MS) is also described. Copyright © 2013 John Wiley & Sons, Ltd. Keywords: Bromo-dragonFLY; amphetamine-type stimulants (ATS); ketone precursors; clandestine synthesis; novel psychoactive substances (NPS) Introduction Bromo-dragonFLY (BDF) or (1-(8-bromobenzo[1,2-b:4,5-b’]difuranyl- 4-yl)-2-aminopropane) 1 is a hallucinogenic benzodifuranyl deriva- tive and an emerging drug of recreational abuse. [1–4] Initially synthesized as a research chemical for mapping the topography of specific serotonin receptors, [5] BDF is a potent 5-HT 2A and 5-HT 2C receptor agonist. [6] Typical doses are in the milligram to sub- milligram range (0.2–1.5 mg) and onset of action is in the range of 20–90 min. Its effects have been known to last for up to 36 h. [1] Rec- reational use of BDF has been associated with a number of adverse effects including vasoconstriction and liver and kidney failure, as well as severe convulsions. [1–4,7,8] There are several documented cases where BDF either contributed to, or was the sole cause of death of the user. [4,9–13] BDF is often branded as a ‘legal-high’ and is currently available for sale online in several forms: powder, liquid, and impreg- nated on paper. [1–4] There have also been reports of BDF being mislabelled as other less potent recreational drugs; a number of these reported cases have resulted in a fatal overdose. [9–13] Drug enforcement agencies have documented seizures of BDF in the United States of America, Australia, and Europe. [2,12–14] In 2006, the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA)–Europol Annual Report flagged BDF as part of its Early Warning System on new psychoactive substances. [2,15] Currently BDF is only controlled under legislation in a limited number of coun- tries worldwide. On 1 November 2011 it was added by name to the Irish Misuse of Drugs Act, 1977. [16] Since the 1990s, the Nichols group has reported numerous syntheses of BDF and its analogues. [5,6,17–21] A selection of these compounds is shown in Figure 1. However, none of these isopropylamines were prepared via a 1-aryl-2-propanone-type intermediate. Analysis of the literature has also revealed no examination of BDF ‘street samples’ for route-specific impurities, thus no data are currently available on the synthetic methods used by clandestine chemists in the manufacture of BDF. It is well known that simple ketones such as 1-phenyl-2-propanone (P2P, BMK) can be utilized as precursors in the production of amphetamine-type stimulants (ATS). [22–25] Accordingly it would be reasonable to assume that novel ketone precursors may potentially be exploited in the clandestine synthesis of BDF and its analogues. This concept is the motivation for our current investigation and incorporates a number of the aims of our research group, some of which are (1) to isolate and characterize precursors, intended products and impurities of novel psychoac- tive substances (NPS), in this instance BDF, (2) to identify route- specific impurities so as to elucidate the possible synthetic routes to NPS including BDF analogues, and (3) to determine the chemical fingerprint for a given route to a given NPS. [26,27] Completed profiles may help to assist authorities in their investi- gations of such compounds. * Correspondence to: John J. Keating, Analytical & Biological Chemistry Research Facility (ABCRF), School of Pharmacy / Department of Chemistry, University College Cork, College Road, Cork, Ireland E-mail: [email protected] Analytical & Biological Chemistry Research Facility (ABCRF), School of Pharmacy/Department of Chemistry, University College Cork, College Road, Cork, Ireland Drug Test. Analysis (2013) Copyright © 2013 John Wiley & Sons, Ltd. Research article Drug Testing and Analysis Received: 10 April 2013 Revised: 29 May 2013 Accepted: 29 May 2013 Published online in Wiley Online Library (www.drugtestinganalysis.com) DOI 10.1002/dta.1504

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Research articleDrug Testing
and Analysis
Received: 10 April 2013 Revised: 29 May 2013 Accepted: 29 May 2013 Published online in Wiley Online Library
(www.drugtestinganalysis.com) DOI 10.1002/dta.1504
Characterization of synthetic routes to‘Bromo-DragonFLY’ and benzodifuranylisopropylamine homologues utilizingketone intermediates. Part 1: Synthesisof ketone precursorsRichard E. O’Connor and John J. Keating*
Bromo-DragonFLY (BDF) and many of its analogues are misused as recreational drugs due to their potency as psychoactive sub-stances. To date, none of the published routes to these designer amphetamines have exploited a ketone intermediate. It is wellknown that benzyl methyl ketone (BMK) can be employed as a precursor in the synthesis of amphetamine. Similarly, it is reason-able to assume that ketone precursors may potentially be utilized in the clandestine synthesis of BDF and its homologues. Thispaper describes the multifaceted synthesis of novel precursor ketones structurally related to BDF, namely benzodifuranylpropanone 16, its tetrahydrobenzodifuranyl homologue 8, and their brominated analogues 12 and 20. Their characterizationby Fourier transform infrared spectroscopy (FTIR), proton nuclear magnetic resonance spectroscopy (1H-NMR), carbon nuclearmagnetic resonance spectroscopy (13C-NMR), high performance liquid chromatography (HPLC), gas chromatography (GC) andmass spectrometry (MS) is also described. Copyright © 2013 John Wiley & Sons, Ltd.
Keywords: Bromo-dragonFLY; amphetamine-type stimulants (ATS); ketone precursors; clandestine synthesis; novel psychoactivesubstances (NPS)
* Correspondence to: John J. Keating, Analytical & Biological Chemistry ResearchFacility (ABCRF), School of Pharmacy / Department of Chemistry, UniversityCollege Cork, College Road, Cork, Ireland E-mail: [email protected]
Analytical & Biological Chemistry Research Facility (ABCRF), School ofPharmacy/Department of Chemistry, University College Cork, College Road,Cork, Ireland
Introduction
Bromo-dragonFLY (BDF) or (1-(8-bromobenzo[1,2-b:4,5-b’]difuranyl-4-yl)-2-aminopropane) 1 is a hallucinogenic benzodifuranyl deriva-tive and an emerging drug of recreational abuse.[1–4] Initiallysynthesized as a research chemical for mapping the topography ofspecific serotonin receptors,[5] BDF is a potent 5-HT2A and 5-HT2Creceptor agonist.[6] Typical doses are in the milligram to sub-milligram range (0.2–1.5mg) and onset of action is in the range of20–90min. Its effects have been known to last for up to 36h.[1] Rec-reational use of BDF has been associated with a number of adverseeffects including vasoconstriction and liver and kidney failure, as wellas severe convulsions.[1–4,7,8] There are several documented caseswhere BDF either contributed to, or was the sole cause of death ofthe user.[4,9–13] BDF is often branded as a ‘legal-high’ and is currentlyavailable for sale online in several forms: powder, liquid, and impreg-nated on paper.[1–4] There have also been reports of BDF beingmislabelled as other less potent recreational drugs; a number ofthese reported cases have resulted in a fatal overdose.[9–13] Drugenforcement agencies have documented seizures of BDF in theUnited States of America, Australia, and Europe.[2,12–14] In 2006, theEuropean Monitoring Centre for Drugs and Drug Addiction(EMCDDA)–Europol Annual Report flagged BDF as part of its EarlyWarning System on new psychoactive substances.[2,15] CurrentlyBDF is only controlled under legislation in a limited number of coun-tries worldwide. On 1 November 2011 it was added by name to theIrish Misuse of Drugs Act, 1977.[16]
Since the 1990s, the Nichols group has reported numeroussyntheses of BDF and its analogues.[5,6,17–21] A selection of these
Drug Test. Analysis (2013)
compounds is shown in Figure 1. However, none of theseisopropylamines were prepared via a 1-aryl-2-propanone-typeintermediate. Analysis of the literature has also revealed noexamination of BDF ‘street samples’ for route-specific impurities,thus no data are currently available on the synthetic methodsused by clandestine chemists in the manufacture of BDF. It is wellknown that simple ketones such as 1-phenyl-2-propanone(P2P, BMK) can be utilized as precursors in the production ofamphetamine-type stimulants (ATS).[22–25] Accordingly it wouldbe reasonable to assume that novel ketone precursors maypotentially be exploited in the clandestine synthesis of BDF andits analogues. This concept is the motivation for our currentinvestigation and incorporates a number of the aims of ourresearch group, some of which are (1) to isolate and characterizeprecursors, intended products and impurities of novel psychoac-tive substances (NPS), in this instance BDF, (2) to identify route-specific impurities so as to elucidate the possible synthetic routesto NPS including BDF analogues, and (3) to determine thechemical fingerprint for a given route to a given NPS.[26,27]
Completed profiles may help to assist authorities in their investi-gations of such compounds.
Copyright © 2013 John Wiley & Sons, Ltd.

Figure 1. A selection of benzodifuranyl & tetrahydrobenzodifuranylisopropylamines.
R. E. O’Connor and J. J. Keating
Drug Testing
and Analysis
Knowledge of the potential impurities associated with a givenNPS may be medically relevant, as it is possible that there are im-purities present in ‘street samples’ which could be responsible forunforeseen side-effects if ingested.[28,29] The work describedherein details the comprehensive synthesis and characterizationof four analogous ketones which may act as precursors to BDFand a number of structurally related derivatives.
