REVIEW OF LITERATURE Colon specific drug delivery Lipid based drug delivery system Drug Profile

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

REVIEW OF LITERATURE
Colon specific drug delivery
Lipid based drug delivery system
Drug Profile

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 17
2. REVIEW OF LITERATURE
2.1. Colon-specific drug delivery system
Oral colon-specific drug delivery may be achieved by a wide variety of controlled release
technologies, which may be divided into the three main approaches. The first approach is the
programmed release. In this approach drug release (often a pre-designed release profile) will
start after a specific lag-time following ingestion of the drug product. A second approach is the
triggered release system, a system from which drug release starts after it encountered a
specific intraluminal condition. A third approach is targeted oral drug delivery, where the aim is
to target the location of disease. The different approaches may be based on the delivery of the
intact molecule or on pro-drugs of which the conversion principle to the active molecule may
add to the specificity of drug release (De et al., 2006). Targeted delivery of the intact drug
substance may also be achieved by formulation of micro- and nanoparticles (Lamprecht et al.,
2001), in a passive or active mode. Passive targeting is achieved by enhanced permeability
and retention-effect for micro- or nanoparticles in certain diseased conditions. The application
of micro- or nanoparticles is based on the uptake of small particulate drug carriers by immune
related cells in the inflammed tissue with the aim to achieve a more local effect through their
accumulation at the site of action (Lamprecht et al., 2001). Negatively charged liposomes have
been investigated for their ability to target inflammed tissues based on specific electrostatic
interaction (Tirosh et al., 2009). In active targeting the drug delivery system holds a ligand
which interacts with a disease-specific molecular target.
With the advancements in colon targeted drug delivery system, various formulation
technologies have been reported. These technologies may be broadly classified based on the
formulation approaches that have been exploited for the development of colon targeted drug
delivery, viz, pH-dependent, time-dependent, microflora-activated, pH- and time-dependent,
and pH- and microflora- activated systems. Amongst them, pH-dependent systems are most
widely used as far as the commercial availability of colonic delivery is concerned (Patel et al.,
2011). In 2007, the first and only FDA-approved once daily oral formulation of mesalamine,
MMS (Multi-Matrix System, Shire Pharmaceuticals Inc., Pennsylvania, USA) was developed for
the induction of remission of mild to moderate ulcerative colitis. Recently developed
technologies and formulations for colon targeted drug delivery system are summarized in Table
5 and Table 6 respectively.

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 18
Table 5: Recently developed technologies for colon targeting
Trade name Description Inventor/Reference
pH-dependent systems
Lialda® Mesalamine (MMX technology; pH-dependent gastro-resistant coating with tablet
core containing hydrophilic and lipophilic excipients)
Shire Pharmaceuticals Pennsylvania, USA
Asacol® Mesalamine (Eudragit
® S 100 coated tablets) Tillots Pharma AG, Rheinfelden, Switzerland
Claversal® Mesalamine (Eudragit
® L 100 coated tablets)
Merckle Recordati GmbH, Germany
Mesasal® GlaxoSmithKline Inc., Ontario, Canada
Calitoflak®
Time-dependent systems
Pentasa® Mesalamine (ethyl cellulose coated microgranules slowly dissolve throughout the
small intestine and colon in time-dependent fashion)
Ferring Pharmaceuticals, Saint-Prex,
Switzerland
Port®
system Captopril (hard gelatin capsule, film coated with semi-permeable membrane
(capsule body), an insoluble plug and an osmotic agent along with drug molecule)
Therapeutic System Research Laboratory
Ann Arbor, MI, USA
Egalet® Quinine (3K form of Egalet technology consists of an impermeable shell with two lag
plugs, enclosing a plug of active drug in middle of unit)
Egalet Ltd., Vaerlose, Denmark
Erodible plug, time-
delayed capsule
Chlorpheniramine (erodible compressed tablet is used in place of the swelling
hydrogel plug)
Krogel et al., 1998
Hydrophilic sandwich
capsule
Paracetamol (capsule-within-capsule system in which the inter-capsular space was
filled with a layer of hydrophilic polymer (HPMC)
Stevens et al., 2000
Contd….

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 19
Time controlled
explosion system
Metoprolol tartrate (expansion of swelling agent by water penetrating through outer
membrane causes destruction of the membrane by stress due to swelling force and
subsequent rapid drug release)
Ueda et al., 1994
Microflora-activated systems
Colazal®
Balsalazide (delivered to the colon intact then bacteria cleave the compound to
release 5-amino salicylic acid)
Salix Pharmaceuticals Inc., North Carolina,
USA
Dipentum® Olsalazine (rapidly converted in the colon to molecules of 5-amino salicylic acid by
bacteria and the colon‘s low prevailing redox potential)
Pharmacia AB Stockholm, Sweden
Salazopyrin® Sulfasalazine (metabolized by intestinal bacteria to 5-amino salicylic acid and
sulfapyridine)
Pfizer Australia Pty Ltd, West Ryde NSW,
Australia
Azulfidine® Sulfasalazine (azulfidine En-Tabs contain a cellulose acetatephthalate coating that
retards disintegration in the stomach)
Pfizer, Inc., New York, USA
Colal-Pred®
Prednisolone sodium metasulfobenzoate (COLAL involves a coating for drug
pellets, tablets or capsules, which is composed of ethyl cellulose and a form of
starch called ‗glassy amylose‘)
Alizyme plc, Cambridge, UK
pH and Time-dependent systems
Time Clock® Salbutamol (inner coating consists of hydrophobic surfactant layer to which a water-
soluble polymer is added to improve adhesion to the core)
Pozzi et al., 1994
Chronotopic® Antipyrine (composed of a drug-containing core and hydrophilic swellable polymeric
coating capable of delaying drug release through slow interaction with aqueous
fluids)
Gazzaniga et al., 1994
Contd……….

