Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1 Riccardo Perbellini a,b , Simona Greco a , Gianluca Sarra-Ferraris a , Rosanna Cardani c,d , Maurizio C. Capogrossi e , Giovanni Meola f , Fabio Martelli e,⇑ a Molecular Cardiology Laboratory, IRCCS-Policlinico San Donato, 20097 Milan, Italy b University of Milan-Hospital “L.Sacco”, Department of Clinical Sciences, 20157 Milan, Italy c Department of Biomolecular Sciences and Biotechnology, University of Milan, 20097 Milan, Italy d CMN-Neuromuscular Disease Center, IRCCS Policlinico San Donato, University of Milan, Milan, Italy e Laboratorio Patologia Vascolare, Istituto Dermopatico dell’Immacolata-IRCCS, 00167 Rome, Italy f University of Milan-IRCCS Policlinico San Donato, Department of Neurology, 20097 Milan, Italy Received 20 September 2010; received in revised form 15 November 2010; accepted 22 November 2010 Abstract Myotonic Dystrophy Type-1 (DM1) is caused by the expansion of a CTG repeat with a peculiar pattern of multisystemic involvement affecting skeletal muscles, the heart, the eye, the central nervous system and the endocrine system. Since microRNA expression is dis- rupted in several myopathies, the expression of 24 candidate microRNAs was analyzed in skeletal muscle biopsies of 15 DM1 patients. Controls were constituted by biopsies without overt pathological features derived from 14 subjects with suspected neuromuscular disor- der of undetermined nature. We found that miR-1 and miR-335 were up-regulated, whereas miR-29b and c, and miR-33 were down- regulated in DM1 biopsies compared to controls. We also found that the cellular distribution of muscle specific miR-1, miR-133b and miR-206 was severely altered in DM1 skeletal muscles. MicroRNA dysregulation was likely functionally relevant, since it impacted on the expression of the predicted miR-1, and miR-29 targets. The observed miRNA dysregulations and myslocalizations may contribute to DM1 pathogenetic mechanisms. Ó 2010 Elsevier B.V. All rights reserved. Keywords: Myotonic dystrophy; microRNAs; Gene expression; Subcellular localization 1. Introduction Myotonic Dystrophy (DM) is an autosomal dominant multisystemic disorder and the most common form of mus- cular dystrophy in adults that affects 1 in 8000 individuals worldwide. DM is characterized by a variety of multisystem- ic features including myotonia (muscle hyperexcitability), muscular dystrophy, dilated cardiomyopathy, cardiac con- duction defects, cataracts, cerebral involvement, insulin- resistance and disease-specific serological abnormalities. Clinical manifestations are highly variable and can range from minimal features, to moderately severe facial and distal limb muscle wasting and weakness, to severe congenital dis- order [1]. Respiratory failure and cardiac arrhythmias are among the major life threatening complications in DM1, being the first two causes of premature death in affected patients [1]. The most common form of the disorder, named type 1 (DM1-Steinert’s disease), is caused by an expanded (CTG)n in the 3 0 untranslated region of the Dystrophia Myotonica Protein Kinase (DMPK) gene [2–4]. A second form of myotonic dystrophy (DM2) has also been described, that closely mimics the DM1 phenotype, with prevalently proximal impairment. The DM2 mutation consists in the expansion of a tetranucleotidic repetition (CCTG)n in the first intron of the Zinc Finger Protein-9 (ZNF9) gene [5]. 0960-8966/$ - see front matter Ó 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.nmd.2010.11.012 ⇑ Corresponding author. Address: Laboratorio Patologia Vascolare, Istituto Dermopatico dell’Immacolata-IRCCS, via dei monti di creta 104, 00167 Rome, Italy. Tel.: +39 0666462431; fax: +39 0666462430. E-mail address: [email protected] (F. Martelli). www.elsevier.com/locate/nmd Available online at www.sciencedirect.com Neuromuscular Disorders 21 (2011) 81–88

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Available online at www.sciencedirect.com
www.elsevier.com/locate/nmd
Neuromuscular Disorders 21 (2011) 81–88
Dysregulation and cellular mislocalization of specific miRNAsin myotonic dystrophy type 1
Riccardo Perbellini a,b, Simona Greco a, Gianluca Sarra-Ferraris a, Rosanna Cardani c,d,Maurizio C. Capogrossi e, Giovanni Meola f, Fabio Martelli e,⇑
a Molecular Cardiology Laboratory, IRCCS-Policlinico San Donato, 20097 Milan, Italyb University of Milan-Hospital “L.Sacco”, Department of Clinical Sciences, 20157 Milan, Italy
