JOURNAL OF CELLULAR PHYSIOLOGY 195:346–355 (2003) REVIEW ARTICLES MHC Class I Antigens, Immune Surveillance, and Tumor Immune Escape ANGEL GARCIA-LORA, 1 IGNACIO ALGARRA, 2 AND FEDERICO GARRIDO 1 * 1 Servicio de Ana ´lisis Clı ´nicos, Hospital Universitario Virgen de las Nieves, Universidad de Granada, Granada, Spain 2 Departamento de Ciencias de La Salud, Universidad de Jae ´n, Jae ´n, Spain Oncogenic transformation in human and experimental animals is not necessarily followed by the appearance of a tumor mass. The immune system of the host can recognize tumor antigens by the presentation of small antigenic peptides to the receptor of cytotoxic T-lymphocytes (CTLs) and reject the nascent tumor. However, cancer cells can sometimes escape these specific T-cell immune responses in the course of somatic (genetic and phenotypic) clonal evolution. Among the tumor immune escape mechanisms described to date, the alterations in the expression of major histocompatibility complex (MHC) molecules play a crucial step in tumor development due to the role of MHC antigens in antigen presentation to T-lympho- cytes and the regulation of natural killer cell (NK) cell function. In this work, we have (1) updated information on the mechanisms that allow CTLs to recognize tumor antigens after antigen processing by transformed cells, (2) described the altered MHC class I phenotypes that are commonly found in human tumors, (3) summarized the molecular mechanisms responsible for MHC class I alteration in human tumors, (4) provided evidence that these altered human leukocyte antigens (HLA) class I phenotypes are detectable as result of a T-cell immunoselection of HLA class I-deficient variants by an immunecompetent host, and (5) presented data indicating the MHC class I phenotype and the immunogenicity of experimental metastatic tumors change drastically when tumors develop in immunodeficient mice. J. Cell. Physiol. 195: 346 – 355, 2003. ß 2003 Wiley-Liss, Inc. Evidence has accumulated in recent years indicating that tumors appear and develop despite an active and sometimes efficient immune response. The appearance of a particular tumor is not due to the existence of a deteriorated immune system, at least in early stages of tumor development, but rather to the acquisition by the cancer cells of new genetic and phenotypic character- istics that allow them to escape anti-tumor immune responses (Mareel et al., 1990; Seymour et al., 1999; Villunger and Strasser, 1999). Somatic evolution of genetically unstable tumor cells leads to the develop- ment of sophisticated immune escape variants in prim- ary tumor lesions that are selected out by T-lymphocyte responses. As a result, different altered tumor pheno- types are produced (Marincola et al., 2000; Garrido and Algarra, 2001; Seliger et al., 2002). These altered tumor phenotypes drastically affect the recognition of tumor antigens, and therefore, immune escape variants ap- pear in the primary tumors and later in metastatic lesions. The combination of somatic evolution versus immune selection in cancer development is a modern view of the clonal expansion of tumor cells, which may also provide an explanation for why the escapee tumor phenotype is a crucial step in the natural evolution of human and experimental cancers. This review will concentrate on describing the antigen processing machinery (APM) that allows tumor anti- gens to be recognized by T-lymphocytes, and on the generation of tumor immune escape variants that are frequently found in a variety of human tumors. In particular, we will analyze the major histocompatibility complex (MHC) class I-altered profiles, which allow tumor cells to blind T-lymphocyte immune responses ß 2003 WILEY-LISS, INC. Abbreviations: MHC, major histocompatibility complex; CTL, cytotoxic T-lymphocyte; NK, natural killer cell; TAP, transporter associated antigen processing; LMP, low molecular mass poly- peptide; TCR, T-cell receptor; KIR, killer cell inhibitory receptor; HLA, human leukocyte antigens; LOH, loss of heterozygosity; IFN, interferon; APM, antigen processing machinery; SCID, severe combined immunodeficiency; HC, heavy chain. Contract grant sponsor: Fondo de Investigaciones Sanitarias; Contract grant sponsor: Plan Andaluz de Investigacion; Contract grant sponsor: Ministerio de Ciencia y Tecnologia, Spain; Contract grant number: BSA 2001-3080; Contract grant sponsor: European Commission (project ESTDAB); Contract grant number: QLRI- CT-2001-01325. *Correspondence to: Federico Garrido, Servicio de Ana ´lisis Clı ´nicos, Hospital Universitario Virgen de las Nieves, Av. de las Fuerzas Armadas 2, 18014 Granada, Spain. E-mail: [email protected] Received 13 November 2002; Accepted 10 December 2002 DOI: 10.1002/jcp.10290

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF CELLULAR PHYSIOLOGY 195:346–355 (2003)
REVIEW ARTICLES
MHC Class I Antigens, Immune Surveillance,and Tumor Immune Escape
ANGEL GARCIA-LORA,1 IGNACIO ALGARRA,2 AND FEDERICO GARRIDO1*1Servicio de Analisis Clınicos, Hospital Universitario Virgen de las Nieves,
Universidad de Granada, Granada, Spain2Departamento de Ciencias de La Salud, Universidad de Jaen, Jaen, Spain
Oncogenic transformation in human and experimental animals is not necessarilyfollowed by the appearance of a tumor mass. The immune system of the host canrecognize tumor antigens by the presentation of small antigenic peptides to thereceptor of cytotoxic T-lymphocytes (CTLs) and reject the nascent tumor.However,cancer cells can sometimes escape these specific T-cell immune responses in thecourse of somatic (genetic and phenotypic) clonal evolution. Among the tumorimmune escape mechanisms described to date, the alterations in the expression ofmajor histocompatibility complex (MHC) molecules play a crucial step in tumordevelopment due to the role of MHC antigens in antigen presentation to T-lympho-cytes and the regulationof natural killer cell (NK) cell function. In thiswork,wehave(1) updated information on the mechanisms that allow CTLs to recognize tumorantigens after antigen processing by transformed cells, (2) described the alteredMHC class I phenotypes that are commonly found in human tumors, (3)summarized the molecular mechanisms responsible for MHC class I alteration inhuman tumors, (4) provided evidence that these altered human leukocyte antigens(HLA) class I phenotypes are detectable as result of a T-cell immunoselection ofHLA class I-deficient variants by an immunecompetent host, and (5) presented dataindicating the MHC class I phenotype and the immunogenicity of experimentalmetastatic tumors change drastically when tumors develop in immunodeficientmice. J. Cell. Physiol. 195: 346–355, 2003. � 2003 Wiley-Liss, Inc.
Evidence has accumulated in recent years indicatingthat tumors appear and develop despite an active andsometimes efficient immune response. The appearanceof a particular tumor is not due to the existence of adeteriorated immune system, at least in early stages oftumor development, but rather to the acquisition by thecancer cells of new genetic and phenotypic character-istics that allow them to escape anti-tumor immuneresponses (Mareel et al., 1990; Seymour et al., 1999;Villunger and Strasser, 1999). Somatic evolution ofgenetically unstable tumor cells leads to the develop-ment of sophisticated immune escape variants in prim-ary tumor lesions that are selected out by T-lymphocyteresponses. As a result, different altered tumor pheno-types are produced (Marincola et al., 2000; Garrido andAlgarra, 2001; Seliger et al., 2002). These altered tumorphenotypes drastically affect the recognition of tumorantigens, and therefore, immune escape variants ap-pear in the primary tumors and later in metastaticlesions. The combination of somatic evolution versusimmune selection in cancer development is a modernview of the clonal expansion of tumor cells, which mayalso provide an explanation for why the escapee tumorphenotype is a crucial step in the natural evolution ofhuman and experimental cancers.