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 20
Pulsincap® Salbutamol sulphate (comprises an impermeable capsule body containing a drug
formulation sealed in the capsule with a hydrogel polymer plug)
R.P. Scherer International Corp., Michigan,
USA
OROS®-CT Aminosalicylates, Corticosteroids (push--pull units encapsulated within a hard
gelatin capsule)
Alza Corp., Palo Alto, California, USA
Osmet™ Anti-inflammatory, anti-hypertensive and receptor blocking agents (a miniature
osmotic pump that can be swallowed, which will pass through the stomach and small
intestine and then deliver its contents (240 μl) more than 8 h in the large bowel)
Chronset™ Acetaminophen (proprietary OROS®
delivery system that reproducibly delivers a
bolus drug dose (> 80% drug release within 15 min).
Alza Corp., Palo Alto, California, USA
Eudracol® Caffeine (multiparticulate, multilayer system that uses aqueous polymethacrylate
dispersion in the design of the release profile)
Evonik Rohm GmbH Pharma Polymers,
Darmstadt, Germany
Entocort® EC Budesonide (granules, coated to protect dissolution in gastric juice, but which
dissolve at pH > 5.5, i.e., normally when the granules reach the duodenum
AstraZeneca, Sodertalje, Sweden
pH and Microflora activated systems
CODES™ 5-Aminosalicylic acid, Insulin, salmon calcitonin (coupled with a pH-sensitive
polymer coating. On entry into the colon, the polysaccharide inside the core tablet
dissolves and diffuses through the coating. Bacteria enzymatically degrade the
polysaccharides into organic acids)
Yamanouchi Pharmaceuticals Co. Ltd., Japan
TARGIT® Budesonide (enteric-coated injection-moulded starch capsule, consists of a mixture
of Eudragit L and S)
West Pharmaceutical Services Drug Delivery
and Clinical Research Centre Ltd.,
Nottingham, UK

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 21
Table 6: Novel drug delivery systems for colon targeting
Delivery system Description Reference(s)
pH-dependent systems
Self micro-emulsifying
drug delivery system
Curcumin (system was filled into a capsular system and coated with Eudragit S 100)
Zhang et al., 2012
Nanogels 5- Fluorouracil (drug entrapment within the polymeric backbone was achieved using solvent
evaporation method)
Kumar et al., 2012
pH and time-dependent systems
Microparticles Celecoxib (dual coated microparticulate system; where Poly- ε- caprolactone was evaluated as time-
dependent coat and Eudragit S100 as pH-dependent coat)
Ghorab et al., 2011
Tablets Meloxicam (dual coated microparticulate system; where inner coat consists of ethyl cellulose
containing polyethylene glycol as time-dependent coat and Eudragit FS 30D as pH-dependent coat)
Patel et al., 2011
Multiparticulate systems
(pellets)
5- Fluorouracil (dual coated multiparticulate pellets; where inner coat consists of Eudragit
NE30D as time-dependent coat and Eudragit FS 30D as pH-dependent coat)
Kulthe et al., 2013
Microbially and/or enzymatically driven drug delivery systems
Mucoadhesive
Microsphere
5- Fluorouracil (assam Bora rise starch has been used due to its efficiency as CoDDS) Ahmad et al., 2012
Multiparticulate
System (Pellets)
5- Fluorouracil (multiparticulate system consisting of pectin as coat and core material whereas Ethyl
cellulose was employed as an in situ intracapsular coating Material)
Elyagoby et al., 2013
Contd……..
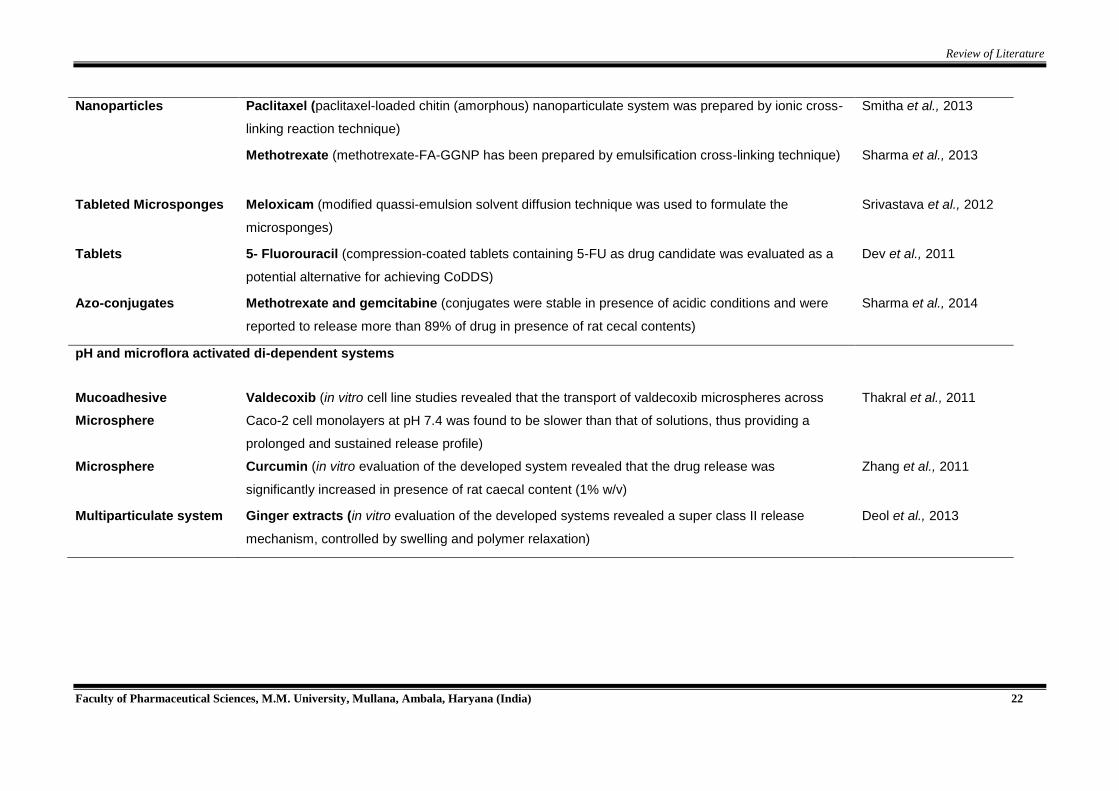
Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 22
Nanoparticles Paclitaxel (paclitaxel-loaded chitin (amorphous) nanoparticulate system was prepared by ionic cross-
linking reaction technique)
Smitha et al., 2013
Methotrexate (methotrexate-FA-GGNP has been prepared by emulsification cross-linking technique) Sharma et al., 2013
Tableted Microsponges Meloxicam (modified quassi-emulsion solvent diffusion technique was used to formulate the
microsponges)
Srivastava et al., 2012
Tablets 5- Fluorouracil (compression-coated tablets containing 5-FU as drug candidate was evaluated as a
potential alternative for achieving CoDDS)
Dev et al., 2011
Azo-conjugates Methotrexate and gemcitabine (conjugates were stable in presence of acidic conditions and were
reported to release more than 89% of drug in presence of rat cecal contents)
Sharma et al., 2014
pH and microflora activated di-dependent systems
Mucoadhesive
Microsphere
Valdecoxib (in vitro cell line studies revealed that the transport of valdecoxib microspheres across
Caco-2 cell monolayers at pH 7.4 was found to be slower than that of solutions, thus providing a
prolonged and sustained release profile)
Thakral et al., 2011
Microsphere Curcumin (in vitro evaluation of the developed system revealed that the drug release was
significantly increased in presence of rat caecal content (1% w/v)
Zhang et al., 2011
Multiparticulate system Ginger extracts (in vitro evaluation of the developed systems revealed a super class II release
mechanism, controlled by swelling and polymer relaxation)
Deol et al., 2013

