



FACULTAD DE CIENCIAS
DEPARTAMENTO DE FÍSICA DE MATERIALES
Sistemas híbridos basados en
grafeno y MoS2-2D para detección
óptica
Memoria para acceder al grado de Doctor realizada por
Sandra Cortijo Campos
Tesis codirigida por
Prof. Alicia de Andrés Miguel
Prof. Carlos A. Prieto de Castro
Instituto de Ciencia de Materiales de Madrid (CSIC)
Tutora: Prof. Luisa E. Bausá López (UAM)
Madrid, septiembre 2021

RESUMEN
Tras la aparición del grafeno, un gran número de materiales bidimensionales
han atraído el interés de la comunidad científica debido a sus potenciales
aplicaciones, que en muchos casos se espera que sean superiores a las del
grafeno. De entre estos materiales bidimensionales destacan los
dicalcogenuros de metales de transición (TMCDs), en particular el disulfuro de
molibdeno.
En este trabajo de Tesis doctoral se ha tenido por objetivo, por una parte, el
diseño y montaje de un sistema apto para el crecimiento y dopado por
distintas técnicas de crecimiento en fase de vapor (CVD, del inglés chemical
vapour deposition y PVT, del inglés physical vapour transport) de
dicalcogenuros de metales de transición, el crecimiento de MoS2
bidimensional por dichas técnicas y el estudio de sus propiedades en función
del método de crecimiento, del número de capas, del tamaño de los copos y
de distintos defectos intrínsecos. También se ha estudiado el efecto que tiene
en la eficiencia de fotoluminiscencia la presencia de nanopartículas de MoO2
en muestras de MoS2 bidimensional crecidas por CVD.
Por otra parte, se ha llevado a cabo el diseño, estudio y desarrollo de
diferentes plataformas basadas en estos dos materiales bidimensionales
(grafeno y MoS2) para su uso en aplicaciones de detección óptica.
El grafeno se ha combinado con nanopartículas de plata para aplicaciones de
detección óptica mediante la amplificación de la señal Raman. La
espectroscopía Raman cumple el requisito de especificidad para la detección
molecular, ya que el espectro de vibración de cada compuesto es único,
pudiéndose utilizar para su identificación. Por esto, durante los últimos años,
la comunidad científica ha hecho un gran esfuerzo para aumentar la señal

Raman, normalmente de intensidad débil, principalmente a través del acoplo
con plasmones localizados resonantes de nanopartículas metálicas, llamado
efecto SERS (del inglés surface enhanced Raman spectroscopy). Sin embargo,
las plataformas SERS presentan serios problemas tales como la estabilidad de
las nanopartículas, su interacción con el analito o la adsorción, distribución y
orientación de las moléculas sobre el sustrato. En este campo, la introducción
de grafeno proporciona varias ventajas como son la estabilización de la
superficie sobre la que se depositan las nanopartículas metálicas, un sustrato
altamente bio-compatible, la modulación de la interacción analito-metal y la
dotación de una vía de desexcitación no-radiativa que elimina la fluorescencia
no deseada de moléculas orgánicas. En concreto, en este trabajo se evalúa la
capacidad para amplificar las señales Raman por parte de NPs de Ag esféricas,
monodispersas, ultra pequeñas (R ~ 2 nm) y libres de impurezas, y se compara
con otras NPs de Ag de mayor tamaño y forma diferente. También se estudia
la relevancia de la distancia entre NPs en el plasmón y en la eficiencia de la
amplificación SERS. Para ello se emplean las señales del grafeno y de una
molécula de referencia como la Rodamina 6G (R6G). Por último, se estudia el
efecto que tiene la combinación los dos tipos de NPs en una misma
plataforma.
La detección de bio-marcadores es de gran importancia para la medicina
preventiva del cáncer y sin embargo es todavía particularmente difícil debido
a su extremadamente baja concentración en sangre y a la complejidad del
medio en el que se encuentran. Por ello, se ha llevado a cabo la
funcionalización tanto del grafeno como del MoS2 para su uso como
componentes clave en aplicaciones de detección de biomarcadores de
proteínas. En el caso del grafeno, se ha se ha diseñado y optimizado una nueva
ruta de síntesis para funcionalizar in-situ grafeno-CVD con grupos carboxílicos
(-COOH) por enlace covalente de una forma controlada. Estos grupos
carboxílicos son necesarios para facilitar la posterior unión con otros grupos

específicos, como las aminas primarias (-NH2), presentes en diferentes
moléculas biológicamente activas (como anticuerpos) y disponer así de
plataformas aptas para sensores ópticos o electrónicos de biomoléculas. Por
otra parte, se ha desarrollado un biosensor en el que se aprovecha la
fotoluminiscencia del MoS2 monocapa para la detección óptica de
biomarcadores a través de los cambios producidos en la fotoluminiscencia tras
la hibridación con el biomarcador correspondiente. En particular, se ha
probado la especificidad para la detección del biomarcador miARN21 del
cáncer de mama.

Índice
1 Introducción...................................................................................... 1
1.1 Materiales 2D ................................................................................... 1
1.1.1 El grafeno: definición, propiedades y caracterización ............. 1
1.1.2 TMDCs: propiedades, producción y caracterización ................ 4
1.2 Plataformas para la detección de moléculas y biomarcadores ..... 17
1.2.1 Nanopartículas: Amplificación de la señal Raman ................. 17
1.2.2 Biosensores basados en materiales bidimensionales ............ 23
1.3 Referencias ..................................................................................... 30
2 Técnicas experimentales.................................................................. 41
2.1 Técnicas de preparación de materiales-2D .................................... 41
2.1.1 Depósito químico en fase de vapor ........................................ 41
2.1.2 Transporte físico de vapor...................................................... 49
2.1.3 Transferencia de materiales bidimensionales crecidos por
CVD/PVT ................................................................................................ 53
2.1.4 Inmovilización de anticuerpos. método carbodiimida ........... 56
2.2 Técnicas de preparación de nanopartículas y láminas delgadas ... 58
2.2.1 Pulverización catódica o sputtering ....................................... 58
2.2.2 Agregación en fase gas para el depósito de nanopartículas .. 60
2.3 Técnicas de caracterización ............................................................ 62
2.3.1 Espectroscopía Raman ........................................................... 62
2.3.2 Espectroscopia de absorción UV-Vis ...................................... 67
2.3.3 Fotoluminiscencia .................................................................. 69
2.3.4 Espectroscopia fotoelectrónica de rayos X ............................ 70
2.3.5 Microscopía de fuerza atómica .............................................. 71
2.3.6 Medidas eléctricas .................................................................. 74

2.4 Referencias ..................................................................................... 76
3 MoS2 bidimensional ........................................................................ 79
3.1 Síntesis de MoS2-2D por técnicas en fase vapor ........................... 81
3.1.1 Síntesis de MoS2-2D por CVD a partir de MoO3 y de S ......... 81
3.1.2 Síntesis de MoS2-2D por PVT a partir de MoS2 en polvo ...... 89
3.2 Estudio de los espectros Raman de MoS2 tridimensional y
bidimensional ............................................................................................. 95
3.2.1 Región I: 100-270 cm-1 ........................................................ 107
3.2.2 Región II: 270-420 cm-1 ....................................................... 116
3.2.3 Región III: 420-500 cm-1 ...................................................... 119
3.2.4 Región IV: 500-650 cm-1 ...................................................... 123
3.2.5 Región V: 650-850 cm-1 ....................................................... 124
3.3 Efectos del tamaño en nanocopos de MoS2 de una y pocas capas ...
...................................................................................................... 126
3.4 Amplificación Raman y de Fotoluminiscencia por Nanopartículas de
MoO2 ...................................................................................................... 138
3.5 Referencias ................................................................................... 149
4 SERS en plataformas de grafeno y nanopartículas de Ag ................. 155
4.1 Tamaño y morfología de las nanopartículas de Ag ultrapequeñas ....
...................................................................................................... 156
4.2 Análisis de los plasmones de las nanopartículas ultrapequeñas:
experimentos y simulaciones ................................................................... 157
4.3 Amplificación Raman originada por las AgNPs de 1.8 nm ........... 165
4.4 Comparación de Películas de Ag Nanoestructuradas y
Nanopartículas de Ag Ultrapequeñas. ..................................................... 172
4.5 Películas de Ag Nanoestructuradas Combinadas con AgNPs de R =
1.8 nm ...................................................................................................... 176
4.6 Referencias ................................................................................... 181

5 Funcionalización de grafeno con grupos carboxílicos para aplicaciones
en biodetección .................................................................................... 185
5.1 Optimización de la funcionalización in-situ de grafeno CVD con
COOH ...................................................................................................... 186
5.2 Caracterización mediante espectroscopía Raman del grafeno
obtenido mediante funcionalización in-situ. ........................................... 188
5.3 Caracterización XPS de grafeno obtenido mediante funcionalización
in-situ. 194
5.4 Caracterización eléctrica del grafeno obtenido mediante
funcionalización in-situ. ........................................................................... 198
5.5 Inmovilización de anticuerpos por el método carbodiimida ....... 203
5.6 Referencias ................................................................................... 208
6 MoS2 monocapa como detector de biomarcadores de cáncer de mama
..................................................................................................... 211
6.1 Caracterización por espectroscopías Raman y de fotoluminiscencia
de plataformas de MoS2 crecidas por PVT sobre zafiro .......................... 212
6.2 Análisis de la fotoluminiscencia del MoS2 monocapa en la detección
de biomarcadores .................................................................................... 215
6.3 Referencias ................................................................................... 225
7 Conclusiones ................................................................................. 229
Anexo .................................................................................................. 237
Publicaciones ....................................................................................... 241

1
1 INTRODUCCIÓN
1.1 MATERIALES 2D
1.1.1 El grafeno: definición, propiedades y caracterización
El grafeno es un material bidimensional formado por átomos de carbono
densamente empaquetados y unidos entre sí por enlaces covalentes con
hibridación sp2 formando una red hexagonal.1 El orbital π restante da lugar a
la carga deslocalizada que le confiere la mayoría de las propiedades
electrónicas1,2 y que hacen de él un material prometedor para su uso en
diversas aplicaciones, entre ellas las de detección.3 Desde su descubrimiento
en 2004, se han realizado observaciones y predicciones teóricas sobre
propiedades como el efecto campo,4 el efecto cuántico Hall a temperatura
ambiente,5,6 la movilidad de portadores y la estructura electrónica.7,8
Figura 1.1. Estructura de bandas del grafeno. Figura adaptada de Ref. 2.
Atendiendo a la estructura electrónica de bandas, el grafeno es considerado
un semiconductor de gap cero. Las bandas de valencia y de conducción
CÁPITULO 1
2
coinciden con el nivel de Fermi en los puntos de alta simetría, K y K’,9 formando
los denominados conos de Dirac, donde la masa de los portadores se puede
considerar nula (Figura 1.1).2 De esta manera, los portadores de carga pueden
moverse a grandes velocidades sin perder energía, consiguiendo movilidades
extraordinarias.
Uno de los métodos más comunes para la producción de grafeno es el
depósito químico en fase de vapor (CVD), ya que, a un precio asequible, es
posible obtener monocapas de grafeno de una alta calidad. Este método
consiste en la descomposición térmica de hidrocarburos, generalmente
metano, dando lugar a la disposición ordenada de los átomos de carbono
sobre un sustrato metálico (Cu o Ru) que se encuentra a altas temperaturas.
A día de hoy, el crecimiento de grafeno por CVD sobre metales es una de las
vías más eficientes para obtener grafeno de alta calidad, aunque presenta el
inconveniente de que para usarlo como componente electrónico es necesaria
su transferencia sobre otro sustrato aislante o semiconductor.
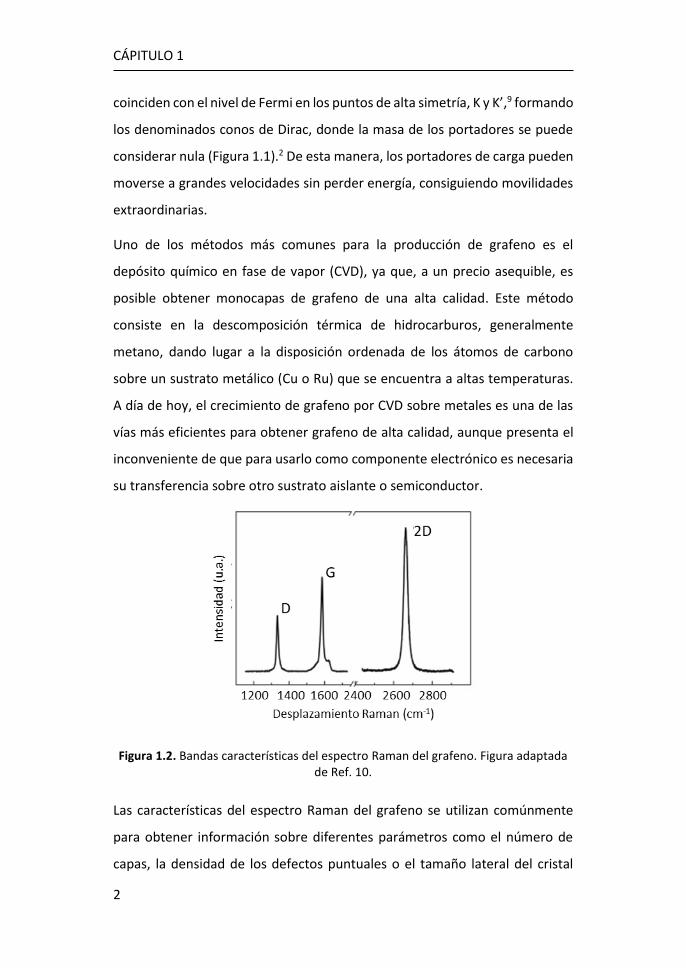
Figura 1.2. Bandas características del espectro Raman del grafeno. Figura adaptada de Ref. 10.
Las características del espectro Raman del grafeno se utilizan comúnmente
para obtener información sobre diferentes parámetros como el número de
capas, la densidad de los defectos puntuales o el tamaño lateral del cristal
Introducción
3
presente en la estructura.11 El espectro Raman de grafeno monocapa presenta
los picos G, situado a ~1580 cm-1, y 2D a ~2700 cm-1. El pico G es el único fonón
Raman de primer orden y está relacionado con la vibración óptica E2g en el
plano. El pico 2D es un proceso de segundo orden doblemente resonante, en
el que intervienen dos fonones de momento opuesto cerca del punto K, y
dispersivo, es decir, su frecuencia varía en función de la energía de excitación
incidente.12 Además, la presencia de defectos en el grafeno induce la aparición
del pico D, aproximadamente a la mitad de la frecuencia del pico 2D (~1350
cm-1), que se debe a los modos de vibración de los anillos de seis átomos y
requiere la presencia de un defecto para su activación (Figura 1.2).10,11
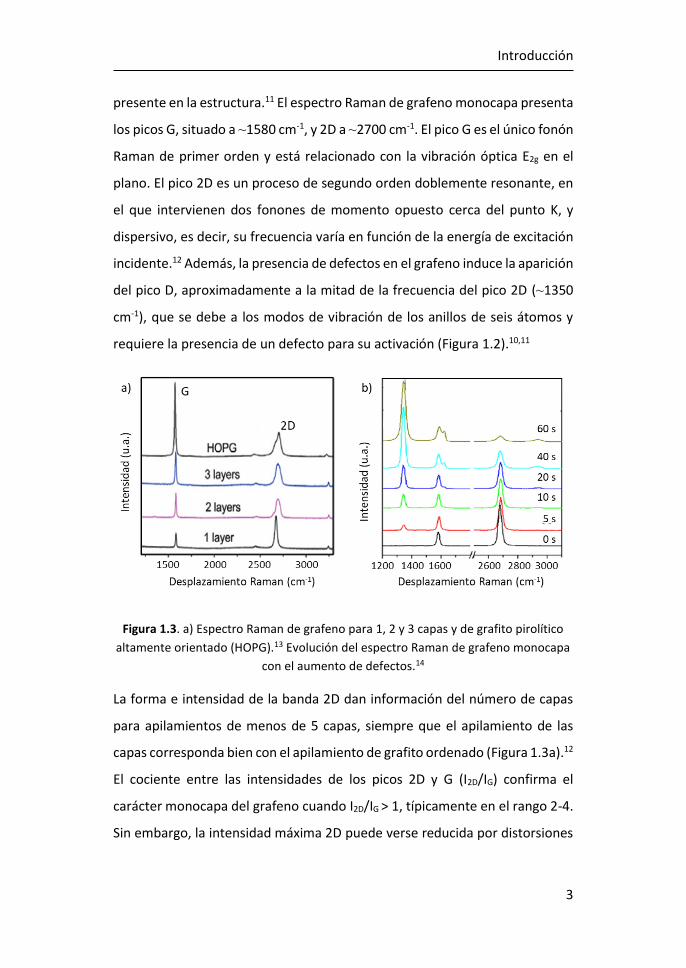
Figura 1.3. a) Espectro Raman de grafeno para 1, 2 y 3 capas y de grafito pirolítico
altamente orientado (HOPG).13 Evolución del espectro Raman de grafeno monocapa
con el aumento de defectos.14
La forma e intensidad de la banda 2D dan información del número de capas
para apilamientos de menos de 5 capas, siempre que el apilamiento de las
capas corresponda bien con el apilamiento de grafito ordenado (Figura 1.3a).12
El cociente entre las intensidades de los picos 2D y G (I2D/IG) confirma el
carácter monocapa del grafeno cuando I2D/IG > 1, típicamente en el rango 2-4.
Sin embargo, la intensidad máxima 2D puede verse reducida por distorsiones

CÁPITULO 1
4
y defectos de la red, de modo que un grafeno monocapa con defectos puede
presentar I2D/IG ≤ 1 (Figura 1.3b).15
La relación de intensidad del pico D al modo G (ID/IG) se utiliza para estimar la
cantidad de defectos del grafeno (Figura 1.3b). Al aumentar el número de
defectos, como por ejemplo la rotura de enlaces sp2, anillos irregulares,
anclaje de grupos funcionales en la red o incluso los bordes de grano de la
propia red, aumenta también la anchura de los picos G y D y la relación ID/IG.15
De este modo, las relaciones I2D/IG y ID/IG proporcionan información sobre la
estructura y la calidad del grafeno a partir de su espectro Raman.
1.1.2 TMDCs: propiedades, producción y caracterización
Después de que el grafeno demostrase que las propiedades de los materiales
bidimensionales (2D) difieren de las de sus equivalentes en volumen, se
impulsó el estudio de más materiales 2D. Como ya se ha dicho anteriormente,
el grafeno tiene gap cero, y esto limita su uso en ciertas aplicaciones. Esto hace
que se necesiten materiales alternativos que presenten intrínsecamente un
gap, como es el caso de los dicalcogenuros de metales de transición (TMDCs,
transition metal dichalcogenides). Estos materiales, al igual que el grafito,
presentan una estructura laminar formada por capas que se mantienen unidas
a través de fuerzas de van der Waals. Su estequiometria es MX2, siendo M un
metal de transición y X un calcógeno. El estudio de los TMDCs monocapa
comenzó alrededor de 2010, principalmente de muestras exfoliadas, ya que la
calidad de las muestras sintetizadas era significativamente menor.16 Sin
embargo, la mayoría de las aplicaciones requieren monocapas con un tamaño
superior a las decenas de micras obtenidas por exfoliación, además de un
método de síntesis eficiente y asequible. Algunos de estos materiales como el
MoS2, MoSe2, WS2 y WSe2 han originado un gran interés debido a sus posibles
aplicaciones en optoelectrónica, como fototransistores,17 transistores de
Introducción
5
efecto campo de alto rendimiento o dispositivos espintrónicos,18 debido a las
importantes modificaciones que se producen en la estructura electrónica de
bandas y en las propiedades ópticas con la variación del número de capas. Esta
amplia gama de aplicaciones puede aumentarse aún más mediante el dopado
o la deformación controlada de los TMDCs. Por ejemplo, el dopado del MoS2
con Mn induce un comportamiento ferromagnético,19 lo que ofrece la
posibilidad de obtener semiconductores 2D ferromagnéticos con un amplio
rango de gaps altamente interesante para la espintrónica. Por otro lado, los
TMDCs también han demostrado tener una excelente biocompatibilidad y una
alta afinidad con ciertas biomoléculas, lo que los convierte en excelentes
soportes para su uso en biosensores.20–24
Figura 1.4. Posibles aplicaciones del MoS2 bidimensional.
CÁPITULO 1
6
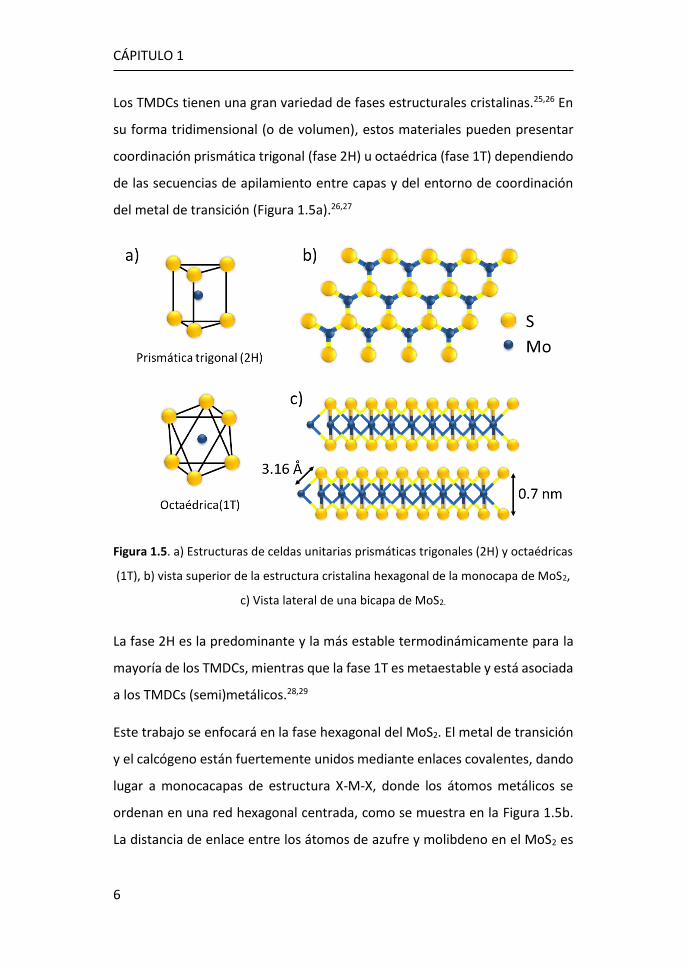
Los TMDCs tienen una gran variedad de fases estructurales cristalinas.25,26 En
su forma tridimensional (o de volumen), estos materiales pueden presentar
coordinación prismática trigonal (fase 2H) u octaédrica (fase 1T) dependiendo
de las secuencias de apilamiento entre capas y del entorno de coordinación
del metal de transición (Figura 1.5a).26,27
Figura 1.5. a) Estructuras de celdas unitarias prismáticas trigonales (2H) y octaédricas
(1T), b) vista superior de la estructura cristalina hexagonal de la monocapa de MoS2,
c) Vista lateral de una bicapa de MoS2.
La fase 2H es la predominante y la más estable termodinámicamente para la
mayoría de los TMDCs, mientras que la fase 1T es metaestable y está asociada
a los TMDCs (semi)metálicos.28,29
Este trabajo se enfocará en la fase hexagonal del MoS2. El metal de transición
y el calcógeno están fuertemente unidos mediante enlaces covalentes, dando
lugar a monocacapas de estructura X-M-X, donde los átomos metálicos se
ordenan en una red hexagonal centrada, como se muestra en la Figura 1.5b.
La distancia de enlace entre los átomos de azufre y molibdeno en el MoS2 es
Introducción
7
de 3.16 Å, mientras que la altura de una capa es de aproximadamente 0.7 nm
(Figura 1.5c).30
Algunos TMDCs son materiales semiconductores con un gap indirecto cuando
forman estructuras tridimiensionales, de 1.3 eV en el caso del MoS2. Sin
embargo, el gap indirecto se transforma gradualmente al reducir el número
de capas (N), hasta obtener un gap directo, de ~1.8 eV para el MoS2, cuando
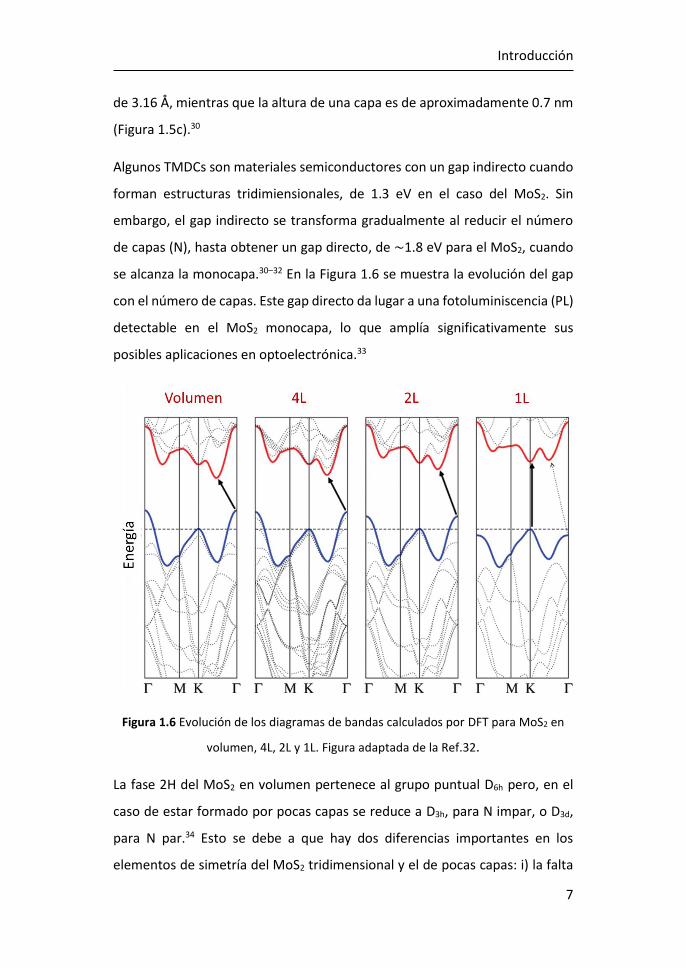
se alcanza la monocapa.30–32 En la Figura 1.6 se muestra la evolución del gap
con el número de capas. Este gap directo da lugar a una fotoluminiscencia (PL)
detectable en el MoS2 monocapa, lo que amplía significativamente sus
posibles aplicaciones en optoelectrónica.33
Figura 1.6 Evolución de los diagramas de bandas calculados por DFT para MoS2 en
volumen, 4L, 2L y 1L. Figura adaptada de la Ref.32.
La fase 2H del MoS2 en volumen pertenece al grupo puntual D6h pero, en el
caso de estar formado por pocas capas se reduce a D3h, para N impar, o D3d,
para N par.34 Esto se debe a que hay dos diferencias importantes en los
elementos de simetría del MoS2 tridimensional y el de pocas capas: i) la falta
CÁPITULO 1
8
de simetría traslacional a lo largo del eje perpendicular a las capas (eje z) y ii)
la presencia o ausencia de un plano de reflexión para N impar o par,
respectivamente.
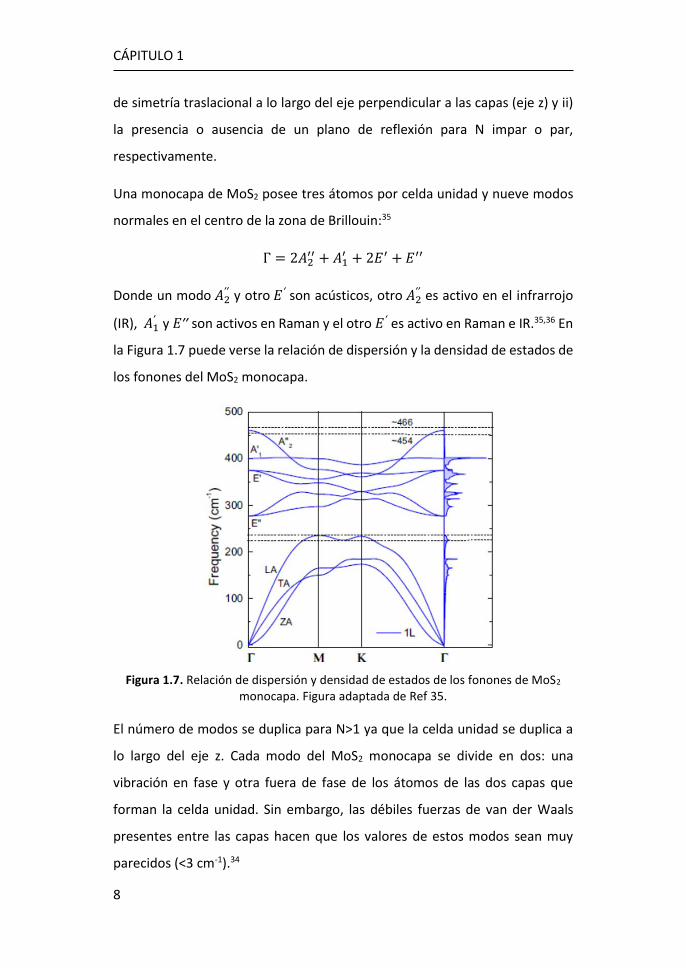
Una monocapa de MoS2 posee tres átomos por celda unidad y nueve modos
normales en el centro de la zona de Brillouin:35
Γ = 2𝐴2′′ + 𝐴1
′ + 2𝐸′ + 𝐸′′
Donde un modo 𝐴2′′ y otro 𝐸′ son acústicos, otro 𝐴2
′′ es activo en el infrarrojo
(IR), 𝐴1′ y 𝐸′′ son activos en Raman y el otro 𝐸′ es activo en Raman e IR.35,36 En
la Figura 1.7 puede verse la relación de dispersión y la densidad de estados de
los fonones del MoS2 monocapa.
Figura 1.7. Relación de dispersión y densidad de estados de los fonones de MoS2 monocapa. Figura adaptada de Ref 35.
El número de modos se duplica para N>1 ya que la celda unidad se duplica a
lo largo del eje z. Cada modo del MoS2 monocapa se divide en dos: una
vibración en fase y otra fuera de fase de los átomos de las dos capas que
forman la celda unidad. Sin embargo, las débiles fuerzas de van der Waals
presentes entre las capas hacen que los valores de estos modos sean muy
parecidos (<3 cm-1).34

Introducción
9
En el caso del MoS2 en volumen (D6h), hay seis átomos por celda unidad y 18
modos normales en el centro de la zona de Brillouin:
Γ = 𝐴1𝑔 + 2𝐴2𝑢 + 2𝐵2𝑔 +𝐵1𝑢 + 𝐸1𝑔 + 2𝐸1𝑢 + 2𝐸2𝑔 + 𝐸2𝑢
Los modos de vibración activos Raman son los que tienen simetría 𝐴1𝑔, 𝐸1𝑔 y
𝐸2𝑔,35,37 dando lugar a cuatro picos Raman (𝐴1𝑔 + 𝐸1𝑔 + 2𝐸2𝑔). Un modo 𝐴2𝑢
y otro 𝐸1𝑢 son acústicos, otro 𝐴2𝑢 y otro 𝐸1𝑢 son activos en IR, y los demás
son ópticamente inactivos.38
Los modos de vibración para N impar coinciden con los descritos para el MoS2
monocapa (D3h), sin embargo, para N par (D3d) los modos activos los modos
normales son:
Γ = 3𝐴1𝑔 + 3𝐴2𝑢 + 3𝐸𝑔 + 3𝐸𝑢
Donde un modo 𝐴2𝑢 y otro 𝐸𝑢 son acústicos, otro 𝐴2𝑢 y otro 𝐸𝑢 son activos
en IR y los modos con simetría 𝐴1𝑔 y 𝐸𝑔 son activos en Raman. Los modos con
simetría E son doblemente degenerados, por lo que se esperan 4 modos para
MoS2 en volumen, 3 modos para N impar y 5 modos para N par.
Las representaciones irreducibles de los modos y el número de modos
normales de vibración para del MoS2 en volumen y para una o dos capas son
diferentes. Esto se debe a que al variar los elementos de simetría que existen
en cada caso, varía también el grupo puntual que describe cada cristal. En la
Figura 1.8 puede verse una representación de los distintos modos de vibración
de los TMDCs en la fase 2H en volumen, bicapa y monocapa. Como a lo largo
de esta memoria se discutirán espectros Raman de muestras con un número
variable de capas, desde la monocapa (1L) hasta una docena de capas, se
utilizará la nomenclatura de los modos de vibración correspondiente al MoS2
en volumen en todos los casos para simplificar y facilitar la lectura, siendo la

CÁPITULO 1
10
correlación entre los modos activos Raman en 1L-MoS2 y el cristal en volumen
la siguiente: A’1 – A1g, E’- E12g, E’’ – E1g.
Figura 1.8. Simetría y desplazamientos normales de cada modo óptico de vibración
para TMDCs en volumen, bicapa y monocapa. Se indican los modos activos en
Raman (R), en infrarrojo (IR), activos en ambos (IR+R) e inactivos (silent). Figura
adaptada de Ref 39.
Los tensores Raman correspondientes a los grupos puntuales del MoS2
monocapa, número de capas impar (D3h) y MoS2 en volumen volumen (D6h)
son los siguientes:

Introducción
11
En el caso de pocas capas con número de capas par (D3d) los tensores son:
El modo Raman de baja frecuencia (E22g ~35 cm-1) está asociado con las
vibraciones de cizallamiento entre capas, de modo que no aparece en el MoS2
monocapa. Su frecuencia varía con el grosor, lo que permite deducir el número
de capas en el rango entre 1 y 12 capas,40 aunque no suele usarse, debido a
que su baja frecuencia no permite que sea detectado en los sistemas Raman
habituales basados en filtros Notch estándar, utilizados para eliminar la
frecuencia del láser de excitación.
Como se observa en la Figura 1.8, el modo de simetría E1g está asociado con
vibraciones en el plano y tiene una frecuencia de aproximadamente 284 cm-1
en volumen.41 El modo E12g también está asociado con vibraciones atómicas
en el plano, donde los átomos de S y de Mo vibran en direcciones opuestas.
Aparece a una frecuencia de aproximadamente 384 cm-1 para el MoS2
monocapa y de 381 cm-1 para el volumen. Por otra parte, el fonón A1g está
relacionado con las vibraciones de los átomos de S fuera del plano, mientras
que los átomos de Mo permanecen en posición de equilibrio. Su frecuencia es
aproximadamente 403 y 408 cm-1 para el MoS2 monocapa y volumen,
respectivamente.34,39,42
CÁPITULO 1
12
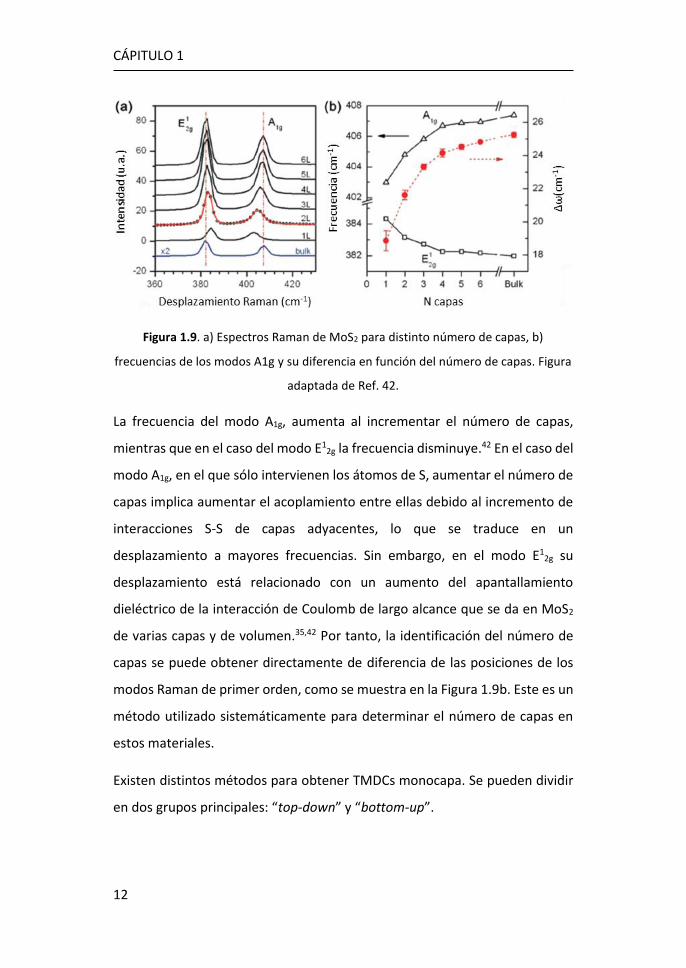
Figura 1.9. a) Espectros Raman de MoS2 para distinto número de capas, b)
frecuencias de los modos A1g y su diferencia en función del número de capas. Figura
adaptada de Ref. 42.
La frecuencia del modo A1g, aumenta al incrementar el número de capas,
mientras que en el caso del modo E12g la frecuencia disminuye.42 En el caso del
modo A1g, en el que sólo intervienen los átomos de S, aumentar el número de
capas implica aumentar el acoplamiento entre ellas debido al incremento de
interacciones S-S de capas adyacentes, lo que se traduce en un
desplazamiento a mayores frecuencias. Sin embargo, en el modo E12g su
desplazamiento está relacionado con un aumento del apantallamiento
dieléctrico de la interacción de Coulomb de largo alcance que se da en MoS2
de varias capas y de volumen.35,42 Por tanto, la identificación del número de
capas se puede obtener directamente de diferencia de las posiciones de los
modos Raman de primer orden, como se muestra en la Figura 1.9b. Este es un
método utilizado sistemáticamente para determinar el número de capas en
estos materiales.
Existen distintos métodos para obtener TMDCs monocapa. Se pueden dividir
en dos grupos principales: “top-down” y “bottom-up”.

Introducción
13
Dentro de los métodos top-down destacan tres técnicas: exfoliación mecánica,
exfoliación en fase líquida, y exfoliación por intercalación química.
La exfoliación mecánica consiste en separar las láminas del TMDC en volumen
utilizando una cinta adhesiva.43,44 La adhesión entre la cinta y el plano basal
del cristal es mucho más fuerte que las fuerzas de van der Waals que hay entre
las capas, lo que permite aislar las láminas. Otra variante es la utilizada por el
grupo de Castellanos-Gómez, donde sustituyen la cinta adhesiva por un sello
viscoelástico, que tiene la ventaja de no dejar restos de adhesivo en las
láminas exfoliadas.45 Este método da como resultado copos de TMDCs de
formas, tamaños y número de capas aleatorios, donde identificar los copos
monocapa es costoso en tiempo, pero de muy alta calidad.
En la exfoliación en fase líquida, los cristales de TMDCs son sometidos a una
fuerte agitación, de modo que las ondas generadas se propagan a través del
disolvente provocando ciclos de alta y baja presión que ayudan a la
exfoliación. Después, los nanocopos de TMDCs que se han exfoliado se
separan del resto de material mediante centrifugación.46–48 En este caso la
forma, tamaño y número de capas de los copos son también muy diversos, y
su calidad es inferior a la obtenida por exfoliación mecánica.
Por último, la exfoliación asistida por intercalación se basa en la inserción de
especies entre las láminas de los TMDCs, ampliando así la distancia entre
capas y debilitando las fuerzas de adhesión entre ellas.49,50 Todos estos
métodos tienen además el inconveniente de dejar posibles restos de reactivos
y disolventes utilizados en el proceso, además de generar copos de tamaños
por lo general inferiores a 10 μm.
En los métodos bottom-up destacan dos técnicas: procesos químicos en
solución, y depósito de vapor, ya sea por vía física (PVT, Physical Vapor
Transport) o química (CVD, Chemical Vapor Deposition).

CÁPITULO 1
14
Entre los procesos en solución se encuentran la síntesis hidrotermal,51 que
utiliza agua como disolvente, y la síntesis solvotermal, que utiliza un disolvente
distinto del agua.52 En ambos casos las reacciones implicadas se llevan a cabo
en un autoclave a temperaturas de aproximadamente 200 ºC y altas presiones.
El resultado suele ser nanocopos multicapa (grosor de unas pocas decenas de
nanómetros) o nanocopos monocapa, con tamaño lateral de también unas
decenas de nanómetros.43
Los métodos PVT se basan en la sublimación del TMDC deseado en su forma
volumen (generalmente en polvo), sometiéndolo a altas temperaturas (800-
970 ºC) y a bajas presiones (<100 mbar).53,54 En los métodos CVD se lleva a
cabo la volatilización y la termólisis de precursores que contienen los
elementos de interés. Los iones de los metales y los calcógenos en fase vapor
reaccionan para formar el monómero del TMDC también en fase vapor, que
es transportado por un gas inerte de para posteriormente depositarse en el
sustrato utilizado.55–57 Después de la nucleación, el crecimiento tiende a ser
bidimensional, lo que da lugar a la formación de islas/copos. El continuo
crecimiento de estas islas puede provocar la coalescencia de las mismas,
formando películas continuas con límites de grano.58 La posibilidad de formar
películas continuas es lo que hace que las técnicas PVT/CVD tomen gran
importancia, ya que la obtención de estos materiales en grandes áreas harían
posible aprovechar todo su potencial en aplicaciones opto-electrónicas.
Lo ideal para la caracterización de materiales es que las técnicas empleadas
sean rápidas, no destructivas, ofrezcan alta resolución espectral y espacial y
ofrezcan información tanto estructural como electrónica. En el caso de los
TMDCs las técnicas más utilizadas son la espectroscopía Raman, la
foluminiscencia (PL) y las espectroscopías ópticas de absorción y reflexión.
La espectroscopía Raman es una de las técnicas más utilizadas para estudiar
los materiales bidimensionales, como se ha visto en el caso del grafeno. Como

Introducción
15
ya se ha mencionado, el espectro Raman del MoS2 da información sobre el
número de capas, pero también puede dar información sobre defectos como
el desorden, la tensión o el dopado.59
Otra de las técnicas más utilizadas para la caracterización de estos materiales
es la fotoluminiscencia. Ésta permite estudiar la calidad cristalina del material,
obtener información fundamental como la determinación del gap, niveles de
impurezas y defectos, además de dar información acerca de los mecanismos
de recombinación. El MoS2 en volumen no emite fotoluminiscencia, sin
embargo, para pocas capas, a medida que el número de capas disminuye,
aparecen dos emisiones pronunciadas a 1.82 eV (680 nm) y 1.98 eV (625
nm) derivadas de las transiciones excitónicas directas en el punto K de la zona
de Brillouin, denominadas excitones A y B, respectivamente.30,32 La diferencia
de energía entre estos dos picos se debe al desdoblamiento espín-órbita de la
energía de la banda de valencia. El MoS2 monocapa es el que presenta una
mayor intensidad de fotoluminiscencia, así como mayor eficiencia cuántica.
La absorción óptica es otra característica de los semiconductores relacionada
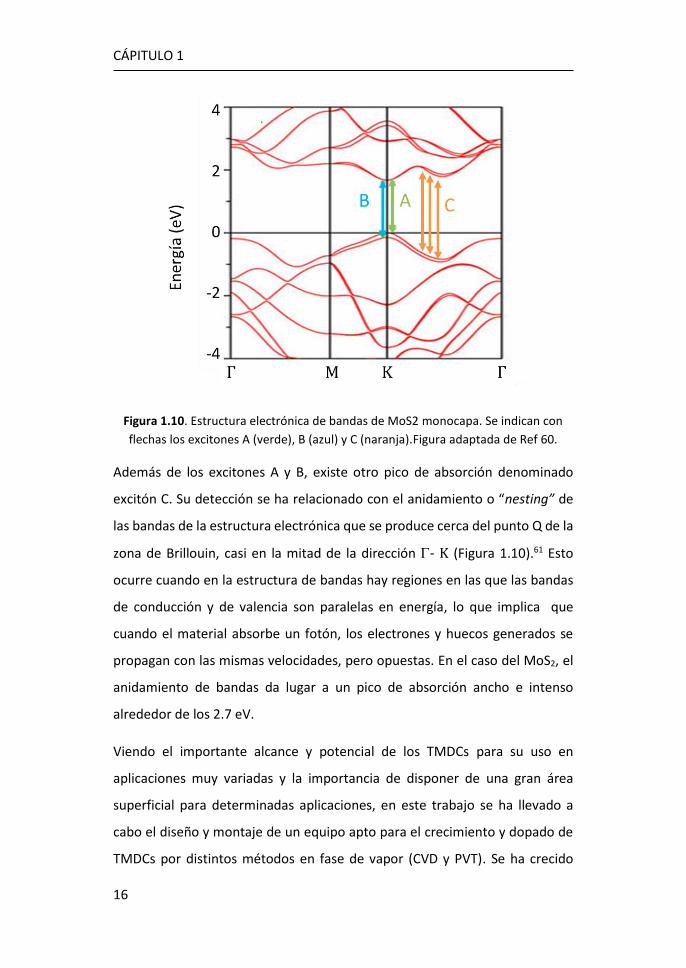
con la estructura de bandas. En el MoS2 se observan dos picos de absorción a
1.89 eV (656 nm) y 2.00 eV (620 nm), que corresponden a las transiciones
excitónicas directas en el punto K de la zona de Brillouin (Figura 1.10).
CÁPITULO 1
16
Figura 1.10. Estructura electrónica de bandas de MoS2 monocapa. Se indican con
flechas los excitones A (verde), B (azul) y C (naranja).Figura adaptada de Ref 60.
Además de los excitones A y B, existe otro pico de absorción denominado
excitón C. Su detección se ha relacionado con el anidamiento o “nesting” de
las bandas de la estructura electrónica que se produce cerca del punto Q de la
zona de Brillouin, casi en la mitad de la dirección - K (Figura 1.10).61 Esto
ocurre cuando en la estructura de bandas hay regiones en las que las bandas
de conducción y de valencia son paralelas en energía, lo que implica que
cuando el material absorbe un fotón, los electrones y huecos generados se
propagan con las mismas velocidades, pero opuestas. En el caso del MoS2, el
anidamiento de bandas da lugar a un pico de absorción ancho e intenso
alrededor de los 2.7 eV.
Viendo el importante alcance y potencial de los TMDCs para su uso en
aplicaciones muy variadas y la importancia de disponer de una gran área
superficial para determinadas aplicaciones, en este trabajo se ha llevado a
cabo el diseño y montaje de un equipo apto para el crecimiento y dopado de
TMDCs por distintos métodos en fase de vapor (CVD y PVT). Se ha crecido

Introducción
17
MoS2 por ambos métodos y se han estudiado en profundidad las
características Raman, de fotoluminiscencia y de absorción de las muestras
obtenidas. También se han estudiado los cambios en el espectro Raman de
muestras de MoS2 con distinto número de capas obtenidas por diferentes
métodos (ME, PVT y CVD) y se ha desarrollado un biosensor óptico basado de
MoS2 crecido por PVT para la detección de moléculas presentes en tejidos
cancerosos.
1.2 PLATAFORMAS PARA LA DETECCIÓN DE MOLÉCULAS Y
BIOMARCADORES
1.2.1 Nanopartículas: Amplificación de la señal Raman
La espectroscopia Raman permite la identificación directa de una especie
específica entre diferentes componentes, ya que el espectro vibracional es
una huella dactilar de la molécula. Sin embargo, su eficiencia (o sección eficaz)
es muy baja, aproximadamente 1010 veces menor que la absorción vibracional,
lo que dificulta la detección a bajas concentraciones. La amplificación de la
radiación electromagnética debida a las resonancias plasmónicas superficiales
localizadas (Localized Surface Plasmon Resonances, LSPR) de las
nanopartículas metálicas (NPs) es muy eficiente y se utiliza comúnmente para
detectar señales Raman débiles de analitos diluidos mediante la técnica de
dispersión Raman amplificada en superficie, SERS (del inglés, Surface
Enhanced Raman Scattering).
Cuando una radiación electromagnética incide sobre una nanopartícula
metálica, sus electrones de conducción se desplazan con respecto a los iones
positivos, induciendo una polarización del sistema. La nanopartícula puede

CÁPITULO 1
18
ejemplificarse entonces como un oscilador simple en el que los electrones
libres oscilan coherentemente sujetos a la fuerza impulsora del campo
eléctrico periódico. Esta oscilación coherente se denomina resonancia de
plasmón superficial localizada (LPSR). El término "localizada" indica que las
oscilaciones de los electrones no se propagan porque están confinadas
espacialmente en tres dimensiones por el tamaño finito de la nanopartícula,
mucho menor que la longitud de onda de la luz.62 La Figura 1.11 ilustra las
oscilaciones colectivas de los electrones en una nanopartícula esférica bajo la
acción del campo eléctrico externo.
Figura 1.11. Oscilación colectiva de electrones en una nanopartícula metálica
esférica bajo la acción del campo eléctrico externo. Figura adaptada de Ref 62.
Esto da lugar a enormes aumentos de las señales Raman de los modos de
vibración de las moléculas o los fonones de los sólidos que se encuentran en
las proximidades de las NPs. Es posible aumentar la intensidad de la señal
Raman a través del acoplamiento a las LSPR de las nanopartículas metálicas,
denominado efecto SERS (Figura 1.12a). Esta técnica óptica presenta varias
características importantes para la detección y la identificación de moléculas,
ya que es rápida, asequible, no destructiva, con una resolución
submicrométrica, e incluso en algunos casos permite la detección de
moléculas individuales.63,64 Algunas de las especies químicas que esta técnica

Introducción
19
puede detectar son cationes/aniones tóxicos o radioactivos, nutrientes
iónicos,65 pesticidas,66 drogas y productos farmacéuticos, o materiales
explosivos.67
Figura 1.12. a) Representación del efecto SERS originado por las nanopartículas
metálicas al estar expuestas a una radiación electromagnética I0 y b) de la variación
del campo eléctrico inducido en las NPs polarizadas por la radiación incidente I0.
El factor de amplificación (EF) del efecto SERS se puede evaluar considerando
dos factores multiplicadores, uno para el campo incidente y otro para el
campo emitido (Raman). Por eso, en general, la amplificación es proporcional
a la cuarta potencia del campo eléctrico local. Por otra parte, en una primera
aproximación, la amplitud del campo eléctrico LSPR, E, decae como E ~d-3,
donde d es la distancia molécula-NP, por lo que para que la molécula se vea
afectada por el efecto SERS debe estar muy cerca de la NP (Figura 1.12b).68
La distribución del aumento del campo eléctrico en la superficie de un sustrato
plasmónico es muy poco homogénea y, bajo ciertas condiciones, se amplifica
aún más en regiones espaciales muy pequeñas denominadas puntos calientes
o " hot spots". Estos puntos calientes suelen localizarse en zonas muy
puntiagudas, en espacios muy pequeños entre NPs (nanogaps de 1-2 nm) o en
las regiones entre una nanopartícula y una superficie metálica,69–72 siendo los
nanogaps notablemente más eficientes que las zonas puntiagudas en la
amplificación de las señales ópticas.

CÁPITULO 1
20
La razón por la que los campos generados en los nanogaps entre NPs son tan
altos puede entenderse observando la Figura 1.13, que muestra el caso de una
molécula colocada entre dos esferas metálicas. En la Figura 1.13a el campo
eléctrico se polariza a lo largo del eje principal del dímero, mientras que la
Figura 13b se polariza perpendicularmente al eje. En la polarización a lo largo
del eje, se observa que, al acercar las nanopartículas entre sí, se reduce la
separación entre las cargas superficiales inducidas y, por tanto, aumenta el
campo eléctrico en esa zona. Además, en la polarización de cada NP, no sólo
contribuye el campo externo, sino que también lo hace el dipolo inducido en
la nanopartícula adyacente. Ambos efectos funcionan en la configuración (a),
pero evidentemente no lo hacen en la configuración (b).73
Figura 1.13. Representación de un dímero formado por dos polarizadas por la acción
de un campo eléctrico externo, E0, con una molécula en el centro del hueco. a) E0 se
polariza a lo largo del eje principal del dímero, b) E0 se polariza perpendicularmente
al eje del dímero. Las flechas azules dentro de las nanopartículas representan los
dipolos inducidos. Figura adaptada de Ref. 73.
Los métodos de preparación de NPs metálicas se pueden clasificar en métodos
químicos y métodos físicos. La mayoría de los métodos químicos se llevan a
cabo en disolución, empleando un precursor metálico, un agente reductor y
un agente estabilizante que impida la aglomeración de las NPs. Uno de los

Introducción
21
métodos químicos más utilizados es el método Creighton,74 que consiste en la
reducción de AgNO3 con el agente reductor NaBH4. Este método es el más
popular en la actualidad y da lugar a la obtención de nanopartículas de plata
de aproximadamente 10 nm y con una distribución estrecha de tamaños
(monodispersas). Entre los métodos físicos destaca la técnica de ablación
láser. Esta técnica consiste en enfocar un láser pulsado en un blanco metálico
sumergido en un líquido. La energía del pulso del láser es absorbida por el
blanco generando un plasma que contiene el material ablacionado, que da
lugar a NPs en suspensión por la condensación de los átomos expulsados del
blanco metálico.75,76
Con las técnicas descritas hasta el momento las NPs obtenidas están en
suspensión, pero existen otras técnicas con las que se pueden obtener NPs
directamente sobre el sustrato deseado, como la litografía o la agregación en
fase gas. Uno de los métodos de nanolitografía más utilizados es la litografía
de haz de electrones. Esta técnica consiste en barrer con un haz de electrones
finamente enfocado un sustrato recubierto por una resina sensible a los
electrones, que tras ser lavada con un revelador químico muestra el patrón en
el que posteriormente se deposita el metal deseado y se elimina la resina,
dejando un patrón nanométrico del metal directamente sobre el sustrato.77
Con esta técnica se pueden llegar a obtener nanoestructuras ordenadas de
unos 10 nm de diámetro, pero requiere numerosos pasos y lavados. Por
último, es de destacar la técnica de agregación en fase gas que se basa en la
formación por “sputtering” de un flujo continuo de nanopartículas, creadas
por colisiones dentro de una cámara de agregación, que se deposita
directamente sobre el sustrato. Mediante esta técnica se obtienen NPs
ultrafinas de tamaño controlado (de 1 a ~10 nm de radio, es decir, hasta 106
átomos por partícula) y con distribuciones de tamaño de partícula estrechas
en un entorno limpio y controlado, ya que puede funcionar en alto o ultra alto

CÁPITULO 1
22
vacío, lo que evita la presencia de tensioactivos o cualquier subproducto del
proceso de fabricación que sí están presentes en el resto de técnicas.78,79
Las propiedades ópticas de las NPs dependen en gran medida de su tamaño,
forma y composición, por lo que existe una intensa actividad para optimizar
estos parámetros.80–83 Por ejemplo, la relación de aspecto de nanorods
metálicos permite controlar las longitudes de onda de resonancia y alcanzar
el rango del infrarrojo, que es útil para diferentes terapias.84
Experimentalmente, el tamaño óptimo, incluso para NPs esféricas, no está
claro. Por ejemplo, para NPs de oro individuales, con radios en el rango de 20-
50 nm, se ha reportado que cuanto mayor es el tamaño de la NP,85 mayor es
la amplificación, mientras que para coloides de Ag con radios en el rango de
10-35 nm, el tamaño óptimo es 25 nm,86 parecido al radio óptimo obtenido
por los cálculos Mie para esferas aisladas.87 Para NPs de plata esféricas de
entre 50-65 nm de diámetro los EFs suelen estar en el rango de 104, y en
algunos casos de hasta 106.86,88 Otra cuestión que no está clara es cómo varía
la amplificación Raman con la distancia entre partículas. Debido a los puntos
calientes mencionados anteriormente, el EF depende en gran medida de la
posición de la molécula. Por ejemplo, recientemente se han sintetizado NPs
de Rh ultrapequeñas (~2.5 nm de radio) sobre ADN que muestran un EF
creciente a medida que se reduce la concentración de ADN, probablemente
debido a un aumento de puntos calientes.89 Algunas aplicaciones, como la
detección de una sola molécula, se basan en estos puntos calientes para
obtener el EF de hasta 109, sin embargo, estos experimentos no son fácilmente
reproducibles.90
En este trabajo se evalúa la capacidad de NPs de Ag monodispersas
ultrapequeñas (R = 1.8 nm), para amplificar la señal Raman del grafeno y de
una molécula de referencia como la Rodamina 6G (R6G). Para aprovechar los
altos valores de resonancia de las NPs de mayor tamaño, y la elevada relación

Introducción
23
superficie/volumen de las NPs ultrapequeñas, así como para fomentar la
formación de puntos calientes, se combinan películas delgadas
nanoestructuradas y NPs ultrapequeñas. También se discuten las simulaciones
de plasmón de campo cercano, que incluyen la dependencia del tamaño y la
distancia media entre las NPs.
1.2.2 Biosensores basados en materiales bidimensionales
En los últimos años, materiales bidimensionales (2D) como el grafeno,
dicalcogenuros de metales de transición (TMDCs) o el nitruro de boro
hexagonal (h-BN), han atraído una gran atención como elementos de
transducción y soporte en una gran variedad de tecnologías de biosensores.
Lo que hace interesantes los materiales 2D es la variación de sus propiedades
con respecto a sus formas en volumen. Los materiales 2D pueden
proporcionar una alta densidad de sitios superficiales activos en un área
extensa, lo que les hace ideales para su uso en aplicaciones de biodetección.
Además, la familia de los materiales 2D puede presentar una amplia gama de
propiedades electrónicas, que van desde las metálicas/semimetálicas
(grafeno), hasta las semiconductoras (MoS2, WS2) y aislantes (h-BN). Del
mismo modo, abarcan una amplia gama de propiedades ópticas, la emisión de
fluorescencia. Además, al modificar la química superficial mediante su
funcionalización o la ingeniería de defectos, los materiales 2D pueden ser
adaptados para responder selectivamente a analitos específicos con una
sensibilidad extremadamente alta. Todas estas propiedades, combinadas con
la capacidad de fabricar heteroestructuras (mediante crecimiento directo o
procesos de transferencia) hacen de ellos unos nanomateriales muy
interesantes para biosensores y otras aplicaciones sanitarias.

CÁPITULO 1
24
Dentro del área de los biosensores, un aspecto muy importante es el
diagnóstico precoz de enfermedades graves, que constituye uno de los pasos
esenciales para aumentar la tasa de supervivencia a la enfermedad. Es por
esto por lo que la comunidad científica debe poner énfasis en la mejora de los
métodos de diagnóstico, especialmente mediante procedimientos no
invasivos, como el uso de biosensores selectivos.
Figura 1.14. Esquema de un biosensor básico y sus componentes.
Los biosensores suelen contar con un biomarcador (molécula diana), un
biorreceptor (elemento de reconocimiento) y un transductor compatible (la
plataforma utilizada para convertir lo biológico en señales medibles), papel
que desempeña el material 2D (Figura 1.14).
1.2.2.1 Biosensores basados en grafeno
Existe un gran interés en la utilización del grafeno en aplicaciones de detección
debido a sus propiedades únicas, como son su gran área específica, y
excelentes resistencias térmica y mecánica y conductividad eléctrica.3 En
general, los sensores electrónicos adecuados para detectar biomoléculas
diana se basan en transistores de efecto campo (FET)91,92 y, por lo tanto, en el
caso de los sensores basados en grafeno, es necesario que se preserve su alta
conductividad y movilidad después de su procesado. El grafeno, debido a su

Introducción
25
aromaticidad es químicamente inerte. Al interaccionar con una biomolécula
se produce un enlace π-π no covalente entre los anillos de carbono de la
biomolécula y los del grafeno, manteniendo su aromaticidad, pero perdiendo
capacidades como biosensor debido a la debilidad del enlace. Por este motivo,
la funcionalización del grafeno con grupos oxigenados resulta de gran interés,
ya que son ionizables y altamente reactivos, lo que los hace imprescindibles a
la hora de establecer enlaces que permitan la inmovilización del biorreceptor.
La inmovilización de la biomolécula se puede llevar a cabo mediante enlaces
físicos o químicos. En la adsorción física, aunque se preserva la estructura sp2
del grafeno, y consecuentemente sus propiedades eléctricas, las fuerzas
involucradas son Van-der-Waals, y aunque es el método más sencillo, las
interacciones electrostáticas pueden romperse fácilmente por cambios de pH
o temperatura. Sin embargo, en un enlace químico con unión covalente la
energía de enlace es mucho mayor y por tanto también lo es su estabilidad.93–
95
Un procedimiento común para desarrollar sensores electrónicos es la
adaptabilidad del óxido de grafeno (GO), una forma oxigenada del grafeno que
contiene distintos grupos funcionales en su superficie, como hidróxidos (-OH),
epóxidos, carbonilos (CO) y ácidos carboxílicos (-COOH), que le dotan de un
carácter hidrofílico y una alta reactividad. De entre estos grupos funcionales,
son los -COOH los que proporcionan la bio-conjugación específica para enlazar
de manera covalente la biomolécula. El GO se sintetiza mediante el método
Hummer’s, que consiste en la oxidación del grafito utilizando una mezcla de
ácido sulfúrico, nitrato sódico y permanganato potásico.96 La presencia de
estos grupos oxigenados cambia la hibridación sp2 original del grafeno,
rompiendo la conjugación y modificando así su estructura electrónica y
propiedades. Esto hace del GO un material más fácil de funcionalizar con los
correspondientes biorreceptores, pero no es válido para aplicaciones
conductoras ya que, al romperse la conjugación, empeora la conductividad. Es

CÁPITULO 1
26
posible reducir el GO mediante métodos térmicos, químicos o
electroquímicos, mejorando así sus propiedades eléctricas.97 Algunos de los
agentes químicos más utilizados son la hidrazina, el borohidruro sódico, el
hidruro de litio y aluminio y diversas soluciones alcalinas.98 Sin embargo,
ninguno de estos métodos permite eliminar de forma selectiva los grupos
oxigenados no deseados de forma que permanezcan únicamente los -COOH,
lo que plantea problemas como la eliminación de reactivos, reproducibilidad
y baja conductividad.
Como ya se ha comentado, para el anclaje de los biorreceptores únicamente
son eficaces los grupos -COOH, por lo que la presencia de los otros grupos sólo
tiene efectos negativos, tanto estéricos como por la disminución de
conductividad y movilidad eléctrica.
Otra opción para obtener grafeno con grupos funcionales consiste en
funcionalizar el grafeno monocapa en etapas posteriores a su obtención. La
técnica que más destaca es la oxidación inducida por fases de plasma reactivo.
En este caso el grafeno se expone a un flujo de gas de oxígeno que es excitado
mediante microondas, generando un plasma de oxígeno ionizado que ataca el
grafeno creando defectos y grupos funcionales oxigenados.99–101
Todos estos métodos requieren más de una etapa, además de que no
permiten controlar la composición de grupos funcionales oxigenados. Por ello,
es de gran importancia el diseño de una metodología capaz de implantar
grupos -COOH, de forma controlada y mayoritaria, en la superficie del grafeno.
Anticuerpos y su inmovilización por el método carbodiimida
Los anticuerpos son una de las posibles biomoléculas capaces de reaccionar
con los grupos -COOH y actuar como biorreceptores. Un anticuerpo
(inmunoglobulina, Ig) es una proteína que reacciona contra un agente extraño

Introducción
27
(antígeno).102 Un anticuerpo típico está formado por tres unidades
estructurales básicas. Dos de ellas son idénticas y están implicadas en la unión
del antígeno (brazos Fab, Fragmentos de unión al antígeno). La tercera unidad
Fc (Fragmento cristalizable) está implicada en la unión a las células efectoras
(las células que actúan en la respuesta inmune frente al antígeno), o en este
caso, a la unión con grupos -COOH del grafeno (transductor del biosensor). La
estructura del anticuerpo también se puede ver como un conjunto de cuatro
cadenas polipeptídicas, que consisten en dos cadenas pesadas idénticas que
contienen Fab y Fc, y dos cadenas ligeras, también idénticas, asociadas
únicamente con el Fab (Figura 1.15).102
Figura 1.15. Estructura de un anticuerpo.
Hay cinco clases de anticuerpos o inmunoglobulinas denominadas
inmunoglobulina G (IgG), IgM, IgA, IgD e IgE. El principal anticuerpo en el suero
es la IgG y, además, es también el mejor comprendido en términos de
estructura y el más utilizado en investigación de diagnóstico.103–107
La inmovilización de los anticuerpos por unión covalente resulta atractiva a
nivel industrial debido a que suelen ser métodos sencillos, económicos y
extrapolables a una gran variedad de anticuerpos. La unión covalente se basa
en una reacción entre grupos reactivos de la biomolécula (-NH2) y los grupos

CÁPITULO 1
28
reactivos del soporte (-COOH). La estrategia de inmovilización mediante unión
covalente comúnmente utilizada es el enlace carboxilo-amino, denominado
acoplamiento carbodiimida, que se utiliza para activar y conjugar un ácido
carboxílico por reacción directa con aminas primarias a través de la formación
de enlaces amida estables.108
Las carbodiimidas más utilizadas son la N,N'-diciclohexilcarbodiimida (DCC) y
la 1-etil-3-(3-dimetilaminopropil)carbodiimida (EDC). Estos dos compuestos
contienen en su estructura el grupo carbodiimida (-N=C=N-), altamente
reactivo, capaz de activar los grupos carboxílicos para que después éstos
puedan conjugarse con las aminas presentes en las proteínas.
Figura 1.16. Esquema de reacción de la formación de un enlace amida mediante el método carbodiimida.
La formación del enlace amida a través del método carbodiimida consta de
dos etapas: i) la activación de los grupos carboxílicos por reacción con la
carbodiimida produciendo el intermedio de reacción o-acilisourea y ii) la bio-
conjugación con los grupos amino de las proteínas dando lugar al enlace amida
(Figura 1.16).
En este trabajo se presenta un procedimiento en el que se lleva a cabo la
implantación de grupos carboxílicos durante la síntesis del grafeno por CVD,
evitando concentraciones significativas de los demás grupos oxigenados, y así
disponer de una plataforma apta para sensores ópticos o electrónicos de
biomoléculas.

Introducción
29
1.2.2.2 Biosensores basados en TMDCs
La creciente demanda de sensores sensibles, fiables, rentables y portátiles ha
promovido una enorme investigación sobre nuevos nanomateriales. Entre
ellos, los TMDCs bidimensionales (2D), como el MoS2, han atraído una intensa
atención como materiales funcionales en una gran variedad de aplicaciones
de biosensores.21 En particular, la elevada relación superficie-volumen hace
que el MoS2 2D sea especialmente sensible a los cambios de su entorno. Al
exponerse a los analitos, pueden producirse cambios de señal en sus
propiedades ópticas y electrónicas como resultado de interacciones físicas y
químicas, como la absorción, la transferencia de carga y el dopaje, los cambios
en la permitividad y las vibraciones de la red. Esto hace del MoS2 2D un
material muy prometedor para sensores con una alta sensibilidad y
selectividad.
Los biosensores actuales basados en MoS2 2D se pueden clasificar en varios
tipos, incluyendo dispositivos basados en detección eléctrica y óptica. Se ha
demostrado que los sensores de MoS2 basados en el efecto campo (FETs) son
capaces de detectar proteínas, ADN y otros componentes bioquímicos.23,109,110
En los sensores electroquímicos, diseñados para detectar especies biológicas
iónicas, el electrodo de trabajo se recubre con MoS2.111–114 Por otro lado, El
MoS2 2D tiene un bandgap directo, en el visible (rojo)115 y una buena
estabilidad química en condiciones ambientales,33,116 lo que lo convierte en un
candidato perfecto para biosensores de fotoluminiscencia (PL). La
fotoluminiscencia del MoS2 2D puede sufrir cambios tras la inmovilización de
biomoléculas en su superficie y/o la interacción con ellos.117,118 Además, tras
la interacción con el biomarcador, los procesos de enlace que ocurren cerca
de la superficie del MoS2 podrían producir perturbaciones en la permitividad
dieléctrica local, que también pueden causar alteraciones en la
fotoluminiscencia.119

CÁPITULO 1
30
Entre los biomarcadores más utilizados se encuentra el miRNA21.120 El
miRNA21 es uno de los primeros genes miRNA humanos cuya regulación ha
sido ampliamente estudiada, ya que se ha encontrado que se sobreexpresa en
muchos tejidos cancerosos, como el cáncer gástrico, el de pulmón, el
colorrectal, el de próstata y el de mama.121 El biomarcador típico utilizado es
el miRNA21 complementario (miRNA21-C) o una versión modificada de éste.
Para inmovilizar el biorreceptor sobre el MoS2 se utiliza una secuencia
específica modificada con grupos tiol (-SH). Las moléculas orgánicas con el
grupo funcional tiol tienden a reparar o eliminar las vacantes de S presentes
en la red de MoS2,33,116,122,123 formando un enlace covalente entre la
biomolécula y las láminas de MoS2 en una disposición que permite la posterior
hibridación con el biomarcador.
Con el objetivo de aprovechar las ventajas que proporciona la
fotoluminiscencia del MoS2 para el desarrollo de nuevos biosensores, en este
trabajo se ha utilizado MoS2 crecido por PVT sobre zafiro, como transductor
para el reconocimiento del biomarcador de cáncer de mama miRNA21. Para
verificar la selectividad del biosensor se han empleado biomarcadores de
miRNA21 complementarias y no complementarias.
1.3 REFERENCIAS
1. Yang, G., Li, L., Lee, W. B. & Ng, M. C. Structure of graphene and its
disorders: a review. Sience Technol. Adv. Mater. 19, 613–648 (2018).
2. Geim, A. K. & MacDonald, A. H. Graphene: Exploring carbon flatland.
Phys. Today 60, 35 (2007).
3. Mao, S. & Chen, J. Graphene-based electronic biosensors. J. Mater. Res.
2017 3215 32, 2954–2965 (2017).
4. Novoselov, K. S. et al. Electric Field Effect in Atomically Thin Carbon

Introducción
31
Films. Science (80-. ). 306, 666–669 (2004).
5. Novoselov, K. S. et al. Unconventional quantum Hall effect and Berry’s
phase of 2π in bilayer graphene. Nat. Phys. 2006 23 2, 177–180 (2006).
6. Novoselov, K. S. et al. Room-Temperature Quantum Hall Effect in
Graphene. Science (80-. ). 315, 1379–1379 (2007).
7. Bolotin, K. I. et al. Ultrahigh electron mobility in suspended graphene.
Solid State Commun. 146, 351–355 (2008).
8. Morozov, S. V. et al. Giant Intrinsic Carrier Mobilities in Graphene and
Its Bilayer. Phys. Rev. Lett. 100, 016602 (2008).
9. EV, C. et al. Biased bilayer graphene: semiconductor with a gap tunable
by the electric field effect. Phys. Rev. Lett. 99, (2007).
10. Malard, L. M., Pimenta, M. A., Dresselhaus, G. & Dresselhaus, M. S.
Raman spectroscopy in graphene. Phys. Rep. 473, 51–87 (2009).
11. Ferrari, A. C. & Basko, D. M. Raman spectroscopy as a versatile tool for
studying the properties of graphene. Nat. Nanotechnol. 2013 84 8,
235–246 (2013).
12. Ferrari, A. C. Raman spectroscopy of graphene and graphite: Disorder,
electron–phonon coupling, doping and nonadiabatic effects. Solid
State Commun. 143, 47–57 (2007).
13. Ni, Z., Wang, Y., Yu, T. & Shen, Z. Raman Spectroscopy and Imaging of
Graphene. (2008) doi:10.1007/s12274-008-8036-1.
14. Felten, A., Eckmann, A., Pireaux, J. J., Krupke, R. & Casiraghi, C.
Controlled modification of mono- and bilayer graphene in O2, H2 and
CF4 plasmas. Nanotechnology 24, (2013).
15. Anemone, G. et al. Quality of graphene on sapphire: long-range order
from helium diffraction versus lattice defects from Raman
spectroscopy. RSC Adv. 6, 21235–21245 (2016).
16. Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. & Kis, A. Single-
layer MoS2 transistors. Nat. Nanotechnol. 6, 147–150 (2011).
17. Yin, Z. et al. Single-Layer MoS2 Phototransistors. ACS Nano 6, 74–80
(2011).
18. Kumar, N. et al. Second harmonic microscopy of monolayer MoS2.
Phys. Rev. B - Condens. Matter Mater. Phys. 87, (2013).

