UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE CENTRO DE CIÊNCIAS EXATAS E DA TERRA PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIA E ENGENHARIA DE MATERIAIS TESE DE DOUTORADO Síntese de catalisadores do tipo LaNi x Fe 1-x O 3 como precursores catalíticos para reação oxidação parcial do metano Daniele de Macedo Henrique Martinelli Orientadora: Prof a . Dr a . Dulce Maria de Araújo Melo Tese N.º 90/PPGCEM Natal 2011

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE
CENTRO DE CIÊNCIAS EXATAS E DA TERRA PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIA E ENGENHARIA D E
MATERIAIS
TESE DE DOUTORADO
Síntese de catalisadores do tipo LaNixFe1-xO3 como precursores
catalíticos para reação oxidação parcial do metano
Daniele de Macedo Henrique Martinelli
Orientadora: Prof a. Dra. Dulce Maria de Araújo Melo
Tese N.º 90/PPGCEM
Natal
2011

DANIELE DE MACEDO HENRIQUE MARTINELLI
Síntese de catalisadores do tipo LaNixFe1-xO3 como precursores
catalíticos para reação oxidação parcial do metano
Tese apresentada ao Programa de Pós-graduação em Ciência e Engenharia de Materiais da Universidade Federal do Rio Grande do Norte como parte dos requisitos para a obtenção do título de doutora em Ciência e Engenharia de Materiais.
Orientadora: Profa. Dra. Dulce Maria de Araújo Melo
Fevereiro de 2011
Natal - RN

Seção de Informação e Referência
Catalogação da Publicação na Fonte. UFRN / Biblioteca Central Zila Mamede
Martinelli, Daniele de Macedo Henrique. Síntese de catalisadores do tipo LaNixFe1-xO3 como precursores
catalíticos para reação oxidação parcial do metano / Daniele de Macedo Henrique Martinelli. – Natal, RN, 2011.
113 f. : il.
Orientadora: Dulce Maria de Araújo Melo.
Tese (Doutorado) – Universidade Federal do Rio Grande do Norte. Centro de Tecnologia. Programa de Pós-Graduação em Ciência e Engenharia de Materiais.
1. Pechini – Tese. 2. Microondas – Tese. 3. LaNixFe1-xO3 – Tese. 4. Oxidação parcial do
metano – Tese. I. Melo, Dulce Maria de Araújo. II. Universidade Federal do Rio Grande do Norte. III. Título.


À Professora Dulce Maria de Araújo Melo, pela sua orientação, por toda
ajuda, não só profissional como pessoal, pela confiança que sempre teve em mim,
pela compreensão, e especialmente, pela sua grande amizade.

AGRADECIMENTOS
A Deus por sempre olhar para mim.
A minha filha por ter me dado razão para viver e procurar ser sempre feliz
A meu esposo Martinelli por todo amor e paciência
A minha família, pelo amor, carinho, compreensão, incentivo e
ensinamentos.
A ANP – PRH-30 pelo apoio financeiro.
Ao programa de pós-graduação em ciência e Engenharia de materiais.
Aos amigos de laboratório: Patrícia Pimentel, Júlio, Bruno, Priscilla,
Kellyanny, Auristela, Renata, Danilo, Diego, Gabi, Maria Luiza, Rodrigo
Melo pela ajuda direta ou indireta em meu trabalho, pela amizade, pelos
momentos de luta e descontração que tivemos durante estes anos.
Agradecimento especial: À João Ribeiro pela grande ajuda durante toda a
realização deste trabalho, principalmente durante a minha gravidez; À
Rodrigo Santiago pelas análises de TG e disponibilidade de tempo,
mesmo sem tê-lo e principalmente à Felipe Oliveira, pela imensa ajuda
nesta reta final do meu trabalho.
Ao CTGAS-ER pelas análises de TPR e pelas reações de oxidação parcial
do metano, em especial a Cláudio e Juan, por toda ajuda
A minha avó por ser tão especial.
A minha mãe por tudo o que sou.

RESUMO
Catalisadores a base de níquel têm sido empregados em diversos tipos de reações de reforma, inclusive na oxidação parcial do metano para obtenção de H2 ou gás de síntese (H2 + CO). Normalmente, altos níveis de conversão são obtidos por estes catalisadores, entretanto, a desativação por deposição de carbono ainda é um problema a ser solucionado. Diversas abordagens têm sido empregadas no intuito de minimizar este problema, dentre as quais tem se destacado nos últimos anos a utilização de óxidos com estrutura perovisquita e/ou estruturas relacionadas. Paralelamente, o uso de metodologias de sínteses mais rápidas, fáceis, aplicáveis em escala industrial e que permitam o controle das características microestruturais destes catalisadores, pode em conjunto, prover a solução para este problema. A nível de comparação perovisquitas do tipo LaNixFe1-xO3 (x=0, x=0,3 e x=0,7) foram preparados por dois métodos: precursores poliméricos (pechini) e autocombustão assistida por microondas. Todas as amostras foram calcinadas a 900 °C/4h para obtenção das fases desejadas. Os pós-obtidos foram caracterizados por análise termogravimétrica, espectroscopia na região do infravermelho, difração de raios-X, medidas de área superficial especifica, redução à temperatura programada. As amostras calcinadas foram testadas na reação de oxidação parcial do metano, sendo avaliados os respectivos níveis de conversão, seletividade e a resistência à deposição de carbono. Após calcinação a fase desejada foi obtida para todas as amostras independente do método de síntese, sugerindo claramente a formação de pós cristalinos. De acordo com o resultados obtidos pelo refinamento Rietveld, observou-se a formação de 70,0% de LaNi0,3Fe0,7O3 e 30,0 % de La2O3 para as amostras LN3F7-900-P e LN3F7-900-M e 41,6% de LaNi0.7Fe0.3O3; 30,7% de La2NiO4 e 27,7 % de La2O3 para as amostras LN7F3-900-P e LN7F3-900-M. Os perfis de redução à temperatura programada da amostra LaNiO3 apresentou pico de redução em torno de 510 °C, já a amostra LaFeO 3 apresentou pico de redução acima de 1000°C. Dentre os catalisadores estudados o que apresentou maior nível de conversão de metano foi LaNiO3 obtido pelo método pechini, de uma maneira geral os catalisadores obtidos pelo método pechini apresentaram melhores resultados de conversão que os catalisadores obtidos pela autocombustão assistida por microondas.
Palavras chaves: Pechini, microondas, LaNixFe1-xO3, oxidação parcial do metano

ABSTRACT
Nickel-bases catalysts have been used in several reform reactions, such as in the
partial oxidation of methane to obtain H2 or syngas (H2 + CO). High levels of
conversion are usually obtained using this family of catalysts, however, their
deactivation resulting from carbon deposition still remains a challenge. Different
approaches have been tested aiming at minimizing this difficulty, including the
production of perovskites and related structures using modern synthesis methods
capable of producing low cost materials with controlled microstructural characteristics
at industrial scale. To establish grounds for comparison, in the present study
LaNixFe1-xO3 (x=0, 0.3 or 0.7) perovskites were prepared following the Pechini
method and by microwave assisted self-combustion. All samples were sub sequently
calcined at 900 °C to obtain the target phase. The resulting ceramic powders were
characterized by thermogravimetric analysis, infrared spectroscopy, X ray diffraction,
specific area and temperature programmed reduction tests. Calcined samples were
also used in the partial oxidation reaction of methane to evaluate the level of
conversion, selectivity and carbon deposition. The results showed that the calcined
samples were crystalline and the target phase was formed regardless of the
synthesis method. According to results obtained by Rietveld refinement, we observed
the formation of 70.0% of LaNi0.3Fe0.7O3 and 30.0% of La2O3 for samples LN3F7-900-
P, LN3F7-900-M and 41,6% of LaNi0.7Fe0.3O3, 30.7% of La2NiO4 and 27.7% of La2O3
for samples LN7F3-900-P and LN7F3-900-M.Temperature-programmed profiles of
the LaNiO3 sample revealed the presence of a peak around 510 °C, whereas the
LaFeO3 sample depicted a peak above 1000°C. The highest l evel of methane
conversion was obtained for LaNiO3 synthesized by the Pechini method. Overall,
catalysts prepared by the Pechini method depicted better conversion levels
compared to those produced by microwave assisted self-combustion.
Key Words: Pechini, microwaves, LaNixFe1-xO3, partial oxidation of methane

LISTA DE FIGURAS
Figura 2.1 Duas diferentes apresentações para a estrutura cúbica ideal da Perovisquita...............................................................................................................17
Figura 2.2 – Distorções (a) ortorrômbica e (b) romboédrica da estrutura perovisquita...................................................................................................................................20
Figura 2.3 – Série de Ruddlesden-Popper. (a) Para n =1 e (b) Para n = 2. ..............29
Figura 2.4 – Reações envolvidas no método Pechini................................................35
Figura 2.5 – Infraestrutura do Hidrogênio para uso com fins energéticos. ................43
Figura 2.6 – Esquema de uma membrana cerâmica não porosa..............................54
Figura 3.1 - Esquema da linha reacional para oxidação parcial do metano. .............68
Figura 4.1– Curva TG do precursor catalítico LN3 – P..............................................73
Figura 4.2 – Curva TG do precursor catalítico LN3-M...............................................73
Figura 4.3 - Curva TG do precursor catalítico LF3-P.................................................74
Figura 4.4 - Curva TG do precursor catalítico LF3-M................................................75
Figura 4.5 - Curva TG do precursor catalítico LN7F3-P ............................................76
Figura 4.6 - Curva TG do precursor catalítico LN7F3-M ...........................................76
Figura 4.7 - Curva TG do precursor catalítico LN3F7-P ............................................77
Figura 4.8 - Curvas TG e DTG do precursor catalítico LN3F7 – M ...........................78
Figura 4.9 – Difratograma de raios-X para as amostras (a) LF9 – P e (b) LF9 – M .79
Figura 4.10 – Difratograma de raios-X para as amostras (a) LN9 – P e (b) LN9-M .80
Figura 4.11 – Difratograma de raios-X para as amostras (a) LN7F3-900-P e (b) LN7F3-900-M ............................................................................................................81
Figura 4.12 – Difratograma de raios-X para as amostras (a) LN3F7-900-P e (b) LN3F7-900-M ............................................................................................................82
Figura 4.13 - Espectro de absorção na região do IV das amostras LN3-P e LN3-M.84
Figura 4.14 - Espectro de absorção na região do IV das amostras LF3-P e LF3-M..85
Figura 4.15 - Espectro de absorção na região do IV das amostras LN7F3-P e LN7F3-M ...............................................................................................................................86
Figura 4.16 - Espectro de absorção na região do IV das amostras LN3F7-P e LN3F7-M ...............................................................................................................................87
Figura 4.17-Perfil de redução por temperatura programada para a amostra LN9-P .90
Figura 4.18-Perfil de redução por temperatura programada para a amostra LN9-M.90
Figura 4.19-Perfil de redução por temperatura programada para as amostras (a) LF9 – P e (b) LF9 – M ...............................................................................................91
Figura 4.20-Perfil de redução por temperatura programada para as amostras (a) LN7F3–900-P e (b) LN7F3–900-M............................................................................92
Figura 4.21-Perfil de redução por temperatura programada para as amostras (a) LN3F7–900-P e (b) LN3F7–900-M............................................................................93
Figura 4.22 – Conversão do metano e seletividade para CO e CO2 usando o catalisador LN9-P......................................................................................................95
Figura 4.23 – Conversão do metano e seletividade para CO e CO2 usando o catalisador LN9-M .....................................................................................................96
Figura 4.24 – Razão entre H2 e CO utilizando o catalisador LN9..............................97
Figura 4.25 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador LN7F3-900-P ..........................................................................................98
Figura 4.26 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador LN7F3-900-M..........................................................................................99

Figura 4.27 - Razão entre H2 e CO utilizando os catalisadores LN7F3 – 900 – M e LN7F3 – 900 – P. ......................................................................................................99
Figura 4.28 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador LN3F7 – 900 – P...................................................................................100
Figura 4.29 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador LN3F7 – 900-M .....................................................................................102
Figura 4.30- Razão entre H2 e CO utilizando os catalisadores LN3F7 – 900 – M e LN3F7 - 900-P.........................................................................................................102
Figura 4.31 - Conversão do metano e seletividade para CO e CO2 utilizando o catalisador LF9-P ....................................................................................................103

LISTA DE TABELAS
Tabela 3.1- Reagentes utilizados na síntese dos óxidos. .........................................59
Tabela 3.2 – Quantidade de moles utilizados para a preparação dos catalisadores LaNiO3 LaFeO3 LaNi0,3Fe0,7O3 LaNi0,7Fe0,3O3 ............................................................61
Tabela 3.3 - Catalisadores obtidos pelo método Pechini e códigos utilizados. .........62
Tabela 3.4 - Catalisadores obtidos pelo método microondas e códigos utilizados....63
Tabela 3.5- Condições operacionais do sistema utilizadas na reação de oxidação parcial do metano......................................................................................................69
Tabela 4.1 – Valores de cristalinidade das amostras estudadas...............................82
Tabela 4.2 - Resultados da área superficial específica para as perovisquitas LN9, LF9, LN7F3 e LN3F7 obtidas pelo método pechini e autocombustão assistida por microondas................................................................................................................88

SUMÁRIO
1 INTRODUÇÃO E OBJETIVOS .......................................................................12
2 REVISÃO BIBLIOGRÁFICA ...........................................................................16
2.1 Óxidos com estrutura perovisquita............................................................16
2.1.1 Análises de transições entre várias estruturas perovisquitas .............18
2.1.2 Papel dos cátions na posição A e B ...................................................21
2.1.3 Estruturas das perovisquitas não estequiométricas ...........................22
2.1.4 Composição superficial das perovisquitas..........................................24
2.1.5 Adsorção de oxigênio nas perovisquitas ............................................25
2.1.6 Estabilidade das perovisquitas em atmosfera redutora......................26
2.1.7 Perovisquitas em camadas ................................................................28
2.1.8 Emprego de Perovisquitas como Catalisador.....................................30
2.2 Métodos de síntese...................................................................................33
2.2.1 Método dos precursores poliméricos (Método Pechini)......................34
2.2.2 Combustão microondas......................................................................36
2.2.3 Método sol-gel....................................................................................40
2.2.4 Método da Co-precipitação ................................................................41
2.3 Produção de H2 para fins Energéticos ......................................................42
2.4 Conversão do Metano a Gás de Síntese ..................................................44
2.5 Reações para Obtenção de Gás de Síntese.............................................44
2.5.1 Reforma a Vapor do Metano ..............................................................44
2.5.2 Reforma a Seco do Metano................................................................47
2.5.3 Oxidação Parcial do Metano ..............................................................49
2.5.4 Reforma autotérmica..........................................................................53
2.5.5 Reforma com membrana....................................................................53
2.6 Deposição do carbono ..............................................................................55
3 PROCEDIMENTO EXPERIMENTAL..............................................................59
3.1 Reagentes utilizados.................................................................................59
3.2 Preparação dos catalisadores...................................................................59
3.3 Codificação das amostras preparadas......................................................62
3.4 Caracterização dos catalisadores .............................................................63
3.4.1 Análise Termogravimétrica (ATG) ......................................................63
3.4.2 Espectroscopia de absorção na região do infravermelho (IV) ............63
3.4.3 Difração de Raios-X (DRX) ................................................................64
3.4.4 Área superficial específica (AS) .........................................................65
3.4.5 Redução à temperatura programada (RTP) .......................................67
3.4.6 Ensaio Catalítico ................................................................................68
4 RESULTADOS E DISCUSSÕES ...................................................................72
4.1 Análise termogravimétrica.........................................................................72
4.2 Difração de Raios-X..................................................................................78
4.3 Espectroscopia de absorção na região do infravermelho .........................83
4.4 Área superficial específica ........................................................................88
4.5 Redução à temperatura programada (RTP)..............................................89
4.6 Reação de oxidação parcial do metano....................................................94
5 CONCLUSÕES ............................................................................................104

12
1 INTRODUÇÃO E OBJETIVOS
O metano é o principal componente das reservas mundiais de gás
natural e quase todas as opções de transformações deste hidrocarboneto, envolvem
sua conversão inicial ao gás de síntese (mistura de CO e H2). Atualmente, o uso
principal do gás de síntese é para fabricação de amônia, metanol, síntese de
hidrocarbonetos via processos de síntese de Fischer-Tropsch, uso de hidrogênio
puro para indústrias petroquímicas e refinarias, na redução de minérios de ferro e
também, para alimentação direta de células a combustível para geração de energia
elétrica alternativa [TRIMM, D. L.,1997; TRIMM, D. L., 1999]
O principal processo industrial de produção de gás de síntese é a
reforma catalítica a vapor do metano. No entanto, este processo é altamente
endotérmico, utiliza reatores em que somente 40 % da energia fornecida é
consumida pela reação e sofre algumas limitações, como baixa seletividade para CO
e alta razão H2/CO. Além disso, as condições reacionais necessárias levam à
desativação do catalisador por deposição de carbono na superfície do mesmo. A
viabilidade econômica de processos como a síntese de Fischer- Tropsch, que
utilizam o H2 e o CO como matéria-prima, depende de rotas mais eficientes para a
obtenção do gás de síntese [Kohei Urasaki, 2005]. Sendo assim, rotas alternativas
têm sido estudadas, tais como: oxidação parcial catalítica do metano, reforma do
metano com CO2 e reforma autotérmica.
Por vários anos, muitos esforços foram direcionados para o
desenvolvimento de catalisadores que mostrassem alta atividade e que
apresentassem resistência à formação de depósitos de carbono. Diversos
catalisadores metálicos foram estudados. Metais nobres do grupo VIII-A, com
exceção do ósmio, e não nobres como Cu, Ni e Co, depositados em diferentes tipos
de suporte, têm sido estudados como catalisadores para a reforma do metano com
CO2 [Bradford, M. e Vannice, M.; 1996].
Dentre estes catalisadores, os metais nobres mostraram-se melhores
quando comparados ao Ni, pois este último se desativa rapidamente devido à
deposição de carbono. No entanto, o alto custo dos metais nobres torna atrativo o
desenvolvimento de catalisadores de Ni para utilização industrial e um dos desafios
consiste em torná-lo mais resistente à deposição de carbono. [LIMA, S. M. e ASSAF,
J. M., 2007].

13
A adição de óxidos de metais alcalinos ou alcalinos terrosos e de
lantanídeos [CAO, L. e colaboradores, 1997], ou a utilização de espécies metálicas
altamente dispersas [BARTHOLOMEW, C. H.; 1982] sobre o suporte tem contribuído
para a redução da formação de carbono, aumentando a estabilidade catalítica. O
uso de precursores com estrutura tipo perovisquita, com fórmula geral ABO3, que
apresentam alta estabilidade térmica, podem ser a resposta para o problema de
desativação [GOLDWASSER , M. R. e colaboradores, 2003; DE ARAÚJO G. C.;
2005].
As perovisquitas tem sido extensamente estudadas por suas
interessantes propriedades catalíticas, óticas, magnéticas e condutoras. Em geral,
essas propriedades são potencialmente influenciadas pelo método de síntese,
condições de calcinação (temperatura, tempo e atmosfera) e substituições dos sítios
A e/ou B. O efeito destas variáveis tem sido estudado com o objetivo de aperfeiçoar
o desempenho catalítico do material. A atividade catalítica de óxidos com estrutura
perovisquita em diferentes reações pode ser modificada pela substituição parcial do
sítio A. Entretanto a atividade catalítica de perovisquitas é principalmente
determinada pelo metal que ocupa o sítio B [TANAKA, H. e MISONO, M., 2001].
Diversos métodos de síntese têm sido propostos para a preparação de
óxidos mistos como as perovisquitas e estruturas relacionadas, dentre outros,
visando à obtenção de materiais com alta homogeneidade química, pequeno
tamanho de partícula e alta pureza. Métodos como sol–gel, Pechini, co-precipitação
e combustão ou autocombustão, têm sido amplamente citados pela literatura. A
autocombustão destaca-se como uma técnica promissora para a preparação destes
óxidos por permitir a obtenção de materiais com todas as características desejadas
e ainda ser um método extremamente fácil e rápido, podendo ser aplicado em
escala industrial. A autocombustão assistida por microondas vem sendo empregada
com grande sucesso na preparação de materiais com propriedades ópticas,
magnéticas e catalíticas. Entre as suas vantagens com relação a outros métodos,
destacam-se a facilidade, rapidez, baixo custo e homogeneidade química dos
produtos obtidos [BARROS, B.S., 2009].
O uso de microondas promove um aquecimento mais homogêneo da
solução precursora da síntese (sais metálicos + combustível), fazendo com que a
ignição da reação ocorra uniformemente ao mesmo tempo para toda a solução. Isto

14
implica maior homogeneidade e maior pureza, além de menor gasto de energia e
tempo para a preparação de pós com características nanométricas [BARROS, B.S.,
2009].
O método dos precursores poliméricos ou método Pechini tem sido
bastante investigado para a síntese de nanopartículas. Utilizam-se amplamente o
ácido cítrico e o etilenoglicol. O ácido cítrico, com três grupos ácido carboxílico e um
grupo alcoólico na molécula, forma quelatos bastante estáveis com vários íons
metálicos e, juntamente com o etilenoglicol, sofre facilmente esterificação em
temperaturas moderadas (~100ºC). O sistema polimérico resultante tem uma
distribuição uniforme de cátions por toda a rede. Assim, o polímero retém
homogeneidade na escala atômica e deve ser calcinado em temperaturas
controladas para produzir óxidos de finas partículas [VOLPE, M.A; 2001].
O método Pechini se destaca em relação aos outros métodos de
síntese química, pois garante uma composição química reprodutível, com
granulometria controlada, estrutura cristalina estável e alta pureza.
Entretanto, os problemas de aglomeração e agregação ainda não são
adequadamente tratados. Outro problema encontrado no método é a remoção
efetiva de grandes quantidades de orgânicos e a grande perda de massa durante o
tratamento térmico.
Uma das aplicações recentes que as perovisquitas a base de Ni e Fe
têm apresentado é como catalisadores promissores para reação de oxidação parcial
do metano.
Este trabalho trata do desempenho de perovisquitas do tipo LaNixFe1-
xO3 obtidas por dois diferentes métodos de síntese: Pechini e combustão assistida
por microondas na reação de oxidação parcial do metano.
Estes três elementos La, Ni, Fe foram escolhidos de acordo com os
dados da literatura: lantânio é um terra rara que ajuda na prevenção da formação de
coque, o níquel é muito ativo frente a reação de oxidação parcial de metano e o ferro
é especialmente interessante porque o raio iônico é próximo ao de Ni.
Os dois metais (Ni, Fe) podem estar envolvidos em perovisquitas de
lantânio: LaNiO3, LaFeO3, mas também em perovisquitas mistas como LaNixFe1-xO3 .
A reatividade das estruturas trimetálicas de La - Ni - Fe na reação de
oxidação parcial de metano tem sido estudada com foco sobre o efeito da presença
de ferro na estabilização da estrutura LaNiO3 em condições redutoras. Um ponto

15
importante é a evolução das estruturas de catalisadores em teste e sua possível
regeneração [H. PROVENDIER e colaboradores, 1999].
Dentro deste contexto este trabalho teve como principais
objetivos: Sintetizar perovisquitas do tipo LaNixFe1-xO3 pelo método pechini e
autocombustão assistida por microondas; verificar a influência do grau de
substituição de Ni por Fe na estrutura perovisquita e a influência do método de
síntese no comportamento catalítico das amostras na reação de oxidação parcial do
metano.

16
2 REVISÃO BIBLIOGRÁFICA
2.1 Óxidos com estrutura perovisquita
Nos últimos anos, a pesquisa de novos óxidos do tipo perovisquita tem
sido intensificada devido ao vasto campo de propriedades e aplicações que estes
materiais oferecem.
O uso de óxidos tipo perovisquita como catalisadores foi estudado pela
primeira vez por MEADOWCROFT, em 1970, na redução eletroquímica de oxigênio.
Os óxidos tipo perovisquitas são caracterizados por possuírem uma
única estrutura capaz de aceitar uma grande faixa de defeitos [TEJUCA, L.G. e
colaboradores, 1989] e por exibirem propriedades do estado sólido que são
relativamente fáceis de controlar e modificar mediante trocas na composição
química, dando lugar a interessantes propriedades catalíticas. Estes sólidos são
altamente resistentes a altas temperaturas, estáveis mecânica e quimicamente em
condições de reação e apresentam interessantes propriedades condutoras e
dielétricas [GOLDWASSER, M.R. e colaboradores, 2000].
Dentre essas propriedades podemos citar estabilidade térmica a altas
temperaturas, supercondutividade, atividade catalítica e eletrocatalítica,
propriedades ópticas, dentre outras.
As perovisquitas são óxidos cerâmicos mistos com estrutura do tipo
ABO3, onde os cátions com raios iônicos maiores têm número de coordenação 12 e
ocupam o sítio A, e os cátions com raios menores apresentam número de
coordenação 6 e localizam-se no sítio B, conforme é apresentado na Figura 2.1. O
oxigênio finaliza o empacotamento da estrutura cúbica, formando também um
octaedro no qual um átomo do sítio B ocupa o centro [ VOLPE, M.A; 2001].
O sítio A é ocupado por um metal de terras raras ou metal alcalino e o
B, por um metal de transição [GOLDWASSER, M.R. e colaboradores, 2005].
Os íons A são, dependendo da aplicação, cataliticamente inativos e os
íons metálicos ativos que ocupam a posição B são colocados a distâncias
relativamente grandes um do outro, fazendo com que uma molécula reagente
interaja somente com um único sítio. Há ainda a possibilidade de substituição parcial

17
de um íon A ou B, produzindo assim uma ampla diversidade de compostos
[CHUFFA, P.A. e ASSAF, J. M.,1999].
Cátions com raio iônico relativamente grande (Ca+2 ou Sr+2) substituem
preferencialmente os cátions do sítio A e cátions com raio iônico pequeno (Co, Ni ou
Mn) ocupam os sítios B.
A estrutura cristalina é função da composição (substituição nos sítios A
e B) e estequiometria do oxigênio, a qual é influenciada pela temperatura e pressão
parcial de oxigênio [FLORIO, D. Z., 2004].
Figura 2.1 Duas diferentes apresentações para a estrutura cúbica ideal da Perovisquita.
Perovisquitas são materiais interessantes para aplicações catalíticas e
de fundamental importância na área da catálise. Muitos estudos têm sido uma
tentativa de esclarecer a relação entre a atividade catalítica e as propriedades
químicas e físicas da superfície e em massa de óxidos mistos. As vantagens do uso
de perovisquitas como catalisadores são listados abaixo: [H. TANAKA e M. MISONO
2001]
1. Ampla variedade de composição e elementos constituintes, mantendo a
estrutura básica inalterada.
2. Sua estrutura pode ser bem caracterizada.
3. Valência, estequiometria e vacâncias podem variar amplamente.
4. Diversas informações sobre as propriedades físicas e do estado sólido.

