
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Conjunto de afecciones con manifestaciones clínicas,
radiológicas y funcionales respiratorias similares, en las
que las principales alteraciones anatomopatológicas
afectan a las estructuras alveolointersticiales.
EPID no describe en realidad el sustrato
anatomopatológico, ya que no afectan sólo a las
estructuras alveolointersticiales, sino también en muchas
ocasiones, las pequeñas vías respiratorias, así como la
vasculatura pulmonar

Enfermedades ambientales 25%
Sarcoidosis 20%
Fibrosis idiopática 15%
Enfermedades del colágeno vascular 10%

Neumonías Intersticiales
Idiopáticas
Fibrosis Pulmonar Idiopática
– Neumonía Intersticial Aguda
– Neumonía Intersticial No Específica
– Bronquiolitis Respiratoria con Enfermedad Pulmonar Intersticial
(BR/EPID)
– Neumonía Intersticial Descamativa (NED)
– Neumonía Organizada Criptogenética (NOC)
– Neumonía Intersticial Linfocítica
De causa conocida o
asociadas :
Asociadas a enfermedades del colágeno
– Causadas por Polvos Inorgánicos (neumoconiosis)
– Inducidas por Fármacos o Radioterapia
– Causadas por Polvos Orgánicos (alveolitis alérgicas extrínsecas)
– Asociadas a enfermedades Hereditarias (Hermansky-Pudlak, etc.)
Primarias o asociadas a
otros procesos no bien
definidos
Sarcoidosis
– Proteinosis Alveolar
– Microlitiasis Alveolar
– Linfangioleiomatosis
– Eosinofilias Pulmonares
– Histiocitosis X (granulomatosis de células de Langerhans)
– Amiloidosis
Clasificación de las enfermedades intersticiales pulmonares

Tos seca
Disnea progresiva
Crepitos bibasales
Independientemente de estos síntomas, las distintaspatologías que engloban este grupo de la patologíaintersticial pulmonar presentan síntomas inherentesa cada enfermedad en particular:
son síntomas comunes quecaracterizan a estas enfermedades.

Órgano Síntoma Enfermedad
Afectación
dérmica
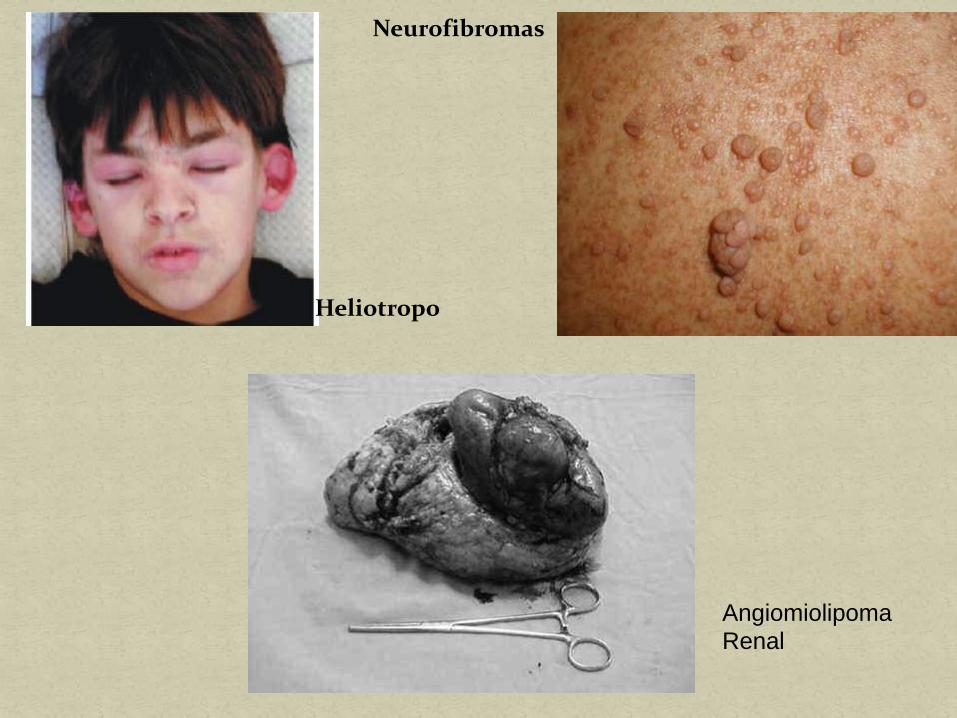
Eritema Nudoso Neurofibromas y
manchas “café con leche”
Nódulos subcutáneos Exantema
Heliotropo
Albinismo
Sarcoidosis,
Enfermedades del
colágeno
Neurofibromatosis
Artritis reumatoide,
neurofibromatosis,
sarcoidosis
Dermatomiositis
Síndrome de Hermansky-
Pudlak
Afectación
ocular
Escleritis
Queratoconjuntivitis seca
Uveítis
Aumento de la glándula lagrimal
Lupus, esclerodermia,
sarcoidosis
Síndrome de Sjögren
Sarcoidosis
Sarcoidosis
Afectación
músculoesquelética
Miositis
Artritis
Afectación ósea
Afectación neurológica
Enfermedades del
colágeno
Sarcoidosis,
enfermedades del
colágeno
Histiocitosis X,
sarcoidosis
Sarcoidosis,
neurofibromatosis,
esclerosis tuberosa,
enfermedades del
colágeno

