The Role of Candidate G-protein Coupled Receptors in Mediating Remote Myocardial Ischemic Preconditioning. by Harinee Surendra A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Laboratory Medicine and Pathobiology University of Toronto © Copyright by Harinee Surendra (2009)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The Role of Candidate G-protein Coupled Receptors
in Mediating Remote Myocardial Ischemic
Preconditioning.
by
Harinee Surendra
A thesis submitted in conformity with the requirements for the degree of
Master of Science
Graduate Department of Laboratory Medicine and Pathobiology
University of Toronto
© Copyright by Harinee Surendra (2009)

i
ACKNOWLEDGEMENTS
I would like to extend my heartfelt gratitude to my family for supporting me through this
rewarding and demanding experience. Their constant encouragement has made this thesis
possible.
Also, my sincere thanks are given to the guidance and support of Dr. Gregory Wilson
(Division of Pathology, Department of Paediatric Laboratory Medicine, Hospital for Sick
Children) and Roberto Diaz (Dr. Wilson‘s Lab, Hospital for Sick Children). Their extensive
knowledge in the field of ischemic preconditioning has provided excellent direction for this
thesis and given me opportunities to progress in my career. Also, tremendous guidance was
provided by members of my graduate committee through Dr. Herman Yeger (Research Institute,
Hospital for Sick Children) and Dr Aleksander Hinek (Research Institute, Hospital for Sick
Children). My genuine appreciation is given to Alina Hinek (Dr. Wilson‘s Lab, Hospital for Sick
Children) and Taneya Hossain (Dr. Wilson‘s Lab, Hospital for Sick Children) for their assistance
in providing training.
I would like to thank Jing Li (Dr. Redington‘s Lab, Research Institute, Hospital for Sick
Children) for providing the dialysate used in this study and Michael Tropak (Dr. Callahan‘s Lab,
Research Institute, Hospital for Sick Children) for measuring substances in the dialysate with
mass spectrometry. Finally, I would like to thank Foundation Leducq, the Heart and Stroke
Foundation of Ontario, and the University of Toronto for providing funding for my research.

ii
ABSTRACT
The Role of Candidate G-protein Coupled Receptors in Mediating Remote Myocardial
Ischemic Preconditioning.
By Harinee Surendra
Degree of Master of Science (2009)
Graduate Department of Laboratory Medicine and Pathobiology
University of Toronto
This study investigated the role of opioid, adenosine, bradykinin, and calcitonin-gene
related peptide (CGRP) receptors, and potential ‗cross-talk‘ among suspected G-protein coupled
receptors in a humoral model of remote ischemic preconditioning (rIPC) cardioprotection.
Compared to Control dialysate (from non-preconditioned donor rabbit blood), rIPC dialysate
(from remotely preconditioned blood) reduced cell death in rabbit cardiomyocytes following
simulated ischemia and reperfusion. Non-selective, δ-, or κ-opioid receptor blockade and non-
selective adenosine receptor blockade abolished rIPC dialysate protection; whereas, bradykinin
B2 and CGRP receptor blockade had no effect. Non-selective adenosine receptor blockade fully
and partially abolished protection by κ- and δ-opioid receptors, respectively. Multiple reaction
monitoring mass spectrometry detected low levels of adenosine, and other preconditioning
substances, in the dialysate. An increase in extracellular adenosine was not detected during
opioid-induced preconditioning to explain this cross-talk. These results suggest that δ-opioid, κ-
opioid, adenosine receptors, and opioid-adenosine cross-talk are involved in rIPC of freshly
isolated cardiomyocytes.

iii
TABLE OF CONTENTS
ABSTRACT ................................................................................................................................... ii
GLOSSARY................................................................................................................................ viii
1. INTRODUCTION......................................................................................................................1
ISCHEMIC PRECONDITIONING (IPC) 1.1 .........................................................................................1
Background in IPC 1.1.a ...........................................................................................................1
Signalling Mechanisms in IPC 1.1.b .........................................................................................2
REMOTE ISCHEMIC PRECONDITIONING (RIPC) 1.2 .........................................................................5
Background in rIPC 1.2.a .........................................................................................................5
Signalling Mechanisms in rIPC 1.2.b .......................................................................................5
INVOLVEMENT OF OPIOID RECEPTORS 1.3 .....................................................................................8
Opioids and Their Receptors 1.3.a ...........................................................................................8
Evidence of Opioid Involvement in IPC1.3.b ............................................................................9
Evidence of Opioid Involvement in rIPC1.3.c.........................................................................10
INVOLVEMENT OF BRADYKININ B2 RECEPTORS 1.4 .....................................................................11
Bradykinin and Their Receptors 1.4.a ....................................................................................11
Evidence of Bradykinin Involvement in IPC 1.4.b ..................................................................12
Evidence of Bradykinin Involvement in rIPC 1.4.c .................................................................13
INVOLVEMENT OF CGRP RECEPTORS 1.5 ....................................................................................14
CGRP and Their Receptors 1.5.a ............................................................................................14
Evidence of CGRP Involvement in IPC 1.5.b..........................................................................15
Evidence of CGRP Involvement in rIPC 1.5.c ........................................................................16
INVOLVEMENT OF ADENOSINE RECEPTORS 1.6 ...........................................................................17

iv
Adenosine and Their Receptors 1.6.a......................................................................................17
Evidence of Adenosine Involvement in IPC 1.6.c....................................................................18
Evidence of Adenosine Involvement in rIPC 1.6.b ..................................................................19
OTHER POTENTIAL PRECONDITIONING SUBSTANCES 1.7 .............................................................20
OPIOID-ADENOSINE RECEPTOR CROSS-TALK 1.8 ........................................................................22
Background 1.8.a ....................................................................................................................22
Adenosine Levels in the Blood 1.8.b .......................................................................................23
Potential Mechanisms of Cross-talk 1.8.c ...............................................................................23
2. RATIONALE ...........................................................................................................................26
3. OBJECTIVES ..........................................................................................................................27
HYPOTHESIS 3.1 ..........................................................................................................................27
SPECIFIC AIMS 3.2 .......................................................................................................................27
4. RESEARCH DESIGN & METHODS ...................................................................................28
ANIMALS AND HUMAN SUBJECTS 4.1 ..........................................................................................28
ISOLATION OF ADULT CARDIOMYOCYTES 4.2 .............................................................................28
Operative Procedure 4.2.a ......................................................................................................28
Digestion Protocol 4.2.b .........................................................................................................29
Comparison of Digestion Protocols 4.2.c ...............................................................................32
DIALYSATE PREPARATION 4.3 .....................................................................................................34
FRESH-CELL EXPERIMENTAL MODEL 4.4 ....................................................................................37
General Protocol 4.4.a ............................................................................................................37
AIM (1) Protocol 4.4.b ............................................................................................................39

v
AIM (2) Protocol 4.4.c ............................................................................................................40
AIM (3) Protocol 4.4.d ............................................................................................................41
TRYPAN BLUE EXCLUSION ASSAY 4.5 .........................................................................................42
Inter-Observer Error Data 4.5.a .............................................................................................44
DRUGS 4.6 ...................................................................................................................................46
OTHER METHODS 4.7 ..................................................................................................................47
Western Blotting 4.7.a .............................................................................................................47
Multiple Reaction Monitoring Mass Spectrometry 4.7.b ........................................................48
Statistics 4.7.c ..........................................................................................................................50
5. CHARACTERIZATION OF THE PROTECTION INDUCED BY RIPC .......................51
RABBIT AND HUMAN PRECONDITIONED DIALYSATE INDUCES PROTECTION 5.1 .........................51
Dialysate Characterization 5.1.a ............................................................................................53
Discussion 5.1.b ......................................................................................................................53
6. THE ROLE OF CELL MEMBRANE RECEPTORS IN RIPC .........................................56
THE ROLE OF OPIOID RECEPTORS 6.1 ..........................................................................................56
Survey of Opioid Receptors in Rabbit Cardiomyocytes 6.1.a .................................................56
Non-selective Opioid Receptor Blockade of Protection 6.1.b .................................................58
δ-Opioid Receptor Blockade of Protection 6.1.c ....................................................................60
κ-Opioid Receptor Blockade of Protection 6.1.d ....................................................................62
Discussion 6.1.e.......................................................................................................................64
THE ROLE OF BRADYKININ B2 RECEPTORS 6.2 ............................................................................66
Bradykinin B2 Receptor Blockage Conserves Protection 6.2.a ..............................................66
Discussion 6.2.b ......................................................................................................................68

vi
THE ROLE OF CGRP RECEPTORS 6.3 ...........................................................................................70
Existence of Calcitonin-like Receptors in Rabbit Cardiomyocytes 6.3.a ................................70
CGRP Receptor Blockage Conserves Protection 6.3.b...........................................................71
Discussion 6.3.c.......................................................................................................................73
THE ROLE OF ADENOSINE RECEPTORS 6.4 ..................................................................................74
Non-selective Adenosine Receptor Blockade of Protection 6.4.a ...........................................74
Discussion 6.4.b ......................................................................................................................76
7. RIPC MEDIATES OPIOID-ADENOSINE CROSS-TALK ................................................78
OPIOID-ADENOSINE CROSS-TALK 7.1 ..........................................................................................78
Adenosine Deamination of the Dialysate Conserves Protection 7.1.a ...................................78
Partial Adenosine Blockade of δ-Opioid Receptor-Induced Protection 7.1.b ........................80
Compete Adenosine Blockade of κ-Opioid Receptor-Induced Protection 7.1.c .....................82
Discussion 7.1.d ......................................................................................................................84
THE INHIBITION OF ADENOSINE KINASE HYPOTHESIS 7.2 ...........................................................86
Exposure to Dynorphin B Does Not Accumulate Extracellular Adenosine 7.2.a ...................86
Discussion 7.2.b ......................................................................................................................89
8. SUMMARY OF FINDINGS ...................................................................................................92
9. GENERAL DISCUSSION ......................................................................................................93
OVERALL PERSPECTIVE 9.1 .........................................................................................................93
OTHER MECHANISMS OF OPIOID-ADENOSINE CROSS-TALK 9.2 ..................................................95
Dimerization of G-protein Coupled Receptors 9.2.a ..............................................................95
FUTURE DIRECTIONS 9.3 .............................................................................................................96

vii
Specific Adenosine Receptor Subtypes in Cross-talk & rIPC 9.3.a ........................................96
Heterodimerization of Opioid and Adenosine Receptors 9.3.b ...............................................98
Cell Signalling in rIPC 9.3.c ...................................................................................................98
LIMITATIONS 9.4 .......................................................................................................................103
CONCLUSIONS 9.5 ......................................................................................................................104
APPENDIX .................................................................................................................................105
APPENDIX I: LIST OF FIGURES ..................................................................................................105
APPENDIX II: LIST OF TABLES ..................................................................................................107
APPENDIX III: RIPC SUMMARY: MYOCARDIUM AS THE TARGET ORGAN .................................108
APPENDIX IV: RIPC SUMMARY: SKELETAL MUSCLE AS THE PRECONDITIONING ORGAN ..........109
APPENDIX V: MRM MASS SPECTROMETRY PLOTS OF STANDARD CONCENTRATIONS ..............110
REFERENCE LIST ...................................................................................................................113

viii
GLOSSARY
TERM DEFINITION
8-SPT 8-(p-sulfophenyl)theophylline
ADA Adenosine deaminase
ADO Adenosine
BNTX 7-benzylidenealtrexone
cAMP cyclic adenosine monophosphate
CAO Coronary artery occlusion
CCPA 2-chloro-N6-cyclopentyladenosine
CGRP (8-37) Human fragment of the calcitonin-gene related peptide
DynB Dynorphin B
FAO Femoral artery occlusion
GNTI 5‘-guanidinyl-17-(cyclopropylmethyl)-6,7-dehyrdo-4,5α-epoxy-3,14-
dihydroxy-6,7-2‘,3‘-indolomorphinan
HOE140 Hoechst 140
IPC Ischemic preconditioning
IR Ischemia-reperfusion
KH Krebs-Henseleit
MAO Mesenteric artery occlusion
ME Met-enkephalin
mKATP Mitochondrial ATP sensitive potassium channels

ix
MPG N-2-mercaptopropionyl glycine
mPTP Mitochondrial permeability transition pore
MRM MS Multiple reaction monitoring mass spectrometry
Nal Naloxone
NO Nitric oxide
NOS nitric oxide synthase
NTI Naltrindole
PGI2 Prostacyclin
PI3 Kinase Phosphatidylinositol 3-kinase
PKC Protein kinase C
RAO Renal artery occlusion
rIPC Remote ischemic preconditioning
ROS Reactive oxygen species
SI Simulated ischemia
sKATP Sarcolemmal ATP sensitive potassium channels
SR Simulated reperfusion
TNF-α Tumour necrosis factor –α
WB Western blotting
Wort Wortmannin

1
- Chapter 1 -
INTRODUCTION
1.1 Ischemic Preconditioning (IPC)
1.1.a Background in IPC:
Ischemic preconditioning (IPC) is one of the most potent strategies, under experimental
conditions, to reduce myocardial infarction to date. One or more periods of brief ischemia (~5
min) and reperfusion (~5 min) render the myocardium protected from a much longer damaging
ischemic stress. This phenomenon, first discovered by Murry et al.1 in 1986, exhibits two distinct
windows of protection. This thesis will focus on the first window of protection (called acute
preconditioning), which occurs within 2 hours of the IPC stimulus, whereas the second window
(called delayed preconditioning) occurs 24-72 hours later and is thought to be gene transcription-
dependent.
Since the discovery of IPC to induce protection in a wide range of tissues and species, the
triggers involved in this phenomenon have been of great interest as a therapeutic intervention for
myocardial infarction. Thornton et al.2 determined that G-protein coupled receptors were vital to
preconditioning when pertussis toxin (an inhibiter of G-protein coupled receptors) administered
in vivo to rabbits blocked myocardial preconditioning. Since then, the major extracellular
molecules involved in IPC at the trigger phase were found to be adenosine, bradykinin and
opioid peptides. Goto et al.3 postulated that receptors for these three peptides exert an additive
effect to reach a hypothetical threshold that is required to induce cardioprotection. Thus,
blockade of any one of these G-protein coupled receptors is enough to abolish protection by IPC.

2
1.1.b Signalling Mechanisms in IPC:
The downstream signalling pathways with respect to classical IPC have been well
established. According to Downey et al. 4
, cell signalling exhibits three distinct phases: the
‗trigger‘ phase, the ‗mediator‘ and the ―end effector‖ phase.
Mocanu et al.5 demonstrated that cardioprotection was abolished by the
phosphatidylinositol 3-kinase (PI3 kinase) inhibitor, wortmannin and LY 294002. The activation
of PI3 kinase results in translocation of Akt to the plasma membrane6 and its subsequent
phosphorylation7. Akt then stimulates nitric oxide synthase (NOS) to generate nitric oxide (NO)
8,
an important molecule that also acts as a trigger in IPC9 as suggested by Lochner et al.
10.
Oldenburg et al.11
also proposed the involvement of guanylyl cyclase (GC) in IPC by abrogating
protection with the GC inhibitor, ODQ. GC then produces cGMP and activates protein kinase G
(PKG).
PKG then induces the opening of mitochondrial potassium channels (mKATP) which
results in swelling of the mitochondria due to potassium influx. This leads to reactive oxygen
species (ROS) generation as shown by Forbes et al.12
when protection from diazoxide (an mKATP
channel opener) was abolished by ROS scavengers. Finally, the target of this downstream
signalling pathway converges on protein kinase C (PKC)13
.
To summarize, the trigger phase involves the release of endogenous substances, such as
opioid peptides and bradykinin, that activate a complex pathway which includes, in the following

3
order: phosphatidylinositol 3-kinase (PI3-kinase), Akt, nitric oxide synthase (NOS), nitric oxide
(NO), guanylyl cyclase, protein kinase G, opening of mitochondrial KATP channels, which in turn
generates reactive oxygen species (ROS) and actives protein kinase C (PKC). In addition, opioid
peptides undergo an additional step of activating an epidermal growth factor receptor (EGFR)
before activating PI3-kinase and Akt. Adenosine, another important trigger in IPC, is thought to
activate PKC directly through adenosine A1 and A3 receptors.4
The ‗mediator‘ phase occurs either during the long ischemia (called the index ischemia)
or early during reperfusion and is characterized by adenosine-dependent activation of adenosine
A2b receptors. PKC can also modulate the activity of this adenosine receptor directly. Adenosine
A2b receptors, in turn, lead to ERK and PI3-kinase/Akt activation. These kinases phosphorylate
GSK-3β, which is thought to inhibit formation of mitochondrial permeability transition pores
(mPTP) (see Figure 1). The mPTP has been proposed to be an end effector of IPC but this a
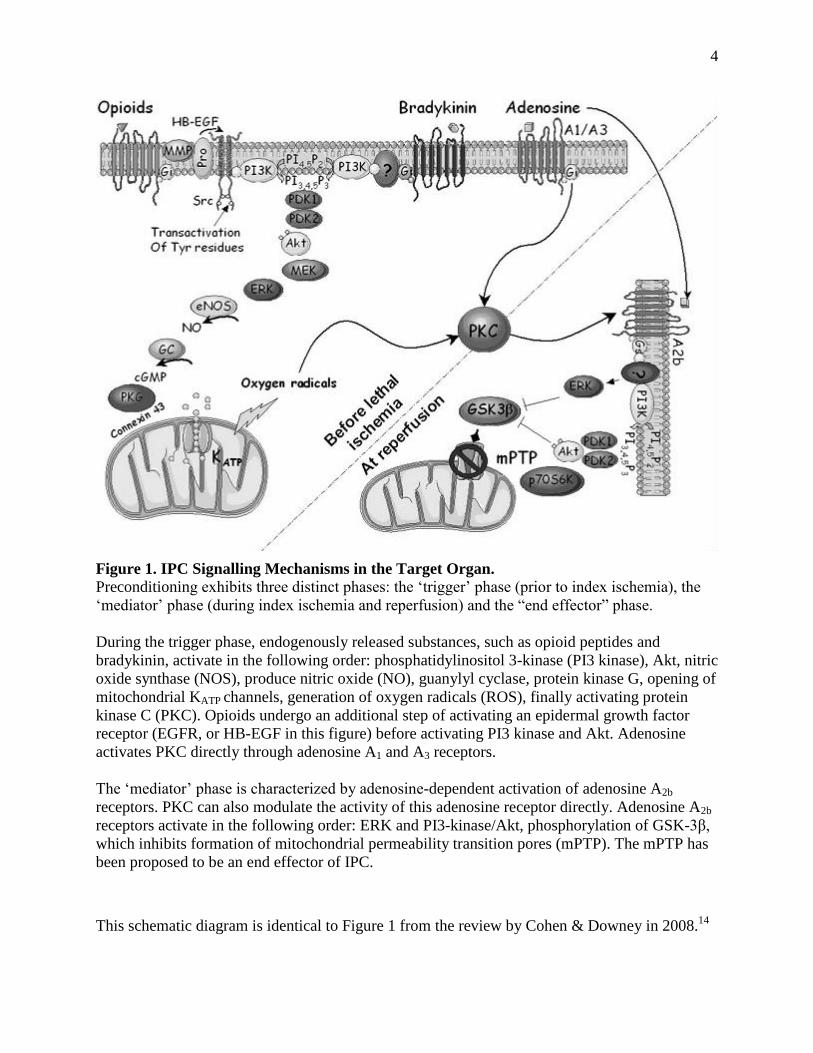
subject of intense debate. 4 (See Figure 1 for an overall summary)
4
14
This schematic diagram is identical to Figure 1 from the review by Cohen & Downey in 2008.14
Figure 1. IPC Signalling Mechanisms in the Target Organ.
Preconditioning exhibits three distinct phases: the ‗trigger‘ phase (prior to index ischemia), the
‗mediator‘ phase (during index ischemia and reperfusion) and the ―end effector‖ phase.
During the trigger phase, endogenously released substances, such as opioid peptides and
bradykinin, activate in the following order: phosphatidylinositol 3-kinase (PI3 kinase), Akt, nitric
oxide synthase (NOS), produce nitric oxide (NO), guanylyl cyclase, protein kinase G, opening of
mitochondrial KATP channels, generation of oxygen radicals (ROS), finally activating protein
kinase C (PKC). Opioids undergo an additional step of activating an epidermal growth factor
receptor (EGFR, or HB-EGF in this figure) before activating PI3 kinase and Akt. Adenosine
activates PKC directly through adenosine A1 and A3 receptors.
The ‗mediator‘ phase is characterized by adenosine-dependent activation of adenosine A2b
receptors. PKC can also modulate the activity of this adenosine receptor directly. Adenosine A2b
receptors activate in the following order: ERK and PI3-kinase/Akt, phosphorylation of GSK-3β,
which inhibits formation of mitochondrial permeability transition pores (mPTP). The mPTP has
been proposed to be an end effector of IPC.

5
1.2 Remote Preconditioning (rIPC):
1.2.a Background in rIPC:
In 1993, Przyklenk et al.15
first conceived the idea of remote ischemic preconditioning
(rIPC) wherein repeated brief episodes of ischemia and reperfusion in a non-local tissue/distant
organ has the ability to render the myocardium protected against ischemia/reperfusion injury.
Remote preconditioning of the myocardium has can be induced by other organs such as the liver,
small intestine, kidneys, and also from skeletal muscle ischemia (see Appendix III & IV).
Similarly, two windows of protection have been confirmed for rIPC16
. However, the focus of my
thesis will be on the first window of protection (called acute preconditioning/rIPC) which occurs
within 2 hours of the preconditioning stimulus and not the second window (called delayed
preconditioning/rIPC).
1.2.b Signalling Mechanisms in rIPC:
There is much controversy over the involvement of either a humoral (via blood) or
neurogenic (via nerves) pathway, or both, as a mechanism of transferring protection from the
preconditioned tissue/organ to the myocardium16
. Coronary effluent from preconditioned donor
rabbit hearts elicited protection in untreated hearts in the study by Dickson et al.17
, suggesting
the involvement of humoral protective factors. However, Gho et al.18
demonstrated that
protection by mesenteric artery occlusion (MAO) was abolished by ganglion blockade, providing
support for a neurogenic pathway in rIPC.
The actual trigger mechanisms involved to induce protection in the myocardium are
unclear. Multiple triggers have been suggested such as opioid peptides, adenosine, bradykinin,

6
and calcitonin-gene related peptide (CGRP). Once G-protein coupled receptors are triggered on
the cardiomyocyte cell surface, there is translocation of PKCε from the cytosol to the
mitochondrial membrane fraction, an event thought to be dependent on mitochondrial ROS
generation. However, it is yet to be determined whether activation of survival kinases during
reperfusion or inhibition of mPTP occurs. 19
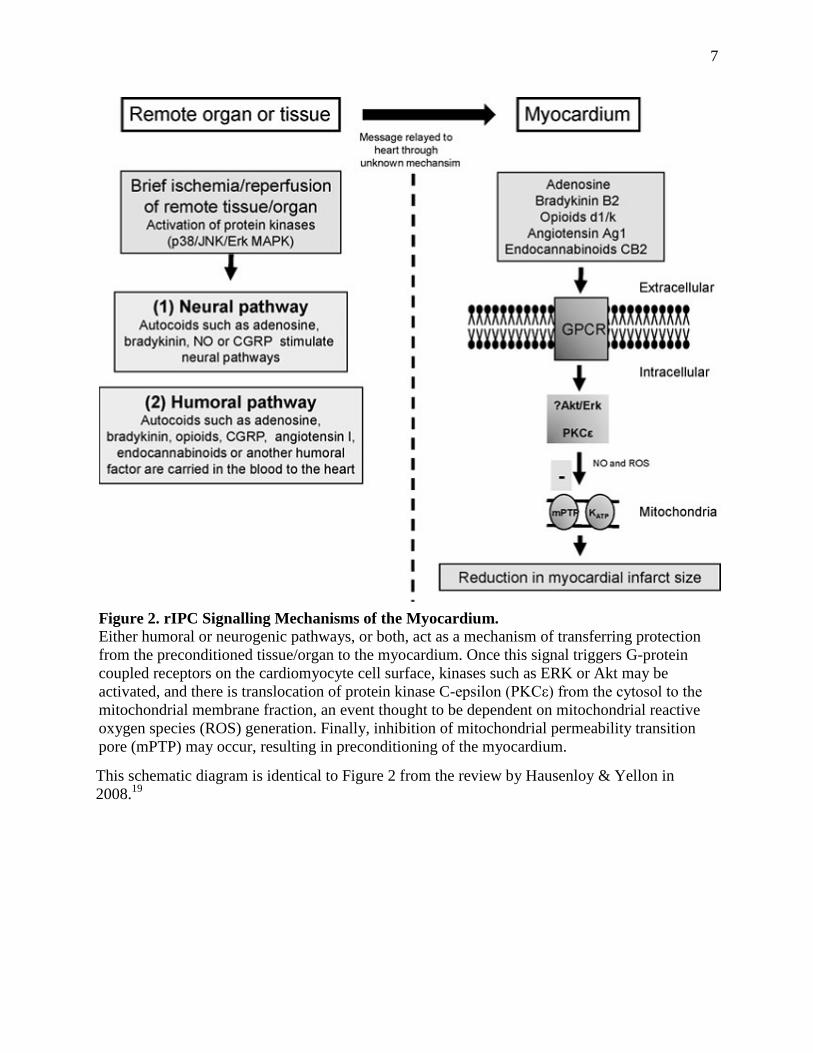
(See Figure 2 for an overall summary)
7
This schematic diagram is identical to Figure 2 from the review by Hausenloy & Yellon in
2008.19
Figure 2. rIPC Signalling Mechanisms of the Myocardium. Either humoral or neurogenic pathways, or both, act as a mechanism of transferring protection
from the preconditioned tissue/organ to the myocardium. Once this signal triggers G-protein
coupled receptors on the cardiomyocyte cell surface, kinases such as ERK or Akt may be
activated, and there is translocation of protein kinase C-epsilon (PKCε) from the cytosol to the
mitochondrial membrane fraction, an event thought to be dependent on mitochondrial reactive
oxygen species (ROS) generation. Finally, inhibition of mitochondrial permeability transition
pore (mPTP) may occur, resulting in preconditioning of the myocardium.