18
O lantânio é um dos principais elementos terras raras e o óxido deste
elemento é comumente utilizado como promotor estrutural e eletrônico em varias
aplicações no campo da catálise ambiental [COLUSSI, S. e colaboradores, 2007].
Muita atenção tem sido dada as perovisquitas à base de lantânio
(LaBO3), B = Ni, Co ou Mn, devido à sua alta atividade catalítica e estabilidade
térmica em reações de oxidação de hidrocarbonetos. O sistema LaNiO3 é um dos
mais conhecidos. O Níquel tem carga 2+, mas assume a carga 3+ em óxidos de
terras raras [NORMAN, A.K.e MORRIS, M.A, 1999].
Perovisquitas deste tipo chegam a apresentar atividades que são
comparáveis àquelas observadas nos metais nobres [TEJUCA, L G. e
colaboradores; 1989]
A substituição parcial dos cátions A, geralmente um metal terra rara,
com elementos que apresentam estados de valência diferente de 3+, tem sido
freqüentemente estudado [ISUPROVA, L. A e colaboradores, 2001; INABA, H. e
colaboradores, 2001].
Os niquelatos de terras raras, puros ou suportados, têm sido testados
como catalisadores industriais potencialmente importantes. As aplicações catalíticas
para esses materiais estão relacionados com a existência de mais de um estado de
oxidação dos metais de transição d, a presença de defeitos e a alta mobilidade dos
íons de oxigênio existentes nestas estruturas [GOMES, D.K.S., 2005].
.
2.1.1 Análises de transições entre várias estruturas perovisquitas
As ligações em perovisquitas do tipo ABO3 são predominantemente
iônicas. Portanto, o raio iônico é usado para pesquisar a respeito de sua estrutura
geométrica. O sistema freqüentemente usado é o publicado por SHANNON e
PREWITT [1969] e por SHANNON [1976].
Os raios iônicos RA e RB dos íons A e B com número de coordenação
12 e 6, respectivamente, e o raio do oxigênio RO, encontram-se nas relações: RB = (
√2 – 1) RO e RA = RO. Portanto, na estrutura de uma perovisquita ideal os íons B
adaptam-se exatamente dentro do sítio octaédrico, ou seja, estão coordenados a
seis átomos oxigênio, enquanto que os íons A, estão coordenados a doze átomos de

19
oxigênio, tal que os ânions e cátions estão tão próximos que entram em contato.
Então a constante de rede αo (equação 2.1) pode ser calculada como:
αo = 2 . √2 . Ro = 2 . (RO + RB) = √2 . (RA + RB) (equação 2.1)
Se o tamanho do raio iônico não permitir a construção de uma rede
perovisquita ideal, ele sofre distorção, o qual está mencionado por um fator de
tolerância (equação 2.2) introduzido por GOLDSCHMIDT [1926].
)(2 OB
OA
RR
RRt
++
= (equação 2.2)
Por definição, o fator de tolerância t é igual a 1 em uma rede de
perovisquita ideal. Um decréscimo de t inicia uma deformação da rede, contudo, a
estrutura pode permanecer sem distorção se t for desprezivelmente menor que 1. De
acordo com GOLDSCHMIDT, a estrutura perovisquita distorcida é estável se 1,0 ≥ t
≥ 0,8.
Além da relação entre os raios iônicos, outra condição fundamental
para a formação da perovisquita é a eletroneutralidade, isto é, a soma das cargas
dos íons A e B deve ser igual ao total de cargas dos ânions X. Isso pode ser obtido,
no caso dos óxidos, pela distribuição de carga da forma A1+B5+O3, A2+B4+O3 ou
A3+B3+O3. Por outro lado, a substituição parcial dos íons A e B para originar óxidos
complexos é possível, desde que se mantenha a estrutura perovisquita. TEJUCA,
FIERRO e TASCÓN [1989] mostraram que a maioria dos elementos podem ser
incluídos no arranjo de perovisquitas, muitos deles em ambas, posições A e B. A
estrutura ideal da perovisquita, com fatores de tolerância muito próximos à unidade,
é muito rara e é obtida a temperaturas de calcinações elevadas. Em outras
condições, aparecem diferentes distorções da estrutura perovisquita. Assim, durante
muitos anos, acreditou-se que o composto CaTiO3 possuía estrutura cúbica, mas
logo se determinou que sua verdadeira simetria era ortorrômbica. São conhecidas

20
estruturas distorcidas com simetria ortorrômbica, romboédrica, tetragonal,
monoclínica e triclínica, mas os três últimos tipos são muito raros e têm sido muito
pouco caracterizados. As distorções mais conhecidas e estudadas são a
ortorrômbica e a romboédrica. No caso da simetria ortorrômbica, os valores de t
devem se situar entre 0,75 e 0,9, produzindo uma distorção cooperativa dos
octaedros dando lugar a este tipo de simetria.
A Figura 2.2 (a) mostra um tipo de distorção que pode ocorrer na
estrutura perovisquita. A distorção é obtida inclinando-se o octaedro de oxigênio de
tal maneira, que os átomos A se deslocam ao longo das direções pseudocúbicas
(110) ou das direções (010). A verdadeira célula ortorrômbica é referida geralmente
como “O-ortorrômbica”, caracterizada por uma relação de parâmetros da rede (c/a) >
√2, que se distingue da estrutura “O”-ortorrômbica”, com (c/a) < √2. Este último
arranjo é o resultado de uma sobreposição da distorção Jahn-Teller da estrutura
perovisquita.
Quando não há nenhum dobramento octaédrico, pode ocorrer uma
pequena deformação da simetria cúbica para romboédrica. Isto ocorre com fatores
de tolerância 0,9 < t < 1,0. A distorção romboédrica, algumas vezes se refere à
“estrutura LaAlO3”, com uma simetria R3c [TEJUCA. FIERRO e TASCÓN. 1989].
Sua relação com a estrutura de estabilidade ideal é mostrada na Figura 2.2 (b)
Figura 2.2 – Distorções (a) ortorrômbica e (b) romboédrica da estrutura perovisquita.
FONTE: KHALIFAH e colaboradores, 2000

21
2.1.2 Papel dos cátions na posição A e B
Os óxidos mistos tipo perovisquita são sistemas ideais para estudar o
modo pelo qual as propriedades do estado sólido, tanto coletivas (elétricas,
magnéticas, funções termodinâmicas) quanto locais (defeitos, campo cristalino),
influenciam nas propriedades catalíticas. Isto se deve, fundamentalmente, ao amplo
conhecimento destas propriedades [GOODNOUGH e LONGO, 1970; NOMURA,
1978] e às suas variações nos sólidos.
De acordo com NITADORI e MISONO [1985], a atividade catalítica do
composto LaBO3 (B = Fe, Co e Mn), na oxidação de hidrocarbonetos e monóxido de
carbono, é afetada pela natureza do cátion na posição B. Para explicar esse fato,
considera-se o estado eletrônico dos elétrons d do cátion na posição B [YAMAZOE e
TERAOKA, 1988], a energia de ligação B-O [WELLER, 1983] e a energia de
estabilização do campo cristalino [GUSHEE; KAZ e WARD, 1988].
Nos óxidos tipo perovisquita, os orbitais 2p do oxigênio e os orbitais s e
p externos do metal B, formam uma banda de valência e uma banda anti-ligante de
alta energia, com os orbitais d do íon B ocupando uma zona intermediária de
energia. Portanto, os orbitais d são, em princípio, os responsáveis pelas
propriedades de transporte eletrônico. Pelo fato da distância entre os dois átomos B
vizinhos ser muito grande (5,5 Ǻ), a superposição entre seus orbitais é muito
pequena e a interação se produz através de oxigênio intermediário. No que diz
respeito à ligação B-O-B, os orbitais d se dividem em orbitais σ (eg) e π (t2g), sendo
esta energia de ligação mais elevada para os orbitais σ que para os π. Isto faz com
que os orbitais t2g sejam localizados, enquanto que os eg tenham um certo caráter
itinerante. Por isso, a geometria de ligação B-O-B determina o caráter localizado ou
itinerante dos elétrons do átomo B.
Quando os óxidos tipo perovisquita são estudados do ponto de vista
catalítico, a atividade do oxigênio da rede, a capacidade de cedê-lo ou captá-lo, sua
estequiometria e as diferentes fases geradas nestes processos, são fatores cruciais.
Um dos parâmetros mais usados em nível teórico, quando são
realizadas correlações entre propriedades catalíticas e este tipo de propriedade do
óxido (atividade do oxigênio da rede), é a energia de ligação do oxigênio com os
cátions da rede, que é dada pela equação (2.3).

22
∆(Me-O) = (Hf – Hs.m – n.Do/2) / (Cn.m) (equação 2.3)
Onde Hf é a entalpia de formação do óxido MemOn, Hs é a energia de sublimação do
metal. Do é a energia de dissociação do oxigênio e Cn é o número de coordenação
do cátion.
Todas as correlações estão baseadas na natureza do cátion na
posição B, que influenciam notavelmente na atividade catalítica do catalisador, tal
como foi demonstrado por diversos autores.
NITADORI e MISONO [1985] estudaram o efeito do cátion na posição
A e encontraram que o efeito do lantanídeo (cátion na posição A) é relativamente
pouco intenso e a atividade esperada dos catalisadores tipo perovisquita é similar
àquela dos óxidos de metais de transição na posição B. Por outro lado, as atividades
catalíticas observadas com os óxidos de terras raras foram mais baixas que nas
perovisquitas correspondentes.
Outros estudos foram conduzidos por PENA [1990], que correlacionou
a natureza dos cátions na posição A com a atividade na oxidação total de isobuteno
e concluiu que na série de perovisquita estudada a atividade catalítica cresce na
seguinte a ordem:
DyCoO3 <<<GdCoO3 < LaCoO3 < SmCoO3 < PrCoO3 < NdCoO3
Estas variações foram inferiores às encontradas quando, fixando-se o
cátion na posição A, varia-se o cátion na posição B. Entretanto, não podem ser
desprezadas.
2.1.3 Estruturas das perovisquitas não estequiométricas
A inexistência de estequiometria em óxidos puros e óxidos mistos
permitem, habitualmente, a estabilização de íons em estados de valência não usuais
e a co-existência de diferentes coordenações. SMITH [1993] definiu a inexistência
de estequiometria, em termos cristalográficos, como sendo o excesso de átomos de

23
oxigênio em relação ao número de sítios na subrede de cátions. A não
estequiometria é definida como sendo o conteúdo de oxigênio em excesso,
necessário para compensar a carga dos cátions constituintes em seu estado de
oxidação normal. Os desvios da estequiometria, devido fundamentalmente a
deficiências catiônicas (nas posições A ou B), deficiências de oxigênio ou excesso
de oxigênio [PEDERSEN e LIBBY, 1972], produzem também um distanciamento da
estrutura ideal da perovisquita. Em catálise heterogênea, os sistemas mais
estudados são os que apresentam um elemento alcalino, alcalino terroso ou
lantanídeo como cátion na posição A e na posição B, um metal de transição da
primeira série. A deficiência nas posições A se produz facilmente, visto que o
esqueleto estrutural de octaedros BO3 permanece inalterado. Um exemplo deste tipo
de não estequiometria é o Cu0,5TaO3 ortorrômbico, com uma distribuição ordenada
dos íons Cu(ll) sobre os sítios A [LONGO e SLEIGTH, 1975].
As vacâncias nos sítios B das perovisquitas não são tão comuns, já
que a deficiência catiônica nas posições B não está energeticamente favorecida e
acontece somente quando os cátions A e o oxigênio (camadas AO3) se estruturam
em um empacotamento hexagonal compacto, o que dá lugar a octaedros BO3 que
dividem as faces. Os empacotamentos intercrescem originando �uperfíci diferentes
segundo sua proporção e ordenamento. Entretanto, se conhecem exemplos com
este tipo de defeito, que apresentam geralmente uma estequiometria complexa, tal
como aquela observada nos compostos BaSm2/3UO6 e Ba2CeSb4/5º6 [RAUSER e
KEMMLER-SACK. 1980].
As vacâncias aniônicas são mais comuns que as não estequiométricas
catiônicas. Desse modo, nos óxidos tipo perovisquita, as vacâncias de oxigênio são
mais comuns [BREARD e colaboradores, 2002]. Nos óxidos ABO3-x, não se
encontram planos cristalográficos de cisalhamento (Crystaligraphic Shear Planes,
CSP), senão uma grande variedade de superestruturas devido ao ordenamento das
vacâncias. Um dos óxidos tipo perovisquita mais conhecidos com vacâncias
aniônicas, é o que exibe a estrutura brownmillerita como os compostos Ca2Fe205 e
Ca2FeAIO5. Estas estruturas podem ser consideradas perovisquitas com deficiências
aniônicas com um sexto dos sítios aniônicos, porém estas vacâncias estão
ordenadas de tal forma, que se alternam os planos BO2 (001) da estrutura
pseudocúbica e as colunas alternantes (110) do oxigênio são perdidas [GRENIER;
POUCHARD e HAGENMÜLLER, 1981].

24
O conteúdo de oxigênio ou o grau de não estequiometria dos óxidos
tipo perovisquita, dependem do método de preparação utilizado e da atmosfera,
temperatura e tempo dos tratamentos térmicos [PEDERSEN e LIBBY, 1972].
2.1.4 Composição superficial das perovisquitas
Os íons, na posição B das perovisquitas ABO3, são os mais ativos em
numerosas reações em fase heterogênea. Visto que estes íons estão situados em
distâncias relativamente grandes uns dos outros (≈0,4nm), cada molécula reagente
pode interagir somente com um centro. A superfície representa uma
descontinuidade do cristal: a simetria e a coordenação dos íons Na+ e Bm+ (n+m=6)
se perde na superfície, pelo fato da superfície apresentar uma grande tendência em
completar sua coordenação através da reação com moléculas gasosas [FIERRO,
1993]. Dessa forma, a análise dos espectros fotoeletrônicos de raios X de muitos
compostos tipo perovisquita ABO3, expostos a umidade ou reduzidos em fluxo de
hidrogênio, indicam a presença de mais de uma espécie de oxigênio. Isto quer dizer
que o oxigênio da rede (O2-) vem acompanhado por outras espécies de oxigênio
menos ricas em elétrons, tais como OH- e CO32-. Quantidades ainda menores de
óxidos individuais AOx e BOy podem não ser completamente incorporadas à
estrutura perovisquita durante a síntese e, por isso, ficarão na superfície. Como
conseqüência deste depósito, a composição superficial se desvia, em muitos casos,
da estequiométrica. É de se esperar, que estas diferenças na composição
superficial, em relação à estequiométrica, modifiquem tanto as propriedades
catalíticas, quanto as propriedades de adsorção das perovisquitas.
FIERRO e TEJUCA [1987] observaram, por meio de um estudo de
espectroscopia fotoeletrônica de raios X (XPS), que os espectros dos níveis internos
B 2p3/2 e La 3d5/2 das perovisquitas LaBO3 (B = Cr, Mn, Co, Ni, Rh), tratadas em ar a
700 e 900°C, não mostraram mudanças significativas das linhas B 2p3/2 e La 3d5/2
com a temperatura.
Os espectros O1s de todos os membros da série exibiram um pico a
529,7 – 530,0 eV, correspondentes a íons O2-, junto a outro pico a 531,4 – 532,6 eV,
que aumentava em intensidade e se deslocava para valores mais elevados de
energia de ligação, quando aumentava o número atômico do metal B. Este último

25
pico foi atribuído ao oxigênio adsorvido (provavelmente O-) de modo similar àqueles
encontrados por ICHIMURA, INOUE e YASUMORI [1980] bem como YAMAZOE,
TERAOKA e SEIYAMA [1981] para a espécie de oxigênio adsorvido das
perovisquitas LaCoO3.
VOORHOEV, REMEIKA e TRIMBLE [1976] encontraram que a
atividade catalítica dos óxidos tipo perovisquitas ABO3, nas reações de oxidação,
dependia da concentração superficial dos íons metálicos em um estado de oxidação
diferente de 3+ e, portanto, de seu caráter não estequiométrico. MILT e
colaboradores [1996] prepararam perovisquitas LaFeO3 e LaCoO3, pelo método de
combustão, e obtiveram perovisquitas altamente cristalinas e monofásicas, mas com
heterogeneidade superficial. Através da análise de �superfície, usando XPS os
autores determinaram que as camadas mais externas destas perovisquitas diferiam
na estrutura e composição mássicas, contendo altas concentrações de espécies
carbonatadas mais elevadas que aquelas encontradas nas perovisquitas preparadas
por métodos mais convencionais.
GUNASEKARAN e colaboradores [1996] ao realizarem a
caracterização superficial, estudaram as propriedades catalíticas na reação de
oxidação de metano com perovisquitas La0,8Sr0,2BO3 (B = Mn, Cr, Fe, Co e Y) e
concluíram que, não apenas a concentração superficial do metal ativo influi na
reação de oxidação, como também a natureza do íon B é um importante parâmetro.
2.1.5 Adsorção de oxigênio nas perovisquitas
A interação do oxigênio nas perovisquitas tem sido estudada,
principalmente, por causa da importância destes materiais como catalisadores em
reações de oxidação – redução.
O desempenho particular destes materiais, como catalisadores de
oxidação, foi explicado em termos da facilidade relativa com que as espécies de
oxigênio podem ser liberadas da superfície do catalisador [PENA e FIERRO, 1981].
Dois tipos de espécies de oxigênio (adsorvido na superfície e oxigênio de rede), com
diferentes forças de ligação, estão presentes na superfície de óxidos tipo
perovisquitas. Admite-se que o oxigênio adsorvido torna-se ativo e reage com os
hidrocarbonetos em temperaturas mais baixas que aquelas em que reagiria com o

26
oxigênio de rede. Estas considerações estão consistentes com a classificação de
VOORHOEV, REMEIKA e TRIMBLE [1976] das reações de oxidação em
perovisquita, nos processos suprafacial e intrafacial, que ocorrem em baixas e altas
temperaturas, respectivamente [FIERRO, 1990].
YAMAZOE, TERAOKA e SEIYAMA [1981] realizaram experimentos de
adsorção de oxigênio, desde a temperatura ambiente até 750°C, e observaram dois
picos de dessorção na curva do composto La1-xSrxCoO3. O pico em temperatura
mais baixa (Tipo α) foi atribuído ao oxigênio adsorvido na superfície, enquanto o pico
em altas temperaturas (Tipo β) foi devido ao oxigênio da rede cristalina. Estas
espécies podem ser detectadas por espectroscopia fotoeletrônica de raios X
combinada com estudos de dessorção de oxigênio a temperatura programada
[TABATA; HIRANO e SUZUKI, 1998]. Foi considerado que o pico a altas
temperaturas (Tipo β) está mais especificamente associado ao cátion B, embora ele
seja também afetado pela substituição do cátion A (normalmente um lantanídeo).
Em uma recente contribuição, YOKOI e UCHIDA [1998] observaram
que a quantidade de oxigênio dessorvido e a temperatura de dessorção do oxigênio
(Tipo β) do LaBO3 (B = Cr, Mn, Fe, Co, Ni) tendem a diminuir com o aumento do
número atômico do metal de transição. O valor da força de ligação B-0 torna-se
menor com o aumento do número atômico de B, sugerindo que a temperatura de
dessorção do oxigênio é afetada pela interação eletrônica entre o oxigênio adsorvido
e o íon metal de transição (B3+).
2.1.6 Estabilidade das perovisquitas em atmosfera redutora
Os estudos a respeito da redutibilidade das perovisquitas podem ser
realizados em atmosfera de monóxido de carbono, porém, devido ao fato de se obter
resíduos carbonáceos, é recomendável conduzir os mesmos em atmosfera de
hidrogênio para eliminar este efeito. Este é o caso do composto LaRhO3 que foi
reduzido em atmosfera de monóxido de carbono, obtendo-se uma perda de peso
equivalente à redução de 75% da espécie Rh3+ a Rh0, a 580°C e, ao se aumentar a
temperatura, obteve-se um aumento no peso, devido à formação de depósitos de
carbonatos ou carvão [TASCÓN; OLIVÁN e TEJUCA, 1984]. FIERRO e TEJUCA
[1984], FIERRO, TASCÓN e TEJUCA [1984, 1985] e TASCÓN e colaboradores

27
[1985, 1986] estudaram a série de óxidos LaMO3 (M = Cr, Mn, Fe, Ni, Rh). Eles
observaram que a facilidade de redução aumentava da espécie Cr3+ à Ni3+, na série
de perovisquitas de LaM03. A mesma tendência foi encontrada com os óxidos
simples de ferro, cobalto e níquel [ANDERSON, 1956]. Por outro lado, encontrou-se
que os óxidos do tipo LaMO3 (M = Fe, Co, Ni) eram mais estáveis em atmosfera de
hidrogênio que os óxidos simples do tipo NiO, Fe203, Co304 [WACHOWSKl; ZlELISKl
e BUREWICZ, 1981], o que indica maior estabilidade dos cátions dos metais de
transição na estrutura perovisquita.
Os experimentos de redução à temperatura programada também
mostraram que a estabilidade dos óxidos tipo perovisquita aumentava com o
aumento no tamanho do íon A. Notou-se, ainda, que as diferentes etapas de
redução correspondiam a diferentes mecanismos de redução. Tais mecanismos
podem ser estudados mediante experimentos de cinética de redução em condições
isotérmicas. Assim, no composto LaNiO3 [FIERRO; TASCÓN e TEJUCA, 1985] a
espécie Ni3+ se reduzia a Ni2+ de acordo com o modelo de contração de esfera e a
espécie Ni2+ a Ni°, de acordo com o modelo de nucleação. A dife rença entre os dois
mecanismos de redução é arbitrária, devido ao fato de que o modelo de contração
de esfera se inicia com uma nucleação muito rápida e o mecanismo de nucleação
termina de acordo com o modelo de contração de esfera. Segundo PIERRÔ,
TASCÓN e TEJUCA [1984] o processo de redução do composto LaMO3 ocorre em
uma etapa única, controlada pela formação e crescimento dos núcleos de redução
da superfície, seguida da diminuição da massa do catalisador.
O cátion na posição A possui um papel importante na redutibilidade
das perovisquitas. Por exemplo, ARAKAWA, OHARA e SHIOKAWA [1986]
mostraram através de análise termogravimétrica que a extensão da redução do
cobalto, em atmosfera de hidrogênio dos óxidos LnCoO3, aumentava a partir do
LaCoO3 até o EuCoO3, com a diminuição do raio iônico do elemento lantanídeo. A
mesma seqüência de redução foi encontrada por FUTAI, YONGHUA e HUI [1986]
por meio de experimentos de redução à temperatura programada. Estes autores
[FUTAI; YONGHUA e Hul, 1986] encontraram uma relação direta entre a
redutibilidade e a soma das energias das ligações Ln-O e Co-O, isto é, a facilidade
de redução aumenta com a diminuição da energia de ligação.
Os processos de redução-oxidação de perovisquitas podem ser
reversíveis quando o ciclo completo for conduzido em temperaturas em que não

28
ocorre a sinterização das espécies oxidadas ou reduzidas. Pode-se dizer que,
mediante os tratamentos de redução ou ciclos redox, o metal na posição B, uma vez
reduzido, se encontra em um estado altamente disperso em uma matriz composta
pelo óxido do metal na posição A. Desse modo, dependendo do grau de sinterização
a que foi submetido à estrutura perovisquita por causa da redução, é possível
reverter este processo e obter, ou não, uma única fase perovisquita após a
reoxidação [CRESPIN e HALL, 1981].
2.1.7 Perovisquitas em camadas
Perovisquitas em camadas consistem infinitas camadas 2D da estrutura tipo
ABO3, que estão separados por camadas. A fórmula geral para as camadas é:
Na+1BnO3n+1. As características que as diferem das perovisquitas convencionais são:
1) a separação por camadas, e 2) a compensação das camadas.
As perovisquitas podem ser vistas como o membro n = da série dos
compostos de Ruddlesden-Popper (RP), com a estrutura K2NiF4 estando no extremo
oposto (n = 1). As fases de RP são constituídas de n camadas consecutivas de
perovisquita (ABO3) alternadas com uma camada de “sal de rocha” (“rock-salt”)
(AO), ao longo da direção cristalográfica c. As fases RP são formadas por blocos de
octaedros BO6 com vértices compartilhados que extendem-se ao infinito no plano xy
e tem uma espessura de n octaedros paralelos ao eixo z. Após as primeiras
observações da magnetoresistência colossal na fase n = , as propriedades
eletrônicas de outros membros da série foram estudadas. Porém, a
magnetoresistência colossal não é observada em amostras do sistema n = 1.
Os exemplos mais claros disto são n = 1 e n = 2, conhecidas como fases de
Ruddleson-Popper (La2NiO4 e La3Ni2O7), Figura 2.3. Para estas fases, o La é o
cátion A e Ni é o cátion B.

