MOLECULAR AND CELLULAR BIOLOGY, Oct. 2008, p. 6496–6509 Vol. 28, No. 20 0270-7306/08/$08.000 doi:10.1128/MCB.00477-08 Copyright © 2008, American Society for Microbiology. All Rights Reserved. Inhibition of Polysome Assembly Enhances Imatinib Activity against Chronic Myelogenous Leukemia and Overcomes Imatinib Resistance Min Zhang, 1 Wuxia Fu, 1 Sharmila Prabhu, 1 James C. Moore, 1 Je Ko, 1 Jung Woo Kim, 1 † Brian J. Druker, 2 Valerie Trapp, 1 John Fruehauf, 1 Hermann Gram, 3 Hung Y. Fan, 1,4,5 and S. Tiong Ong 1,5 * Division of Hematology/Oncology, Department of Medicine, University of California at Irvine, Irvine, California 1 ; Howard Hughes Medical Institute, Oregon Health and Science University Cancer Institute, Portland, Oregon 97239 2 ; Novartis Institutes for Biomedical Research, Basel, Switzerland 3 ; and Department of Molecular Biology and Biochemistry, School of Biological Sciences, 4 and Cancer Research Institute, 5 University of California at Irvine, Irvine, California 92697 Received 22 March 2008/Returned for modification 2 May 2008/Accepted 28 July 2008 Dysregulated mRNA translation is implicated in the pathogenesis of many human cancers including chronic myelogenous leukemia (CML). Because our prior work has specifically implicated translation initiation in CML, we tested compounds that could modulate translation initiation and polysomal mRNA assembly. Here, we evaluated the activity of one such compound, CGP57380, against CML cells and explored its mechanisms of action. First, using polysomal mRNA profiles, we found that imatinib and CGP57380 could independently, and cooperatively, impair polysomal mRNA loading. Imatinib and CGP57380 also synergistically inhibited the growth of Ba/F3-Bcr-Abl and K562 cells via impaired cell cycle entry and increased apoptosis. Mechanistically, CGP57380 inhibited efficient polysomal assembly via two processes. First, it enhanced imatinib-mediated inhibition of eukaryotic initiation factor 4F induction, and second, it independently impaired phosphorylation of ribosomal protein S6 on the preinitiation complex. We also identified multiple substrates of the mTOR, Rsk, and Mnk kinases as targets of CGP57380. Finally, we found a novel negative-feedback loop to the mitogen- activated protein kinase/Mnk pathway that is triggered by CGP57380 and demonstrated that an interruption of the loop further increased the activity of the combination against imatinib-sensitive and -resistant CML cells. Together, this work supports the inhibition of translation initiation as a therapeutic strategy for treating cancers fueled by dysregulated translation. Chronic myelogenous leukemia (CML) is a myeloprolifera- tive disorder characterized by the presence of the Philadelphia chromosome (26), which gives rise to the t(9;22) translocation, and the resulting fusion oncogene Bcr-Abl. The constitutive tyrosine kinase activity of Bcr-Abl is required for the trans- forming properties of Bcr-Abl, and the inhibition of Bcr-Abl kinase by imatinib mesylate (imatinib) has resulted in durable responses in early-stage CML (32). However, patients with late-stage disease respond much less well to imatinib therapy and remain at high risk for developing drug resistance (8, 31) and for death from progressive disease (19, 30). These obser- vations make a strong case for the use of non-cross-resistant drug combinations to combat late-stage disease. There is, thus, a pressing need to understand other pathways and cellular processes that are dysregulated by Bcr-Abl, particularly those that represent “drugable” targets. Because prior work from our group has shown that Bcr-Abl modulates the activity of several molecules which regulate the initiation of cap-dependent mRNA translation (14, 23), we turned our attention to the therapeutic potential of targeting this process in CML. Specifically, we were interested in small molecules that might interfere with translation initiation and with the efficient assembly of polysomal mRNA, since both processes have been implicated in several cancer types (15), in addition to CML (22). In mammalian cells, initiation is both the rate-limiting step for the process of mRNA translation and the convergence point for signaling pathways that convey the message from extracellular stimuli to a distal set of proteins that regulate this process (24). In healthy cells, a critical control point is repre- sented by the amount of eukaryotic initiation factor 4E (eIF4E) available to form the cap-binding complex eIF4F. eIF4F consists of (i) eIF4E, which binds the cap structure present at the 5 end of mRNAs; (ii) eIF4G, a scaffolding protein; and (iii) eIF4A, an RNA-dependent ATPase and RNA helicase. Formation of eIF4F then allows recruitment of the translational machinery to mRNA and includes assembly of the preinitiation complex (PIC) that comprises eIF3, 40S ribosomal subunits, and the ternary complex (eIF2/Met-tRNA/ GTP) (16). The availability of eIF4E is determined primarily by the phosphorylation status of 4E-BP1, a negative regulator of eIF4E. 4E-BP1 is phosphorylated by the serine/threonine kinase mTOR when the latter forms a complex with raptor that is called mTORC1, which is in turn activated by phosphatidyl- inositol 3-kinase (PI3K)/Akt signaling. In addition to regula- tion by the PI3K/Akt/mTOR pathway, mitogen-activated pro- tein kinase (MAPK) signaling also has recently been described as a regulator of the efficiency of polysomal mRNA assembly * Corresponding author. Present address: Duke-NUS Graduate Medical School, 2 Jalan Bukit Merah, Singapore 169547. Phone: 65 6516 7763. Fax: 65 6534 8632. E-mail: [email protected]. † Present address: Paichai Bio-Diagnostic Fusion Technology Cen- ter, Dept. of Life Science and Technology, Pai Chai University, Dae- jeon, Republic of Korea 302-735. Published ahead of print on 11 August 2008. 6496

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

MOLECULAR AND CELLULAR BIOLOGY, Oct. 2008, p. 6496–6509 Vol. 28, No. 200270-7306/08/$08.00�0 doi:10.1128/MCB.00477-08Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Inhibition of Polysome Assembly Enhances Imatinib Activity againstChronic Myelogenous Leukemia and Overcomes Imatinib Resistance�
Min Zhang,1 Wuxia Fu,1 Sharmila Prabhu,1 James C. Moore,1 Je Ko,1 Jung Woo Kim,1†Brian J. Druker,2 Valerie Trapp,1 John Fruehauf,1 Hermann Gram,3
Hung Y. Fan,1,4,5 and S. Tiong Ong1,5*Division of Hematology/Oncology, Department of Medicine, University of California at Irvine, Irvine, California1;
Howard Hughes Medical Institute, Oregon Health and Science University Cancer Institute, Portland, Oregon 972392;Novartis Institutes for Biomedical Research, Basel, Switzerland3; and Department of Molecular Biology and Biochemistry,
School of Biological Sciences,4 and Cancer Research Institute,5 University of California at Irvine,Irvine, California 92697
Received 22 March 2008/Returned for modification 2 May 2008/Accepted 28 July 2008
Dysregulated mRNA translation is implicated in the pathogenesis of many human cancers including chronicmyelogenous leukemia (CML). Because our prior work has specifically implicated translation initiation inCML, we tested compounds that could modulate translation initiation and polysomal mRNA assembly. Here,we evaluated the activity of one such compound, CGP57380, against CML cells and explored its mechanismsof action. First, using polysomal mRNA profiles, we found that imatinib and CGP57380 could independently,and cooperatively, impair polysomal mRNA loading. Imatinib and CGP57380 also synergistically inhibited thegrowth of Ba/F3-Bcr-Abl and K562 cells via impaired cell cycle entry and increased apoptosis. Mechanistically,CGP57380 inhibited efficient polysomal assembly via two processes. First, it enhanced imatinib-mediatedinhibition of eukaryotic initiation factor 4F induction, and second, it independently impaired phosphorylationof ribosomal protein S6 on the preinitiation complex. We also identified multiple substrates of the mTOR, Rsk,and Mnk kinases as targets of CGP57380. Finally, we found a novel negative-feedback loop to the mitogen-activated protein kinase/Mnk pathway that is triggered by CGP57380 and demonstrated that an interruptionof the loop further increased the activity of the combination against imatinib-sensitive and -resistant CMLcells. Together, this work supports the inhibition of translation initiation as a therapeutic strategy for treatingcancers fueled by dysregulated translation.
Chronic myelogenous leukemia (CML) is a myeloprolifera-tive disorder characterized by the presence of the Philadelphiachromosome (26), which gives rise to the t(9;22) translocation,and the resulting fusion oncogene Bcr-Abl. The constitutivetyrosine kinase activity of Bcr-Abl is required for the trans-forming properties of Bcr-Abl, and the inhibition of Bcr-Ablkinase by imatinib mesylate (imatinib) has resulted in durableresponses in early-stage CML (32). However, patients withlate-stage disease respond much less well to imatinib therapyand remain at high risk for developing drug resistance (8, 31)and for death from progressive disease (19, 30). These obser-vations make a strong case for the use of non-cross-resistantdrug combinations to combat late-stage disease. There is, thus,a pressing need to understand other pathways and cellularprocesses that are dysregulated by Bcr-Abl, particularly thosethat represent “drugable” targets.
Because prior work from our group has shown that Bcr-Ablmodulates the activity of several molecules which regulate theinitiation of cap-dependent mRNA translation (14, 23), weturned our attention to the therapeutic potential of targeting
this process in CML. Specifically, we were interested in smallmolecules that might interfere with translation initiation andwith the efficient assembly of polysomal mRNA, since bothprocesses have been implicated in several cancer types (15), inaddition to CML (22).
