Conformational states and association mechanism of Yersinia pestis Caf1 subunits Luigi Vitagliano a , Alessia Ruggiero a , Carlo Pedone a,b , Rita Berisio a, * a Istituto di Biostrutture e Bioimmagini, C.N.R., Via Mezzocannone 16, I-80134 Napoli, Italy b Dipartimento delle Scienze Biologiche, Sezione di Biostrutture, Università degli Studi di Napoli ‘‘Federico II”, Via Mezzocannone 16, I-80134 Napoli, Italy article info Article history: Received 20 May 2008 Available online 4 June 2008 Keywords: Protein–protein interactions Donor-strand exchange Chaperone Molecular dynamics Adhesive proteins abstract Bacterial infectivity often relies on efficient attachment to the host cells through adhesive extensions. Unveiling the structural basis of the formation of these organelles is of paramount importance for both academic and applicative implications. Computational approaches may fruitfully complement experi- mental studies by providing information on specific conformational states whose characterization is dif- ficult. Here, we report molecular dynamics characterizations of Yersinia pestis Caf1 subunit in its monomeric-unbound and dimeric states. Data on the monomeric form indicate that it is highly reactive and evolves toward compact states, which likely hamper subunit–subunit association. In line with recent experimental reports, this finding implies that chaperone release and subunit–subunit association must be simultaneous. MD analysis on Caf1 dimer lead to the formation of a novel assembly endowed with a significant stability in the simulation timescale. Using these data, an end-to-end model of the fiber, which well agrees with available experimental data, was also generated. Ó 2008 Elsevier Inc. All rights reserved. Bacterial infectivity often relies on efficient attachment to the host cells. To this scope, bacteria have developed a variety of dif- ferent adhesive extensions. The chaperone/usher secretion sys- tem represents the prominent machinery devoted to the production of these organelles [1,2]. Based on their structure and morphology, two distinct families have been identified. One includes either thick rigid or thin flexible adhesive pili made of pilin or fimbrin subunit, while the other contains non-pilus organelles with an amorphous or capsule-like morphology [3,4]. The assembly of adhesive pili and non-pilus extensions is assisted by FGS and FGL chaperones, respectively [5,6]. Although these chaperones share a common Ig-like fold, they are charac- terized by a different length of the loop connecting the F1 and G1 strand, which is longer in the FGL family [7]. Very recently, a different classification of these organelles, based on the adhe- sive properties of the structural subunit has been proposed [7]. In this framework, FGL chaperone-assisted adhesive extensions and the majority of FGS chaperone assembled adhesive pili are fimbrial polyadhesins and monoadhesins, respectively [7]. Struc- tural investigations have provided enlightening information on the mechanisms of monoadhesins and fimbrial polyadhesins [2,5–15]. These studies have also highlighted analogies and dif- ferences between these two classes [7,8]. In both cases, their constituting subunits exhibit a non-canonical incomplete immu- no-globulin-like (Ig-like) fold with an N-terminal region which does not interact with the rest of the protein. The association of the subunits is based on the binding of the N-terminus of one subunit to the main body of the adjacent one via a donor- strand complementation mechanism. Chaperones (FGS and FGL) assist the folding of the subunits and are vital for their correct assembly in the final organelles. However, in this common sce- nario, significant differences between the two classes are de- tected in the subunit/chaperone association. In particular, fimbrial polyadhesins/FGL complexes present an enhanced sur- face interaction, compared to monoadhesins/FGS ones. Although these crystallographic studies have provided a solid base for the understanding of these processes at molecular level, some important aspects, essentially related with the structure and the role of transient species, are yet to be clarified. Recent inves- tigations have shown that molecular dynamics simulations are valuable in providing atomic level details of these complex pro- cesses, whose inhibition strongly depotentiate these pathogens [13,16,17]. Here we applied this approach to the Caf1 subunit, which represents the basic element of the capsulae formed by Yersinia pestis, the pathogen responsible for human bubonic and pneumonic plague. We focused our attention on two states of the protein whose structure has not been characterized exper- imentally. In particular, our simulations provide insights into the structural features of the incomplete Ig-like Caf1 domain in its unbound state and into the subunit/subunit association process that leads to the formation of capsulae. 0006-291X/$ - see front matter Ó 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.bbrc.2008.05.145 * Corresponding author. Fax: +39 08 12536642. E-mail address: [email protected] (R. Berisio). Biochemical and Biophysical Research Communications 372 (2008) 804–810 Contents lists available at ScienceDirect Biochemical and Biophysical Research Communications journal homepage: www.elsevier.com/locate/ybbrc

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Biochemical and Biophysical Research Communications 372 (2008) 804–810
Contents lists available at ScienceDirect
Biochemical and Biophysical Research Communications
journal homepage: www.elsevier .com/locate /ybbrc
Conformational states and association mechanism of Yersinia pestisCaf1 subunits
Luigi Vitagliano a, Alessia Ruggiero a, Carlo Pedone a,b, Rita Berisio a,*
a Istituto di Biostrutture e Bioimmagini, C.N.R., Via Mezzocannone 16, I-80134 Napoli, Italyb Dipartimento delle Scienze Biologiche, Sezione di Biostrutture, Università degli Studi di Napoli ‘‘Federico II”, Via Mezzocannone 16, I-80134 Napoli, Italy
a r t i c l e i n f o
Article history:Received 20 May 2008Available online 4 June 2008
Keywords:Protein–protein interactionsDonor-strand exchangeChaperoneMolecular dynamicsAdhesive proteins
0006-291X/$ - see front matter � 2008 Elsevier Inc. Adoi:10.1016/j.bbrc.2008.05.145
* Corresponding author. Fax: +39 08 12536642.E-mail address: [email protected] (R. Berisio).