8
1.3 Involvement of Opioid Receptors:
1.3.a Opioids and Their Receptors:
Opioids exhibit various functions in the heart such as cardiac arrhythmogenesis and
modulation of vasculature20
. Opioid peptides consist of enkephalins, endorphins, dynorphins, and
endomorphins as well as other non-opioid substances, such as nociceptin, that exhibit similar
pharmacological properties20
. Enkephalins are most abundant in cardiac ventricles compared to
any other organ in the body aside from the central nervous system21
, suggesting the important
role of opioids in cardiomyocyte regulation and maintenance during stress. Each opioid class is
derived from a corresponding gene which produces a distinct hormone precursor. Enkephalins
are produced from pro-enkephalin, endorphins from pro-opiomelanocortin (POMC), dynorphins
from pro-dynorphin, and finally the endomorphin precursor has yet to be identified20
. In
particular, opioids in the myocardium exhibit unique features such as larger molecular weighted
peptides (e.g. Met5-enkephalin-Arg
6-Phe
7 or MERF and dynorphin A)
22. Regardless, the
myocardium contains all necessary machinery to generate all peptide forms and receptor types. 20
Minimum concentrations reported to protect rabbit cardiomyocytes from
ischemia/reperfusion damage by common endogenous opioids such as dynorphin B is 10µM152
and 1µM148
for Met-enkephalin. However, this does not include all known and unknown opioids
which may induce protection at lower concentrations.
The above mentioned peptide classes bind with varying affinities to three subtypes of Gi-
protein coupled opioid receptors, which are: delta (δ), kappa (κ), and mu (μ). Endorphins and
enkephalins are known to associate mostly with δ receptors (and to some extent μ), while

9
dynorphins are highly selective for κ opioid receptors. Endomorphins are most potent at μ
receptors. The κ receptor can be divided into κ1 and κ2, and the δ receptor into δ1 and δ2, and all
of these have been identified on rat and human cardiomyocytes20
. However, there is evidence
that the μ subtype (which exists as three isoforms, µ1, µ2, µ3 23
) is not expressed in adult
cardiomyocytes24,25
. The μ receptor is highly abundant in the central nervous system, suggesting
this subtype may be present at nerve ends in the myocardium; however, this requires further
investigation. Nonetheless, the tissue specificity of these opioids and their receptors as well as
their pharmacological functions within the heart is still a subject of study.
1.3.b Evidence of Opioid Involvement in IPC:
During episodes of myocardial ischemic stress, opioid levels were found to be elevated in
humans26
, suggesting an involvement of opioid peptides in preconditioning. A number of studies
have investigated the role of opioids in IPC. Mesenteric artery occlusion (MAO) in rats has
shown that morphine (a non-selective opioid agonist) could mimic IPC and this protection was
abolished by naloxone (a non-selective opioid antagonist)27
. In addition, coronary effluent that
was collected following IPC measured increased levels of endogenous opioid peptides.
There is controversy regarding which opioid receptor subtype is involved in ischemic
preconditioning. The involvement of δ receptors was proposed by Schultz et al.28
when µ and κ
receptor antagonists failed to block protection from 3 cycles of 5 min ischemia-5min reperfusion
in in vivo rat hearts. In addition, the detrimental effects of κ receptors to aggravate ischemia-
reperfusion injury have been reported in κ-opioid perfused isolated rat hearts29
and found to be
proarrhythmic for in vivo κ opioid-induced preconditioning in swine30
. Interestingly, κ receptors

10
have also been reportedly involved in the beneficial effects of IPC in rats by Wang et al. 31
.
Though δ and κ agonists reduced infarct size in isolated perfused hearts, Wang and colleagues
suggested that only the κ opioid agonist mimicked the antiarrhythmic effects of IPC in this study.
Very few studies have investigated the role of opioids during reperfusion. Gross et al.32
in
2004 determined that activation of opioid receptors during the reperfusion phase by morphine
(non-selective opioid agonist) and BW373U86 (selective δ agonist) reduced infarct size in rats.
However, this is an aspect of IPC that requires further exploration but is not the focus of my
thesis.
1.3.c Evidence of Opioid Involvement in rIPC:
The involvement of opioids in rIPC was proposed in a study by Dickson et al.33
in which
pre-treatment with naloxone (a non-selective opioid antagonist) blocked protection from
transferred coronary effluent from one isolated rabbit heart to another. The coronary effluent was
analyzed and found to contain Met- and Leu-enkephalin (endogenous agonists of δ and µ opioid
receptors)33,34
. Patel et al.35
had found that MAO in rats could reduce myocardial infarction, and
this effect was blocked by naloxone. Preconditioning by femoral artery occlusion (FAO) in rats
also reduced infarct size and lactate dehydrogenase levels (a marker of oxidative stress)36
. In a
separate study in isolated rat hearts, Weinbrenner et al.37
found that protection was abolished by
pre-treatment with naloxone and the free radical scavenger N-2-mercaptopropionyl glycine
(MPG), thus suggesting a relationship between opioid signalling in rIPC and ROS generation.
The same study demonstrated the involvement of the δ1 opioid receptor subtype in protection

11
induced by limb ischemia. However, a recent study by Zhang et al.36
has proposed the idea that it
is κ receptors, not δ, that mediate rIPC through femoral artery occlusion in rats.
The exact mechanism of opioid involvement is unclear. Skeletal muscle ischemia with
ganglion blockade by hexamethonium did not affect preconditioning in pigs38
. Thus, it is thought
that opioid peptides enter the blood stream through a humoral pathway and mediate their effects
through receptor binding at the target organ20
. In addition, similarities in signalling pathways
between rIPC and IPC are even less clear. However, there are implications of ROS generation
and inhibition of the mPTP with regards to opioids and rIPC37
.
1.4 Involvement of Bradykinin B2 Receptors:
1.4.a Bradykinin and Their Receptors:
Bradykinin (BK), a major player of the kinin family, is a potent vasodilatory protein that
is released into the blood stream to lower blood pressure following stimuli such as ischemia and
tissue damage. BK is produced when high-molecular-weight kininogen is released into the blood
stream following proteolytic cleavage by serine proteases called kallikreins39,40
. Bradykinin is
also degraded in the blood by angiotensin converting enzyme (ACE) (in this role, ACE is called
kininase II), carboxypeptidase N, and neutral endopeptidase 1.Kinins generally reduce vascular
resistance and increase vascular permeability41
through the release of nitric oxide (NO) and
prostacyclin (PGI2) from the endothelium42
.
The kinin receptors, B1 and B2 were first cloned in the 1990‘s from humans43,44
. The
constitutively active rabbit bradykinin B2 receptor (a Gi- and Gq- protein coupled receptor) was

12
cloned in 199545
and is the target of endogenous kinins such as bradykinin (BK) and Lys-
bradykinin (aka: kallidin). This receptor is also highly expressed in rabbit kidney and duodenum
but mRNA is present in rabbit heart45
. Bradykinin B2 receptors were reported on rat
cardiomyocytes in 199446
. The cloned rabbit B2 receptor is 92% homologous with humans,
indicating the homogeneity of this receptor across species. The inducible B1 receptor is thought
to activate as a result of tissue injury47
. Also, bradykinin is highly selective for the B2 receptor
over B1 in rabbit smooth muscle cells48
. The potent antagonist, Hoechst 140 (HOE140) is highly
selective for B2 receptors such that it is considered a non-competitive antagonist for this
receptor49,50
.
There is also some controversy over the existence of other kinin receptor subtypes such
as B3, B4, and B5 which are species specific51
. In addition, these receptor subtypes may be
expressed as varying homologs in different species52
.
The source of bradykinin in the heart is cardiac endothelium53
. BK levels are ten-fold
higher in tissues, such as the kidneys, heart, and brain, compared to plasma in rats53
and levels of
circulating BK in the blood are low in humans54
at basal conditions. However during ischemia,
kinin levels dramatically increase five-fold in plasma55,56
.
1.4.b Evidence of Bradykinin Involvement in IPC:
The first studies of bradykinin in IPC illustrated the beneficial effects of exogenous BK
to recover cardiac function through increased coronary flow and an improved metabolic profile
following ischemia57
. However, this protection in rats was abolished by the B2 receptor

13
antagonist, HOE140. This finding was recapitulated in dogs58
and rabbits59
when investigating
cardiac necrosis following ischemia. In vivo canine studies in which BK was injected into the
coronary artery showed a decrease in infarct size following coronary artery occlusion (CAO)60
,
which was subsequently abolished by HOE140. With respect to rabbits, ischemic
preconditioning was blocked by the administration of HOE140 and exogenous bradykinin has
been shown to reduce infarct size59
. Studies have also revealed increased kinin release following
local and global ischemia in rats, dogs, and humans61,62,63
.
Ebrahim et al.64
determined that exogenous bradykinin limits infarct size through a
concentration-dependent manner. Male rats were preconditioned on a Langendorff apparatus
with increasing concentrations of bradykinin. Concentrations >0.1µM were able to induce
protection from ischemia-reperfusion injury in rat hearts.
Kinin activation is thought to increase intracellular endothelial cyclic guanine
monophosphate (cGMP) through NO and increase cyclic adenosine monophosphate (cAMP)
through activation of prostacyclin (PGI2)65
. Also, B2 receptors from neonatal rats show G-protein
coupled activation of PI3 kinase66
.
1.4.c Evidence of Bradykinin Involvement in rIPC:
Bradykinin has been implicated in a combined neuronal and humoral pathway in rIPC
through the G-protein coupled B2 receptor. Schoemaker et al.67
abolished rIPC protection with
mesenteric artery occlusion (MAO) in early reperfusion through administration of the bradykinin
B2 receptor antagonist, HOE140. The authors suggested that bradykinin exerts its effects in rIPC

14
by activating the neuronal pathway via stimulatory sensory nerves. In a later finding by Wolfrum
et al.68
, the involvement of PKCε downstream of bradykinin was also implicated.
1.5 Involvement of CGRP Receptors:
1.5.a CGRP and Their Receptors:
Calcitonin gene-related peptide (CGRP) is a vasodilatory neuropeptide released by vagal
and capsaicin-sensitive sensory nerves. CGRP is widely recognized as the most potent
vasodilator in the cardiovascular system69
through its actions on vascular smooth muscle70
. In
addition to these roles, CGRP is thought to be involved in neurogenic inflammation,
nociception71,72
, and vascular hypertrophy73
. Recently, this peptide has been implicated in
protection from myocardial infarction70,74
.
CGRP is a 37 amino acid peptide present in cardiac C-fibres (unmyelinated free nerve
fibres)75
and cardiomyocytes76
, and is highly abundant in central and peripheral neurons77
. Of the
two isoforms, α- and β-CGRP, α-CGRP is known to bind to the CGRP receptor. α-CGRP is a
neuropeptide derived from the calcitonin gene in neural tissues78
. β-CGRP is highly homologous
to the α isoform but is derived from another gene located near the calcitonin gene on
chromosome 1179
. Since the cloning of human CGRP in the early 1980‘s, CGRP is found to be
highly homologous among mammalian species80
.
The binding of CGRP to its receptor depends on the association of the calcitonin-like
receptor with two accessory proteins. Calcitonin-like receptor (CLR) is a rhodopsin-like Gs-
protein couple receptor first discovered in the 1990s81,82
exhibiting an unknown function83
. The

15
association of the CLR to specific receptor activity-modifying proteins (RAMPs) determine
which ligands can bind to the receptor84,85
. RAMPs are single-pass transmembrane proteins
which are 148 amino acids in length. CLR coupled to RAMP1 allows binding of CGRP to the
receptor (now a CGRP receptor). CLR coupled to RAMP2 allows binding of another vasodilator,
adrenomedullin (a peptide associated with the adrenal medulla tumour, pheochromocytoma).
Finally, CLR coupled with RAMP3 produces a dual CGRP and adrenomedullin receptor. When
comparing the different accessory proteins, RAMP2 and RAMP3 are 30% homologous to
RAMP185
. The second accessory protein associated with CLR is the receptor component protein
(RCP) which forms a component of the CLR-RAMP1 complex86
. Both rat and human CLRs
have been cloned and expression of CLR in rat aortic smooth muscle cells has been reported87,88
,
however, no such investigations have been conducted in rabbits.
1.5.b Evidence of CGRP Involvement in IPC
CGRP, adrenomedullin89
, and intermedin90
(another protein that binds to CLR in
association to all three RAMPs) are considered cardioprotective 91,92,93
. With respect to
cardioprotection, coronary effluent from isolated rat hearts displayed elevated CGRP levels
during preconditioning94
. Several clinical studies have also shown elevated CGRP during early
reperfusion following acute myocardial infarction in human patients74
suggesting that CGRP is
an endogenous substance produced by the myocardium95
. In isolated rat hearts, preconditioning
by global ischemia was abolished by CGRP (8-37), a CGRP receptor antagonist91,94
. In vivo
studies with a CGRP antibody also abolished ischemic preconditioning in rats96
(only the
abstract is available, since the article is in Chinese). CGRP has also been implicated in delayed
preconditioning and has been extensively studied in gastrointestinal preconditioning97,98,99
.

16
Chai et al.92
examined the role of CGRP in isolated rat hearts to induce IPC. The authors
measured CGRP release in coronary effluent and determined that CGRP improved left
ventricular pressure and coronary flow in a heart subjected to ischemia and reperfusion. A
concentration of 1µM improved coronary flow and left ventricular pressure, suggesting that
concentrations >1µM will protect isolated rat hearts from ischemia-reperfusion injury.
CGRP downstream of receptors is known to activate protein kinase C, but not ATP-
sensitive K+ (KATP) channels, and inhibits tumour necrosis factor –α (TNF-α)
100,101. Also,
suggestions have been made that CGRP-induced cardioprotection in rats is activated by nitric
oxide release102
.
1.5.c Evidence of CGRP Involvement in rIPC:
A number of remote intestinal preconditioning studies of the myocardium suggest that
CGRP is released by capsaicin sensory nerves into the blood stream97,98
and activates PKCε in
the myocardium103
. Wolfrum et al.103
found that administration of the CGRP receptor antagonist,
CGRP (8-37) abolished this protection. The experiments by Wolfrum et al. demonstrated
increased CGRP plasma levels following preconditioning, thus suggesting a humoral pathway in
rIPC. However, this CGRP protective effect was abolished by ganglion blockade, yet CGRP
levels in the plasma remained unaffected. Also, a clinical study by Li et al.104
has shown that
cardiac ischemic preconditioning improved lung preservation during value replacement
operations through CGRP receptor activation.

17
1.6 Involvement of Adenosine Receptors:
1.6.a Adenosine and Their Receptors:
Adenosine plays several important biological roles, such as energy transfer, an anti-
inflammatory agent, and can act as an inhibitory neurotransmitter. This purine nucleoside is
formed both intracellularly and extracellularly in cardiomyocytes, endothelial cells, and vascular
cells by dephosphorylation of AMP by 5‘-nucleotidase or by hydrolysis of S-adenosyl-
homocysteine105,106
. Degradation of adenosine occurs when extracellular concentrations of
adenosine increase, facilitating diffusion of adenosine into the cell. Adenosine is then broken
down to AMP by adenosine kinase or into inosine by adenosine deaminase (ADA) which is
found in all mammalian tissues107
. Though extracellular adenosine is mostly broken down by
ADA via erythrocytes in the blood stream105
, ventricular cardiomyocytes do not posses
extracellular adenosine deaminase108
.
There are four known G-protein coupled adenosine receptors: A1, A3 (both inhibitory;
coupled to Gi, thus resulting in a decrease in cAMP), A2a and A2b (both stimulatory, coupled to
Gs which increases cAMP; A2b is also coupled to Gq, which mediates phosphoinositide
metabolism)109
. A1 and A3 receptors are involved in the trigger phase of classical IPC, whereas
the A2b subtype has been implicated in the mediator phase14
. The binding affinity of adenosine to
rat A1 receptors is 3-30nM and is >1µM for A3 receptors110
. A2b is considered a low affinity
receptor as adenosine concentrations required to stimulate cAMP levels in the brain are >10µM
compared to A2a (requires 0.1-1µM of adenosine)111
. Only A1, A2b and A3 receptors have been
shown to be definitively expressed in cardiomyocytes112
, while A2a has been found in coronary

18
arterioles113
. With respect to cell signalling, A1 receptors are known to activate phospholipase C,
while A3 receptors activate phospholipase D109
.
1.6.b Evidence of Adenosine Involvement in IPC:
Studies have shown that in vivo infusion of adenosine deaminase through coronary
arteries in swine throughout the preconditioning event and index ischemia abolished IPC
protection114
. Also, in vivo rabbit studies by Thornton et al.115
demonstrate that A1 adenosine
agonists, PIA and 2-chloro-N6-cyclopentyladenosine (CCPA), triggered protection before the
index ischemia (i.e. longer, damaging ischemia), but not activation via A2a receptors by
CGS21680. This finding was later recapitulated in other species such as dogs116
, swine114
,
rabbits117
, and humans118
. In vivo studies with rabbits indicated that 8-(p-
sulfophenyl)theophylline (8-SPT) and PD115,199, non-selective and A2 adenosine blockers
respectively, blocked protection from ischemia by coronary artery occlusion (CAO)119
.
In addition, rabbit hearts perfused with adenosine and the A1 agonist, N6-1-(phenyl-2R-
isopropyl) adenosine (abbreviated: PIA), conferred protection on a level similar to IPC 119
. The
above study determined that saturation of adenosine receptors was required to induce
cardioprotection and short pulses or low concentrations of receptor activation was insufficient to
confer protection. Armstrong et al.120
states that the minimum adenosine concentration required
to protect rabbit cardiomyocyte is 10µM. This is also supported by Peart et al.121
in which 10µM
of adenosine reduced cell necrosis but did not limit contractile dysfunction in isolated perfused
rat hearts. However, 50µM of adenosine was able to reduced cell death and improve contractile
dysfunction.

19
Lui et al.122
demonstrated the involvement of A3 receptors by agonism with N6-2-(4-
aminophenyl)ethyl-adenosine (APNEA) and A3 antagonism with BW-A1433 in isolated rabbit
hearts. Later in 1997, Auchampach et al.123
illustrated that activation of A3 receptors by N6-(3-
iodobenzyl) adenosine-5‘-N-methyluronamide (IB-MECA) mimicked IPC in rabbits as well.
Murry et al.1 discovered that IPC protection could be abolished when 8-SPT, a non-
selective adenosine blocker was administered during reperfusion following the index ischemia.
In addition, the A1/A2 receptor agonist 5‘-(N-ethylcarboxamido) adenosine (NECA) given at
reperfusion induced protection, but was blocked by the A2b blocker MRS1754 in rabbits124
.
MRS1754 administered at reperfusion in classical IPC also abolished protection125
. Recently in
2007, the novel agonist BAY60-6583, that is highly selective for A2b receptors, induced
protection at reperfusion126
. However, this protection was abolished by MRS1754. Thus, the
above studies demonstrate the role of A2b receptors in the mediator phase (i.e. during
reperfusion) of IPC.
1.6.c Evidence of Adenosine Involvement in rIPC:
The mechanism of adenosine activation in rIPC was proposed by Liem et al.127
to act via
a non-humoral pathway that differed from classical IPC. In this study, 4 cycles of 15 min CAO
followed by 2 cycles of 15 min adenosine-dependent CAO induced preconditioning tolerance in
rats. Also, interstitial adenosine initially increased, but rapidly decreased to basal levels.
However, 4 cycles of 15 min CAO followed by 3 cycles of adenosine-independent 3 min
mesenteric artery occlusion (MAO) maintained cardioprotection. Even though tolerance of

20
adenosine-dependent preconditioning can occur in IPC (i.e. repeated cycles of ischemia-
reperfusion can no longer induce protection), rIPC may employ alternate pathways to maintain
this protection. To further support the role of adenosine in rIPC, Pell et al.128
in 1998
demonstrated that protection from renal artery occlusion (RAO) in rabbits was abolished by
treatment with 8-(p-sulfophenyl)theophylline (8-SPT). The same study determined that rIPC
blockade could also be achieved when 8-SPT was administered during reperfusion, suggesting
the role of adenosine in both the trigger and mediator phase.
A study by Pang et al.129
demonstrated that plasma adenosine concentrations increased
with skeletal muscle rIPC, and this effect was partially abolished by reserpine, an inhibitor of the
vesicular monoamine transporter (VMT, a transporter of catecholamines at synaptic nerve
endings). In addition, Addison et al.38
has shown that blockade by 8-SPT and the free radical
scavenger, mercaptopropionyl glycine (MPG) completely abolished skeletal muscle rIPC
cardioprotection in pigs.
1.7 Other Potential Preconditioning Substances:
A number of G-protein coupled and non-G-protein coupled receptors have been
confirmed to play a role in classical IPC and their involvement in rIPC has been suggested.
However, the focus of my thesis will be on G-protein coupled receptors, and in particular, the
major receptors that have been implicated: δ and κ opioid receptors, adenosine receptors, and
bradykinin B2 receptors; as well as the recently proposed CGRP receptor. Unfortunately,
investigating the claims of other receptors (both G-protein and non-G protein coupled) is beyond

21
the scope of this thesis. Nonetheless, this section will bring to light other receptors that may be
involved in rIPC.
With respect to other GPCRs, α1 adrenergic receptors which bind both norepinephrine
and epinephrine have been implicated in myocardial rIPC; however, the results in this area are
conflicting130,131
. Losartin (an angiotensin I receptor blocker) was found to block renal artery
occlusion (RAO) preconditioning in rat myocardium132
. Prostaglandin E2 (PGE2) has also been
implicated in rIPC gastric preconditioning by Brzozowski et al.133
. However, there is yet to be a
study on the role of PGE2 in myocardial preconditioning. The endocannabinoid receptor CB2 has
also been implicated in a humoral role in rIPC. Intestinal ischemia studies have shown that
blockage of the CB2 receptor abolished myocardial protection in mice134
and rats135
. Finally,
studies have demonstrated that carbachol (a muscarinic M2 receptor agonist) mimicked
preconditioning in isolated rabbit whole hearts, suggesting the role of acetylcholine in IPC2. To
date, no studies have been conducted in rIPC involving muscarinic M2 receptors.
A number of substances have been implicated to trigger rIPC. These include nitric oxide
(NO) and the following cytokines: tumour necrosis factor-α (TNF-α), nuclear factor- κB (NF-
κB), interleukin-6 (IL-6), and interleukin-1β (IL-1β). Also, free radicals and heat shock proteins
such as hemoxygenase-1 (HO-1) have been suggested. However, these substances do not activate
G-protein coupled receptors and are beyond the scope of this thesis. That being said, the findings
presented in this section are not conclusive and requires exploration of the mechanisms involved
in rIPC. Consequently, many of the above studies are preliminary and require further testing.19

22
1.8 Opioid-Adenosine Receptor Cross-talk:
1.8.a Background:
A number of non-preconditioning studies have implicated opioid-adenosine cross-talk in
the central nervous system. Interestingly, adenosine A1 and A2 blockers antagonize the
synergistic signalling actions between opioid and dopamine D2 receptors in the rat CNS136
. In
this study, adenosine deaminase and the adenosine blocker, BW A1434U, prevented increased
activation of cAMP downstream of colocalized δ opioid and dopamine D2 receptors in
transfected NG108-15/D2 cells. Also, Eisenach et al.137
conducted a clinical trial in which
patients were administered intrathecal opioid (morphine and fentanyl), which resulted in
increased adenosine release in cerebrospinal fluid. This study provided support for an opioid-
adenosine role in analgesia in humans.
The novel idea of ‗cross-talk‘ between opioid and adenosine receptors in ischemic
preconditioning was first proposed by Peart et al.138
in 2003. Peart et al. found that myocardial
protection induced by either morphine (non-selective opioid receptor agonist) or CCPA (A1
adenosine receptor agonist) was blocked, for both morphine and CCPA, by either δ opioid or A1
adenosine specific blockers. This study was conducted in an in vivo coronary artery occlusion
(CAO) model in rats. Later, Peart et al.139
in 2005, they claimed that adenosine kinase inhibition
reduced infarct size in the same in vivo CAO model in rats. A1 adenosine, A3 adenosine, and δ
opioid receptor antagonists abolished this protection, suggesting that stimulation of these
receptors resulted in adenosine kinase inhibition. Currently, there are no studies of opioid-
adenosine receptor cross-talk in rIPC.

23
1.8.b Adenosine Levels in the Blood:
Concentrations of extracellular adenosine are 30-300nM in tissues and can increase
during ischemia to 10µM when ATP is converted to adenosine111
. An increase in adenosine
during ischemia suggests that adenosine could enter the blood stream to reach the target organ.
However, the half-life of adenosine in the blood stream is 0.6-1.5 seconds due to constant
deamination by adenosine deaminase from erythrocytes, pericytes, and endothelial cells140
. Since
there are no studies demonstrating an inhibition of nucleoside transportation that prevents
adenosine degradation in the blood, it is postulated that adenosine either works entirely through
the neurogenic pathway, or partially activates this pathway to induce the release of humoral
mediators4. Liem et al.
141 provided evidence of a neurogenic pathway in which exogenous
adenosine mimicked MAO rIPC, however this protection was abolished by prior ganglion
blockade.
1.8.c Potential Mechanisms of Cross-talk:
Two mechanisms have been proposed by Peart et al.139
to mediate ‗cross-talk‘ between
opioid and adenosine receptors. One possibility for cross-talk is via adenosine kinase inhibition
in cardiomyocytes (see Figure 3). Adenosine kinase continually converts adenosine to AMP,
effectively maintaining very low concentrations of adenosine within cardiomyocytes. In turn,
adenosine continually diffuses into cardiomyocytes to replenish adenosine levels. However,
Peart et al. postulates that opioid receptor stimulation inactivates adenosine kinase, resulting in a
build-up of adenosine within cardiomyocytes. Since the uptake of adenosine by cells is
dependent on a concentration gradient, there is an accumulation of extracellular adenosine. The
amassing of adenosine around cardiomyocytes activates adenosine receptors on the cell

24
membrane and initiates downstream signalling that ultimately results in ischemic
preconditioning. In support of this mechanism, Deussen et al.142
provides evidence that
inhibition of adenosines deaminase doubles cardiac adenosine release, whereas inhibition of
adenosine kinase causes a 30-fold increase in adenosine. In addition, the role of adenosine kinase
is thought to be important in IPC since a low pH due to hypoxia inhibits adenosine kinase, which
may contribute to increases in adenosine during ischemia142
.
The second mechanism of cross-talk proposed by Peart et al.139
is heterodimerization of
opioid and adenosine receptors. This hypothesis is addressed in detail in the General Discussion
chapter of this thesis (see section 9.2.a Dimerization of G-protein Coupled Receptors).