Órgano Síntoma enfermedad
Afectación renal Angiomiolipomas
Síndrome nefrótico
Glomerulonefritis:
Linfangioleiomiomatosis
Amiloidosis,
lupus
Enfermedades del
colágeno
Afectación
digestiva
Hepatoesplenomegalia
Diarrea crónica
Disfagia
Sarcoidosis, histiocitosis
X,
enfermedades del
colágeno, amiloidosis
Enfermedad inflamatoria
intestinal
Esclerosis sistémica,
dermatomiositis /
polimiositis
Afectación
cardiaca
Miocardio
Pericardio
Sarcoidosis
Enfermedades del
colágeno
Afectación
endocrina
Diabetes insipida Sarcoidosis, histiocitosis
X
Heliotropo
Neurofibromas
Angiomiolipoma
Renal

Se debe realizar un interrogatorio exhaustivo sobre los siguientes aspectos:
• Historia ocupacional y antecedentes de exposición a agentes ambientales orgánicos e inorgánicos, (asbestos, sílice, berilio, proteínas animales, hongos, etc.).
• Preguntar sobre enfermedades similares en la familia. La fibrosis pulmonar idiopática y la sarcoidosis pueden presentarse en miembros de una misma familia.
• Hábito de fumar: la histiocitosis, neumonía intersticial descamativa, bronquiolitis respiratoria asociada a neumonía intersticial y el síndrome de Goodpasture se presentan casi siempre en fumadores.
• Antecedentes de ingestión de drogas: por ejemplo amiodarona, bleomicina, metrotexate, cic1ofosfamida, etc. Algunas drogas como amiodarona y cic1ofosfamida pueden haber sido ingeridas muchos años antes de las manifestaciones clínicas.

Cuando el paciente acude a consulta médica con la sospecha de
una de estas enfermedades nos basaremos en 3 puntos cardinales
para el diagnostico:
-Cuadro clínico compatible (tos seca, disnea de esfuerzo y crepitantes
basales).
– Pruebas funcionales respiratorias que nos confirmen nuestras
sospechas clínicas y que muestren un patrón restrictivo con disminución
de la capacidad de difusión.
– Imágenes radiológicas pulmonares de patrón intersticial, objetivadas
con radiografía de tórax o TAC torácico de alta resolución. Patrón
reticular, micronodular, reticulonodular, alveolar, nodular o en panal de
miel caracterizan a este grupo de enfermedades.

RX torax en panal de abeja – Fibrosis Pulmonar Idiopatica

Una vez tenemos la convicción de que estamos ante una
enfermedad intersticial pulmonar, intentaremos diagnosticar
el tipo de patología en concreto.
Para ello, valoraremos en primer lugar otros síntomas
asociados, ya comentados anteriormente, que nos puedan
ayudar al diagnóstico.
Posteriormente pediremos pruebas analíticas que en
ocasiones nos pueden servir de orientación diagnóstica
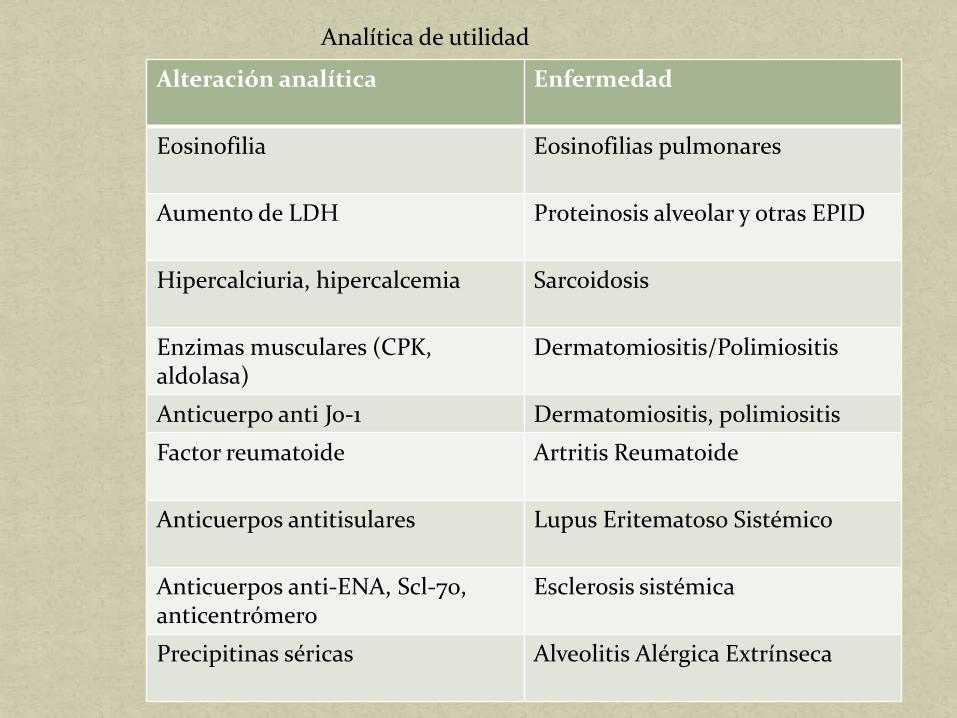
Alteración analítica Enfermedad
Eosinofilia Eosinofilias pulmonares
Aumento de LDH Proteinosis alveolar y otras EPID
Hipercalciuria, hipercalcemia Sarcoidosis
Enzimas musculares (CPK, aldolasa)
Dermatomiositis/Polimiositis
Anticuerpo anti Jo-1 Dermatomiositis, polimiositis
Factor reumatoide Artritis Reumatoide
Anticuerpos antitisulares Lupus Eritematoso Sistémico
Anticuerpos anti-ENA, Scl-70, anticentrómero
Esclerosis sistémica
Precipitinas séricas Alveolitis Alérgica Extrínseca
Analítica de utilidad

