351 Journal of Biomolecular Structure & Dynamics, ISSN 0739-1102 Volume 29, Issue Number 2, (2011) ©Adenine Press (2011) *Phone: +55 21 25467195 Fax: +55 21 25467059 E-mail: [email protected] Aline A. Oliveira a Magdalena N. Rennó b César A. S. de Matos c Morena D. Bertuzzi a Teodorico C. Ramalho d Carlos A. M. Fraga e Tanos C. C. França a * a Laboratory of Molecular Modeling Applied to the Chemical and Biological Defense (LMCBD), Military Institute of Engineering, 22290-270, Rio de Janeiro, RJ, Brazil b Pharmacy Course, Federal University of Rio de Janeiro, Campus Macaé, 27930-560, Macaé, RJ, Brazil c Schlumberger Serviços de Petróleo Ltda, 48100-000, Catu, BA, Brazil d Chemistry Department, Federal University of Lavras, 37200-000, MG, Brazil e Pharmacy Faculty, Federal University of Rio de Janeiro, 21944-971, Rio de Janeiro, RJ, Brazil Molecular Modeling Studies of Yersinia pestis Dihydrofolate Reductase http://www.jbsdonline.com Abstract Considering the risk represented by plague today as a potential biological warfare agent, we propose cytosolic Yersinia pestis dihydrofolate reductase (YpDHFR) as a new target to the design of selective plague chemotherapy. This enzyme has a low homology with the human enzyme and its crystallographic structure has been recently deposited in the Protein Data Bank (PDB). Comparisons of the docking energies and molecular dynamic behaviors of five known DHFR inhibitors inside a 3D model of YpDHFR (adapted from the crystallographic structure) and human DHFR (HssDHFR), revealed new potential interactions and suggested insights into the design of more potent HssDHFR inhibitors as well as selective inhibitors for YpDHFR. Key words: Plague; Yersinia pestis; YpDHFR; Homology Modeling; Docking; Molecular Dynamics; Selective inhibition. Introduction Plague is a zoonosis transmitted mainly by flea bites that affects humans and ani- mals like cow, dogs and rats. This disease is caused by the Gram negative bacillus Yersinia pestis, a microorganism that since its emergence about 1,500–20,000 years ago, as a derivative of Y. pseudotuberculoisis (1), has already been responsible for three big pandemics of plague of devastating magnitude, in centuries VI, XIV and XX, killing more people than any other known infectious agent (2, 3). Y. pestis exists in natural reservoirs like wild and urban rats and can be transmit- ted to humans by fleas of the species Xenopsylla cheopis, X. brasiliensis, X. astia, Nosopsyllus fasciatus, Leptosylla segnis, Ctenocephalides canis and C. felis (4-6). The direct contact with infected animals or, more rarely, secretions from infected people, constitute two additional ways of human infection. The infection by Y. pestis is highly invasive because of its high proteolytic activity which can promote a massive destruction of the host cellular tissue. Degradation provoked by its proteinases usually facilitates the bacterial penetration and dissipa- tion into the tissue barriers, present in the extracellular matrix, until reaching the blood stream (7, 8). When Y. pestis spreads out from the lymph nodes into the blood stream or affect the meninges and the lungs, plague can evolve to its lethal forms: septicemic (in 20% of the cases) or pneumonic (in 10% of the cases) (7, 8). The pneumonic form is of special importance not only because of its lethality but, also, because in this form the plague can be efficiently transmitted from one per- son to another by aerosol spread, triggering devastating pandemics, mainly in over

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

351
Journal of Biomolecular Structure & Dynamics, ISSN 0739-1102 Volume 29, Issue Number 2, (2011) ©Adenine Press (2011)
*Phone: +55 21 25467195Fax: +55 21 25467059E-mail: [email protected]
Aline A. Oliveiraa
Magdalena N. Rennób
César A. S. de Matosc
Morena D. Bertuzzia
Teodorico C. Ramalhod
Carlos A. M. Fragae
Tanos C. C. Françaa*
aLaboratory of Molecular Modeling
Applied to the Chemical and Biological
Defense (LMCBD), Military Institute of
Engineering, 22290-270, Rio de Janeiro,
RJ, BrazilbPharmacy Course, Federal University of
Rio de Janeiro, Campus Macaé,
27930-560, Macaé, RJ, BrazilcSchlumberger Serviços de Petróleo Ltda,
48100-000, Catu, BA, BrazildChemistry Department, Federal
University of Lavras, 37200-000,
MG, BrazilePharmacy Faculty, Federal University
of Rio de Janeiro, 21944-971, Rio de
Janeiro, RJ, Brazil
Molecular Modeling Studies of Yersinia pestis Dihydrofolate Reductase
http://www.jbsdonline.com
Abstract
Considering the risk represented by plague today as a potential biological warfare agent, we propose cytosolic Yersinia pestis dihydrofolate reductase (YpDHFR) as a new target to the design of selective plague chemotherapy. This enzyme has a low homology with the human enzyme and its crystallographic structure has been recently deposited in the Protein Data Bank (PDB). Comparisons of the docking energies and molecular dynamic behaviors of five known DHFR inhibitors inside a 3D model of YpDHFR (adapted from the crystallographic structure) and human DHFR (HssDHFR), revealed new potential interactions and suggested insights into the design of more potent HssDHFR inhibitors as well as selective inhibitors for YpDHFR.
Key words: Plague; Yersinia pestis; YpDHFR; Homology Modeling; Docking; Molecular Dynamics; Selective inhibition.
Introduction
Plague is a zoonosis transmitted mainly by flea bites that affects humans and ani-mals like cow, dogs and rats. This disease is caused by the Gram negative bacillus Yersinia pestis, a microorganism that since its emergence about 1,500–20,000 years ago, as a derivative of Y. pseudotuberculoisis (1), has already been responsible for three big pandemics of plague of devastating magnitude, in centuries VI, XIV and XX, killing more people than any other known infectious agent (2, 3).
Y. pestis exists in natural reservoirs like wild and urban rats and can be transmit-ted to humans by fleas of the species Xenopsylla cheopis, X. brasiliensis, X. astia, Nosopsyllus fasciatus, Leptosylla segnis, Ctenocephalides canis and C. felis (4-6). The direct contact with infected animals or, more rarely, secretions from infected people, constitute two additional ways of human infection.
The infection by Y. pestis is highly invasive because of its high proteolytic activity which can promote a massive destruction of the host cellular tissue. Degradation provoked by its proteinases usually facilitates the bacterial penetration and dissipa-tion into the tissue barriers, present in the extracellular matrix, until reaching the blood stream (7, 8). When Y. pestis spreads out from the lymph nodes into the blood stream or affect the meninges and the lungs, plague can evolve to its lethal forms: septicemic (in 20% of the cases) or pneumonic (in 10% of the cases) (7, 8).
The pneumonic form is of special importance not only because of its lethality but, also, because in this form the plague can be efficiently transmitted from one per-son to another by aerosol spread, triggering devastating pandemics, mainly in over

