



oo4o-4020193 s6oo+oo 1993 Perganon Press Ltd
Heterocycles from Heterocycles. 1,3-Diaryi4,5-irmdazolidinediones from 1,3,5-Tnarylhexahydro-1,3,54nazmes and Oxalyl Chlonde
Giancarlo Verardo, Angelo G. Giumanir& * Faust0 Gorass~~, Manlena Tolazv and
Pa010 Strazzohni
Department of Chemical Sc~cncer and Technologies, Unlverslty of
Udine. Via de1 Cotontficio 108, 33100 Udine. Italy
(Recewed m UK 26 Juiy 1993, accepted 10 September 1993)
Abstract: 1.3-Dlaryl-4,5-imidazclidinedicnes (IS) are easily
synthesized from 1,3,5-trierylhexehydro-1,3,5-triez~nes CL) and
CXClYl chloride <S) L” a reection not likely to involve the
zwltterionlc lntermediste (3) of the N-methylenearyIam~ne dlmer,
but VIewing the sequential pack up of two un1tt of the mcncmer
(1) by oxsly chloride (2) The essential role of ethyl alcohol
added to the reaction mixture 1s recognized Reactlo” conditions
have been cptlmized and scme ten ~midazoi~d~nediones (6-1 were
prepared I” good to excellent yields Geometric parameters of S
were obtolned by X-ray dlffrsctlon snslys~r all the nuclei are
found almost in one plane except for a small twist of the phcnyl
rings about the C-N bond
IRTRODUCTIOR
1,3,5-Tnarylhexahydro-1,3,5-triazines Q), the cychc tnmersi of the elusive N-me-
thylenearylaminesz Q) may be induced to react as 2 or the dxmenc zwittenon3 2 by
interaction with substrates with strongly polarized bonds.4 It is not clear in most cases
whether depolymerization occurs first, either thermally or induced by an added catalyst
or whether the substrate attacks 1 dxectly, causing a reaction sequence all the way
down to the observed products.
The employment of & to yield heterocycks has been recorded
the patented preparation of 1,3-drary1-1,3-(5-thiadiarine)-6-thlones
1 with carbon disulphide.5 On a different front, the rea&on of
documented,6 but not that with bdun&onal compounds of the type
10609
m the literature for
from the rea&on
1 with G-X(=O)Hal
G(X[=OlHal),.
of
is

10610 G VERARDO et al
Ar
I N
(‘1
\-6
4 * Ar-N=Ct$ /
Ar -N
ArHNVNNAr 2 \
CH,Hal
1 t!
Most of the ptinent work has appeared m the form of patents and 4 were usually
poorly characterized rntermedrates of processes leatig to other final products.
RESULTS AND DISCUSSION
The reactin between 1 and oxalyl chlmde 3 has now been mvestigated and
developed into a very convenient way of obtimg N,N’-draryl-4,5rmrdazohdinetines (4)
(Table l), a class of compounds so far unreported.
Table 1. L,3-Dlaryl-4.5-lmldazolldlnedlones (5) Prepared.
Trlazlne (Ar)
Product Yield’ Crystaillzatlon solvent (W g,
A!? (3-He-C,H,)
Ic (4-He-C,H,)
(4-?&H,)
If (L-F-CsH,)
(3-F%,H,)
G! (4-F-C,H,)
li (3-Cl-C,H,)
(4&H,)
llz (3-Br-CGH4)
83
80
90
55
60
90
80
93
90
67
269
217
>290
261
182
251b
266
247
267
268
ethyl
ethyl
ethyl
ethyl
ethyl
ethyl
acetate
acetate/dlchloromethane
acetate/dlchloromethane
acetate
acetate/trlchloromethane
acetate/dlchloromethane
ethyle acetate/dlchlorcmethane
ethyl acetate/dlchloromethane
ethyl acetate/dlchloromethane
ethyl acetate/dlchloromethane
‘Yield of recrystallized product bDecomposltlon temperature.

Hctcrocycles from hetemcycles 10611
The parent molecule itself, an isomer of the well known compound hydantoln (irm-
dasohdine-2,3-lone), is not known and only two 1,3-denvataves, namely 1,3-dimethyl-
-4.5~inudazokdinedlone7 and 1,3-drbenzoyl-4,5_irdasolidinedione,8 were described to
date.
Our synthetic procedure calls for the portionwise addition of 5 to an equlmolecular
amount of 1 in dlethyl ether at OV, immediately followed by a necessary termnation of
the reaction with ethanol. Two different courses may be envisaged to rationahze the
outcome. If the monomer (2), either in fast eqmhbnum with the tnmer or ongmating
from the induced decomposition of 1 is to be involved, pathway A should be at work
(Scheme 1) On the other hand, the drmenc zwittenonic species 1 may be active, thus
facilitating pathway B.
Headspace analysis ruled out the presence of CH,Cl,, but both diethoxymethane (1)
and ethoxymethylchlonde (i3) were detected, the former coming from ethanolysrs of the
latter in solution. This observation pointed to an essential role of the alcohol in
carrying out the reaction and ruled out the rnitzal intervention of 3 In fact, it is
conceivable that 2, d formed, would rapidly evolve to l0, in turn bound to undergo fast
reaction with the naked chlonde ion to produce _6: but, besrde the absence of CH,Cl,,
ethanol was found to play an essential role
The monomer 2 is, on the other hand, expected to react promptly with 5 to form
the intermediate Q and furthermore to give lJ, which, apparently unable to undergo
nng closure to lJ, survives until ethanol rs added and the key intermediate j.J 19
generated. This, in turn, either undergoes ring closure to 6a_k or further competitive
ethanolysis to the constantly observed by-product oxalamrdes 14a_k.
Indirect support for this hypothesis was offered by the reaction between 1,3,5-
-tn(4-anlsyl)hexahydro-1,3,5-tnazin e (le) and oxalyl chlonde 5 in its devious behaviour:
enhanced stabilization of 2 sparked route B,
oxaiyl-di(4-anlsylarno)methane (15), detected
rmxture, together with another side product,
-hydroxymethyl-di(4-anlsylamino)methane (l6)
which did not lead to Se, but to N-ethoxy-
by direct inlet MS analysis on the reaction
tentatively identified as N-chloromethyl-N’-
(~-AII-N=CH~)~
le
I I
0 N-MI +
I I p-An/NvN\p-An
It IS possible
attack of 3
p-in p-in I!?