c Department of Biomolecular Sciences and Biotechnology, University of Milan, 20097 Milan, Italyd CMN-Neuromuscular Disease Center, IRCCS Policlinico San Donato, University of Milan, Milan, Italy
e Laboratorio Patologia Vascolare, Istituto Dermopatico dell’Immacolata-IRCCS, 00167 Rome, Italyf University of Milan-IRCCS Policlinico San Donato, Department of Neurology, 20097 Milan, Italy
Received 20 September 2010; received in revised form 15 November 2010; accepted 22 November 2010
Abstract
Myotonic Dystrophy Type-1 (DM1) is caused by the expansion of a CTG repeat with a peculiar pattern of multisystemic involvementaffecting skeletal muscles, the heart, the eye, the central nervous system and the endocrine system. Since microRNA expression is dis-rupted in several myopathies, the expression of 24 candidate microRNAs was analyzed in skeletal muscle biopsies of 15 DM1 patients.Controls were constituted by biopsies without overt pathological features derived from 14 subjects with suspected neuromuscular disor-der of undetermined nature. We found that miR-1 and miR-335 were up-regulated, whereas miR-29b and c, and miR-33 were down-regulated in DM1 biopsies compared to controls. We also found that the cellular distribution of muscle specific miR-1, miR-133band miR-206 was severely altered in DM1 skeletal muscles. MicroRNA dysregulation was likely functionally relevant, since it impactedon the expression of the predicted miR-1, and miR-29 targets. The observed miRNA dysregulations and myslocalizations may contributeto DM1 pathogenetic mechanisms.� 2010 Elsevier B.V. All rights reserved.
Keywords: Myotonic dystrophy; microRNAs; Gene expression; Subcellular localization
1. Introduction
Myotonic Dystrophy (DM) is an autosomal dominantmultisystemic disorder and the most common form of mus-cular dystrophy in adults that affects 1 in 8000 individualsworldwide. DM is characterized by a variety of multisystem-ic features including myotonia (muscle hyperexcitability),muscular dystrophy, dilated cardiomyopathy, cardiac con-duction defects, cataracts, cerebral involvement, insulin-resistance and disease-specific serological abnormalities.
0960-8966/$ - see front matter � 2010 Elsevier B.V. All rights reserved.
doi:10.1016/j.nmd.2010.11.012
⇑ Corresponding author. Address: Laboratorio Patologia Vascolare,Istituto Dermopatico dell’Immacolata-IRCCS, via dei monti di creta 104,00167 Rome, Italy. Tel.: +39 0666462431; fax: +39 0666462430.
E-mail address: [email protected] (F. Martelli).
Clinical manifestations are highly variable and can rangefrom minimal features, to moderately severe facial and distallimb muscle wasting and weakness, to severe congenital dis-order [1]. Respiratory failure and cardiac arrhythmias areamong the major life threatening complications in DM1,being the first two causes of premature death in affectedpatients [1]. The most common form of the disorder, namedtype 1 (DM1-Steinert’s disease), is caused by an expanded(CTG)n in the 30 untranslated region of the DystrophiaMyotonica Protein Kinase (DMPK) gene [2–4]. A secondform of myotonic dystrophy (DM2) has also been described,that closely mimics the DM1 phenotype, with prevalentlyproximal impairment. The DM2 mutation consists in theexpansion of a tetranucleotidic repetition (CCTG)n in thefirst intron of the Zinc Finger Protein-9 (ZNF9) gene [5].

82 R. Perbellini et al. / Neuromuscular Disorders 21 (2011) 81–88
These genetic lesions cause the accumulation of theexpanded CUG/CCUG transcripts into discrete nuclearRNA foci that trigger a toxic gain of function, leading toaberrant RNA splicing and cell dysfunction [6].
MicroRNAs (miRNAs) are short non-coding RNAsthat regulate the stability and/or the translational efficiencyof target messenger RNAs. A functional role has beenidentified for miRNAs in virtually all cell functions, includ-ing the regulation of differentiation, proliferation andapoptosis [7]. Specifically, miRNA sequences can regulateskeletal and heart muscle function in both developmentand disease, including ischemia, and muscular dystrophy[8–10].
Certain microRNAs are highly enriched in skeletal mus-cles and the expression of muscle specific miR-1, -133a/b, -206 (myomiRs) is regulated by muscle transcriptional net-works involving MyoD, MEF2, SRF, Twist [8]. Interest-ingly, non-muscle specific miRNAs such as miR-221, -222and -181, also regulate skeletal muscle differentiation[11,12] and regeneration after injury [12].