352
Oliveira et al.
populated regions. Bioterrorists could make use of this fact in order to spread out mutant strains of Y. pestis using sprinkling airplanes or other ways of rapid spreading (9).
Y. pestis is susceptible to most antibiotics except penicillin and its derivatives. Drugs usually employed in the treatment are streptomycin, chloramphenicol and the tetracyclines. For prophylaxis tetracyclines and sulphonamides are recommended. However, literature has reported Y. pestis strains with resistance to all these anti-biotics acquired by plasmid-transported genes (10, 11). This characteristic can be transmitted to other strains, turning Y. pestis into a very dangerous bacterium. Besides, the eventual use of this microorganism as a biological warfare agent or in one terrorist action, could also make use of genetically modified Y. pestis strains for which there would not be chemotherapy available. Considering this argument it is of crucial importance a constant search for new antibiotics and new molecular tar-gets to the development of chemotherapy against plague in face of the emergence of multiresistance and possible genetic modifications on Y. pestis strains.
It is well known that inhibitors of the folate metabolism are quite important drugs in the chemotherapy of bacterial infections and cancer (12-14). The effectiveness of antifolates is based on the perturbations they cause in the folate pathways, which rapidly lead to nucleotide imbalances and cell death (13, 14). This turns the enzy-mes involved in this cycle into good targets to chemotherapy. These three enzymes are thymidylate synthase (TS), dihydrofolate reductase (DHFR) and serine hydro-xymethyltransferase (SHMT). Among them, DHFR have been the main target for the drug design mainly because this enzyme shows important active site differences for different species. For example, the similarity between the active sites of human DHFR (HssDHFR) and the Y. pestis DHFR (YpDHFR) is of only 53%, a fact that allows for the development of antifolates, which could be very selective towards the YpDHFR. Besides, the literature has already reported several studies on potent antibiotics designed to inhibit bacterial DHFR like Pneumocistis carini and E. coli DHFRs (EcDHFR and PcDHFR) which active sites are about 90% conserved when related to YpDHFR (15-17). Considering this, together with the low similarity observed between the HssDHFR and YpDHFR active sites, we were stimulated to propose here the enzyme YpDHFR as a new molecular target to the design of selec-tive chemotherapy against plague.
The computer-aided drug design has been a promising strategy for identifying potential lead compounds and molecular structural features that are related to biological activity. Structure-based investigations using docking and molecular dynamics simulations have been widely used to study ligand and receptor inter-action and have been applied in the design of new drugs against biological war-fare agents (18-48). In line with this approach we performed here docking and molecular dynamics (MD) studies of 5 known DHFR inhibitors (49, 50), on human HssDHFR and a 3D model adapted from the crystallographic structure of YpDHFR, in order to probe for insights to the design of YpDHFR inhibitors. Results obtained validated the proposed model as a functional theoretical structure for YpDHFR and, also, suggested potential interactions to be further considered in the design of more potent HssDHFR inhibitors as well as selective inhibitors of YpDHFR.
Methodology
After a visual inspection of the 3D structure of YpDHFR deposited in the Protein Data Bank (PDB) (51, 52) by Maltseva (paper not publish yet) (PDB entry 3Q1H; resolution 5 1.80 Å, R-value 5 0.167) and comparison with the 3D structure of EcDHFR complexed with NADPH and 1,4-bis-{(N-(1-imino-1guanidino-methyl)) sulfanylmethyl}-3,6-dimethyl-benzeno, deposited by Summerfield et al. (PDB entry 2ANQ; resolution 5 2.13 Å, R-value 5 0.235) (53) we realized that the YpDHFR structure correspond to an apoenzyme in a non-functional folding, with a loop

353
Binding to Yersinia pestis Dihydrofolate Reductase
(named here as loop 1) splitting the active site and avoiding the appropriate anchor-ing of the substrates. It was also observed that the residues of loop 1 are 96.30% conserved between the two enzymes while the overall sequential identity (ID) between the two enzymes is 80%. Thus, in order to obtain a functional model, appropriated to the drug design, it was necessary to re-build the loop 1. This task was performed using the multiple alignment of the primary sequence of YpDHFR to its own 3D structure, without the 27 amino acid residues belonging to loop 1, and the 3D structure of EcDHFR as template. The software used to this task was SwissPDBViewer (54) and the refined alignment was submitted to the SWISS-MODEL server (55, 56), in the optimize mode, in order to generate the final model of the monomeric apoenzyme. Figure 1 show the alignments of the sequences of YpDHFR, EcDHFR and HssDHFR.
After building the YpDHFR apoenzyme model, the following step was to build the enzyme/NADPH/folate complex by docking NADPH and folate into the YpDHFR active site. This task was accomplished by the superposition of the enzyme backbone with the crystallographic structure of Pneumocistes carini DHFR (PcDHFR) com-plexed with NADPH and folate (PDB entry 2CD2; resolution 5 1.90 Å, R-value 5 0.196) determined by Cody et al. (57). The coordinates of folate and NADPH were, then, copied into the model. The topological files of these ligands compatible with the format of the GROMACS 4.0 package (58, 59), were generated at the Dundee PRODRG server (60).
The holoenzyme of HssDHFR complexed with folate and NADPH used in the dock-ing and MD studies was built according to the same procedure described above to YpDHFR, when applied to the crystallographic structure of HssDHFR complexed with NADPH and 6-{[5-quinolylamino] methyl}-2,4-diamino-5-mehtylpyrido [2,3-D] pyrimidine deposited in PDB (51, 52) by Klon et al. (PDB entry 1KMS; resolution 5 1.09 Å, R-value 5 0.131) (61).
In order to check for the quality of our model, it was submitted to the validation server of the PDB (51, 52). There were performed the atomic volume analysis by PROVE (62), the full geometric analysis by PROCHECK (63) and all the WHAT IF (64) checks available on the site. Furthermore, the model was also submitted to the Verify3D structure evaluation server (65-67), in order to check for the chemi-cal environment of each residue, and had its Z-scores calculated using the software PROSA 2003 (68).
The structures of ligands used in molecular docking were built and optimized in the PC SPARTAN software (69) and their partial atomic charges calculated using the AM1 semi-empirical molecular orbital method (70).
Figure 1: Alignment of the sequences of YpDHFR, EcDHFR and HssDHFR. Residues belonging to loop 1 are shown in bold while residues belonging to the active sites are colored. The non-matching residues of the active site of HssDHFR are shown in red.

354
Oliveira et al.
The calculation of the docking energies of the ligands inside the actives sites of YpDHFR and HssDHFR were performed using the software Molegro Virtual Docker® (MVD) (71). This program is able to predict the most likely conforma-tion of how a ligand will bind to a macromolecule. The MolDock scoring function (EScore) used by MVD® (71), is defined by the following energy terms:
EScore 5 EInter 1 EIntra [1]
Where EInter is the ligand-protein interaction energy and EIntra is the internal energy of the ligand.
For the molecular docking study all the water molecules co-crystallized with the structure of HssDHFR (61) were removed using the MVD® (71). We considered the flexibility of residues around 6 Å of the ligand and the binding sites of YpDHFR and HssDHFR were restricted to spheres with radius of 7 and 10 Å respectively. Due to the stochastic nature of the docking algorithm, about 20 runs were performed for each compound with 30 poses (conformation and orientation of the ligand) returned to the analysis of the overlap with the folate inside the structures of the enzymes and the ligand-protein interactions. The best conformation of each compound was selected using their greater degree of structural similarity with folate and evaluating the energy of interaction with the enzymes.
MD studies were performed for the selected conformations of each compound by molecular docking. The parameterization and the charges distributions of selected conformations were carried out in the Dundee PRODRG Server (60).
The enzyme/ligands complexes were simulated using the GROMACS 4.0 package (58, 59), in cubic boxes of approximately 265 nm3 containing around 7,600 water molecules for YpDHFR and around 7,900 water molecules for HssDHFR.
The GROMOS 96 (72) force field, was used for the minimization of the systems stud-ied applying four different algorithms: steepest descent with position restrained (PR) of the ligands followed by steepest descent without PR, conjugate gradients and quasi Newton Raphson down to an energy of 1.00 kcal·mol21·Å21. The MD simulations were carried out after the minimization and involved two steps. In the first, 500 ps of MD were performed at 300 K with restricted positions for the system, except for the water molecules, in order to equilibrate them around the protein residues. Then, we performed 10.0 ns of MD at 300 K without any restriction, using 2 fs of integration time and a cutoff of 10 Å for long-distance interactions. A total of 500 conformations were obtained during each simulation. In this step, the lists of pairs (pairlists) were updated every 500 steps, all Arg and Lys residues were assigned with positive charges and the residues Glu and Asp were assigned with negative charges. The GROMOS 96 (72) force field was chosen to perform the MD studies because it’s one of the most appropriated force fields for simulating proteins and also because it’s possible to gener-ate topologies of small molecules for this force field using the PRODRG server (60).
The VMD (73) and SPDBViewer (54) softwares were used to analyze the struc-tures generated by the MD simulations. Graphs of total energy, root mean square deviation (RMSD), distances and H-Bonds were generated in the Origin® software (74). Qualitative spatial RMSD pictures and frames of MD simulations were gener-ated in the MolMol (75) and PyMOL softwares (76), respectively.
Results and Discussion
Homology Modeling
Even considering the availability of the 3D structure of YpDHFR in the PDB (51, 52), it was necessary to build a homology model in order to obtain the functional