This review will concentrate on describing the antigenprocessing machinery (APM) that allows tumor anti-gens to be recognized by T-lymphocytes, and on the
generation of tumor immune escape variants that arefrequently found in a variety of human tumors. Inparticular, we will analyze the major histocompatibilitycomplex (MHC) class I-altered profiles, which allowtumor cells to blind T-lymphocyte immune responses
� 2003 WILEY-LISS, INC.
Abbreviations: MHC, major histocompatibility complex; CTL,cytotoxic T-lymphocyte; NK, natural killer cell; TAP, transporterassociated antigen processing; LMP, low molecular mass poly-peptide; TCR, T-cell receptor; KIR, killer cell inhibitory receptor;HLA, human leukocyte antigens; LOH, loss of heterozygosity;IFN, interferon; APM, antigen processing machinery; SCID,severe combined immunodeficiency; HC, heavy chain.
Contract grant sponsor: Fondo de Investigaciones Sanitarias;Contract grant sponsor: Plan Andaluz de Investigacion; Contractgrant sponsor: Ministerio de Ciencia y Tecnologia, Spain; Contractgrant number: BSA 2001-3080; Contract grant sponsor: EuropeanCommission (project ESTDAB); Contract grant number: QLRI-CT-2001-01325.
*Correspondence to: Federico Garrido, Servicio de AnalisisClınicos, Hospital Universitario Virgen de las Nieves, Av. de lasFuerzas Armadas 2, 18014 Granada, Spain.E-mail: [email protected]
Received 13 November 2002; Accepted 10 December 2002
DOI: 10.1002/jcp.10290

because of the role that these MHC molecules playin antigen presentation. Any alteration affecting theexpression of MHC class I molecules on tumors will havea profound effect on the recognition of tumor antigenpeptides by T-lymphocytes. We will also update thecurrent knowledge about the structure and function ofthe antigen processing machinery of tumor cells, whichproduces a functional MHC class I-tumor peptidecomplex. Additional evidence will be provided indicatingthat T-lymphocytes are responsible for selecting theMHC class I-negative or -deficient tumors by destroyingthe antigenic variants and in some way ‘‘favoring’’ theappearance of the MHC class I tumor escape variants.
ROLE OF MHC CLASS I MOLECULES IN ANTIGENPRESENTATION TO T-CELLS AND INTERACTION
WITH NATURAL KILLER (NK) CELLS
The molecular mechanisms involved in the recogni-tion of tumor cells by cytotoxic T-lymphocytes (CTLs)and NK cells have been partially elucidated in recentyears (Boon et al., 1994; Trowsdale, 2001). The class Imolecules encoded by the MHC are cell surface glyco-proteins that play a fundamental role in the regulationof immune responses. MHC class I molecules arenecessary for the presentation of peptide antigens toCTLs (Townsend et al., 1986) and for the immuneregulatory activity exerted by NK cells (Ljunggren andKarre, 1990).
MHC class I molecules comprise the classical (class Ia)human leukocyte antigens (HLA)-A, -B, and -C antigensin humans and H-2K, D, and L in mice, and thenonclassical (class Ib) E, F, and G, in humans and Qaand Tla antigens in mice (Bjorkman et al., 1987). Theyform a trimolecular complex consisting of a 45-kDaheavy chain (HC), peptide antigen, and the nonpoly-morphic 12-kDa b2-microglobulin (b2-m) light chain.The HLA-A, -B, and -C and the H-2K, D, and L HCs arehighly polymorphic (Bjorkman and Parham, 1990). Inhumans, the class I HCs are encoded by genes locatedwithin the MHC region on chromosome 6, whereas b2-mis encoded by a gene mapped on chromosome 15. In mice,these antigens are encoded in chromosome 17 andchromosome 2, respectively. The classical HLA/H-2class I molecules are expressed on the surface of mostmammalian cells with only a few exceptions (LeBouteiller, 1994). It is estimated that there are up to250,000 of each HLA class I molecule on the surface of asomatic cell (Parham and Ohta, 1996).
Most glandular or squamous epithelia, as well as thesurrounding connective tissue, express MHC class Iantigens. However, the intensity of expression varies be-tween different locations. For instance, skeletal smoothmuscle and gastric mucosa are weakly positive (Ferronet al., 1989; Fernandez et al., 1991), whereas the centraland peripheral nervous system are considered negative(Daar et al., 1984). The nonclassical MHC class I molec-ules have a much more limited degree of polymorphismand a more restricted tissue distribution (Braud et al.,1999).
MHC class I molecules bind antigens in the form ofpeptides that are presented to CTLs on the surface oftumor or virus-infected cells. These antigenic peptidesare generated from degraded endogenous proteins(tumor-associated antigens (TAAs) in tumor cells) by
the APM. This process, known as antigen processing(Fig. 1), is carried out by a large, multicatalytic proteasecomplex called the proteasome (Maffei et al., 1997; VanEndert, 1999). Their different subunits, i.e., low-molec-ular-mass polypeptides (LMPs), are involved in theefficiency of proteosome functions to produce antigenicpeptides capable of binding MHC class I molecules.These peptides are transported into the lumen of theendoplasmatic reticulum (ER) by the transporter asso-ciated with antigen processing-1 and -2 (TAP1 andTAP2) (Abele and Tampe, 1999). The folding andassembly of a complete MHC class I molecule dependson the association of the HC first with b2-m and thenwith the peptide in the ER. This process involves anumber of accessory proteins with a chaperone-likefunction, such as calnexin, calreticulin, Erp57 (endo-plasmic reticulum glycoprotein 57), and tapasin (Fig. 1)(Grandea and Van Kaer, 2001). The exit of the trimolec-ular complex from the ER through the Golgi secretorypathway and its display on the cell surface are referredto as antigen presentation. The correct functioning ofthese antigen processing and presentation machinerycomponents gives rise to cells with normal surfaceexpression of the MHC class I molecules (Koopman et al.,1997; Parmer and Cresswell, 1998). Any defect in theseprocesses will lead to nonexpression of MHC class Imolecules on the cell surface.
MHC class I-bearing cells are essential structuresthat interact with the corresponding recognition mole-cule, the T-cell receptor (TCR), on CD8þ CTL cells(Fig. 2). This interaction triggers a cascade of T-signal-ing events that ultimately lead to cell proliferation,cytokine production, and target cell lysis (Johnsen et al.,1999; Seliger et al., 2000). MHC class I antigens alsoregulate the lytic activity of NK immune cells, which isrelated to their ability to kill target cells lacking MHCclass I expression (Fig. 2). Although NK cells do notexpress TCR receptors, the detection of targets by NKcells is mediated by two major families of receptormolecules belonging to the killer immunoglobulin-likereceptor (KIR) and to the C-type lectin superfamily.These receptors have different specificities and can bedivided into two groups of inhibiting or activatingreceptors (Moretta et al., 1996; Lopez-Botet et al., 2000).
It appears evident that any alteration in the expres-sion of any of the MHC class I subunits can affect normalMHC cell surface expression and alter both T andNK cell-mediated immunity. In human as well as ex-perimental tumors, such alterations may affect thetumorigenic phenotype as well as metastatic capacity(Villunger and Strasser, 1999; Garrido and Algarra,2001).