We and others previously demonstrated miRNAs dereg-ulation in Duchenne Muscular Dystrophy patients and inmdx mice, both lacking a functional dystrophin gene[13,14]. Moreover, Eisenberg et al. described a miRNAexpression profile in muscle tissues from several human pri-mary muscle disorders and identified a series of miRNAsthat are regulated in almost all myopathies analyzed [15].Finally, Gambardella et al. analyzed the expression ofmyomiRs in DM1 and identified miR-206 over-expressionin five out of seven patients [16].
In this study, we assayed whether levels and cellularlocalization of a group of candidate miRNAs were alteredin DM1 patients, potentially contributing to DM1 patho-genetic mechanisms.
2. Materials and methods
2.1. Patients and skeletal muscle biopsies
Human muscle biopsies from biceps brachii were har-vested under sterile conditions and snap-frozen in cooledisopentane. All biopsy specimens were taken after informedconsent disclosing future use for research was obtained.This study was authorized by the Institutional Ethics Com-mittee, and was conducted according to the institutionalregulation and Italian laws and guidelines. Clinical diagno-sis of DM1 (nine males and six females, age = 38 ± 17 -years) and DM2 patients (six males and three females,age = 52 ± 10 years) was based upon the criteria set bythe International Consortium for Myotonic Dystrophies[17]. Genetic analysis was carried out to confirm DM1diagnosis and fluorescence in situ hybridization was per-formed on frozen muscle sections to confirm DM2 diagno-sis according to Cardani et al. [18]. Control biopsies werefrom subjects (eight males and six females, age = 46 ± 9 -years) admitted with suspected neuromuscular disorder ofundetermined nature. Control biopsies did not show overt
signs of muscle pathology on histological and histochemi-cal examination. Age and sex distribution differences vsDM1 (p < 0.114 and p = 0.876, respectively) and vs DM2(p < 0.219 and p = 0.648, respectively) were not significant.All muscle biopsies were processed by the same pathologyteam.
2.2. RNA extraction and quantification
Total RNA was extracted using TRIzol (Invitrogen) andthe TissueLyser system (Qiagen), according to the manu-facturer’s instructions. miRNA levels were analyzed usingthe TaqMan Real Time PCR (qPCR) method (3.4 ng/assay), and quantified with a 7900HT Fast Real-TimePCR System (Applied Biosystems). Primers for miRNAsand the reagents for reverse transcriptase and RT-PCRreactions were all obtained from Applied Biosystems. Rel-ative expression was calculated using the comparative Ctmethod (2�DDCt) [19]. Different samples were normalizedto miR-16 expression.mRNAs levels were analyzed usingthe SYBR-GREEN qPCR method (0.5 ng/assay, AppliedBiosystems) and quantified with 7900HT Real-Time PCRSystem (Applied Biosystems). Specific primers weredesigned using Universal ProbeLibrary software by Roche.Relative expression was calculated using the comparativeCt method [19]. mRNA expression was normalized forGAPDH levels. Each sample was measured in triplicateand values were averaged.
2.3. In situ hybridization and immunostaining
In situ hybridization was performed using locked nucleicacid (LNA) detection probes (Exiqon). Transverse musclecryostat sections (6 lm) were dried for 30 min at room tem-perature and fixed in 2% paraformaldehyde for 30 min at4 �C. Then, sections were washed five times in phosphate-buffered saline, pH 7.4 (PBS) for 2 min at room tempera-ture and incubated with 0.03% H2O2 in PBS for 10 minat room temperature. Sections were then permeabilized inpre-chilled 2% acetone in PBS for 5 min and incubated in30% formamide and 2�SSC for 10 min at room tempera-ture. Then, sections were hybridized with 20 nM digoxi-genin labelled probes for 2 h at 37 �C in 30% formamide,2�SSC, 0.02% BSA, 67 ng/ll yeast tRNA, 2 mM vanadylribonucleoside complex. Sections were washed first in30% formamide and 2�SSC at 45 �C for 30 min and2�SSC at 45 �C for 15 min, then in 1�SSC at 45 �C for15 min and 1�SSC at room temperature for 10 min. Sec-tions were incubated with 5% normal goat serum,2%BSA in PBS for 20 min at room temperature and thenincubated with antibody anti-digoxigenin conjugated withalkaline phosphase (Roche) diluted 1:2000 inPBS + 2%BSA overnight at 4 �C. Hybridized probes werevisualized by color reaction with nitroblue tetrazolium(NBT) and 5-bromo 4-chloro-3-indolyl phosphate (BCIP)overnight at room temperature. To identify fiber marginsand to discriminate between type I and II fibers, sections

Table 1miRNA assayed and their regulation inDM1 skeletal muscle biopsies.
microRNAs assayed Modulation
miR-1 UpmiR-29a No changemiR-29b DownmiR-29c DownmiR-31 No changemiR-33 DownmiR-34a No changemiR-34b No changemiR-34c No changemiR-124a No changemiR-133a No changemiR-133b No changemiR-135a No changemiR-181a No changemiR-181b No changemiR-206 No changemiR-222 No changemiR-223 No changemiR-335 UpmiR-449 No changemiR-452 No changemiR-485-5p No changemiR-494 No changemiR-505 No change
Fig. 1. microRNA modulation in adult skeletal muscle biopsies of DM1patients. RNA was extracted from DM1 and control biopsies and theexpression of the indicated miRNAs was measured by qPCR. The bargraphs represent average fold changes of miRNA expression (DM1 = 15,control = 14; *p < 0.01; **p < 0.05).