15
that for ahphatic 1 to react, decomposition must be induced by direct

10612 G Wmumoetal
Scheme 1
Ar
Route A I N
f‘l
Ar HNvNIAr
la-k \ +5
Ar
b 1 8
b Ar-N&Ii2 + f
2 Ar ON% 2
/- 3 +5
0 0
73
ArMN \ c1 cH,Cl
Ar -N
+ IztOH
0 / -EtOCH,CI
0 +EtOH *
Ar NNACH cH~cl
-EtOCH,Cl
I 2 Cl
Ar f
Cc1,Cl
lo
Ar xNAN'--CH Cl
+f
2
Cl 4 x
0
0 2
N-Ar
14
-CH,Ci,
-
6a-k
0 0
PC + 9
ci -Cl Route B
s
Heterocycles from hctcxocycles 10613
No systematrc work was carrzed out to optrnuxe the yield of 6 in the reactins
between &g and _5, but a few useful expenmental observatrons were made m adlustrnq
the condltrons for the reactin of la, which were essentilly apphed to the other
substrates. Good solvents for 1, hke droxane and chloroform, were detrrmental; the best
results were obtarned when 1 was added to 5. The use of anhydrous solvents and
absolute ethanol gave better yields
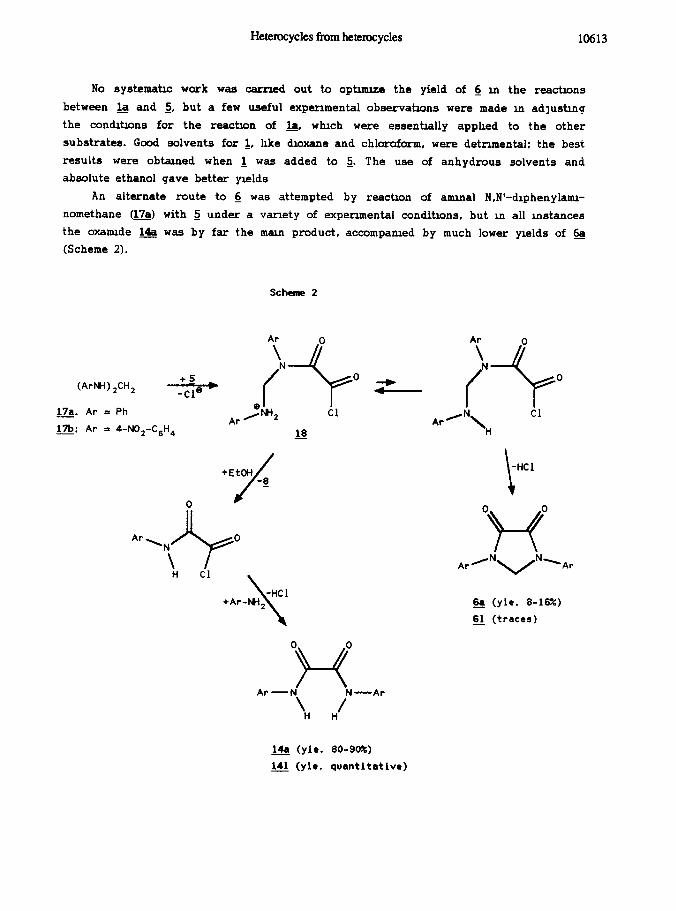
An alternate route to 6 was attempted by reactin of amrnal N,N’-drphenylamr-
nomethane (17a) with 2 under a variety of expenmental conditrons, but m all instances
the oxamrde & was by far the matn product, accompanred by much lower yrelds of &
(Scheme 2).
Schsme 2
(ArW ,CH,
m. Ar = Ph
m: Ar = 4-N02-C6H4
+EtOH
/
-a
0 0 0
7-f ArRNvN-Ar
& (yie. 8-16x)
a (traces)
0 0
Y-3 Ar-N N-Ar
‘H H’
&& (yie. 80-90X;)
141 (yic. quantitative)
10614 G VERARDO et al
In the above scheme the cause for the low yields is to be found m the productin
of acltity which tres up the mtermetite @) ln an unreactive form. In fact, we found
that practically all of & was formed m this reactin before alcohol ad&tin.
All the products 6a_k showed msolubllity u most common solvents and excepbnally
high melting pcunts which are also lndlcatlve of thw extraordinary thermal stablhty
The steadfastness of the crystal structure points to strong intermolecular polar
Interactins. m fact, the isolated molecules present two tides of opposite polanties
Ar
In view of the novelty of the molecules and these propeties of the sohds, we
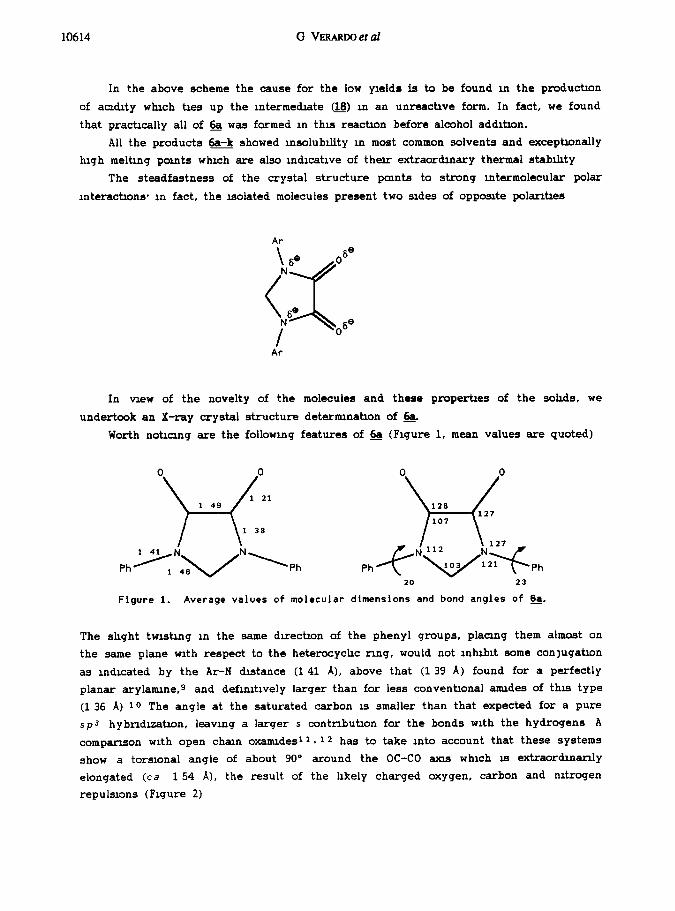
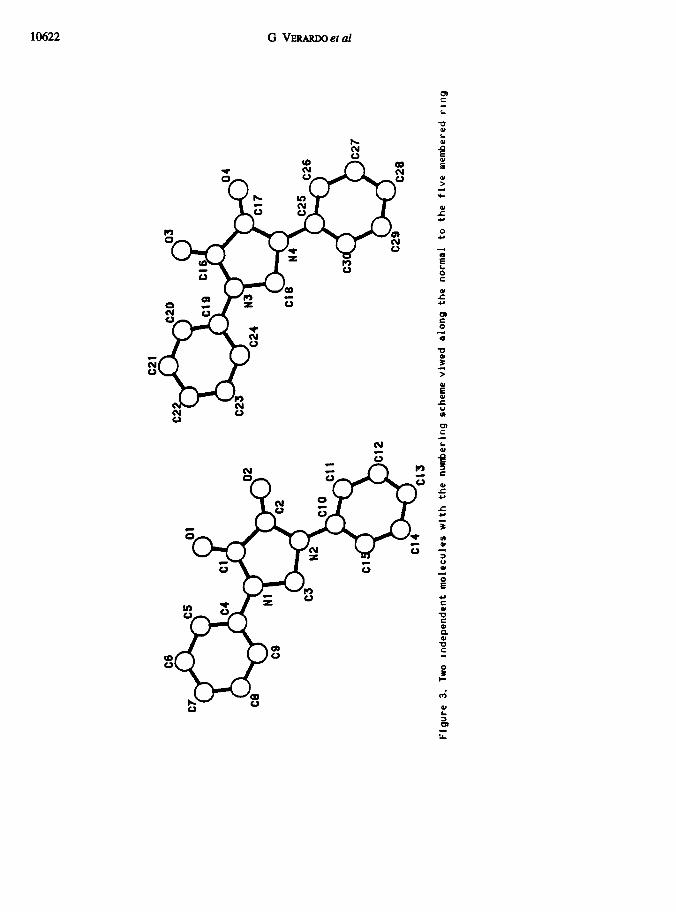
undertook an X-ray crystal structure deternunatin of &.