En caso de no llegar a un diagnóstico con las técnicas anteriores citadas,
el siguiente paso consistirá en realizar pruebas invasivas cuya función es
la de conseguir material adecuado para ser valorado desde el punto de
vista citológico y/o anatómicopatológico. Dos pruebas son fundamentales
a este respecto:
1. Fibrobroncoscopia: Técnica que consiste en introducir a nivel
bronquial un instrumento flexible con una luz fría en su extremo distal
con el objetivo de realizar dos técnicas:
Lavado broncoalveolar (BAL)
Biopsia transbronquial (BTB)

2. Biopsia pulmonar por minitoracotomia o
videotoracoscopia:Son 2 técnicas quirúrgicas que consiguen material pulmonar suficiente para
diagnóstico.
La minitoracotomia en general consigue tejido de língula y puede tener el
inconveniente de no ser diagnóstico si la enfermedad es de distribución
heterogénea y la língula no esta muy afectada.
Por el contrario la videotoracoscopia permite visualizar toda la cavidad torácica y
ver la zona biopsiada, ya que consiste en introducir un tubo rígido en el tórax
con una luz fría distal.

Diversas enfermedades se pueden confundir con este grupo patológico, por cual debemos realizar diagnóstico diferencial con:
Insuficiencia cardiaca izquierda: líneas B de Kerley, derrame pleural e infiltrados perihiliares.
Bronquiectasias: pueden causar un patrón intersticial en la radiografía de tórax. Se diferencian de las EPID por TAC de alta resolución y clínica.
Neumonías: pueden confundirse clínica y radiografía de tórax con las formas agudas de la AEE.
Linfangitis carcinomatosa: patrón intersticial bilateral con líneas Kerley B en la radiografía de tórax. Diagnóstico por BAL y BTB.

Infiltrados pulmonares en inmunodeprimidos: gérmenesoportunistas, a veces patrón intersticial. Se diferencian porantecedentes de inmunodepresión y análisis microbiológico delBAL.
Hemorragias pulmonares difusas: imágenes alveolares oalveolointersticiales en la radiografía. Se diferencian por clínica(anemia, hemoptisis), BAL (líquido hemorrágico yhemosiderófagos) y alteraciones inmunológicas (anticuerposanticitoplasma de neutrófilos, antinucleares, antimembrana basal).
TBC miliar o enfermedad miliar por BCG. El patrón miliar seproduce en el 6% de las TBC. La diseminación hematógena por elbacilo de Calmette-Guérin puede darse en pacientes con cáncer devejiga y tratamientos tópicos con el BCG, y cursa con un patrónmiliar en la Rx.


El pronóstico es muy variable, en dependencia de
la enfermedad que el paciente presente.
Dentro de las enfermedades mas frecuentes
como puedan ser la fibrosis pulmonar idiopática o
la sarcoidosis, podemos encontrar un claro
ejemplo de esta discordancia, ya el pronóstico es
excelente en la sarcoidosis y decepcionante en la
fibrosis pulmonar idiopática.


Es una entidad que también se conoce con el nombre de neumonitis
por hipersensibilidad, y se trata de un proceso inflamatorio de las
zonas más distales del pulmón que se produce como respuesta a la
inhalación de una serie de antígenos (polvos orgánicos).
Inicialmente se produce una inflamación granulomatosa y más tarde
puede evolucionar a fibrosis y a veces a enfermedad pulmonar
obstructiva crónica si el antígeno sigue inhalándose. La prevalencia
varía con el tipo de antígeno inhalado, así por ejemplo en el caso del
cuidador de palomas ocurre entre un 0.3-7% de expuestos.

La mayor parte de los antígenos son el resultado
de exposiciones ocupacionales, en las que suele
haber numerosos tipos de antígenos
involucrados.

Enfermedad Fuente antigeno
Pulmón del granjero Heno enmohecido Termoactinomicesvulgaris, Aspergillus
Pulmón del cuidador de aves
Palomas, periquitos, etc. Proteínas de excrementos y séricas
P. del humidificador doméstico
Agua contaminada Múltiples bacterias, hongos y protozoos
Enfermedad de los graneros
Trigo, y otros cereales contaminados
Sitophilus granarius
P. de los lavadores de queso
Moho de queso Penicilium casei, Acarussiro

Se produce en individuos que están expuestos a la inhalación de
antígenos durante un tiempo prolongado y el antígeno tiene que tener un
tamaño necesario para alcanzar el alveolo (1-3 micras de diámetro), y se
debe producir la sensibilización del paciente, cosa que solo ocurre en
individuos susceptibles.
Dado que la enfermedad solo ocurre en una pequeña proporción de
pacientes es posible que exista una base genética, aunque se desconoce
con certeza.