29
Figura 2.3 – Série de Ruddlesden-Popper. (a) Para n =1 e (b) Para n = 2.
O sistema Lan+1NinO3n+1 tem sido amplamente investigado como um
possível material catódico de temperatura intermediária devido à sua elevada
condutividade iônica e eletrônica [SKINNER, S. J. e KILNER, J.A., 2000; WAN, J.,
2007].
A estrutura cristalina Lan+1NinO3n+1, para n = 1 tem sido estudado por
diversos grupos [VORONINA, V.I., 2001; POLTAVETS, V. V. e colaboradores, 2006].
Essa estrutura, à temperatura ambiente é tetragonal, enquanto que para n = 2 e 3,
as estruturas são ortorrômbicas.
As perovisquitas tridimensionais têm sido muito estudadas; em
particular, as manganitas de camadas com n = 2 tem começado a ganhar
importância devido à sua inerente anisotropia e suas conseqüências para o estudo
da física de baixa dimensionalidade [J. F. MITCHELL e colaboradores, 1999]. A
magnetoresistência colossal pode ser fortemente melhorada em sistemas com
dimensionalidade reduzida [ROSENKRANZ,S. e colaboradores,1998].
Devido ao forte acoplamento entre os graus de liberdade orbital e da rede que
existem nestes sistemas, um entendimento das propriedades magnéticas e de
transporte requer um conhecimento completo dos efeitos da rede cristalina.
(a) (b)

30
2.1.8 Emprego de Perovisquitas como Catalisador
Neste item serão apresentados os resultados de alguns trabalhos
relacionados com o uso da perovisquita como catalisador, com a substituição parcial
de alguns metais nesta estrutura, e trabalhos que têm relação com este trabalho.
É conhecido que a substituição parcial dos metais na estrutura perovisquita
do sistema catalítico pode provocar alterações na matriz original e, portanto, no seu
comportamento catalítico. Este procedimento pode alterar as propriedades da
estrutura, transformando e neutralizando os sítios, modificando a estrutura eletrônica
do metal e a atividade e seletividade catalítica.
Segundo SLAGTERN e OLSBYE [1994], as interações geradas pela
formação de ligações entre óxidos de níquel e terras raras permitem aumentar a
temperatura de redução do óxido de níquel. No entanto, particularmente para
LaNiO3, a estabilidade térmica é baixa sob atmosfera redutora e a formação de
coque ainda é relevante. Nestas condições, os autores concluíram que a adição de
um terceiro metal na estrutura perovisquita pode estabilizar o sistema catalítico e
limitar o crescimento da partícula metálica.
Foi constatada a alta eficiência de precursores de perovisquita tipo
LaNixFe1-xO3 na reforma a seco do metano para produção de gás de síntese. Esse
sistema de perovisquitas foi obtido via o método sol-gel com ácido propiônico, sendo
a calcinação realizada a 750°C por 4 horas, resulta ndo em solução sólida altamente
homogênea de LaFeO3 e LaNiO3. Os autores observaram, depois dos testes
catalíticos, através de espectros de DRX, que para valores de x > 0,1 o Ni foi
totalmente reduzido; observaram também que Ni e NiO foram depositados sobre
La2O3 para perovisquitas inicialmente ricas em Ni (x = 0,9 e 1,0). Para 0,2 ≤ x ≤ 0,7
parte da estrutura ficou presente como uma perovisquita LaFeO3 e La2O3 e Ni
metálico foram então observados [PROVENDIER, H.; 1998].
Lima e Assaf [2006] também realizaram um estudo com perovisquitas tipo
LaNi1-xFexO3, preparadas pelo método de precipitação, como precursores de
catalisadores para a reforma a seco do metano e observaram a formação de uma
solução sólida em todas as amostras. Foi observado pelos autores que a amostra
não substituída (LaNiO3) desativou-se rapidamente com o tempo de reação devido à
formação de carbono na superfície do catalisador, confirmado por análise de
oxidação a temperatura programada depois do teste catalítico. A adição de ferro na

31
estrutura perovisquita resultou em aumento da estabilidade do catalisador e a
formação de carbono não foi mais observada. No entanto, a substituição parcial de
Ni por Fe conduziu ao decréscimo dos valores de conversão do metano, pois o Fe
contribuiu para redução de deposição de carbono, mas substituiu parcialmente os
sítios ativos de Ni para a reação de reforma a seco.
No trabalho de NAM e colaboradores [1998] foi relatado o resultado de
estudos sobre a reforma a seco do metano utilizando catalisadores tipo perovisquita
La1-xSrxNiO3 (x = 0, 0,1), cujas atividade catalítica e a estabilidade foram
comparadas com catalisadores convencionais de Ni suportado, 5%Ni/SiO2 e
5%Ni/Al2O3 calcinados a 700°C. Apesar da atividade inicial da perovisquita LaNiO3 a
700°C ter sido mais baixa que aquelas dos catalisad ores de Ni impregnados, a sua
atividade aumentou consideravelmente com o tempo antes de alcançar um estado
estacionário. Não tendo sido detectado carbono na superfície da perovisquita LaNiO3
depois da reação, os autores concluíram que a desativação pelo coque foi impedida
devido à boa dispersão de partículas de Ni, provenientes da própria redução da
perovisquita de LaNiO3.
Óxidos tipo perovisquita de La1-xSrxNiO3 foram sintetizados pelo
método de auto-combustão e testados como precursores de catalisadores na reação
de reforma do metano com CO2 a 700°C [VALDERRAMA, G. e colaboradores;
2005]. As análises de difração de raios-X mostraram que a redução das
perovisquitas ocorrem através de espécies intermediárias para produzir Ni0, La2O3 e
SrO. A perovisquita LaNiO3 foi a mais ativa dentre as estudadas, enquanto que
La2NiO4 não foi ativa na reação de reforma a seco. Foi observado que a atividade
catalítica depende do conteúdo de estrôncio, seguindo a seguinte seqüência:
LaNiO3 > La0,6Sr0,4NiO3 > La0,9Sr0,1NiO3.
A alta atividade catalítica encontrada para estes precursores sólidos foi
devido à presença de níquel metálico durante o curso da reação e à formação da
fase de La2O2CO3 que permitiu a oxidação do metano e a regeneração da fase de
La2O3, suprimindo a formação de depósitos carbonáceos.
BATIOT-DUPEYRAT e colaboradores [2003], usando óxido tipo perovisquita
LaNiO3 como material de partida para reforma do metano com CO2, estudaram a
conduta do catalisador injetando pulsos de reagentes com razão CH4/CO2 = 1,0.

32
Três picos de redução foram observados na análise de TPR, sendo o principal pico
em 630°C. O primeiro pico de redução (200-500°C) fo i correspondente à formação
de La4Ni3O10 de acordo com a equação 2.4:
4LaNiO3 + 2H2 → La4Ni3O10 + Ni0 + 2H2O (Equação 2.4)
O segundo passo de redução (600-650°C) deveu-se à f ormação da fase
espinélica La2NiO4, de acordo com a equação 2.5:
La4Ni3O10 + 3H2 → La2NiO4 + 2Ni0 + La2O3 + 3H2O (Equação 2.5)
O terceiro pico foi atribuído à redução completa (680-750°C):
La2NiO4 + H2 → Ni0 + La2O3 + H2O (Equação 2.6)
Os autores estudaram a influência da temperatura de reação (700°C e 800°C)
e observaram que depois da reação a 800°C foram det ectadas somente as fases de
Ni0 e La2O3, as mesmas presentes no início da reação; a 700°C, as fases
predominantes foram Ni0 e La2NiO4. Isto levou à sugestão de que esta
transformação de fase resulta da reação de CO2 com La2O3 e Ni, de acordo com a
reação:
Ni + La2O3 + CO2 → CO + La2NiO4 (Equação 2.7)
A formação da fase espinélica envolve a migração de átomos de níquel das
partículas de Ni0 sobre La2O3 ou a presença de partículas de Ni0 ultra dispersas
capazes de reagir facilmente com La2O3 e CO2. Isto mostra que a fase espinélica
pode ser considerada como um intermediário, onde a reação de reforma seca
ocorreria por um mecanismo de dois passos, sendo o primeiro a ativação de CO2
(Equação 2.7) com a formação de CO e La2NiO4, e o segundo, a redução de
La2NiO4 por CH4 da seguinte forma:
CH4 + La2NiO4 → CO + 2H2 + La2O3 + Ni0 (Equação 2.8)
CHOUDHARY e colaboradores [1996], estudaram a oxidação parcial
seletiva do metano para produção de gás de síntese sobre perovisquitas LaNiO3,
La0.8Ca(ou Sr)0.2NiO3 e LaNi1-xCoxO3 (onde x = 0,2 – 1,0), em tempos de contato

33
extremamente baixos (≈ 0,8 ms). Também foram estudadas as reações simultâneas
de oxidação parcial, reforma a vapor e reforma com CO2 do metano para produção
de gás de síntese sobre LaNiO3 em pequenos tempos de contato (≈ 9 ms) e em
diferentes temperaturas e razões de alimentação CH4/O2.
Verificaram que a perovisquita LaNiO3 tem um alto potencial como catalisador
para a oxidação parcial seletiva do metano na produção de gás de síntese a tempos
de contato extremamente baixo (≈ 0,8 ms), uma vez que, durante o processo
catalítico, ela é transformada em Ni0/La2O3, forma ativa do catalisador para a reação
de reforma. No entanto, a perovisquita com substituição parcial ou completa de
níquel por cobalto mostrou desempenho inferior no processo catalítico. Quando as
reações simultâneas foram conduzidas sobre a perovisquita LaNiO3 a 800 – 850°C e
alta velocidade espacial (tempo de contato ≈ 9 ms) o gás de síntese (com razão
H2/CO de 2.0) pôde ser obtido com alta conversão (>90%) e alta seletividade
(aproximadamente 100% para ambos H2 e CO), podendo o processo ser operado de
modo mais eficiente, requerendo pouca ou nenhuma energia externa, em
conseqüência do acoplamento das reações exotérmicas de oxidação e das reações
endotérmicas de reforma com vapor e CO2 sobre o mesmo catalisador.
2.2 Métodos de síntese
Existe um grande número de métodos para a síntese de perovisquitas,
mas para se determinar o melhor é necessário, inicialmente, saber para que fim será
utilizado o óxido obtido. As propriedades destes sistemas dependem em grande
parte do caminho de síntese, especialmente sua textura e superfície específica,
estados de oxidação dos cátions e a estequiometria do oxigênio [DE LIMA, S. M.;
2006].
O método ideal deve dar lugar a óxidos de excelentes propriedades
catalíticas e deve ser altamente reprodutível, de forma que permita comparar
propriedades de diferentes sistemas.
Materiais cerâmicos como os óxidos, são em geral preparados a partir
da purificação de minerais ou por métodos convencionais de preparação a partir de
misturas de constituintes óxidos, hidróxidos e/ou carbonatos. Estes materiais
geralmente possuem um grande tamanho de partículas requerendo, para gerar um

34
material homogêneo e constituído por uma única fase, procedimentos de preparação
que fazem uso de repetidas misturas, aquecimentos prolongados e elevadas
temperaturas. Desvantagens como, baixa área superficial e controle limitado da
microestrutura inerente aos processos a altas temperaturas tendem a ser superadas
através da preparação destes materiais por outros métodos, tais como, co-
precipitação, sol-gel, Pechini, reação de combustão, etc.. Estes métodos garantem a
obtenção de materiais com composição química reprodutível e controlada, alta
pureza, homogeneidade e tamanho de partícula rigidamente controlado.
2.2.1 Método dos precursores poliméricos (Método Pechini)
O método dos precursores poliméricos vem sendo bastante reportado
na literatura, por apresentar alta qualidade das propriedades. Logo, vários materiais
têm sido preparados por este método e com uma variedade de aplicações, tais
como: fotoluminescência (SrSnO3) [UDAWATTE e colaboradores, 2000], pigmento
cerâmico (ZnTiO4) [SOUZA e colaboradores, 2005], catalisador (LaNiO3)
[FERNANDES e colaboradoes, 2002], entre outras.
O método dos precursores poliméricos, comparado com outros
métodos tradicionais, oferece a posibilidade de materiais com uma boa
homogeneidade química, reprodutibilidade e um bom controle estequiométrico
[MELO e colaboradores, 2006].
Este método foi desenvolvido originalmente por Pechini, em 1967
(Pechini, 1967) e se tornou popular por H. Anderson, que originalmente aplicou o
método para fabricar pós de perovisquita [LESSING, 1989]. Atualmente, é utilizado
na síntese de diversos óxidos policatiônicos.
O método Pechini consiste na formação de quelatos entre os cátions
metálicos (dissolvidos como sais numa solução aquosa) com um ácido carboxílico
(preferencialmente o ácido cítrico) e, em seguida, uma reação de poliesterificação
utilizando uma poliálcool (preferencialmente etileno glicol), promovendo, dessa
forma, a polimerização. Vários sais de cátions podem ser usados, tais como,
carbonatos, cloretos, hidróxidos, isopropóxido e nitratos [LESSING, 1989]. O
aquecimento da resina, a aproximadamente 300 ºC, provoca a ruptura do polímero
(combustão de parte da matéria orgânica), resultando na formação do pó precursor,
um material semi-carbonizado, de cor escura. Em seguida, um tratamento térmico

35
adequado é realizado para a eliminação do material orgânico e a obtenção da fase
desejada.
O etileno glicol proporciona uma imobilização do complexo metal–ácido
cítrico (quelato) em uma rígida rede polimérica altamente ramificada, reduzindo a
segregação dos metais durante o processo de pirólise a altas temperaturas,
garantindo uma composição final estequiométrica [UDAWATTE e colaboradores,
2000; POPA e colaboradores, 2002]. Isto é de vital importância para a síntese de
óxidos multicomponentes com composição complexa [KAKIRANA e YOSHIMURA,
1999].
A idéia principal do método é manter a estequiometria dos íons
metálicos na resina polimérica, ou seja, que os íons metálicos estejam distribuídos
atomisticamente por toda a estrutura polimérica, de forma que a ruptura da rede do
polímero não comprometa a distribuição homogênea destes íons metálicos, uma vez
que essa distribuição determina a homogeneidade do óxido final [KAKIHANA, 1996].
Este método recebeu atenção considerável por sua simplicidade.
A Figura 2.4 ilustra as reações envolvidas no método Pechini.
Figura 2.4 – Reações envolvidas no método Pechini
O ácido cítrico é muito utilizado como agente quelante, pela sua
facilidade de formar complexos estáveis com muitos íons metálicos (exceto os

36
monovalentes), enquanto, o etileno glicol é bastante utilizado como agente
polimerizante, pois possui grande afinidade de complexação metal-ácido cítrico.
O ácido cítrico contém três grupos carboxílicos (-COOH), o etileno
glicol dois grupos hidroxílicos (-OH) em cada molécula, logo reações sucessivas de
esterificação podem ocorrer para a formação da resina polimérica [KAKIHANA,
1996].
O método dos precursores poliméricos, em relação a outras técnicas, apresenta
vantagens e desvantagens [KAKIRANA e YOSHIMURA, 1999]:
Vantagens:
• Homogeneidade química dos multicomponentes em escala atômica;
• Temperaturas de calcinações relativamente baixas;
• Controle direto e preciso da estequiometria de sistemas complexos;
• Pós-cerâmicos com partículas muito finas;
• Simplicidade de processamento;
• Maior reprodutibilidade
• Flexibilidade,
Desvantagem:
Grande quantidade de matéria orgânica, que pode levar à formação de carbonato
como fase secundária e de fortes aglomerados.
Apesar de o método já oferecer a vantagem de obter partículas finas, a síntese pode
ser aprimorada com o processo de moagem.
2.2.2 Combustão microondas
A síntese por reação de combustão, também conhecida como síntese
auto-propagante, é uma técnica onde reações exotérmicas são usadas para produzir
uma variedade de pós cerâmicos.
O processo é baseado no principio de que, uma vez iniciada por uma
fonte externa, uma reação exotérmica muito rápida ocorre, tornando-se auto-
sustentável e resultando em um produto final (óxido), dentro de um curto período de
tempo. A técnica é uma maneira fácil, segura e rápida para a produção de pós-
cerâmicos, tendo como principais vantagens: requerer menos energia que os
processos convencionais de síntese de materiais cerâmicos e tempo de
processamento reduzido para poucos minutos.

37
Podemos também destacar como grandes vantagens do método de
combustão, características interessantes como a sua simplicidade (uma vez que não
necessita de múltiplas etapas), custo relativamente baixo e normalmente leva a
produtos com estrutura e composição desejadas, devido à elevada homogeneização
favorecida pela solubilidade dos sais em água.
A base da técnica de síntese através da reação de combustão deriva
dos conceitos termodinâmicos usados na química dos propelentes e explosivos,
envolvendo a reação de uma mistura redox, contendo os íons metálicos de interesse
como reagentes oxidantes, e um combustível, geralmente a uréia (CO(NH2)2) como
agente redutor. Os nitratos metálicos são dentre as fontes de íons, os sais mais
usados por serem solúveis na água, e baixas temperaturas são suficientes para
fundi-los, garantindo uma excelente homogeneização da solução.
Os nitratos metálicos reagem com o combustível redutor, resultando na
formação de um pó óxido fino, seco, e geralmente cristalino. Enquanto as reações
de redução são exotérmicas por natureza e conduzem a uma explosão se não
controlada, a combustão da mistura de nitratos metálicos com a uréia geralmente
ocorre com uma reação exotérmica não explosiva. A grande quantidade de gases
formada pode resultar na aparência de uma chama, que pode alcançar temperaturas
elevadas (superiores a 1000°C).
Além da uréia, vários outros combustíveis têm sido usados na síntese
por combustão de óxidos cerâmicos mistos e puros, tais como, glicina (C2H5NO2),
triazina tetraformol (TFTA, C4H16N6O2), hidrazida maléica (C4H4N2O2) e
carbohidrazida (CO(N2H3)2, etc.. Todos estes combustíveis contêm nitrogênio, mas
diferem na capacidade de “redução de pó” e na quantidade de gases por eles
gerados, fatores que obviamente afetam as características do produto de reação. A
uréia tem a mais baixa redução do pó (valência total +6) e produz um pequeno
volume de gases (4 mol/mol de uréia). As vantagens de se usar a uréia são:
disponibilidade comercial, baixo custo, e o fato de gerar altas temperaturas, as quais
são necessárias para a formação das fases desejadas nos produtos. Segundo
alguns autores, utilizando-se teor de uréia em excesso na reação, os gases gerados
são liberados mais rapidamente e com maior dissipação de energia, ou seja, menor
será a quantidade de energia disponível para a sinterização e cristalização,
evitando-se a formação de aglomerados duros e/ou crescimento das partículas.

38
Segundo MANOHARAN e PATIL, o mecanismo das reações de combustão pode ser
resumido nos seguintes passos:
I. Fusão dos nitratos e da uréia (também chamada carbamida) e eliminação das
águas de hidratação.
II. Decomposição da uréia em biureto e NH3 a 240 °C e do biureto em ácido
cianúrico (HNCO)3 a 360°C e dos nitratos formando óxidos de nitrogên io.
III. Ignição dos produtos de decomposição da uréia e nitratos, formando uma chama
com temperatura de 1200 ± 100 °C, garantindo energe ticamente a formação do
óxido a partir dos precursores gelificados, formados por cadeias poliméricas de
ácido cianúrico e gel hidroxonitroso dos íons metálicos.
As bases da técnica de síntese por reação de combustão provem dos
conceitos termodinâmicos usados no campo dos propelentes e explosivos.
O sucesso deste processo está intimamente ligado à escolha de um combustível ou
agente complexante (ácido cítrico, uréia, glicina, ...) e da reação redox em meio
aquoso entre este e um agente oxidante contendo os íons de interesse (nitratos,
acetatos, ...).
A escolha dos nitratos como agentes oxidantes é conseqüência de sua
excelente solubilidade em meio aquoso, o que proporciona melhor homogeneidade
da solução. Por sua vez, a escolha do combustível é baseada em fatores como,
preço, disponibilidade, quantidade e tipo de gases gerados e o calor liberado durante
a reação.
Os mecanismos envolvidos na reação de combustão são muito
complexos. Além da escolha dos agentes oxidantes e do combustível, diversos
outros parâmetros influenciam a reação, tais como, razão combustível-oxidante, uso
de oxidante em excesso, quantidade de água contida na mistura precursora. Além
destes, a própria forma de aquecimento da solução pode proporcionar a obtenção
de pós com características diferentes. Nos últimos anos tem se destacado o uso das
microondas para prover um aquecimento rápido e homogêneo da mistura
precursora.
As características dos pós, tais como, tamanho de cristalito, área
superficial e natureza de aglomeração (forte ou fraca) são governadas
principalmente pela entalpia e temperatura de chama gerada durante a combustão
que, por sua vez, depende da natureza do combustível e da razão combustível-

39
oxidante empregada. Por outro lado, a rápida evolução de um grande volume de
gases durante a combustão dissipa o calor do processo e limita a temperatura,
reduzindo a possibilidade de sinterização prematura localizada entre as partículas
primarias. A evolução dos gases também ajuda a limitar o contato entre as
partículas, resultando em um produto mais facilmente fragmentável, o que permite a
obtenção de materiais mais porosos e em geral com maior área superficial. Por outro
lado, o excesso de gases pode também promover a formação de fases espúrias nos
produtos obtidos, tais como, hidróxidos, oxicarbonatos e oxinitratos, quando a
reação não é completa [BARROS, B.S., 2009].
Reação de combustão assistida por microondas
A autocombustão assistida por microondas consiste em uma reação de
combustão convencional, onde o calor necessário para a ignição é fornecido através
do aquecimento gerado por moléculas polares quando submetidas à incidência de
microondas. As vantagens deste método de aquecimento em relação aos métodos
normalmente usados na síntese por reação de combustão (placa de aquecimento e
mufla) são: baixo tempo necessário para alcançar a temperatura de ignição e
uniformidade da distribuição de temperatura, que neste caso é gerada dentro da
própria solução precursora. Além disso, o aquecimento via microondas permite um
maior controle das condições de síntese, visto que a intensidade das emissões de
microondas pode ser rapidamente interrompida, diminuída ou aumentada,
permitindo a obtenção de materiais com características bastante especificas.
Dois importantes aspectos da reação de combustão assistida por
microondas são as características de aquecimento instantâneo e volumétrico. Desta
forma, toda a mistura pode reagir uniformemente durante o aquecimento via
microondas.
O aquecimento por microondas também apresenta outras vantagens.
Como o calor é gerado dentro do próprio material, ou seja, do centro para a
superfície podem ser criados gradientes de temperatura inversos. Isto pode levar a
obtenção de diferentes microestruturas [BARROS, B.S., 2009].

40
2.2.3 Método sol-gel
É um método de síntese polimérica ou de partículas em solução. A rota
sol-gel envolve a dispersão de partículas coloidais com diâmetros da ordem de 1-
100 nm num meio líquido para formar um “sol”, o qual é transformado numa rede
sólida similar a uma esponja, o gel polimérico. A gelação, nesse caso, é controlada
por interações eletrostáticas entre as partículas coloidais no sol. Neste método as
interações interpartículas são interações físicas. A transição sol-gel pode ser
observada por um aumento muito rápido na viscosidade [ZANETTI, S. M. 2001;
PERTHUIS, H. e COLOMBAN, P., 1986].
As vantagens apresentadas por este método são [PESSOA, R. C., 2005]:
� Controle da homogeneidade;
� Redução da temperatura de sinterização;
� Controle da porosidade e da cristalinidade;
� Obtenção de pós com alta pureza;
� Reatividade e homogeneidade a nível atômico dos cátions no
composto final;
Como desvantagens, podemos citar [PESSOA, R. C., 2005]:
� Utilização de matérias primas caras e de difícil manipulação;
� Presença de traços residuais de carbono após tratamento térmico;
� Crescimento anormal dos grãos quando a razão água/ alcóxido está
em excesso;

41
� Necessidade de tempo longo de hidrólise/condensação, no caso de
obtenção de monólitos;
2.2.4 Método da Co-precipitação
É o método mais utilizado para a separação de um precursor a partir de
uma solução. Consiste na preparação de soluções homogêneas contendo os cátions
desejados e a precipitação estequiométrica desses cátions em solução,
simultaneamente e estequiometricamente na forma de hidróxidos, oxalatos entre
outros [PERTHUIS, H.; 1986].
Para que ocorra a precipitação simultânea é preciso que os cátions ou
os ânions em solução, estejam em uma concentração que exceda o produto de
solubilidade (Kps), e que não ocorra a precipitação de nenhum dos cátions quando a
solução contendo os sais precursores é preparada. A precipitação simultânea ocorre
em decorrência da mudança do pH, pela adição de um ânion formador de um sal
insolúvel [PESSOA, R. C., 2005].
Algumas das vantagens oferecidas por este método são [PESSOA, R. C.,
2005]:
� Aplicação em grande número de materiais;
� Oportunidade da lavagem de impurezas solúveis antes da calcinação;
� Apresentar pós-estequiométricos, reativos e homogêneos.
As principais desvantagens são [PESSOA, R. C., 2005]:
� Homogeneidade garantida somente para a precipitação de uma única
espécie;
� Concentração, temperatura e o pH de estabilidade desses sais
influenciam na formação do composto;

42
� Dificuldade de se obter o composto desejado na presença de dopantes
em baixas concentrações;
2.3 Produção de H2 para fins Energéticos
O hidrogênio é uma matéria prima extremamente importante e estratégica
devido a sua utilização em uma grande variedade de processos de refino do
petróleo, tais como hidrotratamento e hidrocraqueamento, e petroquímicos como a
produção de amônia e metanol. Além de ser um combustível ideal para as células a
combustíveis.
Hoje em dia, há uma grande preocupação na obtenção de
combustíveis menos poluentes ao meio ambiente, como o H2, que é um combustível
não poluente e altamente energético, tem levado a um aumento crescente na
demanda de sua produção [VASCONCELOS, N., 2006].
Atualmente, o hidrogênio é largamente produzido pela reação
endotérmica de reforma a vapor do metano para produzir H2 e CO. Nos Estados
Unidos 95 % da produção do H2 vêm do metano [ARMOR, J.N. 2005]. No Brasil, a
produção de H2 a partir do metano é bastante interessante em função das grandes
reservas de gás natural existentes.
A catálise tem grandes oportunidades de potencializar o mercado de
hidrogênio. Porém, o maior desafio está em produzir H2 em grandes volumes à baixa
pressão, com alta pureza em um custo baixo.
Como atualmente o H2 é gerado a partir do gás de síntese (SINGÁS),
produtores de H2 estão sempre procurando caminhos para se obter maior
quantidade de H2. Isto é possível, pela remoção do CO do gás de síntese através da
reação de deslocamento da água (equação 2.9) onde o monóxido de carbono reage
com vapor de água gerando CO2 (Reação de Shift).
CO + H2O ↔ CO2 + H2 ∆Hº298 = -41 kJ/mol (Equação 2.9)
O CO2 produzido é removido através de processos de absorção com
solventes químicos e constitui um subproduto do processo.

43
Os catalisadores da reação de shift não estão devidamente adaptáveis para o
uso em células a combustível. Em adição, eles devem estar aptos a resistir
numerosos ciclos de partidas e paradas, passar por procedimentos especiais de
ativação, tolerar condições ricas em oxigênio, e mostrar alta tolerância a compostos
sulforosos. Catalisadores novos e muito mais ativos para a reação de shift
necessitam ser desenvolvidos quando o gás de síntese é parte da corrente do
processo de células a combustível, pois estas células compreenderão um enorme
potencial para o mercado de H2 [VASCONCELOS, N., 2006].
A Figura 2.5 mostra a interação dos passos de geração, estocagem,
transporte, recuperação, produção e finalmente o uso do H2 como combustível. O
hidrogênio utilizado em células a combustível necessita ser purificado, e a
distribuição para milhões de consumidores deve ser realizada de forma prática e
efetiva, minimizando gastos quanto ao transporte. Logo, comprimir o H2 permitirá
transportar um maior número de mols de H2 até os centros consumidores. Mas isto
não será possível sem um meio de produzir H2 economicamente [ARMOR, J.N.
2005].
Figura 2.5 – Infraestrutura do Hidrogênio para uso com fins energéticos.
Fonte: Armor, 2005.

44
2.4 Conversão do Metano a Gás de Síntese
Existem diversas tecnologias para a geração de gás de síntese, as quais
fornecem diferentes razões H2/CO. Sendo assim, é possível aperfeiçoar a produção
de gás de síntese de acordo com a síntese pretendida, como por exemplo, obtenção
de combustíveis líquidos, H2 para células a combustível, síntese de metanol e
amônia, entre outras [VASCONCELOS, N., 2006]. Dentro deste contexto, várias são
as rotas catalíticas para obtenção do gás de síntese a partir do metano, sendo estas
descritas a seguir.
2.5 Reações para Obtenção de Gás de Síntese
2.5.1 Reforma a Vapor do Metano
A Reforma a Vapor do Metano é tipicamente descrita pela reação dada pela
equação 2.10 e é o principal processo industrial para a produção de hidrogênio e
gás de síntese.
CH4 + H2O ↔ CO + 3H2 ∆Hº298 = +206 kJ/mol (Equação 2.10)
O primeiro estudo detalhado da reação catalítica entre metano e vapor
d’água foi publicado em 1924. Durante todos esses anos, muitos avanços na
tecnologia desse processo têm sido alcançados, verificando que muitos metais,
incluindo níquel, cobalto, ferro e platina, podem catalisar esta reação [TSANG, S.C.
e colaboradores; 1995].
Esta reação é altamente endotérmica e realizada a pressões de até 30
atm. Apesar da estequiometria da reação sugerir que é necessário apenas 1,0 mol e
água por mol de metano, o excesso de vapor deve ser usado para reduzir a
formação de carbono no produto.
Por ser uma reação endotérmica, muitas unidades de reforma a vapor
operam em temperaturas acima de 800°C. O processo d e reforma a vapor do
metano na maioria das vezes tem a contribuição de várias reações diferentes,
mostrada pelas equações (2.11) a (2.17).

