



1
Failure of MBNL1-dependent postnatal splicing transitions in myotonic dystrophy
Xiaoyan Lin1, Jill W. Miller2, Ami Mankodi2‡, Rahul N. Kanadia3, Yuan Yuan3,
Richard T. Moxley2, Maurice S. Swanson3, Charles A. Thornton2*
Departments of 1Neuroscience and 2Neurology, University of Rochester Medical Center,
Rochester, New York, 14642, and 3Department of Molecular Genetics and Microbiology and the
Genetics Institute, University of Florida, College of Medicine, Gainesville, FL 32610. ‡Current
address, Department of Neurology, Johns Hopkins University School of Medicine, Baltimore,
MD
*Corresponding Author:
Charles A. Thornton, M.D.
Department of Neurology, Box 673
University of Rochester Medical Center
601 Elmwood Avenue
Rochester, NY 14642
Tel # 585 275 2542
Fax # 585 273 1255
Email: [email protected]
© The Author 2006. Published by Oxford University Press. All rights reserved
HMG Advance Access published May 22, 2006 by guest on February 19, 2016
http://hmg.oxfordjournals.org/
Dow
nloaded from

2
Abstract
In myotonic dystrophy (DM), expression of RNA containing expanded CUG or CCUG repeats
leads to misregulated alternative splicing of pre-mRNA. The repeat-bearing transcripts
accumulate in nuclear foci, together with proteins in the muscleblind family, MBNL1 and
MBNL2. In transgenic mice that express expanded CUG repeats, we show that the splicing
defect selectively targets a group of exons that share a common temporal pattern of
developmental regulation. These exons undergo a synchronized splicing switch between
postnatal day 2 and 20 in wild-type mice. During this postnatal interval, MBNL1 protein
translocates from a predominantly cytoplasmic to nuclear distribution. In the absence of
MBNL1, these physiological splicing transitions do not occur. The splicing defect induced by
expanded CUG repeats in mature muscle fibers is closely reproduced by deficiency of MBNL1
but not by deficiency of MBNL2. A parallel situation exists in human DM type 1 and type 2.
MBNL1 is depleted from the muscle nucleoplasm due to sequestration in nuclear foci, and the
associated splicing defects are remarkably similar to those observed in MBNL1 knockout mice.
These results indicate that MBNL1 participates in the postnatal remodeling of skeletal muscle by
controlling a key set of developmentally-regulated splicing switches. Sequestration of MBNL1,
and failure to maintain these splicing transitions, has a pivotal role in the pathogenesis of muscle
disease in DM.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

3
Introduction
Myotonic dystrophy type 1 (DM1) is the most common degenerative disease of skeletal muscle
in adults. This multi-system disorder is characterized by muscle wasting, myotonia,
degeneration of the cardiac conduction system, cataracts, and neuropsychological dysfunction.
DM1 is caused by expansion of a CTG repeat in the 3' untranslated region (UTR) of the
dystrophia myotonica protein kinase (DMPK) gene (1). A second, less common form of
myotonic dystrophy, DM type 2 (DM2), is caused by expansion of a CCTG repeat in intron 1 of
the zinc finger 9 (ZNF9) gene (2). Both mutations are in non-coding sequences, raising
questions about the mechanism of genetic dominance in DM.
A body of work has accumulated indicating that DMPK mRNA containing an abnormally
expanded CUG repeat has a deleterious effect on muscle fibers. For example, expression of
CUG expansion RNA in transgenic mice reproduces characteristic signs of DM1, either when the
CUG tract is expressed in the natural context of the DMPK 3' UTR (3) or when it is inserted in
the 3' UTR of an unrelated transcript (4). In both types of DM, and in transgenic mouse models,
the mutant RNA forms intranuclear (ribonuclear) foci in muscle fibers (2,4,5).
A core mechanism underlying symptoms of DM1 and DM2 is that expanded poly(CUG) or
(CCUG) RNA interferes with the regulated alternative splicing of certain pre-mRNAs. This
effect was first observed for cardiac troponin T (cTnT) (6). In the case of the insulin receptor
(IR), the predominant splice product expressed in DM1 muscle is the exon 11 skipped (non-
muscle) isoform, which may contribute to the insulin resistance in DM1 muscle fibers (7).
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

4
Misregulated splicing of the muscle-specific chloride ion channel (ClC-1) leads to reduced
chloride conductance and repetitive action potentials (myotonia) in DM muscle fibers (8,9).
CUG binding protein 1 (CUG-BP1) and related RNA-binding proteins in the CELF family are
implicated in the misregulated alternative splicing of DM (6). CUG-BP1 binds to UG-rich
sequences in vitro (10,11), including (CUG)8 (12), but it does not colocalize with ribonuclear
foci in DM1 cells (13-15) (for a contrary view, see reference (19)). The RNA binding motifs in
CUG-BP1 are of the type that recognize single stranded RNA, whereas poly(CUG) is stabilized
in a duplex (hairpin) conformation when the repeat is pathologically expanded (16-18).
Irrespective of whether CUG-BP1 directly interacts with the mutant mRNA, this protein may
have a role in the pathogenesis of splicing abnormalities because it is overexpressed in DM1
muscle cells (7,19,20). The mechanism for this increase has not been determined, but
overexpression of CUG-BP1 in striated muscle has been shown to trigger abnormal splicing of
cTnT and IR similar to that observed in DM1 (6,7).
In contrast, proteins in the muscleblind-like (MBNL) family preferentially recognize CUG or
CCUG repeats when they are pathologically expanded (13,21). MBNL proteins colocalize with
ribonuclear foci in DM1 and DM2 cells (13,22,23), which led to the proposal that symptoms of
DM may result from loss of MBNL activity due to protein sequestration on repeat expansion
RNAs. All three family MBNL members, MBNL1, MBNL2, MBNL3, are able to regulate
alternative splicing of cTnT and IR minigenes (24). In support of a sequestration mechanism,
Mbnl1 knockout mice show myotonia, cataracts, and misregulated splicing of ClC-1 and cTnT
that are similar to DM1 (25).
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

5
One putative mechanism for DM pathogenesis involves increased CUG-BP1 activity, another
involves sequestration of MBNL proteins, but presently it is unclear which mechanism is mainly
responsible for the splicing defect, or whether both must operate in concert. Moreover, as
MBNL proteins are abundant in skeletal muscle, it is uncertain whether expanded poly(CUG)
RNA is actually expressed in muscle fibers at levels that are capable of sequestering these factors.
If DM1 cells achieve such levels of expression, it is unknown which members of the MBNL
family are mainly responsible for the misregulation of splicing. To address these questions, we
have compared the developmental regulation of alternative splicing in skeletal muscle in
transgenic and Mbnl1 knockout models of DM1, and we have derived mice that have reduced
expression of Mbnl2. We found that misregulated splicing in a transgenic model that expresses
expanded poly(CUG) RNA is closely reproduced by deficiency of Mbnl1 but not by deficiency
of Mbnl2. Remarkably, every exon that is misregulated in response to expanded poly(CUG),
among the exons that we examined, shows a similar pattern of developmental regulation. In
WT mice, these exons transition from neonatal to adult splice isoforms within the first 3 weeks
of postnatal life, but in the absence of Mbnl1, these transitions do not occur. In both types of
human DM, MBNL1 is recruited into ribonuclear foci so extensively that free MBNL1 is
depleted from the nucleoplasm, and the splicing defects are strikingly similar to those observed
in Mbnl1 knockout mice. By contrast, splicing defects in mice that express expanded
poly(CUG) are not associated with abnormal accumulation or mislocalization of CUG-BP1.
These results point to a pivotal role for MBNL1 in DM pathogenesis, and indicate that this
splicing factor functions in the postnatal remodeling of skeletal muscle by controlling a key set
of developmentally-regulated splicing switches.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