In mammalian cells, initiation is both the rate-limiting stepfor the process of mRNA translation and the convergencepoint for signaling pathways that convey the message fromextracellular stimuli to a distal set of proteins that regulate thisprocess (24). In healthy cells, a critical control point is repre-sented by the amount of eukaryotic initiation factor 4E(eIF4E) available to form the cap-binding complex eIF4F.eIF4F consists of (i) eIF4E, which binds the cap structurepresent at the 5� end of mRNAs; (ii) eIF4G, a scaffoldingprotein; and (iii) eIF4A, an RNA-dependent ATPase andRNA helicase. Formation of eIF4F then allows recruitment ofthe translational machinery to mRNA and includes assemblyof the preinitiation complex (PIC) that comprises eIF3, 40Sribosomal subunits, and the ternary complex (eIF2/Met-tRNA/GTP) (16). The availability of eIF4E is determined primarilyby the phosphorylation status of 4E-BP1, a negative regulatorof eIF4E. 4E-BP1 is phosphorylated by the serine/threoninekinase mTOR when the latter forms a complex with raptor thatis called mTORC1, which is in turn activated by phosphatidyl-inositol 3-kinase (PI3K)/Akt signaling. In addition to regula-tion by the PI3K/Akt/mTOR pathway, mitogen-activated pro-tein kinase (MAPK) signaling also has recently been describedas a regulator of the efficiency of polysomal mRNA assembly
* Corresponding author. Present address: Duke-NUS GraduateMedical School, 2 Jalan Bukit Merah, Singapore 169547. Phone: 656516 7763. Fax: 65 6534 8632. E-mail: [email protected].
† Present address: Paichai Bio-Diagnostic Fusion Technology Cen-ter, Dept. of Life Science and Technology, Pai Chai University, Dae-jeon, Republic of Korea 302-735.
� Published ahead of print on 11 August 2008.
6496

by activation of extracellular-regulated kinase (ERK)-medi-ated activation of Rsk. This activation is thought to occur viaRsk-dependent phosphorylation of ribosomal protein S6 (rpS6)at Ser235/236 (25), which in turns facilitates recruitment ofrpS6 to the PIC and the efficient assembly of polysomalmRNA. Thus, at least two signaling pathways that are essentialfor Bcr-Abl-mediated transformation, MAPK and PI3K/Akt(6), share a final common path to proteins involved in theregulation of translation initiation.
It was therefore of interest to determine if pharmacologicinterruption of translation initiation and polysomal mRNAassembly might provide evidence to support the hypothesisthat these processes contribute to the transformation by Bcr-Abl. Accordingly, we set out to test a novel small molecule,CGP57380, which had been previously found to impair efficientpolysome assembly (18) in CML cells and to determine itsmechanism of action. First, we found that imatinib andCGP57380 impaired translation initiation via distinct mecha-nisms and that this impairment resulted in synergistic activityagainst CML cells. Next, we uncovered putative targets ofCGP57380 and describe a novel negative-feedback loop toMEK/ERK that is triggered by CGP57380 exposure. Finally,we show that pharmacologic interruption of the MEK/ERKfeedback loop further increases the activity of imatinib andCGP57380 against imatinib-sensitive and -resistant CML cells.Because dysregulated mRNA translation has been implicatedin the pathogenesis of several other cancer types (15), ourresults also have wider application to the therapy of cancer ingeneral.
MATERIALS AND METHODS
Cell culture, cell lines, and patient samples. Parental Ba/F3 cells and Ba/F3cells stably transfected with wild-type p210 (Ba/F3-Bcr-Abl) and imatinib-resis-tant mutants have been previously described (12). K562 cells were obtained fromATCC. Peripheral blood (PB) and bone marrow (BM) samples were obtainedwith appropriate consent and institutional review board approval from patientsat the University of California at Irvine. Primary cells were grown at 37°C inserum-free medium (StemCell Technologies, Vancouver, BC, Canada) supple-mented with growth factors at concentrations found in stroma-conditioned me-dium (5). CGP57380 was obtained from Novartis (Basel, Switzerland) (11).LY294002 and rapamycin were commercially obtained (Sigma, St. Louis, MO).BI-D1870 was purchased from the Division of Signal Transduction Therapy atthe University of Dundee.
Polysome analysis. Ba/F3-Bcr-Abl or K562 cells (2 � 107) were treated withdimethyl sulfoxide (DMSO) or drug for 4 h. Cycloheximide was added to a finalconcentration of 0.1 mg/ml for 5 min before collection. Cells were then lysed inextraction buffer (15 mM Tris-HCl [pH 7.4], 15 mM MgCl2, 200 mM NaCl, 0.5mg/ml heparin, 0.1 mg/ml cycloheximide, 1% Triton X-100, 40 U/�l RNasin[Invitrogen], and 5 mM dithiothreitol). Extracts were fractionated on a 10%to 50% sucrose gradient composed of extraction buffer lacking Triton X-100,RNasin, and dithiothreitol and centrifuged at 35,000 rpm for 150 min in a SW41model rotor at 4°C. The polysome profile was visualized with an ISCO densitygradient fraction system (Lincoln, NE). Areas under the curves corresponding topolysomes and monosomes were calculated using ImageJ software (http://rsb.info.nih.gov/ij/index.html).
Tetrazolium-based proliferation assays. Cells (5 � 103) were plated in tripli-cate with DMSO or inhibitors in a 100-ml volume in a 96-well plate at 37°C. After48 or 72 h, CellTiter 96 AQueous One solution (Promega, Madison, WI) wasadded to each well and processed per the manufacturer’s instructions. Theoptical density for each condition was calculated as a percentage of the untreatedcontrol after subtracting for background absorbance. To assess drug synergy, adose-response analysis was performed according to the method of Chou, usingCalcuSyn software (version 1.1; Biosoft, Ferguson, MO) (4).
Flow cytometric analysis of cell cycle and apoptosis. Cells (1 � 106) wereincubated with DMSO or inhibitors for 48 h and then washed and fixed withice-cold 80% ethanol. After 1 h, the cells were washed twice in phosphate-
buffered saline and stained with 20 mg/ml propidium iodide (PI) in phosphate-buffered saline with 50 mg/ml RNase A, 10% Na citrate, 10% NP-40 at roomtemperature for 30 min. Cells were analyzed by flow cytometry, and cell cycleanalysis was performed using FlowJo software. For apoptosis assays, 1 � 106 cellswere incubated with DMSO or inhibitors for 48 h, stained with annexin V-fluorescein isothiocyanate (BD PharMingen, Carlsbad, CA) and PI per themanufacturer’s instructions and then analyzed by flow cytometry.
Immunoblot and antibodies. Cells (4 � 106) were incubated with an equivalentvolume of DMSO or inhibitor and lysed in Laemmli buffer with protease andphosphatase inhibitors. Lysates were resolved by sodium dodecyl sulfate-poly-acrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidenedifluoride membrane. The following antibodies were used: 4E-BP1, �-actin(Sigma), cyclin D1, cyclin D2 (catalog no. M-20; Santa Cruz Biotechnology,Santa Cruz, CA), cyclin D3 (catalog no. D-7; Santa Cruz Biotechnology), eIF4G(catalog no. N-20; Santa Cruz Biotechnology), activated caspase-3, Bcl-XL,c-Abl, and CDK4. In addition, antibodies to the phosphorylated and total formsof the following proteins were also employed: Mnk1 (Santa Cruz Biotechnology),ERK, eIF4E, S6K1, and rpS6. Antibodies were from Cell Signaling Technology(Beverly, MA) unless otherwise stated. Immunoblotting was performed accord-ing to the manufacturers’ instructions. Immunoreactive bands were visualized bychemiluminescence (Pierce, Rockford, IL) using a FluorChem SP imaging sys-tem (Alpha Innotech, San Leandro, CA).
CD34� cell selection. PB or BM mononuclear cells were isolated by Ficoll-Hypaque (Sigma, St. Louis, MO) density gradient centrifugation. CD34� cellswere then selected by immunomagnetic bead-based column separation accordingto the manufacturer’s instructions (Miltenyi Biotec, Auburn, CA), and a purityvalue of �95% was confirmed by flow cytometry.
Cap-binding assay. The cap-binding assay was performed as previously de-scribed (1, 23). Briefly, after a 4-h incubation with DMSO or inhibitors, cells werelysed by three freeze-thaw cycles, and lysates were incubated with a suspensionof 7-methyl-GTP-Sepharose beads (Amersham, Piscataway, NJ). After 1 h, thebeads were pelleted and washed three times with GTP-containing buffer, andcap-bound proteins were eluted with buffer containing 7-methyl-GTP. Cap-bound material was then subjected to SDS-PAGE and visualized using immu-noblots.