R. Perbellini et al. / Neuromuscular Disorders 21 (2011) 81–88 83
were incubated with mouse primary antibodies against theC-terminus of dystrophin (1:20 in PBS + 2%BSA overnightat 4 �C; Novocastra) or to slow myosin heavy chain (1:300in PBS + 2%BSA, overnight at 4 �C; Sigma–Aldrich),respectively. After incubation with a secondary biotinyla-ted antibody, dystrophin was visualized by ABComplex(Vector) and diamidinobenzidine tetrahydrochloride(DAB). Slow myosin was visualized by secondary antibodyAlexaFluor 488-labeled (Molecular Probes). Nuclei werecounterstained with 40,6 diamidino-2-phenylindole dihy-drochloride hydrate (DAPI). Sections were mounted inVectastain acqueous mounting medium (Vector) and ana-lyzed with a ZEISS Axio imager M1 microscope equippedwith a Axiocam MRc5 camera and Carl ZEISS imagingsystems. In situ and immunoistochemistry analyses werecarried out by two blinded readers with comparable results.
2.4. Statistical analysis
Value distribution was checked for normality by the Kol-mogorov–Smirnov test and variables were analyzed by 2 tailStudent’s t test. Differences in sex distribution between con-trols and DM patients were analyzed using Fisher’s-exacttest. A p 6 0.05 was deemed statistically significant. Valuesare expressed as average +/� standard error.
3. Results
3.1. Differential miRNA expression in DM1 patients
To investigate differences in miRNAs expression inpatients affected by DM1, we measured the expression of24 candidate miRNAs playing an important regulatoryfunction in skeletal muscle or that we previously identifiedas deregulated in both mdx mice and patients affected byDuchenne Muscle Dystrophy (Table 1) [13,14]. TotalRNA was extracted from biceps brachii muscle biopsiesobtained from adult individuals affected by DM1 (n = 15)or age and sex matched controls (n = 14). Relative miR-NAs quantity was measured by qPCR using miR-16 tonormalize values, since preliminary experiments showedthat this miRNA was not affected in the examined disease(not shown).
Fig. 1 shows that miR-1 and -335 were up-regulated,whereas miR-29b, -29c and -33 were down-regulated inDM1 biopsies compared to control subjects. Deregulationof these miRNAs was present in both males and femalesand did not correlate with age. When DM2 patients(n = 9) were examined, the differences observed in miRNAlevels did not reach statistical significance (Table S1).
3.2. MyomiRs are located in internal nuclei and in the
nuclear clumps of DM1 myofibers
The cellular localization of the identified miRNAs wasassayed by in situ hybridization on cryostat muscle sectionsderived from five DM1 and control biopsies using digoxi-