25
(Peart et al., 2005)139
A
Figure 3. The Adenosine Kinase Inhibition Hypothesis.
(A) Adenosine kinase continually converts adenosine to AMP, effectively maintaining very low
concentrations of adenosine within cardiomyocytes. In turn, adenosine continually diffuses into
cardiomyocytes to replenish adenosine levels.
(B) Opioid receptor stimulation inactivates adenosine kinase, resulting in a build-up of adenosine
within cardiomyocytes. Since the uptake of adenosine by cells is dependent on a concentration
gradient, there is an accumulation of extracellular adenosine. The amassing of adenosine around
cardiomyocytes activates adenosine receptors on the cell membrane and initiates downstream
signalling that ultimately results in ischemic preconditioning.
B

26
- Chapter 2 -
RATIONALE
The mechanisms involved in IPC have been studied extensively; however, the exact
triggering molecules involved in rIPC are a subject of controversy. Possible candidates for rIPC
include opioid peptides, bradykinin, adenosine, and CGRP. Even less is known about possible
cross-talk between the G-protein coupled receptors for these molecules in IPC or rIPC. Thus, a
comprehensive study is needed to evaluate the similarities between classical IPC and rIPC in
regard to receptor activation and explore the possibility of receptor cross-talk, a novel concept in
myocardial preconditioning.
The most appealing aspect of rIPC is its clinical applicability. Unlike classical IPC and
the recent phenomenon of post-conditioning (i.e. short periods of re-occlusion during long
reperfusion), which is restricted to specific settings such as cardiac surgery, rIPC does not
require invasive treatment that can initiate harmful side effects. Cheung et al.143
have already
demonstrated the use of rIPC in a clinical setting wherein four cycles of 5 min limb ischemia and
reperfusion was able to reduce myocardial injury in children undergoing cardiac surgery. The
clinical use of rIPC in acute myocardial infarction may prove to be a major medical advance.
Given the practical value of rIPC alone, the elucidation of mechanisms involved in rIPC is of
significant importance.

27
- Chapter 3 -
OBJECTIVES
3.1 Hypothesis
I hypothesize that one or more humoral protective factor(s) released by hind limb rIPC
activate two G-protein coupled receptors on the cardiomyocyte cell surface, adenosine and
opioid receptors, and involves a cross-talk mechanism between these two receptors to induce
protection against cardiomyocyte cell death from ischemia-reperfusion injury.
3.2 Specific Aims
1. To characterize the protection induced by rIPC in freshly isolated rabbit ventricular
cardiomyocytes.
2. To determine the role of G-protein coupled cell membrane receptors in rIPC in
cardiomyocytes.
3. To determine whether rIPC involves cross-talk between adenosine and opioid receptors.

28
- Chapter 4 -
RESEARCH DESIGN & METHODS
4.1 Animals and Human Subjects
Male New Zealand rabbits were used to obtain both preconditioned and control dialysate.
Isolated cardiomyocytes for fresh cell experiments were obtained from either male or female
New Zealand adult rabbits. All rabbits were ~12-14 weeks old, weighed ~3.2-3.5 kg, and had
been designated specific pathogen free. All animals were maintained and used in accordance
with recommendations from the Department of Laboratory Animal Services at the Hospital for
Sick Children.
Human dialysate was obtained from male human volunteers. Prior to obtaining blood,
subjects were instructed not to exercise or consume caffeine and remain calm during the
procedure.
4.2 Isolation of Adult Cardiomyocytes
4.2.a Operative Procedure:
Male or female New Zealand White rabbits were treated with a mild topical anaesthetic
(xylocaine) prior to being anesthetised via the ear vein with pentobarbital (30mg/kg) and heparin
(100U/kg) to prevent coagulation. Once rabbits reached a surgical level of anaesthesia, hearts
were rapidly excised and hung by the aortic root onto a Langendorff apparatus.

29
4.2.b Digestion Protocol:
Buffers were made in 1L of distilled water from a Millipore filtration system according to
specifications in Table 1and Figure 4. This Krebs-Henseleit buffer (contains the following in
mM: 0.5 MgSO4, 4.7KCl, 10.0 NaHCO3, 1.2 KH2PO4, 10.0 Dextrose, 20.0 HEPES, and 128.3
NaCl) was filtered and adjusted to pH 3.7. All buffers were oxygenated at 37°C with 95% O2 and
5% CO2. Rabbit hearts were mounted on a pressure-controlled Langendorff apparatus via the
aorta and immediately perfused with Ca+ Buffer (with 0.99mM CaCl2) for 2 min in order to
expel blood from the heart. Hearts were then perfused with Ca- Buffer containing 9.99mM
ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid (EGTA) for 7 min to chelate
calcium and suppress heart contraction. Enzymatic digestion was conducted via recirculation of
the Collagenase Buffer (with 200U/ml crude collagenase) until the heart was soft. Samples were
taken throughout the digestion process to ensure sufficient perfusion of the heart. Once digestion
was complete, the heart was agitated in the Mincing Buffer to separate cardiomyocytes from
connective tissue.
The isolate then underwent subsequent filtration and wash steps in Wash Buffer (2%
albumin). Dead cells were removed by low centrifugation (at 500g for 2 min, all subsequent
spins occur at 500g for 1min) and removal of the supernatant. Cardiomyocytes in Wash Buffer
were gradually re-introduced to calcium in a step-wise manner until concentrations reached
0.9mM CaCl2. Finally, calcium tolerant cardiomyocytes were re-suspended in Reactive Buffer
(0.1% albumin, 0.99mM CaCl2) to remove excess calcium from the re-introduction mentioned
earlier. Only calcium tolerant cardiomyocyte isolates with <30% cell necrosis were used in fresh
cell experiments.

30
Table 1. Krebs-Henseleit Buffer Composition.
Reagent Concentration (mM) Osmolarity (mOsm)
MgSO4 0.5 1.0
KCl 4.7 9.4
NaHCO3 10.0 20.0
KH2PO4 1.2 4.8
Dextrose 10.0 10.0
HEPES 20.0 20.0
NaCl 128.3 256.6
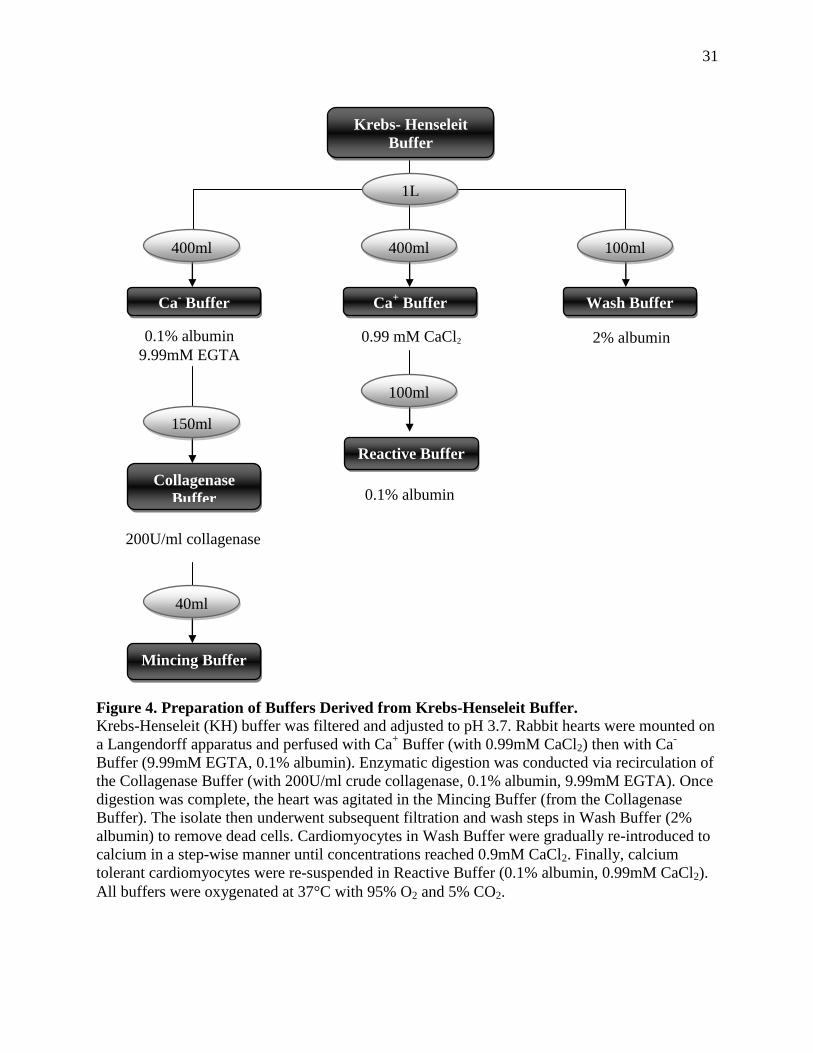
31
Figure 4. Preparation of Buffers Derived from Krebs-Henseleit Buffer.
Krebs-Henseleit (KH) buffer was filtered and adjusted to pH 3.7. Rabbit hearts were mounted on
a Langendorff apparatus and perfused with Ca+ Buffer (with 0.99mM CaCl2) then with Ca
-
Buffer (9.99mM EGTA, 0.1% albumin). Enzymatic digestion was conducted via recirculation of
the Collagenase Buffer (with 200U/ml crude collagenase, 0.1% albumin, 9.99mM EGTA). Once
digestion was complete, the heart was agitated in the Mincing Buffer (from the Collagenase
Buffer). The isolate then underwent subsequent filtration and wash steps in Wash Buffer (2%
albumin) to remove dead cells. Cardiomyocytes in Wash Buffer were gradually re-introduced to
calcium in a step-wise manner until concentrations reached 0.9mM CaCl2. Finally, calcium
tolerant cardiomyocytes were re-suspended in Reactive Buffer (0.1% albumin, 0.99mM CaCl2).
All buffers were oxygenated at 37°C with 95% O2 and 5% CO2.
0.1% albumin
9.99mM EGTA
200U/ml collagenase
0.1% albumin
Ca+ Buffer
0.99 mM CaCl2
Wash Buffer
2% albumin
Ca- Buffer
Reactive Buffer
Mincing Buffer
Collagenase
Buffer
Krebs- Henseleit
Buffer
400ml 400ml 100ml
150ml
100ml
40ml
1L

32
4.2.c Comparison of Digestion Protocols:
A number of modifications to the digestion protocol were studied in an effort to improve
isolate yields. Digestions were modified in two protocols, each supplemented with DNase I
(degrades DNA, 0.02mg/ml; Worthington)144
, hyaluronidase (degrades hyaluronic acid,
chondroitin, and chondroitin sulphates, 0.6mg/ml; Sigma)145
, ovamucoid trypsin inhibitor
(inhibits trypsin, derived from ovamucoid extracts, 0.1mg/ml; Worthington)145
, or soybean
trypsin inhibitor (derived from soybean extracts, 1mg/ml; Worthington)144
and compared to
digestions with only collagenase (200U/ml crude collagenase; Worthington).
In the first protocol, hearts were initially digested with collagenase for 10 min, removed
from the Langendorff system, minced, and re-suspended in collagenase buffer supplemented
with a reagent for a further 10 min (n=2). This protocol was designed to ensure that
cardiomyocytes from the same heart were used in order to avoid heart-to-heart variability. Also,
these were preliminary experiments to confirm the supplemented reagents were non-toxic. The
second protocol involved re-circulation of collagenase with the supplemented reagent in a
Langendorff until digestion was complete (n=1-2). All isolates underwent similar wash and
filtration steps as in a non-supplemented digestion. The results of these modifications indicate
that re-circulation with only collagenase is the most optimal method of digestion. (See Figure 5)

33
Figure 5. Comparison of Digestion Protocols.
Digestions with collagenase were modified in two protocols that were supplemented with DNase
I (degrades DNA, 0.02mg/ml), hyaluronidase (degrades hyaluronic acid, chondroitin, and
chondroitin sulphates, 0.6mg/ml), ovamucoid trypsin inhibitor (inhibits trypsin, derived from
ovamucoid extracts, 0.1mg/ml), or soybean trypsin inhibitor (derived from soybean extracts,
1mg/ml) and compared to digestions with only collagenase (200U/ml crude collagenase).
In the first protocol (Minced Heart), hearts were initially digested with collagenase for 10 min,
removed from the Langendorff system, minced, and re-suspended in collagenase buffer
supplemented with a reagent for a further 10 min. The second protocol (Perfusion) involved re-
circulation of collagenase with the supplemented reagent in a Langendorff until digestion was
complete. All isolates underwent similar wash and filtration steps for a non-supplemented
digestion. The results of these modifications indicate that re-circulation with only collagenase is
the most optimal method of digestion.

34
4.3 Dialysate Preparation
Male New Zealand White rabbits were anaesthetized with akmezine (0.25mg/kg) (a pre-
anaesthetic containing ketamine, acepromazine, and atropine) followed by pentobarbital
(30mg/kg) with heparin (100U/kg) via the ear vein then placed on a ventilator. rIPC rabbits were
preconditioned by 4 cycles of 5 min hind limb ischemia and 5 min reperfusion using a blood
pressure cuff. Control rabbits were only anaesthetized and ventilated for the same time period as
rIPC rabbits. About 100-150ml of blood was quickly drawn from the left carotid artery
immediately after the preconditioning or control protocol. Rabbits were monitored throughout
the procedure to detect any hemodynamic variability. The whole blood obtained was centrifuged
(3000g for 20 min) to obtain 50 or 100ml of plasma which was then dialysed using a 12-14kDa
cut-off membrane against a 10 fold greater volume of water. Thus, the dialysate contents were
limited to <14 kDa. Dialysate was divided into 900µl aliquots and immediately stored at -80°C.
Prior to use, the frozen aliquots were thawed only once and reconstituted with 10µl of 10 fold
concentrated Krebs-Henseleit buffer. Since dialysate was created against water, a concentrated
salt solution was supplemented in order to achieve the same physiological salt levels as in 1x
Krebs-Henseleit buffer. Osmolarity and pH was measured in the dialysate before exposing the
solution to cardiomyocytes.
For human subjects, 100 ml of blood was withdrawn before (Control Human Dialysate)
and after (rIPC Human Dialysate) 4 cycles of 5 min ischemia (>20mmHg above systolic
pressure) and 5 min reperfusion to the upper arm by a blood pressure cuff. Dialysate was
obtained using the identical procedure as described above for rabbits. The protocol for preparing

35
both rabbit and human dialysate is shown on Figure 6 and was previously described by Shimizu
et al.146
. All dialysate used in these studies were prepared by an external laboratory*.
* Dialysate prepared by Jing Li (Dr. Andrew Redington‘s Lab; Research Institute, Hospital for Sick Children).

36
(Shimizu et al., 2009)146
Figure 6. Rabbit and Human Dialysate Preparation.
rIPC rabbits were preconditioned by 4 cycles of 5 min hind limb ischemia and 5 min reperfusion
using a blood pressure cuff. Control rabbits were only anaesthetized and ventilated for the same
time period as rIPC rabbits. About 100-150ml of blood was quickly drawn from the left carotid
artery immediately after the preconditioning or control protocol.
For human subjects, 100 ml of blood was withdrawn before (Control Human Dialysate) and after
(rIPC Human Dialysate) 4 cycles of 5 min ischemia (>20mmHg above systolic pressure) and 5
min reperfusion to the upper arm by a blood pressure cuff. Dialysate was obtained using the
identical procedure as described above for rabbits.
The whole blood obtained was centrifuged (3000g for 20 min) to obtain 50 or 100ml of plasma
which was then dialysed using a 12-14kDa cut-off membrane against a 10 fold greater volume of
water. Thus, the dialysate contents were limited to <14 kDa. Dialysate was divided into 900µl
aliquots and immediately stored at -80°C. Prior to use, the frozen aliquots were thawed only once
and reconstituted with 10µl of 10 fold concentrated Krebs-Henseleit buffer. Osmolarity and pH
was measured in dialysate before exposing the solution to cardiomyocytes.

37
4.4 Fresh-Cell Experimental Model
4.4.a General Protocol:
Cardiomyocyte isolates were evenly divided into Eppendorff tubes such that each group
contained >200µl per group. Cardiomyocytes were re-suspended in 1ml Reactive Buffer and
placed in 12-well plates (2ml total volume) at 37°C and 100% O2. Samples from the Baseline
group were taken after stabilization and at the end of the experiment to ensure that
cardiomyocyte viability remained stable. All cardiomyocytes underwent a 20 min stabilization
period in suspension. Cardiomyocytes were subjected to 20 min of a particular treatment,
depending on the protocol. This was followed by another 20 min wash period in suspension with
fresh Reactive Buffer such that any residual drug(s) in the supernatant was/were removed. All
treatment groups subsequently underwent 45 min damaging simulated ischemia (SI) and 60 min
simulated reperfusion (SR). SI was conducted by low speed centrifugation (500g for 1 min) of
cardiomyocytes into a pellet and leaving only a thin layer of supernatant with an oil layer on top
of cells. Cardiomyocytes then underwent SR through suspension in fresh Reactive Buffer.
Samples from the treatment groups were taken before SI and after SR. The purpose of these fresh
cell protocols was to observe the effect of a treatment on cell survival following the simulation of
normally damaging ischemia and reperfusion.147
(See Figure 7)

38
Figure 7. General Protocol: Isolated Rabbit Cardiomyocytes.
All cardiomyocytes underwent a 20 min stabilization period in suspension. Cardiomyocytes were
subjected to 20 min of a particular treatment, depending on the protocol. This was followed by
another 20 min wash period in suspension with fresh Reactive Buffer such that any residual
drug(s) in the supernatant was/were removed. All treatment groups subsequently underwent 45
min damaging simulated ischemia (SI) and 60 min simulated reperfusion (SR). SI was conducted
by low speed centrifugation (500g for 1 min) of cardiomyocytes into a pellet, leaving only a thin
layer of supernatant with an oil layer on top of cells. Cardiomyocytes then underwent SR through
suspension in Reactive Buffer. Samples from the treatment groups were taken before SI and after
SR (see arrows).

39
4.4.b AIM (1) Protocol:
AIM (1): To characterize the protection induced by rIPC in freshly isolated rabbit
ventricular cardiomyocytes.
Cardiomyocytes freshly isolated from non-preconditioned donor rabbit hearts were
preconditioned by exposing them to10 min ischemic pelleting (IPC treatment group) or rIPC
dialysate, human or rabbit derived (rIPC Dialysate group). Cardiomyocyte groups that were not
preconditioned were subjected to 10 min re-suspension in buffer (IR group) or control human or
rabbit dialysate (Control Dialysate group). Treatment groups underwent a wash period in fresh
buffer for 20 min. All groups were then subjecting to a long period of 45 min simulated ischemia
(SI) and 60 min simulated reperfusion (SR). Samples were taken before and after SI/SR. (See
Figure 8)
Figure 8. AIM (1) Protocol: To Characterize Dialysate Protection.
Cardiomyocytes freshly isolated from non-preconditioned donor rabbit hearts were
preconditioned by exposing them to10 min ischemic pelleting (IPC treatment group) or rIPC
dialysate, human or rabbit derived (rIPC Dialysate group). Cardiomyocyte groups that were not
preconditioned were subjected to 10 min re-suspension in buffer (IR group) or control human or
rabbit dialysate (Control Dialysate group). Treatment groups underwent a wash period in fresh
buffer for 20 min. All groups were then subjecting to a long period of 45 min simulated ischemia
(SI) and 60 min simulated reperfusion (SR). Samples were taken before and after SI/SR.

40
4.4.c AIM (2) Protocol:
AIM (2): To determine the role of G-protein coupled cell membrane receptors in rIPC in
cardiomyocytes.
Freshly isolated cardiomyocytes were either subjected to 10 min IR or rIPC (using
dialysate) in the presence of an antagonist (or its vehicle) for 20 min to prevent specific
activation of each receptor being studied (adenosine, bradykinin B2, and κ opioid receptors,
and CGRP receptors). Treatment groups underwent a wash period in fresh buffer for 20 min. All
groups were then subjecting to a long period of 45 min simulated ischemia (SI) and 60 min
simulated reperfusion (SR). Samples were taken before and after SI/SR. (See Figure 9)
Figure 9. AIM (2) Protocol: The Role of Cell Membrane Receptors.
Freshly isolated cardiomyocytes were either subjected to 10 min IR or rIPC (using dialysate)
in the presence of an antagonist (or its vehicle) for 20 min to prevent specific activation of
each receptor being studied (adenosine, bradykinin B2, and κ opioid receptors, and CGRP
receptors). Treatment groups underwent a wash period in fresh buffer for 20 min. All groups
were then subjecting to a long period of 45 min simulated ischemia (SI) and 60 min
simulated reperfusion (SR). Samples were taken before and after SI/SR.

41
4.4.d AIM (3) Protocol:
AIM (3): To determine whether rIPC involves cross-talk between adenosine and opioid
receptors.
Cardiomyocytes were subjected to 10 min classic IPC, specific opioid receptor agonists
(Ag), such as Met-enkephalin (δ-specific agonist) or dynorphin B (κ-specific agonist) in the
presence of adenosine receptor blockers, such as 8-SPT (non-selective adenosine blocker) for 20
min. Treatment groups underwent a wash period in fresh buffer for 20 min. All groups were then
subjecting to a long period of 45 min simulated ischemia (SI) and 60 min simulated reperfusion
(SR). Samples were taken before and after SI/SR. (See Figure 10)
Figure 10. AIM (3) Protocol: Investigate Receptor Cross-talk.
Cardiomyocytes were subjected to 10 min classic IPC, specific opioid receptor agonists, such as
Met-enkephalin (δ-specific agonist) or dynorphin B (κ-specific agonist) in the presence of
adenosine receptor blockers, such as 8-SPT (non-selective adenosine blocker) for 20 min.
Treatment groups underwent a wash period in fresh buffer for 20 min. All groups were then
subjecting to a long period of 45 min simulated ischemia (SI) and 60 min simulated reperfusion
(SR). Samples were taken before and after SI/SR.

42
4.5 Trypan Blue Exclusion Assay
Cardiomyocyte samples were taken before and after simulated ischemia (SI) and
reperfusion (SR) to assess cell death by a trypan blue exclusion assay. 10µL of cardiomyocytes
were suspended in 13µl of 85mOsm hypotonic Tyrode Buffer (contains the following in mM:
10.7 KCl, 1.5 NaH2PO4, 14.7 NaHCO3, 5.6 glucose, 1.6 MgSO4, 7.2 CaCl2, 3.0 amylobarbitone,
0.5% glutaraldehyde, 0.5% trypan blue; see Table 2 for buffer composition) and 13µl of Reactive
Buffer for 1 min in order to allow dead cardiomyocytes to take up the trypan dye. Samples were
place on a hemocytometer and images were taken with a light microscope at 20x magnification.
Cardiomyocytes unable to exclude the trypan blue dye due to the loss of membrane integrity
were counted as dead cells, whereas live cells that maintained membrane integrity were able to
exclude the dye and were seen as clear (see Figure 11). The percent cell death was counted from
>300 cells per sample. Images were taken by the Micro-Cap software (Spectrum) and ImageJ
software (NIH) was used to count sample images.
Table 2. Tyrode Buffer Composition.
Reagent Concentration (mM) Osmolarity (mOsm)
KCl 10.7 21.4
NaH2PO4 1.5 3.0
NaHCO3 14.7 29.4
Glucose 5.6 5.6
MgSO4 1.6 3.2
CaCl2 7.2 21.6
Total Osmolarity ~85 mOsm
Amylobarbitone 3.0
Glutaraldehyde 0.5%
Trypan Blue 0.5%

43
Figure 11. Trypan Blue Exclusion Assay.
Cardiomyocyte samples were taken before and after ischemia (SI) and reperfusion (SR) to
assess cell death by a trypan blue exclusion assay. Cardiomyocytes unable to exclude the
trypan blue dye due to the loss of membrane integrity were counted as dead cells (Nonviable
Cells), whereas live cells (Viable Cells) that maintained membrane integrity were able to
exclude the dye and were seen as clear

44
4.5.a Inter-Observer Error Data:
Inter-observer measurements were collected to ensure consistency in cell counting from
various observers and experiments. Three experiments (Sept. 17, 2007; Sept 19, 2007; Sept 25,
2007) were conducted using the rabbit dialysate protocol described in Figure 8. Treatment
groups included a Baseline, ischemia-reperfusion (IR), ischemic preconditioning (IPC), control
rabbit dialysate (Control Dialysate), and preconditioned rabbit dialysate (rIPC Dialysate).
Sample pictures were taken before and after simulated ischemia and simulated reperfusion.
Cardiomyocytes were exposed to hypotonic trypan blue and images were counted. Four different
observers (Harinee Surendra, Elena Kuzmin, Sue Omar, and Mohammad Escandarian) counted
cell necrosis from the same images in all three experiments (a total of 45 images). The mean was
calculated for the following parameters: live, dead, and total cardiomyocytes counted and the
percent necrosis for reach observer. Statistical analysis was conducted via correlation
coefficients and correlation p-values. The results indicated that all counts correlated among
observers for a given parameter (coefficients approached 1) and the probability of incorrectly
concluding this correlation was very small among the observers (p<0.0001). Thus, the method of
counting cell necrosis through trypan blue staining showed little variation among observers. (See
Table 3 for results)
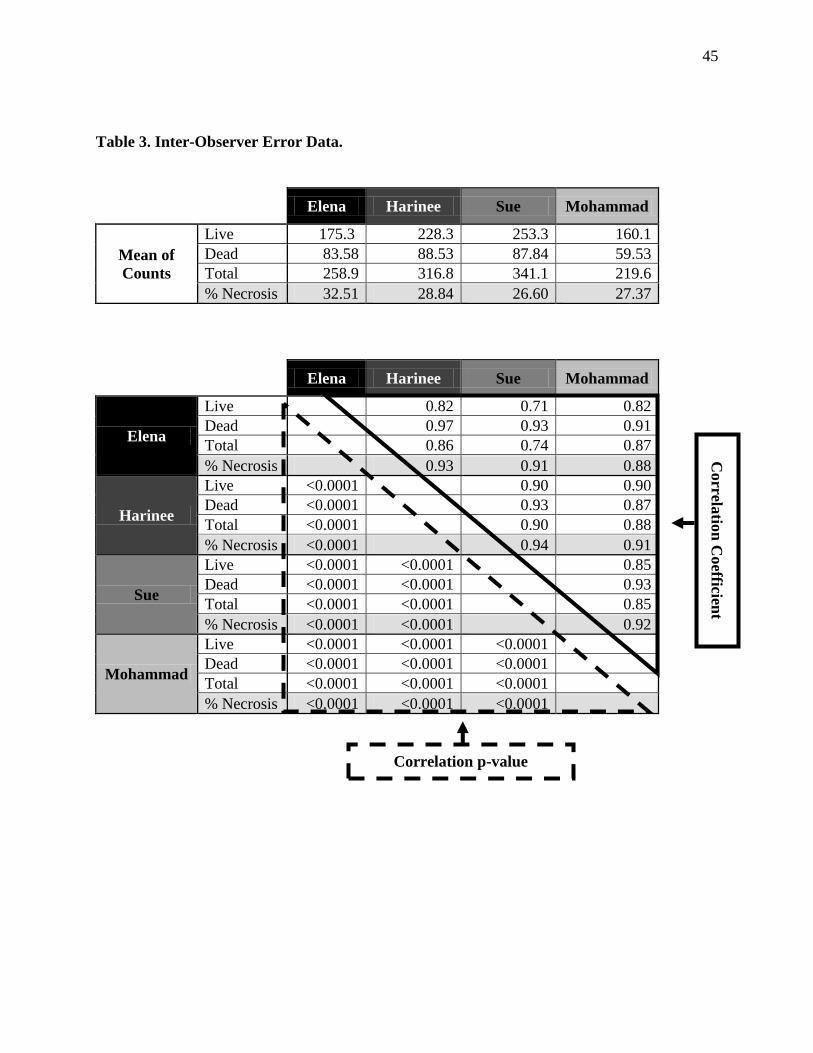
45
Table 3. Inter-Observer Error Data.
Elena Harinee Sue Mohammad
Mean of
Counts
Live 175.3 228.3 253.3 160.1
Dead 83.58 88.53 87.84 59.53
Total 258.9 316.8 341.1 219.6
% Necrosis 32.51 28.84 26.60 27.37
Elena Harinee Sue Mohammad
Elena
Live 0.82 0.71 0.82
Dead 0.97 0.93 0.91
Total 0.86 0.74 0.87
% Necrosis 0.93 0.91 0.88
Harinee
Live <0.0001 0.90 0.90
Dead <0.0001 0.93 0.87
Total <0.0001 0.90 0.88
% Necrosis <0.0001 0.94 0.91
Sue
Live <0.0001 <0.0001 0.85
Dead <0.0001 <0.0001 0.93
Total <0.0001 <0.0001 0.85
% Necrosis <0.0001 <0.0001 0.92
Mohammad
Live <0.0001 <0.0001 <0.0001
Dead <0.0001 <0.0001 <0.0001
Total <0.0001 <0.0001 <0.0001
% Necrosis <0.0001 <0.0001 <0.0001
Correla
tion
Co
efficient
Correlation p-value