6
Results
Expanded poly(CUG) RNA and deficiency of Mbnl1 have equivalent effects on alternative
splicing in mouse skeletal muscle. Alternative splicing of fast troponin T (Tnnt3) is abnormally
regulated in DM1 patients and in transgenic mice that express expanded CUG repeats (25). The
isoform of Tnnt3 expressed in DM1 muscle fibers includes an alternative exon that normally is
included in fetal transcripts. In normal adult muscle, however, this exon is skipped. To further
examine the physiological regulation of this exon during muscle development we examined its
splicing in WT mice from embryonic day 18 onward. The transition from inclusion to exclusion
of the “fetal” exon mainly occurred after birth, between postnatal day 2 and day 16 (Fig. 1A).
In an effort to identify other exons that respond to expanded poly(CUG), and assess their
developmental regulation, we selected 54 exons (from 36 genes) known to be alternatively
spliced in skeletal muscle. For each exon selected, the ratio of alternative splice products was
determined in mouse models of DM1, relative to WT mice of the appropriate background strain.
The models we examined were HSALR transgenic mice that express a human skeletal actin
mRNA containing (CUG)250 in the 3' UTR (4), and Mbnl1∆E3/∆E3 mice homozygous for a targeted
allele of Mbnl1 (25). In the initial RT-PCR splicing assays, eleven exons showed abnormal
regulation in HSALR mice. For example, splice products containing Serca1 exon 22 were
decreased in HSALR mice, whereas inclusion of ZASP exon 11 and titin m-line region exon 5
were increased (Fig. 1B). Re-examination of these 11 exons in an independent group confirmed
the abnormality of splicing regulation (n = 6 WT and HSALR mice per group, p < 0.0001 for each
exon, Supplemental Fig. 1, Table 1). The direction and magnitude of the effect varied among
genes. The most extreme example involved Serca1, encoding the calcium re-uptake pump of
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

7
the sarcoplasmic reticulum. The fraction of Serca1 mRNA skipping exon 22 increased from 3 ±
0.7% in WT to 78 ± 4% in HSALR mice. Abnormal splicing of Serca1 in DM1 has recently been
reported (26). Interestingly, the alternative splicing of Mbnl1 was also misregulated in HSALR
mice, showing an increased frequency of exon 7 inclusion (Fig. 1B). Ten of the 11 misregulated
exons were correctly spliced in disease controls having generalized myotonia (ClC-1 null mice)
or dystrophin deficiency (mdx mice), indicating that these abnormalities did not result from non-
specific effects of repetitive action potentials or dystrophic muscle (Fig. 1C and data not shown,
splicing of m-Titin was abnormally regulated in ClC-1 null mice).
The pattern of misregulated alternative splicing in HSALR transgenic mice was remarkably
concordant with Mbnl1∆E3/∆E3 mice across all 54 exons that we examined (Table 1 and
Supplemental Table 1). Furthermore, among exons that were abnormally regulated in DM1
mouse models, the pattern of splicing in adult HSALR or Mbnl1∆E3/∆E3 mice invariably resembled
that seen in WT mice at post-natal day 2 (Fig. 1B, C). We carried out a more detailed
examination of the postnatal splicing switch for 7 of the most-affected exons (Serca1, ZASP, m-
Titin, z-Titin, Nrap, Capn3, Mbnl1). In WT mice, each of these exons showed a splicing
transition between postnatal day 2 and day 20 (Fig. 2 and data not shown). However,
expression of expanded poly(CUG) or loss of Mbnl1 resulted in complete failure of these
splicing transitions. Integrin beta 1 and CapZb also showed a switch in alternative splicing
during this postnatal interval, however, these transitions were properly executed in HSALR and
Mbnl1∆E3/∆E3 mice (Fig. 2, lower panels). Thus, expanded poly(CUG) RNA has not produced a
global defect of developmentally-regulated alternative splicing, instead it has selectively targeted
exons that show Mbnl1-dependent splicing transitions. Furthermore, the postnatal regulation of
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

8
these exons followed the Tnnt3 fetal exon time course, suggesting that all of these splicing
transitions were coordinated by a common triggering event.
Exons misregulated in HSALR transgenic mice are not affected by disruption of Mbnl2. Mbnl1
and Mbnl2 are both expressed in skeletal muscle, and, when overexpressed, either factor is able
to regulate the alternative splicing of IR and cTnT minigenes (24). As a first step to investigate
the respective contributions of Mbnl1 and Mbnl2 in skeletal muscle, we examined the effects of
Mbnl2 deficiency on alternative splicing in adult muscle fibers. Mbnl2 gene trap (GT) mice
were derived from ES cells that have integrated a retroviral gene trap vector in Mbnl2 intron 4
(Fig. 3A). In mice homozygous for the GT allele (Mbnl2GT4/GT4 mice), Mbnl2 mRNA was
reduced by >90% (Fig. 3B), yet muscle histology was normal at 4 months of age and there was
no myotonia. Each of the exons misregulated in HSALR and Mbnl1∆E3/∆E3 mice showed normal
regulation in Mbnl2GT4/GT4 adult mice (Fig. 3C and data not shown, with the exception that
splicing of Mbnl2 itself could not be assessed due to insertion of the gene trap vector).
Mis-splicing in DM1 and DM2 skeletal muscle is similar to mouse models of DM1. To determine
if transgenic and Mbnl1 knockout mouse models predict misregulated splicing in human DM, we
examined splicing of the equivalent exons in skeletal muscle from patients with DM1 or DM2, as
compared to healthy subjects. Each exon misregulated in HSALR mice showed a similar
alteration in both types of DM (n = 3 per group, representative gels are shown in Fig. 4,
summary data are listed in Table 1) with the exception that splicing of GFAT1 exon 10 was not
consistently increased in DM2. Conversely, among the exons that were normally regulated in
HSALR and Mbnl1∆E3/∆E3 mouse models, the 27 that we examined in human DM also were
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

9
correctly regulated. Thus, both of these mouse models accurately reproduce the splicing defect
in human DM1 and DM2 skeletal muscle.
Mbnl1 is sequestered in DM1 and DM2 myonuclei. While MBNL1 protein has been localized
to nuclear foci in DM1 and DM2 muscle (22), the extent of MBNL1 sequestration (i.e., depletion
from the nucleoplasm) was unclear because antibodies used in the previous studies did not
clearly show MBNL1 in normal myonuclei. To re-examine this question, we raised a
polyclonal antibody against a C-terminal peptide that is conserved in human and murine Mbnl1.
Specificity of the antibody was demonstrated by lack of immunoreactivity on immunoblots or
tissue sections from Mbnl1∆E3/∆E3 muscle (see Fig. 6A and 6B). In sections of normal human
muscle, MBNL1 immunofluorescence (IF) was diffusely distributed in myonuclei. In sections
of DM1 and DM2 muscle, MBNL1 was heavily recruited into RNA foci (Supplemental Fig. 2A)
and the mean intensity of MBNL1 IF in nucleoplasm, exclusive of nuclear foci, was reduced by
≥78% (Fig. 4B). Interestingly, the amount of MBNL1 in nuclear foci was greater in DM2 than
in DM1 (Fig. 4B).
Cumulatively, these results suggest that loss of MBNL1 activity due to sequestration on mutant
DMPK and ZNF9 transcripts is the primary determinant of misregulated splicing in DM1 and
DM2. However, CUG-BP1 also plays a role in the developmental regulation of alternative
splicing in striated muscle (27,28), and increased levels of this protein have been linked to
abnormal splicing regulation in DM1 (6,7). Next, we examined the developmental regulation of
CUG-BP1 expression in skeletal muscle and determined if the splicing defect in HSALR mice and
DM2 patients is also associated with increased levels of CUG-BP1 protein.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