ESCAPE OF TUMOR CELLS FROM THE IMMUNERESPONSE: LOSS OF MHC CLASS I ANTIGENS
The loss of MHC class I molecules is a frequent mech-anism in experimental and spontaneous tumors toescape recognition and destruction by CTLs (Garridoet al., 1993, 1997; Hicklin et al., 1999) (Fig. 3). Accordingto the ‘‘missing self’’ hypothesis (Ljunggren and Karre,1990), these losses should make tumor cells more sus-ceptible to NK immune effector mechanisms, since NKeffector cells monitor MHC class I cell surface expressionthrough their specific receptor (KIRs), and eliminate
TUMOR IMMUNE ESCAPE AND T-LYMPHOCYTES 347

those cells with downregulated HLA/H-2 class I mole-cules (Moretta et al., 1996).
However, despite an active and a priori normal im-mune response in a healthy immune system, tumor cellsgrow, invade, and metastasize in the host (Klein et al.,1960; Boon et al., 1994). Data obtained in differentlaboratories in the past 20 years indicate that this is duein many instances to a highly sophisticated and not yetwell understood selection of MHC class I-deficient tumor
escape variants (Festenstein and Garrido, 1987; Garridoet al., 1997). This escape strategy used widely by tumorcells allows them to behave as stealth targets to immuneeffectors (Ljunggren and Karre, 1990). This is notsurprising, since MHC genes control the synthesis ofmolecules that are at the center of the immune functionmediated by T-lymphocytes and NK cells.
The loss of an MHC antigen associated with the H-2Kk
class I molecule was first described in 1976 in a mouse
Fig. 1. The antigen processing machinery (APM). Endogenous proteins are degraded by themulticatalytic protease complex (the proteosome) to peptides of 9 amino acids. These peptides aretransported to the endoplasmatic reticulum (ER) by the transporter associated antigen processing (TAP)transporter and associated with some major histocompatibility complex (MHC) class I molecules to beexported to the cell membrane for surveillance by T-lymphocytes.
Fig. 3. Natural history of MHC antigens during tumor development. Primary tumors originate fromMHC class I-positive epithelia (green). Human leukocyte antigens (HLA) class I-negative or-deficientvariants that appear during tumor development (red) escape T-cell recognition, invade, and start tometastasize. The metastatic colonies are composed of homogeneous MHC class I-negative tumor cellvariants.
Fig. 4. Expression of HLA antigens in normal colon mucosa and a colorectal carcinoma. Normal colonmucosa reacts with a monoclonal antibody (mAb) directed against a monomorphic determinant of HLAclass I molecules with an immunoperoxidase technique (A). The malignant cells of a colorectal carcinomaare HLA class I-negative (Phenotype I), whereas the surrounding stroma is positive (B).
348 GARCIA-LORA ET AL.
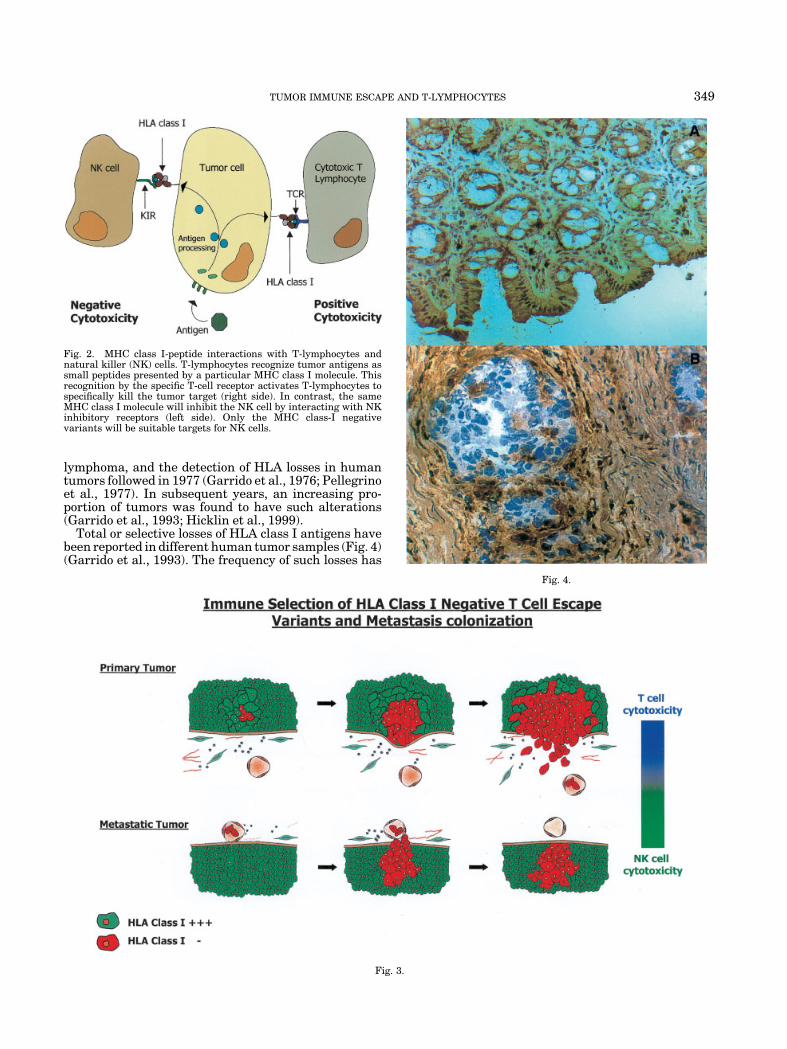
lymphoma, and the detection of HLA losses in humantumors followed in 1977 (Garrido et al., 1976; Pellegrinoet al., 1977). In subsequent years, an increasing pro-portion of tumors was found to have such alterations(Garrido et al., 1993; Hicklin et al., 1999).
Total or selective losses of HLA class I antigens havebeen reported in different human tumor samples (Fig. 4)(Garrido et al., 1993). The frequency of such losses has
Fig. 2. MHC class I-peptide interactions with T-lymphocytes andnatural killer (NK) cells. T-lymphocytes recognize tumor antigens assmall peptides presented by a particular MHC class I molecule. Thisrecognition by the specific T-cell receptor activates T-lymphocytes tospecifically kill the tumor target (right side). In contrast, the sameMHC class I molecule will inhibit the NK cell by interacting with NKinhibitory receptors (left side). Only the MHC class-I negativevariants will be suitable targets for NK cells.
Fig. 3.
Fig. 4.
TUMOR IMMUNE ESCAPE AND T-LYMPHOCYTES 349

been evaluated by studying series of tumor samples withimmunohistological techniques or by flow cytometryin disrupted tumor cell suspensions using monoclonalantibodies (mAbs) directed against HLA class I mono-morphic, HLA-A or -B locus-specific or HLA allelicepitopes (Garrido et al., 1997; Koopman et al., 2000). Therates of HLA class I losses in some tumors are closeto 100%, being reported as 96% in cervical carcino-mas (Koopman et al., 2000), 96% in breast carcinomas(Cabrera et al., 1996), 87% in colorectal carcinomas(Cabrera et al., 1998), and 70% in laryngeal carcino-mas (Cabrera et al., 2000).
It is, therefore, important to define the MHC class Ideficiencies of tumor cells precisely, since heterogeneouspopulations of tumor cells exist in tumor tissues. Ourlaboratory has been developing new strategies andextensively analyzing the MHC altered phenotypesfound in a variety of human tumors (Cabrera et al.,2003). The description of these MHC altered phenotypes(Garrido and Algarra, 2001) has proved to be usefulin establishing additional strategies to analyze themechanisms responsible for such alterations.