Worth notrcrng are the following features of & (Figure 1, mean values are quoted)
Ph
0 0
126 H 117 107
,-c_"+::zyph
20 23
Figure 1. Average values of molecular dimensions and bond angles of &.
The shght twisting m the same dvectron of the phenyl groups, placzng them almost on
the same plane with respect to the heterocychc ring, would not lnhlblt some conlugatin
as indicated by the Ar-N distance (141 A), above that (1 39 A) found for a perfectly
planar arylarmne,g and definitively larger than for less conventinal armdes of this type
(1 36 A) 10 The angle at the saturated carbon EI smaller than that expected for a pure
sp3 hybnduatin, leaving a larger s contnbutlon for the bonds with the hydrogens A
comparison with open ch;lm oxarmdes 11.12 has to take into account that these systems
show a torsional angle of about 90° around the OC-CO anus which 15 extraordmanly
elongated (ca 154 A), the result of the hkely charged oxygen, carbon and nitrogen
repulsions (Figure 2)

Hetemcycles from heterocycles 10615
The only possible response of & is the widening
value of 126’ agatnst 117O for the open charn case which
distance As a consequence the nng NCC bond angles are
107’ from 117’ observed in the open chsun cases.
6’ N x OS9
6’ N 06’ II
of the OCC bond angles to a
would cause a quite short O-O
squeezed to a meager value of
III
Figure 2. Conformations of open oxamides
Planar nng enclosed oxanudes, hke 6+ can only exist in the high energy form II,
where hkely charges are the closest. Different stenc requirements UI open oxanndes may
allow for perpendicular geometnes III more or less approaching the ideal lowest
electrostatic energy configuration I Entropic factors play a role in the equilibrium
positions.
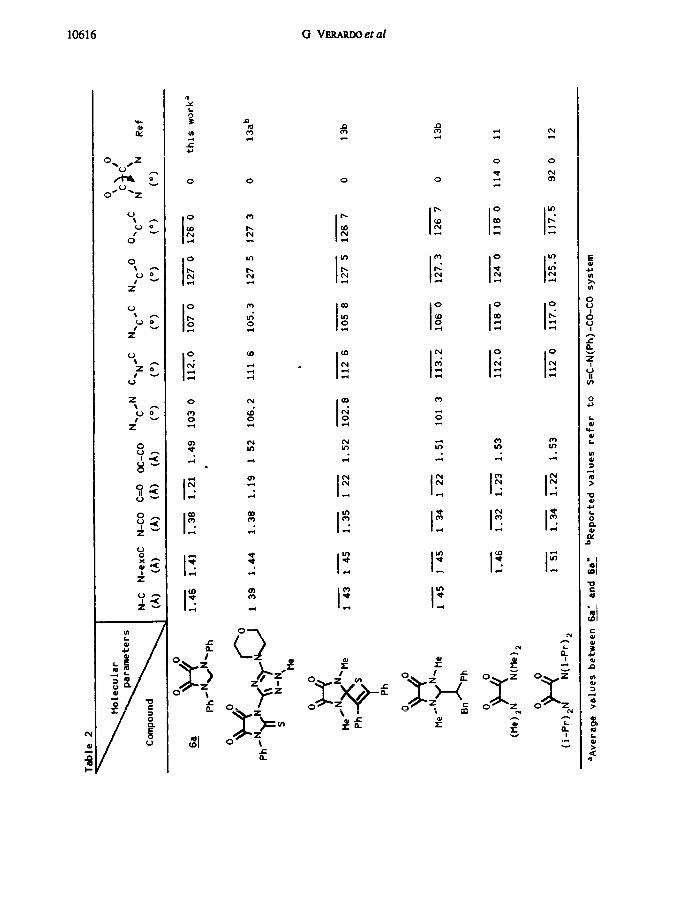
Literature structural data for the heterocycbc system present m & are few and
some were collected for more substituted denvatives (Table 2) Noteworthy is the
contraction by cd. 0 04 A of the C,-C, bond (Figure 3 and Table-a) found 19 @ in
comparison with all other s~~1a.r cases 13 Surnlarly the CC0 angle is strongly widened