10
CUG-BP1 protein levels are not elevated in HSALR or DM2 skeletal muscle. In WT skeletal
muscle, levels of CUG-BP1 decreased sharply between P2 and P20 (Fig. 5A). Postnatal
downregulation of CUG-BP1 occurred to a similar extent in HSALR transgenic mice. In HSALR
transgenic mice, marked accumulation of expanded poly(CUG), and induction of splicing
abnormalities characteristic of DM1, was not associated with increased steady state levels of
CUG-BP1 protein (Fig. 5B). CUG-BP1 did not colocalize with ribonuclear foci in HSALR
muscle, as visualized by immunofluorescence of the endogenous protein or by expression of
GFP-tagged CUG-BP1 in vivo by electroporation (Fig. 5D and 5E). By comparison, GFP-
tagged Mbnl1 colocalized with nuclear RNA foci under these conditions (Fig. 5F). Interestingly,
while abnormalities of alternative splicing in quadriceps muscle biopsy samples tend to be more
pronounced in DM2 than in DM1 (8), we did not find any consistent increase in the levels of
CUG-BP1 in DM2 muscle tissue compared to healthy controls (Fig. 5C). These observations
argue that misregulated splicing in HSALR transgenic mice and human DM2 does not require
elevated levels or mislocalization of CUG-BP1 protein.
Mbnl1 relocalizes to the nucleus during postnatal development. To investigate the mechanism
for the postnatal splicing transitions pertinent to DM, we also examined the developmental
regulation of Mbnl1. Levels of Mbnl1 protein, as determined by immunoblot of whole muscle
lysates, declined in the interval between P2 and P20 (Fig. 6A). A similar decline occurred in
HSALR mice, and expression of poly(CUG) RNA did not result in abnormal accumulation of
Mbnl1 protein in mature muscle fibers (Fig. 6A, right panel). However, the postnatal decline
occurred during a period of rapid muscle fiber hypertrophy, and consequently these results may
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

11
reflect increasing dilution of nuclear contents by proteins in the muscle cytoplasm. In view of
the difficulty in obtaining nuclear fractions from multinucleate muscle fibers (29), we examined
the distribution of Mbnl1 by IF on muscle sections. Surprisingly, the postnatal splicing
transitions coincided with translocation of Mbnl1 from a predominantly cytoplasmic location at
P2 to a predominantly nuclear location at P20 (Fig. 6B). By comparison, neither hnRNP I nor
hnRNP H showed a parallel shift in cellular distribution during this interval (Suppl. Fig. 2B and
2C). However, Mbnl1 is not entirely excluded from the nucleus in neonatal muscle, as indicated
by its presence in nuclear foci of HSALR mice throughout the postnatal interval (Fig. 6C). This
indicates that CUG expansion RNA is expressed, and available to interact with Mbnl1 in the
nucleus, as early as P2 in HSALR muscle.
Mbnl1 pre-mRNA is alternatively spliced at exons 3, 5, 7, and 9, raising the possibility that
postnatal splicing transitions result from a switch to production of Mbnl1 splice products that
have a different cellular distribution or activity. For example, exclusion of exon 3 (E3)
eliminates one of four zinc finger domains and alters the RNA binding activity of Mbnl1 (21,25).
However, the fraction E3– splice products was similar in neonatal and adult WT and HSALR
mice, and the 30 kD protein encoded by this isoform was not detected in skeletal muscle by
immunoblot or immunoprecipitation (not shown). Exon 5 was included in all Mbnl1 transcripts
in neonatal and adult WT or HSALR muscle (not shown). Exon 9 (E9) was alternatively spliced
in muscle, but the ratio of E9+:E9– splice products was similar in neonatal and adult WT or
HSALR muscle (not shown).
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

12
In contrast, inclusion of exon 7 (E7) was increased in HSALR and Mbnl1∆E3/∆E3 mice, and
alternative splicing of this exon was developmentally regulated in WT mice (Fig. 2). The
function of the 18 amino acid domain encoded by this small exon is unknown. However, E7
and 156 nt of the flanking intronic sequence are ultraconserved regions in vertebrate genomes
(30), suggesting that regulation of E7 splicing is functionally important. Also, the analogous
exon of Mbnl2 shows similar dysregulation in DM (Suppl. Fig. 1). E7– and E7+ Mbnl1 both
retain their splicing and RNA binding activities (refs. (21) and (24), and M. Swanson,
unpublished observations). We postulated that postnatal splicing transitions may result from
preferentially localization of E7+ Mbnl1 to the muscle cytoplasm. We examined this possibility
by using electroporation to express different GFP-tagged Mnbl1 isoforms in skeletal muscle in
vivo. Against our prediction, however, we found that nuclear localization was stronger for E7+
than E7– Mbnl1 (Fig. 6D). Results in transfected COS cells were similar, with or without the
inclusion of exon 9 (Supplemental Fig. 2D). Thus, a functional depletion of MBNL1 has
occurred in DM1 nucleoplasm, despite a shift to production of E7+ isoforms that preferentially
localize to the nucleus. Taken together, these results raise the possibility that the postnatal
splicing transitions are triggered, at least in part, by translocation of Mbnl1 from the cytoplasm
to nucleus. However, alternative splicing of Mbnl is not the main factor driving this
redistribution.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

13
Discussion
The disease process in myotonic dystrophy involves trans-interference by repeat-containing
transcripts with regulated alternative splicing of select pre-mRNAs (6). Current evidence
indicates that misregulated splicing results from abnormal activity of poly(CUG)/poly(CCUG)
binding proteins. The first RNA binding protein shown to interact with poly(CUG) was CUG-
BP1 (12), but the link between this protein and DM-related splicing abnormalities remains
unclear. Despite the accumulation of expanded poly(CUG) to high levels in HSALR muscle, we
did not see colocalization of endogenous or GFP-tagged CUG-BP1 with RNA foci (Fig. 5),
consistent with previous observations that this protein is not sequestered in ribonuclear foci in
DM1 cells (13,14). One potential explanation for the non-colocalization of CUG-BP1 is that its
affinity for expanded poly(CUG) is lower than that of MBNL1 (17), so that colocalization is
difficult to observe due to occupancy of binding sites by Mbnl1. However, this cannot be the
sole explanation, because CUG-BP1 also does not colocalize when expanded poly(CUG) RNA is
expressed in Mbnl1 knockout mice (X Lin, C Thornton, unpublished). Furthermore,
misregulated alternative splicing has occurred in HSALR transgenic mice and DM2 patients
without any consistent effect on the levels of CUG-BP1 protein in skeletal muscle (Fig. 5).
These results suggest that perturbations of CUG-BP1 distribution or amount are not required to
produce the defect of splicing regulation in DM. These results are consistent with findings that
siRNA-mediated depletion of CUG-BP1 failed to restore normal patterns of alternative splicing
in DM1 myoblasts (20).
By contrast, we found that splicing derangements in Mbnl1 deficient muscle are remarkably
concordant with those induced by CUG- or CCUG-repeat expansion RNA, whether in human
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