genin labelled LNA probes. Anti-dystrophin counterstain-ing allowed myofiber margin detection. We found thatmiR-29b, -29c, -33 and -335 were barely detectable, possi-bly due to low sensitivity of the in situ hybridization tech-nique (data not shown). Conversely, miR-1 was readilydetectable and its intracellular distribution was disrupted.Specifically, in control samples, miR-1 displayed a peculiarenrichment in the perinuclear area (Fig. 2). In DM1 sec-tions, centrally nucleated myofibers, a hallmark of DM1,also exhibited a centralization of miR-1 localization(Fig. 2A, red arrows). Furthermore, very small fibers withnuclear clumps, a typical histopathological DM1 alter-ation, displayed intense miR-1 staining (Fig. 2B, yellowarrows). Finally, certain myofibers displayed an extremely
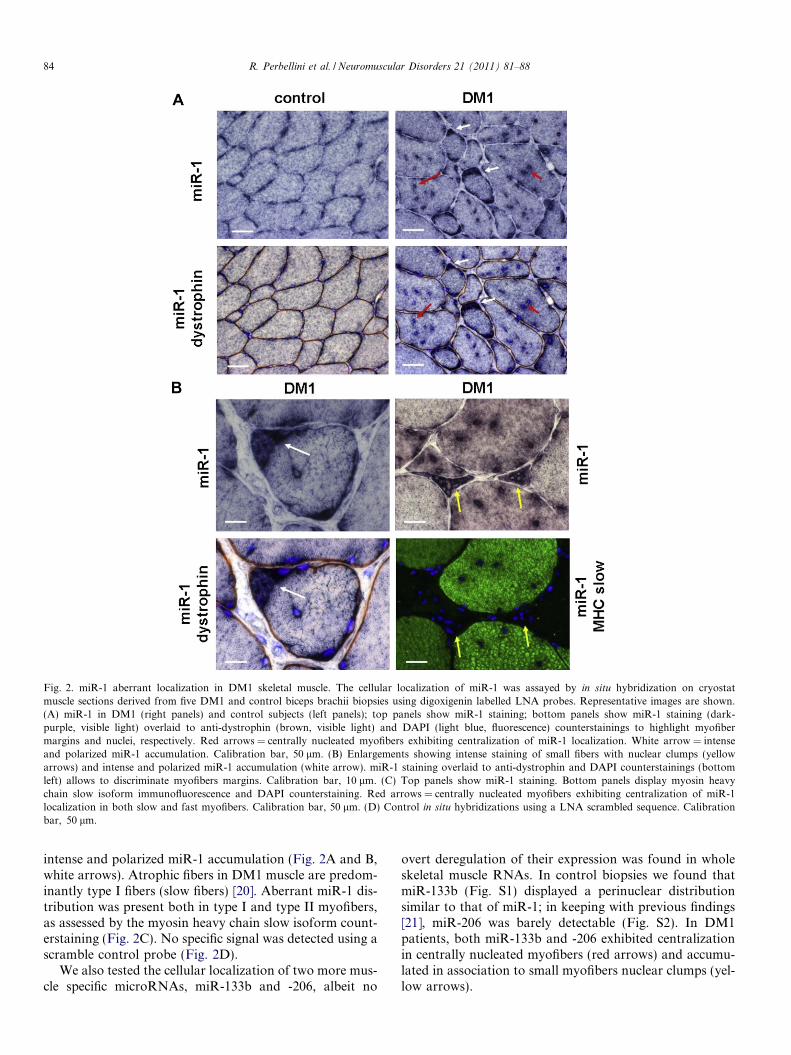
Fig. 2. miR-1 aberrant localization in DM1 skeletal muscle. The cellular localization of miR-1 was assayed by in situ hybridization on cryostatmuscle sections derived from five DM1 and control biceps brachii biopsies using digoxigenin labelled LNA probes. Representative images are shown.(A) miR-1 in DM1 (right panels) and control subjects (left panels); top panels show miR-1 staining; bottom panels show miR-1 staining (dark-purple, visible light) overlaid to anti-dystrophin (brown, visible light) and DAPI (light blue, fluorescence) counterstainings to highlight myofibermargins and nuclei, respectively. Red arrows = centrally nucleated myofibers exhibiting centralization of miR-1 localization. White arrow = intenseand polarized miR-1 accumulation. Calibration bar, 50 lm. (B) Enlargements showing intense staining of small fibers with nuclear clumps (yellowarrows) and intense and polarized miR-1 accumulation (white arrow). miR-1 staining overlaid to anti-dystrophin and DAPI counterstainings (bottomleft) allows to discriminate myofibers margins. Calibration bar, 10 lm. (C) Top panels show miR-1 staining. Bottom panels display myosin heavychain slow isoform immunofluorescence and DAPI counterstaining. Red arrows = centrally nucleated myofibers exhibiting centralization of miR-1localization in both slow and fast myofibers. Calibration bar, 50 lm. (D) Control in situ hybridizations using a LNA scrambled sequence. Calibrationbar, 50 lm.
84 R. Perbellini et al. / Neuromuscular Disorders 21 (2011) 81–88
intense and polarized miR-1 accumulation (Fig. 2A and B,white arrows). Atrophic fibers in DM1 muscle are predom-inantly type I fibers (slow fibers) [20]. Aberrant miR-1 dis-tribution was present both in type I and type II myofibers,as assessed by the myosin heavy chain slow isoform count-erstaining (Fig. 2C). No specific signal was detected using ascramble control probe (Fig. 2D).
We also tested the cellular localization of two more mus-cle specific microRNAs, miR-133b and -206, albeit no
overt deregulation of their expression was found in wholeskeletal muscle RNAs. In control biopsies we found thatmiR-133b (Fig. S1) displayed a perinuclear distributionsimilar to that of miR-1; in keeping with previous findings[21], miR-206 was barely detectable (Fig. S2). In DM1patients, both miR-133b and -206 exhibited centralizationin centrally nucleated myofibers (red arrows) and accumu-lated in association to small myofibers nuclear clumps (yel-low arrows).