355
Binding to Yersinia pestis Dihydrofolate Reductase
form of the loop 1 over the active site considering that the main aim of this work was to propose YpDHFR as a new target to the development of plague chemother-apy. The most desirable model for this type of work is the crystallographic structure of the protein with a known inhibitor inside its active site. In the present case, the use of an identical crystallographic model to build the loop guarantees a very good quality model, as it is necessary for a precise description of the active site.
Figure 2 shows the 3D structures of YpDHFR from the PDB (51, 52) and the func-tional model proposed here, with NADPH and folate docked inside the active site. As can be seen in the structure of the functional model (Figure 2B) the loop 1, shown in orange and pointed out by the arrows, does not split the active site and allows the docking of NADH and folate inside the cavity (shown in green).
Validation of the Model and Determination of the Folate Binding Site
Regarding the main chain properties of the modeled enzyme, the careful examina-tion of the checking results performed at the validation server of the PDB (51, 52), did not show any considerable bad contacts, nor Cα tetrahedron distortion or buried unsatisfied H-bond donors and acceptors. Moreover, the average G-factor (21.04), was well inside the permitted values for homology models (63). The G-factor pro-vides a measure of how “normal”, or alternatively how “unusual”, a given stereo-chemical property is. When applied to a given residue, a low G-factor indicates that the property corresponds to a low-probability conformation. For example, residues falling inside the disallowed regions of the Ramachandran plot will have a low (or very negative) G-factor. Also, the Ramachandran plot (77) of the model (data not shown) has more than 98.00% of the amino acid residues in the favorable regions of the plot for the whole enzyme, which is considered very good for a homology model. Additionally, no distortions were found on the side-chain torsion angles, the 3D-1D averaged score for each residue calculated by the Verify3D (65, 66), ranged between 0.08 and 0.69 and the Z-scores calculated using the software PROSA 2003 (68), showed that YpDHFR and its template are inside the range of a typical native structure. Accordingly to these results, the model was considered satisfactory.
In order the identify the residues of the folate binding site in YpDHFR, we first iden-tified the corresponding residues proposed to HssDHFR by Oefner et al. and Davies et al. ( 78, 79) and then determined the folate binding site in EcDHFR based on the alignment between HssDHFR and EcDHFR proposed by Polshakov et al. (80). Hav-ing the residues of the folate binding site of EcDHFR in hands, we used the alignment
Figure 2: 3D structures of crystallographic YpDHFR (A) and modeled YpDHFR (B). The ligands are shown in yellow and the cavities in green. Loop 1 is shown in orange and pointed by arrows. Note in (A) that the loop 1 split the cavity.
356
Oliveira et al.
between YpDHFR and EcDHFR in Figure 1 to identify the corresponding active site residues in YpDHFR. A sequence identity of 88.9% was found for the folate binding sites between YpDHFR and EcDHFR. Like in EcDHFR, the folate binding site of YpDHFR includes the residues: Ile6, Ala7, Ala8, Met21, Trp23, Asp28, Leu29, Phe32, Thr36, Ser50, Ile51, Leu55, Pro56, Arg58, Leu59, Met95, Tyr101, and Thr114. The only non-matching residues are Leu59 (Lys58 in EcDHFR) and Met95 (Ile94 in EcDHFR) (see Figure 1). Further alignment among the folate binding site residues of YpDHFR and HssDHFR (54) presented 8 non matching residues (shown in red in Figure 1) and a sequence similarity of 57.90% between the two sites. Such result increases the possibility of designing selective inhibitors to YpDHFR.
DHFR Inhibitors Studied
After validation of the YpDHFR 3D structure, the next step was the search for candi-dates to YpDHFR inhibitors and the evaluation of the potential interactions of these molecules with the YpDHFR active site. Considering that we did not find inhibitors of YpDHFR reported in literature, we started this search by analyzing the docking results of five known DHFR inhibitors (49, 50) inside YpDHFR active site and comparing to theoretical and experimental results obtained with HssDHFR and other bacterial DHFRs (15-17, 49, 50, 81). The idea was to perform a preliminary qualitative in silico evaluation of the potential interactions of these inhibitors with YpDHFR compared to folate and, also, their potential as efficient selective inhibitors of YpDHFR. Results from these studies will guide the search for YpDHFR inhibitors by contributing as starting points for further refinements on their structures and so on until the proposi-tion of the best theoretical possible structures for YpDHFR selective inhibitors.
The DHFR inhibitors chosen for this preliminary investigation (Figure 3) were methotre-xate (MTX), p-PT523, trimetoprime (TMP), piritrexim (PTX) and TMP-5′-CBP (50).
Figure 3: Structures of folate and the DHFR inhibitors studied.