14
DM1, DM2, or the HSALR transgenic mouse model. These observations, when coupled with
evidence for depletion of MBNL1 from the nucleoplasm in both types of human DM (Fig. 4),
argue that sequestration of a single splicing factor, MBNL1, is sufficient to explain misregulated
splicing in adult DM skeletal muscle. A similar conclusion was reached in studies of siRNA-
mediated knockdown of MBNL1, MBNL2, or CUG-BP1, individually or in combination, in
myoblasts (20). In the previous studies, knockdown of either MBNL1 or MBNL2 repressed
splicing of IR exon 11 in WT myoblasts, suggesting that IR splicing in myoblasts was
determined by combined action of both MBNL proteins (MBNL3 was not examined because its
expression is mainly restricted to placenta). However, in mature skeletal muscle, we found that
the full extent of poly(CUG)-induced splicing abnormalities, in terms of magnitude and range of
exons affected, was reproduced by loss of Mbnl1 alone, whereas splicing of these same exons in
Mbnl2GT4/GT4 mice remained normal. It is possible that Mbnl2 acts mainly during earlier stages
of muscle development, or that its main function in mature tissue is at the level of cytoplasmic
localization or decay of mRNA (31). Further studies are needed to determine the combined
effects of Mbnl1 and Mbnl2 loss in vivo.
The extent of MBNL1 accumulation in ribonuclear foci was greater in DM2 than in DM1. This
extends our previous finding that levels of repeat expansion RNA in ribonuclear foci are greater
in DM2 than DM1, which likely reflects greater expansion and higher expression of the ZNF9
repeat (15). Together, these results indicate that capacity for MBNL1 sequestration by an
intronic CCUG repeat in DM2 is no less than an exonic CUG repeat in DM1. Despite greater
severity of the muscle degeneration in DM1, our results indicate that depletion of MBNL1 from
nucleoplasm is more extensive in DM2. Thus, while sequestration of MBNL1 evidently has a
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

15
central role in splicing misregulation in both types of DM, it appears likely that CUG-expanded
DMPK mRNA has additional pathogenic consequences in DM1, not shared by CCUG-expanded
ZNF9 RNA or mediated through MBNL1, that lead to greater muscle wasting.
Muscle fibers form during prenatal development but are extensively remodeled after birth. In
rodents, the first three weeks of postnatal life are associated with pruning and stabilization of the
neuromuscular junction, formation of the T tubule system, maturation of excitation-contraction
coupling and the sarcoplasmic reticulum, and restructuring of the sarcomere (32,33). Postnatal
remodeling may require activation of transcriptional programs that were not initially engaged
during muscle differentiation. However, a recent study in cardiac muscle has emphasized the
importance of post-transcriptional regulation, and shown that alternative splicing plays an
essential role in postnatal remodeling. Conditional knockout of splicing factor ASF/SF2 in
cardiac muscle led to failure of a specific set of postnatal alternative splicing transitions (34). In
mice having ASF/SF2 deficiency in heart, persistent expression of neonatal splice isoforms was
closely related to the subsequent development of progressive cardiomyopathy. Thus, a set of
splicing switches during a critical period of postnatal remodeling were required for normal
cardiac function in adults, and these transitions were controlled by a specific splicing factor.
Our results suggest that MBNL1 is such a factor in skeletal muscle, and that symptoms of DM
result, at least in part, from failure to execute or maintain a set of postnatal splicing transitions.
Strikingly, every exon shown to undergo misregulated splicing in DM muscle showed a similar
pattern of developmental regulation between P2 and P20 in mice (Fig. 2). Mbnl1 was required
for these physiological splicing transitions, and the timing of these transitions may depend upon
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

16
translocation of Mbnl1 from the cytoplasm to nucleus, postnatal downregulation of CUG-BP1
(28), or both. The mechanism that controls Mbnl1 translocation to the nucleus has not been
determined, although its presence in the HSALR nucleus at P2 suggests that Mbnl1 shuttles
between cytoplasm and nucleus and has become trapped on the ribonuclear foci. While nuclear
localization is more likely for the E7 inclusion isoforms, alternative splicing is not the main
factor driving the postnatal relocalization of Mbnl1. As yet we have not identified a
posttranslational modification of Mbnl1 that determines its cellular distribution.
The transcripts that undergo Mbnl1-dependent splicing transitions in skeletal muscle encode
proteins involved in excitation and contraction (Serca1, ClC-1, RyR); sarcomere structure (ZASP,
Tnnt2, Tnnt3), and signaling (IR, MTMR1). In the case of ClC-1, the functional implications are
clear: neonatal muscles produce a transcript that encodes a truncated, nonfunctional chloride
channel protein. Reversion to this splicing outcome in adult DM leads to loss of ClC-1 channels
and hyperexcitable muscle fibers (8,9). In the case of other transcripts, the functional
consequences of misregulated splicing are less obvious, but they are likely to impact multiple
pathways. Cumulatively, our results suggest that the complex phenotype of DM may derive, to
a surprising extent, from a single RNA-protein interaction: the recognition of poly(CUG) or
poly(CCUG) RNA by MBNL1. Selective disruption of this interaction may prove to be a
pharmacologically tractable approach for reversing the functional impairments of DM.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

17
Materials and Methods
Mice. Transgenic mice in HSALR line 20b were maintained as homozygotes on an FVB inbred
background (4). Mbnl1∆E3/∆E3, Mbnl1∆E3/+, and Mbnl+/+ littermates were maintained on a mixed
C57Bl6 • SV129 background (25). adr-mto2J (ClC-1 null mutant) mice with recessive
generalized myotonia were obtained from Jackson Laboratories (Bar Harbor, ME). Hindlimb
muscle from adult mdx mice was kindly provided by Dr Paula Clemens, University of Pittsburgh.
Mice were maintained according to guidelines of the Association for Assessment and
Accreditation of Laboratory Animal Care.
DM muscle tissue. Needle biopsies of vastus lateralis muscle were obtained from patients with
genetically confirmed DM1, DM2, or fascioscapulohumeral (FSH) muscular dystrophy, or
healthy volunteers. All study subjects gave informed consent under protocols approved by the
University of Rochester Human Subjects Review Board. DM1 quadriceps muscle tissue also
was obtained at autopsy. Muscle tissue was flash frozen in liquid nitrogen and stored at -70°C.
Mbnl2 genetrap mice. Clone XB595, derived by integration of genetrap vector pGTOpfs in
sv129/Ola ES cells, was obtained from the Mutant Mouse Regional Resource Center (Davis,
CA). The approximate site of pGTOpfs integration in Mbnl2 was confirmed by sequencing the
Mbnl2-pGTOpfs fusion cDNA, obtained from XB595 cells by RT-PCR. C57Bl6 blastocysts
were injected with XB595 cells and implanted into pseudopregnant females. Chimeric progeny
were bred with C57Bl6 mice, and the agouti offspring were screened by PCR. The exact site of
vector integration in Mbnl2 intron 4, and the integrity of the flanking exons, was determined by
cloning and sequencing the integration site from genomic DNA obtained from homozygous
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