Seven major altered HLA class I phenotypes havebeen defined in different tumor tissues (Fig. 5) (Garridoet al., 1997).
* Phenotype I: HLA class I total loss. This phenotype ischaracterized by the absence of any HLA class Iantigen expression in tumor cells and is present to adifferent extent in different tumors.
* Phenotype II: HLA haplotype loss. Tumors canpartially or entirely lose one of the two HLAhaplotypes present in the tumor cell.
* Phenotype III: HLA A, B, or C locus product down-regulation. This altered phenotype is found whenboth products of HLA A, B, or C loci are coordinatelydownregulated. Loss of class I locus expression hasbeen documented in several tumors.
* Phenotype IV: HLA allelic loss. This alteration isdefined as the loss of a single HLA class I allele. Theuse of anti-HLA class I mAbs that define individualHLA class I alleles is required to establish thisdiagnosis in tissues.
* Phenotype V: Compound phenotypes. To produce thisphenotype a combination of two different alterationsis required, for instance, an HLA haplotype loss andan HLA B–C locus downregulation (a combination ofPhenotypes II and III.). The final result is a tumor cellexpressing only one HLA class I allele.
* Phenotype VI: Unresponsiveness to interferons.Some tumor cells express basal levels of HLA class Iantigens but have lost the capacity to upregulatethese molecules in response to different cytokines,including a and g interferons.
* Phenotype VII: Downregulation of classical HLA A–B–C molecules and appearance of HLA-E molecules.
MOLECULAR MECHANISMS LEADINGTO MHC CLASS I LOSSES
Different mechanisms can lead to the total or partialloss of HLA expression. These MHC losses can be prod-
uced at any step required for HLA synthesis, assembly,transport, or expression on the cell surface (Garridoet al., 1997; Ruiz Cabello and Garrido, 1998). Indeed,reports have implicated as major mechanisms in thesedefects (1) the presence of HC and b2-m gene mutations(D’Urso et al., 1991; Browning et al., 1996; Benitez et al.,1998; Perez et al., 1999), (2) alterations in regulatoryfactors induced by the cis–trans mechanism (Blanchetet al., 1991), (3) alterations in glycosylation and tran-sport (Cromme et al., 1994; Johnsen et al., 1999), and (4)HLA gene deletions associated with loss of heterozygos-ity (LOH, Torres et al., 1996).
A variety of tumor cell lines derived from melanoma,colon, head–neck, and breast tumors have been used todefine the molecular mechanisms responsible for MHCclass I alterations (Garrido et al., 1993).These mechan-isms are summarized in Table 1. Additional analyses ofthe different HLA class I altered phenotypes commonlyfound in tumor tissues and described in the previoussection are also shown (Garrido et al., 1997; Ruiz-Cabello and Garrido, 1998).
Phenotype I, HLA class I total loss, can be associatedwith a lack of synthesis or synthesis of a truncated b2-m.There are indications that b2-m mutations are involvedin some cases of HLA class I total loss observed incolorectal carcinomas (Browning et al., 1996), melano-mas (Benitez et al., 1998; Wang et al., 1999), lungadenocarcinomas (Chen et al., 1996), and Burkitt’slymphoma (Rosa et al., 1983). A summary of suchmutations was recently published (Garrido and Algarra,2001). Another mechanism that causes defects in the
Fig. 5. Normal and altered HLA class I phenotypes found in humantumors. Normal cells express 6 HLA class I alleles (2 HLA-A, 2 HLA-Band 2, HLA-C). HLA molecules can be totally or partially absent fromtumor cells (Phenotype I–Phenotype V). In addition, tumor cells maynot respond to INFs (Phenotype VI), or may also express aberrantHLA-E molecules in cells with low expression of HLA A, B, or Cclassical class I antigens (Phenotype VII).
350 GARCIA-LORA ET AL.

assembly and stability of HLA class I molecules involvesinterference with the normal function of APM compo-nents, including the transporter associated with pep-tides (TAP). This interference leads to failure totransport peptides from the cytoplasm to the lumenof the ER, and hence failure of the class I processingpathway (Rosenberg and Bennink, 1993; Vitale et al.,2002). TAP defects can lead to the complete loss of class Imolecules (Phenotype I), and are frequently associatedwith the absence of reactivity to antibodies directedagainst TAP proteins in HLA-negative tumor cells.Other alterations include the coordinated downregula-tion of several APM components (Ritz et al., 2001) andstructural defects in the MHC genes that induce totalloss of HLA class I molecules. These later defects will notbe corrected by cytokine treatment, hence such treat-ment will not restore HLA expression.
Phenotype II, HLA haplotype loss, has been found intumors of different histological origin (Torres et al.,1996; Mendez et al., 2001). The detection of microsa-tellite markers in chromosome 6 using PCR amplifica-tion of these short tandem repeats has provided an easyand effective way to diagnose this altered HLA pheno-type. In a pancreatic adenocarcinoma, HLA haplotypeloss was demonstrated in the fresh tumor and the tumor-derived cell line, indicating that such loss was not due toan in vitro event (Torres et al., 1996). The majority ofthese studies also revealed LOH at other loci of chromo-some 6 by deletion of a full chromosome 6 or a largegenomic region. Current figures for LOH for the HLAregion in chromosome 6 are 46% in cervix carcinomas,36% in head and neck, 17% in colorectal, and 14% inbreast carcinomas (Feenstra et al., 1999; Jimenez et al.,1999; Koopman et al., 2000; Maleno et al., 2002). This isthe most common mechanism of alteration in HLAclass I expression (Ramal et al., 2000).
The mechanism of HLA A, B, or C locus down-regulation, Phenotype III, is associated with alterationsin MHC class I gene transcription, since the mRNAlevels in these tumor cell lines can be upregulated withcytokines, and low expression of transcription factorsthat bind to locus-specific DNA motifs can induce HLA-Blocus downregulation (Soong and Hui, 1992). In mela-
nomas, selective HLA-B locus downregulation cor-relates with increased c-myc transcription whichinterferes with HLA-B transcription at the promoterlevel (Peltenburg and Schrier, 1994).
Phenotype IV, HLA allelic loss, might be the resultof point mutations, partial deletions of HLA class Igenes, chromosomal breakage, or somatic recombina-tion (Brady et al., 2000; Serrano et al., 2000). Thesemechanisms can not be overridden by cytokine treat-ment.
Phenotype V, the compound phenotype observed insome tumors (Ikeda et al., 1997; Real et al., 1998),requires a combination of at least two different altera-tions. For instance, an HLA haplotype loss and HLA-Band -C locus downregulation (combination of Pheno-types II and III) give rise to a tumor cell expressing asingle HLA class I molecule.
Phenotype VI, unresponsiveness to interferons(IFNs), is the result of the downregulation of transcrip-tional factor binding to interferon response sequenceelement (IRSE) (Abril et al., 1996) and altered TAP-1and LMP-2 expression by a defective IFN-g signalingpathway (Dovhey et al., 2000). Phenotype VII, low HLAclass Ia and aberrant expression of HLA class Ib,appears when the tumor drastically reduces the expres-sion of HLA A–B–C molecules and at the same timeexpressed HLA-E, a nonclassical HLA class I moleculethat produces a strong NK inhibition capacity afterinteracting with the CD94/NKG2a inhibitory receptor(Marin et al., 2003).