a b s t r a c t
Bacterial infectivity often relies on efficient attachment to the host cells through adhesive extensions.Unveiling the structural basis of the formation of these organelles is of paramount importance for bothacademic and applicative implications. Computational approaches may fruitfully complement experi-mental studies by providing information on specific conformational states whose characterization is dif-ficult. Here, we report molecular dynamics characterizations of Yersinia pestis Caf1 subunit in itsmonomeric-unbound and dimeric states. Data on the monomeric form indicate that it is highly reactiveand evolves toward compact states, which likely hamper subunit–subunit association. In line with recentexperimental reports, this finding implies that chaperone release and subunit–subunit association mustbe simultaneous. MD analysis on Caf1 dimer lead to the formation of a novel assembly endowed with asignificant stability in the simulation timescale. Using these data, an end-to-end model of the fiber, whichwell agrees with available experimental data, was also generated.
� 2008 Elsevier Inc. All rights reserved.
Bacterial infectivity often relies on efficient attachment to thehost cells. To this scope, bacteria have developed a variety of dif-ferent adhesive extensions. The chaperone/usher secretion sys-tem represents the prominent machinery devoted to theproduction of these organelles [1,2]. Based on their structureand morphology, two distinct families have been identified.One includes either thick rigid or thin flexible adhesive pili madeof pilin or fimbrin subunit, while the other contains non-pilusorganelles with an amorphous or capsule-like morphology[3,4]. The assembly of adhesive pili and non-pilus extensions isassisted by FGS and FGL chaperones, respectively [5,6]. Althoughthese chaperones share a common Ig-like fold, they are charac-terized by a different length of the loop connecting the F1 andG1 strand, which is longer in the FGL family [7]. Very recently,a different classification of these organelles, based on the adhe-sive properties of the structural subunit has been proposed [7].In this framework, FGL chaperone-assisted adhesive extensionsand the majority of FGS chaperone assembled adhesive pili arefimbrial polyadhesins and monoadhesins, respectively [7]. Struc-tural investigations have provided enlightening information onthe mechanisms of monoadhesins and fimbrial polyadhesins[2,5–15]. These studies have also highlighted analogies and dif-ferences between these two classes [7,8]. In both cases, theirconstituting subunits exhibit a non-canonical incomplete immu-
ll rights reserved.
no-globulin-like (Ig-like) fold with an N-terminal region whichdoes not interact with the rest of the protein. The associationof the subunits is based on the binding of the N-terminus ofone subunit to the main body of the adjacent one via a donor-strand complementation mechanism. Chaperones (FGS and FGL)assist the folding of the subunits and are vital for their correctassembly in the final organelles. However, in this common sce-nario, significant differences between the two classes are de-tected in the subunit/chaperone association. In particular,fimbrial polyadhesins/FGL complexes present an enhanced sur-face interaction, compared to monoadhesins/FGS ones. Althoughthese crystallographic studies have provided a solid base forthe understanding of these processes at molecular level, someimportant aspects, essentially related with the structure andthe role of transient species, are yet to be clarified. Recent inves-tigations have shown that molecular dynamics simulations arevaluable in providing atomic level details of these complex pro-cesses, whose inhibition strongly depotentiate these pathogens[13,16,17]. Here we applied this approach to the Caf1 subunit,which represents the basic element of the capsulae formed byYersinia pestis, the pathogen responsible for human bubonicand pneumonic plague. We focused our attention on two statesof the protein whose structure has not been characterized exper-imentally. In particular, our simulations provide insights into thestructural features of the incomplete Ig-like Caf1 domain in itsunbound state and into the subunit/subunit association processthat leads to the formation of capsulae.