RESULTS
Imatinib mesylate and CGP57380 cooperatively inhibitmRNA recruitment to polysomes in Bcr-Abl-containing cells.CGP57380 was originally identified as an inhibitor of Mnk1kinase from an in vitro screen and has a 50% inhibitory con-centration of 2.2 �M against Mnk1 kinase (11). Importantly,CGP57380 also exerts minimal cellular toxicity against un-transformed cells, even at concentrations as high as 30 �M (C.Tschopp and H. Gram, unpublished data). Since then, othershave also demonstrated the ability of CGP57380 to negativelyaffect polysomal mRNA assembly (18). Because of these in-teresting properties and because our prior work had implicateda role for Bcr-Abl-mediated translation initiation in transfor-mation (14, 23), we tested the ability of CGP57380, togetherwith imatinib, to impair polysomal mRNA assembly in CMLcell lines. In the first series of experiments, we analyzed thepolysome profile of drug-exposed Ba/F3-Bcr-Abl cells. Consis-tent with imatinib’s ability to inhibit eIF4F induction (23), wefound that brief (4-h) exposures to imatinib decreased the totalamount of polysome-bound mRNA. This effect is appreciableas a decrease in the amplitude of the peaks representing thepolysome fraction within the sucrose gradient and is associatedwith a reciprocal increase in the amplitude of the 80S mono-some peaks (Fig. 1A). CGP57380 exerted a similar effect, andthe combination of the two drugs was additive. Importantly,these results were replicated in the human CML cell line,K562, demonstrating that the phenomenon is not cell linespecific (Fig. 1B). To quantify these findings, we also calcu-lated the ratio of the area representing the polysome fraction
VOL. 28, 2008 TARGETING TRANSLATION INITIATION IN CML WITH CGP57380 6497
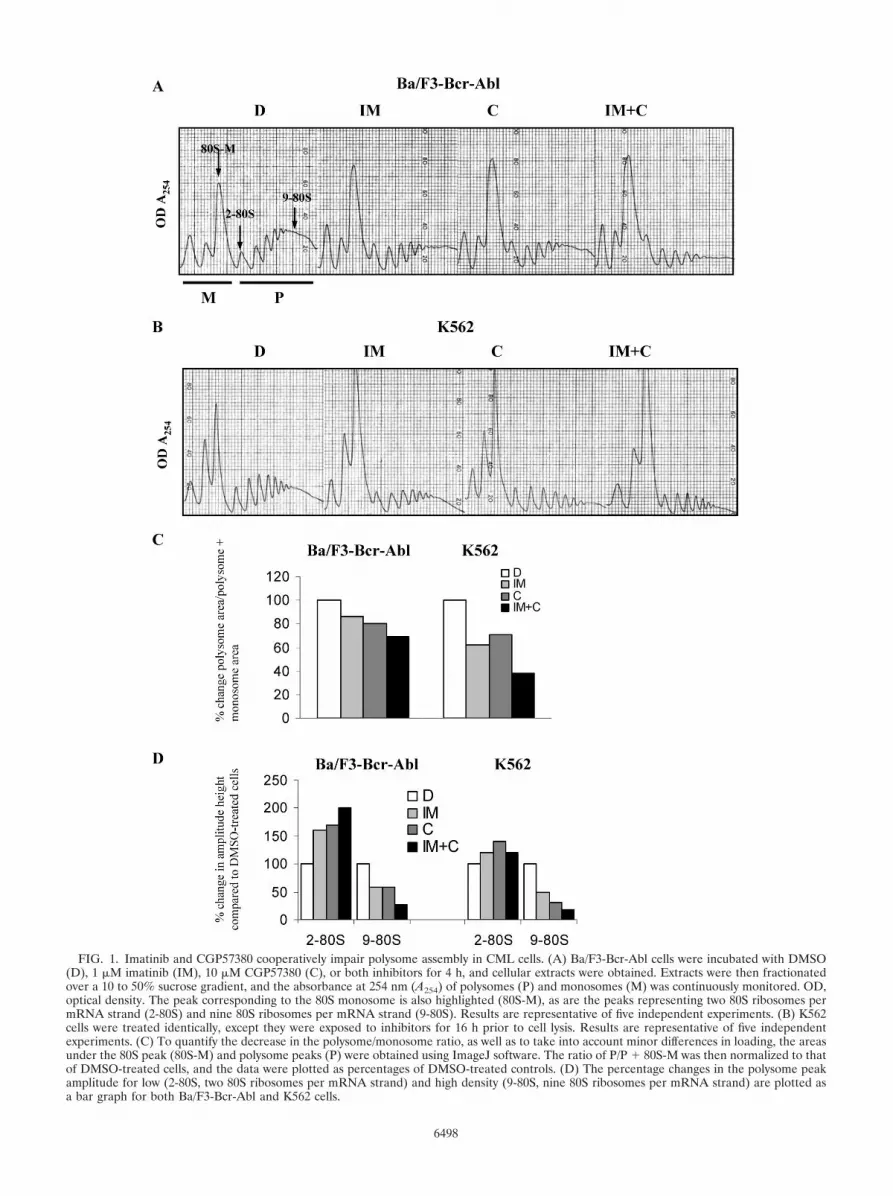
FIG. 1. Imatinib and CGP57380 cooperatively impair polysome assembly in CML cells. (A) Ba/F3-Bcr-Abl cells were incubated with DMSO(D), 1 �M imatinib (IM), 10 �M CGP57380 (C), or both inhibitors for 4 h, and cellular extracts were obtained. Extracts were then fractionatedover a 10 to 50% sucrose gradient, and the absorbance at 254 nm (A254) of polysomes (P) and monosomes (M) was continuously monitored. OD,optical density. The peak corresponding to the 80S monosome is also highlighted (80S-M), as are the peaks representing two 80S ribosomes permRNA strand (2-80S) and nine 80S ribosomes per mRNA strand (9-80S). Results are representative of five independent experiments. (B) K562cells were treated identically, except they were exposed to inhibitors for 16 h prior to cell lysis. Results are representative of five independentexperiments. (C) To quantify the decrease in the polysome/monosome ratio, as well as to take into account minor differences in loading, the areasunder the 80S peak (80S-M) and polysome peaks (P) were obtained using ImageJ software. The ratio of P/P � 80S-M was then normalized to thatof DMSO-treated cells, and the data were plotted as percentages of DMSO-treated controls. (D) The percentage changes in the polysome peakamplitude for low (2-80S, two 80S ribosomes per mRNA strand) and high density (9-80S, nine 80S ribosomes per mRNA strand) are plotted asa bar graph for both Ba/F3-Bcr-Abl and K562 cells.
6498

to that of the polysome fraction plus the 80S monosome (Fig.1C, P/P � 80S). For both cell lines, the data confirmed theadditive effect of both drugs in decreasing the polysome/mono-some-plus-polysome ratio and are consistent with the impair-ment of polysomal mRNA loading in Bcr-Abl-expressing cells.Closer inspection of the profiles also revealed that drug treat-ment resulted in a shift toward smaller polysomes, i.e., a de-crease in the mRNA ribosome density. We quantified thiseffect by plotting the amplitude changes of the peaks corre-sponding to the lowest and highest density polysomes (Fig.1D). In Ba/F3-Bcr-Abl cells, imatinib and CGP57380 treat-ment resulted in a 60% and 70% increase for low-densityribosomes, respectively, compared to a 41% and 41% decreasefor high-density ribosomes, respectively. In K562 cells, ima-tinib and CGP57380 treatment resulted in a 20% and 40%increase for low-density ribosomes, respectively, compared to a50% and 69% decrease, respectively, for high-density ribo-somes. Together, these data demonstrate that both drugs im-pair polysome loading on mRNA, and data are consistent witha negative effect on translation initiation.
CGP57380 acts synergistically with imatinib to impairgrowth of Bcr-Abl-transformed cells via effects on cell cycleentry and apoptosis. To determine if the combination ofCGP57380 and imatinib had activity against Bcr-Abl-trans-formed cells, we evaluated the combined activity of both drugsin the Ba/F3 system and in K562 cells at 72 h by using atetrazolium-based (MTS) assay, which measures cell viability.For both of these cell lines, combination index plots of theMTS assays demonstrated values of �1, indicating synergisticactivity between the two agents (Fig. 2A). The assay was alsorepeated at 48 h and demonstrated similar results (data notshown).
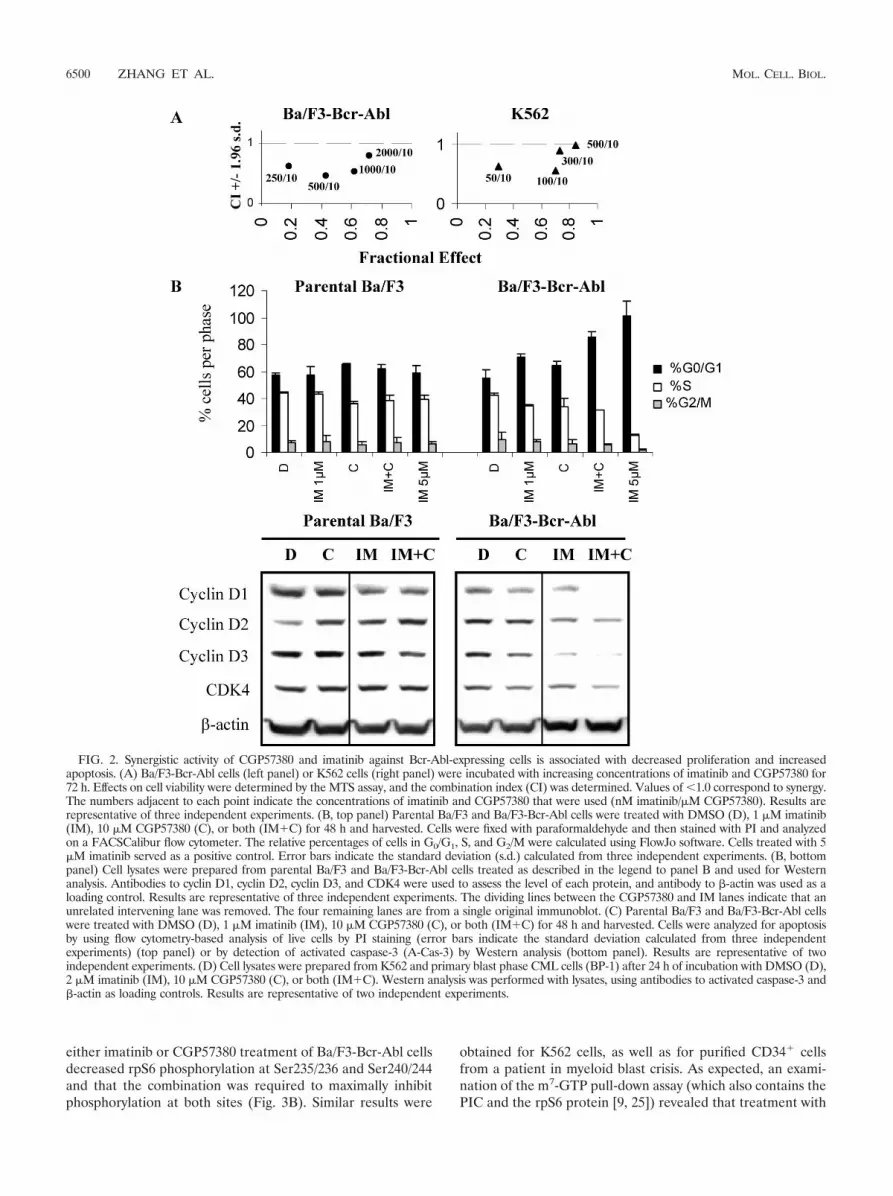
Because the MTS assay reflects changes in both cell prolif-eration and survival, direct measurements of CGP57380’s ef-fects on both of these parameters were carried out. Using flowcytometry and PI staining, we treated the Ba/F3-Bcr-Abl andcontrol parental Ba/F3 cells with inhibitors and performed cellcycle analysis. As expected, imatinib treatment of Ba/F3-Bcr-Abl cells (but not parental Ba/F3 cells) increased the propor-tion of cells at G0/G1, while single-agent treatment withCGP57380 had a small effect on inhibition of cell cycle entry inboth cell lines. However, in combination with imatinib,CGP57380 exposure resulted in an additive effect only in theBa/F3-Bcr-Abl cells (Fig. 2B, top panel). Since the D-typecyclins, together with CDK4, regulate G1/S transition, levels ofeach of these proteins were measured by immunoblotting ofinhibitor-treated cells. In Ba/F3-Bcr-Abl cells, treatment withimatinib alone decreased levels of all three cyclins, whileCGP57380 decreased cyclin D1 and D3 but not D2 (Fig. 2B,lower panel). However, when the drugs were used in combi-nation, the effect was to substantially decrease cyclins D1 andD3 but not D2. A similar but smaller effect of the two drugs onCDK4 expression was also seen.