Experimental procedures
Chemicals, reagents and methods
All commercially available reagents were purchased from SigmaAldrich Ltd. (Arklow, Wicklow, Ireland), (reagent-grade purity orhigher) andwere usedwithout further purification unless otherwiseindicated. Dry methylene chloride was obtained by distillation fromphosphorus pentoxide. Dry THF was obtained by distillation frombenzophenone-sodium under nitrogen immediately before use.Thin-layer chromatography was performed using pre-coated silicagel plates (Merck Silica Gel 60, PF254), visualizing with UV light at254 nm and eluting with 4:1 hexanes-ethyl acetate unlessotherwise noted. Flash column chromatography was performedusing Merck Kieselgel 60 (particle size 0.040–0.063mm) and ‘plugcolumns’ were implemented using the dry column vacuumchromatography (DCVC) technique[30] with 2.5 cm of silica in asintered glass funnel (35mm diameter). All reactions were carriedout under an inert atmosphere of nitrogen unless otherwiseindicated.
Instrumentation
Melting points were determined using a Gallenkamp apparatusand are uncorrected. Infra-red (IR) spectra were recorded on aPerkin Paragon 1000 FT-IR spectrometer. Band positions aregiven in cm�1. Samples were analyzed as potassium bromide(KBr) discs. 1H and 13C nuclear magnetic resonance (NMR) wererecorded at 300 K on either a Bruker Avance 300, 400 or 600spectrometer: 1H-NMR (300.13MHz, 400.13MHz or 600.07MHz);13C-NMR (75.47MHz) in CDCl3 [internal standard tetramethylsilane(TMS)]. For CDCl3,
1H-NMR spectra were assigned relative to theTMS peak at 0.00 d, 13C-NMR spectra were assigned relative tothe middle CDCl3 triplet at 77.00 ppm. Coupling constants arereported in Hertz (Hz). For the 1H-NMR assignments, chemical shiftsare reported as follows: shift value (d) (number of protons, splittingpattern, proton assignment, coupling constant(s)). Splitting patternsin 1H-NMR spectra are designated s (singlet), br s (broad singlet),
wileyonlinelibrary.com/journal/dta Copyright © 20
d (doublet), dd (doublet of doublets), t (triplet), td (triplet ofdoublets), tt (triplet of triplets), tq (triplet of quartets), m (multiplet).For 13C-NMR, assignments are reported in parts per million (ppm).Low resolution mass spectra (LRMS) were acquired on a Waters/Micromass LCT premier Time of Flight spectrometer (ESI) and highresolution mass spectra (HRMS) were acquired on a Waters/Micromass Quattra Micro Triple Quadrupole spectrometer (ESI).Samples were dissolved in HPLC-grade acetonitrile.
Qualitative high performance liquid chromatography (HPLC)was performed on an Agilent Technologies 1260 Infinity system.Separations were performed using a Zorbax SB-C18 column(150mm x 4.6mm, 3.5 mm; Agilent Technologies, Santry, Dublin,Ireland) using a mobile phase of 30:70 acetonitrile/water. Thetemperature of the column was set to 25 �C and the flow ratewas 1.5ml/min. UV detection was recorded at 210 nm and sam-ples were prepared in acetonitrile/water (30:70). Qualitative gaschromatography-mass spectrometry (GC-MS) was performed onan Agilent Technologies 5975C VL MSD with triple-axis detector(MSD; EI mode, 70 eV; m/z 40–350; source temperature 200 �C)coupled to a Hewlett Packard 6890 GC [injector temperature200 �C; 1ml injected in split mode (split ratio 25:1); carrier gas, hydro-gen; flow rate 1.0ml/min] fitted with an Agilent J&W DB-WAX GCColumn (30m� 0.25mm id� 0.25mm film, Agilent Technologies122-7032). The temperature program was as follows: initial temper-ature, 45 �C; solvent delay, 3.0min; ramp rate, 10 �C/min; tempera-ture 250 �C, hold 8.5min; final temperature, 250 �C. Samples weredissolved in HPLC-grade acetonitrile.
Synthesis
General procedure A: Formylation under Friedel-Crafts conditions
A solution of the tetrahydrobenzodifuran (see text) (20mmol) in110ml of anhydrous methylene chloride was stirred under N2
and cooled over an acetone-ice bath at �10 �C. Tin(IV) chloride(3.0ml, 6.68 g, 25.64mmol) was added to the solution, and themixture was stirred for 10min. Dichloromethyl methyl ether(1.81ml, 2.30 g, 20mmol) dissolved in 7ml of anhydrousmethylenechloride was added to the mixture dropwise over a 10 min periodand the mixture was then allowed to stir for 1 h. The reaction wasquenched by the addition of 60ml of cold deionized water, andthe layers were separated. The aqueous phase was extracted with(2� 30ml) of methylene chloride and the organic fractions werecombined. The organic extract was washed with 3M HCl(3� 100ml), 110ml of deionized water, and 110ml of brine, driedover MgSO4, and filtered through a pad of CeliteW and silica gel.The padwas then rinsedwith a further 40ml ofmethylene chloride.The solvent was removed in vacuo to give a yellow solid. If required,the sample was recrystallized from ethyl acetate.
General procedure B: Henry-Knoevenagel condensation
To a stirred solution of the aldehyde (20mmol) in glacial aceticacid (25ml), nitroethane (2.88ml, 3.01 g, 40mmol) was addedfollowed by the dropwise addition of cyclohexylamine (2.29ml,1.99 g, 20mmol). The reaction mixture was heated to 100 �C for6 h and then allowed to cool to room temperature and deionizedwater (35ml) was added. In some cases the nitropropene precipi-tated from solution. In this instance the crude product wascollected via Büchner filtration and then dissolved in methylenechloride (50ml) before the work-up was continued. Alternatively,when the crude product remained in solution, methylene chloride(20ml) was added and the phases were separated. The aqueous
13 John Wiley & Sons, Ltd. Drug Test. Analysis (2013)

Synthesis and characterization of BDF ketone precursors
Drug Testing
and Analysis
phase was extracted withmethylene chloride (3� 10ml). The com-bined organic phases were washed with saturated aqueousNaHCO3 (3� 15ml), deionized water (3� 15ml), dried over MgSO4
and filtered through a pad of silica in a sintered glass funnel.The pad was then rinsed with additional methylene chloride(20ml) and the filtrate concentrated in vacuo to give the crudeproduct as an orange coloured solid, which was recrystallizedfrom methanol.
General procedure C: Iron-mediated reduction of nitropropenesto propanones
A stirred suspension of iron powder (3.35 g, 60mmol) in glacialacetic acid (15ml) was heated to 60 �C. Separately, a solution ofthe nitropropene (10mmol) in glacial acetic acid (15ml) was alsoheated to 60 �C. The nitropropene solution was added dropwiseto the iron suspension over a 20 min period. The reaction mixturewas then heated to 90 �C for 4–6 h and allowed to cool to roomtemperature. Chilled deionized water (60ml) was added slowlyand the mixture filtered through CeliteW in a sintered glass funneland rinsed with methylene chloride (25ml). The phases wereseparated and the aqueous phase was extracted with methylenechloride (3� 20ml). The combined organic phases were washedwith 15% aqueous NaOH (3� 15ml) and deionized water(3� 15ml), dried over MgSO4, filtered and concentrated in vacuoto give the product as a cream coloured solid. The sample wasthen recrystallized from diethyl ether.
General procedure D: Regioselective aromatic bromination usingN-Bromosuccinimide (NBS)
The substrate (10.0mmol) was dissolved in acetonitrile (60ml),and the solution was cooled to 0 �C. NBS (1.78 g, 10.0mmol)was added in five separate portions over 30min. The reactionwas allowed to warm to room temperature and stirred for afurther 3 h. Deionized water (200ml) was added to the solutionresulting in precipitation of the product. The precipitate was thencollected via filtration using a Büchner funnel and dissolved inmethylene chloride (60ml), washed with deionized water (30ml),brine (30ml), dried over MgSO4, filtered and concentrated in vacuoto give the pure product as a white to off-white solid. The samplewas then recrystallized from a suitable solvent (see relevantExperimental data).
General procedure E: Dehydrogenation of tetrahydrobenzodifurans(THBDs) to benzodifurans using 2,3-dichloro-5,6-dicyano-1,4-benzo-quinone (DDQ)
A solution of DDQ (5.0 g, 22.0mmol) in 1,4-dioxane (100ml) wasslowly added to a solution of the THBD (10.0mmol) in 1,4-dioxane(60ml). The resulting solution was then heated at reflux for 60 h, atwhich time TLC indicated that the reaction had reached comple-tion. The mixture was allowed to cool to room temperature andthe solvent evaporated in vacuo. The 4,5-dichloro-3,6-dihydroxy-phthalonitrile (DDP) precipitate that formed was removed byfiltering the crude product mixture through a short pad of silicain a sintered glass funnel under vacuum. The filter cake was thenwashed thoroughly with methylene chloride and the combinedfiltrate transferred to a round bottom flask containing 1.0 g ofCeliteW and evaporated to dryness in vacuo. The sample wassubjected to flash column chromatography to remove anyDDQ and dihydrobenzodifuran intermediates. The productwas then recrystallized from a suitable solvent (see relevantExperimental data).