L. Vitagliano et al. / Biochemical and Biophysical Research Communications 372 (2008) 804–810 805
Materials and methods
Starting models. The starting coordinates of Caf1 subunit werederived from the complex between the chaperone Caf1M andtwo Caf1 subunits (Caf1M–Caf10–Caf100) [5,13]. In the complexthe Caf10 subunit is bound the chaperone Caf1M, while Caf100 bindsthe N-terminal extension donated by Caf10. The highest resolutionstructure of the complex (1.99 Å), corresponding to the mutantAla9Arg was used to build the models used in the simulations(PDB code 1P5U). To simulate the conformational transition occur-ring upon chaperone release, the main body of the chaperone-bound Caf1’ subunit (residues 16–149) was used as starting modelin the analysis of the unbound Caf1 subunit dynamics. The startingarrangement of Caf1 dimer was based on the Caf10–Caf100 organiza-tion observed in the ternary Caf1M–Caf10–Caf00 complex. Also, tosimulate the final fiber, which is made of Caf1 subunits resemblingCaf00, the model of Caf10 was replaced by Caf100. Model building wasused to reconstruct the missing regions, typically loops, of thestarting structure. To reproduce the sequence of the wild-typeform, an Ala residue was built in position 9.
Simulation procedure. MD simulations were performed using theGROMACS software package 3.3 [18] and following the proceduresused in previous studies [16,19,20]. In all MD simulations the mod-el was immersed in a rectangular box filled with water molecules.The dimensions of the box were 7.54 � 5.23 � 4.93 nm3 (numberof water molecules 5538) and 11.20 � 8.07 � 6.25 nm3 (numberof water molecules 17,257) for the runs carried on the monomericand dimeric forms of Caf1, respectively (see above). The GRO-MOS43a1 force field and the SPCE water model were used in thesimulations. The simulations were run with periodic boundaryconditions. Systems were simulated in NPT ensemble by keepingconstant temperature (300 K) and pressure (1 atm).
Before starting the MD simulation, energies were minimizedby fixing the protein atoms and then without restraints. Thesystem temperature was brought to 300 K in a step-wise man-ner. In particular, 20 ps MD runs were carried out at 50, 100,150, 200, 250, and 300 K. For each model, MD simulations werecarried out in a time scale of 30,000 ps. Bond lengths were con-strained by the LINCS procedure. Lennard–Jones interactionswere calculated with a 12 Å twin-range cutoff. A dielectric con-stant of 1 and a time step of 2fs were used. Electrostatic inter-actions were treated using the Particle Mesh Ewald (PME)method with a cutoff of 10.0 Å. Trajectories were checked to as-sess the quality of the simulation using GROMACS routines andthe program VMD [21]. We checked that in all simulations thelowest distance between protein images was always larger thannon-bonded and electrostatic cut-off values (12 Å).
Results and discussion
Dynamics of the unliganded Caf1 reveals a new accessibleconformational state for this subunit
The investigation of the intrinsic conformational behavior of theIg-like Caf1 domain (residues 16–149) in its unbound state wasconducted by MD simulations using the structure of the Caf10 sub-unit, derived from the ternary complex Caf1M–Caf10–Caf100, as astarting model [5](Fig. S1). The analysis of the system evolutionthroughout the MD simulation, carried out using several indica-tors, clearly show that Caf1 undergoes a rapid transition withinthe first ns (Fig. S2A). RMSD deviations from the starting X-raymodel indicate that an early event (<1000 ps) is followed by a sec-ond smoother transition occurring at 7000–8000 ps, which leads toa state that is stable over the last 20,000 ps of the trajectory. Therather large deviations of the trajectory structures from the start-ing X-ray model (�3.3 Å) are indicative of significant variations
occurring during the simulation. The overall transformation ofthe system is associated with a significant decrease of the totalaccessible surface area (Fig. S2B). Analysis of the trajectory struc-tures suggests that the largest modifications affect the regions onthe protein surface (strands A and F) that were initially involvedin the complex with Caf1M and line an empty groove in the start-ing X-ray model. The system reacts to the chaperone dissociationby reducing the extent of the exposed surface. In particular, resi-dues of the strands A and F come closer during the simulation.To quantify these effects we evaluated the evolution, throughoutthe simulation, of distances between Ca atoms of residues facingeach other in the groove (Fig. S1). As shown in Fig. 1, a significantdecrease is observed for all distances taken into account. However,the entity of the movements is diversified along the groove. In par-ticular, the distance between residues involved in the P1 site (19–22 and 143–145) evolves to values similar to those detected in the‘‘closed” Caf1’’ state of the ternary complex Caf1M–Caf10–Caf100 [5].A similar trend is observed in an alternative 30,000 ps MD run car-ried out starting the simulation directly at 300 K without the step-wise temperature increase pre-equilibration procedure (Fig. S3).This finding is in agreement with a previous short (<1 ns) MD sim-ulation on the same system [13] and supports the hypothesis thatthis spontaneous transformation of the subunit upon chaperonedissociation provides the energy required for the fiber formationprocess [5,13]. However, our simulations show that other regionsof the groove, specifically those corresponding to the site P5 (resi-dues 26–29 and residues 136–138), exhibit a more complicateddynamic behavior. Indeed, the initial closure of the structure ob-served in the first 4000–7000 is followed by a second transition.As a consequence, in the plateau region of the trajectory(10,000–30,000), the approaching of facing residues of the groovegoes beyond that observed in the ‘‘closed” Caf100 form (Fig. 1 and2) and leads to the formation of H-bonds between residues of thestrands A and F (Fig. 1E and F). The analysis of the distances be-tween atoms involved in these H-bonds interactions clearly indi-cates that, once formed, these interactions are fairly stable.Notably, the P5 site is the recognition region that initiates the do-nor-strand exchange in the subunit–subunit association that leadsto the formation of the capsulae. Therefore, the spontaneous clo-sure of this site upon chaperone dissociation makes the subunit–subunit association process more problematic. There are severalindications that in chaperone/usher secretion systems the chaper-one release and donor-strand complementation between subunitsare concerted processes [6,22]. It has also been shown that not onlyare chaperones important for subunit folding but they also favorthe fiber formation [6]. In this framework, present data provide arationale to these observations. Indeed, chaperones are importantfor preventing a collapse of the subunit that would obstruct thegroove at the P5 side and, therefore, hamper strand exchangeand subunit–subunit association. Since analogue effects are ob-served in chaperone (FGS family) release by pilins [16], these find-ings suggest that unliganded monoadhesins and fimbrialpolyadhesins share a similar tendency to collapse despite a ratherdifferent interaction with their chaperones. This quick and sponta-neous structural transition can be considered as a manifestation ofthe high reactivity of these chains. Their tendency to bury residuesthat are exposed in their unbound state may be an important factorthat favors the polymerization process.
