“FORMULATION AND DEVELOPMENT OF REPAGLINIDE MICROPARTICLES BY IONOTROPIC GELATION TECHNIQUE” by SACHIN E. BHADKE B.PHARM. Dissertation Submitted to the Rajiv Gandhi University Of Health Sciences, Karnataka, Bangalore. In partial fulfillment of the requirements for the award of degree of MASTER OF PHARMACY In PHARMACEUTICS Under the guidance of Shri. S. P. HIREMATH SELECTION GRADE LECTURER Department of Pharmaceutics K.L.E.Society’s College of Pharmacy, Hubli - 580 031. Karnataka (India) JUNE-2006 I

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

“FORMULATION AND DEVELOPMENT OF REPAGLINIDE
MICROPARTICLES BY IONOTROPIC GELATION TECHNIQUE”
by
SACHIN E. BHADKE B.PHARM.
Dissertation Submitted to the
Rajiv Gandhi University Of Health Sciences, Karnataka, Bangalore.
In partial fulfillment of the requirements for the award of degree of
MASTER OF PHARMACY
In
PHARMACEUTICS
Under the guidance of Shri. S. P. HIREMATH
SELECTION GRADE LECTURER Department of Pharmaceutics
K.L.E.Society’s College of Pharmacy, Hubli - 580 031. Karnataka
(India)
JUNE-2006
I

DECLARATION BY THE CANDIDATE
I hereby declare that this dissertation / thesis entitled “FORMULATION
AND DEVELOPMENT OF REPAGLINIDE MICROPARTICLES BY
IONOTROPIC GELATION TECHNIQUE” is a bonafied and genuine
research work carried out by me under the guidance of Shri. S. P.
HIREMATH Selection grade lecturer, Department of Pharmaceutics,
K.L.E. Society’s College of Pharmacy, Hubli.
SACHIN E. BHADKE Date: Place: Hubli
RAJIV GANDHI UNIVERSITY OF HEALTH SCIENCES, KARNATAKA, BANGALORE
II

CERTIFICATE BY THE GUIDE
This is to certify that the dissertation entitled “FORMULATION AND
DEVELOPMENT OF REPAGLINIDE MICROPARTICLES BY
IONOTROPIC GELATION TECHNIQUE” is a bonafied research work
done by SACHIN E. BHADKE in partial fulfillment of the requirement for the
award of degree of MASTER OF PHARMACY IN PHARMACEUTICS.
Shri. S.P. HIREMATH Selection Grade Lecturer
Date: Place: Hubli
Department of PharmaceuK.L.E. Society’s College o Hubli - 580 031, Karnatak(India)
III
tics, f Pharmacy,a,

ENDORSEMENT BY THE HOD, PRINCIPAL/HEAD OF THE
INSTITUTION
This is to certify that the dissertation entitled “FORMULATION AND
DEVELOPMENT OF REPAGLINIDE MICROPARTICLES BY
IONOTROPIC GELATION TECHNIQUE” is a bonafied research work
done by SACHIN E. BHADKE under the guidance of Shri. S.P. HIREMATH
Selection Grade Lecturer, Department of Pharmaceutics, K.L.E. Society’s
College of Pharmacy, Hubli.
Dr. B.M.PATIL M. Pharm, Ph.D.
Principal K.L.E. Society’s College of Pharmacy, Hubli - 580 031, Karnataka, (India)
Shri. V.G. JAMAKANDI M. Pharm, Head of the Department Department of Pharmaceutics, K.L.E. Society’s College of Pharmacy, Hubli - 580 031, Karnataka (India)
Date: Place: Hubli
IV

COPYRIGHT
Declaration by the Candidate
I hereby declare that the Rajiv Gandhi University of Health Sciences, Karnataka
shall have the rights to preserve, use and disseminate this dissertation / thesis in
print or electronic format for academic / research purpose.
© Rajiv Gandhi University of Health Sciences, Karnataka
SACHIN E.BHADKE
Date: Place: Hubli
V

DEDICATED TO MY BELOVED
PARENTS AND
SPECIAL THANKS
TO MY
BROTHER
VI

VII VII

VIII

Acknowledgement
I consider myself most lucky to work under the able guidance of Shri. S. P.
Hiremath, Selection Grade Lecturer, Department of Pharmaceutics, K.L.E.S’s College of
Pharmacy, Hubli. I take this opportunity to express my heartfelt gratitude to my reverend
guide. His discipline, principles, simplicity, caring attitude and provision of fearless work
environment will be cherished in all walks of my life. I am very much greatful to him for
his invaluable guidance and ever-lasting encouragement throughout my course.
I am immensely thankful to Dr. V. I. Hukkeri, Ex-Principal and Dr.B.M.Patil,
Principal, K.L.E.S’s College of Pharmacy, Hubli.
I owe my warmest and humble thanks to Mr. Umesh Zope, Research and
Development (EMCURE PHARMACEUTICALS) Pune for providing complete details
regarding drug.
I owe my warmest and humble thanks to Mr. V. G. Jamakandi, Associate
Professor, HOD of Pharmaceutics, and Mr. S. A. Sreenivas, Mrs. Fatima, Mr. S .S.
Biradar, Mr. S .T. Bhagawati, Mr. Jameel S. Mulla, Mrs. K. R. Praveena, K.L.E.S’s
College of Pharmacy, Hubli, for their timely help, encouragement, boosting my
confidence in the progress of my academics.
I also take this opportunity to express my sincere thanks to teaching and non-
teaching staff of K.L.E.S’s College of Pharmacy, Hubli, for their kind co-operation and
help throughout my course.
I express my deepest and very special thanks to my batch mates Nilesh, Anupam,
Siddu, Ashwin, Manoj, Rohan, Prashant, Shrikant, Muzzaffar, Saurabh, Prashant,
Sanjay and afrin, siddhart and my colleagues Nilesh, Sanjay, Rahul, Meena, Ramdas,

Tushar, for their kind co-operation, help and encouragement throughout my post-
graduation.
My heartfelt thanks to my seniors especially Rajiv, Alok, Manish, Pranshu,
Gauda, Kiran, Johnson, Sandeep, Mangesh, Yadvindar, Adesh, Faheem, Pasha, and
Zakir for their help and encouragement.
I am thankful to all my juniors, especially, Mahesh, Nitesh, Ashish, Ratan,
Vinay, Avinash, Nagraj, Ashwini, Sandip, Ajit, Ram, Abdulla, Umesh, Anant, and
Rashmi, and others who have contributed directly or indirectly during my dissertation.
I am really thankful to my family members Nilesh, Mom, Anna, Narendra and
others for constant encouragement, moral support throughout my life.
It is with deep gratitude and humbleness; I express my thanks to Mr. Akshay
Baheti, Mr.V.P Chowdhary, and Mr. Dayanand Kunnur for their constant
encouragement and moral support throughout my dissertation work, with out him
experiments would not have come to reality.
The completion of this dissertation is not only fulfillment of my dreams but also the
dreams of my Parents who have taken lots of pain for me in completion of my higher
studies.
Lastly I thank ‘God’ the Almighty, to show the path to the ladder of success.
Thankful I ever remain....
Sachin E. Bhadke

CONTENTS
No. Particulars Page No.
1. INTRODUCTION …………………………………………. 1-14
2. NEED FOR STUDY……………………………………….. 15-17
3. REVIEW OF LITERATURE………………….…………... 18-37
4. MATERIALS AND METHODS…………………………… 38-53
5. RESULTS AND DISCUSSION …………………………. 54-70
6. SUMMARY ………………………………………………… 71-72
7. CONCLUSION …………………………………………….. 73
8. BIBLIOGRAPHY……………….………………………….. 74-80

LIST OF TABLES Sl.No. PARTICULARS OF THE TABLES Page No.
1 Microencapsulation processes & their Applicabilities 11
2 Pharmacokinetic data of Repaglinide 25
3 Materials were used as supplied by manufacturers 38
4 Equipments used for experimental work 38
5 Formulation Design of Microparticles
45
6 Common techniques for measuring fine particles of various sizes 46
7 Designations and Dimensions of IP Specification Sieves 48
8 Relation Between Angle of Repose and Flow property of the
microparticles
50
9 Standard Calibration Curves of Repaglinide 59
10 Comparison of I.R. Spectra of Repaglinide and in
Combination with Polymers
60
11 Percentage weight remained on various sieve size
60
12 Angle of Repose of Microparticles 61
13 Drug entrapment Efficiency of Microparticles
61
14 In-vitro Dissolution Profile for Formulation F1 62
15 In-vitro Dissolution Profile for Formulation F2 63
16 In-vitro Dissolution Profile for Formulation F3 64
17 In-vitro Dissolution Profile for Formulation F4 65
18 In-vitro Dissolution Profile for Formulation F5 66
19 In-vitro Dissolution Profile for Formulation F6 67
20 In-vitro Dissolution Profile for Formulation F7 68
21 Values of Correlation-coefficient (r) of Repaglinide 69
22 Curve Fitting Data of the Release Profile for Repaglinide 69
23 Results of assay of Formulations F1 & F4 after Accelerated
Stability Studies
70

Abstract
DEPT. OF Pharmaceutics KLESCOP, HUBLI
ABSTRACT
Repaglinide is an anti-diabetic, oral blood-glucose lowering drug of the
meglitinide class used in the management of type-II diabetes mellitus.
The present investigation involves formulation and evaluation of microparticles
with repaglinide as model drug for prolongation of drug release time. An attempt was
made to prepare microparticles of repaglinide by ionotropic gelation technique, with a
view to deliver the drug at sustained or controlled manner in gastrointestinal tract and
consequently into systemic circulation.
The microparticles were formulated by calcium chloride cross-linking method
using various concentration of Hydroxy Propyl Methyl Cellulose and Chitosan by
dropping the Drug-Polymer solution along with sodium alginate in calcium chloride
solution.
The prepared microparticles were evaluated for Flow behavior, Compatibility
study, Drug Entrapment Efficiency, In-vitro Dissolution, Scanning Electron Microscopy
and Sieving method. Among the seven formulations prepared and evaluated F1 and F4 are
found to show satisfactory results.
The Prepared Microparticles shows entrapment efficiency of 78.62% to 91.25%
Fourier transform Infrared spectroscopy confirmed the absence of any Drug-Polymer
interaction. In-vitro release studies carried out in 1.2 pH and 7.2 pH phosphate buffer
solution shows 93.78% and 91.78% of drug was released from F1 and F4 respectively.
The results of Flow behavior and Particle size analysis were found to be in the
official limits.

Abstract
DEPT. OF Pharmaceutics KLESCOP, HUBLI
The In-vitro release studies shows that formulation F1, F2 and F3 releases 91.99 %,
81.66 % and 71.66 % respectively after 12 hours.
Formulation F4, F5 and F6 shows release of 92.11%, 81.93% and 81.76%
respectively.
Formulation F7 which is a combination of hydroxy propyl methyl cellulose and
Chitosan shows drug release of 71.88%.

Chapter-I
Introduction
“ That is good study, which is opened with expectation and closed with delight ”

Chapter-II
Need for study
“ The real act of discovering is not a finding in new lands, but in seeing with new eyes ”

Chapter-III
Review of Literature

Chapter-IV
Materials and Methods
“ Art and science have their meeting point in method ”

Chapter-V
Results and
Discussion “ He, who is not open to conviction , is not qualified for discussion ”

Chapter-VI
Summary

Chapter-VII
Conclusion

Chapter-VIII
Bibliography

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
1
INTRODUCTION
DIABETES MELLITUS1 :
It is a metabolic disorder characterized by hyperglycaemia, glycosuria, negative
nitrogen balance and sometimes ketonemia. A wide-spread pathological change is
thickening of capillary basement membrane, increase in vessel wall matrix and cellular
proliferation resulting in vascular complications like lumen narrowing, early
atherosclerosis, sclerosis of glomerular capillaries, retinopathy, neuropathy and
peripheral vascular insufficiency.
Two major types of diabetes mellitus are:
Type I Insulin dependent diabetes mellitus (IDDM), juvenile onset diabetes mellitus:
There is a β-cell destruction in pancreatic islets; majority of cases autoimmune (type
I A) antibodies that destroy β-cells are detectable in blood, but some are idiopathic (type I
B)-no β-cell antibody is found. In all type I cases circulating insulin levels are low, and
patients are more prone to ketosis. This type is less common and has low degree of
genetic predisposition.
Type II Non insulin dependent diabetes mellitus (NIDDM), maturity onset diabetes
mellitus:
There is no loss or moderate reduction in β-cell mass; insulin in circulation is low,
normal or even high, no anti- β-cells antibody is demonstrable; has a high degree of
genetic predisposition; generally has a late onset (past middle age). Over 90% cases are
type II DM.