To determine if the potentiation of imatinib’s effects onCML cells was also associated with enhanced apoptosis, West-ern- and flow cytometry-based analyses of apoptosis were per-formed. In Ba/F3-Bcr-Abl cells, the addition of CGP57380enhanced the apoptotic effect of imatinib across a wide doserange but, in contrast, had little effect on imatinib-treatedparental cells (Fig. 2C, upper panel). In addition, immunoblot-
ting using antibody to activated caspase-3 demonstrated thatCGP57380 administered alone had minimal effects on the Ba/F3-Bcr-Abl cells (Fig. 2C, lower panel). However, in cellstreated with the combination, levels of activated caspase-3were increased over that of cells exposed to imatinib alone.The effect of CGP57380 on K562 and primary blast phase (BP)CML cells exhibited similar characteristics, in that CGP57380produced a minimal proapoptotic effect by itself but greatlyenhanced the ability of imatinib to activate caspase-3 (Fig. 2D,upper panel). Together, these data suggest that CGP57380induces G0/G1 arrest by decreasing D-type cyclin expressionand, in cooperation with imatinib, promotes cell death viaactivating caspase-3.
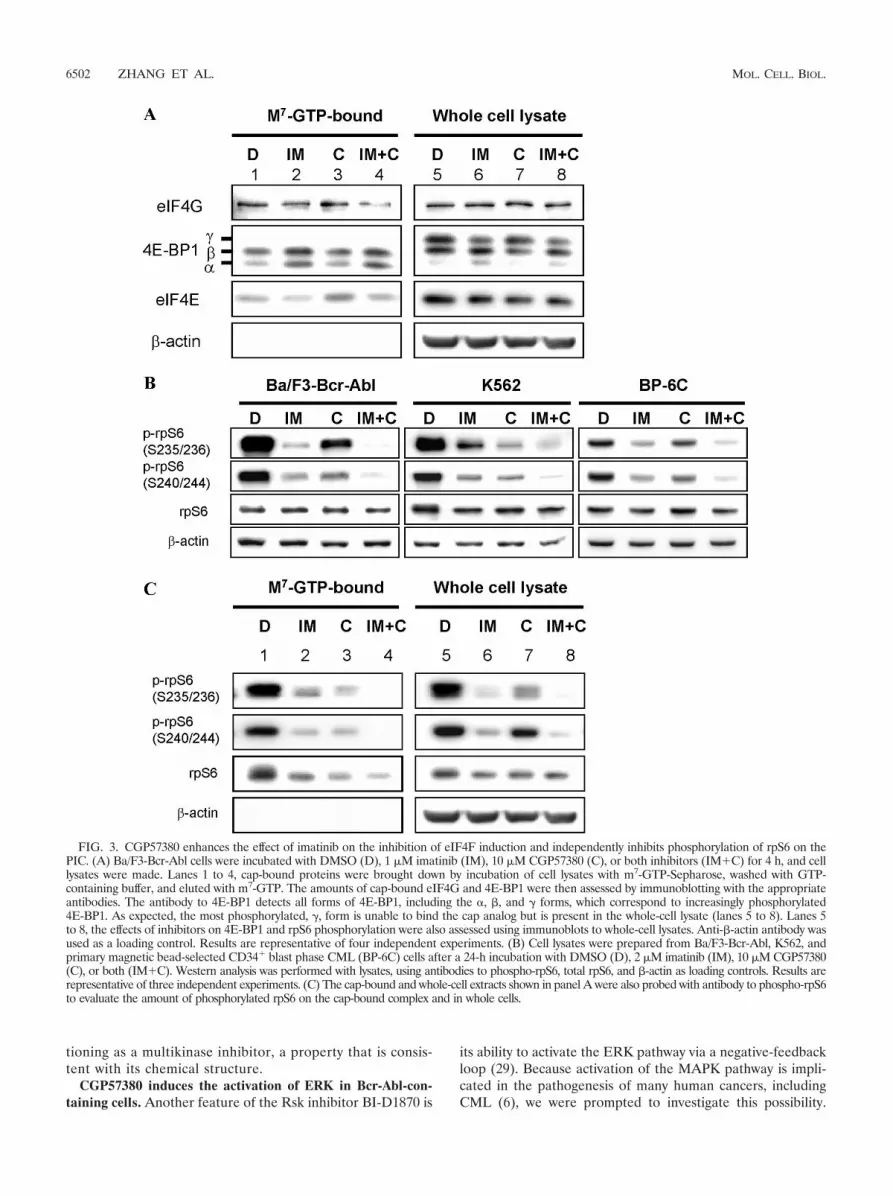
CGP57380 independently inhibits phosphorylation of rpS6on the PIC and cooperates with imatinib to inhibit eIF4Finduction. The ability of both imatinib and CGP57380 to im-pair polysome assembly (Fig. 1) suggested that these agentswere interfering with cap-dependent mRNA translation at thepoint of translation initiation. Because induction of eIF4F isessential for translation initiation and the subsequent recruit-ment of the 43S subunit to the mRNA strand, we assessed theintegrity of the eIF4F 7-methylguanosine-cap complex in Ba/F3-Bcr-Abl cells treated with imatinib and/or CGP57380.Here, we used the cap analog, m7-GTP-Sepharose, to captureeIF4E and its binding partners from whole-cell lysates (1, 23).Following a GTP wash and elution with m7-GTP, cap-boundfractions were assessed for the presence of 4E-BP1 and eIF4Gby using immunoblotting, and the integrity of eIF4F was notedas an increase in the ratio of eIF4G to 4E-BP1 in the cap-bound fractions. In Ba/F3-Bcr-Abl cells, imatinib treatmentresulted in an increase in cap-bound 4E-BP1 and a corre-sponding decrease in cap-bound eIF4G, as previously de-scribed (Fig. 3A, lane 2 versus 1) (1, 23). Interestingly, despitethe equivalent effects shown by CGP57380 on the inhibition ofpolysome assembly (Fig. 1A), CGP57380 treatment had littleeffect on eIF4F integrity compared to that in imatinib-treatedcells (Fig. 3A, lane 3 versus 1). Consistent with these observa-tions, we found that imatinib exerted a greater ability thanCGP57380 to inhibit 4E-BP1 phosphorylation in whole-celllysates (Fig. 3A, lane 6 versus 7). However, when both agentswere combined, we found that eIF4F induction was almostcompletely inhibited, which was appreciable as a disappear-ance of cap-bound eIF4G, as well as a corresponding increasein cap-bound 4E-BP1 (Fig. 3A, lane 4). Together, these resultssuggested two features of CGP57380’s effect on the cap-bind-ing complex. First, CGP57380 was modulating polysome as-sembly via a mechanism that was independent of an effect oneIF4F integrity. Second, CGP57380 was also able to cooperatewith imatinib to inhibit eIF4F induction more completely.
To further explore the mechanism by which CGP57380might inhibit efficient polysome assembly, we took note of arecent finding by Roux and colleagues, who demonstrated thatRsk-mediated phosphorylation of rpS6 at Ser235/236 promotescap-dependent mRNA translation (25). Specifically, theyfound that phosphorylation of rpS6 at this residue increasedthe affinity of rpS6 for the 7-methylguanosine-cap complex andpromoted polysome assembly and cap-dependent translation.Accordingly, we assessed the phosphorylation status of rpS6 atSer235/236, as well as at Ser240/244, in cells treated with ima-tinib and/or CGP57380. In whole-cell lysates, we found that
VOL. 28, 2008 TARGETING TRANSLATION INITIATION IN CML WITH CGP57380 6499
either imatinib or CGP57380 treatment of Ba/F3-Bcr-Abl cellsdecreased rpS6 phosphorylation at Ser235/236 and Ser240/244and that the combination was required to maximally inhibitphosphorylation at both sites (Fig. 3B). Similar results were
obtained for K562 cells, as well as for purified CD34� cellsfrom a patient in myeloid blast crisis. As expected, an exami-nation of the m7-GTP pull-down assay (which also contains thePIC and the rpS6 protein [9, 25]) revealed that treatment with
FIG. 2. Synergistic activity of CGP57380 and imatinib against Bcr-Abl-expressing cells is associated with decreased proliferation and increasedapoptosis. (A) Ba/F3-Bcr-Abl cells (left panel) or K562 cells (right panel) were incubated with increasing concentrations of imatinib and CGP57380 for72 h. Effects on cell viability were determined by the MTS assay, and the combination index (CI) was determined. Values of �1.0 correspond to synergy.The numbers adjacent to each point indicate the concentrations of imatinib and CGP57380 that were used (nM imatinib/�M CGP57380). Results arerepresentative of three independent experiments. (B, top panel) Parental Ba/F3 and Ba/F3-Bcr-Abl cells were treated with DMSO (D), 1 �M imatinib(IM), 10 �M CGP57380 (C), or both (IM�C) for 48 h and harvested. Cells were fixed with paraformaldehyde and then stained with PI and analyzedon a FACSCalibur flow cytometer. The relative percentages of cells in G0/G1, S, and G2/M were calculated using FlowJo software. Cells treated with 5�M imatinib served as a positive control. Error bars indicate the standard deviation (s.d.) calculated from three independent experiments. (B, bottompanel) Cell lysates were prepared from parental Ba/F3 and Ba/F3-Bcr-Abl cells treated as described in the legend to panel B and used for Westernanalysis. Antibodies to cyclin D1, cyclin D2, cyclin D3, and CDK4 were used to assess the level of each protein, and antibody to �-actin was used as aloading control. Results are representative of three independent experiments. The dividing lines between the CGP57380 and IM lanes indicate that anunrelated intervening lane was removed. The four remaining lanes are from a single original immunoblot. (C) Parental Ba/F3 and Ba/F3-Bcr-Abl cellswere treated with DMSO (D), 1 �M imatinib (IM), 10 �M CGP57380 (C), or both (IM�C) for 48 h and harvested. Cells were analyzed for apoptosisby using flow cytometry-based analysis of live cells by PI staining (error bars indicate the standard deviation calculated from three independentexperiments) (top panel) or by detection of activated caspase-3 (A-Cas-3) by Western analysis (bottom panel). Results are representative of twoindependent experiments. (D) Cell lysates were prepared from K562 and primary blast phase CML cells (BP-1) after 24 h of incubation with DMSO (D),2 �M imatinib (IM), 10 �M CGP57380 (C), or both (IM�C). Western analysis was performed with lysates, using antibodies to activated caspase-3 and�-actin as loading controls. Results are representative of two independent experiments.