EVIDENCE FOR T-CELL IMMUNOSELECTIONOF MHC CLASS I-DEFICIENT TUMOR VARIANTS
Immune surveillance against cancer was postulatedmany years ago (Ehrlich, 1909; Thomas, 1959; Burnet,1970), but clinical and experimental evidence for a directrole of the immune system in protecting against spont-aneous malignancies has been slow to appear. Never-theless, the idea seemed to fit some clinical observations.For example, cancer is rare in children and youngadults, but appears more frequently with advancingage, when the efficiency of the immune system isdeclining. Also, some solid tumors were found to contain
TABLE 1. Mechanism of MHC class I downregulation
MHC phenotype Mechanisms References
Phenotype I Defects in b2-m synthesis and mutations Benitez et al. (1998)Loss or downregulation of APM components (LMP or TAP) Seliger et al. (2000)Impaired transcriptional factor Sanda et al. (1995)MHC class I genes hypermethylation Serrano et al. (2001)
Phenotype II LOH, gene loss at 6p21 Torres et al. (1996)Phenotype III Regulatory defect (transcriptional activator or suppressors) Mendez et al. (2001)Phenotype IV HLA class I gene mutations
Somatic recombination within genes Browning et al. (1996)Nonsense mutations Koopman et al. (2000)Missense mutations Wang et al. (1999)Deletions and insertions Koopman et al. (2000)
Phenotype V LOH at 6p21þdownregulation of A-, B-locus Real et al. (1998), Lehmann et al. (1995)LOH at 6p21þmutation Koopman et al. (2000)
Phenotype VI Downregulation of transcriptional factor binding to IRSE Abril et al. (1996)Altered TAP-1 and LMP2 expression by a defective IFN-g signaling pathway Dovhey et al. (2000)
Phenotype VII Aberrant expression of HLA-Eþ low HLA la expression Marin et al. (2003)
MHC, major histocompatibility complex; b2-m, b2 microglobulin; APM, antigen processing machinery; TAP, transporter associated antigen processing; LMP, lowmolecular mass polypeptide; LOH, loss of heterozygosity; HLA, human leukocyte antigens; IRSE, interferon response sequence element; IFN, interferon.
TUMOR IMMUNE ESCAPE AND T-LYMPHOCYTES 351

large numbers of lymphocytes, suggesting that thecellular immune system was recognizing the malignantneoplasm as foreign and fighting it.
It has mostly been assumed that both innate (NK cell)and acquired (CTL cell) immune mechanisms maycontribute to the host’s anti-tumor immunity by destroy-ing transformed cells or by creating a local environmentthat suppresses tumor growth. These mechanismshave received significant support with the discovery ofthe pathway of antigen presentation to T-lymphocytes(Townsend et al., 1986), the identification of tumor anti-gens recognized by T-lymphocytes (Boon et al., 1994),and the partial elucidation of the molecular mechanismsthat allow NK cells to perform their function (Morettaet al., 1996).
MHC class I total or partial loss is a widespreadmechanism used by tumor cells to evade the immunesystem (Garrido et al., 1993; Marincola et al., 2000). Ithas been proposed that the major force contributing tothe appearance of these MHC class I-negative tumorclones is T-cell immunoselection (Kaklamanis and Hill,1992; Lehmann et al., 1995; Jager et al., 1997). Thishypothesis implies that T-cells can recognize tumorantigens presented by HLA class I-positive tumor cells,thereby performing effective immune surveillance.But when HLA class I-defective tumor variants appear,T-cells cannot ‘‘see’’ these targets, and these tumorclones acquire a growth advantage that allows them totake over the other clonal tumor populations. Never-theless, the origin of these MHC-negative tumorvariants remains unknown, and there is thus far noreported experimental or clinical data to substantiatethis hypothesis (Tomlinson and Bodmer, 1999; Villun-ger and Strasser, 1999).
Our laboratory recently obtained direct evidence thata T-cell immune mechanism is responsible for theselection of tumor cells with a specific MHC class Iphenotype (Garcia-Lora et al., 2001). The data indicatethat a particular tumor can produce MHC class I-negative or -positive metastatic colonies depending onthe immune status of the host (Fig. 6). The studies wereperformed with an H-2 class I negative fibrosarcomatumor clone (Garrido et al., 1986; Perez et al., 1990;Algarra et al., 1991) that generated H-2 class I-negativespontaneous lung metastases in immunocompetentBALB/c mice. In contrast, the same tumor clone pro-duced MHC class I-positive metastatic nodes in athymicnu/nu mice (Garcia-Lora et al., 2001) (Fig. 6). Thisphenomenon was observed in metastatic nodules gen-erated after a period of in vivo growth, but not in theprimary tumors growing locally in the footpad. Ananalysis of the molecular mechanisms implicated in theorigin of these MHC class I-deficient metastatic nodesin immunocompetent mice suggested the coordinatedsuppression of multiple components of the MHC class IAPM (Garcia-Lora et al., 2003). Treatment with IFN-gtranscriptionally induces the expression of these down-regulated APM components, thereby also enhancingMHC class I surface expression. Such deficiencies arenot present in metastases obtained from immunodefi-cient nu/nu BALB/c mice. In this connection, it has beenshown that inoculation of immunocompetent C57BL/6mice with mixtures of TAP1-positive and TAP1-negativecells produced tumors composed exclusively of TAP1-
negative cells, indicating selection and evasion ofimmune surveillance in cells with the TAP deficiency(Johnsen et al., 1999).
These findings support the hypothesis that the MHCphenotype of metastatic nodes is influenced by the T-cellrepertoire of the host, since in the absence of this T-cellpressure, the metastatic nodes ‘‘recovered’’ not only H-2class I expression but also the APM functioningnecessary to produce stable MHC class I molecules onthe cell surface. These results showed that in this tumormodel the major factor that contributed to the appear-ance of MHC class I-deficient tumor variants is T-cellimmunoselection.
It was also recently shown that lymphocytes andIFN-g play a central role in providing an immunocom-petent host with a mechanism of tumor surveillanceagainst carcinogen-induced sarcomas and spontaneoustumors, a finding that suggest a role for the immunesystem in editing tumors with low immunogenicity(tumor escape variants, Shankaran et al., 2001). Thesestudies used a strain of mice that completely lackedfunctional lymphocytes. Lymphocyte depletion wasaccomplished by inactivating a lymphocyte-specificgene called RAG2. When the mice were injected withthe chemical carcinogen MCA, more than 50% devel-oped tumors, compared to only 19% of the wild-type
Fig. 6. The MHC class I phenotype of a lung metastasis originatedfrom a tumor clone is dependent on the immune status of the tumor-bearing mouse. H-2 class I-negative cells in an immune-competenthost. H-2 class I-positive cells in nude/nude T-cell-deficient animals.Metastases originated in immunodeficient animals are rejected byimmunocompetent syngeneic mice.
352 GARCIA-LORA ET AL.

animals. Similar results were obtained in mice thatlacked either receptor for IFN-g or one of the proteins(Stat1) required for the receptor to function (Kaplanet al., 1998). It was also reported that these tumor-suppressor systems prevented the formation of sponta-neous tumors. Our findings of greater production ofmetastases in nude mice (5–7 per mouse) than in wild-type animals (1 per mouse) indicate that an effectiveimmune system also acts during metastatic evolution ofthe tumor (Garcia-Lora et al., 2001).
It might be predicted that tumors arising in immuno-deficient patients such as kidney transplant recipientsor HIV-infected individuals, who have received long-term treatment with immunosuppressive drugs, willnot require the production of HLA-deficient clonesto escape T-cell responses, since T-cell function willpresumably be markedly decreased (Garrido andAlgarra, 2001).