45
CO + H2O(g) ↔ CO2 + H2 ∆Hº298= -40 kJ/mol (Equação 2.11) CO2 + H2 ↔ CO + H2O(g) ∆Hº298= +40 kJ/mol (Equação 2.12) 2CO ↔ C + CO2 ∆Hº298 = -171 kJ/mol (Equação 2.13) CH4 → C + 2H2 ∆Hº298 = +75 kJ/mol (Equação 2.14) CnHm + vapor ↔ nCO + m/2H2 (Equação 2.15) CO2 + 2CH4 + H2O → 3CO + 5H2 (Equação 2.16) CO2 + CH4 ↔ 2CO + 2H2 ∆Hº298 = +247 kJ/mol (Equação 2.17)
O processo de reforma a vapor do metano é usado para converter
metano em CO e H2 como descrito nas equações (2.11) a (2.17). O que realmente
ocorre é um número de reações competitivas que incluem a reforma com CO2 e
vapor, em como a reação de deslocamento gás-água (3 – “shift”), metanação e
reações de formação de carbono. O catalisador mais utilizado é o Ni suportado em
material como alfa alumina contendo uma variedade de aditivos [ARMOR, J.N,
1999].
Apesar da reforma a vapor do metano ser o principal processo para produção
de hidrogênio e gás de síntese, esta apresenta algumas desvantagens, são elas:
� Necessidade de uma grande quantidade de energia;
� O vapor superaquecido (em excesso) a alta temperatura tem um custo
elevado;
� A reação de deslocamento gás-água produz concentrações significativas de
CO2 no produto gasoso;
� Tem baixa seletividade para CO;
� Alta razão H2/CO, que é inadequada para a síntese do metanol e de Fischer-
Tropsch.

46
Dessa forma, as reações de oxidação parcial do metano usando
oxigênio e de reforma do metano com dióxido de carbono (reforma seca)
apresentam-se como alternativas promissoras para a produção de gás de síntese
[BHARADWAJ, S.S. e SCHMIDT, L.D.; 1995].
2.1.1.1. Mecanismo da Reação de Reforma a Vapor do Metano
Alguns autores propuseram o seguinte mecanismo para a reforma a vapor [QIN, D. e
colaboradores,1996 e DIAS, J. A. C.; 2002]:
CH4 + 2M → CH3-M + H-M (Equação 2.18) CH3-M + 2M → CH-M + 2H-M (Equação 2.19) CH-M + M → C-M + H-M (Equação 2.20) H2O + 3M → O-M + 2H-M (Equação 2.21) CHx-M + O-M + (x-1)M → CO-M + xH-M (Equação 2.22) CO-M + O-M → CO2 + 2M (Equação 2.23) CO-M → CO + M (Equação 2.24) 2H-M → H2 + 2M (Equação 2.25) 2CO + 2M → C-M + CO2-M (Equação 2.26)
em que M é o sítio ativo.
As reações dadas pelas equações (2.18), (2.19) e (2.20) representam a
ativação do metano e a reação dada pela equação (2.21) consiste na decomposição
da água. Por outro lado, a reação da equação (2.22) consiste na recombinação das
espécies adsorvidas formando as espécies de CO-M e xH-M, que são dessorvidos
como mostra as equações (2.23) e (2.24) para a obtenção dos produtos CO e H2.
Estas reações estão envolvidas no processo de formação de carbono, que se
deposita sobre o catalisador, levando a formação de coque e conseqüentemente à
desativação do catalisador.
A formação do carbono se dá na superfície do catalisador através da
dissociação do metano sobre a superfície do metal, produzindo espécies altamente

47
reativas, que são provavelmente carbono atômico. Existem dificuldades em impedir
a formação de carbono e muitos esforços têm sido realizados para diminuí-lo.
Atualmente, nas indústrias, contorna-se o problema através da alimentação de
excesso de vapor, embora exista uma tendência a se diminuir a razão de
alimentação H2O/CH4 para reduzir o consumo de vapor e, conseqüentemente, o de
energia. Para isso, é evidente a necessidade do desenvolvimento de catalisadores
mais estáveis para o processo [DIAS, J. A. C.; 2002].
2.5.2 Reforma a Seco do Metano
A reação de reforma de metano com dióxido de carbono foi estudada pela
primeira vez por Fischer e Tropsch [1928] e cálculos indicaram que esta reação era
termodinamicamente favorável acima de 640ºC. Esse tipo de reforma consiste em
uma rota alternativa para a produção de gás síntese.
Do ponto de vista ambiental, a reforma com CO2 (equação 2.27) é um
processo interessante, pois consome gases parcialmente responsáveis pelo efeito
estufa, porém, estudos recentes mostraram que em um balanço global, a utilização
da reforma com CO2 não seria capaz de reduzir a quantidade de CO2 a ponto de
minimizar o aquecimento global do planeta [ARMOR, J.N,1999].
CH4 + CO2 ↔ 2CO + 2H2 ∆Hº298 = +247 kJ/mol (Equação 2.27)
Este processo de reforma é endotérmico, produzindo uma razão H2/CO
igual a 1, sendo adequado à produção de compostos oxigenados e monóxido de
carbono com alta pureza. Entretanto, há um maior risco de depósito de carbono
tanto sobre o suporte quanto da fase ativa quando comparado à reforma a vapor
[ROSTRUP-NIELSEN, J.R., 1984; LERCHER, J.A . e colaboradores, 1999; CHENG,
Z.X. e colaboradores, 2001]
Além disso, este processo tem um grande potencial termoquímico para
recuperação, estocagem e transmissão de energia solar e outras fontes de energia
renovável através do uso do alto calor de reação e a reversibilidade deste sistema
reacional [FERNANDES, J. D. G. e colaboradores, 2002].
Das muitas reações químicas reversíveis que vêm sendo estudadas
para este propósito, a reforma CO2/CH4 e a reação reversa de metanação CO/H2

48
são consideradas pioneiras em aplicações no armazenamento e na transmissão de
energia solar.
Do ponto de vista industrial, não é fácil fornecer CO2 puro e
concentrado para o reformador sob pressões operacionais típicas. A maior
dificuldade para realizar a reforma do metano com CO2 é a formação de carbono,
termodinamicamente favorecida, que desativa os catalisadores. Os catalisadores de
Ni são os mais propensos à desativação. Apesar destes problemas, a reforma seca
está entre as reações catalíticas mais elaboradas, o que é confirmado por
publicações de numerosos trabalhos na literatura especializada [RYNKOWSKI, J.;
2004].
Segundo EDWARDS E MAITRA [1995], além da reação da equação (2.27), o
processo de reforma do metano com CO2 pode estar acompanhado, em certas
condições de operação, de outras reações paralelas termodinamicamente possíveis.
Dentre elas, podem-se citar as reações de formação de carbono, representadas
pelas equações (2.13) e (2.14). Além disso, as equações (2.12) e (2.28), podem
também ter importante influência nos resultados finais do processo de reforma.
C + H2O(g) → CO + H2 ∆H = +131 kJ/mol (Equação 2.28)
A reação da equação (2.27) pode ser constituída da equação (2.13) e da
reversa da reação da equação (2.14). Idealmente, o carbono formado na reação da
equação (2.13) deveria ser consumido parcialmente pela reação reversa da equação
(2.14) e, em menor escala, pela reação da equação (2.20). Esta pode ter um papel
importante quanto à retirada do carbono formado, mas, no contexto da reação, o
vapor é quase sempre formado via reação reversa de deslocamento gás-água
(equação 2.12). Se a reação da equação (2.13) for mais rápida do que a taxa de
remoção do carbono, haverá sérios problemas quanto à formação de coque, com
conseqüente desativação do catalisador e bloqueio do reator pelo carbono formado,
ou seja, a quantidade de coque aumenta impedindo o fluxo dos reagentes [Edwards
e Maitra, 1995].
É interessante notar que a reação da equação (2.12), que consome
hidrogênio, representa uma desvantagem, a menos que ambas as reações,
(equação 2.12) e (equação 2.20), ocorram mantendo a estequiometria constante.
Portanto, o catalisador adequado para esta reação seria aquele que não somente

49
acelerasse a reação e apresentasse uma alta conversão inicial, mas também
evitasse a formação de depósitos carbonáceos e de água [Edwards e Maitra, 1995].
2.5.3 Oxidação Parcial do Metano
A oxidação parcial do metano para a produção de gás de síntese foi
investigada pela primeira vez por volta dos anos 30 e esquecida por mais de 50
anos, certamente por causa do aumento no interesse pela reforma a vapor do
metano e pelo fato de não haver, na época, catalisadores que evitassem a formação
de carbono, uma vez que essa deposição não podia ser evitada pelo aumento da
razão O2/CH4 ou pelo aumento da temperatura de reação, sem que houvesse o
perigo de explosão, problemas de separação e diminuição na seletividade do gás de
síntese. Depois deste período, a constatação de que alguns metais nobres
poderiam, em escala laboratorial, catalisar a oxidação parcial do metano com pouca
ou nenhuma deposição de carbono reascendeu o interesse industrial e acadêmico
pela oxidação parcial catalítica do metano para a produção de gás de síntese
[TSANG, S.C. e colaboradores, 1995].
Estudos mostraram que sob certas condições e com a utilização de
certos catalisadores, a formação direta do gás de síntese a partir do metano e do
oxigênio é possível [FATHI, M. e colaboradores, 2000].
O processo de oxidação parcial do metano é o ideal para a tecnologia
GTL no que se refere à produção de gás de síntese, uma vez que fornece uma
razão H2/CO = 2, perfeita para os catalisadores atuais da síntese de Fischer-
Tropsch.
CH4 + ½º2 → CO + 2H2 ∆Hº298 = -36 kJ/mol (Equação 2.29)
Este processo (equação 2.29) opera em altas temperaturas e altas
pressões (150 atm) e apresenta algumas vantagens sobre a reforma a vapor do
metano convencional, uma vez que, este último é um processo altamente
endotérmico e produz gás de síntese com razão H2/CO ≥ 3 [TAKEHIRA, K. e
colaboradores, 1995]. Estudos mostraram que sob certas condições e com a

50
utilização de certos catalisadores, a formação direta do gás de síntese a partir do
metano e do oxigênio é possível.
A reação de oxidação parcial é exotérmica, o que significa uma
economia de energia, ao mesmo tempo em que a presença de O2 reduz o depósito
de carbono a altas temperaturas aumentando o tempo de vida do catalisador
[FATHI, M. e colaboradores, 2000]. Porém é um processo desvantajoso, por
necessitar da utilização de O2 puro. Isto eleva os custos da planta, pois requer uma
unidade de separação criogênica do ar. A utilização direta de ar acarretaria em
equipamentos de maior volume devido à presença de N2 [PEÑA, M.A. e
colaboradores,1996]. Uma tentativa envolvendo a eliminação da planta de O2 inclui a
concepção de um reator com adição de oxigênio através de membranas seletivas
[ROSTRUP NIELSEN e colaboradores, 2002].
Assim, a oxidação parcial do metano é considerada uma alternativa
promissora para substituir a reforma a vapor, pois apresenta a razão H2/CO
desejada e também é suavemente exotérmica [DE LIMA, S. M.; 2006].
A reação do metano com oxigênio a altas temperaturas resulta
principalmente em CO, CO2, H2O, e H2. A composição do efluente gasoso depende
da temperatura, pressão, composição do gás na entrada, e também de fatores
cinéticos. Na literatura têm sido propostos dois mecanismos para esta reação,
dependendo do catalisador. Em um deles, considera-se que a oxidação parcial do
metano ocorre via um mecanismo de dois passos, no qual ocorre primeiramente a
oxidação total de parte do metano a CO2 e vapor, seguida pelas reformas a vapor e
a seco do metano restante; enquanto que o outro sugere a oxidação em um único
passo, ou melhor, envolve a dissociação do metano e a reação deste com átomos
de oxigênio adsorvidos na superfície do catalisador [QIN, D. e colaboradores,1996].
Mecanismo da Reação de Oxidação Parcial do Metano
Como já citado, existem duas propostas de mecanismos para a reação de
oxidação parcial do metano: a oxidação parcial direta (via um passo) e o mecanismo
via dois passos.
O mecanismo sugerido para a oxidação parcial via um passo (assim como
ocorre nas reações de reforma a seco e a vapor) envolve a ativação do metano e a

51
reação deste com átomos de oxigênio adsorvidos na superfície do catalisador [QIN,
D. e colaboradores,1996].
Devido ao fato do metano ter uma estrutura molecular estável, a sua ativação
requer altas temperaturas, em muitos casos. No entanto, os metais nobres são
eficientes neste processo em temperaturas relativamente baixas.
Assim, a reação de oxidação parcial direta [QIN, D. e colaboradores,1996]
segue o mesmo mecanismo que a reforma a seco e a vapor, mostrado
anteriormente pelas equações 2.18 - 2.25.
Enquanto que a oxidação parcial via dois passos ocorre através das
seguintes reações:
CH4 + 2º2 → CO2 + 2H2O ∆Hº298 = -802 kJ/mol (Equação 2.30) CH4 + H2O ↔ CO + 3H2 ∆Hº298 = +206 kJ/mol (Equação 2.10) CH4 + CO2 ↔ 2CO + 2H2 ∆Hº298 = +247 kJ/mol (Equação 2.27) CO + H2O(g) ↔ CO2 + H2 ∆Hº298 = -40 kJ/mol (Equação 2.11)
Inicialmente, ocorre a oxidação total de uma parte do metano (equação 2.30)
formando dióxido de carbono e vapor de água. Estes produtos reagem com o
metano remanescente (equações 2.10 e 2.27) para produção de gás de síntese.
Também acontece a reação de deslocamento gás-água (equação 2.11) que
acompanha as reações de reforma. A partir destas reações é possível verificar que a
oxidação parcial via dois passos depende do progresso da oxidação total e das
reformas para produção de gás de síntese.
Neste processo, a deposição de carbono, que é termodinamicamente
favorável, deve ser considerada e pode se dar pelo desproporcionamento do CO
(equação 13) (reação de Boudouard), ou pela decomposição catalítica do metano
(equação 14) [TSANG, S.C. e colaboradores; 1995].
Catalisadores Empregados na Oxidação Parcial do Metano
Os catalisadores de metais nobres, tais como Ir, Pt, Pd, Rh e Ru
apresentam elevada atividade na oxidação parcial do metano [ELMASIDES, C. e
colaboradores, 2001; GRUNWALDT, J.-D. e colaboradores; 2001; PANTU, P e

52
colaboradores, 2002; NAKAGAWA, K., 2001], mas têm um custo muito elevado.
Portanto, vários tipos de catalisadores baseados em metais não-nobres como Co,
Fe, Ni, Mo, V ou W têm sido estudados [TSANG, S.C. e colaboradores; 1995; JIN, R.
e colaboradores; 2000; WANG, H.Y. e RUCKENSTEIN, E.,2001; ERDOHELYI, A. e
colaboradores, 2001]
Os catalisadores de Ni têm sido extensivamente usados devido a sua
alta atividade e custo relativamente baixo. No entanto, para inibir a formação de
coque, eles têm sido modificados com a presença de óxidos de metais alcalinos ou
de terras raras [LIU, S. e colaboradores, 2001; MIAO, Q. e colaboradores,1997] ou
suportados sobre óxidos do tipo perovisquitas [TAKEHIRA, K. e colaboradores;
2002].
MIAO e colaboradores [1997] estudaram catalisadores de Ni modificados com
óxidos de metais alcalinos e óxidos de terras raras e verificaram que esta
modificação não somente aumentava a atividade na oxidação parcial do metano
para produção de gás de síntese como também a estabilidade térmica dos
catalisadores a altas temperaturas, além de aumentar a resistência dos
catalisadores à deposição de carbono.
Catalisadores de níquel suportados em La2O3 e MgO foram altamente
ativos na reação de oxidação parcial do metano [REQUIES, J. e colaboradores;
2005]. Sobre o suporte de óxido de lantânio, o óxido de níquel interagiu, através de
uma reação do estado sólido, com as camadas próximas da superfície das
partículas do suporte e formou a estrutura LaNiO3, sendo esta a fase com maior
quantidade de Ni. Esta fase, quando ativada em fluxo de H2 se reduz, gerando
partículas metálicas finamente dispersas sobre a matriz La2O3. Estas partículas de
Ni é que são responsáveis pelo desempenho do catalisador na produção de gás de
síntese.
Os catalisadores Ni/MgO mostraram-se mais ativos e mais estáveis
que os catalisadores Ni/La2O3. Esta maior atividade dos catalisadores suportados
em MgO foi atribuída ao fato da solução sólida NiO/MgO produzir partículas
metálicas de Ni mais dispersas sobre a superfície do suporte do que os
catalisadores suportados em La2O3.

53
2.5.4 Reforma autotérmica
O processo de reforma autotérmica do metano consiste em uma
combinação entre os processos de reforma a vapor e oxidação parcial, no qual a
reforma do metano com vapor é realizada em presença de oxigênio [ARMOR, J.N,
1999; CHAN, S. H. e WANG, H. M., 2001; KRUMPELT, M. e colaboradores, 2002;
AYABE, S. e colaboradores; 2003].
O termo autotérmico é utilizado uma vez que neste processo são
realizadas reações exotérmicas e endotérmicas. Sendo assim, o calor gerado pela
oxidação parcial é utilizado pela reforma a vapor, otimizando os custos energéticos
da unidade industrial [KRUMPELT, M. e colaboradores, 2002]. Isto constitui uma
grande vantagem deste processo em comparação aos outros, uma vez que, nos
reatores convencionais há a utilização de combustão externa de outros combustíveis
para a geração de calor [AYABE, S. e colaboradores; 2003].
As reações envolvidas no processo são descritas pela equação 2.31,
além das equações 2.10 e 2.11.
CH4 + ½ O2 ↔ CO + 2H2O ∆Hº298 = -520 kJ/mol (Equação 2.31)
A principal vantagem da reforma autotérmica é que a razão H2/CO no
gás de síntese produzido pode ser facilmente ajustada através da reação
CH4/O2/H2O na alimentação, fazendo com que haja um direcionamento para a
síntese do produto desejado. Sendo assim, para aplicações GTL baseadas na
síntese de Fischer-Tropsch, a reforma autotérmica produz um gás de síntese com
razão H2/CO próxima a dois a partir de uma baixa razão H2O/CH4 [VASCONCELOS,
N., 2006].
No entanto, assim como em outros processos de geração de gás de
síntese, os catalisadores utilizados, geralmente à base de níquel, sofrem
desativação pela formação de coque em sua superfície [PALM, C.; 2002] e
colaboradores.
2.5.5 Reforma com membrana
Outra tecnologia, menos conhecida, que visa a redução nos custos de
produção do gás de síntese, utiliza membranas cerâmicas não porosas que podem
ser feitas de uma grande variedade de materiais, sendo os mesmos óxidos

54
metálicos multicomponentes e que, em altas temperaturas (maiores que 700ºC),
conduzam tanto elétrons, quanto íons oxigênio. Essas membranas são chamadas
membranas de transporte de íons e desperta um especial interesse, pois os íons
oxigênio as permeiam com uma alta vazão e com infinita seletividade. O oxigênio
pode ser separado do ar por um lado da membrana, a pressão ambiente, e assim
reagir na outra superfície da membrana com o metano a uma alta pressão e formar
dessa forma, H2 e CO. Em função da alta vazão através da membrana, o reator é
muito compacto e deve ser de baixo custo. O esquema da membrana cerâmica não-
porosa é ilustrado na Figura 2.6 [DYER, P. N.; e colaboradores,1999].
Figura 2.6 – Esquema de uma membrana cerâmica não porosa.
Apesar das vantagens que a membrana seletiva ao O2 trás para o processo
de gás de síntese, existe ainda a necessidade de um grande trocador de calor entre
a alimentação e o efluente no processo de reforma catalítica com membrana, o que
aumenta o custo dessa tecnologia.
Sendo assim, estudos mostraram que a difusão de O2 através de
membranas em uma unidade de produção de gás de síntese é possível, porém a
viabilidade econômica do processo ainda está para ser comprovada [AASBERG-
PETERSEN, K. e colaboradores, 2001; ROSTRUP NIELSEN, J.R e colaboradores;
2002].

55
2.6 Deposição do carbono
A dissociação do metano (reações 2.19, 2.20 e 2.21) e a reação de
desproporcionamento de CO ou reação de Boudouard (reação 4) são provavelmente
as duas principais rotas da deposição de carbono [EDWARDS e MAITRA, 1995].
A reação de desproporcionamento de CO é exotérmica e a constante
de equilíbrio diminui com o aumento da temperatura da reação, enquanto que a
reação de decomposição do metano é endotérmica e a constante de equilíbrio
aumenta com o aumento da temperatura. REITMEIER e colaboradores [1948]
publicaram que para algumas reações com uma mistura de H2, CO, CH4, CO2 e
H2O, em equilíbrio termodinâmico, a deposição de carbono grafite diminuía com o
aumento da temperatura, evidenciando que a principal causa para a deposição de
carbono era o desproporcionamento de CO. Em outro estudo, foi observado que
com o aumento da temperatura de reação, a quantidade de carbono diminuía em
ambas atmosferas de CO2/ CH4/N2 e CO/N2, mas aumentava em atmosfera de
CH4/N2. As imagens de microscopia eletrônica de transmissão mostraram formações
de carbono filamentoso tipo “whisker” nas atmosferas de CO2/CH4/N2 e CO/N2 e
carbono encapsulante na atmosfera de CH4/N2. Desde que a morfologia do carbono
depositado sobre o catalisador em atmosfera de CO2/CH4/N2 seja similar ao
depositado em atmosfera de CO/N2, foi sugerido que o desproporcionamento de CO
é um dos principais passos para a geração de carbono na decomposição de CO2
[LUO e colaboradores, 2000].
Estudos realizados com metais de transição mostraram que entre os
metais níquel, rutênio e platina, a seguinte ordem crescente de deposição de
carbono foi encontrada: Ru < Pt < Ni. Portanto, o níquel é muito mais sensível à
deposição de carbono do que os metais nobres. Este depósito ocorre de várias
formas: carbono atômico adsorvido, que é altamente reativo; carbono amorfo;
carbono filamentoso e carbeto.
O carbono atômico adsorvido é aquele cuja quantidade formada não
varia com o tempo de reação, sendo proporcional à atividade, o que sugere sua
participação como intermediário da reação.
O carbono amorfo se forma encapsulando a partícula metálica,
resultando em desativação. Sua deposição ocorre através da formação de um

56
polímero –CH2-, o qual é lentamente transformado em depósitos poli aromáticos
menos reativos, tendendo a grafite.
O carbono filamentoso, também chamado de carbono “whisker”, é
formado no processo de dissociação de hidrocarbonetos. Esta dissociação ocorre de
um lado do cristal do metal, enquanto sua nucleação ocorre no lado oposto, o que
ocasiona o crescimento de um filamento de carbono, sem desativação do sítio.
No entanto, esta fibra pode crescer e se romper, causando a remoção da partícula
metálica do catalisador.
A fase carbeto surge da reação do carbono com o metal, arrastando-o
para fora da estrutura, provocando corrosão por “pitting” [DIAS, 2002].
A formação de carbono sobre a superfície de catalisadores de níquel é
bem conhecida e o seu mecanismo consiste em dois passos: o primeiro é a
formação de átomos de carbono por um composto intermediário tipo carbeto e o
segundo é a formação da fase grafítica. Na dissociação dos hidrocarbonetos ocorre
a produção do carbono monoatômico, conhecido como Cα que é altamente reativo e
facilmente gaseificado e, por isso, é um intermediário da reação. No entanto, se o
Cα existe em excesso, a polimerização é favorecida formando a fase grafítica, ou
Cβ, que é bem menos reativa que a fase carbeto, o que levará ao seu acúmulo e à
dissolução no níquel [TRIMM, 1999].
O crescimento de filamentos de carbono se inicia em partículas
metálicas de 50 a 500 Ǻ de diâmetro. O passo principal do crescimento do filamento
é a difusão do átomo de carbono através da partícula. As faces do cristal de níquel
apresentam propriedades e funções diferentes na formação de carbono: as faces
(100) e (110) iniciam cataliticamente a geração de átomos de carbono a partir do
metano, enquanto que a face (111) é responsável pela construção da estrutura
grafítica [CHESNOKOV e colaboradores,1995].
A dissolução do carbono no níquel é essencial para o crescimento de
carbono tipo “whisker”. O processo se inicia aparentemente pela formação de
carbeto de níquel, embora isto não esteja claro, pois o carbeto é instável sob as
condições onde a amostra pode ser analisada. No entanto, traços de carbeto têm
sido observados [TRIMM, 1977; ROSTRUP-NIELSEN e TRIMM,1977].
Uma vez que o carbono tenha sido dissolvido e formado um composto
de níquel, a difusão através da partícula metálica para as partículas vizinhas começa
a ocorrer. O carbono levanta a partícula de níquel na extremidade de um filamento.

57
O níquel permanece como um catalisador ativo, mas o acúmulo de filamentos
bloqueia o leito catalítico e aumenta a pressão até níveis inaceitáveis [ROSTRUP-
NIELSEN e TRIMM,1977]. A gaseificação destes filamentos ocorre via o processo
reverso, com o carbono difundindo-se através da partícula de níquel para ser
gaseificado na superfície catalítica [BERNARDO e TRIMM, 1979]. Nem todo carbono
formado na superfície dissolve no níquel. Parte do carbono permanece na superfície
e encapsula o níquel. Uma vez formado, o encapsulamento desativa o catalisador e
é dificilmente gaseificado [TRIMM, 1977; ROSTRUP-NIELSEN e TRIMM,1977].
Em geral, a deposição de carbono pode ser reduzida se o níquel é
suportado sobre óxidos de metais com alta basicidade, pois o aumento na
basicidade do material do suporte promove a quimissorção do CO2 e o aumento na
concentração de CO2 adsorvido retarda a formação de carbono via a reação reversa
de desproporcionamento de CO. Por outro lado, a deposição de carbono também
depende da estrutura do catalisador. Para desenvolver um catalisador com alto
desempenho, é necessário esclarecer o mecanismo da reação e identificar os
passos determinantes da formação de carbono [LUO e colaboradores., 2000]. Os
efeitos dos suportes e aditivos sobre a formação de carbono e sobre a estabilidade
dos catalisadores baseados em níquel tem sido investigado extensivamente.
BRADFORD e VANNICE [1996] examinaram catalisadores de níquel e platina e
verificaram que a deposição de carbono foi suprimida quando Ti02 e Zr02 foram
usados como suporte. HORIUCHI e colaboradores [1996] e YAMAZAKI e
colaboradores [1992] mostraram que a deposição de carbono diminuiu quando o
metal foi suportado em óxido metálico com forte basicidade de Lewis, favorecendo
então a adsorção de CO2 e suprimindo o desproporcionamento de CO. Porém,
ZHANG e VERYKIOS [1994] reportaram que a adição de um promotor básico como
CaO no catalisador de Ni/Al2O3 resultou em aumento, tanto da estabilidade catalítica
como da deposição de carbono. Alternativamente, é plausível que a deposição de
carbono esteja relacionada com a estrutura do catalisador. Por exemplo, CHEN e
REN [1994] estudaram a reforma do metano com C02 sobre catalisadores de
Ni/Al2O3 e observaram que a deposição de carbono foi marcadamente suprimida
quando a estrutura espinélica NiAl204 era formada durante o pré-tratamento. Tem
sido sugerido que a forte interação entre o Ni e Al2O3 resulta na formação de
pequenos cristais de níquel, que são relativamente mais resistentes à sinterização e
à formação de carbono. Além disso, a acidez do suporte também deve ser um fator

58
importante para a estrutura cristalina do metal. Depois de investigar a natureza de
Pd, Pt e Rh sobre um número de suportes utilizando medidas de NH3, MASAI e
colaboradores [1988] reportaram que havia uma correlação entre a dispersão do
metal e a acidez de Lewis do suporte. Esta relação deve ser devido ao fato que os
átomos metálicos preferem residir nos sítios ácidos de Lewis. Em outras palavras, a
atividade catalítica e a deposição de carbono são fortemente dependentes de
parâmetros como estrutura cristalina do metal, interação metal-suporte, acidez do
suporte e a possibilidade da basicidade do suporte.