46
4.6 Drugs
All drugs used as agonists or antagonists for these studies competitively bound to cell-
membrane receptors and the concentrations used have previously been published in peer-
reviewed preconditioning literature to pharmacologically precondition (agonist) or inhibit the
protection of classical or remote IPC (antagonists).
Antagonists used for opioid receptors were naloxone (Nal), naltrindole (NTI), and GNTI.
Naloxone (chemical name: (5a)- 4,5-Epoxy-3,14-dihydro-17-(2-propenyl)morphinan-6-one
hydrochloride; from Sigma) is a highly potent, non-selective, competitive inhibitor of opioid
receptors used at 100µM148,149,150,151
. Naltrindole (chemical name: 17-(cyclopropylmethyl)-6,7-
dehydro-4,5a-epoxy-3,14-dihyd roxy-6,7-2',3'-indolomorphinan hydrochloride; Sigma) is a δ
opioid blocker used at 10nM152
with a 223- and 346-fold greater selectivity for δ over µ and κ
opioid receptors, respectively153,154,155
. GNTI (chemical name: 5'-Guanidinyl-17-
(cyclopropylmethyl)-6,7-dehydro-4,5a-ep oxy-3,14-dihydroxy-6,7-2',3'-indolomorphinan
dihydrochloride; Tocris Biosciences) is a κ-opioid blocker used at 1nM152
with a 208- and 799-
fold selectively for κ receptors over µ and δ opioid receptors, respectively156,157,158
.
Agonists used for opioid receptors were Met-enkephalin (ME) and dynorphin B (DynB).
Met-enkephalin is an endogenous ligand of δ opioid receptors used at 100µM152
that is highly
selective for δ receptors, and to a lesser extent µ, over κ receptors20
. The minimum concentration
of Met-enkephalin required to protect rabbit cardiomyocytes from ischemia/reperfusion damage
is 1µM148
. Dynorphin B (aka: rimorphin; Phoenix Pharmaceuticals) is an endogenous peptide
that is highly selective for κ opioid receptors over µ and δ receptors20
. DynB was used at 100µM

47
and the minimum concentration reported to protect rabbit cardiomyocytes from
ischemia/reperfusion damage is 10µM152
.
This study used Hoechst 140 (HOE140) as a blocker of bradykinin B2 receptors and
calcitonin gene-related peptide 8-37 (CGRP (8-37)) as a blocker for CGRP receptors. Hoechst
140 (aka: icatibant; Sigma) is a highly selective inhibitor of bradykinin B2 receptors and is
negligibly active against B1 receptors49,49 ,159
when used at 5µM160
. CGRP (8-37) (calcitonin
gene-related peptide, fragment 8-37; Sigma) is a fragment of the endogenous α-CGRP human
peptide and inhibits CGRP receptors and not calcitonin receptors at 5nM103,161,162
.
Finally, the antagonist used for adenosine receptors was 8-SPT. 8-SPT (chemical name:
8-(p-sulfophenyl)theophylline; Sigma) is non-selective blocker of adenosine receptors used at
100µM120,163
.
4.7 Other Methods
4.7.a Western Blotting:
Western blots were performed to determine the presence of cell-membrane receptors on
rabbit cardiomyocytes. For µ opioid receptors, a synthetic rabbit monoclonal antibody
corresponding to amino acid residues 220–250 of the human µ opioid receptor sequence was
used (Santa Cruz Biotechnology). This human epitope is 100% homologous with both the cloned
rat and mouse µ-opioid receptor sequence over residues 220-250. The δ opioid receptor was
detected with a rabbit polyclonal synthetic antibody which corresponded to amino acids 3-17 of
both the mouse and rat δ opioid receptor (Abcam). The antibody used for κ opioid receptors was

48
a goat polyclonal antibody corresponding to amino acids 330 – 380 of the human κ-opioid
receptor (Santa Cruz Biotechnology). This fragment is 88% homologous to the rat and mouse κ-
opioid receptor sequence over these residues.
The calcitonin-like receptor in rabbits was detected using a mouse polyclonal antibody of
human origin, corresponding to amino acids 23-133 (Abnova). This immunogen has a 92%
homology to the mouse calcitonin-like receptor over these residues.
4.7.b Multiple Reaction Monitoring Mass Spectrometry:
Multiple reaction monitoring (MRM) mass spectrometry was used to identify molecules
in the rabbit dialysate. MRM mass spectrometry is a form of tandem mass spectrometry which
ionizes chemical compounds into multiple fragment ions. Once the particular fragments are
detected by a magnetic and/or electric field, the mass-to-charge ratio is calculated and data
analysis allows identification of the unknown compound. The purpose of utilizing MRM mass
spectrometry, compared to other methods such as chromatography, is the high degree of
sensitivity (can detect substances at concentrations as low as 1-3nM) and selectivity (is able to
distinguish adenosine from other purines, such as inosine which differs by 1Da). In order to
measure accurate concentrations, all substances were compared to known standard
concentrations prepared in water (see Figure 28 in Appendix V for a plot of the standard
concentrations).
In order to investigate the role of adenosine kinase inhibition in cross-talk, MRM mass
spectrometry was also used to measure levels of adenosine in supernatant samples. Supernatant

49
was obtained from freshly isolated rabbit cardiomyocytes after 20 min stabilization, after 10 min
of exposure to dynorphin B (κ-opioid receptor agonist) in fresh Reactive Buffer, and after a 20
min wash period in which cardiomyocytes were suspended in fresh Reactive Buffer (see Figure
12). Measured adenosine concentrations were compared to known standard concentrations
prepared in Krebs-Henseleit buffer (see Figure 29 in Appendix V for a plot of the standard
concentrations).
All mass spectrometry measurements were conducted on an API 4000 Triple Quadrupole
with Agilent LC by an external laboratory†.
† MRM mass spectrometry was conducted by Michelle Young (AIMS Lab; Department of Chemistry, University of
Toronto) and Michael Tropak (Dr. Callahan‘s Lab, Hospital for Sick Children).
Figure 12. Adenosine Kinase Inhibition Protocol.
Supernatant was obtained from freshly isolated rabbit cardiomyocytes after 20 min stabilization,
after 10 min of exposure to dynorphin B (κ-opioid receptor agonist) in fresh Reactive Buffer, and
after a 20 min wash period in which cardiomyocytes were suspended in fresh Reactive Buffer.
MRM mass spectrometry was used to measure levels of adenosine in supernatant samples in
order to investigate the role of adenosine kinase inhibition in cross-talk.

50
4.7.c Statistics:
Averages and standard error of the mean were reported for all results. For all experiment
sets, the N (number of different rabbit dialysates used) and n (number of times experiments were
replicated for a particular dialysate) were stated. All treatment groups were statistically assessed
by an Analysis of Variance (ANOVA) Scheffé post hoc test for multiple comparisons. For
groups that were not distributed normally, an ANOVA Kruskal-Wallis post hoc test was
conducted. Statistics was conducted with Statview (Abacus Corporation).

51
- Chapter 5 -
CHARACTERIZATION OF THE PROTECTION INDUCED BY RIPC
5.1 Rabbit and Human Preconditioned Dialysate Induces Protection
Cardiomyocytes were subjected to 10 min. exposure to either preconditioned rIPC
dialysate or non-preconditioned control dialysate. Results indicate that remotely preconditioned
dialysate, whether rabbit (31.9% ±3.7 vs. 48.0%±2.7 IR, p=0.0002; N=6, n=1 each) or human
derived (32.4%±2.9 vs. 48.0%±2.7 IR, p=0.0001; N=1, n=6), reduces SI/SR induced cell death
similar to classical IPC (30.5%±3.1 vs. 48.0%±2.7 IR, p=0.0006; n=6). (See Figure 13 and Table
4).

52
Table 4. Rabbit and Human Dialysate Results (Mean ± SEM).
Stabilization End
Baseline 26.8 ± 1.0 32.3 ± 2.3
Before SI After SR
IR 33.1 ± 2.9 48.0 ± 2.7
IPC 30.1 ± 1.6 30.5 ± 3.1
Control Rabbit Dialysate 29.5 ± 2.0 49.1 ± 4.8
rIPC Rabbit Dialysate 29.5 ± 1.4 31.9 ± 3.7
Control Human Dialysate 31.0 ± 1.7 47.4 ± 1.9
rIPC Human Dialysate 28.9 ± 1.5 32.4 ± 2.9
Figure 13. Rabbit and Human Dialysate Administered Prior to Ischemia-Reperfusion.
Cardiomyocytes were subjected to 10 min. exposure to either preconditioned rIPC dialysate or
non-preconditioned control dialysate. Results indicate that remotely preconditioned dialysate,
whether rabbit (31.9% ±3.7 vs. 48.0%±2.7 IR, p=0.0002; N=6, n=1 each) or human derived
(32.4%±2.9 vs. 48.0%±2.7 IR, p=0.0001; N=1, n=6), reduces SI/SR induced cell death similar to
classical IPC (30.5%±3.1 vs. 48.0%±2.7 IR, p=0.0006; n=6).

53
5.1.a Dialysate Characterization:
Substances in rabbit control and rIPC dialysate were identified using MRM mass
spectrometry. Opioids such as Met-enkephalin and dynorphin B had no detectable peaks and
Met5-enkephalin-Arg
6-Phe
7 (MEAP) had a concentration of <0.033 µM in control dialysate and
<0.031 µM in rIPC dialysate. Similarly, acetylcholine and angiotensin II were undetectable in
the dialysate. Adenosine in control dialysate was <0.035µM and <0.80 µM in rIPC dialysate.
Inosine was detected at maximum concentrations of 0.583µM in control dialysate and 0.456µM
in rIPC. Bradykinin was <0.0034 µM in control dialysate and was at levels below measurement
in rIPC dialysate. Norepinephrine was <0.021 µM in control dialysate and <0.019 µM in rIPC
dialysate. The above substances were at either undetectable or at minuscule levels that remained
unaffected in either the control or rIPC dialysate (with the exception of adenosine). (See Table 5
for results; see Figure 28 in Appendix V for a plot of the standard concentrations).
Table 5. Characterization of Rabbit Dialysate Using MRM Mass Spectrometry.
Control Rabbit Dialysate (µM) rIPC Rabbit Dialysate (µM)
Met-Enkephalin No peak No peak
Dynorphin B No peak No peak
MEAP No peak – 0.033 No peak – 0.031
Adenosine 0.001-0.035 0.003-0.80
Inosine 0.103-0.583 0.056-0.456
Bradykinin No peak-0.0034 <0
Norepinephrine 0.0087-0.021 0.0080-0.019
Acetylcholine No peak No peak
Angiotensin II No peak No peak
5.1.b Discussion:
Remotely preconditioned (rIPC) rabbit dialysate conferred protection to rabbit
cardiomyocytes on a level similar to IPC. Non-preconditioned (Control) dialysate and
cardiomyocytes subjected to ischemia-reperfusion alone did not provide protection (see Figure

54
13). Thus, there is a protective factor in rIPC dialysate that travels to distant organs through a
humoral route.
Literature on rIPC suggests two mechanisms of transferring protection from one organ to
another. The first is a neuronal pathway, in which substances are released from the stimulus
organ and activate local nerves to release protective factors at the target organ. As evidence for
this pathway, Gho et al.18
demonstrated that ganglion blockade could abolish protection from
mesenteric artery occlusion (MAO). Another method of transferring protection is the humoral
pathway and is the focus of this thesis. Coronary effluent from preconditioned donor hearts
provided protection to untreated virgin hearts in a study by Dickson et al17
. For many substances
implicated in rIPC, there is evidence for a dual neurogenic and humoral pathway which may
work simultaneously in concert or independent of each other16
. However, our model does not
involve the role of the neurogenic pathway, since there is no neuronal involvement in isolated
rabbit cardiomyocytes, and the dialysate used to elicit protection is derived from blood plasma.
Therefore, the protection from rabbit rIPC dialysate indicates that a humoral mechanism alone
can confer protection to distant organs.
Human dialysate can also protect rabbit cardiomyocytes, indicating cross-species
reactivity of the preconditioned dialysate. Though preconditioning has been shown in many
species, very few papers have investigated if protection from one species can elicit protection in
another, i.e. inter-species rIPC, except Shimizu et al. 146
(our data regarding human and rabbit
dialysate to induced protection was presented in this paper) . Since rIPC mechanisms in rabbits
may also apply to humans, cross-species reactivity of preconditioned dialysate is clinically

55
relevant for humans. However, human dialysate from different volunteers are needed to
determine variability in cardioprotection, since control conditions are difficult to enforce in
humans compared to rabbits.
MRM-MS analysis has shown various G-protein coupled receptor ligands are either
undetectable (Met-enkephalin, dynorphin B, acetylcholine, and angiotensin II) or well below
levels that protect in tissues (MEAP, adenosine, bradykinin, and norepinephrine) (see Table 5).
However, this list is a sample survey of molecules that have been suggested in rIPC. For
example, there are a diverse number of opioids and opioid-like molecules that bind to opioid
receptors. To further complicate the matter, Pugsley20
points out that many opioid peptides have
yet to be discovered. Similarly, a number of molecules can bind to a single GPCR.
Unfortunately, an exhaustive survey of substances that bind to G-protein coupled receptors is
beyond the scope of this thesis. However, we did investigate the most common agonist for a
particular receptor through MRM-MS. Considering the above mentioned limitations in
characterizing the dialysate, a better approach would be to investigate the particular receptors
involved in rIPC by blocking their activation.

56
- Chapter 6 -
THE ROLE OF CELL MEMBRANE RECEPTORS IN RIPC
6.1 The Role of Opioid Receptors
6.1.a Survey of Opioid Receptors in Rabbit Cardiomyocytes:
In order to determine the existence of the δ, κ, and µ opioid receptors in cardiomyocytes,
western blotting (n=2) revealed the presence of all three receptors in rabbit whole heart tissues.
The δ and κ subtype were found in rabbit cardiomyocytes, δ was also located in rabbit and rat
brain. The µ subtype was in rabbit brain tissue but absent in rabbit cardiomyocytes, suggesting
the µ subtype is present in neuronal tissue within rabbit whole hearts. (See Figure 14).

57
Figure 14. Western Blot Analysis of Opioid Receptors in Rabbit Tissues.
In order to determine the existence of the δ, κ, and µ opioid receptors in cardiomyocytes, western
blotting (n=2) revealed the presence of all three receptors in rabbit whole heart tissues. The δ and
κ subtype were found in rabbit cardiomyocytes, δ was also located in rabbit and rat brain. The µ
subtype was in rabbit brain tissue but absent in rabbit cardiomyocytes, suggesting the µ subtype
is present in neuronal tissue within rabbit whole hearts.
Survey of Opioid Receptors in Rabbit Cardiomyocytes

58
6.1.b Non-selective Opioid Receptor Blockade of Protection:
The effect of naloxone (a non-selective opioid blocker; 100µM concentration was used;
Takasaki, 1999148
) in conjunction with rabbit dialysate was studied. Blockers of opioid receptor
activity abolished the protection induced by the preconditioned dialysate (43.5%±1.5 vs. 31.3%
± 2.0, p=0.001; N=2, n=4,1). (See Figure 15 and Table 6).

59
Table 6. Naloxone Results (Mean ± SEM).
Group Stabilization End
Baseline 27.4 ± 1.4 28.2 ± 1.5
Baseline + Nal 26.8 ± 0.9 31.0 ± 1.7
Before SI After SR
IR 27.9 ± 2.3 43.0 ± 1.1
IR + Nal 26.7 ± 2.8 45.0 ± 1.6
rIPC Rabbit Dialysate 28.1 ± 1.9 31.3 ± 2.0
rIPC Rabbit Dialysate + Nal 28.7 ± 1.6 43.5 ± 1.5
Figure 15. Naloxone Administered Prior to Rabbit Dialysate.
The effect of naloxone (a non-selective opioid blocker; 100µM concentration was used) in
conjunction with rabbit dialysate was studied. Blockers of opioid receptor activity abolished the
protection induced by the preconditioned dialysate (43.5%±1.5 vs. 31.3% ± 2.0, p=0.001; N=2,
n=4,1).

60
6.1.c δ-Opioid Receptor Blockade of Protection:
The effect of naltrindole (NTI; a δ opioid blocker with a 350 fold selectivity for δ
receptors over κ receptors; 10nM concentration was used on cardiomyocytes; Cao, 2003152
) in
conjunction with rabbit dialysate was studied. Blockers of δ opioid receptor activity completely
abolished the protection induced by the preconditioned dialysate (43.2%±2.0 vs. 29.6%±1.6,
p=0.0003; N=2, n=3,2). (See Figure 16 and Table 7).
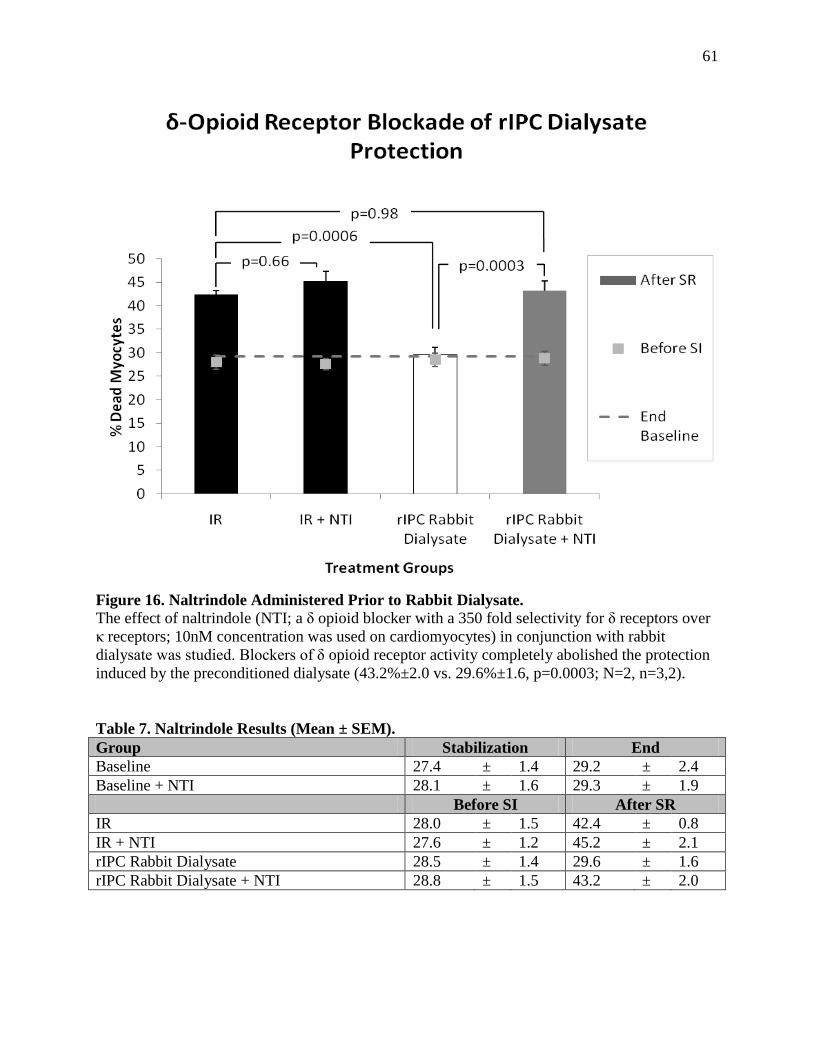
61
Table 7. Naltrindole Results (Mean ± SEM).
Group Stabilization End
Baseline 27.4 ± 1.4 29.2 ± 2.4
Baseline + NTI 28.1 ± 1.6 29.3 ± 1.9
Before SI After SR
IR 28.0 ± 1.5 42.4 ± 0.8
IR + NTI 27.6 ± 1.2 45.2 ± 2.1
rIPC Rabbit Dialysate 28.5 ± 1.4 29.6 ± 1.6
rIPC Rabbit Dialysate + NTI 28.8 ± 1.5 43.2 ± 2.0
Figure 16. Naltrindole Administered Prior to Rabbit Dialysate.
The effect of naltrindole (NTI; a δ opioid blocker with a 350 fold selectivity for δ receptors over
κ receptors; 10nM concentration was used on cardiomyocytes) in conjunction with rabbit
dialysate was studied. Blockers of δ opioid receptor activity completely abolished the protection
induced by the preconditioned dialysate (43.2%±2.0 vs. 29.6%±1.6, p=0.0003; N=2, n=3,2).

62
6.1.d κ-Opioid Receptor Blockade of Protection:
The effect of GNTI (a κ selective opioid blocker with an 800 fold selectivity for κ
receptors over δ receptors; 1nM concentration was used; Cao, 2003152
) in conjunction with rabbit
dialysate was studied. Blockers of κ-opioid receptor activity completely abolished the protection
induced by the preconditioned dialysate (46.8%±2.7 vs. 29.6%±1.6, p=0. 0002; N=2, n=3, 2).
(See Figure 17 and Table 8).

63
Table 8. GNTI Results (Mean ± SEM).
Group Stabilization End
Baseline 27.4 ± 1.4 29.2 ± 2.4
Baseline + GNTI 30.9 ± 0.4 32.0 ± 1.3
Before SI After SR
IR 28.0 ± 1.5 42.4 ± 0.8
IR + GNTI 26.5 ± 0.9 45.0 ± 2.3
rIPC Rabbit Dialysate 28.0 ± 1.5 29.6 ± 1.6
rIPC Rabbit Dialysate + GNTI 29.3 ± 1.3 46.8 ± 2.7
Figure 17. GNTI Administered Prior to Rabbit Dialysate.
The effect of GNTI (a κ selective opioid blocker with an 800 fold selectivity for κ
receptors over δ receptors; 1nM concentration was used) in conjunction with rabbit
dialysate was studied. Blockers of κ-opioid receptor activity completely abolished the
protection induced by the preconditioned dialysate (46.8%±2.7 vs. 29.6%±1.6, p=0.0002;
N=2, n=3, 2).

64
6.1.e Discussion:
Literature regarding µ opioid receptors confirms that this receptor subtype is not present
in rabbit cardiomyocytes24,25
. Also, there is indirect evidence that denies the involvement of µ
receptors in preconditioning. Fentanyl (a selective µ agonist) administered during
preconditioning was unable to abolish protection in Langendorff perfused rabbit hearts164
.
There is little surprise regarding the involvement of opioids in rIPC. Multiple studies
have suggested that naloxone (a non-selective opioid antagonist) can abolish protection across
species and in different organs. Dickson et al. 33
used naloxone to blocked protection from
preconditioned coronary effluent in untreated rabbit hearts. Patel et al.35
determined that MAO-
induced cardioprotection was blocked by naloxone in rats. Weinbrenner et al.37
attests to a
similar findings when failing to precondition isolated rat hearts in the presence of naloxone.
However, controversy lies in which receptor subtype is involved in cardioprotection. In
our isolated rabbit cardiomyocyte model, both δ and κ opioid receptors are involved in a humoral
mechanism of rIPC (see Figure 16 and Figure 17). Interestingly, Cao et al.152
has also shown that
both Met-enkephalin (δ opioid agonist) and dynorphin B (κ opioid agonist) administered before
the index ischemia can protect isolated rabbit cardiomyocytes. Naltrindole (δ receptor blocker)
and GNTI (κ receptor blocker) administered during classical IPC abolished this protection. Also,
Weinbrenner et al.37
has observed the involvement of δ1 in isolated rat hearts. More recently,
Zhang et al.36
has reported the involvement of only κ receptors in rIPC when δ receptor blockers
failed to abolish protection from femoral artery occlusion (FAO). However, the authors

65
attributed the controversy of their findings to variations with the experimental models and
species used when compared to previous studies.
Another reason for the discrepancies regarding opioid receptor subtype involvement is
promiscuity of opioid peptides and heterodimerization of receptors. Opioids have the ability to
bind to multiple receptor subtypes20
. Also, many endogenous peptides that are subtype-specific
can operate as non-selective agonists at higher concentrations. The δ-specific Met-enkephalin
and the µ-specific morphine are such examples. The versatility of endogenous opioid peptides to
bind to multiple receptors can be explained by the high degree of homology among receptor
subtypes. The µ subtype is 58% and 67% homologous to δ and κ receptively, whereas δ and κ
share 61% homology165
. Another explanation for the involvement of both δ and κ opioid
receptors is the evidence of dimerization across and within different subtypes. There is evidence
of hybrid δ and µ receptors that have unique structures, pharmacology, and functions upon
activation by either δ or µ-specific agonists166
.
The results so far suggest the involvement of δ and κ opioid receptors in mediating a
humoral mechanism of rIPC protection. However, dialysate characterization using MRM-MS
was unable to detect Met-enkephalin, dynorphin B, or significant levels of the more common
endogenous opioid peptide in coronary effluent, MEAP. Therefore, at least one of the trigger
molecule(s) in dialysate is/are opioid-like in nature. However, since the factor(s) is/are not these
common endogenous peptides, this provides room for discovering a novel/unfamiliar opioid
involved in both δ and κ receptor activation in rIPC.