Dynamics of the Caf1 dimer provides insight into subunit–subunitassociation within the fiber
The simultaneous presence in the ternary complex Caf1M–Caf10–Caf100 of two Caf1 chains provides the opportunity to evalu-ate preferred association modes of these subunits that may berelevant for the fiber formation process. However, in the crystal
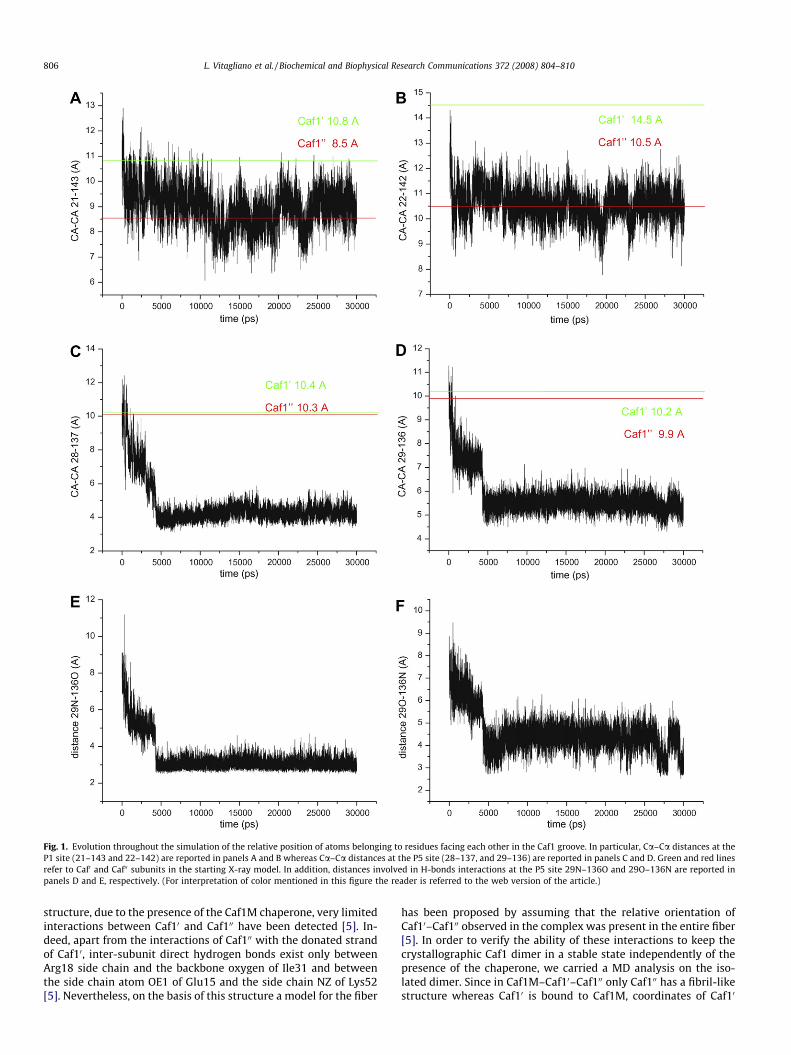
Fig. 1. Evolution throughout the simulation of the relative position of atoms belonging to residues facing each other in the Caf1 groove. In particular, Ca–Ca distances at theP1 site (21–143 and 22–142) are reported in panels A and B whereas Ca–Ca distances at the P5 site (28–137, and 29–136) are reported in panels C and D. Green and red linesrefer to Caf0 and Caf00 subunits in the starting X-ray model. In addition, distances involved in H-bonds interactions at the P5 site 29N–136O and 29O–136N are reported inpanels D and E, respectively. (For interpretation of color mentioned in this figure the reader is referred to the web version of the article.)