59
3 PROCEDIMENTO EXPERIMENTAL
3.1 Reagentes utilizados
Na Tabela 3.1 são listados os reagentes utilizados na preparação das
amostras estudadas, pelos métodos dos precursores poliméricos e de
autocombustão assistida por microondas.
Os materiais listados na Tabela 3.1 foram utilizados para se obter os
óxidos com estrutura perovisquita do tipo LaNixFe1-xO3, onde x=0; x=0,3; x=0,7 e
x=1 de composições: LaNiO3, LaFeO3 , LaNi0,7Fe0,3O3 e LaNi0,3Fe0,7O3 .
Tabela 3.1- Reagentes utilizados na síntese dos óxidos.
Reagentes Procedência Pureza (%)
Nitrato de Níquel II ALDRICH 99,9
Nitrato de Lantânio III ALDRICH 99,9
Nitrato de Ferro III ALDRICH >=98
Uréia VETEC 98
Ácido cítrico MERCK 99,9
Etileno glicol VETEC 99,5
3.2 Preparação dos catalisadores
Método Pechini
Inicialmente, o ácido cítrico foi adicionado a um becker contendo água
deionizada sob agitação constante a uma temperatura de 60 ºC por 20 minutos.
Após esse tempo, foi adicionado nitrato de níquel, para obter LaNiO3 ou nitrato de
ferro, para obter LaFeO3, à solução a uma temperatura de 60 ºC durante 40 minutos.
Adicionou-se nitrato de lantânio e o sistema foi homogeneizado por mais 20 minutos.
No caso dos óxidos substituídos, adicionou-se nitrato de ferro por mais 30 minutos.
Em seguida foi adicionado etilenoglicol a esta solução obtida numa proporção de
60:40 (ácido cítrico: etilenoglicol) e o novo sistema foi mantido a 90 0C sob agitação
constante durante 1 hora, resultando em uma resina, a qual foi pré calcinada a
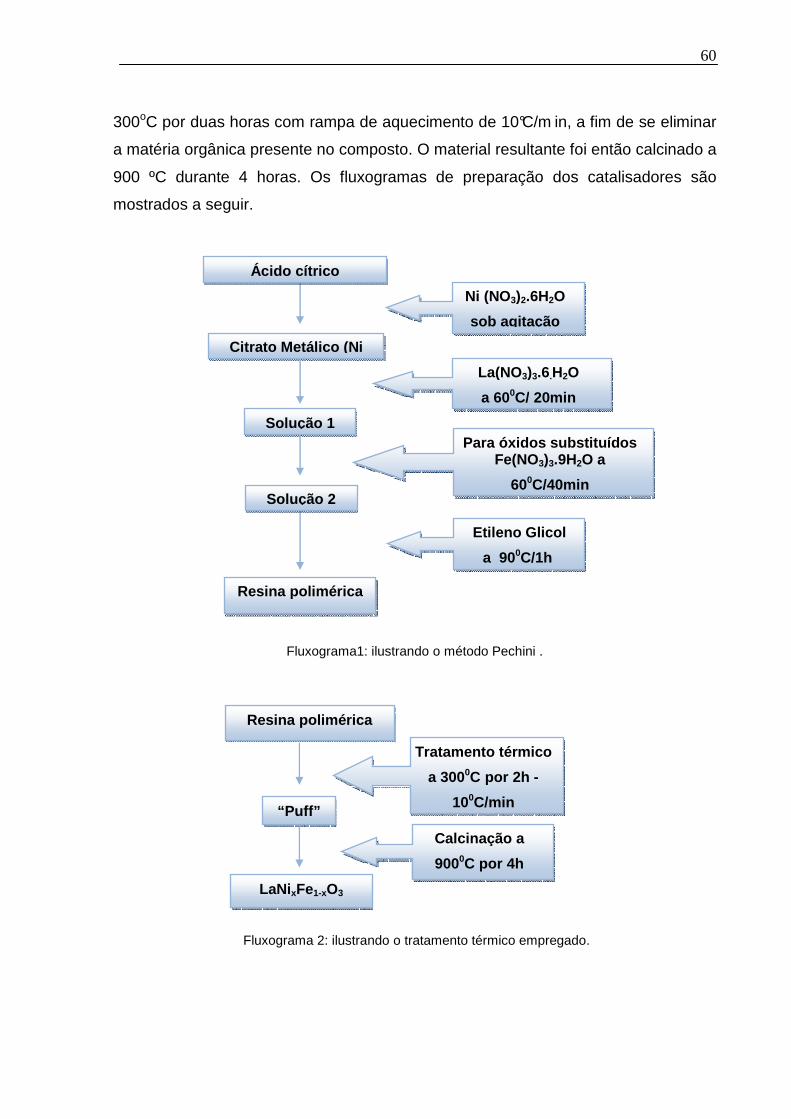
60
300oC por duas horas com rampa de aquecimento de 10°C/m in, a fim de se eliminar
a matéria orgânica presente no composto. O material resultante foi então calcinado a
900 ºC durante 4 horas. Os fluxogramas de preparação dos catalisadores são
mostrados a seguir.
Fluxograma1: ilustrando o método Pechini .
Fluxograma 2: ilustrando o tratamento térmico empregado.
Citrato Metálico (Ni
Ácido cítrico
Solução 1
Ni (NO3)2.6H2O
sob agitação
La(NO3)3.6.H2O
a 600C/ 20min
Para óxidos substituídos Fe(NO3)3.9H2O a
600C/40min
Resina polimérica
Etileno Glicol
a 900C/1h
Solução 2
Resina polimérica
Tratamento térmico
a 3000C por 2h -
100C/min “Puff”
LaNi xFe1-xO3
Calcinação a
9000C por 4h

61
Autocombustão assistida por microondas
As amostras foram preparadas pelo método de autocombustão
assistida por microondas utilizando-se uma solução aquosa de nitratos que foi
misturada com uréia. Os respectivos reagentes foram misturados em um Becker
pirex e submetidos ao aquecimento diretamente em uma placa quente à temperatura
aproximada de 100 °C sob constante agitação por apr oximadamente 10 minutos
para garantir uma boa homogeneização da mistura. Em seguida, o Becker com a
mistura foi colocado em um forno de microondas a uma potência de 650 W e
freqüência de 2.45 GHz até a auto-ignição ocorrer (aproximadamente entre 5 e 7
minutos).
A quantidade em massa de cada reagente foi calculada em função dos
respectivos pesos molares. Os pós-obtidos foram então calcinados a 900 °C por 4h.
A Tabela 3.2 apresenta a quantidade de moles usados para a
preparação dos catalisadores LaNiO3, LaFeO3, LaNi0,3Fe0,7O3, LaNi0,7Fe0,3O3.
Tabela 3.2 – Quantidade de moles utilizados para a preparação dos catalisadores LaNiO3 LaFeO3 LaNi0,3Fe0,7O3 LaNi0,7Fe0,3O3
Número de moles
Catalisadores La Ni Fe Uréia
LaNiO3 1 1 0 4
LaFeO3 1 0 1 4
LaNi0,3Fe0,7O3 1 0,3 0,7 4
LaNi0,7Fe0,3O3 1 0,7 0,3 4
O fluxograma de preparação dos catalisadores é mostrado a seguir:
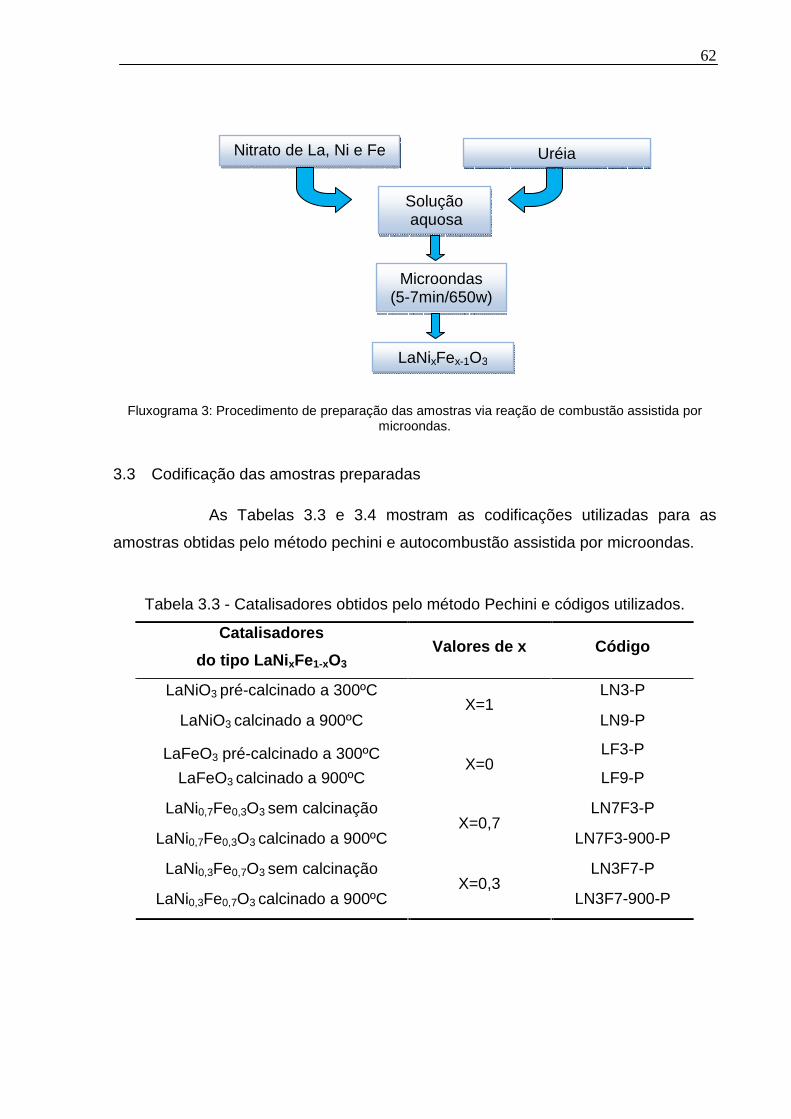
62
Fluxograma 3: Procedimento de preparação das amostras via reação de combustão assistida por microondas.
3.3 Codificação das amostras preparadas
As Tabelas 3.3 e 3.4 mostram as codificações utilizadas para as
amostras obtidas pelo método pechini e autocombustão assistida por microondas.
Tabela 3.3 - Catalisadores obtidos pelo método Pechini e códigos utilizados.
Catalisadores
do tipo LaNi xFe1-xO3 Valores de x Código
LaNiO3 pré-calcinado a 300ºC LN3-P
LaNiO3 calcinado a 900ºC X=1
LN9-P
LaFeO3 pré-calcinado a 300ºC LF3-P
LaFeO3 calcinado a 900ºC X=0
LF9-P
LaNi0,7Fe0,3O3 sem calcinação LN7F3-P
LaNi0,7Fe0,3O3 calcinado a 900ºC X=0,7
LN7F3-900-P
LaNi0,3Fe0,7O3 sem calcinação LN3F7-P
LaNi0,3Fe0,7O3 calcinado a 900ºC X=0,3
LN3F7-900-P
Nitrato de La, Ni e Fe Uréia
Solução aquosa
Microondas (5-7min/650w)
LaNixFex-1O3

63
Tabela 3.4 - Catalisadores obtidos pelo método microondas e códigos utilizados.
Catalisadores
do tipo LaNi xFe1-xO3 Valores de x Código
LaNiO3 como obtido LN3-M
LaNiO3 calcinado a 900ºC X=1
LN9-M
LaFeO3 como obtido LF3-M
LaFeO3 calcinado a 900ºC X=0
LF9-M
LaNi0,7Fe0,3O3 sem calcinação LN7F3-M
LaNi0,7Fe0,3O3 calcinado a 900ºC X=0,7
LN7F3-900-M
LaNi0,3Fe0,7O3 sem calcinação LN3F7-M
LaNi0,3Fe0,7O3 calcinado a 900ºC X=0,3
LN3F7-900-M
3.4 Caracterização dos catalisadores
3.4.1 Análise Termogravimétrica (ATG)
Análise térmica foi realizada em uma termobalança da Shimadzu
TG/DTA-60H sob atmosfera de ar para as amostras sem ferro e sob atmosfera de
nitrogênio para as amostras contendo ferro, na faixa de temperatura entre 30 e 900 0C a uma razão de aquecimento de 10 oC min-1, usando-se uma massa de 10mg, o
cadinho uttilizado foi de platina.
3.4.2 Espectroscopia de absorção na região do infravermelho (IV)
Os espectros de absorção na região do infravermelho foram
registrados na faixa espectral de 4000 a 400 cm-1 em um espectrômetro ABB
Bomem, modelo MB104, usando-se janelas de KBr para análise e os catalisadores
óxidos na forma de pastilhas dispersos em KBr. Para a preparação das pastilhas foi
pesado 0,004 g de catalisador e 0,196g de KBr.

64
3.4.3 Difração de Raios-X (DRX)
A identificação das fases cristalinas presentes na amostra tratada
termicamente e nas amostras calcinadas foram realizadas através da analise de
difração de raios-X pelo método do pó. Os difratogramas de raios-X foram obtidos
em um difratômetro XRD-6000 da Shimadzu utilizando CuKα como fonte de
radiação, filtro de níquel e 2θ na faixa de 10 a 80°, passo de 0,02 e tempo de
contagem por passo de 2°/min.
As fases cristalinas foram identificadas usando o banco de dados do Centro
Internacional para Dados de Difração (ICDD).
A avaliação da cristalinidade é tecnologicamente importante, uma vez que
permite a definição da temperatura mínima necessária para a obtenção de amostras
com elevada cristalinidade. A cristalinidade nas amostras analisadas foi obtida com
base na relação entre a área integrada dos picos cristalinos e a área integrada da
fase amorfa.
A cristalinidade da amostra pode ser calculada a partir da Equação descrita
abaixo:
%100(%) xAA
ACR
ac
c
+
=
Onde, CR é a cristalinidade da amostra, Ac a área integrada dos picos
cristalinos e Aa a área integrada da fase amorfa.
Método de Rietveld
O percentual das fases LaNi0,3Fe0,7O3 e LaNi0,7Fe0,3O3 foi determinado
mediante refinamento pelo método Rietveld [RIETVELD, 1969]
O método de Rietveld consiste na construção de um padrão de difração
calculado, de acordo com o modelo estrutural. O padrão calculado é obtido pela
introdução direta dos dados cristalográficos, como: Simetria do grupo espacial,
posições atômicas, posições de ocupação e parâmetros de rede.
O método tem como principio realizar um ajuste do padrão de difração,
refinando os fatores instrumentais e os parâmetros estruturais característicos da
(Equação 3.1)

65
amostra a ser analisada (parâmetro de rede, posições atômicas, anisotropia,
parâmetros de alargamento relacionados com tensões da rede e tamanho de
cristalitos).
É fundamental ter o cuidado de verificar se o padrão de difração
calculado está suficientemente próximo do observado. A aproximação entre os
padrões calculados e observados é feita pelo método dos mínimos quadrados. Este
método apresenta algumas vantagens como rapidez de resolução e determinação
dos erros estatísticos em cada ciclo do refinamento. A quantidade minimizada
utilizada no refinamento é o resíduo (R). A diferença entre o difratograma
experimental e um difratograma baseado num modelo estrutural de partida resulta
em uma função que expressa o resíduo (R), de acordo com a equação abaixo:
( )2icioi yywR −= ∑
onde :
wi = 1/yi
yio = intensidade observada no i-ésimo passo do difratograma;
yic = intensidade calculada no i-ésimo passo do difratograma;
As análises realizadas por refinamento neste trabalho puderam ser avaliadas
pela observação da plotagem dos padrões calculados e observado. Neste trabalho,
o refinamento foi realizado no programa Philips X'Pert HighScore e, então, os
resultados foram tratados em uma tabela de cálculo do Laboratório de Análises
Térmicas e Materiais da UFRN (que ainda está em desenvolvimento).
3.4.4 Área superficial específica (AS)
A área superficial especifica dos catalisadores foi medida pelo método
desenvolvido por Brunauer-Emmett-Teller (BET). Este método baseia-se na
determinação do volume de N2 adsorvido a diversas pressões relativas, na
temperatura do nitrogênio líquido, a pressões de até 2 atm e pressões relativas
(P/P0) inferiores a 0,3. Foi utilizado um medidor de área superficial Quanta Chrome,
modelo NOVA modelo 2000.
(Equação 3.2)

66
O nitrogênio adsorvido fisicamente em cada pressão produz uma
alteração na pressão interna do porta-amostra. Esta informação é registrada e, por
calibração, transformada em volume adsorvido. Com o aquecimento da amostra,
devido à perda de contato do nitrogênio líquido com a célula de amostragem, o gás
é dessorvido. A adsorção e a dessorção geram sinais que são registrados em
gráficos na forma de picos. A área dos picos é proporcional à massa de nitrogênio
dessorvido. Então, a partir do volume de nitrogênio e da Equação 3.3, conhecida
como equação de B.E.T., determina-se o volume de nitrogênio necessário para
recobrir a superfície adsorvente com uma monocamada.
Onde, P é a pressão de operação, V é o volume de gás adsorvido, Psat
é a pressão de saturação do gás, c é uma constante que envolve entalpias de
adsorção e condensação e Vm é o volume da monocamada de adsorção por massa
de catalisador.
Assim, conhecendo-se o volume de nitrogênio adsorvido na
monocamada (VM), a área superficial específica da amostra pode ser calculada pela
Equação 3.4.
Onde, S é a área superficial específica da amostra, α a área de
projeção da molécula de N2 para a monocamada (16 Å2), NA o número de Avogadro,
V o volume molar do nitrogênio e M a massa da amostra.
(Equação 3.3)
(Equação 3.4)

67
3.4.5 Redução à temperatura programada (RTP)
A técnica de redução à temperatura programada (RTP) permite
determinar o intervalo de temperatura em que ocorre a redução do metal (Ni, Fe). As
análises de redução a temperatura programada (RTP) foram realizadas em um
equipamento de RTP da Micromeritics, modelo AutoChem II 2920.
Os ensaios foram realizados em um reator de quartzo na forma de “U”
com diâmetro interno de 0.97 mm. A atmosfera redutora do reator consistiu de uma
mistura de 10%H2-Argônio, com fluxo total de 50 mL.min-1. A taxa de aquecimento
desde 50 a 1000 oC foi de 10 oC.min-1. Um detector de condutividade térmica foi
utilizado para analisar o gás após trapeamento da água, permitindo assim quantificar
o consumo de hidrogênio.
O Fluxograma 4 mostra de forma resumida todas as caracterizações
realizadas.
Fluxograma 4: Etapas de Caracterização dos Catalisadores
DRX
IV
LaNi xFe1-xO3
ATG
AS RTP

68
3.4.6 Ensaio Catalítico
Catalisadores do tipo LaNixFe1-xO3 foram submetidos a ensaio catalítico
para verificação do comportamento do mesmo quanto a estabilidade frente a reação
de oxidação parcial do metano.
Equipamento Utilizado
O teste catalítico foi realizado em uma unidade de avaliação catalítica para produção do gás de síntese, o esquema esta ilustrado na Figura 3.1.
Figura 3.1 - Esquema da linha reacional para oxidação parcial do metano.
Onde, HV 8015 e 8013 são válvulas; V 8000 e V 8001 são vasos
condensadores; PI 8016 e um indicador de pressão; PCV 8016 e uma válvula para
controle de pressão e CG e o cromatográfico.
A operação da linha de ensaio catalítico pode ser resumidamente
descrita da seguinte forma: utilizou-se um pequeno reator tubular de leito fixo,

69
construído em aço inoxidável colocando em sua base lã de quartzo para evitar a
passagem do material para a parte inferior do reator. Foram pesados 100mg do
catalisador a ser testado, o qual foi diluído em quartzo com a proporção 10:1
(quartzo: catalisador), sendo utilizado o quartzo com granulometria retida na malha
de 100 Mesh. Na Tabela 3.5 é mostrada as condições de operação do reator.
Tabela 3.5- Condições operacionais do sistema utilizadas na reação de oxidação
parcial do metano.
Condições Valores
Temperatura de ativação 900°C
Temperatura de reação 800°C
Pressão na cabeça do reator 1 atm
Fluxo de alimentação do metano 334 mL/min
Fluxo de alimentação do oxigênio 166 mL/min
Tempo de reação 18h
Oxidação Parcial do Metano
Inicialmente, o sistema foi purgado com nitrogênio com vazão de 200
mL/min durante aproximadamente 30 minutos para a retirada de oxigênio no
sistema. O monitoramento cromatográfico era feito a cada 15 minutos. Logo após,
mudou-se o fluxo para hidrogênio na mesma vazão anterior com velocidade espacial
de 120 SL/hr/g-cat) com rampa de aquecimento de 10°C/min até a temperatura de
900°C, mantendo o patamar de ativação de 2 horas. P osteriormente, mudou-se a
corrente gasosa de hidrogênio para nitrogênio mantendo-se a mesma vazão e
reduzindo a temperatura do sistema para 800°C. Após a estabilização da
temperatura em 800°C e comprovada a retirada do hid rogênio por análise
cromatográfica iniciou-se o procedimento de ajuste da carga (CH4: O2) sem passar
pelo reator para proteger o catalisador do contato com o oxigênio. Uma vez que as
vazões da carga estava correta, o mesmo foi alinhado ao reator. O ensaio foi
realizado por um tempo de 18 horas.

70
O cálculo de conversão total do metano foi realizado por balanço de
carbono, considerando desprezíveis as quantidades retidas na forma de coque,
sendo verificado através da análise termogravimétrica. Desta forma, o cálculo se
baseou no balanço de carbono no reator, no qual todo o carbono admitido ao
sistema como metano saia como dióxido de carbono, monóxido de carbono ou
metano não reagido, o que leva a fórmula de conversão abaixo.
onde: X%= conversão total de metano em porcentagem;
CH4s= número de mols de metano que sai do reator;
CH40= número de mols de metano que entra no reator, que por balanço de carbono
pode ser determinada pela equação 3.6:
onde: COs= numero de mols de monóxido de carbono que sai do reator;
CO2s= numero de mols de dióxido de carbono que sai do reator.
De acordo com o balanço de carbono, as seletividades dos produtos para CO e CO2
podem ser representadas pelas equações que seguem abaixo.
(Equação 3.5)
(Equação 3.7)
(Equação 3.6)

71
De acordo com o balanço de H2, a seletividade dos produtos para H2 e H2O pode ser
descrito a seguir pelas equações 3.9 e 3.10.
onde, nH2
S= numero de mols de hidrogênio que sai do reator; nH2O
S= numero de mols de água que sai do reator
(Equação 3.8)
(Equação 3.9)
(Equação 3.10)

72
4 RESULTADOS E DISCUSSÕES
4.1 Análise termogravimétrica
A curva TG do precursor catalítico LN3-P, Figura 4.1, obtida sob
atmosfera de ar, foi obtida com a finalidade de se determinar a melhor faixa de
temperatura para calcinação de todos os precursores envolvidos nesse trabalho. A
curva termogravimétrica do LN3-P mostra uma quantidade razoável de perda de
massa referente à eliminação de água residual que envolve a faixa de temperatura
de 30 a 180°C; uma outra faixa de perda de massa referente a decomposição de
Ni(OH)2 e La(OH)3 ocorreu entre 180 °C e 600 °C. Na faixa de 800 a 9 00 °C é
possível observar um ligeiro ganho de massa devido a reações de oxidação do Ni2+
para Ni3+.
A curva TG do precursor catalítico LN3–M (Figura 4.2) não apresentou
perda de massa considerável, isso ocorreu devido a boa interação que o
combustível uréia tem com o metal níquel, ocorrendo assim a reação de combustão
completa, atingindo elevadas temperatura (acima de 700 °C) e eliminando o máximo
de matéria orgânica do material.
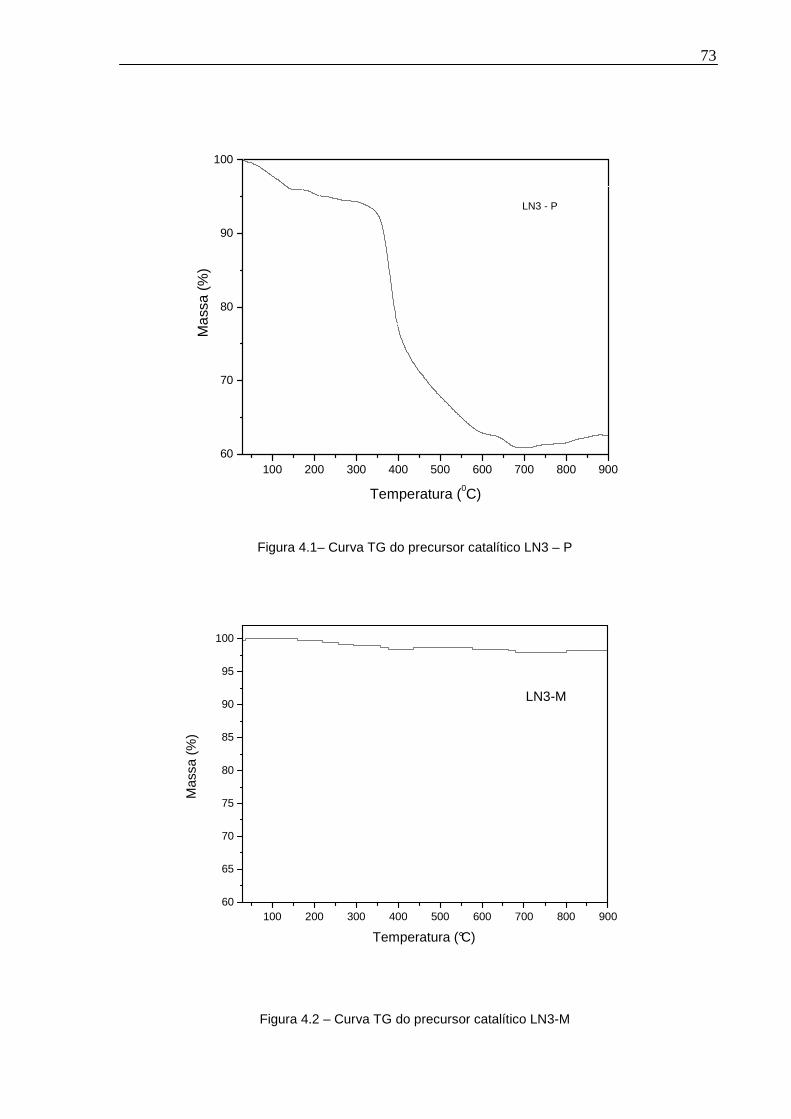
73
100 200 300 400 500 600 700 800 90060
70
80
90
100
Mas
sa (
%)
Temperatura (0C)
LN3 - P
Figura 4.1– Curva TG do precursor catalítico LN3 – P
100 200 300 400 500 600 700 800 90060
65
70
75
80
85
90
95
100
Mas
sa (
%)
Temperatura (°C)
LN3-M
Figura 4.2 – Curva TG do precursor catalítico LN3-M