The tumor metastases generated in our model ap-peared to be immunoselected depending on the immunestatus of the host. Those metastases that were notimmunoselected should be more immunogenic thanthose that were immunoselected. In fact, the metastas-tic colonies generated in immunodeficient nu/nu mice(H-2 positive) were more immunogenic than the metas-tases generated in immunocompetent mice (H-2 nega-tive, Garcia-Lora et al., 2003). In this connection, it hasbeen reported that chemically induced sarcomas pro-duced in nude and severe combined immunodeficiency(SCID) mice were more immunogenic than similarsarcomas induced in congenic immunocompetent mice(Svane et al., 1996; Engel et al., 1997), and that tumorsoriginated in RAG2 �/� knockout mice were moreimmunogenic that those originated in wild-type animals(Shankaran et al., 2001).
At this point, it is important to consider that MHCclass I downregulation is not the only escape mechanismavailable to tumors to avoid T-cell responses. Othersmechanisms, such as downregulation of the tumorantigens (Jager et al., 1997), alteration of the apoptosisprogram (Hahne et al., 1996), expression of inhibitorycytokines (Chouaib et al., 1997), or immunological ignor-ance (Ochsenbein et al., 1999) have also been described.
Taken together, the studies reviewed above providenew evidence that support the original concept of cancerimmunosurveillance. In addition, the identification ofa particular immune escape mechanism in human ormouse tumors also implies the existence of activeimmune surveillance.
FUTURE PERSPECTIVES
We are just starting to understand that immunesur-veillance against cancer cells may be more than anattractive theory. Evidence obtained in different labora-tories, including ours indicates that the tumor escapefrom cell immune responses is a frequent finding inclinical tumor samples and can be detected when theHLA class I antigens are analyzed with immunohisto-logical techniques in fresh tumor sections or tumor celllines. Another important consequence of these findingsis that the immunogenicity of tumors can be inferred todepend on the immune status of the host, i.e., tumorsinduced in immunodeficient mice are rejected byimmunocompetent individuals.
The identification of a particular tumor immuneescape mechanism will no doubt have a profoundinfluence on T-cell-based immunotherapy protocolscurrently being used for cancer patients, and will helpto select particular individuals suitable for such thera-pies. But the cancer cell can evolve to fight back theimmune system, generating new escape variants. Weare, therefore, facing the very difficult task of trying todestroy all possible metastatic variants growing inparticular individual. Restoration of the normal tumorMHC class I phenotype may be a new way to restore anefficient immune response in cancer patients, but atpresent remains hypothetical, and no such procedureshave been tested thus far.
ACKNOWLEDGMENTS
We thank all the members of the research group of theDepartamento de Analisis Clınicos, Hospital Universi-tario Virgen de las Nieves, Granada for their contribu-tion to this work, and K. Shashok for checking the use ofEnglish in the manuscript.
LITERATURE CITED
Abele R, Tampe R. 1999. Function of the transport complex TAP incellular immune recognition. Biochim Biophys Acta 1461:405–419.
Abril E, Mendez RE, Garcia A, Serrano A, Cabrera T, Garrido F, Ruiz-Cabello F. 1996. Characterization of a gastric tumor cell linedefective in MHC class I inducibility by both alpha- and gamma-interferon. Tissue Antigens 47:391–398.
Algarra I, Gaforio JJ, Garrido A, Mialdea MJ, Perez M, Garrido F.1991. Heterogeneity of MHC-class I antigens in clones of methyl-cholanthrene induced tumors. Implications for local growth andmetastasis. Int J Cancer 6:73–81.
Benitez R, Godelaine D, Lopez-Nevot MA, Brasseur F, Marchand M,Cabrera T, Van Baren N, Andry G, Jimenez P, Andry C, Ruiz-Cabello F, Boon T, Garrido F. 1998. Mutations of the b2-microglobulin gene result in a lack of HLA class I molecules onmelanoma cells of two patients immunized with MAGE peptides.Tissue Antigens 52:520–524.
Bjorkman PJ, Parham P. 1990. Structure, function, and diversityof class I major histocompatibility complex molecules. Annu RevBiochem 59:253–288.
Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL,Wiley DC. 1987. Structure of the human class I histocompatibilityantigen, HLA-A2. Nature 329:506–512.
Blanchet O, Bourge JF, Zinszner H, Tatari Z, Degos L, Paul P. 1991.DNA binding of regulatory factor interacting with MHC-class I geneenhancer correlates with MHC class I transcriptional level in class Idefective cell lines. Int J Cancer 6:138–145.
Boon T, Cerottini JC, Van den Eynde B, van der Bruggen P, Van Pel A.1994. Tumor antigens recognized by T lymphocytes. Annu RevImmunol 12:337–365.
Brady CS, Burt DJ, Bartholomew JS, Duggan-Keen MF, Glenville S,Telford N, Litlee AM, Davison J, Jimenez P, Ruiz-Cabello F, GarridoF, Stern PL. 2000. Multiple mechanism underlie HLA dysregulationin cervical cancer: Implications for immunotherapy. Tissue Anti-gens 55:401–411.
Braud VM, Allan DS, McMichael AJ. 1999. Functions of nonclassicalMHC and non-MHC-encoded class I molecules. Curr Opin Immunol11(1):100–108.
Browning M, Petronzelli F, Bicknell D, Kraus P, Rowan A, Tonks S,Murray N, Bodmer J, Bodmer W. 1996. Mechanism of loss of HLAclass I expression on colorectal tumor cells. Tissue Antigens 47:364–371.
Burnet FM. 1970. The concept of immunological surveillance. ProgExp Tumor Res 13:1–27.
Cabrera T, Sierra A, Fernandez MA, Herruzo A, Fabra A, Garrido F.1996. High frequency of altered HLA class I phenotypes in invasivebreast carcinomas. Human Immunol 50:127–134.
Cabrera T, Collado A, Fernandez MA, Ferron A, Sancho J, Ruiz-Cabello F, Garrido F. 1998. High frequency of altered HLA class Iphenotypes in invasive colorectal carcinomas. Tissue Antigens 52:114–123.
TUMOR IMMUNE ESCAPE AND T-LYMPHOCYTES 353

Cabrera T, Salinero J, Fernandez MA, Garrido A, Esquivias J, GarridoF. 2000. High frequency of altered HLA class I phenotypes inlaryngeal carcinomas. Human Immunol 61:499–506.
Cabrera T, Lopez-Nevot MA, Gaforio JJ, Ruiz-Cabello F, Garrido F.2003. The analysis of HLA expression in human tumor tissues.Cancer Immunol Immunother 52(1):1–9.
Chen HL, Gabrilovich D, Virmani A, Ratnani I, Girgis KR, Nadaf-Rahrov S, Fernandez-Vina M, Carbone DP. 1996. Structural andfunctional analysis of beta2 microglobulin abnormalities in humanlung and breast cancer. Int J Cancer 67:756–763.
Chouaib S, Asselin-Paturel C, Mami-Chouaib F, Caignard A, Blay JY.1997. The host-tumor immune conflict: From immunosuppression toresistance and destruction. Immunol Today 18:493–497.
Cromme FV, Airey J, Heemels MT, Ploegh HL, Ketaing PJ, Stern PL,Meijer CJLM, Walboomers JMM. 1994. Loss of transporter protein,encoded by the TAP-1 gene, is highly correlated with loss of HLAexpression in cervical carcinomas. J Exp Med 179:335–340.