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
2
Causes may be:
• Abnormality in gluco-receptor of β-cells so that they respond at higher glucose
concentration.
• Reduced sensitivity of peripheral tissues to insulin: reduction in number of insulin
receptors, ‘down regulation’of insulin receptors. Many hypertensives are
hyperinsulinemic but normoglycaemic; exhibit insulin resistance.
Hyperinsulinemia perse has been implicated in causing angiopathy.
• Excess of hyperglycemic hormones (glucagons etc.)/obesity: cause relative
insulin deficiency—the β-cells lag behind.
ORAL HYPOGLYCAEMIC DRUGS
These drugs are blood-glucose lowering agents are effective orally. The chief draw
back of insulin is—it must be given by injection. Orally active drugs have always been
searched. The early sulfonamides tested in 1940s produced hypoglycaemia as side effect.
Taking this lead, the first clinically acceptable sulfonylurea tolbutamide was introduced
in 1957. Others followed soon after. In the 1970s many so called ‘second generation’
sulfonylureas have been developed which are 20-100 times more potent. A diguanidine
synthalin was found to be hypoglycaemic in the 1920s, but was toxic. Clinically useful
biguanide phenformin was developed parallel to sulfonylureas in 1957s. Recently 3
newer classes of drugs viz. α-glucosidase inhibitors, meglitinide analogues and
thiazolidinediones have been inducted.
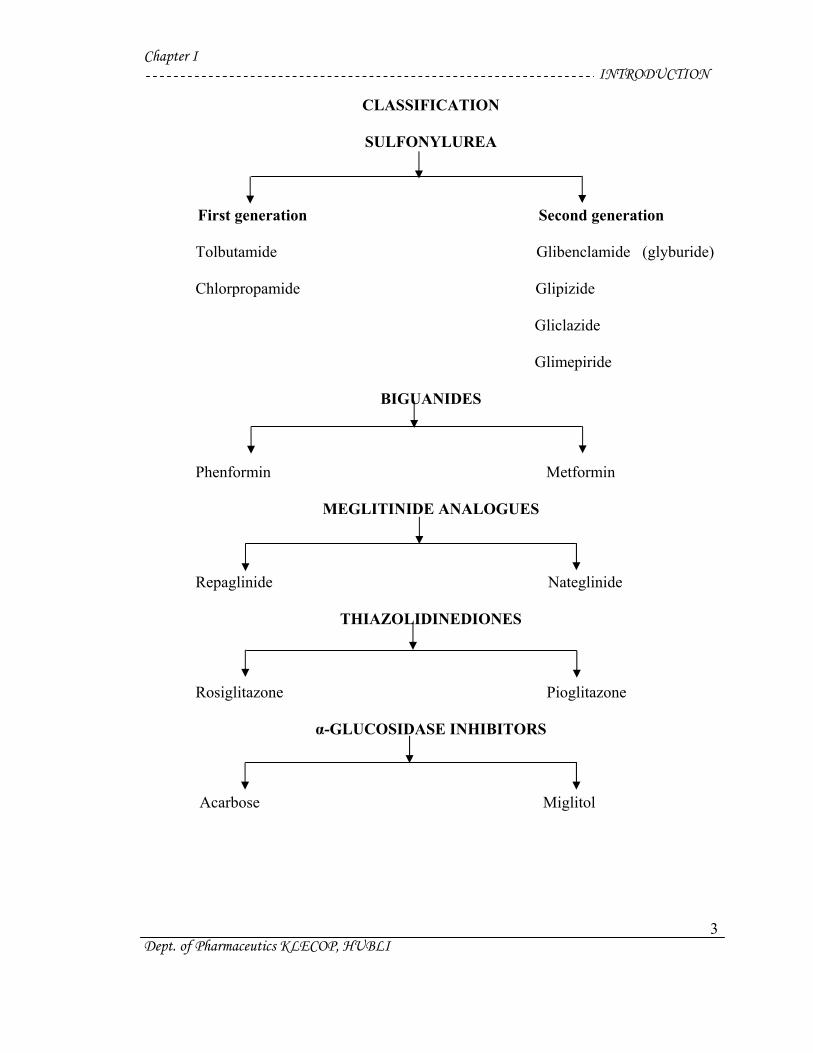
Chapter I INTRODUCTION
CLASSIFICATION
SULFONYLUREA
First generation Second generation
Tolbutamide Glibenclamide (glyburide)
Chlorpropamide Glipizide
Gliclazide
Glimepiride
BIGUANIDES
Dept. of Pharmaceutics KLECOP, HUBLI
3
Phenformin Metformin
MEGLITINIDE ANALOGUES
Repaglinide Nateglinide
THIAZOLIDINEDIONES
Rosiglitazone Pioglitazone
α-GLUCOSIDASE INHIBITORS
Acarbose Miglitol

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
4
MEGLITINIDE ANALOGUES
These are recently developed quick and short acting insulin releases.
Repaglinide:
It is the first member of new class of oral hypoglycaemics designed to normalize the
meal time glucose excursions. Though not a sulfonylurea, it acts in an analogous manner
by binding to sulfonylurea as well as to other distinct receptors –closer of ATP dependent
K+ channels – depolarization --- insulin release.
Repaglinide induces rapid onset short lasting insulin release. It is administered
before each major meal to control postprandial hyperglycaemia; the dose may be omited
if a meal is missed. Because of short lasting action it may have a lower risk of serious
hypoglycemia. Side effects are mild headache, dyspepsia, arthralgia, and weight gain.
Repaglinide is indicated only in type II DM as an alternative to sulfonylureas, or to
supplement metformin/long acting insulin. It should be avoided in the liver disease.
Nateglinide:
Another nonsulfonylurea drug which principally stimulates the 1st phase insulin
secretion resulting in rapid onset and shorter duration of hypoglycaemic action than
repaglinide. Ingested 10-30 min. before meal, it limits postprandial hyperglycaemia in
type II DM without producing late phase hypoglycaemia. There is effect on fasting blood
glucose level. Episodes of hypoglycaemia are less frequent than with sulfonylureas. Side
effects are dizziness, nausea, flu like symptoms and joint pain. It is used in the type II
DM along with other antidiabetics, to control postprandial rise in blood glucose.

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
5
CONTROLLED DRUG DELIVERY SYSTEM
For many decades, medication of an acute disease or a chronic illness has been
accomplished by delivering drugs to the patients via various pharmaceutical dosage
forms like tablets, capsules, pills, creams, ointments, liquids, aerosols, injectables
and suppositories as carriers. To achieve and then to maintain the concentration of
drug administered within the therapeutically effective range needed for medication, it is
often necessary to take this type of drug delivery systems several times a day. This results
in a fluctuated drug level and consequently undesirable toxicity and poor efficiency. This
factor as well as other factors such as repetitive dosing and unpredictable absorption lead
to the concept of controlled drug delivery systems.2, 3
The objectives of controlled release drug delivery includes two important aspects
namely spatial placement and temporal delivery of drug.
Spatial placement relates to targeting a drug to a specific organ or tissue, while
temporal delivery refers to controlling the rate of drug delivery to the target tissue.
An approximately designed controlled release drug delivery system can be a major
advance towards solving these two problems. It is this reason that the science and
technology responsible for development of controlled release pharmaceuticals have been
and continue to be the focus of a great deal of attention in both industrial and academic
laboratories.
Controlled release systems includes any drug delivery system that “achieves slow
release of the drug over an extended period of time.” If the system can provide some
control weather this is of a temporal or spatial nature, in other words, if the system is

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
6
successful in maintaining predictable and reproducible kinetics in the target tissue or cell,
it is considered as a controlled release system.
If the system only extends the duration of release without reproducible kinetics it is
considered as a prolong release system.
The objectives in designing a controlled release system is to deliver the drug at a
rate necessary to achieve and maintain a constant drug blood level. This rate should be
analogous to that achieved by continuous intravenous infusion where a drug is provided
to the patient at a rate just equal to its rate of elimination. This implies that the rate of
delivery must be independent of the amount of drug remaining in the dosage form and
constant over time. That is release from the dosage form should follow zero-order
kinetics.4
The several advantages of a controlled drug delivery system over a conventional
dosage forms:
• Improved patient convenience and compliance due to less frequent drug
administration.
• Reduction in fluctuation in steady-state levels and therefore better control of
disease condition and reduced intensity of a local or systemic side effects.
• Increased safety margin of high potency drugs due to better control of plasma
levels.
• Maximum utilization of drug enabling reduction in total amount of dose
administered.
• Reduction in health care costs through improved therapy, shorter treatment

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
7
period, less frequency of dosing and reduction in personnel time to dispense,
administer and monitor patients5.
Microencapsulation :
Microencapsulation is a rapidly expanding technology. As a process, it is a means of
applying relatively thin coatings to small particles of solids or droplets of liquids and
dispersions. Microencapsulation is arbitrarily differentiated from macrocoating
techniques in that the former involves the coating of particles ranging dimensionally from
several tenths of a micron to 5000 microns in size.6
Microencapsulation provides the means of converting liquids to solids, of altering
colloidal and surface properties, of providing environmental protection, and of
controlling the release characteristics or availability of coated materials.
Microenacapsulation is a process where by small discrete solid particles or small
liquid droplets are surrounded or enclosed, by an intact shell. Two major classes of
microencapsulation methods have evolved i.e. chemical and physical.
The first class of encapsulation method involves polymerization during the process
of preparing the microcapsules. The second type involves the controlled precipitation of a
polymeric solution where in physical changes usually occur.7, 8

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
8
Microencapsulation process:
Basic microencapsulation processes can be divided into chemical and mechanical.
Chemical processes involved :-
Complex coacervation
Polymer-polymer compatibility
Interfacial polymerization in liquid media
In-situ polymerization
In-liquid drying
Thermal and ionic gelation in liquid media
Mechanical processes involved:-
Spray drying
Spray coating
Fluidized bed coating
Electrostatic deposition
Centrifugal extrusion
Spinning disk or rotational suspension separation
Polymerization at liquid-gas or solid-gas interface
Pressure extraction or spraying into solvent extraction bath.9, 10

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
9
Ideal characteristics of drug for microencapsulation5 :
• Particle size requirement.
The lower the molecular weight, faster and complete is the absorption of the drug.
The drugs having size 150-600 daltons they can easily diffuse through the membrane but
diffusivity (the ability of drug to diffuse through the membrane) is inversely related to
molecular size.
• The drug or the protein should not be adversely affected by the process.
• Reproducibility of the release profile and the method.
• No stability problem.
Drugs unstable in GI environment cannot be administered as oral controlled release
formulation because of bioavailability problems e.g. Nitroglycerine.
• There should be no toxic product associated with the final product.
• Therapeutic range:
A candidate drug for controlled delivery system should have a therapeutic range
wide enough such that variations in the release rate do not result in a concentration
beyond this level.
• Therapeutic index:
The ratio of maximum safe concentration to the minimum effective concentration of
drug is called as therapeutic index. The release rate of a drug with narrow therapeutic
index should be such that the plasma concentration is attained between the therapeutically
safe and effective range. It is necessary because such drugs have toxic concentration
nearer to their therapeutic range.