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 23
2.2. Lipid based drug delivery system
Most of the investigations described so far have evaluated the pharmacokinetics of the drug in
SEFs and very few reports demonstrates pharmacodynamic efficacy. Although pharmacokinetic
studies are sufficient to establish the proof of concept for SEFs, the result of the investigation
should be preferably corroborated by pharmacodynamic studies. This is particularly important
for drugs such as simvastatin, atorvastatin and ezetimibe, which do not show pharmacokinetic-
pharmacodynamic correlation. Although the potential of SEFs in improving oral bioavailability of
lipophilic drugs has been established, an increase in the drug bioavailability need not be
translated into an increase in the pharmacodynamic effects of these drugs. Such aspects
should be carefully considered while planning investigations on the SEFs. The key
investigations that describe the potential of SEFs in oral drug delivery are listed in Table 7 and
some of them have been discussed in the subsequent sections.
2.2.1. Self-emulsifying formulations
Self-emulsifying tablets (SE tablets)
Incorporation of lipid formulation into a solid dosage form combines the advantages of lipid-
based drug delivery systems with those of solid dosage forms. Attama, 2003 formulated a solid
self-emulsifying formulation using goat fat and tween for the delivery of diclofenac. Fatty
material was melted and mixed with surfactant and the drug incorporated into this mixture. This
wet mass was poured into plastic molds and cooled to form a tablet. During the processing of
this formulation it was observed that agitation during fabrication of tablets reduced the
liquification time, resulting in faster emulsification. These results demonstrated that different
formulation ratios possess varying dissolution profiles at constant speed/agitation, and the
optimized formulation showed good release profiles with acceptable tablet properties.
Nazzal and Khan 2002, evaluated the effect of some parameters (colloidal silicates, magnesium
stearate mixing time, and compression force) on coenzyme Q10 (CoQ10) dissolution from
tablets of eutectic-based SMEFs. The optimized conditions were achieved by a face centred
cubic design. In order to significantly reduce the amount of solidifying excipients required for
transformation of SEFs into solid dosage forms, gelled SEFs have been developed by Patil,
2004. In this study, colloidal silicon dioxide (Aerosil 200) was selected as a gelling agent for the
oil-based systems. Colloidal silicon dioxide served a dual purpose: (i) reducing the amount of
solidifying excipients required; and (ii) aiding in reducing drug release.
In a clinical study, it was found that SE tablets may be of use in reducing adverse effects
(Schwarz, 2003). The incorporation of indomethacin (or other hydrophobic NSAIDs) in SE
tablets was found to increase the penetration efficacy of the drug through the GI mucosal
membranes, potentially reducing GI bleeding. The SEF in this study composed of glycerol
monolaurate and Tyloxapol TM (a copolymer of alkyl phenol and formaldehyde). The tablets

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 24
consistently maintained a higher active ingredient concentration in blood plasma over the same
time period compared with a non-emulsifying tablet.
Self-emulsifying powder formulation (SE powder formulation)
Arida et al. 2007 formulated an SE powder formulation in order to enhance the dissolution and
absorption of the poorly water-soluble drug griseofulvin. Capmul GMO- 50, poloxamer and
myvacet were used as surfactants and co-surfactants. A significant enhancement in dissolution
(without ultra-micronisation) and bioavailability of griseofulvin was observed. Balakrishnan et al.,
2009, developed a novel solid SEF of dexibuprofen using spray drying. Aerosil 200 was used as
an inert solid carrier. Both in vitro and in vivo studies were carried out. The optimization of the
SEF composition was carried out by assessing solubility, preparation of phase diagram, particle
size analysis, drug release studies etc. The study showed that Labrafil M 1944 CS, Labrafil M
2125, Labrasol, Capryol 90 and Lauroglycol FCC could enhance the solubility of CoQ10 and
provide the desired drug loading.
Self-emulsifying pellets (SE pellets)
Oral pellets are known to overcome the poor and variable GIT absorption of drugs and have
shown the ability to reduce or eliminate the influence of food on bioavailability. Thus, it appears
highly appealing to combine the advantages of pellets with those of SETs by formulating SE
pellets. Kang et al., 2004 as part of their study to develop a self-emulsifying drug delivery
system, have reported considerable differences in the solubility of simvastatin in a range of
surfactants. The authors suggest that the properties of surfactants need to be considered when
selecting them for the formulation of SE pellets.
Franceschinis et al., 2005 developed a new method for preparing self-emulsifying pellets by wet
granulation consisting of a binder solution containing an oil (mono and diglycerides),
polysorbate 80 and nimesulide as a model drug. The oil surfactant mixture was added to water
to obtain binder solution. The prepared binder solutions were sprayed onto the granules
(prepared from microcrystalline cellulose and lactose) to give pellets. In vivo studies indicated
significantly higher bioavailability with the prepared pellets in comparison to the corresponding
emulsions.
Tuleu, 2004 conducted a comparative bioavailability study of progesterone from SE pellet
formulation, SE solution, capsule and an aqueous suspension in dogs. Complete drug release
was seen within 30 min of capsule administration and within 5 min of administration of the self-
emulsifying system. However, in the case of aqueous suspension the drug release was very low
(~50% of the dose in 60 min). Plasma drug concentration was significantly higher when the drug
was orally administered from self-emulsifying pellets and self-emulsifying solution when
compared to aqueous suspension at the same dose. Abdalla and Mader, 2007, prepared three
self-emulsifying pellet formulations by melting cithrol GMS (mono and diglycerides) and solutol