357
Binding to Yersinia pestis Dihydrofolate Reductase
MTX is a known DHFR inhibitor widely used in the treatment of cancer and malaria and p-PT523 is a structural analogue of the anticancer drug aminopterin, 10 to 100-fold more potent than MTX (49). TMP is widely used in the treatment of several bacterial infections and in some cases is replaced clinically by PTX (50). TMP is reported by Roth et al. as being 40,000 times more selective towards bacterial DHFR while PTX is not selective at all (15-17). TMP-5′-CBP is a TMP deriva-tive with a carboxyl group at the end of a tail composed of 05 carbons, 55 times more potent than TMP as inhibitor of EcDHFR (17, 50). Inspection of Figure 3 reveals that each inhibitor, selected to this preliminary study, show an individual fea-ture, like the peculiar tail on TMP-5′-CBP or the extra aromatic ring in p-PT523. This structural diversity was chosen in order to explore most of the binding possibilities inside the enzymes actives sites when performing the docking studies.
Docking Studies of Folate and the DHFR Inhibitors Inside the Active Sites of YpDHFR and HssDHFR
After optimization with PC SPARTAN 2008® (69), folate and the DHFR inhibitors were docked, using MVD® (71), inside the active sites of YpDHFR and HssDHFR, in order to evaluate their interactions on each active site, searching for insights to the design of selective inhibitors to YpDHFR.
For the docking energies calculations we used the .pdb files of HssDHFR proposed by Klon et al. (61) and the .pdb file of YpDHFR proposed in this work. All docked with folate and NADPH and after energy minimization with GROMACS 4.0 (58, 59) down to 1 kcal·mol21·Å21, as described in the methodology section.
The docking energies were calculated according to the procedure described in the methodology section and the conformations of each ligand were analyzed in order to choose the one that best reproduces the folate conformation in the active sites after optimization. Moreover we considered the most favorable values of MolDock Score and hydrogen bond energy. Once selected the best conformation for each ligand according to this procedure, we investigated all the residues involved in interactions with that conformation.
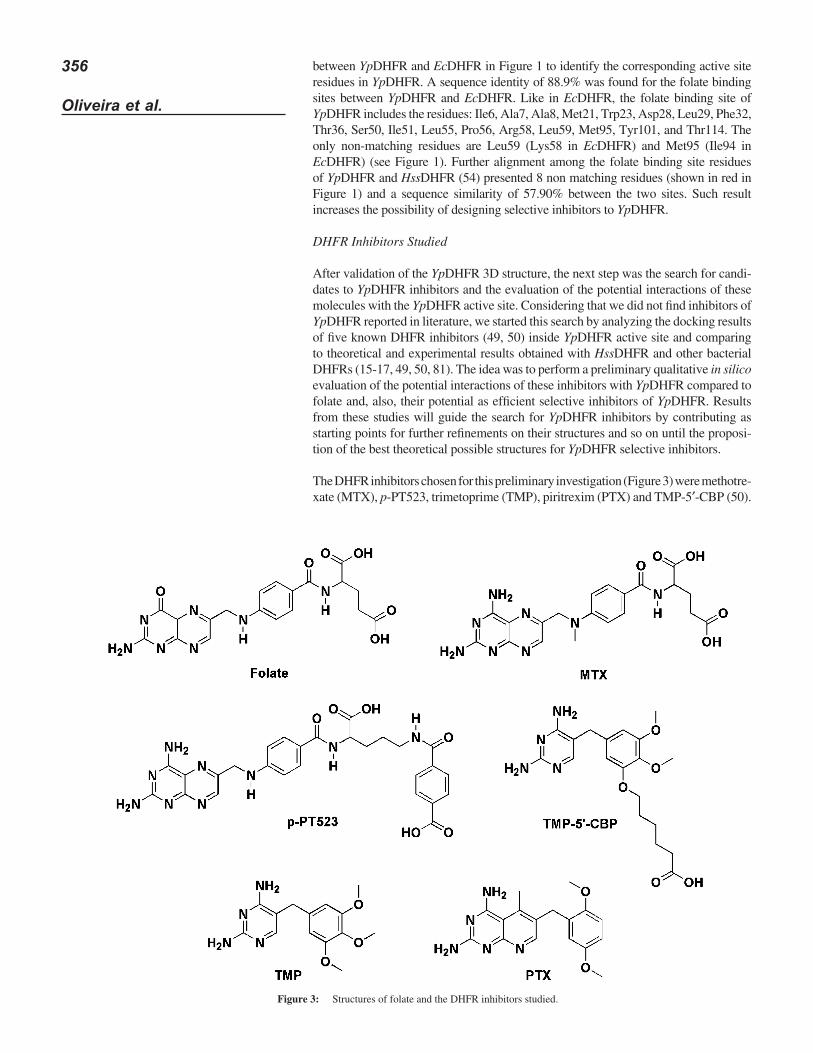
Table I shows the values of MolDock Score and hydrogen bonding for folate, as well as the RMSD generated by the docking protocol used in this work. The RMSDs of the superposition of the non-hydrogen atoms of folate (re-docking) were of 1.699 Å for YpDHFR and 1.515 Å for HssDHFR, respectively (Figure 4). Keeping in mind that a RMSD value under 2.000 Å is considered acceptable (82-84), these results validate the docking protocol used.
The MolDock Score and hydrogen bond energy values of each ligand are presented in Table II. Besides Tables I and II in the supplementary material relate the resi-dues interacting with each ligand and their distances and H-Bond energies respec-tively. The best conformation for each ligand inside the active sites of YpDHFR and HssDHFR and the interactions with the active site residues are represented in Figures 5 and 6. Analyzing the MolDock Score values we can see that most of the inhibitors presented values similar to folate. This result suggests that the affinities
Table IMolDock Score (kcal·mol21), H-Bond energy values (kcal·mol21) and the root mean square deviation (RMSD) calculated for folate inside YpDHFR and HssDHFR.
YpDHFR HssDHFR
MolDock Score H-Bond RMSD MolDock Score H-Bond RMSD
2142.851 211.235 1.699 2170.933 213.170 1.515
358
Oliveira et al.
of the inhibitors to the enzymes active sites are similar to the natural substrate of DHFR, corroborating them as good DHFR inhibitors as already reported in literature (49, 50). Besides, this result also suggests that these drugs could be lead compounds for the development of YpDHFR inhibitors.
The MolDock Scores obtained for MTX and p-PT523 with HssDHFR (2134.301 and 2170.255 kcal·mol21, respectively) were better than with YpDHFR, corrobo-rating experimental results pointing these compounds as better inhibitors of DHFRs of mammals and p-PT523 as a more potent anticancer drug than MTX (49, 50). In fact Wright et al., reported the Kis of p-PT523 and MTX with HssDHFR as 0.3 6 0.036 and 3.7 6 0.35 pM respectively (81).
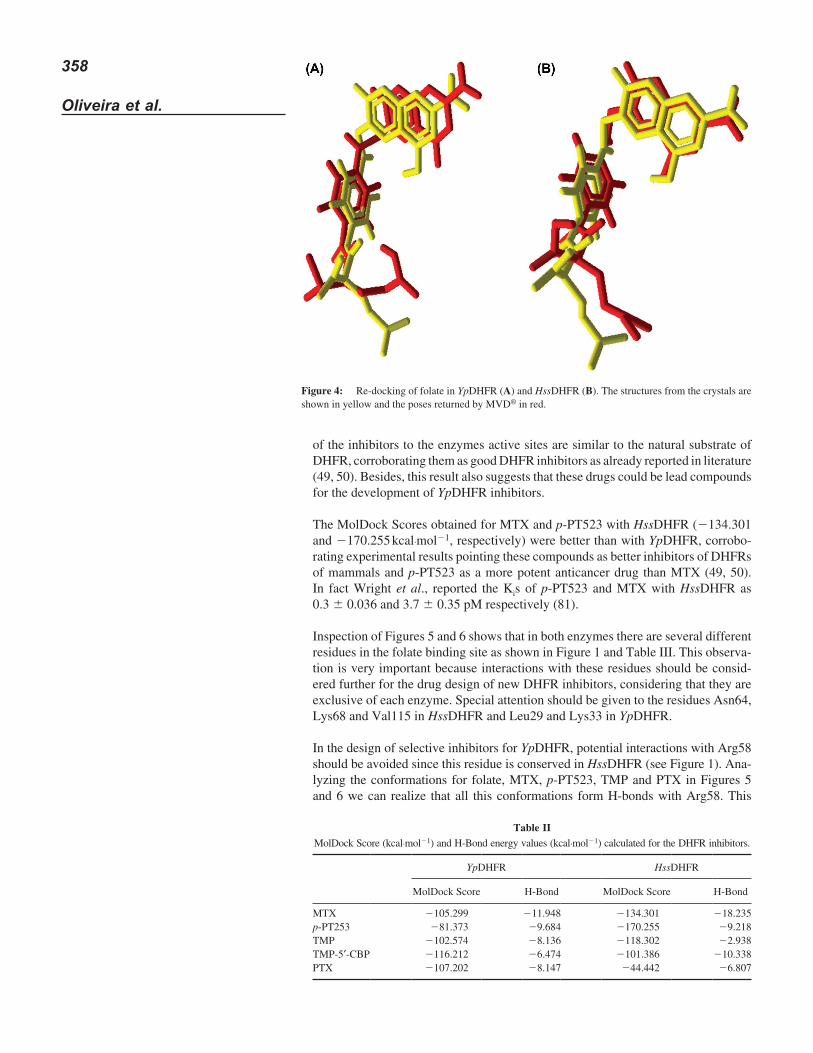
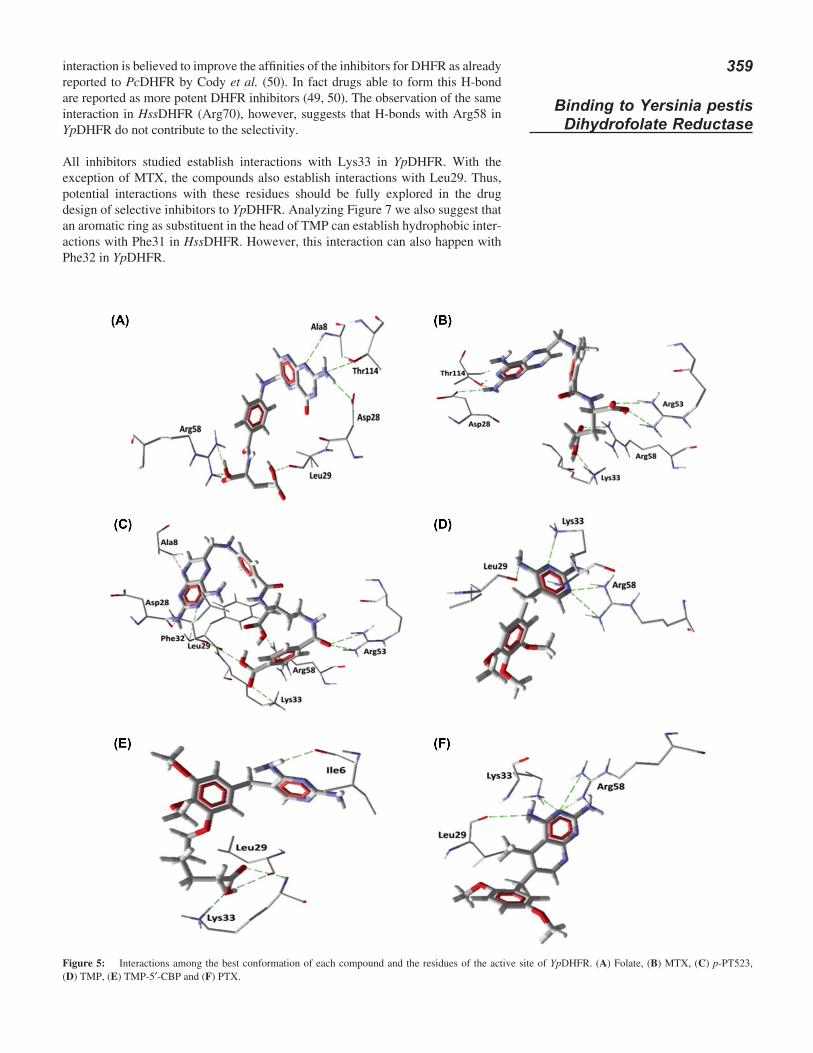
Inspection of Figures 5 and 6 shows that in both enzymes there are several different residues in the folate binding site as shown in Figure 1 and Table III. This observa-tion is very important because interactions with these residues should be consid-ered further for the drug design of new DHFR inhibitors, considering that they are exclusive of each enzyme. Special attention should be given to the residues Asn64, Lys68 and Val115 in HssDHFR and Leu29 and Lys33 in YpDHFR.
In the design of selective inhibitors for YpDHFR, potential interactions with Arg58 should be avoided since this residue is conserved in HssDHFR (see Figure 1). Ana-lyzing the conformations for folate, MTX, p-PT523, TMP and PTX in Figures 5 and 6 we can realize that all this conformations form H-bonds with Arg58. This
Figure 4: Re-docking of folate in YpDHFR (A) and HssDHFR (B). The structures from the crystals are shown in yellow and the poses returned by MVD® in red.
Table IIMolDock Score (kcal·mol21) and H-Bond energy values (kcal·mol21) calculated for the DHFR inhibitors.
YpDHFR HssDHFR
MolDock Score H-Bond MolDock Score H-Bond
MTX 2105.299 211.948 2134.301 218.235p-PT253 281.373 29.684 2170.255 29.218TMP 2102.574 28.136 2118.302 22.938TMP-5′-CBP 2116.212 26.474 2101.386 210.338PTX 2107.202 28.147 244.442 26.807
359
Binding to Yersinia pestis Dihydrofolate Reductase
interaction is believed to improve the affinities of the inhibitors for DHFR as already reported to PcDHFR by Cody et al. (50). In fact drugs able to form this H-bond are reported as more potent DHFR inhibitors (49, 50). The observation of the same interaction in HssDHFR (Arg70), however, suggests that H-bonds with Arg58 in YpDHFR do not contribute to the selectivity.
All inhibitors studied establish interactions with Lys33 in YpDHFR. With the exception of MTX, the compounds also establish interactions with Leu29. Thus, potential interactions with these residues should be fully explored in the drug design of selective inhibitors to YpDHFR. Analyzing Figure 7 we also suggest that an aromatic ring as substituent in the head of TMP can establish hydrophobic inter-actions with Phe31 in HssDHFR. However, this interaction can also happen with Phe32 in YpDHFR.
Figure 5: Interactions among the best conformation of each compound and the residues of the active site of YpDHFR. (A) Folate, (B) MTX, (C) p-PT523, (D) TMP, (E) TMP-5′-CBP and (F) PTX.
360
Oliveira et al.
Molecular Dynamics Studies
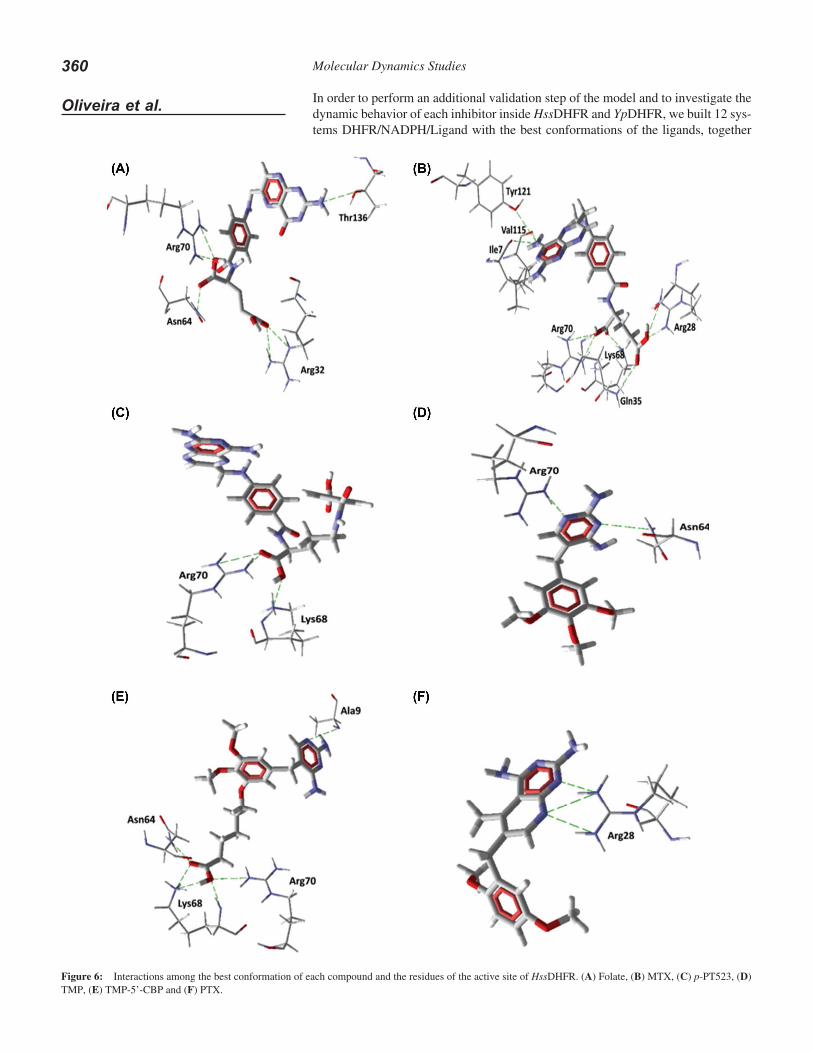
In order to perform an additional validation step of the model and to investigate the dynamic behavior of each inhibitor inside HssDHFR and YpDHFR, we built 12 sys-tems DHFR/NADPH/Ligand with the best conformations of the ligands, together
Figure 6: Interactions among the best conformation of each compound and the residues of the active site of HssDHFR. (A) Folate, (B) MTX, (C) p-PT523, (D) TMP, (E) TMP-5’-CBP and (F) PTX.