806 L. Vitagliano et al. / Biochemical and Biophysical Research Communications 372 (2008) 804–810
structure, due to the presence of the Caf1M chaperone, very limitedinteractions between Caf10 and Caf100 have been detected [5]. In-deed, apart from the interactions of Caf100 with the donated strandof Caf10, inter-subunit direct hydrogen bonds exist only betweenArg18 side chain and the backbone oxygen of Ile31 and betweenthe side chain atom OE1 of Glu15 and the side chain NZ of Lys52[5]. Nevertheless, on the basis of this structure a model for the fiber
has been proposed by assuming that the relative orientation ofCaf10–Caf100 observed in the complex was present in the entire fiber[5]. In order to verify the ability of these interactions to keep thecrystallographic Caf1 dimer in a stable state independently of thepresence of the chaperone, we carried a MD analysis on the iso-lated dimer. Since in Caf1M–Caf10–Caf100 only Caf100 has a fibril-likestructure whereas Caf10 is bound to Caf1M, coordinates of Caf10
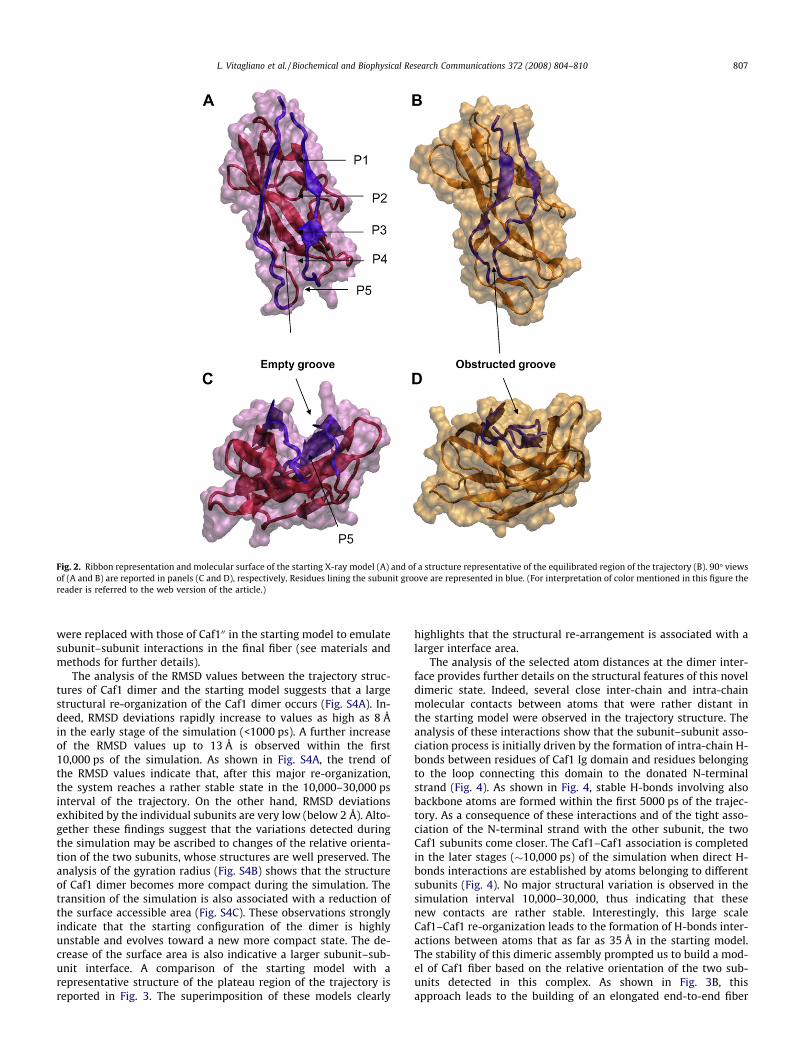
Fig. 2. Ribbon representation and molecular surface of the starting X-ray model (A) and of a structure representative of the equilibrated region of the trajectory (B). 90� viewsof (A and B) are reported in panels (C and D), respectively. Residues lining the subunit groove are represented in blue. (For interpretation of color mentioned in this figure thereader is referred to the web version of the article.)
L. Vitagliano et al. / Biochemical and Biophysical Research Communications 372 (2008) 804–810 807
were replaced with those of Caf100 in the starting model to emulatesubunit–subunit interactions in the final fiber (see materials andmethods for further details).
The analysis of the RMSD values between the trajectory struc-tures of Caf1 dimer and the starting model suggests that a largestructural re-organization of the Caf1 dimer occurs (Fig. S4A). In-deed, RMSD deviations rapidly increase to values as high as 8 Åin the early stage of the simulation (<1000 ps). A further increaseof the RMSD values up to 13 Å is observed within the first10,000 ps of the simulation. As shown in Fig. S4A, the trend ofthe RMSD values indicate that, after this major re-organization,the system reaches a rather stable state in the 10,000–30,000 psinterval of the trajectory. On the other hand, RMSD deviationsexhibited by the individual subunits are very low (below 2 Å). Alto-gether these findings suggest that the variations detected duringthe simulation may be ascribed to changes of the relative orienta-tion of the two subunits, whose structures are well preserved. Theanalysis of the gyration radius (Fig. S4B) shows that the structureof Caf1 dimer becomes more compact during the simulation. Thetransition of the simulation is also associated with a reduction ofthe surface accessible area (Fig. S4C). These observations stronglyindicate that the starting configuration of the dimer is highlyunstable and evolves toward a new more compact state. The de-crease of the surface area is also indicative a larger subunit–sub-unit interface. A comparison of the starting model with arepresentative structure of the plateau region of the trajectory isreported in Fig. 3. The superimposition of these models clearly
highlights that the structural re-arrangement is associated with alarger interface area.