74
A perda de massa apresentada pela amostra LF3–P (Figura 4.3) é
observada em quatro etapas entre 25 e 700 °C, aprox imadamente. A primeira etapa
ocorre entre 25 e 300 °C, sendo atribuído a perda d e água adsorvida, a inflexão da
curva mostra uma perda de 3%. A decomposição de material orgânico ocorre entre
300 e 500 °C, a perda de massa pode estar associado à combustão de carbono
residual, a inflexão da curva mostra uma perda de 7,5%. Entre 500 e 700 °C têm-se
duas outras perdas de massa de 6 e 4%, devido à formação de cristais LaFeO3. A
Perda de massa total apresentada pelo catalisador foi 20%.
Diferente da amostra LF3-P a amostra LF3 – M (Figura 4.4) apresentou
apenas três eventos de perda de massa. O primeiro evento em aproximadamente 90
°C, com inflexão da curva mostrando perda de 11 %, o segundo apresenta perda de
massa entre 250°C e 400°C, com 15% de perda. E fina lmente o terceiro evento
entre 500 e 700 °C. A perda de massa total apresent ada pelo catalisador LF3-M foi
de 42%.
100 200 300 400 500 600 700 800 90060
65
70
75
80
85
90
95
100
Mas
sa (
%)
Temperatura (°C)
LF3-P
Figura 4.3 - Curva TG do precursor catalítico LF3-P

75
100 200 300 400 500 600 700 800 90040
50
60
70
80
90
100
M
assa
(%
)
Temperatura (°C)
LF3-M
Figura 4.4 - Curva TG do precursor catalítico LF3-M
Para a amostra LN7F3 – P (Figura 4.5) observa-se perda de massa
entre 25 e 200 °C, sendo atribuída a perda de água adsorvida. A partir de 330 até
460 °C a curva apresenta uma maior inclinação, indi cando que outro processo esta
associado a perda de massa (27 %) neste intervalo de temperatura. Este processo é
a decomposição dos oxinitratos de lantânio (LaONO3 e La5O7NO3), formando La2O3.
Entre 460 e 700 °C observa-se uma perda de massa de aproximadamente 15 %
referente provavelmente à cristalização da estrutura do material. A perda de massa
total apresentada pelo catalisador LN7F3 – P foi de aproximadamente 51%. Já a
amostra LN7F3-M (Figura 4.6) apresentou perda de massa total de 30%.
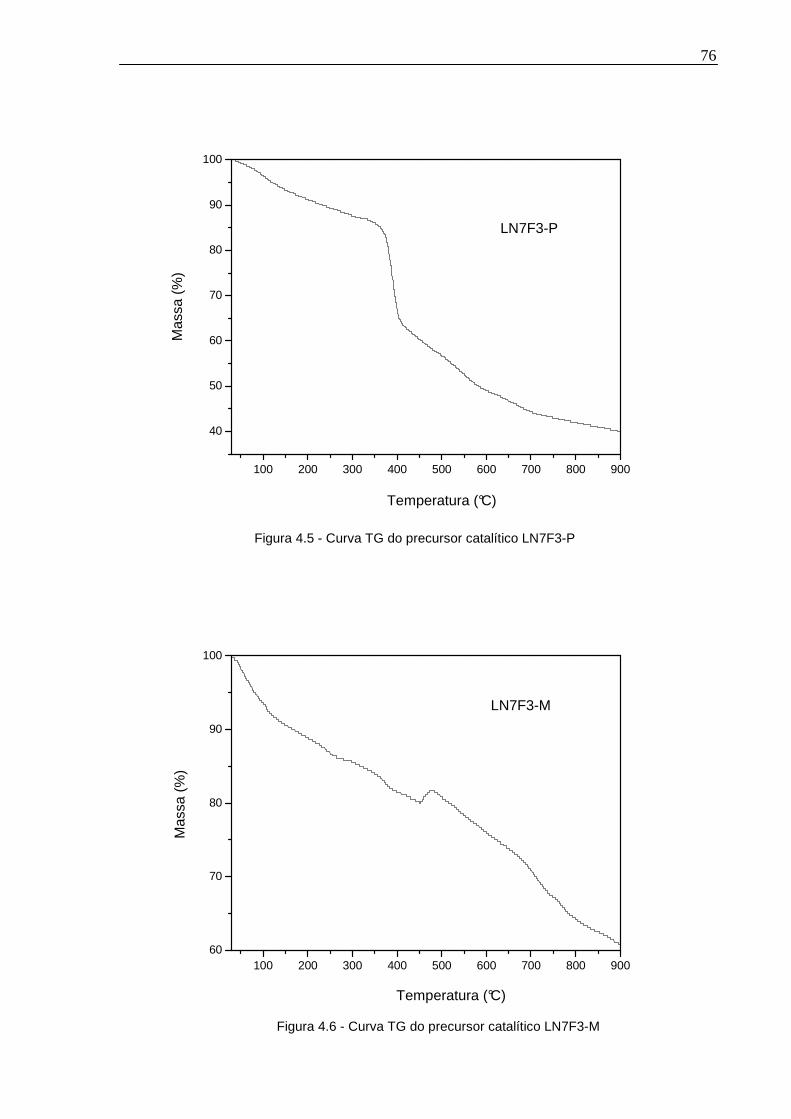
76
100 200 300 400 500 600 700 800 900
40
50
60
70
80
90
100
Mas
sa (
%)
Temperatura (°C)
LN7F3-P
Figura 4.5 - Curva TG do precursor catalítico LN7F3-P
100 200 300 400 500 600 700 800 90060
70
80
90
100
Mas
sa (
%)
Temperatura (°C)
LN7F3-M
Figura 4.6 - Curva TG do precursor catalítico LN7F3-M
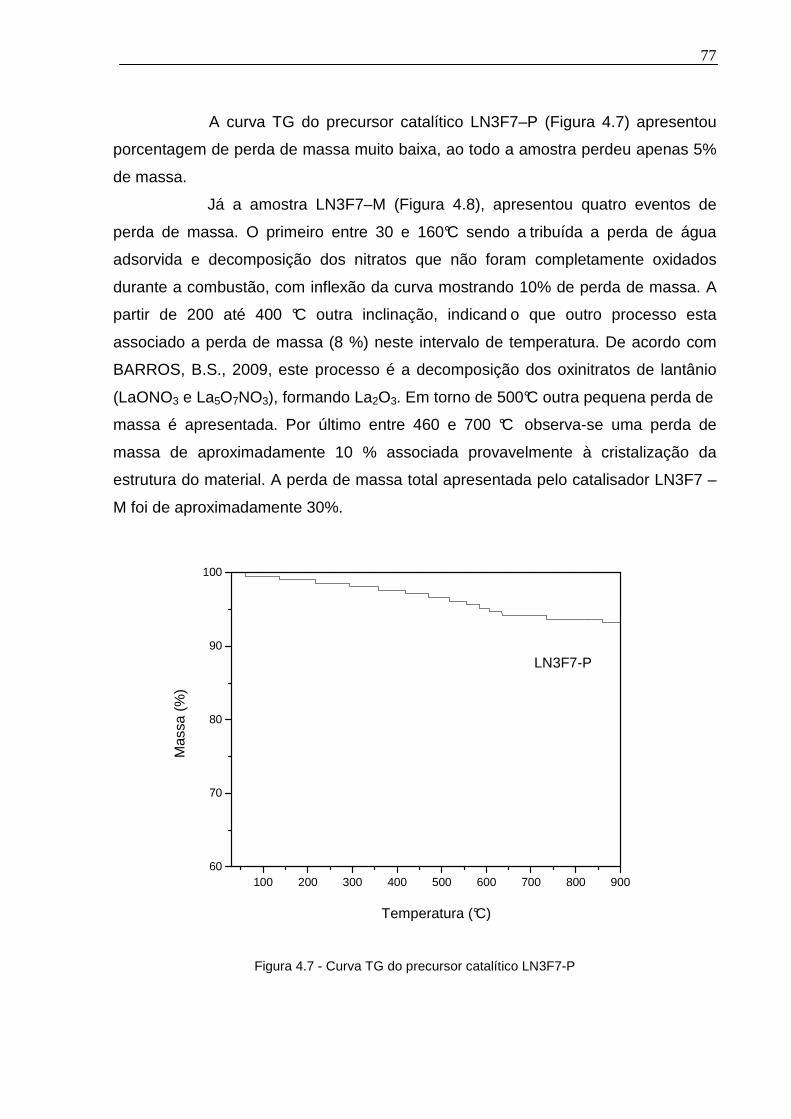
77
A curva TG do precursor catalítico LN3F7–P (Figura 4.7) apresentou
porcentagem de perda de massa muito baixa, ao todo a amostra perdeu apenas 5%
de massa.
Já a amostra LN3F7–M (Figura 4.8), apresentou quatro eventos de
perda de massa. O primeiro entre 30 e 160°C sendo a tribuída a perda de água
adsorvida e decomposição dos nitratos que não foram completamente oxidados
durante a combustão, com inflexão da curva mostrando 10% de perda de massa. A
partir de 200 até 400 °C outra inclinação, indicand o que outro processo esta
associado a perda de massa (8 %) neste intervalo de temperatura. De acordo com
BARROS, B.S., 2009, este processo é a decomposição dos oxinitratos de lantânio
(LaONO3 e La5O7NO3), formando La2O3. Em torno de 500°C outra pequena perda de
massa é apresentada. Por último entre 460 e 700 °C observa-se uma perda de
massa de aproximadamente 10 % associada provavelmente à cristalização da
estrutura do material. A perda de massa total apresentada pelo catalisador LN3F7 –
M foi de aproximadamente 30%.
100 200 300 400 500 600 700 800 90060
70
80
90
100
Mas
sa (
%)
Temperatura (°C)
LN3F7-P
Figura 4.7 - Curva TG do precursor catalítico LN3F7-P

78
100 200 300 400 500 600 700 800 90040
50
60
70
80
90
100
Mas
sa (
%)
Temperatura (°C)
LN3F7-M
Figura 4.8 - Curvas TG e DTG do precursor catalítico LN3F7 – M
4.2 Difração de Raios-X
Com a intenção de promover a cristalização das fases desejadas, as
amostras preparadas tanto pelo método pechini quanto pelo método microondas
foram calcinadas a 900°C por 4h e os respectivos di fratogramas são apresentados
nas Figuras 4.9, 4.10, 4.11 e 4.12.
As amostras LF9 – P e LF9-M (Figura 4.9) apresentaram apenas a fase
perovisquita LaFeO3, sem traços de fases secundárias. Todos os picos foram
indexados e estão em acordo com a ficha ICSD n° 74- 2203 da perovisquita LaFeO3
com estrutura Ortorrômbica e grupo espacial Pbnm.

79
10 20 30 40 50 60 70 80
(116
)
(040
)(204
)
(004
)
(022
)
(112
)(a)
(b)
Inte
nsid
ade
(u.a
.)
2 θ (graus)
(002
)
(116
)
(040
)
(204
)
(004
)
(022
)
(002
)
(112)
Figura 4.9 – Difratograma de raios-X para as amostras (a) LF9 – P e (b) LF9 – M
Após calcinação a 900 oC por 4 h das amostras LN9 –P e LN9 –M (Figura
4.10) todos os picos foram indexados e estão em acordo com a ficha ICSD n°33-
0711 da perovisquita LaNiO3 com estrutura romboédrica e grupo espacial R-3m.
Uma inspeção cuidadosa destes perfis de difração revela algumas
características: (i) uma mudança muito pequena para maiores ângulos 2�, devido à
contração da microestrutura, é observado após a substituição dos íons Fe3+ na
estrutura por Ni3+ íons e (ii) metade das linhas de difração aumenta a sua largura
total após a substituição de Fe3+ por Ni3 +. Esta discussão está de acordo com a
tendência geral de que o grau de substituição do níquel está relacionada a esses
efeitos e corresponde a cerca de modificação da estrutura. O deslocamento do pico
para ângulos 2� maiores, em função do aumento da quantidade de Níquel, sugere
uma diminuição na distância interplanar, ou seja, o volume da célula unitária diminui
devido a presença de Ni3 +. PROVENDIER e colaboradores [2006] atribuem a
dependência linear dos parâmetros de rede com o grau de substituição.

80
10 20 30 40 50 60 70 80
(223
)
(012
)
(223
)
(312
)(0
24)
(122
)
(211
)
(202
)
(003
)(0
21)
(110
)
Inte
nsid
ade
(u.a
.)
2 θ (graus)
(b)
(a)
(101
)
(312
)
(024
)
(122
)
(211
)(202
)
(101
)
(110)
Figura 4.10 – Difratograma de raios-X para as amostras (a) LN9 – P e (b) LN9-M
As curvas de DRX das amostras LN7F3-900-P e LN7F3-900-M são
mostradas na Figura 4.11. É possível observar que além da formação da fase
desejada LaNi0,7Fe0,3O3 (ICSD n° 88-0636) com estrutura romboédrica e grupo
espacial R-3c, foram também obtidas as fases La2NiO4 (ICSD n° 33-0712) com
estrutura tetragonal e grupo espacial I4/mmm e La2O3 (ICSD n° 05-0602) com
estrutura hexagonal e grupo espacial P-3m1.
Segundo CUI H. e colaboradores [2002] a progressiva substituição de
íons de ferro na perovisquita LaFeO3 por íons de níquel provoca uma distorção no
alinhamento do octaédro para a estrutura romboédrica da fase LaNiO3. Aumentando
mais a quantidade de íons Ni pode ocorrer a formação de três fases: La2NiO4, La2O3
e NiO. Em concentrações ainda maiores de íons Ni no composto, a estrutura LaFeO3
não pode estabilizar os íons Ni 3+ instáveis levando à formação de NiO divalentes e
La2NiO4.
Durante a reação de combustão o ambiente redutor gerado pela
decomposição do combustível impede ou pelo menos dificulta a oxidação do Ni2+,
estado de oxidação original do níquel em solução aquosa, em Ni3+ necessário para a

81
cristalização da estrutura perovisquita LaNiO3. Como conseqüência são formados
preferencialmente La2NiO4 e/ou NiO.
O refinamento dos difratogramas confirmou a presença de fases secundárias
observadas nas amostras LN7F3-900-P e LN7F3-900-M. De acordo com o dados
obtidos pelo refinamento, observa-se a presença de 41,6% de LaNi0,7Fe0,3O3; 30,7%
de La2NiO4 e 27,7 % de La2O3. Portanto, observa-se uma boa correlação entre os
difratogramas experimentais e refinados, indicando boa reprodutibilidade e
confiabilidade, isto é, o modelo de parâmetros foram representativos de estrutura
cristalina (baixos RBragg) e das características da amostra.
10 20 30 40 50 60 70 80
*# o
###
*
#
ooooo
o
o
*
(a)
(b)
Inte
nsid
ade
(u.a
.)
2 θ (graus)
*o#
* LaNi0,7Fe0,3O3o La2O3# La2NiO4
*# o
###
*
#
ooooo
o
o
*
(a)
*o#
* LaNi0,7Fe0,3O3o La2O3# La2NiO4
*# o
###
*
#
ooooo
o
o
*
*o# *# o
###
*
#
ooooo
o
o
*
*o#
Figura 4.11 – Difratograma de raios-X para as amostras (a) LN7F3-900-P e (b) LN7F3-900-M
As curvas de DRX das amostras LN3F7-900-P e LN3F7-900-M são
mostradas na Figura 4.12. Observa-se que além da formação da fase desejada
LaNi0,3Fe0,7O3 (ICSD n° 88-0639) com estrutura Ortorrômbica e grup o espacial
Pbnm, foi também obtida a fase La2O3 (ICSD n° 05-0602) com estrutura hexagonal
e grupo espacial P-3m1 .
De acordo com o dados obtidos pelo refinamento, observa-se a
formação de 70,0% de LaNi0,3Fe0,7O3 e 30,0 % de La2O3.
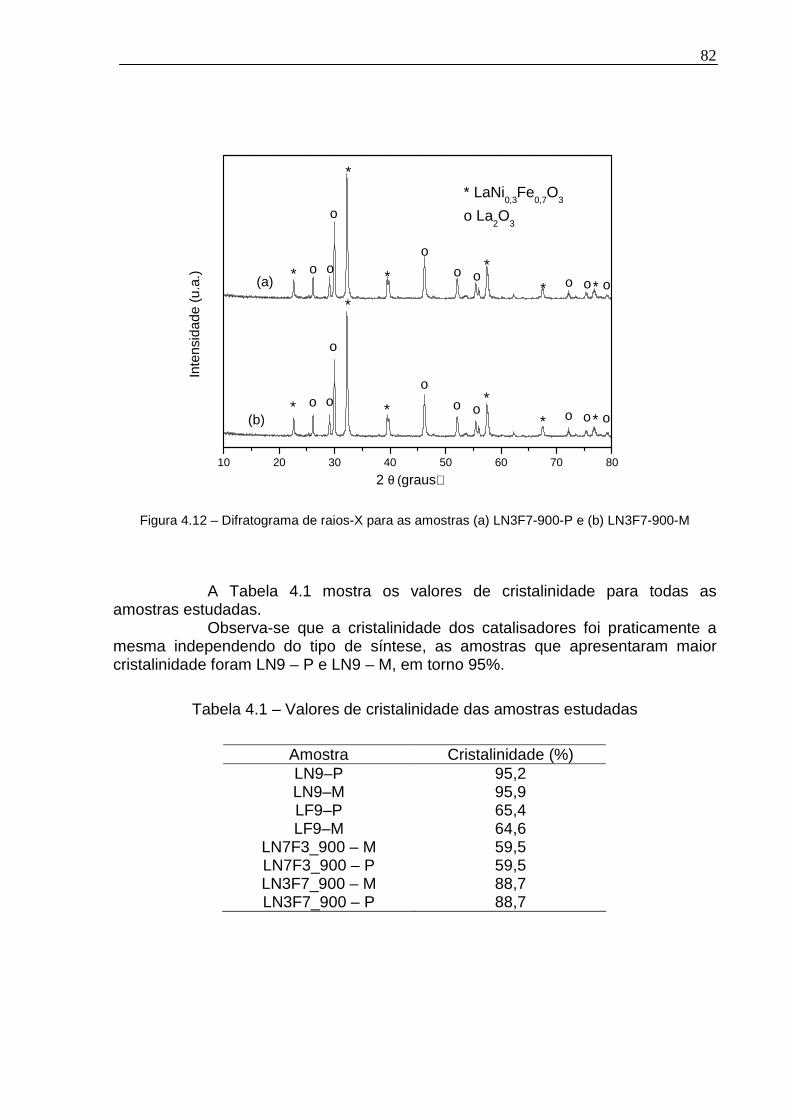
82
10 20 30 40 50 60 70 80
o *o o*
*oo
o
*
*
o
o(a)
(b)
Inte
nsid
ade
(u.a
.)
2 θ (graus)
* o
* LaNi0,3
Fe0,7
O3
o La2O
3
o *o o*
*oo
o
*
*
o
o* o
Figura 4.12 – Difratograma de raios-X para as amostras (a) LN3F7-900-P e (b) LN3F7-900-M
A Tabela 4.1 mostra os valores de cristalinidade para todas as
amostras estudadas. Observa-se que a cristalinidade dos catalisadores foi praticamente a
mesma independendo do tipo de síntese, as amostras que apresentaram maior cristalinidade foram LN9 – P e LN9 – M, em torno 95%.
Tabela 4.1 – Valores de cristalinidade das amostras estudadas
Amostra Cristalinidade (%) LN9–P 95,2 LN9–M 95,9 LF9–P 65,4 LF9–M 64,6
LN7F3_900 – M 59,5 LN7F3_900 – P 59,5 LN3F7_900 – M 88,7 LN3F7_900 – P 88,7

83
4.3 Espectroscopia de absorção na região do infravermelho
Os espectros de absorção na região do infravermelho podem ser vistos
nas Figuras 4.13, 4.14, 4.15 e 4.16. Ao analisar os espectros FTIR do material LN3-
P (Figura 4.13) observa-se uma banda larga entre 3700 e 3000 cm-1 a qual pode ser
atribuída às bandas de estiramento simétrico O-H do citrato e da água (material
remanescente da síntese). As bandas observadas na região de 1538-1350 cm-1
nestes espectros podem ser relacionadas à coordenação dos cátions Ni3+ pelos
grupos carboxílicos na forma de um complexo bidentado. Em aproximadamente 642
cm-1 é observado outra banda de vibração.
Para a amostra LN3 – M foram observadas as mesmas bandas
apresentadas por LN3 – P, porém foi identificada a presença de CO2 com
deformação axial assimétrica em 2350 cm -1.
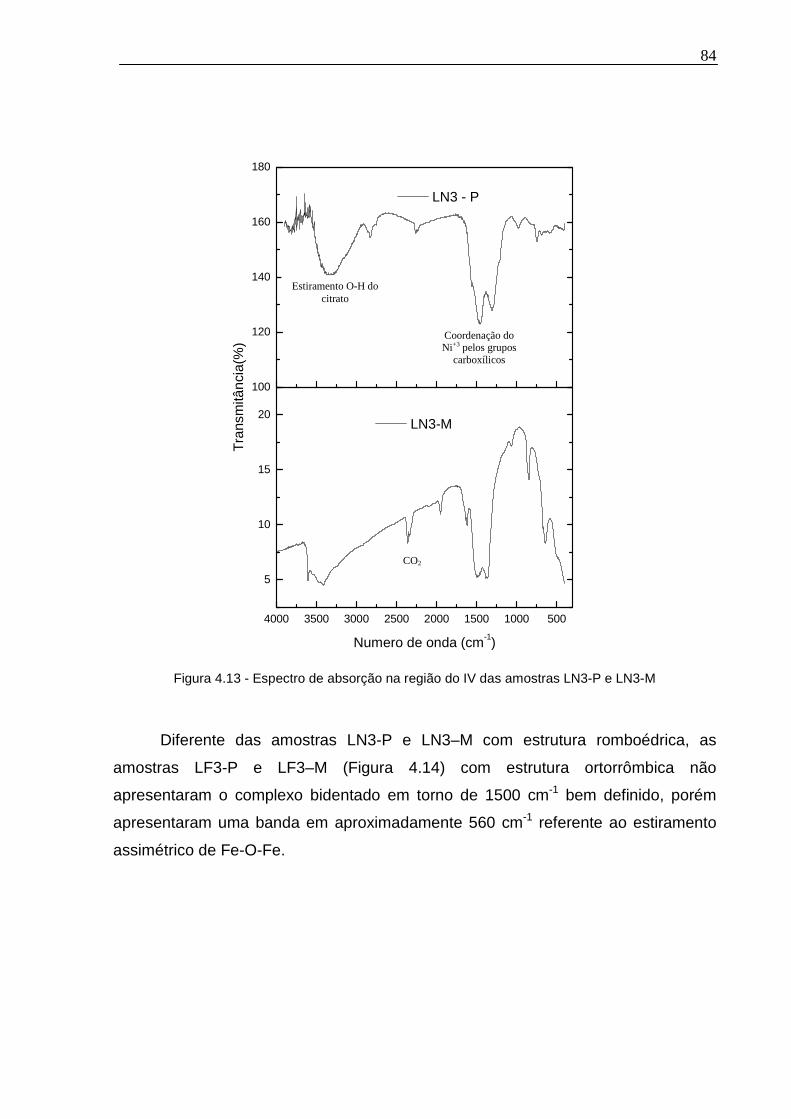
84
4000 3500 3000 2500 2000 1500 1000 500
5
10
15
20
100
120
140
160
180
Tra
nsm
itânc
ia(%
)
Numero de onda (cm-1)
LN3-M
LN3 - P
Figura 4.13 - Espectro de absorção na região do IV das amostras LN3-P e LN3-M
Diferente das amostras LN3-P e LN3–M com estrutura romboédrica, as
amostras LF3-P e LF3–M (Figura 4.14) com estrutura ortorrômbica não
apresentaram o complexo bidentado em torno de 1500 cm-1 bem definido, porém
apresentaram uma banda em aproximadamente 560 cm-1 referente ao estiramento
assimétrico de Fe-O-Fe.
Coordenação do Ni+3 pelos grupos
carboxílicos
Estiramento O-H do citrato
CO2