Fase aguda: Inflamación alveolointersticial con aumento
de linfocitos, células plasmáticas y macrófagos activados
con citoplasma espumoso (ALVEOLITIS LINFOCITARIA),
y también pueden observarse signos de bronquiolitis
obliterante.
Fase subaguda: Granulomas no caseificantes
semejantes a la sarcoidosis.
Fase crónica: Los granulomas van desapareciendo a
medida que se instaura fibrosis, asociada con enfisema
compensador. La fibrosis afecta más a las zonas
superiores.

Aguda: Se produce tras exposiciones intensas pero
intermitentes al antígeno y suele ser reversible. Se
produce un cuadro clínico consistente en tos, fiebre,
escalofríos, disnea y malestar general de 4-12 horas
después de la inhalación del antígeno y puede estar
sintomático durante 24 horas. La exploración física puede
revelar crepitantes.
Crónica: Disnea progresiva, astenia, pérdida de peso, a
veces febrícula y en ocasiones síntomas de bronquitis
crónica como única manifestación. En la exploración
crepitantes y raramente acropaquias. Si la exposición no
cesa puede evolucionar hacia la insuficiencia respiratoria y
cor pulmonale crónico

Valoración clínica entre los síntomas y la exposición
a antígenos que sabemos pueden producir la
enfermedad, la mejoría al retirar al paciente de la
exposición patológica y la recidiva de síntomas al
volver a exponer al paciente al antígeno.
Nos podemos apoyar en los hallazgos de
laboratorio antes descritos y si hay dudas realizar
una prueba de provocación.

Retirar al paciente de la exposición
Corticoides en algunos casos.

Aspergilosisbroncopulmonar alérgica
neumonía por hipersensibilidad (alveolitis alérgica extrínseca)



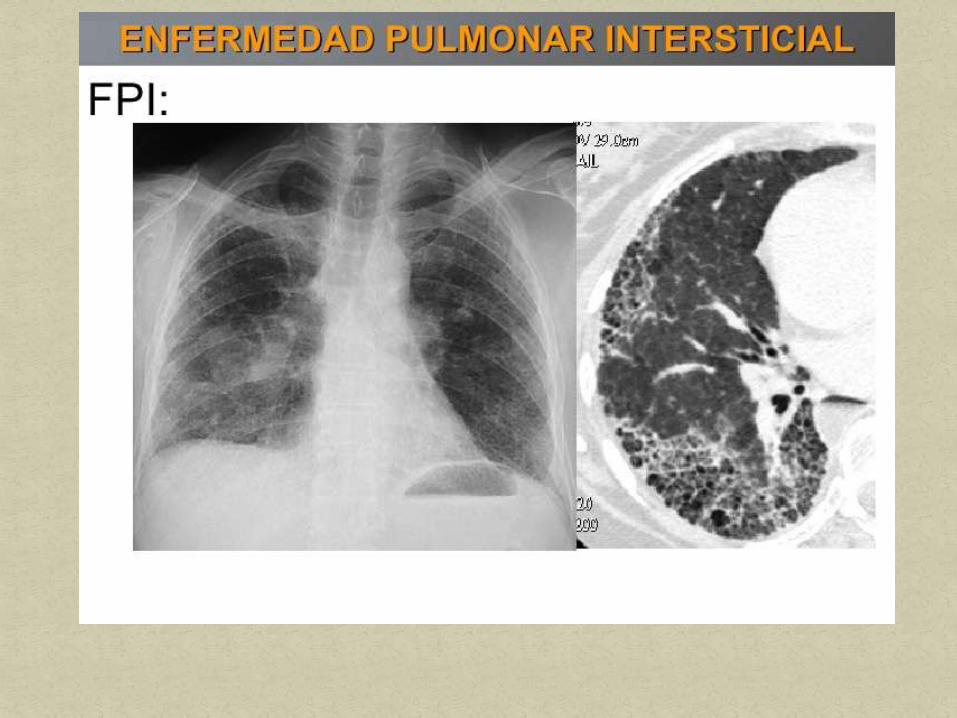
La fibrosis pulmonar idiopática (FPI) es una de las
enfermedades intersticiales más frecuentes. La prevalencia
es desconocida y se estima en 3-5/100.000 habitantes.
La vida media es de 4-6 años y tiene un curso clínico muy
variable. Se caracteriza por presentar cianosis
(manifestación tardía), hipertensión arterial pulmonar y cor
pulmonale.
Se pueden asociar neumotórax espontáneo,
sobreinfecciones respiratorias, tromboembolismo pulmonar
(TEP) y cáncer de pulmón.

Es más frecuente en la edad media de la vida (40 a 70
años). Se presenta con un cuadro de tos seca no
productiva, disnea de esfuerzo progresiva, pérdida de
peso, fiebre, fatiga, mialgias y artralgias. Ocasionalmente,
el comienzo sugiere un proceso virus-like. Se han
documentado casos de comienzo con infecciones víricas y
por mycoplasma.
La exploración raramente es normal: crepitantes finos al
final de la inspiración (rales velcro), de predominio en
bases y dedos en palillo de tambor (40-75% de los casos).

PATOGÉNESIS
La etiología de la fibrosis pulmonar es desconocida pero sehan involucrado en su origen causas infecciosas,inmunológicas y genéticas.
ORIENTACIÓN DIAGNÓSTICA
Habitualmente el diagnóstico se realiza por la clínica y laradiología. Además de ello, podemos utilizar las siguientespruebas complementarias:
• Lavado broncoalveolar (BAL), en el que encontraremos unaumento de neutrófilos.
• Pruebas funcionales respiratorias (PFR), que demuestran unpatrón restrictivo con disminución de la difusión.
• Biopsia transbronquial (BTB) y Biopsia pulmonar, únicasformas de diagnóstico de seguridad.