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
10
• Elimination half life:
Smaller the half life larger the amount of drug to be incorporated in the controlled
release dosage form. Drugs with t1/2 in the range of 2 to 4 hours make a good candidates
for such a system e.g. propranolol.
• Plasma Concentration-Response Relationship:
Drugs whose pharmacologic activity is independent of its concentration are poor
candidates for controlled release systems.
Microencapsulation of pharmaceuticals is undertaken for the following
applications:
Microencapsulation has been employed to provide protection to the core material
against atmospheric effects. The separation of incompatible substances, for example
pharmaceutical eutectics, has been achieved by encapsulation. Toxic chemicals such as
insecticides may be microencapsulated to reduce hazards. Also the hygroscopic
properties of many core materials such as sodium chloride may be reduced by
microencapsulation.
Many drugs have been microencapsulated to reduce the gastric and other
gastrointestinal tract irritation. The local irritation and release properties of a number of
topically applied products can be altered by microencapsulation. This process is also used
to mask the taste of bitter drugs.
Microencapsulation has been widely employed in the design of controlled release
and sustained release dosage forms. It is the most recent addition to oral prolonged
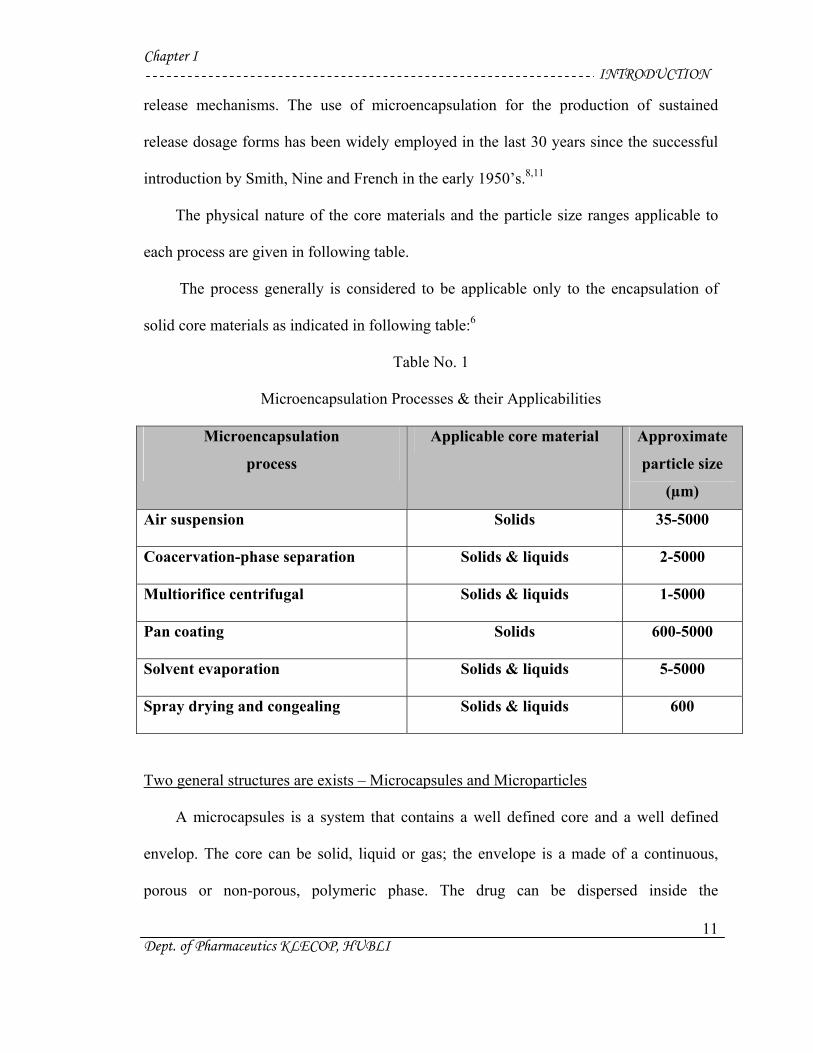
Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
11
release mechanisms. The use of microencapsulation for the production of sustained
release dosage forms has been widely employed in the last 30 years since the successful
introduction by Smith, Nine and French in the early 1950’s.8,11
The physical nature of the core materials and the particle size ranges applicable to
each process are given in following table.
The process generally is considered to be applicable only to the encapsulation of
solid core materials as indicated in following table:6
Table No. 1
Microencapsulation Processes & their Applicabilities
Microencapsulation
process
Applicable core material Approximate
particle size
(µm)
Air suspension Solids 35-5000
Coacervation-phase separation Solids & liquids 2-5000
Multiorifice centrifugal Solids & liquids 1-5000
Pan coating Solids 600-5000
Solvent evaporation Solids & liquids 5-5000
Spray drying and congealing Solids & liquids 600
Two general structures are exists – Microcapsules and Microparticles
A microcapsules is a system that contains a well defined core and a well defined
envelop. The core can be solid, liquid or gas; the envelope is a made of a continuous,
porous or non-porous, polymeric phase. The drug can be dispersed inside the

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
12
microcapsule as solid particulates with regular or irregular shapes, pure or dissolved
solution, suspension, emulsion or a combination of suspension and emulsion.
A microparticle is a structure made of a continuous phase of one or more miscible
polymers in which particulate drug is dispersed at either the macroscopic or molecular
levels. However, difference between the two system is the nature of the microparticle
matrix in which no well defined wall or envelop exists.7, 8
On the basis of classification these microcapsules and microspheres are prepared by
following techniques:12
Single emulsion technique
Double emulsion technique
Polymerization technique
Normal polymerization technique
Interfacial polymerization technique
Phase separation coacervation technique
Spray drying & spray congealing technique
Solvent extraction technique
Solvent evaporation technique
Solvent diffusion technique
Ionotropic gelation technique

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
13
Release characteristics of microparticles :
Release of core material from a non-erodible microparticle can occur in several
ways. Non-erodible spherical microparticles releases the encapsulated material by
steady-state diffusion through a coating of uniform thickness. The rate of release remains
constant as long as the internal and external concentration of core material and
concentration gradient through the membrane are constant.
If some of the encapsulated material migrates through the microcapsule membrane
during storage a burst effect occurs. If the microcapsule acts as inert matrix particle in
which core material is dispersed (microparticles) the Higuchi model is valid up to 60%
release.7
Microencapsulation by Ionotropic Gelation Technique:
In this method strong spherical beads with a narrow particle size distribution and
low friability could be prepared with high yield and a drug content approaching 91.25 %.
The flow properties of micronized or needle like drug crystals were significantly
improved by this agglomeration technique when compared with non-agglomerated drug
crystals. The ionic character of the polymers results from pH dependent disintegration of
the beads.
One of the most important and useful properties of alginates is the ability to form
gels by reaction with calcium salts. Alginic acid is composed of D-mannuronic acid and
L-gluronic acid residues at varying proportions of GG-, MM- and MG-blocks.
Crosslinking takes place only between the carboxylate residue of GG-blocks and Ca+2

Chapter I INTRODUCTION
Dept. of Pharmaceutics KLECOP, HUBLI
14
ions via egg-box model to give a tight gel network structure. These gels resembles a solid
in retaining their shape and resisting stress and consist of almost 100% water.
A gel in classical colloidal terminology is defined as a system which owes its
characteristic properties to a cross-linked network of polymer chains which form at the
gel point. A considerable amount of research has been carried out in recent years to
elucidate the nature of the cross-links and determine the structure of alginate gels.
It has been suggested that the cross-links were caused either by simple ionic
bridging of two carboxyl groups on adjacent polymer chains via calcium ions or by
chelating of a single calcium ions by hydroxyl and carboxyl groups on each of a pair of
polymer chains.13

Chapter III Research Investigated
Dept. of Pharmaceutics KLECOP, HUBLI
15
Need for study:
Microparticles have been widely accepted as a means to achieve oral and parenteral
controlled release. The microsphere requires a polymeric substance as a coat material or
carrier. A number of different substances both biodegradable as well as non-
biodegradable have been investigated for the preparation of microparticles17.
It not only reduces the dose of the drug, reaching to the effective biological sites
rapidly but also results in reduced toxicity of the targeting. But in the past few years,
pharmacists have been focused their research in colloidal drug delivery system/colloidal
carriers, like liposomes, microspheres and nanoparticles as a targeting carriers, which has
given selective targeting41.
Diabetes mellitus is a group of syndrome characterized by hyperglycaemia, altered
metabolism of lipids, carbohydrates, proteins and an increased risk of complication from
the vascular disease42.
Repaglinide is a nonsulfonylurea oral hypoglycaemic agent of the meglitinide
class, is mainly used in the management of type II diabetes mellitus. Chemically it is (S)-
2-ethoxy-4-{2-[3-methyl-1-[2-(1-piperidinyl) phenyl] butyl] amino]-2-oxoethyl} benzoic
acid. It has short biological half-life of less than one hour and rapidly eliminated from
body43.
The main aim of the present work is to develop the repaglinide microparticles by
using chitosan and hydroxyl propyl methyl cellulose as a polymer for prolonged,
relatively constant effective level of repaglinide and improve patient compliance.

Chapter III Research Investigated
Dept. of Pharmaceutics KLECOP, HUBLI
16
OBJECTIVES
Repaglinide is antidiabetic agent used in the treatment of Diabetes Mellitus. It is
used orally in the dose of 0.5-8 mg 3-4 times a day.
The present study “Repaglinide microparticles by ionotropic gelation technique”
was meticulously designed to improve the therapeutic efficacy and to prolong its release.
The following experimental protocol was therefore designed to allow a systematic
approach to the study:
1) Compatibility study:
Compatibility of drug with various polymers by using IR.
2) Preparation of standard curve for Repaglinide in acid buffer (pH 1.2),
and alkaline buffer (pH 7.2).
3) To prepare microparticles of Repaglinide using polymers Chitosan and
Hydroxy Propyl Methyl Cellulose by using Ionotropic Gelation Technique.
4) The following evaluation parameters were carried out based on
laboratory experiments:
i) Drug Entrapment Efficiency.
ii) Flow property of prepared microparticles.
iii) In-vitro dissolution studies by using dissolution tester USP (XXIII)
basket method release.
iv) Analysis by Scanning Electron Microscopy (SEM).
v) Particle size analysis by Sieving Method.

Chapter III Research Investigated
Dept. of Pharmaceutics KLECOP, HUBLI
17
The objectives of the proposed study are as follows:
• To overcome the rapid elimination of drug and to develop the oral controlled drug
delivery system.
• To increase the biological half-life of the drug or to maintain the constant drug
concentration in the body.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
17 Dept. of Pharmaceutics KLESCOP, HUBLI

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
18 Dept. of Pharmaceutics KLESCOP, HUBLI
Review of Literature :
A great deal of work has been done by the scientists about microspheres or
microparticles.
Bhalla H. L. et. al., (1988) worked on the development of sustained release tablets,
beads and coated pellets of Indomethacin using guar gum, sodium alginate and acrylic
resins. In comparison with the marketed slow release product of Indomethacin, the
prepared formulations showed considerable in-vitro drug release patterns.18
Chowdary K.P.R. et al., (1989) carried out microencapsulation of aspirin, diazepam
and nitrofurantoin by calcium alginate. The microparticles were found to be discrete,
spherical and free flowing. The release mechanism was found to be of diffusion type and
release depended on solubility of the core material in the dissolution fluid.19
Udupa N. et. al., (1994), developed implantable formulations of Flubiprofen using
biodegradable aliphatic polyesters, hydrophilic polymers like alginates and HPMC by
ionotropic gelation technique, in the form of films, microspheres and pellets for treating
chronic inflamed conditions associated with arthritis.20
Kakkar A.P. (1995) developed and characterized Ibuprofen loaded microcapsules
with sodium alginate and calcium chloride by ionotropic gelation technique. Spherical,
smooth surfaced alginate microcapsules of Ibuprofen were obtained by this method. The
preparation was based on dispersion of sodium alginate-Ibuprofen matrix in liquid
paraffin followed by coating process by calcium chloride.21
M. Guzman, J. Molepeceres et. al., (1996) have Prepared, characterized poly-e-
caprolactone and hydroxyl propyl methyl cellulose phathalate ketoprofen loaded