85
4000 3500 3000 2500 2000 1500 1000 5000
10
20
30
0
10
20
30
40
Numero de onda (cm-1)
LF3-M
Tra
nsm
itânc
ia (
%)
LF3 - P
Figura 4.14 - Espectro de absorção na região do IV das amostras LF3-P e LF3-M
Em relação as amostras LN7F3 – P e LN7F3 – M (Figura 4.15), como a
concentração de níquel é bem maior que a de ferro (x=0,7), os espectros na região
do infravermelho se assemelham bastante as amostras LN3 – P e LN3 – M onde o
níquel se encontra em uma concentração de 100 %, isto é x=1.
A banda de absorção de 560 cm -1 da amostra LF3 é deslocada para
592 cm -1 no espectro da amostra LN7F3, devido à substituição íons de níquel por
ferro.
Já para as amostras LN3F7 – P e LN3F7 – M (Figura 4.16) como a
concentração do ferro é bastante elevada em relação a concentração do níquel
Estiramento O-H do citrato
CO2 Estiramento Fe-O-Fe
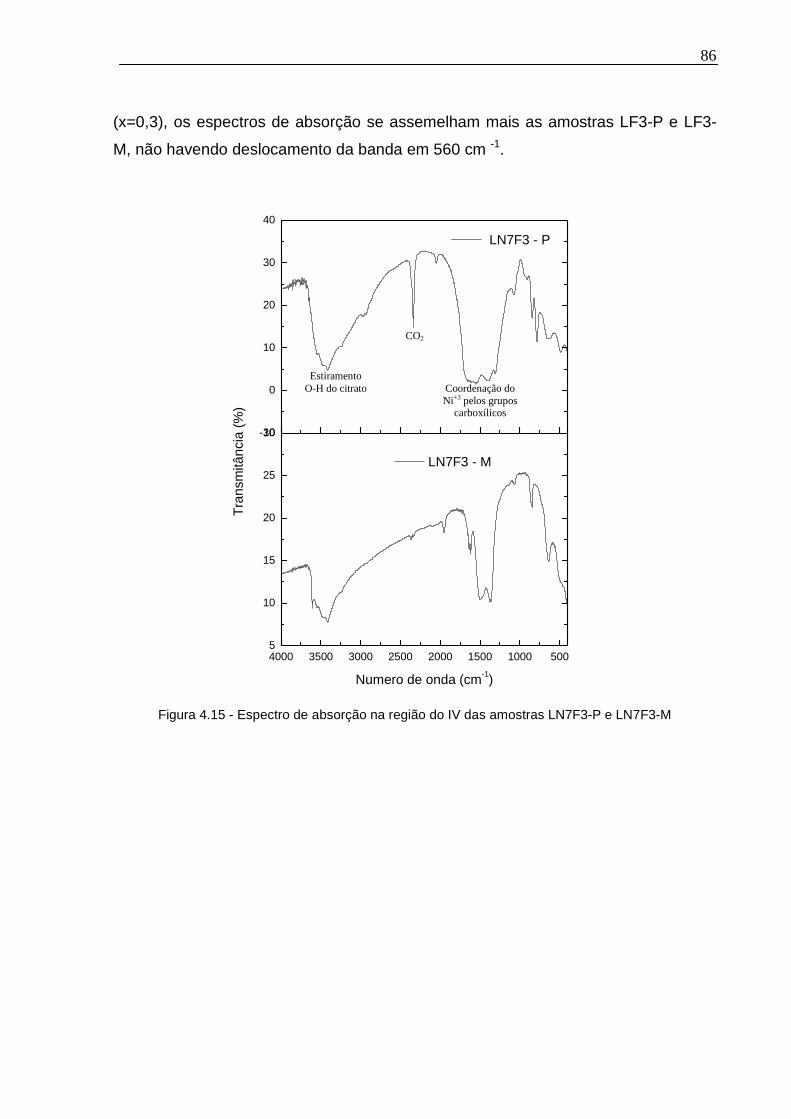
86
(x=0,3), os espectros de absorção se assemelham mais as amostras LF3-P e LF3-
M, não havendo deslocamento da banda em 560 cm -1.
4000 3500 3000 2500 2000 1500 1000 5005
10
15
20
25
30-10
0
10
20
30
40
Numero de onda (cm-1)
LN7F3 - M
Tra
nsm
itânc
ia (
%)
LN7F3 - P
Figura 4.15 - Espectro de absorção na região do IV das amostras LN7F3-P e LN7F3-M
Coordenação do Ni+3 pelos grupos
carboxílicos
Estiramento O-H do citrato
CO2
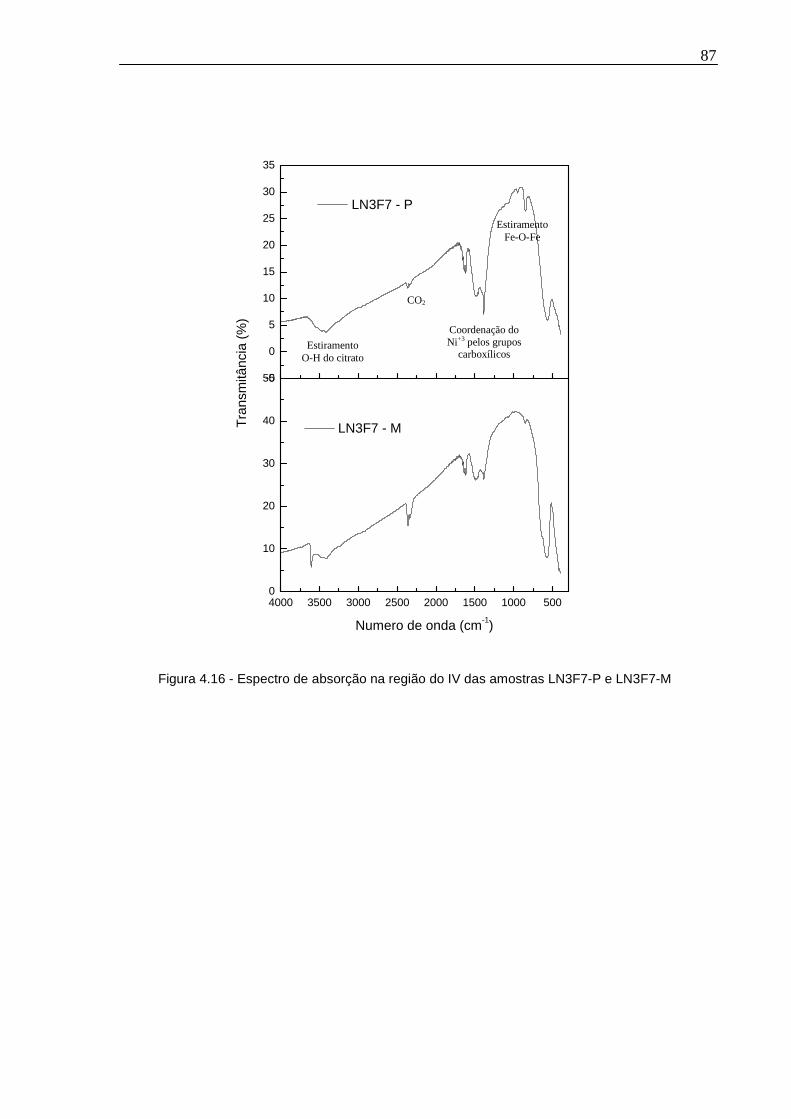
87
4000 3500 3000 2500 2000 1500 1000 5000
10
20
30
40
50-5
0
5
10
15
20
25
30
35
Tra
nsm
itânc
ia (
%)
Numero de onda (cm-1)
LN3F7 - M
LN3F7 - P
Figura 4.16 - Espectro de absorção na região do IV das amostras LN3F7-P e LN3F7-M
Estiramento Fe-O-Fe
Coordenação do Ni+3 pelos grupos
carboxílicos
Estiramento O-H do citrato
CO2

88
4.4 Área superficial específica
A Tabela 4.3 apresenta os resultados da área BET das amostras
estudadas.
Observa-se que todas as amostras apresentaram baixos valores de
área superficial (< 5,3 m2/g) provavelmente como conseqüência da alta temperatura
de calcinação como também da elevada temperatura obtida durante a
autocombustão. Estes resultados estão de acordo com o que foi estudado por LIMA
E ASSAF [2002], G. PECCHI e colaboradores [2008], PROVENDIER e
colaboradores [1999].
As amostras obtidas pelo método pechini apresentaram valores de
área superficial ligeiramente maiores que as obtidas pela autocombustão assistida
por microondas.
Observa-se também a influência da substituição parcial de Ni por Fe
nos valores de área superficial específica da perovisquita LaNixFex-1O3. Em todas as
amostras a substituição de Ni por Fe (x=1; x=0,7, x=0,3 e x=0) aumentou a área de
superfície da perovisquita. Um resultado semelhante foi descrito por Lima e Assaf
(2002).
Tabela 4.2 - Resultados da área superficial específica para as perovisquitas LN9,
LF9, LN7F3 e LN3F7 obtidas pelo método pechini e autocombustão assistida por
microondas.
Catalisadores Área BET (m 2 g-1)
LN9-P 5,3 LN9-M 4,7
LF9-P 2,3 LF9-M 1,2
LN7F3-P 4,7 LN7F3-M 4,4
LN3F7-P 3,8 LN3F7-M 3,2

89
4.5 Redução à temperatura programada (RTP)
As Figuras 4.17 e 4.18 mostram os perfis de redução por temperatura
programada para as amostras LN9-P e LN9-M. De acordo com a curva RTP da
amostra LN9 – P podemos observar dois picos de redução um a aproximadamente
355 ºC e outro a 467 ºC. Estes picos correspondem a sucessivas mudanças na
estrutura perovisquita: o primeiro pico pode ser atribuído a formação da estrutura
La4Ni3O10 de acordo com a seguinte reação:
4LaNiO3 + 2H2 → La4Ni3O10 + Ni0 + 2H2O
A segunda etapa corresponde a formação de uma fase espinélio
La2NiO4 e logo após ocorre a redução desta fase para níquel metálico entre 400 e
530 ºC:
La4Ni3O10 + 3H2 → La2NiO4 + 2Ni0 + La2O3 + 3H2O
La2NiO4 + H2 → Ni0 + La2O3 + H2O
De acordo com estes resultados de perfil RTP a completa redução do
LaNiO3 em La2O3 e níquel metálico ocorrem a aproximadamente 510 ºC.
Para a amostra LN9 – M, apresentada na Figura 4.18 observa-se um
pequeno deslocamento no pico de redução de 510 °C p ara 540 °C.
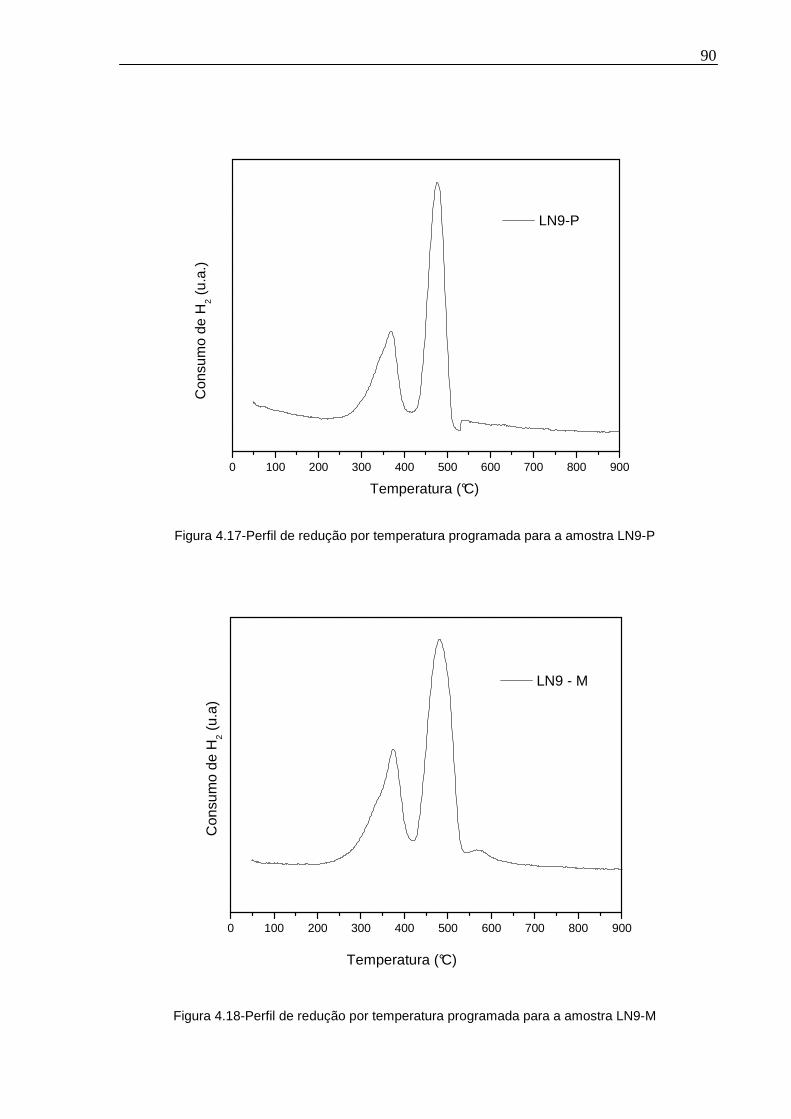
90
0 100 200 300 400 500 600 700 800 900
Temperatura (°C)
LN9-P
Con
sum
o de
H2 (
u.a.
)
Figura 4.17-Perfil de redução por temperatura programada para a amostra LN9-P
0 100 200 300 400 500 600 700 800 900
Temperatura (°C)
Con
sum
o de
H2
(u.a
)
LN9 - M
Figura 4.18-Perfil de redução por temperatura programada para a amostra LN9-M

91
A Figura 4.19 mostra perfis de TPR das amostras LF9 – P e LF9-M.
Diferente das perovisquitas a base de níquel (LN9 – P e LN9-M), a perovisquitas a
base de ferro (LF9 – P e LF9-M) apresentaram apenas um pico de redução.
Na Figura 4.19 nota-se que a perovisquita LaFeO3 é quase irredutível
em nossas condições de análise de TPR. A redução da LaFeO3 começa a 750 °C e
é não se completa a 1000 °C. Resultados similares f oram reportados por Provendier
e colaboradores (1999) e Ciambelli e colaboradores (2001).
100 200 300 400 500 600 700 800 900 1000
Con
sum
o de
H2
(u.a
)
(b)
Temperatura (°C)
(a)
Figura 4.19-Perfil de redução por temperatura programada para as amostras (a) LF9 – P e (b) LF9 –
M
Os perfis de redução das amostras LN7F3 – 900 – P e LN7F3 – 900 –
M; LN7F3 – 900 – P e LN7F3 – 900 – M estão apresentados nas Figuras 4.20 e
4.21. Observa-se que o primeiro pico de redução é semelhante para todos os óxidos
contendo níquel (LN9 (x=1), LN7F3 (x=0,7) e LN3F7(x=0,3)), apesar de a
temperatura de consumo máximo de hidrogênio diminuir com a redução da
concentração de níquel (420 °C, 400°C e 380 °C), re spectivamente.
Para as amostras LN7F3 (x=0,7) e LN3F7(x=0,3), o segundo pico
apresentou uma ampla faixa de redução. De acordo com PROVENDIER e
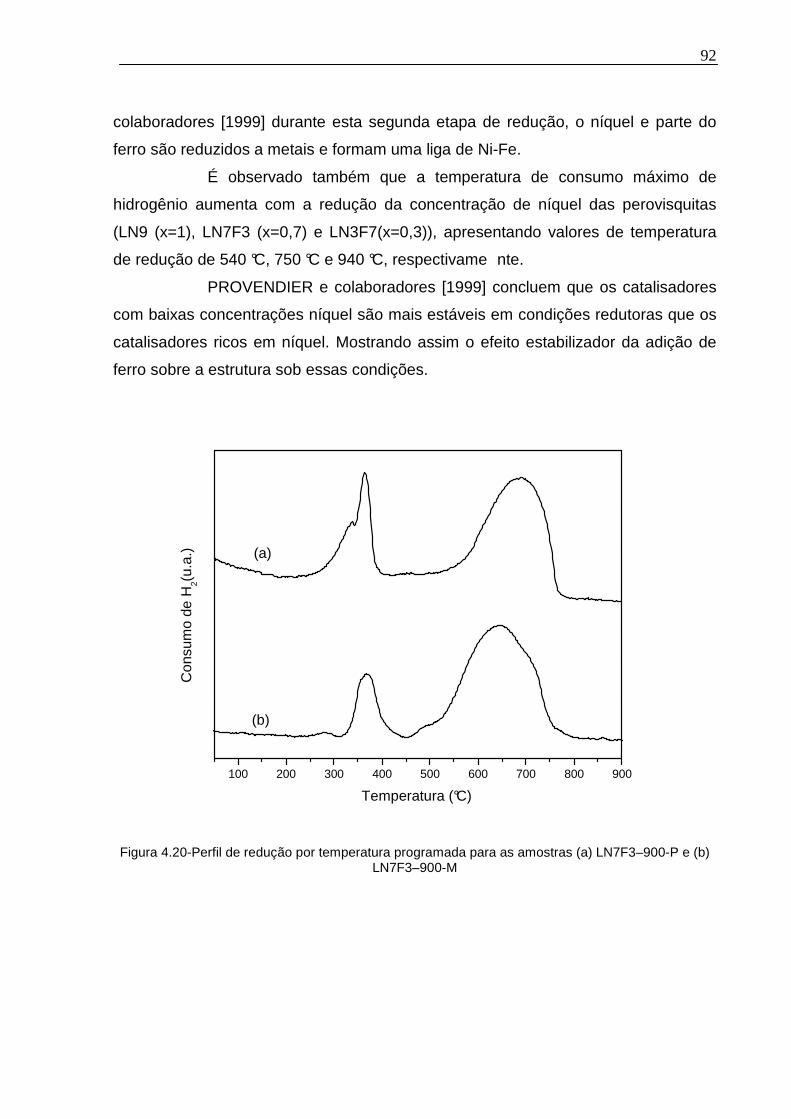
92
colaboradores [1999] durante esta segunda etapa de redução, o níquel e parte do
ferro são reduzidos a metais e formam uma liga de Ni-Fe.
É observado também que a temperatura de consumo máximo de
hidrogênio aumenta com a redução da concentração de níquel das perovisquitas
(LN9 (x=1), LN7F3 (x=0,7) e LN3F7(x=0,3)), apresentando valores de temperatura
de redução de 540 °C, 750 °C e 940 °C, respectivame nte.
PROVENDIER e colaboradores [1999] concluem que os catalisadores
com baixas concentrações níquel são mais estáveis em condições redutoras que os
catalisadores ricos em níquel. Mostrando assim o efeito estabilizador da adição de
ferro sobre a estrutura sob essas condições.
100 200 300 400 500 600 700 800 900
Con
sum
o de
H2(u
.a.)
Temperatura (°C)
(a)
(b)
Figura 4.20-Perfil de redução por temperatura programada para as amostras (a) LN7F3–900-P e (b) LN7F3–900-M

93
100 200 300 400 500 600 700 800 900 1000
Con
sum
o de
H2 (u
.a.)
Temperatura (°C)
(a)
(b)
Figura 4.21-Perfil de redução por temperatura programada para as amostras (a) LN3F7–900-P e (b) LN3F7–900-M

94
4.6 Reação de oxidação parcial do metano
Os catalisadores LN9, LF9, LN7F3 e LN3F7 obtidos pelos métodos
pechini e microondas calcinados a 900 °C por 4h for am testados frente à reação de
oxidação parcial do metano a temperatura de 800 °C. Conversão de CH4,
seletividade a CO e CO2 e razão H2/CO foram analisados em função do tempo de
reação (18h). Os resultados são apresentados nas Figuras de 4.22 a 4.31.
Na Figura 4.22 têm-se os resultados da amostra LN9 – P é possível
observar a conversão de CH4 diminuiu com o tempo de reação até atingir um estado
estacionário, onde se manteve constante em aproximadamente 70% durante todo o
teste catalítico. Esta diminuição sugere que o catalisador continua sendo ativado
durante as primeiras horas de reação.
Os resultados de seletividade para a formação de CO, CO2 e a razão
H2/CO podem ser explicados pelo mecanismo da reação de oxidação parcial do
metano, conforme sugerido por [MATTOS e colaboradores, 2002] e apresentado nas
equações 45, 46 e 47.
Inicialmente, ocorre a combustão completa do metano com formação
de CO2 e H2O. Na segunda etapa, ha a formação do gás de síntese a partir das
reações de reforma com CO2 e a reforma a vapor do metano não reagido na
primeira etapa do processo. Por isso que ao longo do tempo, a quantidade de CO
aumenta até estabilizar e a de CO2 apresenta o perfil oposto.
(Equação 45)
(Equação 46)
(Equação 47)

95
Havendo formação excessiva de coque, a segunda etapa do processo
é inibida ocorrendo apenas à formação de dióxido de carbono e H2O. Desta maneira,
quando altos valores de seletividade para a formação de CO2 são obtidos, significa
que a segunda etapa da reação está sendo prejudicada. No caso da amostra em
estudo, verificou-se alta seletividade para a formação de CO enquanto que para CO2
foram encontrados baixos valores, indicando que não ocorreu desativação por
formação de coque na amostra.
Com base nestes resultados sugerimos que o catalisador LN9 – P
apresentou alta atividade em termos de conversão de metano, assim como uma
maior resistência a uma possível formação de depósitos de carbono.
0 2 4 6 8 10 12 14 16 180
10
20
30
40
50
60
70
80
90
100
Conversao do CH4
Seletividade para CO Seletividade para CO
2
%
Tempo (h)
Figura 4.22 – Conversão do metano e seletividade para CO e CO2 usando o catalisador LN9-P
Para a perovisquita LaNiO3 obtida pelo método de autocombustão
assistida por microondas (LN9-M) a seletividade para CO e CO2 também se
estabilizou em torno de 75% e 25%, respectivamente, já a conversão do CH4
diminuiu durante as três primeiras horas de reação e depois se estabilizou em torno
de 50%.
Na Figura 4.24 tem-se o gráfico com as curvas de H2/CO para os
catalisadores LN9 – P e LN9 –M, observa-se que para o catalisador LN9 – P a razão

96
H2/CO se estabilizou em torno de 2, já o catalisador LN9 – M em torno de 1,5,
indicando que o catalisador obtido pelo método pechini tem razão adequada para
ser insumo na reação de síntese de Fischer-Tropsh.
Neste período de tempo sob reação, os catalisadores foram estáveis
não sendo observada variações consideráveis nos indicadores de performance ou
indícios de desativação.
0 2 4 6 8 10 12 14 16 180
10
20
30
40
50
60
70
80
90
100
Conversao do CH4
Seletividade para CO Seletividade para CO
2
%
Tempo (h)
Figura 4.23 – Conversão do metano e seletividade para CO e CO2 usando o catalisador LN9-M

97
0 2 4 6 8 10 12 14 16 180
1
2
3
4
5
LN9 - M LN9 - P
H2/C
O
Tempo (h)
Figura 4.24 – Razão entre H2 e CO utilizando o catalisador LN9
É possível observar na Figura 4.25 que a amostra LN7F3–900-P
apresentou um comportamento muito irregular, a conversão de CH4, por exemplo,
apresentou valor mínimo em 5% e valor máximo em 31%. Comparando-se o
resultado de conversão de metano da amostra LN9 – P (Figura 4.22), onde (x=1) e o
resultado da amostra LN7F3–900-P, onde x=0,7, esta perdeu bastante atividade, isto
ocorre devido ao maior numero de espécies ativas de níquel disponível na amostra
LN9 – P, onde a concentração de Ni é maior.
A redução parcial dos óxidos contendo níquel pode promover melhor
dispersão e controle do tamanho de partícula da espécie ativa. É de se esperar que
os níveis de conversão de metano obtidos durante a reforma catalítica estejam
associados ao teor de níquel reduzido. Entretanto, teores elevados de níquel
reduzido podem facilitar a sinterização e crescimento de partícula conduzindo a
formação de carbono e conseqüentemente a desativação do catalisador.
Os valores máximos de seletividade em CO e CO2 foram de
aproximadamente 60 e 70%, respectivamente.

98
0 2 4 6 8 10 12 14 16 180
10
20
30
40
50
60
70
80
90
100
%
Tempo (h)
Conversao do CH4
Seletividade para CO Seletividade para CO
2
Figura 4.25 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador LN7F3-900-P
Na Figura 4.26 têm-se os resultados da amostra LN7F3-900-M é
possível observar que diferente da amostra obtida pelo método pechini (Figura 4.25),
a conversão de CH4 se manteve em torno de 7% durante todo o período de reação.
Esta baixa conversão de metano pode estar associada à formação de fase
secundárias como: La2NiO4 e La2O3, fases estas que foram confirmadas por DRX.
Os resultados de seletividade para CO e CO2 foram bem próximos, em
torno 50%.
A razão H2/CO (Figura 4.27) tanto para a amostra LN7F3-900-P quanto
para a amostra LN7F3-900-M, foi inferior a 2.
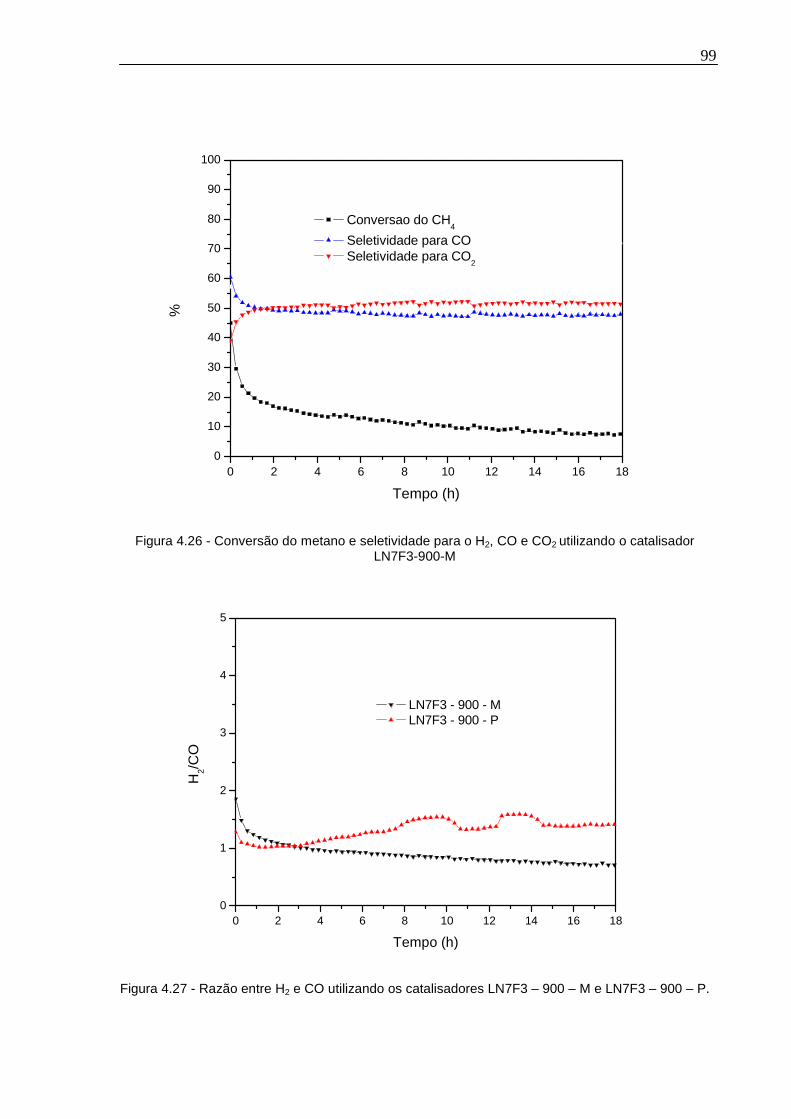
99
0 2 4 6 8 10 12 14 16 180
10
20
30
40
50
60
70
80
90
100
Conversao do CH4
Seletividade para CO Seletividade para CO
2
%
Tempo (h)
Figura 4.26 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador
LN7F3-900-M
0 2 4 6 8 10 12 14 16 180
1
2
3
4
5
LN7F3 - 900 - M LN7F3 - 900 - P
H2/
CO
Tempo (h)
Figura 4.27 - Razão entre H2 e CO utilizando os catalisadores LN7F3 – 900 – M e LN7F3 – 900 – P.

100
Os catalisadores LN3F7-900-P (Figura 4.28) e LN3F7-900-M (Figura
4.29) apresentaram baixos valores de conversão do CH4, 8% e 2%,
respectivamente. Como foi mostrado nos resultados de TPR, a 900°C estas
amostras não foram reduzidas completamente, como a temperatura de reação de
oxidação parcial foi de 800°C, nem tudo foi reduzid o a esta temperatura, logo estes
catalisadores têm menos fases ativas, resultando em baixa porcentagem de
conversão do CH4.
Podemos verificar que o perfil das curvas de seletividade em CO e CO2
são opostos, com aumento da seletividade de CO ocorre uma diminuição da
seletividade de CO2. Esta relação entre as seletividades de CO e CO2 sugere a
ocorrência de reações que propiciem a obtenção de um deles em função do
consumo do outro. Desta forma é presumível a ocorrência da reação de
deslocamento gás-água (CO + H2O ↔ CO2 + H2, equação 11) e/ou da reação de
Boudouard (2CO ↔ C + CO2, equação 13).
0 2 4 6 8 10 12 14 16 180
10
20
30
40
50
60
70
80
90
100
%
Tempo (h)
Conversao do CH4
Seletividade para CO Seletividade para CO
2
Figura 4.28 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador LN3F7 – 900 – P

101
É possível estabelecer uma relação entre os níveis observados de
conversão de metano e o teor de níquel reduzido e conseqüentemente entre a
estabilidade da estrutura perovisquita e o método de preparação. Maior quantidade
de níquel reduzido pode significar maior quantidade de sítios ativos, mas também
maior possibilidade de sinterização levando ao crescimento das partículas da
espécie ativa.
A Figura 4.30 mostra as razões molares H2/CO para os dois
catalisadores LN3F7-900-P e LN3F7-900-M foram inferiores a 1.
Razões molares H2/CO menores que 1 e conversão de CO2 maior que
de CH4 como é o caso dos dois catalisadores LN3F7-900-P e LN3F7-900-M,
indicam a ocorrência da reação reversa de deslocamento gás-água “RWGS” (CO2 +
H2 ↔ CO + H2O, Equação 12), enquanto que razões molares H2/CO maiores que 1 e
níveis de conversão de CH4 maior que de CO2 podem estar relacionadas com a
reação de craqueamento do metano (CH4 ↔ C + 2H2, Equação 14).
De uma maneira geral os catalisadores obtidos pelo método pechini
apresentaram melhores resultados de conversão que os catalisadores obtidos pelam
autocombustão assistida por microondas.