CÁPITULO 1
32
19. Ramasubramaniam, A. Large excitonic effects in monolayers of
molybdenum and tungsten dichalcogenides. Phys. Rev. B - Condens.
Matter Mater. Phys. 86, 1–6 (2012).
20. Wang, T. et al. Direct detection of DNA below ppb level based on
thionin-functionalized layered MoS2 electrochemical sensors. Anal.
Chem. 86, 12064–12069 (2014).
21. Kalantar-Zadeh, K. & Ou, J. Z. Biosensors Based on Two-Dimensional
MoS2. ACS Sensors vol. 1 5–16 (2016).
22. Zhu, X. et al. Recent advances in synthesis and biosensors of two-
dimensional MoS2. Nanotechnology 30, (2019).
23. Sarkar, D. et al. MoS2 field-effect transistor for next-generation label-
free biosensors. ACS Nano 8, 3992–4003 (2014).
24. Gan, X., Zhao, H. & Quan, X. Two-dimensional MoS2: A promising
building block for biosensors. Biosens. Bioelectron. 89, 56–71 (2017).
25. Voiry, D., Mohite, A. & Chhowalla, M. Phase engineering of transition
metal dichalcogenides. Chemical Society Reviews vol. 44 2702–2712
(2015).
26. Chhowalla, M. et al. The chemistry of two-dimensional layered
transition metal dichalcogenide nanosheets. Nature Chemistry vol. 5
263–275 (2013).
27. Wilson, J. A. & Yoffe, A. D. The transition metal dichalcogenides
discussion and interpretation of the observed optical, electrical and
structural properties. Adv. Phys. 18, 193–335 (1969).
28. Wypych, F. & Schöllhorn, R. 1T-MoS2, a new metallic modification of
molybdenum disulfide. J. Chem. Soc. Chem. Commun. 1386–1388
(1992) doi:10.1039/C39920001386.
29. Tang, Q. & Jiang, D.-E. Stabilization and Band-Gap Tuning of the 1T-MoS
2 Monolayer by Covalent Functionalization. Chem. Mater. 27, 3743–
3748 (2015).
30. Huang, X., Zeng, Z. & Zhang, H. Metal dichalcogenide nanosheets:
Preparation, properties and applications. Chem. Soc. Rev. 42, 1934–
1946 (2013).
31. Ho, W., Yu, J. C., Lin, J., Yu, J. & Li, P. Preparation and photocatalytic

Introducción
33
behavior of MoS2 and WS 2 nanocluster sensitized TiO2. Langmuir 20,
5865–5869 (2004).
32. Splendiani, A. et al. Emerging photoluminescence in monolayer MoS2.
Nano Lett. 10, 1271–1275 (2010).
33. Wang, Q. H., Kalantar-Zadeh, K., Kis, A., Coleman, J. N. & Strano, M. S.
Electronics and optoelectronics of two-dimensional transition metal
dichalcogenides. Nat. Nanotechnol. | 7, (2012).
34. Pimenta, M. A., Del Corro, E., Carvalho, B. R., Fantini, C. & Malard, L. M.
Comparative Study of Raman Spectroscopy in Graphene and MoS 2-
type Transition Metal Dichalcogenides. Acc. Chem. Res 48, 2021 (2015).
35. Carvalho, B. et al. Intervalley scattering by acoustic phonons in two-
dimensional MoS2 revealed by double-resonance Raman
spectroscopy. Nat. Commun. 8, 14670 (2017).
36. Carvalho, B. R., Malard, L. M., Alves, J. M., Fantini, C. & Pimenta, M. A.
Symmetry-dependent exciton-phonon coupling in 2D and bulk MoS2
observed by resonance Raman scattering. Phys. Rev. Lett. 114, (2015).
37. Molina-Sánchez, A. & Wirtz, L. Phonons in single-layer and few-layer
MoS2 and WS2. Phys. Rev. B - Condens. Matter Mater. Phys. 84, (2011).
38. Gołasa, K., Grzeszczyk, M., … R. B.-S. state & 2014, undefined.
Resonant Raman scattering in MoS2—From bulk to monolayer.
Elsevier.
39. Zhang, X. et al. Phonon and Raman scattering of two-dimensional
transition metal dichalcogenides from monolayer, multilayer to bulk
material †. Chem. Soc. Rev 44, 2757 (2015).
40. Zhao, Y. et al. Interlayer breathing and shear modes in few-trilayer
MoS2 and WSe2. Nano Lett. 13, 1007–1015 (2013).
41. Sekine, T., Nakashizu, T., Toyoda, K., … K. U.-S. S. & 1980, undefined.
Raman scattering in layered compound 2H-WS2. Elsevier.
42. Lee, C. et al. Anomalous lattice vibrations of single- and few-layer
MoS2. ACS Nano 4, 2695–2700 (2010).
43. Brent, J. R., Savjani, N. & O’Brien, P. Synthetic approaches to two-
dimensional transition metal dichalcogenide nanosheets. Progress in
Materials Science vol. 89 411–478 (2017).

CÁPITULO 1
34
44. Novoselov, K. S. et al. Two-dimensional atomic crystals. Proc. Natl.
Acad. Sci. U. S. A. 102, 10451–10453 (2005).
45. Castellanos-Gomez, A., Agrat, N. & Rubio-Bollinger, G. Optical
identification of atomically thin dichalcogenide crystals. Appl. Phys.
Lett. 96, (2010).
46. Varrla, E. et al. Large-scale production of size-controlled MoS2
nanosheets by shear exfoliation. Chem. Mater. 27, 1129–1139 (2015).
47. Coleman, J. N. et al. Two-dimensional nanosheets produced by liquid
exfoliation of layered materials. Science 331, 568–571 (2011).
48. Štengl, V. & Henych, J. Strongly luminescent monolayered MoS2
prepared by effective ultrasound exfoliation. Nanoscale 5, 3387–3394
(2013).
49. Cha, J. J. et al. Two-dimensional chalcogenide nanoplates as tunable
metamaterials via chemical intercalation. Nano Lett. 13, 5913–5918
(2013).
50. Whittingham, M. S. & Gamble, F. R. The lithium intercalates of the
transition metal dichalcogenides. Mater. Res. Bull. 10, 363–371 (1975).
51. Zhou, X., Xu, B., Lin, Z., Shu, D. & Ma, L. Hydrothermal synthesis of
flower-like MoS2nanospheres for electrochemical supercapacitors. J.
Nanosci. Nanotechnol. 14, 7250–7254 (2014).
52. Feng, X. et al. Novel mixed-solvothermal synthesis of MoS 2 nanosheets
with controllable morphologies. Cryst. Res. Technol. 48, 363–368
(2013).
53. Najmaei, S. et al. Vapour phase growth and grain boundary structure
of molybdenum disulphide atomic layers. Nat. Mater. 12, 754–759
(2013).
54. Garg, S., Mollah, A. S., Waters, J. L., Kim, S. M. & Kung, P. (Invited)
Transition Metal Dichalcogenide Semiconductor Growth and Large
Area Devices for Optoelectronics and Sensing. ECS Trans. 80, 1–11
(2017).
55. Chang, Y. H. et al. Monolayer MoSe2 grown by chemical vapor
deposition for fast photodetection. ACS Nano 8, 8582–8590 (2014).
56. Wang, S. et al. Shape evolution of monolayer MoS2 crystals grown by

Introducción
35
chemical vapor deposition. Chem. Mater. 26, 6371–6379 (2014).
57. Lee, Y.-H. et al. Synthesis of Large-Area MoS 2 Atomic Layers with
Chemical Vapor Deposition. Adv. Mater. 24, 2320–2325 (2012).
58. Zhou, W. et al. Intrinsic structural defects in monolayer molybdenum
disulfide. Nano Lett. 13, 2615–2622 (2013).
59. Iqbal, M. W., Shahzad, K., Akbar, R. & Hussain, G. A review on Raman
finger prints of doping and strain effect in TMDCs. Microelectron. Eng.
219, (2020).
60. Kim, J. G., Yun, W. S., Jo, S., Lee, J. & Cho, C. H. Effect of interlayer
interactions on exciton luminescence in atomic-layered MoS 2 crystals.
Sci. Rep. 6, 1–7 (2016).
61. Carvalho, A., Ribeiro, R. M. & Castro Neto, A. H. Band nesting and the
optical response of two-dimensional semiconducting transition metal
dichalcogenides. Phys. Rev. B - Condens. Matter Mater. Phys. 88, 1–6
(2013).
62. Pilot, R. et al. A review on surface-enhanced Raman scattering.
Biosensors vol. 9 57 (2019).
63. Ambrose, W. P., Goodwin, P. M., Martin, J. C. & Keller, R. A. Single
molecule detection and photochemistry on a surface using near-field
optical excitation. Phys. Rev. Lett. 72, 160–163 (1994).
64. Xie, W. & Schlücker, S. Rationally designed multifunctional plasmonic
nanostructures for surface-enhanced Raman spectroscopy: A review.
Reports on Progress in Physics vol. 77 116502 (2014).
65. Mosier-Boss, P. A. & Lieberman, S. H. Detection of Anionic Nutrients by
SERS of Cationic-Coated Silver Substrates. Effect of Chloride Ion. Appl.
Spectrosc. 55, 1327–1336 (2001).
66. Kim, A., Barcelo, S. J. & Li, Z. SERS-based pesticide detection by using
nanofinger sensors. Nanotechnology 26, (2015).
67. Fierro-Mercado, P. M. & Hernández-Rivera, S. P. Highly Sensitive Filter
Paper Substrate for SERS Trace Explosives Detection. Int. J. Spectrosc.
2012, 1–7 (2012).
68. Jensen, L., Aikens, M. & Schatz, G. C. Electronic structure methods for
studying surface-enhanced Raman scattering. (2008)

CÁPITULO 1
36
doi:10.1039/b706023h.
69. Ding, S. Y. et al. Nanostructure-based plasmon-enhanced Raman
spectroscopy for surface analysis of materials. Nature Reviews
Materials vol. 1 1–16 (2016).
70. Kleinman, S. L., Frontiera, R. R., Henry, A. I., Dieringer, J. A. & Van
Duyne, R. P. Creating, characterizing, and controlling chemistry with
SERS hot spots. Physical Chemistry Chemical Physics vol. 15 21–36
(2013).
71. Li, W., Camargo, P. H. C., Lu, X. & Xia, Y. Dimers of Silver Nanospheres :
Facile Synthesis and Their Use as Hot Spots for Surface-Enhanced
Raman Scattering 2009. 18–23 (2009).
72. Hao, E. & Schatz, G. C. Electromagnetic fields around silver
nanoparticles and dimers. J. Chem. Phys. 120, 357–366 (2004).
73. Moskovits, M. Surface-enhanced Raman spectroscopy: A brief
retrospective. J. Raman Spectrosc. 36, 485–496 (2005).
74. Creighton, J. A., Blatchford, C. G. & Albrecht, M. G. Plasma resonance
enhancement of Raman scattering by pyridine adsorbed on silver or
gold sol particles of size comparable to the excitation wavelength. J.
Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 75, 790–798 (1979).
75. Amendola, V. et al. Laser generation of iron-doped silver nanotruffles
with magnetic and plasmonic properties. Nano Res. 8, 4007–4023
(2015).
76. Tsuji, T., Iryo, K., Watanabe, N. & Tsuji, M. Preparation of silver
nanoparticles by laser ablation in solution: Influence of laser
wavelength on particle size. Appl. Surf. Sci. 202, 80–85 (2002).
77. Kahl, M., Voges, E., Kostrewa, S., Viets, C. & Hill, W. Periodically
structured metallic substrates for SERS. Sensors Actuators, B Chem. 51,
285–291 (1998).
78. Haberland, H., Karrais, M. & Mall, M. A new type of cluster and cluster
ion source. Zeitschrift für Phys. D Atoms, Mol. Clust. 20, 413–415
(1991).
79. Wegner, K., Piseri, P., Tafreshi, H. V. & Milani, P. Cluster beam
deposition: A tool for nanoscale science and technology. Journal of

Introducción
37
Physics D: Applied Physics vol. 39 439 (2006).
80. Khoury, C. G., Vo-dinh, T., V, D. U. & Carolina, N. Gold Nanostars For
Surface-Enhanced Raman Scattering : Synthesis , Characterization and
Optimization. 18849–18859 (2008).
81. Kelly, K. L., Coronado, E., Zhao, L. L. & Schatz, G. C. The optical
properties of metal nanoparticles: The influence of size, shape, and
dielectric environment. J. Phys. Chem. B 107, 668–677 (2003).
82. Noguez, C. Surface plasmons on metal nanoparticles: The influence of
shape and physical environment. J. Phys. Chem. C 111, 3606–3619
(2007).
83. Chapus, L. et al. Tunable SERS Platforms from Small Nanoparticle 3D
Superlattices: A Comparison between Gold, Silver, and Copper.
ChemPhysChem 18, 3066–3075 (2017).
84. Zhang, J., Langille, M. R. & Mirkin, C. A. Synthesis of Silver Nanorods by
Low Energy Excitation of Spherical Plasmonic Seeds. 2495–2498
(2011).
85. Benz, F. et al. SERS of Individual Nanoparticles on a Mirror: Size Does
Matter, but so Does Shape. J. Phys. Chem. Lett. 7, 2264–2269 (2016).
86. Stamplecoskie, K. G., Scaiano, J. C., Tiwari, V. S. & Anis, H. Optimal Size
of Silver Nanoparticles for Surface-Enhanced Raman Spectroscopy. J.
Phys. Chem. C 115, 1403–1409 (2011).
87. Craig F. Bohren; Donald R. Huffman. Absorption and Scattering of Light
by Small Particlesitle. (1988).
88. Cassar, R. N., Graham, D., Larmour, I., Wark, A. W. & Faulds, K.
Synthesis of size tunable monodispersed silver nanoparticles and the
effect of size on SERS enhancement. Vib. Spectrosc. 71, 41–46 (2014).
89. Sangeetha, K. et al. Synthesis of ultra-small Rh nanoparticles
congregated over DNA for catalysis and SERS applications. Colloids
Surfaces B Biointerfaces 173, 249–257 (2019).
90. Michaels, A. M., Nirmal, M. & Brus, L. E. Surface Enhanced Raman
Spectroscopy of Individual Rhodamine 6G Molecules on Large Ag
Nanocrystals. 9932–9939 (1999) doi:10.1021/ja992128q.
91. Zhan, B. et al. Graphene Field-Effect Transistor and Its Application for

CÁPITULO 1
38
Electronic Sensing. 4042–4065 (2014) doi:10.1002/smll.201400463.
92. He, Q., Wu, S., Yin, Z. & Zhang, H. Graphene-based electronic sensors.
Chem. Sci. 3, 1764–1772 (2012).
93. Niyogi, S. et al. Covalent Chemistry for Graphene Electronics. J. Phys.
Chem. Lett. 2, 2487–2498 (2011).
94. Criado, A., Melchionna, M., Marchesan, S. & Prato, M. The Covalent
Functionalization of Graphene on Substrates Angewandte. 10734–
10750 (2015) doi:10.1002/anie.201501473.
95. Rösicke, F. et al. Functionalization of any substrate using covalently
modified large area CVD graphene. Chem. Commun. 53, 9308–9311
(2017).
96. Chen, J., Yao, B., Li, C. & Shi, G. An improved Hummers method for eco-
friendly synthesis of graphene oxide. Carbon N. Y. 64, 225–229 (2013).
97. Pei, S. & Cheng, H. M. The reduction of graphene oxide. Carbon N. Y.
50, 3210–3228 (2012).
98. Dreyer, D. R., Park, S., Bielawski, C. W. & Ruoff, R. S. The chemistry of
graphene oxide. Chemical Society Reviews vol. 39 228–240 (2010).
99. Hadish, F., Jou, S., Huang, B., … H. K.-J. of T. & 2017, undefined.
Functionalization of CVD Grown Graphene with Downstream Oxygen
Plasma Treatment for Glucose Sensors. iopscience.iop.org 164, B336–
B341 (2017).
100. Bianco, G. V. et al. Engineering graphene properties by modulated
plasma treatments. Carbon N. Y. 129, 869–877 (2018).
101. Childres, I., Jauregui, L. A., Tian, J. & Chen, Y. P. Effect of oxygen plasma
etching on graphene studied using Raman spectroscopy and electronic
transport measurements. New J. Phys. 13, 25008 (2011).
102. Delves, P. J., Martin, S., Burton, R. D. & Roitt, I. M. Roitt. Inmunología.
Fundamentos (12a edición). (Editorial Médica Panamericana, 2014).
103. Chen, M., Song, Z., Han, R., Li, Y. & Luo, X. Low fouling electrochemical
biosensors based on designed Y-shaped peptides with antifouling and
recognizing branches for the detection of IgG in human serum. Biosens.
Bioelectron. 178, 113016 (2021).
104. Wang, Q., Jing, J. Y. & Wang, B. T. Highly Sensitive SPR Biosensor Based

Introducción
39
on Graphene Oxide and Staphylococcal Protein A Co-Modified TFBG for
Human IgG Detection. IEEE Trans. Instrum. Meas. (2018)
doi:10.1109/TIM.2018.2875961.
105. Dancil, K. P. S., Greiner, D. P. & Sailor, M. J. A porous silicon optical
biosensor: Detection of reversible binding of IgG to a protein A-
modified surface. J. Am. Chem. Soc. 121, 7925–7930 (1999).
106. Liu, Q. et al. Electrochemiluminescent biosensor with DNA link for
selective detection of human IgG based on steric hindrance. Talanta
194, 745–751 (2019).
107. Wang, B. T. & Wang, Q. An interferometric optical fiber biosensor with
high sensitivity for IgG/anti-IgG immunosensing. Opt. Commun. 426,
388–394 (2018).
108. Sehgal, D. & Vijay, I. K. A method for the high efficiency of water-
soluble carbodiimide-mediated amidation. Anal. Biochem. 218, 87–91
(1994).
109. Zhu, C. et al. Single-layer MoS2-based nanoprobes for homogeneous
detection of biomolecules. J. Am. Chem. Soc. 135, 5998–6001 (2013).
110. Wang, L. et al. Functionalized MoS2 nanosheet-based field-effect
biosensor for label-free sensitive detection of cancer marker proteins
in solution. Small 10, 1101–1105 (2014).
111. Loo, A. H., Bonanni, A., Ambrosi, A. & Pumera, M. Molybdenum
disulfide (MoS2) nanoflakes as inherently electroactive labels for DNA
hybridization detection. Nanoscale 6, 11971–11975 (2014).
112. Wu, S. et al. Electrochemically reduced single-layer MoS2 nanosheets:
Characterization, properties, and sensing applications. Small 8, 2264–
2270 (2012).
113. Wang, T. et al. Direct detection of DNA below ppb level based on
thionin-functionalized layered MoS2 electrochemical sensors. Anal.
Chem. 86, 12064–12069 (2014).
114. Zhu, D. et al. Label-Free Electrochemical Sensing Platform for
MicroRNA-21 Detection Using Thionine and Gold Nanoparticles Co-
Functionalized MoS2 Nanosheet. ACS Appl. Mater. Interfaces 9, 35597–
35603 (2017).

CÁPITULO 1
40
115. Splendiani, A. et al. Emerging photoluminescence in monolayer MoS2.
Nano Lett. 10, 1271–1275 (2010).
116. Jariwala, D., Sangwan, V. K., Lauhon, L. J., Marks, T. J. & Hersam, M. C.
Emerging device applications for semiconducting two-dimensional
transition metal dichalcogenides. ACS Nano vol. 8 1102–1120 (2014).
117. Lin, T. et al. Visual detection of blood glucose based on peroxidase-like
activity of WS2 nanosheets. Biosens. Bioelectron. 62, 302–307 (2014).
118. Wang, Y. & Ni, Y. Molybdenum disulfide quantum dots as a
photoluminescence sensing platform for 2,4,6-trinitrophenol
detection. Anal. Chem. 86, 7463–7470 (2014).
119. Lin, Y. et al. Dielectric screening of excitons and trions in single-layer
MoS2. Nano Lett. 14, 5569–5576 (2014).
120. Mittal, S., Kaur, H., Gautam, N. & Mantha, A. K. Biosensors for breast
cancer diagnosis: A review of bioreceptors, biotransducers and signal
amplification strategies. Biosens. Bioelectron. 88, 217–231 (2017).
121. Iorio, M. V. & Croce, C. M. MicroRNA dysregulation in cancer:
Diagnostics, monitoring and therapeutics. A comprehensive review.
EMBO Molecular Medicine vol. 4 143–159 (2012).
122. Chen, X., McGlynn, C. & McDonald, A. R. Two-Dimensional MoS2
Catalyzed Oxidation of Organic Thiols. Chem. Mater. 30, 6978–6982
(2018).
123. Tuxen, A. et al. Size threshold in the dibenzothiophene adsorption on
MoS2 nanoclusters. ACS Nano 4, 4677–4682 (2010).

41
2 TÉCNICAS EXPERIMENTALES
2.1 TÉCNICAS DE PREPARACIÓN DE MATERIALES-2D
2.1.1 Depósito químico en fase de vapor
La técnica de depósito químico en fase vapor, llamada CVD por sus siglas en
inglés (Chemical Vapor Deposition) es la principal alternativa a la exfoliación
para la síntesis escalada de materiales bidimensionales.1,2 Este es el caso tanto
del grafeno como de los dicalcogenuros de metales de transición (TMDCs).
Esta técnica presenta varias ventajas con respecto a otras técnicas de
recubrimiento. Algunas de ellas son el control estequiométrico de la
composición de los materiales, un recubrimiento uniforme de los sustratos
independientemente de su forma, o la posibilidad de realizar el depósito de
los materiales en un amplio rango de temperaturas, disminuyendo la
temperatura cuando se utilizan catalizadores.
En su representación más simple, la técnica CVD implica el flujo de uno o más
gases precursores al interior de un reactor, donde tiene lugar una reacción
química y transformación de esos gases en un depósito sólido. Las reacciones
químicas se producen en las superficies calientes y cerca de ellas en un espacio
que se denomina capa límite, dando lugar al depósito de una película del
material sobre dichas superficies. Otra variante consiste en calentar el
producto de partida (sólido o líquido). En este caso, el producto de partida se
puede calentar dentro del reactor donde tendrá lugar la reacción química o en
un contenedor adyacente desde donde se pueda introducir el vapor formado.
Todo este proceso reactivo va acompañado de la producción de subproductos
químicos volátiles. Estos subproductos, así como los gases precursores que no

CÁPITULO 2
42
han reaccionado, son expulsados de la cámara por medio de un flujo de gas
que es extraído por un sistema de vacío.
La secuencia de acontecimientos que tienen lugar durante una reacción CVD
puede resumirse en las siguientes etapas:
a) Transporte de los reactivos desde la entrada de gases por flujo
forzado a la zona de reacción.
b) Reacciones en fase de vapor que forman los precursores gaseosos de
la película y los subproductos.
c) Transporte de los reactivos y sus productos desde la fase gaseosa al
sustrato a través de la capa límite.
d) Adsorción de estas especies en la superficie del sustrato.
e) Difusión en superficie, reacciones químicas e incorporación de estas
especies en diferentes sitios de nucleación y crecimiento.
f) Desorción de los subproductos volátiles de las reacciones en
superficie, a través de la capa límite.
g) Transporte de los subproductos fuera de la zona de reacción.
Las técnicas CVD pueden clasificarse en función de la presión de trabajo,1 ya
que influye enormemente en la termodinámica del proceso. La técnica se
denomina AP-CVD (Atmospheric Pressure Chemical Vapour Deposition)
cuando se trabaja a presión atmosférica o LP-CVD (Low Pressure Chemical
Vapour Deposition) cuando la presión está por debajo de los 10 Torr.1 En
función de la presión se distinguen dos tipos de reacciones: homogéneas o
heterogéneas. En las reacciones homogéneas se forman partículas que
precipitan sobre el sustrato generando puntos de nucleación. Estas reacciones
se ven favorecidas a alta presión. Por otro lado, las reacciones heterogéneas
tienen lugar en la cercanía del sustrato o en la misma superficie, dando lugar
a la formación de películas bidimensionales. En el caso tanto del grafeno como
de los TMDCs se debe favorecer la reacción heterogénea, evitando la

Técnicas experimentales
43
formación de puntos de nucleación y favoreciendo el crecimiento
bidimensional de los materiales, aunque en la mayoría de los casos de
crecimiento de TMDCs por CVD presentes en la literatura emplean presiones
atmosféricas.3–5
En cuanto a la temperatura, la técnica CVD se puede realizar en reactores de
pared caliente o en reactores de pared fría. En estos últimos, el calentamiento
de los sustratos se realiza directamente, ya sea mediante un horno situado en
el interior del reactor o por inducción, mientras que las paredes permanecen
frías. Por otro lado, en los reactores de pared caliente tanto la cámara como
el sustrato se calientan a la misma temperatura. Esto tiene el inconveniente
de la formación de depósitos en las paredes del reactor, sin embargo, la
homogeneidad de la temperatura en una amplia zona del reactor facilita el
depósito del material sobre un gran número de muestras.1
El ajuste de parámetros como la temperatura, la presión, el flujo de gases o la
duración del proceso, permite controlar características como el grosor del
revestimiento, la morfología, cristalinidad, forma o tamaño del producto final.
A continuación, se describen los sistemas CVD empleados a lo largo de este
trabajo.

CÁPITULO 2
44
2.1.1.1 Sistema CVD para el crecimiento de grafeno funcionalizado
Figura 2.1. Esquema del sistema CVD utilizado en la síntesis y funcionalización de
grafeno.
En la Figura 2.1 se muestra un esquema general del sistema de CVD térmico
de pared caliente diseñado y puesto a punto en nuestros laboratorios para la
síntesis y funcionalización de grafeno. El equipo se divide en tres partes:
- Entrada de reactivos, que se compone del sistema de entrada de
gases, en este caso H2 y CH4, regulados por controladores de flujo
másico, y de un horno auxiliar adyacente donde se calienta el
precursor que dará lugar a la funcionalización con grupos carboxílicos
(-COOH). En este caso el precursor es ácido dicloroacético
(Cl2CHCOOH, DCA), cuyo punto de ebullición es 194 °C a presión
atmosférica, por lo que se calienta aparte para generar el vapor que
posteriormente es introducido en el reactor. Su flujo se controla con
una válvula reguladora.
- El reactor, formado por un horno tubular comercial (Hobersal, modelo
ST-145045 monofásico) de 900 mm de longitud y con una
temperatura máxima de trabajo de 1450 °C, y por un tubo de cuarzo
de 50 mm de diámetro, donde se introducirán los reactivos y las
muestras.
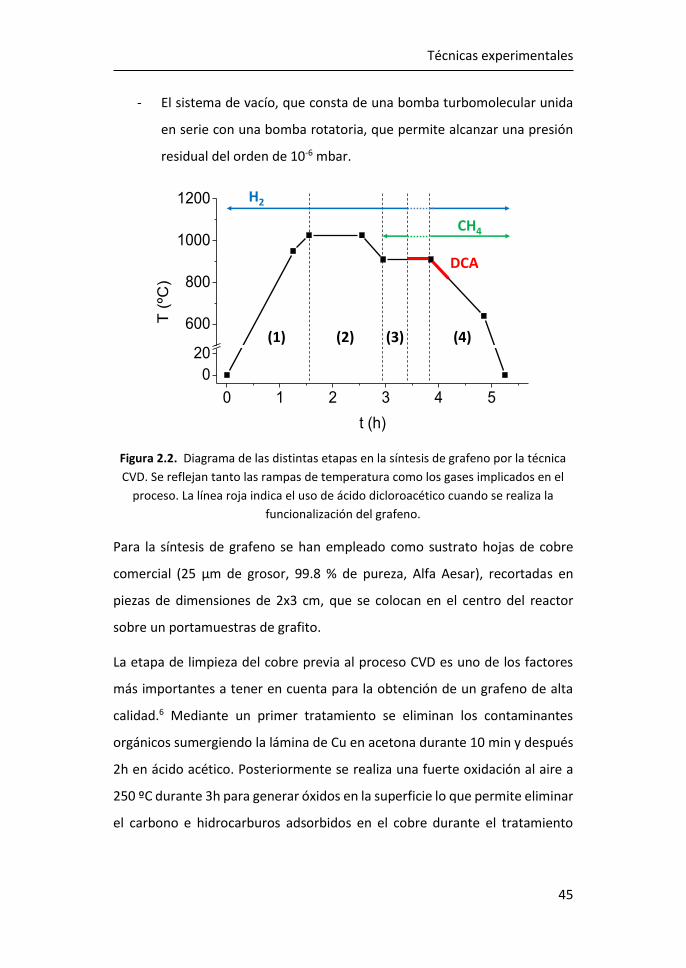
Técnicas experimentales
45
- El sistema de vacío, que consta de una bomba turbomolecular unida
en serie con una bomba rotatoria, que permite alcanzar una presión
residual del orden de 10-6 mbar.
Figura 2.2. Diagrama de las distintas etapas en la síntesis de grafeno por la técnica
CVD. Se reflejan tanto las rampas de temperatura como los gases implicados en el
proceso. La línea roja indica el uso de ácido dicloroacético cuando se realiza la
funcionalización del grafeno.
Para la síntesis de grafeno se han empleado como sustrato hojas de cobre
comercial (25 μm de grosor, 99.8 % de pureza, Alfa Aesar), recortadas en
piezas de dimensiones de 2x3 cm, que se colocan en el centro del reactor
sobre un portamuestras de grafito.
La etapa de limpieza del cobre previa al proceso CVD es uno de los factores
más importantes a tener en cuenta para la obtención de un grafeno de alta
calidad.6 Mediante un primer tratamiento se eliminan los contaminantes
orgánicos sumergiendo la lámina de Cu en acetona durante 10 min y después
2h en ácido acético. Posteriormente se realiza una fuerte oxidación al aire a
250 ºC durante 3h para generar óxidos en la superficie lo que permite eliminar
el carbono e hidrocarburos adsorbidos en el cobre durante el tratamiento
0 1 2 3 4 50
20
600
800
1000
1200
T (º
C)
t (h)
(2) (3) (4)(1)
H2
CH4
DCA

CÁPITULO 2
46
posterior a alta temperatura. Con estos dos tratamientos la lámina de Cu está
lista para introducirse en el reactor.
La síntesis de grafeno llevada a cabo consta de las siguientes etapas:
(i) Calentamiento. En esta etapa se calienta el sistema de reacción desde
temperatura ambiente hasta la temperatura de reducción (1015 ºC)
con una rampa de 11 °C/min. El gas utilizado durante esta etapa es H2,
con un flujo de 110 sccm y una presión de 8.5 10-1 mbar.
(ii) Acondicionado del cobre. Se realiza a 1015 °C y tiene una duración de
1h. Se mantiene el flujo de H2 de la etapa anterior para
eliminar/reducir la capa de óxido de cobre formada durante el
tratamiento previo. También se activa la superficie del metal,
aumentando su tamaño de grano y eliminando posibles defectos
estructurales existentes en la superficie.
(iii) Crecimiento. Tiene lugar a 910 °C y su duración es de 30 minutos para
obtener un recubrimiento completo de grafeno monocapa. En el caso
del grafeno funcionalizado esta etapa tendrá distintas duraciones en
función de la muestra. Durante esta etapa se introduce el gas que
actúa como fuente de carbono, en este caso CH4, y se mantiene un
flujo constante de una mezcla H2/CH4 en relación 10:1, obteniendo
una presión dentro de la cámara de 9 10-1 mbar.
(iv) Enfriamiento. El reactor se enfría, manteniendo la atmósfera de la
etapa anterior, a un ritmo de -4.5 ºC hasta 650 ºC, temperatura a la
que la muestra de grafeno es extraída desde el centro del horno hasta
un extremo donde la temperatura es cercana a la ambiente.
En el caso de la síntesis de grafeno funcionalizado in situ con grupos –COOH
tiene lugar una etapa extra:

Técnicas experimentales
47
Funcionalización con ácido dicloroacético (DCA). Esta etapa tiene lugar
inmediatamente después del crecimiento del grafeno y a la misma
temperatura (910 °C). Su duración dependerá de la muestra, variando entre
los 15 y los 30 minutos. En los 20 minutos previos a esta etapa se calienta el
DCA en el horno auxiliar hasta alcanzar los 200 °C a un ritmo de 10 °C/min con
la válvula de paso cerrada, transformándolo en vapor. Cuando comienza la
etapa de funcionalización se introduce un flujo del vapor de DCA en el reactor.
Dependiendo de la muestra se mantendrá, o no, la mezcla de gases H2/CH4.
En algunos casos esta etapa de funcionalización se puede ver extendida
abarcando parte del enfriamiento, manteniendo el flujo de DCA durante los
primeros minutos de la rampa de bajada de temperatura. Esta etapa viene
marcada en el diagrama de rampas de temperatura de la Figura 2.2 por una
línea gruesa roja.
2.1.1.2 Equipo CVD para la síntesis de TMDCs
En la síntesis de dicalcogenuros de metales de transición (TMDCs)
bidimensionales vía CVD se produce la sulfurización del óxido del metal de
transición. Para llevar a cabo el dopado de estos materiales, es necesario
introducir además un reactivo que contenga el elemento con el que nos
interesa dopar. Los productos de partida son sólidos en condiciones normales,
y se utiliza argón como gas para transportar los materiales vaporizados a lo
largo del reactor.
En primer lugar, se realizó el montaje de un sistema sencillo para la síntesis de
MoS2 por CVD utilizando como productos de partida óxido de molibdeno (VI)
y azufre (Figura 2.3), ya que es el método de síntesis más referenciado en la
literatura.
Este primer sistema consta de un tubo de cuarzo de 38 mm de diámetro
(reactor) y un horno tubular de 400 mm de longitud. En ambos extremos se

CÁPITULO 2
48
han acoplado dos piezas metálicas rodeando el tubo con dos o-ring haciendo
presión entre dichas piezas y el reactor, lo que permite obtener una presión
base de vacío del orden de 10-4 mbar. Dichas piezas tienen una terminación en
forma de brida estándar KF40, lo que permite anclar por un extremo el sistema
de extracción de gases y por el otro un adaptador con tres salidas. En estas
tres salidas se han acoplado un medidor de vacío, la entrada de gases, y una
barra de transferencia que permite manipular la barquilla que contiene el
azufre una vez haya comenzado el proceso. La entrada de Ar consiste en un
tubo flexible conectado a un controlador de flujo másico (MFC) que regula el
flujo de gas.
Durante el proceso de síntesis, es necesario que el azufre se caliente a 200-
250 °C, pero su punto de fusión a presión atmosférica es 115 °C, por lo que se
utiliza una barra de transferencia para colocarlo en la zona a 200-250 °C en el
momento en que el óxido de molibdeno haya alcanzado su temperatura
correspondiente, evitando así que el azufre sea transportado antes de que
pueda tener lugar la reacción.
El sistema de extracción de gases consta de una bomba turbomolecular
descargada por una bomba primaria. Se utiliza el sistema completo para la
limpieza del reactor previa a la síntesis (presión residual 10-4 mbar). Durante
la síntesis se desconecta la bomba turbomolecular y se trabaja únicamente
con la bomba primaria. Además, se instaló una válvula regulable entre el
reactor y el sistema de vacío para variar la presión dentro del reactor sin tener
que modificar el flujo del gas de transporte y viceversa.

Técnicas experimentales
49
Figura 2.3. Esquema del sistema CVD empleado en la síntesis de MoS2 a partir de
MoO3 y S.
En este trabajo se ha sintetizado MoS2 sobre sustratos de silicio (100), sílice
vítrea y zafiro (0001). Se han empleado como reactivos óxido de molibdeno
(VI) (Alfa Aesar, 99.95%) y azufre (Puratronic, 99.999%). El óxido de molibdeno
se calentó a temperaturas entre 680-715 ºC, mientras que el azufre se calienta
a 200 ºC. Se emplearon flujos de Ar entre 20 y 100 sccm y presiones entre 10-
2-1 mbar.
2.1.2 Transporte físico de vapor
La técnica de transporte físico de vapor, llamada PVT por sus siglas en inglés
(Physical Vapor Transport) es una alternativa al CVD para el crecimiento de
materiales bidimensionales. En este caso el material evaporado tiene la misma
composición que el material a depositar, por lo que no se produce ninguna
reacción química durante el proceso. El material que se evapora es
transportado a lo largo del reactor por un gas inerte, en nuestro caso Ar, hasta
alcanzar los sustratos donde se deposita formando el material bidimensional.
Aunque en algunos trabajos de la literatura se refieren a este método también
como CVD,7,8 aquí se ha querido diferenciarlos ya que los mecanismos de
formación del MoS2 son muy diferentes.

CÁPITULO 2
50
2.1.2.1 Sistema PVT para la síntesis de TMDCs utilizado en la
Universidad de Alabama
El sistema consta de un tubo de cuarzo de 2 m de longitud y 2.5 mm de
diámetro, que pasa a través de dos hornos tubulares independientes, de 30 y
100 cm de longitud (Figura 2.4). Este último puede abrirse, lo que permite el
enfriamiento rápido tanto de los sustratos como de los reactivos que se
encuentren en su interior. Durante el proceso el gas transportador se
introduce por un extremo del reactor mediante un controlador de flujo
másico, y se extrae por el otro extremo a través de una bomba primaria. En
este sistema, la limpieza previa al crecimiento de las muestras se realiza
mediante tres purgas consecutivas utilizando primero nitrógeno y después
argón.
Figura 2.4. Esquema del sistema PVT empleado en la Universidad de Alabama para la
síntesis de MoS2 a partir de MoS2 en polvo.
Las muestras de MoS2 obtenidas en este equipo se crecieron sobre sustratos
de zafiro (0001) utilizando como producto de partida MoS2 en polvo (Alfa
Aesar, 99%). El MoS2 en polvo se calienta a 970 ºC y los sustratos a 800 ºC
aproximadamente. El flujo de Ar durante el crecimiento fue 20 sccm y la
presión 10 mbar.

Técnicas experimentales
51
2.1.2.2 Sistema PVT para síntesis y dopado de TMDCs (ICMM-CSIC)
Se diseñó un sistema similar al utilizado en la Universidad de Alabama para
poder llevar a cabo la síntesis y el dopado de TMDCs tanto por CVD como por
PVT (Figura 2.5). Para ello es necesario contar con un sistema que nos permita
controlar la temperatura de manera precisa en distintos puntos del horno, ya
que todos los productos de partida son sólidos a temperatura ambiente y cada
uno tiene un punto de fusión distinto. Por ello, se diseñó un sistema que
consta de un único reactor y de tres zonas calefactoras móviles e
independientes. Durante el proceso, el gas transportador se introduce por un
extremo del reactor mediante un controlador de flujo másico y se extrae por
el otro extremo junto con los productos no deseados a través de una bomba
primaria.
Figura 2.5. Esquema del sistema CVD/PVT diseñado para la síntesis y dopado del
MoS2.
Horno CVD
El sistema consta de un tubo de cuarzo de 120 cm de longitud y 3.8 mm de
diámetro, denominado reactor, que pasa a través de tres hornos tubulares
independientes unidos a un rail paralelo al reactor, lo que permite
desplazarlos horizontalmente mientras el tubo de cuarzo permanece fijo. Que
sean móviles tiene principalmente dos ventajas. Por un lado, permite ver las
posiciones de los reactivos y los sustratos dentro del reactor, para que sean
las deseadas. Por otro lado, permite llevar a cabo un enfriamiento rápido de

CÁPITULO 2
52
las muestras cuando sea necesario, ya que se puede dejar la parte del reactor
que interese expuesta a la temperatura ambiente.
La temperatura de los tres hornos se regula a través de tres controladores
programables PID eurotherm modelo 3216, que permiten el diseño de las
rampas necesarias para llevar a cabo las distintas síntesis. La temperatura de
cada horno se mide con un termopar tipo K, colocado en el centro de cada
zona.
Los extremos del reactor están unidos a dos piezas metálicas interiores con
dos O-ring en cada una de ellas situados entre el tubo de cuarzo y la pieza
metálica, obteniendo una presión residual del orden de 10-5 mbar. Estas dos
piezas tienen una terminación de brida estándar KF40, permitiendo la unión
del sistema de entrada de gases en un extremo y del sistema de vacío en el
otro. La introducción de los reactivos y los sustratos en el reactor se realiza
por cualquiera de los dos extremos hasta las posiciones adecuadas. En la parte
exterior de los extremos del reactor se han fijado otras dos piezas metálicas
que lo fijan al rail, sosteniendo el peso de todo el sistema y evitando que
recaiga sobre el tubo de cuarzo.
Sistemas de introducción y extracción de gases
Un extremo del reactor está unido al sistema de entrada del gas de transporte,
Ar. Su flujo está regulado por un controlador de flujo másico (MFC) de la marca
Bronkhorst mediante el software suministrado por la misma marca. El
controlador tiene una capacidad de flujo máxima de 750 sccm. El otro
extremo está unido al sistema de vacío, compuesto por una bomba
turbomolecular descargada por una bomba primaria, que se utiliza como
medio de limpieza previa a cada síntesis, alcanzando una presión residual de
10-5 mbar. Durante la síntesis la presión requerida es mayor (1-10 mbar), por
lo que se aísla la bomba turbomolecular con un bypass para evitar que esté

Técnicas experimentales
53
sometida a dicha presión y que se contamine, de modo que el gas es extraído
únicamente con la bomba rotatoria.
Aunque este equipo se ha diseñado para crecer MoS2 tanto por CVD como por
PVT, durante este trabajo, sólo se han crecido muestras por el método PVT.
Para ello se ha empleado como producto de partida MoS2 en polvo (Alfa Aesar,
99%). Se ha depositado sobre sustratos de zafiro (0001) y sobre obleas de Si
con una capa de SiO2 de 300 nm. El flujo de Ar empleado es 20 sccm y la
presión 1 mbar.
2.1.3 Transferencia de materiales bidimensionales crecidos por
CVD/PVT
2.1.3.1 Transferencia de grafeno y grafeno funcionalizado
Una vez crecida la capa de grafeno sobre la lámina de Cu suele ser necesario
transferirla a sustratos aislantes para su posterior caracterización o estudio de
sus aplicaciones. En muchas de las aplicaciones del grafeno, el cobre no es un
sustrato adecuado, ya que al ser un material conductor enmascara e interfiere
con algunas de las propiedades del grafeno. Existen diferentes protocolos para
llevar a cabo la transferencia de grafeno crecido por CVD, siendo el más
utilizado el método que emplea el compuesto polimérico PMMA
(polimetilmetacrilato) como soporte.
Figura 2.6. Etapas del proceso de transferencia de grafeno.

CÁPITULO 2
54
La transferencia de grafeno utilizando PMMA llevada a cabo en nuestro
laboratorio (Figura 2.6) puede dividirse en las siguientes etapas:
(i) Recubrimiento con PMMA: se prepara una disolución de PMMA en
anisol de 2 ó 6% y se deja en agitación durante 3 días. El recubrimiento
de la muestra se lleva a cabo mediante la técnica de “spin-coating”,
para ello se deposita una gota de 8.5 μl sobre la muestra a transferir y
se somete a 1500 rpm durante un minuto. Por último, se calientan las
muestras sobre una placa calefactora a 130 ºC durante 15 min para
curar el polímero y después se dejan reposar al aire durante al menos
24 h para asegurar el secado completo. En el caso de las muestras de
grafeno se utiliza la disolución de PMMA del 2%. En las muestras de
grafeno funcionalizado, la presencia de defectos las hace más frágiles,
por lo que se emplea una disolución del 6%.
(ii) Eliminación del cobre: para poder eliminar el cobre de forma
satisfactoria es necesario eliminar los posibles restos de PMMA en la
cara posterior del cobre utilizando acetona. Después se coloca la
muestra PMMA/Gr/Cu en un vidrio de reloj y se añaden 0.5 ml de una
solución ácida de FeCl3 (2.8 M) que ataca el cobre, agitando
suavemente durante 5 minutos a 40 °C. Este proceso se repite tres
veces hasta haber eliminado el Cu por completo, enjuagando la
muestra con agua desionizada entre cada repetición. En el caso de las
muestras funcionalizadas suele ser necesario repetirlo más de tres
veces ya que suelen quedar más restos de PMMA en la parte posterior
de la muestra.
(iii) Lavados con H2O desionizada y HCl: con la muestra PMMA/Gr aun
sobre el vidrio de reloj se realizan varios enjuagues a temperatura
ambiente de 30 min con agua destilada y un enjuague de 10 min con
una solución de HCl al 10% para eliminar posibles impurezas.

Técnicas experimentales
55
(iv) “Pesca” con sustrato: la muestra (PMMA/Gr) se encuentra flotando
sobre agua destilada en el vidrio de reloj. a continuación, se sumerge
el sustrato sobre el que se va a transferir la muestra en un ángulo de
30° y se va eliminando el agua con una pipeta, de modo que a medida
que el nivel de agua disminuye el PMMA/Gr se fija sobre el sustrato.
Por último, se seca la muestra a 90 °C durante 2h y después se somete
a vacío (10-2 mbar) para asegurar su adherencia al sustrato.
(v) Eliminación del PMMA: se coloca la muestra (PMMA/Gr/sustrato)
sobre un vidrio de reloj, se añade acetona hasta cubrirla y se calienta
a 50 °C mientras se agita suavemente para favorecer la disolución del
PMMA. Este proceso se repite varias veces enjuagando la muestra
entre cada repetición con más acetona. Después se calienta a 45 °C
durante 20 minutos para eliminar posibles restos de acetona. Para
finalizar, se calienta la muestra (Gr/sustrato) a 250 °C en condiciones
de vacío (10-4 mbar) durante 3 horas para eliminar posibles residuos
de PMMA.
2.1.3.2 Transferencia de MoS2-2D
El método de transferencia que se describe a continuación es el utilizado en el
grupo del Dr. Kung en la Universidad de Alabama durante mi estancia. Al igual
que en el caso del grafeno se emplea el polímero PMMA. Las etapas son las
siguientes:
(i) Recubrimiento con PMMA: Se coloca la muestra a transferir sobre
una placa calefactora a 100 ºC. A continuación, recubren la muestra
con una solución de PMMA en anisol y se deja secar durante 10 min.
(ii) Retirada del sustrato original: Se enfría la muestra a temperatura
ambiente y después se sumerge en agua destilada a durante dos

CÁPITULO 2
56
minutos. Con ayuda de unas pinzas se levanta la capa de PMMA y con
ella el MoS2 que hay debajo (PMMA/MoS2).
(iii) Transferencia sobre el nuevo sustrato: se retira el exceso de agua del
PMMA/MoS2 con una pistola de N2 y se coloca sobre el nuevo sustrato
(PMMA/MoS2/Sust). Después se calienta a 100 ºC durante 10 min.
(iv) Eliminación del PMMA: se cubre la muestra con acetona y se deja
reposar durante 5 min. Se repite este proceso hasta haber eliminado
por completo el PMMA.
2.1.4 Inmovilización de anticuerpos. método carbodiimida
El método carbodiimida se utiliza para la inmovilización covalente de
anticuerpos en sustratos con grupos -COOH mediante la formación de enlaces
amida entre esos grupos carboxilo y los grupos amina de los anticuerpos.9,10
la inmovilización de los anticuerpos mediante el método carbodiimida consta
de dos etapas: (i) la activación de los grupos carboxílicos via enlaces
carbodiimida y (ii) la bio-conjugación con los grupos amino de las proteínas.
(i) Activación de los grupos carboxilo via enlace carbodiimida. En primer
lugar se utiliza la carbodiimida N‐(3‐dimetilaminopropil)‐N’‐
etilcarbodiimida (EDC) que forma un intermedio activo con los grupos
carboxílicos, susceptible de ser atacado por aminas, denominado O-
acilisourea. Este compuesto es altamente inestable debido a que se
hidroliza fácilmente en ausencia de grupos amino. Por ello esta
reacción se realiza en presencia de N‐hidroxisuccinimida (NHS), que
reacciona con el intermedio formado dando lugar a otro intermedio,
denominado éster-NHS, más estable y que favorecerá la reacción de
inmovilización. En las Figura 2.7 y 2.8 se han representado las etapas
que tienen lugar y las especies intermedias y subproductos que se
producen.
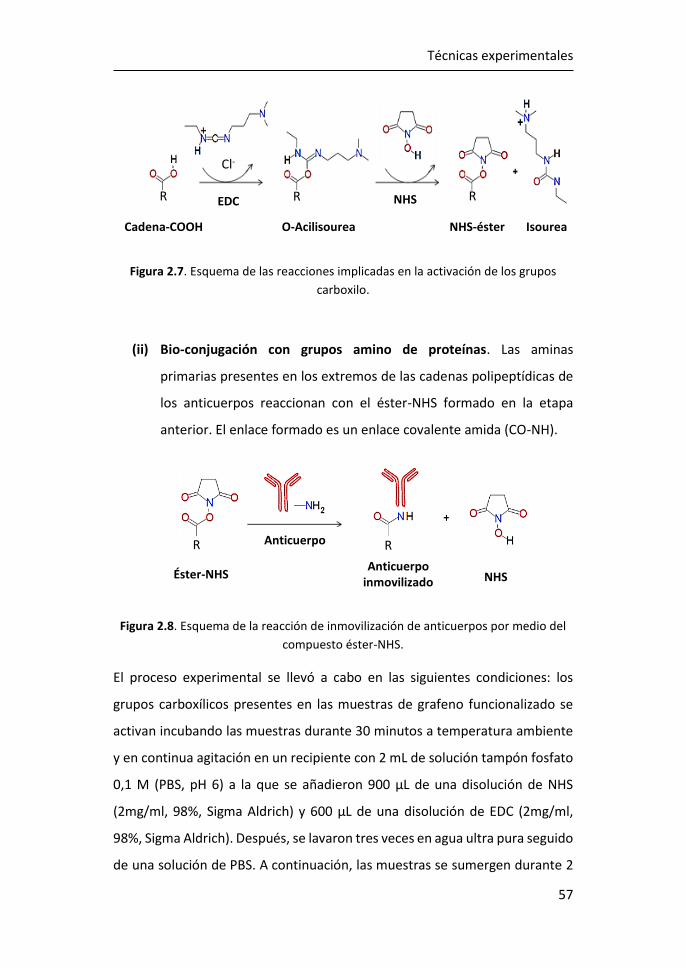
Técnicas experimentales
57
Figura 2.7. Esquema de las reacciones implicadas en la activación de los grupos
carboxilo.
(ii) Bio-conjugación con grupos amino de proteínas. Las aminas
primarias presentes en los extremos de las cadenas polipeptídicas de
los anticuerpos reaccionan con el éster-NHS formado en la etapa
anterior. El enlace formado es un enlace covalente amida (CO-NH).
Figura 2.8. Esquema de la reacción de inmovilización de anticuerpos por medio del
compuesto éster-NHS.
El proceso experimental se llevó a cabo en las siguientes condiciones: los
grupos carboxílicos presentes en las muestras de grafeno funcionalizado se
activan incubando las muestras durante 30 minutos a temperatura ambiente
y en continua agitación en un recipiente con 2 mL de solución tampón fosfato
0,1 M (PBS, pH 6) a la que se añadieron 900 µL de una disolución de NHS
(2mg/ml, 98%, Sigma Aldrich) y 600 µL de una disolución de EDC (2mg/ml,
98%, Sigma Aldrich). Después, se lavaron tres veces en agua ultra pura seguido
de una solución de PBS. A continuación, las muestras se sumergen durante 2
Cadena-COOH NHS-ésterO-Acilisourea
NHS EDC
Cl-
Isourea
R R R
Anticuerpo
NHS
R R
Éster-NHSAnticuerpo
inmovilizado

CÁPITULO 2
58
horas a temperatura ambiente en una solución de anticuerpo diluida (2 mL
PBS + 100 L anticuerpo, pH 7.4) en continua agitación. Una vez finalizado el
proceso, las muestras se lavan con agua ultra-pura y se almacenan a
temperaturas entre 2 - 8 °C en una solución de PBS 0.1 M.
El anticuerpo utilizado es el comercial de Sigma-Aldrich “Monoclonal Anti-
Human IgG1 Clone 8c/6-39 FITC Conjugate Purified Mouse Immunoglobulin”
producido en ratones. Dicho anticuerpo (anticuerpo primario) está marcado
con FITC (Fluorescein Isothiocyanate), que es un fluorocromo derivado de la
fluoresceína capaz de emitir fluorescencia en 518 nm (color verde).
2.2 TÉCNICAS DE PREPARACIÓN DE NANOPARTÍCULAS Y LÁMINAS
DELGADAS
2.2.1 Pulverización catódica o sputtering
La técnica de pulverización catódica o sputtering1,11 es uno de los métodos más
utilizados para la formación de láminas delgadas con excelentes propiedades
de densidad y adherencia, abarcando una gran variedad de materiales. Su uso
está ampliamente extendido en las industrias de metalización de
componentes y de preparación de materiales semiconductores fotovoltaicos,
así como en otras áreas más específicas como la fabricación de sensores o
sistemas ópticos, debido principalmente a la simplicidad del fenómeno físico
y versatilidad de la técnica.
En la técnica de sputtering se produce la vaporización de los átomos de un
material sólido provocado por el bombardeo de este por iones energéticos
acelerados por un campo eléctrico, por lo que al material sólido se denomina
“blanco” o “diana”.

Técnicas experimentales
59
Figura 2.9. Esquema del proceso de pulverización catódica.
Según se observa en la Figura 2.9, el electrodo negativo actúa como cátodo y
está formado por el material que se va a evaporar, mientras que el electrodo
positivo (ánodo) lo forma la cámara de vacío. Al aplicar un potencial elevado,
se produce una descarga eléctrica que ioniza el gas inerte introducido en la
cámara, generalmente Ar, de modo que los iones positivos bombardean el
blanco de sputtering. La descarga eléctrica para la ionización del gas se puede
producir por corriente continua (DC) para pulverizar materiales conductores,
o alterna en el rango de radio frecuencias (RF) en el caso de querer depositar
materiales aislantes).
En la actualidad, esta técnica consigue grandes rendimientos al utilizar
campos magnéticos (sputtering con magnetrón) mediante la implementación
de imanes debajo del cátodo. La disposición de las líneas del campo magnético
paralelas a la superficie del cátodo hace que los electrones secundarios
emitidos describan trayectorias helicoidales alrededor de ellas, aumentando
la densidad del plasma en las proximidades de la superficie del cátodo y
quedando confinado en esta región, alejándolo así de los sustratos. De este
modo se consigue aumentar la velocidad de depósito, reduciendo la presión

CÁPITULO 2
60
de gas necesaria para el crecimiento y dando lugar a láminas con baja
rugosidad.
El equipo de sputtering (Pfeiffer-PLS500) utilizado en este trabajo consta de
una precámara de vacío (10-3 - 10-2 mbar) por donde se introducen los
sustratos a la cámara de vacío, donde tiene lugar el crecimiento. Los sustratos
se fijan en un soporte monitorizado situado en el interior de la cámara que
permite colocarlos sobre diferentes magnetrones con los materiales a
depositar. La presión residual de la cámara de vacío está en el rango bajo de
10-6 mbar.
En este trabajo, esta técnica se ha utilizado en la fabricación de películas
nanoestructuradas de Ag con grosores nominales de 1 a 4 nm, así como para
el depósito de láminas delgadas de Al y Al2O3. La presión base de vacío es del
orden de 10-6 mbar. Las condiciones de depósito fueron las siguientes:
- Películas de Ag: intensidad de corriente de 0.01 A y presión de 6·10-3
mbar a una velocidad de depósito de 1.5 nm/min.
- Lámina de Al: intensidad de corriente de 0.04 A y presión de 6·10-3
mbar y velocidad de depósito de 3.4 nm/min.
- Lámina de Al2O3: intensidad de corriente de 0.37 A, a una presión de
1·10-2 mbar en atmósfera 50% Ar y 50% O2, con una velocidad de
depósito de 0.63 nm/min.
2.2.2 Agregación en fase gas para el depósito de nanopartículas
La agregación en fase gas es un caso particular de sputtering para la formación
de nanopartículas o agregados por medio de la vaporización de un material
sólido y su posterior condensación, que da lugar a su agregación antes de
llegar al sustrato.

Técnicas experimentales
61
Esta técnica de fabricación de agregados presenta varias ventajas únicas
frente a otras, entre ellas el control de la densidad del depósito y del tamaño
de los agregados formados. Además, el depósito se realiza en condiciones de
alto vacío, obteniendo nanoestructuras de alta pureza y libres de
contaminantes.
El equipo empleado en la síntesis de nanopartículas por agregación en fase
gas está basado en el concepto desarrollado por Haberland,12 y consta de una
precámara y de dos cámaras independientes, denominadas de agregación y
de depósito (Figura 2.10).
Figura 2.10. Esquema del equipo de agregación empleado en la síntesis de
nanopartículas.
En la cámara de agregación tiene lugar la formación de partículas en fase gas,
que viene dada por procesos de nucleación homogénea y de condensación por
colisiones entre átomos del blanco y entre átomos del blanco con átomos de
un gas inerte. Los gases inertes empleados en este trabajo son argón y helio.
El He disminuye el tamaño promedio de los agregados, ya que cuanto menor
es la masa de los iones del gas inerte, menos efectivos son los iones para el
proceso de sputtering. La cámara de agregación contiene un magnetrón

CÁPITULO 2
62
cubierto por una campana de acero con un pequeño orificio de 3 mm de
diámetro que comunica con la bomba de vacío, alcanzando una presión de
trabajo dentro de la cámara de agregación de 10-1 mbar. De esta manera se
consiguen las condiciones de vacío diferencial adecuadas para favorecer la
agregación del material arrancado y formar agregados de tamaño
nanométrico. Los agregados se extraen de la zona de agregación a través de
un diafragma hacia la cámara de depósito, donde se encuentra el sustrato a
una presión inferior a la de la cámara de agregación (10-3 mbar). La menor
presión en el interior de esta cámara durante el depósito permite a los
agregados viajar desde la cámara de agregación hasta el sustrato.
El tamaño y la distribución de los agregados se pueden controlar modificando
diferentes parámetros como el flujo de los gases de agregación, la longitud del
camino de vuelo, la potencia de sputtering o la temperatura.
En este trabajo, esta técnica se ha utilizado en la fabricación de nanopartículas
ultrapequeñas de Ag. Se han depositado sobre distintos sustratos. La presión
base de vacío fue del orden de 10-7 mbar. Las condiciones de depósito fueron
las siguientes: distancia de vuelo de 13 cm, una intensidad de corriente de 0.04
A, y una relación Ar/He 1:2 (30 sccm y 60 sccm respectivamente), lo que se
traduce en una presión de trabajo de 2.5·10-1 mbar en la cámara de agregación
y de 2·10-3 mbar en la cámara de depósito. La densidad de nanopartículas se
controló variando los tiempos de depósito.
2.3 TÉCNICAS DE CARACTERIZACIÓN
2.3.1 Espectroscopía Raman
La espectroscopia Raman13 estudia las transiciones entre niveles energéticos
vibracionales de la materia. Es una técnica óptica de alta resolución capaz de

Técnicas experimentales
63
proporcionar información química y estructural de casi cualquier material o
compuesto, ya sea orgánico o inorgánico, permitiendo así su identificación.
Sus principales ventajas radican en que no es necesario ningún tipo de
preparación especial del material a analizar y que además es una técnica no
destructiva, a excepción de que el material se pueda degradar con su
exposición a la luz.
El análisis mediante espectroscopía Raman se basa en el estudio de la luz
dispersada por un material al incidir sobre él un haz de luz monocromático de
frecuencia ν0. La mayor parte de la luz es dispersada de forma elástica,
presentando la misma frecuencia que la luz incidente y conocida como
dispersión Rayleigh, pero una pequeña porción del haz (del orden de 1 fotón
de cada 1011 incidentes) es dispersado inelásticamente experimentando
ligeros cambios de frecuencia, +νr y -νr, resultado de la interacción de la luz
con la materia. Estos cambios frecuencia son conocidos como
desplazamientos Raman que son característicos del material analizado, e
independientes de la frecuencia de la luz incidente.
Las variaciones de frecuencia observadas están asociadas a los continuos
movimientos vibracionales y rotacionales de los átomos en las moléculas o
redes cristalinas (fonones). A cada uno de estos movimientos le corresponde
un valor determinado de energía, representados en la Figura 2.11.

CÁPITULO 2
64
Figura 2.11. Diagrama energético en el que las líneas horizontales representan los
distintos niveles vibracionales y que muestra transiciones entre estados energéticos
para diferentes interacciones luz-materia.
En el diagrama se observa como el fotón incidente puede producir una
transición electrónica temporal a un nivel de energía excitado no permitido
(virtual). A través de su interacción con el medio, puede generar o absorber
un fonón de la red o de la molécula, y se relaja rápidamente para pasar a uno
de los niveles de energía permitidos emitiendo un fotón. Si el resultado de la
interacción fotón-materia es un fotón dispersado a la misma frecuencia que el
fotón incidente, se dice que el choque es elástico (dispersión Rayleigh). Por
otro lado, si el fotón dispersado tiene una frecuencia menor a la del incidente,
se produce una transferencia de energía del fotón al medio generando un
fonón, volviendo a un estado de energía permitido mayor al que tenía
inicialmente (dispersión Raman Stokes). Sin embargo, si el fotón dispersado
tiene una frecuencia mayor a la del fotón incidente, se produce una
transferencia de energía del medio al fotón mediante un fonón del medio, esto
significa que inicialmente se encontraba en un estado vibracional superior al
fundamental, volviendo a este tras el choque (dispersión Rama anti-Stokes).
Según la ley de distribución de Maxwell-Boltzman, a temperatura ambiente la
mayoría de las moléculas se encuentra en el estado vibracional de menor

Técnicas experimentales
65
energía, y por tanto la probabilidad de se den transiciones energéticas que
den lugar a la dispersión Raman Stokes es mucho mayor que la de dispersión
Raman anti-Stokes. Esto hace que habitualmente se trabaje midiendo sólo el
efecto Stokes, aunque por comodidad se sitúa el resultado en la parte positiva
del eje.
Debido a que el nivel excitado es virtual, su vida media es extremadamente
corta, y el proceso Raman es muy ineficiente. Una forma de aumentar su
eficiencia es utilizar una energía de excitación que coincida con una transición
electrónica real del material estudiado, lo que se denomina efecto Raman
resonante, aumentando varios órdenes de magnitud la eficiencia del proceso
y por tanto la sensibilidad de la técnica. Este efecto ocurre también en sólidos
cuando la energía del láser empleado es superior a la energía de la banda
prohibida (bandgap) del material analizado, aunque su eficiencia a una energía
de excitación dada depende de la estructura electrónica. Este es el caso tanto
del grafeno como de muchos TMDCs cuando se excita con láseres en el rango
del visible pues el grafeno es un semimetal y, en numerosos casos, los bandgap
de los TMDCs monocapa tienen energías inferiores al rojo (695, 673 nm para
el MoS2 y WS2 respectivamente).

CÁPITULO 2
66
Figura 2.12. Esquema del equipo Raman y de fotoluminiscencia empleado. La línea
azul indica la trayectoria del láser.
En la Figura 2.12 se presenta el equipo empleado para las medidas Raman.
Todos los experimentos han sido realizados a temperatura ambiente
empleando la línea en 488 nm de un láser de Ar+ con un rango de potencia
incidente entre 1 - 10 mW, que se reduce cuando es necesario mediante la
utilización de filtros de diferente densidad óptica (DO=0.4-3.0). El haz
incidente se focaliza con un microscopio Olympus BX60M, con varios objetivos
(x4, x20, x50 y x100). Con el objetivo x100 de alta apertura numérica NA =
0.95, la resolución espacial es inferior a 1 μm a 488 nm (según el límite de
difracción, es alrededor de 0.5 μm). El portamuestras se coloca sobre una
mesa monitorizada con una precisión de 10 nm/step en XY y de 2 nm/step en
z. El equipo cuenta con filtros holográficos “super-notch-plus” de marca Kaiser
para eliminar la señal Rayleigh. Finalmente, la luz dispersada se analiza
inicialmente con un monocromador Jobin-Yvon HR-460 y posteriormente con
un Horiba iHR-320 acoplados a un detector Synapse CCD refrigerado por
efecto Peltier (T = 70 ºC). El monocromador Horiba cuenta con tres redes de

Técnicas experimentales
67
difracción intercambiables de 600, 1200 y 1800 líneas/mm. A medida que
aumenta el número de líneas por milímetro la resolución aumenta en la misma
relación que la del número de líneas por milímetro y en consecuencia el rango
espectral que abarca la CCD disminuye.
2.3.2 Espectroscopia de absorción UV-Vis
Esta técnica espectroscópica consiste en hacer pasar un haz de luz
monocromática de intensidad 𝐼0(𝜆) a través de un material de grosor d.14 La
intensidad de la luz al atravesar la muestra decrece según la Ley de Lambert-
Beer:
𝐼(𝜆) = 𝐼0(𝜆)𝑒−𝛼𝑑 (Ec. 2.1)
Donde α es el coeficiente de absorción del material y d, la distancia que
recorre la luz atravesando la muestra. El coeficiente de absorción está
relacionado con la parte imaginaria del índice de refracción (κ) como:
𝛼(𝜆) = 4𝜋𝜅(𝜆)/𝜆 (Ec. 2.2)
Una de las magnitudes de medida comúnmente utilizadas es la transmitancia
(T):
𝑇(𝜆) =𝐼𝑇𝐼0
(Ec. 2.3)
Donde 𝐼𝑇 es la intensidad transmitida. La absorbancia o densidad óptica (OD,
absorbancia por unidad de longitud) es otra de las magnitudes de medida
utilizadas, y está relacionada con la transmitancia como:
𝑂𝐷(𝜆) = log (1
𝑇(𝜆)) (Ec. 2.4)

CÁPITULO 2
68
Despreciando las pérdidas debidas a las reflexiones en las intercaras, la OD
medida está directamente relacionada con el coeficiente de absorción:
𝛼(𝜆) = 2.3𝑂𝐷(𝜆)
𝑑 (Ec. 2.5)
Esta relación permite obtener información relevante del material a partir de
su densidad óptica, como su estructura electrónica, si las transiciones
involucradas están en el rango visible-UV del espectro electromagnético.
El equipo empleado es un espectrofotómetro Varian modelo Cary 4000, que
permite realizar medidas de absorción tanto en sólidos como en líquidos,
obteniendo espectros con una alta resolución y muy buena relación señal-
ruido en el rango espectral de 180 - 900 nm. Es un espectrofotómetro UV-Vis
de doble haz, equipado con dos lámparas, una de deuterio (185 - 340 nm) y
otra halógena (340 - 780 nm), con un monocromador Littrow doble y un
detector fotomultiplicador R928 que abarca toda la región UV-Vis.
Para las medidas de microrreflexión se utilizaron una lámpara de luz blanca,
un microscopio Olympus (objetivo x100) y un espectrógrafo con una rejilla de
150 l/mm acoplado a una cámara CCD refrigerada. Se utilizó un pin-hole de
500 nm para el filtrado espacial. En la Figura 2.13 se muestra una imagen del
equipo de microrreflexión. En todos los experimentos se mide la reflectancia
del sustrato en una posición cercana a la de la muestra y se utiliza para obtener
la reflectancia de las películas: R(muestra) = Rmedida /Rsustrato.

Técnicas experimentales
69
Figura 2.13. Equipo empleado en las medidas de microrreflexión/microreflectancia.
La línea amarilla representa el camino del haz de luz reflejado.
2.3.3 Fotoluminiscencia
La técnica de fotoluminiscencia (PL) consiste en excitar la muestra en una de
sus bandas de absorción y detectar la intensidad de luz emitida por el material
a causa de la desexcitación. La representación de la intensidad de luz emitida
en función de la longitud de onda proporciona el espectro de emisión para una
longitud de onda de excitación fija. La fotoluminiscencia estudia diversas vías
de recombinación de los pares electrón-hueco fotoexcitados. Es una técnica
de caracterización no destructiva utilizada para estudiar propiedades
intrínsecas y extrínsecas de los semiconductores. Mediante el análisis del
espectro de fotoluminiscencia en función de diversos parámetros
(temperatura, energía de excitación, etc.) es posible obtener información
general sobre la calidad del material.