18
Mbnl2GT4/GT4 mice. Splicing of Mbnl2 exon 4 to the engrailed splice acceptor in pGTOpfs was
confirmed in RT-PCR products obtained from Mbnl2GT4/GT4 muscle. The Mbnl2GT4 allele was
maintained on a mixed C57Bl6/129 background. Reduction of Mbnl2 mRNA in skeletal muscle
of Mbnl2GT4/GT4 homozygotes, as compared to WT littermates, was confirmed by Northern blot
using 1.5 ug of polyadenylated RNA probed with a 500 bp fragment of the Mbnl2 3' UTR (nt
2292 – 2793, accession # NM_175341).
Antibodies. CUG-BP1 was detected using monoclonal antibody 3B1 (12). Polyclonal antibody
A2764 directed against the C-terminus of Mbnl1 was raised by immunization of rabbits with
peptide PIISAEHLTSHKYVTQM conjugated to KLH carrier. This sequence is identical in
human and mouse Mbnl1 but not conserved in other Mbnl proteins. Other antibodies used were
anti-GADPH mouse monoclonal (Biogenesis, Brentwood, NH), anti-hnRNP H rabbit polyclonal
(J Wilusz, Ft. Collins, CO), anti-hnRNP I (M Garcia-Blanco, Durham, NC), and secondary
antibodies conjugated with Alexa 488, Alexa 680, or horse radish peroxidase (HRP) (Invitrogen
Molecular Probes, Carlsbad, CA).
RT-PCR analysis of alternative splicing. Total cellular RNA was extracted from tissue using
Tri-reagent (Molecular Research Center). For fetal and neonatal mice, RNA was extracted from
hindlimb muscle. For adult mice, RNA was extracted from vastus (quadriceps) muscle.
Alternative splicing in DM1 muscle was assessed using autopsy samples, because myopathic
abnormality and misregulated splicing is more pronounced than in biopsy tissue (8). All other
analyses of human splicing or tissue sections were performed on quadriceps biopsy samples.
The genes and exons selected for alternative splicing analysis are listed in Supplemental Table 1.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

19
We selected 54 exons based on (1) previous evidence for alternative splicing in skeletal muscle;
and (2) splicing patterns that were suitable for RT-PCR analysis (alternative exon cassettes or
alternative 5' or 3' splice sites). Primers for these assays are listed in Supplemental Table 2.
RT-PCR splicing assays were carried out using procedures we have previously described (8). In
brief, cDNA synthesis was primed with oligo(dT) (mouse) or oligo(dT) plus random hexamers
(human). All cDNAs were treated with RNase H at 37°C for 35 min. Primers were selected to
give a length difference of >10% and <25% for exon inclusion versus exon exclusion products.
PCR amplification was carried out for 20-24 cycles, within the linear range of amplification for
each gene. PCR products were resolved on agarose gels, stained with SybrGreenII, and scanned
on a laser fluorimager. The density of each band was quantified using ImageQuant software.
Because of the low variability of the RT-PCR splicing assays in inbred mice (within-group
coefficient of variation less than 10%), the initial analysis to identify splicing differences was
carried out on muscle RNA from individual mice in each of the following categories (ages):
wild-type FVB (day 2 and 6 months), HSALR (6 months), and Mbnl1∆E3/∆E3, Mbnl1∆E3/+, or
Mbnl1+/+ littermates (6 months). Apparent differences in alternative splicing were confirmed
and quantified in an independent group of HSALR and WT mice (n = 6 per group, age 6 months).
FISH (fluorescence in situ hybridization) and IF (immunofluorescence). FISH combined with
IF was performed on frozen sections of quadriceps muscle as previously described (22). The
probe for FISH was a CAG-repeat 2-O-methyl oligoribonucleotide, 20 nt in length, conjugated at
the 3' end with Texas Red.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

20
IF for nuclear Mbnl1. Frozen sections of human or mouse muscle were fixed in 3%
paraformaldehyde for 15 min at room temperature, permeabilized in 2% pre-chilled acetone for 5
min and then soaked in primary antibody (A2764 at 1:10,000) at 4°C overnight. After washing
with PBS, sections were stained with secondary antibody and DAPI at 20°C for 30min. For
quantification of nuclear Mbnl1 in human biopsy samples, sections were stained on the same
slide, imaged at 1000X magnification under identical illumination and exposure settings, and
analyzed using uniform threshold settings. A z-plane stack (15 images, 0.25 μm steps) centered
on the region of maximal MBNL1 staining was deconvolved using Autoquant software v9.3
(Watervliet, NY). The plane with maximum signal intensity was selected for quantification.
The nucleus (defined by DAPI staining), nucleoli, and foci were outlined manually. To estimate
the amount of MBNL1 in foci, the IF signal (area x intensity) in nuclear foci was determined
using Metaview software (Universal Imaging Corp., Downington, PA). We also used Metaview
software to determine the average MBNL1 signal intensity in the nucleoplasm, exclusive of
nuclear foci. Multiple nuclei (42 to 65) were examined in each of the following groups (number
of individuals per group): DM1 (2), DM2 (3), healthy controls (3), FSH (2).
Immunoblot. Mouse quadriceps muscle was pulverized under liquid nitrogen and then
homogenized in lysis buffer (10mM Tris pH 7.6, 2% SDS, 2mM DTT, 1mM PMSF, 2mM
Benzamidine and 1x Protease Arrest). For human biopsy muscles, ten consecutive 10 µm
frozen sections were homogenized in the same lysis buffer. Equal amounts of protein were
resolved on 10% SDS-PAGE gels and then transferred to nitrocellulose (Bio-Rad Laboratories).
After incubation with primary antibody at 4°C overnight, membranes were incubated with
secondary antibodies at room temperature for 1 h. The primary antibodies were anti-MBNL1
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

21
polyclonal A2764 (1:10000), anti-CUG-BP1 mouse monoclonal 3B1 (1:500), or anti-GADPH
mouse monoclonal (1:10,000). Secondary antibodies were conjugated either with HRP or Alexa
680.
Localization of Mbnl1 and CUG-BP1 fusion proteins. MBNL1 cDNAs, including all four
possible combinations of exon 7 or 9 inclusion/exclusion, were cloned in pEGFP-C1 (Clontech)
and verified by sequencing. cDNA constructs were expressed in vivo by electroporation of
muscle tissue. Under general anesthesia, tibialis anterior muscle was prepared for
electroporation by injection of hyaluronidase (25 µl of 0.4 U/µl). Two hours later, 15µl of
pEGFP-CUG-BP1 or pEGFP-MBNL1-41 plasmid (3 µg/µl in normal saline) were injected into
the pretreated muscle followed by electrical field stimulation. The voltage used for
electrotransfer was 175V/cm in eight 20ms square wave pulses at 1 sec intervals (Electro Square
Porator ECM 830, BTX A Division of Genetronics, Inc.). Electrode jelly was applied on the 7
mm circular electrodes to ensure good electrical contact. Muscle was dissected five to seven
days after electroporation and prepared for frozen sectioning. The subcellular localization of
EGFP-Mbnl1 fusion proteins also was determined in COS7 cells by transfection of cDNA
expression constructs using SuperFect (Qiagen, Valencia, CA). Cells were observed 1 or 2 days
following transfection.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