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
19 Dept. of Pharmaceutics KLESCOP, HUBLI
microspheres by encapsulating the ketoprofen within poly-e-caprolactone and hydroxyl
propyl methyl cellulose phathalate22.
M. Cuna, M. L. Lorenzo-Lamosa et. al., (1997) have prepared pH dependent
cellulosic microspheres containing Cefuroxime Axetil: Stability and In-vitro release
behavior by using CAT (cellulose acetate trimellitate) and two types of hydroxyl propyl
methyl cellulose phathalate, HPMCP-55 and HPMCP-50 were obtained by solvent
extraction procedure.23
Propranolol hydrochloride was incorporated into formed calcium alginate beads or
incorporated simultaneously with the gelation of alginate beads by calcium ions, and the
interaction of the drug with alginate molecular chains in the beads was studied by Lim-Ly
and Wan-LS, (1997)24.
Lim-Ly et. al., (1997), carried out the preparation of chitosan microspheres by an
emulsion phase separation technique and ionotropic gelation technique. They reported
that the microspheres, so prepared were spherical and free flowing and had smooth
surfaces25.
Pillay V., Dangor C.M. et. al., (1998) worked on encapsulation of indomethacin in
calcium alginate gel disks by ionotropic gelation technique.26
A. Manna, I. Ghosh, et. al., (1999) prepared and evaluated of an oral controlled
release microparticulate drug delivery system of Nimesulide by ionotropic gelation
technique and statistical optimization by factorial analysis by using HPLC and sodium
alginate as a polymer and calcium chloride as a crosslinking agent. Finally they have
done statistical optimization with two-way analysis of varience (ANOVA) and linear
regression analysis27.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
20 Dept. of Pharmaceutics KLESCOP, HUBLI
Alf Lamprecht, Ulrich Schafer, et. al., (2000) demonstrated the potential of
confocal laser scanning microscopy as a characterization tool for different types of
microparticles which are prepared by various methods including complex coacervation,
spray drying, double emulsion solvent evaporation technique and ionotropic gelation
technique.28
Patil V. B. and Pokharkar Varsha B., (2001) prepared and evaluated sustained
release nimesulide microspheres from sodium alginate. In this investigation a 23 full
factorial design was used to study the joint influence of three variables. The polymer
concentration, Drug concentration, and cross-linker concentration and also various
dependent variables like percent efficiency, spericity, particle size, and drug release.29
Choi B.Y., Park H. J., et. al., (2002) prepared alginate beads for floating drug
delivery system in which they prepared floating beads by adding the sodium alginate
drop by drop in calcium chloride solution and the floating properties were investigated.
Bead porosity and volume average pore size, as well as surface and cross sectional
morphology of beads were examined with mercury porosimetry and scanning electron
microscopy30.
Chowdary K.P.R. et al., (2003) developed indomethacin microcapsules with a coat
consisting of alginate and Mucoadhesive polymers such as HPMC, MC etc. by an
emulsification-ionic gelation process and were investigated to develop Mucoadhesive
microcapsules. The resulting microcapsules were discrete, large, spherical and free
flowing.31
Jelena Filipovie-Grcie, et. al., (2003) have evaluated Spray-dried Carbamazepine-
loaded Chitosan and HPMC microspheres and characterized by X-ray powder

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
21 Dept. of Pharmaceutics KLESCOP, HUBLI
diffractometry (XRD) and differential scanning calorimetry (DSC), were also studied
with particle size distribution, drug content analysis and drug release.32
Nikhil O. Dhoot et. al., (2003) studied the release of fluorescein isothiocynate
labled bovine serum albumin from alginate-microencapsulated liposomes and evaluated
the properties of the system for controlled drug delivery.33
Mukherjee A. et. al., (2003) have conducted a nebulization technique for
nanocapsulation of methotrexate using calcium alginate as a coating material.
Nanocapsules formed were compared with those produced by in-situ gelling technique,
which showed that by controlling various process parameters superior drug nanocapsules
were produced by pneumatic nebulization technique34.
Rajesh K.S., Khanrah A. and Biswanath Sa, (2003) studied preparation of
ketoprofen-loaded microparticles by dropping alginate-drug suspension, with or without
aqueous colloidal polymer dispersions, into calcium chloride solution. The effect of
various formulation variables on the physical characteristics of the microparticles were
studied. In-vitro release showed that drug release from the microparticles depended only
on the concentration of alginate and the nature of aqueous colloidal polymer dispersion13.
Arul B., Kothai R., Sangameswaran B., et. al., (2003) have conducted formulation
and evaluation of chitosan microspheres containing isoniazid in this they have prepared
microspheres by glutaraldehyde cross-linking method using various concentration of
chitosan15.
Pralhad T. Tayade, et. al., (2004) they have done Encapsulation of water-insoluble
drug by a cross-linking technique: Effect of process and formulation variables on

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
22 Dept. of Pharmaceutics KLESCOP, HUBLI
encapsulation efficiency, particle size, and in vitro dissolution rate they have prepared
Ibuprofen-gelation micropellets by cross-linking technique formaldehyde.35
Mingshi Yanga, Fudea et. al., (2004) have conducted a novel pH dependent
gradient-release delivery system for nitrendipin microspheres. By using Eudragit E-100,
Hydroxy propyl methyl cellulose phthalate and Hydroxy propyl methyl cellulose acetate
succinate which dissolve at an acid condition, the pH of ≥ 5.5 and ≤ 6.5, respectively.36
Chowdary K.P.R., Koteswara Rao N., et. al., (2004) prepared ethyl cellulose
microspheres of glipizide. In this study glipizide was incorporated into Ethyl cellulose
and were investigated by in-vitro and in-vivo methods.14
Govindrajan G., Muttusamy K., et. al., (2004) have formulated and developed
lansoprazole floating microspheres in which they have studied improvement of half-life
and bioavailability of lansoprazole by using chitosan as polymer in which it acts as a
carrier and characterized by scanning electron microscope (SEM)37.
Jamagondi L. N., Patil V. B. et. al., (2004) studied the microencapsulation of
metformin hydrochloride using ethyl cellulose as a polymer. By this they have tried to
improve glucose tolerance in patients with type-II diabetes38.
Dandagi P. M., Manvi F.V., et. al., (2004) worked on “Microencapslulation of
verapamil hydrochloride by ionotropic gelation technique”, By using hydroxyl propyl
methyl cellulose and hydroxyl propyl cellulose as a polymer and characterized by
scanning electron microscopy.39
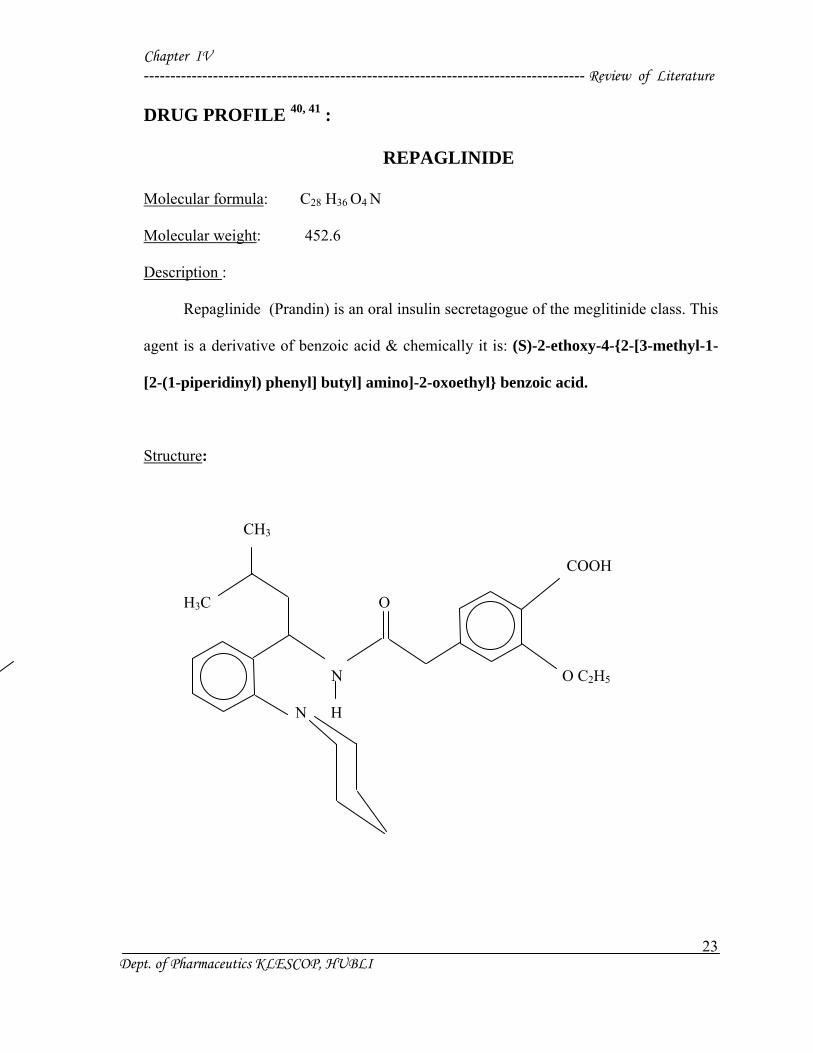
Chapter IV ----------------------------------------------------------------------------------- Review of Literature
23 Dept. of Pharmaceutics KLESCOP, HUBLI
DRUG PROFILE 40, 41 :
REPAGLINIDE
Molecular formula: C28 H36 O4 N
Molecular weight: 452.6
Description :
Repaglinide (Prandin) is an oral insulin secretagogue of the meglitinide class. This
agent is a derivative of benzoic acid & chemically it is: (S)-2-ethoxy-4-{2-[3-methyl-1-
[2-(1-piperidinyl) phenyl] butyl] amino]-2-oxoethyl} benzoic acid.
Structure:
CH3
COOH
H3C O
N O C2H5
N H