CÁPITULO 2
70
En las medidas de fotoluminiscencia de las muestras de MoS2-2D, el sistema
de medida empleado es el mismo sistema que para el Raman. Se usa en
general la red de 600 l/mm ya que no suele ser necesaria una mayor
resolución.
Para realizar las medidas de epifluorescencia de las muestras de grafeno
funcionalizado con anticuerpos se excitó con la línea de 488 nm del láser Ar+
desenfocado sobre las muestras y se recogió la emisión como imágenes de
fluorescencia a través del microscopio. Se utilizó un filtro de paso alto de 500
nm (permite pasar las longitudes de onda por encima de 500 nm) para eliminar
la señal proveniente del láser.
2.3.4 Espectroscopia fotoelectrónica de rayos X
La espectroscopia de fotoelectrones emitidos por rayos X (XPS, X-ray
Photoelectron Spectroscopy), es una técnica de caracterización de la
superficie que proporciona información cualitativa y cuantitativa de los
elementos presentes en la muestra (a excepción del hidrógeno) hasta unos 1-
2 nm de profundidad (para las energías habituales de los rayos X utilizados en
equipos de laboratorio).15 Se basa en el efecto fotoeléctrico, excitando los
niveles internos de los átomos mediante un haz de rayos-X se provoca la
emisión de fotoelectrones que proporcionan información sobre la energía de
cada nivel y, por tanto, sobre la naturaleza de cada átomo emisor. Midiendo
la energía cinética de los electrones emitidos (EK) es posible determinar la
energía de enlace del orbital atómico del que proviene el electrón (El)
aplicando la ecuación:
𝐸𝑙 = ℎ𝜈 − 𝐸𝑘 − 𝜙 (Ec. 2.6)
Donde hν es la energía de la radiación incidente y φ es la función de trabajo,
o lo que es lo mismo, la energía mínima necesaria para que un electrón pueda

Técnicas experimentales
71
salir al exterior del sólido. En general, es necesario trabajar en condiciones de
ultra alto vacío debido a que los fotoelectrones han de viajar desde la muestra
hasta el detector sin colisionar con ningún átomo.
En este trabajo se ha empleado esta técnica para analizar el contenido de los
distintos grupos funcionales oxigenados (carboxílicos, hidroxilos, carbonilos y
epoxi) presentes en las muestras de grafeno funcionalizado.
2.3.5 Microscopía de fuerza atómica
La Microscopía de Fuerza Atómica (AFM) basa su funcionamiento en la
interacción atractiva-repulsiva entre los átomos de una sonda, una punta muy
afilada, y los átomos de la superficie de la muestra a estudiar. Esta interacción
se controla ajustando la posición relativa entre la punta y la muestra. Las
fuerzas entre la punta y la superficie estudiada pueden ser de corto (repulsiva)
y largo (atractiva) alcance y se dan en todos los materiales, por lo que el AFM
es aplicable a una gran variedad de materiales, ya sean conductores o
aislantes. Además, estas medidas pueden llevarse a cabo bajo diversas
condiciones (ambiente seco o líquido) y sin la necesidad de una preparación
previa especial de las muestras. Por otro lado, una de las principales
desventajas que presenta el AFM es su baja resolución lateral debido al
tamaño de la punta (15 nm para puntas estándar), sin embargo, presenta
una excelente resolución en altura ( 0.1 nm).

CÁPITULO 2
72
Figura 2.14. (a) Esquema del sistema de detección óptico por desviación del
cantiléver, (b) Microscopio empleado en las medidas de AFM.
Para cuantificar y controlar la magnitud de las fuerzas de interacción entre
muestra y punta, esta última se coloca en el extremo de un fleje, denominado
cantiléver. Las fuerzas generadas sobre la punta modifican el comportamiento
del cantiléver produciendo variaciones de la deflexión, amplitud de oscilación,
etc. De este modo se pueden obtener imágenes topográficas de la muestra,
así como mapas superficiales de propiedades como la hidrofobicidad, la
fricción o la adhesión en función del tipo de interacción que se mida.
El modo de medida en un microscopio de fuerza atómica consiste en que una
luz láser incide sobre el cantiléver por la cara opuesta a la de la punta, y es
reflejada al centro de un detector fotodiodo dividido en cuatro secciones. Los
cambios en la superficie de la muestra producen doblamientos del cantiléver,
que provocan cambios en el ángulo de la reflexión del láser en el fotodiodo, lo
cual se traduce en variaciones en la intensidad de la luz del láser reflejada. Esta
diferencia de intensidad se transforma en una señal eléctrica con la que se
pueden cuantificar tanto los movimientos verticales de la punta debidos a las
fuerzas de atracción o de repulsión, como los movimientos laterales y de

Técnicas experimentales
73
torsión del cantiléver provocados por la interacción entre la punta y la
superficie de la muestra (Figura 2.14a).
El microscopio de fuerza atómica puede operar en varios modos según la
distancia entre la punta y la muestra:
- Modo contacto. La distancia entre la punta y la muestra es de unos
pocos angstroms (Å) y las fuerzas que actúan son de repulsión
electrostática. En este modo pueden obtenerse imágenes de altura
(topografía de la muestra), o imágenes de desviación del cantilever,
que muestran con mayor sensibilidad los detalles de la superficie.
- Modo dinámico. La distancia es relativamente mayor (10-100 Å) y las
fuerzas interatómicas son de naturaleza atractiva como resultado de
las fuerzas de van der Waals de largo alcance. En este caso las
imágenes obtenidas pueden ser de altura; de amplitud, donde se
acentúan finas estructuras de la superficie a expensas de la
información general de la topografía; o de fase, que proporcionan
información sobre la composición de la muestra, así como de sus
propiedades elásticas, viscoelásticas y de adhesión.
Para las medidas de AFM se ha utilizado un sistema AFM de Nanotec
Electronica S.L. Se empleó el modo de operación dinámico, utilizando la
amplitud de oscilación como señal de control. Se utilizaron puntas comerciales
(Nanosensors PPP-NCH-w) con una constante de fuerza nominal de 34 N/m y
una frecuencia de resonancia de 270 kHz. Para el análisis de las imágenes se
utilizaron los software WSxM16 y Gwyddion.17

CÁPITULO 2
74
2.3.6 Medidas eléctricas
El método utilizado comúnmente para medir las propiedades de transporte de
una lámina delgada es el método de Van der Pauw.18 Con este método se
pueden obtener propiedades como la resistividad del material, el tipo de
dopado, la densidad de portadores del portador mayoritario, y su movilidad.
Figura 2.15. Diagrama de las medidas de a) Resistencia para cálculo de la resistividad
y b) Coeficiente Hall para cálculo de densidad de portadores mayoritarios y
movilidad.
Para poder emplear este método la muestra debe ser homogénea e isotrópica,
plana y de grosor uniforme, no debe tener agujeros aislados, los cuatro
electrodos deben estar situados en el borde de la muestra y sus áreas de
contacto deben ser al menos un orden de magnitud menor que el área total
de la muestra.
Resistividad
La resistividad de una muestra viene dada por la siguiente expresión:
𝜌 = 𝑅𝑠 · 𝑑 (Ec. 2.7)
Donde Rs es la resistencia laminar, definida a continuación, y d es el grosor de
la muestra (que debe ser caracterizado independientemente por otra técnica).
Van der Pauw muestra que la resistencia laminar, Rs, puede ser determinada a
partir de dos resistencias (Ra y Rb) siguiendo la siguiente ecuación:

Técnicas experimentales
75
𝑅𝑠 =𝜋
𝑙𝑛2·𝑅𝑎+𝑅𝑏2
· 𝑓 (Ec. 2.8)
Donde f es un parámetro cuyo valor viene dado por el cociente Ra/Rb y que se
encuentra entre 0.8 y 1. Las medidas de resistencia se realizan haciendo pasar
una corriente eléctrica a lo largo de un borde de la muestra (I12) y midiendo el
voltaje a través del borde opuesto (V34). Es necesario medir una resistencia a
lo largo del borde vertical (Ra) y otra a lo largo del borde horizontal (Rb) (Figura
2.15a).
Movilidad
Las medidas de efecto Hall permiten obtener el número de portadores
mayoritarios presentes en la muestra y su movilidad.19 Para ello, mientras se
aplica un campo magnético (B) perpendicular a la superficie, se hace pasar una
corriente a través de dos contactos con disposición diagonal (I13) y se mide el
voltaje en los contactos opuestos (V24) (Figura 2.15b). De este modo se calcula
la densidad de portadores mayoritarios (N), que sigue la expresión:
𝑁 =𝐼 · Δ𝐵
Δ𝑉𝐻 · 𝑑 · 𝑒 (Ec. 2.9)
Donde I es la intensidad de corriente aplicada, ΔB/ΔVH es el cociente entre el
campo magnético aplicado y el incremento de voltaje obtenido, d es el grosor
de la muestra y e la carga del electrón.
Con estos valores es posible calcular la movilidad de los portadores
mayoritarios (μ) de la muestra:
𝜇 = (𝑒 · 𝜌 · 𝑁)−1 (Ec. 2.10)
Donde e es la carga del electrón, ρ es la resistividad del material y N la
densidad de portadores mayoritarios.

CÁPITULO 2
76
En este trabajo se han medido las propiedades eléctricas de las muestras de
grafeno y grafeno funcionalizado transferidas sobre sustratos aislantes. Las
medidas eléctricas se hicieron usando una fuente de intensidad de corriente
(Keithley 2400) y un multímetro de alta precisión para la medida de voltaje
(Keithley 1700). Se colocaron los electrodos con pintura de plata de acuerdo
con la geometría de Van der Pauw para determinar la resistencia y la
conductividad laminares de las muestras. Se realizaron mediciones de efecto
Hall empleando un electroimán con el campo magnético perpendicular
(variable hasta 0.8 T) a la muestra para obtener la densidad y la movilidad de
los portadores de carga mayoritarios.
2.4 REFERENCIAS
1. Albella, J. M., Sánchez, O. & Jiménez, I. Láminas Delgadas y
Recubrimientos. Preparación, propiedades y aplicaciones. (2003).
2. Vescan, L. Chemical Vapour Deposition. in Handbook of thin films
process technology (eds. Glocker, D. A. & Shah, S. I.) (The Institute of
Physics, 1995).
3. Zeng, Y. et al. Highly Enhanced Photoluminescence of Monolayer MoS2
with Self-Assembled Au Nanoparticle Arrays. Adv. Mater. Interfaces 4,
1–7 (2017).
4. Deb, S., Chakrabarti, P., Mohapatra, P. K., Barick, B. K. & Dhar, S.
Tailoring of defect luminescence in CVD grown monolayer MoS 2 film.
Appl. Surf. Sci. 445, 542–547 (2018).
5. Ozden, A., Ay, F., Sevik, C. & Perkgöz, N. K. CVD growth of monolayer
MoS2: Role of growth zone configuration and precursors ratio. Jpn. J.
Appl. Phys. 56, (2017).

Técnicas experimentales
77
6. Nemanich, R. J., Lucovsky, G. & Solin, S. A. Infrared active optical
vibrations of graphite. Solid State Commun. 23, 117–120 (1977).
7. Catalán-Gómez, S. et al. Photoluminescence enhancement of
monolayer MoS2 using plasmonic gallium nanoparticles. Nanoscale
Adv. 1, 884–893 (2019).
8. Kwon, H. et al. Monolayer MoS2 field-effect transistors patterned by
photolithography for active matrix pixels in organic light-emitting
diodes. npj 2D Mater. Appl. 3, 1–9 (2019).
9. Rich, D. H. & Singh, J. The carbodiimide method" The peptides. Analysis,
synthesis, biology. (1979).
10. Sehgal, D. & Vijay, I. K. A method for the high efficiency of water-
soluble carbodiimide-mediated amidation. Anal. Biochem. 218, 87–91
(1994).
11. Shah, S. I. & Glocker, D. A. Sputtering. in Handbook of thin films process
technology (eds. Glocker, D. A. & Shah, S. I.) (The Institute of Physics,
1995).
12. Haberland, H., Karrais, M. & Mall, M. A new type of cluster and cluster
ion source. Zeitschrift für Phys. D Atoms, Mol. Clust. 20, 413–415
(1991).
13. Schrader, B. & Bougeard, D. Infrared and Raman spectroscopy :
methods and applications. (VCH, 1995).
14. Andrés, A. de, Jiménez-Villacorta, F. & Prieto, C. The Compromise
Between Conductivity and Transparency. in Transparent Conductive
Materials 1–30 (John Wiley & Sons, Ltd, 2018).
doi:10.1002/9783527804603.CH1.
15. Wagner, C. D., Riggs, W. M., Davis, L. E. & Moulder, J. F. Handbook of
X-ray Photoelectron Spectroscopy. Surface and Interface Analysis
(Wiley, 1979). doi:10.1002/sia.740030412.
16. Horcas, I. et al. WSXM: A software for scanning probe microscopy and
a tool for nanotechnology. Rev. Sci. Instrum. 78, (2007).

CÁPITULO 2
78
17. Nečas, D. & Klapetek, P. Gwyddion: An open-source software for SPM
data analysis. Central European Journal of Physics vol. 10 181–188
(2012).
18. van der PAUW, L. J. A METHOD OF MEASURING SPECIFIC RESISTIVITY
AND HALL EFFECT OF DISCS OF ARBITRARY SHAPE. in Semiconductor
Devices: Pioneering Papers 174–182 (WORLD SCIENTIFIC, 1991).
doi:10.1142/9789814503464_0017.
19. van der PAUW, L. J. A method of measuring the resistivity and Hall
coefficient on lamellae of arbitrary shape. Philips Tech. Rev. 20, 220–
224 (1958).

79
3 MOS2 BIDIMENSIONAL
En este capítulo se exponen los resultados más relevantes de las muestras
obtenidas en los equipos diseñados e instalados en el laboratorio de
preparación de láminas delgadas del ICMM-CSIC descritos en el Capítulo 2, así
como de las muestras obtenidas durante mi estancia en el laboratorio del Prof.
Patrick Kung (Universidad de Alabama).
En la primera parte (3.1) se analizan los factores que influyen en el crecimiento
del MoS2 bidimensional (MoS2-2D). Este estudio se ha realizado para MoS2
crecido en los equipamientos experimentales del ICMM: CVD (partiendo de
MoO3 y azufre) y PVT (partiendo de MoS2 en polvo) sobre distintos sustratos.
En la segunda parte (3.2) se analiza en detalle las variaciones en los espectros
Raman, focalizando en aspectos aún no explorados, como los modos
prohibidos de MoS2 en función del número de capas, del sustrato empleado
para el crecimiento y del método de obtención: exfoliación mecánica (ME) y
distintos métodos de crecimiento por fase vapor (PVT y CVD).
La tercera parte (3.3) se dedica al estudio de nanocopos de MoS2 crecidos por
PVT y CVD. Mediante estas técnicas de depósito se han obtenido copos de
MoS2, monocapa y de pocas capas, con tamaños laterales que varían desde
unos 10 nm hasta decenas de micras. Los nanocopos de MoS2 son un campo
de reciente interés con posibles aplicaciones de detección, electrocatálisis o
bio-imagen,1 por lo que se ha estudiado el efecto del tamaño de los copos en
las características del MoS2-2D en función del número de capas mediante
espectroscopías Raman y de fotoluminiscencia.
En la cuarta parte (3.4) se demuestra el efecto de amplificación de las señales
Raman y de fotoluminiscencia de los copos de MoS2 por efecto SERS mediante

CÁPITULO 3
80
la formación de nanopartículas de MoO2 en el proceso de crecimiento por
CVD, así como el efecto del oxígeno adsorbido en el MoS2.
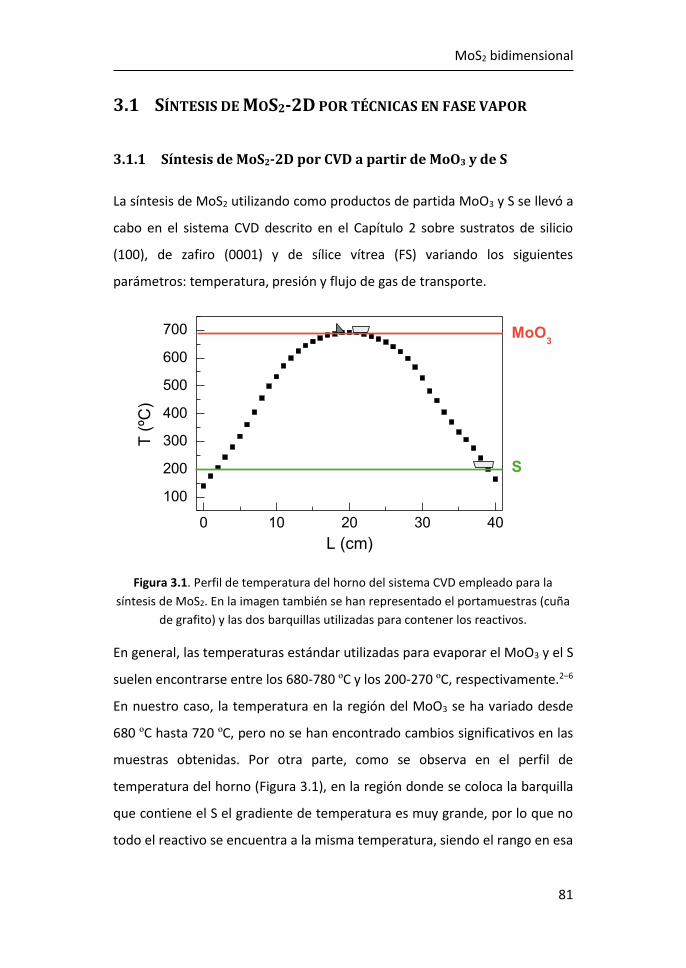
MoS2 bidimensional
81
3.1 SÍNTESIS DE MOS2-2D POR TÉCNICAS EN FASE VAPOR
3.1.1 Síntesis de MoS2-2D por CVD a partir de MoO3 y de S
La síntesis de MoS2 utilizando como productos de partida MoO3 y S se llevó a
cabo en el sistema CVD descrito en el Capítulo 2 sobre sustratos de silicio
(100), de zafiro (0001) y de sílice vítrea (FS) variando los siguientes
parámetros: temperatura, presión y flujo de gas de transporte.
Figura 3.1. Perfil de temperatura del horno del sistema CVD empleado para la
síntesis de MoS2. En la imagen también se han representado el portamuestras (cuña
de grafito) y las dos barquillas utilizadas para contener los reactivos.
En general, las temperaturas estándar utilizadas para evaporar el MoO3 y el S
suelen encontrarse entre los 680-780 ºC y los 200-270 ºC, respectivamente.2–6
En nuestro caso, la temperatura en la región del MoO3 se ha variado desde
680 ºC hasta 720 ºC, pero no se han encontrado cambios significativos en las
muestras obtenidas. Por otra parte, como se observa en el perfil de
temperatura del horno (Figura 3.1), en la región donde se coloca la barquilla
que contiene el S el gradiente de temperatura es muy grande, por lo que no
todo el reactivo se encuentra a la misma temperatura, siendo el rango en esa
0 10 20 30 40
100
200
300
400
500
600
700 MoO3
T (º
C)
L (cm)
S

CÁPITULO 3
82
zona de 200-250 ºC. Más adelante se discutirá sobre los posibles efectos de
este gradiente.
La mayoría de las muestras crecidas por este método presentan copos
triangulares de MoS2 de tamaño de entre 100-500 nm aunque en algunos
casos, se han obtenido nanocopos en torno a 10 nm.
A continuación, se presentan los resultados más relevantes de las muestras de
MoS2 crecidas sobre sustratos de zafiro variando distintos parámetros. Para
comparar las intensidades Raman todos los espectros que se presentan han
sido corregidos por la intensidad y posición de un silicio de referencia.
Efecto de la presión
En la técnica CVD, las presiones bajas favorecen el crecimiento bidimensional,
sin embargo, en este trabajo se ha comprobado que a presiones de
aproximadamente 0.01 mbar los copos de MoS2 crecen incompletos y rotos,
mientras que, para presiones más altas, de aproximadamente 1 mbar, los
copos formados presentan una estructura triangular completa.
Figura 3.2. Imágenes de topografía medidas por AFM de a) una muestra de MoS2
crecida a 10-2 mbar y b) crecida a 1 mbar.
La Figura 3.2 muestra imágenes de AFM de dos muestras crecidas con
diferentes presiones. En la Figura 3.2a, se pueden distinguir fácilmente las
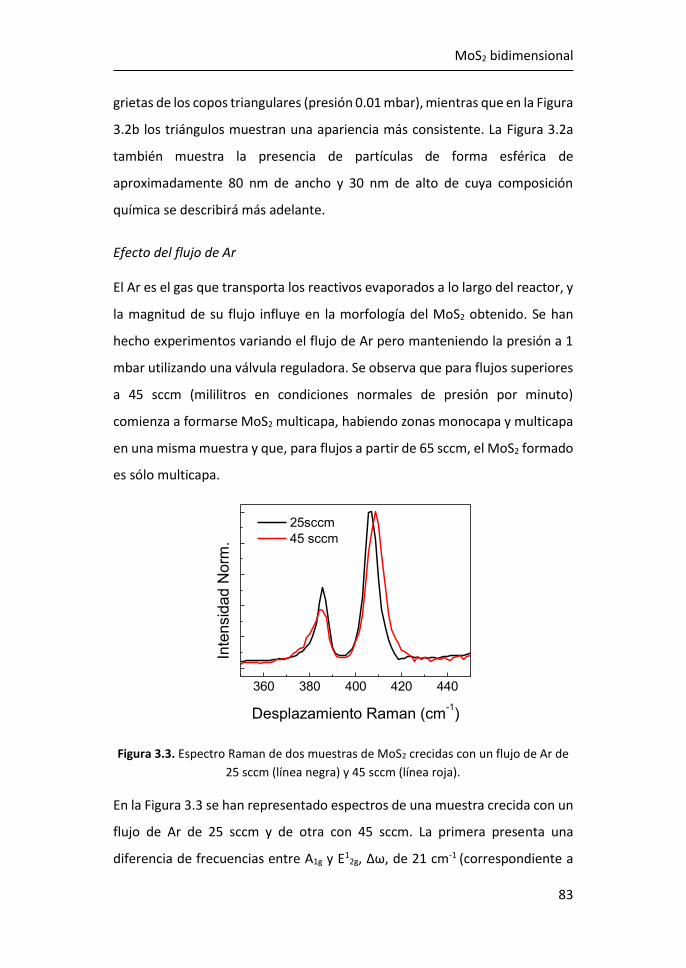
MoS2 bidimensional
83
grietas de los copos triangulares (presión 0.01 mbar), mientras que en la Figura
3.2b los triángulos muestran una apariencia más consistente. La Figura 3.2a
también muestra la presencia de partículas de forma esférica de
aproximadamente 80 nm de ancho y 30 nm de alto de cuya composición
química se describirá más adelante.
Efecto del flujo de Ar
El Ar es el gas que transporta los reactivos evaporados a lo largo del reactor, y
la magnitud de su flujo influye en la morfología del MoS2 obtenido. Se han
hecho experimentos variando el flujo de Ar pero manteniendo la presión a 1
mbar utilizando una válvula reguladora. Se observa que para flujos superiores
a 45 sccm (mililitros en condiciones normales de presión por minuto)
comienza a formarse MoS2 multicapa, habiendo zonas monocapa y multicapa
en una misma muestra y que, para flujos a partir de 65 sccm, el MoS2 formado
es sólo multicapa.
Figura 3.3. Espectro Raman de dos muestras de MoS2 crecidas con un flujo de Ar de
25 sccm (línea negra) y 45 sccm (línea roja).
En la Figura 3.3 se han representado espectros de una muestra crecida con un
flujo de Ar de 25 sccm y de otra con 45 sccm. La primera presenta una
diferencia de frecuencias entre A1g y E12g, Δω, de 21 cm-1 (correspondiente a
360 380 400 420 440
Inte
nsid
ad N
orm
.
Desplazamiento Raman (cm-1)
25sccm 45 sccm

CÁPITULO 3
84
una capa), mientras que en la segunda Δω es aproximadamente 23 cm-1
(correspondiente a 2 ó 3 capas).
Formación de nanopartículas de MoO2
Haciendo referencia a las nanopartículas que se observan en la Figura 3.2a, en
un principio su composición es desconocida, ya que en los espectros Raman
no se detectaban más señales que las del MoS2 y el sustrato empleado. Se
intentó minimizar la densidad de impurezas aumentando la distancia entre el
MoO3 y los sustratos (Figura 3.4a, distancia 1 cm y Figura 3.4b, distancia 5 cm),
consiguiendo buenos resultados en los primeros experimentos. Sin embargo,
con el crecimiento consecutivo de muestras la densidad de impurezas fue
aumentando, hasta llegar a detectarse picos Raman característicos de MoO2
en algunas de ellas, además de cambiar en gran medida su aspecto en el
microscopio óptico. En la Figura 3.4 se ven las imágenes ópticas de la cuarta
(Figura 3.4c), de la décima (Figura 3.4d) y de la decimoquinta (Figura 3.4e)
muestra crecida, quedando reflejado el aumento de la densidad de partículas
con el crecimiento consecutivo de muestras. Se comprobó que es posible
frenar esta tendencia limpiando el horno mediante un tratamiento a vacío a
750 ºC durante 60 min.
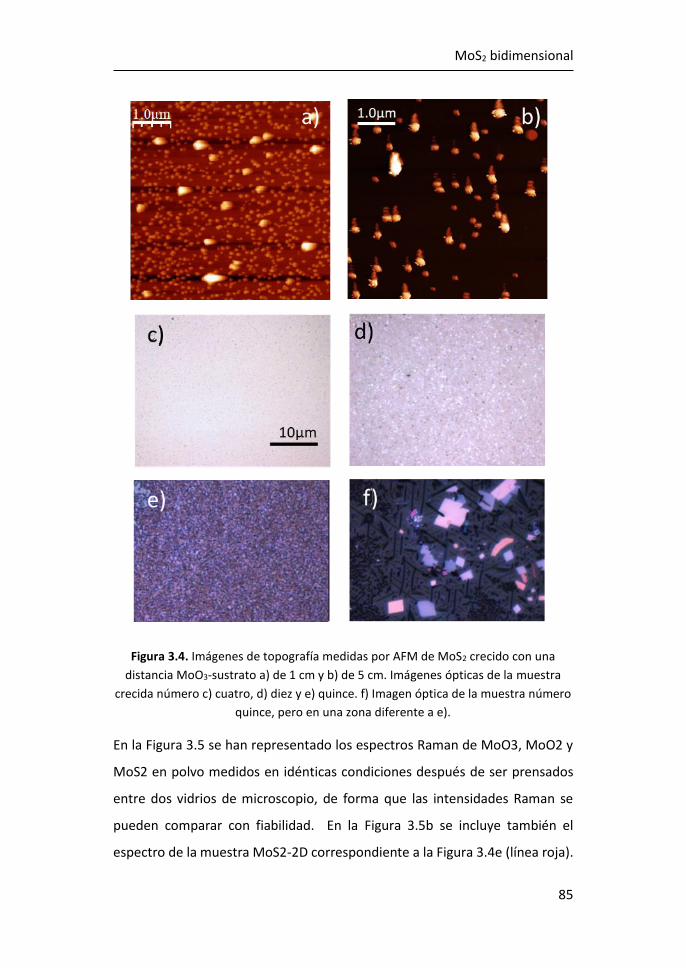
MoS2 bidimensional
85
Figura 3.4. Imágenes de topografía medidas por AFM de MoS2 crecido con una
distancia MoO3-sustrato a) de 1 cm y b) de 5 cm. Imágenes ópticas de la muestra
crecida número c) cuatro, d) diez y e) quince. f) Imagen óptica de la muestra número
quince, pero en una zona diferente a e).
En la Figura 3.5 se han representado los espectros Raman de MoO3, MoO2 y
MoS2 en polvo medidos en idénticas condiciones después de ser prensados
entre dos vidrios de microscopio, de forma que las intensidades Raman se
pueden comparar con fiabilidad. En la Figura 3.5b se incluye también el
espectro de la muestra MoS2-2D correspondiente a la Figura 3.4e (línea roja).
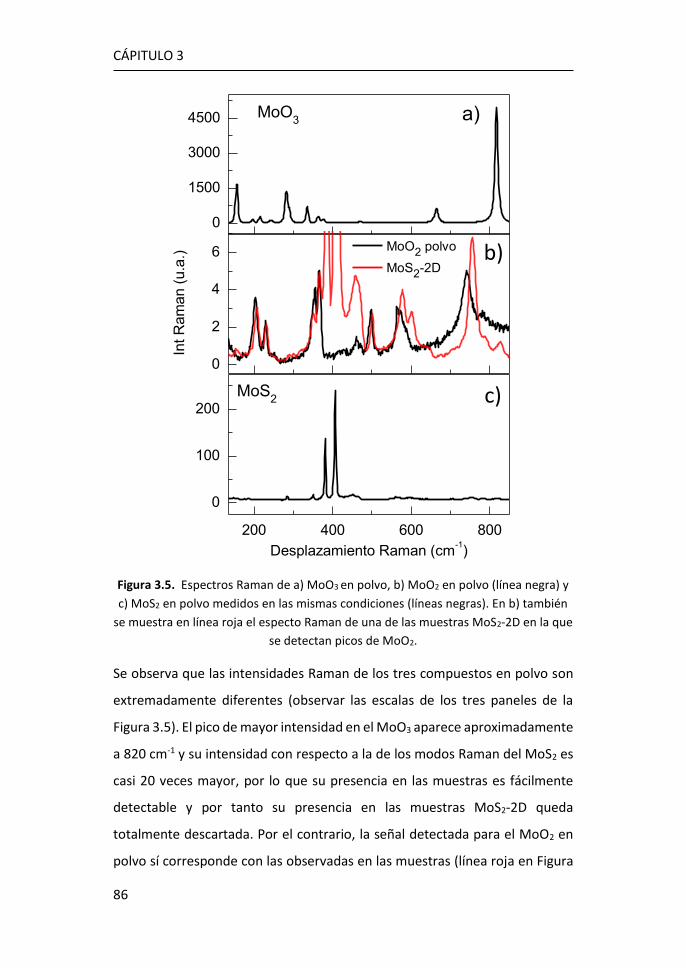
CÁPITULO 3
86
Figura 3.5. Espectros Raman de a) MoO3 en polvo, b) MoO2 en polvo (línea negra) y
c) MoS2 en polvo medidos en las mismas condiciones (líneas negras). En b) también
se muestra en línea roja el especto Raman de una de las muestras MoS2-2D en la que
se detectan picos de MoO2.
Se observa que las intensidades Raman de los tres compuestos en polvo son
extremadamente diferentes (observar las escalas de los tres paneles de la
Figura 3.5). El pico de mayor intensidad en el MoO3 aparece aproximadamente
a 820 cm-1 y su intensidad con respecto a la de los modos Raman del MoS2 es
casi 20 veces mayor, por lo que su presencia en las muestras es fácilmente
detectable y por tanto su presencia en las muestras MoS2-2D queda
totalmente descartada. Por el contrario, la señal detectada para el MoO2 en
polvo sí corresponde con las observadas en las muestras (línea roja en Figura
0
1500
3000
4500 a)MoO3
b)
c)
200 400 600 800
0
100
200
0
2
4
6
Desplazamiento Raman (cm-1)
MoS2
MoO2 polvo MoS2-2D
Int R
aman
(u.a
.)

MoS2 bidimensional
87
3.5b). Las frecuencias de los modos Raman del MoO2 en polvo corresponden
bien con los datos publicados7: 203, 228, 363 y 495 cm-1 aunque con un
desplazamiento sistemático importante a mayores frecuencias en las
muestras crecidas por CVD (207, 230, 366 y 500 cm-1) mostrando una fuerte
compresión de la red cristalina en las nanopartículas tal vez asociada a una
estequiometría defectuosa. Por otra parte, la intensidad Raman del MoO2 en
comparación con la del MoS2 (ambos en polvo) es del orden de 50 veces
menor, por lo que, las impurezas presentes en el resto de las muestras MoS2-
2D muy probablemente también correspondan a nanopartículas de MoO2 y
que, dada su baja señal Raman no se detecten en los espectros.
Se ha reportado la formación de nanoislas de MoO2 con forma romboide por
CVD a partir de MoO3 y S,8 los mismos reactivos que los utilizados
normalmente para la síntesis de MoS2,2–6 pero calentando el azufre a una
temperatura inferior a la estándar (normalmente 200-270 ºC). La altura de las
islas depende de dicha temperatura, siendo de 20 y 9 nm para temperaturas
120 ºC y 90 ºC, respectivamente.8 La forma romboide de estas islas es similar
a la forma de los cristales que manifiestan algunas muestras (Figura 3.4f). En
los experimentos llevados a cabo en nuestro laboratorio la zona del horno en
la que se coloca el azufre presenta un importante gradiente de temperatura
(Figura 3.1), dándose la posibilidad de que parte del azufre se caliente a
temperaturas menores a la deseada, y dando lugar al depósito de MoO2 en las
muestras. En nuestro caso, los cristales tienen una altura de
aproximadamente 35 nm y sobre éstos crecen copos triangulares de
MoS2monocapa (Figura 3.6).

CÁPITULO 3
88
Figura 3.6. a) Imagen de topografía medida por AFM de uno de los cristales de MoO2
presentes en las muestras de MoS2 crecido por CVD y b) perfiles del cristal y de los
copos de MoS2 que crecen sobre el cristal.
En todos los experimentos de crecimiento de MoS2 por CVD se utilizaron
sustratos de zafiro, silicio y sílice vítrea a la vez. Sin embargo, a pesar de las
variaciones de los parámetros que se fueron haciendo, el MoS2 crecido sobre
los sustratos de silicio y de sílice vítrea es siempre multicapa. En la Figura 3.7
se han representado espectros Raman y de fotoluminiscencia de MoS2-2D
crecido sobre FS (línea roja) y sobre Si (línea negra). También se han
representado los espectros de una de las muestras de MoS2 monocapa crecida
sobre zafiro (línea azul). Se observa que la diferencia entre la energía de los
fonones A1g y E12g (Δω) es mayor en las muestras sobre Si y FS. El MoS2 crecido
sobre FS no presenta fotoluminiscencia y el crecido sobre Si presenta una
señal muy débil que podría asociarse con MoS2 de aproximadamente 3-4
capas.
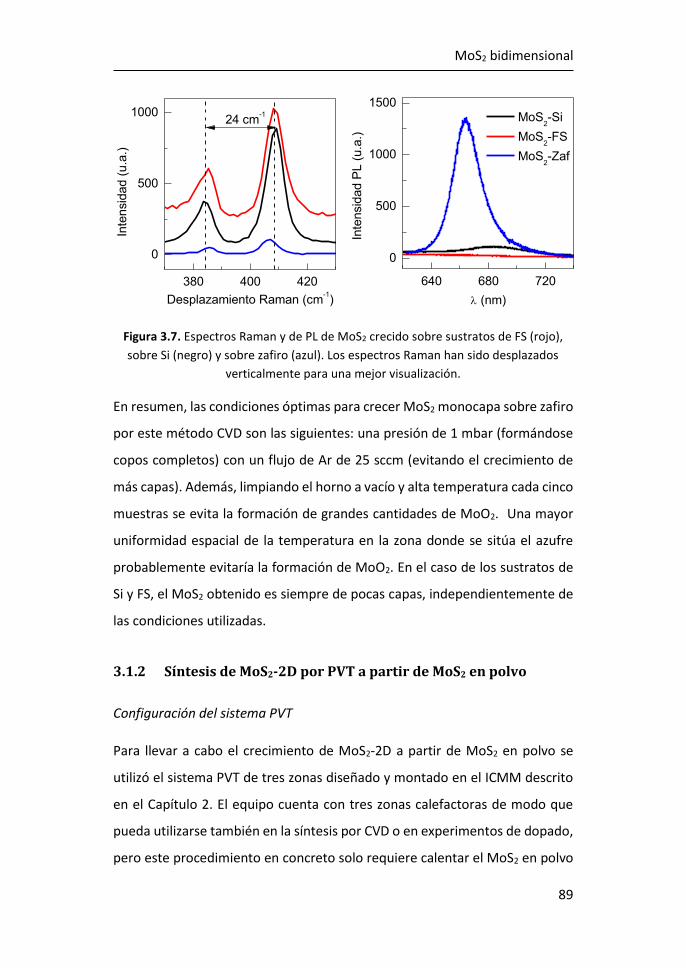
MoS2 bidimensional
89
Figura 3.7. Espectros Raman y de PL de MoS2 crecido sobre sustratos de FS (rojo),
sobre Si (negro) y sobre zafiro (azul). Los espectros Raman han sido desplazados
verticalmente para una mejor visualización.
En resumen, las condiciones óptimas para crecer MoS2 monocapa sobre zafiro
por este método CVD son las siguientes: una presión de 1 mbar (formándose
copos completos) con un flujo de Ar de 25 sccm (evitando el crecimiento de
más capas). Además, limpiando el horno a vacío y alta temperatura cada cinco
muestras se evita la formación de grandes cantidades de MoO2. Una mayor
uniformidad espacial de la temperatura en la zona donde se sitúa el azufre
probablemente evitaría la formación de MoO2. En el caso de los sustratos de
Si y FS, el MoS2 obtenido es siempre de pocas capas, independientemente de
las condiciones utilizadas.
3.1.2 Síntesis de MoS2-2D por PVT a partir de MoS2 en polvo
Configuración del sistema PVT
Para llevar a cabo el crecimiento de MoS2-2D a partir de MoS2 en polvo se
utilizó el sistema PVT de tres zonas diseñado y montado en el ICMM descrito
en el Capítulo 2. El equipo cuenta con tres zonas calefactoras de modo que
pueda utilizarse también en la síntesis por CVD o en experimentos de dopado,
pero este procedimiento en concreto solo requiere calentar el MoS2 en polvo
380 400 420
0
500
1000In
tens
idad
(u.a
.)
Desplazamiento Raman (cm-1)
24 cm-1
640 680 720
0
500
1000
1500
Inte
nsid
ad P
L (u
.a.)
(nm)
MoS2-Si MoS2-FS MoS2-Zaf
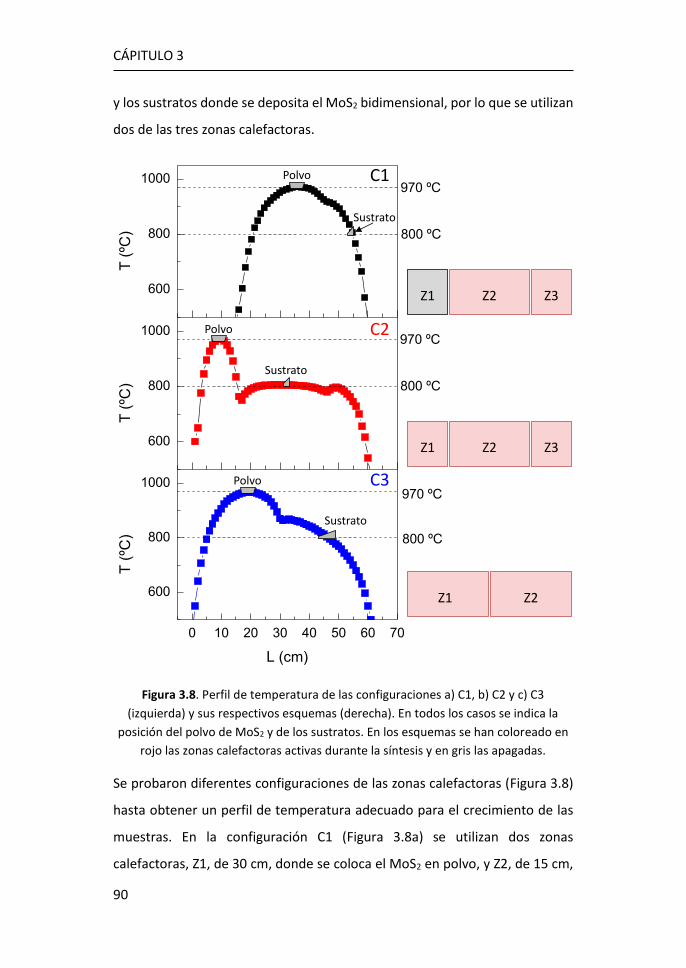
CÁPITULO 3
90
y los sustratos donde se deposita el MoS2 bidimensional, por lo que se utilizan
dos de las tres zonas calefactoras.
Figura 3.8. Perfil de temperatura de las configuraciones a) C1, b) C2 y c) C3
(izquierda) y sus respectivos esquemas (derecha). En todos los casos se indica la
posición del polvo de MoS2 y de los sustratos. En los esquemas se han coloreado en
rojo las zonas calefactoras activas durante la síntesis y en gris las apagadas.
Se probaron diferentes configuraciones de las zonas calefactoras (Figura 3.8)
hasta obtener un perfil de temperatura adecuado para el crecimiento de las
muestras. En la configuración C1 (Figura 3.8a) se utilizan dos zonas
calefactoras, Z1, de 30 cm, donde se coloca el MoS2 en polvo, y Z2, de 15 cm,
600
800
1000
600
800
1000
0 10 20 30 40 50 60 70
600
800
1000
T (º
C)
970 ºC
800 ºC
800 ºC
970 ºC
970 ºC
800 ºC
T (º
C)
T (º
C)
L (cm)
C1
C2
C3
Polvo
Polvo
Polvo
Sustrato
Sustrato
Sustrato
Z1 Z3Z2
Z1 Z2
Z1 Z3Z2

MoS2 bidimensional
91
donde se sitúan los sustratos. En esta configuración, aunque la temperatura
de la posición central de Z2 se controla con precisión, no se consigue una zona
lo suficientemente amplia donde la temperatura sea constante. Tener el
gradiente de temperatura reproducido en la Figura 3.8a, hace difícil saber cuál
es la temperatura a la que se encuentran los sustratos y, por tanto, saber qué
efecto está teniendo en las muestras obtenidas. Por ello, en la configuración
C2 se opta por usar tres zonas calefactoras, situando el MoS2 en polvo en Z1
(15 cm) a 970 ºC y el portamuestras con los sustratos en la zona central de Z2
(30 cm), calentando también Z3 para hacer que Z2 tenga una zona lo más
amplia posible donde la temperatura sea constante. Para esta configuración,
en la intersección entre Z1 y Z2 la temperatura desciende en gran medida
(Figura 3.8b), depositándose en esa zona la mayor parte del MoS2 evaporado
y apenas llegando a los sustratos colocados en Z2. Finalmente se opta por usar
de nuevo dos únicas zonas calefactoras, pero empleando tanto en Z1 como en
Z2 piezas calefactoras de 30 cm de longitud (Figura 3.8c). De este modo el
gradiente de temperatura en el centro de Z2 es más suave de lo que se
consigue en la configuración C1. Además, se ha diseñado un portamuestras de
grafito de 5 cm de longitud, que permite colocar sustratos a distintas
temperaturas en un mismo experimento.
Muestras de MoS2 obtenidas con la configuración C3
Todas las muestras han sido crecidas a una presión de 1 mbar, con un flujo de
20 sccm y manteniendo el MoS2 en polvo a 970 ºC durante 20 min. Las
condiciones son las mismas que las empleadas en el grupo del Prof. Kung de
la Universidad de Alabama, salvo por la presión, que en su caso es de 10 mbar.
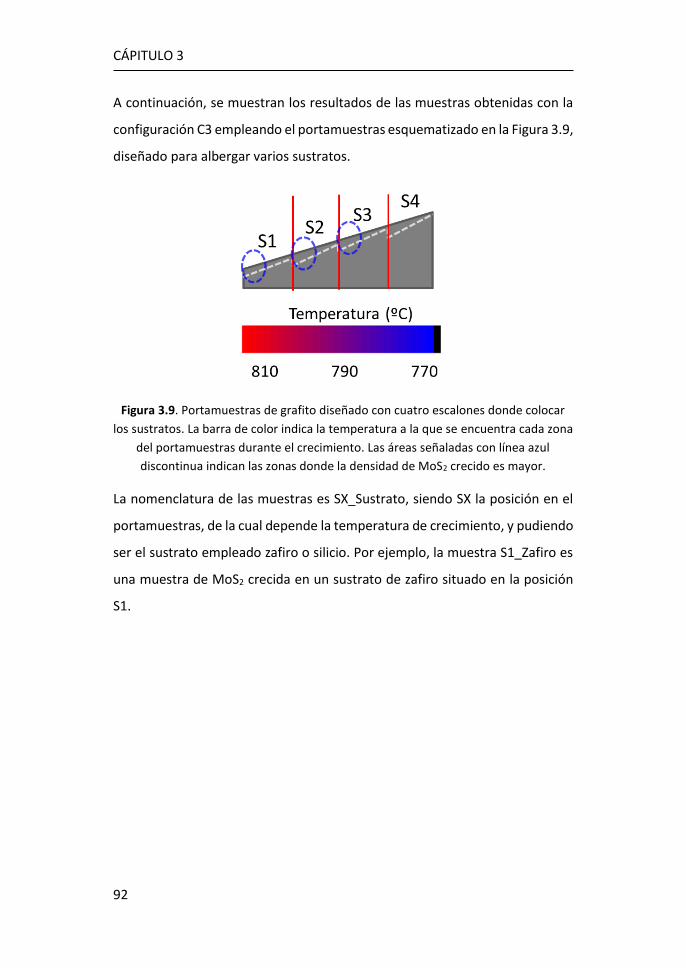
CÁPITULO 3
92
A continuación, se muestran los resultados de las muestras obtenidas con la
configuración C3 empleando el portamuestras esquematizado en la Figura 3.9,
diseñado para albergar varios sustratos.
Figura 3.9. Portamuestras de grafito diseñado con cuatro escalones donde colocar
los sustratos. La barra de color indica la temperatura a la que se encuentra cada zona
del portamuestras durante el crecimiento. Las áreas señaladas con línea azul
discontinua indican las zonas donde la densidad de MoS2 crecido es mayor.
La nomenclatura de las muestras es SX_Sustrato, siendo SX la posición en el
portamuestras, de la cual depende la temperatura de crecimiento, y pudiendo
ser el sustrato empleado zafiro o silicio. Por ejemplo, la muestra S1_Zafiro es
una muestra de MoS2 crecida en un sustrato de zafiro situado en la posición
S1.
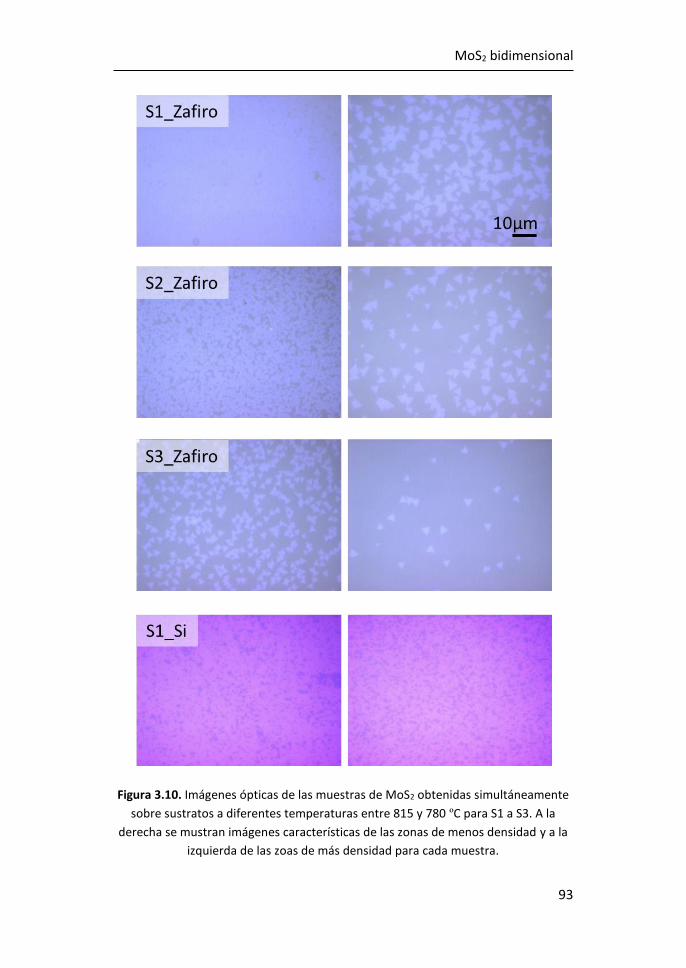
MoS2 bidimensional
93
Figura 3.10. Imágenes ópticas de las muestras de MoS2 obtenidas simultáneamente
sobre sustratos a diferentes temperaturas entre 815 y 780 ºC para S1 a S3. A la
derecha se mustran imágenes características de las zonas de menos densidad y a la
izquierda de las zoas de más densidad para cada muestra.

CÁPITULO 3
94
En la Figura 3.10 se muestran imágenes ópticas de las zonas con mayor
densidad de copos de MoS2 (a la izquierda) y con menor densidad (derecha)
de muestras crecidas sobre zafiro y sobre silicio. Se observa cómo a medida
que la temperatura a la que se encuentra el sustrato disminuye, la densidad y
el tamaño de los copos es cada vez menor (5 μm en S1 y 3 μm en S3), llegando
a no depositarse en S4_Zafiro. A lo largo de cada una de las muestras se
distinguen zonas con distinta densidad de copos. En la muestra S1_Zafiro
(crecida aproximadamente a 810 ºC) hay zonas macroscópicas donde los copos
conectan formando una capa cuasi-continua. Estas zonas de mayor densidad
se forman en la parte baja del sustrato (las zonas señaladas con línea
discontinua azul en la Figura 3.9) lo que podría estar relacionado con una
acumulación del MoS2 evaporado en los rincones formados por el escalón. En
el caso de los sustratos de silicio, sólo creció MoS2 en el sustrato de la posición
S1 (S1_Si). La distribución y tamaño de los copos es constante a lo largo de la
muestra.
Es importante remarcar que, aunque esta configuración es la más eficiente en
cuanto al control de la temperatura, la reproducibilidad en el crecimiento de
las muestras está todavía por optimizar. En la literatura se han encontrado
numerosos casos en los que, aun empleando los mismos parámetros, los
copos de MoS2 crecidos presentan distintas formas, tamaños y defectos.9,10

MoS2 bidimensional
95
3.2 ESTUDIO DE LOS ESPECTROS RAMAN DE MOS2
TRIDIMENSIONAL Y BIDIMENSIONAL
Antes de acometer el análisis detallado de las características de las muestras
2D obtenidas en nuestros equipos, realizamos un estudio comparativo de dos
tipos de muestras 2D de alta calidad de diferente número de capas (N) con
una muestra tridimensional (en volumen, polvo de MoS2 prensado). Las
muestras MoS2-2D corresponden a una muestra obtenida por exfoliación
mecánica y a muestras crecidas por PVT durante mi estancia en el laboratorio
de la Universidad de Alabama.
Se estudian, por tanto, los efectos inducidos por el método de crecimiento, el
sustrato y el número de capas y su correlación con la estructura electrónica,
los defectos y la posible tensión inducida por el sustrato. Se ha empleado una
excitación de 2.54 eV, lejos de la condición de resonancia Raman con los
excitones A y B (con transiciones K-K en torno a 1.89 y 2 eV) pero cercana a la
de la banda de absorción presente en torno a 2.6-2.8 eV. La estructura
electrónica en función del número de capas y los excitones están descritos en
el Capítulo 1. Esta banda de absorción, denominada excitón C, se ha
relacionado con el anidamiento o “nesting” de las bandas de la estructura
electrónica que se produce cerca del punto Q de la zona de Brillouin, casi en
la mitad de la dirección - K.11
Para llevar a cabo el análisis de los resultados de los estudios realizados se ha
elaborado una revisión exhaustiva de la bibliografía sobre los factores que
influyen en los espectros Raman y en la fotoluminiscencia del MoS2-2D.
Se ha comprobado que el bombardeo con iones de monocapas de MoS2
genera defectos provocando un confinamiento fonónico que, debido a la
pendiente de las relaciones de dispersión de las bandas de los modos A1g y E12g
en torno a (Figura 3.11a), produce un aumento de la frecuencia de A1g y una
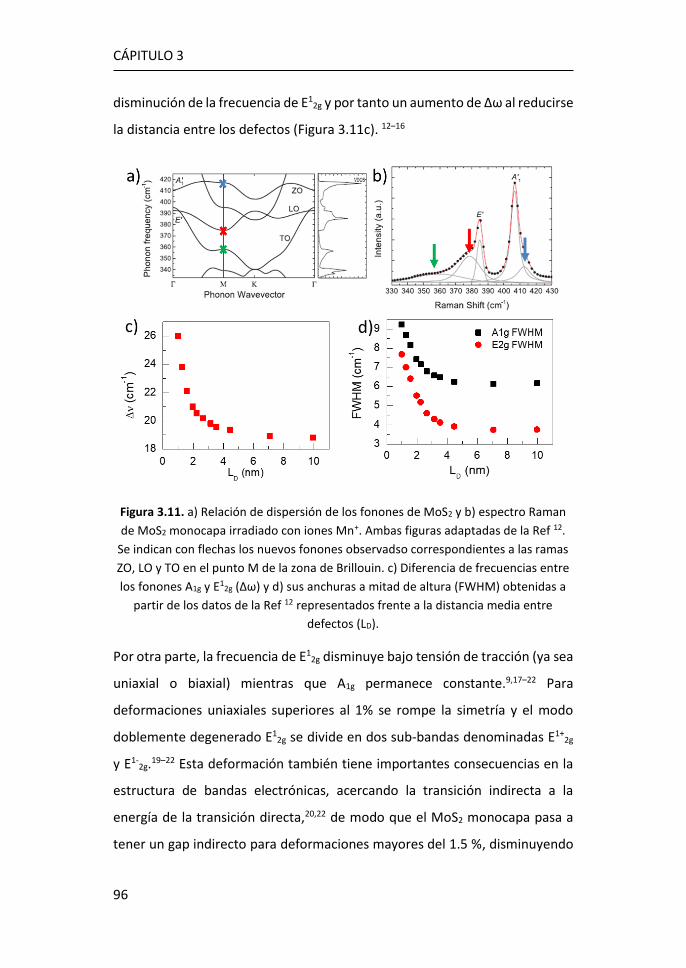
CÁPITULO 3
96
disminución de la frecuencia de E12g y por tanto un aumento de Δω al reducirse
la distancia entre los defectos (Figura 3.11c). 12–16
Figura 3.11. a) Relación de dispersión de los fonones de MoS2 y b) espectro Raman
de MoS2 monocapa irradiado con iones Mn+. Ambas figuras adaptadas de la Ref 12.
Se indican con flechas los nuevos fonones observadso correspondientes a las ramas
ZO, LO y TO en el punto M de la zona de Brillouin. c) Diferencia de frecuencias entre
los fonones A1g y E12g (Δω) y d) sus anchuras a mitad de altura (FWHM) obtenidas a
partir de los datos de la Ref 12 representados frente a la distancia media entre
defectos (LD).
Por otra parte, la frecuencia de E12g disminuye bajo tensión de tracción (ya sea
uniaxial o biaxial) mientras que A1g permanece constante.9,17–22 Para
deformaciones uniaxiales superiores al 1% se rompe la simetría y el modo
doblemente degenerado E12g se divide en dos sub-bandas denominadas E1+
2g
y E1-2g.19–22 Esta deformación también tiene importantes consecuencias en la
estructura de bandas electrónicas, acercando la transición indirecta a la
energía de la transición directa,20,22 de modo que el MoS2 monocapa pasa a
tener un gap indirecto para deformaciones mayores del 1.5 %, disminuyendo

MoS2 bidimensional
97
la intensidad de la banda del excitón A (~1.82 eV) de la fotoluminiscencia y
desplazándose a menores energías.22 También se ha visto que es posible
generar tensión en copos de MoS2 crecidos por CVD durante la etapa de
enfriamiento debido a las diferencias en los coeficientes de expansión térmica
del MoS2 y el sustrato.9
Por otro lado, el dopado, sin sustitución iónica, modifica únicamente la
frecuencia A1g, aumentando o disminuyendo para el dopado p y n,
respectivamente. 23–29 Por ejemplo, el dopado tipo-n con una concentración
de portadores de 2 ⨯ 1013cm-2 da lugar a una disminución de la frecuencia de
A1g de 4 cm-1 y a un aumento de su anchura (FWHM) de hasta 6 cm-1.23–26 En el
espectro de fotoluminiscencia, la intensidad del excitón A disminuye y se
desplaza a menores energías. Esto se debe a la aparición de triones debido al
exceso de electrones provocado por la transferencia de carga al MoS2 a
expensas de los excitones neutros. El dopado tipo-p producido por adsorción
de moléculas o átomos en zonas de defectos aumenta la frecuencia del modo
A1g.23,27–31 Sin embargo, cuando se produce por la sustitución de un átomo de
S por un átomo más pequeño (N, O), se induce una tensión compresiva en la
estructura del MoS2 debido a la distorsión generada en la red, que hace que
también se vea afectado el modo E12g.27,29,32,33 En este caso se da un
desplazamiento de ambos fonones a frecuencias mayores: el desplazamiento
del modo A1g debido al dopado tipo-p, y el del modo E12g debido a la tensión
generada por los nuevos enlaces Mo-X. Por otra parte, el dopado tipo-p baja
el nivel de Fermi y, por tanto, incrementa la energía de la
fotoluminiscencia.27,29,30,33 En la Figura 3.12 se ha representado un esquema
que resume los efectos de la tensión, el desorden y el dopado sobre los modos
A1g y E12g y sobre la fotoluminiscencia.

CÁPITULO 3
98
Figura 3.12. Esquema en el que se resumen los efectos de la tensión, el desorden y el
dopado en los modos Raman A1g y E12g y en la fotoluminiscencia (PL).
Con el fin de visualizar de una manera clara cómo se ven afectadas las
frecuencias de los picos A1g y E12g como resultado de la tensión, el dopado o el
desorden, se han recopilado datos de algunos de los trabajos a los que se han
hecho referencia y se han representado las frecuencias de A1g frente a las de
E12g para cada trabajo. La digitalización de los datos se llevó a cabo usando el
software de WebPlotDigitizer (https://automeris.io/WebPlotDigitizer) y la
representación gráfica se hizo utilizando el lenguaje de programación R, la
librería ggplot2 y la función ggplot(). Los diferentes tipos de defectos
representados son: desorden,12 dopado tipo-n,34 dopado tipo-p con oxígeno,27
dopado tipo-p con N (distorsión de la red),29 tensión de tracción uniaxial21 y
vacantes de S con adsorción de oxígeno.32 Hay que resaltar que las frecuencias
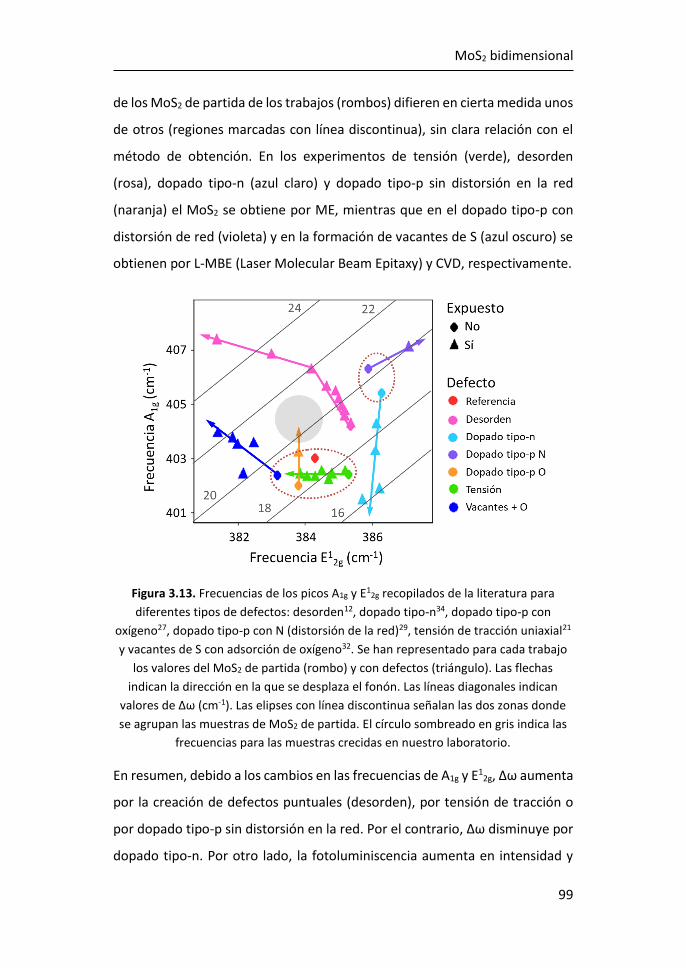
MoS2 bidimensional
99
de los MoS2 de partida de los trabajos (rombos) difieren en cierta medida unos
de otros (regiones marcadas con línea discontinua), sin clara relación con el
método de obtención. En los experimentos de tensión (verde), desorden
(rosa), dopado tipo-n (azul claro) y dopado tipo-p sin distorsión en la red
(naranja) el MoS2 se obtiene por ME, mientras que en el dopado tipo-p con
distorsión de red (violeta) y en la formación de vacantes de S (azul oscuro) se
obtienen por L-MBE (Laser Molecular Beam Epitaxy) y CVD, respectivamente.
Figura 3.13. Frecuencias de los picos A1g y E12g recopilados de la literatura para
diferentes tipos de defectos: desorden12, dopado tipo-n34, dopado tipo-p con
oxígeno27, dopado tipo-p con N (distorsión de la red)29, tensión de tracción uniaxial21
y vacantes de S con adsorción de oxígeno32. Se han representado para cada trabajo
los valores del MoS2 de partida (rombo) y con defectos (triángulo). Las flechas
indican la dirección en la que se desplaza el fonón. Las líneas diagonales indican
valores de Δω (cm-1). Las elipses con línea discontinua señalan las dos zonas donde
se agrupan las muestras de MoS2 de partida. El círculo sombreado en gris indica las
frecuencias para las muestras crecidas en nuestro laboratorio.
En resumen, debido a los cambios en las frecuencias de A1g y E12g, Δω aumenta
por la creación de defectos puntuales (desorden), por tensión de tracción o
por dopado tipo-p sin distorsión en la red. Por el contrario, Δω disminuye por
dopado tipo-n. Por otro lado, la fotoluminiscencia aumenta en intensidad y

CÁPITULO 3
100
energía con dopado tipo-p, y disminuye con la tensión de tracción y con el
dopado tipo-n (donde se incluyen las vacantes de S). La Figura 3.14 resume la
dirección de los desplazamientos de las frecuencias de A1g y de E12g según el
tipo de defecto.
Figura 3.14. Variación de las frecuencias Raman de los modos A1g y E12g según los
datos obtenidos en la literatura.
Para estudiar en profundidad todos los modos Raman del MoS2, incluidas las
señales más débiles, se transfirieron dos muestras de MoS2-2D obtenidas por
ME y por PVT sobre plataformas de amplificación Raman, basadas en la
optimización de la interferencia óptica en el rango de longitudes de onda de
interés, desarrolladas en nuestro laboratorio.35 La plataforma está formada
por una capa de aluminio altamente reflectante depositada sobre un sustrato
de Si (100) y una capa dieléctrica transparente de Al2O3 del grosor adecuado
en la parte superior. Para una excitación de 488 nm, como es el caso, el grosor
óptimo para la capa de Al2O3 es de 60 nm.35 Parte del sustrato de Si se
enmascara durante el depósito de Al y de Al2O3 para poder comparar los
espectros Raman de zonas adyacentes con y sin amplificación. Puede verse un
esquema de la plataforma en la Figura 3.15a.

MoS2 bidimensional
101
Figura 3.15. a) Esquema de MoS2-2D sobre la frontera Si/Plataforma amplificadora,
b) Espectros Raman de 1L-PVT sobre Si y sobre la plataforma. El espectro sobre Si ha
sido multiplicado por un factor 100 y la señal intensa a 520 cm-1 se corresponde al
sustrato de Si. Imágenes ópticas de MoS2-2D c) obtenido por ME y d) obtenido por
PVT transferidas sobre la plataforma amplificadora en la frontera con el Si. e) Imagen
óptica de MoS2-PVT sobre zafiro sin transferir.
Los nombres de las muestras utilizados aquí indican: el número de capas, el
método de fabricación y el sustrato en el que se han crecido o transferido:
Nº Capas-Método de obtención-Sustrato
Los sustratos utilizados son: la plataforma de amplificación “Plat”, obleas de
silicio (100) “Si” y sustratos de zafiro (0001) “Zaf”. Los métodos de obtención
son transporte físico de vapor (PVT) y exfoliación mecánica (ME). Por ejemplo,
la muestra 1L-PVT-Plat se corresponde con MoS2 monocapa crecida por PVT y
transferida sobre la plataforma de amplificación. También se estudia MoS2
comercial en polvo (Alfa Aesar, 99%) prensado entre dos portamuestras de
vidrio etiquetado como “bulk-MoS2”. En todos los casos se han restado los
espectros de los sustratos medidos en las mismas condiciones que sus
respectivas muestras.