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 25
HS 15. To this molten blend, the drug (diazepam) and dry microcrystalline cellulose (MCC) were
added to obtain a suitable mass for extrusion. A dye was incorporated for assessment of self-
emulsification and spin probe was added to assess the release kinetics and microenvironment
of pellets. The results from the release study, with higher load of diazepam and lower volume of
the dissolution media, indicated that the formulation was able to create and maintain a state of
supersaturation for the poorly water-soluble diazepam. Nearly 90% of the drug was released
within an hour while only 55% was released from the GMS/MCC pellets.
Wang et al., 2010 demonstrated that the extrusion/ spheronization technique is a large-scale
production method for preparing solid SE pellets from the liquid SEF to improve oral absorption.
SE pellets of a hydrophobic drug (nitrendipine) were prepared. Formulation stability and
solubilisation capacity were noted. The system was optimized on the basis of equilibrium
solubility, pseudo-ternary phase diagram and supersaturation studies. The liquid SEFs were
solidified using adsorbents (porous silicon dioxide), MCC and lactose to form fine flowable
powder. Crospovidone was added to the formulation. The AUC of nitrendipine from the SE
pellets was two-fold greater than the conventional tablets and was comparable with the liquid
SEFs.
Controlled release self-emulsifying pellets
Serratoni and Newton, 2007, observed that the release of methyl paraben (MP) and propyl
parabens (PP) from pellet formulations could be controlled by incorporating them into self-
emulsifying systems containing water soluble plasticiser and talc. Oil and surfactant were mixed
and added to the damp mass of MCC and lactose monohydrate. Extrusion spheronization of the
wet mass was carried out. The pellets obtained were initially coated with ethyl cellulose and
subsequently with an aqueous solution of hydroxy propyl methyl cellulose in a fluid bed coater.
Results obtained from the in vitro study revealed that the presence of self-emulsifying system
enhanced drug release of MP and PP while the film coating considerably reduced the drug
release from pellets.
Iosio et al., 2008 prepared two types of pellets containing vinpocetine (model insoluble drug)
where Type I pellets contained a self-emulsifying system internally and an inert matrix
externally, whereas Type II contained an inert matrix internally and a self-emulsifying system
externally. Formulations were prepared in two steps. In the first step, the oil-surfactant mixture
was added to water to form self-emulsifying systems whereas in the next stage this mixture was
loaded onto MCC and lactose to form extrusion-spheronization mass for pellets. Results indica-
ted that Type I pellets released 90% of vinpocetine within 30 min while the same quantity was
released within 20 min from Type II pellets. The physical mixture of the excipients with drug was
able to release around 25% of the drug in 60 min. Although both types of pellets demonstrated
adequate morphological and technological characteristics, type II pellets showed better drug

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 26
solubility and in vivo bioavailability. The above investigations suggest that a solid dosage form
containing a self-emulsifying system is a promising approach for the formulation of drug
compounds with poor aqueous solubility.
Self-emulsifying beads (SE beads)
Self-emulsifying beads can be formulated as a solid dosage form using smaller amounts of
different excipients. Patil and Paradkar formulated an isotropic formulation of loratadine
consisting of Captex 200, Cremophore EL and Capmul MCM. The SE mixture was loaded onto
poly propylene beads (PPB) using the solvent evaporation method. Formulations were optimi-
zed for loading efficiency and in vitro drug release by evaluating their geometrical features such
as bead size and pore architecture. Results indicated that the poly propylene beads are
potential carriers for solidification of SE mixture, with sufficiently high SE mixture to PPB ratios
for the solid form. The results indicated that self-emulsifying beads can be formulated as a solid
dosage form with a minimal amount of solidifying agents.
Self-emulsifying sustained-release microspheres
You et al., 2006 prepared solid SE sustained-release microspheres of zedoary turmeric oil (oil
phase) using the quasi-emulsion-solvent-diffusion method involving spherical crystallization.
The release behaviour of zedoary turmeric oil from the formulation was found to be dependent
upon the hydroxyl propyl methylcellulose acetate succinate to aerosil 200 ratio. The plasma
concentration time profiles after oral administration in rabbits showed a bioavailability of 135.6%
compared with the conventional liquid SEFs.
Self-emulsifying implants (SE implants)
Research in the field of implants has greatly increased the use and application of solid self-
emulsifying formulation (S-SEF). Carmustine (BCNU) is a chemotherapeutic agent used to treat
malignant brain tumours. However, its effectiveness is hindered by its short half-life. In order to
enhance its stability, the SEF of carmustine was formulated using tributyrin, Cremophor RH 40
(polyoxyl 40 hydrogenated castor oil) and Labrafil 1944 (polyglycolyzed glyceride). The self-
emulsified BCNU was fabricated into wafers with a flat and smooth surface by compression
moulding. The release profile was compared with a wafer implant fabricated using poly (d, l-
lactide-co-glycolide) acetic acid. It was found that SEF increased the in vitro half-life of BCNU to
130 min compared with 45 min with intact BCNU. The in vitro release of BCNU from self-
emulsifying PLGA wafers was prolonged up to 7 days and was found to have higher in vitro
anti-tumor activity (Chae et al., 2005).

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 27
2.2.2. Self-microemulsifying formulations
Self-micro emulsifying formulations (SMEFs) have attracted great attention recently. In an
attempt to combine the advantages of SMEFs with those of solid dosage forms and overcome
the shortcomings of liquid formulations, increasing attention has been focused on solid self-
(micro) emulsifying formulations. The thermotropic stability of SMEFs and their high drug
loading efficiency make them a promising system for low aqueous soluble drugs (Jannin et al.,
2007). SMEFs are usually placed in soft gelatin capsules, but can also be transformed into
granules, pellets, powders for dry filled capsules or tablet preparations (Nazzal, Khan, 2006;
Serratoni, Newton, 2007; Abdalla et al., 2008; Tan et al., 2009). The commercial success of the
SMEF, Neoral® drew greater attention to the development of SMEFs. Many poorly water-soluble
drugs such as acyclovir, atorvastatin, and fenofibrate have been reported to offer improved oral
bioavailability by SMEFs (Wang et al., 2006; Shen, Zhong, 2006; Patel, Vavia, 2007).
Postolache et al., 2002 compared the bioavailability of two cyclosporine capsule products with
different pharmaceutical formulations. Results showed that the test cyclosporine non-SMEFs
formulation was not bioequivalent to the cyclosporine SMEFs formulation due to a statistically
significantly lower absorption rate. These authors demonstrated that the non-self
microemulsifying capsules are not totally interchangeable with the self microemulsifying
capsules unless validated clinical and laboratory conversion protocols for each kind of organ
transplantation are enforced.
Zvonar et al., 2010 suggested that, SMEFs possessing a composition similar to microcapsules
with Ca-pectinate shell and a drug loaded SMEFs as the core phase, would be a potential
approach for enhancing low permeability and solubility of BCS class II drugs.
2.2.3. Self nanoemulsifying formulations (SNEFs)
The classical lipid nanoparticles that have been proposed for drug delivery are composed of
solid lipids. A distinct advantage of SNEFs over polymeric nanoparticles is that the lipid matrix is
made from physiologically tolerated lipid components, which decreases potential acute and
chronic toxicity. Nazzal et al., 2002 developed a SNEF based on the eutectic properties of
ubiquinone (CoQ10) and also studied the progress of emulsion formation and drug release
mechanisms by turbidimetry and droplet size analysis. Results obtained from study revealed
that eutectic-based semisolid SEFs can overcome the drawbacks of the traditional emulsified
systems such as low solubility and irreversible precipitation of the active drug in the vehicle with
time.
Cyclosporine lipid nanoparticles (lipospheres) consisting of phospholipids, Span 80, Tween 80,
Tricaprin, and Cremophor RH 40 were prepared (Bekerman et al., 2004). The CsA dispersion
systems prepared had a particle size ranging from 25 nm to 400 nm. Particles with a size of 25
nm showed maximum oral bioavailability. In a study surfactant–co-surfactant blend (Witepsol®