6500 ZHANG ET AL. MOL. CELL. BIOL.

either drug also decreased phosphorylation of cap-bound rpS6at Ser235/236 and Ser240/244 (Fig. 3C). In addition, we foundthat either imatinib or CGP57380 treatment resulted in a slightdecrease in the amount of cap-bound rpS6 but that the effectson the phosphorylation of cap-bound rpS6 were more promi-nent.
These results demonstrate that CGP57380’s effect onpolysome assembly is associated with the inhibition of rpS6phosphorylation at Ser235/236. Since single-agent CGP57380therapy is able to impair polysome assembly without signifi-cantly influencing eIF4F formation (Fig. 1A and 3A), thesedata indicate that phosphorylation of cap-bound rpS6 may alsobe important for efficient functioning of the PIC, following itsinduction.
CGP57380 inhibits phosphorylation of multiple substratesthat regulate cap-dependent translation within the MAPK andmTOR signaling pathways. To date, CGP57380 has been em-ployed as an inhibitor of the Mnk kinases (11, 18). However,the results shown in Fig. 3B demonstrated that CGP57380 wasalso functioning to inhibit rpS6 phosphorylation. In addition,we noticed that there was a high degree of similarity betweenthe molecular structure of CGP57380 and that of anothercompound, BI-D1870, that was recently shown to be a specific
inhibitor of the p90 S6 kinase (Rsk) (Fig. 4A) (29). Bothcompounds consist of fluoroanilines connected to a heterobi-cyclic core. Indeed, since it has very few functional groups,CGP57380 might be expected to be more promiscuous thanother N-arylaniline kinase inhibitors (like BI-D1870 or ge-fitinib). These observations suggested that CGP57380 mightactually be functioning as a multikinase inhibitor. We there-fore tested the ability of CGP57380 to inhibit substrates of themajor signaling pathways known to regulate cap-dependenttranslation, including those of the mTORC1, Rsk, and Mnkkinases (Fig. 4B). Using LY294002, rapamycin, and BI-D1870as positive controls for PI3K, mTORC1, and Rsk inhibition,respectively, we found that CGP57380 was also able to inhibita similar range of substrates including S6K1 and rpS6 at bothSer235/236 and Ser240/244 (Fig. 4B). In addition, unlike theother inhibitors in the panel, CGP57380 was also able toinhibit Mnk kinase activity, as determined by eIF4E phos-phorylation (34). Interestingly, CGP57380 was able to dif-ferentiate between two mTOR substrates, S6K1 and 4E-BP1, suggesting that their phosphorylation might bedifferentially regulated (Fig. 4C). Together, these resultsdemonstrate that CGP57380 inhibits the phosphorylation ofa broad range of kinase substrates. CGP57380 is, thus, func-
FIG. 2—Continued.
VOL. 28, 2008 TARGETING TRANSLATION INITIATION IN CML WITH CGP57380 6501
tioning as a multikinase inhibitor, a property that is consis-tent with its chemical structure.
CGP57380 induces the activation of ERK in Bcr-Abl-con-taining cells. Another feature of the Rsk inhibitor BI-D1870 is
its ability to activate the ERK pathway via a negative-feedbackloop (29). Because activation of the MAPK pathway is impli-cated in the pathogenesis of many human cancers, includingCML (6), we were prompted to investigate this possibility.
FIG. 3. CGP57380 enhances the effect of imatinib on the inhibition of eIF4F induction and independently inhibits phosphorylation of rpS6 on thePIC. (A) Ba/F3-Bcr-Abl cells were incubated with DMSO (D), 1 �M imatinib (IM), 10 �M CGP57380 (C), or both inhibitors (IM�C) for 4 h, and celllysates were made. Lanes 1 to 4, cap-bound proteins were brought down by incubation of cell lysates with m7-GTP-Sepharose, washed with GTP-containing buffer, and eluted with m7-GTP. The amounts of cap-bound eIF4G and 4E-BP1 were then assessed by immunoblotting with the appropriateantibodies. The antibody to 4E-BP1 detects all forms of 4E-BP1, including the �, �, and forms, which correspond to increasingly phosphorylated4E-BP1. As expected, the most phosphorylated, , form is unable to bind the cap analog but is present in the whole-cell lysate (lanes 5 to 8). Lanes 5to 8, the effects of inhibitors on 4E-BP1 and rpS6 phosphorylation were also assessed using immunoblots to whole-cell lysates. Anti-�-actin antibody wasused as a loading control. Results are representative of four independent experiments. (B) Cell lysates were prepared from Ba/F3-Bcr-Abl, K562, andprimary magnetic bead-selected CD34� blast phase CML (BP-6C) cells after a 24-h incubation with DMSO (D), 2 �M imatinib (IM), 10 �M CGP57380(C), or both (IM�C). Western analysis was performed with lysates, using antibodies to phospho-rpS6, total rpS6, and �-actin as loading controls. Results arerepresentative of three independent experiments. (C) The cap-bound and whole-cell extracts shown in panel A were also probed with antibody to phospho-rpS6to evaluate the amount of phosphorylated rpS6 on the cap-bound complex and in whole cells.
6502 ZHANG ET AL. MOL. CELL. BIOL.

FIG. 4. CGP57380 inhibits phosphorylation of multiple substrates in the MAPK and mTOR signaling pathways that regulate cap-dependenttranslation. (A) Chemical structures of CGP57380 and BI-D1870. (B) Signaling diagram of known regulators of cap-dependent translation in theMAPK and mTOR pathways. Gray boxes indicate substrates that were evaluated by the immunoblotting shown in panel C. (C) Ba/F3-Bcr-Abl andK562 cells were treated with DMSO (D), 10 �M LY294002 (LY), 10 ng/ml rapamycin (R), 10 �M CGP57380 (C), 2 �M imatinib (IM), or 20 �Mof BI-D1870 (BI) for 2 (Ba/F3-Bcr-Abl) and 8 (K562) h. Western analyses were performed with cell lysates using antibodies to phosphorylated4E-BP1, S6K1, rpS6, and eIF4E. Loading was assessed using antibodies for total 4E-BP1, S6K1, rpS6, eIF4E, and �-actin. Results are represen-tative of three independent experiments.
VOL. 28, 2008 TARGETING TRANSLATION INITIATION IN CML WITH CGP57380 6503

FIG. 5. CGP57380 induces the activation of ERK and Mnk in Bcr-Abl-containing cells. (A, left panel) K562 cells were exposed to increasingdoses of CGP57380, after which immunoblots of cell lysates were probed with antibodies to the phosphorylated and total forms of ERK, Mnk, andeIF4E. Antibody to �-actin was used as a loading control. (Right panel) K562 cells were exposed to 10 �M CGP57380 over a time course, andcell lysates were analyzed as described in the legend to panel A, above. (B) Parental Ba/F3 and Ba/F3-Bcr-Abl cells were exposed to 10 �MCGP57380 over a time course, and cell lysates were analyzed as described in the legend above. (C) Primary CML cells from a patient in blast phase(BP-1) were treated with DMSO, 2 �M imatinib, 10 �M CGP57380, or both for 24 h. Immunoblots of cell lysates were probed as described in thelegend to panel A. (D) K562 cells were incubated with a panel of pharmacologic inhibitors or DMSO (D) for 24 h, as follows: 200 nM imatinib(IM/I), 10 �M U0126 (U), 10 �M SB203580 (S), 10 �M CGP57380 (C), or combinations of inhibitors. Immunoblots of cell lysates were thenprobed as described above. Results are representative of at least four independent experiments.
6504 ZHANG ET AL. MOL. CELL. BIOL.

Here, we found that CGP57380 treatment did indeed result inincreased ERK and Mnk phosphorylation in a dose- and time-dependent manner in K562 cells (Fig. 5A). Activation of thisloop was first evident at 2.5 �M CGP57380 (Fig. 5A, left panel)and was associated with an early transient phosphorylation ofboth ERK and Mnk, beginning within 30 min of treatment,followed by a second late phosphorylation beginning at 24 hand persisting for 48 to 72 h (Fig. 5A, right panel). Consistentwith an increase in Mnk kinase activity, we also observedrecrudescent eIF4E phosphorylation, although levels of phos-phorylation did not return to baseline. Interestingly, by repeat-ing the studies of the Ba/F3 system, we also show that certainfeatures of the negative-feedback loop were dependent onthe presence of Bcr-Abl. These features included the earlyCGP57380-mediated ERK phosphorylation, as well as sus-tained Mnk phosphorylation, since these effects were seen withBa/F3-Bcr-Abl cells but not with parental cells (Fig. 5B). Toconfirm that the feedback loop was present in primary cells,similar experiments were conducted in primary BP CML cells.Here, we also observed the phenomenon of CGP57380-medi-ated ERK and Mnk activation (Fig. 5C).