22
Acknowledgements
The authors thank Matt Krym, Don Henderson, and Bharati Shah for technical assistance and Dr
Thurman Wheeler for assistance with electroporation of skeletal muscle. This work comes from
the University of Rochester Senator Paul D. Wellstone Muscular Dystrophy Cooperative
Research Center (NIH/NS48843) and Wayne C. Gorell Jr. Laboratory with support from
NIH/NIAMS (AR46806, AR48143), the Muscular Dystrophy Association, and the Saunders
Family Neuromuscular Research Fund. Dr Thornton is supported by a Mid-Career Investigator
Award in Patient-Oriented Research (AR48143). Patients were enrolled in the study with the
assistance from the National Registry of Myotonic Dystrophy and Facioscapulohumeral
Dystrophy Patients and Family Members (AR02250).
Conflicts of Interest
No conflict of interest to report.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

23
References
1. Brook,J.D., McCurrach,M.E., Harley,H.G., Buckler,A.J., Church,D., Aburatani,H.,
Hunter,K., Stanton,V.P., Thirion,J.P., Hudson,T., et al. (1992) Molecular basis of
myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a
transcript encoding a protein kinase family member. Cell, 68, 799-808.
2. Liquori,C.L., Ricker,K., Moseley,M.L., Jacobsen,J.F., Kress,W., Naylor,S.L., Day,J.W.,
and Ranum,L.P. (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in
intron 1 of ZNF9. Science, 293, 864-867.
3. Seznec,H., Agbulut,O., Sergeant,N., Savouret,C., Ghestem,A., Tabti,N., Willer,J.C.,
Ourth,L., Duros,C., Brisson,E., et al. (2001) Mice transgenic for the human myotonic
dystrophy region with expanded CTG repeats display muscular and brain abnormalities.
Hum.Mol.Genet., 10, 2717-2726.
4. Mankodi,A., Logigian,E., Callahan,L., McClain,C., White,R., Henderson,D., Krym,M.,
and Thornton,C.A. (2000) Myotonic dystrophy in transgenic mice expressing an
expanded CUG repeat. Science, 289, 1769-1773.
5. Taneja,K.L., McCurrach,M., Schalling,M., Housman,D., and Singer,R.H. (1995) Foci of
trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J.Cell
Biol., 128, 995-1002.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

24
6. Philips,A.V., Timchenko,L.T., and Cooper,T.A. (1998) Disruption of splicing regulated
by a CUG-binding protein in myotonic dystrophy. Science, 280, 737-741.
7. Savkur,R.S., Philips,A.V., and Cooper,T.A. (2001) Aberrant regulation of insulin
receptor alternative splicing is associated with insulin resistance in myotonic dystrophy.
Nat.Genet., 29, 40-47.
8. Mankodi,A., Takahashi,M.P., Jiang,H., Beck,C.L., Bowers,W.J., Moxley,R.T.,
Cannon,S.C., and Thornton,C.A. (2002) Expanded CUG repeats trigger aberrant splicing
of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in
myotonic dystrophy. Mol.Cell, 35-44.
9. Charlet-B,N., Savkur,R.S., Singh,G., Philips,A.V., Grice,E.A., and Cooper,T.A. (2002)
Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to
misregulated alternative splicing. Mol.Cell, 10, 45-53.
10. Takahashi,N., Sasagawa,N., Suzuki,K., and Ishiura,S. (2000) The CUG-binding protein
binds specifically to UG dinucleotide repeats in a yeast three-hybrid system.
Biochem.Biophys.Res.Commun., 277, 518-523.
11. Faustino,N.A., Cooper,T.A. (2005) Identification of putative new splicing targets for
ETR-3 using sequences identified by systematic evolution of ligands by exponential
enrichment. Mol.Cell Biol., 25, 879-887.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

25
12. Timchenko,L.T., Miller,J.W., Timchenko,N.A.X.D.D., Datar,K.V., Lin,L., Roberts,R.,
Caskey,C.T., and Swanson,M.S. (1996) Identification of a (CUG)n triplet repeat RNA-
binding protein and its expression in myotonic dystrophy. Nucleic Acids Res., 24, 4407-
4414.
13. Miller,J.W., Urbinati,C.R., Teng-umnuay,P., Stenberg,M.G., Byrne,B.J., Thornton,C.A.,
and Swanson,M.S. (2000) Recruitment of human muscleblind proteins to (CUG)(n)
expansions associated with myotonic dystrophy. EMBO J., 19, 4439-4448.
14. Fardaei,M., Larkin,K., Brook,J.D., and Hamshere,M.G. (2001) In vivo co-localisation of
MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res., 29, 2766-
2771.
15. Mankodi,A., Teng-umnuay,P., Krym,M., Henderson,D., Swanson,M., and Thornton,C.A.
(2003) Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2.
Ann.Neurol., 54, 760-768.
16. Napierala,M., Krzyzosiak,W.J. (1997) CUG repeats present in myotonin kinase RNA
form metastable slippery hairpins. J.Biol.Chem., 272, 31079-31085.
17. Michalowski,S., Miller,J.W., Urbinati,C.R., Paliouras,M., Swanson,M.S., and Griffith,J.
(1999) Visualization of double-stranded RNAs from the myotonic dystrophy protein
kinase gene and interactions with CUG-binding protein. Nucleic Acids Res., 27, 3534-
3542.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

26
18. Tian,B., White,R., Xia,T., Welle,S., Turner,D., Mathews,M., and Thornton,C. (2000)
Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-
dependent protein kinase PKR. RNA, 6, 79-87.
19. Timchenko,N.A., Cai,Z.J., Welm,A.L., Reddy,S., Ashizawa,T., and Timchenko,L.T.
(2001) RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of
CUGBP1. J.Biol.Chem., 276, 7820-7826.
20. Dansithong,W., Paul,S., Comai,L., and Reddy,S. (2005) MBNL1 is the primary
determinant of focus formation and aberrant insulin receptor splicing in DM1.
J.Biol.Chem., 280, 5773-5780.
21. Kino,Y., Mori,D., Oma,Y., Takeshita,Y., Sasagawa,N., and Ishiura,S. (2004)
Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats.
Hum.Mol.Genet., 13, 495-507.
22. Mankodi,A., Urbinati,C.R., Yuan,Q.P., Moxley,R.T., Sansone,V., Krym,M.,
Henderson,D., Schalling,M., Swanson,M.S., and Thornton,C.A. (2001) Muscleblind
localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2.
Hum.Mol.Genet., 10, 2165-2170.
23. Fardaei,M., Rogers,M.T., Thorpe,H.M., Larkin,K., Hamshere,M.G., Harper,P.S., and
Brook,J.D. (2002) Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

27
nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum.Mol.Genet., 11,
805-814.
24. Ho,T.H., Charlet,B., Poulos,M.G., Singh,G., Swanson,M.S., and Cooper,T.A. (2004)
Muscleblind proteins regulate alternative splicing. EMBO J., 23, 3103-3112.
25. Kanadia,R.N., Johnstone,K.A., Mankodi,A., Lungu,C., Thornton,C.A., Esson,D.,
Timmers,A.M., Hauswirth,W.W., and Swanson,M.S. (2003) A muscleblind knockout
model for myotonic dystrophy. Science, 302, 1978-1980.
26. Kimura,T., Nakamori,M., Lueck,J.D., Pouliquin,P., Aoike,F., Fujimura,H., Dirksen,R.T.,
Takahashi,M.P., Dulhunty,A.F., and Sakoda,S. (2005) Altered mRNA splicing of the
skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-
ATPase in myotonic dystrophy type 1. Hum.Mol.Genet., 14, 2189-2200.
27. Ladd,A.N., Charlet,N., and Cooper,T.A. (2001) The CELF family of RNA binding
proteins is implicated in cell-specific and developmentally regulated alternative splicing.
Mol.Cell Biol., 21, 1285-1296.
28. Ladd,A.N., Stenberg,M.G., Swanson,M.S., and Cooper,T.A. (2005) Dynamic balance
between activation and repression regulates pre-mRNA alternative splicing during heart
development. Dev.Dyn., 233, 783-793.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