66
6.2 The Role of Bradykinin B2 Receptors
6.2.a Bradykinin B2 Receptor Blockage Conserves Protection:
The effect of Hoechst (HOE140) (a bradykinin B2 receptor blocker; 5µM concentration
was used; Schoemaker, 200067
) in conjunction with rabbit dialysate was studied. Blockers of
bradykinin B2 receptor activity conserved the protection induced by preconditioned rIPC
dialysate (31.2%±3.0 vs. 26.9%±2.2, p=0.60; N=2, n=4, 1). (See Figure 18 and Table 9).

67
Table 9. HOE140 Results (Mean ± SEM).
Group Stabilization End
Baseline 28.9 ± 3.1 26.5 ± 1.7
Baseline + HOE140 26.9 ± 2.0 28.3 ± 2.1
Before SI After SR
Control Rabbit Dialysate 24.5 ± 1.3 41.3 ± 1.1
Control Rabbit Dialysate + HOE140 26.1 ± 2.2 44.6 ± 1.9
rIPC Rabbit Dialysate 25.1 ± 1.5 26.9 ± 2.2
rIPC Rabbit Dialysate + HOE140 26.2 ± 1.6 31.2 ± 3.0
Figure 18. HOE140 Administered Prior to Rabbit Dialysate.
The effect of Hoechst (HOE140) (a bradykinin B2 receptor blocker; 5µM concentration was
used) in conjunction with rabbit dialysate was studied. Blockers of bradykinin B2 receptor
activity conserved the protection induced by preconditioned rIPC dialysate (31.2%±3.0 vs.
26.9%±2.2, p=0.60; N=2, n=4, 1).

68
6.2.b Discussion:
Blockage with HOE140 did not abolish rIPC dialysate protection, suggesting that
bradykinin B2 receptors are not involved (see Figure 18). To reiterate this finding, levels of
bradykinin were detected at minuscule levels using MRM-MS (see Table 5).
According to Schoemaker et al.67
, MAO-induced rIPC of the myocardium was abolished
when HOE140 (bradykinin B2 receptor blocker) was administered during early reperfusion in in
vivo rats. Also, infusion of bradykinin into the mesenteric artery resulted in cardioprotection, but
was subsequently abolished by the ganglion blocker, hexamethonium. Thus, bradykinin is
thought to exert its effects on distant organs through a dual neurogenic and humoral (since
plasma bradykinin levels increase following ischemia55
) pathway. Once the neurogenic pathway
is blocked, there may be insufficient bradykinin from the humoral pathway to evoke protection at
the target organ. Knocking out one pathway may be enough to abolish protection entirely, as
protection from ischemia-reperfusion injury is commonly an all-or-none phenomenon that
requires a threshold to be met in order to induce protection according to Goto et al.3. To confirm
that dialyzing blood plasma did not dilute bradykinin concentrations (dialysate is 10x diluted
compared to plasma), dialysate was lyophilized into a powder and reconstituted with 1x Krebs-
Henseleit buffer such that dialysate concentrations were comparable to rabbit plasma. This
reconstituted dialysate still protected cardiomyocytes from ischemia-reperfusion damage and
HOE140 did not abolish this protection (N=1, n=1; see Table 10). Thus, in isolated rabbit
cardiomyocytes, the bradykinin B2 receptor and bradykinin molecule do not play a role in
humorally-mediated rIPC nor cross-talk with other receptors.

69
Table 10. 1x Dialysate with HOE140 Results (% Cardiomyocyte Death).
Group Stabilization End
Baseline 24.3 26.4
Before SI After SR
1x Control Rabbit Dialysate 24.9 40.8
1x Control Rabbit Dialysate + HOE140 25.3 34.5
1x rIPC Rabbit Dialysate 25.5 24.4
1x rIPC Rabbit Dialysate + HOE140 24.7 27.3

70
6.3 The Role of CGRP Receptors
6.3.a Existence of Calcitonin-like Receptors in Rabbit Cardiomyocytes:
The presence of calcitonin-like receptors (CLRs) in rabbit cardiomyocytes was
investigated using western blotting (n=1). CLRs were found in rabbit whole hearts, isolated
cardiomyocytes, and cardiomyocyte particulate fractions (i.e. the mitochondrial membrane was
excluded). (See Figure 19).
Figure 19. Western Blot Analysis of Calcitonin-Like Receptors in Rabbit Tissues.
The presence of calcitonin-like receptors (CLRs) in rabbit cardiomyocytes was investigated
using western blotting (n=1). CLRs were found in rabbit whole hearts, isolated cardiomyocytes,
and cardiomyocyte particulate fractions (i.e. the mitochondrial membrane was excluded).
Existence of Calcitonin-Like Receptors in Rabbit Cardiomyocytes

71
6.3.b CGRP Receptor Blockage Conserves Protection:
The effect of CGRP (8-37) (a CGRP receptor blocker; 5nM concentration was used;
Wolfrum, 2005103
) in conjunction with rabbit dialysate was studied. Blockers of CGRP receptor
activity conserved the protection induced by the preconditioned dialysate (24.8%±1.9 vs.
26.0%±3.3, p=0.96; N=2, n=2, 2). (See Figure 20 and Table 11).

72
Table 11. CGRP (8-37) Results (Mean ± SEM).
Group Stabilization End
Baseline 23.4 ± 2.2 23.3 ± 2.6
Baseline + CGRP (8-37) 22.6 ± 7.7 21.8 ± 4.0
Before SI After SR
IR 21.7 ± 2.3 41.6 ± 2.0
rIPC Rabbit Dialysate 22.4 ± 3.3 26.0 ± 3.3
rIPC Rabbit Dialysate + CGRP (8-37) 23.4 ± 2.7 24.8 ± 1.9
Figure 20. CGRP (8-37) Administered Prior to Rabbit Dialysate.
The effect of CGRP (8-37) (a CGRP receptor blocker; 5nM concentration was used) in
conjunction with rabbit dialysate was studied. Blockers of CGRP receptor activity conserved the
protection induced by the preconditioned dialysate (24.8%±1.9 vs. 26.0%±3.3, p=0.96; N=2,
n=2, 2).

73
6.3.c Discussion:
Calcitonin-like receptors (CLRs) have been identified in humans and rat smooth muscle
cells87,
88
. However, calcitonin-like receptors in rabbit tissues and in particular, rabbit
cardiomyocytes have not been studied. Western blotting indicates that CLRs are present in rabbit
whole heart, cardiomyocyte lysate, and the particulate fraction (with mitochondria excluded) (see
Figure 19). Even though this is a significant finding, there are limitations to concluding that
CGRP receptors exist on rabbit cardiomyocytes. The CGRP receptor is a receptor complex that
consists of the CLR and associated receptor activity-modifying proteins (RAMPs). CLR
association with RAMP1 and RAMP3 allows binding of CGRP to the receptor, resulting in CLR
becoming a CGRP receptor85
. CLR association with RAMP2 permits the binding of another
cardioprotective factor, adrenomedullin84
. Thus, in order to conclusively determine the existence
of CGRP receptors on rabbit cardiomyocytes, it is important to establish that RAMP1 and
RAMP3 associate with this CLR.
CGRP (8-37) blocked CGRP receptors and did not affect rIPC dialysate protection,
suggesting that the CGRP receptor is not involved in humoral rIPC (see Figure 20). In addition,
CGRP (8-37) has been used to abolish the protective effects of adrenomedullin167
in rat
cardiomyocytes. Therefore, the results presented in Figure 20 used CGRP (8-37) to block all
CLRs on rabbit cardiomyocytes. CGRP (8-37) was also administered to isolated rabbit
cardiomyocytes under basal conditions (i.e. the Baseline + CGRP (8-37) treatment group). Since
the CGRP blocker did not increase cell necrosis, this antagonist was deemed as non-toxic in
rabbit cardiomyocytes.

74
The claim by Wolfrum et al.103
regarding CGRP and rIPC cardioprotection suggests the
involvement of a dual neurogenic and humoral pathway. The authors determined that infusion of
CGRP could mimic MAO-preconditioned cardioprotection in rats. MAO preconditioning with
CGRP (8-37) abolished protection. Also, CGRP-induced protection was abolished by
hexamethonium (a ganglion blocker).Throughout MAO preconditioning, the level of CGRP was
found to be elevated by radioimmunoassay regardless of ganglion blockade. However, Wolfrum
et al. did not investigate the role of CGRP in a humoral model alone. Since CGRP is released by
caspasin sensitive sensory nerves, most likely this peptide‘s mechanism of action occurs through
a neurogenic pathway. Thus, there may be insufficient CGRP circulating in the blood following
preconditioning to induce cardioprotection. In addition, the work regarding CGRP is in remote
preconditioning of the intestine97,103
in rats to induced cardioprotection, suggesting that CGRP
may play a unique role when the intestine as the preconditioning organ but not in skeletal muscle
ischemia.
6.4 The Role of Adenosine Receptors
6.4.a Non-selective Adenosine Receptor Blockade of Protection:
The effect of 8-SPT (a non-selective adenosine receptor blocker; 100µM concentration
was used; Armstrong, 1995120
) in conjunction with rabbit dialysate was studied. Blockers of
adenosine receptor activity abolished the protection induced by the preconditioned dialysate
(41.6%±2.2 vs. 28.1%±1.7, p=0 0002; N=2, n=5,1). (See Figure 21 and Table 12).

75
Table 12. 8-SPT Results (Mean ± SEM).
Group Stabilization End
Baseline 26.9 ± 2.3 27.3 ± 0.9
Baseline + SPT 25.2 ± 1.3 26.7 ± 2.2
Before SI After SR
Control Rabbit Dialysate 25.6 ± 1.2 43.7 ± 1.4
Control Rabbit Dialysate + SPT 24.5 ± 1.4 42.3 ± 0.8
rIPC Rabbit Dialysate 25.3 ± 1.4 28.1 ± 1.7
rIPC Rabbit Dialysate + SPT 26.5 ± 1.5 41.6 ± 2.2
Figure 21. 8-SPT Administered Prior to Rabbit Dialysate.
The effect of 8-SPT (a non-selective adenosine receptor blocker; 100µM concentration
was used) in conjunction with rabbit dialysate was studied. Blockers of adenosine
receptor activity abolished the protection induced by the preconditioned dialysate
(41.6%±2.2 vs. 28.1%±1.7, p=0.0002; N=2, n=5,1).

76
6.4.b Discussion:
My results indicate the involvement of adenosine in rIPC since 8-SPT (a non-selective
adenosine blocker) was able to abolish protection from rIPC dialysate (see Figure 21). To
support this finding, Pell et al.128
suggested the involvement of adenosine in rabbit renal artery
occlusion (RAO). Also, Liem et al.127
confirmed this in rat mesenteric artery occlusion (MAO)
to induce cardioprotection. However, these studies have suggested a neurogenic model for rIPC
in renal and mesenteric preconditioning. Pang et al.129
demonstrated that skeletal muscle
ischemia increased plasma adenosine concentrations and inhibition of vesicular monoamine
transport (inhibition of catecholamines at synaptic nerve endings) partially abolished
preconditioning. Thus, it is possible that skeletal muscle ischemia releases adenosine that
activates local nerves, which in turn affect distant organs like the myocardium through a dual
neurogenic/humoral pathway. However, the protective factor in rIPC dialysate is humoral in
nature and this alone can protect isolated cardiomyocytes without the need of neuronal
intervention (see Figure 13).
According to Pang et al.129
, levels of adenosine in the plasma were elevated during
skeletal muscle ischemia, hinting at this purine‘s involvement in a humoral pathway. However,
the half-life of adenosine is very short (0.6-1.5 sec in plasma) due to continual breakdown by
circulating adenosine deaminase140
. To support this finding, the level of adenosine was
determined to be <0.80µM by MRM-MS in rIPC dialysate (see Table 5). However, adenosine
concentrations are required to be >10uM to protect isolated rabbit cardiomyocytes according to
Armstrong et al.120
. Based on the above evidence, the involvement of the adenosine molecule in

77
a humoral route is unlikely. Therefore, the blockage of dialysate protection with 8-SPT (Figure
21) suggests the involvement of the adenosine receptor and not the adenosine molecule.
A number of controversies exist with the role of adenosine in IPC and rIPC. Auchampach
et al.168
demonstrated that administration of three A1 antagonists (DPCPX, BG9719, BG9928) in
dogs throughout preconditioning until before index ischemia did not block protection. Mice with
knockouts for adenosine receptors also show contradictory results. Lankford et al.169
could not
precondition A1 knockouts, however Guo et al.170
demonstrated that A3 knockout mice are still
capable of preconditioning, thus illustrating the importance of A1 receptors in IPC. However, a
study conducted by Eckle et al.171
demonstrated that A1, A3, and A2a knockouts had preserved
protection following ischemia. It was A2b receptors alone that could not be preconditioned
through IPC. To explain these controversies, a review by Cohen and Downey14
suggests the
involvement of multiple receptor agonists acting in parallel during IPC. This also suggests the
involvement of other receptors in the form of cross-talk.

78
- Chapter 7 -
RIPC MEDIATES OPIOID-ADENOSINE CROSS-TALK
7.1 Opioid-Adenosine Cross-talk
7.1.a Adenosine Deamination of the Dialysate Conserves Protection:
The effect of adenosine deamination of dialysate was studied. Rabbit dialysate was
passed through a column containing adenosine deaminase (ADA) coupled to beads‡. Levels of
adenosine and inosine before and after deamination were measured using MRM mass
spectrometry to ensure complete elimination of adenosine from the dialysate. The adenosine
deamination of the dialysate conserved the protection induced by rIPC dialysate§ (34.9% vs.
33.3% and 30.1% vs. 31.8%, n=2). (See Table 13).
NOTE: These experiments were originally conducted with an n=3.However, adenosine in
rIPC dialysate from one experiment was not deaminated, thus this experiment was excluded.
‡ Dialysate deamination was conducted by Amy Yeung (Dr. John Callahan‘s Lab; Department of Paediatric
Laboratory Medicine, Hospital for Sick Children). § These experiments were conducted by Alina Hinek (Dr. Wilson‘s Lab, Hospital for Sick Children).

79
Table 13. ADA Results (% Cardiomyocyte Death).
Experiment A Stabilization End
Baseline 24.1 35.1
Before SI After SR
Control Rabbit Dialysate 24.3 42.6
rIPC Rabbit Dialysate 28.4 34.9
rIPC Rabbit Dialysate + ADA 31.1 33.3
Pre-ADA (µM) Post-ADA (µM)
Adenosine in rIPC Rabbit Dialysate 0.01 No peak
Inosine in rIPC Rabbit Dialysate 0.22 0.39
Experiment B Stabilization End
Baseline 26.6 33.6
Before SI After SR
Control Rabbit Dialysate 24.0 45.0
rIPC Rabbit Dialysate 29.5 30.1
rIPC Rabbit Dialysate + ADA 30.1 31.8
Pre-ADA (µM) Post-ADA (µM)
Adenosine in rIPC Rabbit Dialysate 0.01 No peak
Inosine in rIPC Rabbit Dialysate 0.26 0.20

80
7.1.b Partial Adenosine Blockade of δ-Opioid Receptor-Induced Protection:
The effect of 8-SPT (a non-selective adenosine receptor blocker; 100µM concentration
was used; Armstrong, 1995120
) in conjunction with Met-enkephalin (a δ-opioid receptor agonist,
100µM; Cao, 2003152
) was studied. The adenosine receptor blocker partially abolished the
protection induced by δ -opioid receptor activation (41.8%±1.5 vs. 32.8%±1.0 compared to ME,
p=0.0007, and vs. 50.7%±1.1 compared to IR, p=0.0008; n=5). (See Figure 22 and Table 14).

81
Table 14. Met-Enkephalin with 8-SPT Results (Mean ± SEM).
Group Stabilization End
Baseline 29.3 ± 0.8 31.2 ± 0.7
Baseline + ME 23.9 ± 0.0 21.7 ± 0.0
Before SI After SR
IR 28.6 ± 1.3 50.7 ± 1.1
ME 30.6 ± 1.9 32.8 ± 1.0
ME + 8-SPT 29.4 ± 0.8 41.8 ± 1.5
ME + NTI 33.4 ± 0.0 51.7 ± 0.0
Figure 22. 8-SPT Administered Prior to Met-Enkephalin.
The effect of 8-SPT (a non-selective adenosine receptor blocker; 100µM concentration was
used) in conjunction with Met-enkephalin (a δ-opioid receptor agonist, 100µM) was studied. The
adenosine receptor blocker partially abolished the protection induced by δ -opioid receptor
activation (41.8%±1.5 vs. 32.8%±1.0 compared to ME, p=0.0007, and vs. 50.7%±1.1 compared
to IR, p=0.0008; n=5).

82
7.1.c Complete Adenosine Blockade of κ -Opioid Receptor-Induced Protection:
The effect of 8-SPT (a non-selective adenosine receptor blocker; 100µM concentration
was used; Armstrong, 1995120
) in conjunction with dynorphin B (a κ-opioid receptor agonist,
100µM; Cao, 2003152
) was studied. The adenosine receptor blocker abolished the protection
induced by κ-opioid receptor activation (44.3%±2.6 vs. 31.8%±2.1 compared to DynB, p=0.02,
and vs. 51.3%±5.0 compared to IR, p=0.17; n=5). (See Figure 23 and Table 15).

83
Table 15. Dynorphin B with 8-SPT Results (Mean ± SEM).
Group Stabilization End
Baseline 26.4 ± 1.1 27.2 ± 2.4
Baseline + DynB 19.2 ± 0.0 22.4 ± 0.0
Before SI After SR
IR 29.1 ± 2.1 51.3 ± 5.0
DynB 28.7 ± 1.8 31.8 ± 2.1
DynB + 8-SPT 30.5 ± 1.9 44.3 ± 2.6
DynB + GNTI 29.1 ± 1.6 48.3 ± 3.1
Figure 23. 8-SPT Administered Prior to Dynorphin B.
The effect of 8-SPT (a non-selective adenosine receptor blocker; 100µM concentration was
used) in conjunction with dynorphin B (a κ-opioid receptor agonist, 100µM) was studied. The
adenosine receptor blocker abolished the protection induced by κ-opioid receptor activation
(44.3%±2.6 vs. 31.8%±2.1 compared to DynB, p=0.02, and vs. 51.3%±5.0 compared to IR,
p=0.17; n=5).

84
7.1.d Discussion:
Preconditioned dialysate was exposed to adenosine deaminase (ADA) conjugated beads
in order to remove any trace of adenosine. The results of our study confirm that adenosine
removal from the rIPC dialysate maintains protection, indicating with certainty that the
protective factor(s) is/are not adenosine (see Table 13). ADA-conjugated beads were utilized in
order to remove ADA prior to cardiomyocyte exposure, thus avoiding deamination of vital
adenosine released during the mediator phase of preconditioning. To summarize the results so
far: the half-life of adenosine in plasma is 0.6-1.5sec140
, MRM-MS detected low levels of
adenosine in the dialysate (see Table 5), and rIPC protection was conserved even in the absence
of adenosine through adenosine deamination. However, rIPC dialysate protection was abolished
by the adenosine receptor blocker, 8-SPT (see Figure 21). Based on this accumulating evidence,
there is involvement of adenosine receptors in rIPC but not the adenosine molecule itself.
ADA also increases inosine concentrations during the breakdown of adenosine. Jin et
al.172
has reported that inosine increases mast cell degranulation during ischemia or
inflammation. The same study demonstrated that inosine can bind only to adenosine A3 receptors
and not A1 or A2a. Recently, Shen et al.173
suggested that inosine preconditions rats from cerebral
brain injury via adenosine A3 receptors. Litsky et al.174
found inosine protects mouse neuronal
and glial cell cultures from hypoxia. The authors constructed a concentration response curve for
inosine and determined the minimum concentration to protect neuronal cells is 500µM in rats.
However, the level of inosine in rIPC dialysate is <0.26µM and upon deamination of adenosine,
inosine levels elevated to <0.39µM (see Table 13). Therefore, the protection seen after adenosine
deamination is not due to inosine.

85
The non-selective adenosine blocker 8-SPT partially abolished protection from Met-
enkephalin (δ-specific opioid agonist) (see Figure 22). The activation of κ receptors by
dynorphin B (κ-specific opioid agonist) was fully abolished by 8-SPT since there was no
statistical significance between ischemia-reperfusion alone and administration of dynorphin B
with 8-SPT (see Figure 23). However, increased number of experiments may result in dynorphin
B exhibiting partial protection similar to Met-enkephalin. Regardless, these studies have
confirmed cross-talk between δ and κ opioid receptors with adenosine receptors during the
trigger phase of classical IPC.
Interestingly, Peart et al.138
suggests that cross-talk between opioid and adenosine
receptors work both ways. However, our rIPC dialysate model does not involve adenosine due to
its deamination in the blood. Also, preliminary data from our lab indicates that cross-talk is
unidirectional (n=2, see Table 16). This may be due to species differences or due to experimental
models since Peart et al. utilized an in vivo rat model.

86
Table 16. Adenosine with Naloxone Results (% Dead Cardiomyocytes).
Experiment A Stabilization End
Baseline 19.2 22.1
Baseline + Adenosine 19.3 22.1
Before SI After SR
IR 26.8 52.9
Adenosine 25.1 29.0
Adenosine + Nal 23.3 31.0
Adenosine + SPT 25.3 45.1
Experiment B Stabilization End
Baseline 26.9 29.0
Before SI After SR
IR 26.2 36.2
Adenosine 28.4 36.2
Adenosine + Nal 26.9 37.6
One explanation, suggested by Peart et al.139
, of opioid-adenosine cross-talk is the
adenosine kinase inhibition hypothesis. Adenosine kinase degrades adenosine into AMP within
cardiomyocytes. According to the authors, activation of opioid receptors inhibits this enzyme,
which leads to an accumulation of intracellular adenosine. Since diffusion of adenosine across
the cell membrane is dependent on a concentration gradient, extracellular adenosine remains
outside cardiomyocytes. The build-up of adenosine activates adenosine receptors and results in
ischemic preconditioning. Therefore, a vital component of the adenosine kinase inhibition
hypothesis is the amassing of extracellular adenosine.
7.2 The Inhibition of Adenosine Kinase Hypothesis
7.2.a Exposure to Dynorphin B Does Not Accumulate Extracellular Adenosine:
The inhibition of adenosine kinase to increase levels of extracellular adenosine was
measured using MRM mass spectrometry. Supernatants from freshly isolated rabbit
cardiomyocytes were obtained after stabilization, dynorphin B (a κ-opioid receptor agonist,

87
100µM; Cao, 2003152
) exposure, and after a wash period. Levels of adenosine did not change
significantly (contained the following in µM: 1.54±0.5 after stabilization, 0.96±0.1 after DynB
exposure, 1.14±0.1 after wash; n=5) and were well below 10µM (the minimum concentration of
adenosine required to protect cardiomyocytes; Armstrong, 1995120
). (See Figure 24 and Table 17
for results; see Figure 29 in Appendix V for a plot of the standard concentrations).
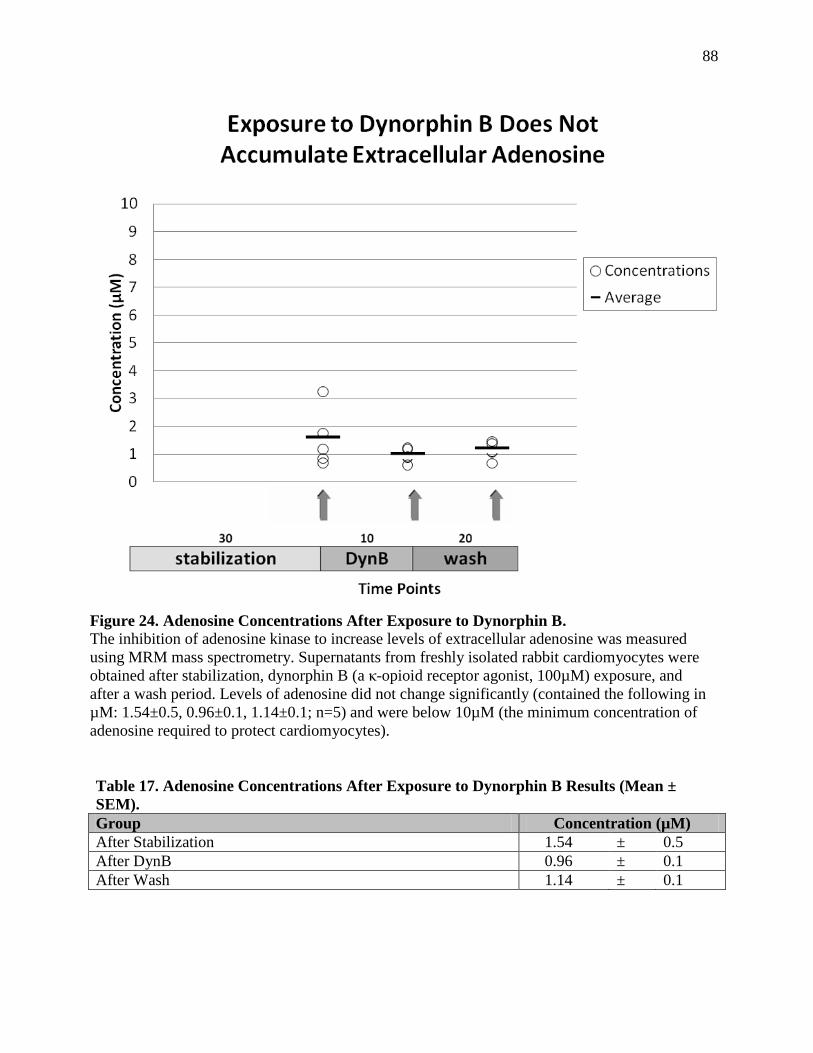
88
Table 17. Adenosine Concentrations After Exposure to Dynorphin B Results (Mean ±
SEM).
Group Concentration (µM)
After Stabilization 1.54 ± 0.5
After DynB 0.96 ± 0.1
After Wash 1.14 ± 0.1
Figure 24. Adenosine Concentrations After Exposure to Dynorphin B.
The inhibition of adenosine kinase to increase levels of extracellular adenosine was measured
using MRM mass spectrometry. Supernatants from freshly isolated rabbit cardiomyocytes were
obtained after stabilization, dynorphin B (a κ-opioid receptor agonist, 100µM) exposure, and
after a wash period. Levels of adenosine did not change significantly (contained the following in
µM: 1.54±0.5, 0.96±0.1, 1.14±0.1; n=5) and were below 10µM (the minimum concentration of
adenosine required to protect cardiomyocytes).