CÁPITULO 3
102
En la Figura 3.15c y d se muestran las imágenes ópticas de las muestras
obtenidas por ME y por PVT transferidas sobre las plataformas en las regiones
fronterizas plataforma/Si. En ambas se distingue un copo de MoS2 en la
interfase entre la plataforma de amplificación y el Si. En la Figura 3.15b se han
representado los espectros Raman obtenidos para la muestra ME-Plat y ME-
Si. Se observa un factor de amplificación ~50 y ~250 para las muestras ME y
PVT, respectivamente, lo que permite estudiar en mayor detalle los modos de
menor intensidad del MoS2 con la ventaja, además, de no tener presente la
señal Raman del Si, que dificulta en gran medida el análisis de los espectros.
Además de los picos asociados a los fonones de primer orden, A1g y E12g,
habitualmente empleados en la caracterización de los TMCDS y descritos en
la introducción, los espectros Raman de MoS2 presentan numerosas
características debidas a: i) procesos Raman de segundo orden y ii) modos
prohibidos activados por la presencia de defectos. Los modos que no
corresponden al centro de la zona de Brillouin (Γ) se nombran utilizando su
nomenclatura en Γ, su vector de onda y su carácter longitudinal o transversal
(L o T). Por ejemplo, E12g(LOM) se refiere al modo longitudinal óptico E1
2g en el
punto M.
Es importante recordar que las frecuencias de los picos Raman varían según
cual sea la temperatura local en el punto de incidencia del láser. Por ello,
primero se optimizan las condiciones de medida de modo que la temperatura
local sea la misma en todas las muestras, 300 K. La temperatura local se puede
obtener analizando los espectros Stokes y anti-Stokes (AS). La intensidad de
los modos Raman Stokes (𝐼𝑆 ) y anti-Stokes (𝐼𝐴𝑆 ) son proporcionales a la
población de esos modos de la siguiente forma:
𝐼𝑆 ∝ 𝑛(𝜔, 𝑇) + 1 (Ec. 3.1)
𝐼𝐴𝑆 ∝ 𝑛(𝜔, 𝑇) (Ec. 3.2)

MoS2 bidimensional
103
Donde n es la población del modo de vibración, que depende a su vez de la
frecuencia (ω) y la temperatura (T) según la distribución estadística de Bose-
Einstein:
𝑛(𝜔, 𝑇) =1
𝑒ℏ𝜔𝐾𝑇 − 1
(Ec. 3.3)
Siendo ℏ la constante de Planck y K la constante de Boltzmann. Conociendo
las intensidades 𝐼𝑆 y 𝐼𝐴𝑆 de los espectros medidos es posible calcular la
temperatura local (T). Para ello se ha corregido 𝐼𝐴𝑆 hasta hacerla coincidir con
𝐼𝑆 :
𝐼𝑆 = 𝐼𝐴𝑆
(
1
𝑒ℏ𝜔𝐾𝑇 − 1
+ 1
1
𝑒ℏ𝜔𝐾𝑇 − 1 )
(Ec. 3.4)
En la Figura 3.16 se han representado los espectros Raman Stokes, Anti-Stokes
y Anti-Stokes corregido por la expresión de Bose-Einstein (AS corr BE) de la
muestra bulk-MoS2 incidiendo con una potencia de 3.4 mW (Figura 3.16a) y
de 0.3 mW (Figura 3.16b). Se ha comprobado que la temperatura local en el
caso de las muestras MoS2-2D no se modifica con una potencia incidente de
3.4 mW.
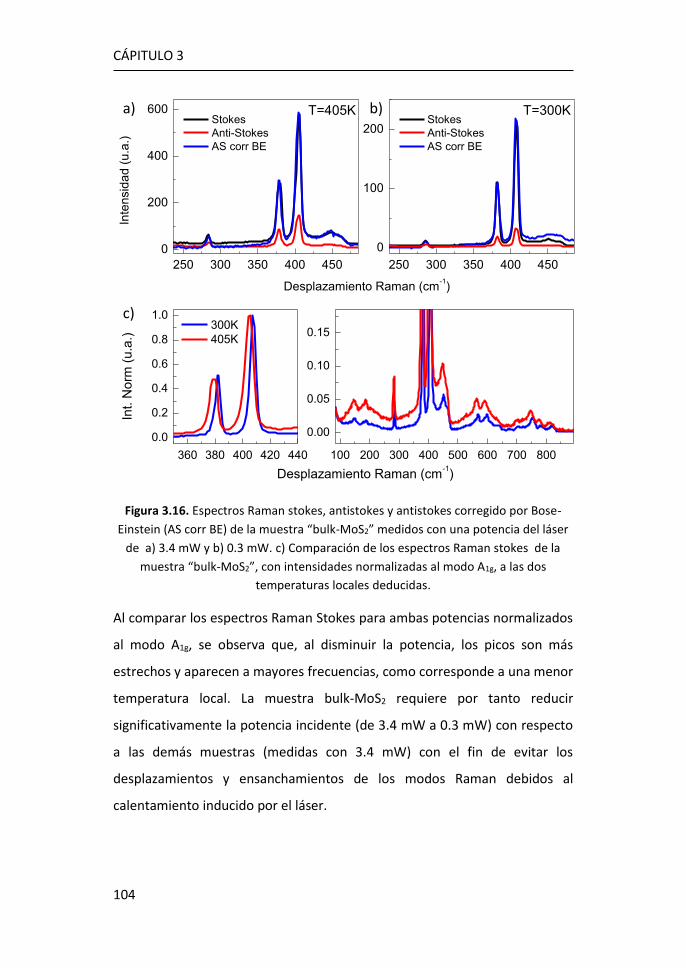
CÁPITULO 3
104
Figura 3.16. Espectros Raman stokes, antistokes y antistokes corregido por Bose-
Einstein (AS corr BE) de la muestra “bulk-MoS2” medidos con una potencia del láser
de a) 3.4 mW y b) 0.3 mW. c) Comparación de los espectros Raman stokes de la
muestra “bulk-MoS2”, con intensidades normalizadas al modo A1g, a las dos
temperaturas locales deducidas.
Al comparar los espectros Raman Stokes para ambas potencias normalizados
al modo A1g, se observa que, al disminuir la potencia, los picos son más
estrechos y aparecen a mayores frecuencias, como corresponde a una menor
temperatura local. La muestra bulk-MoS2 requiere por tanto reducir
significativamente la potencia incidente (de 3.4 mW a 0.3 mW) con respecto
a las demás muestras (medidas con 3.4 mW) con el fin de evitar los
desplazamientos y ensanchamientos de los modos Raman debidos al
calentamiento inducido por el láser.
100 200 300 400 500 600 700 800
0.00
0.05
0.10
0.15
360 380 400 420 4400.0
0.2
0.4
0.6
0.8
1.0
Desplazamiento Raman (cm-1)
Int.
Nor
m (u
.a.)
300K 405K
250 300 350 400 4500
200
400
600
250 300 350 400 4500
100
200In
tens
idad
(u.a
.)
Desplazamiento Raman (cm-1)
Stokes Anti-Stokes AS corr BE
T=405K Stokes Anti-Stokes AS corr BE
T=300Ka)
c)
b)
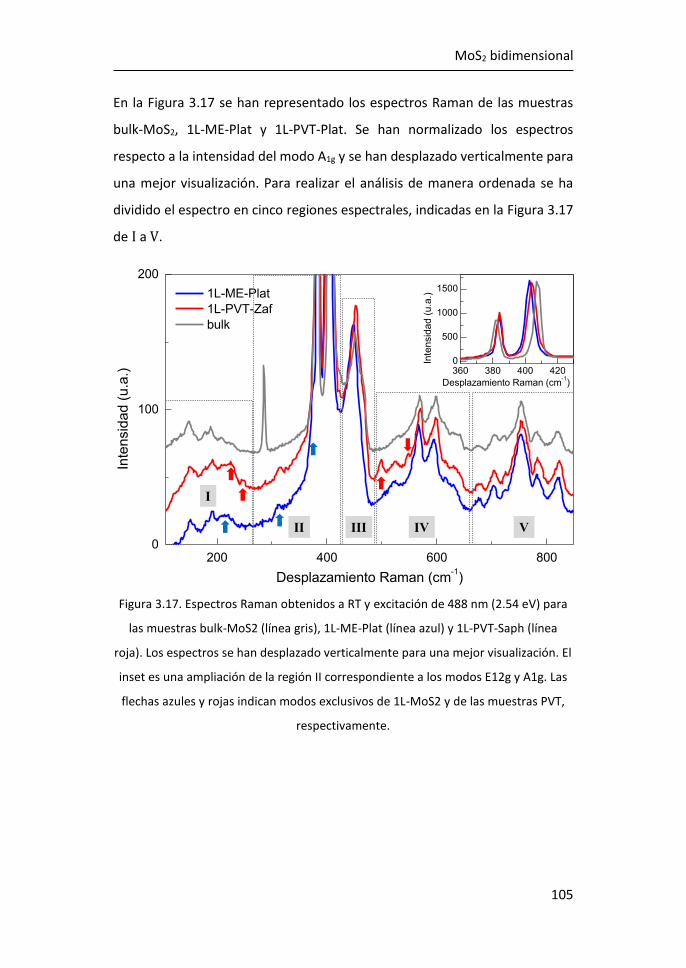
MoS2 bidimensional
105
En la Figura 3.17 se han representado los espectros Raman de las muestras
bulk-MoS2, 1L-ME-Plat y 1L-PVT-Plat. Se han normalizado los espectros
respecto a la intensidad del modo A1g y se han desplazado verticalmente para
una mejor visualización. Para realizar el análisis de manera ordenada se ha
dividido el espectro en cinco regiones espectrales, indicadas en la Figura 3.17
de I a V.
Figura 3.17. Espectros Raman obtenidos a RT y excitación de 488 nm (2.54 eV) para
las muestras bulk-MoS2 (línea gris), 1L-ME-Plat (línea azul) y 1L-PVT-Saph (línea
roja). Los espectros se han desplazado verticalmente para una mejor visualización. El
inset es una ampliación de la región II correspondiente a los modos E12g y A1g. Las
flechas azules y rojas indican modos exclusivos de 1L-MoS2 y de las muestras PVT,
respectivamente.
200 400 600 8000
100
200
360 380 400 4200
500
1000
1500
Inte
nsid
ad (u
.a.)
Desplazamiento Raman (cm-1)
1L-ME-Plat 1L-PVT-Zaf bulk
Inte
nsid
ad (u
.a.)
Desplazamiento Raman (cm-1)
I
II III IV V
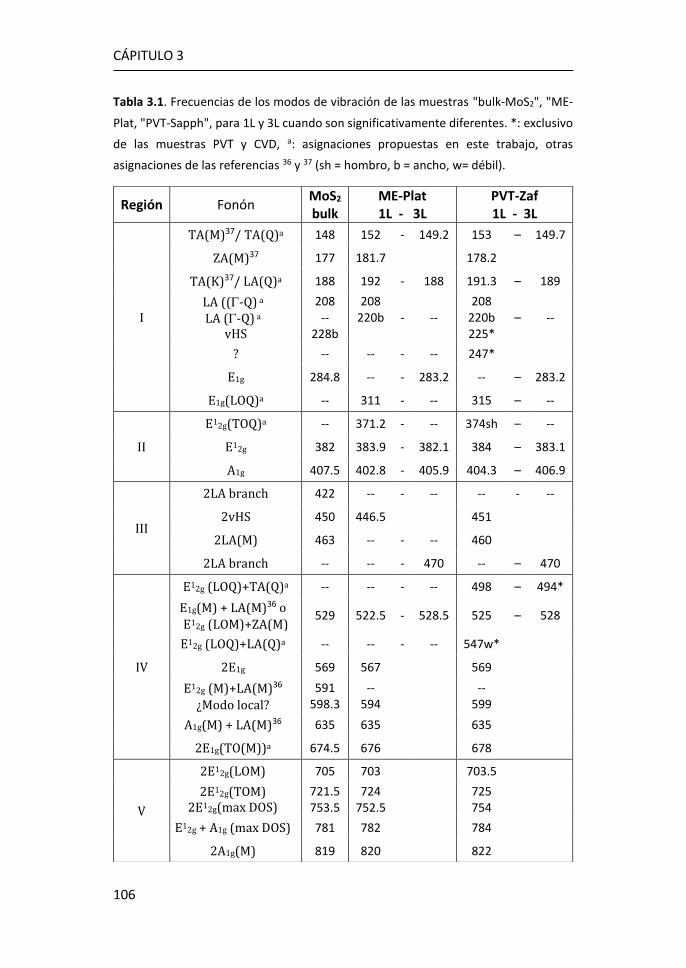
CÁPITULO 3
106
Tabla 3.1. Frecuencias de los modos de vibración de las muestras "bulk-MoS2", "ME-
Plat, "PVT-Sapph", para 1L y 3L cuando son significativamente diferentes. *: exclusivo
de las muestras PVT y CVD, a: asignaciones propuestas en este trabajo, otras
asignaciones de las referencias 36 y 37 (sh = hombro, b = ancho, w= débil).
Región Fonón MoS2 bulk
ME-Plat 1L - 3L
PVT-Zaf 1L - 3L
I
TA(M)37/ TA(Q)a 148 152 - 149.2 153 – 149.7
ZA(M)37 177 181.7 178.2
TA(K)37/ LA(Q)a 188 192 - 188 191.3 – 189
LA ((-Q) a LA (-Q) a
vHS
208 --
228b
208 220b
- --
208 220b 225*
– --
? -- -- - -- 247*
E1g 284.8 -- - 283.2 -- – 283.2
E1g(LOQ)a -- 311 - -- 315 – --
II
E12g(TOQ)a -- 371.2 - -- 374sh – --
E12g 382 383.9 - 382.1 384 – 383.1
A1g 407.5 402.8 - 405.9 404.3 – 406.9
III
2LA branch 422 -- - -- -- - --
2vHS 450 446.5 451
2LA(M) 463 -- - -- 460
2LA branch -- -- - 470 -- – 470
IV
E12g (LOQ)+TA(Q)a -- -- - -- 498 – 494*
E1g(M) + LA(M)36 o
E12g (LOM)+ZA(M) 529 522.5 - 528.5 525 – 528
E12g (LOQ)+LA(Q)a -- -- - -- 547w*
2E1g 569 567 569
E12g (M)+LA(M)36
¿Modo local?
591 598.3
-- 594
--
599
A1g(M) + LA(M)36 635 635 635
2E1g(TO(M))a 674.5 676 678
V
2E12g(LOM) 705 703 703.5
2E12g(TOM) 2E12g(max DOS)
721.5 753.5
724 752.5
725 754
E12g + A1g (max DOS) 781 782 784
2A1g(M) 819 820 822

MoS2 bidimensional
107
La Tabla 3.1 recoge las frecuencias de los picos detectados para bulk-MoS2, 1L-
ME-Plat y 1L-PVT-Zaf en este trabajo. También se incluyen algunas
asignaciones de picos de la literatura y las que aquí se proponen. Las
frecuencias para 3L-ME-Plat y 3L-PVT-Zaf se indican cuando son
significativamente diferentes de las de 1L. La Tabla 3.1 expone de una manera
clara los casos en los que los picos sólo se detectan para 1L, para N>1 o para
las muestras PVT/CVD.
3.2.1 Región I: 100-270 cm-1
Todos los picos de esta región corresponden a modos Raman prohibidos. La
detección de modos Raman prohibidos se suele relacionar con la presencia de
defectos que provocan una ruptura de la simetría. Por ejemplo, al bombardear
1L-MoS2 con iones para inducir altas densidades de defectos puntuales (en el
rango 1012 - 1014 cm-2)12 se ha observado que, a medida que aumenta la
densidad de defectos, las anchuras de los modos E12g y A1g aumentan y además
se activan otros modos (Figura 3.11). De entre estos nuevos modos, los
autores explican que la banda ancha alrededor de 227 cm-1 (asignada a LA(M))
es adecuada para la estimación de la concentración de defectos (Figura
3.18).12–16
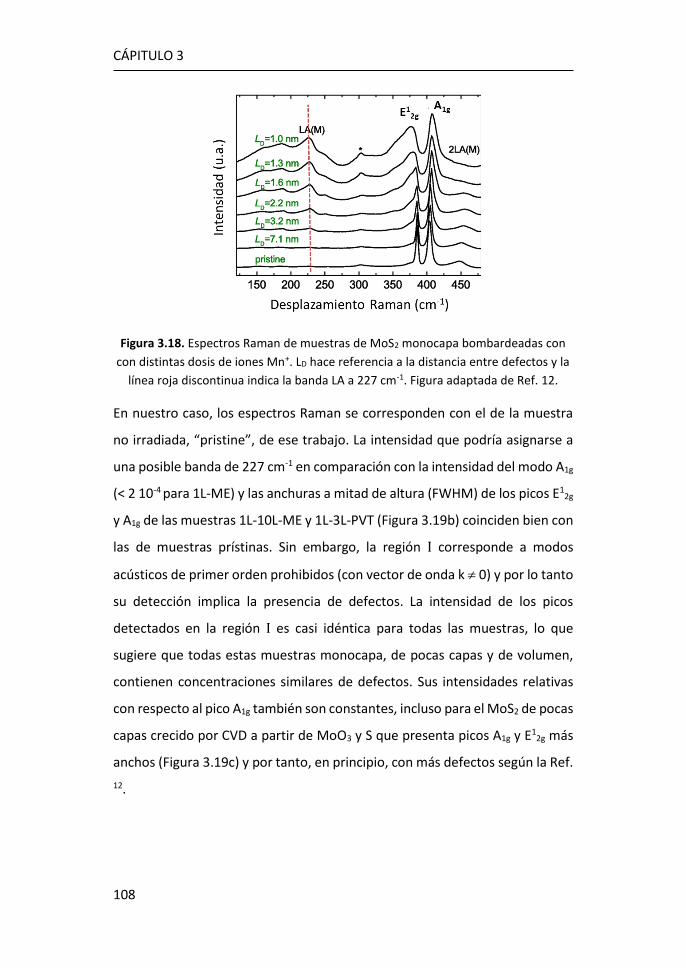
CÁPITULO 3
108
Figura 3.18. Espectros Raman de muestras de MoS2 monocapa bombardeadas con
con distintas dosis de iones Mn+. LD hace referencia a la distancia entre defectos y la
línea roja discontinua indica la banda LA a 227 cm-1. Figura adaptada de Ref. 12.
En nuestro caso, los espectros Raman se corresponden con el de la muestra
no irradiada, “pristine”, de ese trabajo. La intensidad que podría asignarse a
una posible banda de 227 cm-1 en comparación con la intensidad del modo A1g
(< 2 10-4 para 1L-ME) y las anchuras a mitad de altura (FWHM) de los picos E12g
y A1g de las muestras 1L-10L-ME y 1L-3L-PVT (Figura 3.19b) coinciden bien con
las de muestras prístinas. Sin embargo, la región I corresponde a modos
acústicos de primer orden prohibidos (con vector de onda k 0) y por lo tanto
su detección implica la presencia de defectos. La intensidad de los picos
detectados en la región I es casi idéntica para todas las muestras, lo que
sugiere que todas estas muestras monocapa, de pocas capas y de volumen,
contienen concentraciones similares de defectos. Sus intensidades relativas
con respecto al pico A1g también son constantes, incluso para el MoS2 de pocas
capas crecido por CVD a partir de MoO3 y S que presenta picos A1g y E12g más
anchos (Figura 3.19c) y por tanto, en principio, con más defectos según la Ref.
12.
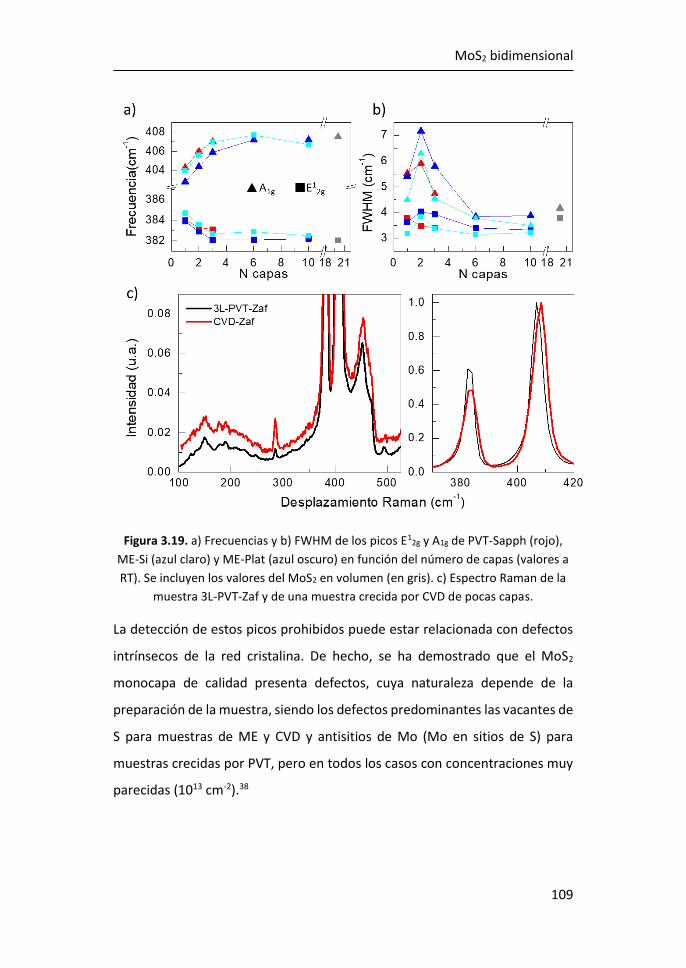
MoS2 bidimensional
109
Figura 3.19. a) Frecuencias y b) FWHM de los picos E12g y A1g de PVT-Sapph (rojo),
ME-Si (azul claro) y ME-Plat (azul oscuro) en función del número de capas (valores a
RT). Se incluyen los valores del MoS2 en volumen (en gris). c) Espectro Raman de la
muestra 3L-PVT-Zaf y de una muestra crecida por CVD de pocas capas.
La detección de estos picos prohibidos puede estar relacionada con defectos
intrínsecos de la red cristalina. De hecho, se ha demostrado que el MoS2
monocapa de calidad presenta defectos, cuya naturaleza depende de la
preparación de la muestra, siendo los defectos predominantes las vacantes de
S para muestras de ME y CVD y antisitios de Mo (Mo en sitios de S) para
muestras crecidas por PVT, pero en todos los casos con concentraciones muy
parecidas (1013 cm-2).38
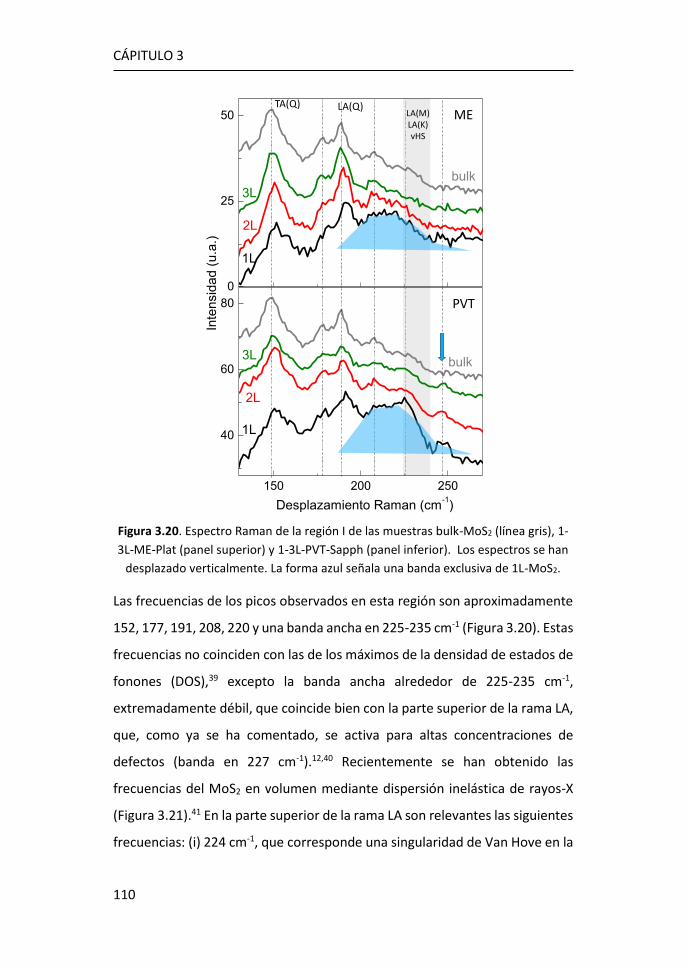
CÁPITULO 3
110
Figura 3.20. Espectro Raman de la región I de las muestras bulk-MoS2 (línea gris), 1-
3L-ME-Plat (panel superior) y 1-3L-PVT-Sapph (panel inferior). Los espectros se han
desplazado verticalmente. La forma azul señala una banda exclusiva de 1L-MoS2.
Las frecuencias de los picos observados en esta región son aproximadamente
152, 177, 191, 208, 220 y una banda ancha en 225-235 cm-1 (Figura 3.20). Estas
frecuencias no coinciden con las de los máximos de la densidad de estados de
fonones (DOS),39 excepto la banda ancha alrededor de 225-235 cm-1,
extremadamente débil, que coincide bien con la parte superior de la rama LA,
que, como ya se ha comentado, se activa para altas concentraciones de
defectos (banda en 227 cm-1).12,40 Recientemente se han obtenido las
frecuencias del MoS2 en volumen mediante dispersión inelástica de rayos-X
(Figura 3.21).41 En la parte superior de la rama LA son relevantes las siguientes
frecuencias: (i) 224 cm-1, que corresponde una singularidad de Van Hove en la
0
25
50
150 200 250
40
60
80
25
50
75
100
280 30020
40
60
80
100
120
Inte
nsid
ad (u
.a.)
3L
2L
Desplazamiento Raman (cm-1)
1L
bulk
bulk3L
2L
1L
LA(M)LA(K)vHS
ME
PVT
TA(Q) LA(Q)
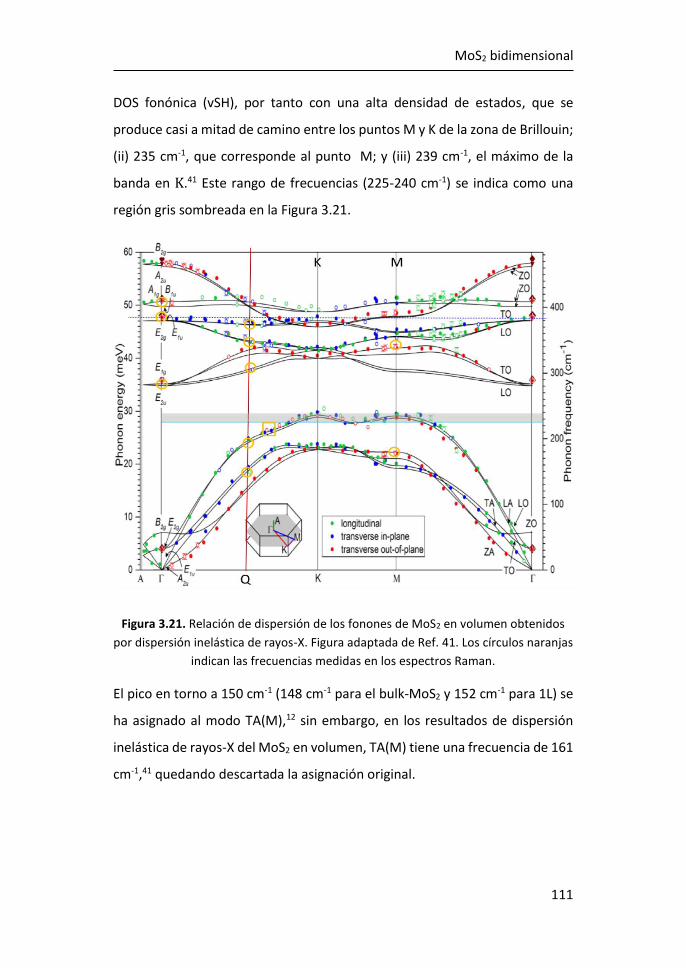
MoS2 bidimensional
111
DOS fonónica (vSH), por tanto con una alta densidad de estados, que se
produce casi a mitad de camino entre los puntos M y K de la zona de Brillouin;
(ii) 235 cm-1, que corresponde al punto M; y (iii) 239 cm-1, el máximo de la
banda en K.41 Este rango de frecuencias (225-240 cm-1) se indica como una
región gris sombreada en la Figura 3.21.
Figura 3.21. Relación de dispersión de los fonones de MoS2 en volumen obtenidos
por dispersión inelástica de rayos-X. Figura adaptada de Ref. 41. Los círculos naranjas
indican las frecuencias medidas en los espectros Raman.
El pico en torno a 150 cm-1 (148 cm-1 para el bulk-MoS2 y 152 cm-1 para 1L) se
ha asignado al modo TA(M),12 sin embargo, en los resultados de dispersión
inelástica de rayos-X del MoS2 en volumen, TA(M) tiene una frecuencia de 161
cm-1,41 quedando descartada la asignación original.
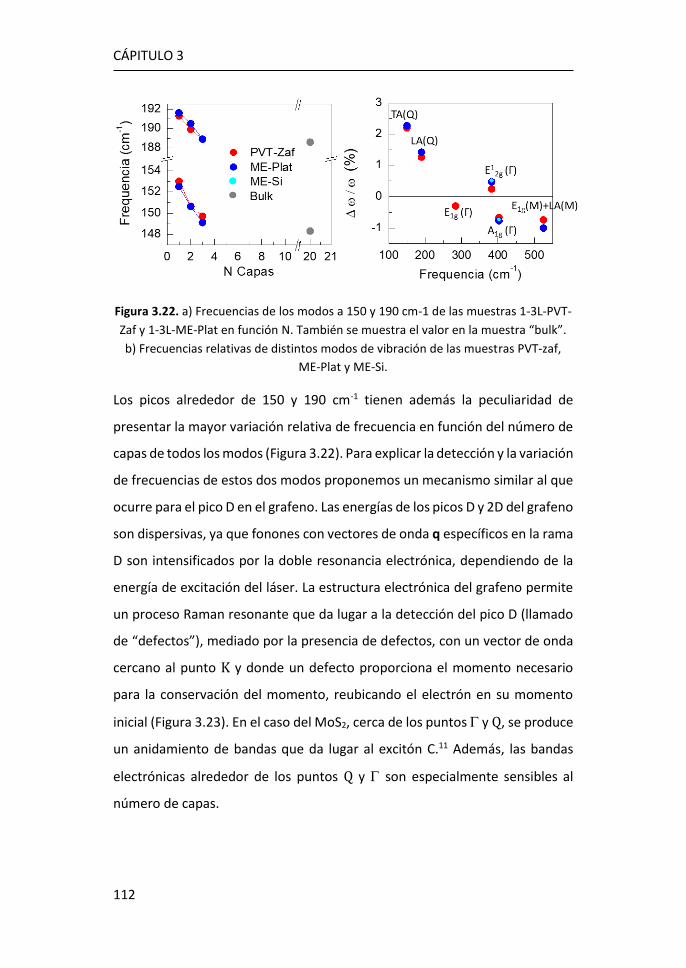
CÁPITULO 3
112
Figura 3.22. a) Frecuencias de los modos a 150 y 190 cm-1 de las muestras 1-3L-PVT-
Zaf y 1-3L-ME-Plat en función N. También se muestra el valor en la muestra “bulk”.
b) Frecuencias relativas de distintos modos de vibración de las muestras PVT-zaf,
ME-Plat y ME-Si.
Los picos alrededor de 150 y 190 cm-1 tienen además la peculiaridad de
presentar la mayor variación relativa de frecuencia en función del número de
capas de todos los modos (Figura 3.22). Para explicar la detección y la variación
de frecuencias de estos dos modos proponemos un mecanismo similar al que
ocurre para el pico D en el grafeno. Las energías de los picos D y 2D del grafeno
son dispersivas, ya que fonones con vectores de onda q específicos en la rama
D son intensificados por la doble resonancia electrónica, dependiendo de la
energía de excitación del láser. La estructura electrónica del grafeno permite
un proceso Raman resonante que da lugar a la detección del pico D (llamado
de “defectos”), mediado por la presencia de defectos, con un vector de onda
cercano al punto K y donde un defecto proporciona el momento necesario
para la conservación del momento, reubicando el electrón en su momento
inicial (Figura 3.23). En el caso del MoS2, cerca de los puntos y Q, se produce
un anidamiento de bandas que da lugar al excitón C.11 Además, las bandas
electrónicas alrededor de los puntos Q y son especialmente sensibles al
número de capas.

MoS2 bidimensional
113
Figura 3.23. Esquema de doble resonancia que explica el proceso de dispersión que
da origen de la banda D en el grafeno. 1) La excitación con el láser (hω0) crea un par
electrón(punto)-hueco(círculo), 2) el electrón se dispersa inelásticamente con un
fonón de vector de onda Qf, 3) ocurre una dispersión elástica por un defecto de
vector de onda QD, teniendo Qf y QD el mismo valor, pero de signo opuesto y 4) se
produce la recombinación entre el electrón y el hueco.
En la Figura 3.24 se ve cómo la banda de valencia varía enormemente con el
número de capas, variando la energía del excitón C entre 2.6 y 2.8 eV, de modo
que una energía de excitación constante producirá cambios en el vector de
onda que cumple el mecanismo de resonancia al variar el número de capas,
en un proceso similar al pico D en grafeno, modificando así la frecuencia del
fonón. La excitación empleada (2.54 eV), está cerca de la condición de
anidamiento de bandas y por tanto de la resonancia del excitón C (Figura 3.24).
Consecuentemente, un mecanismo resonante asistido por defectos
intrínsecos podría explicar la detección y las variaciones de las frecuencias con
el número de capas observadas en los modos cercanos a 150 y 190 cm-1. Según
las bandas de dispersión medidas y calculadas por dispersión inelástica de
rayos-X,41 el vector de onda de los fonones puede estar cerca de Q (Figura
3.21), por lo que proponemos las asignaciones TA(Q) y LA(Q) para las
frecuencias 150 y 190 cm-1, respectivamente.
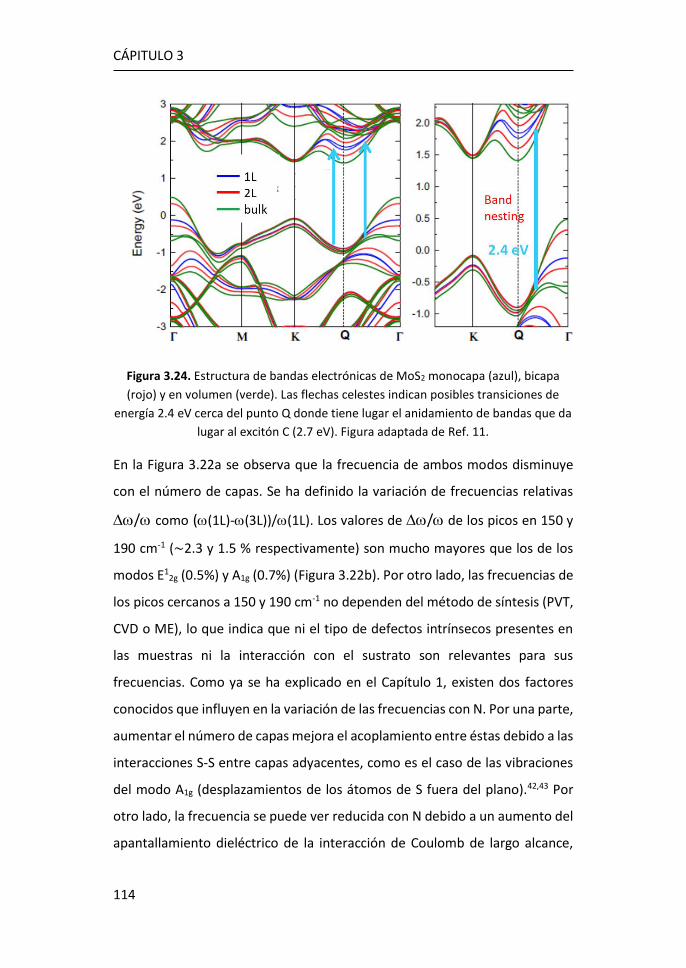
CÁPITULO 3
114
Figura 3.24. Estructura de bandas electrónicas de MoS2 monocapa (azul), bicapa
(rojo) y en volumen (verde). Las flechas celestes indican posibles transiciones de
energía 2.4 eV cerca del punto Q donde tiene lugar el anidamiento de bandas que da
lugar al excitón C (2.7 eV). Figura adaptada de Ref. 11.
En la Figura 3.22a se observa que la frecuencia de ambos modos disminuye
con el número de capas. Se ha definido la variación de frecuencias relativas
/ como ((1L)-(3L))/(1L). Los valores de / de los picos en 150 y
190 cm-1 (~2.3 y 1.5 % respectivamente) son mucho mayores que los de los
modos E12g (0.5%) y A1g (0.7%) (Figura 3.22b). Por otro lado, las frecuencias de
los picos cercanos a 150 y 190 cm-1 no dependen del método de síntesis (PVT,
CVD o ME), lo que indica que ni el tipo de defectos intrínsecos presentes en
las muestras ni la interacción con el sustrato son relevantes para sus
frecuencias. Como ya se ha explicado en el Capítulo 1, existen dos factores
conocidos que influyen en la variación de las frecuencias con N. Por una parte,
aumentar el número de capas mejora el acoplamiento entre éstas debido a las
interacciones S-S entre capas adyacentes, como es el caso de las vibraciones
del modo A1g (desplazamientos de los átomos de S fuera del plano).42,43 Por
otro lado, la frecuencia se puede ver reducida con N debido a un aumento del
apantallamiento dieléctrico de la interacción de Coulomb de largo alcance,
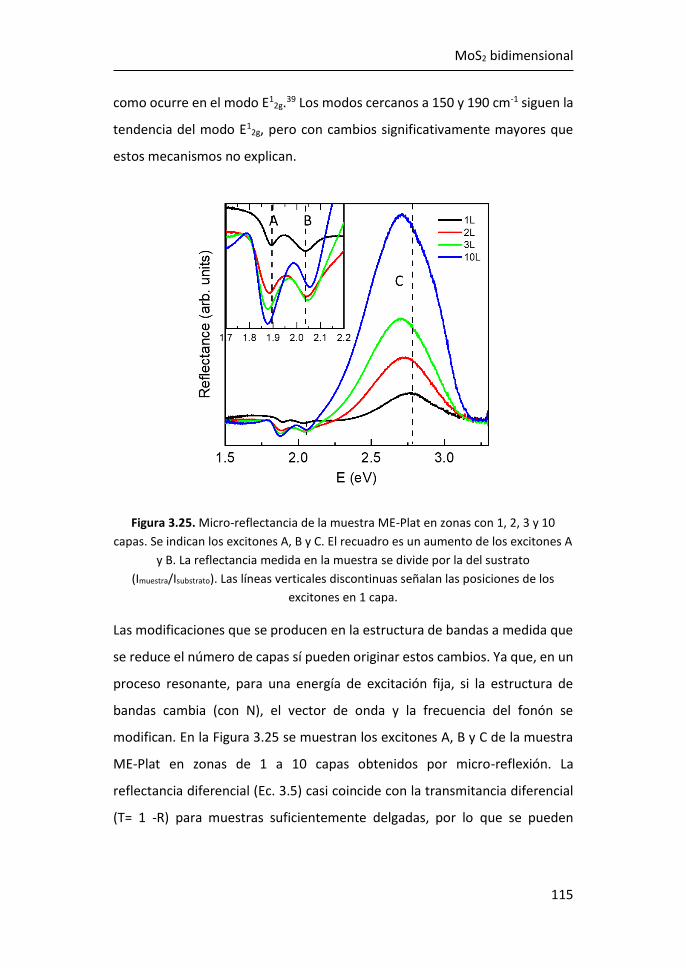
MoS2 bidimensional
115
como ocurre en el modo E12g.39 Los modos cercanos a 150 y 190 cm-1 siguen la
tendencia del modo E12g, pero con cambios significativamente mayores que
estos mecanismos no explican.
Figura 3.25. Micro-reflectancia de la muestra ME-Plat en zonas con 1, 2, 3 y 10
capas. Se indican los excitones A, B y C. El recuadro es un aumento de los excitones A
y B. La reflectancia medida en la muestra se divide por la del sustrato
(Imuestra/Isubstrato). Las líneas verticales discontinuas señalan las posiciones de los
excitones en 1 capa.
Las modificaciones que se producen en la estructura de bandas a medida que
se reduce el número de capas sí pueden originar estos cambios. Ya que, en un
proceso resonante, para una energía de excitación fija, si la estructura de
bandas cambia (con N), el vector de onda y la frecuencia del fonón se
modifican. En la Figura 3.25 se muestran los excitones A, B y C de la muestra
ME-Plat en zonas de 1 a 10 capas obtenidos por micro-reflexión. La
reflectancia diferencial (Ec. 3.5) casi coincide con la transmitancia diferencial
(T= 1 -R) para muestras suficientemente delgadas, por lo que se pueden

CÁPITULO 3
116
comparar fácilmente. Aquí se presenta la reflectancia dividida por la señal del
sustrato (Rmuestra/Rsustrato).
𝑅𝑚𝑢𝑒𝑠𝑡𝑟𝑎 − 𝑅𝑠𝑢𝑠𝑡𝑟𝑎𝑡𝑜𝑅𝑠𝑢𝑠𝑡𝑟𝑎𝑡𝑜
=𝑅𝑚𝑢𝑒𝑠𝑡𝑟𝑎
𝑅𝑠𝑢𝑠𝑡𝑟𝑎𝑡𝑜 − 1 (Ec. 3.5)
En la Figura 3.25 se ve que se produce un desplazamiento de energía del
excitón C casi completo de 1L a 3L. Esto indica que en este rango de 1 a 3 capas
tienen lugar modificaciones importantes de las bandas de valencia y de
conducción (implicadas en los procesos resonantes), provocando así los
desplazamientos de frecuencia de los fonones observados.
3.2.2 Región II: 270-420 cm-1
La Región II corresponde a los modos Raman de primer orden: E1g, E12g y A1g
con frecuencias 284 cm-1, 382 cm-1 y 408 cm-1, respectivamente, para MoS2 en
volumen.
El modo de simetría E1g es una flexión en el plano con una frecuencia de
aproximadamente 284 cm-1 en volumen y, como se explica a continuación, no
es detectable en el caso de MoS2 monocapa. Las representaciones irreducibles
de este modo para MoS2-2D de pocas capas con número (N) par o impar de
capas son E’’ y Eg respectivamente. Los tensores Raman de E1g, E’’ y Eg son las
siguientes:
(Ec. 3.6)

MoS2 bidimensional
117
(Ec. 3.7)
Este modo no debería detectarse en MoS2-2D con N impar, ya que los tensores
Raman en este caso sólo tienen componentes xz e yz no nulas (Ec. 3.6) y la
intensidad Raman es proporcional a |Ed R Ei|, donde Ed es la polarización
dispersada y Ei la polarización incidente. En espectroscopía Raman, la
configuración geométrica experimental accesible para materiales 2D es la de
retrodispersión con las direcciones incidente y dispersada perpendiculares a
la muestra, es decir, paralelas al eje z en el caso de MoS2-2D, de modo que
sólo accede a componentes en el plano (xx, yy y xy). En efecto, el fonón E’’ no
se detecta en 1L- MoS2 (monocapa), independientemente del modo de
obtención, pero sí se detecta una señal débil para N>1. Para MoS2 con N par,
E1g tiene componentes xx e yy distintos de cero (Ec. 3.7), pero su valor debe
ser pequeño ya que la vibración a 285 cm-1 es probablemente idéntica a la del
fonón con N impar, siendo la intensidad para N=2 ligeramente mayor que para
N=3 (Figura 3.26). La detección de una señal débil de este fonón puede estar
originada por la apertura de los objetivos de los microscopios usados en micro-
Raman. El haz incidente proveniente del láser está bien colimado, pero se
enfoca sobre la muestra con el objetivo del microscopio y después se recoge
la luz dispersada con un ángulo sólido finito. Esto hace que en la medida se
incluyan campos eléctricos con pequeñas componentes fuera del plano de la
muestra, lo que permite detectar elementos del tensor Raman fuera del plano
(xz e yz). Como es de esperar, las intensidades del fonón E1g detectadas en
MoS2 de pocas capas son mucho más débiles que para el bulk-MoS2. Sin
embargo, no está claro por qué no se detecta para 1L-MoS2, ya que son válidos
los mismos argumentos geométricos.
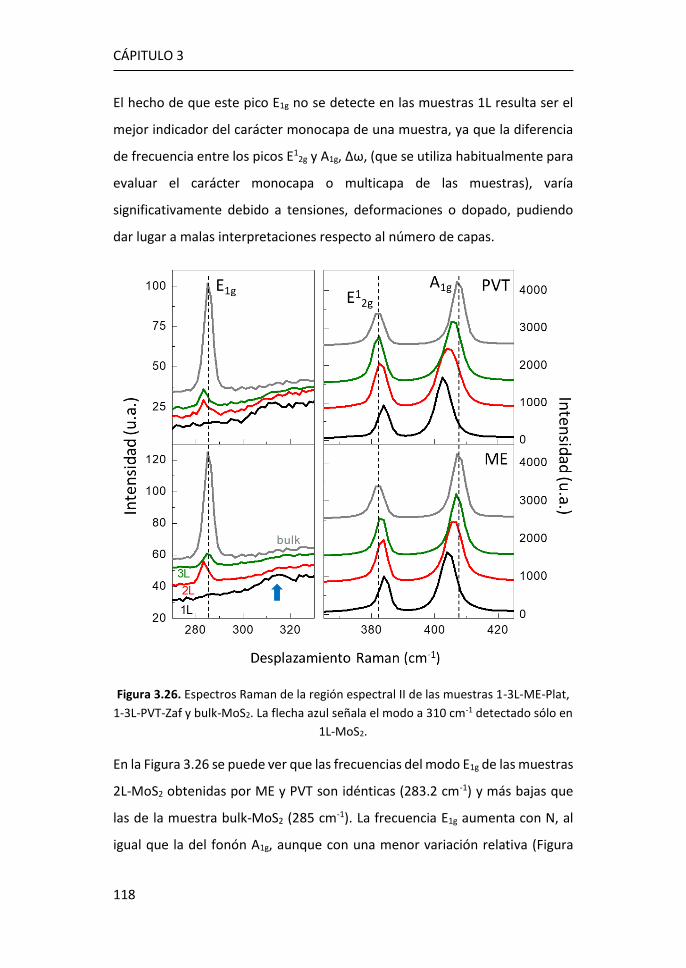
CÁPITULO 3
118
El hecho de que este pico E1g no se detecte en las muestras 1L resulta ser el
mejor indicador del carácter monocapa de una muestra, ya que la diferencia
de frecuencia entre los picos E12g y A1g, Δω, (que se utiliza habitualmente para
evaluar el carácter monocapa o multicapa de las muestras), varía
significativamente debido a tensiones, deformaciones o dopado, pudiendo
dar lugar a malas interpretaciones respecto al número de capas.
Figura 3.26. Espectros Raman de la región espectral II de las muestras 1-3L-ME-Plat,
1-3L-PVT-Zaf y bulk-MoS2. La flecha azul señala el modo a 310 cm-1 detectado sólo en
1L-MoS2.
En la Figura 3.26 se puede ver que las frecuencias del modo E1g de las muestras
2L-MoS2 obtenidas por ME y PVT son idénticas (283.2 cm-1) y más bajas que
las de la muestra bulk-MoS2 (285 cm-1). La frecuencia E1g aumenta con N, al
igual que la del fonón A1g, aunque con una menor variación relativa (Figura

MoS2 bidimensional
119
3.22b). Esta es la tendencia esperada ya que los iones de Mo no participan en
el modo de vibración, al igual que pasa en el modo A1g, y por tanto el
acoplamiento de corto alcance entre capas es dominante.39
El caso contrario ocurre con un pico alrededor de 310 cm-1 que se detecta
exclusivamente en las muestras monocapa, 1L-MoS2, a 311 cm-1 para ME y 315
cm-1 para PVT (flecha azul en la Figura 3.26). Este pico puede asignarse a
E1g(LOQ) (círculo naranja en Figura 3.21). Este pico y la banda azul de la Figura
3.20 en torno a 220 cm-1 (que puede asignarse a LA cerca de Q) son exclusivos
de 1L-MoS2, por lo que pueden estar relacionados con una amplificación de la
intensidad Raman debida a características específicas de la estructura de
bandas del 1L-MoS2. También pueden estar originados por un proceso de
doble resonancia mediado por un defecto, como ocurría con los picos de 150
y 190 cm-1.
En la Figura 3.19a, se muestra la variación en función de N para los modos A1g
y E12g de las muestras 1-10L-ME-Si, 1-10L-ME-Plat, 1-3L-PVT-Zaf y de bulk-
MoS2. Se observa que el comportamiento es muy similar en todos los casos,
independientemente del método de síntesis y del sustrato.
3.2.3 Región III: 420-500 cm-1
En la región III se encuentra una banda compleja situada en el rango 440-480
cm-1 que ha sido estudiada recientemente con gran detalle,40,44
relacionándose con tres procesos Raman de segundo orden (2vHS, 2LA(M) y
2LA(K)), para excitaciones cerca del excitón B (2 eV).40,44 Estas frecuencias
coinciden muy razonablemente con el doble de los valores obtenidos para el
MoS2 en volumen según los datos obtenidos por dispersión inelástica de rayos
X (vHS = 224 cm-1, LA(M) = 235 cm-1 y LA(K) = 239 cm-1).41
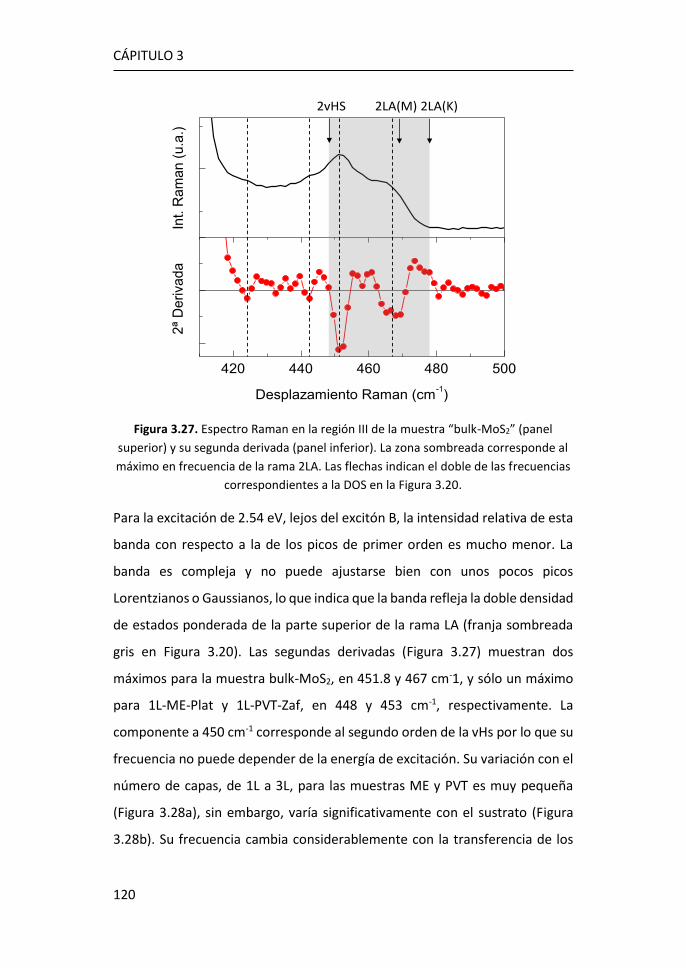
CÁPITULO 3
120
Figura 3.27. Espectro Raman en la región III de la muestra “bulk-MoS2” (panel
superior) y su segunda derivada (panel inferior). La zona sombreada corresponde al
máximo en frecuencia de la rama 2LA. Las flechas indican el doble de las frecuencias
correspondientes a la DOS en la Figura 3.20.
Para la excitación de 2.54 eV, lejos del excitón B, la intensidad relativa de esta
banda con respecto a la de los picos de primer orden es mucho menor. La
banda es compleja y no puede ajustarse bien con unos pocos picos
Lorentzianos o Gaussianos, lo que indica que la banda refleja la doble densidad
de estados ponderada de la parte superior de la rama LA (franja sombreada
gris en Figura 3.20). Las segundas derivadas (Figura 3.27) muestran dos
máximos para la muestra bulk-MoS2, en 451.8 y 467 cm-1, y sólo un máximo
para 1L-ME-Plat y 1L-PVT-Zaf, en 448 y 453 cm-1, respectivamente. La
componente a 450 cm-1 corresponde al segundo orden de la vHs por lo que su
frecuencia no puede depender de la energía de excitación. Su variación con el
número de capas, de 1L a 3L, para las muestras ME y PVT es muy pequeña
(Figura 3.28a), sin embargo, varía significativamente con el sustrato (Figura
3.28b). Su frecuencia cambia considerablemente con la transferencia de los
420 440 460 480 500
Int.
Ram
an (u
.a.)
2ª D
eriv
ada
Desplazamiento Raman (cm-1)
2vHS 2LA(M) 2LA(K)
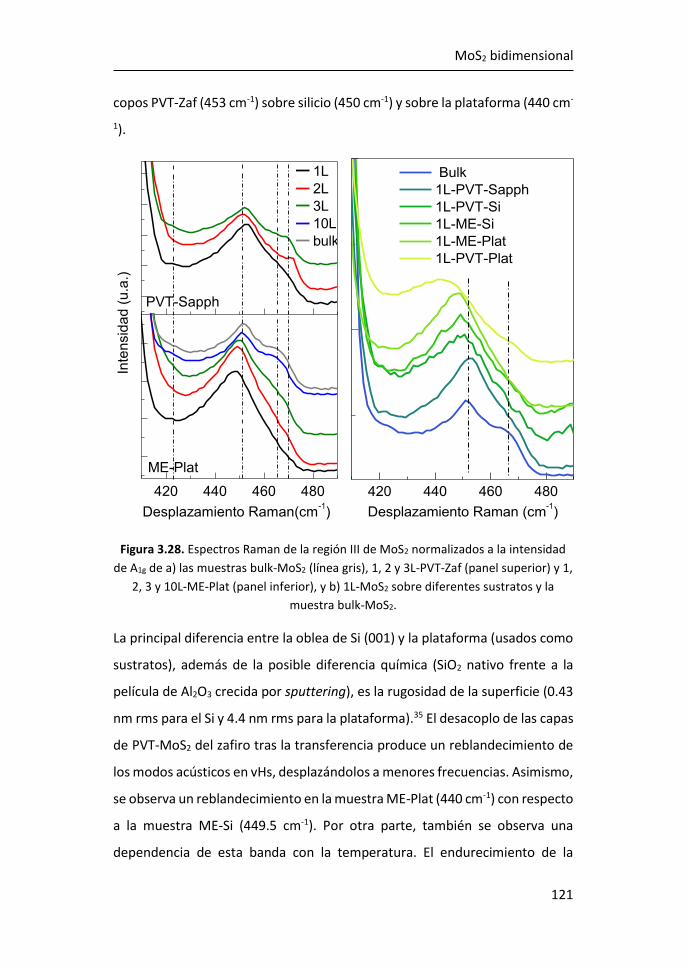
MoS2 bidimensional
121
copos PVT-Zaf (453 cm-1) sobre silicio (450 cm-1) y sobre la plataforma (440 cm-
1).
Figura 3.28. Espectros Raman de la región III de MoS2 normalizados a la intensidad
de A1g de a) las muestras bulk-MoS2 (línea gris), 1, 2 y 3L-PVT-Zaf (panel superior) y 1,
2, 3 y 10L-ME-Plat (panel inferior), y b) 1L-MoS2 sobre diferentes sustratos y la
muestra bulk-MoS2.
La principal diferencia entre la oblea de Si (001) y la plataforma (usados como
sustratos), además de la posible diferencia química (SiO2 nativo frente a la
película de Al2O3 crecida por sputtering), es la rugosidad de la superficie (0.43
nm rms para el Si y 4.4 nm rms para la plataforma).35 El desacoplo de las capas
de PVT-MoS2 del zafiro tras la transferencia produce un reblandecimiento de
los modos acústicos en vHs, desplazándolos a menores frecuencias. Asimismo,
se observa un reblandecimiento en la muestra ME-Plat (440 cm-1) con respecto
a la muestra ME-Si (449.5 cm-1). Por otra parte, también se observa una
dependencia de esta banda con la temperatura. El endurecimiento de la
420 440 460 480Desplazamiento Raman (cm-1)
Bulk 1L-PVT-Sapph 1L-PVT-Si 1L-ME-Si 1L-ME-Plat 1L-PVT-Plat
420 440 460 480
Inte
nsid
ad (u
.a.)
ME-Plat
Desplazamiento Raman(cm-1)
PVT-Sapph
420 440 460 480 500
Inte
nsid
ad R
aman
(u.a
.)
1L 2L 3L 10L bulk
ME-Plat
Desplazamiento Raman (cm-1)
PVT-Zaf
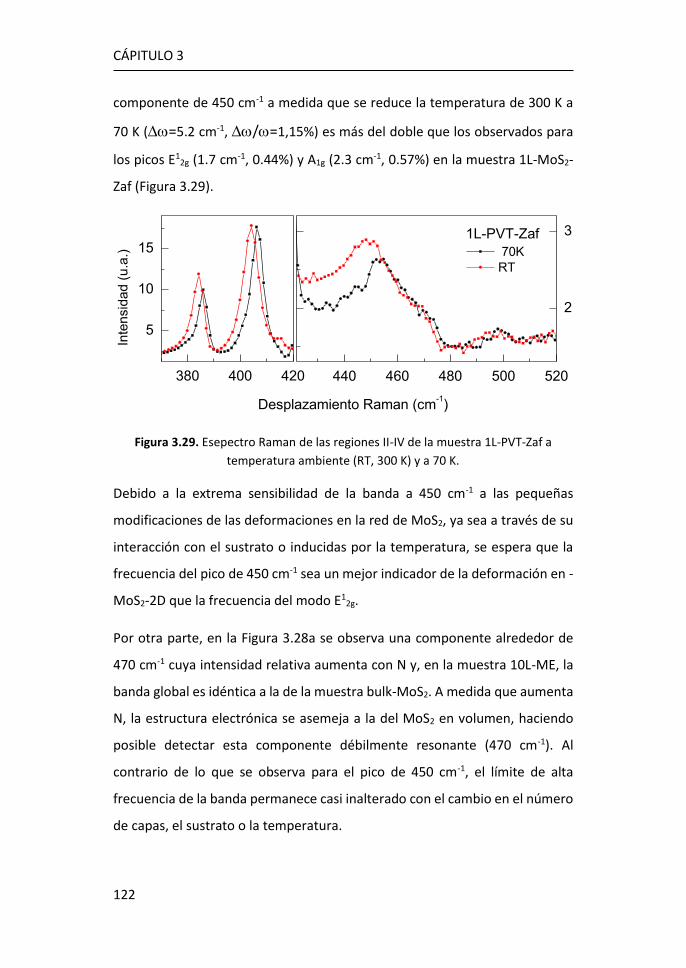
CÁPITULO 3
122
componente de 450 cm-1 a medida que se reduce la temperatura de 300 K a
70 K (=5.2 cm-1, /=1,15%) es más del doble que los observados para
los picos E12g (1.7 cm-1, 0.44%) y A1g (2.3 cm-1, 0.57%) en la muestra 1L-MoS2-
Zaf (Figura 3.29).
Figura 3.29. Esepectro Raman de las regiones II-IV de la muestra 1L-PVT-Zaf a
temperatura ambiente (RT, 300 K) y a 70 K.
Debido a la extrema sensibilidad de la banda a 450 cm-1 a las pequeñas
modificaciones de las deformaciones en la red de MoS2, ya sea a través de su
interacción con el sustrato o inducidas por la temperatura, se espera que la
frecuencia del pico de 450 cm-1 sea un mejor indicador de la deformación en -
MoS2-2D que la frecuencia del modo E12g.
Por otra parte, en la Figura 3.28a se observa una componente alrededor de
470 cm-1 cuya intensidad relativa aumenta con N y, en la muestra 10L-ME, la
banda global es idéntica a la de la muestra bulk-MoS2. A medida que aumenta
N, la estructura electrónica se asemeja a la del MoS2 en volumen, haciendo
posible detectar esta componente débilmente resonante (470 cm-1). Al
contrario de lo que se observa para el pico de 450 cm-1, el límite de alta
frecuencia de la banda permanece casi inalterado con el cambio en el número
de capas, el sustrato o la temperatura.
380 400 420
5
10
15
440 460 480 500 520
2
3 70K RT
Inte
nsid
ad (u
.a.)
1L-PVT-Zaf
Desplazamiento Raman (cm-1)
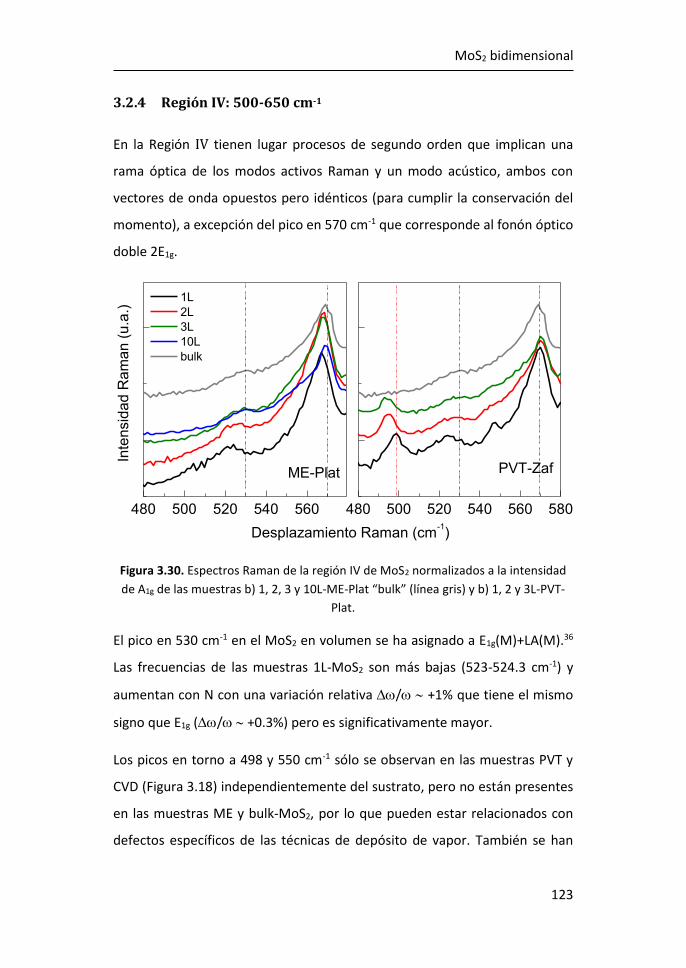
MoS2 bidimensional
123
3.2.4 Región IV: 500-650 cm-1
En la Región IV tienen lugar procesos de segundo orden que implican una
rama óptica de los modos activos Raman y un modo acústico, ambos con
vectores de onda opuestos pero idénticos (para cumplir la conservación del
momento), a excepción del pico en 570 cm-1 que corresponde al fonón óptico
doble 2E1g.
Figura 3.30. Espectros Raman de la región IV de MoS2 normalizados a la intensidad
de A1g de las muestras b) 1, 2, 3 y 10L-ME-Plat “bulk” (línea gris) y b) 1, 2 y 3L-PVT-
Plat.
El pico en 530 cm-1 en el MoS2 en volumen se ha asignado a E1g(M)+LA(M).36
Las frecuencias de las muestras 1L-MoS2 son más bajas (523-524.3 cm-1) y
aumentan con N con una variación relativa / +1% que tiene el mismo
signo que E1g (/ +0.3%) pero es significativamente mayor.
Los picos en torno a 498 y 550 cm-1 sólo se observan en las muestras PVT y
CVD (Figura 3.18) independientemente del sustrato, pero no están presentes
en las muestras ME y bulk-MoS2, por lo que pueden estar relacionados con
defectos específicos de las técnicas de depósito de vapor. También se han
480 500 520 540 560 480 500 520 540 560 580
Inte
nsid
ad R
aman
(u.a
.)
1L 2L 3L 10L bulk
ME-Plat
Desplazamiento Raman (cm-1)
PVT-Zaf

CÁPITULO 3
124
reportado picos con frecuencias similares en nanopartículas de MoS2 tipo
fullereno y plaquetas.36
Observando la relación de dispersión de los fonones obtenida por dispersión
inelástica de rayos X41 (Figura 3.21) es posible relacionar el pico de 498 cm-1
con combinaciones de modos acústicos y Raman con vectores de onda Q,
como LA(Q) + E1g (LOQ) o TA(Q) + E12g(LOQ). Alternativamente, estos dos picos
podrían deberse a modos locales relacionados con defectos resultantes de la
síntesis de MoS2 que no están presentes en el MoS2 obtenido por exfoliación.
3.2.5 Región V: 650-850 cm-1
En la Región V, se encuentran los modos de segundo orden originados por
combinaciones de los modos activos Raman. La banda más interesante es la
etiquetada como 2A1g, cuya frecuencia, sin embargo, no corresponde al doble
de la frecuencia de A1g() en ninguno de los casos. En el caso de la muestra
bulk-MoS2 aparece en 819 cm-1, una frecuencia mayor que el doble de A1g()
(2 x 407.5 = 815 cm-1). Probablemente su frecuencia esté relacionada con el
máximo del fonón en DOS, que coincide con el valor de A1g(M) a 410 cm-1
reportado por dispersión inelástica de rayos X para el MoS2 en volumen.41 En
el caso de 1L-MoS2, este pico se detecta a una frecuencia casi idéntica
(ligeramente superior) a la de la muestra bulk-MoS2, 820 cm-1 en ME y 822 cm-
1 en PVT, mientras que la frecuencia de A1g es significativamente inferior (402.8
y 404.3 cm-1, para ME y PVT respectivamente). Esto indica que la frecuencia
de la rama A1g se ve afectada de forma diferente por las capas adyacentes
cuando el vector de onda está en o en un borde de zona.
En resumen, las muestras estudiadas encajan dentro de los estándares de
muestras sin defectos, pero presentan picos prohibidos de primer orden que
corresponden a ramas acústicas activados por la presencia de defectos
intrínsecos. Dependiendo del proceso Raman que da lugar a cada uno de los

MoS2 bidimensional
125
fonones, éstos son sensibles a diferentes variables como el número de capas,
el método de crecimiento o el sustrato. En el caso de los picos de 150 y 190
cm-1, los cambios de frecuencia observados con el número de capas pueden
relacionarse con las modificaciones en la estructura de la banda electrónica, a
través de un proceso de doble resonancia mediado por defectos intrínsecos,
que activa los fonones alrededor del punto Q de la zona de Brillouin. Se
observa un comportamiento opuesto para la banda compleja en el rango 420-
470 cm-1 (procesos de dos fonones), que es resonante para energías de
excitación cercanas al excitón B (2 eV), y que, fuera de esta resonancia, para
una excitación en 2.54 eV, es especialmente sensible a la interacción MoS2-2D
con el sustrato y a los cambios en la temperatura. Algunos picos son
específicos de las técnicas de depósito de vapor y también se han detectado
en nanopartículas, lo que indica que estas muestras pueden contener defectos
que no están presentes en las muestras exfoliadas mecánicamente o en el
MoS2 en volumen. Otros picos sólo se detectan en 1L-MoS2,
independientemente de su origen. En este trabajo se proponen nuevas
asignaciones para varios picos (Tabla 3.1) basándonos en los resultados aquí
expuestos y respaldados por la relación de dispersión de fonones obtenida
experimentalmente publicadas recientemente para el MoS2 en volumen.