Because of the marked phosphorylation and activation ofMnk that were observed after CGP57380 exposure, we deter-mined if the feedback loop was mediated by MEK/ERK and/orp38 MAPK activation, since both of these pathways are knownto phosphorylate and activate Mnk (35). We therefore treatedK562 cells with either the MEK inhibitor U0126 or the p38MAPK inhibitor SB203580 and performed immunoblotting ofcell lysates. In these experiments, we found that U0126 wasable to completely inhibit ERK phosphorylation and to atten-uate Mnk and eIF4E phosphorylation (Fig. 5D, compare lane3 to lane 1), while SB203580 actually increased ERK phos-phorylation and had little effect on either Mnk or eIF4E phos-phorylation (Fig. 5D, compare lane 4 to lane 1). In CGP57380-treated cells, MEK inhibition prevented ERK, Mnk, andeIF4E phosphorylation (Fig. 5D, compare lane 7 to lane 1),while in SB203580-treated cells, ERK/Mnk/eIF4E phosphory-lation persisted (Fig. 5D, compare lane 8 to lane 1). Also,consistent with a role for Bcr-Abl in mediating this phenome-non, imatinib prevented the CGP57380-ERK/Mnk activation(Fig. 5D, compare lane 6 to 5). Taken together, these resultsdemonstrate that CGP57380 induces a negative-feedback loopvia MEK that regulates ERK and Mnk activation in Bcr-Abl-containing cells.
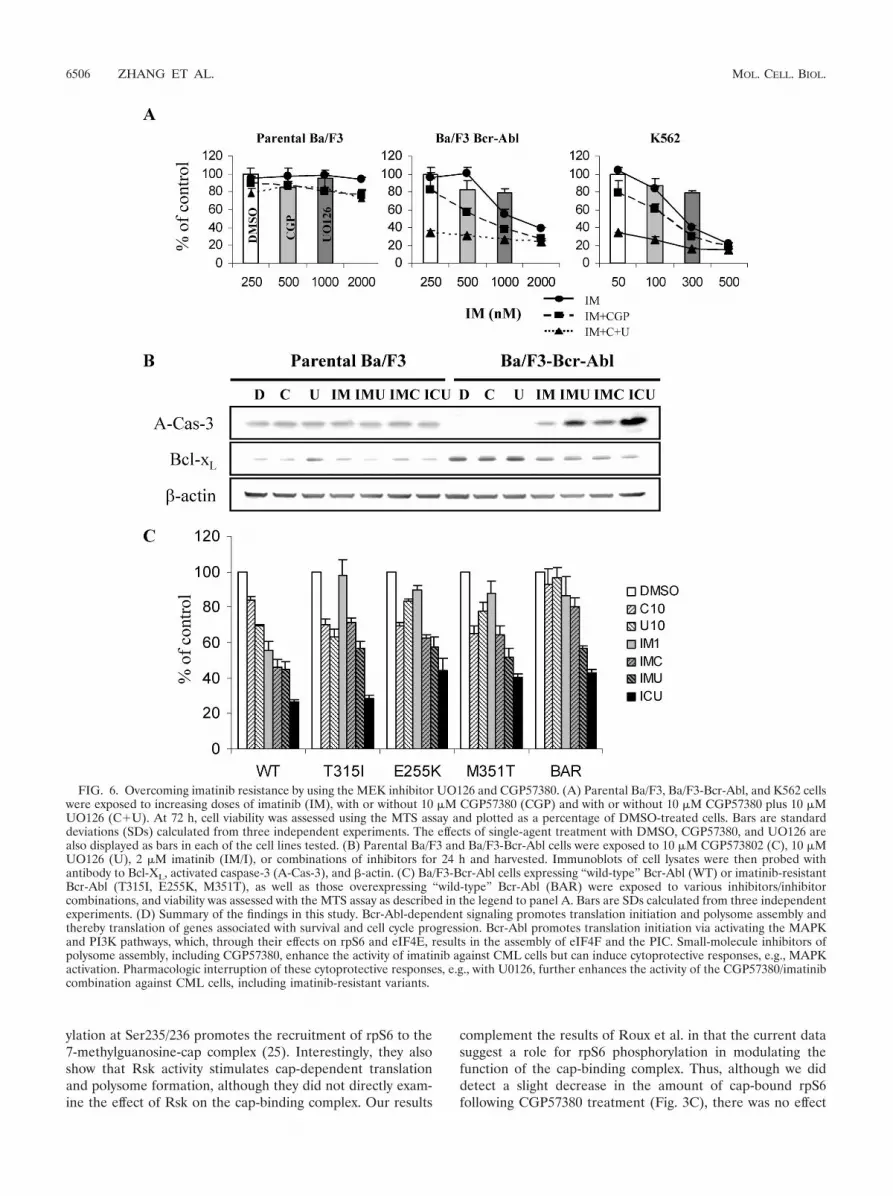
Inhibition of the MEK/ERK feedback loop enhances activityof CGP57380 and imatinib against Bcr-Abl-containing cellsand imatinib-resistant cell lines. Prior work with CML hasshown that the unintended activation of mitogenic pathways bykinase inhibitors, including activation by imatinib itself, can beturned to therapeutic advantage through the inhibition of thepathways concerned (5, 39). It was of interest, therefore, todetermine if the cytotoxic effect of CGP57380 might be en-hanced by concurrent MEK inhibition. Accordingly, U0126was added to the imatinib/CGP57380 combination, and thethree-drug combination was tested against parental Ba/F3, Ba/F3-Bcr-Abl, and K562 cells, using the MTS assay. The additionof U0126 to the imatinib/CGP57380 combination greatly en-hanced the effect of the combination on Bcr-Abl-bearing cells,while it left parental Ba/F3 cells unaffected (Fig. 6A). Remark-ably, the addition of the MEK inhibitor U0126 sensitized Ba/
F3-Bcr-Abl and K562 cells to noncytotoxic doses of imatinib(250 nM and 50 nM, respectively). Immunoblot analysis ofidentically treated cells demonstrated that the three-drug com-bination also augmented the effect of any two-drug combina-tion in activating caspase-3, as well as in decreasing the over-expression of the antiapoptotic protein Bcl-XL, in Ba/F3-Bcr-Abl cells but not in parental cells (Fig. 6B).
Because of this encouraging find, we went on to determinethe efficacy of this combination against cells bearing imatinib-resistant mutants of Bcr-Abl, as well as those overexpressing“wild-type” Bcr-Abl, which together represent the majority ofcases of clinical imatinib resistance (8, 31). We found that theimatinib-CGP57380 combination decreased the viability of allresistant cells compared to that of either agent alone. In eachcase, we also found an additional effect with the triple combi-nation (Fig. 6C).
DISCUSSION
The role of aberrant cap-dependent translation in cancer hasbeen the subject of several recent reviews (15, 27). The impor-tance of cap-dependent translation has also been highlightedby the potential for therapeutic targeting, using novel com-pounds (3, 10, 13, 17, 28), including a small molecule inhibitorof the eIF4E-eIF4G interaction which was found to have ac-tivity against Bcr-Abl-positive cell lines (17). Indeed, FDAapproval of temsirolimus, an inhibitor of the translation regu-lator mTOR, underlines the therapeutic potential of suchdrugs. However, the discovery of compounds which can targetnormal cellular processes with acceptable therapeutic indicesremains a challenging task. Nevertheless, as a result of ourprior work describing a direct effect of Bcr-Abl in the forma-tion of the cap-binding complex, eIF4F, and mRNA transla-tion, we began a series of experiments to screen for smallmolecules that could inhibit the process of translation initia-tion in a Bcr-Abl-specific manner. Here, we describe the ac-tivity of one such compound, CGP57380, against CML cellsand uncover its mechanism of action.
First, using polysomal mRNA profiling, we confirmed that anovel function of Bcr-Abl is to promote polysome assembly, aswould be predicted by the ability of Bcr-Abl to induce eIF4Fformation (23). An important point to note in these experi-ments is that very brief (4-h) exposures to the drug were suf-ficient to elicit gross changes in the polysome profile, thusruling out the possibility that the inhibition of translation wasdue to either imatinib-mediated caspase-3 activation and/orcell cycle arrest, both of which occur several hours after thepolysome changes shown in Fig. 1 (23). Next, we showed thatCGP57380 was also able to inhibit polysome assembly as asingle agent and, furthermore, that it could act cooperativelywith imatinib in this manner. Although the effect of CGP5780on polysomal mRNA assembly had been observed previously(18), the mechanism underlying this phenomenon had beenunclear, given the inability of CGP57380 to inhibit 4E-BP1phosphorylation and/or inhibit eIF4F induction (18). Our find-ing that CGP57380 also functions to inhibit rpS6 phosphory-lation may provide an explanation for this phenomenon (Fig. 3and 4), particularly in light of recent work by Roux et al. (25).In that study, the authors showed that phosphorylation atSer235/236 is largely Rsk kinase-dependent and that phosphor-
VOL. 28, 2008 TARGETING TRANSLATION INITIATION IN CML WITH CGP57380 6505
ylation at Ser235/236 promotes the recruitment of rpS6 to the7-methylguanosine-cap complex (25). Interestingly, they alsoshow that Rsk activity stimulates cap-dependent translationand polysome formation, although they did not directly exam-ine the effect of Rsk on the cap-binding complex. Our results
complement the results of Roux et al. in that the current datasuggest a role for rpS6 phosphorylation in modulating thefunction of the cap-binding complex. Thus, although we diddetect a slight decrease in the amount of cap-bound rpS6following CGP57380 treatment (Fig. 3C), there was no effect
FIG. 6. Overcoming imatinib resistance by using the MEK inhibitor UO126 and CGP57380. (A) Parental Ba/F3, Ba/F3-Bcr-Abl, and K562 cellswere exposed to increasing doses of imatinib (IM), with or without 10 �M CGP57380 (CGP) and with or without 10 �M CGP57380 plus 10 �MUO126 (C�U). At 72 h, cell viability was assessed using the MTS assay and plotted as a percentage of DMSO-treated cells. Bars are standarddeviations (SDs) calculated from three independent experiments. The effects of single-agent treatment with DMSO, CGP57380, and UO126 arealso displayed as bars in each of the cell lines tested. (B) Parental Ba/F3 and Ba/F3-Bcr-Abl cells were exposed to 10 �M CGP573802 (C), 10 �MUO126 (U), 2 �M imatinib (IM/I), or combinations of inhibitors for 24 h and harvested. Immunoblots of cell lysates were then probed withantibody to Bcl-XL, activated caspase-3 (A-Cas-3), and �-actin. (C) Ba/F3-Bcr-Abl cells expressing “wild-type” Bcr-Abl (WT) or imatinib-resistantBcr-Abl (T315I, E255K, M351T), as well as those overexpressing “wild-type” Bcr-Abl (BAR) were exposed to various inhibitors/inhibitorcombinations, and viability was assessed with the MTS assay as described in the legend to panel A. Bars are SDs calculated from three independentexperiments. (D) Summary of the findings in this study. Bcr-Abl-dependent signaling promotes translation initiation and polysome assembly andthereby translation of genes associated with survival and cell cycle progression. Bcr-Abl promotes translation initiation via activating the MAPKand PI3K pathways, which, through their effects on rpS6 and eIF4E, results in the assembly of eIF4F and the PIC. Small-molecule inhibitors ofpolysome assembly, including CGP57380, enhance the activity of imatinib against CML cells but can induce cytoprotective responses, e.g., MAPKactivation. Pharmacologic interruption of these cytoprotective responses, e.g., with U0126, further enhances the activity of the CGP57380/imatinibcombination against CML cells, including imatinib-resistant variants.