by the oxygen repulsion to a value of 126“ in all these systems as well as in &. The
N-CO bond of &a is some 0 02 A larger than in formamide and ca. 0.04 A larger than
that of the 1,2,3-tnsubstituted 1,3-rnndazohdme-4.5dlones mvestigated,lz differences
which are too small to evince any special conclusion. The practrcally mvanant C=O
titance, actually coincident with that of formaldehyde, * 6 though, point to an imperfect
NC0 amide conlugation. Oddly enough, the N-atom seems to be more conlugated with the
ring. But, as we shall see, the 1 H NMR data will introduce a contradiction here
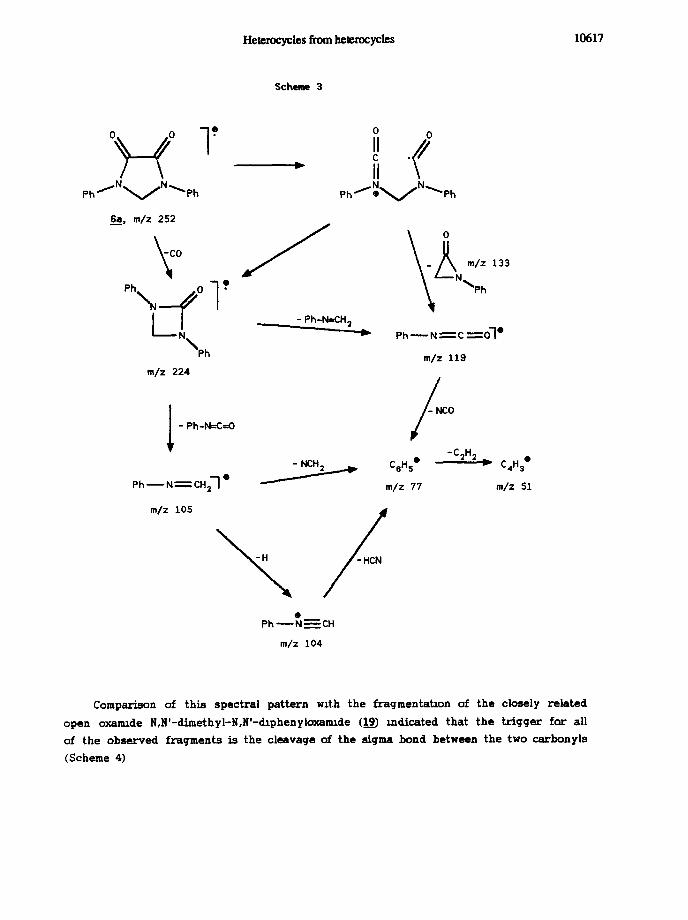
The nature of the obtained products 6 was confirmed by elemental analysis and
their MS and spectroscopic properties. Mass spectra of 6a_k exhibited parent ions of
medium intensity with the exception of the low intensity observed for the
chloro-derivatives &i and 6-J. The common features of the ion decompositions are shown
in scheme 3, which specifically refers to the ion derived from @; the composition of the
ion at m/z 105 was secured by high resolution mass spectrometry whch ruled out the
presence of PhCOe
The peak formally ascribed to the N-methylenearylamine radical cation was
consistently the base peak in all spectra of 6a_k. Of interest is the formal loss of an
azindone (133 mu) from the parent ion of Q to produce what is likely the molecular ion
of an aryhsocyanate molecule (Table 3)
N-C
N
-exo
C
N-C
O C
=O
CC
-CO
N. C
.N
C.
cc
N
N.
.C
C
(A)
(A)
(A)
(A)
(“)
(“)
(“)
co)
(“)
(“)
Ref
0 0
!?!I
9-t
1.4(
i 1.4
1
IPh’
N”N
NPh
’ 3!
3 1-
44
Me
” N
N
NM
e
c Ph
- \
S 14
3 14
5
10
0
W
Me .
-N
N’t
le
x E
)n
Ph
175
145
0 0
Y-t
(M
e) ,N
N
W)
z
1.46
0 0
Y-4
(i-
Pr)
jlN
N(i
-Pr)
, 11
1.38
L
.21
1.49
10
3 0
.
1.38
1.
19
1 52
10
6.2
111
6
1.35
12
2 1.
52
102.
8 11
2 6
105
8
113.
2 10
6 0
118
0
117.
0
127
126
127
5 12
7 3
0 13
ab
124
118
0
0 th
is
wor
k=
0 13
b
13b
‘Ave
rage
va
lues
be
twee
n 6a
’ an
d x
bRep
orte
d va
lues
re
fer
to
S=C
-N(P
h)-C
O-C
O
syst
em
Hctc~les from heterocycles 10617
6a, m/z 252
m/z 224
I - Ph-N=C=O
Ph-N- -CH21e
m/z 105
- Ph-N&l,
*
\
0
P m/z 133
NxPh
Ph-N=C- -01.