89
7.2.b Discussion:
Supernatant was collected before, during and after cardiomyocytes were exposed to
dynorphin B (DynB, κ-selective agonist) and immediately placed on ice to prevent deamination
of adenosine. The same MRM-MS method used to detect adenosine in the dialysate (see Table 5)
was used to measure adenosine concentrations in supernatant samples (see Figure 24). The
results indicate that adenosine concentrations do not change after DynB exposure and are well
below protective levels (<10µM). Therefore, there is no increase in extracellular adenosine as a
result of opioid receptor stimulation during the trigger phase of IPC.
Another method of increasing interstitial adenosine concentrations is through inhibition
of adenosine deaminase which is normally situated outside cardiomyocytes. Silva et al.175
examined the effects of an adenosine deaminase inhibitor (pentostatin) on myocardial infarction
in dogs. Pentostatin increased plasma adenosine levels 3.5-fold above basal conditions prior to
ischemia and sustained these levels until early reperfusion. However, this attenuation of
adenosine had no effect on infarct size and did not precondition myocardium.
Peart et al.138
in 2003 first noted a relationship between opioid and adenosine receptors in
in vivo cardioprotection in rats. Rats were pre-treated with morphine (a non-selective opioid
agonist) and 2-chloro-cyclopentyladenosine (CCPA, adenosine A1 agonist) throughout the
experiment and this mimicked preconditioning by coronary artery occlusion. This protection was
abolished by the δ1 opioid blocker, 7-benzylidenealtrexone (BNTX). Based on these findings,
Peart et al. concluded receptor cross-talk and/or parallel signalling pathways downstream of
opioid and adenosine receptors. Later in 2005, Peart et al.139
postulated that the opioid-adenosine

90
cross-talk seen in 2003 was attributed to inhibition of adenosine kinase in cardiomyocytes (see
Figure 3). In this study, rats were treated in vivo with the adenosine kinase inhibitor, 5-
iodotubercidin which protected myocardium from damaging CAO. 8-Cyclopentyl-1,3-
dipropylxanthine (DPCPX, adenosine A1 blocker) and MRS1523 (adenosine A3 blocker)
abolished this protection. In addition, the δ1 opioid blocker, BNTX, also eliminated protection.
However, my results measuring adenosine in cardiomyocyte supernatant samples do not
support this hypothesis. This may be due to differences in experimental protocols. For example,
our isolated rabbit cardiomyocyte model was not in vivo and therefore, independent of many
factors such as neuronal tissue in the whole heart. Also, Peart et al. administered adenosine
kinase inhibitors throughout the experimental protocol which may have allowed time for
adenosine kinase inhibition and accumulation of extracellular adenosine, whereas my
cardiomyocyte model only treated cells with an opioid agonist during the trigger phase of IPC.
Most importantly, Peart et al.139
determined that δ1 opioid receptor inhibitors abolished
protection induced by adenosine kinase inhibition. However, this finding contradicts their
proposed hypothesis. The model undergoes the following steps: (1) activation of opioid
receptors, (2) which inhibits adenosine kinase, (3) leading to extracellular adenosine
accumulation, (4) resulting in cardioprotection. Inhibition of opioid receptors eliminates step (1),
however, since an adenosine kinase inhibitor is still present throughout the procedure, steps (2) –
(4) are still possible and should result in cardioprotection. Nonetheless, Peart et al. states that this
is only a tentative hypothesis that requires further investigation and proposes other possibilities

91
such as heterodimerization of opioid and adenosine receptor or parallel signalling pathways
downstream of these receptors.

92
- Chapter 8 -
SUMMARY OF FINDINGS
The data presented confirms these major findings:
1. Remotely preconditioned dialysate (both human and rabbit derived) protect
cardiomyocytes against ischemia-reperfusion injury to the same extent as classical IPC.
2. Compounds that precondition in IPC are either undetectable (Met-enkephalin, dynorphin
B, bradykinin, acetylcholine, angiotensin II) or at orders of magnitude below the
threshold to confer protection (MEAP, adenosine, inosine, norepinephrine) in rIPC
dialysate.
3. Cardioprotection induced by rIPC dialysate requires activation of δ or κ opioid receptors
or adenosine receptors.
4. Cardioprotection induced by rIPC dialysate does not require the activation of bradykinin
B2 or CGRP receptors.
5. The adenosine molecule does not play a role in rIPC dialysate protection, but the
adenosine receptor does.
6. Protection induced by κ or δ opioid receptors is fully or partially abolished by non-
selective adenosine blockade. This indicates cross-talk between these receptors.
7. The cross-talk between opioid and adenosine receptors is not due to accumulation of
extracellular endogenous adenosine.
These findings support the hypothesis that δ or κ opioid receptors, through cross-talk with
adenosine receptors, act as possible triggers in remote ischemic preconditioning.

93
- Chapter 9 -
GENERAL DISCUSSION
9.1 Overall Perspective
The investigations conducted in this thesis have determined that rIPC can be mediated
from rabbit or human skeletal muscle to rabbit cardiomyocytes through a humoral pathway (see
Figure 13). Also, there is involvement of δ and κ opioid receptors33
(see Figures 15, 16, & 17) in
the target organ, but this is not mediated by molecules (Met-enkephalin, dynorphin B, MEAP,
adenosine, inosine, bradykinin, acetylcholine, norepinephrine, and angiotensin II) that have been
widely involved in classical IPC (see Table 5). In addition, claims have been made regarding
bradykinin B267
(see Figure 18) and CGRP103
(see Figure 20) receptors, but these do not play a
role in our isolated cardiomyocyte model. Interestingly, adenosine receptors128
have also been
implicated in rIPC (see Figure 21). However, the level of adenosine in rIPC dialysate and
subsequent deamination (Table 13), excludes the involvement of the adenosine molecule to
explain these results. However, I determined there is cross-talk between δ and κ opioid receptors
with adenosine receptors (see Figures 22 & 23) to explain these results. However, this
relationship is not due to the inhibition of adenosine kinase139
increasing extracellular adenosine
concentrations (see Figure 24).Thus, this cross-talk may occur through heterodimerization of
opioid and adenosine receptors (See Figure 25)
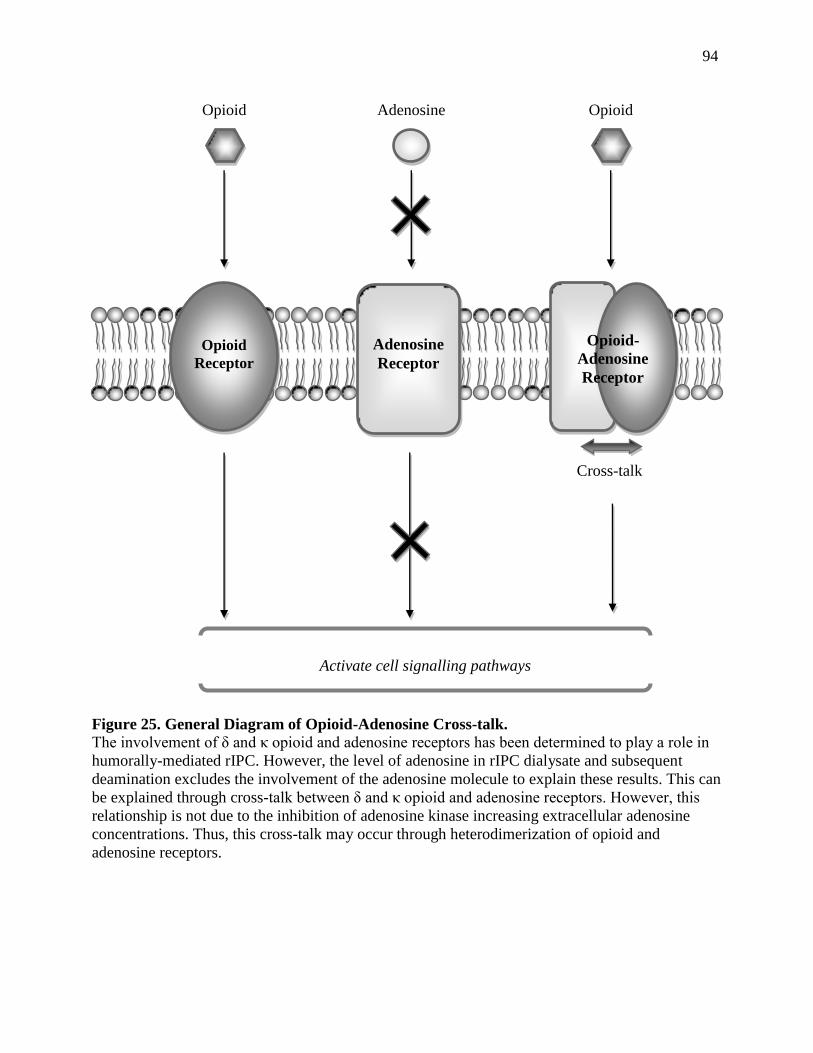
94
Figure 25. General Diagram of Opioid-Adenosine Cross-talk.
The involvement of δ and κ opioid and adenosine receptors has been determined to play a role in
humorally-mediated rIPC. However, the level of adenosine in rIPC dialysate and subsequent
deamination excludes the involvement of the adenosine molecule to explain these results. This can
be explained through cross-talk between δ and κ opioid and adenosine receptors. However, this
relationship is not due to the inhibition of adenosine kinase increasing extracellular adenosine
concentrations. Thus, this cross-talk may occur through heterodimerization of opioid and
adenosine receptors.
Cross-talk
Adenosine
Receptor
Opioid
Receptor
Opioid-
Adenosine
Receptor
Opioid Opioid Adenosine
Activate cell signalling pathways

95
9.2 Other Mechanisms of Opioid-Adenosine Cross-talk
9.2.a Dimerization of G-protein Coupled Receptors:
Heterodimerization among G-protein coupled receptors has been implicated in opioid-
adenosine ‗cross-talk‘138
. Heterodimerization between G-protein coupled receptors is a well
documented phenomenon that can lead to unique pharmacological properties such as altered
receptor sensitivity, endogenous adenosine release, ligand binding and signalling. Also, these
uniquely dimerized receptors may bind to orphan peptides that have yet to be identified.166,176,177
In addition, there is evidence that opioid receptors dimerize with β2-adrenergic and
somatostatin receptors. Jordan et al.178
studied oligomerization between these distant GPCR
family members: δ and κ opioid receptors with β2-adrenergic receptors. Human embryonic
kidney (HEK)-293 cells were cotransfected with anti-myc tagged opioid receptors and anti-Flag
tagged β2-adrenergic receptors. Immunoprecipitation determined that these receptors associate
with each other. Though these receptors did not display altered ligand binding, there were
changes in protein trafficking such as endocytosis. Coimmunoprecipitation of sst(2A)
somatostatin and µ1 opioid receptors were found to heterodimerize in HEK-293 cells by Pfeiffer
et al. 179
. Binding of either sst(2A) or µ ligands altered receptor phosphorylation and
desensitization of this hybrid receptor.
Adenosine A1 receptors can form dimers with P2Y1 and dopamine D1 receptors. Gines et
al. 180
cotransfected cultured mouse Ltk- fibroblasts and rat embryonic cortical neurons with
adenosine A1, dopamine D1, and dopamine D2 human cDNA. Coimmunoprecipitation and
colocalization with immunofluorescence determined that A1 heterodimerized with D1 receptors

96
but not D2. The authors determined that heterodimerization of these receptors resulted in receptor
desensitization. Yoshioka et al.181
extensively investigated in vivo heterodimerized P2Y1 and
adenosine A1 receptors in rat brain tissues and cortical neurons using immunofluorescence and
coimmunoprecipitation. In addition, adenosine A2 receptors exist as naturally occurring
oligomers with dopamine D2 receptors182
. Though GPCR heterodimerization has been described
for both adenosine receptors and opioid receptors individually, there is currently no study that
investigates heterodimerization of opioid and adenosine receptors with each other.
9.3 Future Directions
The further directions of these studies will be to further investigate the mechanism of
‗cross-talk‘ between opioid and adenosine receptors. Studies will involve investigating another
mechanism of opioid-adenosine cross-talk, i.e. heterodimerization of G-protein protein receptors.
In addition, cell signalling downstream of G-protein coupled cardiomyocyte cell membrane
receptors in rIPC and opioid-adenosine cross-talk is another logical avenue for further
investigation.
9.3.a Specific Adenosine Receptor Subtypes in Cross-talk & rIPC:
Before determining if opioid and adenosine receptors heterodimerize, it is important to
elucidate which adenosine receptor subtype(s) cross-talk(s) with δ and κ opioid receptors. To
investigate this, cardiomyocytes will be subjected to 10 min classic IPC, specific opioid receptor
agonists, such as Met-enkephalin (δ-specific agonist) or dynorphin B (κ-specific agonist) in the
presence of adenosine receptor subtype blockers, such as DPCPX (A1 adenosine blocker),
MRS1523 (A3 adenosine blocker), or MRS1754 (A2b adenosine blocker) for 20 min. Treatment

97
groups will undergo a wash period in fresh buffer for 20 min. All groups will then be subjected
to a long period of 45 min simulated ischemia (SI) and 60 min simulated reperfusion (SR).
Samples will be taken before and after SI/SR. (See Figure 26).
Similar experiments will then be conducted with rIPC dialysate and the above mentioned
adenosine blockers in order to confirm which adenosine receptor subtype is involved in rIPC.
Figure 26. Protocol of Adenosine Receptor Subtypes in Opioid-Adenosine Cross-talk.
Cardiomyocytes will be subjected to 10 min classic IPC, specific opioid receptor agonists, such
as Met-enkephalin (δ-specific agonist) or dynorphin B (κ-specific agonist) in the presence of
adenosine receptor subtype blockers, such as DPCPX (A1 adenosine blocker), MRS1523 (A3
adenosine blocker), or MRS1754 (A2b adenosine blocker) for 20 min. Treatment groups will
undergo a wash period in fresh buffer for 20 min. All groups will then be subjected to a long
period of 45 min simulated ischemia (SI) and 60 min simulated reperfusion (SR). Samples will
be taken before and after SI/SR.

98
9.3.b Heterodimerization of Opioid and Adenosine Receptors:
Heterodimerization of GPCRs in cardiomyocytes can be investigated using
coimmunoprecipitation of the opioid and adenosine receptors. In addition, immunofluorescence
of the two receptors may also be examined. While these techniques provide information
regarding receptor association, they cannot prove functionally significant heterodimerization.
A technique that can conclusively state the existence of dimerization is fluorescence
energy transfer (FRET) and bioluminescence energy transfer (BRET) to study protein-protein
interactions in living cells. Hebert et al.183
provides a highly detailed overview of these
techniques. FRET involves fluorescence energy transfer between two adjacent proteins, one
being the donor and the other the acceptor. When these proteins are dissociated, only the donor
emission of fluorescence is detected. However, if they are in close proximity, there is significant
energy transfer between the two proteins and the acceptor‘s emission is detected. BRET is
similar to FRET, yet utilizes bioluminescent luciferase. This technique does not require external
illumination to initiate fluorescence transfer like in FRET, but instead uses luciferase which has
less background noise. Currently, these techniques study interactions between receptors and
downstream signalling molecules, however, Gandia et al.184
describes the potential of the FRET
and BRET techniques to study GPCR oligomerization.
9.3.c Cell Signalling in rIPC:
Signalling downstream of receptors has been studied in rIPC. However, the mechanisms
involved are unclear compared to IPC. Similar to IPC, activation of PKC68
, nitric oxide185
,
reactive oxygen species186
, mitochondrial KATP channels128
, and the reperfusion injury salvage

99
kinase (RISK) pathway146
have been implicated in rIPC. However, inhibition of the
mitochondrial permeability transition pore (mPTP) in rIPC is still unclear.
As a reminder, signalling in IPC involves ligand binging to cell membrane receptors that
result in activation of kinases (e.g. PI3 kinase, Akt, and ERK) and nitric oxide (NO) synthesis,
causing mitochondrial (mKATP) channel opening that produces reactive oxygen species (ROS),
which leads to PKC translocation. During the mediator phase, pro-survival kinases187
of the
reperfusion injury salvage kinase (RISK) pathway are activated and ultimately lead to inhibition
of the mitochondrial permeability transition pore (mPTP) from opening188
.
Nitric oxide (NO) is very important in early and delayed classical IPC, however, the role
of NO in rIPC is still unclear. Tokuno et al.185
proposed that cerebral preconditioning could
induce cardioprotection, however, this protection was absent in nitric oxide synthase (NOS)
knockout mice. In addition, NFκβ and NOS have been implicated in delayed limb rIPC in rat
hearts. Li et al.189
showed that rIPC increased translocation of NFκβ to the nucleus and increased
NOS mRNA. Inactivation of the NFκβ protein or the NOS gene abolished rIPC protection.
Phosphatidylinositol 3-kinase (PI3) is known to be involved in classical IPC. However,
there are no current studies that have investigated the role of PI3 in rIPC. To address this
question, I investigated the effect of wortmannin (a PI3 kinase inhibitor; 100nM concentration
was used; Oldenburg, 2003160
) in conjunction with rabbit dialysate. Blockers of PI3 kinase
activity abolished the protection induced by the preconditioned dialysate (41.0%±4.9 vs.
24.3%±4.0, p=0.01; N=2, n=2,2). (See Figure 27 and Table 18).
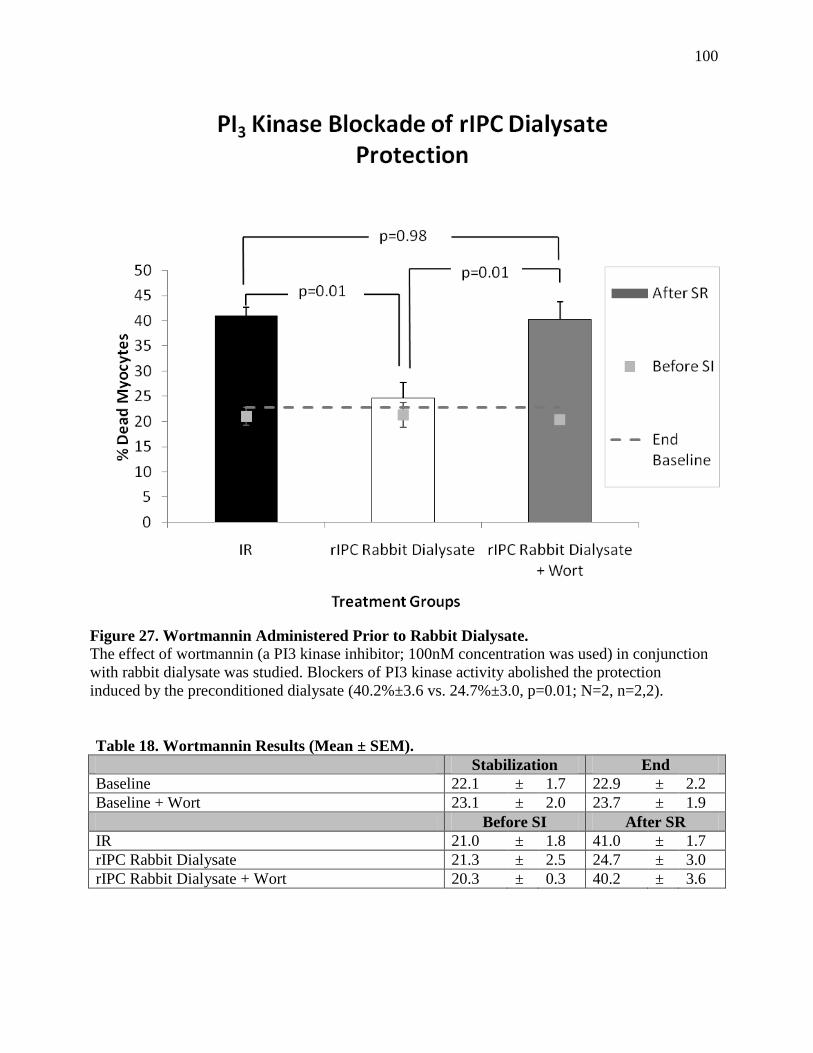
100
Table 18. Wortmannin Results (Mean ± SEM).
Stabilization End
Baseline 22.1 ± 1.7 22.9 ± 2.2
Baseline + Wort 23.1 ± 2.0 23.7 ± 1.9
Before SI After SR
IR 21.0 ± 1.8 41.0 ± 1.7
rIPC Rabbit Dialysate 21.3 ± 2.5 24.7 ± 3.0
rIPC Rabbit Dialysate + Wort 20.3 ± 0.3 40.2 ± 3.6
Figure 27. Wortmannin Administered Prior to Rabbit Dialysate.
The effect of wortmannin (a PI3 kinase inhibitor; 100nM concentration was used) in conjunction
with rabbit dialysate was studied. Blockers of PI3 kinase activity abolished the protection
induced by the preconditioned dialysate (40.2%±3.6 vs. 24.7%±3.0, p=0.01; N=2, n=2,2).

101
The activation of mitochondrial KATP has been proposed in early and delayed rIPC. KATP
channel opening is thought to reduce calcium overload and result in inhibition of the
mPTP190,191,192
. As evidence, rat cardioprotection by limb ischemia was blocked by
glibenclamide (non-selective KATP blocker) and 5-HD (a mitochondrial KATP blocker) but not by
HMR 1098 (a sarcolemmal KATP blocker)193,194,195
. However, studies in pigs show that both
sKATP and mKATP are involved in early and delayed limb ischemia196,197
. In these studies, BMS
191095, a mKATP channel opener, mimicked rIPC by limb ischemia. However, 5-HD removed
this protection.
Reactive oxygen species (ROS) generation plays a role in both detrimental lethal
reperfusion injury and in the protective effects of preconditioning187
and post-conditioning198
. As
evidence, Weinbrenner et al.37
determined that a free radical scavenger (MPG) was able to
abolish in vivo cardioprotection from renal artery occlusion (RAO) in rats.
In rIPC, the role of protein kinase C (PKC) has been confirmed in infrarenal aortic
occlusion186
and MAO194
, when chelerythrine (a PKC blocker) abolished cardioprotection in rats.
A number of studies investigating other triggers have suggested the role of PKC in rIPC, such as
adenosine128
, bradykinin68
, and opioids16
. In these studies, pharmacological preconditioning with
bradykinin B268
and CGRP103
receptor activation is thought to induce PKCε translocation in a
manner similar to rIPC cardioprotection.
With respect to the RISK pathway, Heidbreder et al.199
suggest that mitogen-activated
protein kinases (MAPKs), p38, and ERK1/2 are activated in the small intestine during

102
mesenteric rIPC to protect the myocardium. In addition, inhibition of these kinases reduced rIPC
protection. Also, Shimizu et al.146
determined that these MAPK kinases from the RISK pathway
were activated in the heart itself during in vivo skeletal muscle rIPC in rabbits.
To further complicate the role of adenosine in rIPC, cardiomyocytes not only utilize
adenosine as a trigger, but also a mediator in IPC with its claimed involvement of adenosine A2b
receptors during reperfusion14
. Thus, it is important to investigate the role of adenosine as a
mediator in rIPC, and potentially, a mediator in opioid-adenosine cross-talk. However, the role
of adenosine receptors in the studies described by this dissertation has only been investigated in
the trigger phase.
The role of mitochondrial permeability transition pore (mPTP) as an end effector in
classical IPC is a subject of great interest, but to date, there are no published studies of the role of
mPTP in rIPC. The mPTP is located on the inner mitochondrial membrane and opened during
the beginning of reperfusion. Activation of the mPTP results in uncoupling of oxidative
phosphorylation, ATP depletion, mitochondrial swelling, and ultimately results in cell death.
However, preconditioning is thought to inhibit mPTP opening200
. Zhang et al.36
has shown that a
κ-opioid agonist (U-50,488H) induced mPTP opening, as seen by fluorescent calcein, in rat
cardiomyocytes. In the same study, the mPTP activator (atractyloside) abolished femoral artery
occlusion (FAO)-induced cardioprotection in rats.
Infiltration, adhesion and activation of neutrophils have also been proposed as an end
effector in early and late rIPC201
. Cardioprotection by limb ischemia in mice has shown down-

103
regulation of proinflammatory genes and up-regulation of genes involved in cytoprotection and
protection from oxidative stress202
. This suggests that rIPC suppresses neutrophils from releasing
proinflammatory cytokines and from expressing adhesion markers. However, the role of
neutrophils in rIPC requires an in vivo model and is not suitable to be studied by our isolated
cardiomyocyte model.
9.4 Limitations of the Study:
There are some limitations to the model described in this dissertation. Unfortunately,
utilizing an isolated cardiomyocyte model excludes the ability to study a neuronal pathway in
protection. Though my results indicate that a humoral mediator is sufficient to induce rIPC
cardioprotection (see Figure 13), this does not exclude a neurogenic involvement in vivo. An in
vivo model is also useful in investigating the involvement of an inflammatory pathway via
neutrophils in rIPC. Finally, my study did not measure apoptosis as an indicator of myocardial
reperfusion injury. Such an investigation is more suitable for cultured cardiomyocytes since
apoptosis takes several hours to develop. Regardless, it is important to note that an isolated
cardiomyocyte model is more efficient (since many treatment groups can be derived from a
single heart) and reduces heart-to-heart variability.
In addition, my studies only investigated rIPC during the trigger phase of acute
preconditioning. A comprehensive study would involve the role of rIPC dialysate to induce
protection during the reperfusion phase and in delayed rIPC. Fortunately, these investigations
can be readily conducted either in isolated or cultured cardiomyocytes.