Existen algunos criterios para establecer el diagnóstico en ausencia de biopsia los cuales son:
Mayores1. Excluir otras causas de como ambientales,
medicamentosas y enfermedades de tejido conectivo.
2. Alteración de pruebas de función pulmonar con disminución de la capacidad vital
3. Biopsia transbronquial y lavadobroncoalveolar que no apoyen undiagnóstico alterno.
Menores1. Mayores de 50 años.
2. Inicio insidiosos de disnea con losesfuerzos.
3. Enfermedad mayor de 3 meses.
4. Estertores en velcro inspiratorios

Corticosteroides, que es el tratamiento de elección: 1 mg/kg/día.
Ciclofosfamida, Azatioprina, Penicilamina o Colchicinaen los casos en fracasen los corticoides.
Transplante pulmonar, en caso de fracaso de los anteriores esquemas



Es una enfermedad granulomatosa multiorgánica de origen
desconocido. La mayoría de las ocasiones presenta una resolución
espontánea o mediante tratamiento con corticoides.
Es más frecuente en negros americanos, escandinavos e irlandeses.
La presentación es más frecuente en invierno y a principios de
primavera. Más frecuente en gemelos homocigotos que heterocigotos.

La patogenia de la sarcoidosis es desconocida pero se cree que
algunos factores genéticos pueden jugar un cierto papel en su
patogénesis. También hay evidencias que relacionan sarcoidosis e
inmunología, como son los siguientes hechos:
1. Los antígenos (Ag) o agentes causantes de la sarcoidosis son
desconocidos, pero hay evidencias que sugieren que existen
receptores de Ag de superficie específicos en los linfocitos T
pulmonares, que al estimularse, pueden iniciar el proceso.
2. Los linfocitos T activados se unen a los macrófagos alveolares,
formando rosetas y liberando linfocinas, iniciándose la respuesta
pulmonar
3. Los macrófagos alveolares activados pueden segregar interleukina 1
(IL-1) que atrae linfocitos, factor de necrosis tumoral, protaglandina
E2 (más de 150 substancias), que promueven el desarrollo de una
alveolitis.

En muchos pacientes es asintomática, en los cuales
habitualmente se descubre casualmente por radiografías.
Puede clínica de afectación respiratoria, con tos no
productiva, disnea de esfuerzo, hiperreactividad bronquial.
También puede darse un síndrome tóxico, con astenia,
anorexia y pérdida de peso, y síntomas multisistémicos.
Existe cierta variación entre blancos y negros.

La sarcoidosis es una enfermedad no solo
respiratoria sino que puede afectar a múltiples
órganos y, consiguientemente dar clínica a
múltiples niveles:

1. CLÍNICA RESPIRATORIA
La clínica respiratoria se produce en algún momento de la evolución, en
casi todos los pacientes con sarcoidosis (tos, disnea de esfuerzo, dolor
torácico retroesternal). La mayoría de los pacientes están sintomáticos y
presentan síntomas sistémicos (astenia, anorexia, perdida de peso, fiebre).
Entre un 20-50% de los pacientes tienen una presentación aguda, que se
manifiesta como síndrome de Lofgren.
Más del 50% de las biopsias bronquiales muestran granulomas, y más del
10% muestran estenosis bronquiales que producen limitación al flujo aéreo,
sibilantes o estridor localizado que pueden minimizar asma. En la
exploración se encuentra un patrón restrictivo + disminución DLCO, pero
puede haber un patrón obstructivo por afectación endobronquial e
hiperreactividad bronquial.

2. CLINICA DE OTROS ÓRGANOS
Suele haber afectación linfática (más del 90% presentan evidencias radiológicas de nódulos linfáticos hiliares)
se detectan entre un 5-10% de disfunciones cardiacas graves, pudiendo producir muerte súbita (patología cardiaca no diagnosticada)
Lesiones cutáneas en cerca del 25% (lupus pernio, eritema nodoso, etc.).
Afectación ocular en el 25%, con conjuntivitis con nódulos pequeños de color amarillo pálido, uveitis.
Afectación del sistema nervioso en un 5%, afectando al VII par (facia).
Hígado: más de 1/3 de pacientes tienen hepatomegalia
Pueden afectarse otros órganos y sistemas como el sistema gastrointestinal, páncreas, tiroides, hipotálamo, músculos, médula ósea, mama y sistema reproductor.

El diagnóstico se realiza por la historia clínica y la radiología
simple de tórax y TAC torácica de alta resolución, niveles
elevados de ECA y PFR con DLco. Se puede recurrir a
realizar biopsias accesibles (piel y adenopatías), biopsia de
Daniel s (ganglio escaleno), broncoscopia con BAL, biopsia
bronquial y biopsia transbronquial (BTB), mediastinoscopia
y tóracoscopia con biopsia abierta.