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 28
H35 and Solutol® HS15) at a ratio of 1:4 led to sufficient reduction in free energy of the system
to resist thermodynamic instability of the nano-emulsion as well as providing a sufficient
mechanical barrier to coalescence oil droplets (Nepal et al., 2010). Koynova et al., 2010
suggested the use of nanosized self-emulsifying lipid vesicles as carriers for the inclusion of
lipophilic dietary supplements. These were proposed as good alternatives to liposomal
preparations which pose problems in stability, sterilization, and non-reproducibility between
batches.
2.2.4. Supersaturable self-emulsifying formulation
Supersaturation represents a potent technique for enhancing absorption by generating and
maintaining a supersaturated state in the intestine. Such formulations contain both a reduced
amount of surfactant(s) and a polymeric precipitation inhibitor (e.g., water-soluble cellulosic
polymers, such as HPMC). These maintain a suggested, directly supersaturating a system with
a drug during manufacture adds to the risk of recrystallization of the product. Various ways of
inhibiting recrystallization have been identified. Thermodynamic ―freezing‖ inside a polymer is
one such option. Under storage conditions, the drug is mobilized by thermodynamic changes in
the polymeric structure. To avoid risk of direct super saturation, several strategies can be
employed, such as, evaporation of a solvent from the system, activation of thermodynamically
―frozen‖ drug-supersaturated islands by hydration.
However, attaining full knowledge of these processes, especially in a multi- component
formulation, requires extensive research. Recently, authors investigated the mechanism
responsible for the enhanced intestinal absorption of hydrophobic drugs from supersaturable
SEFs containing HPMC (Gao et al., 2008). This effect could be attributed to enhanced
permeation of drug to the enterocyte brush border region through the aqueous pathway by
mimicking, or equilibrating with, the bile acid /bile acid mixed micelle pathway.
2.2.5. Marketed formulations
The successful commercialization of oral lipid- and surfactant-based formulations of poorly
soluble drugs in the market has encouraged researchers to explore the field further.
Sandimmune®, Sandimmune Neoral®, Norvir® (ritonavir), and Fortovase® (saquinavir) have
been formulated as SEFs. Sandimmune® and Sandimmune Neoral® formulations of CsA are
perhaps the best known examples of marketed lipid and surfactant based systems and the
pharmacokinetic has been studied and reviewed extensively (Ritschel, 1996). When diluted with
water, these form a polydispersed oil-in-water macro/microemulsion. Table 8 lists selected
commercially available self-emulsifying formulations along with their characteristics.

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 29
Table 7: Different categories of drugs, formulations and excipients used in self-emulsifying formulations
Drug (s) Formulation type
Excipients
Comments References
Halofantrine SEF (Powder) Soybean oil: Maisine
Cremophor EL
Absolute ethanol
Developed formulation improved the oral bioavailability significantly (~6-8
fold) relative to previous data of the solid Halofantrine HCl tablet formulation.
Khoo et al., 1999
Loratadine SEF (Beads) Captex 200
Cremophore EL
Capmul MCM
SEF migrated to the surface of PPB to form a fine oil droplet that readily
dispersed in the bulk to form oil-in-water microemulsion.
Patil et al., 2006
Itraconazole SMEFs Tocopherol acetate
Pluronic L64, Transcutol
Greatly enhanced bioavailability of itraconazole. Hong et al., 2006
Griseofulvin SEF (Powder) Castor oil
Capmul GMO-50,
Myvacet 945
The mean AUC and Cmax after oral administration of GRIS-PEG formulation
in rats were 1.28 and 1.15 fold higher, respectively, compared to SEFS.
Arida et al., 2009
Probucol SNEF Sesame oil
Cremophor RH40
Ethanol
The bioavailability from the surfactant solution and the oil solution were
slightly lower compared to the self-nanoemulsifying drug delivery system.
Nielsen, Gibault
2007
Paclitaxel Super
saturable SEF
Glyceryldioleate ,
Cremophor EL
Cremophor EL
Ethanol, PEG 400
The paclitaxel S-SEFS formulation shows 10-fold higher Cmax and 5-fold
higher oral bioavailability compared to orally dosed Taxol formulation.
Gao et al., 2006
Acyclovir SMEF Sunflower oil
Tween 60, Glycerol
SMEFs increased the oral bioavailability of acyclovir by 3.5-fold compared
with the pure drug solution.
Patel et al., 2007
Diazepam SEF (pellets) C18 mono and di-
glycerides, Solutol HS15
Significant improvement in the in vitro dissolution of diazepam compared to
the release from the non-emulsifying formulation.
Abdalla, Mader
2007
Contd….

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 30
Drug (s) Formulation type
Excipients
Comments References
Coenzyme
Q10 (CoQ10)
SNEFs Witepsol H35
Solutol HS15
Lauroglycol
Result observed from SNEDDS vs reported SEFS were AUC (4.6 fold vs 2.4
fold), Cmax (5.5 fold vs 1.7 fold) and reduction in Tmax (2.0 fold).
Nepal et al., 2010
Lemon oil,
Cremophor EL,
Capmul MCM-C8
The extent of dissolution for the samples stored at 40 °C/75% RH was
comparable.
Nazzal et al., 2002
SEF (Tablet) Lemon oil
Cremophor EL,
Capmul MCM-C8
Cumulative percent of CoQ10 released within 8 h ranged from 40.6% to 90%. Nazzal, Khan 2006
Nitrendipine
(NTD)
SEF (pellets) Miglyol 812
Cremophor® RH40 and
Tween80 (2:1)
Transcutol P
AUC of NTD of SE pellets was 1.6-fold greater than the conventional tablets
and were comparable with the liquid SEFs.
Wang et al., 2010
Nimodipine SMEF Ethyl Oleate, Labrasol
Cremophor RH 40
AUC and Cmax after oral administration of the solid SMEFs were 2.6 and 6.6
fold higher, respectively, compared with those of the conventional tablet.
Yi et al., 2008
Furosemide SMEF Mygliol 812®
Caprylocaproyl macrogol
glycerides, Labrasol®
polyglyceryl-6 dioleate
Plurol Oleique®
Self-microemulsifying cores with completely solubilized drug (SMEFs with 1
and 5% furosemide) exhibited the fastest release profiles with pronounced
initial release.
Zvonar et al., 2010
Ezetimibe SNEF Capryol 90
Cremophor EL
Lauroglycol FCC
The SNGs filled into hard gelatin capsules showed 2-3 fold increase in the
dissolution rate as compared to plain drug filled capsules.
Dixit, Nagarsenker
2008
Contd……