The analysis of the selected atom distances at the dimer inter-face provides further details on the structural features of this noveldimeric state. Indeed, several close inter-chain and intra-chainmolecular contacts between atoms that were rather distant inthe starting model were observed in the trajectory structure. Theanalysis of these interactions show that the subunit–subunit asso-ciation process is initially driven by the formation of intra-chain H-bonds between residues of Caf1 Ig domain and residues belongingto the loop connecting this domain to the donated N-terminalstrand (Fig. 4). As shown in Fig. 4, stable H-bonds involving alsobackbone atoms are formed within the first 5000 ps of the trajec-tory. As a consequence of these interactions and of the tight asso-ciation of the N-terminal strand with the other subunit, the twoCaf1 subunits come closer. The Caf1–Caf1 association is completedin the later stages (�10,000 ps) of the simulation when direct H-bonds interactions are established by atoms belonging to differentsubunits (Fig. 4). No major structural variation is observed in thesimulation interval 10,000–30,000, thus indicating that thesenew contacts are rather stable. Interestingly, this large scaleCaf1–Caf1 re-organization leads to the formation of H-bonds inter-actions between atoms that as far as 35 Å in the starting model.The stability of this dimeric assembly prompted us to build a mod-el of Caf1 fiber based on the relative orientation of the two sub-units detected in this complex. As shown in Fig. 3B, thisapproach leads to the building of an elongated end-to-end fiber

Fig. 3. Superimposition of the starting model and a representative structure of the equilibrated region of the trajectory (A). A close-up at the subunit–subunit interface regionis shown in B. (C) Two views of the fiber model generated by using the relative orientation of Caf1 subunits derived from the simulation.
808 L. Vitagliano et al. / Biochemical and Biophysical Research Communications 372 (2008) 804–810
model. The cross section of this nearly linear model is in the range2–2.5 nm, a value which is in agreement with experimental esti-mates of FGL fibers thickness. Also, the buried surface upon associ-ation of Caf1 subunits main bodies (residues 16–149) is about400 Å2. The limited interface area between subunits [23], whoseassociation is primarily stabilized by interactions mediated by N-terminal strands, is consistent with the observed flexibility ofresulting fibers [7,24]. Although other major re-arrangements inthe subunit–subunit organization in later stages of fiber formationprocess may occur, the compatibility of our model with the avail-able experimental data indirectly suggest that Caf1 association at adimer level plays an important role in determining the structuralfeatures of the fiber.
Conclusions
The atomic level definition of the process that leads to theformation of external organelles essential for the pathogenicityof several bacteria is of paramount importance for both aca-demic and applicative implications [25]. The recent discovery