Fig. 2 (continued)
R. Perbellini et al. / Neuromuscular Disorders 21 (2011) 81–88 85
3.3. Dysregulation of miR-1 and miR-29 affects the levels of
their predicted targets
miRNAs have been shown not only to inhibit proteintranslation, but also to induce mRNA degradation, at leastfor certain targets [22]. Thus, in order to assess whethermiRNA deregulation was functionally relevant, we exam-ined the impact of the identified miRNAs deregulationon the expression of their potential target genes in DM1patients. Specifically, we focused on miR-29, that displayedthe strongest deregulation, and miR-1, that plays a crucialrole in muscle differentiation [23,30]. Search of the poten-tial targets was performed using Pictar and Targetscan pre-diction algorithms, given their reported specificity [24].Indeed, to maximize the accuracy, only targets identifiedby both softwares were considered. A sub-pool of the iden-tified targets was analyzed, selected among these with a
potential link to DM1 physio-pathology. Specifically,selected genes were previously demonstrated to beexpressed in skeletal muscle and to be involved in eventssuch as muscle development, atrophy, arrhythmia andsplicing. Potential targets were assayed by qPCR andFig. 3 shows that both miR-29 and miR-1 targets were sig-nificantly up-regulated in DM1 patients.
4. Discussion
Elucidating the molecular mechanisms activated by trip-let expansions in the DMPK gene is of crucial importancefor our understanding of DM1 disease. In this study, biop-sies derived from DM1 patients were used to investigateskeletal muscle functions regulated by miRNAs. Ourapproach was not intended to provide a comprehensivedescription of the disease, but it was aimed at the identifi-

Fig. 3. miR-1 and miR-29 predicted targets are up-regulated in DM1patients. RNA was extracted from DM1 and control biopsies and theexpression of the indicated genes was measured by qPCR. (A) Bar graphrepresenting average fold increase of miR-29 putative target mRNAs(DM1 = 5; controls = 5; *p < 0.01; **p < 0.05). (B) Bar graph representingaverage fold increase of miR-1 putative targets mRNAs (DM1 = 7;controls = 5; *p < 0.01; **p < 0.05).
86 R. Perbellini et al. / Neuromuscular Disorders 21 (2011) 81–88
cation of a miRNAs subset whose expression patternsmight indicate a potential role in the pathological pro-
Fig. 4. Molecular model summarizing the results and their potential relevanceand -335 induction, to miR-29b, -29c and -33 down-modulation, as well as to tevents, in turn, affect the expression of the respective target gene. Target genes eimpact on DM1 pathological mechanisms are indicated. Genes in red are miinduced in DM1 patients by Gambardella et al. [16].
cesses. We identified five miRNA whose expression waseither up-or down-regulated in DM1 patients. The similartrend in miRNA modulation observed in DM2 patientsdid not reach statistical significance, possibly due to thelow number of patients examined. Alternative explanationsare the different genetic lesion causing DM1 and 2 [2,5] orthe relatively milder DM2 phenotype [6].
Intriguingly, deregulation of the miRNAs identified inDM1 has been also observed in Duchenne Muscular Dys-trophy and other muscular disorders [13,15]. The onlyexception was constituted by miR-33 that seems to bedown-modulated in DM1, specifically. While miR-29 fam-ily and miR-335 were similarly dysregulated in DM1 andDuchenne Muscular Dystrophy, miR-1 was induced inDM1 and decreased in Duchenne Muscular Dystrophy.In a recent paper, Gambardella et al. found that miR-206expression was induced in DM1 patients [16]. Differenceswith our study may be attributable to the low patient num-erosity of both studies or to the different skeletal muscleanalyzed (vastus lateralis vs biceps brachii). Furthermore,while we did not find a significant increase of miR-206 inour cohort, we observed that miR-206 cellular localizationwas disrupted.
While the potential involvement of miRNA deregula-tions in DM1 pathogenesis needs to be evaluated in futurestudies, we found that the expression of miR-29 predictedtargets was induced as expected, suggesting that miR-29family decrease is functionally relevant. Intriguingly,among miR-29 predicted targets, TRIM63/MURF1, DIA-BLO, RET and TGFB3 were all induced in atrophic mus-
to DM1 pathology. (CTG)n expansion in the DMPK gene leads to miR-1he aberrant cellular localization of myomiR miR-1, -133b and -206. Thesexperimentally found to be de-regulated in DM1 patients and their potentialR-1 targets; genes in blue are miR-29 targets. *miR-206 was found to be

R. Perbellini et al. / Neuromuscular Disorders 21 (2011) 81–88 87
cles as well, pointing to the possible involvement of miR-29decrease in DM1 myofiber atrophy [25–28]. Moreover, VanRooij et al. found that miR-29 family acts as an inhibitorof cardiac fibrosis in a mouse model of myocardial infarc-tion [29]. However, we did not find a clear correlationbetween miR-29 levels and fibrosis in DM1 patients (RC,FM and GM, unpublished).