D’Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. 1991. Lackof HLA class I antigen expression by cultured FO-1 melanoma cellsdue to a defect in b2 m gene expression. J Clin Invest 87:284–292.
Daar AS, Fuggle SV, Fabre JV, Ting A, Morris PJ. 1984. The detaileddistribution of HLA ABC antigens in normal human organs.Transplantation 38:287–292.
Dovhey SE, Ghosh NS, Wright KL. 2000. Loss of interferon-ginducibility of TAP1 and LMP2 in a renal cell carcinoma cell line.Cancer Res 60:5789–5796.
Ehrlich P. 1909. Ueber den jetzigen Stand der Karzinomforschung.(Title translation: About the current state of the art of cancerresearch). Ned Tijdschr Geneeskd 5(Pt1):273–290.
Engel AM, Svane IM, Rigaard J, Werdenlin O. 1997. MCA sarcomasinduced in scid mice are more immunogenic than MCA sarcomasinduced in congenic, immunocompetent mice. Scand J Immunol 45:463–470.
Feenstra M, Veltkamp M, van Kuik J, Wiertsema S, Slootweg P,van den Tweel J, de Weger R, Tilanus M. 1999. HLA class I expres-sion and chromosomal deletions at 6p and 15q in head and necksquamous cell carcinomas. Tissue Antigens 54:235–425.
Fernandez JE, Concha A, Aranega A, Ruiz-Cabello F, Cabrera T,Garrido F. 1991. HLA class I and II expression in rhabdomyosarco-mas. Immunobiology 182:440–448.
Ferron A, Perez-Ayala M, Concha A, Cabrera T, Redondo M, OlivaMR, Ruiz-Cabello F, Garrido F. 1989. MHC class I and II antigenson gastric carcinomas and autologous mucosa. J Immunogenet 16:413–423.
Festenstein H, Garrido F. 1987. MHC antigens and malignancy.Nature 332:502–504.
Garcia-Lora A, Algarra I, Gaforio JJ, Ruiz-Cabello F, Garrido F. 2001.Immunoselection by T lymphocytes generates repeated MHC class Ideficient metastatic tumor variants. Int J Cancer 91:109–119.
Garcia-Lora A, Martinez M, Algarra I, Gaforio JJ, Garrido F. 2003.MHC class I-deficient metastatic tumor variants immunoselected byT lymphocytes originate from the coordinated downregulation ofAPM components. Int J Cancer (in press).
Garrido F, Algarra I. 2001. MHC antigens and tumor escape fromimmune surveillance. Adv Cancer Res 83:117–158.
Garrido F, Festenstein H, Schirrmacher V. 1976. Further evidence forderepression of H-2 and Ia like specificities of foreign haplotypes inmouse tumor cell lines. Nature 261:228–230.
Garrido A, Perez M, Delgado C, Garrido ML, Rojano J, Algarra I,Garrido F. 1986. Influence of class I H-2 gene expression on localtumor growth. Exp Clin Imnunogenet 13:98–100.
Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL.1993. Natural history of HLA expression during tumor develop-ment. Immunol Today 14:491–493.
Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar J, Lopez-Botet M,Duggan-Keen M, Stern PL. 1997. Implications for immunosurveil-lance of altered class I phenotypes in humans tumors. ImmunolToday 18:89–95.
Grandea AG, Van Kaer L. 2001. Tapasin: An ER chaperone thatcontrols MHC class I assembly with peptide. Trends Immunol22:194–199.
Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French LE,Schneider P, Bornand T, Fontana A, Lienard D, Cerottini JC,Tschopp J. 1996. Melanoma cell expression of Fas (Apo-1/CD95)ligand: Implications for tumor immune escape. Science 274:1363–1366.
Hicklin DJ, Marincola FM, Ferrone S. 1999. HLA class I antigendownregulation in human cancers: T cell immunotherapy revives anold story. Mol Med Today 5:178–186.
Ikeda H, Lethe B, Lehmann F, van Baren N, Baurain JF, De Smet C,Chambost H, Vitale M, Moretta A, Boon T, Coulie PG. 1997. Char-acterization of an antigen that is recognized on a melanomaexhibiting partial HLA loss by CTL expressing an NK inhibitoryreceptor. Immunity 6:199–208.
Jimenez P, Canton J, Collado A, Cabrera T, Serrano A, Real LM,Garcıa A, Ruiz-Cabello F, Garrido F. 1999. Chromosome loss is themost frequent mechanism contributing to HLA haplotype loss inhuman tumors. Int J Cancer 83:91–97.
Johnsen A, Templeton DJ, Sy MS, Harding CV. 1999. Deficiency oftransporter for antigen presentation (TAP) in tumor cells allowsevasion of immune surveillance and increases tumorigenesis.J Immunol 163:4224–4231.
Jager E, Ringhoffer M, Altmannsberger M, Arand M, KarbackJ, Jager D, Oesch F, Knuth A. 1997. Immunoselection in vivo:Independent loss of MHC class I and melanocyte differentiationantigen expression in metastatic melanoma. Int J Cancer 71:142–147.
Kaklamanis L, Hill A. 1992. MHC loss in colorectal tumours: Evidencefor immunoselection. Cancer Surv 13:155–171.
Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ,Scheriber RD. 1998. Demonstration of an interferon gammadependent tumor surveillance system in immunocompetent mice.Proc Natl Acad Sci USA 95:7556–7561.
Klein G, Sjogren HO, Klein E, Hellstrom KE. 1960. Demonstration ofresistance against methylcholanthrene-induced sarcomas in theprimary autocthonous host. Cancer Res 20:1561–1572.
Koopman JO, Hammerling GJ, Moburg F. 1997. Generation, intra-cellular transport, and loading of peptides associated with MHCclass I molecules. Curr Opin Immunol 9:80–88.
Koopman LA, Corver WE, van der Slik AR, Giphart MJ, Fleuren GJ.2000. Multiple genetic alterations cause frequent and heteroge-neous human histocompatibility leukocyte antigen class I loss incervical cancer. J Exp Med 20:961–976.
Le Bouteiller P. 1994. HLA class I chromosomal region, genes, andproducts: Facts and questions. Crit Rev Immunol 14:89–129.
Lehmann F, Marchand M, Hainaut P, Pouillart P, Sastre X, Ikeda H,Boon T, Coulie PG. 1995. Differences in the antigens recognizedby cytolytic T cells on two successive metastases of a melanomapatients are consistent with immune selection. Eur J Immunol 25:340–347.
Ljunggren HG, Karre K. 1990. In search of the ‘‘missing self’’: MHCmolecules and NK cell recognition. Immunol Today 11:237–244.
Lopez-Botet M, Llano M, Navarro F, Bellon T. 2000. Nk cellrecognition of non-classical HLA class I molecules. Semin Immunol12:109–119.
Maffei A, Papadopoulos K, Harris PE. 1997. MHC class I antigenprocessing pathways. Hum Immunol 54:91–103.
Maleno I, Lopez-Nevot MA, Cabrera T, Salinero J, Garrido F. 2002.Multiple mechanisms generate HLA class I altered phenotypes inlaryngeal carcinomas: High frequency of HLA haplotype lossassociated with loss of heterozygosity in chromosome region 6p21.Cancer Immunol Immunother 51(7):389–396.
Mareel M, van Roy FM, de Baetseiller P. 1990. The invasivephenotypes. Cancer Metastasis Rev 9:45–62.
Marin R, Ruiz-Cabello F, Pedrinaci S, Mendez R, Geraghty DE,Garrido F. 2003. Analysis of HLA-E in human tumours. Immuno-genetics 54(11):767–775.