that chaperone/usher secretion system does not require anyexternal agent to generate the external fibrils [26] suggests thata full definition of the structural bases of this system is achiev-able. Over the last decade, insightful crystallographic and bio-chemical investigations have provided illuminating data on therole played by some components of the system. There is increas-ing evidence that computational approaches may fruitfully com-plement these studies by providing information on theproperties of specific conformational states of monoadhesineand polyadhesine systems whose experimental characterizationis difficult. In this scenario, we have here reported a detailedMD characterization of Caf1 subunit in its monomeric unboundstate and in its dimeric state. Data on the monomeric form indi-cate that the unliganded form of the protein is highly reactiveand evolves toward more compact states. Notably, this closureof the structure likely hampers the subunit–subunit associationprocess. Therefore, to preserve this individual subunit’s highreactivity, that is important for their tight association throughthe donor-complementation mechanism, and to prevent thecollapse of the unliganded Caf1, chaperone release and sub-
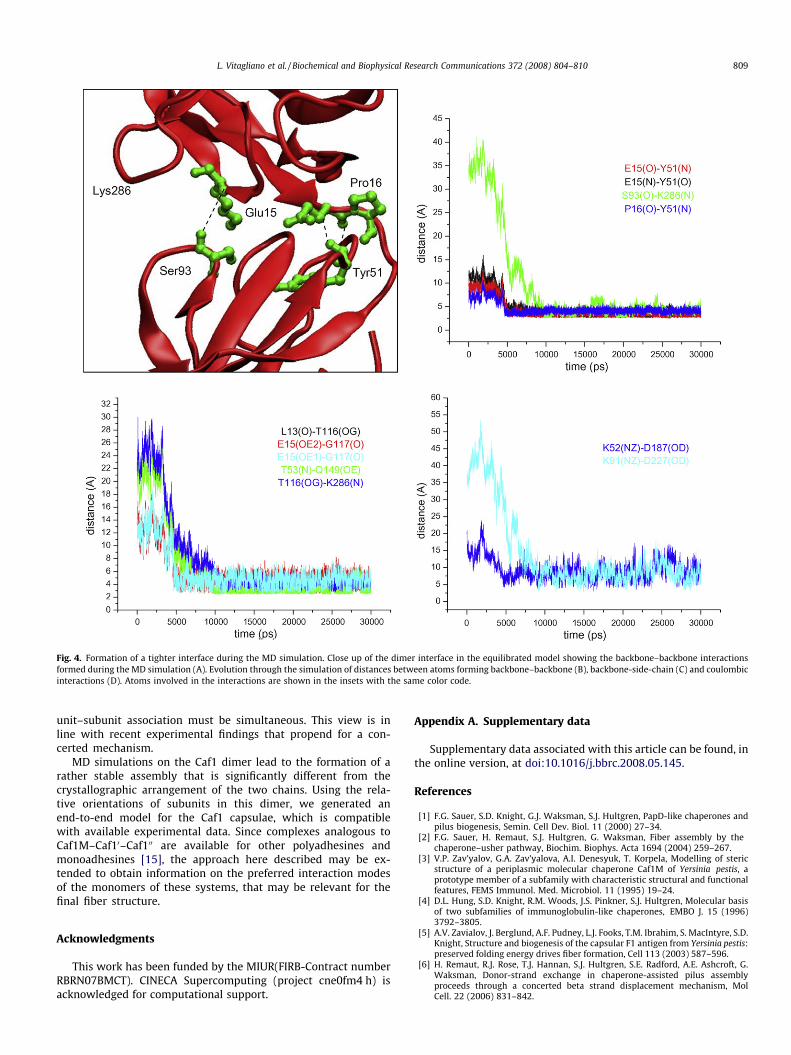
Fig. 4. Formation of a tighter interface during the MD simulation. Close up of the dimer interface in the equilibrated model showing the backbone–backbone interactionsformed during the MD simulation (A). Evolution through the simulation of distances between atoms forming backbone–backbone (B), backbone-side-chain (C) and coulombicinteractions (D). Atoms involved in the interactions are shown in the insets with the same color code.
L. Vitagliano et al. / Biochemical and Biophysical Research Communications 372 (2008) 804–810 809
unit–subunit association must be simultaneous. This view is inline with recent experimental findings that propend for a con-certed mechanism.
MD simulations on the Caf1 dimer lead to the formation of arather stable assembly that is significantly different from thecrystallographic arrangement of the two chains. Using the rela-tive orientations of subunits in this dimer, we generated anend-to-end model for the Caf1 capsulae, which is compatiblewith available experimental data. Since complexes analogous toCaf1M–Caf10–Caf100 are available for other polyadhesines andmonoadhesines [15], the approach here described may be ex-tended to obtain information on the preferred interaction modesof the monomers of these systems, that may be relevant for thefinal fiber structure.
Acknowledgments
This work has been funded by the MIUR(FIRB-Contract numberRBRN07BMCT). CINECA Supercomputing (project cne0fm4 h) isacknowledged for computational support.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.bbrc.2008.05.145.
References
[1] F.G. Sauer, S.D. Knight, G.J. Waksman, S.J. Hultgren, PapD-like chaperones andpilus biogenesis, Semin. Cell Dev. Biol. 11 (2000) 27–34.
[2] F.G. Sauer, H. Remaut, S.J. Hultgren, G. Waksman, Fiber assembly by thechaperone–usher pathway, Biochim. Biophys. Acta 1694 (2004) 259–267.
[3] V.P. Zav’yalov, G.A. Zav’yalova, A.I. Denesyuk, T. Korpela, Modelling of stericstructure of a periplasmic molecular chaperone Caf1M of Yersinia pestis, aprototype member of a subfamily with characteristic structural and functionalfeatures, FEMS Immunol. Med. Microbiol. 11 (1995) 19–24.
[4] D.L. Hung, S.D. Knight, R.M. Woods, J.S. Pinkner, S.J. Hultgren, Molecular basisof two subfamilies of immunoglobulin-like chaperones, EMBO J. 15 (1996)3792–3805.
[5] A.V. Zavialov, J. Berglund, A.F. Pudney, L.J. Fooks, T.M. Ibrahim, S. MacIntyre, S.D.Knight, Structure and biogenesis of the capsular F1 antigen from Yersinia pestis:preserved folding energy drives fiber formation, Cell 113 (2003) 587–596.
[6] H. Remaut, R.J. Rose, T.J. Hannan, S.J. Hultgren, S.E. Radford, A.E. Ashcroft, G.Waksman, Donor-strand exchange in chaperone-assisted pilus assemblyproceeds through a concerted beta strand displacement mechanism, MolCell. 22 (2006) 831–842.