m/z 119
/
-NC0
v? -Cd+, * we m/z 77 m/z 51
9 Ph-N=CH
m/z 104
Comparison of this spectral pattern with the fragmentataon of the closely related
open oxarmde N,N’-dimethyl-N,N’-lphenyloxarmde (19) indicated that the trigger for all
of the observed fragments is the cleavage of the sigma bond between the two carbonyls
(Scheme 4)
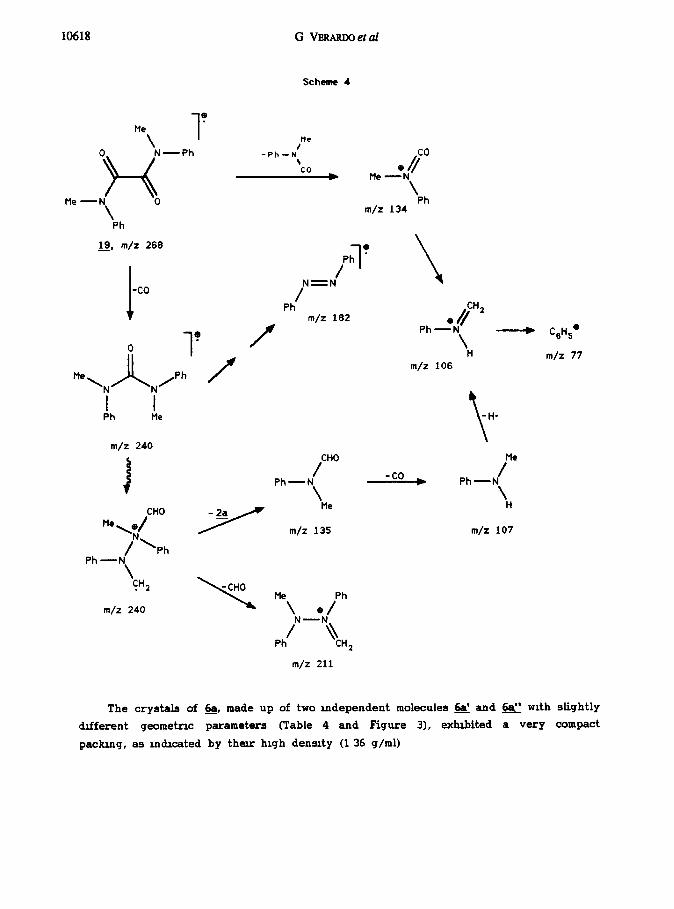

Table 3 Propertles of 1,3-Dlaryl-4,5-imldazolidinediones (5)
Compound
IRa (cm-l)
6a
1725~8, 159Os, 1490vs,
14556, 1405V8, 12908,
1275s, 745~s. 680s
!&
1715~8, 14858, 1400~8,
1290s. 118Om, 79010,
685111
Sr
1735~8, 16108, 151Os,
14OOs, 13008, 128Os,
815s
2930m, 1725vs, 15lOm,
1430m, 13858, 1300m,
1270m. 830s
1740~8, 1500vs, 14606,
1410~9, 13108, 12708,
1230s. 8108, 755~s
1730~8, 16108, 15908,
149ove, 14608, 14208,
14008, 11908, 770s
173OV8, 171OV8, 15006,
14009, 1240~11, 116Om,
1090m. 830s
1H NMRb (b , ppm; J, Hz)
5.66(s, 2H), 7.27-7.35(1n, 2H).
7.47-7.57(m, 4H), 7.93(d, 4H,
Js8.00)
2.43(s, 6H), 5.46(s, 2H), 7.10-
7.63(88, a)
2.36(s, 6H), 5.40(s, 2H), 7.27
(d, 4H Jx9.00). 7.65(d, 4H,
Jl9.00)
1.33(s, 18H). 5.44(s, ZH), 7.48
(d, 4H, Jn9.00). 7.68(d, 4H,
J=9.00)
5.48(s, 2H). 7.19-7.45(m, 6H),
7.70-7.80(m,
2H)
5.49(s, 2H), 6.99-7.12(m, 2H),
7.40-7.55(m, 4X), 7.66-7,75(m,
2H)
5.47(s, 2H), 7.15-7.30(m, 4H),
7.70-7.85(m, 4H)
MS
C
(m/z
, rel%)
252(M+, 23), 119(6), 106(7),
105(100), 77(38), 64(4), 51
(16)
280(M+, 69). 133(13), 120
(22), 119(100), 118(57), 104
(6), 91(60), 77(8), 65(21),
53(6)
280(M+, 60), 133(21), 120
(22), 119(100), 104(7), 91
(70)) 77(8), 65(23), 51(8)
364(M+, 32). 349(42), 175
(9), 167(20), 161(47), 160
(80), 146(100), 132(18),
118(19), 106(6), 91(7), 77
(7), 44(21)
288(M', 59), 137(11), 124
(17), 123(100), 122(85),
109(7), 95(38), 75(18),
57(5)
288(M+, 31), 137(6), 123
(loo), 122(45), 96(5), 95
(29), 75(10)
288(M+, 26), 137(g), 123
(loo), 122(47), 95(29), 75
(9), 57(3)
(continued)

Table 3. (continuation)
Compound
IRa (cm-l)
IH NMRb (6
t ppm;
J,
Hz)
si
1725~8, 1590s. 14708,
141Os, 1270m, 1105m.
5.49(s, 2H), 7.25-7.48(m, 4H),
870m, 775s. 670m
7.70-7.81(m, 4H)
MS
c (m/z, rel%)
324(M+, l), 322(M+, 8), 320
14), 155(l)
153(5)
%32),
139(1OOj 138(31j
111(21), 77(7), 75(12), Si
(7)
5i
1735vs, 14958, 1395vs,
1270m, 1090m, 825vs,
5.48(s, 2H), 7.47(d, 4H. J=9.00)
810111
7.72-7.80(d, 4H. J=9.00)
324(H+, 3), 322(M+, 17), 320
:!;i42), 139(1OOj
138(45j,
27). 155(5)
153(15)
P
111(27), 77(6), 7&15)
6k
1725vs, 15808, 1470~8,
1415s, 14oovs, 13056,
5.50(s, 2H), 7.32-7.47(m, 4H),
7.75-7.93(m, 4H)(H+, lo), 199(5), 1::;:::' 9), 410(M+, 21), 408
12608, 1090s, 760vs,
665s
185(99), 183(100),157(19),
W;l9),
90(13), 77(26), 51
ft
aSpectra were recorded in KBr. CSpectra recorded in CDC13 eolution using TM
as Internal standard.
Upectra recorded via direct inlet.