Drug Test. Analysis (2013) Copyright © 2013 John Wiley
2,3,6,7-Tetrahydrobenzo[1,2-b:4,5-b0]difuran 5[6]
The title compound was synthesized via the method outlined byChambers et al.[6] to afford a tan solid which was thenrecrystallized from diethyl ether to yield colourless crystals; Rf0.87 (1:1 hexane/ethyl acetate); m.p. 153 - 154 �C (diethyl ether),(lit.[17] 154 - 156 �C); IRnmax (KBr): 2896, 1488, 1448, 1312, 1226,1140, 980 cm�1; 1H-NMR d (400MHz, CDCl3): 3.13 (4H, t, H-3,H-7, J = 8.5 Hz), 4.53 (4H, t, H-2, H-6, J = 8.5 Hz), 6.63 (2H, s, H-4,H-8); 13C-NMR ppm (75.5MHz, CDCl3): 30.26 (C-3, C-7), 71.45(C-2, C-6), 105.83 (C-4, C-8) 125.88 (C-2b, C-5b’), 154.09 (C-1b, C-4b’); m/z (ES+) 163.1 (M+H+); HRMS (ES+): Exact mass calculatedfor C10H11O2 (M+H+) 163.0759. Found 163.0751 (M+H+).
4-Formyl-2,3,6,7-tetrahydrobenzo[1,2-b:4,5-b0]difuran 6
The title compound was prepared from THBD 5 using generalprocedure A and isolated (98%) without the need for recrystalliza-tion; Rf 0.71 (1:1 hexane/ethyl acetate); m.p. 87 - 89 �C (chloro-form-hexane), (lit.[17] m.p. 86 - 87 �C); IRnmax (KBr): 2911, 1677,1610, 1473, 1291, 1221, 1034, 985 cm�1; 1H-NMR d (600MHz,CDCl3): 3.17 (2H, tq, H-7, J = 8.7, 1.3Hz), 3.46 (2H, tt, H-3 J= 8.9Hz,1.4Hz), 4.59 (2H, t, H-2, J = 8.9Hz), 4.68 (2H, t, H-6, J = 8.7Hz), 6.87(1H, br s, H-8), 10.27 (1H, s, CHO). 13C-NMR ppm (75.5MHz, CDCl3):29.18 (C-7), 30.38 (C-3), 72.25 (C-2), 72.70 (C-6), 105.79 (C-4),112.56 (C-8), 125.05, 128.27 (C-2b, C-5b’), 154.59, 157.43 (C-1b,C-4b’), 188.92 (CHO); m/z (ES+) 191.1 (M +H+); HRMS (ES+): Exactmass calculated for C11H11O3 (M+H+) 191.0708. Found 191.0709(M+H+).
4-(2-Nitro-1-propenyl)-2,3,6,7-tetrahydrobenzo[1,2-b:4,5-b0]difuran 7
The title compound was prepared from aldehyde 6 using generalprocedure B and recrystallized frommethanol (92%); Rf 0.76 (1:1 hex-ane/ethyl acetate); m.p. 94 - 96 �C (methanol), (lit.[17] 93 - 95 �C);IRnmax (KBr): 2969, 2899, 2848, 1716, 1663, 1518, 1448, 1295, 1170,985 cm�1; 1H-NMR d (600MHz, CDCl3): 2.23 (3H, d, H-g, J = 1.0Hz),3.09 (2H, t, H-3, J = 8.6Hz), 3.19 (2H, td, H-7, J = 8.7, 1.0Hz), 4.59 (4H,t, H-2, H-6, J = 8.7Hz), 6.72 (1H, s, H-8), 7.83 (1H, br s, H-a). 13C-NMRppm (75.5MHz, CDCl3): 15.07 (C-g), 29.58 (C-3), 30.20 (C-7), 71.62,71.78 (C-2/C-6), 107.87 (C-8), 111.80 (C-4), 125.22 (C-2b), 127.40(C-5b’), 127.46 (C-a), 149.45 (C-b), 151.44 (C-1b), 154.16 (C-4b’);m/z (ES+) 248.1 (M+H+); HRMS (ES+): Exact mass calculated forC13H13NO4 (M +H+) 248.0923. Found 248.0911 (M +H+).
1-(2,3,6,7-Tetrahydrobenzo[1,2-b:4,5-b0]difuran-4-yl)propan-2-one 8
The title compound was prepared from nitropropene 7 usinggeneral procedure C and isolated without the need for recrystal-lization (94%); Rf 0.60 (1:1 hexane/ethyl acetate); m.p. 81 - 83 �C(chloroform); IRnmax (KBr): 2952, 2881, 1715, 1480, 1452, 1327,1214, 1166, 1039, 983 cm�1; 1H-NMR d (600MHz, CDCl3): 2.18(3H, s, H-g), 3.04 (2H, t, H-3, J = 8.6 Hz), 3.16 (2H, td, H-7 J = 8.6,1.0 Hz), 3.60 (2H, s, H-a), 4.53 (2H, t, H-2 or H-6 J = 8.6 Hz), 4.54(2H, t, H-2 or H-6, J = 8.6 Hz) 6.57 (1H, s, H-8); 13C-NMR ppm(75.5MHz, CDCl3): 29.21 (C-g), 28.93 (C-3), 30.64 (C-7), 42.95 (C-a),71.38, 71.45 (C-2, C-6), 104.85 (C-8), 112.86 (C-4), 125.72, 125.81(C-2b, C-5b’), 152.38 (C-4b’), 154.06 (C-1b), 205.57 (C-b); m/z (ES+)219.1 (M+H+); HRMS (ES+): Exact mass calculated for C13H15O3
(M+H+) 219.1021. Found 219.1029 (M+H+).
4-Bromo-2,3,6,7-tetrahydrobenzo[1,2-b:4,5-b’]difuran 9
The title compound was prepared from THBD 5 using general pro-cedure D (91%); Rf 0.62 (4:1 hexane/ethyl acetate); m.p. 79 - 81 �C(methanol); IRnmax (KBr): 2968, 2914, 2850, 1594, 1452, 1302, 1233,
& Sons, Ltd. wileyonlinelibrary.com/journal/dta

R. E. O’Connor and J. J. Keating
Drug Testing
and Analysis
1171, 983, 868, 844 752 cm�1; 1H-NMR d (400MHz, CDCl3): 3.16 (2H,tt, H-3, J = 8.7, 1.1Hz), 3.24 (2H, tq, H-7, J = 8.7, 1.1Hz), 4.58 (2H, t H-2,J = 8.7Hz), 4.63 (2H, t, H-6, J = 8.7Hz), 6.56 (1H, s, H-8); 13C-NMR ppm(75.5MHz, CDCl3): 31.23 (C-7), 31.42 (C-3), 71.22 (C-2), 71.77 (C-6),99.50 (C-4), 104.86 (C-8), 126.74, 126.89 (C-2b, C-5b’), 151.20,154.21 (C-1b, C-4b’); m/z (ES+) 241.0 (M+H+).
8-Bromo-2,3,6,7-tetrahydrobenzo[1,2-b:4,5-b’]difuran-4-carbaldehyde 10
The title compound was prepared from THBD 5 using generalprocedure A (86%) and was also prepared from aldehyde 6 usinggeneral procedure D (27 %); Rf 0.56 (4:1 hexane/ethyl acetate); m.p. 186 - 188 �C (ethyl acetate); IRnmax (KBr): 2918, 1678, 1593,1482, 1297, 1218, 1042, 992 cm�1; 1H-NMR d (600MHz, CDCl3):3.21 (2H, tt, H-7, J = 8.7, 1.4 Hz), 3.56 (2H, tt, H-3, J = 8.9 Hz,1.4 Hz), 4.69 (2H, t, H-2, J = 8.9 Hz), 4.74 (2H, t, H-6, J = 8.7 Hz),10.23 (1H, s, CHO); 13C-NMR ppm (75.5MHz, CDCl3): 30.60, 31.48(C-3, C-7), 72.53, 72.67 (C-2, C-6), 107.29 (C-8), 115.04 (C-4), 125.86,129.12 (C-2b, C-5b’), 151.95, 157.00 (C-1b, C-4b’), 188.28 (CHO);m/z (ES+) 271.0 (M+H+); HRMS (ES+): Exact mass calculated forC11H10
81BrO3 (M+H+) 270.9793. Found 270.9780 (M+H+).
4-Bromo-8-(2-nitroprop-1-en-1-yl)-2,3,6,7-tetrahydrobenzo[1,2-b:4,5-b’]difuran 11
The title compound was prepared from aldehyde 10 using gen-eral procedure B (90%) and was also prepared from nitropropene7 using general procedure D (84%); Rf 0.59 (4:1 hexane/ethylacetate); m.p. 147 - 149 �C (methanol); IRnmax (KBr): 2973, 2912,2820, 1661, 1602, 1520, 1417, 1301, 1223, 991, 862 cm�1;1H-NMR d (600MHz, CDCl3): 2.21 (3H, d, H-g, J = 1.0 Hz),3.19-3.24 (4H, m, H-3, H-7), 4.65 (2H, tt, H-2 or H-6,J = 8.7Hz), 4.69 (2H, tt, H-2 or H-6, J = 8.7Hz), 7.75 (1H, br s, H-a);13C-NMR ppm (75.5MHz, CDCl3): 15.10 (C-g), 30.62, 31.50 (C-3, C-7), 71.58, 71.94 (C-2, C-6), 101.89 (C-8), 110.93 (C-4), 126.06 (C-2bor C-5b’), 126.64 (C-a), 128.20 (C-2b or C-5b’), 149.69 (C-b), 151.38,151.43 (C-1b, C-4b’); m/z (ES+) 326.0 (M+H+); HRMS (ES+): Exactmass calculated for C13H13
79BrNO4 (M+H+) 326.0028. Found326.0001 (M+H+).