810 L. Vitagliano et al. / Biochemical and Biophysical Research Communications 372 (2008) 804–810
[7] A. Zavialov, G. Zav’yalova, T. Korpela, V. Zav’yalov, FGL chaperone-assembledfimbrial polyadhesins: anti-immune armament of Gram-negative bacterialpathogens, FEMS Microbiol. Rev. 31 (2007) 478–514.
[8] S.D. Knight, Structure and assembly of Yersinia pestis F1 antigen, Adv. Exp. Med.Biol. 603 (2007) 74–87.
[9] F.G. Sauer, M.A. Mulvey, J.D. Schilling, J.J. Martinez, S.J. Hultgren, Bacterialpili: molecular mechanisms of pathogenesis, Curr. Opin. Microbiol. 3 (2000)65–72.
[10] A. Holmgren, C.I. Branden, Crystal structure of chaperone protein PapD revealsan immunoglobulin fold, Nature 342 (1989) 248–251.
[11] F.G. Sauer, J.S. Pinkner, G. Waksman, S.J. Hultgren, Chaperone priming of pilussubunits facilitates a topological transition that drives fiber formation, Cell111 (2002) 543–551.
[12] F.G. Sauer, K. Futterer, J.S. Pinkner, K.W. Dodson, S.J. Hultgren, G. Waksman,Structural basis of chaperone function and pilus biogenesis, Science 285(1999) 1058–1061.
[13] A.V. Zavialov, V.M. Tischenko, L.J. Fooks, B.O. Brandsdal, J. Aqvist, V.P.Zav’yalov, S. Macintyre, S.D. Knight, Resolving the energy paradox ofchaperone/usher-mediated fibre assembly, Biochem. J. 389 (2005) 685–694.
[14] D. Choudhury, A. Thompson, V. Stojanoff, S. Langermann, J. Pinkner, S.J.Hultgren, S.D. Knight, X-ray structure of the FimC–FimH chaperone–adhesincomplex from uropathogenic Escherichia coli, Science 285 (1999) 1061–1066.
[15] D. Verger, E. Bullitt, S.J. Hultgren, G. Waksman, Crystal structure of the P pilusrod subunit PapA, PLoS Pathog. 3 (2007) e73.
[16] L. Vitagliano, A. Ruggiero, C. Pedone, R. Berisio, A molecular dynamics studyof pilus subunits: insights into pilus biogenesis, J. Mol. Biol. 367 (2007)935–941.
[17] R.J. Rose, T.S. Welsh, G. Waksman, A.E. Ashcroft, S.E. Radford, E. Paci,Donor-strand exchange in chaperone-assisted pilus assembly revealed
in atomic detail by molecular dynamics, J. Mol. Biol. 375 (2008) 908–919.
[18] D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A.E. Mark, H.J. Berendsen,GROMACS: fast, flexible, and free, J. Comput. Chem. 26 (2005) 1701–1718.
[19] L. Esposito, C. Pedone, L. Vitagliano, Molecular dynamics analyses of cross-beta-spine steric zipper models: beta-sheet twisting and aggregation, Proc.Natl. Acad. Sci. USA 103 (2006) 11533–11538.
[20] A. De Simone, L. Vitagliano, R. Berisio, Role of hydration in collagen triple helixstabilization, Biochem. Biophys. Res. Commun. 372 (2008) 121–125.
[21] W. Humphrey, A. Dalke, K. Schulten, VMD: visual molecular dynamics, J. Mol.Graph. 14 (1996) 33–38. 27–38.
[22] M. Vetsch, D. Erilov, N. Moliere, M. Nishiyama, O. Ignatov, R. Glockshuber,Mechanism of fibre assembly through the chaperone–usher pathway, EMBORep. (2006).
[23] S. Jones, J.M. Thornton, Principles of protein–protein interactions, Proc. Natl.Acad. Sci. USA 93 (1996) 13–20.
[24] K.L. Anderson, J. Billington, D. Pettigrew, E. Cota, P. Simpson, P. Roversi, H.A.Chen, P. Urvil, L. du Merle, P.N. Barlow, M.E. Medof, R.A. Smith, B. Nowicki,C. Le Bouguenec, S.M. Lea, S. Matthews, An atomic resolution model forassembly, architecture, and function of the Dr adhesins, Mol. Cell 15 (2004)647–657.
[25] J.S. Pinkner, H. Remaut, F. Buelens, E. Miller, V. Aberg, N. Pemberton, M.Hedenstrom, A. Larsson, P. Seed, G. Waksman, S.J. Hultgren, F. Almqvist,Rationally designed small compounds inhibit pilus biogenesis inuropathogenic bacteria, Proc. Natl. Acad. Sci. USA 103 (2006) 17897–17902.
[26] M. Nishiyama, T. Ishikawa, H. Rechsteiner, R. Glockshuber, Reconstitution ofpilus assembly reveals a bacterial outer membrane catalyst, Science 320(2008) 376–379.
Related Documents