6506 ZHANG ET AL. MOL. CELL. BIOL.

of CGP57380 on the induction of eIF4F (Fig. 3A). Despite thislack of activity against eIF4F, CGP57380 impaired polysomeassembly to the same degree as imatinib (Fig. 1), suggesting aneffect on eIF4F function. Indeed, a role for rpS6 phosphory-lation in eIF4F function would explain the cooperative effect ofimatinib and CGP57380 to inhibit polysome assembly, sinceboth drugs were required to maximally inhibit rpS6 phosphor-ylation (Fig. 3B). Because CGP57380 also inhibits rpS6 phos-phorylation at Ser240/244, our findings also provide a rationalefor investigating the role of phosphorylation at Ser240/244 inpolysome assembly. Because both Ser235/236 and Ser240/244are preferentially phosphorylated by two different kinases, Rskand p70S6K1, respectively (25), it will be important to definethe contribution that each residue makes to polysome assemblyand cap-dependent translation, since it may be possible toselectively target these kinases in the clinic (29).
The results of inhibiting polysome assembly in CML cells aredecreased proliferation and survival (Fig. 2). For either ofthese cellular processes, modulation of polysome assembly pre-sumably decreases the translation of genes that positively reg-ulate proliferation and survival. In the case of the decreasedG1/S transition that was observed following drug treatment,this is likely a result of the decreased expression of the D-typecyclins and CDK4 (Fig. 2B), which together control this check-point. Prior work from our group and others has demonstratedthat in the Ba/F3 system, Bcr-Abl regulates both cyclin D2 andD3 expression but does so at the transcriptional and transla-tional levels, respectively (20, 23). In this respect, the ability ofa single agent, CGP57380, to inhibit cyclin D3 expression whileleaving cyclin D2 unaffected (Fig. 2B) is consistent with thegreater dependence of cyclin D3 expression on translationversus transcription, although this needs to be shown formally.At a global level, both imatinib and CGP57380 decrease thepolysome density of mRNAs and, thus, the translational effi-
ciency of mRNA (Fig. 1). This result implies differential effectsof these drugs on efficiently versus inefficiently translatedmRNAs. Factors within the 5� untranslated region (UTR) ofmRNAs that influence translational efficiency include the de-gree of secondary structure and the length of the 5� UTR, withmRNAs possessing higher levels of secondary structure havinga greater requirement for eIF4F function than those with lessstructure (33). More specifically, the degree of secondary struc-ture proximal to the 5� cap (within 37 nucleotides of the cap)seems to be particularly critical to the binding of translationinitiation factors (21). This fact, coupled with our findings thatCGP57380 modulates functioning of the cap-binding complex(Fig. 3), suggests that an analysis of the secondary structure ofthe first 30 to 40 nucleotides of genes that are prevented frombeing recruited to polysomes by CGP57380 might yield inter-esting information regarding the existence of motifs in theproximal 5� UTR of mRNAs that play an important role in theregulation of mRNA translation.
CGP57380 was initially isolated from a screen for Mnk ki-nase inhibitors (11) and has been employed by several groupsto investigate the interplay between eIF4E phosphorylationand translational regulation. Our results now reveal thatCGP57380 inhibits the phosphorylation of multiple substratesof the MAPK/Mnk/Rsk and PI3K/mTOR/S6K1 axes in vivo.Importantly, since this work was performed, Bain et al. haveshown that CGP57380 is indeed able to inhibit multiple kinasesin vitro, including Rsk1, with the same potency with which itinhibits the Mnk kinases (2). CGP57380 thus joins the ranks ofseveral small-molecule multikinase inhibitors that include anumber of FDA-approved cancer therapies: imatinib (38), sor-afenib, and sunitinib. Recent reviews of these drugs have high-lighted their abilities to target multiple kinases and have un-derlined the theoretical advantages of multitargeting (7, 37),including a broader spectrum of activity to more effectivelycombat the multifaceted nature of tumor growth, a reducedlikelihood of drug resistance, improved patient compliance,and a reduction in the drug interactions and toxicities associ-ated with monopharmacy versus polypharmacy. An importantdisadvantage is that the search for biomarkers for effectiveclinical decision making will be more complicated, and, per-haps more importantly, the critical molecular targets that fuelthe cancer will be more difficult to uncover. In this respect,while we have shown that CGP57380 impairs polysome assem-bly and targets multiple components of the translation machin-ery, we cannot exclude an effect mediated primarily via oneparticular protein, e.g., inhibition of rpS6 phosphorylation byRsk1. Indeed, since Rsk1 has a documented role in cap-de-pendent translation regulation (25), our initial efforts at fur-ther elucidating the mechanism of action of CGP57380 inCML will be focused on this kinase.
The interactions between CGP57380 and imatinib were alsonoteworthy in several respects, including their synergism andability to overcome imatinib resistance, as well as effects onMAPK signaling. The increased MAPK activity in CGP57380-treated CML cells suggests that this may represent a cytopro-tective mechanism by which Bcr-Abl-containing cells respondto the stress of CGP57380 exposure. Indeed, pharmacologicinterruption of this response is associated with enhanced kill-ing of CML cells by the imatinib/CGP57380 combination, in-cluding the potentiation of the lethal effects of extremely low
FIG. 6—Continued.
VOL. 28, 2008 TARGETING TRANSLATION INITIATION IN CML WITH CGP57380 6507

concentrations (50 nM) of imatinib. Such activity may be par-ticularly useful in the instance of suboptimal imatinib re-sponses associated with low intracellular imatinib concentra-tions and reduced OCT-1-mediated drug influx (36). We alsofound that cells with various forms of imatinib-resistant muta-tions were sensitive to the combination. The response of cellswith the M351T and E255K mutations and Bcr-Abl amplifica-tion was now comparable to sensitive cells treated with ima-tinib alone. It is also noteworthy that cells with the T315Imutation, which confers extreme imatinib resistance, re-sponded with a 71% decrease in cell viability (Fig. 6C).
In conclusion, our studies demonstrate that pharmacologicinhibitors of polysome assembly represent a novel group ofcompounds which may be useful in the therapy for CML,including overcoming resistance to tyrosine kinase inhibitors(Fig. 6D). Such an approach may also be useful against othercancers associated with dysregulated translation.
ACKNOWLEDGMENTS
We thank Bert Semler for helpful advice regarding obtaining and an-alyzing polysomal mRNA profiles, David Van Vranken for discussionsregarding the chemical structure of CGP57380, and Parimal Sarayu forcalculating the areas under the curves for the polysome profiles.
This work was supported by Public Health Service grants RO1CA107041, R21CA112936, and R33CA105514 from the National CancerInstitute.
REFERENCES
1. Avdulov, S., S. Li, V. Michalek, D. Burrichter, M. Peterson, D. M. Perlman,J. C. Manivel, N. Sonenberg, D. Yee, P. B. Bitterman, and V. A. Polunovsky.2004. Activation of translation complex eIF4F is essential for the genesis andmaintenance of the malignant phenotype in human mammary epithelialcells. Cancer Cell 5:553–563.
2. Bain, J., L. Plater, M. Elliott, N. Shpiro, C. J. Hastie, H. McLauchlan, I.Klevernic, J. S. Arthur, D. R. Alessi, and P. Cohen. 2007. The selectivity ofprotein kinase inhibitors: a further update. Biochem. J. 408:297–315.
3. Bordeleau, M. E., J. Matthews, J. M. Wojnar, L. Lindqvist, O. Novac, E.Jankowsky, N. Sonenberg, P. Northcote, P. Teesdale-Spittle, and J. Pelletier.2005. Stimulation of mammalian translation initiation factor eIF4A activityby a small molecule inhibitor of eukaryotic translation. Proc. Natl. Acad. Sci.USA 102:10460–10465.
4. Chou, T. C. 1994. Assessment of synergistic and antagonistic effects ofchemotherapeutic agents in vitro. Contrib. Gynecol Obstet. 19:91–107.
5. Chu, S., M. Holtz, M. Gupta, and R. Bhatia. 2004. BCR/ABL kinase inhi-bition by imatinib mesylate enhances MAP kinase activity in chronic my-elogenous leukemia CD34� cells. Blood 103:3167–3174.
6. Deininger, M. W., J. M. Goldman, and J. V. Melo. 2000. The molecularbiology of chronic myeloid leukemia. Blood 96:3343–3356.