1-(8-Bromo-2,3,6,7-tetrahydrobenzo[1,2-b:4,5-b’]difuran-4-yl)propan-2-one 12
The title compound was prepared from nitropropene 11 usinggeneral procedure C (93%) and also prepared from ketone 8 usinggeneral procedure D (87%); Rf 0.20 (4:1 hexane/ethyl acetate); m.p.119 - 121 �C (chloroform); IRnmax (KBr): 2970, 2905, 2854, 1709,1482, 1425, 1356, 1217, 1164, 1044, 985, 846 cm�1; 1H-NMRd (600MHz, CDCl3): 2.19 (3H, s, H-g), 3.14 (2H, tt, H-3, J = 8.7,1.2Hz), 3.19 (2H, tt, H-7, J = 8.7, 1.2Hz), 3.56 (2H, s, H-a), 4.59 (2H,t, H-6, J = 8.7Hz), 4.64 (2H, t, H-2, J = 8.7Hz); 13C-NMR ppm(75.5MHz, CDCl3): 29.36 (C-g), 29.93 (C-3), 31.75 (C-7), 42.49 (C-a),71.17 (C-6), 71.77 (C-2), 98.30 (C-8), 111.98 (C-4), 126.51 (C-5b’),126.79 (C-2b), 151.07 (C-1b), 152.47 (C-4b’), 205.01 (C-b); m/z (ES+)299.0 (M+H+); HRMS (ES+): Exact mass calculated for C13H14
79BrO3
(M+H+) 297.0126. Found 297.0112 (M+H+); exact mass calculatedfor C13H14
81BrO3 (M+H+) 299.0106. Found 299.0104 (M+H+).
Benzo[1,2-b:4,5-b’]difuran 13
The title compound was prepared from THBD 5 using generalprocedure E (95%) and recrystallized from hexane to yield whitecrystals; Rf 0.90 (1:1 hexane/ethyl acetate); m.p 109 - 111 �C(hexane), (lit.[31] m.p. 111 �C); IRnmax (KBr): 3148, 3106, 2926, 1712,
wileyonlinelibrary.com/journal/dta Copyright © 20
1627, 1547, 1433, 1376, 1305, 1161, 1096, 1026, 853 cm�1;1H-NMR d (400MHz, CDCl3): 6.83 (2H, dd, H-3, H-7, J=2.2Hz,0.5Hz) 7.64 (2H, d, H-2, H-6, J= 2.2Hz), 7.65 (2H, d, H-4, H-8,J =0.5Hz); 13C-NMR ppm (75.5MHz, CDCl3): 102.06 (C-3, C-7), 106.71(C-4, C-8), 125.43 (C-2b, C-5b’), 145.63 (C-2, C-6), 151.99 (C-1b, C-4b’).
Benzo[1,2-b:4,5-b’]difuran-4-carbaldehyde 14
The title compound was prepared from aldehyde 6 using gen-eral procedure E (78%); Rf 0.63 (4:1 hexane/ethyl acetate); m.p.105 - 107 �C (chloroform); IRnmax (KBr): 3136, 3081, 2832, 1674,1584, 1486, 1394, 1303, 1224, 1011, 947, 769 cm�1; 1H-NMR d(400MHz, CDCl3): 6.92 (1H, d, H-3, J = 2.3Hz), 7.60 (1H, dd, H-7,J = 2.2, 0.8Hz), 7.76 (1H, d, H-2, J = 2.3Hz), 7.82, (1H, d, H-6,J = 2.2Hz), 7.92 (1H, d, H-8, J = 0.8Hz), 10.83 (1H, s, CHO); 13C-NMRppm (75.5MHz, CDCl3): 106.71 (C-3), 107.16 (C-7), 109.25 (C-8),111.89 (C-4), 124.58, 125.92 (C-2b, C-5b’), 146.22 (C-2), 148.74(C-6), 152.02, 153.70 (C-1b, C-4b’), 187.46 (CHO); m/z (ES+)187.0 (M+H+); HRMS (ES+): Exact mass calculated for C11H7O3
(M+H+) 187.0395. Found 187.0392 (M+H+).
4-(2-Nitroprop-1-en-1-yl)benzo[1,2-b:4,5-b’]difuran 15
The title compound was prepared from aldehyde 14 using gen-eral procedure B (89%) and also prepared from nitropropene 7using general procedure E (68%); Rf 0.55 (4:1 hexane/ethylacetate); m.p. 140 - 142 �C (methanol); IRnmax (KBr): 3120, 2943,1738, 1657, 1615, 1398, 1384, 1303, 1129, 1040, 938, 722 cm�1;1H-NMR d (400MHz, CDCl3): 2.35 (3H, d, H-g, J = 0.9 Hz), 6.81(1H, dd, H-7, J = 2.3, 0.9 Hz), 6.90 (1H, d, H-3, J = 2.3 Hz), 7.71 (1H,d, H-2, J = 2.3 Hz), 7.75 (1H, d, H-6, J = 2.3 Hz), 7.76 (1H, d, H-8,J = 0.9Hz), 8.43 (1H, s, H-a); 13C-NMR ppm (75.5MHz, CDCl3): 15.41(C-g), 104.59 (C-8), 105.71 (C-7), 107.11 (C-3), 107.86 (C-4), 125.14,125.78 (C-2b, C-5b’), 126.53 (C-a), 145.90 (C-2), 146.77 (C-6),149.17, 151.75 (C-1b, C-4b’), 149.66 (C-b); m/z (ES+) 243.2 (M+);HRMS (ES+): Exact mass calculated for C13H10NO4 (M+H+)244.0610. Found 244.0615 (M+H+).
1-(Benzo[1,2-b:4,5-b’]difuran-4-yl)propan-2-one 16
The title compound was prepared from nitropropene 15 usinggeneral procedure C (94 %) and also prepared from ketone8 using general procedure E (76%); Rf 0.47 (4:1 hexane/ethylacetate); m.p. 67 - 69 �C (ethyl acetate); IRnmax (KBr): 3135, 3112,2903, 1725, 1552, 1417, 1358, 1214, 1159, 1045, 1018, 852, 760,700 cm�1; 1H-NMR d (600MHz, CDCl3): 2.17 (3H, s, H-g), 4.17 (2H,s, H-a), 6.82 (1H, dd, H-7, J = 2.3, 1.0Hz), 6.84 (1H, d, H-3,J = 2.2 Hz), 7.60 (1H, br s, H-8), 7.65 (1H, d, H-6, J = 2.3 Hz) 7.66(1H, d, H-2, J = 2.2 Hz); 13C-NMR ppm (75.5MHz, CDCl3): 29.18(C-g), 42.60 (C-a), 101.35 (C-8), 104.97, 107.14 (C-3, C-7), 108.76(C-4), 125.25, 125.46 (C-2b, C-5b’), 145.59, 145.84 (C-2, C-6),150.29, 151.88 (C-1b, C-4b’), 205.32 (C-b); m/z (ES+) 215.0(M+H+); HRMS (ES+): Exact mass calculated for C13H11O3
(M+H+) 215.0708. Found 215.0700 (M+H+).
4-Bromobenzo[1,2-b:4,5-b’]difuran 17
The title compound was prepared from THBD 9 using general pro-cedure E (89%); Rf 0.77 (4:1 hexane/ethyl acetate); m.p. 107 - 109 �C(hexane); IRnmax (KBr): 3146, 3125, 1578, 1546, 1416, 1370,1298, 1177, 1122, 1029, 861, 847, 758, 695 cm�1; 1H-NMR d(400MHz, CDCl3): 6.90 (1H, dd, H-7, J = 2.3, 0.8Hz), 6.91 (1H, d, H-3,J = 2.2Hz), 7.61 (1H, d, H-8, J = 0.8Hz), 7.69 (1H, d, H-2 or H-6,J = 2.3Hz), 7.71 (1H, d, H-2 or H-6, J = 2.2Hz); 13C-NMR ppm(75.5MHz, CDCl3): 94.28 (C-4), 101.50 (C-8), 106.50, 107.58 (C-3,
13 John Wiley & Sons, Ltd. Drug Test. Analysis (2013)

Synthesis and characterization of BDF ketone precursors
Drug Testing
and Analysis
C-7), 125.95, 126.73 (C-2b, C-5b’), 146.04, 146.10 (C-2, C-6),149.21, 151.53 (C-1b, C-4b’); m/z (ES+) 237.0 (M+H+); HRMS (ES+): Exact mass calculated for C10H6
79BrO2 (M +H+) 236.9551.Not found, accurate parent molecular ion (PMI) absent understandard conditions used.