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 31
Drug (s) Formulation type
Excipients
Comments References
Cyclosporine A SNEFs Phospholipids
Chremophor RH 40,
Tween 80, Span 80
Higher AUC and Cmax with lipospheres of small diameter. Bekerman et al.,
2004
SMEFs Hydrogenated castor oil,
medium chain
triglycerides
Polyethylene glycol
Sucrose monolaurate
Solid micellar solution exhibited significant higher Cmax bioavailability (141%
and 139% of Sandimmune, respectively).
Drewe et al., 1992
Silymarin
SEF (Pellets) Migliol®812, Tween80
Propylene glycol
Developed formulation containing (phototherapeutic extract of silymarin)
enhanced the oral bioavailability of its main active compounds.
Iosio et al., 2010
Diclofenac SEF (Tablet) Goat fat
Tween 65
Batches with higher Tween 65: goat fat content ratios yielded better release
rates.
Attama 2003
Dexibuprofen SEF (Powder) Transcutol P, Labrasol
Labrafac CC, Capryol 90
AUC of solid SEFS was about two-fold higher than that of dexibuprofen
powder.
Balakrishnan et al.,
2009
Nimesulide SEF (Pellets) Mono and diglycerides
Polysorbate 80
Bioavailability: Pellets>Emulsions. Franceschinis et
al., 2005
Piroxicam SEF (Pellets) Lauroglycol 90
Cremophor EL
Transcutol HP
Piroxicam release was significantly enhanced with respect to pure drug. Franceschinis et
al., 2010
Curcumin SMEFs Ethyl oleate, Cremorphor
EL, Poloxamer 188
Propylene glycol 400,
Tween 80
Solubility: SMEFs>curcumin suspension. The solubility of curcumin in SMEFs
was found as 21mg/g.
Cui et al., 2009
Contd……

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 32
Drug (s) Formulation type
Excipients
Comments References
Ligusticum-
chuanxiong oil
(VOC)
SMEF Chuanxiong oil, Tween-80
Propylene glycol
The absorption rate of VOC-SMEFs capsules was 2.53 and 1.59 times higher
than that of VOC and VOC/β-Cyclodextrin inclusion (β-CD), and the per cent
absorption was 1.55 and 28.19 times higher than that of VOC and VOC/β-
CD, respectively.
Yao et al., 2010
Curcumin SMEF Labrafac PG and Capryol
90, Cremophor EL,
Labrasol, Propylene
glycol, polyethylene glycol
400
Bioavailability of curcumin from liquid SMEFs and SMEFs pellets was about
16-fold higher than that of unformulated curcumin.
Setthacheewakul et
al., 2010
Progesterone SEF (Pellets) Captex 355, Capmul
MCM Solutol HS 15
Solubilization capacity strongly depends on the concentration of
endogenously secreted materials such as bile salts and phospholipids.
Abdalla et al., 2008
Exemestane SMEF Capryol 90, Transcutol P
Cremophore EL
The relative bioavailability of exemestane of SMEFs was enhanced 2.9 fold. Singh et al., 2009
Vitamin E SEF (Powder) Palm oil, Tween, Span AUC: SEFS> soft gelatin capsule. Julianto et al., 2000
Methyl and
Propyl Paraben
SEF
(Controlled
release
Pellets)
Mono- and diglycerides of
capric and caprylic acids
Tween 80, Ethanol
and glycerol
Water-soluble polymer can refine the control of the in vitro release of drug
from such pellets.
Serratoni, Newton
2007
Vinpocetine Peanut oil, mono- and di-
glycerides
Croscarmellose Sodium,
Microcrystalline Cellulose
Polysorbate 80
Bi-layered pellets resulted in plasma levels 2.4 fold higher than the physical
mixture.
Iosio et al., 2008
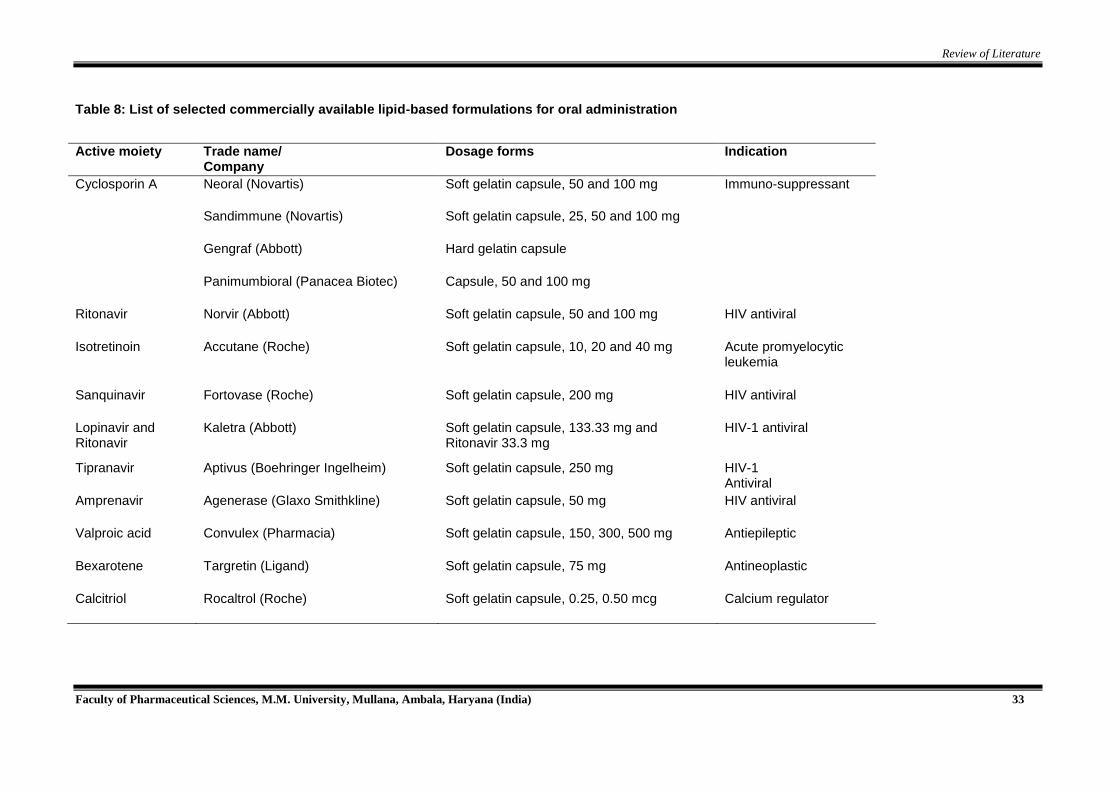
Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 33
Table 8: List of selected commercially available lipid-based formulations for oral administration
Active moiety Trade name/
Company Dosage forms Indication
Cyclosporin A Neoral (Novartis) Soft gelatin capsule, 50 and 100 mg Immuno-suppressant
Sandimmune (Novartis) Soft gelatin capsule, 25, 50 and 100 mg
Gengraf (Abbott) Hard gelatin capsule
Panimumbioral (Panacea Biotec) Capsule, 50 and 100 mg
Ritonavir Norvir (Abbott) Soft gelatin capsule, 50 and 100 mg HIV antiviral
Isotretinoin Accutane (Roche) Soft gelatin capsule, 10, 20 and 40 mg Acute promyelocytic leukemia
Sanquinavir Fortovase (Roche) Soft gelatin capsule, 200 mg HIV antiviral
Lopinavir and Ritonavir
Kaletra (Abbott) Soft gelatin capsule, 133.33 mg and Ritonavir 33.3 mg
HIV-1 antiviral
Tipranavir Aptivus (Boehringer Ingelheim) Soft gelatin capsule, 250 mg HIV-1 Antiviral
Amprenavir Agenerase (Glaxo Smithkline) Soft gelatin capsule, 50 mg HIV antiviral
Valproic acid Convulex (Pharmacia) Soft gelatin capsule, 150, 300, 500 mg Antiepileptic
Bexarotene Targretin (Ligand) Soft gelatin capsule, 75 mg Antineoplastic
Calcitriol Rocaltrol (Roche) Soft gelatin capsule, 0.25, 0.50 mcg Calcium regulator