361
Binding to Yersinia pestis Dihydrofolate Reductase
with the NADPH from the crystallographic structure of PcDHFR reported by Cody et al. (57). After minimization these systems were submitted to 500 ps of PRMD followed by 10.0 ns of MD simulations with no restriction, as described in the meth-odology, and following the same procedure employed in former works (85-92).
Calculations of the temporal RMSD have been carried out on all the atoms in all the systems for 500 frames generated at each 20 ps of MD simulation. In this case the result is a unique general value for each enzyme monitored throughout the whole simulation time. Taking into account that the complexes could float inside the water box, each frame was adjusted to the former by the minimum squares method when calculating the standard deviation. It was observed that the systems DHFR/folate equilibrate along the first few picoseconds of the MD simulation. This behavior was common to all 12 MD simulations, with deviations never passing 0.52 nm (5.2 Å) and 0.42 nm (4.2 Å) for YpDHFR and folate, respectively and 0.40 nm (4.0 Å) and 0.35 nm (3.5 Å) for HssDHFR and folate, respectively. We also observed that, for all simulations, the temporal RMSD of the DHFRs as apoenzymes, practically fits the temporal RMSD of the complexes. Also, as expected, the ligands display a greater fluctuation than for the whole complexes but with a smaller RMSD, thus confirming the stability of the MD simulations. These results can be seen for the systems YpDHFR/Folate and HssDHFR/Folate in Figure 1 of the supplementary material.
The spatial RMSD (RMSF) on each amino acid residue was also calculated in the time range from 100 to 10,000 ps, at each 10 ps, totalizing 900 frames. The qualita-tive illustrations in the tube representation of the RMSF for the systems YpDHFR/Folate and HssDHFR/Folate are shown in the Figrue 2 of the supplementary mate-rial. This picture shows that the most mobile regions along the MD simulations
Table IIIResidues involved in H-bonds with the ligands inside YpDHFR and HssDHFR active sites. The most relevant residues are shown in bold.
Compound YpDHFR HssDHFR
Folate Ala8, Asp28, Leu29, Arg58, Thr114
Arg32, Asn64, Arg70, Thr136
MTX Asp28, Lys33, Arg53, Arg58, Thr114
Ile7, Arg28, Gln35, Lys68, Arg70, Val115, Tyr121
p-PT253 Ala8, Asp28, Leu29, Phe32, Lys33, Arg53, Arg58
Lys68, Arg70
TMP Leu29, Lys33, Arg58 Asn64, Arg70TMP-5′-CBP Ile6, Leu29, Lys33 Ala9, Asn64, Lys68, Arg70PTX Leu29, Lys33, Arg58 Arg28
Figure 7: Possible hydrophobic interactions between TMP and Phe31 in HssDHFR (A) and Phe32 in YpDHFR (B).