La pauta de estudio suele comprender lo siguiente:
– Analítica general, calcemia, calciuria 24 h, LDH e
isoenzimas, Ac. antitejido
– Gammagrafía con galio
– Broncoscopia: BAL, BTB, biopsias bronquiales
– Exploración oftalmológica
– PFR con DLco
– TAC de alta resolución
– Pruebas cutáneas de hipersensibilidad retardada

La resolución espontánea se da en 60-80% del estadio I;
en 50-60% del estadio II, en menos del 30% del estadio III.
Son signos de mal pronostico: raza negra, comienzo de la
enfermedad después de los 40 años, más de 6 meses de
síntomas, esplenomegalia, afectación de más de 3
órganos, estadio III y ausencia de eritema nodoso.
Aún con tratamiento con corticoides, un 25-30% de
pacientes en estadio II y III recidivan al retirar la
medicación. La mortalidad oscila entre 1-10%.

Fármacos utilizados:
Antiinflamatorios no esteroideos
Corticoides
Inmunosupresores
Cloroquina
Pentoxifilina


Las colagenosis son un grupo heterogéneo de
enfermedades caracterizado por una inflamación
secundaria a procesos de origen inmunológico, que
pueden afectar a diversos órganos, siendo frecuente la
afectación pulmonar.

La patogenia de estos tipos de enfermedad es
desconocida. Afectan predominantemente al
intersticio pulmonar y las características clínicas,
radiológicas y funcionales, la anatomía patológica
y las pruebas de laboratorio son indistinguibles de
la fibrosis pulmonar idiopática.
El diagnóstico se realiza por la sintomatología
extrapulmonar de la enfermedad causal.

Las colagenosis que con mayor frecuencia se asocian a
fibrosis pulmonar son:
• Lupus eritematoso sistémico: solamente a un 1-6% de
los pacientes se les asocia fibrosis intersticial. Son posibles
los derrames pleurales y las atelectasias laminares.
• Artritis reumatoidea: usando criterios funcionales se
encuentra afectación intersticial en un 50% de los casos,
pero usando solo la radiografía se afectan un 20% de los
pacientes. Cursa con pocas alteraciones de laboratorio y
su curso es más tórpido.

Esclerodermia: la afectación intersticial ronda el 40%de los pacientes. En las autopsias se ha llegado aencontrar hasta en un 90%. Puede haber alveolitis yfibrosis, predominando una u otra.
• Dermatomiositis: solo se afectan el 5% de lospacientes. Las manifestaciones clínicas y la evolución essuperponible a la FPI.
• Síndrome de Sjögren: solo hay fibrosis pulmonar en un3% de los pacientes. En las pruebas de laboratorio se sueleencontrar neutrofilia y linfocitosis.



Son enfermedades ocupacionales pulmonares causadas por inhalación, retensión y reacción tisular de polvos inorgánicos.
Clasificación etiológica de las enfermedades profesionales
Por polvos inorgánicos
Por polvos orgánicos
Por vapores y emanaciones

Por polvos inorgánicosMineral InorgánicosSilice SilicosisCarbón AntracosisAsbesto Asbestosis o AmiantosisHierro SiderosisBerilio BeriliosisCalcio CalcinosisEstaño EstañosisTalco Talcosis o EsteatosisAluminio AluminosisDuros: Diamante, Cobalto, Titanio, Tungsteno, (son los más
duros)Mercurio, Oro, Plata, Mica

Polvos orgánicos
Bisinosis
Bagasosis
Pulmón del granjero
Vapores y emanaciones
Ácido sulfúrico
Anhídrido sulfúrico
Amoniaco

Etiología Se requieren tres factores: Medio ambiente Con la partícula Con el huesped
Factores relacionados con el medio ambiente Ventilación inadecuada Espacios reducidos
Factores relacionadas con las partículas Cantidad Tamaño Forma Composición química Propiedades

Factores relacionados con el huesped
Susceptibilidad
Antecedentes:
* Fumar
* Enfermedades respiratorias previa

• Enfermedad fibrótica de los pulmones causada porla inhalación, retención y reacción pulmonar alsílice cristalino.
• El sílice o dióxido de silicio es el componentepredominante de la corteza terrestre.
• El contenido en sílice de las rocas, piedrasareniscas, granito y pizarra, varía entre 20 y casi100%.

• Enfermedad pulmonar profesional atribuible a la inhalaciónde dióxido de silicio (sílice) en formas cristalinas, comocuarzo, cristobalita y tridimita (sílice libre, paradiferenciarlas de los silicatos). Es una de las principalesenfermedades pulmonares deshabilitantes a nivel mundial.
La exposición ocupacional a partículas de sílice de tamañorespirable (diámetro aerodinámico de 0.5-5µm) se asocia a:
Minería.
Canteras.
Perforación.
Construcción de túneles.
Limpieza con abrasión con materiales con cuarzo (arena).
Trabajadores de cerámica.
Fundiciones.

Existen las formas:
• Crónica: Suele seguir a 1 o más décadas de exposición a polvorespirable que contiene cuarzo. Puede evolucionar a FMP, aundespués de haber cesado la exposición. Puede ser asintomático otener disnea de esfuerzo o tos, insidiosamente progresivas
• Acelerada: En exposiciones mas breves (5-10 años) e intensas, yevoluciona con mayor rapidez.Muchos trabajadores pueden desarrollaruna infección por micobacterias. A menudo se observan enfermedadesautoinmunitarias como esclerodermia y esclerosis sistémica.
• Aguda: En exposiciones intensas y breves a niveles elevados. Sedesarrolla entre los 2 meses y los 2 años de una exposición masiva al sílice.Los síntomas de presentación son disnea, debilidad y perdida de pesoespectaculares.