8-Bromobenzo[1,2-b:4,5-b’]difuran-4-carbaldehyde 18
The title compound was prepared from aldehyde 10 usinggeneral procedure E (67%); Rf 0.65 (4:1 hexane/ethyl acetate);m.p. 212 - 214 �C (chloroform); IRnmax (KBr): 3153, 3130, 2839,1675, 1576, 1426, 1365, 1307, 1226, 1144, 1023, 968, 764 cm�1;1H-NMR d (400MHz, CDCl3): 6.99 (1H, d, H-3, J = 2.3 Hz), 7.68(1H, d, H-7, J = 2.1 Hz), 7.81 (1H, d, H-2, J = 2.3 Hz), 7.88, (1H, d,H-6, J = 2.1 Hz), 10.79 (1H, s, CHO); 13C-NMR ppm (75.5MHz,CDCl3): 102.79 (C-8), 106.76, 108.16 (C-3, C-7), 111.28 (C-4),124.91, 127.48 (C-2b, C-5b’), 146.72, 149.17 (C-2, C-6), 149.69,153.01 (C-1b, C-4b’), 188.95 (C-a); m/z (ES+) 265.0 (M+H+); HRMS(ES+): Exact mass calculated for C11H6
79BrO3 (M+H+) 264.9500.Found 264.9500 (M+H+).
4-Bromo-8-(2-nitroprop-1-en-1-yl)benzo[1,2-b:4,5-b’]difuran 19
The title compound was prepared from aldehyde 18 usinggeneral procedure B (61%) and also prepared from nitropropene11 using general procedure E (69%); Rf 0.67 (4:1 hexane/ethylacetate); m.p. 168 - 170 �C (methanol); IRnmax (KBr): 3147, 2925,1717, 1656, 1606, 1518, 1312, 1219, 1142, 1029, 957, 845, 758,693 cm�1; 1H-NMR d (400MHz, CDCl3): 2.33 (3H, d, H-g,J = 0.9 Hz), 6.89 (1H, d, H-3 or H-7, J = 2.3 Hz), 6.98 (1H, d, H-3or H-7, J = 2.2 Hz), 7.76 (1H, d, H-2 or H-6, J = 2.2 Hz), 7.82 (1H, d,H-2 or H-6, J = 2.3Hz), 8.36 (1H, d H-a, J = 0.9Hz); 13C-NMR ppm(75.5MHz, CDCl3): 15.47 (C-g), 97.33 (C-8), 106.63, 107.04 (C-3, C-7),
Figure 2. Synthetic route summary to ketones 8, 12, 16 and 20.
Drug Test. Analysis (2013) Copyright © 2013 John Wiley
107.38 (C-4), 125.52, 127.17 (C-2b, C-5b’), 125.81 (C-a), 146.40,147.19 (C-2, C-6), 148.62, 149.94 (C-1b, C-4b’), 149.20 (C-b);m/z (ES+) 324.0 (M+H+); HRMS (ES+): Exact mass calculated forC13H9
81BrNO4 (M+H+) 323.9694 Found 323.9699 (M +H+).
1-(8-Bromobenzo[1,2-b:4,5-b’]difuran-4-yl)propan-2-one 20
The title compound was prepared from nitropropene 19 usinggeneral procedure C (92%) and also prepared from ketone 12using general procedure E (63%); Rf 0.42 (4:1 hexane/ethylacetate); m.p. 107 - 109 �C (diethyl ether); IRnmax (KBr): 3140, 3113,1703, 1544, 1496, 1402, 1364, 1325, 1208, 1138, 1033, 844, 766,695, 612 cm�1; 1H-NMR d (600MHz, CDCl3): 2.19 (3H, s, H-g), 4.14(2H, s, H-a), 6.88 (1H, d, H-3, J = 2.2Hz), 6.92 (1H, d, H-7, J = 2.2Hz),7.71 (1H, d, H-6, J = 2.2Hz) 7.72 (1H, d, H-2, J = 2.2Hz); 13C-NMRppm (75.5MHz, CDCl3): 29.20 (C-g), 42.16 (C-a), 93.34 (C-8), 105.91(C-3), 106.90 (C-7), 108.42 (C-4), 126.08 (C-2b), 126.45 (C-5b’),146.03, 146.19 (C-2, C-6), 149.12 (C-1b), 149.91 (C-4b’), 204.36(C-b); m/z (ES+) 293.0 (M+H+); HRMS (ES+): Exact mass calculatedfor C13H10
79BrO3 (M +H+) 292.9813. Found 292.9816 (M+H+);exact mass calculated for C13H10
81BrO3 (M+H+) 294.9793. Found294.9781 (M +H+).
Results and discussion
Chemistry
The multi-layered reaction scheme for the synthesis of ketones 8,12, 16 and 20 is summarized in Figure 2. The starting materialTHBD 5 was prepared via the method outlined by Chamberset al.[6] The formation of aldehyde 6 and nitropropene 7 wasadapted from Monte et al.[17] and the subsequent reduction to
& Sons, Ltd. wileyonlinelibrary.com/journal/dta

R. E. O’Connor and J. J. Keating
Drug Testing
and Analysis
ketone 8 followed the method outlined by McNamara et al.[32]
and Shulgin and Shulgin.[33] Our investigation of these proce-dures resulted in several alterations which led to improvedreaction conditions and higher product yields.
Friedel-Crafts formylation of 5
Monte et al.[17] reported an 82% conversion of THBD 5 to alde-hyde 6 with an isolated yield of 71%. It was noted that Monteet al.[17] also carried out the formylation of the methyl substitutedTHBD with a much higher isolated yield of 94%. A comparison ofthe reaction conditions showed that the use of a salt-ice bath(ca. �15 �C) was employed for the methyl-substituted-THBD withonly a 10 min reaction time, whereas the formylation of THBDwas carried out at 0 �C in an ice bath for 35min. This inferred thatlowering the temperature below 0 �C might be more suitable forformylation. Thus it was decided to carry out the formylation ofTHBD in an ice-acetone bath (ca. �10 �C) for a period of 1 h andcompare the result with performing the reaction in an ice bathfor 1 h. It was found that the lower temperature (�10 �C) led toa significantly higher yield of 98% compared with that of 73%at 0 �C. Additionally, upon completion of the work-up, the purityof the product was such that there was no need forrecrystallization.
Henry-Knoevenagel condensation of 6 to nitropropene 7
Monte et al.[17] previously achieved a 60% yield of 7 by heatingaldehyde 6 with 1 equivalent of ammonium acetate in 11.6 equiv-alents of nitroethane at 90 �C for 15 h. Optimization was attemptedby varying temperature, solvent and reagent equivalents. Byreplacing ammonium acetate with 1 equivalent of cyclohexylaminein this work, increasing the reaction temperature to 100 �C andusing acetic acid as solvent, nitroethane could now be reduced toa stoichiometric amount of just 2 equivalents. These modificationsresulted in a shorter reaction time of 6h and a significantly higheryield of 89% for nitropropene 7.
Reduction of nitropropene 7 to propanone 8
Several methods have been reported in the literature for thereduction of conjugated nitrostyrenes to their respective ketones.Some conditions and reagents reported include; Fe/AcOH,[32,33]
SnCl2 in THF under microwave irradiation,[34] Fe/HCl,[35,36] or amixture of Raney-nickel and sodium hypophosphite.[37] Thechosen procedure for the reduction of nitropropenes to 1-aryl-2-propanones was adapted from a method employed by Shulginand Shulgin in the book PiHKAL where a mixture of iron powderand glacial acetic acid was used to reduce nitrostyrenes to theirrespective ketones.[33] This method is known to be popularamong clandestine chemists as iron powder is an easy to use,readily available, cheap reducing agent that provides aconvenient and efficient way of preparing simple precursorketones in clandestine laboratories.[27] The endpoint may also bedetermined visually by the loss of the characteristic red colour ofthe nitropropenes. Following a basic-workup, high yields of pureketone were obtained without the need for further purification.
Derivatization of ketone 8 and its precursors
Once an efficient route to our principal ketone precursor 8 wasestablished, it was found that direct functionalization could beapplied to generate its derivatives as seen in Figure 2. In termsof synthetic strategy, bromination of the aromatic ring couldtheoretically be performed at any stage (5–8) during the multi-step
wileyonlinelibrary.com/journal/dta Copyright © 20
synthesis of BDF. However, the choice of which intermediate tobrominate could have a significant effect on the overall yield. Whilethe individual yield of brominating each intermediate may onlyvary to a small degree, the reactivity of these bromo-intermediatesin subsequent steps may differ significantly compared to their non-brominated counterparts. Additional factors affecting the overallyield include the ease with which the product may be isolatedand the potential for impurity formation will also change consider-ably with the reactivity towards bromination. A similar rationalemay also be applied when deciding which step in the synthesis ofBDF should the THBD core be oxidized to benzodifuran.