362
Oliveira et al.
are the regions with major thickness of the tubes. These regions correspond to the residues near to the two terminus of each monomer and to the loops regions. On the other hand, the residues at the active site regions, as well as those at the α-helix and β-sheet regions, present lower RMSF values, revealing to be the most stable regions of the system.
Dynamical Behavior of the Inhibitors Inside the Active Sites of YpDHFR and HssDHFR
The MD and docking results corroborate in several aspects, mainly regarding the stabilization of the inhibitors inside the active sites. MTX and p-PT523 for instance showed less stability inside YpDHFR than inside HssDHFR, especially consider-ing the dynamical behavior of their tails. This result is in accordance with the best MolDock Score values observed for these compounds inside HssDHFR and with the experimental results in literature, pointing them as better inhibitors of DHFRs from mammals (49).
MD results permitted us to observe that MTX was able to establish, in average, 1–4 H-bonds with YpDHFR and 4–6 with HssDHFR along the simulated time. Inside YpDHFR, MTX interacted with Lys33, Asn38, Arg53 and Arg58 but was not able to anchor appropriately its tail. On the other hand, inside HssDHFR, the MTX behavior was much more stable along the simulation showing interactions with residues Ile7, Phe31, Val115 and Tyr121, as already predicted by the docking studies, with its tail well anchored and establishing H-bonds with residues Arg28, Asn64 and Arg70. We believe that this behavior explains the better experimental results of MTX as inhibitor of DHFR from mammals (49, 50, 81). The plots of the H-bonds observed between MTX inside YpDHFR and HssDHFR along the 10.0 ns of MD simulation are shown in Figure 3 of the supplementary material.
p-PT523 was able to establish up to 3 H-bonds inside YpDHFR and 2–5 inside HssDHFR along the MD simulation. The head of this inhibitor interacted with Asp28 and Tyr101 while its tail was able to interact with Lys33 and Arg53 inside YpDHFR. Inside HssDHFR the head of p-PT523 established hydrophobic interac-tions with Phe34 and H-bond with Thr136. The tail stabilized well among residues Arg28, Arg32, Gln35 and Lys 63. The corresponding residues in YpDHFR (Pro26, Ala30, Lys33) were not observed interacting with p-PT523. Regarding Lys63, there is no corresponding residue in YpDHFR. In the same way as for MTX, the p-PT523 tail anchored better inside HssDHFR than inside YpDHFR, showing a bet-ter dynamical behavior and justifying the better results for this compound as inhibi-tor of DHFR from mammals (49, 50, 81). The frames of p-PT523 inside YpDHFR and HssDHFR extracted at each 20 ps of MD simulation are shown in Figure 8.
MD results for TMP showed that this inhibitor established in average only one H-Bond inside both active sites along the simulated time. Interactions with Leu29 and Lys33, the H-Bond with Arg53 and hydrophobic interactions with Phe32 were observed inside YpDHFR along the simulation as suggested in the docking stud-ies. Inside HssDHFR, however, the hydrophobic interactions were observed with Phe31 and the H-Bond with Arg70. The frames of TMP inside HssDHFR and YpDHFR extracted at each 20 ps of MD simulation are shown in Figure 9.
PTX was the inhibitor that presented the most stable behavior inside YpDHFR, also corroborating the docking results. This compound forms up to 2 H-Bonds in both active sites along the simulation. Inside YpDHFR, PTX formed H-Bond with Lys33 and Arg58. The residue corresponding to Arg58 in HssDHFR, Arg70, was not observed to form H-Bond with PTX along the MD simulation. The H-Bonds of PTX inside HssDHFR were formed with residues Asn64 and Lys68. Also, residues Phe31 and Phe34 were able to establish hydrophobic interactions with PTX inside

363
Binding to Yersinia pestis Dihydrofolate Reductase
HssDHFR. The frames of PTX inside HssDHFR and YpDHFR extracted at each 20 ps of MD simulation are shown in Figure 10.
Despite the better MolDock Score docking results inside YpDHFR, the docking and MD simulations for TMP-5′-CBP showed that this inhibitor presented more H-Bonds with HssDHFR. Inside YpDHFR the H-Bonds of TMP-5′-CBP were observed with Asp28 and Lys33. Also, hydrophobic interactions were observed
Figure 8: Frames of p-PT523 (in licorice) in the active site of YpDHFR (A) and HssDHFR (B) along the 10.0 ns of MD simulation.
Figure 9: Frames of TMP (in licorice) in the active site of YpDHFR (A) and HssDHFR (B) along the 10.0 ns of MD simulation.
with Trp23 and Phe31. Phe32 and Phe34 in HssDHFR were, also, able to establish hydrophobic interactions with this inhibitor. The tail of TMP-5′-CBP establishes many interactions with the active site of HssDHFR, involving residues Gln35, Asn64, Lys68, and Arg70 (Figure 11). MD results showed that the TMP-5′-CBP tail behaves better inside HssDHFR, in the same way as MTX and p-PT523. These results suggest that a long tail is not a desirable feature for selective inhibitors of YpDHFR. The plots of the H-bonds observed for TMP-5′-CBP inside YpDHFR and HssDHFR along the 10.0 ns of MD simulation are shown in Figure 4 of the supplementary material.

364
Oliveira et al.
structure of YpDHFR, deposited in the PDB (51, 52), and the crystallographic structure of EcDHFR as templates. The evaluation of this model using the packages PROCHECK (63), WHAT IF (64) and PROSA (68) followed by energy minimiza-tion with the software GROMACS 4.0 (58, 59) validated the model and suggested its equivalence to the experimentally resolved 3D structure.
The further step of the work consisted in a comparative and qualitative preliminary evaluation of folate (natural DHFR substrate), and 5 known DHFR inhibitors, with different structural features, docked in the active sites of YpDHFR and HssDHFR, in order to check out the affinities of the inhibitors related to folate and, also, to look for insights to the design of more potent and selective DHFR inhibitors. Results showed that most of the 5 DHFR inhibitors presented similar values of docking
Figure 11: Frames of TMP-5′-CBP in the active site of YpDHFR (A) and HssDHFR (B) along the 10.0 ns of MD simulation.
Figure 10: Frames of PTX (in licorice) in the active site of YpDHFR (A) and HssDHFR (B) along the 10.0 ns of MD simulation.
Conclusions
In the first step of this work we proposed YpDHFR as an additional target to the development of antiplague chemotherapy and used homology modeling, in a simi-lar approach already employed in former studies (88, 89, 91-93), to propose a func-tional 3D structure for YpDHFR built by single alignment to the crystallographic