CÁPITULO 3
126
3.3 EFECTOS DEL TAMAÑO EN NANOCOPOS DE MOS2 DE UNA Y
POCAS CAPAS
El diseño y la fabricación de nanodots, o nanocopos de materiales 2D,
inicialmente de grafeno y más recientemente de TMDCs, es un tema de
creciente interés debido a las nuevas posibilidades que ofrecen estos
materiales. Los nanodots presentan una emisión en el visible cuya longitud de
onda depende del tamaño, aunque su relación no está del todo clara. Por
ejemplo, se ha reportado que la emisión varía de 480 a 500 nm para diámetros
de 2 a 26 nm,45 pero también se ha publicado una variación mucho mayor (430
- 610 nm) para un rango de tamaño de partícula más estrecho (2-7 nm).46 Esta
sintonización de la fotoluminiscencia con el tamaño hace que los nanodots de
MoS2 sean de gran interés.
La reducción del tamaño de MoS2 monocapa es un objetivo y también un reto,
ya que el control necesario para obtener monocapas reales y copos
nanométricos de alta calidad no es sencillo. La caracterización del MoS2
monocapa de tamaño nanométrico también puede verse comprometida, ya
que los procedimientos estándar pueden no funcionar. Por ejemplo, el
parámetro más utilizado para la estimación del número de capas, Δω, puede
no ser adecuado si las muestras crecidas por CVD o PVT presentan defectos,
ya que las frecuencias de E12g y A1g pueden verse alteradas (ver Figuras 3.12 y
3.13). En la Figura 3.13 se observa que las muestras de partida de cada trabajo
muestran una gran variabilidad de los valores E12g, A1g y Δω
independientemente del método de obtención.
Aquí se van a estudiar las características de copos de MoS2 de una y pocas
capas con tamaños que van de 10 nm a 4 μm, combinando la espectroscopía
Raman con la de reflectancia y la fotoluminiscencia, lo que permite discernir
entre los efectos en los espectros Raman originados por los diferentes tipos
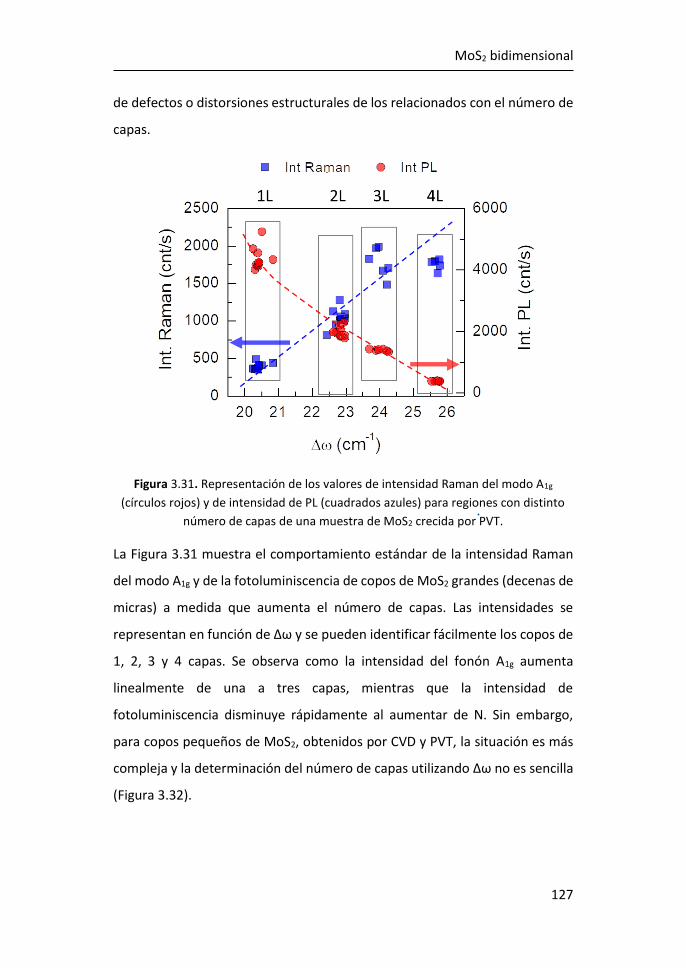
MoS2 bidimensional
127
de defectos o distorsiones estructurales de los relacionados con el número de
capas.
Figura 3.31. Representación de los valores de intensidad Raman del modo A1g
(círculos rojos) y de intensidad de PL (cuadrados azules) para regiones con distinto
número de capas de una muestra de MoS2 crecida por PVT.
La Figura 3.31 muestra el comportamiento estándar de la intensidad Raman
del modo A1g y de la fotoluminiscencia de copos de MoS2 grandes (decenas de
micras) a medida que aumenta el número de capas. Las intensidades se
representan en función de Δω y se pueden identificar fácilmente los copos de
1, 2, 3 y 4 capas. Se observa como la intensidad del fonón A1g aumenta
linealmente de una a tres capas, mientras que la intensidad de
fotoluminiscencia disminuye rápidamente al aumentar de N. Sin embargo,
para copos pequeños de MoS2, obtenidos por CVD y PVT, la situación es más
compleja y la determinación del número de capas utilizando Δω no es sencilla
(Figura 3.32).
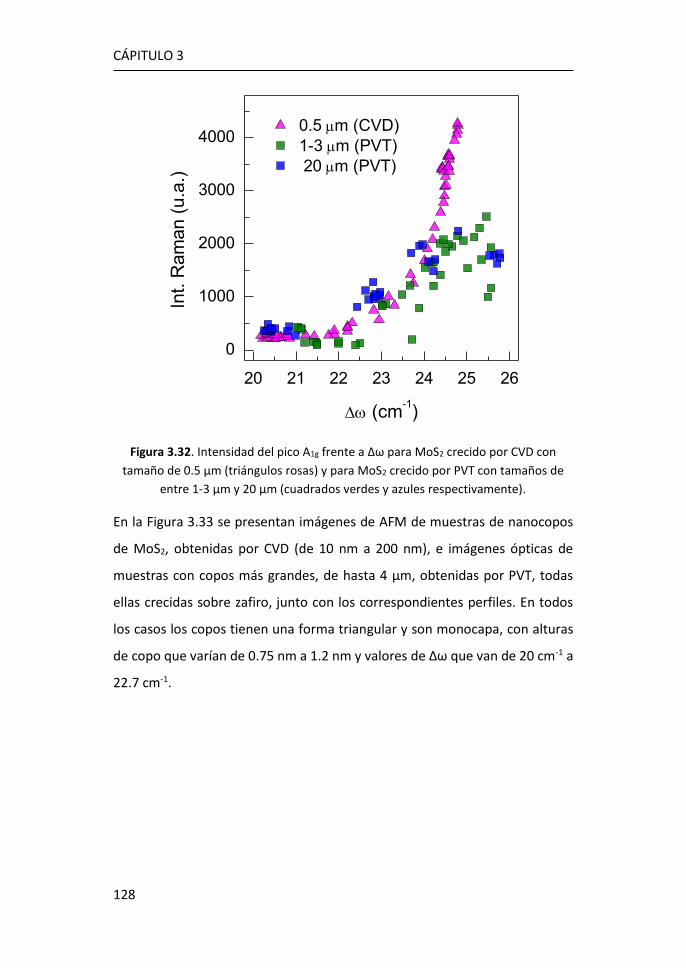
CÁPITULO 3
128
Figura 3.32. Intensidad del pico A1g frente a Δω para MoS2 crecido por CVD con
tamaño de 0.5 µm (triángulos rosas) y para MoS2 crecido por PVT con tamaños de
entre 1-3 µm y 20 µm (cuadrados verdes y azules respectivamente).
En la Figura 3.33 se presentan imágenes de AFM de muestras de nanocopos
de MoS2, obtenidas por CVD (de 10 nm a 200 nm), e imágenes ópticas de
muestras con copos más grandes, de hasta 4 µm, obtenidas por PVT, todas
ellas crecidas sobre zafiro, junto con los correspondientes perfiles. En todos
los casos los copos tienen una forma triangular y son monocapa, con alturas
de copo que varían de 0.75 nm a 1.2 nm y valores de Δω que van de 20 cm-1 a
22.7 cm-1.
20 21 22 23 24 25 26
0
1000
2000
3000
4000
0.5 m (CVD)1-3 m (PVT) 20 m (PVT)
Int.
Ram
an (u
.a.)
(cm-1)
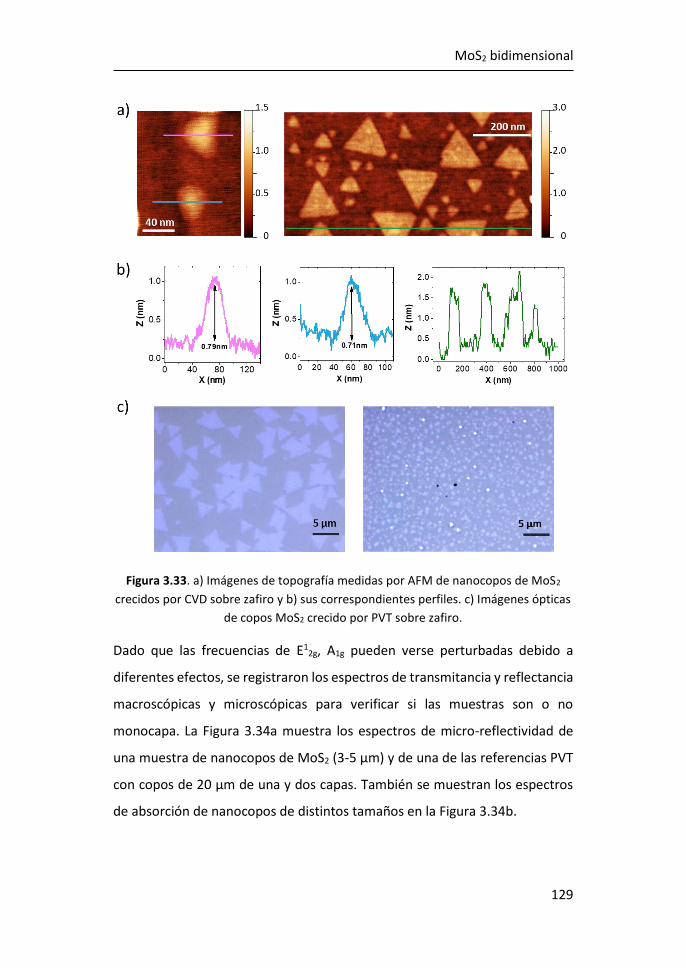
MoS2 bidimensional
129
Figura 3.33. a) Imágenes de topografía medidas por AFM de nanocopos de MoS2
crecidos por CVD sobre zafiro y b) sus correspondientes perfiles. c) Imágenes ópticas
de copos MoS2 crecido por PVT sobre zafiro.
Dado que las frecuencias de E12g, A1g pueden verse perturbadas debido a
diferentes efectos, se registraron los espectros de transmitancia y reflectancia
macroscópicas y microscópicas para verificar si las muestras son o no
monocapa. La Figura 3.34a muestra los espectros de micro-reflectividad de
una muestra de nanocopos de MoS2 (3-5 μm) y de una de las referencias PVT
con copos de 20 μm de una y dos capas. También se muestran los espectros
de absorción de nanocopos de distintos tamaños en la Figura 3.34b.
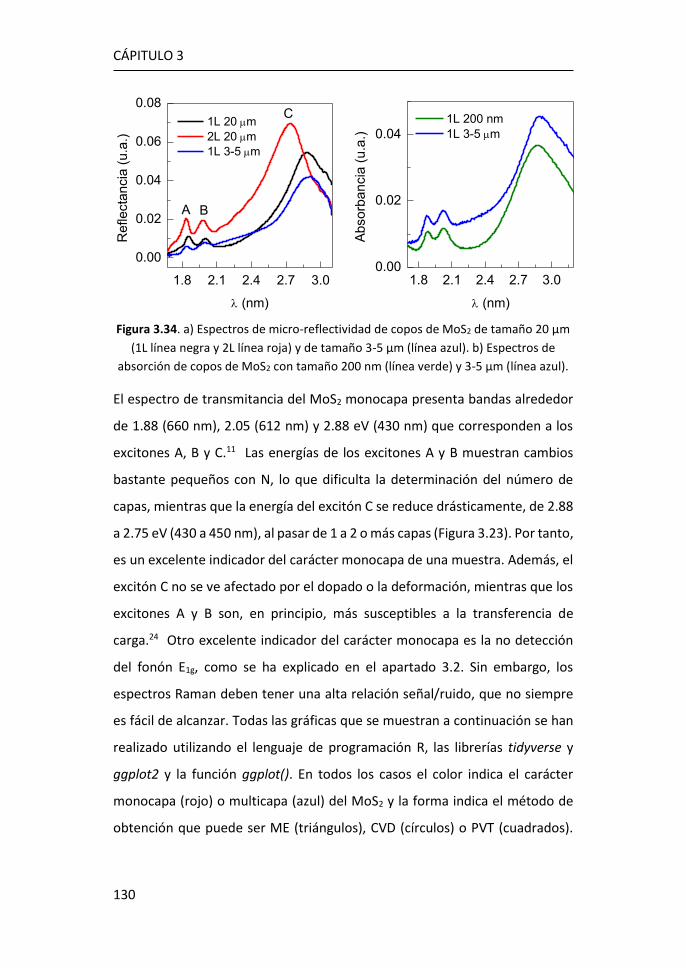
CÁPITULO 3
130
Figura 3.34. a) Espectros de micro-reflectividad de copos de MoS2 de tamaño 20 μm
(1L línea negra y 2L línea roja) y de tamaño 3-5 μm (línea azul). b) Espectros de
absorción de copos de MoS2 con tamaño 200 nm (línea verde) y 3-5 μm (línea azul).
El espectro de transmitancia del MoS2 monocapa presenta bandas alrededor
de 1.88 (660 nm), 2.05 (612 nm) y 2.88 eV (430 nm) que corresponden a los
excitones A, B y C.11 Las energías de los excitones A y B muestran cambios
bastante pequeños con N, lo que dificulta la determinación del número de
capas, mientras que la energía del excitón C se reduce drásticamente, de 2.88
a 2.75 eV (430 a 450 nm), al pasar de 1 a 2 o más capas (Figura 3.23). Por tanto,
es un excelente indicador del carácter monocapa de una muestra. Además, el
excitón C no se ve afectado por el dopado o la deformación, mientras que los
excitones A y B son, en principio, más susceptibles a la transferencia de
carga.24 Otro excelente indicador del carácter monocapa es la no detección
del fonón E1g, como se ha explicado en el apartado 3.2. Sin embargo, los
espectros Raman deben tener una alta relación señal/ruido, que no siempre
es fácil de alcanzar. Todas las gráficas que se muestran a continuación se han
realizado utilizando el lenguaje de programación R, las librerías tidyverse y
ggplot2 y la función ggplot(). En todos los casos el color indica el carácter
monocapa (rojo) o multicapa (azul) del MoS2 y la forma indica el método de
obtención que puede ser ME (triángulos), CVD (círculos) o PVT (cuadrados).
1.8 2.1 2.4 2.7 3.0
0.00
0.02
0.04
0.06
0.08C
B
1L 20 m 2L 20 m 1L 3-5 m
Ref
lect
anci
a (u
.a.)
(nm)
A
1.8 2.1 2.4 2.7 3.00.00
0.02
0.04
Abso
rban
cia
(u.a
.)
(nm)
1L 200 nm 1L 3-5 m
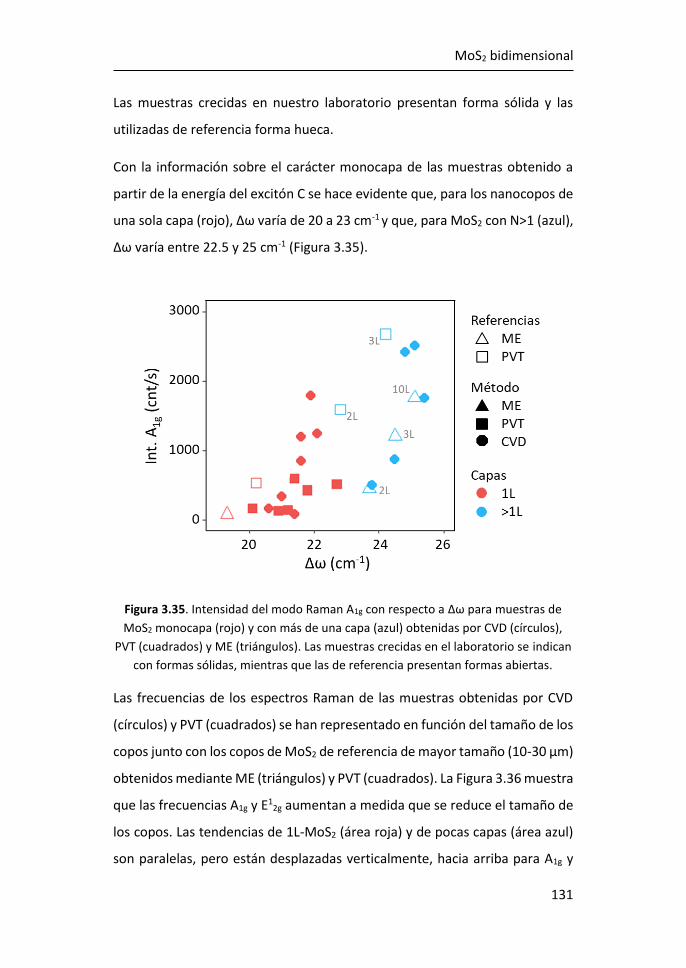
MoS2 bidimensional
131
Las muestras crecidas en nuestro laboratorio presentan forma sólida y las
utilizadas de referencia forma hueca.
Con la información sobre el carácter monocapa de las muestras obtenido a
partir de la energía del excitón C se hace evidente que, para los nanocopos de
una sola capa (rojo), Δω varía de 20 a 23 cm-1 y que, para MoS2 con N>1 (azul),
Δω varía entre 22.5 y 25 cm-1 (Figura 3.35).
Figura 3.35. Intensidad del modo Raman A1g con respecto a Δω para muestras de
MoS2 monocapa (rojo) y con más de una capa (azul) obtenidas por CVD (círculos),
PVT (cuadrados) y ME (triángulos). Las muestras crecidas en el laboratorio se indican
con formas sólidas, mientras que las de referencia presentan formas abiertas.
Las frecuencias de los espectros Raman de las muestras obtenidas por CVD
(círculos) y PVT (cuadrados) se han representado en función del tamaño de los
copos junto con los copos de MoS2 de referencia de mayor tamaño (10-30 μm)
obtenidos mediante ME (triángulos) y PVT (cuadrados). La Figura 3.36 muestra
que las frecuencias A1g y E12g aumentan a medida que se reduce el tamaño de
los copos. Las tendencias de 1L-MoS2 (área roja) y de pocas capas (área azul)
son paralelas, pero están desplazadas verticalmente, hacia arriba para A1g y
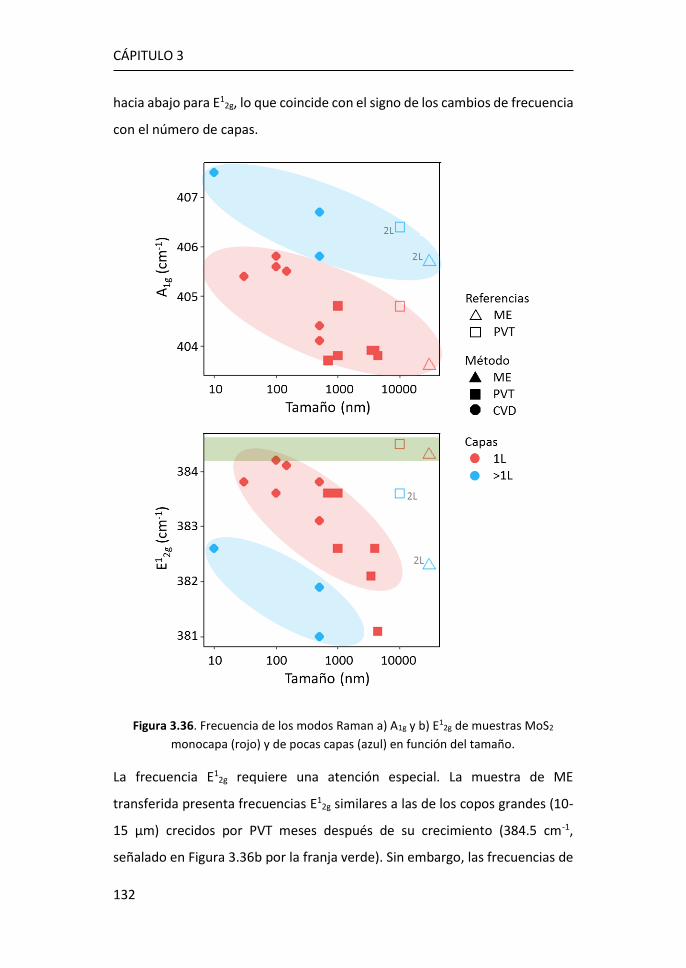
CÁPITULO 3
132
hacia abajo para E12g, lo que coincide con el signo de los cambios de frecuencia
con el número de capas.
Figura 3.36. Frecuencia de los modos Raman a) A1g y b) E12g de muestras MoS2
monocapa (rojo) y de pocas capas (azul) en función del tamaño.
La frecuencia E12g requiere una atención especial. La muestra de ME
transferida presenta frecuencias E12g similares a las de los copos grandes (10-
15 μm) crecidos por PVT meses después de su crecimiento (384.5 cm-1,
señalado en Figura 3.36b por la franja verde). Sin embargo, las frecuencias de
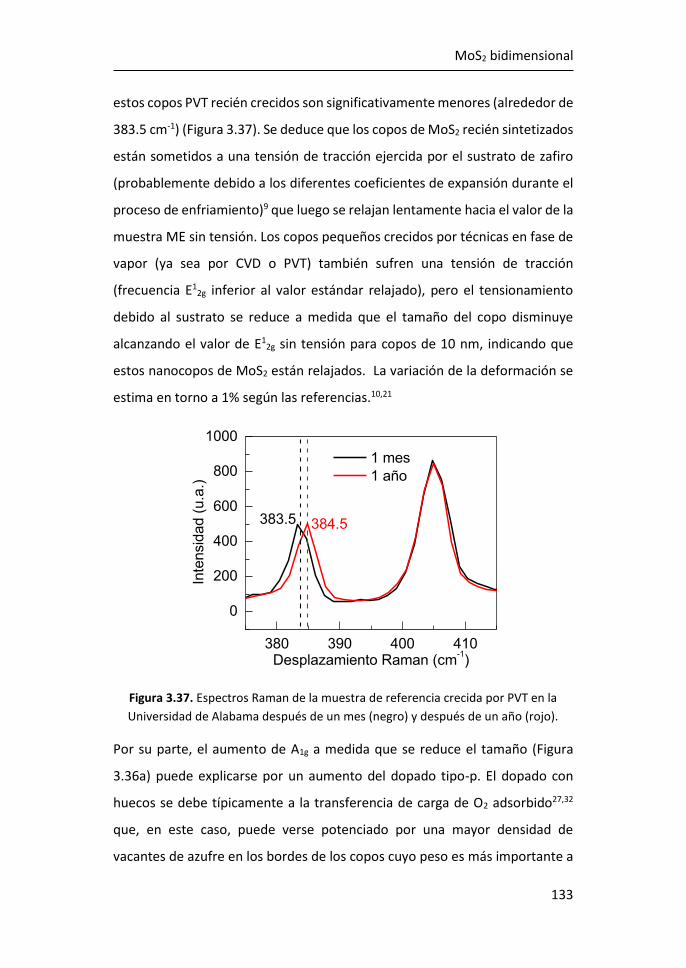
MoS2 bidimensional
133
estos copos PVT recién crecidos son significativamente menores (alrededor de
383.5 cm-1) (Figura 3.37). Se deduce que los copos de MoS2 recién sintetizados
están sometidos a una tensión de tracción ejercida por el sustrato de zafiro
(probablemente debido a los diferentes coeficientes de expansión durante el
proceso de enfriamiento)9 que luego se relajan lentamente hacia el valor de la
muestra ME sin tensión. Los copos pequeños crecidos por técnicas en fase de
vapor (ya sea por CVD o PVT) también sufren una tensión de tracción
(frecuencia E12g inferior al valor estándar relajado), pero el tensionamiento
debido al sustrato se reduce a medida que el tamaño del copo disminuye
alcanzando el valor de E12g sin tensión para copos de 10 nm, indicando que
estos nanocopos de MoS2 están relajados. La variación de la deformación se
estima en torno a 1% según las referencias.10,21
Figura 3.37. Espectros Raman de la muestra de referencia crecida por PVT en la
Universidad de Alabama después de un mes (negro) y después de un año (rojo).
Por su parte, el aumento de A1g a medida que se reduce el tamaño (Figura
3.36a) puede explicarse por un aumento del dopado tipo-p. El dopado con
huecos se debe típicamente a la transferencia de carga de O2 adsorbido27,32
que, en este caso, puede verse potenciado por una mayor densidad de
vacantes de azufre en los bordes de los copos cuyo peso es más importante a
380 390 400 410
0
200
400
600
800
1000
384.5
Inte
nsid
ad (u
.a.)
Desplazamiento Raman (cm-1)
1 mes 1 año
383.5
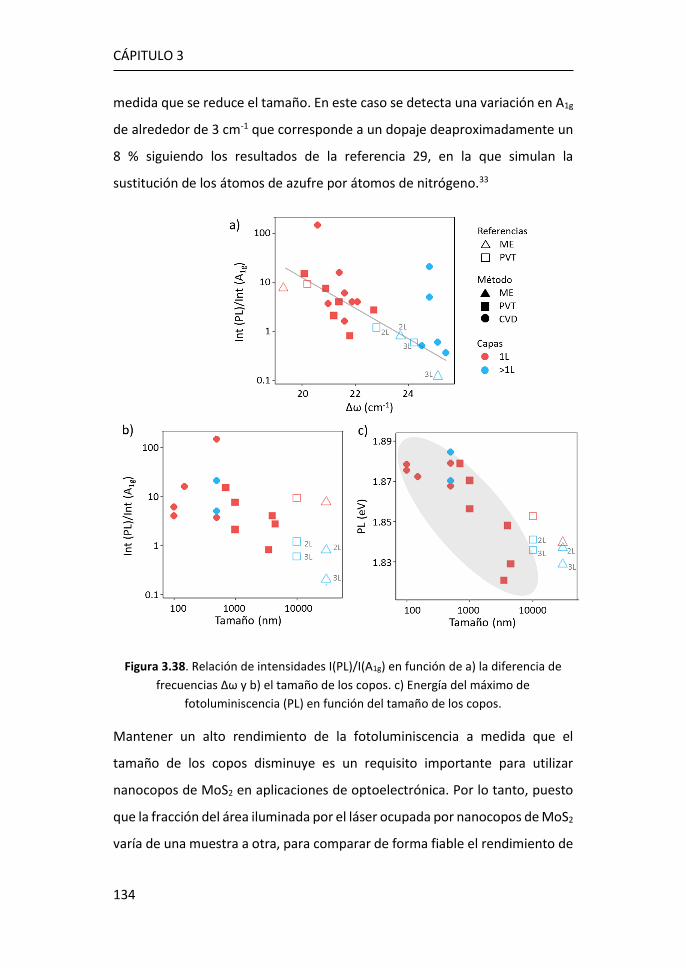
CÁPITULO 3
134
medida que se reduce el tamaño. En este caso se detecta una variación en A1g
de alrededor de 3 cm-1 que corresponde a un dopaje deaproximadamente un
8 % siguiendo los resultados de la referencia 29, en la que simulan la
sustitución de los átomos de azufre por átomos de nitrógeno.33
Figura 3.38. Relación de intensidades I(PL)/I(A1g) en función de a) la diferencia de
frecuencias Δω y b) el tamaño de los copos. c) Energía del máximo de
fotoluminiscencia (PL) en función del tamaño de los copos.
Mantener un alto rendimiento de la fotoluminiscencia a medida que el
tamaño de los copos disminuye es un requisito importante para utilizar
nanocopos de MoS2 en aplicaciones de optoelectrónica. Por lo tanto, puesto
que la fracción del área iluminada por el láser ocupada por nanocopos de MoS2
varía de una muestra a otra, para comparar de forma fiable el rendimiento de

MoS2 bidimensional
135
la fotoluminiscencia de las diferentes muestras, se ha empleado la relación de
intensidades entre la banda de fotoluminiscencia y el pico A1g, I(PL)/I(A1g),
utilizando la señal Raman para normalizar y hacer comparables los datos. La
Figura 3.38 presenta la dependencia de esta relación, I(PL)/I(A1g), con el
tamaño de los copos (Figura 3.38b) y con la diferencia de frecuencias, Δω
(Figura 3.38a). I(PL)/I(A1g) no parece estar correlacionada con el tamaño
(figura 3.38b) de las muestras monocapa (color rojo), pero sí con Δω (figura
3.38a). La dependencia exponencial (nótese la escala logarítmica para el
cociente de intensidad) se observa en todo el rango de Δω, de 19 a 25 cm-1,
que incluye muestras de 1L (color rojo) y de pocas capas (color azul), así como
las muestras de referencia ME y PVT (formas abiertas). La Figura 3.38 indica
que el aumento de Δω, independientemente de su origen, disminuye la
eficiencia de la fotoluminiscencia. También se observa un aumento de hasta
50 meV en la energía de la banda de fotoluminiscencia a medida que el
tamaño de los copos disminuye (Figura 3.38c). Además, la Figura 3.39 muestra
que las anchuras (FWHM) de los picos A1g y E12g aumentan con Δω. La
presencia de defectos da lugar a un ensanchamiento de los picos Raman y a
un incremento de Δω (Figura 3.11).12 Por lo tanto, la reducción de la eficiencia
de fotoluminiscencia, asociada a un incremento de Δω, debe estar relacionada
con un aumento del desorden estructural, y no con el tamaño de los copos.
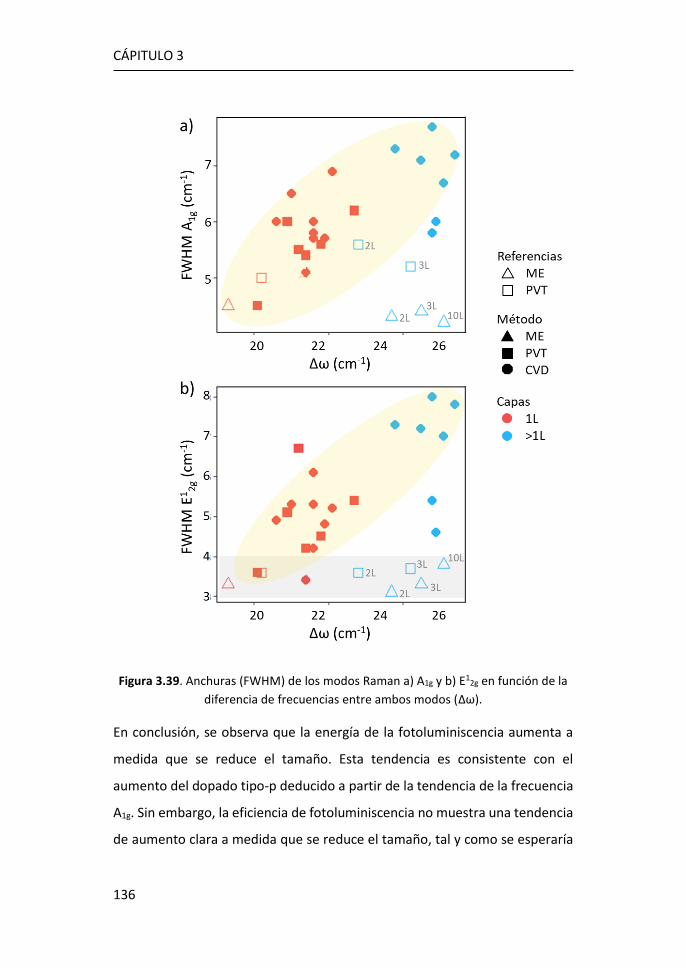
CÁPITULO 3
136
Figura 3.39. Anchuras (FWHM) de los modos Raman a) A1g y b) E12g en función de la
diferencia de frecuencias entre ambos modos (Δω).
En conclusión, se observa que la energía de la fotoluminiscencia aumenta a
medida que se reduce el tamaño. Esta tendencia es consistente con el
aumento del dopado tipo-p deducido a partir de la tendencia de la frecuencia
A1g. Sin embargo, la eficiencia de fotoluminiscencia no muestra una tendencia
de aumento clara a medida que se reduce el tamaño, tal y como se esperaría

MoS2 bidimensional
137
para el dopado tipo-p. Esto podría deberse al desorden estructural
relacionado con la reducción de tamaño, compensando el aumento esperado
en la fotoluminiscencia y dando lugar a intensidades dentro de la dispersión
de valores que se registran para las monocapas de MoS2 de mayor tamaño sin
defectos. Por tanto, se han obtenido nanocopos monocapa de MoS2 en un
amplio rango de tamaños, desde 100 nm hasta varias micras, con una
excelente eficiencia de emisión.
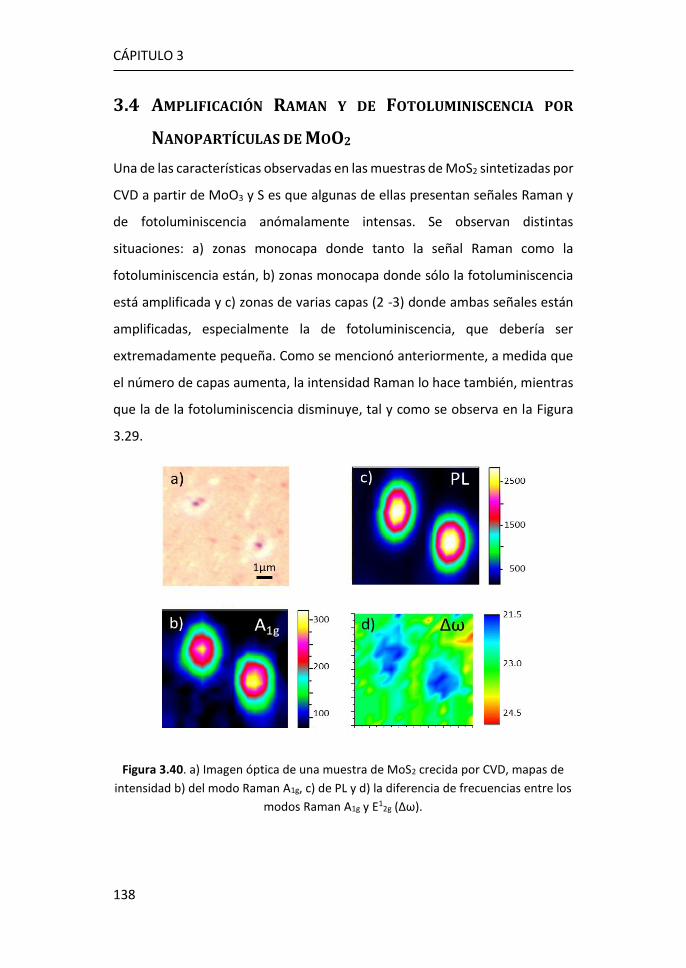
CÁPITULO 3
138
3.4 AMPLIFICACIÓN RAMAN Y DE FOTOLUMINISCENCIA POR
NANOPARTÍCULAS DE MOO2
Una de las características observadas en las muestras de MoS2 sintetizadas por
CVD a partir de MoO3 y S es que algunas de ellas presentan señales Raman y
de fotoluminiscencia anómalamente intensas. Se observan distintas
situaciones: a) zonas monocapa donde tanto la señal Raman como la
fotoluminiscencia están, b) zonas monocapa donde sólo la fotoluminiscencia
está amplificada y c) zonas de varias capas (2 -3) donde ambas señales están
amplificadas, especialmente la de fotoluminiscencia, que debería ser
extremadamente pequeña. Como se mencionó anteriormente, a medida que
el número de capas aumenta, la intensidad Raman lo hace también, mientras
que la de la fotoluminiscencia disminuye, tal y como se observa en la Figura
3.29.
Figura 3.40. a) Imagen óptica de una muestra de MoS2 crecida por CVD, mapas de
intensidad b) del modo Raman A1g, c) de PL y d) la diferencia de frecuencias entre los
modos Raman A1g y E12g (Δω).
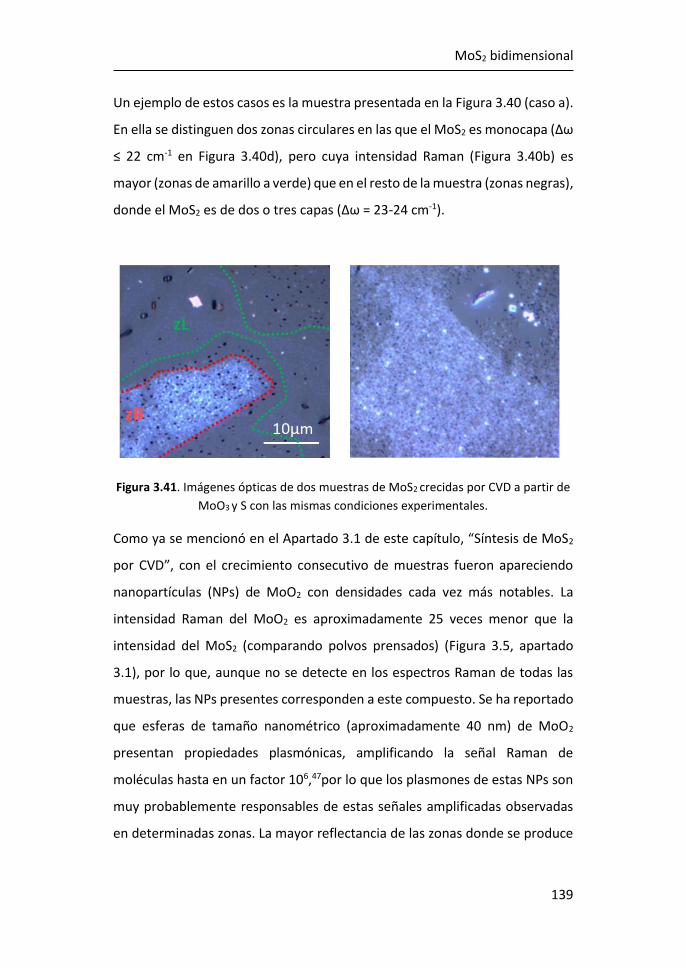
MoS2 bidimensional
139
Un ejemplo de estos casos es la muestra presentada en la Figura 3.40 (caso a).
En ella se distinguen dos zonas circulares en las que el MoS2 es monocapa (Δω
≤ 22 cm-1 en Figura 3.40d), pero cuya intensidad Raman (Figura 3.40b) es
mayor (zonas de amarillo a verde) que en el resto de la muestra (zonas negras),
donde el MoS2 es de dos o tres capas (Δω = 23-24 cm-1).
Figura 3.41. Imágenes ópticas de dos muestras de MoS2 crecidas por CVD a partir de
MoO3 y S con las mismas condiciones experimentales.
Como ya se mencionó en el Apartado 3.1 de este capítulo, “Síntesis de MoS2
por CVD”, con el crecimiento consecutivo de muestras fueron apareciendo
nanopartículas (NPs) de MoO2 con densidades cada vez más notables. La
intensidad Raman del MoO2 es aproximadamente 25 veces menor que la
intensidad del MoS2 (comparando polvos prensados) (Figura 3.5, apartado
3.1), por lo que, aunque no se detecte en los espectros Raman de todas las
muestras, las NPs presentes corresponden a este compuesto. Se ha reportado
que esferas de tamaño nanométrico (aproximadamente 40 nm) de MoO2
presentan propiedades plasmónicas, amplificando la señal Raman de
moléculas hasta en un factor 106,47por lo que los plasmones de estas NPs son
muy probablemente responsables de estas señales amplificadas observadas
en determinadas zonas. La mayor reflectancia de las zonas donde se produce
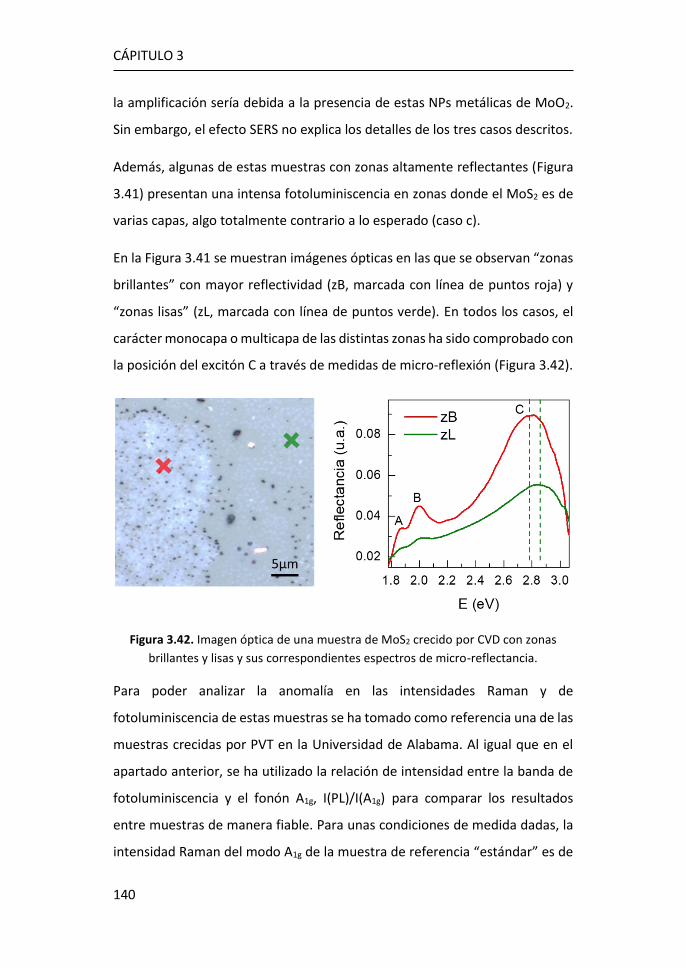
CÁPITULO 3
140
la amplificación sería debida a la presencia de estas NPs metálicas de MoO2.
Sin embargo, el efecto SERS no explica los detalles de los tres casos descritos.
Además, algunas de estas muestras con zonas altamente reflectantes (Figura
3.41) presentan una intensa fotoluminiscencia en zonas donde el MoS2 es de
varias capas, algo totalmente contrario a lo esperado (caso c).
En la Figura 3.41 se muestran imágenes ópticas en las que se observan “zonas
brillantes” con mayor reflectividad (zB, marcada con línea de puntos roja) y
“zonas lisas” (zL, marcada con línea de puntos verde). En todos los casos, el
carácter monocapa o multicapa de las distintas zonas ha sido comprobado con
la posición del excitón C a través de medidas de micro-reflexión (Figura 3.42).
Figura 3.42. Imagen óptica de una muestra de MoS2 crecido por CVD con zonas
brillantes y lisas y sus correspondientes espectros de micro-reflectancia.
Para poder analizar la anomalía en las intensidades Raman y de
fotoluminiscencia de estas muestras se ha tomado como referencia una de las
muestras crecidas por PVT en la Universidad de Alabama. Al igual que en el
apartado anterior, se ha utilizado la relación de intensidad entre la banda de
fotoluminiscencia y el fonón A1g, I(PL)/I(A1g) para comparar los resultados
entre muestras de manera fiable. Para unas condiciones de medida dadas, la
intensidad Raman del modo A1g de la muestra de referencia “estándar” es de
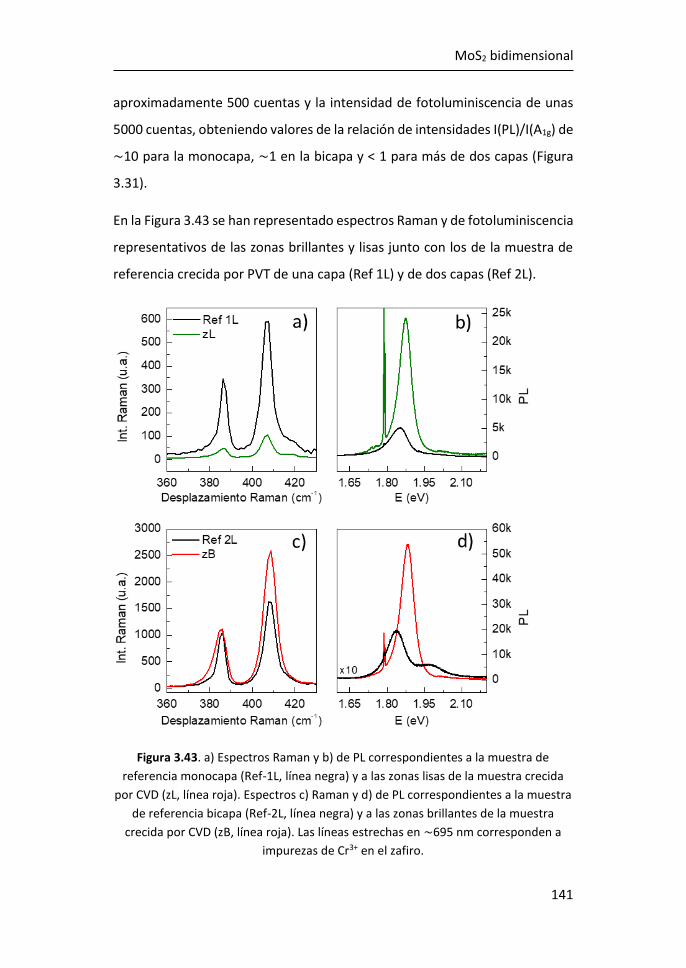
MoS2 bidimensional
141
aproximadamente 500 cuentas y la intensidad de fotoluminiscencia de unas
5000 cuentas, obteniendo valores de la relación de intensidades I(PL)/I(A1g) de
~10 para la monocapa, ~1 en la bicapa y < 1 para más de dos capas (Figura
3.31).
En la Figura 3.43 se han representado espectros Raman y de fotoluminiscencia
representativos de las zonas brillantes y lisas junto con los de la muestra de
referencia crecida por PVT de una capa (Ref 1L) y de dos capas (Ref 2L).
Figura 3.43. a) Espectros Raman y b) de PL correspondientes a la muestra de
referencia monocapa (Ref-1L, línea negra) y a las zonas lisas de la muestra crecida
por CVD (zL, línea roja). Espectros c) Raman y d) de PL correspondientes a la muestra
de referencia bicapa (Ref-2L, línea negra) y a las zonas brillantes de la muestra
crecida por CVD (zB, línea roja). Las líneas estrechas en ~695 nm corresponden a
impurezas de Cr3+ en el zafiro.

CÁPITULO 3
142
El espectro Raman de la zona lisa (zL) indica que se trata de MoS2 monocapa
(Δω=20.5 cm-1) y su ratio I(PL)/I(A1g) es de aproximadamente 150, unas 15
veces mayor que el correspondiente a la muestra de referencia monocapa. En
este caso no se da una amplificación de la señal Raman, pero sí un gran
incremento (x15) de la fotoluminiscencia (caso b).
En la zona brillante (zB), los espectros Raman obtenidos muestran que el MoS2
es de unas pocas capas (Δω ~24 cm-1) y el cociente I(PL)/I(A1g) es ~25 (caso c).
Como ya se ha mencionado, normalmente para MoS2 de más de una capa
I(PL)/I(A1g) suele ser igual o inferior a 1. Sin embargo, en esta muestra es
sorprendentemente alto (x25), incluso mayor que el obtenido para el MoS2
monocapa de referencia. Además de la anomalía observada con respecto a la
intensa fotoluminiscencia que presentan las zonas de pocas capas (zB), la
intensidad Raman de estas zonas también es mayor a la esperada. La
intensidad Raman es proporcional, en general, al volumen de muestra que
origina la señal, por lo que en el MoS2 de pocas capas (entre 2-3L según la
diferencia de frecuencias entre los picos A1g y E12g) debería observarse una
intensidad Raman entre 2 y 3 veces mayor que en las zonas monocapa, como
se muestra en la Figura 3.31. Sin embargo, la intensidad es aproximadamente
20 veces mayor. Desgraciadamente, no es posible calcular el plasmón del
MoO2 en función del tamaño, como se ha hecho en el Capítulo 4 para las NPs
de plata, ya que en este caso no ha sido posible encontrar en la literatura el
índice de refracción (o la constante dieléctrica), ni siquiera para longitudes de
onda concretas. Hay que señalar que la obtención de películas de MoO2 con
fase pura no es sencilla y que hay pocos datos acerca de este compuesto.
Tampoco se han encontrado medidas experimentales del plasmón,
seguramente porque las películas o NPs generadas suelen contener varias
fases MoOx (en muchos casos con MoO3 y otras estequiometrías).
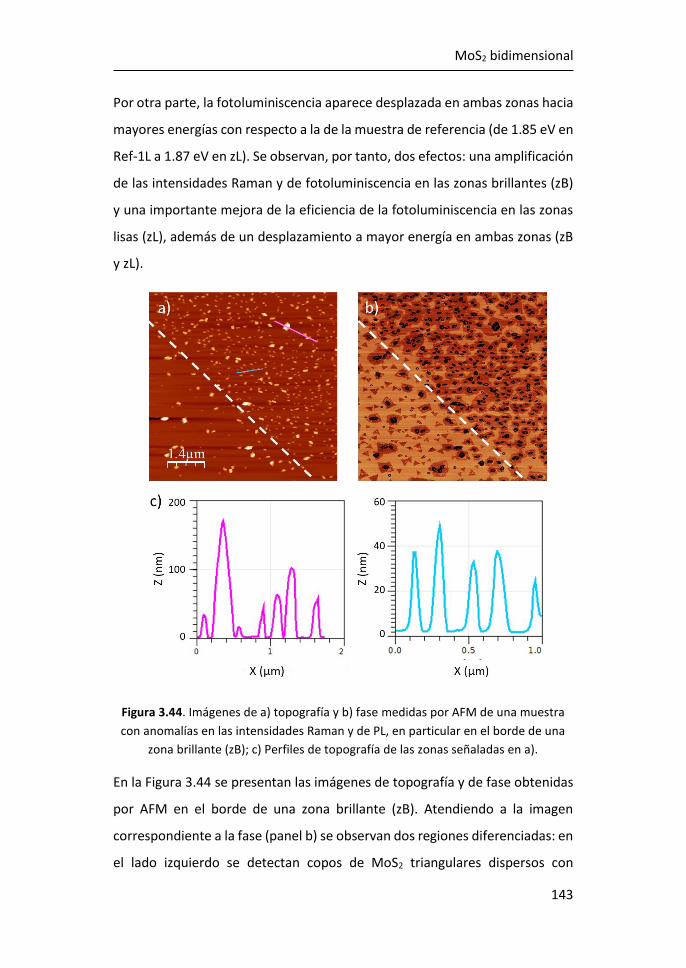
MoS2 bidimensional
143
Por otra parte, la fotoluminiscencia aparece desplazada en ambas zonas hacia
mayores energías con respecto a la de la muestra de referencia (de 1.85 eV en
Ref-1L a 1.87 eV en zL). Se observan, por tanto, dos efectos: una amplificación
de las intensidades Raman y de fotoluminiscencia en las zonas brillantes (zB)
y una importante mejora de la eficiencia de la fotoluminiscencia en las zonas
lisas (zL), además de un desplazamiento a mayor energía en ambas zonas (zB
y zL).
Figura 3.44. Imágenes de a) topografía y b) fase medidas por AFM de una muestra
con anomalías en las intensidades Raman y de PL, en particular en el borde de una
zona brillante (zB); c) Perfiles de topografía de las zonas señaladas en a).
En la Figura 3.44 se presentan las imágenes de topografía y de fase obtenidas
por AFM en el borde de una zona brillante (zB). Atendiendo a la imagen
correspondiente a la fase (panel b) se observan dos regiones diferenciadas: en
el lado izquierdo se detectan copos de MoS2 triangulares dispersos con
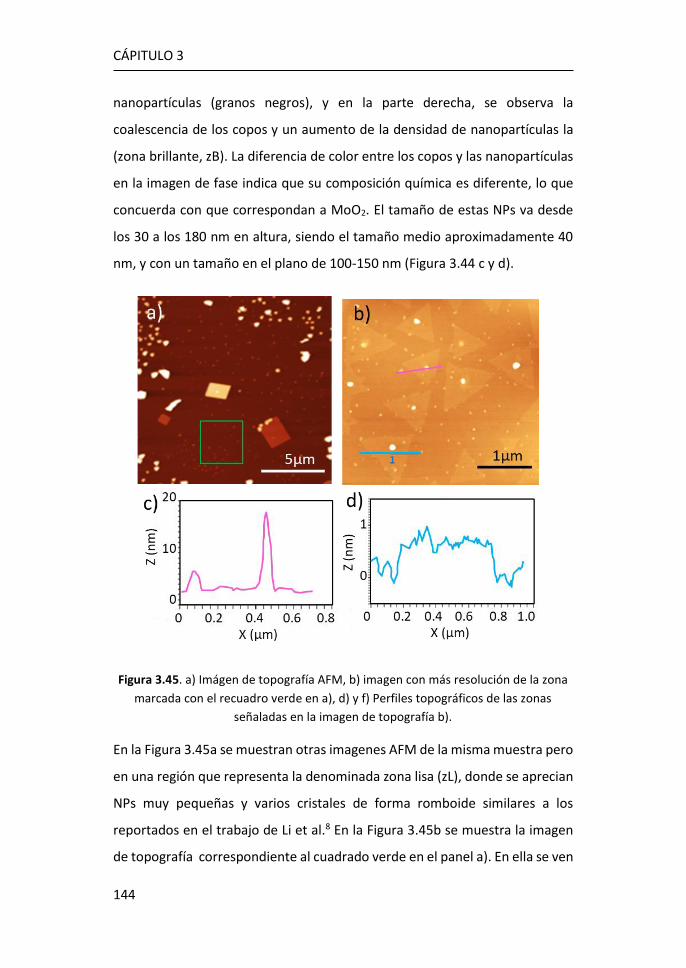
CÁPITULO 3
144
nanopartículas (granos negros), y en la parte derecha, se observa la
coalescencia de los copos y un aumento de la densidad de nanopartículas la
(zona brillante, zB). La diferencia de color entre los copos y las nanopartículas
en la imagen de fase indica que su composición química es diferente, lo que
concuerda con que correspondan a MoO2. El tamaño de estas NPs va desde
los 30 a los 180 nm en altura, siendo el tamaño medio aproximadamente 40
nm, y con un tamaño en el plano de 100-150 nm (Figura 3.44 c y d).
Figura 3.45. a) Imágen de topografía AFM, b) imagen con más resolución de la zona
marcada con el recuadro verde en a), d) y f) Perfiles topográficos de las zonas
señaladas en la imagen de topografía b).
En la Figura 3.45a se muestran otras imagenes AFM de la misma muestra pero
en una región que representa la denominada zona lisa (zL), donde se aprecian
NPs muy pequeñas y varios cristales de forma romboide similares a los
reportados en el trabajo de Li et al.8 En la Figura 3.45b se muestra la imagen
de topografía correspondiente al cuadrado verde en el panel a). En ella se ven

MoS2 bidimensional
145
copos triangulares de MoS2 de aproximadamente 1 μm con baja densidad de
NPs .Los copos son monocapa (altura de aproximadamente 0.7 nm según el
perfil de Fig. 41e) y las NPs son de un tamaño mucho menor (altura de 5 - 15
nm) al observado en la Figura 3.44.
Se observan, por tanto, dos morfologías distintas que se pueden asociar a las
zonas brillantes y zonas lisas. Las zonas brillantes muestran una mayor
densidad de NPs de MoO2 y de mayor tamaño, mientras que las zonas lisas
presentan menor densidad de NPs mucho más pequeñas.
A partir de estas observaciones se deduce que la presencia de MoO2 da lugar
a dos efectos distintos. Por un lado, causa la amplificación de las señales
Raman y de emisión de las zonas brillantes (zB), que puede explicarse por el
efecto SERS asociado al plasmón localizado de nanopartículas de MoO2. Por
otro lado, origina una mejora en la eficiencia de la fotoluminiscencia, además
de su desplazamiento a mayores energías, que puede explicarse por un efecto
de dopado tipo-p del MoS2. Es conocido que el O2 adsorbido sobre MoS2
produce un dopado tipo-p, que da lugar a un incremento de la energía de la
fotoluminiscencia y a un aumento de su intensidad debido al cambio en la
recombinación: de triones a excitones.27,29,32,48 En este caso, se deduce que el
MoO2 también induce una transferencia de huecos hacia el MoS2,
intensificando la fotoluminiscencia y desplazándola a mayores energías. Las
zonas brillantes, con NPs de mayor tamaño, parecen verse afectadas por
ambos efectos (SERS y dopado tipo-p), mientras que las zonas lisas, con NPs
más pequeñas, solo se ven afectadas por el dopado tipo-p.
3.4.1. Estabilidad temporal del MoO2 en las muestras de MoS2
Se ha estudiado la estabilidad temporal de estas muestras con señales
atípicas. También se ha estudiado el impacto que tiene el oxígeno en la
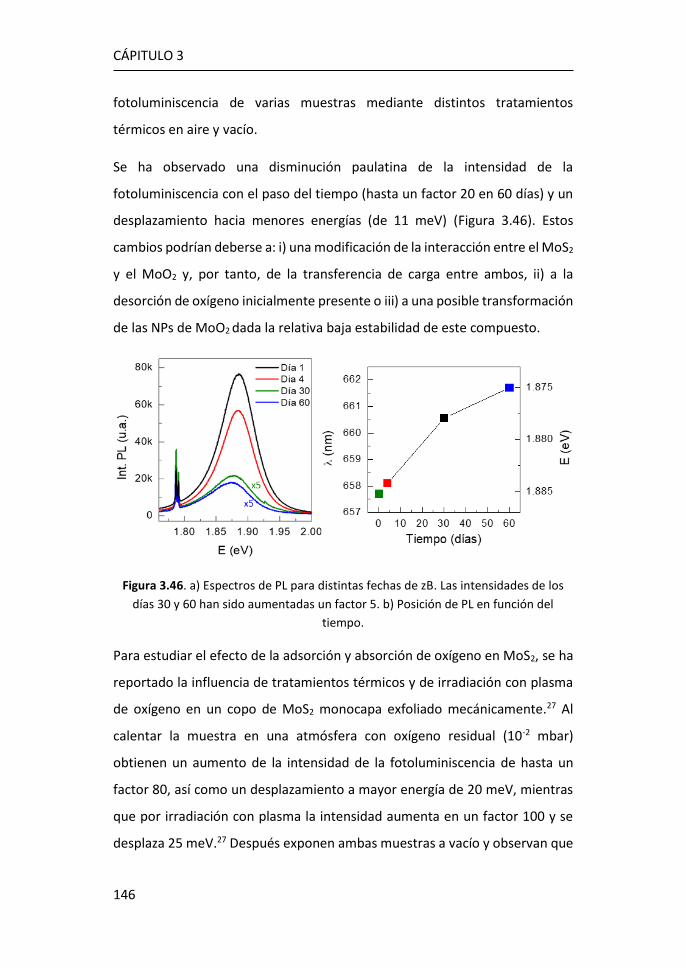
CÁPITULO 3
146
fotoluminiscencia de varias muestras mediante distintos tratamientos
térmicos en aire y vacío.
Se ha observado una disminución paulatina de la intensidad de la
fotoluminiscencia con el paso del tiempo (hasta un factor 20 en 60 días) y un
desplazamiento hacia menores energías (de 11 meV) (Figura 3.46). Estos
cambios podrían deberse a: i) una modificación de la interacción entre el MoS2
y el MoO2 y, por tanto, de la transferencia de carga entre ambos, ii) a la
desorción de oxígeno inicialmente presente o iii) a una posible transformación
de las NPs de MoO2 dada la relativa baja estabilidad de este compuesto.
Figura 3.46. a) Espectros de PL para distintas fechas de zB. Las intensidades de los
días 30 y 60 han sido aumentadas un factor 5. b) Posición de PL en función del
tiempo.
Para estudiar el efecto de la adsorción y absorción de oxígeno en MoS2, se ha
reportado la influencia de tratamientos térmicos y de irradiación con plasma
de oxígeno en un copo de MoS2 monocapa exfoliado mecánicamente.27 Al
calentar la muestra en una atmósfera con oxígeno residual (10-2 mbar)
obtienen un aumento de la intensidad de la fotoluminiscencia de hasta un
factor 80, así como un desplazamiento a mayor energía de 20 meV, mientras
que por irradiación con plasma la intensidad aumenta en un factor 100 y se
desplaza 25 meV.27 Después exponen ambas muestras a vacío y observan que
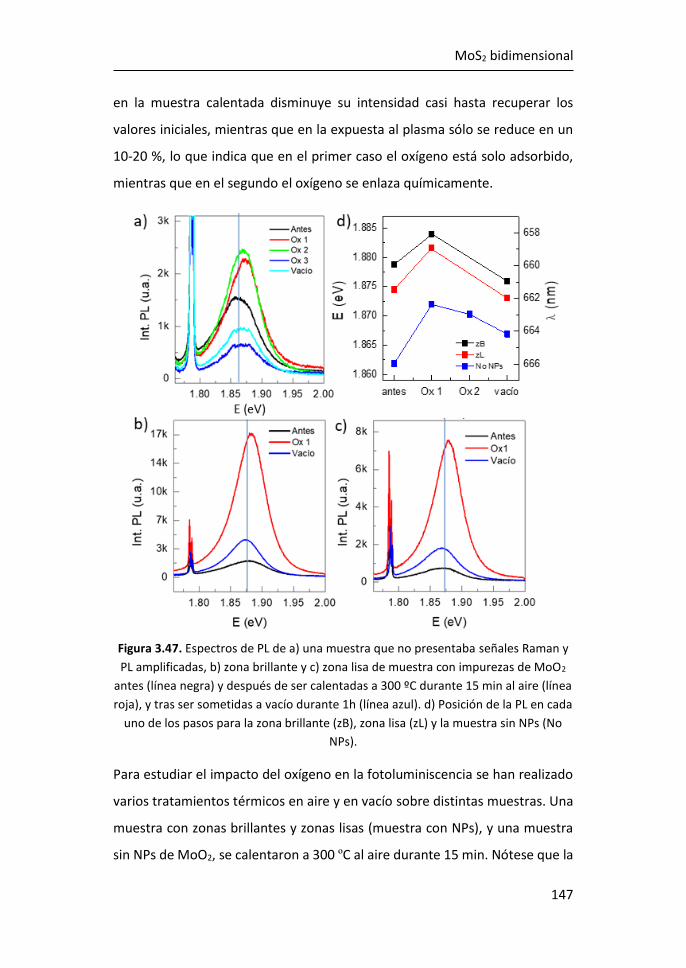
MoS2 bidimensional
147
en la muestra calentada disminuye su intensidad casi hasta recuperar los
valores iniciales, mientras que en la expuesta al plasma sólo se reduce en un
10-20 %, lo que indica que en el primer caso el oxígeno está solo adsorbido,
mientras que en el segundo el oxígeno se enlaza químicamente.
Figura 3.47. Espectros de PL de a) una muestra que no presentaba señales Raman y
PL amplificadas, b) zona brillante y c) zona lisa de muestra con impurezas de MoO2
antes (línea negra) y después de ser calentadas a 300 ºC durante 15 min al aire (línea
roja), y tras ser sometidas a vacío durante 1h (línea azul). d) Posición de la PL en cada
uno de los pasos para la zona brillante (zB), zona lisa (zL) y la muestra sin NPs (No
NPs).
Para estudiar el impacto del oxígeno en la fotoluminiscencia se han realizado
varios tratamientos térmicos en aire y en vacío sobre distintas muestras. Una
muestra con zonas brillantes y zonas lisas (muestra con NPs), y una muestra
sin NPs de MoO2, se calentaron a 300 ºC al aire durante 15 min. Nótese que la

CÁPITULO 3
148
energía de la fotoluminiscencia de esta última muestra antes de ser expuesta
al calentamiento (1.86 eV) es menor que la que presentan las zonas lisas y
brillantes de la muestra con impurezas (1.88 eV). Para la muestra sin NPs de
MoO2 se llevaron a cabo tres oxidaciones consecutivas (líneas roja, verde y
azul oscuro en Figura 3.47a) con la intención de saturar la adsorción de
oxígeno en la muestra. La relación I(PL)/I(A1g) aumenta ligeramente (un factor
~2) con la primera y segunda oxidación, además de aumentar su energía en
13 meV. Sin embargo, a partir de la tercera oxidación el MoS2 empieza a
degradarse, disminuyendo en gran medida tanto las intensidades Raman
como de fotoluminiscencia. En el caso de la muestra con NPs de MoO2, tras el
calentamiento se observa que la relación I(PL)/I(A1g) aumenta hasta un factor
10 tanto en las zonas brillantes (Figura 3.47b) como en las lisas (Figura 3.47c)
y la fotoluminiscencia aumenta su energía en 15 meV.
Todas las muestras fueron después calentadas a 300 ºC en vacío (10-5 mbar)
durante 1h para eliminar el posible oxígeno suministrado en los tratamientos
anteriores. Tras el calentamiento en vacío, se observa una disminución de la
intensidad de la fotoluminiscencia en un 70% en la muestra con NPs de MoO2,
además de un desplazamiento a menores energías, lo que podría asociarse
con una desorción de una parte del oxígeno, mientras que otra parte podría
haber reaccionado haciendo el proceso irreversible, de ahí que no se alcance
el valor inicial. Por otro lado, la muestra que no presenta NPs parece haber
sufrido daños irreparables, ya que su intensidad después del tratamiento a
vacío es menor que la inicial, al igual que su intensidad Raman. Haciendo
referencia únicamente a la primera oxidación parece que, de algún modo,
aunque los desplazamientos en energía de la fotoluminiscencia son similares
en las dos muestras, la fotoluminiscencia de la muestra con NPs de MoO2
muestra una mayor sensibilidad a la oxidación.

MoS2 bidimensional
149
En resumen, una suma de las contribuciones del efecto SERS originado por las
NPs de MoO2, de la transferencia de carga proporcionada por dichas NPs,
además de una posible adsorción de oxígeno en zonas con defectos podrían
justificar las diferentes intensidades Raman y de fotoluminiscencia
observadas, así como la activación de la fotoluminiscencia en zonas donde el
MoS2 es de pocas capas.
3.5 REFERENCIAS
1. Arul, N. S. & Nithya, V. D. Molybdenum disulfide quantum dots: synthesis and applications. RSC Adv. 6, 65670–65682 (2016).
2. Ozden, A., Ay, F., Sevik, C. & Perkgöz, N. K. CVD growth of monolayer MoS2: Role of growth zone configuration and precursors ratio. Jpn. J. Appl. Phys. 56, (2017).
3. Wang, L., Chen, F. & Ji, X. Shape consistency of MoS2 flakes grown using chemical vapor deposition. Appl. Phys. Express 10, 2–6 (2017).
4. Deb, S., Chakrabarti, P., Mohapatra, P. K., Barick, B. K. & Dhar, S. Tailoring of defect luminescence in CVD grown monolayer MoS 2 film. Appl. Surf. Sci. 445, 542–547 (2018).
5. Zeng, Y. et al. Highly Enhanced Photoluminescence of Monolayer MoS2 with Self-Assembled Au Nanoparticle Arrays. Adv. Mater. Interfaces 4, 1–7 (2017).
6. Wen, Y. Y. et al. Synthesis of Monolayer MoS2 by CVD Approach. 1034–1039 (2016) doi:10.2991/AME-16.2016.167.
7. Spevack, P. A. & McIntyre, N. S. Thermal reduction of MoO3. J. Phys. Chem. 96, 9029–9035 (1992).
8. Li, J., Yin, J., Li, X., Zhou, J. & Guo, W. Chemical vapor deposition of ultra-thin molybdenum dioxide nanosheets. Mater. Lett. 174, 188–191 (2016).

CÁPITULO 3
150
9. Luo, S., Cullen, C. P., Guo, G., Zhong, J. & Duesberg, G. S. Investigation of growth-induced strain in monolayer MoS2 grown by chemical vapor deposition. Appl. Surf. Sci. 508, 145126 (2020).
10. Pollmann, E. et al. Apparent differences between single layer molybdenum disulphide fabricated via chemical vapour deposition and exfoliation. Nanotechnology 31, (2020).
11. Carvalho, A., Ribeiro, R. M. & Castro Neto, A. H. Band nesting and the optical response of two-dimensional semiconducting transition metal dichalcogenides. Phys. Rev. B - Condens. Matter Mater. Phys. 88, 1–6 (2013).
12. Mignuzzi, S. et al. Effect of disorder on Raman scattering of single-layer Mo S2. Phys. Rev. B - Condens. Matter Mater. Phys. 91, 195411 (2015).
13. Maguire, P. et al. Defect sizing, separation, and substrate effects in ion-irradiated monolayer two-dimensional materials. Phys. Rev. B 98, 1–11 (2018).
14. He, Z. et al. Defect Engineering in Single-Layer MoS 2 Using Heavy Ion Irradiation. ACS Appl. Mater. Interfaces 10, 42524–42533 (2018).
15. Aryeetey, F., Ignatova, T. & Aravamudhan, S. Quantification of defects engineered in single layer MoS2. RSC Adv. 10, 22996–23001 (2020).
16. Fujisawa, K. et al. Quantification and Healing of Defects in Atomically Thin Molybdenum Disulfide: Beyond the Controlled Creation of Atomic Defects. ACS Nano 15, 9658–9669 (2021).
17. Chang, C. H., Fan, X., Lin, S. H. & Kuo, J. L. Orbital analysis of electronic structure and phonon dispersion in MoS 2, MoSe2, WS2, and WSe2 monolayers under strain. Phys. Rev. B - Condens. Matter Mater. Phys. 88, 195420 (2013).
18. Zhang, X., Tan, Q. H., Wu, J. Bin, Shi, W. & Tan, P. H. Review on the Raman spectroscopy of different types of layered materials. Nanoscale vol. 8 6435–6450 (2016).
19. Wang, Y., Cong, C., Qiu, C. & Yu, T. Raman Spectroscopy Study of Lattice Vibration and Crystallographic Orientation of Monolayer MoS 2 under Uniaxial Strain. Small 9, 2857–2861 (2013).
20. Zhu, C. R. et al. Strain tuning of optical emission energy and polarization in monolayer and bilayer MoS2. Phys. Rev. B - Condens. Matter Mater. Phys. 88, 1–5 (2013).
21. Rice, C. et al. Raman-scattering measurements and first-principles

MoS2 bidimensional
151
calculations of strain-induced phonon shifts in monolayer MoS2. Phys. Rev. B - Condens. Matter Mater. Phys. 87, (2013).
22. Conley, H. J. et al. Bandgap engineering of strained monolayer and bilayer MoS2. Nano Lett. 13, 3626–3630 (2013).
23. Iqbal, M. W., Shahzad, K., Akbar, R. & Hussain, G. A review on Raman finger prints of doping and strain effect in TMDCs. Microelectron. Eng. 219, (2020).
24. Dhakal, K. P. et al. Confocal absorption spectral imaging of MoS2: Optical transitions depending on the atomic thickness of intrinsic and chemically doped MoS2. Nanoscale 6, 13028–13035 (2014).
25. Chakraborty, B. et al. Symmetry-dependent phonon renormalization in monolayer MoS 2 transistor. Phys. Rev. B - Condens. Matter Mater. Phys. 85, 161403 (2012).
26. Tarasov, A. et al. Controlled Doping of Large-Area Trilayer MoS2 with Molecular Reductants and Oxidants. Adv. Mater. 27, 1175–1181 (2015).
27. Nan, H. et al. Strong photoluminescence enhancement of MoS2 through defect engineering and oxygen bonding. ACS Nano 8, 5738–5745 (2014).
28. Shi, Y. et al. Selective decoration of Au nanoparticles on monolayer MoS 2 single crystals. Sci. Rep. 3, (2013).
29. Hu, C. et al. Work function variation of monolayer MoS2 by nitrogen-doping. Appl. Phys. Lett. 113, 1–6 (2018).
30. Shakya, J., Kumar, S. & Mohanty, T. Role of oxygen adsorption in modification of optical and surface electronic properties of MoS2. J. Appl. Phys. 123, 165103 (2018).
31. Kim, J. H. et al. Work function variation of MoS2 atomic layers grown with chemical vapor deposition: The effects of thickness and the adsorption of water/oxygen molecules. Appl. Phys. Lett. 106, 251606 (2015).
32. Wu, K. et al. Controllable defects implantation in MoS2 grown by chemical vapor deposition for photoluminescence enhancement. Nano Res. 11, 4123–4132 (2018).
33. Azcatl, A. et al. Covalent Nitrogen Doping and Compressive Strain in MoS2 by Remote N2 Plasma Exposure. Nano Lett. 16, 5437–5443 (2016).

CÁPITULO 3
152
34. Chakraborty, B. et al. Symmetry-dependent phonon renormalization in monolayer MoS 2 transistor. Phys. Rev. B - Condens. Matter Mater. Phys. 85, 1–5 (2012).
35. Alvarez-Fraga, L. et al. Efficient heterostructures for combined interference and plasmon resonance raman amplification. ACS Appl. Mater. Interfaces 9, 4119–4125 (2017).
36. Frey, G. L., Tenne, R., Matthews, M. J., Dresselhaus, M. S. & Dresselhaus, G. Raman and resonance Raman investigation of MoS2 nanoparticles. Phys. Rev. B - Condens. Matter Mater. Phys. 60, 2883–2892 (1999).
37. Mignuzzi, S. et al. Effect of disorder on Raman scattering of single-layer Mo S2. Phys. Rev. B - Condens. Matter Mater. Phys. 91, 15–19 (2015).
38. Hong, J. et al. Exploring atomic defects in molybdenum disulphide monolayers. Nat. Commun. 6, (2015).
39. Molina-Sánchez, A. & Wirtz, L. Phonons in single-layer and few-layer MoS2 and WS2. Phys. Rev. B - Condens. Matter Mater. Phys. 84, (2011).
40. Carvalho, B. R. & Pimenta, M. A. Resonance Raman spectroscopy in semiconducting transition-metal dichalcogenides: Basic properties and perspectives. 2D Mater. 7, (2020).
41. Tornatzky, H., Gillen, R., Uchiyama, H. & Maultzsch, J. Phonon dispersion in MoS2. Phys. Rev. B 99, 144309 (2019).
42. Zhang, X. et al. Phonon and Raman scattering of two-dimensional transition metal dichalcogenides from monolayer, multilayer to bulk material. Chem. Soc. Rev. 44, 2757–2785 (2015).
43. Lee, C. et al. Anomalous lattice vibrations of single- and few-layer MoS2. ACS Nano 4, 2695–2700 (2010).
44. Carvalho, B. R. et al. Intervalley scattering by acoustic phonons in two-dimensional MoS2 revealed by double-resonance Raman spectroscopy. Nat. Commun. 8, 6–13 (2017).
45. Mukherjee, S., Maiti, R., Katiyar, A. K., Das, S. & Ray, S. K. Novel Colloidal MoS2 Quantum Dot Heterojunctions on Silicon Platforms for Multifunctional Optoelectronic Devices. Sci. Rep. 6, 1–11 (2016).
46. Lin, H. et al. Colloidal synthesis of MoS2 quantum dots: Size-dependent tunable photoluminescence and bioimaging. New J. Chem. 39, 8492–8497 (2015).

MoS2 bidimensional
153
47. Zhang, Q. et al. Plasmonic MoO2 Nanospheres as a Highly Sensitive and Stable Non-Noble Metal Substrate for Multicomponent Surface-Enhanced Raman Analysis. Anal. Chem. 89, 11765–11771 (2017).
48. Yang, G. et al. A comprehensive study of enhanced photoluminescence on monolayer MoS2 with Ag nano-ridge structures. Appl. Surf. Sci. 508, 144794 (2020).