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
24 Dept. of Pharmaceutics KLESCOP, HUBLI
Clinical Pharmacology:
Mechanism of action:
It is the first member of new class of oral hypoglycaemics designed to normalize the
meal time glucose excursions. Though not a sulfonylurea, it acts in an analogous manner
by binding to sulfonylurea as well as to other distinct receptors –closer of ATP dependent
K+ channels ---- depolarization ------ insulin release.
Repaglinide induces rapid onset short lasting insulin release. It is administered
before each major meal to control postprandial hyperglycaemia; the dose may be omited
if a meal is missed. Because of short lasting action it may have a lower risk of serious
hypoglycemia. Side effects are mild headache, dyspepsia, arthralgia, and weight gain.
Repaglinide is indicated only in type II DM as an alternative to sulfonylureas, or to
supplement metformin/long acting insulin. It should be avoided in the liver disease.
Pharmacokinetic data of Repaglinide:
After oral administration, Repaglinide is rapidly and completely absorbed from the
gastrointestinal tract. After single and multiple oral doses inn healthy subjects or in
patients, peak plasma drug levels (Cmax) occurs within 1 hour (Tmax). Repaglinide is
rapidly eliminated from the blood stream with a half life of approximately 1-hour. The
mean absolute bioavailability is 56%. When repaglinide was given with food.
Repaglinide is 98% bound to plasma proteins, primarily to albumin. The drug is
completely metabolized by oxidative biotransformation and direct conjugation with
glucuronic acid after either an intravenous or oral dose. Metabolites do not contribute to
the glucose-lowering effect of repaglinide.
Chapter IV ----------------------------------------------------------------------------------- Review of Literature
25 Dept. of Pharmaceutics KLESCOP, HUBLI
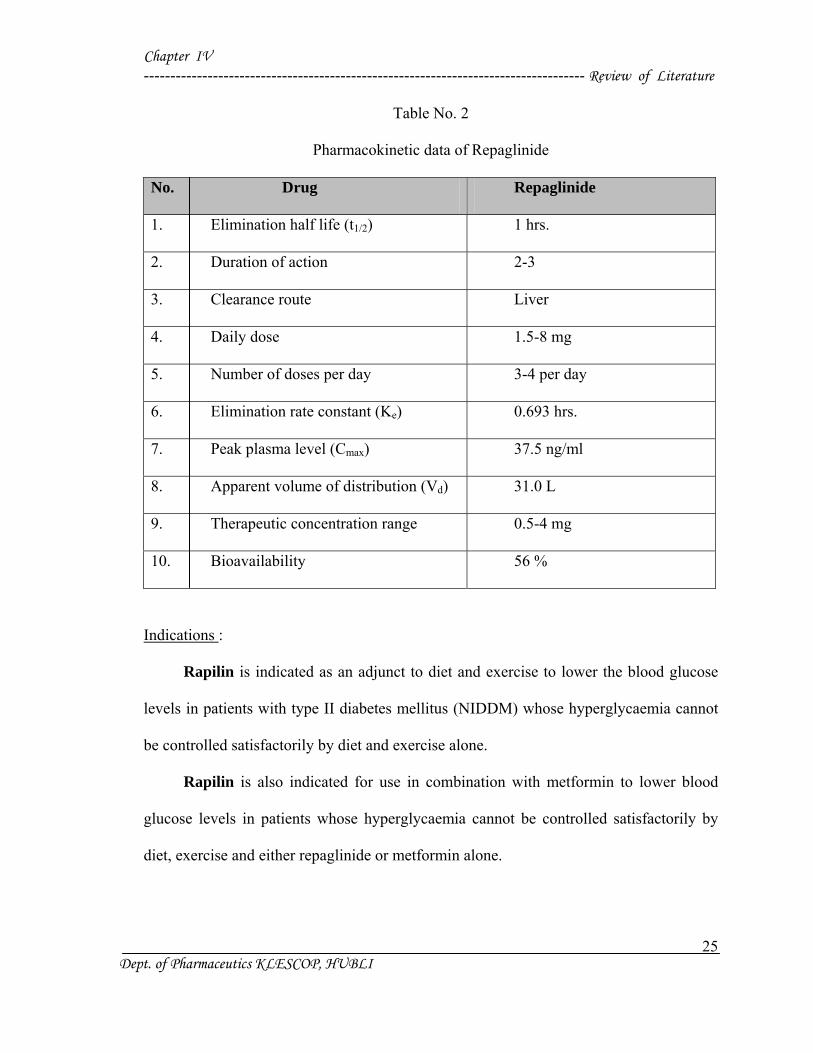
Table No. 2
Pharmacokinetic data of Repaglinide
No. Drug Repaglinide
1. Elimination half life (t1/2) 1 hrs.
2. Duration of action 2-3
3. Clearance route Liver
4. Daily dose 1.5-8 mg
5. Number of doses per day 3-4 per day
6. Elimination rate constant (Ke) 0.693 hrs.
7. Peak plasma level (Cmax) 37.5 ng/ml
8. Apparent volume of distribution (Vd) 31.0 L
9. Therapeutic concentration range 0.5-4 mg
10. Bioavailability 56 %
Indications :
Rapilin is indicated as an adjunct to diet and exercise to lower the blood glucose
levels in patients with type II diabetes mellitus (NIDDM) whose hyperglycaemia cannot
be controlled satisfactorily by diet and exercise alone.
Rapilin is also indicated for use in combination with metformin to lower blood
glucose levels in patients whose hyperglycaemia cannot be controlled satisfactorily by
diet, exercise and either repaglinide or metformin alone.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
26 Dept. of Pharmaceutics KLESCOP, HUBLI
Drug interactions:
Invitro data indicate that antifungal agents like ketoconazole and miconazole may
inhibit repaglinide metabolism, and antibacterial agents like erythromycin. Drugs that
induce the cytochrome P-450 enzyme system 3A4 may increase repaglinide metabolism;
such drugs include troglitazone, rifampin, barbiturates, and carbamazepine.
Repaglinide had no clinically relevant effect on the pharmacokinetics properties of
digoxin, theophylline, or warfarin,. Thus no dosage adjustment is required for digoxin,
theophylline, or warfarin on co-administration of cimetidine with repaglinide did not
significantly alter the absorption and disposition of repaglinide.
Side effects:
In various clinical trials, the most common side effects leading to withdrawal were
hyperglycaemia, hypoglycaemia and related symptoms. Other commonly reportedly side
effects were upper respiratory tract infections, nausea, vomiting, arthralgia, backpain and
headache. The incidence of serious cardiovascular side effects added together, including
ischaemia was slightly higher for repaglinide (4 %) than for sulfonylurea drug (3 %) in
controlled comparator clinical trials.
Overdosage:
There were few adverse effects other those associated with the intended effect of
lowering blood glucose in the patients who received increasing doses of repaglinide upto
80 mg a day for 14 days. Hypoglycaemia did not occur when meals were given with
these high doses. Hypoglycaemia symptoms without loss of consciousness or neurologic
findings should be treated aggressively with oral glucose and adjustment in drug dosage
and / or meal pattern.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
27 Dept. of Pharmaceutics KLESCOP, HUBLI
If hypoglycaemic reactions with coma is diagnosed or suspected, the patients
should be given a rapid intravenous injection of concentrated (50%) glucose solution.
This should be followed by a continuous infusion of more dilute (10%) glucose solution
at a rate that will maintain the blood glucose at a level above 100 mg/dl.
Dosage and Administration :
There is no fixed-dosage regimen for the management of type II diabetes with
repaglinide. Short-term duration of repaglinide may be sufficient during periods of
transient loss of control in patient’s usually well-controlled on diet. Repaglinide doses are
usually taken within 15 minutes of the meal but time may vary from immediately
preceding the meal to as long as 30 minutes before the meal.
Starting dose :
For patients not previously treated or whose glycosylated haemoglobin is < 8 % the
starting dose should be 0.5 mg with each meal. For patients previously treated with blood
glucose-lowering drug and whose glycosylated haemoglobin is < 8% the initial dose is 1
or 2 mg with each meal preprandially.
Dose adjustment:
Dosage adjustment should be determined by blood glucose response, usually
fasting blood glucose. The preprandial dose should be doubled upto 4 mg with each meal
until satisfactory blood glucose response is achieved. At least one week should elapse to
asses response after each dose adjustment.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
28 Dept. of Pharmaceutics KLESCOP, HUBLI
The recommended dose range is 0.5 mg to 4 mg taken with meals. Repaglinide
may be preprandial 2, 3, or 4 times a day in response to changes in the patient’s meal
pattern. The maximum dose recommended daily dose is 16 mg.
No dosage adjustments are required for the elderly.
Combination Therapy :
The starting dose adjustments for repaglinide combination therapy is the same as
for repaglinide monotherapy. The dose of each drug should be carefully adjusted to
determine the minimal dose required to achieve the desired pharmacological effects.
Appropriate monitoring of fasting plasma glucose and glycosylated haemoglobin
measurements should be used to ensure that the patient is not subjected to excessive drug
exposure or increased probability of secondary drug failure.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
29 Dept. of Pharmaceutics KLESCOP, HUBLI
POLYMER REVIEW
HYDROXY PROPYL METHYL CELLULOSE42, 43 :
Hydroxy propyl methyl cellulose (HPMC) is one of the cellulose ether, commonly
used in the formulation of controlled release dosage forms. These polymers hydrates in
water, forming a gel layer at the matrix periphery. Drug is released by a combination of
diffusion through and erosion of gel.
HPMC offers the advantage of being non-toxic and relatively inexpensive, it can be
directly compressed into matrices and the many grades available allow a wide latitude in
the ability to tailor desired drug release profiles.
It is believed that provided the drug and the resin are oppositely charged they will
bind together in-situ within the HPMC matrix, leading to reduced drug release rates.
Once the drug is released sufficient ions are available to displace it from the binding site.
It was not surprising, to find that the ionic strength of the dissolution fluid affected the
action of the resin. Nevertheless, the HPMC matrix has shown to have an inherent
buffering capacity, which allowed a virtually pH independent release profile to be
obtained.
Structural Formula of HPMC :
CH2OCH3 H OCH3 H OH HO H H O O H OCH3 H H OCH3 H H OH H H H OH H O O O CH2OCH3 H OCH2CHCH3
OH

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
30 Dept. of Pharmaceutics KLESCOP, HUBLI
Empirical formula :
C8H15O6 - (C10H18O6)N - C8H15O5
Synonyms :
Methyl hydroxypropyl cellulose, propylene glycol ether of methyl cellulose.
Solubility :
Soluble in cold water forming a viscous colloidal solution, insoluble in alcohol,
ether and chloroform but soluble in mixtures of methyl alcohol and methylene chloride.
Functional categories :
USP: Suspending and or viscosity increasing agent, tablet binder, coating agent.
BP: Viscosity increasing agent, adhesive anhydrous ointment ingredient.
Others: Film former, emulsion stabilizer.
Method of manufacture :
Cellulose fibers, obtained from cotton linters or wood pulp, are treated with caustic
solution. The alkali cellulose thus obtained is in turn treated with methyl chloride and
propylene oxide to produce methyl hydroxyl propyl; ethers of cellulose. The fibrous
reaction products is then purified and ground to a fine, uniform power.
Stability and storage condition :
Very stable in dry condition, solutions are stable at pH 3.0-11.0. Aqueous solutions
are liable to be affected by microorganisms.
Safety :
Human and animal feeding studies have shown hydroxypropyl methyl cellulose to
be safe.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
31 Dept. of Pharmaceutics KLESCOP, HUBLI
Application in Pharmaceutical formulation :
Film former in tablet film coating (perhaps the most commonly used film forming
agent). Lower viscosity grades are used in aqueous film coating and higher viscosity
grades are used in solvent film coating. The concentration varies from 2 to 10 %
depending on the viscosity grade of polymer. High viscosity grade are used to retard the
release of water soluble drugs.
Review of past work on HPMC :
Microparticulates drug delivery system of nimesulide was prepared with HPMC as
a polymer. HPMC microparticles are sufficiently hard, spherical in shape, capable of
loading drug upto 90%. Drug leaching from the surface of microparticles is negligible
and capable of releasing drug up to 12 hrs.28
Microencapsulation of drug with aqueous colloidal polymer was studied with
HPMC, sodium alginate, and chitosan as a polymers. About 80% to 90% encapsulation
efficiency was achieved with Ibuprofen, Theophylline, Guaifenesin. Water soluble drugs
with HPMC shows highest drug loading capacity.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
32 Dept. of Pharmaceutics KLESCOP, HUBLI
SODUM ALGINATE :44, 45
Sodium alginate consists of mainly of the sodium salt of alginic acid which is a
mixture of polyuronic acids [(C6H8O6) n] composed of β-D-mannuronic acid and residue
linked so that the carboxyl group of each unit is free while the aldehyde group is shielded
by a glycosidic linkage.
Empirical formula :
(C6H7O6Na) n
Structural formula : H COOH H H H O O OH OH HO O O H H O
H COONa n
Description :
Sodium alginate occurs as a white or buff powder which is odourless and tasteless.
Sodium alginate produces aqueous solutions that forms gel on the addition of small
amount of soluble calcium salt.
Grades:
Various grades of sodium alginate are available yielding aqueous solutions of
varying viscosities within a range of 20 to 400 centipoises in 1% solution at 200C.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
33 Dept. of Pharmaceutics KLESCOP, HUBLI
Solubility :
Sodium alginate is very slowly soluble in water forming a viscous colloidal
solution practically it is insoluble in alcohol, chloroform and ether and in hydroalcoholic
solutions in which alcohol content is greater than 30% by weight.
Stability and Storage Conditions :
Sodium alginate is hygroscopic, the moisture content at equilibrium is a function of
relative humidity. Dry storage stability is excellent when the powder is stored in a well
closed container at temperature of 250C or less.
Incompatibility :
Depending on the concentration, sodium alginate is incompatible with phenols and
parabens.
Uses :
Alginic acid and alginates such as propylene glycol alginate and sodium alginate
are used as suspending and thickening agents, aid in the preparation of water miscible
pastes, creams and gels. They may be used as stabilizers for oil in water emulsions and as
binding and disintegrating agents in tablets. Alginic acid and alginates (ammonium
alginate, calcium alginate, potassium alginate, propylene glycol alginate and sodium
alginate) are also employed as emulsifiers and stabilizers in food industry.