Marincola MFM, Jaffe EM, Hicklin DJ, Ferrone S. 2000. Escape ofhuman solid tumors from T-cell recognition: Molecular mechanismsand functional significance. Adv Immunol 74:181–273.
Mendez R, Serrano A, Jaeger E, Maleno I, Ruiz-Cabello F, Knuth A,Garrido F. 2001. Analysis of HLA class I expression in differentmetastases from two melanoma patients undergoing peptideimmunotherapy. Tissue Antigens 257:508–519.
Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC,Moretta L. 1996. Receptors for HLA class-I molecules in humannatural killer cells. Annu Rev Immunol 14:619–648.
Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M,Hengartner H, Zinkernagel RM. 1999. Immune surveillance againsta solid tumor fails because of immunological ignorance. Proc NatlAcad Sci USA 96:2233–2238.
Parham P, Ohta T. 1996. Population biology of antigen presentationby MHC class I molecules. Science 272:67–74.
Parmer E, Cresswell P. 1998. Mechanisms of MHC class I restrictedantigen processing. Annu Rev Immunol 16:323–358.
Pellegrino MA, Ferrone S, Reisfeld MA, Irie RF, Golub SH. 1977.Expression of histocompatibility (HLA) antigens on tumor cells andnormal cells from patients with melanoma. Cancer 40:36–41.
354 GARCIA-LORA ET AL.

Peltenburg LT, Schrier PI. 1994. Transcriptional suppression of HLA-B expression by c-Myc is mediated through the core promoterelements. Immunogenetics 40:54–61.
Perez M, Algarra I, Ljunggren HG, Caballero A, Mialdea MJ, GaforioJJ, Garrido F. 1990. A weakly tumorigenic phenotype with highMHC class I expression is associated with high metastatic potentialafter surgical removal of the primary murine fibrosarcoma. IntJ Cancer 46:258–261.
Perez B, Benitez R, Fernandez MA, Oliva MR, Soto JL, Serrano S,Lopez-Nevot MA, Garrido F. 1999. A new b2 microglobulin mutationfound in a melanoma tumor cell line. Tissue Antigens 53:569–572.
Ramal LM, Maleno I, Cabrera T, Collado A, Ferron A, Lopez-NevotMA, Garrido F. 2000. Molecular strategies to define HLA haplotypeloss in microdissected tumor cells. Hum Immunol 61:1001–1012.
Real LM, Cabrera T, Canton J, Kirkin AF, Garcia A, Abril E, ZeuthenJ, Ruiz-Cabello F, Garrido F. 1998. In vivo and in vitro generation ofa new altered HLA phenotype in melanoma tumor cell variantsexpressing a single HLA class I allele. Int J Cancer 75:317–323.
Ritz U, Momburg F, Pilch H, Huber C, Maeurer MJ, Seliger B. 2001.Deficient expression of components of the MHC class I antigenprocessing machinery in human cervical carcinoma. Int J Oncol19(6):1211–1220.
Rosa F, Berissi H, Weissenbach J, Maroteaux L, Fellous M, Revel M.1983. The beta2-microglobulin mRNA in human Daudi cells has amutated initiation codon but is still inducible by interferon. EMBO J2:239–243.
Rosenberg SA, Bennink JR. 1993. Identification of human cancersdeficient in antigen processing. J Exp Med 177:265–272.
Ruiz Cabello F, Garrido F. 1998. HLA and cancer: From research toclinical impact. Immunol Today 19:539–542.
Sanda MG, Restifo NP, Walsh JC, Kawakami Y, Nelson WG,Pardoll DM, Simona JW. 1995. Molecular characterization ofdefective antigen processing in human prostate cancer. J NatlCancer Inst 87(4):280–285.
Seliger B, Maurer MJ, Ferrone S. 2000. Antigen-processing machin-ery breakdown and tumor growth. Immunol Today 21:455–464.
Seliger B, Cabrera T, Garrido F, Ferrone S. 2002. HLA class I antigenabnormalities and immune escape by malignant cells. Semin CancerBiol 12:3–13.
Serrano A, Brady CS, Jimenez P, Duggan-Keen MF, Mendez R, SternPL, Garrido F, Ruiz-cabello F. 2000. A mutation determining theloss of HLA-A2 antigen expression in a cervical carcinoma revealsnovel splicing of human MHC class I classical transcripts in bothtumoral and normal cells. Immunogenetics 51(12):1047–1052.
Serrano A, Tanzarella S, Lionello I, Mendez R, Traversari C, Ruiz-Cabello F, Garrido F. 2001. Re-xpression of HLA class I antigens
and restoration of antigen-specific CTL response in melanoma cellsfollowing 5-aza-20-deoxycytidine treatment. Int J Cancer 94(2):243–251.
Seymour K, Pettit S, O’Flaherty E, Charnley RM, Kirby JA. 1999.Selection of metastatic tumor phenotypes by host immune system.Lancet 353:1989–1991.
Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ,Scheriber RD. 2001. IFNg and lymphocytes prevent primarytumour development and shape tumour immunogenicity. Nature26:1107–1111.
Soong TW, Hui KM. 1992. Locus-specific transcriptional control ofHLA genes. J Immunol 149:2008–2020.
Svane IM, Engel AM, Nielsen MB, Ljunggren HG, Rygaard J,Werdelin O. 1996. Chemically induced sarcomas from nude miceare more immunogenic than similar sarcomas from congenic normalmice. Eur J Immunol 26:1844–1850.
Thomas L. 1959. Discussion of cellular and humoral aspects of thehypersensitivity states. In: Lawrence HS, editor. New York: Hoeber-Harper. pp 529–532.
Tomlinson I, Bodmer W. 1999. Selection, the mutation rate andcancer: Ensuring that the tail does not wag the dog. Nat Med 5:11–12.
Torres MJ, Ruiz-Cabello F, Skoudy A, Berrozpe G, Jimenez P, SerranoA, Real FX, Garrido F. 1996. Loss of an HLA haplotype in pancreascancer tissue and its corresponding tumor derived cell line. TissueAntigens 47:372–381.
Townsend AR, Rothbard J, Gotch FM, Bahadur G, Wraith D,McMichael AJ. 1986. The epitopes of influenza nucleoproteinrecognized by cytotoxic T lymphocytes can be defined with shortsynthetic peptides. Cell 28:959–968.
Trowsdale J. 2001. Genetic and functional relationships betweenMHC and NK receptor genes. Immunity 15(3):363–374.
Van Endert P. 1999. Genes regulating MHC class I processing ofantigen. Curr Opin Immunol 11:82–88.
Villunger A, Strasser A. 1999. The great escape: Is evasion requiredfor tumour progression? Nat Med 5:874–875.
Vitale M, Zimmer J, Castriconi R, Hanau D, Donato L, Bottino C,Moretta L, de la Salle H, Moretta A. 2002. Analysis of naturalkiller cells in TAP2-deficient patients: Expression of functionaltriggering receptors and evidence for the existence of inhibitoryreceptor(s) that prevent lysis of normal autologous cells. Blood 1:1723–1729.
Wang Z, Marincola FM, Rivoltini L, Parmiani G, Ferrone S. 1999.Selective histocompatibility leukocyte antigen (HLA)-A2 loss causedby aberrant pre-mRNA splicing in 624MEL28 melanoma cells. J ExpMed 190:205–215.
TUMOR IMMUNE ESCAPE AND T-LYMPHOCYTES 355
Related Documents