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 34
Another formulation marketed as an amorphous, semi-solid dispersion was the hard gelatin
capsule of ritonavir (Norvir®). However, unexpected precipitation of amorphous ritonavir as a
less soluble crystalline form in the excipient matrix negatively impacted both the drug dissolution
rate and bioavailability, leading to a temporary withdrawal of the product from the market in
1998. Norvir® was reintroduced in 1999 after reformulation as a thermodynamically stable
solution containing 100 mg of ritonavir solubilized in a self-emulsifying excipient delivered in soft
gelatin capsules. Saquinavir was first introduced in 1996 as a solid oral dosage form (Invirase®)
and subsequently, as a self-emulsifying lipid-based formulation in a soft gelatin capsule
(Fortovase®) containing 200 mg of saquinavir. In 2006, Fortovase® was removed from the
market due to lack of demand. Saquinavir is still available as 200 mg and 500 mg Invirase hard
gelatin capsules.
2.3. Drug Profile
The growing public interest in traditional medicine, particularly plant based medicines, has led to
extensive research on the potentials of natural substances (Wadhwa et al., 2013). Hundreds of
studies were conducted to investigate the effects of natural compounds on human health and
prevention and treatment of chronic diseases (Schmidt et al., 2007). Among studied
compounds, polyphenols appear as one of the most promising groups. These have recently
received much attention in disease prevention and treatment due to their proven anticancer
capabilities (Zern and Fernandez, 2005). Polyphenols are mainly derived from human food
including peanuts, dark chocolate, green and black tea and turmeric. Among polyphenols,
curcumin is currently one of the most studied substances.
Curcumin [1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione] is a major chemical
component of turmeric powder, produced from the rhizome of the plant Curcuma longa
(Jayaprakasha et al., 2002), and is also known as diferyloylmethane. It contains two
parahydroxyl groups responsible for antioxidant activity, two keto groups and two double bonds
responsible for anti-inflammatory, anticancer and antimutagenic activity, two methoxy groups
and an active methylene group (
Figure 3) (Priyadarsini et al., 2003). Physicochemical properties of CUR are enlisted in Table 9.
CH
H3CO
OH
CH C
O
CH2 C
O
CH
CH
OH
OCH3
1
1
33
22
Figure 3: Structure of curcumin; 1. parahydroxyl groups; 2. double bonds; 3. keto groups

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 35
Table 9: Physicochemical properties of CUR
Parameter Specification Reference
Appearance Orange-yellow crystalline powder
Aggarwal et al., 2003 Molecular formula
(Molecular weight)
C21H20O6 (368.37 Daltons)
pKa 7.8, 8.5, and 9.0 Tonnesen et al.,1985
Melting point 183˚C Sharma et al., 2005
Solubility Ethanol (10 mg/ml)
Acetone (20 mg/ml)
Water (0.6 µg/ml)
Tonnesen et al.,2002
Log P 3.29 US 2009/0326275A1
BCS Class IV Tonnesen et al., 2002
Stability Unstable in neutral or alkaline conditions,
pH dependent
Tonnesen et al.,1985
2.3.1. Pharmacological aspects of selected drug
The pharmacodynamics and pharmacokinetic profile of CUR are described below:
Pharmacodynamics
CUR has been reported as one of the most promising candidates of natural origin (Aggarwal
and Sung, 2009) exerting a fascinating array of pharmacological effects in cells in vitro at
physiologically attainable and supra physiological concentrations. Studies indicated that
curcumin exerts hepato and nephroprotective, thrombosis suppressing and myocardial
infarction-protective properties. Figure 4 highlights some of these activities.
Figure 4: Highlights of therapeutic potentials of CUR

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 36
Additionally, its strong antioxidant, antimicrobial, anti-carcinogenic and anti-inflammatory
activities were also reported (Aggarwal and Harikumar, 2009). The mechanisms for anti-
inflammatory potential of CUR may include:
Suppression of the activation of the transcription factor (NF–κB), which regulates the
expression of pro-inflammatory gene products (Singh et al.,1995)
Down-regulation of the expression of cyclooxygenase-2 (COX-2), an enzyme linked with
most types of inflammations (Kawamori et al., 1999)
Decrease the activity and protein levels of inducible nitric oxide synthase (iNOS) enzymes
through reducing the expression of iNOS genes (Ben et al., 2011)
Inhibition of arachidonic acid metabolism via lipoxygenase and scavenging the free
radicals generated in this pathway (Menon and Sudheer, 2007)
Down-regulation of the expression of various cell surface adhesion molecules that have
been linked with inflammation
Decrease in the expression of various inflammatory cytokines, including TNF-α, IL-1, IL-6,
IL-8, and chemokines
All these effects lead to lowering the formation of inflammatory compounds and suppressing the
inflammatory response. This outcome is considered to be beneficial in many abnormal
conditions such as autoimmune diseases (Jagetia and Aggarwal, 2007). Furthermore, there are
growing evidences linking many of the targets mentioned above with tumor promotion. Studies
have shown that overexpression of enzymes such as COX-2 and iNOS have been implicated in
carcinogenesis of many tumors. Although it has not any direct effect on the human cells, it
should be noted that curcumin is potentially chemopreventive (Hasima et al., 2012).
Pharmacokinetics
Pharmacokinetic parameters of CUR are listed in Table 10. It has poor absorption, low
biodistribution, high metabolism and low bioavailability. Cmax is achieved after about 1-3 h.
Protein binding is approximately 97%. It is metabolized predominantly by demethylation and
conjugation, and is eliminated mainly as metabolites in the bile and faeces.
Table 10: Pharmacokinetic parameters of oral CUR (Anand et al., 2007)
Pharmacokinetic parameters* Value
AUC (ng.hr/ml) 461.86
Peak plasma concentration (Cmax) 149.8 ng/ml
Time to reach Cmax (Tmax) 1-3 h
Elimination half-life (t1/2) 2.3 h
Elimination rate constant 0.296 h-1