Heterocycles from heterocycles 10621
Table 4. Most Significant Gecmetrlcal Characteristics of the Refined Molecules
O(1) - C(1) O(2) - C(2) O(3) - C(16) O(4) - C(17) N(1) - C(1) N(1) - C(4) N(1) - C(3) N(2) - C(2) N(2) - C(3)
C(4) - N(1) - C(3) C(1) - N(1) - C(3) C(1) - N(1) - C(4) C(3) - N(2) - C(10) C(2) - N(2) - C(10) C(2) - N(2) - C(3) C(18) - N(3) - C(19) C(l6) - N(3) - C(19) C(16) - N(3) - C(18) C(18) - N(4) - C(17) C(25) - N(4) - C(17) C(25) - N(4) - C(18) O(3) - C(16) - N(3) N(3) - C(18) - C(17) O(3) - C(16) - C(17) N(4) - C(25) - C(30) N(4) - C(26) - C(26) O(1) - C(l)- - N(i) N(1) - C(1) - C(2) O(l) - C(1) - C(2) N(2) - C(2) - C(1)
Bond Lengths (A)
1.203(7) N(2) - C(10) 1 396(g) 1.216(7) N(3) - C(16) 1.375(8) 1 222(7) N(3) - C(18) 1 458(7) 1 205(8) N(3) - C(19) 1 409(8) 1.377(8) N(4) - C(25) 1 424(8) 1 422(8) N(4) - C(18) 1 452(7) l-458(7) N(4) - C(17) 1 380(8) 1.379(8) C(16) - C(17) 1.502(9) 1.467(8) C(1) - C(2) 1.485(8)
Bond Angles (“)
119.8(S) 111 7(S) 128.6(S) 120.8(S) 127.1(S) 112.1(S) 121.1(S) 127.3(S) 111.6(S) 112.9(S) 126.8(S) 120.3(S) 127.5(S) 107.3(S) 125.2(6) 119.9(6) 120.4(6) 126.5(6) 107 4(S) 126.2(6) 106.2(S)
O(2) - C(2) - C(1) 126.3(6) O(2) - C(2) - N(2) 127 S(5) N(3) - C(l8) - N(4) 102 7(4) N(4) - C(17) - C(16) 105.2(S) O(4) - C(17) - C(16) 126.2(8) O(4) - C(17) - N(4) 128.6(6) N(l) - C(4) - C(9) 121.3(6) N(1) - C(4) - C(5) 117.9(5) N(l) - C(3) - N(2) 102.6(4) N(3) - C(19) - C(24) 119.1(S) N(3) - C(19) - C(20) 121.1(S) N(2) - C(10) - C(15) 120.1(6) N(2) - C(10) - C(11) 122.2(6)
Selected Torsion Angles (")
C(1) - N(1) - C(4) - C(5) 21.0(9) C(3) - N(2) - C(10) - C(15) -24.0(9) C(l8) - N(3) - C(19) - C(24) 19.3(8) C(l8) - N(4) - C(25) - C(30) -22.0(8)
The molecules are stacked m the cryatala like parallel columns; the columns,
contactig each other by a shght overlapping of the me ta-Poe&on of a phenyl nng and
the pars-posrtron of a phenyl nng of the adjacent column, are made up of piles of
alternatig molecules &ia/ and K, where the heterocychc nng of one 19 almoat parallel
to the second phenyl nng of the other. The relative posrtrons of theae molecular
sectxnns are mdrcatrve of some electrostatrc interactin between the negatively charged
oxygens and the electron lmpovenshed phenyl nng.
1 H NMR spectra of 6 III CDCl, showed the expected pattern with a sharp singlet for
the methylene group locahzed at 6 values between 5.40 and 5.66 ppm from the standard
tetramethylsllane (Table 3). Whereas the sryl protona appeared I.II three well separated
regrons, para-substituted 5 showed two types of resonances: all of them were located at
between 7.10 and 8.00 ppm (Table 3), posrtions definitively at lower field than for
aruhne,l7 N,N_drmethylanrhne18 and N-methyl-N-formylanAnel e and the correapondlng
denvatlvea These data are an mdlcatron of a somewhat strong electron depletxon away
G VERARLW et al
or __

Heterocycles from hetemycles
from the aromatic nng into the amrde fun&on.
An lndrrect confrrmatron of the poslbve polandion of thr aromatic rugs came
10623
from
the packing of the molecules &’ and +&’ m the crystals with the oxygens of one lust
on top of the carbons of phenyl ring, two such Interaction bag active for every single
molecule with shghtly different posltiolllngs This appears to be due to the strong polar
intermolecular attractron playmg an important part m holtig the molecules together 111
the crystals.
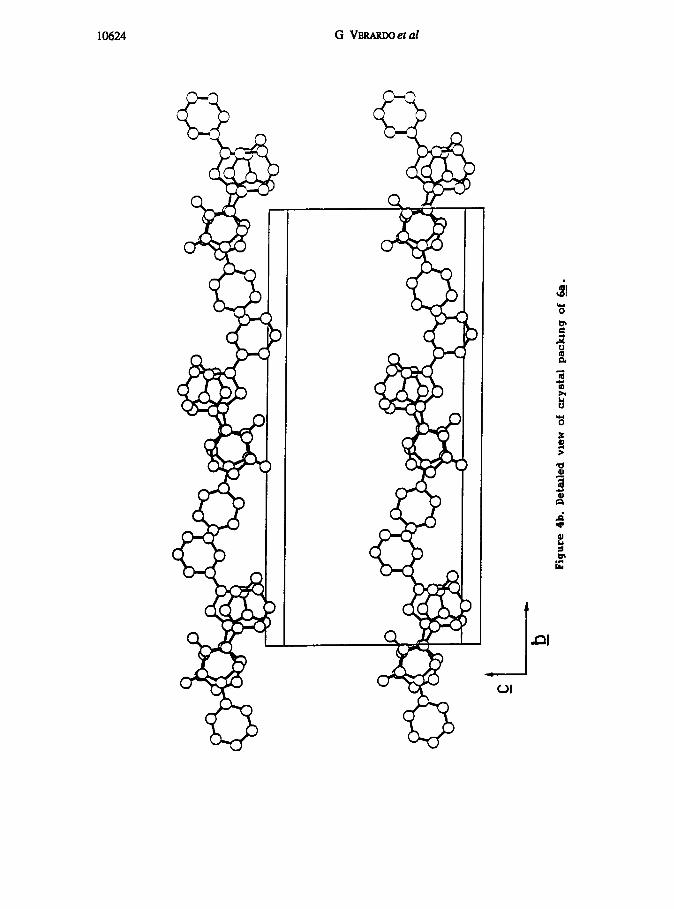
A full view of the crystal packrng rs offered by figure 4a and 4b, where a side by
side columnar paclung of the stacked molecules, with any two columns barely “touchmg”
with the free phenyls, 15 evidenced. The closest distance between the phenyl nng of
one molecule 6a’ and the related heterocyclic nng of the other (6a”) was found to be -
3 6 A, well beyond any charge transfer Interactin.