CÁPITULO 3
154

155
4 SERS EN PLATAFORMAS DE GRAFENO Y
NANOPARTÍCULAS DE AG
Es sabido que la amplificación de la radiación electromagnética debida a las
resonancias plasmónicas superficiales localizadas (LSPR) de nanopartículas
metálicas (NPs) es muy eficiente, por lo que se utiliza comúnmente para
detectar señales Raman débiles de analitos diluidos. Es lo que se denomina
técnica SERS (del inglés surface enhancement Raman scattering).1–4 Por otro
lado, la distribución del aumento del campo eléctrico en la superficie de un
sustrato plasmónico es muy poco homogénea, localizándose principalmente
en puntos calientes o " hot spots". El Capítulo 1 recoge una explicación más
amplia y detallada de los conceptos aquí nombrados. Las plataformas SERS
más comunes pueden clasificarse en tres clases: i) nanopartículas metálicas en
suspensión, normalmente fabricadas por el método Creighton,5 que se basa
en la reducción de AgNO3 y cuyas NPs de Ag resultantes tienen un tamaño
medio de ~10 nm, o sintetizadas mediante la técnica de ablación láser en
medio líquido;6–9 ii) nanopartículas metálicas inmovilizadas en sustratos
sólidos,10 como cuarzo,11 papel;10,12 o filtros de alúmina;8 y iii) nanoestructuras
fabricadas directamente sobre sustratos sólidos, por ejemplo mediante
distintas técnicas de litografía,13–15 que puede utilizarse para producir
conjuntos periódicos 2D bien ordenados de nanopartículas.
Las plataformas desarrolladas en este trabajo pertenecen a la tercera
categoría, ya que las nanopartículas se depositan directamente sobre
sustratos sólidos (sílice vítrea o grafeno/cuarzo) mediante la técnica de
agregación en fase gas.16 Se trata de un método físico para generar un flujo
continuo de partículas ultrafinas de tamaño controlado (de 1 a ~10 nm de
radio, es decir, desde 103 hasta 106 átomos por partícula) con distribuciones

CÁPITULO 4
156
de tamaño de partícula estrechas, en un entorno limpio y controlado, en alto
vacío, lo que evita la presencia de cualquier subproducto durante el proceso
de fabricación, como tensioactivos utilizados en métodos de preparación de
NPs por disolución.
El objetivo de este trabajo es evaluar la capacidad de NPs esféricas de Ag
monodispersas ultra pequeñas (R = 1.8 nm) para amplificar las señales Raman
y compararla con otras NPs, de mayor tamaño y forma aplanada (similar a
semiesferoides cuya altura es menor que el radio), así como la relevancia de
la distancia inter-NP en la eficiencia de la amplificación SERS. Para ello se
emplean las señales del grafeno y de una molécula de referencia como la
Rodamina 6G (R6G). Para el análisis de los resultados se realizan y discuten las
simulaciones de campo cercano de los plasmones de NPs individuales y de
redes de NPs, que incluyen la dependencia del tamaño y la distancia media
entre las NPs. Las simulaciones han sido llevadas a cabo por el Dr. Rafael
Ramírez Jiménez (Universidad Carlos III de Madrid). Se comparan los
plasmones y amplificaciones originados por las NPs ultrapequeñas con las
debidas a películas de plata nanoestructuradas con granos de mayor tamaño
(7-15 nm), y se estudia el efecto que tiene la combinación de ambas
estructuras en una misma plataforma.17
4.1 TAMAÑO Y MORFOLOGÍA DE LAS NANOPARTÍCULAS DE AG
ULTRAPEQUEÑAS
Los parámetros descritos en el Capítulo 2 dan lugar a nanopartículas esféricas
monocristalinas, como se observa en las imágenes HRTEM (Figura 4.1a y b).
En la Figura 4.1c se ha representado la distribución de tamaños obtenida del
análisis de aproximadamente 100 NPs. De las medidas se obtiene un radio
medio de ~1.8 nm con una distribución muy estrecha (FWHM = 0.8 nm).
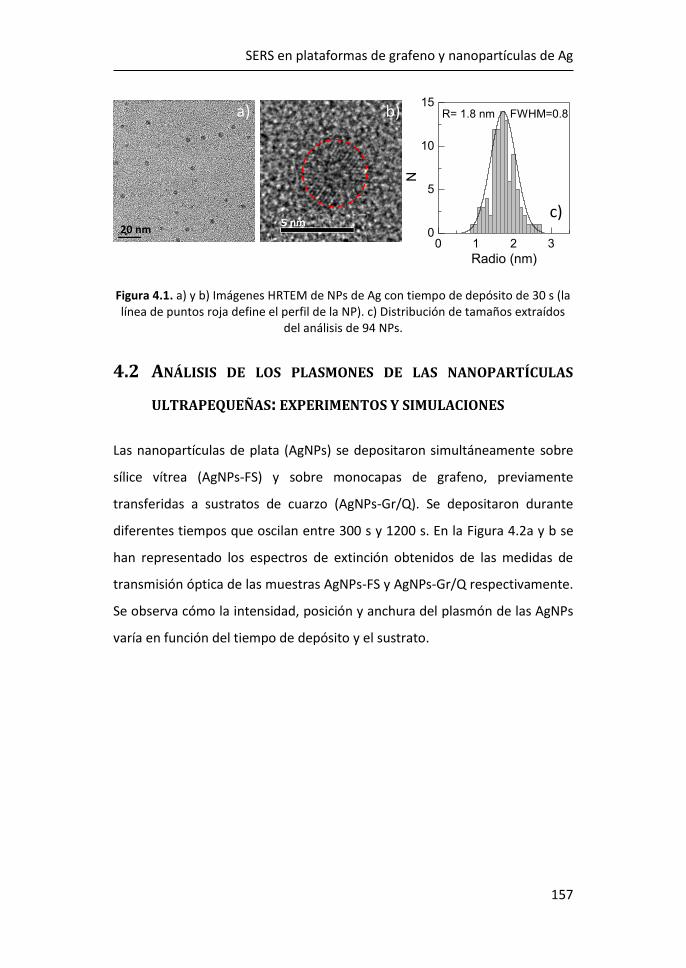
SERS en plataformas de grafeno y nanopartículas de Ag
157
Figura 4.1. a) y b) Imágenes HRTEM de NPs de Ag con tiempo de depósito de 30 s (la línea de puntos roja define el perfil de la NP). c) Distribución de tamaños extraídos
del análisis de 94 NPs.
4.2 ANÁLISIS DE LOS PLASMONES DE LAS NANOPARTÍCULAS
ULTRAPEQUEÑAS: EXPERIMENTOS Y SIMULACIONES
Las nanopartículas de plata (AgNPs) se depositaron simultáneamente sobre
sílice vítrea (AgNPs-FS) y sobre monocapas de grafeno, previamente
transferidas a sustratos de cuarzo (AgNPs-Gr/Q). Se depositaron durante
diferentes tiempos que oscilan entre 300 s y 1200 s. En la Figura 4.2a y b se
han representado los espectros de extinción obtenidos de las medidas de
transmisión óptica de las muestras AgNPs-FS y AgNPs-Gr/Q respectivamente.
Se observa cómo la intensidad, posición y anchura del plasmón de las AgNPs
varía en función del tiempo de depósito y el sustrato.
0 1 2 30
5
10
15
N
Radio (nm)
R= 1.8 nm FWHM=0.8
5 nm20 nm
a) b)
c)
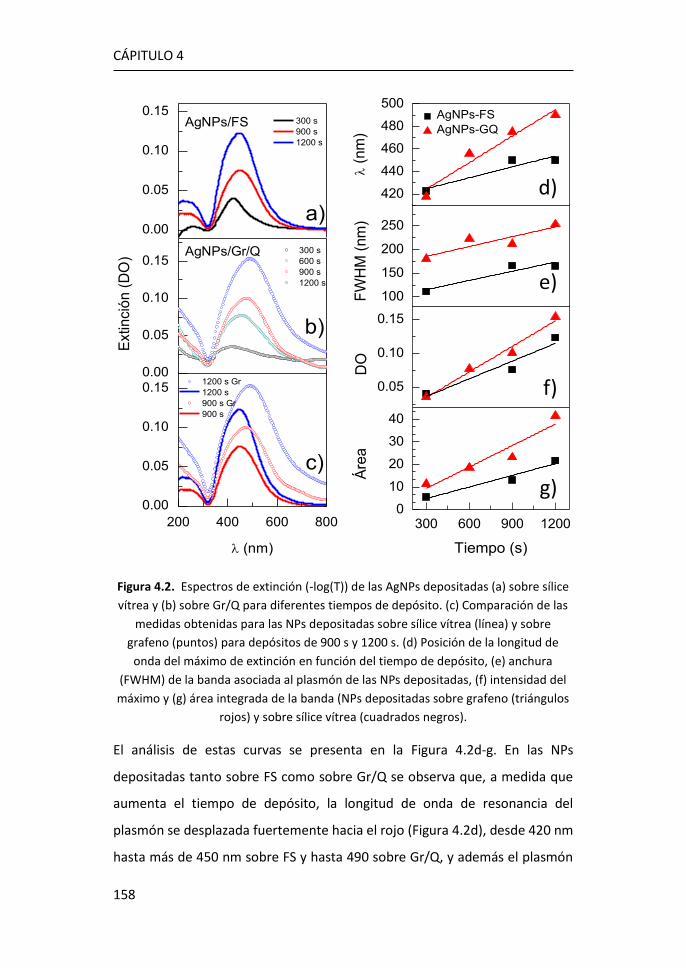
CÁPITULO 4
158
Figura 4.2. Espectros de extinción (-log(T)) de las AgNPs depositadas (a) sobre sílice
vítrea y (b) sobre Gr/Q para diferentes tiempos de depósito. (c) Comparación de las
medidas obtenidas para las NPs depositadas sobre sílice vítrea (línea) y sobre
grafeno (puntos) para depósitos de 900 s y 1200 s. (d) Posición de la longitud de
onda del máximo de extinción en función del tiempo de depósito, (e) anchura
(FWHM) de la banda asociada al plasmón de las NPs depositadas, (f) intensidad del
máximo y (g) área integrada de la banda (NPs depositadas sobre grafeno (triángulos
rojos) y sobre sílice vítrea (cuadrados negros).
El análisis de estas curvas se presenta en la Figura 4.2d-g. En las NPs
depositadas tanto sobre FS como sobre Gr/Q se observa que, a medida que
aumenta el tiempo de depósito, la longitud de onda de resonancia del
plasmón se desplazada fuertemente hacia el rojo (Figura 4.2d), desde 420 nm
hasta más de 450 nm sobre FS y hasta 490 sobre Gr/Q, y además el plasmón
420440460480500
100
150
200
250
0.05
0.10
0.15
300 600 900 12000
10203040
AgNPs-FS AgNPs-GQ
(n
m)
FWH
M (n
m)
DO
Área
Tiempo (s)
0.00
0.05
0.10
0.15
0.00
0.05
0.10
0.15
c)
a)AgNPs/Gr/Q
Extin
ción
(DO
) 300 s 900 s 1200 s
AgNPs/FS
b)
300 s 600 s 900 s 1200 s
200 400 600 8000.00
0.05
0.10
0.15 1200 s Gr 1200 s 900 s Gr 900 s
(nm)
d)
e)
f)
g)

SERS en plataformas de grafeno y nanopartículas de Ag
159
se ensancha significativamente (Figura 4.2e). Esto ocurre a pesar de que el
radio de las NPs (R = 1.8 nm) es constante en todas las series. Los valores
máximos de la extinción (Figura 4.2f) y de la intensidad integrada del plasmón
(Figura 4.2g) son proporcionales al tiempo de depósito.
En un primer paso, se ha utilizado la teoría de Mie para calcular la extinción
de esferas de Ag independientes (suficientemente alejadas para que su
interacción sea despreciable) teniendo en cuenta el cambio en la constante
dieléctrica debido a la amortiguación adicional en la superficie de las NPs que
tiene un peso importante en el límite de tamaño pequeño.18,19 En la Figura 3
se muestra la dependencia del espectro de extinción del plasmón en función
del radio de la NP, normalizado a la masa de la NP. La resonancia del plasmón
presenta un pequeño desplazamiento hacia el rojo, de 8 nm a medida que
aumenta el radio de 2 a 20 nm (recuadro en Figura 4.3). Al aumentar el radio
a valores mayores que 20 nm, se produce un importante desplazamiento hacia
el rojo y un ensanchamiento, así como una drástica disminución de la
intensidad del plasmón. El plasmón más eficiente para las partículas de Ag sin
interacción está alrededor de R = 20 nm, sin embargo, se encuentra en el UV
(365 nm) lo cual no es conveniente para las aplicaciones, siendo preferibles
plasmones en el rango del visible en el que los láseres y los equipos Raman
son más comunes.
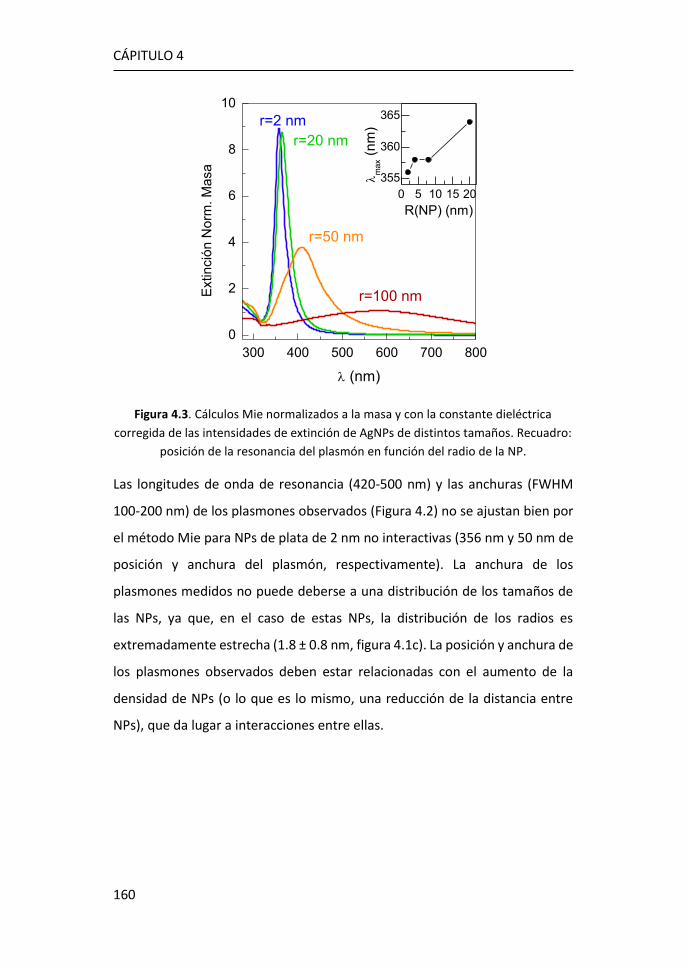
CÁPITULO 4
160
Figura 4.3. Cálculos Mie normalizados a la masa y con la constante dieléctrica
corregida de las intensidades de extinción de AgNPs de distintos tamaños. Recuadro:
posición de la resonancia del plasmón en función del radio de la NP.
Las longitudes de onda de resonancia (420-500 nm) y las anchuras (FWHM
100-200 nm) de los plasmones observados (Figura 4.2) no se ajustan bien por
el método Mie para NPs de plata de 2 nm no interactivas (356 nm y 50 nm de
posición y anchura del plasmón, respectivamente). La anchura de los
plasmones medidos no puede deberse a una distribución de los tamaños de
las NPs, ya que, en el caso de estas NPs, la distribución de los radios es
extremadamente estrecha (1.8 ± 0.8 nm, figura 4.1c). La posición y anchura de
los plasmones observados deben estar relacionadas con el aumento de la
densidad de NPs (o lo que es lo mismo, una reducción de la distancia entre
NPs), que da lugar a interacciones entre ellas.
300 400 500 600 700 8000
2
4
6
8
10r=2 nm
r=20 nm
Extin
ción
Nor
m. M
asa
(nm)
r=50 nm
r=100 nm
300 350 400 450
0 5 10 15 20355
360
365
m
ax (n
m)
R(NP) (nm)
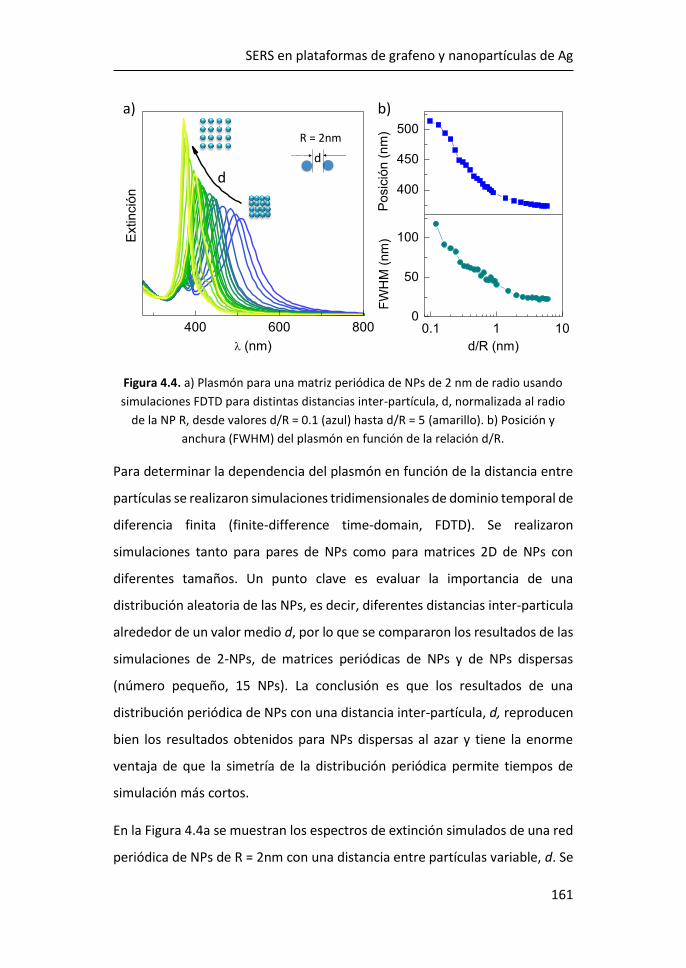
SERS en plataformas de grafeno y nanopartículas de Ag
161
Figura 4.4. a) Plasmón para una matriz periódica de NPs de 2 nm de radio usando
simulaciones FDTD para distintas distancias inter-partícula, d, normalizada al radio
de la NP R, desde valores d/R = 0.1 (azul) hasta d/R = 5 (amarillo). b) Posición y
anchura (FWHM) del plasmón en función de la relación d/R.
Para determinar la dependencia del plasmón en función de la distancia entre
partículas se realizaron simulaciones tridimensionales de dominio temporal de
diferencia finita (finite-difference time-domain, FDTD). Se realizaron
simulaciones tanto para pares de NPs como para matrices 2D de NPs con
diferentes tamaños. Un punto clave es evaluar la importancia de una
distribución aleatoria de las NPs, es decir, diferentes distancias inter-particula
alrededor de un valor medio d, por lo que se compararon los resultados de las
simulaciones de 2-NPs, de matrices periódicas de NPs y de NPs dispersas
(número pequeño, 15 NPs). La conclusión es que los resultados de una
distribución periódica de NPs con una distancia inter-partícula, d, reproducen
bien los resultados obtenidos para NPs dispersas al azar y tiene la enorme
ventaja de que la simetría de la distribución periódica permite tiempos de
simulación más cortos.
En la Figura 4.4a se muestran los espectros de extinción simulados de una red
periódica de NPs de R = 2nm con una distancia entre partículas variable, d. Se
400 600 800
Extin
ción
(nm)
R = 2nm
d
d400
450
500
0.1 1 100
50
100
Posi
ción
(nm
)
FWH
M (n
m)
d/R (nm)
a) b)
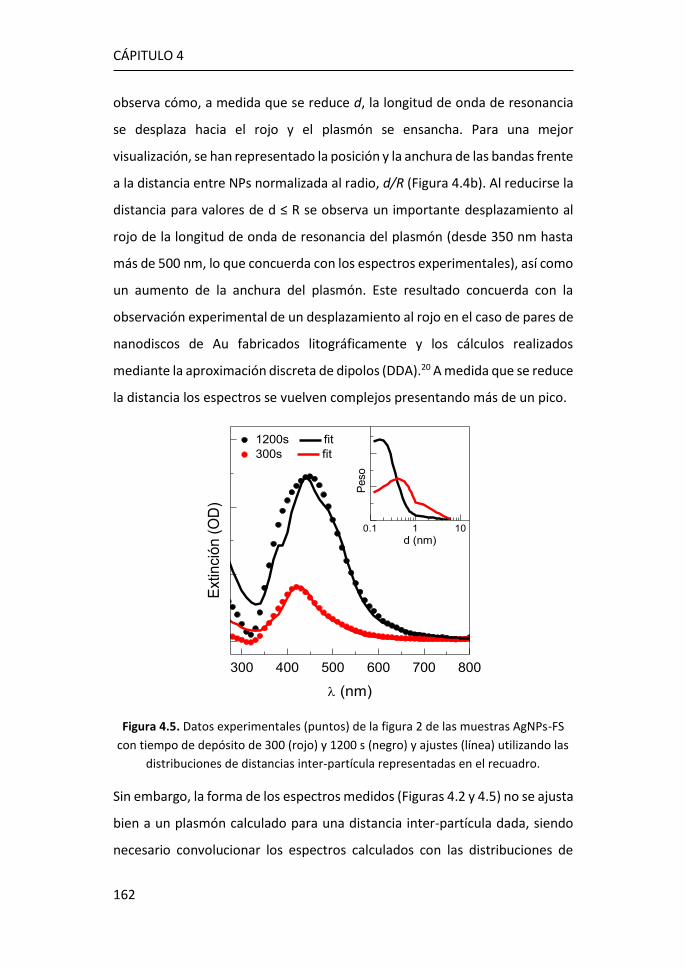
CÁPITULO 4
162
observa cómo, a medida que se reduce d, la longitud de onda de resonancia
se desplaza hacia el rojo y el plasmón se ensancha. Para una mejor
visualización, se han representado la posición y la anchura de las bandas frente
a la distancia entre NPs normalizada al radio, d/R (Figura 4.4b). Al reducirse la
distancia para valores de d ≤ R se observa un importante desplazamiento al
rojo de la longitud de onda de resonancia del plasmón (desde 350 nm hasta
más de 500 nm, lo que concuerda con los espectros experimentales), así como
un aumento de la anchura del plasmón. Este resultado concuerda con la
observación experimental de un desplazamiento al rojo en el caso de pares de
nanodiscos de Au fabricados litográficamente y los cálculos realizados
mediante la aproximación discreta de dipolos (DDA).20 A medida que se reduce
la distancia los espectros se vuelven complejos presentando más de un pico.
Figura 4.5. Datos experimentales (puntos) de la figura 2 de las muestras AgNPs-FS
con tiempo de depósito de 300 (rojo) y 1200 s (negro) y ajustes (línea) utilizando las
distribuciones de distancias inter-partícula representadas en el recuadro.
Sin embargo, la forma de los espectros medidos (Figuras 4.2 y 4.5) no se ajusta
bien a un plasmón calculado para una distancia inter-partícula dada, siendo
necesario convolucionar los espectros calculados con las distribuciones de
300 400 500 600 700 800
1200s fit 300s fit
Extin
ción
(OD
)
(nm)
0.1 1 10
Peso
d (nm)
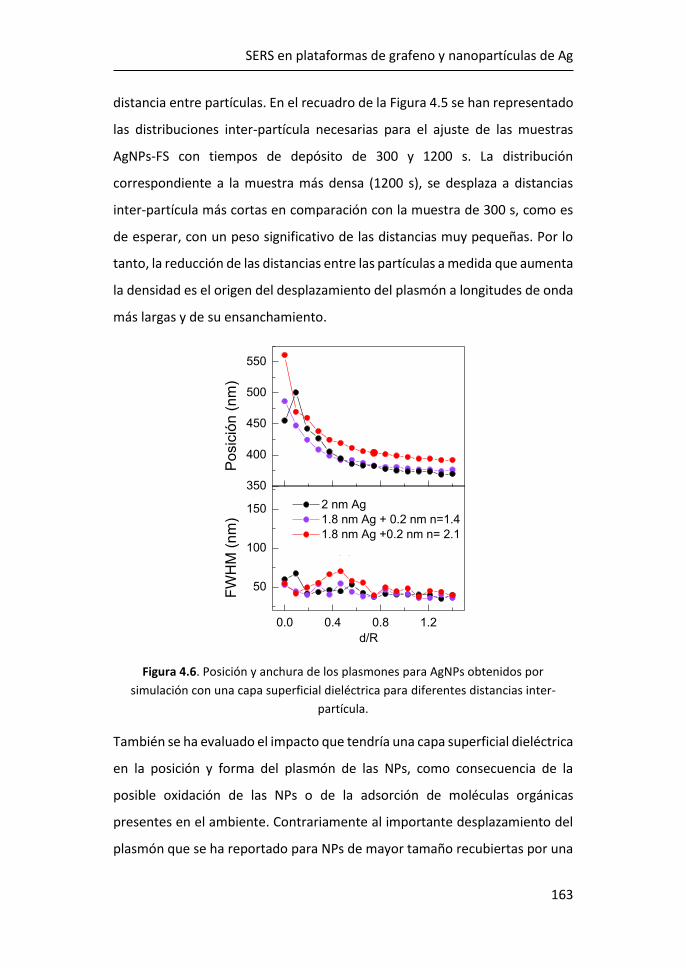
SERS en plataformas de grafeno y nanopartículas de Ag
163
distancia entre partículas. En el recuadro de la Figura 4.5 se han representado
las distribuciones inter-partícula necesarias para el ajuste de las muestras
AgNPs-FS con tiempos de depósito de 300 y 1200 s. La distribución
correspondiente a la muestra más densa (1200 s), se desplaza a distancias
inter-partícula más cortas en comparación con la muestra de 300 s, como es
de esperar, con un peso significativo de las distancias muy pequeñas. Por lo
tanto, la reducción de las distancias entre las partículas a medida que aumenta
la densidad es el origen del desplazamiento del plasmón a longitudes de onda
más largas y de su ensanchamiento.
Figura 4.6. Posición y anchura de los plasmones para AgNPs obtenidos por
simulación con una capa superficial dieléctrica para diferentes distancias inter-
partícula.
También se ha evaluado el impacto que tendría una capa superficial dieléctrica
en la posición y forma del plasmón de las NPs, como consecuencia de la
posible oxidación de las NPs o de la adsorción de moléculas orgánicas
presentes en el ambiente. Contrariamente al importante desplazamiento del
plasmón que se ha reportado para NPs de mayor tamaño recubiertas por una
350
400
450
500
550
Posi
ción
(nm
)
0.0 0.4 0.8 1.2
50
100
150
FWH
M (n
m)
d/R
C C C % (4)
2 nm Ag1.8 nm Ag + 0.2 nm n=1.41.8 nm Ag +0.2 nm n= 2.1

CÁPITULO 4
164
capa superficial de SiOx,21 en este caso de Nps muy pequeñas, el impacto sólo
es notable para valores muy altos del índice de refracción de la capa
dieléctrica. Por ejemplo, para una NP con una capa dieléctrica exterior cuyo
grosor es del orden del 10 % del radio de la NP, y un índice de refracción de
2.1, la banda del plasmón se desplaza al rojo en un 5 % y se produce un cierto
ensanchamiento cuando la distancia entre las partículas es corta (<1 nm)
(Figura 4.6). Con todo esto se puede concluir que este factor no juega ningún
papel relevante en la dependencia observada del plasmón con el tiempo de
depósito.
El papel del grafeno se revela cuando se comparan los espectros de extinción
de las AgNPs depositadas sobre FS y sobre Gr/Q. Se aprecian algunas
modificaciones, especialmente en las regiones espectrales del rojo y el IR
(Figura 4.2b y c)). Los aspectos más evidentes son: i) un aumento de los valores
de extinción y de la extinción integrada (Figuras 4.2f y g), ii) un
ensanchamiento (mayor peso del rojo y del IR) y iii) un desplazamiento de la
resonancia hacia el rojo (Figuras 4.2e y 4.d). El aumento de los valores de
extinción probablemente está relacionado con la mayor adherencia de las NPs
durante el depósito al grafeno que a la sílice vítrea. El mayor peso espectral
del IR se debe a la intervención del grafeno en la interacción entre las NPs,
proporcionando un canal para la conexión eléctrica sin producir una
deslocalización total de los electrones oscilantes. Se realizaron simulaciones
para las NPs sobre grafeno, pero no se consiguió reproducir la cola del IR
debido a que la superficie de contacto entre las NPs y el grafeno es muy
pequeña. Además, el efecto del grafeno no se corresponde con el debido a
una variación de la constante dieléctrica del medio en el que las NPs
estuvieran incrustadas. Se ha reportado que en simulaciones DDA de
partículas de plata parcialmente incrustadas en esferas de mica se produce un
desplazamiento al rojo,20 pero no se ha informado de un aumento de la cola
IR como el que se observa para las NPs sobre grafeno.

SERS en plataformas de grafeno y nanopartículas de Ag
165
4.3 AMPLIFICACIÓN RAMAN ORIGINADA POR LAS AGNPS DE 1.8
NM
Para evaluar el comportamiento de la amplificación Raman de las NPs
ultrapequeñas, se ha calculado el cuadrado del campo eléctrico, E2, para un
conjunto periódico de NPs con diferentes tamaños, 1.2 < R < 10 nm, en función
de la distancia inter-partícula. El comportamiento en este rango de radios es
similar, por lo que de nuevo se normalizan los resultados por R (z/R, siendo z
la posición del plano horizontal en el que se calcula E). En la Figura 4.7 se
muestra E2 en el plano ecuatorial (z=0) de las NPs para dos distancias, d = 1.6R
y d = 0.3R. La onda incidente es perpendicular al sustrato y la polarización se
indica en la Figura 4.7a mediante una flecha blanca. Se observa la formación
de puntos calientes (hot spots) en la superficie de las NPs en la dirección de la
polarización incidente y un aumento de su intensidad a medida que las NPs se
acercan. En la Figura 4.7d se muestran los perfiles de intensidad de E2 de las
NPs a lo largo de la línea tangente a la NP (línea negra vertical en la Figura 4.7c)
así como del 10% hacia dentro (línea azul) y hacia fuera (línea roja). Se observa
cómo a una distancia del 10% del radio el campo eléctrico aumenta
significativamente. Los puntos calientes están muy localizados en ciertas
zonas de la superficie de las NPs, pero su extensión aumenta
significativamente a medida que se reduce la distancia inter-partícula. Estos
cálculos muestran que la amplificación Raman a lo largo de la superficie de las
NPs es muy poco homogénea.
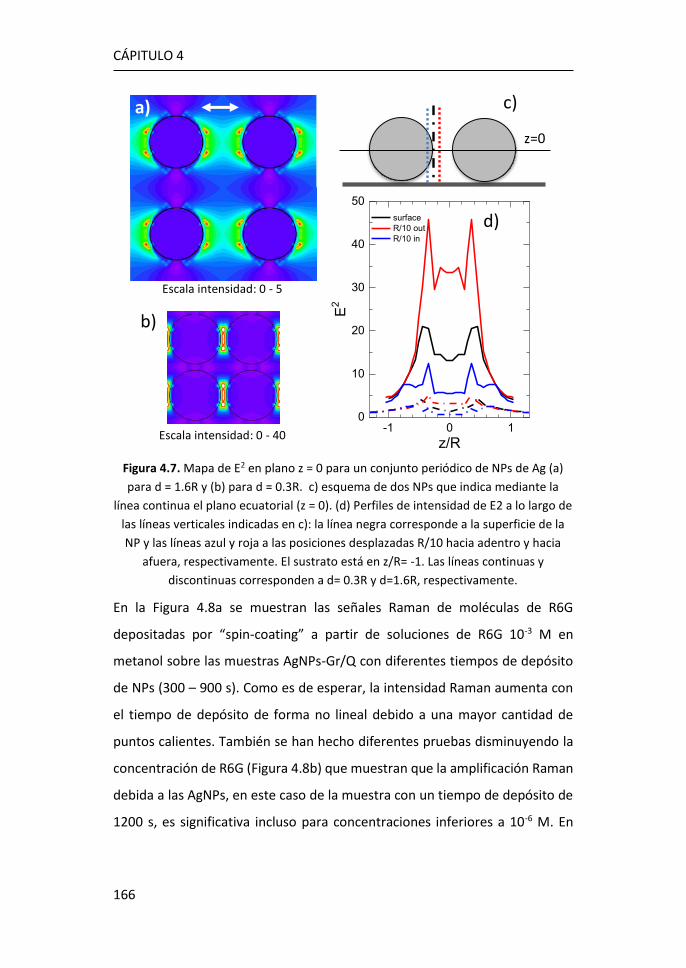
CÁPITULO 4
166
Figura 4.7. Mapa de E2 en plano z = 0 para un conjunto periódico de NPs de Ag (a)
para d = 1.6R y (b) para d = 0.3R. c) esquema de dos NPs que indica mediante la
línea continua el plano ecuatorial (z = 0). (d) Perfiles de intensidad de E2 a lo largo de
las líneas verticales indicadas en c): la línea negra corresponde a la superficie de la
NP y las líneas azul y roja a las posiciones desplazadas R/10 hacia adentro y hacia
afuera, respectivamente. El sustrato está en z/R= -1. Las líneas continuas y
discontinuas corresponden a d= 0.3R y d=1.6R, respectivamente.
En la Figura 4.8a se muestran las señales Raman de moléculas de R6G
depositadas por “spin-coating” a partir de soluciones de R6G 10-3 M en
metanol sobre las muestras AgNPs-Gr/Q con diferentes tiempos de depósito
de NPs (300 – 900 s). Como es de esperar, la intensidad Raman aumenta con
el tiempo de depósito de forma no lineal debido a una mayor cantidad de
puntos calientes. También se han hecho diferentes pruebas disminuyendo la
concentración de R6G (Figura 4.8b) que muestran que la amplificación Raman
debida a las AgNPs, en este caso de la muestra con un tiempo de depósito de
1200 s, es significativa incluso para concentraciones inferiores a 10-6 M. En
Escala intensidad: 0 - 5
Escala intensidad: 0 - 40
a)
b)
d)
c)
z=0
-1 0 10
10
20
30
40
50
E2
z/R
surface R/10 out R/10 in
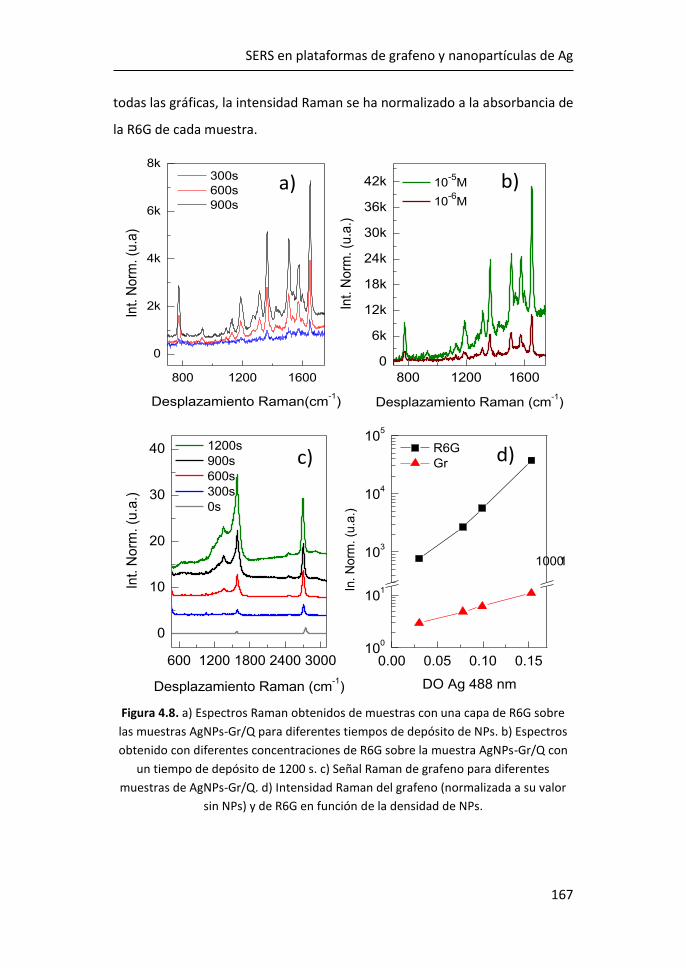
SERS en plataformas de grafeno y nanopartículas de Ag
167
todas las gráficas, la intensidad Raman se ha normalizado a la absorbancia de
la R6G de cada muestra.
Figura 4.8. a) Espectros Raman obtenidos de muestras con una capa de R6G sobre
las muestras AgNPs-Gr/Q para diferentes tiempos de depósito de NPs. b) Espectros
obtenido con diferentes concentraciones de R6G sobre la muestra AgNPs-Gr/Q con
un tiempo de depósito de 1200 s. c) Señal Raman de grafeno para diferentes
muestras de AgNPs-Gr/Q. d) Intensidad Raman del grafeno (normalizada a su valor
sin NPs) y de R6G en función de la densidad de NPs.
800 1200 1600
0
2k
4k
6k
8k
In
t. No
rm. (
u.a)
Desplazamiento Raman(cm-1)
300s 600s 900s
0.00 0.05 0.10 0.15100
101
103
104
105
R6G Gr
In. N
orm
. (u.
a.)
DO Ag 488 nm
800 1200 16000
6k
12k
18k
24k
30k
36k
42k 10-5M 10-6M
Int.
Norm
. (u.
a.)
Desplazamiento Raman (cm-1)
10001500200025003000
600 1200 1800 2400 3000
0
10
20
30
40 1200s 900s 600s 300s 0s
Int.
Nor
m. (
u.a.
)
Desplazamiento Raman (cm-1)
a) b)
c) d)

CÁPITULO 4
168
La Figura 4.8c muestra la señal Raman del grafeno de las muestras AgNPs-Gr/Q
con diferentes tiempos de depósito de NPs (0 - 1200 s). En la Figura 4.8d se ha
representado la dependencia de la amplificación de la señal Raman del
grafeno (calculada después de normalizar a su intensidad sin NPs) y de la señal
Raman las moléculas de R6G en función de la extinción del plasmón de las
AgNPs (relacionada con el tiempo de depósito de las NPs como se muestra en
la Figura 4.2) a la longitud de onda del láser de excitación (488 nm). No es
posible normalizar la intensidad Raman de la R6G a la señal sin NPs ya que su
intensidad está por debajo del límite de detección. Hay dos observaciones
importantes que discutir: i) la amplificación medida para los fonones de
grafeno es mucho más baja que para las moléculas de R6G; ii) la amplificación
para las vibraciones de R6G no es lineal con la intensidad del plasmón
(estrechamente relacionada con la masa total) mientras que para el grafeno
es casi lineal.
La diferencia entre la amplificación Raman para el grafeno y para las moléculas
de R6G se origina principalmente por sus diferencias en las configuraciones
geométricas: el grafeno es una capa plana que contacta con cada NP sólo en
un punto tangencial, mientras que las moléculas R6G envuelven a las NPs
(Figura 4.9).
Figura 4.9. Esquema de la configuración geométrica del grafeno y de la R6G con
respecto a las NPs.
Para estudiar estas diferencias geométricas se han realizados simulaciones de
la distribución de E2 en el plano ecuatorial (z=0), en la posición del sustrato
(z=-R), que se corresponde con la ubicación del grafeno, y en un plano
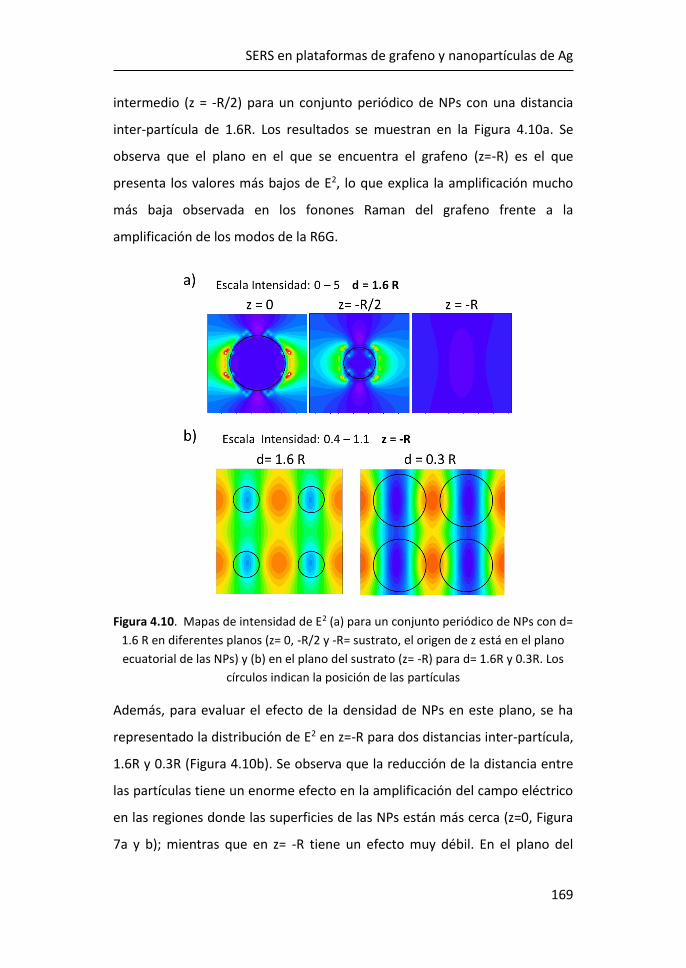
SERS en plataformas de grafeno y nanopartículas de Ag
169
intermedio (z = -R/2) para un conjunto periódico de NPs con una distancia
inter-partícula de 1.6R. Los resultados se muestran en la Figura 4.10a. Se
observa que el plano en el que se encuentra el grafeno (z=-R) es el que
presenta los valores más bajos de E2, lo que explica la amplificación mucho
más baja observada en los fonones Raman del grafeno frente a la
amplificación de los modos de la R6G.
Figura 4.10. Mapas de intensidad de E2 (a) para un conjunto periódico de NPs con d=
1.6 R en diferentes planos (z= 0, -R/2 y -R= sustrato, el origen de z está en el plano
ecuatorial de las NPs) y (b) en el plano del sustrato (z= -R) para d= 1.6R y 0.3R. Los
círculos indican la posición de las partículas
Además, para evaluar el efecto de la densidad de NPs en este plano, se ha
representado la distribución de E2 en z=-R para dos distancias inter-partícula,
1.6R y 0.3R (Figura 4.10b). Se observa que la reducción de la distancia entre
las partículas tiene un enorme efecto en la amplificación del campo eléctrico
en las regiones donde las superficies de las NPs están más cerca (z=0, Figura
7a y b); mientras que en z= -R tiene un efecto muy débil. En el plano del
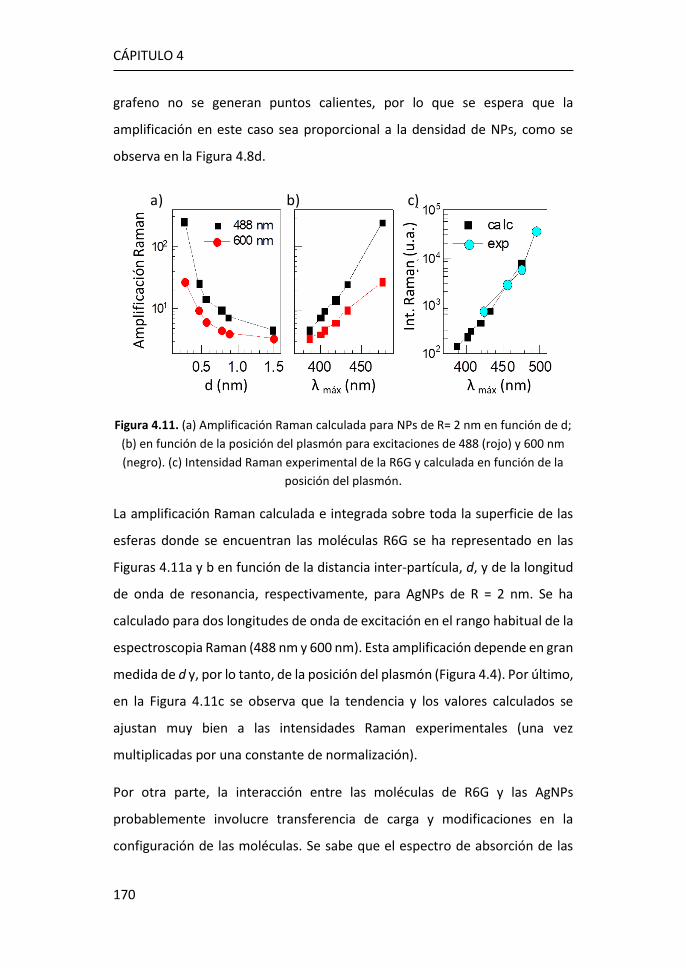
CÁPITULO 4
170
grafeno no se generan puntos calientes, por lo que se espera que la
amplificación en este caso sea proporcional a la densidad de NPs, como se
observa en la Figura 4.8d.
Figura 4.11. (a) Amplificación Raman calculada para NPs de R= 2 nm en función de d;
(b) en función de la posición del plasmón para excitaciones de 488 (rojo) y 600 nm
(negro). (c) Intensidad Raman experimental de la R6G y calculada en función de la
posición del plasmón.
La amplificación Raman calculada e integrada sobre toda la superficie de las
esferas donde se encuentran las moléculas R6G se ha representado en las
Figuras 4.11a y b en función de la distancia inter-partícula, d, y de la longitud
de onda de resonancia, respectivamente, para AgNPs de R = 2 nm. Se ha
calculado para dos longitudes de onda de excitación en el rango habitual de la
espectroscopia Raman (488 nm y 600 nm). Esta amplificación depende en gran
medida de d y, por lo tanto, de la posición del plasmón (Figura 4.4). Por último,
en la Figura 4.11c se observa que la tendencia y los valores calculados se
ajustan muy bien a las intensidades Raman experimentales (una vez
multiplicadas por una constante de normalización).
Por otra parte, la interacción entre las moléculas de R6G y las AgNPs
probablemente involucre transferencia de carga y modificaciones en la
configuración de las moléculas. Se sabe que el espectro de absorción de las
a) b) c)
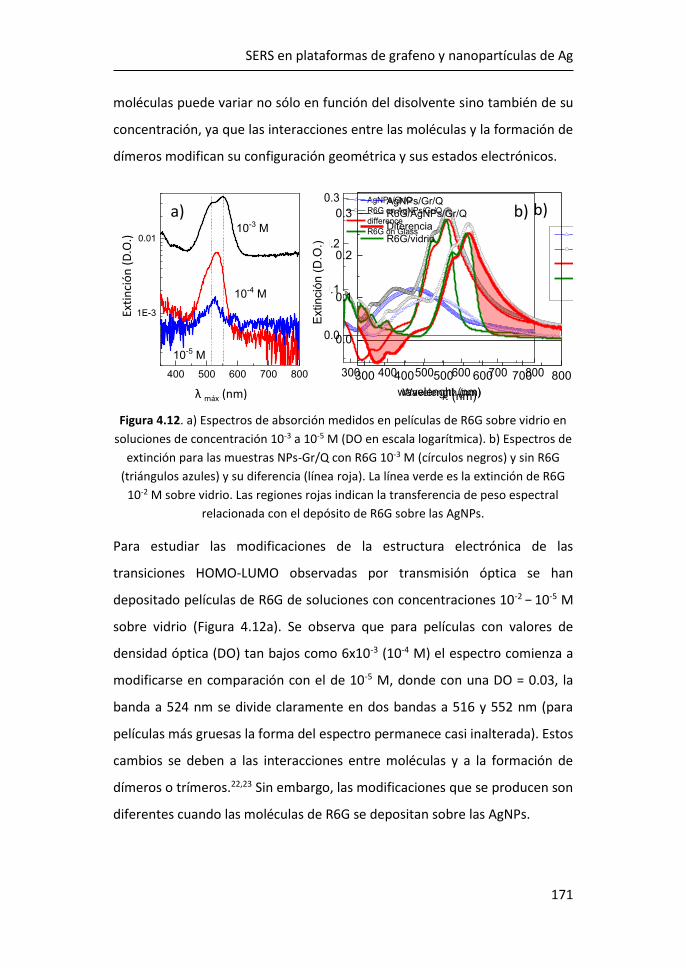
SERS en plataformas de grafeno y nanopartículas de Ag
171
moléculas puede variar no sólo en función del disolvente sino también de su
concentración, ya que las interacciones entre las moléculas y la formación de
dímeros modifican su configuración geométrica y sus estados electrónicos.
Figura 4.12. a) Espectros de absorción medidos en películas de R6G sobre vidrio en
soluciones de concentración 10-3 a 10-5 M (DO en escala logarítmica). b) Espectros de
extinción para las muestras NPs-Gr/Q con R6G 10-3 M (círculos negros) y sin R6G
(triángulos azules) y su diferencia (línea roja). La línea verde es la extinción de R6G
10-2 M sobre vidrio. Las regiones rojas indican la transferencia de peso espectral
relacionada con el depósito de R6G sobre las AgNPs.
Para estudiar las modificaciones de la estructura electrónica de las
transiciones HOMO-LUMO observadas por transmisión óptica se han
depositado películas de R6G de soluciones con concentraciones 10-2 − 10-5 M
sobre vidrio (Figura 4.12a). Se observa que para películas con valores de
densidad óptica (DO) tan bajos como 6x10-3 (10-4 M) el espectro comienza a
modificarse en comparación con el de 10-5 M, donde con una DO = 0.03, la
banda a 524 nm se divide claramente en dos bandas a 516 y 552 nm (para
películas más gruesas la forma del espectro permanece casi inalterada). Estos
cambios se deben a las interacciones entre moléculas y a la formación de
dímeros o trímeros.22,23 Sin embargo, las modificaciones que se producen son
diferentes cuando las moléculas de R6G se depositan sobre las AgNPs.
300 400 500 600 700 800
0.0
0.1
0.2
0.3 AgNPs/Gr/Q R6G on AgNPs/Gr/Q difference R6G on Glass
Ext
intio
n (O
.D.)
wavelenght (nm)
QGAg900 QGAg900R6G resta2 vidrioR6G % (5)
vidrio con pelicula de 10-2MQ/G/Ag900 sec con y sin R6G (10-3)resta: Q/G/Ag+ R&G - vidrio con R6G
300 400 500 600 700 8000.0
0.1
0.2
400 500 600 700 800
1E-3
0.01
10-5 M10-4 M
10-3 M
O.D
.
nm
gssR6G-2 gssR6G-2_2 gssR6G-3_1 gssR6G-3_2 gssR6G-4 gssR6G-4_2 gssR6G-5 gssR6G-5_2
R6G spin coated on glass
10-2 M
10-5 M
10-4 M
10-3 M
Extin
tion
(O.D
.)
wavelenght (nm) gssR6G-2 gssR6G-2_2 gssR6G-3_1 gssR6G-3_2 gssR6G-4 gssR6G-4_2 gssR6G-5 gssR6G-5_2
R6G spin coated on glass
a) b)
Wavelength (nm) Wavelength (nm)λ máx (nm)
Exti
nci
ón
(D
.O.)
300 400 500 600 700 800
0.0
0.1
0.2
0.3 AgNPs/Gr/Q R6G/AgNPs/Gr/Q Diferencia R6G/vidrio
Extin
ción
(D.O
.)
(nm)
b)

CÁPITULO 4
172
En la Figura 4.12b se muestra el espectro de las moléculas de R6G depositadas
sobre la muestra de AgNPs-Gr/Q con un tiempo de depósito de 900 s (círculos
negros) y el de la misma muestra antes de depositar las moléculas (triángulos
azules), que muestra el típico plasmón de Ag. La línea verde traza el espectro
para una película de R6G sobre vidrio con DO similar y la línea roja es la
diferencia entre los espectros de la muestra de AgNPs-Gr/Q con y sin R6G. Este
espectro de diferencia difiere significativamente del de R6G sobre vidrio (línea
verde), lo que evidencia la interacción entre la R6G y las AgNPs. También se
produce un importante desplazamiento del peso espectral del UV (<400 nm)
al rojo (>600 nm) (zonas sombreadas en rojo). En el grupo de J. Zhao se reportó
un desplazamiento del plasmón de Ag a causa de las moléculas de R6G para
determinadas longitudes de onda de plasmón,22 sin embargo, para las
longitudes de onda de estos plasmones no se espera ningún desplazamiento.
Además, las simulaciones del espectro de extinción de AgNPs de recubiertas
con R6G, como una capa con índice de refracción distinto al del aire, no
explican el efecto observado experimentalmente, lo que confirma el efecto
por transferencia de carga.
4.4 COMPARACIÓN DE PELÍCULAS DE AG NANOESTRUCTURADAS
Y NANOPARTÍCULAS DE AG ULTRAPEQUEÑAS.
Se depositaron mediante sputtering una serie de películas delgadas de Ag
sobre sílice vítrea con un grosor nominal de 1, 2 y 4 nm (TF1, TF2 y TF4). En la
Figura 4.13a se muestran las imágenes topográficas de AFM y los
correspondientes perfiles característicos de cada muestra. Las películas
presentan un aspecto granular y una relación de aspecto que disminuye,
pasando de 10 (para TF1) a 3 (TF4) a medida que aumenta el grosor nominal.
El promedio de los radios en el plano aumenta de 7 (TF1) a 15 nm (TF4)
(valores después de la deconvolución del tamaño de la punta del AFM) así
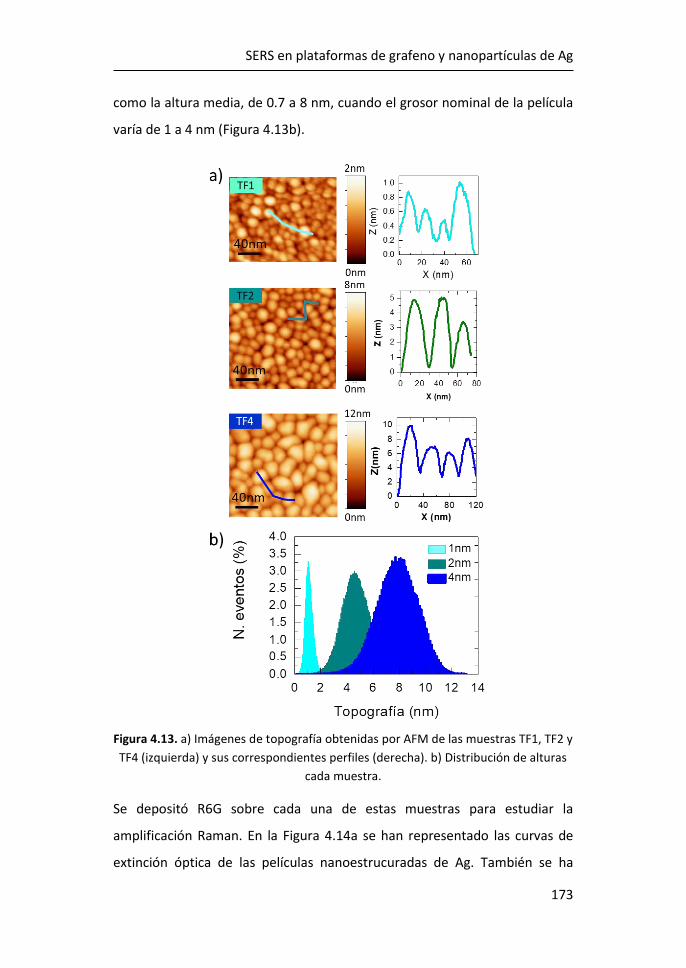
SERS en plataformas de grafeno y nanopartículas de Ag
173
como la altura media, de 0.7 a 8 nm, cuando el grosor nominal de la película
varía de 1 a 4 nm (Figura 4.13b).
Figura 4.13. a) Imágenes de topografía obtenidas por AFM de las muestras TF1, TF2 y
TF4 (izquierda) y sus correspondientes perfiles (derecha). b) Distribución de alturas
cada muestra.
Se depositó R6G sobre cada una de estas muestras para estudiar la
amplificación Raman. En la Figura 4.14a se han representado las curvas de
extinción óptica de las películas nanoestrucuradas de Ag. También se ha
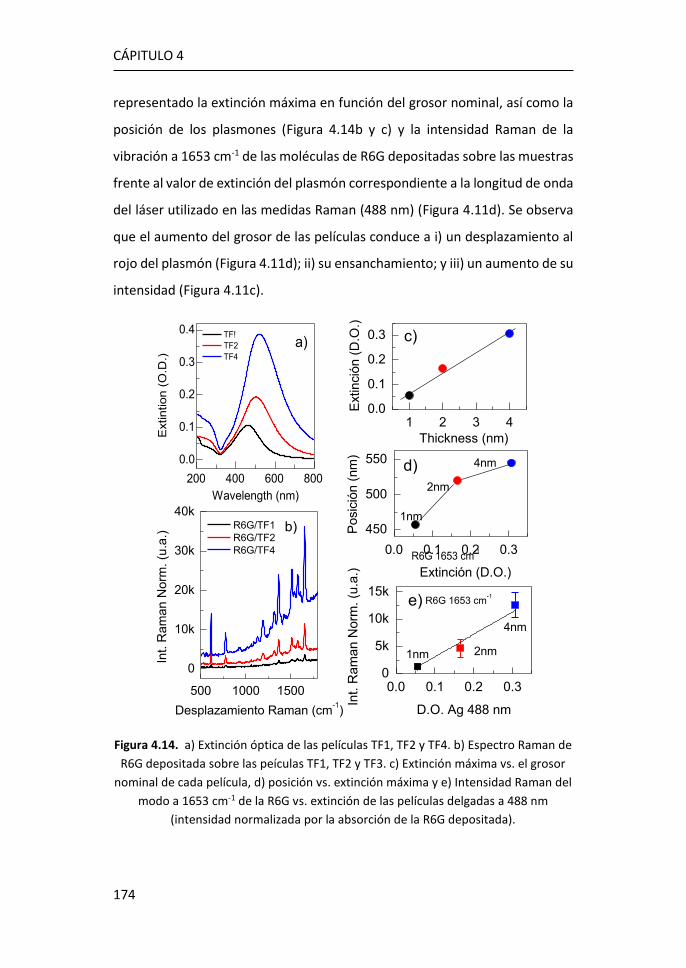
CÁPITULO 4
174
representado la extinción máxima en función del grosor nominal, así como la
posición de los plasmones (Figura 4.14b y c) y la intensidad Raman de la
vibración a 1653 cm-1 de las moléculas de R6G depositadas sobre las muestras
frente al valor de extinción del plasmón correspondiente a la longitud de onda
del láser utilizado en las medidas Raman (488 nm) (Figura 4.11d). Se observa
que el aumento del grosor de las películas conduce a i) un desplazamiento al
rojo del plasmón (Figura 4.11d); ii) su ensanchamiento; y iii) un aumento de su
intensidad (Figura 4.11c).
Figura 4.14. a) Extinción óptica de las películas TF1, TF2 y TF4. b) Espectro Raman de
R6G depositada sobre las peículas TF1, TF2 y TF3. c) Extinción máxima vs. el grosor
nominal de cada película, d) posición vs. extinción máxima y e) Intensidad Raman del
modo a 1653 cm-1 de la R6G vs. extinción de las películas delgadas a 488 nm
(intensidad normalizada por la absorción de la R6G depositada).
0.0 0.1 0.2 0.30
5k
10k
15kR6G 1653 cm-1
Int.
Ram
an N
orm
. (u.
a.)
D.O. Ag 488 nm
1nm
4nm
2nm
e)
500 1000 1500
0
10k
20k
30k
40k
Int.
Ram
an N
orm
. (u.
a.)
Desplazamiento Raman (cm-1)
R6G/TF1 R6G/TF2 R6G/TF4
b)
200 400 600 8000.0
0.1
0.2
0.3
0.4
0.1 0.2 0.30
5k
10k
1 2 3 40.0
0.1
0.2
0.3
0.0 0.1 0.2 0.3450
500
550 d)
Extin
ción
(D.O
.)
Thickness (nm)
c)
Posi
ción
(nm
)
Extinción (D.O.)
1nm
4nm
2nm200 400 600 800
0.0
0.1
0.2
0.3
0.4
0.1 0.2 0.30
5k
10k
1 2 3 40.0
0.1
0.2
0.3
0.0 0.1 0.2 0.3
450
500
550
Wavelength (nm)
Ext
intio
n (O
.D.)
TF! TF2 TF4
a)
R6G 1653 cm-1
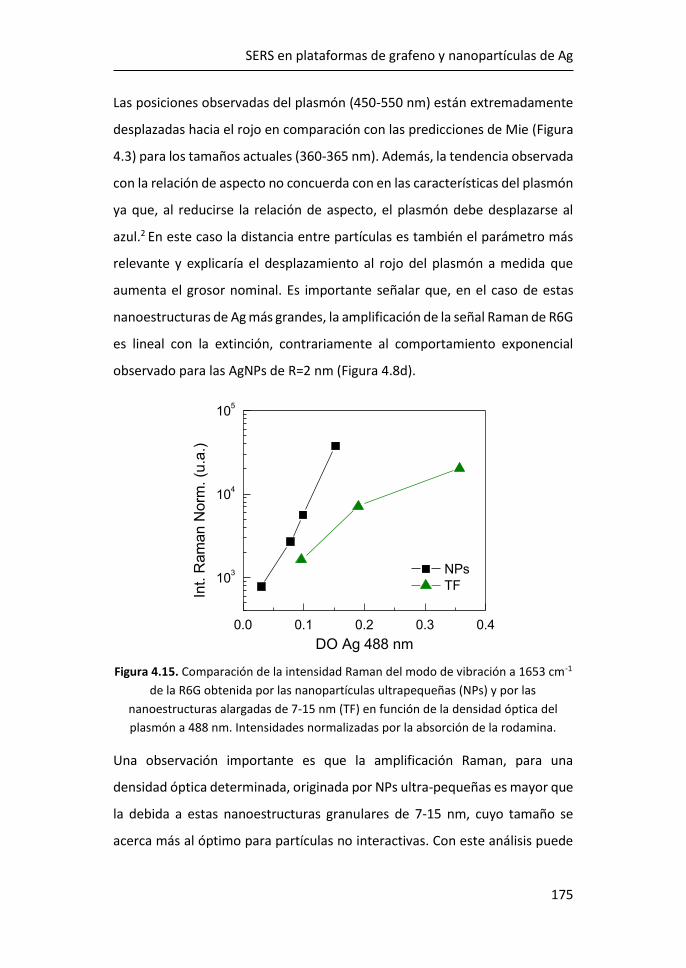
SERS en plataformas de grafeno y nanopartículas de Ag
175
Las posiciones observadas del plasmón (450-550 nm) están extremadamente
desplazadas hacia el rojo en comparación con las predicciones de Mie (Figura
4.3) para los tamaños actuales (360-365 nm). Además, la tendencia observada
con la relación de aspecto no concuerda con en las características del plasmón
ya que, al reducirse la relación de aspecto, el plasmón debe desplazarse al
azul.2 En este caso la distancia entre partículas es también el parámetro más
relevante y explicaría el desplazamiento al rojo del plasmón a medida que
aumenta el grosor nominal. Es importante señalar que, en el caso de estas
nanoestructuras de Ag más grandes, la amplificación de la señal Raman de R6G
es lineal con la extinción, contrariamente al comportamiento exponencial
observado para las AgNPs de R=2 nm (Figura 4.8d).
Figura 4.15. Comparación de la intensidad Raman del modo de vibración a 1653 cm-1
de la R6G obtenida por las nanopartículas ultrapequeñas (NPs) y por las
nanoestructuras alargadas de 7-15 nm (TF) en función de la densidad óptica del
plasmón a 488 nm. Intensidades normalizadas por la absorción de la rodamina.
Una observación importante es que la amplificación Raman, para una
densidad óptica determinada, originada por NPs ultra-pequeñas es mayor que
la debida a estas nanoestructuras granulares de 7-15 nm, cuyo tamaño se
acerca más al óptimo para partículas no interactivas. Con este análisis puede
0.0 0.1 0.2 0.3 0.4
103
104
105
NPs TFIn
t. R
aman
Nor
m. (
u.a.
)
DO Ag 488 nm

CÁPITULO 4
176
concluirse que, en la amplificación Raman, el peso de los puntos calientes es
especialmente importante para las NPs extremadamente pequeñas en los que
la relación superficie/volumen es muy alta.
4.5 PELÍCULAS DE AG NANOESTRUCTURADAS COMBINADAS CON
AGNPS DE R = 1.8 NM
Para evaluar la posibilidad de mejorar aún más la amplificación de la señal, se
fabricaron sistemas más complejos combinando las películas delgadas
nanoestructuradas de granos con R=10 nm con NPs de R= 1.8 nm. De este
modo se pretende aumentar la amplificación Raman relacionada con una
mayor masa de Ag de las películas nanoestructuradas, y promover la
formación de puntos calientes debidos a las NPs ultra-pequeñas.
Figura 4.16. Esquema de las tres zonas de las que consta cada muestra:
nanopartículas ultrapequeñas (NPs, zona A), película delgada nanoestructurada (TF,
zona B), y una combinación de ambas (TF+NPs, zona C).
Cada muestra presenta tres regiones: NPs (zona A), película delgada
nanoestructurada (TF) (zona B) y NPs y TF combinados (zona C) depositados
en sustratos de sílice vítrea (Figura 4.16). De esta manera es posible comparar
de manera fiable las características del plasmón de las diferentes zonas, así
como la amplificación Raman de los modos vibracionales de las moléculas de
R6G depositadas posteriormente por inmersión.
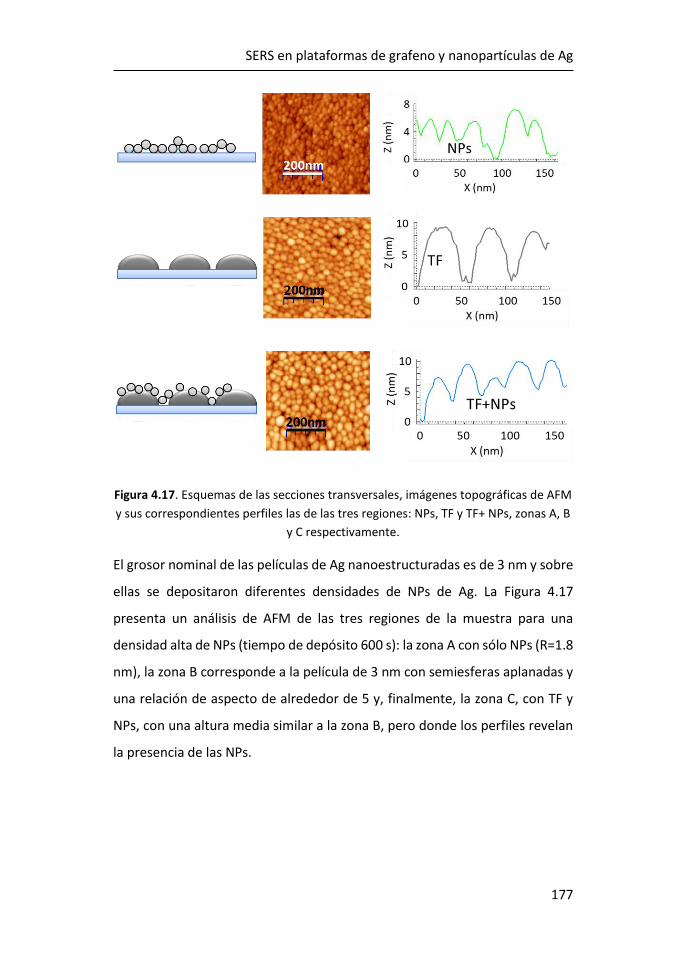
SERS en plataformas de grafeno y nanopartículas de Ag
177
Figura 4.17. Esquemas de las secciones transversales, imágenes topográficas de AFM
y sus correspondientes perfiles las de las tres regiones: NPs, TF y TF+ NPs, zonas A, B
y C respectivamente.
El grosor nominal de las películas de Ag nanoestructuradas es de 3 nm y sobre
ellas se depositaron diferentes densidades de NPs de Ag. La Figura 4.17
presenta un análisis de AFM de las tres regiones de la muestra para una
densidad alta de NPs (tiempo de depósito 600 s): la zona A con sólo NPs (R=1.8
nm), la zona B corresponde a la película de 3 nm con semiesferas aplanadas y
una relación de aspecto de alrededor de 5 y, finalmente, la zona C, con TF y
NPs, con una altura media similar a la zona B, pero donde los perfiles revelan
la presencia de las NPs.
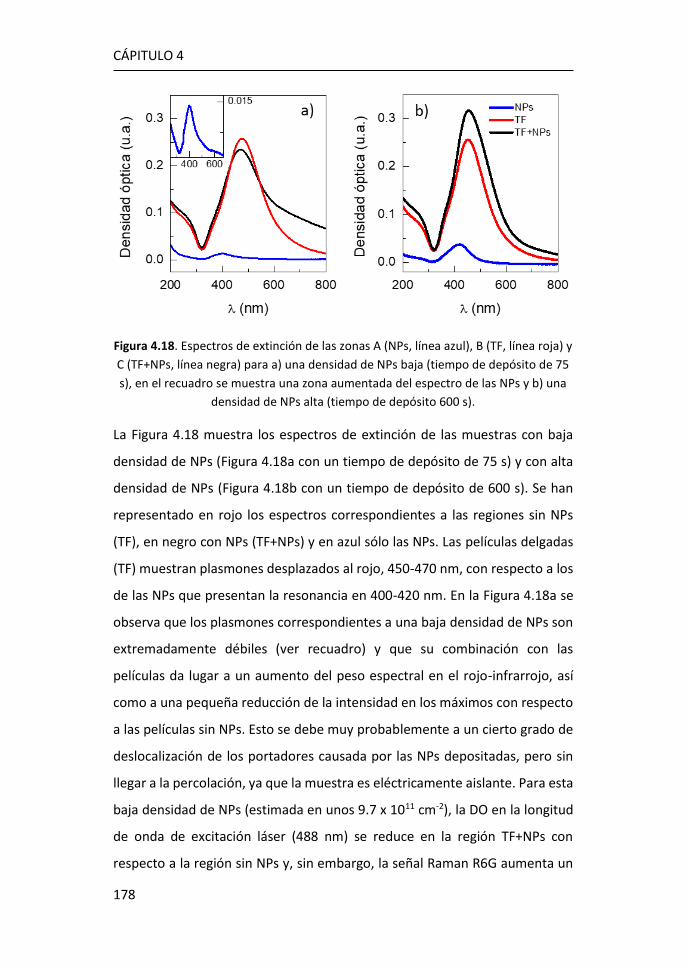
CÁPITULO 4
178
Figura 4.18. Espectros de extinción de las zonas A (NPs, línea azul), B (TF, línea roja) y
C (TF+NPs, línea negra) para a) una densidad de NPs baja (tiempo de depósito de 75
s), en el recuadro se muestra una zona aumentada del espectro de las NPs y b) una
densidad de NPs alta (tiempo de depósito 600 s).
La Figura 4.18 muestra los espectros de extinción de las muestras con baja
densidad de NPs (Figura 4.18a con un tiempo de depósito de 75 s) y con alta
densidad de NPs (Figura 4.18b con un tiempo de depósito de 600 s). Se han
representado en rojo los espectros correspondientes a las regiones sin NPs
(TF), en negro con NPs (TF+NPs) y en azul sólo las NPs. Las películas delgadas
(TF) muestran plasmones desplazados al rojo, 450-470 nm, con respecto a los
de las NPs que presentan la resonancia en 400-420 nm. En la Figura 4.18a se
observa que los plasmones correspondientes a una baja densidad de NPs son
extremadamente débiles (ver recuadro) y que su combinación con las
películas da lugar a un aumento del peso espectral en el rojo-infrarrojo, así
como a una pequeña reducción de la intensidad en los máximos con respecto
a las películas sin NPs. Esto se debe muy probablemente a un cierto grado de
deslocalización de los portadores causada por las NPs depositadas, pero sin
llegar a la percolación, ya que la muestra es eléctricamente aislante. Para esta
baja densidad de NPs (estimada en unos 9.7 x 1011 cm-2), la DO en la longitud
de onda de excitación láser (488 nm) se reduce en la región TF+NPs con
respecto a la región sin NPs y, sin embargo, la señal Raman R6G aumenta un
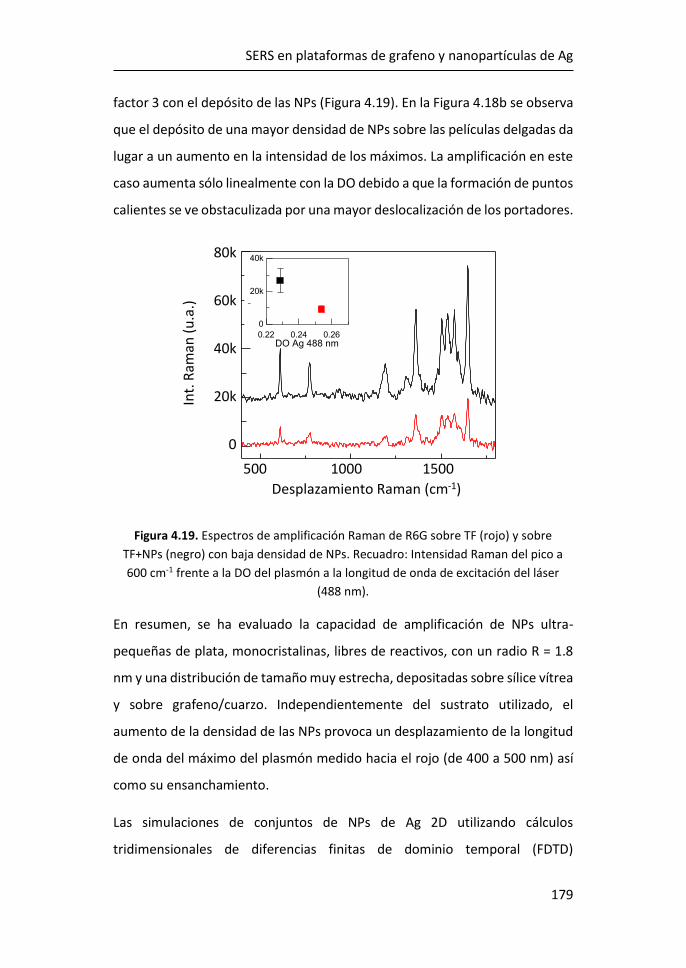
SERS en plataformas de grafeno y nanopartículas de Ag
179
factor 3 con el depósito de las NPs (Figura 4.19). En la Figura 4.18b se observa
que el depósito de una mayor densidad de NPs sobre las películas delgadas da
lugar a un aumento en la intensidad de los máximos. La amplificación en este
caso aumenta sólo linealmente con la DO debido a que la formación de puntos
calientes se ve obstaculizada por una mayor deslocalización de los portadores.
Figura 4.19. Espectros de amplificación Raman de R6G sobre TF (rojo) y sobre
TF+NPs (negro) con baja densidad de NPs. Recuadro: Intensidad Raman del pico a
600 cm-1 frente a la DO del plasmón a la longitud de onda de excitación del láser
(488 nm).
En resumen, se ha evaluado la capacidad de amplificación de NPs ultra-
pequeñas de plata, monocristalinas, libres de reactivos, con un radio R = 1.8
nm y una distribución de tamaño muy estrecha, depositadas sobre sílice vítrea
y sobre grafeno/cuarzo. Independientemente del sustrato utilizado, el
aumento de la densidad de las NPs provoca un desplazamiento de la longitud
de onda del máximo del plasmón medido hacia el rojo (de 400 a 500 nm) así
como su ensanchamiento.
Las simulaciones de conjuntos de NPs de Ag 2D utilizando cálculos
tridimensionales de diferencias finitas de dominio temporal (FDTD)
500 1000 1500
0
20k
40k
60k
80k
0.20 0.25
20k
40k
Int. Ra
man (
u.a.)
(nm)
TF TF + NPs
DO Ag 514 nm
500 1000 1500
0
20k
40k
60k
80k
0.22 0.24 0.260
20k
40k
Int. Ra
man (
u.a.)
(nm)
DO Ag 488 nm
500 1000 1500
80k
60k
40k
20k
0
500 1000 1500
0
20k
40k
60k
80k
0.20 0.25
20k
40k
Int.
Ram
an (u
.a.)
(nm)
TF TF + NPs
DO Ag 514 nm
500 1000 1500
0
20k
40k
60k
80k
0.22 0.24 0.260
20k
40k
Int.
Ram
an (u
.a.)
(nm)
DO Ag 488 nm
Desplazamiento Raman (cm-1)
Int.
Ram
an (
u.a
.)