Feng et al.[38] converted 5 to 9 without isolation in a 72% yieldusing bromine in acetic acid at 0 �C. However, this technique isinconvenient for small-scale preparatory synthesis as it requiresthe use of an overhead stirrer due to the relatively high freezingpoint of glacial acetic acid. Considering the efficient brominationof THBD-analogues demonstrated by Chambers et al.,[6] it wasdecided to attempt the bromination of each intermediate 5–8 inacetic acid with 1 equivalent of bromine at room temperaturewithout a catalyst to determine the feasibility of this method inproducing brominated precursors for BDF. Unfortunately thismethod was not regioselective; over-bromination occurred andoxidized benzodifuran analogues were also detected, a phenome-non alluded to previously by Chambers et al.[6] In particular, directbromination of ketone 8 with bromine in acetic acid producednumerous side products, with bromination occurring at multiplesites (including benzylic and furanyl), resulting in a complex mix-ture of products. Consequently the bromine/acetic acid systemwas not pursued further. Examination of the literature showedpromising results for the regioselective monobromination ofaromatic rings using 1 equivalent of N-bromosuccinimide (NBS) inacetonitrile.[39–42] This milder, more selective technique wasadapted for the regiospecific aromatic bromination of THBD’swithin our group. Using this method the desired ketone 12 precip-itated from the acetonitrile solution andwas easily isolated by filtra-tion without the need for further purification in excellent yield(87%). Based on this evidence, lower temperatures, short reactiontimes and choice of brominating reagent are undoubtedly crucialto preventing impurity formation. Generally this NBS procedureprovedmore successful than the initial bromine/acetic acid system.All NBS-mediated brominations were regioselective giving highyields of the desired product except for aldehyde 6 which onlyyielded 27% of the corresponding product 10.
A number of dehydrogenation methods reported in theliterature were considered for the oxidation/aromatization of2,3-dihydrobenzofuran derivatives to their benzodifuran homo-logues. One technique involves refluxing the hydrobenzofuranwith 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in either1,4-dioxane or toluene.[5,6] Alternatively the hydrobenzofurancould be refluxed with NBS and either dibenzoyl peroxide orazabisisobutyronitrile (AIBN) in CCl4.
[43,44] However, the lattermay be susceptible to bromination under certain conditions.DDQ was chosen as the oxidant considering that a clandestinechemist would simply refer to Nichols et al.,[5,6] who have previ-ously successfully aromatized a range of THBDs to benzodifuransvia this procedure. Prior to aromatizing ketone 12, an attemptwas made to optimize the dehydrogenation process by examin-ing the conversion of THBD 5 to benzodifuran 13. It was foundthat benzodifuran 13 could be produced in high yield (95%)using only 2.2 equivalents of DDQ refluxing in 1,4-dioxane for28 h. The isolation of the product from DDQ / 4,5-dichloro-3,6-dihydroxy-phthalonitrile (DDP) was easily achieved using dry
13 John Wiley & Sons, Ltd. Drug Test. Analysis (2013)

Figure 3. Generic numbering scheme for 4-substituted benzodifuranyl &tetrahydrobenzodifuranyl derivatives.
Figure 4. 1H-NMR spectra of ketones 8, 12, 16 and 20.
Synthesis and characterization of BDF ketone precursors
Drug Testing
and Analysis
column vacuum chromatography (DCVC)[30] with hexane as theeluent. Subsequently, each of the intermediates 6–12 weresubjected to this DDQ-aromatization procedure. However, asthe rate of oxidation to benzodifuran is influenced by the proper-ties of the substituents attached to the THBD core,[45] the reac-tion progress was followed by TLC and 1H-NMR to determineconsumption of starting material. It was found that with THBDderivatives 6–12, the reaction time had to be extended signifi-cantly, thus an unoptimized reaction time of 60 h was chosenand the results summarized in Figure 2. In particular, THBD ke-tones 8 and 12 were oxidized to benzodifuranyl ketones 16and 20 in moderate yields of 76% and 63%, respectively.
Typically, the crude product mixture consisted of traceamounts of the starting material and mono-aromatizeddihydrobenzodifuran intermediates as well as a stoichiometricamount of DDP and unreacted DDQ. In the oxidations describedhere only 10% excess of DDQ was used, while up to 50% excessDDQ was employed for similar reactions in the literature.[5,6,45]
Although using such an excess of DDQ might be consideredwasteful, excess DDQ can potentially lead to an increased rateof oxidation resulting in shorter reaction times and higher yields.
Expansion and development of synthetic route map
The multifaceted route map shown in Figure 2 also illustrates thefunctional group interconversions of aldehydes 10, 14 and 18 totheir corresponding nitropropene and ketone derivatives.Friedel-Crafts formylation of 9 was achieved in high yield (86%)using the same conditions described to formylate its unsubstitutedanalogue 5. It is worth noting that bromination of 5 followed byformylation of 9 is a much more efficient synthesis of aldehyde10 (78% overall yield) when compared to formylation of 5 withsubsequent bromination of aldehyde 6 (26% overall yield). Thesame formylation conditions (general procedure A) were alsoapplied to benzodifurans 13 and 17, however no aldehyde productwas recovered. Interestingly, direct formylation of thesebenzodifurans appears not to occur via this method. Instead solidswhich were insoluble in water, chloroform and methanol wereobtained, presumably the result of a polymerization reaction. Whilethis outcome warrants further investigation, it is beyond the scopeof this paper but may be considered in terms of future impurityprofiling. The conditions used for the Henry-Knoevenagelcondensation of 6 to 7 (general procedure B)were reproducedwithaldehyde derivatives 10, 14 and 18 to yield the three nitropropenes11, 15, and 19, respectively as shown in Figure 2. Excellentpercentage conversions were achieved in most cases except for19 which showed only moderate conversion (61%) after 6 h.Recrystallization from methanol was sufficient to isolate and purifyeach of the nitropropene products. Similarly, the reactionconditions used for the iron-mediated reduction of 7 to 8 (GeneralProcedure C) were reproduced with nitropropene derivatives 11,15, and 19 to yield the three ketones 12, 16 and 20 as shown inFigure 2. Upon work-up, pure ketones were obtained without theneed for recrystallization. Notably the standard reaction time forthis type of reduction in the literature[32] is only 2 h and the averagerequired time for 7, 11, and 15was 4h with the conversion of 19 to20 being the longest at 6 h.
A large number of variables exist when performing eachsynthetic step including reaction scale, temperature, stirring rateand work-up techniques, etc. In clandestine laboratories these vari-ables may not be adequately controlled, resulting in unoptimizedconditions. Without the use of adequate purification techniques it
Drug Test. Analysis (2013) Copyright © 2013 John Wiley
is feasible that individual starting materials/intermediates may becarried forward into subsequent synthetic steps. Thus it is evidentfrom Figure 2 that each intermediate 5–19 could possibly bepresent in a crude sample of ketone 20 regardless of the path takento reach the desired ketone. Consequently, if a ketone intermediatewas used in the clandestine synthesis of BDF, then compounds5–20 may be potential impurities.
1H-NMR analysis
In order to prevent any uncertainty as to which signals are beingreferred to, an illustration of the systematic numbering schemefor THBD and benzodifuranyl derivatives is shown in Figure 3.There are a number of similarities as well as key differencesbetween the 1H-NMR spectra of the four ketones 8, 12, 16, and20 shown in Figure 4 (600MHz is the preferred frequency forrecording this 1H-NMR data, see Figure S1 for details). The methylsignal (H-g) is a singlet in each ketone spectrum and its chemicalshift is unchanged by derivatization (Figure 4(a)-(d)). The additionof a bromine atom to the THBD ketone 8 results in the methylenetriplets (H-3 and H-7) being shifted downfield in ketone 12Figure 4(b). The methylene singlet (H-a), is relatively unaffectedby the addition of the bromine atom to (C-8). However, it canbe clearly seen for the benzodifuranyl ketones 16 and 20(Figures 4(c) and 4(d)) that H-a is shifted downfield by approxi-mately 0.6 ppm when compared to THBD ketones 8 and 12. InFigure 4(a) the heterocyclic H-2 and H-6 methylene tripletscentred at 4.53 ppm for ketone 8 almost entirely overlap with
& Sons, Ltd. wileyonlinelibrary.com/journal/dta
R. E. O’Connor and J. J. Keating
Drug Testing
and Analysis
only 0.01 ppm separating the two signals. In contrast the corre-sponding H-2 and H-6 methylene triplets for ketone 12 are fullyresolved due to the deshielding effect of the bromine atomsubstituted on C-8. Bromine addition to 8 also results in the lossof H-8 at 6.57 ppm (Figures 4(a) and 4(b)). As the aromatic regionsfor the benzodifuranyl ketones 16 and 20 are more intricate theyhave been magnified in supplementary information (Figure S2)
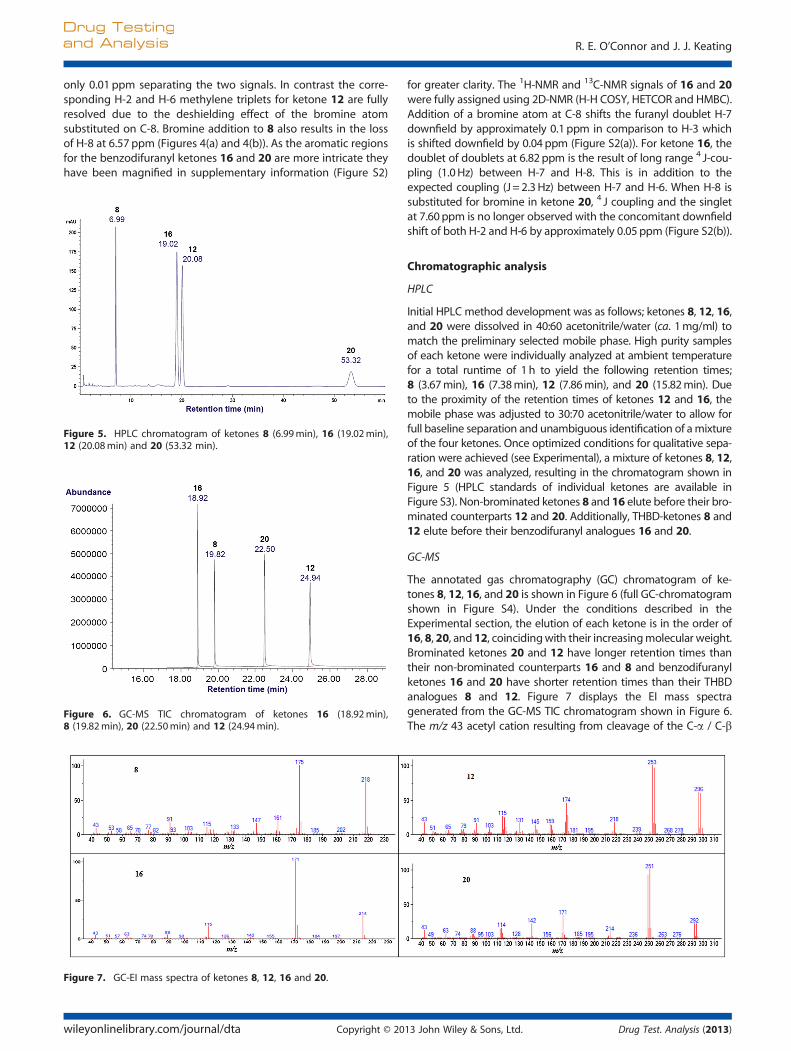
Figure 5. HPLC chromatogram of ketones 8 (6.99min), 16 (19.02min),12 (20.08min) and 20 (53.32 min).