Figure 4a. General view of crystal packing of &.
The huge hypsochrormc shift of the carbonyl stretching frequency (1725 cm-l) of
& and the range 1715-1740 cm- 1 for 6b_k (Table 3) compared with the pratrcally
cmncrdental values for N-methylacetamrde (20) (1656 cm-l) and N,N’-lmethyl-N,N’-
-drphenyloxamzde Clg) (1650-1665, doublet) rs due to the combined effect of enhanced
carbonyl character and, therefore lesser delocalizatin of electrons from the nrtrogen
atom, and nng strm 20
Thrs may be simply a descreenlng effect by the nitrogen lone pans or an
overwhelming field electron wlthdrawlng o-effect. Thrs effect overlaps with some much
less effectrve u-electron transfer from the nrtrogen to the nng.
10624 G VMARDO~~ al
b
. ,
. 9

Heterocycles from heterocycles 10625
EXPERIMENTAL SECTION
llaterials. Oxalyl chlonde (3) and pnmary aromatic amines (2) were commercially
available (Aldrich, Milano, Italy). they were conveniently purified before use and used to
prepare the 1,3,5-triarylhexahydro-1,3,5-tnananes (1) accord.ing to the amine
paraformaldehyde method.1’ Diarylarmnomethanes (17) were prepared according to a
described procedure.1 b Dry solvents were obtained following standard procedures.2 *
TLC plates (neutral alumina on alumuuum plates) were obtied from Merk, Italy.
Equ ~pment. lhgh pressure liquid chromatography analyses were performed with a
Waters Milhpore instrument, equipped with an inverse phase C,, Bondapak column
(lengh 30 cm, 1.d 3.9 mm) and a &ted wavelength (240 nm) uv detector. The system
uses two independent pumps and a processing unit enabling eluent composition control.
Water-acetonitrile mixtures were found su&able for our analysea operating at a flow rate
of cd. 1 ml/min.
Infrared spectra were recorded with a Jasco Mod. DS-702G spectrophotometer by
the KBr pellet technique
Electron impact (70 eV) mass spectra were obtsnned from a Finnegan MAT 1020 with
automatic continuous data recording. During direct inlet vaponxataon of the whole
sample rnto the ion source, the full recordmg was carefully inspected ~II order to detect
any side product and check sample punty. Headspace analyses were performed by
injecting the vapours over the solutions kept in inert atmosphere into a
gaschromatograph pnor to the electron impact with continuous ms mo&onng of the
eluate for the detection of gaseous products. The most intense peaks with therr relative
intensrty (%) are reported for each product.
ill NMR data were secured from a Bruker Mod. AC-F 200 spectrometer using
tetramethylsllane as internal standard. The high insolubility of 6a_k presented a
practical difficulty in recording of the 1 3 C NMR spectra.
Elemental analyses were obtained with a Carlo Frba Mod. 1106 elemental analyzer
for all isolated compounds and were satifactory.
X-ray diffraction analyses were obtatned fmm a crystal of @ cd. 0.2 x 0.2 x 0.5
mm that was mounted on a CAD4 single crystal drffractometer with graphite
monochmmatrzed MO Ka radiation, 25 reflections with 9 in range 10 s s s 16’ used for
measuring lattace constants (Table 5).
For data collection 3 5 s 5 26O (-8 J; h s 8, 0 5 k s 32, 0 s 1 5 14). o - 29
scans, o-scan width (0.80 + 0.35 tana); intensities of three reflections monitored every
2h of exposure time showed no slgruficant variation 4537 unique reflections were
collected; 1095 with I 2 3s(I). The structure was solved with MULTAN 8022 m default
setting and refined with SEELX 76.2 3 At convergence R = 0.059 for 1095 observed data.
Hydrogen atoms were located at calculated positions. Atomrc scattering factors were

10626 G. VERARDO et al
taken from Cromer & Mann.24
Table 5. Crystal Data and Experimental Details
Formula
M W.
Space Group
a/A
b/A
c/A
B/”
v/A3
z
D ..,,fg cmm3
~(Mo Ka)A
p/cm-’
F(OO0)
C15H1202N2
252.3
p2,/c
7.259(3)
27.466(2)
12.525(2)
99.23(3)
2464.6(6)
8
1.36
0.71069
0.86
1024
Cryst. size /mn
0 range /”
h range
k range
1 range
scan mode
Measd. Reflections
Solution of Structure
Ref lnement
Final R factor
Final R, factor
Room temperature
0.2 x 0 2 x 0.5
3.26
-8.8
0.32
0 14
0 - 28
2402
MuLTAN80
SHELX76
0.058
0.059
General Procedure for the Preparation of ga_k. A suspension of the
appropriate 1,3,5-trxarylhexahydro-1,3,5-triazine (l, 10 mmol) III anhydrous ether (ca. 50
ml) was slowly added to neat oxalyl chlonde (5, 30 mmol) kept at O°C under efficient
stxnng in an atmosphere of Argon. About 10 nunutes after the end of the addltin
anhydrous ethanol (30 ml) EI added at O°C slowly, wUe hydrogen chltide 19 evolved
and a sohd separates. The preupltatin is completed at rcom temperature by addition of
ether. The preupltate, separated by filtratin, IZI recrystallized from a sortable solvent.
Headspace analyw was performed m the case of the syntheu of &, by sampling the
atmosphere over the reactron mxture after the additin of ethanol and analysrng It by
GC-MS. The procedure with inverted order of addition of the ~~taal reagents or not
using ethanol to end the process yielded products 6 m substantially lower yields
The above procedure IS IJI part the result of the study of the variatxon of a
number of reactin parameters, when & was used as a substrate (Table 6)
Reaction between N,N’-Diarylaminomethane (l7) and 2. The react&on was
carned out according to the optamal procedure described for the preparation of 6. When
G was the substrate only ca 10% yield of & was obhned; N,N’-dlphenyloxalanude
(&) bexng the other observed product.