365
Binding to Yersinia pestis Dihydrofolate Reductase
energy to folate inside the two enzymes. These results corroborate literature reports of these compounds as potent DHFR inhibitors and also suggest that they could work as well as YpDHFR inhibitors.
An additional analysis of the residues afforded by MVD® (71) as interacting with the best conformations of each ligand, revealed several residues not reported yet as part of the folate binding sites in YpDHFR and HssDHFR. Interactions with these residues in HssDHFR, for example, should be kept in mind when designing more potent DHFR inhibitors for cancer chemotherapy. Besides, the residues observed for YpDHFR should be considered in the design of selective inhibitors of YpDHFR.
MD simulations performed in the GROMACS 4.0 package (58, 59) allowed us to evaluate the dynamic behavior of the systems studied and corroborate the docking results, as well as identify key residues in the enzyme-inhibitor interaction. Results of these studies will guide the design of potent selective inhibitors of YpDHFR in order to strengthen the therapeutic arsenal available today against the dangerous biological warfare agent represented by Y. pestis.
Acknowledgements
The Authors wish to thank the Brazilian financial agencies CNPq, FAPERJ and CAPES/MD (Edital PRODEFESA 2008) for financial support and the Military Institute of Engineering for the physical infrastructure and working space.
References
M. Achtman, K. Zurth, G. Morelli, G. Torrea, A. Guiyoule, and E. Carniel. 1. Proc Natl Acad Sci USA 96, 14043-14048 (1999). Erratum in: Proc Natl Acad Sci USA 97, 8192 (2000).R. D. Perry and J. D. Fetherston. 2. Clin Microbiol Rev 10, 35-66 (1997).M. Kukkonen, K. Lahteenmaki, M. Suomalainen, N. Kalkkinen, L. Emody, H. Lang, and 3. T. K. Korhonen. Mol Microbiol 40, 1097-1111 (2001).I. Bitam, B. Baziz, J. Rolain, M. Belkaid, and D. Raoult. 4. Emerg Infect Dis 12, 1975-1976 (2006).I. Bitam, K. Dittmar, P. Parola, M. F. Whiting, and D. Raoult. 5. Int J Infect Dis 14, e667-e676 (2010).R. J. Eisen, J. N. Borchert, J. L. Holmes, G. Amatre, K. V. Wyk, R. E. Enscore, N. Babi, 6. L. A. Atiku, A. P. Wilder, S. M. Vetter, S. W. Bearden, J. A. Montenieri, and K. L. Gage. Am J Trop Med Hyg 78, 949-956 (2008).K. Lahteenmaki, R. Virkola, A. Saren, L. Emody, and T. K. Korhonen. 7. Infect Immun 66, 5755-5762 (1998).K. Lahteenmaki, P. Kuuselam, and T. K. Korhonen. 8. FEMS Microbiol Rev 25, 531-552 (2001).M. Achtman, G. Morelli, P. Zhu, T. Wirth, I. Diehl, B. Kusecek, A. J. Vogler, D. M. Wagner, 9. C. J. Allender, W. R. Easterday, V. Chenal-Francisque, P. Worsham, N. R. Thomson, J. Parkhill, L. E. Lindler, E. Carniel, and P. Keim. Proc Natl Acad Sci USA 101, 17837-17842 (2004).M. Galimand, A. Guiyoule, G. Gerbaud, B. Rasoamanana, S. Chanteau, E. Carniel, and 10. P. Courvalin. New Eng J of Med 337, 677-680 (1997).T. J. Welch, W. F. Fricke, P. F. McDermott, D. G. White, M. L. Rosso, D. A. Rasko, 11. M. K. Mammel, M. Eppinger, M. J. Rosovitz, D. Wagner, L. Rahalison, J. E. LeClerc, J. M. Hinshaw, L. E. Lindler, T. A. Cebula, E. Carniel, and J. Ravel. PloS ONE 2, e309 (2007).F. M. Huennekens, T. H. Duffy, and K. S. Vitols. 12. NCI Monogr 5, 1-8 (1987).H. A. Ingraham, L. Dickey, and M. Goulian. 13. Biochemistry 25, 3225- 3230 (1986).A. Yoshida, S. Tanaka, O. Hiraoka, Y. Koyama, Y. Hirota, D. Ayusawa, T. Seno, C. Garrett, 14. and Y. Wataya. J Biol Chem 262, 8235- 8241 (1987).B. Roth, B. S. Rauckman, R. Ferone, D. P. Baccanari, J. N. Champness, and R. M. Hyde. 15. J Med Chem 30, 348-356 (1987).A. Rosowsky, R. A. Forsch, and S. F. Queener. 16. J Med Chem 45, 233-241 (2002).L. F. Kuyper, B. Roth, D. P. Baccanari, R. Ferone, C. R. Beddell, J. N. Champness, 17. D. K. Stammers, J. Dann, F. E. Norrington, D. J. Baker, and P. J. Goodford. J Med Chem 28, 303-311 (1985).E. D. Akten, S. Cansu, and P. Doruker. 18. J Biomol Struct Dyn 27, 13-25 (2009).A. M. Andrianov. 19. J Biomol Struct Dyn 26, 445-454 (2009).A. M. Andrianov and I. V. Anishchenko. 20. J Biomol Struct Dyn 27, 179-193 (2009).