7. Faivre, S., G. Demetri, W. Sargent, and E. Raymond. 2007. Molecular basisfor sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov.6:734–745.
8. Gorre, M. E., M. Mohammed, K. Ellwood, N. Hsu, R. Paquette, P. N. Rao,and C. L. Sawyers. 2001. Clinical resistance to STI-571 cancer therapycaused by BCR-ABL gene mutation or amplification. Science 293:876–880.
9. Holz, M. K., B. A. Ballif, S. P. Gygi, and J. Blenis. 2005. mTOR and S6K1mediate assembly of the translation preinitiation complex through dy-namic protein interchange and ordered phosphorylation events. Cell 123:569–580.
10. Kentsis, A., I. Topisirovic, B. Culjkovic, L. Shao, and K. L. Borden. 2004.Ribavirin suppresses eIF4E-mediated oncogenic transformation by physicalmimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA101:18105–18110.
11. Knauf, U., C. Tschopp, and H. Gram. 2001. Negative regulation of proteintranslation by mitogen-activated protein kinase-interacting kinases 1 and 2.Mol. Cell. Biol. 21:5500–5511.
12. La Rosee, P., A. S. Corbin, E. P. Stoffregen, M. W. Deininger, and B. J.Druker. 2002. Activity of the Bcr-Abl kinase inhibitor PD180970 againstclinically relevant Bcr-Abl isoforms that cause resistance to imatinib mesy-late (Gleevec, STI571). Cancer Res. 62:7149–7153.
13. Low, W. K., Y. Dang, T. Schneider-Poetsch, Z. Shi, N. S. Choi, W. C.Merrick, D. Romo, and J. O. Liu. 2005. Inhibition of eukaryotic transla-tion initiation by the marine natural product pateamine A. Mol. Cell20:709–722.
14. Ly, C., A. F. Arechiga, J. V. Melo, C. M. Walsh, and S. T. Ong. 2003. Bcr-Ablkinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target ofrapamycin. Cancer Res. 63:5716–5722.
15. Mamane, Y., E. Petroulakis, O. LeBacquer, and N. Sonenberg. 2006. mTOR,translation initiation and cancer. Oncogene 25:6416–6422.
16. Mamane, Y., E. Petroulakis, L. Rong, K. Yoshida, L. W. Ler, and N. Sonen-berg. 2004. eIF4E—from translation to transformation. Oncogene 23:3172–3179.
17. Moerke, N. J., H. Aktas, H. Chen, S. Cantel, M. Y. Reibarkh, A. Fahmy, J. D.Gross, A. Degterev, J. Yuan, M. Chorev, J. A. Halperin, and G. Wagner.2007. Small-molecule inhibition of the interaction between the translationinitiation factors eIF4E and eIF4G. Cell 128:257–267.
18. Morley, S. J., and S. Naegele. 2002. Phosphorylation of eukaryotic initiationfactor (eIF) 4E is not required for de novo protein synthesis followingrecovery from hypertonic stress in human kidney cells. J. Biol. Chem. 277:32855–32859.
19. Ottmann, O. G., B. J. Druker, C. L. Sawyers, J. M. Goldman, J. Reiffers,R. T. Silver, S. Tura, T. Fischer, M. W. Deininger, C. A. Schiffer, M. Bac-carani, A. Gratwohl, A. Hochhaus, D. Hoelzer, S. Fernandes-Reese, I. Gath-mann, R. Capdeville, and S. G. O’Brien. 2002. A phase 2 study of imatinibin patients with relapsed or refractory Philadelphia chromosome-positiveacute lymphoid leukemias. Blood 100:1965–1971.
20. Parada, Y., L. Banerji, J. Glassford, N. C. Lea, M. Collado, C. Rivas, J. L.Lewis, M. Y. Gordon, N. S. Thomas, and E. W. Lam. 2001. BCR-ABL andinterleukin 3 promote haematopoietic cell proliferation and survival throughmodulation of cyclin D2 and p27Kip1 expression. J. Biol. Chem. 276:23572–23580.
21. Pelletier, J., and N. Sonenberg. 1985. Photochemical cross-linking of capbinding proteins to eucaryotic mRNAs: effect of mRNA 5� secondary struc-ture. Mol. Cell. Biol. 5:3222–3230.
22. Perrotti, D., F. Turturro, and P. Neviani. 2005. BCR/ABL, mRNA transla-tion and apoptosis. Cell Death Differ. 12:534–540.
23. Prabhu, S., D. Saadat, M. Zhang, L. Halbur, J. P. Fruehauf, and S. T. Ong.2007. A novel mechanism for Bcr-Abl action: Bcr-Abl-mediated induction ofthe eIF4F translation initiation complex and mRNA translation. Oncogene26:1188–1200.
24. Richter, J. D., and N. Sonenberg. 2005. Regulation of cap-dependent trans-lation by eIF4E inhibitory proteins. Nature 433:477–480.
25. Roux, P. P., D. Shahbazian, H. Vu, M. K. Holz, M. S. Cohen, J. Taunton, N.Sonenberg, and J. Blenis. 2007. RAS/ERK signaling promotes site-specificribosomal protein S6 phosphorylation via RSK and stimulates cap-depen-dent translation. J. Biol. Chem. 282:14056–14064.
26. Rowley, J. D. 1973. A new consistent chromosomal abnormality in chronicmyelogenous leukaemia identified by quinacrine fluorescence and Giemsastaining. Nature 243:290–293.
27. Ruggero, D., and P. P. Pandolfi. 2003. Does the ribosome translate cancer?Nat. Rev. Cancer 3:179–192.
28. Sabatini, D. M. 2006. mTOR and cancer: insights into a complex relation-ship. Nat. Rev. Cancer 6:729–734.
29. Sapkota, G. P., L. Cummings, F. S. Newell, C. Armstrong, J. Bain, M.Frodin, M. Grauert, M. Hoffmann, G. Schnapp, M. Steegmaier, P. Cohen,and D. R. Alessi. 2007. BI-D1870 is a specific inhibitor of the p90 RSK(ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 401:29–38.
30. Sawyers, C. L., A. Hochhaus, E. Feldman, J. M. Goldman, C. B. Miller, O. G.Ottmann, C. A. Schiffer, M. Talpaz, F. Guilhot, M. W. Deininger, T. Fischer,S. G. O’Brien, R. M. Stone, C. B. Gambacorti-Passerini, N. H. Russell,J. J. Reiffers, T. C. Shea, B. Chapuis, S. Coutre, S. Tura, E. Morra, R. A.Larson, A. Saven, C. Peschel, A. Gratwohl, F. Mandelli, M. Ben-Am, I.Gathmann, R. Capdeville, R. L. Paquette, and B. J. Druker. 2002. Ima-tinib induces hematologic and cytogenetic responses in patients withchronic myelogenous leukemia in myeloid blast crisis: results of a phaseII study. Blood 99:3530–3539.
31. Shah, N. P., J. M. Nicoll, B. Nagar, M. E. Gorre, R. L. Paquette, J. Kuriyan,and C. L. Sawyers. 2002. Multiple BCR-ABL kinase domain mutationsconfer polyclonal resistance to the tyrosine kinase inhibitor imatinib(STI571) in chronic phase and blast crisis chronic myeloid leukemia. CancerCell 2:117–125.
32. Simonsson, B., on behalf of the International Randomized IFN versusSTI571 Study Group. 2005. Beneficial effects of cytogenetic and molecularresponse on long-term outcome in patients with newly diagnosed chronicmyeloid leukemia in chronic phase (CML-CP) treated with imatinib (IM):update from the IRIS study (ASH Annual Meeting Abstracts). Blood 106:166.
33. Svitkin, Y. V., A. Pause, A. Haghighat, S. Pyronnet, G. Witherell, G. J.Belsham, and N. Sonenberg. 2001. The requirement for eukaryotic initiationfactor 4A (elF4A) in translation is in direct proportion to the degree ofmRNA 5� secondary structure. RNA 7:382–394.
34. Ueda, T., R. Watanabe-Fukunaga, H. Fukuyama, S. Nagata, and R. Fuku-naga. 2004. Mnk2 and Mnk1 are essential for constitutive and inducible
6508 ZHANG ET AL. MOL. CELL. BIOL.

phosphorylation of eukaryotic initiation factor 4E but not for cell growth ordevelopment. Mol. Cell. Biol. 24:6539–6549.
35. Waskiewicz, A. J., A. Flynn, C. G. Proud, and J. A. Cooper. 1997. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 andMnk2. EMBO J. 16:1909–1920.
36. White, D. L., V. A. Saunders, P. Dang, J. Engler, A. Venables, S. Zrim, A.Zannettino, K. Lynch, P. W. Manley, and T. Hughes. 2007. Most CMLpatients who have a suboptimal response to imatinib have low OCT-1 activ-ity. Higher doses of imatinib may overcome the negative impact of lowOCT-1 activity. Blood 110:4064–4072.
37. Wilhelm, S., C. Carter, M. Lynch, T. Lowinger, J. Dumas, R. A. Smith, B. Schwartz,
R. Simantov, and S. Kelley. 2006. Discovery and development of sorafenib: amultikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5:835–844.
38. Wong, S., J. McLaughlin, D. Cheng, C. Zhang, K. M. Shokat, and O. N.Witte. 2004. Sole BCR-ABL inhibition is insufficient to eliminate all myelo-proliferative disorder cell populations. Proc. Natl. Acad. Sci. USA 101:17456–17461.
39. Yu, C., G. Krystal, L. Varticovksi, R. McKinstry, M. Rahmani, P. Dent, andS. Grant. 2002. Pharmacologic mitogen-activated protein/extracellular sig-nal-regulated kinase kinase/mitogen-activated protein kinase inhibitors in-teract synergistically with STI571 to induce apoptosis in Bcr/Abl-expressinghuman leukemia cells. Cancer Res. 62:188–199.
VOL. 28, 2008 TARGETING TRANSLATION INITIATION IN CML WITH CGP57380 6509
Related Documents