104
9.5 Conclusions:
This dissertation provides important insights into the role of particular G-protein coupled
receptors in a humoral model of remote preconditioning. Also, there are benefits of using this
model over others in investigating this area. For example, characterization of the dialysate (see
Table 5) using MRM-MS excluded the possibility of certain substances to mediate protection in
the rIPC dialysate. However, this method does not encompass all endogenous ligands that can
bind to a particular GPCR. Considering this, selective inhibition of receptors is a better method
of determining which receptors are involved even if the stimulatory agent is not present.
Also, our model can be used to determine cross-talk between receptors on a functional
level. MRM-MS also did not exclude the possibility of certain receptors involved in cross-talk
with opioid receptors. However, our model is able to determine which receptors can participate
in cross-talk (i.e. opioid with adenosine receptors) and which possibilities can be excluded (i.e.
opioid cross-talk with bradykinin B2 and CGRP receptors).
In conclusion, remote preconditioning is a clinically relevant strategy to reduce
myocardial infarction. However, the mechanistic pathways in remote precondition are still
unclear, with some evidence resulting to conflicting results. Thus, it is important to compare the
pathways in classical and rIPC in order to create beneficial therapeutic interventions.

105
APPENDIX
APPENDIX I: List of Figures
Figure 1. IPC Signalling Mechanisms in the Target Organ. ........................................................... 4
Figure 2. rIPC Signalling Mechanisms of the Myocardium. .......................................................... 7
Figure 3. The Adenosine Kinase Inhibition Hypothesis. .............................................................. 25
Figure 4. Preparation of Buffers Derived from Krebs-Henseleit Buffer. ..................................... 31
Figure 5. Comparison of Digestion Protocols............................................................................... 33
Figure 6. Rabbit and Human Dialysate Preparation. .................................................................... 36
Figure 7. General Protocol: Isolated Rabbit Cardiomyocytes. ..................................................... 38
Figure 8. AIM (1) Protocol: To Characterize Dialysate Protection. ............................................. 39
Figure 9. AIM (2) Protocol: The Role of Cell Membrane Receptors. .......................................... 40
Figure 10. AIM (3) Protocol: Investigate Receptor Cross-talk..................................................... 41
Figure 11. Trypan Blue Exclusion Assay. .................................................................................... 43
Figure 12. Adenosine Kinase Inhibition Protocol......................................................................... 49
Figure 13. Rabbit and Human Dialysate Administered Prior to Ischemia-Reperfusion............... 52
Figure 14. Western Blot Analysis of Opioid Receptors in Rabbit Tissues. .................................. 57
Figure 15. Naloxone Administered Prior to Rabbit Dialysate. ..................................................... 59
Figure 16. Naltrindole Administered Prior to Rabbit Dialysate. .................................................. 61
Figure 17. GNTI Administered Prior to Rabbit Dialysate. ........................................................... 63
Figure 18. HOE140 Administered Prior to Rabbit Dialysate. ...................................................... 67
Figure 19. Western Blot Analysis of Calcitonin-Like Receptors in Rabbit Tissues. ................... 70
Figure 20. CGRP (8-37) Administered Prior to Rabbit Dialysate. ............................................... 72

106
Figure 21. 8-SPT Administered Prior to Rabbit Dialysate. .......................................................... 75
Figure 22. 8-SPT Administered Prior to Met-Enkephalin. ........................................................... 81
Figure 23. 8-SPT Administered Prior to Dynorphin B. ................................................................ 83
Figure 24. Adenosine Concentrations After Exposure to Dynorphin B. ...................................... 88
Figure 25. General Diagram of Opioid-Adenosine Cross-talk. .................................................... 94
Figure 26. Protocol of Adenosine Receptor Subtypes in Opioid-Adenosine Cross-talk. ............. 97
Figure 27. Wortmannin Administered Prior to Rabbit Dialysate. .............................................. 100
Figure 28. Standard Plot of Substances in Water. ...................................................................... 110
Figure 29. Standard Plot of Adenosine in Krebs-Henseleit Buffer. ........................................... 112

107
APPENDIX II: List of Tables
Table 1. Krebs-Henseleit Buffer Composition. ............................................................................ 30
Table 2. Tyrode Buffer Composition. ........................................................................................... 42
Table 3. Inter-Observer Error Data. .............................................................................................. 45
Table 4. Rabbit and Human Dialysate Results (Mean ± SEM). ................................................... 52
Table 5. Characterization of Rabbit Dialysate Using MRM Mass Spectrometry. ........................ 53
Table 6. Naloxone Results (Mean ± SEM). .................................................................................. 59
Table 7. Naltrindole Results (Mean ± SEM). ............................................................................... 61
Table 8. GNTI Results (Mean ± SEM). ........................................................................................ 63
Table 9. HOE140 Results (Mean ± SEM). ................................................................................... 67
Table 10. 1x Dialysate with HOE140 Results (% Cardiomyocyte Death). .................................. 69
Table 11. CGRP (8-37) Results (Mean ± SEM). .......................................................................... 72
Table 12. 8-SPT Results (Mean ± SEM). ..................................................................................... 75
Table 13. ADA Results (% Cardiomyocyte Death). ..................................................................... 79
Table 14. Met-Enkephalin with 8-SPT Results (Mean ± SEM). .................................................. 81
Table 15. Dynorphin B with 8-SPT Results (Mean ± SEM). ....................................................... 83
Table 16. Adenosine with Naloxone Results (% Dead Cardiomyocytes). ................................... 86
Table 17. Adenosine Concentrations After Exposure to Dynorphin B Results (Mean ± SEM). . 88
Table 18. Wortmannin Results (Mean ± SEM). ......................................................................... 100
Table 19. Summary of Literature in which Myocardium was the Target Organ. ....................... 108
Table 20. Summary of Literature in which Skeletal Muscle was the Preconditioning Organ.... 109
Table 21. Results of Standard Concentrations of Substances in Water. ..................................... 111
Table 22. Results of Standard Concentrations of Adenosine in Krebs-Henseleit Buffer. .......... 112

108
APPENDIX III: rIPC Summary: Myocardium as the Target Organ
Table 19. Summary of Literature in which Myocardium was the Target Organ.
This table is identical to Table 1 from the review by Hausenloy & Yellon in 2008.19

109
APPENDIX IV: rIPC Summary: Skeletal Muscle as the Preconditioning Organ
203
Table 20. Summary of Literature in which Skeletal Muscle was the Preconditioning Organ.
This table is identical to Table 5 from the review by Tapuria et al. in 2008.203

110
APPENDIX V: MRM Mass Spectrometry Plots of Standard Concentrations
Figure 28. Standard Plot of Substances in Water.
Known standard concentrations of all substances were prepared in water and measured by MRM
mass spectrometry in order to measure accurate concentrations.

111
Table 21. Results of Standard Concentrations of Substances in Water.
Standard
Concentrations
(nM)
Measured Concentrations (nM)
Met
-en
kep
hali
n
Dyn
orp
hin
B
ME
AP
Ad
enosi
ne
Inosi
ne
Bra
dyk
inin
Nore
pin
eph
rin
e
Ace
tylc
holi
ne
An
gio
ten
sin
II
Water --- --- --- --- --- --- --- --- ---
2.5 2.64 --- --- 2.45 0.068
3.4 3.57
3.5 3.77
5.0 4.46 --- --- 5.08 4.94
5.9 7.20
6.9 6.79
10.0 9.9 --- --- 10.5 10.3
14.2 12.4
14.3 14.4
15.0 17.1
17.0 15.8
25.0 24.8 27.0 27.6 24.7 25.1
50.0 50.7 --- 40.7 48.5 47.2
56.6 57.1
57.4 51.3
59.0 55.6
68.0 74.4
100 104 54.3 96.8 101 99.1
113 114
115 108
150 149 155
250 252 296 233 241 255
296 295
342 329
500 517 680 578 521 493
566 751
574 566
592 593
685 687
1000 981 823 959
NOTE: --- represents ‗No Peak‘.

112
Table 22. Results of Standard Concentrations of Adenosine in Krebs-Henseleit Buffer.
Standard Concentrations (nM) Measured Concentrations (nM)
Water ---
100 96.3
1000 1070
10000 9940
10000 10800
NOTE: --- represents ‗No Peak‘.
Figure 29. Standard Plot of Adenosine in Krebs-Henseleit Buffer.
Known standard concentrations of adenosine was prepared in Krebs-Henseleit buffer and
measured by MRM mass spectrometry in order to measure accurate concentrations.

113
REFERENCE LIST
1. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell
injury in ischemic myocardium. Circulation. 1986; 74:1124-1136.
2. Thornton JD, Liu GS, Downey JM. Pretreatment with pertussis toxin blocks the protective
effects of preconditioning: evidence for a G-protein mechanism. J Mol Cell Cardiol. 1993 Mar;
25(3):311-20.
3. Goto M, Liu Y, Yang X-M, Ardell JL, Cohen MV, Downey JM. Role of bradykinin in
protection of ischemic preconditioning in rabbit hearts. Mol Cell Biochem. 1998; 186:3-12.
4. Downey JM, Davis AM, Cohen MV. Signalling pathways in ischemic preconditioning. Heart
Fail Rev. 2007; 12:181-188.
5. Mocanu MM, Bell RM, Yellon DM. PI3 kinase and not p42/p44 appears to be implicated in
the protection conferred by ischemic preconditioning. J Mol Cell Cardiol. 2002; 34:661–668
6. Andjelković M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P,
Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase
B. J Biol Chem. 1997 Dec 12; 272(50):31515-24.
7. Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney
PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that
mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B.
Science. 1998 Jan 30; 279(5351):710-4.
8. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric
oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999; 399:601–
605

114
9. Qin Q, Yang XM, Cui L, Critz SD, Cohen MV, Browner NC, Lincoln TM, Downey JM.
Exogenous NO triggers preconditioning via a cGMP and mitoKATP-dependent mechanism. Am
J Physiol Heart Circ Physiol. 2004 Aug; 287(2):H712-8. Epub 2004 Mar 25.
10. Lochner A, Marais E, Genade S, Moolman JA. Nitric oxide: a trigger for classic
preconditioning? Am J Physiol. 2000; 279:H2752–H2765
11. Oldenburg O, Qin Q, Krieg T, Yang XM, Philipp S, Critz SD, Cohen MV, Downey JM.
Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP
channel opening and leads to cardioprotection. Am J Physiol Heart Circ Physiol. 2004 Jan;
286(1):H468-76. Epub 2003 Sep 4.
12. Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires
signaling through a redox-sensitive mechanism. Circ Res. 2001; 88:802–809
13. Korichneva I, Hoyos B, Chua R, Levi E, Hammerling U. Zinc release from protein kinase C
as the common event during activation by lipid second messenger or reactive oxygen. J Biol
Chem. 2002; 277:44327–44331
14. Cohen MV, Downey JM. Adenosine: trigger and mediator of Cardioprotection. Basic Res
Cardiol. 2008; 103:203–215.
15. Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P. Regional ischemic
‗preconditioning‘ protects remote virgin myocardium from subsequent sustained coronary
occlusion. Circulation. 1993; 87:893.
16. Kanoria S, Jalan R, Seifalian AM, Williams R, Davidson BR. Protocols and mechanisms for
remote ischemic preconditioning: a novel method for reducing ischemia reperfusion injury.
Transplantation. 2007; 84:445-458.

115
17. Dickson EW, Reinhardt CP, Renzi FP, Becker RC, Porcaro WA, Heard SO. Ischemic
preconditioning may be transferable via whole blood transfusion: preliminary evidence. J
Thromb Thrombolysis. 1999; 8:123-129.
18. Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD. Myocardial
protection by brief ischemia in noncardic tissue. Circulation. 1996; 94(9):2193-2200.
19. Hausenloy DJ, Yellon DM. Remote ischaemic preconditioning: underlying mechanisms and
clinical application. Cardiovascular Research. 2008; 79:377–386.
20. Pugsley MK. The diverse molecular mechanisms responsible for the actions of opioids on the
cardiovascular system. Pharmacology & Therapeutics. 2002; 93:51-75.
21. Howells RD, Kilpatrick DL, Bailey LC, Noe M, Udenfriend S. Proenkephalin mRNA in rat
heart. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:1960– 1963.
22. Barron BA. Opioid peptides and the heart. Cardiovasc Res. 1999; 43:13-16
23. Cadet P, Mantione KJ, Stefano GB. Molecular identification and functional expression of μ3,
a novel alternatively spliced variant of the human μ opiate receptor gene. J Immunol. 2003; 170
(10): 5118–23.
24. Krumins SA, Faden AE, Feuerstein G. Opiate binding in rat hearts: modulation of binding
after hemorrhagic shock. Biochem Biophys Res Commun. 1985; 127:120-128.
25. Wittert G, Hope P, Pyle D. Tissue distribution of opioid receptor gene expression in the rat.
Biochem Biophys Res Commun. 1996; 218:877-881.
26. Eliasson T, Mannheimer C, Waagstein F, Andersson B, Bergh CH, Augustinsson LE, Hedner
T, Larson G. Myocardial turnover of endogenous opioids and calcitonin-gene-related peptide in

116
the human heart and the effects of spinal cord stimulation on pacing-induced angina pectoris.
Cardiology. 1998; 89:170–177.
27. Zhang Y, Wu YX, Hao YB, Dun Y, Yang SP. Role of endogenous opioid peptides in
protection of ischemic preconditioning in rat small intestine. Life Sci. 2001; 68:1013.
28. Schultz JE, Hsu AK, Gross GJ. Ischemic preconditioning in the intact rat heart is mediated
by delta1- but not mu- or kappa-opioid receptors. Circulation. 1998 Apr 7; 97(13):1282-9.
29. Aitchison KA, Baxter GF, Awan MM, Smith RM, Yellon DM, Opie LH. Opposing effects
on infarction of delta and kappa opioid receptor activation in the isolated rat heart: implications
for ischemic preconditioning. Basic Res Cardiol. 2000; 95:1–10.
30. Coles JA, Sigg DC, Iaizzo PA. Role of kappa-opioid receptor activation in pharmacological
preconditioning of swine. Am J Physiol Heart Circ Physiol. 2003; 84:H2091–H2099.
31. Wang GY, Wu S, Pei JM, Yu XC, Wong TM. Kappa- but not delta-opioid receptors mediate
effects of ischemic preconditioning on both infarct and arrhythmia in rats. Am J Physiol Heart
Circ Physiol. 2001; 280:H384– H391.
32. Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase
kinase beta inhibition during reperfusion in intact rat hearts. Circ Res. 2004; 94:960– 966.
33. Dickson EW, Blehar DJ, Carraway RE, Heard SO, Steinberg G, Przyklenk K. Naloxone
blocks transferred preconditioning in isolated rabbit hearts. J Mol Cell Cardiol. 2001; 33:1751.
34. Dickson EW, Tubbs RJ, Porcaro WA, Lee WJ, Blehar DJ, Carraway RE, Darling CE,
Przyklenk K. Myocardial preconditioning factors evoke mesenteric ischemic tolerance via opioid
receptors and K(ATP) channels. Am J Physiol Heart Circ Physiol. 2002; 283:H22.

117
35. Patel HH, Moore J, Hsu AK, Gross GJ. Cardioprotection at a distance: Mesenteric artery
occlusion protects the myocardium via an opioid sensitive mechanism. J Mol Cell Cardiol. 2002;
34:1317.
36. Zhang SZ, Wang NF, Xu J, Gao Q, Lin GH, Bruce IC, Xia Q. κ-opioid receptors mediate
cardioprotection by remote preconditioning. Anesthesiology. 2006; 105:550-556.
37. Weinbrenner C, Schulz F, Sarvary L, Strasser RH. Remote preconditioning by infrarenal
aortic occlusion is operative via δ1-opioid receptors and free radicals in vivo in rat heart.
Cardiovasc Res. 2004; 61:591-599.
38. Addison PD, Neligan PC, Ashrafpour H, Khan A, Zhong A, Moses M, Forrest CR, Pang CY.
Noninvasive remote ischemic preconditioning for global protection of skeletal muscle against
infarction. Am J Physiol Heart Circ Physiol. 2003; 285:H1435.
39. Regoli D, Rizzi A, Perron SI, Gobeil F. Classification of Kinin Receptors. Biol Chem. 2001;
382:31– 35.
40. Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins, kallikreins, kininogens, and
kininases. Pharmacol Rev. 1992 Mar; 44(1):1-80. Review.
41. Regoli D, Barabé J. Pharmacology of bradykinin and related kinins. Pharmacol Rev. 1980;
32:1–46.
42. Wiemer G, Schölkens BA, Linz W. Endothelial protection by converting enzyme inhibitors.
Cardiovasc Res. 1994 Feb; 28(2):166-72. Review.
43. Hess JF, Borkowski JA, Young GS, Strader CD, Ransom RW. Cloning and pharmacological
characterization of a human bradykinin (BK-2) receptor. Biochem Biophys Res Comm. 1992;
184:260 – 268.

118
44. Menke JC, Borkowski JA, Bierilo KK, MacNeil T, Derrick AW, Schneck KA, Ransom RW,
Strader CD, Linemeyer DL, Hess JF. Expression cloning of a human B1 bradykinin receptor. J
Biol Chem. 1994; 269: 21583 – 21586.
45. Bachvarov DR, Saint-Jacques E, Larrivée JF, Levesque L, Rioux F, Drapeau G, Marceau F.
Cloning and pharmacological characterization of the rabbit bradykinin B2 receptor. J Pharmacol
Exp Ther. 1995 Dec; 275(3):1623-30.
46. Minshall RD, Miletich DJ, Vogel SM, Becker RP, Erdös EG, Rabito ST. Existence of
bradykinin _BK. binding sites in rat cardiomyocytes. FASEB Meeting. 1994; Abstract 5122.
47. Marceau F, Larrivée JF, Saint-Jacques E, Bachvarov DR. The kinin B1 receptor: an inducible
G protein coupled receptor. Can J Physiol Pharmacol. 1997 Jun; 75(6):725-30. Review.
48. Regoli D, Barabe J, Park WK. Receptors for bradykinin in rabbit aortae. Can J Physiol
Pharmacol. 1977; 55:855-867.
49. Hock FJ, Wirth K, Albus U, Linz W, Gerhards HJ, Wiemer G, Henke S, Breipohl G, König
W, Knolle J, Schölkens BA. HOE 140, a new potent and long acting bradykinin-antagonist: in
vitro studies. Br J Pharmacol. 1991 Mar; 102(3):769-773.
50. Rhaleb NE, Rouissi N, Jukic D, Regoli D, Henke S, Breipohl G, Knolle J. Pharmacological
characterization of a new highly potent B 2 receptor antagonist HOE 140: D-Arg-[Hyp3,Thi5,D-
Tic7,Qic8]bradykinin). Eur J Pharmacol. 1992; 210:115-120.
51. Regoli D, Jukic D, Gobeil F, Rhaleb NE. Receptors for bradykinin and related kinins: a
critical analysis. Can J Physiol Pharmacol. 1993; 71:556-557.
52. Wirth K.J, Wiemer G. Production of cyclic GMP via activation of B l and B2 kinin receptors
in cultured bovine aortic endothelial cells. J Pharmacol Exp Ther. 1992; 262:729-733.

119
53. Campbell DJ, Kladis A, Duncan AM. Bradykinin peptides in kidney, blood, and other tissues
of the rat. Hypertension. 1993 Feb; 21(2):155-65.
54. Pellacani A, Brunner HR, Nussberger J. Plasma kinins increase after angiotensin-converting
enzyme inhibition in human subjects. Clin Sci. 1994; 87:567-74
55. Linz W, Martorana PA, Wiemer G, Wirth K, Schölkens BA. Role of kinins in myocardial
ischemia. EXS. 1996; 76:231-41. Review.
56. Linz W, Wiemer G, Schölkens BA. Role of kinins in the pathophysiology of myocardial
ischemia. In vitro and in vivo studies. Diabetes. 1996 Jan; 45 Suppl 1:S51-8. Review.
57. Schölkens BA, Linz W, König W. Effects of the angiotensin converting enzyme inhibitor,
ramipril, in isolated ischaemic rat heart are abolished by a bradykinin antagonist. J Hypertens.
1988; 6(Suppl. 4):S25-8.
58. Vegh A, Szekeres L, Parratt JR. Local intracoronary infusions of bradykinin profoundly
reduce the severity of ischaemia-induced arrhythmias in anesthetized dogs. Br J Pharmacol.
1991; 104:294-5.
59. Wall TM, Sheehy R, Hartman JC. Role of bradykinin in myocardial preconditioning. J
Pharmacol Exper Ther. 1994; 270(2):681-9.
60. Martorana PA, Kettenbach B, Breipohl G, Linz W, Schölkens BA. Reduction of infarct size
by local angiotensin-converting enzyme inhibition is abolished by a bradykinin antagonist. Eur J
Pharmacol. 1990; 182:395-6.
61. Hashimoto K, Hamamoto H, Honda Y, Hirose M, Furukawa S, Kimura E. Changes in
components of kinin system and hemodynamics in acute myocardial infarction. Am Heart J.
1978; 95:619-626.

120
62. Noda K, Sasaguri M, Ideishi M, Ikeda M, Arakawa K. Role of locally formed angiotensin II
and bradykinin in the reduction of myocardial infarct size in dogs. Cardiovasc Res. 1993;
27:334-340.
63. Linz W, Wiemer G, Gohlke P, Unger T, Schölkens BA. Contribution of kinins to the
cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol Rev. 1995;
47(1):25-49.
64. Ebrahim Z, Yellon DM, Baxter GF. Attenuated cardioprotective response to bradykinin, but
not classical ischaemic preconditioning, in DOCA-salt hypertensive left ventricular hypertrophy.
Pharmacol Res. 2007 Jan; 55(1):42-8. Epub 2006 Oct 31.
65. Hoffmann G, Düsing R. ACE-inhibition, kinins, and vascular PGI2 synthesis. Eicosanoids.
1992; 5 Suppl:S60-2.
66. Minshall RD, Nakamura F, Becker RP, Rabito SF. Characterization of bradykinin B2
receptors in adult myocardium and neonatal rat cardiomyocytes. Circ Res. 1995 May; 76(5):773-
80.
67. Schoemaker RG, van Heijningen CL. Bradykinin mediates cardiac preconditioning at a
distance. Am J Physiol Heart Circ Physiol. 2000; 278:H1571.
68. Wolfrum S, Schneider K, Heidbreder M, Nienstedt J, Dominiak P, and Dendorfer A. Remote
preconditioning protects the heart by activating myocardial PKCε-isoform. Cardiovasc Res.
2002; 55:583-589.
69. Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide
is a potent vasodilator. Nature. 1985; 313:54–56.

121
70. Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin.
Physiol Rev. 2004; 84:903–934.
71. Muff R, Born W, Lutz TA, Fischer JA. Biological importance of the peptides of the
calcitonin family as revealed by disruption and transfer of corresponding genes. Peptides. 2004;
25:2027–2038.
72. Durham PL. CGRP receptor antagonists: a new choice for acute treatment of migraine? Curr
Opin Invest Drugs. 2004; 5:731–735.
73. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;
362:801–809.
74. Lechleitner P, Genser N, Mair J, Dienstl A, Haring C, Wiedermann CJ, Puschendorf B, Saria
A, Dienstl F. Calcitonin gene-related peptide in patients with and without early reperfusion after
acute myocardial infarction. Am Heart J. 1992; 124:1433–1439.
75. Mulderry PK, Ghatei MA, Rodrigo J, Allen JM, Rosenfeld MG, Polak J, Bloom SR. CGRP
in cardiovascular tissues of the rat. Neuroscience. 1985; 14: 947–954.
76. Bick RJ, Poindexter BJ, Schiess MC. Localization of calcitonin gene-related peptide in
cardiomyocytes: comparison of neonatal and dedifferentiating cell to adult myocytes. Peptides.
2005; 26: 331–336.
77. Rosenfeld MG, Mermod JJ, Amara SG, Swanson LW, Sawchenko PE, Rivier J, Vale WW,
Evans RM. Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific
RNA processing. Nature. 1983; 304 (5922):129–135.

122
78. Amara SG, Jonas V, Rosenfeld MG, Ong ES, Evans RM. Alternative RNA processing in
calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature.
1982; 298:240–244.
79. Amara SG, Arriza JL, Leff SE, Swanson LW, Evans RM, Rosenfeld MG. Expression in
brain of a messenger RNA encoding a novel neuropeptide homologous to calcitonin gene-related
peptide. Science. 1985; 229:1094– 1097.
80. Morris HR, Panico M, Etienne T, Tippins J, Girgis SI, MacIntyre I. Isolation and
characterization of human calcitonin gene-related peptide. Nature. 1984; 308:746–748.
81. Dennis T, Fournier A, Cadieux A, Pomerleau F, Jolicoeur FB, Pierre S, Quirion R. hCGRP8-
37, a calcitonin gene-related peptide antagonist revealing calcitonin gene-related peptide receptor
heterogeneity in brain and periphery. J Pharmacol Exp Ther. 1990; 254:123– 128.
82. Gardiner SM, Compton AM, Kemp PA, Bennett T, Bose C, Foulkes R, Hughes B.
Antagonistic effect of human alpha- CGRP [8– 37] on the in vivo regional haemodynamic
actions of human alpha-CGRP. Biochem Biophys Res Commun. 1990; 171:938– 943.
83. McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG,
Foord SM. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like
receptor. Nature. 1998; 393:333–339.
84. Kamitani S, Asakawa M, Shimekake Y, Kuwasako K, Nakahara K, Sakata T. The
RAMP2/CRLR complex is a functional adrenomedullin receptor in human endothelial and
vascular smooth muscle cells. FEBS Lett. 1999; 448 (1):111–4.

123
85. Flahaut M, Rossier BC, Firsov D. Respective roles of calcitonin receptor like receptor
(CRLR) and receptor activity-modifying proteins (RAMP) in cell surface expression of
CRLR/RAMP heterodimeric receptors. J Biol Chem. 2002; 277:14731–14737.
86. Evans BN, Rosenblatt MI, Mnayer LO, Oliver KR, Dickerson IM. CGRPRCP, a novel
protein required for signal transduction at calcitonin gene-related peptide and adrenomedullin
receptors. J Biol Chem. 2000; 275:31438–31443.
87. Kubota M, Moseley JM, Butera L, Dusting GJ, MacDonald PS, Martin TJ. Calcitonin gene-
related peptide stimulates cyclic AMP formation in rat aortic smooth muscle cells. Biochem
Biophys Res Commun. 1985; 132:88–94.
88. Hirata Y, Takagi Y, Takata S, Fukuda Y, Yoshimi H, Fujita T. Calcitonin gene-related
peptide receptor in cultured vascular smooth muscle and endothelial cells. Biochem Biophys Res
Commun. 1988; 151:1113–1121.
89. Hamid SA, Baxter GF. A critical cytoprotective role of endogenous adrenomedullin in acute
myocardial infarction. J Mol Cell Cardiol. 2006; 41:360–363.
90. Yang JH, Jia YX, Pan CS, Zhao J, Ouyang M, Yang J, Chang JK, Tang CS, Qi YF. Effects
of intermedin1-53 on cardiac function and ischemia/ reperfusion injury in isolated rat hearts.
Biochem Biophys Res Commun. 2005; 327:713–719.
91. Li YJ, Xiao ZS, Peng CF, Deng HW. Calcitonin gene-related peptide-induced
preconditioning protects against ischemia-reperfusion injury in isolated rat hearts. Eur J
Pharmacol. 1996; 311:163–167.