366
Oliveira et al.
A. Borkar, I. Ghosh, and D. Bhattacharyya. 21. J Biomol Struct Dyn 27, 695-712 (2010).M. T. Cambria, D. Di Marino, M. Falconi, S. Garavaglia, and A. Cambria. 22. J Biomol Struct Dyn 27, 501-509 (2010).E. F. F. da Cunha, E. F. Barbosa, A. A. Oliveira, and T. C. Ramalho. 23. J Biomol Struct Dyn 27, 619-625 (2010).C. Meynier, F. Guerlesquin, and P. Roche. 24. J Biomol Struct Dyn 27, 49-57 (2009).S. Mohan, J. J. P. Perry, N. Poulose, B. G. Nair, and G. Anilkumar. 25. J Biomol Struct Dyn 26, 455-464 (2009).T. C. Ramalho, M. S. Caetano, E. F. F. da Cunha, T. C. S. Souza, and M. V. J. Rocha. 26. J Biomol Struct Dyn 27, 195-207 (2009).J. Sille and M. Remko. 27. J Biomol Struct Dyn 26, 431-444 (2009).H. R. Bairagya, B. P. Mukhopadhyay, and K. Sekar. 28. J Biomol Struct Dyn 27, 149-158 (2009).B. Jin, H. M. Lee, and S. K. Kim. 29. J Biomol Struct Dyn 27, 457-464 (2010).C. Koshy, M. Parthiban, and R. Sowdhamini. 30. J Biomol Struct Dyn 28, 71-83 (2010).F. Mehrnejad and M. Zarei. 31. J Biomol Struct Dyn 27, 551-559 (2010).S. Roy and A. R. Thakur. 32. J Biomol Struct Dyn 27, 443-455 (2010).A. Sharadadevi and R. Nagaraj. 33. J Biomol Struct Dyn 27, 541-550 (2010).S. Sharma, U. B. Sonavane, and R. R. Joshi. 34. J Biomol Struct Dyn 27, 663-676 (2010).Y. Tao, Z. H. Rao, and S. Q. Liu. 35. J Biomol Struct Dyn 28, 143-157 (2010).N. A. Timofeyeva, V. V. Koval, D. G. Knorre, D. O. Zharkov, M. K. Saparbaev, A. A. Ishchenko, 36. and O. S. Fedorova. J Biomol Struct Dyn 26, 637-652 (2009).J. F. Varughese, J. M. Chalovich, and Y. Li. 37. J Biomol Struct Dyn 28, 159-173 (2010).J. P. Zhang. 38. J Biomol Struct Dyn 27, 159-162 (2009).L. H. Zhong and J. M. Xie. 39. J Biomol Struct Dyn 26, 525-533 (2009).A. Cordomi and J. J. Perez. 40. J Biomol Struct Dyn 27, 127-147 (2009).L. I. D. Hage-Melim, C. H. T. D. da Silva, E. P. Semighini, C. A. Taft, and S. V. Sampaio. 41. J Biomol Struct Dyn 27, 27-35 (2009).A. K. Kahlon, S. Roy, and A. Sharma. 42. J Biomol Struct Dyn 28, 201-210 (2010).C. Y. C. Chen. 43. J Biomol Struct Dyn 27, 271-282 (2009).C. Y. Chen. 44. J Biomol Struct Dyn 27, 627-640 (2010).C. Y. Chen, Y. H. Chang, D. T. Bau, H. J. Huang, F. J. Tsai, C. H. Tsai, and C. Y. C. Chen. 45. J Biomol Struct Dyn 27, 171-178 (2009).H. J. Huang, K. J. Lee, H. W. Yu, C. Y. Chen, C. H. Hsu, H. Y. Chen, F. J. Tsai, and 46. C. Y. C. Chen. J Biomol Struct Dyn 28, 23-37 (2010).H. J. Huang, K. J. Lee, H. W. Yu, H. Y. Chen, F. J. Tsai, and C. Y. Chen. 47. J Biomol Struct Dyn 28, 187-200 (2010).G. N. Likhatskaya, T. F. Solov’eva, O. D. Novikova, M. P. Issaeva, K. V. Gusev, I. B. Kryzhko, 48. E. V. Trifonov, and E. A. Nurminski. J Biom Struc Dyn 23, 163-174 (2005).E. F. F. da Cunha, T. C. Ramalho, E. R. Maia, and R. B. Alencastro. 49. Expert Opin Ther Patents 15, 967-986 (2005).V. Cody, J. Pace, K. Chisum, and A.Rosowsky. 50. Prot Struc Func Bioinf 65, 959-969(2006).F. C. Bernstein, T. F. Koetzle, G. J. Williams, E. E. Meyer, M. D. Brice, J. R. Rodgers, 51. O. Kennard, T. Shimanouchi, and M. Tasumi. J Mol Biol 112, 535-542 (1977).H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, 52. and P. E. Bourne. Nucleic Acids Res 28, 235-242 (2000).R. L. Summerfield, D. M. Daigle, S. Mayer, D. Mallik, D. W. Hughes, S. G. Jackson, 53. M. Sulek, M. G. Organ, E. D. Brown, and M. S. Junop. J Med Chem 49, 6977-6986 (2006).N. Guex and M. C. Peitsch. 54. Electrophoresis 18, 2714-2723 (1997).T. Schwede, J. Kopp, N. Guex, and M. C. Peitsch. 55. Nucleic Acid Res 31, 3381-3385 (2003).K. Arnold, L. Bordoli, J. Kopp, and T. Schwede. 56. Bioinformatics 22, 195-201 (2006).V. Cody, N. Galitsky, D. Rak, J. R. Luft, W. Pangborn, and S. F. Queener. 57. Biochemistry 38, 4303-4312 (1999).D. van der Spoel, E. Lindahl, B. Hess, A. R. van Buuren, E. Apol, P. J. Meulenhoff, 58. D. P. Tieleman, A. L. T. M. Sijbers, K. A. Feenstra, R. van Drunen, and H. J. C. Berendsen. GROMACS user manual version 4.0. Groningen: University of Groningen, Department of Biophysical Chemistry. (2005).H. J. C. Berendsen, D. Van der Spoel, and R. Van Drunen. 59. Comput Phys Commun 91, 43-56 (1995).A. W. Schuettelkopf and D. M. F. van Aalten. 60. Acta Crystallogr, SectD: BiolCrystallogr 60, 1355-1363 (2004).A. E. Klon, A. Heroux, L. J. Ross, V. Pathak, C. A. Johnson, J. R. Piper, and D. W. Borhani. 61. J Mol Biol 320, 677-693 (2002).J. Pontius, J. Richelle, and S. J. Wodak. 62. J Mol Biol 264, 121-136 (1996).R. A. Laskowski, M. W. Macarthur, D. S. Moss, and J. M. Thornton. 63. J Appl Crystallogr 26, 283-291 (1993).G. J. Vriend. 64. Mol Graph 8, 52-56 (1990).J. U. Bowie, R. Luthy, and D. Eisenberg. 65. Science 253, 164-170 (1991).R. Luthy, J. U. Bowie, and D. Eisenberg. 66. Nature 356, 83-85 (1992).

367
Binding to Yersinia pestis Dihydrofolate Reductase
http://nihserver.mbi.ucla.edu/Verify_3D/ accessed in May 2008.67. M. J. Sippl. 68. Proteins 17, 355-362 (1993).W. J. Hehre, B. J. Deppmeier, and P. E. Klunzinger. 69. PC SPARTAN Pro. Irvine, CA: Wavefunction (1999).M. J. S. Dewar, E. G. Zoebisch, E. F. Healy, and J. J. P. Stewart. 70. J Am Chem Soc 107, 3902-3909 (1985).R. Thomsen and M. H. Christensen. 71. J Med Chem 49, 3315-3332 (2006).C. Oostenbrink, A. Villa, A. E. Mark, and W. F. van Gunsteren. 72. J Comput Chem 25, 1656-1676 (2004).W. Humphrey, A. Dalke, and K. Schulten. 73. J Mol Graph 14, 33-38 (1996).P. M. Edwards. 74. J Chem Inf Comput Sci 42, 1270- 1271 (2002).R. Koradi, M. Billeter, and K. Wüthrich. 75. J Mol Graph 14, 51-55 (1996).D. Warren. 76. The PyMOL molecular graphics system, DeLano Scientific, San Carlos, CA (2002).G. N. Ramachandran, and V. Sasisekharan. 77. Adv Protein Chem 23, 283-437 (1968).C. Oefner, A. D’arcy, and F. K. Winkler. 78. Eur J Biochem 174, 377-385 (1988).J. F. Davies, T. J. Delcamp, N. J. Prendergad, V. A. Ashford, J. H. Freisheim, and J. Kraut. 79. Biochemistry 29, 9467-9479 (1990).V. I. Polshakov. 80. Russ Chem Bull Int Ed50, 1733-1752 (2001).J. E. Wright, G. K. Yurasek, Y. Chen 81. et al.: Further studies on the interaction of nonpolyglu-tamatable aminopterin analogs with dihydrofolate reductase and the reduced folate carrier as determinants of in vitro antitumor activity. Biochem Pharmacol 65, 1427-1433 (2003).G. L. Warren, A. C. Webster, A. M. Capelli, B. Clarke, J. LaLonde, M. H. Lambert, 82. M. Lindvall, N. Nevins, S. F. Semus, S. Senger, G. Tedesco, I. D. Wall, J. M. Woolven, C. E. Peishoff, and M. S. Head. J Med Chem 49, 5912-5931 (2006).A. R. Leach, B. K. Shoichet, and C. E. Peishoff. 83. J Med Chem 49, 5851-5855 (2006).M. Kontoyanni, L. M. McClellan, and G. S. Sokol. 84. J Med Chem 47, 558-565 (2004).A. S. Gonçalves, T. C. C. França, J. D. F. Villar, and P. G. Pascutti. 85. J Braz Chem Soc 22, 155-165 (2011).A. P. Guimarães, A. A. Oliveira, E. F. F. Cunha, T. C. Ramalho, and T. C. C. França. 86. J Biom Struc Dyn 28, 455-469 (2011).A. P. Guimarães, A. A. Oliveira, E. F. F. Cunha, T. C. Ramalho, and T. C. C. França. 87. J Mol Model 17, 1-13 (2011).M. L. da Silva, A. G. da Silva, P. R. Batista, J. D. Figueroa-Villar, P. G. Pascutti, and 88. T. C. C. França. Mol Sim 36, 5-14 (2010).T. C. C. França, M. R. M. Rocha, B. M. Reboredo, M. N. Renno, L. W. Tinoco, and 89. J. D. Figueroa-Villar. J Braz Chem Soc 19, 64-73 (2008).A. S. Gonçalves, T. C. C. França, A. Wilter, and J. D. F. Villar. 90. J Braz Chem Soc 17, 968-975 (2006).T. C. C. França, A. Wilter, P. G. Pascutti, T. C. Ramalho, and J. D. Figueroa-Villar. 91. J Braz Chem Soc 17, 1383-1392 (2006).T. C. C. França, P. G. Pascutti, T. C. Ramalho, and J. D. Figueroa-Villar. 92. Biophys Chem 115, 1-10 (2005).T. C. C. França, A. L. Medeiros, O. A. Santos Filho, E. C. P. Santos, and J. D. Figueroa-Villar. 93. J Braz Chem Soc 15, 450-454 (2004).
Date Received: April 15, 2011
Communicated by the Editor Ramaswamy H. Sarma

Related Documents