• Cuadro clinico
• DisneaEl síntoma primario es la Disnea, al inicio con la actividad o elejercicio y luego en reposo. En ausencia de otra enfermedadpulmonar, puede estar ausente, como una presentación de untrabajador asintomático con una Rx anormal.
TOSA menudo la presentan, generalmente secundaria a labronquitis crónica por exposición ocupacional al polvo,consumo de tabaco o ambos. también puede ser por la presiónde grandes masas de ganglios linfáticos silicóticos sobre latraquea o los bronquiolos principales.
HEMOPTISISEs infrecuente y se debe sospechar que hay complicación conotro proceso.

SIBILANCIAS/OPRESION TORAXICA
Frecuentemente forman parte de una enfermedad obstructivade la vías aéreas o de una bronquitis.
SINTOMAS SISTEMICOS
Como la fiebre y la perdida de peso sugieren complicación porinfección o neoplasia.

ASBESTOS: Término general aplicado a varios tipos de silicatosminerales que cristalizan en fibras largas y delgadas defácil separación y que poseen gran resistencia a la tensióny al calor.
USOS: Productos amianto – cemento Aislante térmico para calderas, tubos. Frenos, embragues. Incorporado a metales para aumentar la resistencia a
temperaturas En la industria del algodón (industria contra incendio,
cortinas). Fabricación de baquelita. Industria de caucho amianto (juntas-hilos). Fabricación de papel, cartón, planchas.
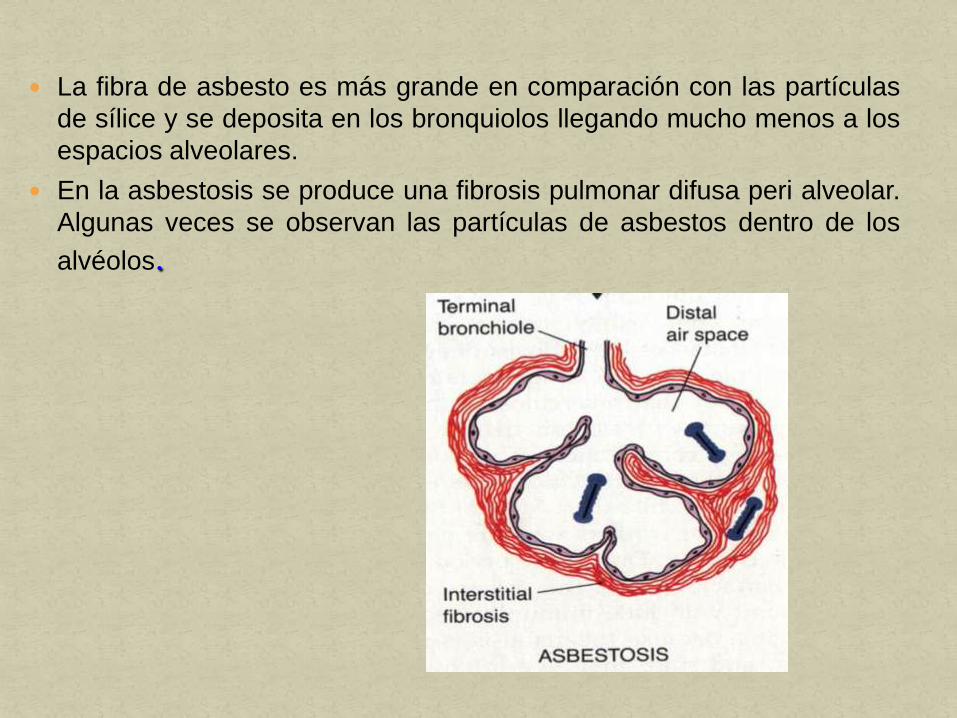
La fibra de asbesto es más grande en comparación con las partículas
de sílice y se deposita en los bronquiolos llegando mucho menos a los
espacios alveolares.
En la asbestosis se produce una fibrosis pulmonar difusa peri alveolar.
Algunas veces se observan las partículas de asbestos dentro de los
alvéolos.
Cuadro clínico: Semejante a la silicosis,
comienzo insidioso con tos productiva, disnea, insuficiencia respiratoria, estertores crepitantes.
La TBC no es complicación notable como en la silicosis
Predispone al desarrollo de Mesoteliomas pleurales y peritoneales.


Son muchos los farmacos com potencial para inducir EPIdifusa ,que casi siempre se manifiesta por disnea y tos noproductiva.
Es necesaria una anamnesis detallada de los medicamentosque ha consumido el paciente para identificar este tipo deenfermedad, incluidos los que no necesitan receta, gotasnasales oleosas y derivados del petroleo (aceite mineral).

En gran parte de los casos se desconoce la patogenia,aunque es probable una combinacion de efectostoxicos directos del farmaco.
La presentacion de la enfermedad puede ser brusca yfulminante o bien gradual a lo largo de varias semanaso meses. Es posible haber consumido el farmaco añosantes de a aparicion.

Se conocen muchos medicamentos como causas de enfermedadpulmonar en algunas personas, entre ellos:
Ciertos antibióticos, como nitrofurantoína y sulfamidas.
Ciertos medicamentos para el corazón, como amiodarona.
Fármacos para quimioterapia, como bleomicina,ciclofosfamida y metotrexato.
Drogas ilícitas.