28
29. Edelman,J.C., Edelman,P.M., Kniggee,K.M., and Schwartz,I.L. (1965) Isolation of
skeletal muscle nuclei. J.Cell Biol., 27, 365-377.
30. Bejerano,G., Pheasant,M., Makunin,I., Stephen,S., Kent,W.J., Mattick,J.S., and
Haussler,D. (2004) Ultraconserved elements in the human genome. Science, 304, 1321-
1325.
31. Adereth,Y., Dammai,V., Kose,N., Li,R., and Hsu,T. (2005) RNA-dependent integrin
alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat.Cell
Biol., 7, 1140-1147.
32. Luff,A.R., Atwood,H.L. (1971) Changes in the sarcoplasmic reticulum and transverse
tubular system of fast and slow skeletal muscles of the mouse during postnatal
development. J Cell Biol, 51, 369-383.
33. Slater,C.R. (1982) Postnatal maturation of nerve-muscle junctions in hindlimb muscles of
the mouse. Dev.Biol., 94, 11-22.
34. Xu,X., Yang,D., Ding,J.H., Wang,W., Chu,P.H., Dalton,N.D., Wang,H.Y.,
Bermingham,J.R., Jr., Ye,Z., Liu,F., et al. (2005) ASF/SF2-regulated CaMKIIdelta
alternative splicing temporally reprograms excitation-contraction coupling in cardiac
muscle. Cell, 120, 59-72.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

29
Legends to Figures
Figure 1. Patterns of misregulated alternative splicing in HSALR transgenic and Mbnl1 knockout
mice recapitulate the patterns observed in WT mice at postnatal day 2 (P2) A. In WT mice,
RT-PCR analysis of fast skeletal muscle troponin T (Tnnt3) mRNA showed alternative splicing
of the fetal (F) exon as well as exons 4 through 8. Transition from inclusion to exclusion of the
fetal exon occurred mainly between P2 and P16. Lower panel shows RT-PCR products after
digestion with BsrBI, which cleaved within the fetal exon. B. Compared to WT adult mice of
the same age and strain background, HSALR transgenic and Mbnl1∆E3/ ∆E3 mice had reduced
inclusion of Serca1 exon 22 and increased inclusion of Zasp exon 11, m-line region of titin exon
5 (Mex5), and Mbnl1 exon 7. For each of these exons, the pattern of splicing in DM1 models
was similar to neonatal WT mice at P2. C. Effects on alternative splicing of Serca1 and Zasp
did not occur in adr (ClC-1 null) mice that had severe myotonia or in mdx mice that had
dystrophin deficiency.
Figure 2. Selective failure of Mbnl1-dependent, postnatal splicing transitions in HSALR
transgenic mice. Alternative splicing of Serca1, Zasp, m-Titin, Mbnl1, Itgb1, and CapZb was
developmentally regulated in the interval between P2 and P20. Postnatal splicing transitions for
Serca1, Zasp, and m-Titin failed in Mbnl1∆E3/ ∆E3 mice, whereas transitions for Itgb1 and CapZb
occurred on schedule. Postnatal downregulation of Mbnl1 exon 7 also failed in Mbnl1 knockout
mice, indicating that splicing of this exon was autoregulated. HSALR transgenic mice showed
selective failure of the splicing transitions that were Mbnl1-dependent (Serca1, Zasp, m-Titin,
Mbnl1). Quantification of splicing shows the mean ± standard deviation for fractional inclusion
or exclusion of the specified exon in triplicate assays. Ad indicates adult mice of age 6 months.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

30
Figure 3. Alternative splicing in mice homozygous for the Mbnl2 genetrap (GT) allele. A. The
gene trap vector is a promoterless construct containing a splice acceptor (SA) site from
engrailed-2, the coding sequence for β-galactosidase fused to neomycin phosphotransferase (β-
Geo), and the SV40 poly-adenylation (pA) signal. In the Mbnl2GT4 allele, the gene trap vector is
integrated in the middle of Mbnl2 intron 4. Transcripts from the Mbnl2GT4 allele encode the N-
terminal and two C3H zinc fingers of Mbnl2, fused in-frame to β-Geo. B. Northern blot of
polyadenylated muscle RNA. Probe from the 3' untranslated region of Mbnl2 shows loss of the
normal Mbnl2 transcript in homozygous Mbnl2GT4/GT4 mice. C. Alternative splicing of indicated
exons in Serca1, Zasp, m-titin, and Mbnl1 is normally regulated in homozygous Mbnl2GT4/GT4
mice.
Figure 4. Splicing defect and nuclear sequestration of MBNL1 protein in human DM1 and
DM2. A. SERCA1, ZASP, m-Titin, and MBNL1 alternative splicing in human DM1 and DM2
skeletal muscle showed abnormal regulation similar to that observed in HSALR transgenic and
Mbnl1 knockout mice. The respective ages of subjects are 47, 55, and 50 years for DM1; 68, 23,
and 24 years for healthy volunteers; and 36, 46, and 33 years for DM2. B. Distribution of
MBNL1 (green) in the nucleus (blue) shown in deconvolved images by immunofluorescence
using anti-MBNL1 polyclonal antibody A2764 on frozen sections of quadriceps biopsy tissue.
In contrast to HSALR transgenic mice (see Fig. 6C), the CUG expansion RNA in DM muscle
nuclei was typically consolidated in a single nuclear focus (see also Supplemental Fig. 2A). In
sections stained on same slide and imaged under the same exposure and threshold settings, the
intensity of MBNL1 staining in nucleoplasm (NP), exclusive of nuclear foci, was reduced in
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

31
DM1 and DM2 compared to healthy controls and patients with fascioscapulohumeral (FSH)
muscular dystrophy. For quantification of MBNL1 in nucleoplasm, the mean
immunofluorescence intensity in the nucleus, exclusive of nuclear foci, was determined on
deconvolved images. The MBNL1 signal within nuclear foci (area x intensity) was 3-fold greater
in DM2 than DM1. Bars indicate mean ± standard deviation. 1000x magnification, bar
indicates 5 µm, sample # indicates number of individuals examined in each category.
Figure 5. Developmental regulation and cellular distribution of CUG-BP1 in skeletal muscle.
A-C. Immunoblot of muscle lysates using anti-CUG-BP1 monoclonal antibody 3B1, with
GAPDH serving as loading control. A. Postnatal downregulation of CUG-BP1 occurred to a
similar extent in hindlimb muscle from WT and HSALR transgenic mice. B. CUG-BP1 protein
did not show a consistent increase in adult skeletal muscle from HSALR transgenic mice as
compared to gender- and age-matched WT mice (representative results from 4 independent
experiments). C. CUG-BP1 protein did not show a consistent increase in quadriceps biopsy
tissue from DM2 patients compared to healthy subjects. D-F. FISH detection of CUG
expansion RNA (left panels, red) combined with fluorescence detection of RNA-binding proteins
(green, center panels) in muscle nuclei (blue, right panels) from HSALR transgenic mice. D.
Endogenous CUG-BP1, revealed by immunofluorescence with antibody 3B1, did not colocalize
with nuclear foci of CUG expansion RNA. GFP-tagged CUG-BP1 also did not colocalize with
nuclear foci of CUG expansion RNA, when expressed by electroporation of skeletal muscle in
vivo (E), whereas MBNL1-GFP did colocalize under these same conditions (F). 1000x
magnification, bar indicates 5 µm.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