Figure 6. GC-MS TIC chromatogram of ketones 16 (18.92min),8 (19.82min), 20 (22.50min) and 12 (24.94min).
Figure 7. GC-EI mass spectra of ketones 8, 12, 16 and 20.
wileyonlinelibrary.com/journal/dta Copyright © 20
for greater clarity. The 1H-NMR and 13C-NMR signals of 16 and 20were fully assigned using 2D-NMR (H-H COSY, HETCOR and HMBC).Addition of a bromine atom at C-8 shifts the furanyl doublet H-7downfield by approximately 0.1 ppm in comparison to H-3 whichis shifted downfield by 0.04 ppm (Figure S2(a)). For ketone 16, thedoublet of doublets at 6.82 ppm is the result of long range 4 J-cou-pling (1.0Hz) between H-7 and H-8. This is in addition to theexpected coupling (J= 2.3Hz) between H-7 and H-6. When H-8 issubstituted for bromine in ketone 20, 4 J coupling and the singletat 7.60 ppm is no longer observed with the concomitant downfieldshift of both H-2 and H-6 by approximately 0.05 ppm (Figure S2(b)).
Chromatographic analysis
HPLC
Initial HPLC method development was as follows; ketones 8, 12, 16,and 20 were dissolved in 40:60 acetonitrile/water (ca. 1mg/ml) tomatch the preliminary selected mobile phase. High purity samplesof each ketone were individually analyzed at ambient temperaturefor a total runtime of 1h to yield the following retention times;8 (3.67min), 16 (7.38min), 12 (7.86min), and 20 (15.82min). Dueto the proximity of the retention times of ketones 12 and 16, themobile phase was adjusted to 30:70 acetonitrile/water to allow forfull baseline separation and unambiguous identification of amixtureof the four ketones. Once optimized conditions for qualitative sepa-ration were achieved (see Experimental), a mixture of ketones 8, 12,16, and 20 was analyzed, resulting in the chromatogram shown inFigure 5 (HPLC standards of individual ketones are available inFigure S3). Non-brominated ketones 8 and 16 elute before their bro-minated counterparts 12 and 20. Additionally, THBD-ketones 8 and12 elute before their benzodifuranyl analogues 16 and 20.
GC-MS
The annotated gas chromatography (GC) chromatogram of ke-tones 8, 12, 16, and 20 is shown in Figure 6 (full GC-chromatogramshown in Figure S4). Under the conditions described in theExperimental section, the elution of each ketone is in the order of16, 8, 20, and 12, coincidingwith their increasingmolecular weight.Brominated ketones 20 and 12 have longer retention times thantheir non-brominated counterparts 16 and 8 and benzodifuranylketones 16 and 20 have shorter retention times than their THBDanalogues 8 and 12. Figure 7 displays the EI mass spectragenerated from the GC-MS TIC chromatogram shown in Figure 6.The m/z 43 acetyl cation resulting from cleavage of the C-a / C-b
13 John Wiley & Sons, Ltd. Drug Test. Analysis (2013)

Figure 8. Major ions from EI-MS of ketone 8.
Synthesis and characterization of BDF ketone precursors
Drug Testing
and Analysis
bond is common to all four ketones, producing similar benzylic-typeheterocycles in each ketone’s fragmentation pattern exemplified bym/z 175 of ketone 8 (Figure 8).[46] This benzylic-type fragment is alsothe base peak in each ketone’s spectra i.e. 8 (175), 12 (253), 16 (171),and 20 (251). The presence of 79Br and 81Br isotopes are clearlyevident by an increased number of peaks (separated by 2 units) inthe spectra of ketones 12 and 20. Additionally, once the bromineatom has been removed from a fragment, the subsequentfragments are similar to those found in their non-brominatedcounterparts. As each ketone has a different molecular weight,it is relatively easy to distinguish these homologues by theirPMIs alone.
Conclusions
The successful synthesis of ketones 8, 12, 16, and 20 and theircharacterization by IR, 1H-NMR, 13C-NMR, HLPC, GC, and massspectrometry represents the first profile of these potentialprecursors of benzodifuranyl isopropylamine analogues. Asindicated in Figure 9, these ketones may be potential precursorsto hallucinogenic amphetamine derivatives such as BDF and itsstructural analogues. The methodology employed for each ofthe synthetic steps outlined here is already available in the
Figure 9. Benzodifuranyl isopropylamine homologues and their ketoneprecursors.
Drug Test. Analysis (2013) Copyright © 2013 John Wiley
literature and accessible to a clandestine chemist. The majorityof procedures described are relatively straightforward and thuscould certainly be implemented by an experienced clandestinechemist with sufficient access to reagents, materials andequipment. Indeed, it is possible that these ketones may alreadybe in use by clandestine chemists. Consequently, ‘Part II’ willinvestigate a number of common synthetic protocols for theconversion of these ketones to their analogous amphetamineswith a focus on their associated impurities.
Acknowledgements
Grateful thanks are extended to Dr. Lorraine Bateman (NMR),Prof. Anita Maguire and Dr. Catherine Slattery (GC-MS), Dr. NualaMaguire (HPLC), Dr. Florence McCarthy and Mr. Michael O’Shea(HRMS). The authors would also like to thank ‘The University CollegeCork O’Connor Scholarship’ endowed by Mr. Dennis J. Doherty(current funding of ROC) and the Higher Education Authority(HEA) Ireland for initial partial funding of ROC.
References[1] Psychonaut WebMapping Research Group. Bromo-Dragonfly Report.
Institute of Psychiatry, King’s College London, London UK, 2009.[2] EMCDDA. Technical profile of bromodragonfly. Available at: http://
ewsd.wiv-isp.be/Publications%20on%20new%20psychoactive%20substances/Bromo-dragonfly/BDF_Tech_Prof_EMCDDA_Mar_2010.pdf [8 April 2013].
[3] O. Corazza, F. Schifano, M. Farre, P. Deluca, Z. Davey, C. Drummond,et al. Designer drugs on the internet: A phenomenon out-of-control?The emergence of hallucinogenic drug bromo-dragonfly. Curr. Clin.Pharmacol. 2011, 6, 125.
[4] M. Coppola, R. Mondola. Bromo-dragonFly: Chemistry, pharmacol-ogy and toxicology of a benzodifuran derivative producing LSD-like effects. J. Addict. Res. Ther. 2012, 3, 133.
[5] M.A. Parker, D. Marona-Lewicka, V.L. Lucaites, D.L. Nelson, D.E. Nichols.A novel (benzodifuranyl)aminoalkanewith extremely potent activity atthe 5-HT2A receptor. J. Med. Chem. 1998, 41, 5148.
[6] J.J. Chambers, D.M. Kurrasch-Orbaugh, M.A. Parker, D.E. Nichols.Enantiospecific synthesis and pharmacological evaluation of a seriesof super-potent, conformationally restricted 5-HT2A/2C receptoragonists. J. Med. Chem. 2001, 44, 1003.
[7] Bromo-dragonfly – life-threatening drug. Can cause tissue necrosisas demonstrated by the first described case. Available at: http://www.lakartidningen.se/engine.php?articleId=9254 [21 May 2013].
[8] D.M. Wood, J.J. Looker, L. Shaikh, J. Button, M. Puchnarewicz, S. Davies,et al. Delayed onset of seizures and toxicity associated with recrea-tional use of bromo-dragonFLY. J. Med. Toxicol. 2009, 5, 226.
[9] M.F. Andreasen, R. Telving, R.I. Dupont Birkler, B. Schumacher,M. Johannsen. A fatal poisoning involving Bromo-Dragonfly. ForensicSci. Int. 2009, 183, 91.