CÁPITULO 4
180
demostraron que la reducción de la distancia inter-partícula, d, conduce a un
desplazamiento hacia el rojo y a un ensanchamiento del plasmón. Estos
efectos son generales para las NPs con radios, R, hasta 10 nm, en términos de
distancia normalizada d/R, y son importantes cuando d<R.
Se ha calculado la influencia de una posible capa de recubrimiento de las NPs
(debido a la oxidación o a la adsorción de especies derivadas del carbono), el
resultado indica una pequeña variación. Los plasmones medidos se originan
por distribuciones de distancias inter-partícula que se estrechan y se
desplazan a distancias más pequeñas a medida que la densidad de NPs
aumenta.
El papel del grafeno como sustrato para el depósito de NPs es deslocalizar
parcialmente los portadores libres, así como favorecer la adherencia de las
NPs sobre el sustrato.
La amplificación de las señales Raman del grafeno y de una película de R6G
depositada por “spin-coating” varía de manera muy diferente al aumentar la
densidad de las NPs. En el caso del grafeno, la amplificación aumenta
linealmente con la extinción del plasmón, mientras que en el caso de la R6G la
amplificación es casi exponencial. Esta diferencia se debe a la diferente
configuración geométrica de ambos con respecto a las NPs. El campo eléctrico
en la posición del sustrato (la posición del grafeno) presenta una amplificación
débil y no se ve significativamente afectada por la reducción de la distancia
entre las partículas. Por el contrario, las moléculas R6G, que envuelven las
NPs, se ven afectadas por los altos campos creados en los espacios entre las
NPs (hot-spots) a medida que la distancia entre estas disminuye. La variación
observada de la intensidad Raman en función de la intensidad del plasmón se
corresponde muy bien con la amplificación calculada al reducir la distancia
media entre las partículas. El comportamiento exponencial demuestra la alta
eficiencia de los puntos calientes que se forman en los espacios inter-partícula.

SERS en plataformas de grafeno y nanopartículas de Ag
181
También se ha comparado la amplificación de las NPs ultra-pequeñas con la
de granos más grandes (7<R<15 nm) de películas delgadas de plata. El
desplazamiento hacia el rojo y el ensanchamiento del plasmón que se observa
a medida que el grosor de la película aumenta también se deben
principalmente a la reducción de la distancia entre las partículas, sin embargo,
la amplificación de R6G en este caso es sólo proporcional a la extinción del
plasmón. Es importante señalar que, para el mismo valor de extinción, la
amplificación proporcionada por las NPs ultra-pequeñas es mayor que la de
los granos más grandes de las películas, debido principalmente a los puntos
calientes entre las NPs ultra-pequeñas.
Desde el punto de vista de las aplicaciones, resulta relevante la posibilidad de
aumentar eficientemente la amplificación obtenida con películas de plata
depositando sobre ellas bajas densidades de NPs ultra-pequeñas, de manera
que se crean puntos calientes adicionales.
Una vez normalizada la intensidad de los respectivos plasmones debido a la
mayor densidad de puntos calientes que se pueden generar para una misma
masa de plata, el presente trabajo demuestra que las NPs ultra-pequeñas son
más eficientes para el SERS que las NPs más grandes comúnmente utilizadas.
4.6 REFERENCIAS
1. Khoury, C. G., Vo-dinh, T., V, D. U. & Carolina, N. Gold Nanostars For Surface-Enhanced Raman Scattering : Synthesis , Characterization and Optimization. J. Phys. Chem. C 112, 18849–18859 (2008).
2. Kelly, K. L., Coronado, E., Zhao, L. L. & Schatz, G. C. The optical properties of metal nanoparticles: The influence of size, shape, and dielectric environment. J. Phys. Chem. B 107, 668–677 (2003).
3. Noguez, C. Surface plasmons on metal nanoparticles: The influence of shape and physical environment. J. Phys. Chem. C 111, 3606–3619

CÁPITULO 4
182
(2007).
4. Chapus, L. et al. Tunable SERS Platforms from Small Nanoparticle 3D Superlattices: A Comparison between Gold, Silver, and Copper. ChemPhysChem 18, 3066–3075 (2017).
5. Creighton, J. A., Blatchford, C. G. & Albrecht, M. G. Plasma resonance enhancement of Raman scattering by pyridine adsorbed on silver or gold sol particles of size comparable to the excitation wavelength. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 75, 790–798 (1979).
6. Chumanov, G., Neddersen, J. & Cotton, T. M. Laser Ablation of Metals: A New Method for Preparing SERS Active Colloids. Appl. Spectrosc. Vol. 47, Issue 12, pp. 1959-1964 47, 1959–1964 (1993).
7. Vinod, M. & Gopchandran, K. G. Au, Ag and Au: Ag colloidal nanoparticles synthesized by pulsed laser ablation as SERS substrates. Prog. Nat. Sci. Mater. Int. 24, 569–578 (2014).
8. Muniz-Miranda, M. et al. Nanostructured films of metal particles obtained by laser ablation. Thin Solid Films 543, 118–121 (2013).
9. Condorelli, M. et al. Plasmon sensing and enhancement of laser prepared silver colloidal nanoplates. Appl. Surf. Sci. 475, 633–638 (2019).
10. Pena-Pereira, F., Duarte, R. M. B. O. & Duarte, A. C. Immobilization strategies and analytical applications for metallic and metal-oxide nanomaterials on surfaces. TrAC - Trends in Analytical Chemistry vol. 40 90–105 (2012).
11. Péron, O., Rinnert, E., Lehaitre, M., Crassous, P. & Compère, C. Detection of polycyclic aromatic hydrocarbon (PAH) compounds in artificial sea-water using surface-enhanced Raman scattering (SERS). Talanta 79, 199–204 (2009).
12. He, S., Chua, J., Tan, E. K. M. & Kah, J. C. Y. Optimizing the SERS enhancement of a facile gold nanostar immobilized paper-based SERS substrate. RSC Adv. 7, 16264–16272 (2017).
13. John C. Hulteen, † et al. Nanosphere Lithography: Size-Tunable Silver Nanoparticle and Surface Cluster Arrays. J. Phys. Chem. B 103, 3854–3863 (1999).
14. Mahajan, S. et al. Tuning plasmons on nano-structured substrates for NIR-SERS. Phys. Chem. Chem. Phys. 9, 104–109 (2006).
15. Hou, X. et al. Periodic silver nanocluster arrays over large-area silica

SERS en plataformas de grafeno y nanopartículas de Ag
183
nanosphere template as highly sensitive SERS substrate. Appl. Surf. Sci. 437, 92–97 (2018).
16. Haberland, H., Karrais, M. & Mall, M. A new type of cluster and cluster ion source. Zeitschrift für Phys. D Atoms, Mol. Clust. 20, 413–415 (1991).
17. Cortijo-Campos, S. et al. Raman amplification in the ultra-small limit of Ag nanoparticles on SiO2 and graphene: Size and inter-particle distance effects. Mater. Des. 192, 108702 (2020).
18. Schatz, G. C., Young, M. A. & Duyne, R. P. Van. Electromagnetic Mechanism of SERS. 46, 19–46 (2006).
19. Garcia, M. A. Surface plasmons in metallic nanoparticles : Fundamentals and Applications. J. Phys. D Appl. Physics. 44, 283001 (2011).
20. Jain, P. K., Huang, W. & El-sayed, M. A. On the Universal Scaling Behavior of the Distance Decay of Plasmon Coupling in Metal Nanoparticle Pairs : A Plasmon Ruler Equation. Nano Lett. 7, 2080–2088 (2007).
21. Haynes, C. L. & Duyne, R. P. Van. Nanosphere Lithography: A Versatile Nanofabrication Tool for Studies of Size-Dependent Nanoparticle Optics. J. Phys. Chem. B 105, 5599–5611 (2001).
22. Zhao, J. et al. Interaction of Plasmon and Molecular Resonances for Rhodamine 6G Adsorbed on Silver Nanoparticles. J. Am. Chem. Soc. 129, 7647–7656 (2007).
23. F. López Arbeloa, *, V. Martínez Martínez, J. Bañuelos Prieto, and & Arbeloa, I. L. Adsorption of Rhodamine 3B Dye on Saponite Colloidal Particles in Aqueous Suspensions. Langmuir 18, 2658–2664 (2002).

185
5 FUNCIONALIZACIÓN DE GRAFENO CON GRUPOS
CARBOXÍLICOS PARA APLICACIONES EN
BIODETECCIÓN
En los últimos años, los materiales de grafeno propuestos para aplicaciones de
detección de biomoléculas se han basado habitualmente en la adaptación del
óxido de grafeno (GO) mediante procesos químicos de reducción,1–4 lo que
plantea algunos problemas de eliminación de reactivos, de reproducibilidad y
de baja conductividad debido a defectos inherentes a ese material. El GO
presenta distintos grupos funcionales oxigenados como grupos carboxilo (-
COOH), hidroxilo (-OH), epoxi y carbonilo (CO), que permiten una gran
variedad de procesos de funcionalización.5 La funcionalización covalente para
el reconocimiento de antígenos suele basarse en el anclaje de un anticuerpo
a través de los grupos carboxilo, mientras que la presencia del resto de grupos
oxigenados interrumpe la conjugación y aumentan la resistividad del grafeno,
convirtiéndolo en un material aislante y dificultando su aplicabilidad en
dispositivos electrónicos.
En este trabajo se ha diseñado una nueva ruta de síntesis para funcionalizar
in-situ grafeno-CVD con grupos -COOH de una forma controlada. Esta
funcionalización pretende facilitar la posterior unión con otros grupos
específicos, como las aminas primarias (-NH2) vía enlaces carbodiimida,
presentes en diferentes moléculas biológicamente activas y disponer así de
plataformas aptas para sensores ópticos o electrónicos de biomoléculas.6
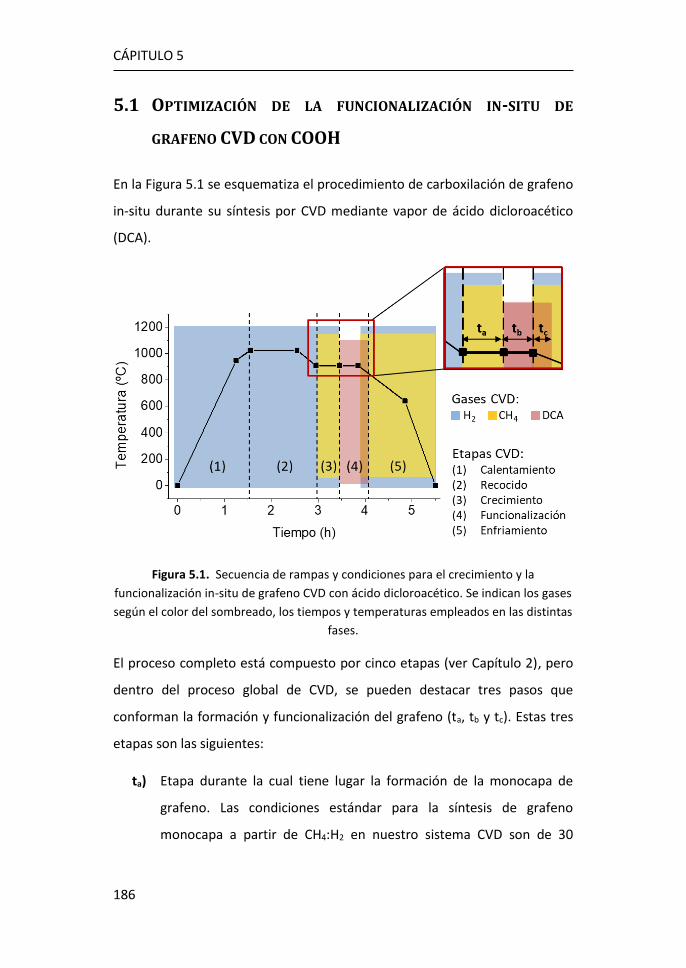
CÁPITULO 5
186
5.1 OPTIMIZACIÓN DE LA FUNCIONALIZACIÓN IN-SITU DE
GRAFENO CVD CON COOH
En la Figura 5.1 se esquematiza el procedimiento de carboxilación de grafeno
in-situ durante su síntesis por CVD mediante vapor de ácido dicloroacético
(DCA).
Figura 5.1. Secuencia de rampas y condiciones para el crecimiento y la
funcionalización in-situ de grafeno CVD con ácido dicloroacético. Se indican los gases
según el color del sombreado, los tiempos y temperaturas empleados en las distintas
fases.
El proceso completo está compuesto por cinco etapas (ver Capítulo 2), pero
dentro del proceso global de CVD, se pueden destacar tres pasos que
conforman la formación y funcionalización del grafeno (ta, tb y tc). Estas tres
etapas son las siguientes:
ta) Etapa durante la cual tiene lugar la formación de la monocapa de
grafeno. Las condiciones estándar para la síntesis de grafeno
monocapa a partir de CH4:H2 en nuestro sistema CVD son de 30

Funcionalización de grafeno con grupos carboxílicos…
187
minutos a 910 ºC. Para la obtención de muestras funcionalizadas se
escogieron tiempos de 15 y 30 min.
tb) En esta etapa se introduce el vapor de DCA. Dependiendo de la
muestra se mantienen o se eliminan los flujos de H2 y CH4 y se lleva a
cabo a la misma temperatura que la etapa anterior (910 ºC). Esta etapa
tiene duraciones diferentes en función de la muestra (0-30 min).
tc) En algunas muestras se mantiene el flujo de DCA durante los primeros
minutos (tc) de la etapa de enfriamiento.
Se crecieron sucesivas muestras variando los tiempos y flujos de gases de
estos tres pasos de manera sistemática con el fin de optimizar el proceso y
obtener grafeno monocapa con el máximo grado de funcionalización. Se han
resumido en la Tabla 5.1 las muestras representativas obtenidas con
diferentes combinaciones de tiempos en ta, tb y tc etiquetadas como
'FGr_ta.tb.tc'. Por ejemplo, la etiqueta 'FGr_30.15.0' se refiere a una muestra
obtenida con un tiempo de crecimiento de grafeno de 30 min con CH4:H2 (ta =
30 min) a 910 ºC seguido de 15 min a 910 ºC con vapor de DCA (tb = 15 min), y
sin vapor de DCA durante el enfriamiento (tc = 0 min). SLG y MLG hacen
referencia a grafeno monocapa y multicapa respectivamente, y su
terminación -F implica que está funcionalizado. En algunas muestras el tipo de
grafeno no es homogéneo a lo largo de la muestra, por lo que el etiquetado
indica las diferentes características.
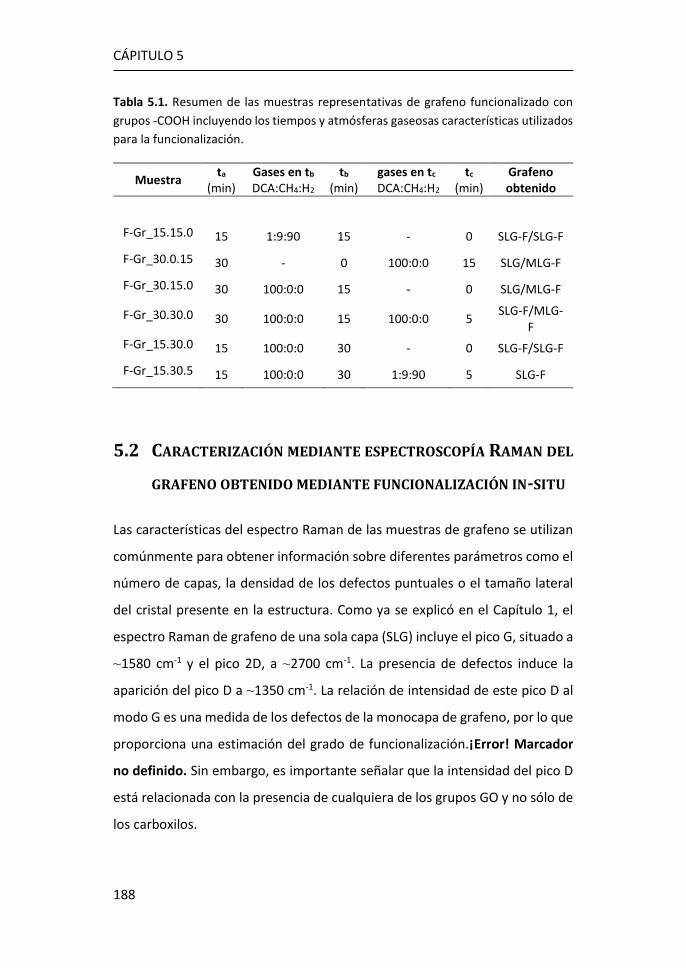
CÁPITULO 5
188
Tabla 5.1. Resumen de las muestras representativas de grafeno funcionalizado con
grupos -COOH incluyendo los tiempos y atmósferas gaseosas características utilizados
para la funcionalización.
Muestra ta
(min) Gases en tb DCA:CH4:H2
tb (min)
gases en tc DCA:CH4:H2
tc (min)
Grafeno obtenido
F-Gr_15.15.0 15 1:9:90 15 - 0 SLG-F/SLG-F
F-Gr_30.0.15 30 - 0 100:0:0 15 SLG/MLG-F
F-Gr_30.15.0 30 100:0:0 15 - 0 SLG/MLG-F
F-Gr_30.30.0 30 100:0:0 15 100:0:0 5 SLG-F/MLG-
F
F-Gr_15.30.0 15 100:0:0 30 - 0 SLG-F/SLG-F
F-Gr_15.30.5 15 100:0:0 30 1:9:90 5 SLG-F
5.2 CARACTERIZACIÓN MEDIANTE ESPECTROSCOPÍA RAMAN DEL
GRAFENO OBTENIDO MEDIANTE FUNCIONALIZACIÓN IN-SITU
Las características del espectro Raman de las muestras de grafeno se utilizan
comúnmente para obtener información sobre diferentes parámetros como el
número de capas, la densidad de los defectos puntuales o el tamaño lateral
del cristal presente en la estructura. Como ya se explicó en el Capítulo 1, el
espectro Raman de grafeno de una sola capa (SLG) incluye el pico G, situado a
~1580 cm-1 y el pico 2D, a ~2700 cm-1. La presencia de defectos induce la
aparición del pico D a ~1350 cm-1. La relación de intensidad de este pico D al
modo G es una medida de los defectos de la monocapa de grafeno, por lo que
proporciona una estimación del grado de funcionalización.¡Error! Marcador
no definido. Sin embargo, es importante señalar que la intensidad del pico D
está relacionada con la presencia de cualquiera de los grupos GO y no sólo de
los carboxilos.

Funcionalización de grafeno con grupos carboxílicos…
189
En la Figura 5.2 se han representado espectros de las zonas más
representativas de cada una de las muestras. Inicialmente, la funcionalización
se llevó a cabo introduciendo el vapor de DCA en la segunda mitad de la etapa
de crecimiento estándar, es decir, ta tuvo una duración de 15 min, y durante
los siguientes 15 minutos se introdujo DCA manteniendo también la mezcla
H2:CH4 (FGr_15.15.0, Fig 5.2a). La muestra obtenida fue de grafeno monocapa
con una funcionalización débil en un 30 % de la muestra (con ID/IG en el rango
0.1 - 0.2). Se observó que la presión de trabajo en la etapa tb fue la misma que
en la etapa ta a pesar de introducir el DCA. Esto se debe a que el flujo
conseguido al evaporar el DCA es muy pequeño en comparación al flujo de
H2:CH4 y por ello la funcionalización es débil. En base a este resultado y con la
intención de conseguir un mayor grado de funcionalización, en las siguientes
muestras se introdujo el DCA en ausencia de los gases H2:CH4 después del paso
de crecimiento del grafeno estándar (ta de 30 minutos), y se hizo de tres
maneras diferentes: i) en los primeros 15 minutos de la rampa de enfriamiento
('FGr_30.0.15', Fig 5.2b), ii) durante 15 y iii) 30 minutos a la temperatura de
crecimiento 910 °C ('FGr_30.15.0', Fig 5.2c y 'FGr_30.30.0', Fig 5.2d,
respectivamente). En los dos primeros casos la funcionalización no es
homogénea y, curiosamente, se puede observar que en la región
funcionalizada (con una intensidad D significativa) se forma grafeno multicapa
(MLG-F), mientras que para las regiones no funcionalizadas se registra el
espectro de grafeno monocapa sin defectos (SLG). La confirmación de que el
grafeno es monocapa viene dada por la relación de intensidad I2D/IG > 1,
típicamente en el rango 2-4. Sin embargo, la intensidad 2D máxima puede
reducirse por las distorsiones y defectos de la red, de modo que un SLG con
defectos puede presentar I2D/IG ≤ 1. Por otro lado, un mayor tiempo tb
(‘FGr_30.30.0’), da lugar a grafeno monocapa y multicapa funcionalizados
(SLG-F y MLG-F) de manera homogénea en toda la muestra.
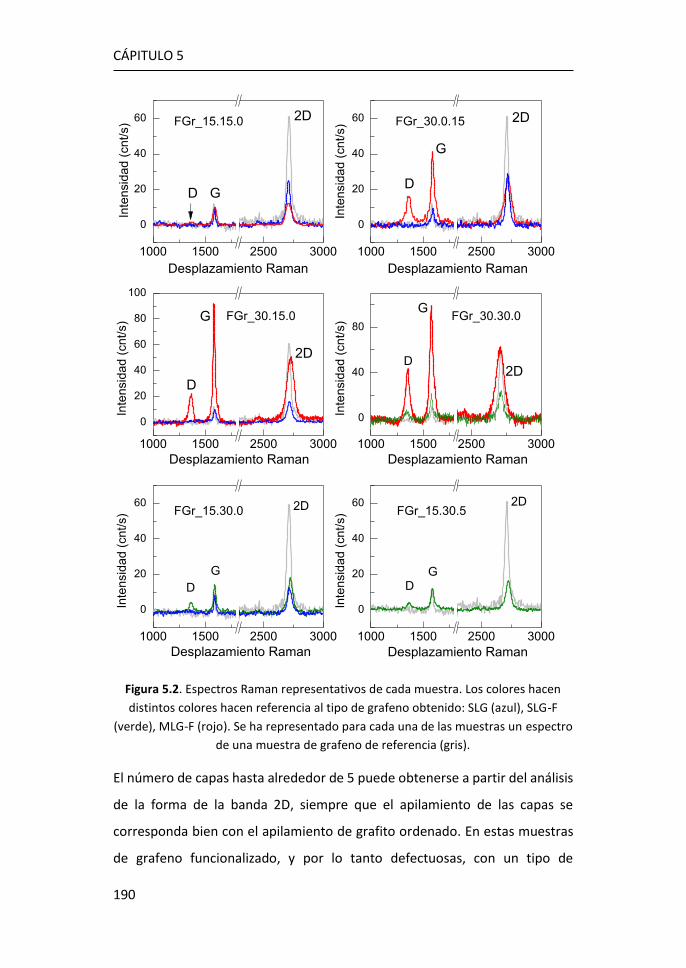
CÁPITULO 5
190
Figura 5.2. Espectros Raman representativos de cada muestra. Los colores hacen
distintos colores hacen referencia al tipo de grafeno obtenido: SLG (azul), SLG-F
(verde), MLG-F (rojo). Se ha representado para cada una de las muestras un espectro
de una muestra de grafeno de referencia (gris).
El número de capas hasta alrededor de 5 puede obtenerse a partir del análisis
de la forma de la banda 2D, siempre que el apilamiento de las capas se
corresponda bien con el apilamiento de grafito ordenado. En estas muestras
de grafeno funcionalizado, y por lo tanto defectuosas, con un tipo de
1000 1500 2500 3000
0
40
80
1000 1500 2500 3000
0
20
40
60
1000 1500 2500 3000
0
20
40
60
1000 1500 2500 3000
0
20
40
60
80
100
1000 1500 2500 3000
0
20
40
60
1000 1500 2500 3000
0
20
40
60
FGr_30.30.0
Inte
nsid
ad (c
nt/s
)In
tens
idad
(cnt
/s)
Inte
nsid
ad (c
nt/s
)
Inte
nsid
ad (c
nt/s
)In
tens
idad
(cnt
/s)
Desplazamiento Raman
Inte
nsid
ad (c
nt/s
)
Desplazamiento Raman
D
FGr_15.15.0
Desplazamiento Raman Desplazamiento Raman
Desplazamiento Raman Desplazamiento Raman
G
2D
D
G
2D
D
G
2DFGr_30.0.15
FGr_30.15.0
2DFGr_15.30.0
DG
2D FGr_15.30.5
DG
D
G
2D

Funcionalización de grafeno con grupos carboxílicos…
191
apilamiento desconocido, se ha utilizado la intensidad del pico G para obtener
una estimación del número de capas.7 Como aproximación razonable, el
número de capas se obtiene de la relación entre la intensidad del pico G de la
muestra y la de un grafeno monocapa sin defectos de referencia. De esta
manera, el número de capas para las muestras con ta=30 min es de entre 2 y
15, dependiendo de la zona de la muestra, y el cociente ID/IG está en el rango
0.25 – 0.35.
Dado que ta= 30 min da como resultado películas de grafeno funcionalizadas
pero multicapa, se probó a utilizar un tiempo ta más corto, 15 min, y tb más
largo, 30 min, con el objetivo de obtener grafeno funcionalizado monocapa.
Como resultado se obtuvieron muestras de grafeno monocapa/bicapa (I2D/IG
≥ 1), funcionalizado de manera heterogénea, con valores de ID/IG en el rango
0-0.3 (‘FGr_15.30.0’, Fig 5.2e). Introduciendo el DCA también durante los
primeros minutos de enfriamiento es posible obtener un grafeno
monocapa/bicapa funcionalizado homogéneamente ('FGr_15.30.5', Fig 5.2f).
Para comprobar la uniformidad de las películas de grafeno funcionalizado se
realizaron mapas Raman de 15x15 μm. En la Figura 5.3 se representan las
imágenes ópticas y sus correspondientes mapas que muestran las relaciones
de intensidad ID/IG (azul) y I2D/IG (verde) de los cuatro tipos de muestras
representativos, así como de una muestra de referencia de grafeno CVD
monocapa sin funcionalizar. En las imágenes Raman I2D/IG las zonas oscuras
representan las zonas multicapa, MLG, y las zonas claras las zonas monocapa,
SLG, mientras que en las imágenes ID/IG las regiones oscuras corresponden al
grafeno no funcionalizado (ID débil) y las claras al grafeno funcionalizado (ID
más intenso). Observando estas imágenes Raman se hace evidente que las
regiones de grafeno multicapa están siempre funcionalizadas.
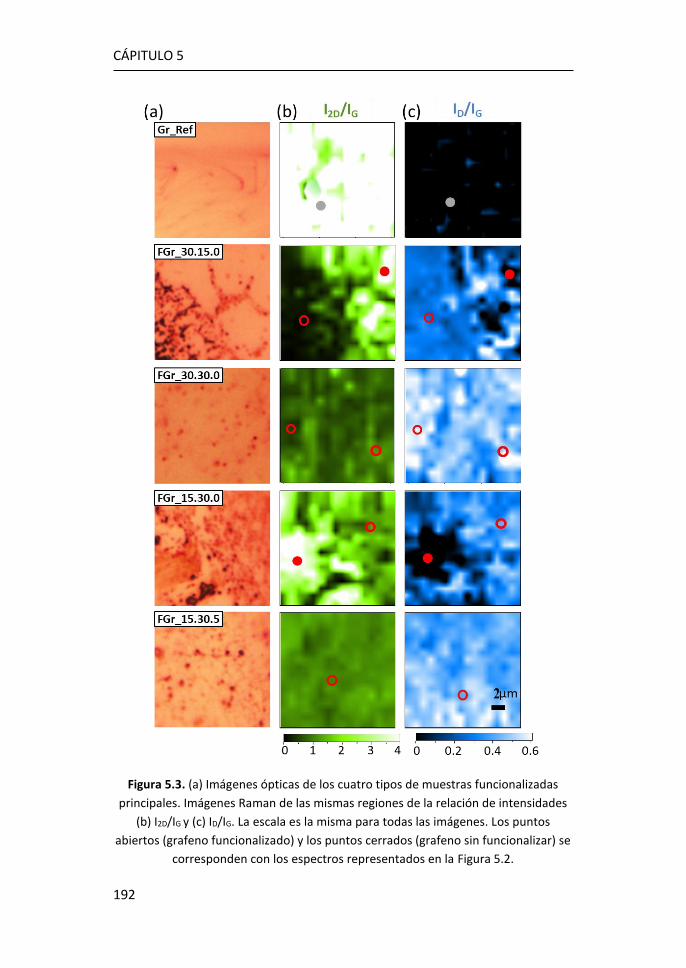
CÁPITULO 5
192
Figura 5.3. (a) Imágenes ópticas de los cuatro tipos de muestras funcionalizadas
principales. Imágenes Raman de las mismas regiones de la relación de intensidades
(b) I2D/IG y (c) ID/IG. La escala es la misma para todas las imágenes. Los puntos
abiertos (grafeno funcionalizado) y los puntos cerrados (grafeno sin funcionalizar) se
corresponden con los espectros representados en la Figura 5.2.

Funcionalización de grafeno con grupos carboxílicos…
193
Como ya se ha mencionado, las muestras crecidas con ta=30 min son
multicapa, lo que indica que el DCA en la etapa tb promueve la formación de
grafeno, ya que para este mismo tiempo de crecimiento, pero sin DCA, se
obtiene una lámina continua de grafeno monocapa sin defectos. Debido a esta
contribución al crecimiento de grafeno por parte del DCA, se probó crecer
grafeno funcionalizado usando DCA como única fuente de carbono (sin CH4)
con la intención de obtener una muestra de grafeno fuertemente
funcionalizado. Sin embargo, se observó que, sin haber crecido previamente
una capa de grafeno con CH4, el DCA corroe totalmente la lámina de Cu hasta
su desaparición. Para confirmarlo, se sumergieron una lámina de Cu con
grafeno (Cu/Gr) y otra sin grafeno en DCA a 150 ºC.
Figura 5.4. Imagen de las láminas de Cu y Cu/Gr sumergidas en DCA a 150ºC a)
durante 10 min.
En la Figura 5.4 se observa cómo la lámina que no tiene grafeno se corroe a
una velocidad mayor. Esto demuestra el efecto protector del grafeno como
barrera contra la corrosión.

CÁPITULO 5
194
Figura 5.5. Esquema con los tipos de muestra obtenidos en función de los tiempos
de cada etapa ta, tb y tc.
En resumen, en función de los tiempos de crecimiento y funcionalización se
obtienen cuatro tipos de muestra (Figura 5.5): i) muestras con regiones de
grafeno monocapa no funcionalizado y regiones de grafeno de varias capas
funcionalizado (SLG/MLG-F), ii) grafeno monocapa y multicapa ambos con
funcionalización homogénea (SLG-F/MLG-F), iii) grafeno monocapa con
funcionalización no homogénea (SLG/SLG-F) y iv) grafeno monocapa con
funcionalización homogénea (SLG-F).
5.3 CARACTERIZACIÓN XPS DE GRAFENO OBTENIDO MEDIANTE
FUNCIONALIZACIÓN IN-SITU
La espectroscopia Raman demuestra la funcionalización del grafeno pero no
proporciona información selectiva de los grupos funcionales presentes. Sin
embargo, mediante el análisis de los espectros de XPS del carbono (C1s) en
muestras de grafeno u óxidos de grafeno es posible obtener información sobre
el contenido de cada grupo funcional. Las medidas XPS así como los ajustes
correspondientes fueron llevados a cabo por el Instituto de Ciencia y
Tecnología del Carbono. En la Figura 5.6 se muestra el espectro XPS del nivel
1s del C de tres muestras con diferente grado de funcionalización. Se observa
un pico principal correspondiente al enlace C-C con hibridación sp2 a 284.4 eV.
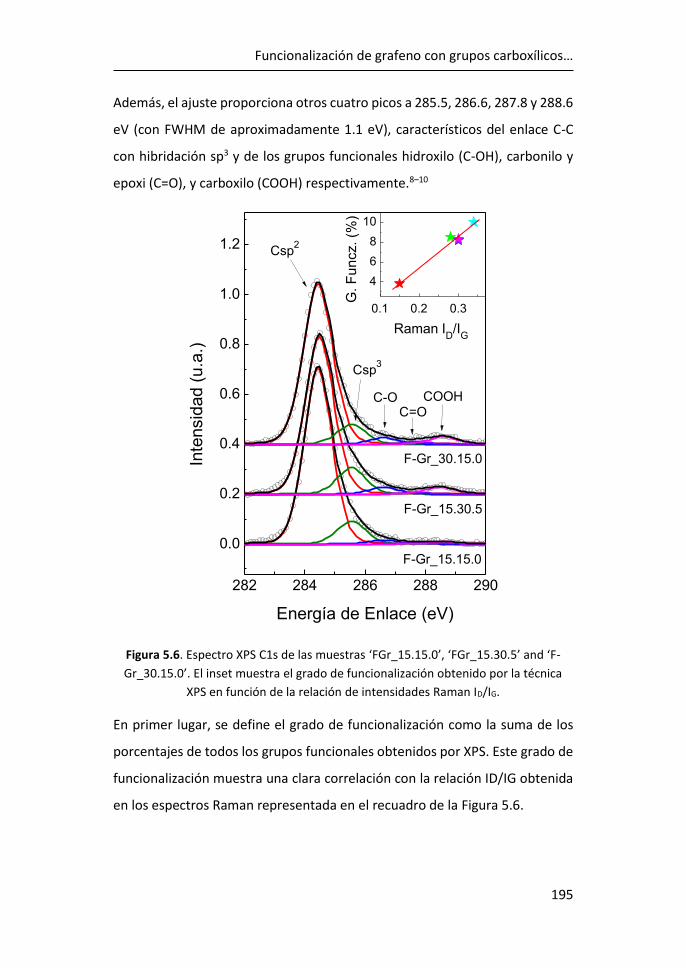
Funcionalización de grafeno con grupos carboxílicos…
195
Además, el ajuste proporciona otros cuatro picos a 285.5, 286.6, 287.8 y 288.6
eV (con FWHM de aproximadamente 1.1 eV), característicos del enlace C-C
con hibridación sp3 y de los grupos funcionales hidroxilo (C-OH), carbonilo y
epoxi (C=O), y carboxilo (COOH) respectivamente.8–10
Figura 5.6. Espectro XPS C1s de las muestras ‘FGr_15.15.0’, ‘FGr_15.30.5’ and ‘F-
Gr_30.15.0’. El inset muestra el grado de funcionalización obtenido por la técnica
XPS en función de la relación de intensidades Raman ID/IG.
En primer lugar, se define el grado de funcionalización como la suma de los
porcentajes de todos los grupos funcionales obtenidos por XPS. Este grado de
funcionalización muestra una clara correlación con la relación ID/IG obtenida
en los espectros Raman representada en el recuadro de la Figura 5.6.
282 284 286 288 290
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0.1 0.2 0.3
468
10
F-Gr_15.15.0
F-Gr_15.30.5
In
tens
idad
(u.a
.)
Energía de Enlace (eV)
COOHC=O
C-O
Csp3
Csp2
F-Gr_30.15.0
G. F
uncz
. (%
)Raman ID/IG
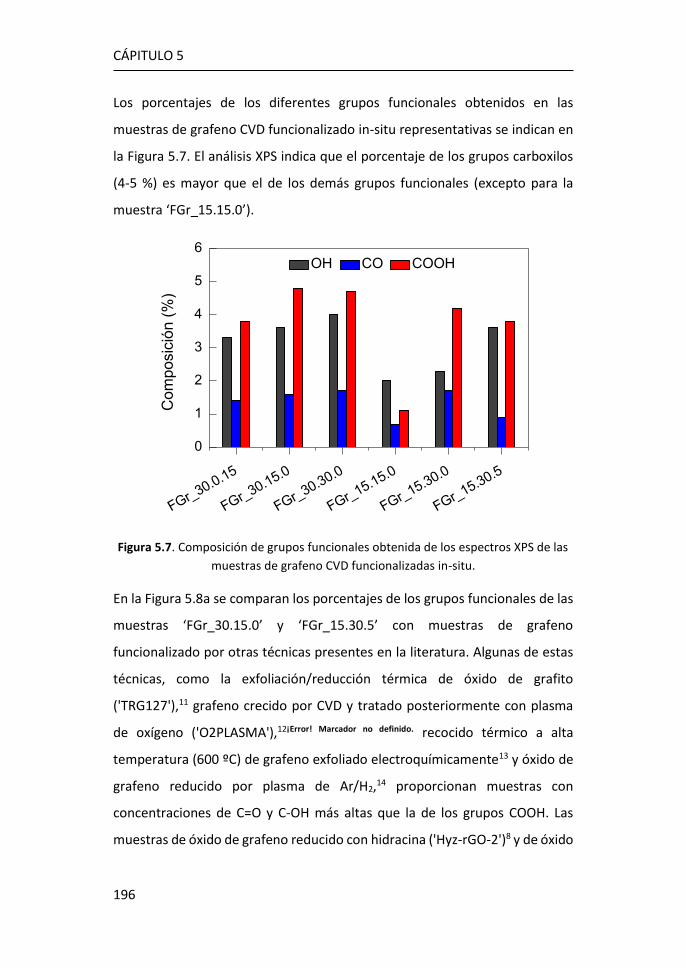
CÁPITULO 5
196
Los porcentajes de los diferentes grupos funcionales obtenidos en las
muestras de grafeno CVD funcionalizado in-situ representativas se indican en
la Figura 5.7. El análisis XPS indica que el porcentaje de los grupos carboxilos
(4-5 %) es mayor que el de los demás grupos funcionales (excepto para la
muestra ‘FGr_15.15.0’).
Figura 5.7. Composición de grupos funcionales obtenida de los espectros XPS de las
muestras de grafeno CVD funcionalizadas in-situ.
En la Figura 5.8a se comparan los porcentajes de los grupos funcionales de las
muestras ‘FGr_30.15.0’ y ‘FGr_15.30.5’ con muestras de grafeno
funcionalizado por otras técnicas presentes en la literatura. Algunas de estas
técnicas, como la exfoliación/reducción térmica de óxido de grafito
('TRG127'),11 grafeno crecido por CVD y tratado posteriormente con plasma
de oxígeno ('O2PLASMA'),12¡Error! Marcador no definido. recocido térmico a alta
temperatura (600 ºC) de grafeno exfoliado electroquímicamente13 y óxido de
grafeno reducido por plasma de Ar/H2,14 proporcionan muestras con
concentraciones de C=O y C-OH más altas que la de los grupos COOH. Las
muestras de óxido de grafeno reducido con hidracina ('Hyz-rGO-2')8 y de óxido
FGr_30.0.15
FGr_30.15.0
FGr_30.30.0
FGr_15.15.0
FGr_15.30.0
FGr_15.30.5
0
1
2
3
4
5
6
Com
posi
ción
(%)
OH CO COOH
0
5
10
15
20
TRG127
O2-Plas
ma
AEHOPG600
H2-Plas
ma
Hyz-rG
O-2
ERGO2C
F-Gr_3
0.15.0
F-Gr_1
5.30.5
0.0
0.2
0.4
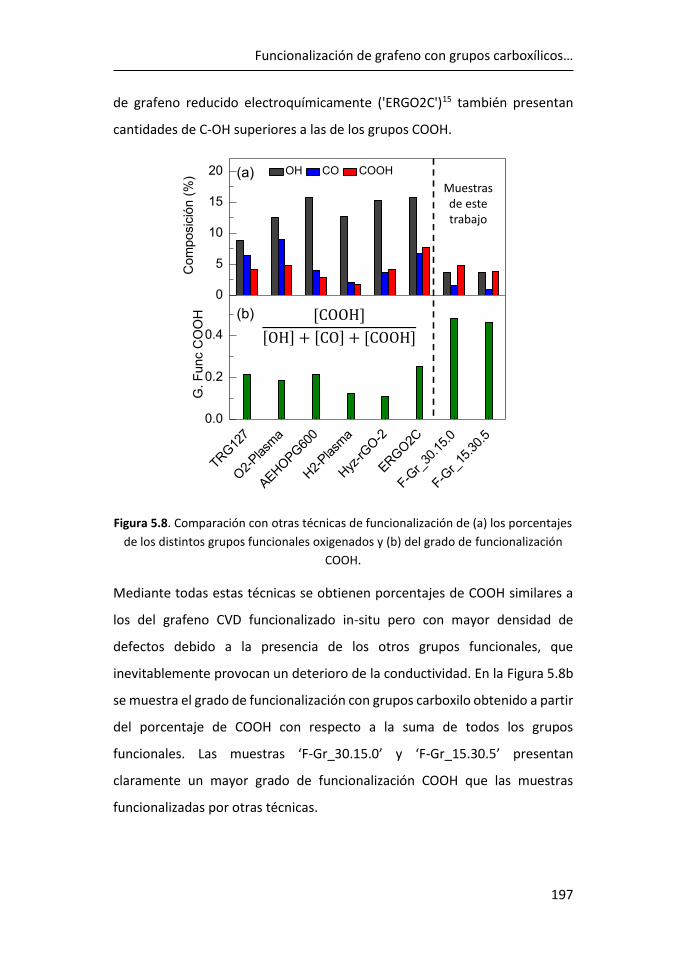
Funcionalización de grafeno con grupos carboxílicos…
197
de grafeno reducido electroquímicamente ('ERGO2C')15 también presentan
cantidades de C-OH superiores a las de los grupos COOH.
Figura 5.8. Comparación con otras técnicas de funcionalización de (a) los porcentajes
de los distintos grupos funcionales oxigenados y (b) del grado de funcionalización
COOH.
Mediante todas estas técnicas se obtienen porcentajes de COOH similares a
los del grafeno CVD funcionalizado in-situ pero con mayor densidad de
defectos debido a la presencia de los otros grupos funcionales, que
inevitablemente provocan un deterioro de la conductividad. En la Figura 5.8b
se muestra el grado de funcionalización con grupos carboxilo obtenido a partir
del porcentaje de COOH con respecto a la suma de todos los grupos
funcionales. Las muestras ‘F-Gr_30.15.0’ y ‘F-Gr_15.30.5’ presentan
claramente un mayor grado de funcionalización COOH que las muestras
funcionalizadas por otras técnicas.
F-Gr_3
0.0.15
F-Gr_3
0.15.0
F-Gr_1
5.15.0
F-Gr_1
5.30.0
F-Gr_1
5.30.5
0123456
0
5
10
15
20 OH CO COOH
Com
posi
ción
(%)
TRG127
O2-Plas
ma
AEHOPG600
H2-Plas
ma
Hyz-rG
O-2
ERGO2C
F-Gr_3
0.15.0
F-Gr_1
5.30.5
0.0
0.2
0.4
(b)
(a)
G. F
unc
CO
OH
Muestrasde estetrabajo

CÁPITULO 5
198
5.4 CARACTERIZACIÓN ELÉCTRICA DEL GRAFENO OBTENIDO
MEDIANTE FUNCIONALIZACIÓN IN-SITU
Dado que una de las posibilidades de detección de antígenos es medir la
modificación de la conductividad después del anclaje del anticuerpo, las
propiedades de transporte eléctrico de estas plataformas de grafeno
funcionalizado con COOH son extremadamente relevantes. Para este
propósito, tanto el grafeno funcionalizado in-situ como el grafeno CVD sin
defectos como referencia, se transfirieron sobre sustratos de sílice vítrea
(aislante eléctrico) para realizar la caracterización eléctrica.
La metodología de transferencia de grafeno CVD se ha descrito en el Capítulo
2, sin embargo, las muestras de grafeno funcionalizado requieren modificar el
protocolo. Esto es debido a que las muestras funcionalizadas tienen defectos,
lo que las hace más frágiles, siendo necesaria una capa de PMMA de mayor
grosor. En las disoluciones de PMMA cuanto mayor es su concentración más
densa es la disolución y más difícil es que la capa depositada sea uniforme,
siendo la del 6 % el límite para obtener un recubrimiento homogéneo. Sin
embargo, la capa depositada con la disolución del 6% sigue sin ser lo
suficientemente gruesa, por lo que finalmente se concluyó que es necesario
depositar dos capas al 6 %, respetando los tiempos de curado y secado entre
cada una de ellas. Por otra parte, las muestras MLG-F presentan una mayor
cantidad de defectos, en parte debidos a los grupos funcionales y en parte a
la rotura del grafeno en pequeños dominios, por lo que sólo ha sido posible la
transferencia de las muestras SLG-F.
Resistencia laminar y movilidad de grafeno obtenido mediante
funcionalización in-situ.
En la Figura 5.9a se han representado las curvas I-V obtenidas según la
configuración de cuatro puntas de Van der Pauw de las muestras transferidas,
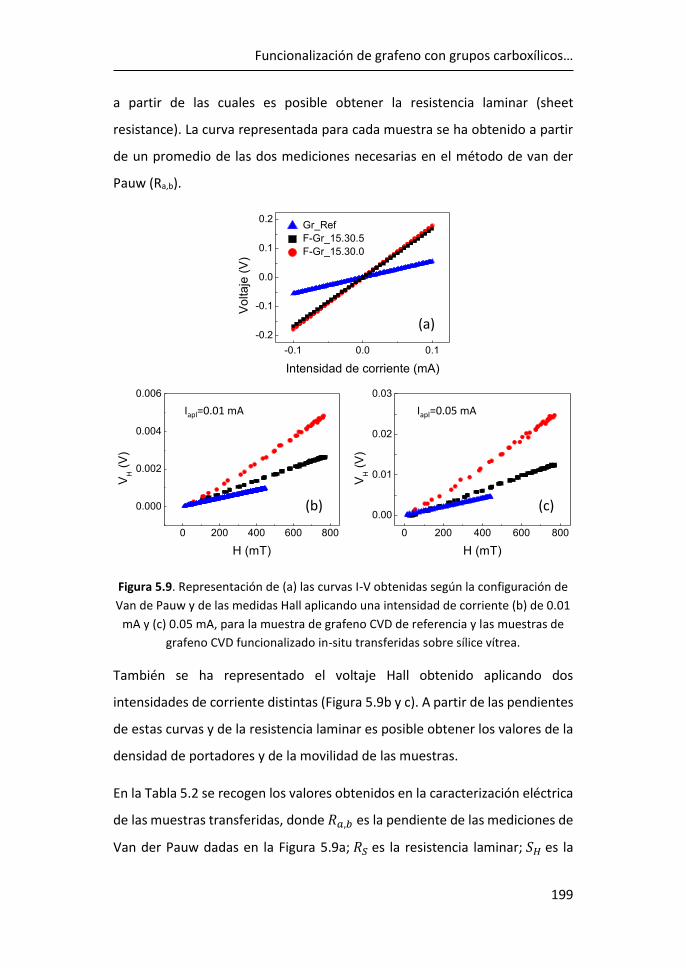
Funcionalización de grafeno con grupos carboxílicos…
199
a partir de las cuales es posible obtener la resistencia laminar (sheet
resistance). La curva representada para cada muestra se ha obtenido a partir
de un promedio de las dos mediciones necesarias en el método de van der
Pauw (Ra,b).
Figura 5.9. Representación de (a) las curvas I-V obtenidas según la configuración de
Van de Pauw y de las medidas Hall aplicando una intensidad de corriente (b) de 0.01
mA y (c) 0.05 mA, para la muestra de grafeno CVD de referencia y las muestras de
grafeno CVD funcionalizado in-situ transferidas sobre sílice vítrea.
También se ha representado el voltaje Hall obtenido aplicando dos
intensidades de corriente distintas (Figura 5.9b y c). A partir de las pendientes
de estas curvas y de la resistencia laminar es posible obtener los valores de la
densidad de portadores y de la movilidad de las muestras.
En la Tabla 5.2 se recogen los valores obtenidos en la caracterización eléctrica
de las muestras transferidas, donde 𝑅𝑎,𝑏 es la pendiente de las mediciones de
Van der Pauw dadas en la Figura 5.9a; 𝑅𝑆 es la resistencia laminar; 𝑆𝐻 es la
0 200 400 600 800
0.000
0.002
0.004
0.006
0 200 400 600 800
0.00
0.01
0.02
0.03
V H (V
)
H (mT)
V H (V
)
H (mT)
-0.1 0.0 0.1-0.2
-0.1
0.0
0.1
0.2 Gr_Ref F-Gr_15.30.5 F-Gr_15.30.0
Volta
je (V
)
Intensidad de corriente (mA)
Iapl=0.01 mA Iapl=0.05 mA
(a)
(b) (c)

CÁPITULO 5
200
pendiente de las mediciones de Hall representadas en la Figura 5.9b y c; 𝑅𝐻𝑎𝑙𝑙
es la resistencia Hall obtenida a partir de la expresión la Ec. 5.1, 𝑁𝑆 es la
densidad de portadores de carga (Ec. 5.2), donde t es el grosor de la muestra
y e la carga del electrón) y µ la movilidad (Ec. 5.3).
𝑅𝐻𝑎𝑙𝑙 =𝑉𝐻𝑎𝑙𝑙(𝐼𝑎𝑝𝑙 𝐻)
Ec. 5.1
𝑁𝑆 = 𝑡(𝑅𝐻𝑎𝑙𝑙 𝑒)−1 Ec. 5.2
𝜇 =𝑆𝐻
𝑅𝑆 𝐼𝑎𝑝𝑙 Ec. 5.3
Tabla 5.2. Resumen de la caracterización eléctrica de las muestras sintetizadas
transferidas sobre sílica fundida.
Muestra Ra,b
()
RS
()
Iappl
(mA)
SH
(V/T)
RHall
(V/A T)
NS
(cm-2)
µ
(cm2/V s)
‘Gr_Ref’ 5.5·102 2.5·103 0.01
0.05
2.0·10-3
1.0·10-2
2.0·102
2.0·102 3.1×1012 80020
‘FGr_15.30.0’ 1.8·103 8.1·103 0.01
0.05
6.6·10-3
3.3·10-2
6.6·102
6.7·102 9.5×1011 90020
‘F-Gr_15.30.5’ 1.7·103 7.6·103 0.01
0.05
3.1·10-3
1.6·10-2
3.1·102
3.1·102 2.0×1012 40020
Los valores de resistencia laminar obtenidos para las muestras 'FGr_15.30.0' y
'FGr_15.30.5' (SLG-F) son de aproximadamente 7-8 kΩ/sq, valores más que
adecuados para que las plataformas sean utilizadas en sensores electrónicos.
Por otra parte, los valores de movilidad eléctrica son de 400 y 900 cm2V-1s-1,
respectivamente. El nivel de dopado y la movilidad del grafeno transferido
dependen en cierta medida del proceso de transferencia y de posibles
residuos de PMMA. Esta puede ser la causa de que la muestra “FGr_13.30.0”

Funcionalización de grafeno con grupos carboxílicos…
201
presente una movilidad mayor a la del grafeno de referencia. En general, los
valores de resistencia y movilidad obtenidos para las muestras funcionalizadas
son comparables a los obtenidos para el grafeno CVD de referencia
transferido.
En la Tabla 5.3 se resumen las características eléctricas y el grado de
funcionalización COOH de las muestras obtenidas en nuestro laboratorio junto
con los obtenidos por otras técnicas de funcionalización. Vemos cómo los
valores de resistencia laminar de las muestras de grafeno CVD funcionalizado
in-situ son inferiores a los obtenidos por rGO reducido por plasma de Ar/H214
y a los del grafeno obtenido mediante la exfoliación de grafito pirolítico
altamente orientado (HOGP) y posteriormente oxidado por plasma de O2.16
Son similares a los del grafeno funcionalizado mediante oxidación por plasma
de oxígeno,17,18 a los de HOGP exfoliado electroquímicamente sometido a un
recocido térmico a alta temperatura (600 ºC),13 y a los de grafeno
funcionalizado por vía electroquímica.19 En cuanto a los valores de la
movilidad, la comparación es difícil pues no son valores que se encuentren
habitualmente en las publicaciones, pero se observa que lo sobtenidos en este
trabajo son superiores a los reportados, como es el caso de grafeno oxidado
por plasma.16,18 Además, el porcentaje de grupos carboxilos en relación con el
total de grupos funcionales obtenidos mediante estas técnicas es inferior, en
todos los casos, al logrado por el crecimiento y la funcionalización in-situ en
CVD que se presenta en este trabajo.
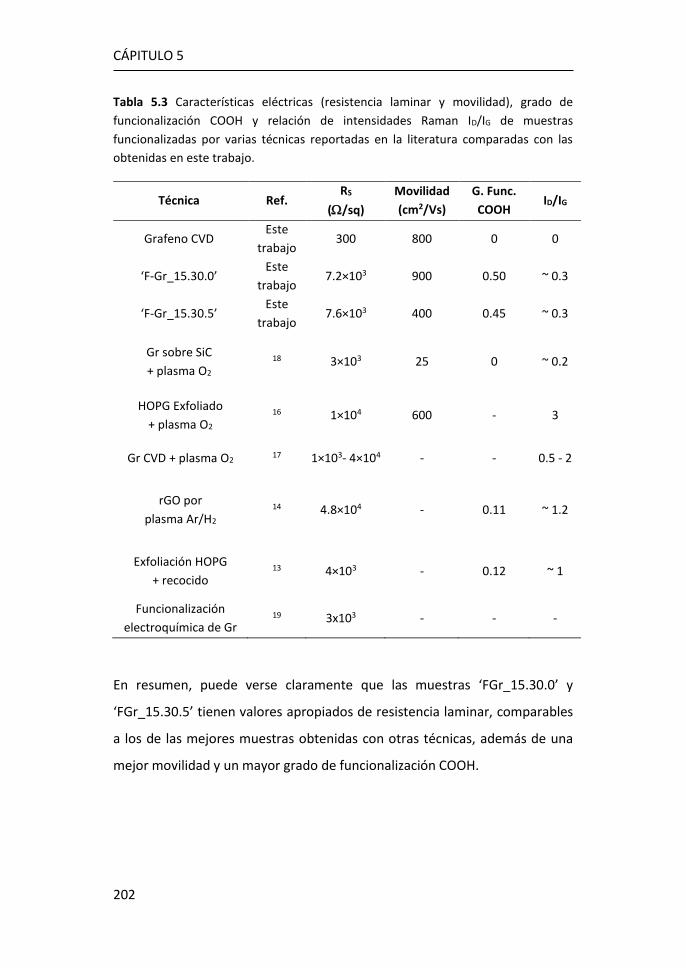
CÁPITULO 5
202
Tabla 5.3 Características eléctricas (resistencia laminar y movilidad), grado de
funcionalización COOH y relación de intensidades Raman ID/IG de muestras
funcionalizadas por varias técnicas reportadas en la literatura comparadas con las
obtenidas en este trabajo.
Técnica Ref. RS
(/sq)
Movilidad
(cm2/Vs)
G. Func.
COOH ID/IG
Grafeno CVD Este
trabajo 300 800 0 0
‘F-Gr_15.30.0’ Este
trabajo 7.2×103 900 0.50 ~ 0.3
‘F-Gr_15.30.5’ Este
trabajo 7.6×103 400 0.45 ~ 0.3
Gr sobre SiC
+ plasma O2 18 3×103 25 0 ~ 0.2
HOPG Exfoliado
+ plasma O2 16 1×104 600 - 3
Gr CVD + plasma O2 17 1×103- 4×104 - - 0.5 - 2
rGO por
plasma Ar/H2 14 4.8×104 - 0.11 ~ 1.2
Exfoliación HOPG
+ recocido 13 4×103 - 0.12 ~ 1
Funcionalización
electroquímica de Gr 19 3x103 - - -
En resumen, puede verse claramente que las muestras ‘FGr_15.30.0’ y
‘FGr_15.30.5’ tienen valores apropiados de resistencia laminar, comparables
a los de las mejores muestras obtenidas con otras técnicas, además de una
mejor movilidad y un mayor grado de funcionalización COOH.

Funcionalización de grafeno con grupos carboxílicos…
203
5.5 INMOVILIZACIÓN DE ANTICUERPOS POR EL MÉTODO
CARBODIIMIDA
Una cuestión importante para poder usar de manera fiable las plataformas
basadas en grafeno para la detección de antígenos es la inmovilización
efectiva de un anticuerpo específico que tiene que ser unido covalentemente
al grafeno. Por lo tanto, tras el crecimiento y la funcionalización in-situ del
grafeno, varias muestras se sometieron al anclaje del anticuerpo por el
método carbodiimida,20,21 según se esquematiza en la Figura 5.10. En este
método, el clorhidrato de N-(3-dimetilaminopropil)-N′-etilcarbodiimida (EDC)
reacciona con los grupos de ácido carboxílico de las muestras formando un
estado intermedio inestable que, tras añadir N-hidroxisuccinimida (NHS),
deriva en un éster-NHS de mayor estabilidad. Después, la amina primaria del
anticuerpo IgG1-FITC reacciona con el éster-NHS para formar un enlace fuerte
amida. El procedimiento completo se ha descrito en el Capítulo 2.
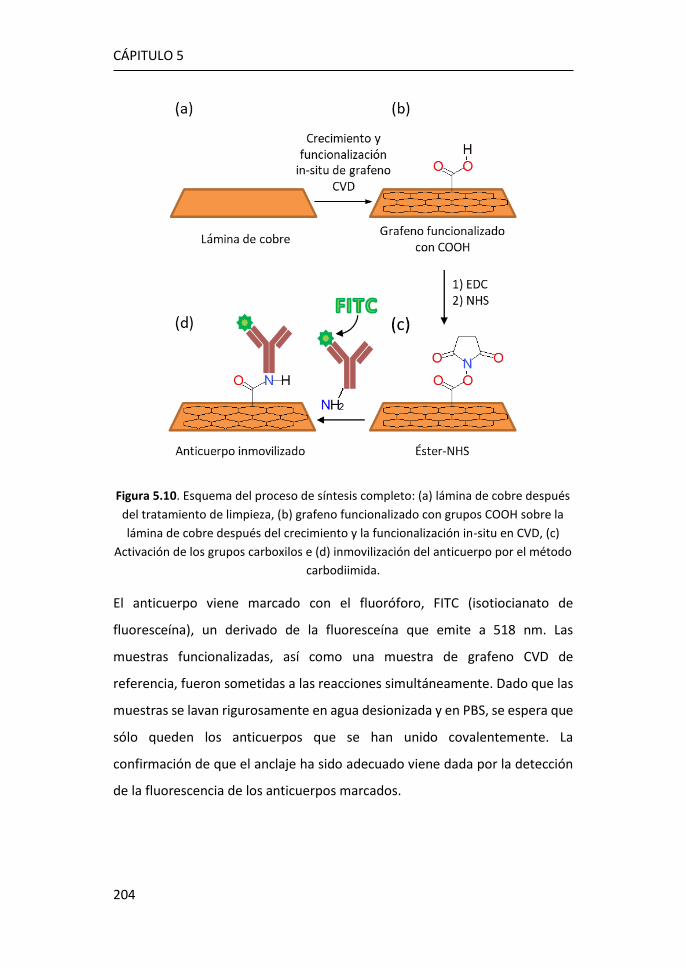
CÁPITULO 5
204
Figura 5.10. Esquema del proceso de síntesis completo: (a) lámina de cobre después
del tratamiento de limpieza, (b) grafeno funcionalizado con grupos COOH sobre la
lámina de cobre después del crecimiento y la funcionalización in-situ en CVD, (c)
Activación de los grupos carboxilos e (d) inmovilización del anticuerpo por el método
carbodiimida.
El anticuerpo viene marcado con el fluoróforo, FITC (isotiocianato de
fluoresceína), un derivado de la fluoresceína que emite a 518 nm. Las
muestras funcionalizadas, así como una muestra de grafeno CVD de
referencia, fueron sometidas a las reacciones simultáneamente. Dado que las
muestras se lavan rigurosamente en agua desionizada y en PBS, se espera que
sólo queden los anticuerpos que se han unido covalentemente. La
confirmación de que el anclaje ha sido adecuado viene dada por la detección
de la fluorescencia de los anticuerpos marcados.
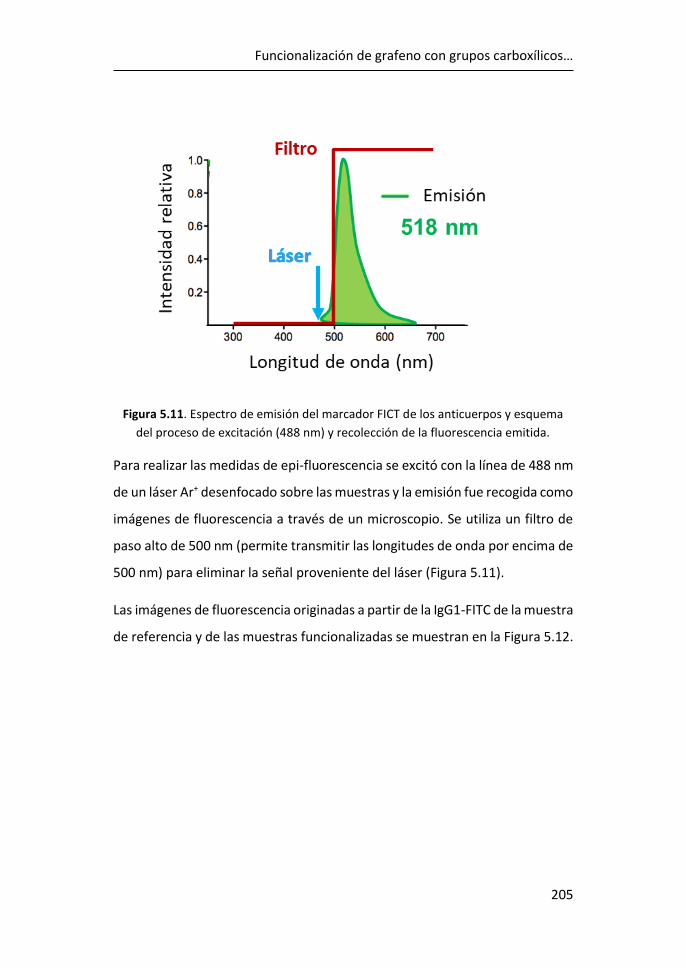
Funcionalización de grafeno con grupos carboxílicos…
205
Figura 5.11. Espectro de emisión del marcador FICT de los anticuerpos y esquema
del proceso de excitación (488 nm) y recolección de la fluorescencia emitida.
Para realizar las medidas de epi-fluorescencia se excitó con la línea de 488 nm
de un láser Ar+ desenfocado sobre las muestras y la emisión fue recogida como
imágenes de fluorescencia a través de un microscopio. Se utiliza un filtro de
paso alto de 500 nm (permite transmitir las longitudes de onda por encima de
500 nm) para eliminar la señal proveniente del láser (Figura 5.11).
Las imágenes de fluorescencia originadas a partir de la IgG1-FITC de la muestra
de referencia y de las muestras funcionalizadas se muestran en la Figura 5.12.
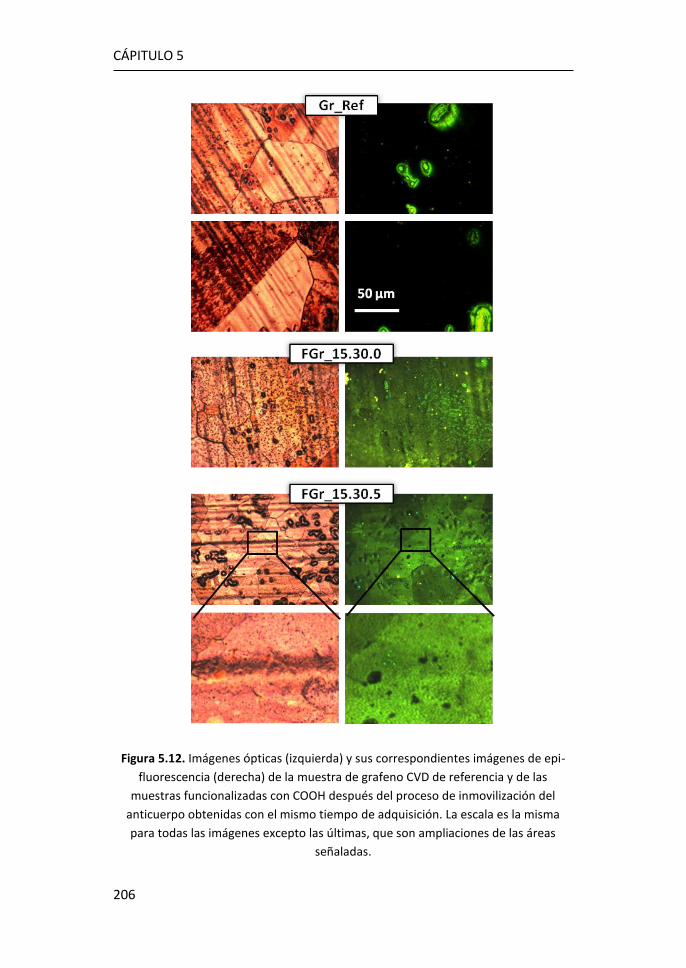
CÁPITULO 5
206
Figura 5.12. Imágenes ópticas (izquierda) y sus correspondientes imágenes de epi-
fluorescencia (derecha) de la muestra de grafeno CVD de referencia y de las
muestras funcionalizadas con COOH después del proceso de inmovilización del
anticuerpo obtenidas con el mismo tiempo de adquisición. La escala es la misma
para todas las imágenes excepto las últimas, que son ampliaciones de las áreas
señaladas.