32
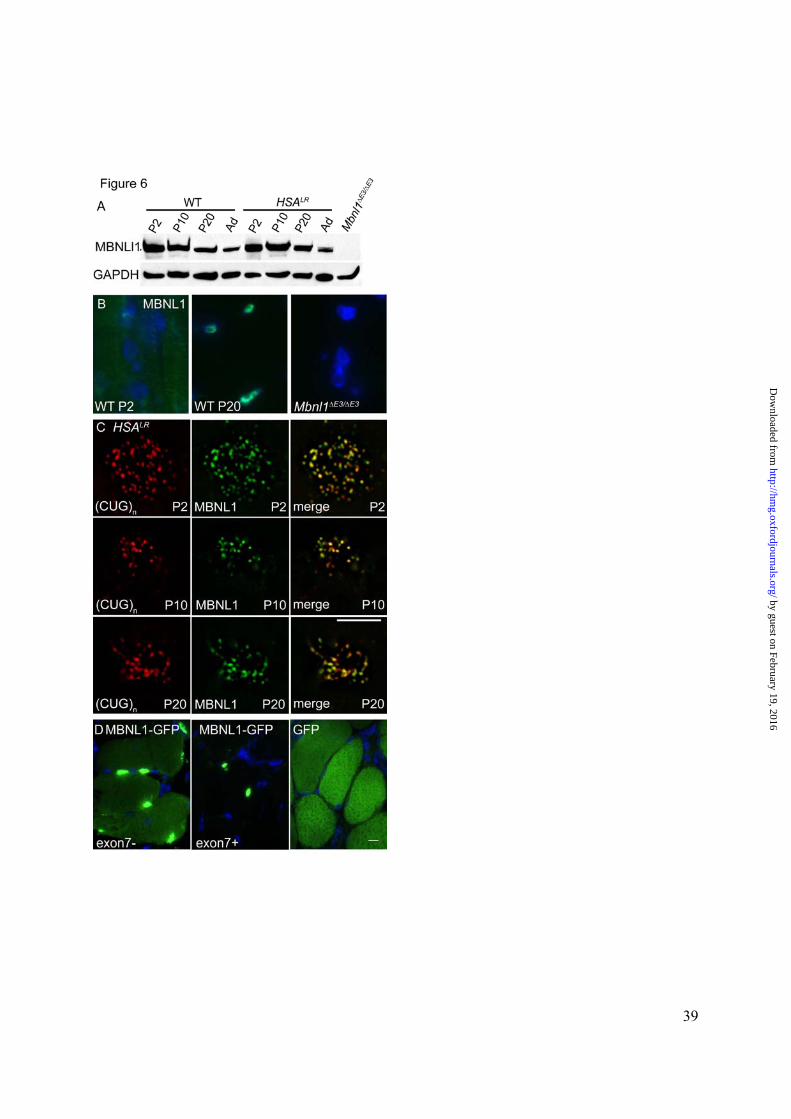
Figure 6. Developmental regulation and cellular distribution of Mbnl1 protein in murine
skeletal muscle. A-B. Immunodetection using polyclonal antibody A2764, directed against a
C-terminal peptide of Mbnl1. Specificity of antibody A2764 was demonstrated by lack of
reactivity on immunoblots (A) or tissue sections (B) from Mbnl1∆E3/ ∆E3 mice. A. Immunoblot
of hindlimb muscle showed a slight postnatal reduction of Mbnl1 protein in WT and HSALR
transgenic mice. Levels of Mbnl1 in adult (Ad) muscle were similar in WT and HSALR
transgenic mice. Note that alternative splice isoforms of Mbnl1, including exon 7 exclusion (40
kD), exon 7 inclusion (41kD), and exons 7 + 9 inclusion (42 kD) isoforms, were not resolved on
these gels, but all were recognized by antibody A2764. B. On frozen sections of WT muscle
stained on the same slide and imaged under the same exposure settings, the distribution Mbnl1
was predominantly cytoplasmic at P2 and predominantly nuclear at P20. C. High power views
(1000X) of individual myonuclei showing FISH detection of nuclear foci of CUG expansion
RNA (left panels, red) combined with immunofluorescence detection of Mbnl1 (green, center
panels) in muscle nuclei (blue, right panels) from HSALR transgenic mice. In the transgenic
model, Mbnl1 was not excluded from the nucleus in neonatal mice, as indicated by its presence
in nuclear foci as early as P2. D. Distribution of Mbnl1-GFP fusion protein in vivo after
electroporation of expression construct in WT vastus muscle. As compared to the cytoplasmic
distribution of GFP, the exon 7 exclusion isoform of Mbnl1-GFP was distributed to cytoplasm
and nucleus (left panel), whereas the exon 7 inclusion isoform localized entirely to the nucleus
(middle panel). Scare bar indicates 5 µm in B and C, 10 µm in D.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

33
Aberrant splicing in DM Cmouse Dhuman
E% neonatal isoform
Gene
accession #
Alt. spliced exon #
ANeonatalisoform
BPost-natal
splicing switch HSALR Mbnl1-/- DM1 DM2 WT HSALR
Serca1 NM_007504 22 exon22- + + + + + 3±0.7 78±3.8
ZASP AY206013 11 exon11+ + + + + + 11±1.6 66±6.3
z-Ttn NT_039207.3 Zr4 Zr4+5+ + + + + + 21±3.7 65±3.2
“ XM_130322 Zr5
m-Ttn XM_130312 Mex5 Mex5+ + + + + + 31±1.2 86±5.4
Nrap AY177622 12 exon12- + + + + + 34±6 59±4
Capn3 X92523 16 exon16- + + + + + 5±0.9 15±0.7
Alp AF002283 5a exon5a+5b- + + + + + 4±0.5 8±0.8
“ NT_039460.3 5b
Fhos NT_078575.3 11a exon11a- + + + + + 6±0.9 18±2
Gfat1 AF334736 10 exon10- + + + + - 7±2.3 22±3.0
Mbnl1 BC060031 7 exon7+ + + + + + 8±0.7 42±2.4 Mbnl2 NM_175341 7 exon7+ + + + + + 31±0.01 41±0.01
Table1. Misregulated alternative splicing in mouse models of DM1 was concordant with human
DM1 and DM2. A indicates the isoform that is preferentially expressed in neonatal muscle at P2
compared to adult WT muscle. B “+” denotes exons that show postnatal splicing transition
between P2 and P20 in WT hindlimb muscle. C “+” denotes exons that show misregulated
alternative splicing in adult (6 month) HSALR transgenic or Mbnl1-/- mice, compared to WT mice
of appropriate background strain. D “+” denotes exons that show misregulated alternative
splicing in quadriceps muscle from DM1 or DM2 patients compared to healthy individuals. E
fraction of splice products, expressed as the % of isoform that was preferentially expressed in
WT neonatal muscle, mean ±S.D for n = 6 per group, see also Suppl. Fig. 1.
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

34
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

35
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

36
Fig 3
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

37
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

38
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from
39
by guest on February 19, 2016http://hm
g.oxfordjournals.org/D
ownloaded from





