We also found that potential miR-1 targets were signif-icantly up-regulated in DM1 patients. Since miR-1 levelswere increased in DM1 patients, one may expect that thelevels of its targets decreased accordingly. However, wealso found that miR-1 subcellular localization was severelydisrupted, likely impairing its function as well. Thus, miR-1increase seems to be futile and its targets are indeed de-repressed. Since miR-1 is a crucial regulator, not only ofmyogenic differentiation, but also of muscle cell excitability[30], it is tempting to speculate that miR-1 deregulationobserved in DM1 may be linked to muscle myotonia andcardiac arrhythmia. In keeping with this hypothesis,DM1 muscles displayed increased levels of both KCNE1[31], the modulatory subunit of the potassium channelKv7.1, responsible for the slow delayed rectifier current,and of KCNJ2 [32] encoding Kir2.1, responsible for theinward rectifier K+ current. Furthermore, deregulationof another miR-1 target, Calmodulin2 [33], may affect Cal-cium handling.
The fact that many of the miRNAs identified in DM1areencoded in introns (www.mirbase.org), suggests a linkbetween pre-mRNA splicing and miRNA processing.Indeed, it has been shown that miRNAs residing in intronsare processed before splicing is completed [34], combiningpre-mRNA splicing and miRNA processing in time andspace. Given that widespread splicing alterations are a hall-mark of DM1 [6], it is tempting to speculate that the bio-synthesis of specific miRNA may be disrupted as well [35].
In conclusion, we identified a small subset of miRNAwhose expression and/or localization were deregulated inDM1. These findings may improve our understanding ofthe molecular mechanisms linking (CTG)n expansion inthe DMPK gene to disease (Fig. 4).
Acknowledgments
This work was partly supported by Ministero della Sa-lute and Association Francaise contre les Myopathies(AFM), MIUR-PRIN and Centro per lo Studio delle Mal-attie Neuromuscolari – CMN. We thank Dr. PasqualeFasanaro, Istituto Dermopatico dell’Immacolata-IRCCSfor critical reading of the manuscript.
References
[1] Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis andmanagement. J Neurol Neurosurg Psychiatry 2010;81(4):358–67.
[2] Brook JD, McCurrach ME, Harley HG, et al.. Molecular basis ofmyotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the30 end of a transcript encoding a protein kinase family member. Cell1992:799–808.
[3] Fu YH, Pizzuti A, Fenwick Jr RG, et al.. An unstable triplet repeat ina gene related to myotonic muscular dystrophy. Science1992;255(5049):1256–8.
[4] Mahadevan M, Tsilfidis C, Sabourin L, et al.. Myotonic dystrophymutation: an unstable CTG repeat in the 30 untranslated region of thegene. Science 1992;255(5049):1253–5.
[5] Liquori CL, Ricker K, Moseley ML, et al.. Myotonic Dystrophy type2 caused by a CCTG expansion in intron 1 of ZNF9. Science2001;293:864–8.
[6] Johanna E, Lee, Cooper Thomas A. Pathogenic mechanisms ofmyotonic dystrophy. Biochem Soc Trans 2009;37:1281–6.
[7] Flynt AS, Lai EC. Biological principles of microRNA-mediatedregulation: shared themes amid diversity. Nat Rev Genetics2008;9(11):831–42.
[8] Van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles.Trends Genet 2008;24(4):159–66.
[9] Fasanaro P, Greco S, Ivan M, Capogrossi MC, Martelli F.MicroRNA: emerging therapeutic targets in acute ischemic diseases.Pharmacol Ther 2010;125(1):92–104.
[10] Latronico MV, Condorelli G. MicroRNAs and cardiac pathology.Nat Rev Cardiol 2009;6(6):419–29.
[11] Cardinali B, Castellani L, Fasanaro P, et al.. Microrna-221 andmicrorna-222 modulate differentiation and maturation of skeletalmuscle cells. PLoS One 2009;4(10):e7607.
[12] Naguibneva I, Ameyar-Zazoua M, Polesskaya A, et al.. ThemicroRNA miR-181 targets the homeobox protein Hox-A11 duringmammalian myoblast differentiation. Nat Cell Biol 2006;8(3):278–84.
[13] Greco S, De Simone M, Colussi C, et al.. Common micro-RNAsignature in skeletal muscle damage and regeneration induced byDuchenne muscular dystrophy and acute ischemia. FASEB J2009;23(10):3335–46.