Síntomas
Expectoración hemoptoica
Dolor torácico
Tos
Fiebre
Dificultad para respirar
Sibilancias

Bleomicina
AINE
Griseofulvina
Mitomicina
Dantrolene
Metildopa
Busulfan
Hidralazina
Practolol
Ciclofosfamida
Hidroclorotiazida
Prazocina
Melfalan
Imipramina
Primidona
Alcaloides de la vinca
Isoniacida
Procainamida
Metotrexato
Metilfenidato
AzatioprinaNadololReserpinaAmiodaronaProcarbacinaTrimetadionaLidocaína ClorambucilClorpropamidaTocaidinaMetisergidaMetilfenidatoNitrofurantoínaMitomicinaAmitriptilinaSulfasalazinaInterleucina-2 ClordiacepóxidoSulfonamidas Anticonceptivos orales Adrenalina
Tetraciclinas Carbonato de litioFenilbutazonaDifenilhidantoínaClorpromacinaMetadonaCarbamazepinaEstreptomicina Sulfato de protaminaPenicilaminaEtosuximidaTerbutalinaritodrineSales de oroFenilbutazonaTocainida

Forma específica de neumonia intersticial fibrosante crónica limitada al pulmón y asociada histológicamente a EPI.
Prevalencia
14-43 x 100 M
Incidencia
7-16 x 100 M
Género
H>M

Fibrosis pulmonar idiopática
Enfermedades del colágeno (AR)
Toxicidad por drogas
Neumonitis crónica por hipersensibilidad
Asbestosis
Síndrome Hermansky-Pudlak

Su nombre se originó al pensar que había descamación del epitelio alveolar; sin embargo, lo que realmente se observa es el acúmulo de los macrófagos intraalveolares lo cual simula al grupo anterior.
Es similar a la bronquiolitis respiratoria asociada a neumonía intersticial no sólo en los hallazgos histológicos sino por la asociación con el cigarrillo; sin embargo, se ve también en los no fumadores

Esta entidad afecta principalmente a las personas entre la 4a. y 5a. décadas, fumadores y más a hombres que a mujeres (2:1). Consultan por una enfermedad insidiosa de disnea, tos seca que puede llegar a progresar a falla respiratoria.
Hay estertores inspiratorios bilaterales y en más de la mitad de los pacientes hay hipocratismo digital

El dejar de fumar usualmente lleva a la resolución del cuadro, con igual sobreviva a la RB-ILD. Aunque pueden recaer al reiniciar el tabaquismo.
En aquellos con síntomas severos y alteraciones del intercambio se le administran esteroides. No hay beneficios demostrados en el uso de inmunosupresores

Inicialmente pertenecía al grupo donde no habíancaracterísticas que definieran un patrón claro eidentificable sin embargo ya se reconoce como unaforma específica de EPI idiopática.
Es una enfermedad que se presenta entre los 40 a 50años e incluso se puede presentar en niños. No tieneun predominio por sexo aunque algunas seriesdescriben ser más frecuente en mujeres

Se presenta usualmente de forma gradual pero sepuede presentar en una forma aguda.
Los principales síntomas son la disnea con losesfuerzos, la tos y la fatiga. A diferencia de laanteriores en esta hay pérdida de peso enpromedio de 6 kg pero hay síntomasconstitucionales hasta en un 10 a 35%.
Al examen físico hay estertores de predominiobasales que luego se hacen difusos.

Es un tipo de neumonía intersticial la cual esrápidamente progresiva y se describe histológicamentecomo daño alveolar difuso donde hay afección tantodel endotelio como el epitelio alveolar.
Se presenta alrededor de los 50 años en ambos sexos ysin una causa clara, para diferenciarlo del síndrome dedificultad respiratorio del adulto el cual usualmente seasocia a un evento precipitante aunque hay algunasformas idiopáticas, pero con una fuerte relación con elcigarrillo

Los pacientes suelen consultar con infecciónrespiratoria viral previa seguida de fiebre, escalofríos,artralgias, mialgias y malestar general. Posteriormentemanifiestan disnea de esfuerzos que se vuelve severaconsultando en menos de 3 semanas de la instauracióndel cuadro.
El examen físico sugiere la presencia de consolidación yestertores difusos

Esta enfermedad tiene un curso malo, sin untratamiento claramente establecido, con mortalidaddel 60% a 6 meses, siendo mayor en los primeros dosmeses y con recurrencia y/o progresión a enfermedadcrónica.

Es una manifestación clínica de una enfermedadintersticial pero con patología de bronquiolitisrespiratoria. Ésta tiene una clara relación con eltabaquismo de forma que mejora al cesar suconsumo y se asocia a la presencia de enfisemacentroacinar

Esta enfermedad rara vez produce síntomas y se asocia amínima disfunción de la vía aérea salvo cuando en algunoscasos se hace severa.
Se observa entre la 4a. y 5a. décadas de la vida, enfumadores usualmente de 30 paquetes año o más, y esmayor en hombres a mujeres con una relación de 2:1 sinverse hipocratismo digital
Mortalidad inferior al 5% en 5 años. El cesar el tabaquismoinduce remisión y no hay reportes
de progresión a fibrosis.

Varón de 69 años, que trabajaba en la construccion ,que acude a consulta por presentar cuadro catarralpersistente, tos productiva, disnea y se le realizo una rxde tórax para evaluar el cuadro respiratorio

Gracias
Related Documents