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
34 Dept. of Pharmaceutics KLESCOP, HUBLI
CHITOSAN 46:
Chitosan is the term applied to deacetylated chitins in various stages of
deacetylation and depolarization and it is therefore not easily defined in terms of its exact
chemical composition. Partial deacetylation of chitin results in the production of chitosan,
which is a polysaccharide comprising copolymers of glucosamine and N-
acetylglucosamine.
Molecular Weight :
Chitosan is commercially available in several types and grades that vary in
molecular weight between 10,000 and 1,000000, and vary in degree of deacetylation and
viscosity.
Synonyms :
2-amino-2-deoxy-(1,4)- β-D-glucopyranan; deacetylated chitin; deacetylchitin;
β-1,4-poly-D-glucosamine; poly-D-glucosamine; poly-(1,4- β-D-glucopyranosamine ).
Chemical name :
Poly- β-(1,4) -2-amino-2-deoxy-D-glucose.
Description :
Chitosan occurs as odorless, white or creamy-white powder or flakes. Fibre
formation is quite common during precipitation and the chitosan may look ‘cotton like.’
Structural formula : CH2OH CH3 O OH O CH3 NHR n R = H or COCH3

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
35 Dept. of Pharmaceutics KLESCOP, HUBLI
Functional category :
Coating agent; disintegrant; film-forming agent; mucoahesive; tablet binder;
viscosity-increasing agent.
Typical properties :
Acidity :
pH = 4.0-6.0 (1% W/V aqueous solution)
Density :
1.35-1.40 g/cm3
Glass transition temperature : 2030C
Moisture content :
Chitosan absorbs moisture from the atmosphere, the amount of water absorbed
depending upon the initial moisture content and the temperature and relative humidity of
the surrounding air.
Particle size distribution : < 30 µm
Solubility :
Sparingly soluble in water; practically insoluble in ethanol (95%), other organic
solvents, and neutral or alkali solutions at pH above 6.5.
Incompatibility :
Chitosan is incompatible with strong oxidizing agents.
Method of manufacturing :
Chitosan is manufactured commercially by chemically treating the shells of
crustaceans such as shrimps and crabs. The basic manufacturing process involves the
removal of proteins by treatment with alkali and of minerals such as calcium carbonate

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
36 Dept. of Pharmaceutics KLESCOP, HUBLI
and calcium phosphate by treatment with acid. Before these treatment the shells are
ground to make as accessible and then deproteinized by treatment with aqueous sodium
hydroxide 3-5 % solution. The resulting product is neutralized and calcium is removed by
treatment of hydrochloric acid at room temperature to precipitate chitin. The chitin is
dried and by deacetylation of this chitin forms chitosan.
N-acetylation of chitin is achieved by treatment with an aqueous sodium hydroxide
at elevated temperature (1100C), the precipitate is washed with water. The crude sample
is dissolved in acetic acid 2% and insoluble part is removed and the resulting clear
supernatant solution is neutralized with sodium hydroxide solution to give purified white
precipitate of chitosan.
Safety :
Chitosan is investigated widely for use as an excipient in oral and other
pharmaceutical formulations. It is biocompatible with both healthy and infected skin.
Chitosan has been shown to be biodegradable.
Stability and storage conditions :
Chitosan powder is a stable material at room temperature, although it is
hygroscopic after drying. Chitosan should be stored in tightly closed container in a cool,
dry place and it should be stored at a temperature of 2-80C.
Applications in Pharmaceutical Formulation or Technology :
The suitability and performance of chitosan as a component of pharmaceutical
formulations for drug delivery applications has been investigated in numerous studies.
These include controlled drug delivery applications, used as a component of
mucoadhesive dosage forms, rapid release dosage forms, improved peptide delivery,

Chapter IV ----------------------------------------------------------------------------------- Review of Literature
37 Dept. of Pharmaceutics KLESCOP, HUBLI
colonic drug delivery systems, and use for gene delivery. Chitosan has been processed in
to several pharmaceutical dosage forms, including gels, films, beads, microspheres,
tablets and coating for liposomes. Furthermore, chitosan may be processed into drug
delivery systems using several techniques including spray-drying, coacervation, direct
compression, and conventional granulation processes.