Funcionalización de grafeno con grupos carboxílicos…
207
Se observa cómo las imágenes correspondientes a la muestra de grafeno CVD
de referencia (Gr_Ref en la Figura 5.12) son casi negras, a excepción de algunas
regiones brillantes donde la solución de anticuerpos está confinada
probablemente debido a aspectos topológicos de la superficie de cobre. Por
el contrario, en las imágenes de emisión recogidas de las muestras
funcionalizadas con COOH (FGr_15.30.5 y FGr_15.30.0 en la Figura 5.12) se
observa una distribución verde homogénea en toda la superficie lo que
demuestra su capacidad para inmovilizar el anticuerpo. Estas imágenes
respaldan que el grado de funcionalización alcanzado con esta técnica, en el
que alrededor del 5 % de los átomos de carbono está enlazado a un grupo
COOH, es suficiente, ya que según este porcentaje se obtiene una distancia
media de 7 Å entre los grupos COOH, una separación adecuada para detección
molecular.
En conclusión, se ha demostrado que un método basado en el crecimiento del
grafeno por vía CVD y el flujo del vapor de ácido dicloroacético in-situ da como
resultado grafeno funcionalizado con grupos COOH.
En función de los tiempos característicos de las distintas etapas del proceso,
se han obtenido muestras funcionalizadas de grafeno monocapa o multicapa.
La optimización de los tiempos de las etapas de crecimiento y funcionalización
de grafeno da lugar a grafeno funcionalizado con grupos COOH, con un
porcentaje de carboxilos del orden del 5 %, superior al de los demás grupos
funcionales oxigenados presentes. La funcionalización carboxílica obtenida en
relación con el total de los grupos funcionales es superior a la obtenida por
otros métodos.
Se transfirieron varias muestras de grafeno funcionalizado a sustratos de sílice
vítrea para evaluar las propiedades de transporte electrónico, obteniéndose
valores de resistencia laminar similares a los de las mejores muestras

CÁPITULO 5
208
reportadas con otras técnicas y con valores de movilidad significativamente
más altos.
Por último, se ha utilizado el método carbodiimida para formar derivados
estables éster NHS, creados en los lugares donde el grafeno ha sido
funcionalizado con grupos -COOH, lo que permite la formación de fuertes
enlaces con las aminas primarias presentes en los anticuerpos monoclonales
antihumanos IgG1. El eficaz enlace covalente de la IgG1 se pone de manifiesto
en la distribución homogénea de la epi-fluorescencia que se observa en las
imágenes de emisión del marcador isotiocianato de fluoresceína.
El carácter covalente del enlace se verifica mediante la comparación con las
imágenes obtenidas del grafeno de referencia sometido simultáneamente al
mismo proceso, en el que no se detecta ninguna emisión significativa.
El conjunto de todos estos resultados lleva a la conclusión de que el método
que se presenta en este trabajo para la carboxilación in-situ del grafeno es
adecuado para la fabricación de plataformas funcionales que pueden
explorarse como sensores ópticos o electrónicos conductores.
5.6 REFERENCIAS
1. Yu, Y. et al. Electrical and Label-Free Quantification of Exosomes with a Reduced Graphene Oxide Field Effect Transistor Biosensor. Anal. Chem. 91, 10679–10686 (2019).
2. Gonçalves, G. et al. Nano-graphene oxide: A potential multifunctional platform for cancer therapy. Adv. Healthc. Mater. 3, 956–956 (2014).
3. Yang, K., Feng, L., Shi, X., Reviews, Z. L.-C. S. & 2013, undefined. Nano-graphene in biomedicine: theranostic applications. Chem. Soc. Rev. 42, 530–547 (2013).
4. Kurkina, T. et al. Self-assembled electrical biodetector based on

Funcionalización de grafeno con grupos carboxílicos…
209
reduced graphene oxide. ACS Nano 6, 5514–5520 (2012).
5. Dreyer, D. R., Park, S., Bielawski, C. W. & Ruoff, R. S. The chemistry of graphene oxide. Chem. Soc. Rev. 39, 228–240 (2010).
6. Cortijo-Campos, S. et al. In-situ carboxylation of graphene by chemical vapor deposition growth for biosensing. Carbon N. Y. 141, 719–727 (2019).
7. Wang, Y. Y. et al. Interference enhancement of Raman signal of graphene Interference enhancement of Raman signal of graphene. 043121, (2015).
8. Blanco, C. et al. RSC Advances Tailored graphene materials by chemical reduction of graphene oxides of different atomic structure. 9643–9650 (2012) doi:10.1039/c2ra21447d.
9. Blanco, M. et al. Graphene-NHC-iridium hybrid catalysts built through -OH covalent linkage Dedicated to the lasting Memory of Prof. Dr. María Pilar García for her valuable talent as human, teacher and chemist. Carbon N. Y. 83, 21–31 (2015).
10. Yang, D. et al. Chemical analysis of graphene oxide films after heat and chemical treatments by X-ray photoelectron and Micro-Raman spectroscopy. Carbon N. Y. 47, 145–152 (2009).
11. Blanco, C. et al. Critical temperatures in the synthesis of graphene-like materials by thermal exfoliation-reduction of graphite oxide. Carbon N. Y. 52, 476–485 (2013).
12. Hadish, F., Jou, S., Huang, B., Kuo, H. & Tu, C. Functionalization of CVD Grown Graphene with Downstream Oxygen Plasma Treatment for Glucose Sensors. J. Electrochem. Soc. 164, B336–B341 (2017).
13. Marković, Z. et al. Semi-transparent, conductive thin films of electrochemical exfoliated graphene. RSC Adv. 6, 39275–39283 (2016).
14. Lee, S. W., Mattevi, C., Chhowalla, M. & Sankaran, R. M. Plasma-assisted reduction of graphene oxide at low temperature and atmospheric pressure for flexible conductor applications. J. Phys. Chem. Lett. 3, 772–777 (2012).
15. Calvo, A. S. et al. Comparative Study of Screen-Printed Electrodes Modified with Graphene Oxides Reduced by a Constant Current Comparative Study of Screen-Printed Electrodes Modified with Graphene Oxides Reduced by a Constant Current. J. Electrochem. Soc. 162, B282–B290 (2015).

CÁPITULO 5
210
16. Childres, I., Jauregui, L. A., Tian, J. & Chen, Y. P. Effect of oxygen plasma etching on graphene studied using Raman spectroscopy and electronic transport measurements. New J. Phys. 13, 25008 (2011).
17. Bianco, G. V. et al. Engineering graphene properties by modulated plasma treatments. Carbon N. Y. 129, 869–877 (2018).
18. Hernández, S. C. et al. Plasma-based chemical modification of epitaxial graphene with oxygen functionalities. Surf. Coatings Technol. 241, 8–12 (2014).
19. Xia, Z. et al. Electrochemical Functionalization of Graphene at the Nanoscale with Self-Assembling Diazonium Salts. ACS Nano 10, 7125–7134 (2016).
20. Rich, D. H. & Singh, J. The carbodiimide method. in Major Methods of Peptide Bond Formation (eds. Gross, E., Meienhofer, J. & Press, A.) 241–261 (Elsevier, 1979).
21. Jung, Y., Jeong, J. Y. & Chung, B. H. Recent advances in immobilization methods of antibodies on solid supports. Analyst 133, 697–701 (2008).

211
6 MOS2 MONOCAPA COMO DETECTOR DE
BIOMARCADORES DE CÁNCER DE MAMA
En la búsqueda de nuevas estrategias para biosensores basados en materiales
2D, se ha llevado a cabo una colaboración con los grupos de los Drs. Andrés
Redondo y Encarnación Lorenzo de la Universidad Autónoma de Madrid, en la
que se estudiaron distintas aproximaciones basadas en MoS2 monocapa de las
cuales fue efectiva la que aquí se describe.
Como ya se comentó en el Capítulo 1, en los últimos años los materiales
bidimensionales han surgido como una vía prometedora para su uso en
biosensores.1,2 Entre ellos, los TMDCs han atraído una intensa atención como
materiales funcionales para la detección del gen miARN21 y de ADN.3–7 Un
micro-ARN (miARN) es un ARN monocatenario no codificante, de una longitud
de entre 21 y 25 nucleótidos, que participa en la regulación de la expresión
génica. Hasta ahora, muchas de las estrategias de detección se han centrado
en sensores basados en electrodos, que pueden ser electroquímicos, dónde
se detectan especies iónicas generadas en reacciones rédox y el MoS2 recubre
el electrodo de trabajo, o en sensores basados en transistores de efecto
campo (FET), donde la hibridación de la sonda, adsorbida sobre el MoS2, con
el biomarcador da lugar a un desplazamiento de la tensión umbral y a un
cambio de la corriente de drenaje.8 Los biosensores ópticos basados en TMDCs
están todavía en desarrollo9 y la mayoría están basados en FRET (del inglés,
fluorescence resonance energy transfer), en los que la sonda está marcada
con un fluoróforo, y al reaccionar con el biomarcador, el MoS2 inhibe
fluorescencia de la sonda.10 En este trabajo, se pretende aprovechar las
posibles modificaciones de la fotoluminiscencia (PL) del MoS2 monocapa para
el desarrollo de nuevos biosensores. Para ello, se ha utilizado MoS2 monocapa

CÁPITULO 6
212
crecido por PVT sobre zafiro como transductor en un nuevo tipo de biosensor
para la detección de biomarcadores de cáncer de mama. En el diagnóstico de
cáncer de mama, el gen miRNA21 posee una alta sensibilidad (87.6%) y
especificidad (87.3%) en las fases iniciales.11 Además, este gen se
sobreexpresa en muchos tejidos cancerosos y su regulación ha sido
ampliamente estudiada,12 por lo que se ha escogido como biorreceptor del
biosensor desarrollado en este trabajo. En la mayoría de los casos, el
biomarcador típico es el miRNA21 complementario o una versión modificada
de éste. En este trabajo se ha utilizado el miRNA21 complementario
(miRNA21-C) y otro no complementario (miRNA21-NC) con el fin de demostrar
la selectividad del biosensor.13
6.1 CARACTERIZACIÓN POR ESPECTROSCOPÍAS RAMAN Y DE
FOTOLUMINISCENCIA DE PLATAFORMAS DE MOS2 CRECIDAS
POR PVT SOBRE ZAFIRO
En un biosensor óptico, la calidad de la plataforma es crucial para la posterior
comprensión del procedimiento de transducción. La Figura 6.1a muestra la
imagen óptica de una de estas muestras, en la que se distingue la morfología
triangular de los copos de MoS2, con dimensiones de decenas de micras. Las
muestras de MoS2 monocapa se sintetizaron en el sistema PVT del grupo del
Dr. Kung descrito en el Capítulo 2 gracias a la estancia en la Universidad de
Alabama. En la Figura 6.1a se identifican diferentes copos triangulares aislados
(A-D), que se caracterizaron mediante espectroscopia Raman y de
fotoluminiscencia para valorar la calidad y homogeneidad de estos.
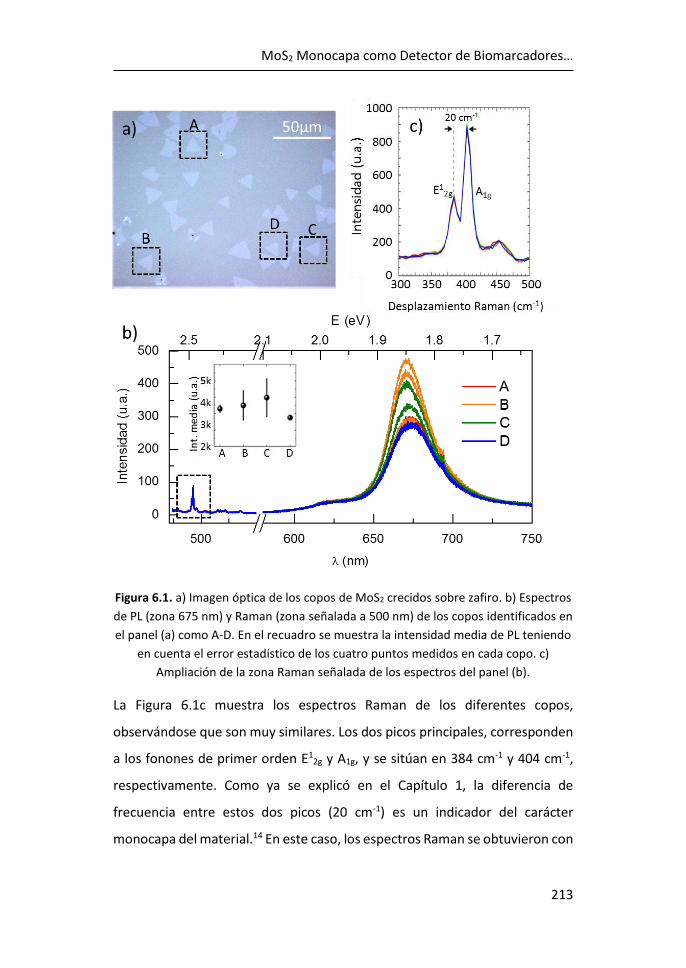
MoS2 Monocapa como Detector de Biomarcadores…
213
Figura 6.1. a) Imagen óptica de los copos de MoS2 crecidos sobre zafiro. b) Espectros
de PL (zona 675 nm) y Raman (zona señalada a 500 nm) de los copos identificados en
el panel (a) como A-D. En el recuadro se muestra la intensidad media de PL teniendo
en cuenta el error estadístico de los cuatro puntos medidos en cada copo. c)
Ampliación de la zona Raman señalada de los espectros del panel (b).
La Figura 6.1c muestra los espectros Raman de los diferentes copos,
observándose que son muy similares. Los dos picos principales, corresponden
a los fonones de primer orden E12g y A1g, y se sitúan en 384 cm-1 y 404 cm-1,
respectivamente. Como ya se explicó en el Capítulo 1, la diferencia de
frecuencia entre estos dos picos (20 cm-1) es un indicador del carácter
monocapa del material.14 En este caso, los espectros Raman se obtuvieron con
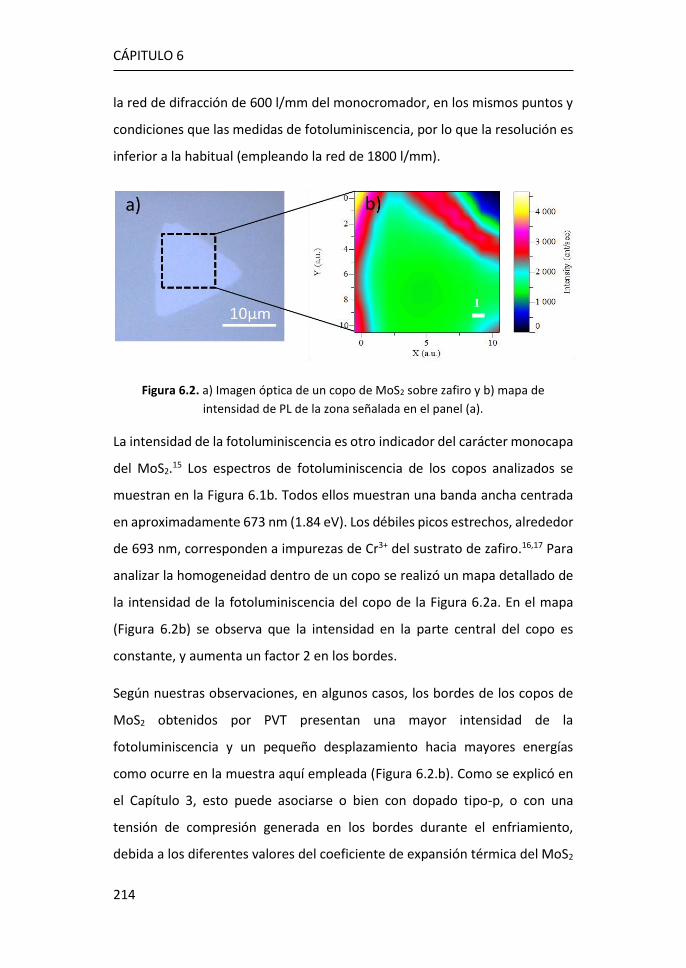
CÁPITULO 6
214
la red de difracción de 600 l/mm del monocromador, en los mismos puntos y
condiciones que las medidas de fotoluminiscencia, por lo que la resolución es
inferior a la habitual (empleando la red de 1800 l/mm).
Figura 6.2. a) Imagen óptica de un copo de MoS2 sobre zafiro y b) mapa de
intensidad de PL de la zona señalada en el panel (a).
La intensidad de la fotoluminiscencia es otro indicador del carácter monocapa
del MoS2.15 Los espectros de fotoluminiscencia de los copos analizados se
muestran en la Figura 6.1b. Todos ellos muestran una banda ancha centrada
en aproximadamente 673 nm (1.84 eV). Los débiles picos estrechos, alrededor
de 693 nm, corresponden a impurezas de Cr3+ del sustrato de zafiro.16,17 Para
analizar la homogeneidad dentro de un copo se realizó un mapa detallado de
la intensidad de la fotoluminiscencia del copo de la Figura 6.2a. En el mapa
(Figura 6.2b) se observa que la intensidad en la parte central del copo es
constante, y aumenta un factor 2 en los bordes.
Según nuestras observaciones, en algunos casos, los bordes de los copos de
MoS2 obtenidos por PVT presentan una mayor intensidad de la
fotoluminiscencia y un pequeño desplazamiento hacia mayores energías
como ocurre en la muestra aquí empleada (Figura 6.2.b). Como se explicó en
el Capítulo 3, esto puede asociarse o bien con dopado tipo-p, o con una
tensión de compresión generada en los bordes durante el enfriamiento,
debida a los diferentes valores del coeficiente de expansión térmica del MoS2

MoS2 Monocapa como Detector de Biomarcadores…
215
y del sustrato.18 En el caso de tratarse de dopado tipo-p puede ser debido bien
a una mayor densidad de vacantes de S en los bordes, que favorece la
adsorción de oxígeno y la transferencia de huecos al MoS2, o podría deberse a
vacantes de Mo. Las vacantes de Mo han sido estudiadas desde un punto de
vista teórico, pero sólo se ha reportado su detección en muestras sometidas a
irradiación con iones o electrones.19
6.2 ANÁLISIS DE LA FOTOLUMINISCENCIA DEL MOS2 MONOCAPA
EN LA DETECCIÓN DE BIOMARCADORES
El desarrollo del biosensor basado en MoS2 se muestra esquemáticamente en
la Figura 6.3. El proceso de biofuncionalización de los copos de MoS2 fue
llevado a cabo por los grupos de A. Redondo y E. Lorenzo de la Universidad
Autónoma de Madrid. La Figura 6.3a muestra la estructura del MoS2 con una
vacante de azufre, defecto que servirá como punto de anclaje para la sonda
(Figura 6.3b). Una secuencia de ADN modificada en el extremo 5′ con un
hexalquiltiol (ss-DNAp-SH) puede inmovilizarse sobre MoS2 a través del grupo
tiol (-SH). Las moléculas orgánicas con el grupo -SH tienden a reparar o
eliminar las vacantes de S de la red de MoS2, dando lugar a la funcionalización
molecular.20–23 Para llevar a cabo este paso se depositaron 10 µl de una
solución 10 nM de sonda (ss-DNAp-SH) sobre las muestras de MoS2 y se
mantuvieron a 4 °C durante 24 h. Después, las muestras de MoS2
funcionalizado (plataforma de detección, ss-DNAp-SH-MoS2) se lavaron con
agua destilada para eliminar la sonda adsorbida de forma inespecífica y se
secaron con nitrógeno. Después de este paso, la plataforma funcionalizada se
hibrida con las secuencias complementaria (miRNA21-C) y no complementaria
(miRNA21-NC), esta última utilizada como control. Para ello, la plataforma de
detección, ss-DNAp-SH-MoS2, se hibridó con una solución de analito mediante
la adición de 10 µl de una secuencia complementaria (miRNA21-C) o no
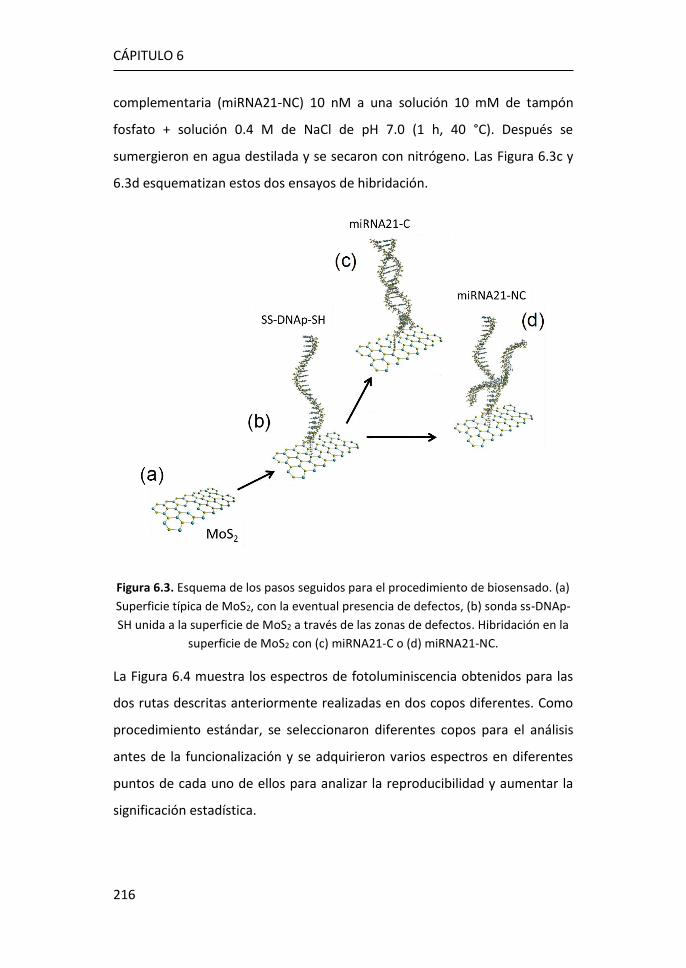
CÁPITULO 6
216
complementaria (miRNA21-NC) 10 nM a una solución 10 mM de tampón
fosfato + solución 0.4 M de NaCl de pH 7.0 (1 h, 40 °C). Después se
sumergieron en agua destilada y se secaron con nitrógeno. Las Figura 6.3c y
6.3d esquematizan estos dos ensayos de hibridación.
Figura 6.3. Esquema de los pasos seguidos para el procedimiento de biosensado. (a)
Superficie típica de MoS2, con la eventual presencia de defectos, (b) sonda ss-DNAp-
SH unida a la superficie de MoS2 a través de las zonas de defectos. Hibridación en la
superficie de MoS2 con (c) miRNA21-C o (d) miRNA21-NC.
La Figura 6.4 muestra los espectros de fotoluminiscencia obtenidos para las
dos rutas descritas anteriormente realizadas en dos copos diferentes. Como
procedimiento estándar, se seleccionaron diferentes copos para el análisis
antes de la funcionalización y se adquirieron varios espectros en diferentes
puntos de cada uno de ellos para analizar la reproducibilidad y aumentar la
significación estadística.
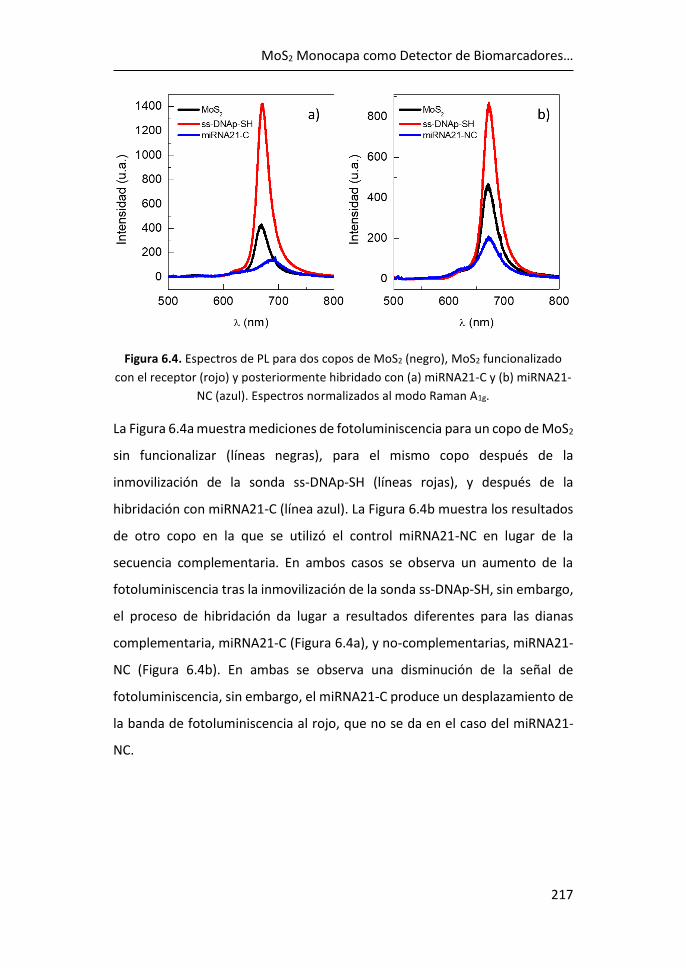
MoS2 Monocapa como Detector de Biomarcadores…
217
Figura 6.4. Espectros de PL para dos copos de MoS2 (negro), MoS2 funcionalizado
con el receptor (rojo) y posteriormente hibridado con (a) miRNA21-C y (b) miRNA21-
NC (azul). Espectros normalizados al modo Raman A1g.
La Figura 6.4a muestra mediciones de fotoluminiscencia para un copo de MoS2
sin funcionalizar (líneas negras), para el mismo copo después de la
inmovilización de la sonda ss-DNAp-SH (líneas rojas), y después de la
hibridación con miRNA21-C (línea azul). La Figura 6.4b muestra los resultados
de otro copo en la que se utilizó el control miRNA21-NC en lugar de la
secuencia complementaria. En ambos casos se observa un aumento de la
fotoluminiscencia tras la inmovilización de la sonda ss-DNAp-SH, sin embargo,
el proceso de hibridación da lugar a resultados diferentes para las dianas
complementaria, miRNA21-C (Figura 6.4a), y no-complementarias, miRNA21-
NC (Figura 6.4b). En ambas se observa una disminución de la señal de
fotoluminiscencia, sin embargo, el miRNA21-C produce un desplazamiento de
la banda de fotoluminiscencia al rojo, que no se da en el caso del miRNA21-
NC.

CÁPITULO 6
218
Para cuantificar este efecto, en la Figura 6.5 se ha representado la longitud de
onda del pico de fotoluminiscencia para los diferentes ensayos realizados:
miRNA21-C (Figura 6.5a) y el miRNA21-NC (Figura 6.5b). Se produce un
desplazamiento de la banda hacia el rojo de ~16 nm (43 meV) para la
secuencia complementaria, miRNA21-C, mientras que, para la no
complementaria, miRNA21-NC, la posición permanece prácticamente
constante. Se han realizado estas pruebas en cuatro copos diferentes y varios
puntos para cada copo para garantizar una buena reproducibilidad de los
datos dentro de los errores estadísticos (Figura 6.5c y 6.5d). Aunque la
intensidad de la fotoluminiscencia varía de un copo a otro (en el caso la
funcionalización con la sonda, ss-DNAp-SH, la intensidad varía hasta en un
factor 2), los valores de la posición de la fotoluminiscencia para cada ensayo
son muy similares, encontrándose los errores estadísticos dentro de los
puntos del gráfico (Figura 6.5c y 6.5d). Estos resultados demuestran la
especificidad del biosensor y la viabilidad del reconocimiento del biomarcador
miRNA21 justificados por los cambios de longitud de onda de la emisión del
MoS2 monocapa.
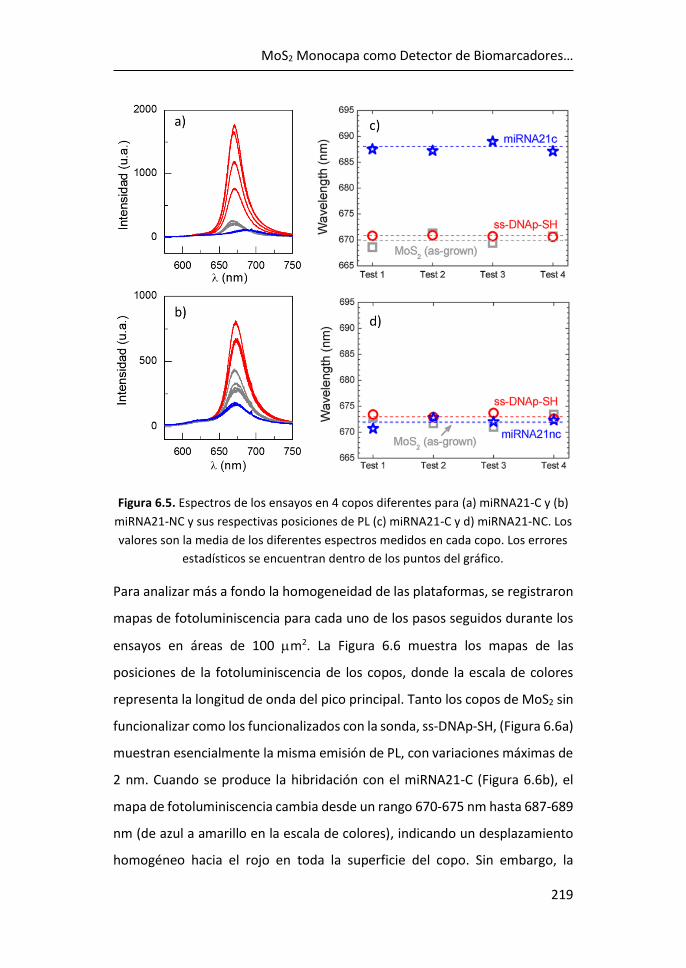
MoS2 Monocapa como Detector de Biomarcadores…
219
Figura 6.5. Espectros de los ensayos en 4 copos diferentes para (a) miRNA21-C y (b)
miRNA21-NC y sus respectivas posiciones de PL (c) miRNA21-C y d) miRNA21-NC. Los
valores son la media de los diferentes espectros medidos en cada copo. Los errores
estadísticos se encuentran dentro de los puntos del gráfico.
Para analizar más a fondo la homogeneidad de las plataformas, se registraron
mapas de fotoluminiscencia para cada uno de los pasos seguidos durante los
ensayos en áreas de 100 m2. La Figura 6.6 muestra los mapas de las
posiciones de la fotoluminiscencia de los copos, donde la escala de colores
representa la longitud de onda del pico principal. Tanto los copos de MoS2 sin
funcionalizar como los funcionalizados con la sonda, ss-DNAp-SH, (Figura 6.6a)
muestran esencialmente la misma emisión de PL, con variaciones máximas de
2 nm. Cuando se produce la hibridación con el miRNA21-C (Figura 6.6b), el
mapa de fotoluminiscencia cambia desde un rango 670-675 nm hasta 687-689
nm (de azul a amarillo en la escala de colores), indicando un desplazamiento
homogéneo hacia el rojo en toda la superficie del copo. Sin embargo, la
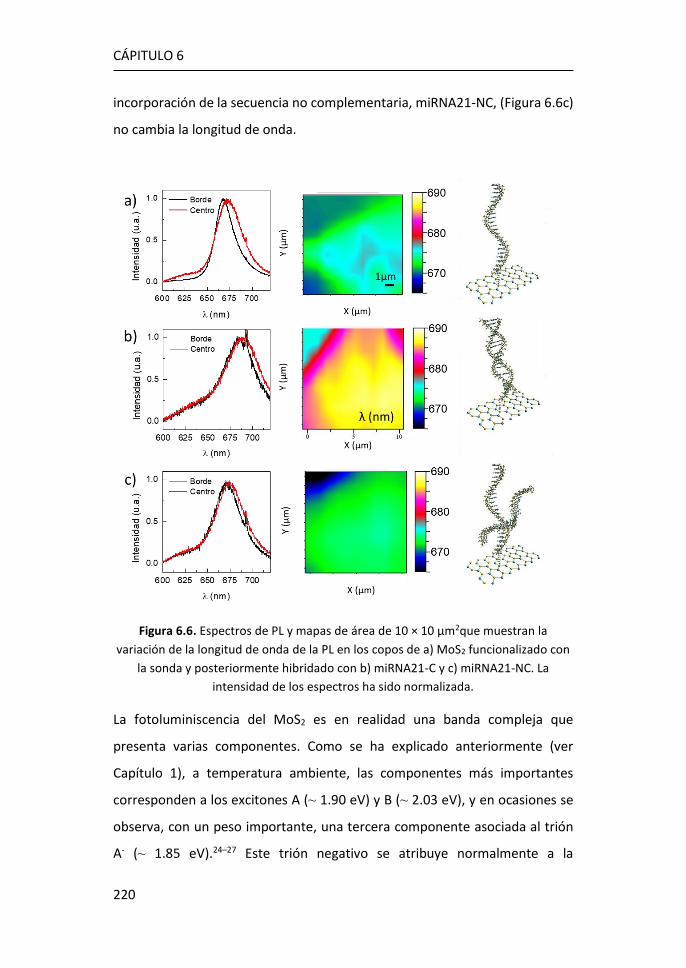
CÁPITULO 6
220
incorporación de la secuencia no complementaria, miRNA21-NC, (Figura 6.6c)
no cambia la longitud de onda.
Figura 6.6. Espectros de PL y mapas de área de 10 × 10 μm2que muestran la
variación de la longitud de onda de la PL en los copos de a) MoS2 funcionalizado con
la sonda y posteriormente hibridado con b) miRNA21-C y c) miRNA21-NC. La
intensidad de los espectros ha sido normalizada.
La fotoluminiscencia del MoS2 es en realidad una banda compleja que
presenta varias componentes. Como se ha explicado anteriormente (ver
Capítulo 1), a temperatura ambiente, las componentes más importantes
corresponden a los excitones A (~ 1.90 eV) y B (~ 2.03 eV), y en ocasiones se
observa, con un peso importante, una tercera componente asociada al trión
A- (~ 1.85 eV).24–27 Este trión negativo se atribuye normalmente a la
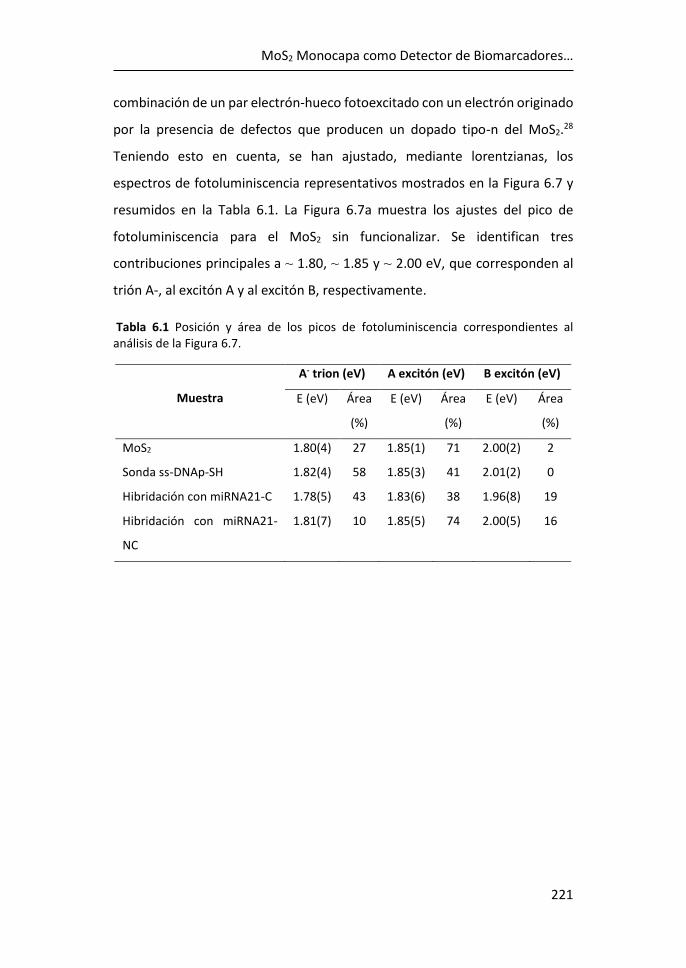
MoS2 Monocapa como Detector de Biomarcadores…
221
combinación de un par electrón-hueco fotoexcitado con un electrón originado
por la presencia de defectos que producen un dopado tipo-n del MoS2.28
Teniendo esto en cuenta, se han ajustado, mediante lorentzianas, los
espectros de fotoluminiscencia representativos mostrados en la Figura 6.7 y
resumidos en la Tabla 6.1. La Figura 6.7a muestra los ajustes del pico de
fotoluminiscencia para el MoS2 sin funcionalizar. Se identifican tres
contribuciones principales a ~ 1.80, ~ 1.85 y ~ 2.00 eV, que corresponden al
trión A-, al excitón A y al excitón B, respectivamente.
Tabla 6.1 Posición y área de los picos de fotoluminiscencia correspondientes al análisis de la Figura 6.7.
Muestra
A- trion (eV) A excitón (eV) B excitón (eV)
E (eV) Área
(%)
E (eV) Área
(%)
E (eV) Área
(%)
MoS2 1.80(4) 27 1.85(1) 71 2.00(2) 2
Sonda ss-DNAp-SH 1.82(4) 58 1.85(3) 41 2.01(2) 0
Hibridación con miRNA21-C 1.78(5) 43 1.83(6) 38 1.96(8) 19
Hibridación con miRNA21-
NC
1.81(7) 10 1.85(5) 74 2.00(5) 16
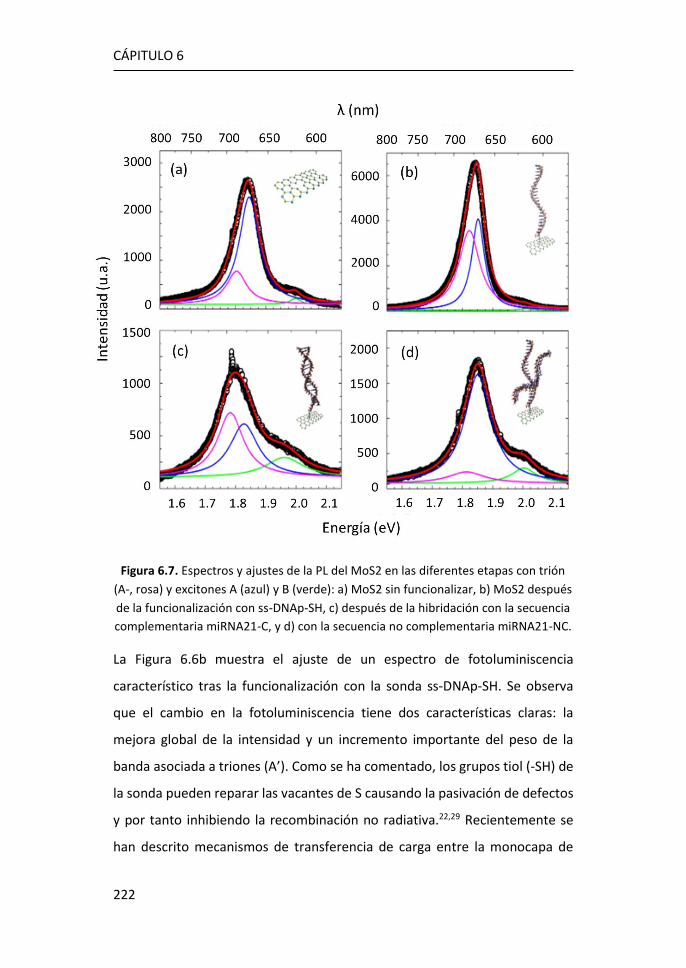
CÁPITULO 6
222
Figura 6.7. Espectros y ajustes de la PL del MoS2 en las diferentes etapas con trión
(A-, rosa) y excitones A (azul) y B (verde): a) MoS2 sin funcionalizar, b) MoS2 después
de la funcionalización con ss-DNAp-SH, c) después de la hibridación con la secuencia
complementaria miRNA21-C, y d) con la secuencia no complementaria miRNA21-NC.
La Figura 6.6b muestra el ajuste de un espectro de fotoluminiscencia
característico tras la funcionalización con la sonda ss-DNAp-SH. Se observa
que el cambio en la fotoluminiscencia tiene dos características claras: la
mejora global de la intensidad y un incremento importante del peso de la
banda asociada a triones (A’). Como se ha comentado, los grupos tiol (-SH) de
la sonda pueden reparar las vacantes de S causando la pasivación de defectos
y por tanto inhibiendo la recombinación no radiativa.22,29 Recientemente se
han descrito mecanismos de transferencia de carga entre la monocapa de
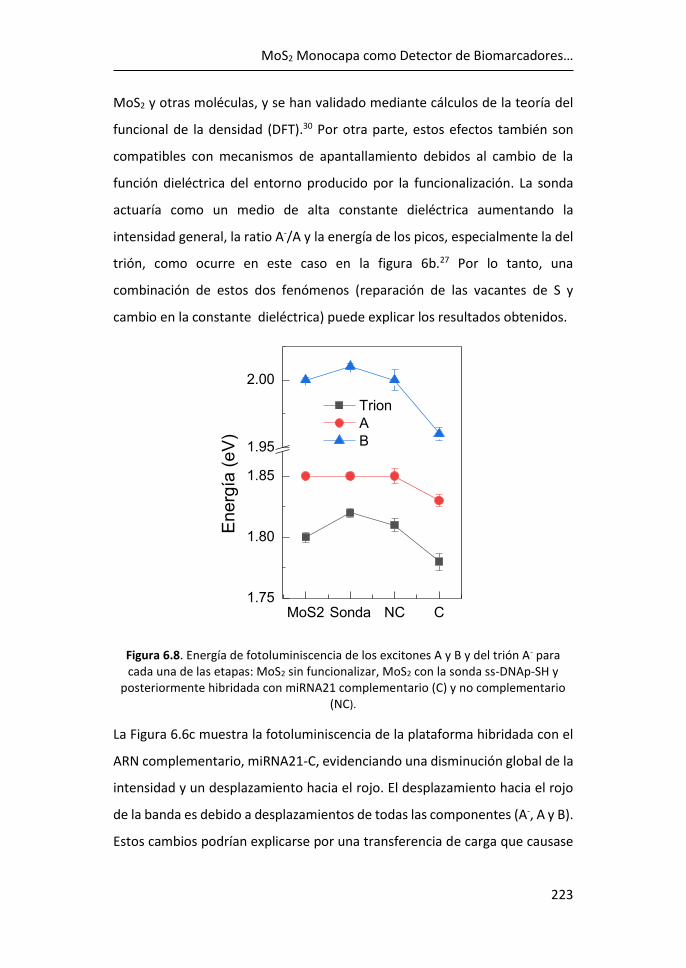
MoS2 Monocapa como Detector de Biomarcadores…
223
MoS2 y otras moléculas, y se han validado mediante cálculos de la teoría del
funcional de la densidad (DFT).30 Por otra parte, estos efectos también son
compatibles con mecanismos de apantallamiento debidos al cambio de la
función dieléctrica del entorno producido por la funcionalización. La sonda
actuaría como un medio de alta constante dieléctrica aumentando la
intensidad general, la ratio A-/A y la energía de los picos, especialmente la del
trión, como ocurre en este caso en la figura 6b.27 Por lo tanto, una
combinación de estos dos fenómenos (reparación de las vacantes de S y
cambio en la constante dieléctrica) puede explicar los resultados obtenidos.
Figura 6.8. Energía de fotoluminiscencia de los excitones A y B y del trión A- para cada una de las etapas: MoS2 sin funcionalizar, MoS2 con la sonda ss-DNAp-SH y
posteriormente hibridada con miRNA21 complementario (C) y no complementario (NC).
La Figura 6.6c muestra la fotoluminiscencia de la plataforma hibridada con el
ARN complementario, miRNA21-C, evidenciando una disminución global de la
intensidad y un desplazamiento hacia el rojo. El desplazamiento hacia el rojo
de la banda es debido a desplazamientos de todas las componentes (A-, A y B).
Estos cambios podrían explicarse por una transferencia de carga que causase
MoS2 Sonda NC C1.75
1.80
1.85
1.95
2.00
Ener
gía
(eV)
Trion A B

CÁPITULO 6
224
un dopado tipo-n del MoS2, disminuyendo la señal de fotoluminiscencia, al
aumentar los canales no radiativos de relajación de los pares e-h, y
produciendo un desplazamiento hacia menores energías.31,32 No se puede
descartar por completo la existencia de un pico adicional (ya que también es
posible un buen ajuste con 4 picos para el caso de la plataforma hibridada con
miRNA21-C). Por el contrario, no se observa un desplazamiento relevante de
las energías de los excitones al incorporar el ARN no-complementario de
control, miRNA21-NC, (Figura 6.6d y 6e) y la intensidad del pico del trión
disminuye significativamente en el espectro. Los cambios observados en las
distintas etapas son complejos pues compiten varios mecanismos y, a pesar
de los recientes avances,24,25,33 las publicaciones actuales no permiten
determinar de forma unívoca su origen. En la Figura 6.7 se han representado
las energías de los tres picos (excitones A y B y trión A-) para cada etapa y se
observa cómo para la sonda hibridada con el miRNA21-C la energía de los tres
picos disminuye drásticamente con respecto a los demás casos.
En resumen, en este trabajo en colaboración con la UAM se ha desarrollado
un biosensor óptico para el biomarcador miRNA21 del cáncer de mama
basado en la modificación de la fotoluminiscencia de MoS2 monocapa. Este
sensor se ha fabricado en tres pasos: (1) crecimiento del MoS2 monocapa por
PVT sobre zafiro, (2) funcionalización con una sonda de ADN tiolada (ss-DNAp-
SH), y (3) hibridación con secuencias complementarias y no complementarias
de miRNA21 (esta última utilizada como control).
La modificación con el biomarcador ss-DNAp-SH (la sonda) aumenta la
intensidad de la fotoluminiscencia del MoS2, que posteriormente disminuye
tras la hibridación. Se observa un desplazamiento de 16 nm en la banda de
fotoluminiscencia exclusivamente para la hibridación con el ARN
complementario, miRNA21-C, pero no para la secuencia de control miRNA21-
NC, demostrando la especificidad del biosensor y la viabilidad del

MoS2 Monocapa como Detector de Biomarcadores…
225
reconocimiento a través de la fotoluminiscencia del MoS2 monocapa. La
reproducibilidad de los resultados se verificó con espectros de
fotoluminiscencia en varios copos.
El análisis detallado de los espectros revela cambios en la relación
trión/excitón, A-/A, atribuyéndose a ambos picos el desplazamiento al rojo
observado tras la hibridación debido a una de transferencia de electrones
hacia el MoS2.
Estos resultados indican las ventajas en términos de selectividad de los
biosensores ópticos basados en MoS2 monocapa. El método de transducción
a través del cambio de longitud de onda de la fotoluminiscencia, en lugar de
la intensidad, es novedoso y un logro significativo para el desarrollo de
biosensores comerciales en el futuro.
6.3 REFERENCIAS
1. Chen, Y., Tan, C., Zhang, H. & Wang, L. Two-dimensional graphene analogues for biomedical applications. Chem. Soc. Rev. 44, 2681–2701 (2015).
2. Tan, C. et al. Recent Advances in Ultrathin Two-Dimensional Nanomaterials. Chem. Rev. 117, 6225–6331 (2017).
3. Su, S. et al. Dual-mode electrochemical analysis of microRNA-21 using gold nanoparticle-decorated MoS2 nanosheet. Biosens. Bioelectron. 94, 552–559 (2017).
4. Pumera, M. & Loo, A. H. Layered transition-metal dichalcogenides (MoS2 and WS2) for sensing and biosensing. TrAC - Trends Anal. Chem. 61, 49–53 (2014).
5. Zhu, C. et al. Single-layer MoS2-based nanoprobes for homogeneous detection of biomolecules. J. Am. Chem. Soc. 135, 5998–6001 (2013).
6. Wang, T. et al. Direct detection of DNA below ppb level based on thionin-functionalized layered MoS2 electrochemical sensors. Anal.

CÁPITULO 6
226
Chem. 86, 12064–12069 (2014).
7. Majd, S. M., Salimi, A. & Ghasemi, F. An ultrasensitive detection of miRNA-155 in breast cancer via direct hybridization assay using two-dimensional molybdenum disulfide field-effect transistor biosensor. Biosens. Bioelectron. 105, 6–13 (2018).
8. Lee, D.-W. et al. Field-effect transistor with a chemically synthesized MoS 2 sensing channel for label-free and highly sensitive electrical detection of DNA hybridization. Nano Res. 8, 2340-32–50 (2015).
9. Oudeng, G., Au, M., Shi, J., Wen, C. & Yang, M. One-Step in Situ Detection of miRNA-21 Expression in Single Cancer Cells Based on Biofunctionalized MoS2 Nanosheets. ACS Appl. Mater. Interfaces 10, 350–360 (2018).
10. Yan, L., Shi, H., Sui, X., Deng, Z. & Gao, L. MoS2-DNA and MoS2 based sensors. RSC Adv. 7, 23573 (2017).
11. Mittal, S., Kaur, H., Gautam, N. & Mantha, A. K. Biosensors for breast cancer diagnosis: A review of bioreceptors, biotransducers and signal amplification strategies. Biosens. Bioelectron. 88, 217–231 (2017).
12. Iorio, M. V. & Croce, C. M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 4, 143–159 (2012).
13. Catalán-Gómez, S. et al. Breast cancer biomarker detection through the photoluminescence of epitaxial monolayer MoS2 flakes. Sci. Reports 2020 101 10, 1–9 (2020).
14. Li, H. et al. From bulk to monolayer MoS 2: Evolution of Raman scattering. Adv. Funct. Mater. 22, 1385–1390 (2012).
15. Joo, P. et al. Functional polyelectrolyte nanospaced MoS2 multilayers for enhanced photoluminescence. Nano Lett. 14, 6456–6462 (2014).
16. Lee, K. C. J. et al. Plasmonic gold nanorods coverage influence on enhancement of the photoluminescence of two-dimensional MoS2 monolayer. Sci. Rep. 5, 1–9 (2015).
17. Chung-Che Huang et al. Scalable high-mobility MoS 2 thin films fabricated by an atmospheric pressure chemical vapor deposition process at ambient temperature. Nanoscale 6, 12792–12797 (2014).
18. Luo, S., Cullen, C. P., Guo, G., Zhong, J. & Duesberg, G. S. Investigation of growth-induced strain in monolayer MoS2 grown by chemical vapor deposition. Appl. Surf. Sci. 508, 145126 (2020).

MoS2 Monocapa como Detector de Biomarcadores…
227
19. Feng, L. ping et al. Influence of Mo-vacancy concentration on the structural, electronic and optical properties of monolayer MoS2: A first-principles study. Mater. Chem. Phys. 209, 146–151 (2018).
20. Chen, X., McGlynn, C. & McDonald, A. R. Two-Dimensional MoS2 Catalyzed Oxidation of Organic Thiols. Chem. Mater. 30, 6978–6982 (2018).
21. Tuxen, A. et al. Size threshold in the dibenzothiophene adsorption on MoS2 nanoclusters. ACS Nano 4, 4677–4682 (2010).
22. Makarova, M., Okawa, Y. & Aono, M. Selective adsorption of thiol molecules at sulfur vacancies on MoS 2(0001), followed by vacancy repair via S-C dissociation. J. Phys. Chem. C 116, 22411–22416 (2012).
23. Chou, S. S. et al. Ligand conjugation of chemically exfoliated MoS2. J. Am. Chem. Soc. 135, 4584–4587 (2013).
24. Pandey, J. & Soni, A. Unraveling biexciton and excitonic excited states from defect bound states in monolayer MoS 2. Appl. Surf. Sci. 463, 52–57 (2019).
25. Lee, H. S., Kim, M. S., Kim, H. & Lee, Y. H. Identifying multiexcitons in Mo S2 monolayers at room temperature. Phys. Rev. B 93, 1–6 (2016).
26. Yang, X. & Li, B. Monolayer MoS2 for nanoscale photonics. Nanophotonics 9, 1557–1577 (2020).
27. Lin, Y. et al. Dielectric screening of excitons and trions in single-layer MoS2. Nano Lett. 14, 5569–5576 (2014).
28. Nan, H. et al. Strong photoluminescence enhancement of MoS2 through defect engineering and oxygen bonding. ACS Nano 8, 5738–5745 (2014).
29. Ding, Q. et al. Basal-Plane Ligand Functionalization on Semiconducting 2H-MoS2 Monolayers. ACS Appl. Mater. Interfaces 9, 12734–12742 (2017).
30. Jing, Y., Tan, X., Zhou, Z. & Shen, P. Tuning electronic and optical properties of MoS2 monolayer via molecular charge transfer. J. Mater. Chem. A 2, 16892–16897 (2014).
31. Dhakal, K. P. et al. Confocal absorption spectral imaging of MoS2: Optical transitions depending on the atomic thickness of intrinsic and chemically doped MoS2. Nanoscale 6, 13028–13035 (2014).
32. Chakraborty, B. et al. Symmetry-dependent phonon renormalization in

CÁPITULO 6
228
monolayer MoS 2 transistor. Phys. Rev. B - Condens. Matter Mater. Phys. 85, 1–5 (2012).
33. Wang, W. et al. Studying of the Biexciton Characteristics in Monolayer MoS2. J. Phys. Chem. C 124, 1749–1754 (2020).

229
7 CONCLUSIONES
MoS2 Bidimensional
• Se han determinado las condiciones óptimas para crecer MoS2 monocapa
sobre zafiro por CVD a partir de MoO3 y S en polvo en el sistema puesto
a punto en el laboratorio: una presión de 1 mbar y un flujo de Ar de 25
sccm, con temperaturas de 700 ºC y 200 ºC para el MoO3 y el S
respectivamente. En el caso de los sustratos de Si y FS, el MoS2 obtenido
es de pocas capas, independientemente de las condiciones utilizadas.
• Se ha observado la formación de nanopartículas de MoO2 en muestras
de MoS2-2D cuya morfología depende de la temperatura del azufre. Un
adecuado control de esta temperatura y una limpieza del horno a vacío y
alta temperatura evita la formación de las NPs de MoO2.
• Se ha diseñado, montado y optimizado en el laboratorio del ICMM un
sistema apto para el crecimiento en fase de vapor de TMDCs. El MoS2
obtenido en este sistema por PVT a partir MoS2 en polvo sobre sustratos
de zafiro y Si es siempre monocapa. Los copos crecidos tienen forma
triangular y tamaños entre 1 y 5 μm, formándose zonas donde los copos
conectan creando capas cuasi-continuas. Las condiciones empleadas son:
temperaturas de 970 ºC y 800 ºC para el MoS2 y los sustratos,
respectivamente, presión de 1 mbar y flujo de 20 sccm.
• Se ha hecho un estudio exhaustivo de los espectros Raman de muestras
de MoS2 en volumen y MoS2 bidimensional de distinto número de capas
y obtenido por diferentes técnicas: exfoliación mecánica (ME), PVT y CVD.
Las medidas se llevaron a cabo a temperatura ambiente empleando la
línea en 488 nm (2.54 eV) de un láser de Ar+. Se han detectado picos

230
prohibidos de primer orden correspondientes a ramas acústicas
activados por la presencia de defectos intrínsecos. Dependiendo del
proceso Raman que da lugar a cada uno de los fonones, éstos son
sensibles a diferentes variables como el número de capas, el método de
crecimiento o el sustrato. Para dos picos detectados a 150 y 190 cm-1, la
frecuencia varía con el número de capas debido a las modificaciones en
la estructura de la banda electrónica, a través de un proceso de doble
resonancia mediado por defectos intrínsecos, que activa los fonones
alrededor del punto Q de la zona de Brillouin. Esto se debe a que, en el
MoS2, se produce un anidamiento de las bandas electrónicas cerca de los
puntos y Q, que da lugar al excitón C a 2.7 eV, muy cerca de la energía
empleada para la excitación (2.4 eV). Además, las bandas electrónicas
alrededor de los puntos Q y son especialmente sensibles al número de
capas. También se observa una banda compleja en el rango 420-470 cm-
1 (procesos de dos fonones), que es resonante para energías de excitación
cercanas al excitón B (2 eV), y que es sensible a la interacción del MoS2
con el sustrato y a la temperatura. También se han encontrado picos
específicos de las técnicas de depósito de vapor a 247, 498 y 550 cm-1 y
otros que sólo se detectan en MoS2 monocapa a 220 y 310 cm-1,
independientemente del método de obtención.
• Se han obtenido nanocopos de MoS2 monocapa y de 2-3 capas por CVD
(tamaños entre 10 nm y 500 nm) y copos monocapa crecidos por PVT
(tamaños entre 1-5 μm). Mediante el análisis de los espectros Raman, de
fotoluminiscencia y de micro-reflectancia, combinado con imágenes de
AFM (topología y fase), se han podido determinar los efectos inducidos
por la reducción del tamaño en las propiedades de los nanocopos de
MoS2. Por una parte, el crecimiento sobre zafiro por PVT de MoS2-2D de
varias micras da lugar a una expansión biaxial (en torno al 1%) del MoS2.

231
A medida que el tamaño de los copos se reduce, esta tensión va
desapareciendo, siendo inapreciable para nanocopos de tamaño inferior
a 100 nm. Por otra parte, al reducirse el tamaño de los copos, se produce
un incremento del dopado tipo p, probablemente originado por la
transferencia de huecos debido a la adsorción de O2. Se propone que el
incremento de la relación borde/superficie (al disminuir el tamaño)
proporciona una mayor densidad de vacantes de azufre que facilita la
adsorción de O2. Este dopaje tipo p da lugar un incremento de la energía
de la emisión a medida que el tamaño de reduce (50 meV) que también
suele implicar un aumento en su intensidad. En este caso, la intensidad
permanece constante (dentro de la dispersión de valores registrados para
MoS2 monocapa, de mayor tamaño y sin defectos) debido al desorden
estructural. Esta distorsión e incorporación de defectos es muy moderada
en todo el rango estudiado, hasta 10 nm. Estos efectos son similares en
nanocopos monocapa y en los de 2-3 capas.
• Se ha demostrado que la presencia de nanopartículas (NPs) de MoO2 en
muestras de MoS2 sintetizadas por CVD, da lugar a una amplificación de
la señal Raman y de fotoluminiscencia debido a distintos efectos. Por una
parte, el efecto SERS originado por las NPs de MoO2 de mayor tamaño da
lugar a una amplificación de ambas señales. Por otra parte, la
transferencia de electrones del MoS2 a las NPs de MoO2 da lugar a un
dopado tipo-p que provoca un aumento de la intensidad y de la energía
de la fotoluminiscencia, pero no de la intensidad Raman. Además, se ha
obtenido una señal de fotoluminiscencia anómalamente intensa en copos
de 2-3 capas.
• Los tratamientos de recocido en aire y en vacío de distintas muestras de
MoS2-2D han confirmado la relevancia de la adsorción y el enlace de

232
oxígeno en la intensidad de la fotoluminiscencia y su capacidad de
transferir huecos.
Amplificación Raman por nanopartículas de Ag
ultrapequeñas: efectos de tamaño y distancia entre
partículas
• Se ha evaluado la capacidad de amplificación SERS de NPs de Ag
ultrapequeñas, de radio 1.8 nm, depositadas por la técnica de agregación
en fase gas sobre sustratos de sílice vítrea y sobre monocapas de grafeno,
previamente transferidas sobre cuarzo. Se han empleado simulaciones de
conjuntos de NPs de plata de distintos tamaños utilizando cálculos
tridimensionales de diferencias finitas de dominio temporal (FDTD) para
analizar los resultados experimentales. Se demuestra que, el
desplazamiento de la longitud de onda del plasmón hacia el rojo (de 400
a 500 nm) y su ensanchamiento se originan por distribuciones de
distancias inter-partícula que se estrechan y se desplazan a distancias
más pequeñas a medida que la densidad entre NPs aumenta. La
reducción de la distancia inter-partícula media es determinante en la
formación de puntos calientes (hot-spots) y por tanto en los valores de
amplificación SERS.
• Se ha observado que el papel del grafeno como sustrato para el depósito
de NPs consiste en deslocalizar parcialmente los portadores libres, así
como favorecer la adherencia de las NPs sobre el sustrato.
• Se ha observado que la configuración geométrica del analito con respecto
a las NPs es determinante para la magnitud de la amplificación de las
señales Raman. Por ejemplo, en el caso del grafeno, la amplificación
aumenta linealmente con la extinción del plasmón, mientras que en el

233
caso de la R6G la amplificación es casi exponencial. Este comportamiento
exponencial en el caso de la R6G demuestra la alta eficiencia de los
puntos calientes que se forman en los espacios entre partículas.
• Para granos de plata de mayor tamaño (7<R<15 nm) depositados por
sputtering también se observa un desplazamiento hacia el rojo y el
ensanchamiento del plasmón a medida que el grosor de la película
aumenta (de 1 a 4 nm). En este caso, la amplificación de R6G es sólo
proporcional a la extinción del plasmón. Es importante señalar que, para
el mismo valor de extinción, la amplificación proporcionada por las NPs
ultra-pequeñas es mayor que la de los granos más grandes de las
películas, debido principalmente a los puntos calientes formados entre
las NPs ultra-pequeñas.
• Desde el punto de vista de las aplicaciones, resulta relevante la
posibilidad de aumentar eficientemente la amplificación obtenida con
películas de plata depositando sobre ellas bajas densidades de NPs ultra-
pequeñas, y creando así puntos calientes adicionales.
Funcionalización de grafeno con grupos carboxílicos
para aplicaciones en biodetección
• Se ha demostrado que un nuevo método de funcionalización de grafeno
basado en el crecimiento del grafeno por vía CVD con un flujo de vapor
de ácido dicloroacético (DCA) in-situ da como resultado grafeno
funcionalizado con grupos COOH.
• En función de los tiempos característicos de las distintas etapas del
proceso ta (tiempo de la etapa de crecimiento) y tb (tiempo de la etapa de
funcionalización) es posible obtener muestras funcionalizadas de grafeno
monocapa o multicapa: i) un ta largo (30 min) y tb corto (15 min) da lugar

234
a muestras con regiones de grafeno monocapa no funcionalizado y
regiones de grafeno de varias capas funcionalizado (SLG/MLG-F); ii) para
ta y tb largos (ambos 30 min) se obtiene grafeno monocapa y multicapa
ambos con funcionalización homogénea (SLG-F/MLG-F); iii) para un ta
corto (15 min) y tb largo se obtiene grafeno monocapa con
funcionalización no homogénea (SLG/SLG-F) y iv) si al último caso se le
añaden 5 min de flujo de DCA durante la etapa de enfriamiento se
obtiene grafeno monocapa con funcionalización homogénea (SLG-F).
• La optimización de los tiempos de las etapas de crecimiento y
funcionalización de grafeno da lugar a grafeno funcionalizado con grupos
COOH, con un porcentaje de carboxilos del orden del 5 %, que es superior
al de los demás grupos funcionales oxigenados presentes. La
funcionalización carboxílica obtenida en relación con el total de los
grupos funcionales es superior a la obtenida por otros métodos.
• Se ha optimizado el proceso de transferencia de grafeno, haciendo
posible la transferencia de las muestras de grafeno funcionalizado
monocapa para la realización de medidas eléctricas.
• En la evaluación de las propiedades de transporte electrónico de las
muestras funcionalizadas, se obtienen valores de resistencia laminar de
7-8 kΩ/sq, similares a los de las mejores muestras reportadas con otras
técnicas, y valores de movilidad de hasta 900 cm2V-1s-1, significativamente
más altos que los reportados en la literatura.
• Por último, se utiliza el método carbodiimida para anclar anticuerpos en
los lugares donde el grafeno ha sido funcionalizado con grupos -COOH. El
enlace entre el grafeno funcionalizado y el anticuerpo IgG1 se pone de
manifiesto en la distribución homogénea de la epi-fluorescencia que se
observa en las imágenes de emisión del marcador isotiocianato de

235
fluoresceína. El carácter covalente del enlace se verifica mediante la
comparación con las imágenes obtenidas del grafeno de referencia
sometido simultáneamente al mismo proceso, en el que no se detecta
ninguna emisión significativa.
• El conjunto de todos estos resultados lleva a la conclusión de que el
método que se presenta en este trabajo para la carboxilación in-situ del
grafeno es adecuado para la fabricación de plataformas funcionales que
pueden explorarse como sensores ópticos o electrónicos conductores.
MoS2 monocapa como detector de biomarcadores de
cáncer de mama a través de cambios en la
fotoluminiscencia
• Se ha desarrollado un biosensor óptico para el biomarcador del cáncer de
mama miRNA21 basado en la modificación de la fotoluminiscencia del
MoS2 monocapa. Este sensor se ha fabricado en tres pasos: (1)
crecimiento del MoS2 monocapa por PVT sobre sustratos de zafiro a partir
de MoS2 en polvo, (2) funcionalización con una sonda de ADN tiolada (ss-
DNAp-SH), y (3) hibridación con secuencias complementarias y no
complementarias de miRNA21 (esta última utilizada como control).
• Tras la hibridación con el ARN complementario se observa un
desplazamiento de 16 nm en la banda de fotoluminiscencia que no se da
para la hibridación con el ARN no complementario, demostrando así la
especificidad del biosensor y la viabilidad del reconocimiento a través de
la fotoluminiscencia del MoS2 monocapa.
• Se ha verificado la reproducibilidad de los resultados se registrando
espectros de fotoluminiscencia en varios copos.

236
• El análisis detallado de los espectros revela cambios en la relación
trión/excitón, A-/A, atribuyéndose a ambos picos el desplazamiento al
rojo observado tras la hibridación debido a una de transferencia de
electrones hacia el MoS2.
• Estos resultados muestran las ventajas en términos de selectividad de los
biosensores ópticos basados en MoS2 monocapa. El método de
transducción a través del cambio de longitud de onda de la
fotoluminiscencia, en lugar de la intensidad, es novedoso además de
significativo para el desarrollo de biosensores comerciales en el futuro.

237
ANEXO
SIMULACIONES POR DIFERENCIAS FINITAS EN EL DOMINIO DEL
TIEMPO (FDTD)
El método de las Diferencias Finitas en el Dominio del Tiempo (FDTD, del inglés
finite-difference time-domine) fue desarrollado para resolver las ecuaciones
de Maxwell y actualmente se utiliza para resolver problemas
electromagnéticos transitorios.1–4 En este trabajo se ha utilizado para llevar a
cabo las simulaciones del Capítulo 4.
Para llevar a cabo las simulaciones tridimensionales se ha utilizado el software
Lumerical FDTD (https://www.lumerical.com/fdtd/), que permite calcular el
campo eléctrico y los espectros de extinción de matrices de nanopartículas en
cualquier punto. Los cálculos de los campos eléctricos y los espectros de
extinción para una única nanopartícula (ya sean esferas o elipsoides) se han
realizado utilizando la teoría de Mie,5 y han utilizado como primer paso para
comprobar los resultados de las simulaciones FDTD.
Los parámetros FDTD utilizados en las simulaciones dependen en gran medida
de la simetría de los conjuntos de nanopartículas considerados. En el caso de
esferas aisladas y de esferas distribuidas de forma aleatoria, se han utilizado
fuentes de campo disperso total (TFSF, del inglés Total-field Scattered-field).
Se han utilizado condiciones de contorno simétricas y antisimétricas en
función de la polarización de las direcciones x e y (en el plano del sustrato) y
condiciones de capas perfectamente acopladas (PML, del inglés Perfectly
Matched Layers) para la dirección z (perpendicular al sustrato). Para las PML
distribuidas aleatoriamente se han utilizado condiciones de contorno. En el
caso de matrices periódicas de nanopartículas, se utilizaron fuentes de ondas

238
planas y condiciones de contorno periódicas para las direcciones x e y, y PML
en la dirección z.
Se han utilizado monitores de potencia para medir la absorción y la
transmisión; y monitores de campo eléctrico en diferentes capas para
encontrar la amplificación del campo. La malla utilizada fue R/200 para
2<R<10 nm, siendo R el radio de la nanopartícula.
REFERENCIAS
1. Yee, K. Numerical solution of initial boundary value problems involving
maxwell’s equations in isotropic media. IEEE Trans. Antennas Propag.
3, 302–307 (1966).
2. Alvarez-Fraga, L. et al. Efficient heterostructures for combined
interference and plasmon resonance raman amplification. ACS Appl.
Mater. Interfaces 9, 4119–4125 (2017).
3. Aguilar-Pujol, M. et al. Supported ultra-thin alumina membranes with
graphene as efficient interference enhanced raman scattering
platforms for sensing. Nanomaterials 10, (2020).
4. Ramírez-Jíménez, R. et al. Interference enhanced Raman effect in
graphene bubbles. Carbon N. Y. 105, 556–565 (2016).
5. Craig F. Bohren; Donald R. Huffman. Absorption and Scattering of Light
by Small Particles. (WILEY‐VCH Verlag, 1988).

241
PUBLICACIONES
Como resultado del trabajo descrito en esta Tesis Doctoral, se han publicado
los siguientes artículos científicos:
• In-situ carboxylation of graphene by chemical vapor deposition
growth for biosensing. Sandra Cortijo-Campos, Leo Álvarez-Fraga, Gil
Gonçalves, Mercedes Vila, Patricia Álvarez, Rosa Menéndez, Alicia de
Andrés, Carlos Prieto. Carbon 141, 719 (2019)
• Raman amplification in the ultra-small limit of Ag nanoparticles on
SiO2 and graphene: size and inter-particle distance effects Sandra
Cortijo-Campos, Rafael Ramírez-Jiménez, Esteban Climent-Pascual,
Montserrat Aguilar- Pujol, Félix Jiménez-Villacorta, Lidia Martínez,
Rafael Jiménez-Riobóo, Carlos Prieto and Alicia de Andrés. Mat. &
Design, 192, 108702 (2020)
• Breast cancer biomarker detection through the photoluminescence
of epitaxial monolayer MoS2 flakes. Sergio Catalán-Gómez, María
Briones, Sandra Cortijo-Campos, Tania García-Mendiola, Alicia de
Andrés, Sourav Garg, Patrick Kung, Encarnación Lorenzo, Jose Luis
Pau, Andrés Redondo-Cubero. Scientific Rep., 10, 16039 (2020)
• Raman and Fluorescence Enhancement Approaches in Graphene-
Based Platforms for Optical Sensing and Imaging. Sandra Cortijo-
Campos, Rafael Ramírez-Jiménez and Alicia de Andrés. Review:
Nanomaterials, 11, 644 (2021)

242
Otras publicaciones en diferentes áreas científicas:
• Anomalous shift of graphene 2D Raman band and grain selective Cu
oxidation in the graphene-Cu system. Javier Bartolomé, Leo Alvarez-
Fraga, Montserrat Aguilar-Pujol, Sandra Cortijo-Campos, Ana
Cremades, Carlos Prieto, Alicia de Andrés. 2D Materials, 6, 015023
(2019)
• Supported alumina membranes with graphene as efficient
interference enhanced Raman scattering platforms for sensing.
Montserrat Aguilar-Pujol, Rafael Ramírez-Jiménez, Elisabet Xifre-
Pérez, Lluis F. Marsal, Sandra Cortijo-Campos, Javier Bartolomé and
Alicia de Andrés. Nanomaterials, 10, 830 (2020)
• A route to detect H2 in ambient conditions using a sensor based on
reduced graphene oxide. G. Tabares, A. Redondo-Cubero, L. Vázquez,
M. Revenga, S. Cortijo-Campos, E. Lorenzo A. de Andrés E. Ruiz and J.
L. Pau. Sensors and Actuators A, 304, 111884 (2020)
• Enhanced stability and efficiency in inverted perovskite solar cells
through graphene doping of PEDOT:PSS hole transport layer. C.
Redondo-Obispo, T. S. Ripolles, S. Cortijo-Campos, A. L. Álvarez, E.
Climent-Pascual, A. de Andrés and C. Coya. Mat. & Design, 191,
108587 (2020)
• Effect of the linkage position on the conjugation length of truxene-
based porous polymers: implications on their sensing performance
of nitroaromatics. M. Echeverri, S. Gámez, R. Cano, J. Guadalupe, S.
Cortijo-Campos, J. Navarrete, M. Iglesias, M.C. Ruiz Delgado, B.
Gómez-Lor, Chemistry of Materials., 31, 17, 6971 (2019).





