[10] S.L. Hill, S.H. Thomas. Clinical toxicology of newer recreational drugs.Clin. Toxicol. 2011, 49, 705.
[11] 2CB-FLY Warning. Pill Reports. Available at: http://www.pillreports.com/index.php?page=display_pill&id=19118 [8 April 2013].
[12] Information on reported deaths related to 2C-B-Fly. Erowid. Avail-able at: http://www.erowid.org/chemicals/2cb_fly/2cb_fly_death1.shtml [8 April 2013].
[13] Bromodragonfly mislabeled as 2-CE caused two deaths in Oklahoma.The Posion Review. Available at: http://www.thepoisonreview.com/2011/11/10/bromodragonfly-mislabeled-as-2c-e-caused-two-deaths-in-oklahoma/ [8 April 2013].
[14] U.S. Department of Justice Drug Enforcement Administration.‘Bromo dragonfly’ in Queensland, Australia.Microgr. Bull. 2008, 41(2), 16.
[15] European Monitoring Centre for Drugs and Drug Addiction(EMCDDA)–Europol. Annual report on the implementation ofCouncil decision 2005/387/JHA. In accordance with Article 10 ofCouncil decision 2005/387/JHA on the information exchange, riskassessment and control of new psychoactive substances. Lisbon,Portugal. 2006, 7.
& Sons, Ltd. wileyonlinelibrary.com/journal/dta

R. E. O’Connor and J. J. Keating
Drug Testing
and Analysis
[16] Misuse of Drugs Act 1977. Controlled Drugs, Declaration. Order S.I.551/2011, Ireland, 2011.
[17] A.P. Monte, D. Marona-Lewicka, M.A. Parker, D.B. Wainscott, D.L. Nelson,D.E. Nichols. Dihydrobenzofuran analogues of hallucinogens. 3. Modelsof 4-substituted (2,5-dimethoxyphenyl)alkylamine derivatives withrigidified methoxy Groups. J. Med. Chem. 1996, 39, 2953.
[18] D.E. Nichols, S.E. Snyder, R. Oberlender, M.P. Johnson, X. Huang.2,3-Dihydrobenzofuran analogues of hallucinogenic phenethylamines.J. Med. Chem. 1991, 34, 276.
[19] A.P. Monte, S.R. Waldman, D. Marona-Lewicka, D.B. Wainscott,D.L. Nelson, E. Sanders-Bush, et al. Dihydrobenzofuran analogues ofhallucinogens. 4. mescaline derivatives. J. Med. Chem. 1997, 40, 2997.
[20] D.M. Schultz, J.A. Prescher, S. Kidd, D. Marona-Lewicka, D.E. Nichols,A. Monte. ‘Hybrid’ benzofuran-benzopyran congeners as rigid analogsof hallucinogenic phenethylamines. Bioorg. Med. Chem. 2008, 16, 6242.
[21] M.A. Parker, D.M. Kurrasch, D.E. Nichols. The role of lipophilicity indetermining binding affinity and functional activity for 5-HT2Areceptor ligands. Bioorg. Med. Chem. 2008, 16, 4661.
[22] S. Di Giovanni, A. Varriale, V.M. Marzullo, G. Ruggiero, M. Staiano,A. Secchi, et al. Determination of benzyl methyl ketone – acommonly used precursor in amphetamine manufacture. Anal.Method 2012, 4, 3558.
[23] M. Collins. Some new psychoactive substances: Precursor chemicalsand synthesis-driven end-products. Drug Test. Anal. 2011, 3, 404.
[24] A. Allen, T.S. Cantrell. Synthetic reductions in clandestine amphet-amine and methamphetamine laboratories - a review. Forensic Sci.Int. 1989, 42, 183.
[25] A.B.E. Theeuwen, A.M.A. Verweij. Impurities in illicit amphetamine. 7.Identification of benzyl methyl ketone phenylisopropylimine andbenzyl methyl ketone benzylimine in amphetamine. Forensic Sci.Int. 1980, 15, 237.
[26] J.J. Keating. Studies in the synthesis and impurity profiling ofmethylenedioxy and alkythioamphetamines. PhD thesis. TrinityCollege, Dublin, 2001.
[27] D.M. Griffin. Studies in the synthesis and impurity profiling of2,5-dimethoxy amphetamines. PhD thesis. University CollegeCork, Cork, 2009.
[28] S.M. Cloonan, J.J. Keating, D. Corrigan, J.E. O’Brien, P.V. Kavanagh,D.C. Williams, et al. Synthesis and in vitro toxicity of 4-MTA, itscharacteristic clandestine synthesis byproducts and related sulfursubstituted a-alkylthioamphetamines. Bioorg. Med. Chem. 2010,18, 4009.
[29] A. Siwinska-Ziolkowska, D. Blachut, E. Widecka-Deptuch. Identifica-tion of impurities in biological material remaining after amphet-amine synthesis. Probl. Forensic Sci. 2004, 60, 130.
[30] D.S. Pedersen, C. Rosenbohm. Dry column vacuum chromatography.Synthesis 2001, 16, 2431.
[31] L. Rene, J.P. Buisson, R. Royer, D. Averbeck. Difurobenzene andfurochromene analogs of psoralen and pseudopsoralen. Eur. J.Med. Chem. 1977, 12, 31.
wileyonlinelibrary.com/journal/dta Copyright © 20
[32] Y.M. McNamara, S.M. Cloonan, A.J.S. Knox , J.J. Keating , S.G. Butler,G.H. Peters, et al. Synthesis and serotonin transporter activity of1,3-bis(aryl)-2-nitro-1-propenes as a new class of anticanceragents. Bioorg. Med. Chem. 2011, 19, 1328.
[33] A.T. Shulgin, A. Shulgin. PiHKAL: A Chemical Love Story. TransformPress, California, 1991.
[34] D.D. Das, A. Nayak, B. Nanda, N.B. Das. Microwave-assisted transfor-mation of, a,b- and, b,g-unsaturated nitroalkenes into carbonylcompounds. J. Chem. Research 2006, 8, 481.
[35] P.K. Pradhan, S. Dey, P. Jaisankar, V.S. Giri. Fe-HCl: An efficientreagent for deprotection of oximes as well as selective oxidativehydrolysis of nitroalkenes and nitro-alkanes to ketones. SyntheticCommun. 2005, 35, 913.
[36] H.B. Hass, A.G. Susie, R.L. Heidler. Nitro alkene derivatives. J. Org.Chem. 1950, 15, 8.
[37] D. Monti, P. Gramatica, G. Speranza, P. Manito. Reaction ofnitroolefins with Raney nickel and sodium hypophosphite. A mildmethod for converting nitroolefins into ketones (or aldehydes).Tetrahedron Lett. 1983, 24, 417.
[38] Z. Feng, S. Mohapatra, P.G. Klimko, M.R. Hellberg, J.A. May, C. Kelly,et al. Novel benzodifuran analogs as potent 5-HT2A receptoragonists with ocular hypotensive activity. Bioorg. Med. Chem. Lett.2007, 17, 2998.
[39] I. Pravst, M. Zupa, S. Stavber. Directed regioselectivity of brominationof ketones with NBS: Solvent-free conditions versus water. TetrahedronLett. 2006, 47, 4707.
[40] M.C. Carreno, J.L. Garcia Ruano, G. Sanz, M.A. Toledo, A. Urbano, N-Bromosuccinimide in acetonitrile: A mild and regiospecific nuclearbrominating reagent for methoxybenzenes and naphthalenes.J. Org. Chem. 1995, 60, 5328.
[41] E. Zysman-Colman, K. Arias, J.S. Siegel. Synthesis of arylbromides fromarenes and N-bromosuccinimide (NBS) in acetonitrile - A convenientmethod for aromatic bromination. Can. J. Chem. 2009, 87, 440.
[42] M.M. Pedemonte, I.N. Munoz, M.S. Lopez, D.M. Julian, M. Rosol,A.L. Soldevila, J.A. Bofarull. Crystax Pharmaceuticals. New compoundsas HSP90 inhibitors. WO Patent No. 2009/007399, 2009.
[43] M. Saitoh, J. Kunitomo, E. Kimura, Y. Hayase, H. Kobayashi,N. Uchiyama, et al. Design, synthesis and structure–activityrelationships of 1,3,4-oxadiazole derivatives as novel inhibitors ofglycogen synthase kinase-3b. Bioorg. Med. Chem. 2009, 17, 2017.
[44] T. Lu, R. Alexander, R.W. Connors, M.D. Cummings, R.A. Galemmo,H.R. Hufnagel, et al. Janssen Pharmaceutica. Triazolopyridazinesas tyrosine kinase modulators. WO Patent No. 2007075567,2007.
[45] J.C. Gonzalez-Gomez, L. Santana, E. Uriarte. A furan ring expansionapproach to the synthesis of novel pyridazino-psoralen derivatives.Tetrahedron 2005, 61, 4805.
[46] T. Awad, J. DeRuiter, C.R. Clark. Gas chromatography–massspectrometry analysis of regioisomeric ring substituted methoxymethyl phenylacetones. J. Chromatogr. Sci. 2007, 45, 458.
13 John Wiley & Sons, Ltd. Drug Test. Analysis (2013)
Related Documents