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 37
2.3.2 Research findings of the selected drug
More than 6000 articles published within the past two decades, researching the molecular basis
for antioxidant, anti-inflammatory, antibacterial, antiviral, antifungal, and anticancer activities of
curcumin (Figure 5). Over one hundred clinical trials conducted on this molecule, prove its
potential in various chronic conditions, including autoimmune, cardiovascular, neurological, and
psychological diseases, as well as diabetes and cancer.
Figure 5: Trends in scientific publication(s) on CUR formulations over last 13 years (2000-2013)
2.2.3. Limitations in formulating dosage forms and delivery systems
Despite the demonstrated efficacy of curcumin its poor systemic bioavailability after oral dosing
compromises the therapeutic potential. Major reasons contributing to low bioavailability of
curcumin include poor absorption and rapid systemic elimination (Strimpakos and Sharma,
2008). Numerous previously reported studies have aimed at improving its poor aqueous
solubility, low bioavailability, poor alkaline stability and/or rapid intestinal metabolism of
curcumin. These include novel formulation containing curcumin impregnated soluble dietary
fibres dispersion, with enhanced bioavailability (20 times) than unformulated curcumin
(Rodriguez et al., 2008). Curcumin may also be combined with piperine, which inhibits
enzymatic conjugation and allows enhanced absorption of unchanged curcuminoids into the
portal blood (Chuah et al., 2013). Table 12 enlist various system developed to enhance the
bioavailability of CUR. In addition, various strategies have been undertaken to deliver CUR in
colon cancer with enhanced systemic bioavailability. Table 13 enlist various system developed
to deliver CUR in colon cancer.

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 38
However, these aforementioned systems have poor localization efficacy due to rapid drug
absorption into the systemic circulation. A review of literature suggests that the application of
carrier technology is not limited to scientific interest in such formulations, but underlines the
potential and versatility in addressing the problems associated with poorly aqueous soluble
drugs for localized delivery (Sylvester et al., 2013, Barrias et al., 2005). Newer approaches,
such as self-emulsifying drug delivery system, have also found its way in enhancing the
solubility of CUR in colonic conditions and have several advantages over the existing ones
(Zhang et al., 2012, Huang et al., 2013).
Table 11: Recent patents of curcumin (grant in 2014 only)
Patent Number Details
US20140193533 Formulation of curcuminoids with enhanced bioavailability of curcumin, demethoxycurcumin, bisdemethoxycurcumin and method of preparation and uses thereof
WO 2013016257 A8
Botanical antioxidant compositions and methods of preparation and use thereof
US 20140161915 A1
Solubilization of cucurminoid compounds and products thereof
US20140127179 Natural killer cell formulations
US20140099390 Formulation of curcumin with enhanced bioavailability of curcumin and method of preparation and treatment thereof
US20140093594 Composition to enhance the bioavailability of curcumin
US20140065061 Curcumin, a liposomal-PLGA sustained release nanocurcumin for minimizing the prolongation for cancer therapy
US20140051742 Lipophilic curcumin analogs and methods of inhibiting HIV-1, treating latent HIV in the brain, and preventing HIV-mediated cognitive decline and HIV dementia
US20140039031 Pharmaceutical formulations of acetyl-11-keto-b-boswellic acid, diindolylmethane and curcumin for pharmaceutical applications
US20140010903 Curcuminoid composition with enhanced bioavailability and process for its preparation

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 39
Table 12: Systems developed to enhance CUR bioavailability
Formulation Details Reference
Nanoparticles Significant delay in progression of diabetic cataract by nanocurcumin attributed to its ability to intervene the biochemical pathways of disease
Grama et al., 2013
Nanoemulsion
Increase in hydrophilicity, bioaccessibility and protection from degradation Sari et al., 2014
Phosphatidylcholine (EPC) liposomes
Curcumin formulated with phosphatidylcholine led to higher systemic levels of parent agent than unformulated curcumin.
Marczylo et al., 2007
Chitosan chloride liposomes
Enhanced bioavailability, compared with curcumin encapsulated by uncoated liposomes and curcumin suspension.
Chen et al., 2012
Self-emulsifying drug delivery system(SEDDS)
Castor oil-(tween-80) -ethanol = 28: 55: 20 (w/ w/w) was selected for optimum curcumin SEDDS.
Wang et al., 2010
Curcumin-SMEDDS in liquid and pellet formulations rapidly formed fine oil-in-water microemulsions
Setthacheewakul et al., 2010
Absorption of curcumin in SMEDDS was via passive transfer by diffusion across the lipid membranes.
Cui et al., 2009
β-cyclodextrin nanoparticle
Formulation increased the dissolution rate of curcumin upto10-fold (p < 0.01). Rachmawati et al., 2013

Review of Literature
Faculty of Pharmaceutical Sciences, M.M. University, Mullana, Ambala, Haryana (India) 40
Table 13: Various systems developed to deliver CUR to intestine in colon cancer
Formulations Description References
PLGA nanoparticles Nanoparticulate curcumin was more bioavailable and had a longer half-life than
native curcumin as revealed from pharmacokinetics study.
Mohanty et al., 2010
N,O-carboxy methyl chitosan
nanoparticles
Improved plasma half-life of curcumin and 5-Fluorouracil up to 72 h. Anitha et al., 2014
pH-sensitive nanoparticles Formulation significantly decreased neutrophil infiltration and TNF-α secretion
while maintaining the colonic structure
Beloqui et al., 2014
Synergistic action of the curcumin-celecoxib drug combination, provide enhanced
efficacy for mitigating ulcerative colitis.
Gugulothu et al., 2014
Lyophilised egg phosphatidylcholine
(EPC) liposomes
Egg phosphatidylcholine liposomal formulation improved cytotoxic activity versus
free curcumin against colorectal cancer cell lines
Pandelidou et al., 2011
Calcium pectin microsphere Eudragit coated calcium pectinate microsphere formulation effectively protected
curcumin in the upper gastrointestinal tract, and then curcumin could be released
specifically in colon
Zhang et al., 2011
Related Documents