102
0 2 4 6 8 10 12 14 16 180
10
20
30
40
50
60
70
80
90
100
%
Tempo (h)
Conversao do CH4
Seletividade para CO Seletividade para CO
2
Figura 4.29 - Conversão do metano e seletividade para o H2, CO e CO2 utilizando o catalisador LN3F7 – 900-M
0 2 4 6 8 10 12 14 16 180
1
2
3
4
5
H2/
CO
Tempo (h)
LN3F7 - 900 - M LN3F7 - 900 - P
Figura 4.30- Razão entre H2 e CO utilizando os catalisadores LN3F7 – 900 – M e LN3F7 - 900-P
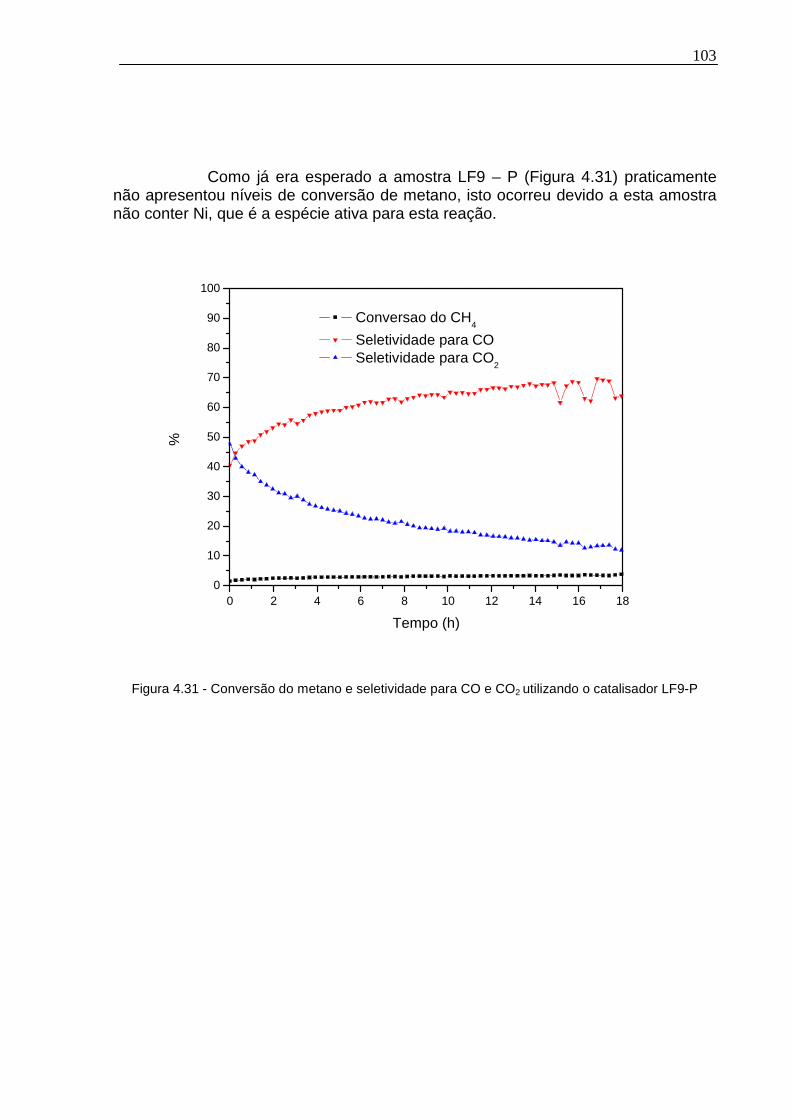
103
Como já era esperado a amostra LF9 – P (Figura 4.31) praticamente não apresentou níveis de conversão de metano, isto ocorreu devido a esta amostra não conter Ni, que é a espécie ativa para esta reação.
0 2 4 6 8 10 12 14 16 180
10
20
30
40
50
60
70
80
90
100
%
Tempo (h)
Conversao do CH4
Seletividade para CO Seletividade para CO
2
Figura 4.31 - Conversão do metano e seletividade para CO e CO2 utilizando o catalisador LF9-P

104
5 CONCLUSÕES
• Os resultados mostrados por termogravimetria indicam que a perda de
massa entre 500 e 700 °C de todos os catalisadores está associada
provavelmente à cristalização da estrutura do material. Isto é, a
calcinação pode ser realizada a partir de 700 °C pa ra formação da fase
desejada.
• Os resultados de DRX mostraram que após calcinação, a fase
desejada foi obtida independente do tipo de síntese para as amostras
LN9 e LF9, sugerindo claramente a formação de pós cristalinos, o qual
consiste de materiais monofásicos com estrutura perovisquita.
• A cristalinidade dos catalisadores em estudo foi praticamente a mesma
independendo do tipo de síntese, as amostras que apresentaram maior
cristalinidade foram LN9 – P e LN9 – M, em torno 95%.
• Para os catalisadores LN3F7-900-P e LN3F7-900-M observou-se que
além da formação da fase desejada LaNi0,3Fe0,7O3 , foi também obtida
a fase La2O3 . De acordo com o dados obtidos pelo refinamento,
observou-se a formação de 70,0% de LaNi0,3Fe0,7O3 e 30,0 % de
La2O3.
• De acordo com o dados obtidos pelo refinamento, observou-se a
presença de 41,6% de LaNi0,7Fe0,3O3; 30,7% de La2NiO4 e 27,7 % de
La2O3 para as amostras LN7F3-900-P e LN7F3-900-M.
• Os resultados de área superficial específica mostraram que todas as
amostras apresentaram baixos valores de área superficial (< 5,3 m2/g)
provavelmente como conseqüência da alta temperatura de calcinação
como também da elevada temperatura obtida durante a
autocombustão, no caso das amostras obtidas por este método.
• De acordo com os resultados de perfil RTP a completa redução do
catalisador LaNiO3 obtido pelo método pechini em La2O3 e níquel
metálico ocorrem a aproximadamente 510 ºC.
• A perovisquita LaFeO3 é quase irredutível em nossas condições de
análise de TPR. A redução da LaFeO3 começa a 750 °C e não se
completa a 1000 °C.

105
• Os resultados de atividade catalítica mostraram que o catalisador LN9
– P apresentou maior valor de conversão de CH4 em aproximadamente
70% e se manteve estável durante todo o teste catalítico. Os valores de
seletividade para CO e CO2 foram de 75% e 25%, respectivamente.
• Para a perovisquita LaNiO3 obtida pelo método de autocombustão
assistida por microondas (LN9-M) a seletividade para CO e CO2
também se estabilizou em torno de 75% e 25%, respectivamente, já a
conversão do CH4 diminuiu durante as três primeiras horas de reação e
depois se estabilizou em torno de 50%.
• As curvas de H2/CO para os catalisadores LN9 – P e LN9 –M,
mostraram que para o catalisador LN9 – P a razão H2/CO se
estabilizou em torno de 2, já o catalisador LN9 – M em torno de 1,5,
indicando que o catalisador obtido pelo método pechini tem razão
adequada para ser insumo na reação de síntese de Fischer-Tropsh.
• De uma maneira geral os catalisadores obtidos pelo método pechini
apresentaram melhores resultados de conversão que os catalisadores
obtidos pelam autocombustão assistida por microondas.

106
REFERÊNCIAS BIBLIOGRÁFICAS
AASBERG-PETERSEN, K.; HANSEN, J. H. B.; CHRISTENSEN, T. S.; DYBKJAER,
I.; CHRISTENSEN, P. S.; NIELSEN, C. S.; MADSEN, S. E. L. W.; ROSTRUP-
NIELSEN, J. R. Applied Catalysis A: General. n. 221, p. 379-387, 2001.
ANDERSON, R. B., in: Catalysis. Ed. Emmettt. P. H., Reinhold, New York. vol. 4. p. 19, 1956.
ARAKAWA, T.; OHARA, N. and SHIOKAWA, J, Journal of Material Science 21, p. 1824, 1986.
ARMOR, J.N. Catalysis letter .Vol. 101, n 3-4, p.131-135, 2005.
ARMOR, J.N; Applied Catalysis A: General, v. 176, p. 156-176, 1999.
AYABE, S.; OMOTO, H.; UTAKA T.; KIKUCHI R.; SASAKI K.; TERAOKA
Y.;VEGUCHI K. Applied Catalysis A: General. n. 241, p. 261-269, 2003.
BARROS, B.S., Tese de doutorado, UFRN, Natal-RN, 2009. BARTHOLOMEW, C. H.; Catal. Rev.-Sci. Eng., v. 24, p. 67, 1982.
BATIOT-DUPEYRAT, C.; VALDERRAMA, G.; MENESES, A.; MARTINEZ, F.;
BARRAULT, J.; TATIBOUËT, J.M.; Applied Catalysis A: General, v. 248, p. 143-151,
2003.
BHARADWAJ, S.S. e SCHMIDT, L.D.; Fuel Processing Technology, v. 42, p. 109-
127, 1995.
BRADFORD, M. & VANNICE, M.; Applied Catalysis A: General, v. 142, p. 73, 1996.
BREARD, Y.; MICHEL, C.; HERVIEL, M.; STUDER, F.; MAIGNAN, A.; and RAVEAU, B. B., Chemistry of Materials, 14, p. 3128, 2002.
CAO, L.; CHEN, Y.; LI, W.; Stud. Surf. Sci. Catal., v. 107, p. 467, 1997.
CHAN, S. H.; WANG, H. M. Journal of Power Sources. n. 101, p. 188-195, 2001.
CHENG, Z.X.; ZHAO, J.L.; LI, J.L.; ZHU, Q,M. Applied Catalysis A: General. n . 205,
p. 31-36, 2001.
CHOUDHARY, V. R.; UPHADE, B. S.; BELHEKAR, A. A.; Journal of Catalysis, v.
163, p.312-318, 1996.

107
CRESPIN, W. M. and HALL, W. K.. Journal of Catalysis, 69. p. 359. 1981.
DE ARAÚJO G. C.; LIMA, S.; RANCEL, M. C.; LA PAROLA, V.; PEÑA, M. A.;
FIERRO, J. L. G.; Catal. Today, v. 107-108, p. 906, 2005.
DE LIMA, S. M. e ASSAF, J. M.; Quim. Nova, v. 30, No. 2, p. 298, 2007.
DE LIMA, S. M.; Tese (Doutorado em Engenharia Química) – Universidade Federal
de São Carlos, 222p., 2006.
DIAS, J. A. C.; Dissertação de Mestrado, São Carlos-SP, DEQ-UFSCar, 2002.
DYER, P. N.; CHEN, C. M.; BENNETT, D. L. Fuels. In: U.S DOE Hydrogen Program
Review NREL/CP-570- 26938, 1999.
EDWARDS, J. H.; MAITRA, A. M.; Fuel Processing Technology, v. 42, p. 269-289,
1995.
ELMASIDES, C.; KONDARIDES, D.I.; NEOPHYTIDES, S.G.; VERYKIOS, X.E.;
Journal of Catalysis, v. 198, p. 195, 2001.
ERDOHELYI, A.; NEMETH, R.; HANCZ, A.; OAZKO, A.; Applied Catalysis A:
General, v. 211, p. 109, 2001.
FATHI, M.; BJORGUM, E.; VIIG, T.; ROKSTAD, O.A. Catalysis Today. n. 63, p. 489-
497, 2000.
FERNANDES, J. D. G.; MELO, D. M. A.; ZINNER, L. B.; SALUSTIANO, C. M.; SILVA, Z. R.; MARTINELLI, A. E.; CERQUEIRA, M.; ALVES, J. C.; LONGO, E.; BERNARDI, M. I. B. Material Letters, v. 53, p. 122, 2002.
FIERRO, J. L. G. and TEJUCA, L. G., Studies in Surface Science and Catalysis, 27, p.453, 1987.
FIERRO, J. L. G. and TEJUCA. L. G., Journal of Catalysis. 87, p. 126, 1984.
FIERRO, J. L. G., Mareei Dekker, NewYork, p. 145, 1993.
FIERRO. J. L. G.; TASCÓN. J. M. D. and TEJUCA, L. G, Journal of Catalysis, 93, p. 83, 1985.
FLORIO, D. Z., FONSECA, F. C., MUCILLO, E. N. S., MUCILLO, R., cerâmica, 50.
275-290, 2004.
GOLDSCHMIDT, V. M.; BARTH, T.;LUNDE, S.; LACHARIASEN, M., J. Math.-Nat. Klasse, 2, p.97, 1926.

108
GOLDWASSER , M. R.; RIVAS, M. E.; PIETRI, E.; PÉREZ-ZURITA, M. J.;
CUBEIRO, M. L.; GINGEMBRE, L.; LECLERCQ, L.; LECLERCQ, G.; Applied
Catalysis A: General, v. 255, p. 46, 2003.
GOLDWASSER, M.R., RIVAS, M.E., PIETRI, E., PÉREZ-ZURITA, M.J., CUBEIRO,
M.L., GRIVOBAL-CONSTANT, A., LECLERCQ, G., Journal of Molecular Catalysis, v.
228, pp. 325-331, Nov. 2005.
GOLDWASSER, M.R.; PIETRI, E.; BARRIOS, A.; GONZÁLEZ, O.; PÉREZ-ZURITA,
M.J.; CUBEIRO, M.L.; Actas do XVII Simpósio Ibero-Americano de Catálise, Porto,
Portugal, p. 51, julho 2000.
GOMES, D.K.S., Dissertação de mestrado, Programa de Pós-Graduação em
Química, UFRN, Natal, 2005.
GOODENOUGH, J. B. and LONGO, J. M., in: Landolt-Bõrnstein New Series. Springer-VerIag, Berlin, 4, Part. A. p. 126, 1970.
GRENIER, J. C.; POUCHARD, M. and HAGENMÜLLER, P., Structure Bonding, 47, p.1, 1981.
GRUNWALDT, J.-D.; BASINI, L.; CLAUSEN, B.S.; Journal of Catalysis, v. 200, p.
321, 2001.
GUNASEKARAN, N.; BAKSHl, N.; ALCOCK, C. B. and CARBERRY, J. J., Journal State lonic, 83, p. 145, 1996.
GUSHEE. B. E.; KAZ, L. and WARD, R., Bulletin of Chemical Society Japanese. 61, p. 3831, 1988.
H. PROVENDIER, C. PETIT, C. ESTOURNES, S. LIBS, A. KIENEMANN, Appl. Catal. A: Gen. 180 (1999) 163. H. TANAKA, M. MISONO - Advances in designing perovskite catalysts. Current
Opinion in Solid State and Materials Science 5 (2001) 381–387.
HIRATSUKA, R. S.; SANTILLI, C. V.; PULCINELLI, S. H. Química Nova, 1995, 18(2), 171-180. HONGTAO CUI, MARCOS ZAYAT, DAVID LEVY, Materials Research, Vol. 5, No. 3,
329-335, 2002.
ICHIMURA, K.; INOUE. Y. and YASUMORI, Y., Bulletin of Chemical Society Japanese, 53, p. 3044, 1980.

109
INABA, H.; HAYASASHI, H.; SUZUKI, M.; Solid State Ionics, 144, p.99, 2001.
ISUPROVA, L. A.; TSYBULYA, S. V.; KRYUKOYA, G. N.; ALIKINA, G. M.;
BOLDYREVA, N. N.; YAKOVLEVA, I. S.; IVANOV, V. P.; SADYKOV, V. A.; Solid
State Ionics, 141, p.417, 2001.
J. F. MITCHELL, J. E. MILLBURN, M. MEDARDE, DIMITRI N. ARGYRIOU, AND J.
D. JORGENSEN. J. Appl. Phys., 85: 4352, (1999).
JIN, R.; CHEN, Y.; LI, W.; CUI, W.; JI, Y.; YU, C.; JIANG, Y.; Applied Catalysis A:
General, v. 201, p. 71-72, 2000.
KAKIHANA, M, Journal of Sol-Gel Science and Technology, 6: 7-55, 1996. KAKIRANA, M., YOSHIMURA, M., Journal Bulletin Chemical Society Japan, 72: 1427-1443, 1999. KOHEI URASAKI, YASUSHI SEKINE, SHO KAWABE, EIICHI KIKUCHI, MASAHIKO
MATSUKATA; Applied Catalysis A: General, v. 286, p.23, 2005.
KRUMPELT, M.; KRAUSE, T.; KOPASZ, J.; CARTER, D. AHMED, S., American
Institute of Chemical Engineering, 2002.
LEITE, E. R.; CERQUEIRA, M.; PERAZOLLI, L. A.; LONGO, E.; VARELA, J. A.
Journal of the American Ceramic Society, v. 79, n. 6, p. 1563-1568, 1995.
LERCHER, J.A .; BITTER, J.H.; STEGHUIS, A.G.; VAN OMMEN, J.G.; SESHAN, K.
Environmental Catalysis. Catalytic Science Series. v. 1, 1999.
LESSING, P. A., Ceramic Bulletin, 68:1002-1007, 1989.
LIMA, S.M. & ASSAF, J.M.; Catalysis Letters, in press, 2006.
LIU, S.; XU, L.; XIE, S.; WANG, Q.; XIONG, G.; Applied Catalysis A: General, v. 211,
p. 145, 2001.
LONGO, J. M. and SLEIGTH, A. W, Material Research Bulletin, 10, p. 1273. 1975.
MELO, D. S., MARINHO, E. P., SOLEDADE, L. B., MELO, D. M. A., LONGO, E., LIMA, S. J. G., SOUZA, A. G., SANTOS, I. M. G.J. Journal of Materials. Science, Material Chemistry, v.9, p.2505, 1999.
MIAO, Q.; XIONG, G.; SHENG, S.; CUI, W.; XU, L.; GUO, X.; Applied Catalysis A: General, v. 154, p. 17-27, 1997.

110
MILT, V. G.; SPRETZ, R.; ULLA, M. A.; LOMBARDO. E. A. and FIERRO, J. L. G., Catalysis Letter. 42. p. 57, 1996.
NAKAGAWA, K.; IKENAGA, N.; KOBAYASHI, T.; SUZUKI, T.; Catalysis Today, v.
64, p. 31, 2001.
NAM, J.W.; CHAE, H.; LEE, S.H.; JUNG, H.; LEE, K.Y.; N atural Gas Conversion V,
119, p. 843-848, 1998.
NATILE, M. M.; UGEL, E.; MACCATO, C., GLISENTI, A. Applied Catalysis B: Environmental, v.72, p. 351, 2007.
NITADORI, T. and MISONO, M., Journal of Catalysis, 93, p. 459. 1985.
NOMURA, S., in: Landolt-Bõrnstein New Series, Springer-Veriag, Berlin, 12, Part A, p. 368. 1978.
NORMAN, A. K.; MORRIS, M. A. Journal of Material Processing Technology, v.92-93, p 91, 1999. P. CIAMBELLI, S. CIMINO, L. LISI, M. FATICANTI, G. MINELLI, I. PETTITI, P. PORTA, Appl. Catal. B: Environ. 33 (2001) 193. PALM, C.; CREMER, P.; PETERS, R.; STOLTEN, D., Journal of Power Sources. n.
106, p. 231-237, 2002.
PANTU, P & GAVALAS, G.R.; Applied Catalysis A: General , v. 223, p. 253, 2002.
PEDERSEN, L. A. and LIBBY. W. F.. Science, 176, p. 1355, 1972.
PEDERSEN, L. A. and LIBBY. W. F.. Science, 176, p. 1355, 1972.
PENA, M. A. and FIERRO, J. L. G., Chemical Review, 101, p. 1981, 2001.
PEÑA, M.A.; GÓMEZ, J.P.; FIERRO, J.L.G. Applied Catalysis A: General. n. 144, p.
7-57, 1996.
PERTHUIS, H.; COLOMBAN, P. Ceramics International, v. 12, n. 1, p. 39-52, 1986.
PESSOA, R. C. 2005, 83 f. Dissertação (Mestrado em Química) - Departamento de
Química. Programa de Pós-Graduação em Química. Universidade do Rio Grande do
Norte, Natal, 2005.
POLTAVETS, V. V., LOKSHIN, K. A., EGAMI, T. AND GREENBLATT, M. 2006.
Mater. Res. Bull. 41 : 955-960.
POPA M., FRANTTI, J., KAKIHANA, M., Solid State Ionics, 154- 155: 437-445, 2002.

111
PROVENDIER, H.; PETIT, C.; ESTOURNES, C.; KIENNEMANN, A.; Studies in
Surface Science and Catalysis: Natural Gas Conversion V, v. 119, p. 741, 1998.
PROVENDIER, H.; PETIT, C.; KIENNEMANN, A.; Chemistry, v. 4, p. 57, 2001.
QIN, D.; LAPSZEWICZ, J.; JIANG, X.; Journal of Catalysis, v. 159, p. 140-149, 1996.
RAUSER. R. and KEMMLER-SACK, S., Journal of Solid State Chemistry, 33, p. 135, 1980.
ROSTRUP NIELSEN, J.R; SEHESTED, J; NORSKOV, J. Advanced Catalysis. n. 47,
p. 65-139, 2002.
ROSTRUP-NIELSEN, J.; HANSEN, J. H. B.; Journal of Catalysis, v. 144, p. 38, 1993.
ROSTRUP-NIELSEN, J.R. In: Catalysis, Science and Technology (Anderson, J.R.;
Boudart, M., eds.). Springer, Berlin Heidelberg, New York, .vol.5, p. 1-117, 1984.
RYNKOWSKI, J.; SAMULKIEWICZ, P.; LADAVOS, A.K.; POMONIS, P.J.; Applied
Catalysis A: General, v. 263, p. 1-9, 2004.
S. ROSENKRANZ, R. OSBORN, J. F. MITCHELL, L. VASIKIU-DOLOC, J. W. LYNN,
S. K. SINHA, AND D. N. ARGYRIOU. J. Appl. Phys., 83: 7348, (1998).
SANIA MARIA DE LIMA, JOSÉ MANSUR ASSAF, Journal of Solid State Chemistry
181 (2008) 905–912.
SHANNON, R. D., Acta Cryst. A: 32, p.751, 1976.
SHANNON, R. D.; PREWITT, C. T., Acta Cryst. B: 25, p.925, 1969.
SKINNER, S. J. AND KILNER, J.A., 2000. Solid State Ionics 135 : 709-712.
SLAGTERN, A.; OLSBYE, U.; Applied Catalysis A: General, v. 110, p. 99, 1994.
SMITH, D. M., Marcel Dekker, Inc., New York, p. 47. 1993.
SOUZA, M.A.F., CANDEIA, R.A., LIMA, S.J.G., SANTOS, M.R.C., SANTOS, I.M.G., LONGO, E. AND SOUZA, A.G., Journal of Thermal Analysis and Calorimetry, 79: 407-410, 2005. SUZUKI, K.; SHIMIZU, M.; Catalysis Today, v. 24, p.237-242, 1995.
TABATA, K.; HJRANO, Y. and SUZUKI. E., Applied Catalysis A: General. 170, p. 245,1998.

112
TAKEHIRA, K.; SHISHIDO, T.; KONDO, M; Journal of Catalysis, v. 207, p. 307,
2002.
TANAKA, H., MISONO, M., Solid State & Materials Science, v. 5, pp. 381-387, 2001.
TASCÓN, J. M. D.; OLIVÂN, A. M. O.; TEJUCA, L. G- and BELL. A. T.. Journal of Physical Chemistry, 90, p. 791, 1986.
TASCÓN, J. M. D.; OLIVÂN. A. M. O. and TEJUCA, L. G., Journal of Chemical Technological Biotechnology, 36, p. 136, 1984.
TEJUCA, L G.; FIERRO, J. L. G. and TASCÓN. J. M. D., Advances in Catalysis, 36,
p. 237, 1989.
TEJUCA, L.G.; BELL, A.T.; CORTÉS-CORBERÁN, V.; Applied Surface Science, v.
37, p. 353, 1989.
TRIMM, D. L.; Catalysis Today, v. 37, p. 233, 1997.
TRIMM, D. L.; Catalysis Today, v. 49, p. 3, 1999.
TSANG, S.C.; CLARIDGE, J.B.; GREEN, M.L.H.; Catalysis Today, v. 23, p. 3, 1995.
TWU, J. & GALLAGHER, P.K.; in: Properties and Applications of Perovskite-Type
Oxides, MARCEL DEKKER, INC., p. 1-2, 1993.
UDAWATTE, C.P., KAKIHANA, M., YOSHIMURA, M., Solid State Ionics, 128: 217-226, 2000. VALDERRAMA, G.; GOLDWASSER, M.R.; NAVARRO, C.U.; TATIBOUËT, J.M.;
BARRAULT, J.; BATIOT-DUPEYRAT, C.; MARTINEZ, F.; Catalysis Today, v. 107-
108, p. 785-791, 2005.
VASCONCELOS, NICE; Dissertação (Mestrado em Química) - Universidade Federal
Fluminense, 94p., 2006.
VOLPE, M.A; Applied Catalysis A: General, v. 210, p. 355, 2001.
VOORHOEV. R. J. H.; REMEIKA, J. P. and TRIMBLE. L. E., Ann. New York
Academic Science, 272, p. 3, 1976.
VORONINA, V.I., BERGERB, I.F., CHEREPANOVC, V. A., GAVRILOVAC, L. YA.,
PETROVC, A.N., ANCHAROVD, A.I., TOLOCHKOD, B. P. AND NIKITENKOE, S.G.
2001. Nuc. Instrum. Methods Phys. Res. A 470 : 202-209.

113
WACHOWSKI, L.; ZIEU.SKI, S. and BUREWICZ. A., Acta Chemical Academic Science Hung. 106, p. 217. 1981.
WAN, J., GOODENOUGH, J. B. AND ZHU, J. H. 2007. Solid State Ionics 178 : 281-
286.
WANG, H.Y. & RUCKENSTEIN, E.; Journal of Catalysis, v. 199, p.309, 2001.
WELLER, S. W., Academic Chemical Research, 16, p. 101, 1983.
YAMAZOE, N. and TERAOKA. Y., Bulletin Chemical Society Japanese. 61, p. 3831, 1988.
YAMAZOE, N.; TERAOKA, Y. and SEIYAMA, T., Chemical Letter, p. 1767. 1981.
YOKOI, Y. and UCHIDA, H., Catalysis Today, 42, p. 167, 1998.
ZANETTI, S. M. 2001, 115 f. Tese (Doutorado em Ciências) - Universidade Federal
de São Carlos, São Carlos.
ZHANG, Z.; VERYKIOS, Applied Catalysis A: General, v. 138, p. 109, 1996.
Related Documents