[14] Eisenberg I, Alexander MS, Kunkel LM. MiRNAS in normal anddiseased skeletal muscle. J Cell Mol Med 2009;13(1):2–11.
[15] Eisenberg I, Eran A, Nishino I, et al.. Distinctive patterns ofmicroRNA expression in primary muscular disorders. Proc NatlAcad Sci USA 2007;104:17016–21.
[16] Gambardella S, Rinaldi F, Lepore S M, et al.. Overexpression ofmicroRNA-206 in the skeletal muscle from myotonc dystrophy type 1patients. J Transl Med 2010;8:48.
[17] Moxley RT 3rd, Meola G, Udd B, Ricker K. Report of the 84thENMC workshop: PROMM (proximal myotonic myopathy) andother myotonic dystrophy-like syndromes: 2nd workshop. 13-15thOctober, 2000, Loosdrecht, The Netherlands. Neuromuscul Disord2002; 12(3): 306-17.
[18] Cardani R, Mancinelli E, Sansone V, Rotondo G, Meola G.Biomolecular identification of (CCTG)n mutation in myotonicdystrophy type 2 (DM2) by FISH on muscle biopsy. Eur J Histochem2004;48(4):437–42.
[19] Livak KJ, Schmittgen TD. Analysis of relative gene expression datausing real-time quantitative PCR and the 2(-Delta Delta C(T))method. Methods 2001;25(4):402–8.
[20] Vihola A, Bassez G, Meola G, et al.. Histopathological differences ofmyotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology2003;60(11):1854–7.
[21] Yuasa K, Hagiwara Y, Ando M, Nakamura A, Takeda S, HijikataT. MicroRNA-206 is highly expressed in newly formed musclefibers: implications regarding potential for muscle regeneration andmaturation in muscular dystrophy. Cell Struct Funct2008;33(2):163–9.
[22] Parker R, Sheth U. P bodies and the control of mRNA translationand degradation. Mol Cell 2007;25(5):635–46.
[23] Williams AH, Liu N, van Rooij E, Olson EN. MicroRNA control ofmuscle development and disease. Curr Opin Cell Biol2009;21(3):461–9.
[24] Hendrickson DG, Hogan DJ, Herschlag D, Ferrell JE, Brown PO.Systematic identification of mRNAs recruited to argonaute 2 byspecific microRNAs and corresponding changes in transcript abun-dance. PLoS One 2008;3(5):e2126.

88 R. Perbellini et al. / Neuromuscular Disorders 21 (2011) 81–88
[25] Murton AJ, Constantin D, Greenhaff PL. The involvement of theubiquitin proteasome system in human skeletal muscle remodellingand atrophy. Biochim Biophys Acta 2008;1782(12):730–43.
[26] Tews DS, Behrhof W, Schindler S. SMAC-expression in denervatedhuman skeletal muscle as a potential inhibitor of coexpressedinhibitor-of-apoptosis proteins. Appl Immunohistochem Mol Mor-phol 2008;16(1):66–70.
[27] Bergman E, Kullberg S, Ming Y, Ulfhake B. Upregulation ofGFRalpha-1 and c-ret in primary sensory neurons and spinalmotoneurons of aged rats. J Neurosci Res 1999;57(2):153–65.
[28] Hirose T, Nakazato K, Song H, Ishii N. TGF-beta1 and TNF-alphaare involved in the transcription of type I collagen alpha2 gene insoleus muscle atrophied by mechanical unloading. J Appl Physiol2008;104(1):170–7.
[29] Van Rooij E, Sutherland LB, Thatcher JE, et al.. Dysregulationof microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA2008;105(35):13027–32.
[30] Latronico MV, Condorelli G. MicroRNAs and cardiac conduction.Curr Drug Targets 2010;11(8):907–12.
[31] Luo X, Xiao J, Lin H, et al.. Transcriptional activation by stimulatingprotein 1 and post-transcriptional repression by muscle-specificmicroRNAs of IKs-encoding genes and potential implications inregional heterogeneity of their expressions. Cell Physiol2007;212(2):358–67.
[32] Yang B, Lin H, Xiao J, et al.. The muscle-specific microRNA miR-1regulates cardiac arrhythmogenic potential by targeting GJA1 andKCNJ2. Nat Med 2007;13(4):486–91.
[33] Ikeda S, He A, Kong SW, et al.. MicroRNA-1 negatively regulatesexpression of the hypertrophy-associated calmodulin and Mef2agenes. Mol Cell Biol 2009;29(8):2193–204.
[34] Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I,Proudfoot NJ. Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 2008;15(9):902–9.
[35] Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy.Biochem Soc Trans 2009;37(Pt 6):1281–6.
Related Documents