124
92. Chai W, Mehrota S, Danser AHJ, Schoemaker RG. The role of calcitonin gene-related
peptide (CGRP) in ischemic postconditioning in isolated rat hearts. Eur J Pharmacol. 2006;
531:246–253.
93. Wang L, Wang DH. TRPV1 gene knockout impairs postischemic recovery in isolated
perfused heart in mice. Circulation. 2005; 112:3617–3623.
94. Lu R, Li YJ, Deng HW. Evidence for calcitonin gene-related peptide-mediated ischemic
preconditioning in the rat heart. Regul Pept. 1999; 82:53– 57.
95. Lu EX, Peng CF, Li YJ, Cheng SX. Calcitonin gene-related peptide-induced preconditioning
improves preservation with cardioplegia. Ann Thorac Surg. 1996; 62:1748– 1751.
96. Ou-Yang W, Qian XX, Li ZL, Fu XY, Wang SH. The roles of endogenous calcitonin gene-
related peptide on myocardial ischemic preconditioning in intact rat. Chin J Arterioscler. 1999;
7:24–26. [Abstract only].
97. Xiao L, Lu R, Hu CP, Deng HW, Li YJ. Delayed cardioprotection by intestinal
preconditioning is mediated by calcitonin gene-related peptide. Eur J Pharmacol. 2001; 427:131.
98. Tang ZL, Dai W, Li YJ, Deng HW. Involvement of capsaicin-sensitive sensory nerves in
early and delayed cardioprotection induced by a brief ischaemia of the small intestine. Naunyn
Schmiedebergs Arch Pharmacol. 1999; 359:243.
99. Pajdo R, Brzozowski T, Konturek PC, Kwiecien S, Konturek SJ, Sliwowski Z, Pawlik M,
Ptak A, Drozdowicz D, Hahn EG. Ischemic preconditioning, the most effective gastroprotective
intervention: involvement of prostaglandins, nitric oxide, adenosine and sensory nerves. Eur J
Pharmacol. 2001; 427:263–276.

125
100. Meldrum DR, Dinarello CA, Shames BD, Cleveland JC, Cain BS, Banerjee A, Meng X,
Harken AH. Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-
alpha production. Potential ultimate effector mechanism of preconditioning. Circulation. 1998;
98(19 Suppl.):II214–II218.
101. Peng J, Xiao J, Ye F, Deng HW, Li YJ. Inhibition of cardiac tumor necrosis factor-alpha
production by calcitonin gene-related peptide-mediated ischemic preconditioning in isolated rat
hearts. Eur J Pharmacol. 2000; 407:303– 308.
102. Hu CP, Li YJ, Deng HW. The cardioprotective effects of nitroglycerin-induced
preconditioning are mediated by calcitonin gene-related peptide. Eur J Pharmacol. 1999;
369:189–194.
103. Wolfrum S, Nienstedt J, Heidbreder M, Schneider K, Dominiak P, Dendorfer A. Calcitonin
gene related peptide mediates cardioprotection by remote preconditioning. Regul Pept. 2005;
127:217–224.
104. Li G, Chen S, Lu E, Luo W. Cardiac ischemic preconditioning improves lung preservation
in valve replacement operations. Ann Thorac Surg. 2001; 71:631–635.
105. Fredholm BB, Ijzerman AP, Jacobson KA, Klotz K-N, Linden J. International Union of
Pharmacology. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;
53:527–552.
106. Deussen A. Metabolic flux rates of adenosine in the heart. Naunyn Schmiedebergs Arch
Pharmacol. 2000 Nov; 362(4-5):351-63. Review.

126
107. Moriwaki Y, Yamamoto T, Higashino K. Enzymes involved in purine metabolism--a
review of histochemical localization and functional implications. Histol Histopathol. 1999 Oct;
14(4):1321-40. Review.
108. Geisbuhler T, Altschuld RA, Trewyn RW, Ansel AZ, Lamka K, Brierley GP. Adenine
nucleotide metabolism and compartmentalization in isolated adult rat heart cells. Circ Res. 1984;
54:536-46.
109. Mubagwa K, Flameng W. Adenosine, adenosine receptors and myocardial protection: an
updated overview. Cardiovasc Res. 2001; 52:25–39.
110. Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P,
Williams M. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994; 46:143-
56.
111. Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein
kinases. Cell Signal. 2003; 15:813–827.
112. Kilpatrick EL, Narayan P, Mentzer RM, Lasley RD. Cardiac myocyte adenosine A2a
receptor activation fails to alter cAMP or contractility: role of receptor localization. Am J
Physiol. 2002; 282:H1035–H1040.
113. Hein TW, Wang W, Zoghi B, Muthuchamy M, Kuo L. Functional and molecular
characterization of receptor subtypes mediating coronary microvascular dilation to adenosine. J
Mol Cell Cardiol. 2001; 33:271–282.
114. Schulz R, Rose J, Post H, Heusch G. Involvement of endogenous adenosine in ischaemic
preconditioning in swine. Pflugers Arch. 1995; 430:273–282.

127
115. Thornton JD, Liu GS, Olsson RA, Downey JM. Intravenous pretreatment with A1-selective
adenosine analogues protects the heart against infarction. Circulation. 1992; 85:659–665.
116. Auchampach JA, Gross GJ. Adenosine A1 receptors, KATP channels, and ischemic
preconditioning in dogs. Am J Physiol. 1993; 264:H1327–H1336.
117. Tsuchida A, Miura T, Miki T, Shimamoto K, Iimura O. Role of adenosine receptor
activation in myocardial infarct size limitation by ischaemic preconditioning. Cardiovasc Res.
1992; 26:456–461.
118. Walker DM, Walker JM, Pugsley WB, Pattison CW, Yellon DM. Preconditioning in
isolated superfused human muscle. J Mol Cell Cardiol. 1995; 27:1349–1357.
119. Liu GS, Thornton J, Van Winkle DM, Stanley AWH, Olsson RA, Downey JM. Protection
against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit
heart. Circulation. 1991; 84:350–356.
120. Armstrong S, Ganote CE. In vitro ischaemic preconditioning of isolated rabbit
cardiomyocytes: effects of selective adenosine receptor blockade and calphostin C. Cardiovasc
Res. 1995 May; 29(5):647-52.
121. Peart J, Headrick JP. Adenosine-mediated early preconditioning in mouse: protective
signaling and concentration dependent effects. Cardiovasc Res. 2003 Jun 1; 58(3):589-601.
122. Liu GS, Richards SC, Olsson RA, Mullane K, Walsh RS, Downey JM. Evidence that the
adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated
rabbit heart. Cardiovasc Res. 1994; 28:1057–1061.
123. Auchampach JA, Rizvi A, Qiu Y, Tang XL, Maldonado C, Teschner S, Bolli R. Selective
activation of A3 adenosine receptors with N6-(3-iodobenzyl) adenosine-5‘-N-methyluronamide

128
protects against myocardial stunning and infarction without hemodynamic changes in conscious
rabbits. Circ Res. 1997; 80:800–809.
124. Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects
rabbit hearts through a protein kinase C adenosine A2b receptor cascade. Cardiovasc Res. 2006;
70:308–314.
125. Solenkova NV, Solodushko V, Cohen MV, Downey JM. Endogenous adenosine protects
preconditioned heart during early minutes of reperfusion by activating Akt. Am J Physiol. 2006;
290:H441–H449.
126. Kuno A, Critz SD, Cui L, Solodushko V, Yang X-M, Krahn T, Albrecht B, Philipp S,
Cohen MV, Downey JM. Protein kinase C protects preconditioned rabbit hearts by increasing
sensitivity of adenosine A2bdependent signaling during early reperfusion. J Mol Cell Cardiol.
2007; 43:262–271.
127. Liem DA, te Lintel Hekkert M, Manintveld OC, Boomsma F, Verdouw PD, Duncker DJ.
Myocardium tolerant to an adenosine-dependent ischemic preconditioning stimulus can still be
protected by stimuli that employ alternative signalling pathways. Am J Physiol Heart Circ
Physiol. 2005; 288:H1165.
128. Pell TJ, Baxter GF, Yellon DM, Drew GM. Renal ischemia preconditions myocardium: role
of adenosine receptors and ATP-sensitive potassium channels. Am J Physiol. 1998; 275:H1542.
129. Pang CY, Neligan P, Zhong A, He W, Xu H, Forrest CR. Effector mechanism of adenosine
in acute ischemic preconditioning of skeletal muscle against infarction. Am J Physiol. 1997;
273:R887.

129
130. Oxman T, Arad M, Klein R, Avazov N, Rabinowitz B. Limb ischemia preconditions the
heart against reperfusion tachyarrhythmia. Am J Physiol. 1997; 273:H1707–H1712.
131. Dickson EW, Lorbar M, Porcaro WA, Fenton RA, Reinhardt CP, Gysembergh A, Przyklenk
K. Rabbit heart can be ‗preconditioned‘ via transfer of coronary effluent. Am J Physiol. 1999;
277:H2451–H2457.
132. Singh D, Chopra K. Evidence of the role of angiotensin AT(1) receptors in remote renal
preconditioning of myocardium. Methods Find Exp Clin Pharmacol. 2004; 26:117–122.
133. Brzozowski T, Konturek PC, Pajdo R, Kwiecień S, Sliwowski Z, Drozdowicz D, Ptak-
Belowska A, Pawlik M, Konturek SJ, Pawlik WW, Hahn GG. Importance of brain-gut axis in the
gastroprotection induced by gastric and remote preconditioning. J Physiol Pharmacol. 2004;
55:165.
134. Di Filippo C, Rossi F, Rossi S, D‘Amico M. Cannabinoid CB2 receptor activation reduces
mouse myocardial ischemia-reperfusion injury: involvement of cytokine/chemokines and PMN.
J Leukoc Biol. 2004; 75:453–459.
135. Hajrasouliha AR, Tavakoli S, Ghasemi M, Jabehdar-Maralani P, Sadeghipour H, Ebrahimi
F, Dehpour AR. Endogenous cannabinoids contribute to remote ischemic preconditioning via
cannabinoid CB(2) receptors in the rat heart. Eur J Pharmacol. 2008; 579:246–52.
136. Yao L, Fan P, Jiang Z, Mailliard WS, Gordon AS, Diamond I. Addicting drugs utilize a
synergistic molecular mechanism in common requiring adenosine and Gi-beta gamma dimers.
Proc Natl Acad Sci USA. 2003; 100:14379–14384.
137. Eisenach JC, Hood DD, Curry R, Sawynok J, Yaksh TL, Li X. Intrathecal but not
intravenous opioids release adenosine from the spinal cord. J Pain. 2004; Feb; 5(1):64-8.

130
138. Peart JN, Gross GJ. Adenosine and opioid receptor-mediated cardioprotection in the rat:
evidence for cross-talk between receptors. Am J Physiol Heart Circ Physiol. 2003 Jul;
285(1):H81-9.
139. Peart JN, Gross GJ. Cardioprotection following adenosine kinase inhibition in rat hearts.
Basic Res Cardiol. 2005 Jul; 100(4):328-36.
140. Horie Y, Ishii H. Liver dysfunction elicited by gut ischemia-reperfusion. Pathophysiology.
2001; 8:11.
141. Liem DA, Verdouw PD, Ploeg H, Kazim S, Duncker DJ. Sites of action of adenosine in
inter-organ preconditioning of the heart. Am J Physiol Heart Circ Physiol. 2002; 283:H29.
142. Deussen A, Stappert M, Schäfer S, Kelm M. Quantification of extracellular and intracellular
adenosine production: understanding the transmembranous concentration gradient. Circulation.
1999; 99:2041–2047.
143. Cheung MM, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, Holtby HM,
Cox PN, Smallhorn JF, Van Arsdell GS, Redington AN. Randomized controlled trial of the
effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical
application in humans. J Am Coll Cardiol. 2006; 47:2277-2282.
144. Ives HE, Schultz GS, Galardy RE, Jamieson JD. Preparation of functional smooth muscle
cells from the rabbit aorta. J Exp Med. 1978 Nov 1; 148(5):1400-13.
145. ter Welle HF, Baartscheer A, Fiolet JW, Schumacher CA. The cytoplasmic free energy of
ATP hydrolysis in isolated rod-shaped rat ventricular myocytes. J Mol Cell Cardiol. 1988 May;
20(5):435-41.

131
146. Shimizu M, Tropak M, Diaz RJ, Suto F, Surendra H, Kuzmin E, Li J, Gross G, Wilson GJ,
Callahan JW, Redington AN. Transient limb ischemia remotely preconditions through a humoral
mechanism acting directly on the myocardium: evidence suggesting cross-species protection.
Clin Sci (Lond). 2009; Jan 28. [Epub ahead of print]
147. Vander Heide RS, Rim D, Hohl CM, Ganote CE. An in vitro model of myocardial ischemia
utilizing isolated adult rat myocytes. J Mol Cell Cardiol. 1990 Feb; 22(2):165-81.
148. Takasaki Y, Wolff RA, Chien GL, Van Winkle DM. Met5-enkephalin protects isolated
adult rabbit cardiomyocytes via δ-opioid receptors. Am J Physiol Heart Circ Physiol. 1999;
277:H2442-H2450.
149. Olson GA, Olson RD, Kastin AJ. Endogenous opiates: 1995. Peptides. 1996; 17(8):1421-
66. Review.
150. Spivak CE, Beglan CL, Seidleck BK, Hirshbein LD, Blaschak CJ, Uhl GR, Surratt CK.
Naloxone activation of mu-opioid receptors mutated at a histidine residue lining the opioid
cavity. Mol Pharmacol. 1997; 52 983.
151. Pinelli A, Trivulzio S. Quantitative evaluation of opioid withdrawal signs in rats repeatedly
treated with morphine and injected with naloxone, in the absence or presence of the anti-
abstinence agent clonidine. J Pharmacol Toxicol Methods. 1997 Nov; 38(3):117-31.
152. Cao Z, Liu L and Van Winkle DM. Activation of δ- and κ-opioid receptors by opioid
peptides protects cardiomyocytes via KATP channels. Am J Physiol Heart Circ Physiol. 2003;
285:H1032-H1039.

132
153. Portoghese PS, Sultana M, Takemori AE. Design of peptidomimetic δ opioid receptor
antagonists using the message-address concept. J Med Chem. 1990 Jun; 33(6):1714-20. Erratum
in: J Med Chem. 1990 Dec; 33(12):3229.
154. Ossipov MH, Kovelowski CJ, Vanderah T, Porreca F. Naltrindole, an opioid δ antagonist,
blocks the enhancement of morphine-antinociception induced by a CCKB antagonist in the rat.
Neurosci Lett. 1994 Nov 7; 181(1-2):9-12.
155. Shippenberg TS, Heidbreder C. The delta-opioid receptor antagonist naltrindole prevents
sensitization to the conditioned rewarding effects of cocaine. Eur J Pharmacol. 1995 Jun 23;
280(1):55-61
156. Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. Mutational evidence for a common
kappa antagonist binding pocket in the wild type kappa and mutant mu[K303E] opioid receptors.
J Med Chem. 1998 Dec 3; 41(25):4911-4.
157. Jones RM, Portoghese PS. 5‘-Guanidinonaltrindole, a highly selective and potent kappa-
opioid receptor antagonist. Eur J Pharmacol. 2000 May 12; 396(1):49-52.
158. Jewett DC, Grace MK, Jones RM, Billington CJ, Portoghese PS, Levine AS. The kappa-
opioid antagonist GNTI reduces U50,488-, DAMGO-, and deprivation-induced feeding, but not
butorphanol- and neuropeptide Y-induced feeding in rats. Brain Res. 2001 Aug 3; 909(1-2):75-
80.
159. Félétou M, Germain M, Thurieau C, Fauchère JL, Canet E. Agonistic and antagonistic
properties of the bradykinin B2 receptor antagonist, Hoe 140, in isolated blood vessels from
different species. Br J Pharmacol. 1994 Jun; 112(2):683-9.

133
160. Oldenburg O, Critz SD, Cohen MV, Downey JM. Acetylcholine-induced production of
reactive oxygen species in adult rabbit ventricular myocytes is dependent on phosphatidylinositol
3- and Src-kinase activation and mitochondrial K(ATP) channel opening. J Mol Cell Cardiol.
2003 Jun; 35(6):653-60.
161. Poyner DR. Pharmacology of receptors for calcitonin gene-related peptide and amylin.
Biochem Soc Trans. 1997 Aug; 25(3):1032-6.
162. Chiba T, Yamaguchi A, Yamatani T, Nakamura A, Morishita T, Inui T, Fukase M, Noda T,
Fujita T. Calcitonin gene-related peptide receptor antagonist human CGRP-(8-37). Am J Physiol.
1989 Feb; 256(2 Pt 1):E331-5.
163. Bruns RF, Daly JW, Snyder SH. Adenosine receptors in brain membranes: binding of N6-
cyclohexyl[3H]adenosine and 1,3-diethyl-8-[3H]phenylxanthine. Proc Natl Acad Sci U S A.
1980 Sep; 77(9):5547-51.
164. Romano MA, McNish R, Seymour EM, Traynor JR, Bolling SF. Differential effects of
opioid peptides on myocardial ischemic tolerance. J Surg Res. 2004 Jun 1; 119(1):46-50.
165. Reisine T, Bell GI. Molecular biology of opioid receptors. Trends Neurosci. 1993; 16:506–
510.
166. Jordan BA, Devi LA. G-protein coupled receptor heterodimerization modulates receptor
function. Nature. 1999; 399:697–700.
167. Yin H, Chao L, Chao J. Adrenomedullin protects against myocardial apoptosis after
ischemia/reperfusion through activation of Akt-GSK signaling. Hypertension. 2004 Jan;
43(1):109-16. Epub 2003 Dec 8.

134
168. Auchampach JA, Jin X, Moore J, Wan TC, Kreckler LM, Ge ZD, Narayanan J, Whalley E,
Kiesman W, Ticho B, Smits G, Gross GJ. Comparison of three different A1 adenosine receptor
antagonists on infarct size and multiple cycle ischemic preconditioning in anesthetized dogs. J
Pharmacol Exp Ther. 2004; 308:846–856.
169. Lankford AR, Yang J-N, Rose‘Meyer R, French BA, Matherne GP, Fredholm BB, Yang Z.
Effect of modulating cardiac A1 adenosine receptor expression on protection with ischemic
preconditioning. Am J Physiol. 2006; 290:H1469–H1473.
170. Guo Y, Bolli R, Bao W, Wu WJ, Black RG, Murphree SS, Salvatore CA, Jacobson MA,
Auchampach JA. Targeted deletion of the A3 adenosine receptor confers resistance to
myocardial ischemic injury and does not prevent early preconditioning. J Mol Cell Cardiol.
2001; 33:825–830.
171. Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H,
Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5‘-nucleotidase (CD73) and
A2B adenosine receptors. Circulation. 2007; 115:1581–1590.
172. Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and
stimulates mast cell degranulation. J Clin Invest. 1997 Dec 1; 100(11):2849-57.
173. Shen H, Chen GJ, Harvey BK, Bickford PC, Wang Y. Inosine reduces ischemic brain injury
in rats. Stroke. 2005 Mar; 36(3):654-9. Epub 2005 Feb 3. Erratum in: Stroke. 2005 May;
36(5):1106.
174. Litsky ML, Hohl CM, Lucas JH, Jurkowitz MS. Inosine and guanosine preserve neuronal
and glial cell viability in mouse spinal cord cultures during chemical hypoxia. Brain Res. 1999
Mar 13; 821(2):426-32.

135
175. Silva PH, Dillon D, Van Wylen DG. Adenosine deaminase inhibition augments interstitial
adenosine but does not attenuate myocardial infarction. Cardiovasc Res. 1995 May; 29(5):616-
23.
176. George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, O'Dowd BF. Oligomerization of mu-
and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;
275:26128–26135.
177. Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA. Heterodimerization of mu
and delta opioid receptors: A role in opiate synergy. J Neurosci. 2000; 20:RC110.
178. Jordan BA, Trapaidze N, Gomes I, Nivarthi R, Devi LA. Oligomerization of opioid
receptors with beta 2- adrenergic receptors: a role in trafficking and mitogen-activated protein
kinase activation. Proc Natl Acad Sci USA. 2001; 98: 343–348.
179. Pfeiffer M, Koch T, Schroder H, Laugsch M, Hollt V, Schulz S. Heterodimerization of
somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and
desensitization. J Biol Chem. 2002; 277:19762–19772.
180. Ginés S, Hillion J, Torvinen M, Le Crom S, Casadó V, Canela EI, Rondin S, Lew JY,
Watson S, Zoli M, Agnati LF, Verniera P, Lluis C, Ferré S, Fuxe K, Franco R. Dopamine D1 and
adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci
USA. 2000; 97:8606–8611.
181. Yoshioka K, Hosoda R, Kuroda Y, Nakata H. Hetero-oligomerization of adenosine A1
receptors with P2Y1 receptors in rat brains. FEBS Lett. 2002; 531:299–303.
182. Kamiya T, Saitoh O, Yoshioka K, Nakata H. Oligomerization of adenosine A2A and
dopamine D2 receptors in living cells. Biochem Biophys Res Commun. 2003; 306:544–549.

136
183. Hébert TE, Galés C, Rebois RV. Detecting and imaging protein-protein interactions during
G protein-mediated signal transduction in vivo and in situ by using fluorescence-based
techniques. Cell Biochem Biophys. 2006; 45(1):85-109. Review.
184. Gandía J, Lluís C, Ferré S, Franco R, Ciruela F. Light resonance energy transfer-based
methods in the study of G protein-coupled receptor oligomerization. Bioessays. 2008 Jan;
30(1):82-9. Review.
185. Tokuno S, Hinokiyama K, Tokuno K, Lowbeer C, Hansson LO, Valen G. Spontaneous
ischemic events in the brain and heart adapt the hearts of severely atherosclerotic mice to
ischemia. Arterioscler Thromb Vasc Biol. 2002; 22:995–1001.
186. Weinbrenner C, Nelles M, Herzog N, Sarvary L, Strasser RH. Remote preconditioning by
infrarenal occlusion of the aorta protects the heart from infarction: a newly identified non-
neuronal but PKC-dependent pathway. Cardiovasc Res. 2002; 55:590–601.
187. Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to
clinical cardiology. Physiol Rev. 2003; 83:1113–1151.
188. Costa AD, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD. The mechanism by
which the mitochondrial ATP-sensitive Kþ channel opening and H2O2 inhibit the mitochondrial
permeability transition. J Biol Chem. 2006; 281:20801–20808.
189. Li G, Labruto F, Sirsjö A, Chen F, Vaage J, Valen G. Myocardial protection by remote
preconditioning: The role of nuclear factor kappa-B p105 and inducible nitric oxide synthase.
Eur J Cardiothorac Surg. 2004; 26:968.
190. O‘Rourke B. Myocardial K(ATP) channels in preconditioning. Circ Res. 2000; 87:845.

137
191. Murata M, Akao M, O‘Rourke B, Marban E. Mitochondrial ATP sensitive potassium
channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: Possible
mechanism of cardioprotection. Circ Res. 2001; 89:891.
192. Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of
opening the ATP-sensitive K(+) channel of heart mitochondria. Am J Physiol Heart Circ
Physiol. 2001; 280:H649.
193. Takaoka A, Nakae I, Mitsunami K, Yabe T, Morikawa S, Inubushi T, Kinoshita M. Renal
ischemia/reperfusion remotely improves myocardial energy metabolism during myocardial
ischemia via adenosine receptors in rabbits: effects of ―remote preconditioning‖. J Am Coll
Cardiol. 1999; 33:556.
194. Wang YP, Maeta H, Mizoguchi K, Suzuki T, Yamashita Y, Oe M. Intestinal ischemia
preconditions myocardium: Role of protein kinase C and mitochondrial K(ATP) channel.
Cardiovasc Res. 2002; 55:576-82.
195. Kristiansen SB, Henning O, Kharbanda RK, Nielsen-Kudsk JE, Schmidt MR, Redington
AN, Nielsen TT, Bøtker HE. Remote preconditioning reduces ischemia-reperfusion injury in the
explanted heart by a KATP channel-dependent mechanism. Am J Physiol Heart Circ Physiol.
2005 Mar; 288(3):H1252-6. Epub 2004 Oct 21.
196. Moses MA, Addison PD, Neligan PC, Ashrafpour H, Huang N, Zair M, Rassuli A, Forrest
CR, Grover GJ, Pang CY. Mitochondrial KATP channels in hind limb remote ischemic
preconditioning of skeletal muscle against infarction. Am J Physiol Heart Circ Physiol. 2005
Feb; 288(2):H559-67. Epub 2004 Sep 30.

138
197. Moses MA, Addison PD, Neligan PC, Ashrafpour H, Huang N, McAllister SE, Lipa JE,
Forrest CR, Pang CY. Inducing late phase of infarct protection in skeletal muscle by remote
preconditioning: Efficacy and mechanism. Am J Physiol Regul Integr Comp Physiol. 2005 Dec;
289(6):R1609-17. Epub 2005 Sep 22.
198. Vinten-Johansen J. Postconditioning: a mechanical maneuver that triggers biological and
molecular cardioprotective responses to reperfusion. Heart Fail Rev. 2007; 12:235–244.
199. Heidbreder M, Naumann A, Tempel K, Dominiak P, Dendorfer A. Remote vs. ischaemic
preconditioning: the differential role of mitogen-activated protein kinase pathways. Cardiovasc
Res. 2008; 78:108–15.
200. Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: its fundamental
role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003; 35:339–
341.
201. Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and
preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003; 284:G15.
202. Konstantinov IE, Arab S, Li J, Coles JG, Boscarino C, Mori A, Cukerman E, Dawood F,
Cheung MM, Shimizu M, Liu PP, Redington AN. The remote ischemic preconditioning stimulus
modifies gene expression in mouse myocardium. J Thorac Cardiovasc Surg. 2005; 130:1326.
203. Tapuria N, Kumar Y, Habib MM, Amara MA, Seifalian AM, Davidson BR. Remote
Ischemic Preconditioning: A Novel Protective Method From Ischemia Reperfusion Injury—A
Review. J Surg Res. 2008; 150:304–330.
Related Documents