Hetcrocyclesfromhetcxocycles 10627
A slrmlar result was obtained when N,N'-&(4-r&.rophenyl)armnomethane (E) was
used, but the cyclic product &I was not present at all.
Table 6
mol z/m01 &
6
3
12
Reaction
time
10 min
10 min
10 min
Quenching
reagent’
Et,Ob
EtDH
EtQH
Reaction
solvent=
-_
__
-_
Yield
(W
5oc
40o
2Sd
1 10 min EtCtl Et,0 45
3 10 min EtDH Et,0 75=
6 10 min EtDH Et,0 58
3 10 min EtDH Dioxane mixe
3 10 mln EtDH CHCl s mixP
6 16 hours EtCH Hexane 65
‘Anhydrous materials were used. bComnercial diethyl ether was used as received ‘DI-MS analysis of mother liquors obtained after filtration of & showed the presence of N,N’-diphenyloxamlde [l&q MS (m/z): 240 (M@, 43), 121(30), 120(31). 105(12), 93(100), 92(22). 77(58)] dDI-MS analysis of mother liquours obtained after filtration of & showed the presence of 14a and bis(N-ethoxyoxalyl-N-phcnyl)diaminomethane [MS (m/e)* 398(<1), 325(18). 252(4). 206(28), 176(100). 134(6). 120(10), 106(83). 93(34). 77(38)]. ‘A very complex mixture was obtained which was not worked up.
Acknowledgements This work was supported in part by grants to AGG (CNR
86.01649.03, CNR 8903765.03, MPI 1987-1989 40% and 60%) and to GV (MPI 1987 60% and
1990 40%). The autors are grateful to Mr. P. Padovam for expert instrumental
mntenance
REFERENCES
(a) Glumamm, A G , Verardo, G, Randacuo, L., Bresciaru-Pahor, N.; Traldr, P. J
Prakt Chem 1985, 327, 739-748. (b) Glumanini, A G., Verardo, G.; Zangrando, E.;
Lasslam, L. J. Prakt Ches 1987, 329, 1087-1103.
(a) Drstefano, G, GUXM~.UU, A. G., Modelh, A.; Poggr, G. J Chea sot. Perkin
Trans 2 1985, 1623-1627. (b) GUXMIUIU, A. G., Verardo, G ; Poiana, M. J Prakt
Chem 1988, 330, 161-174.
Bertoluzza, A.: Bonora, S., Fim, G., J Prakt Chem 1990, 332, 595604.
(a) Verardo, G., Giumanml, A. G.; Cauci, S Actd Chim Hung. 1986, 122, 3-5. (b)

10628 G VERARDO et al
Giumanm A. G.; Verardo, G.; Caucz, S. J. Prakt. Chest 1387, 329, 417-432.
5 &=I=, R., Hllghetag, G.: Mti, A.; NeJedly, 0.; Schlegel, J. Arch. Pharm. 1960,
293, 957-967
6. (a) Ulnch, H.: Richter. R.; Wlutman, P J.; Saylgh, A. A. R. J Org Chem. 1974, 39,
2897-2899. (b) Gronowltz, S.; Lldert, 2. Syn thesl s 1979, 810-814. (c) Rathgeb, P.;
Tournayre, J. C., Vogel, C. GP 2306274/1972 (Chem Abs tr. 1973, 79, 115359x).
7 Hanafin, J; Ben-Ishsu, D. J. Heterocycl Chem. 1976, 13, 889-890.
8. Isaksson, R.: ~l~efors, T., Sandstrom, J J Chem Res. Synop 1981, 43.
9. Giumaxunl. A G., Randacclo, L.; Zangrando, E.; Ga, M. Ii. J. Prak t Chem. 1987,
329, 187-194.
10. Allen, F H.; Kennard, 0 ; Watson, D. G., Brammer, L.; Orpen, A. G., Taylor, R. J
Chest. Sot. Per&in Trans. 2 1987, 1-19.
11. Toda, F.; Tagarm, Y., Mak, T. C. W. Bull. Chem Sot. Jpn. 1977, 59, 1189-1194.
12. A&w.tdlala, G.; Voss, J. Chem. Ber 1977, 110, 1159-1166.
13. (a) Ried, W.; Broft, G. W.; Bats, J. W. Chem Ber. 1983, 116, 1547-1563. (b)
Gotthardt, V H.; Huss, 0. M. Acts Cryst. sect. 8 1982, 38, 875-879.
14. Lide, D. R. Tetrahedron 1962, 17, 229-233.
15. Fryer, R. I.; Earley, J. V.; Blount, J. F. J Org. Chem. 1977, 42, 2212-2219.
16 Kwa, G. II.; Curl, R. F. J. Chem, Phys. 1960, 32, 1592-1594.
17. Pouchert, J. C.; Campbell, J. R. The Aldrich Library of NtlR Spectra, Aldnch
Chesucal Company Inc., Milwaukee, Wisconsm, 1974, V, 37A.
18. Pouchert. J. C.; Campbell, J. R. The Aldrich Library of NMR Spectra, Al&h
Chesucal Company Inc., Milwaukee, Wisconlun, 1974, Y, 40C.
19. Pouchert, J. C.; Campbell, J. R. The Aldrfch Library of NUR Spectra, Aldrich
Chemical Company Inc., Milwaukee, Wisconsin, 1974, VI I, 65C.
20. Zahn, H.; Kunde, J. Chen. Ber. 1961, 94, 2470-2475.
21. Perrin, D. D.; Armarego, W. L. F.; Pernn, D. R. Purification of Laboratory
Chemicals, A. Wheaton 61 Co. Ltd., Exeter, Great Britaxn, 1980, 2nd Ed..
22. Main, P.; Fiske, S. J.; Hull, S. E.; Lesssnger, L.; Germ, G.; Declercq J. P.;
Woolfson, M. M. MIJLTANIO, A System of Computer Programs for the
Automatic Solution of Crystal Structures from X-ray Diffraction Data,
Unrversrty of York, England, 1980.
23. Sheldrick, G. M. SHELX-7s. Program for Crystal Structure Determination,
Unlvcrslty of Cambndge, England, 1976.
24. Cromer, D. T.; Mann, J. B. Acta Cryst. se c t B 1968, A24, 321-322.






