Chapter V -------------------------------------------------------------------------------------------Methodology
37
Dept. of Pharmaceutics, KLECOP, Hubli
Chapter V -------------------------------------------------------------------------------------------Methodology
38
MATERIALS AND METHODS
The following materials were used as supplied by the manufacturers without
further purification:
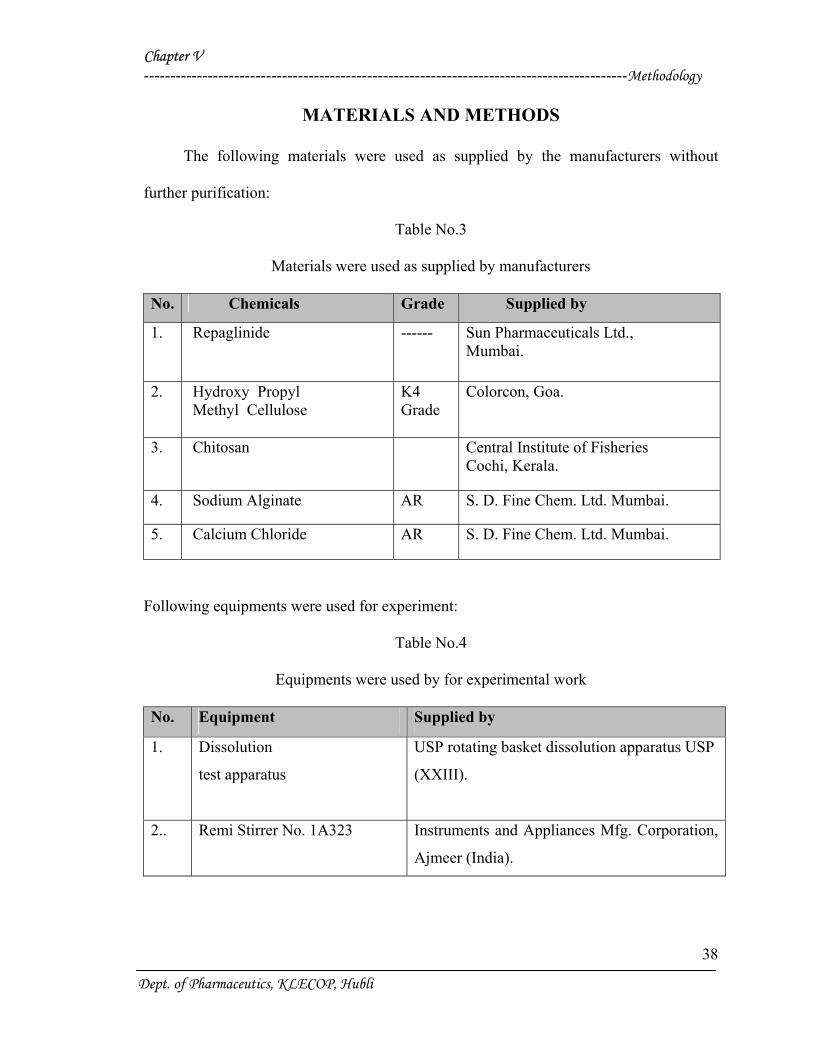
Table No.3
Materials were used as supplied by manufacturers
No. Chemicals Grade Supplied by
1. Repaglinide ------ Sun Pharmaceuticals Ltd., Mumbai.
2. Hydroxy Propyl Methyl Cellulose
K4 Grade
Colorcon, Goa.
3. Chitosan
Central Institute of Fisheries Cochi, Kerala.
4. Sodium Alginate AR S. D. Fine Chem. Ltd. Mumbai.
5. Calcium Chloride AR S. D. Fine Chem. Ltd. Mumbai.
Following equipments were used for experiment:
Table No.4
Equipments were used by for experimental work
No. Equipment Supplied by
1. Dissolution
test apparatus
USP rotating basket dissolution apparatus USP
(XXIII).
2.. Remi Stirrer No. 1A323 Instruments and Appliances Mfg. Corporation,
Ajmeer (India).
Dept. of Pharmaceutics, KLECOP, Hubli
Chapter V -------------------------------------------------------------------------------------------Methodology
39
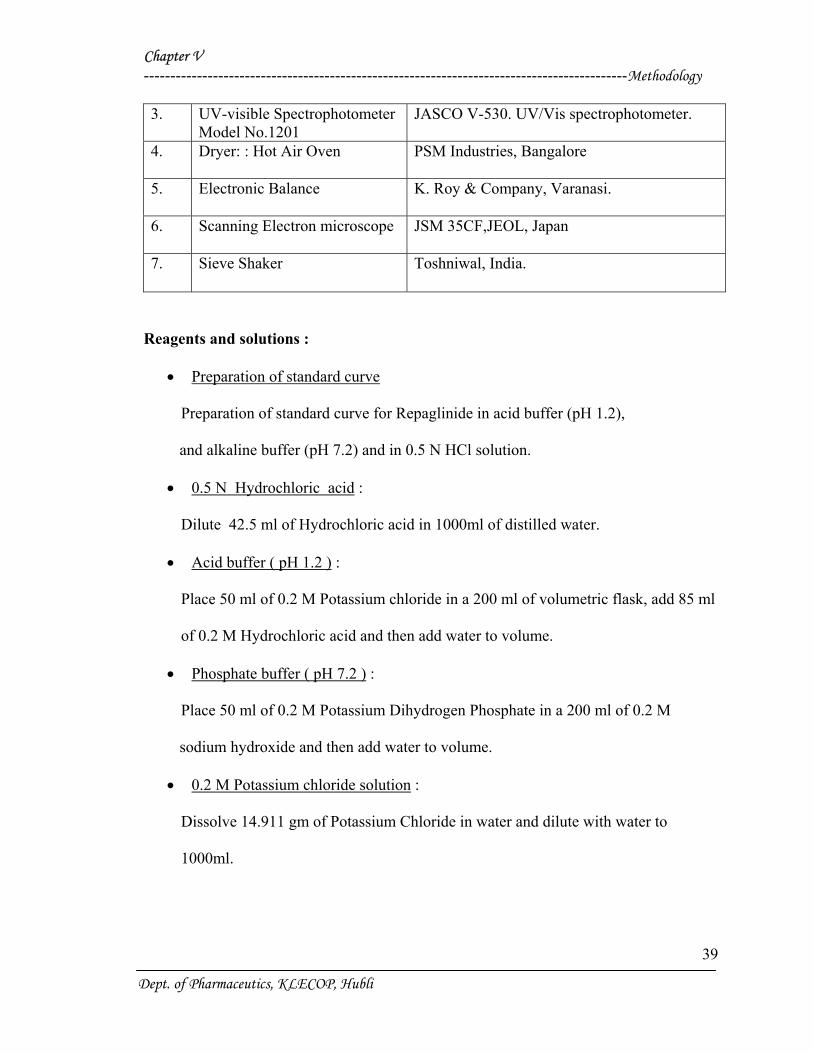
3. UV-visible Spectrophotometer Model No.1201
JASCO V-530. UV/Vis spectrophotometer.
4. Dryer: : Hot Air Oven
PSM Industries, Bangalore
5. Electronic Balance
K. Roy & Company, Varanasi.
6. Scanning Electron microscope
JSM 35CF,JEOL, Japan
7. Sieve Shaker
Toshniwal, India.
Reagents and solutions :
• Preparation of standard curve
Preparation of standard curve for Repaglinide in acid buffer (pH 1.2),
and alkaline buffer (pH 7.2) and in 0.5 N HCl solution.
• 0.5 N Hydrochloric acid :
Dilute 42.5 ml of Hydrochloric acid in 1000ml of distilled water.
• Acid buffer ( pH 1.2 ) :
Place 50 ml of 0.2 M Potassium chloride in a 200 ml of volumetric flask, add 85 ml
of 0.2 M Hydrochloric acid and then add water to volume.
• Phosphate buffer ( pH 7.2 ) :
Place 50 ml of 0.2 M Potassium Dihydrogen Phosphate in a 200 ml of 0.2 M
sodium hydroxide and then add water to volume.
• 0.2 M Potassium chloride solution :
Dissolve 14.911 gm of Potassium Chloride in water and dilute with water to
1000ml.
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
40
• 0.2 M Hydrochloric acid :
Dilute 17 ml of hydrochloric acid in 1000ml of distilled water.
• 0.2 M Potassium Dihydrogen Phosphate :
Dissolve 27.218gm of Potassium Dihydrogen Phosphate in water and dilute with
water to 1000ml.
• 0.2 M Sodium Hydroxide solution 31:
Dissolve sodium hydroxide in water to produce 40% to 60% w/v solution and allow
to stand. Taking precautions to avoid absorption of carbon dioxide, siphone off the
clear supernatant liquid and dilute with carbon dioxide free water a suitable volume
liquid to contain 8gm of sodium hydroxide in 1000 ml.
Formulation and preparation of microparticles
Formulation and preparing microparticles of Repaglinide using polymers Hydroxy
Propyl Methyl Cellulose by using Ionotropic Gelation Technique.
• Sodium alginate solution ( 2% ) :
Accurately weighed amount of sodium alginate (2 gm) mixed in the 100 ml of
distilled water.
• Calcium chloride solution ( 4% ) :
Accurately weighed amount of calcium chloride (2 gm) mixed in the 100 ml of
distilled water.
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
41
• Hydroxy Propyl Methyl Cellulose ( 1% ) :
Accurately weighed amount of Hydroxy Propyl Methyl Cellulose (1 gm) mixed in
the 100 ml of cold water.
• Hydroxy Propyl Methyl Cellulose (1.5%) :
Accurately weighed amount of Hydroxy Propyl Methyl Cellulose (1.5 gm) mixed
in the 100 ml of cold water.
• Hydroxy Propyl Methyl Cellulose ( 2%) :
Accurately weighed amount of Hydroxy Propyl Methyl Cellulose (2 gm) mixed in
the 100 ml of cold water.
• Chitosan ( 1%) :
Accurately weighed amount of Chitosan (1 gm) mixed in the 100 ml of 1% Acetic
acid solution.
• Chitosan (1.5%) :
Accurately weighed amount of Chitosan (1.5 gm) mixed in the 100 ml of 1%
Acetic acid solution.
• Chitosan (2%) :
Accurately weighed amount of Chitosan (2 gm) mixed in the 100 ml of 1% Acetic
acid solution.
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
42
METHODS :
1) Preformulation study13 :
Prior to the development of the dosage forms the preformulation study was carried
out. Hence Infrared spectra of the physical mixture of the drug and the polymers chosen
were taken. The infra-red spectra of the drug57 and polymers were also taken.
The application of infra-red spectroscopy lies more in the qualitative identification
of substances either in pure form or in the mixtures and as a tool in establishment of the
structure. Since I.R. is related to covalent bonds, the spectra can provide detailed
information about the structure of molecular compounds. In order to establish this point,
comparisons can be made between the spectrum of the substance and the drug.
The above discussions imply that infra-red data is helpful to confirm the identity of
the drug and to detect the interaction of the drug with the carriers.
2) Standard Plot for Repaglinide48 :
a) Standard Graph by using 0.5N HCl :
Accurately weighed 10 mg of Repaglinide was dissolved in 100 ml of 0.5 N HCl
buffer solution to form 100 µg/ml stock solution.
From this stock solution aliquots of 2.5 ml, 5 ml, 7.5 ml, 10 ml, 12.5 ml, 15 ml,
17.5 ml, 20 ml, 22.5 ml, 25 ml, were pipetted out into a series of 50 ml volumetric
flask and volume was made up to 50 ml in order to get a concentration ranging from
5-50µg/ml.
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
43
The absorbance of the resulting solution was then measured at 247nm using UV
spectrophotometer against respective parent solvent as a blank. The standard curve was
obtained by plotting absorbance V/s. concentration in µg/ml.
b) Phosphate Buffer (pH 7.2):
Accurately weighed 10 mg of Repaglinide was dissolved in 100 ml of 7.2 pH
buffer solution to form 100 µg/ml stock solution.
From this stock solution aliquots of 2.5 ml, 5 ml, 7.5 ml, 10 ml, 12.5 ml, 15 ml,
17.5 ml, 20 ml, 22.5 ml, 25 ml, were pipetted out into a series of 50 ml volumetric
flask and volume was made up to 50 ml in order to get a concentration ranging from
5-50µg/ml.
The absorbance of the resulting solution was then measured at 247nm using UV
spectrophotometer against respective parent solvent as a blank. The standard curve was
obtained by plotting absorbance V/s. concentration in µg/ml.
c) Acid Buffer (pH 1.2):
The above procedure was followed but instead of (pH7.2), phosphate buffer (pH
1.2) was used.
Preparation of Microparticles28:
In the present study, microparticles of Repaglinide were prepared by ionotropic
gelation technique. In this method weighed quantity of Repaglinide was added to 50 ml
sodium alginate solution and thoroughly mixed with a stirrer at 500 rpm. For the
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
44
formation of microparticles, 50 ml of this solution was extruded dropwise from a needle
into 100 ml aqueous calcium chloride solution and stirred at 100 rpm. After stirring for
10 minutes the obtained microparticles were washed with water and dried at 700 C for 2
hrs in an oven.
Three sets of microparticles were prepared. In the first set microparticles of
repaglinide were prepared using only hydroxyl propyl methyl cellulose in different
concentrations.
In the second set, microparticles of the drug were prepared by using only chitosan
in a different concentrations.
In the third set, microparticles of the drug were prepared in a combination of
polymers like hydroxyl propyl methyl cellulose and chitosan.
Sodium alginate + Drug + Polymer
Calcium chloride
Microparticles
Dept. of Pharmaceutics, KLECOP, Hubli
Chapter V -------------------------------------------------------------------------------------------Methodology
45
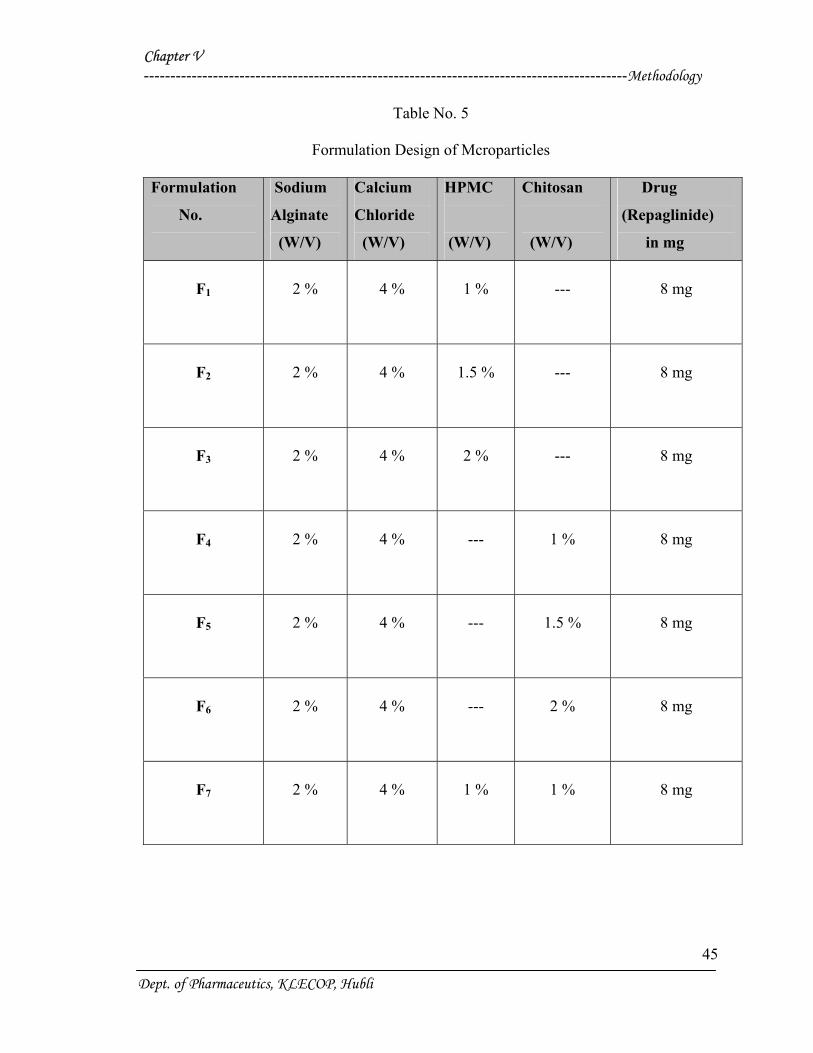
Table No. 5
Formulation Design of Mcroparticles
Formulation
No.
Sodium
Alginate
(W/V)
Calcium
Chloride
(W/V)
HPMC
(W/V)
Chitosan
(W/V)
Drug
(Repaglinide)
in mg
F1
2 %
4 %
1 %
---
8 mg
F2
2 %
4 %
1.5 %
---
8 mg
F3
2 %
4 %
2 %
---
8 mg
F4
2 %
4 %
---
1 %
8 mg
F5
2 %
4 %
---
1.5 %
8 mg
F6
2 %
4 %
---
2 %
8 mg
F7
2 %
4 %
1 %
1 %
8 mg
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
46
EVALUATION PARAMETERS :
Particle size determination49, 50 :
The particle size of a pharmaceutical substance is strictly maintained in order to get
optimal biological activity.
Methods to estimate particle size are :
a. Optical Microscopy
b. Sieving Method
c. Sedimentation Method
d. Elutriation Method
e. Centrifugal defractometry
f. Permeability Method
g. Light scattering Method
Table No.6
Common techniques for measuring fine particles of various sizes
No. Technique Particles sizes in (µm)
1. Optical Microscopy 1-100 µm
2. Sieving >50 µm
3. Sedimentation >1 µm
4. Elutriation 1-50 µm
5. Centrifugal <50 µm
6. Permeability >1 µm
7. Light scattering 0.5-50 µm
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
47
Sieving Method :
Particles having size range between 50 and 1500 µm are estimated by sieving
method. In this method the size is expressed as dsieve , which describes the diameter of a
sphere that passes through the sieve aperture as the assymmetric particle. This method
directly gives weight distribution. The sieving method finds application in dosage form
development of tablets and capsules.
Sieves for pharmaceutical testing are constructed from wire cloth with square
meshes, woven from wire of brass, bronze, stainless steel or any other suitable material.
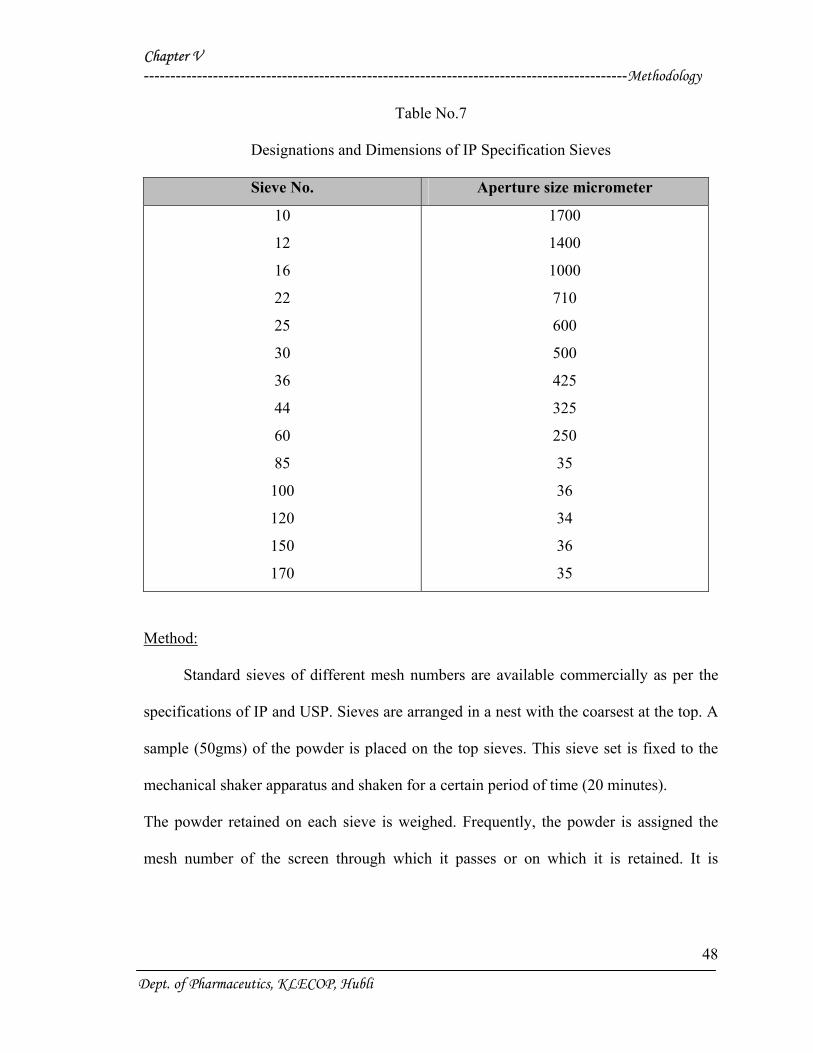
Standard sieves and their dimensions as per IP are given as follows :
Dept. of Pharmaceutics, KLECOP, Hubli
Chapter V -------------------------------------------------------------------------------------------Methodology
48
Table No.7
Designations and Dimensions of IP Specification Sieves
Sieve No. Aperture size micrometer
10
12
16
22
25
30
36
44
60
85
100
120
150
170
1700
1400
1000
710
600
500
425
325
250
35
36
34
36
35
Method:
Standard sieves of different mesh numbers are available commercially as per the
specifications of IP and USP. Sieves are arranged in a nest with the coarsest at the top. A
sample (50gms) of the powder is placed on the top sieves. This sieve set is fixed to the
mechanical shaker apparatus and shaken for a certain period of time (20 minutes).
The powder retained on each sieve is weighed. Frequently, the powder is assigned the
mesh number of the screen through which it passes or on which it is retained. It is
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
49
expressed in terms of arithmetic or geometric mean of the two sieves.
Flow Properties49, 50:
Irregular flow of powder from the hopper produces tablets and capsules with
nonuniform weights. Flow property depends on particle size, shape, porosity and density
of the powder.
Angle of Repose :
The flow characteristics are measured by angle of repose. Improper flow is due to
frictional forces between the particles. These forces are quantified by angle of repose.
Angle of repose is defined as the maximum angle possible between the surface of the pile
of the powder and the horizontal plane. The flow of powder and the angle of repose is
depicted in following fig. By definition :
tan θ = h / r
θ = tan-1 (h / r)
Where, h = height of pile
r = radius of the base of the pile
θ = angle of repose
The lower the angle of repose, the better the flow property. Rough and irregular
surface of particles gives higher angle of repose. Decreased in the particle size leads to a
higher angle of repose.
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
50
Method :
A glass funnel is held in place with a clamp on a ring support over a glass plate.
The glass plate is placed on a stand. Approximately 100 g of particles is transfered into
funnel keeping the orifice of the funnel blocked by the lower thumb. As the thumb is
removed, the particles are emptied from funnel, and the angle of repose is determined by
above mentioned formula.
Table No.8
Relation Between Angle of Repose and Flow of the Particles
Angle of repose (θ)
(degrees)
Flow
<25
25-30
30-40
>40
Excellent
Good
Passable
Very poor
Drug Entrapment Efficiency28, 56 :
Drug entrapment efficiency of repaglinide was performed by accurately weighing
100 mg of microparticles and suspended in 100 ml of simulated intestinal fluid of pH
7.2±0.1 and it was kept for 24 hrs. Next day it was stirred for 15 mins, and subjected for
filtration. After suitable dilution, Repaglinide content in the filtrate was analyzed
spectrophotometrically at 247 nm using Shimadzu 1201 UV-visible spectrophotometer.
The absorbance found from the UV-spectrophotometer was plotted on the standard
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
51
curve to get the concentration of the entrapped drug. Calculating this concentration with
the dilution factor we get the percentage drug encapsulated in microparticles.
In-vitro Dissolution Studies :
A drug is expected to release from the solid dosage forms (granules, tablets,
capsules etc) and immediately go into molecular solution. This process is called as
Dissolution50.
Drug Release Studies :
The method specified in USP for the drug release study was followed.
Apparatus :
USP XXIII dissolution test apparatus employing the round bottom dissolution
vessel and rotating basket assembly.
Acid Stage :
900 ml of simulated gastric fluid TS (acid buffer, pH 1.2 without enzymes).
Buffer stage :
900 ml of pH 7.2 and intestinal fluid Ts (phosphate buffer).
Time :
Acid stage : pH : 1.2 -2 hrs.
Buffer stage : pH :7.2: 6 hrs.
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
52
Procedure :
In-vitro release profile of the microparticles was evaluated using rotating basket
dissolution apparatus. 900 ml of acid buffer (pH1.2), and phosphate buffer (pH7.2)
maintained at 37±0.5 0C were used as dissolution medias respectively, and the basket was
rotated at a constant speed of 50 rpm. Accurately weighed amount of microparticles
equivalent to 200 mg of drug were placed in the baskets.
Aliquotes of samples were withdrawn at the interval of 1 hour for pH 1.2 and 2 hrs
for 7.2 pH. The samples withdrawn were filtered, diluted suitably and analyzed at 247 nm
spectrophotometrically for drug release51.
Kinetic treatment :
The data obtained from the in-vitro dissolution studies was subjected for kinetic
treatment to obtain the order of release and best fit model for the formulations by using
PCP-Disso-V2 software.
Scanning electron microscopy:
Procedure :
Morphology details of the specimens were determined by using a scanning electron
microscope (SEM), Model JSM 35CF, JEOL, Japan.
The samples were dried thoroughly in vacuum desicator before mounting on brass
specimen studies. The samples were mounted on specimen studies using double sided
adhesive tape, and gold-palladium alloy of 120Ao kness was coated on the sample using
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V -------------------------------------------------------------------------------------------Methodology
53
sputter coating unit (Model E5 100 Polaron U.K.) in an Argon ambient of 8-10 pascal
with plasma voltage about 20 MA. The sputtering was done for nearly 3 minutes to
obtain uniform coating on the sample to enable good quality SEM images. The SEM was
operated at low accelerating voltage of about 15 KV with load current of about 80 MA.
The condenser lens position was maintained between 4.4--5.1. The objectives lens
aperture has a diameter of 240 microns and the working distance WD = 39 mm52, 53.
Accelerated Stability Studies:
The formulations were stored in a oven at 37 ± 10C and 60 ± 10C for a period of six
weeks. The samples were analyzed for drug content every week by spectrophotometer at
247 nm54, 55.
Dept. of Pharmaceutics, KLECOP, Hubli

Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
53

Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
54
Results and Discussion:
Microparticles of repaglinide were prepared by ionotropic gelation technique and
various evaluation parameters were assessed, with a view to obtain oral controlled release
of Repaglinide.
In the present work, total seven formulations were prepared and the detailed
composition is shown in Table No. 5. The prepared microparticles were then subjected to
granulometric study, angle of repose, scanning electron microscopy, drug entrapment
efficiency, in-vitro dissolution and stability studies.
1. Standard curve of Repaglinide:
A standard calibration curve for the drug was obtained by measuring absorbance at
247 nm, and by plotting the graph of absorbance V/s concentration. Table No.9 shows the
absorbance readings of repaglinide in triplicate 0.5 N HCl, pH 1.2, pH 7.2 between 5-50
µg/ml concentrations. The standard plots of repaglinide are shown in graph No.1, 2, and 3.
Estimation of drug content and in-vitro drug release studies are based on this
standard curve.
2. Preformulation Study:
To check the compatibility of the drug with various polymers, IR spectra of drugs,
polymers and combination of the drug and polymers were taken. The IR spectra of the
drug, polymers and their combinations are shown in Spectra No.1 to 5.
The characteristics absorption peaks of repaglinide were obtained at1687.3cm-1,
2935.03 cm-1, 1217.12 cm-1, and 3308.38cm-1. The IR spectras of the drug and polymer

Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
55
combinations were compared with the spectra of pure drug and individual polymers. The
principle peaks obtained for the combinations were almost similar to that of the drug.
The details of IR spectra are mentioned in Table No.10.
The IR spectra of the Drug-HPMC, Drug –chitosan, and Drug-Sodium alginate, did
not show any changes. The possibility of interaction was ruled out as there was no major
shift in the absorption bands of drug and the formulations as shown in Spectra No.1 to 5.
Evaluation parameters:
Granulation study:
In the granulometric study it is observed from the Table No.11 that about 64 % to
90 % and 40 % to 55 % percent of microparticles were of 16 and 20 mesh size, which
proves the flexibility of the method. It is observed that, with the increase in the
concentration of HPMC maximum amount of microparticles of desired size were
obtained and with the increase in the concentration of chitosan the distribution of particle
size shifts to the higher sieve size due to increase in viscosity of the medium.
Flow Property:
The flow property of the prepared formulations was checked by the method, angle
of repose. Acceptable range of angle of repose is 22061' to 31060'. All the formulations
showed an angle of repose within the range as shown in Table No.12.
Formulations F1 to F7 showed an angle of repose in the acceptable range, which
indicates a good flow property.

Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
56
Drug Entrapment Efficiency:
The drug entrapment efficiency of all the formulations were in the range between
78.62 % to 91.25 %. The results of drug entrapment efficiency are shown in Table No.13.
Drug entrapment efficiency of microparticles increases with increase in
concentration of HPMC and chitosan.
In-vitro Dissolution Studies:
Dissolution studies of all the formulations were carried out using dissolution tester
USP XXIII.
The dissolution studies were conducted by using two different dissolution medias,
PH 1.2 and PH 7.2.
The data obtained in the in-vitro dissolution studies were grouped according to
modes of data treatment as follows :-
Cumulative percent drug release V/s. Time (Zero-order).
Cumulative percent drug retained V/s. Square root of Time (Higuchi Matrix Model).
Log Cumulative percent drug retained V/s. Time (First-order).
Cumulative percent drug release in (mg) V/s. Time (Krosmeyer-Peppas Model).
The results of the in-vitro dissolution studies of formulations F1 to F7 are shown in
table 14 to 20. The plots of Cumulative percentage drug release V/s. Time, Cumulative
percent drug retained V/s. Time, Log Cumulative percent drug retained V/s. Time and
Cumulative percent drug release in (mg) V/s. Time were drawn and represented
graphically as shown in Graph No. 4 to 15, respectively.
The formulation F1, F2 and F3 Containing 1%, 1.5%, and 2% Hydroxy Propyl
Methyl Cellulose respectively showed a release of 91.99%, 81.66% and 71.66% after 12

Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
57
hours. This shows that more sustained release was observed with the increase in
percentage of Hydroxy Propyl Methyl Cellulose.
The formulation F4, F5 and F6 Containing 1%, 1.5%, and 2% Chitosan respectively
showed a release of 92.11%, 81.93% and 81.79% after 12 hours. This shows that more
sustained release was observed with the increase in percentage of Chitosan.
The formulation F7 containing both 1% HPMC and 1% chitosan showed a release
of 71.88%. This shows that the particles formulated with HPMC and Chitosan prolongs
the release but without satisfactory surface characteristics.
The formulations F1, F2 and F3 containing 1%, 1.5%, and 2% Hydroxy Propyl
Methyl Cellulose respectively showed a release that more sustained release was observed
with the increase in percentage of HPMC after 12 hours. This indicates that the release
rate is further retarded due to addition and in percentage of Hydroxy Propyl Methyl
Cellulose because of the strong bonds between the HPMC and sodium alginate. As the
percentage of HPMC increased the release was further sustained.
The formulations F4, F5 and F6 containing 1%, 1.5%, and 2% chitosan respectively
showed a release that more sustained release was observed with the increase in
percentage of Chitosan after 12 hours. This indicates that the release rate is further
retarded due to addition and in percentage of chitosan because of the strong bonds
between the chitosan and sodium alginate. As the percentage of chitosan increased the
release was further sustained.
Further these drug releases were subjected for mathematical treatment to check
weather the release is following first order or zero-order kinetics.
The co-efficient of correlation values are shown in table No.13

Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
58
The values of co-efficient of correlation were found to be best fitted to Krosmeyer-
Peppas model and Higuchi model.
The calculated values of various kinetic models are shown in table No. 22.
The values of diffusion co-efficient (n) for formulations F1 to F7 are shown to be
0.453, 0.499, 0.547, 0.418, 0.515, 0.469, 0.596 respectively which indicates that the
release of drug occurs by diffusion following Fickian transport and Anomalous
mechanism.
Scanning Electron Microscopy:
Morphology of the microparticles were investigated by Scanning electron
microscopy. The photographs of formulations taken by scanning electron microscope
are shown in the figure No.1 and 2.
Microparticles of formulation F1 were approximately spherical and their surface
was rough giving them a sandy appearance.
The surface characteristics observed for Formulation F4 shows oval or slightly
spherical structure with smooth appearance.
From the photographic observation it can be stated that bridging and dense nature
of formulation indicated to retard the release of Repaglinide.
Stability study :
Among the seven formulations prepared F1 and F4 were used, which showed the
best release from in-vitro dissolution data selected for stability studies.
Stability study was carried out for the formulations F1 and F4 at 400C ± 10C RH
for a period of 45 days. The samples were analyzed for drug content at different time
intervals, and it is evident that there were slight changes in the content of drug as shown
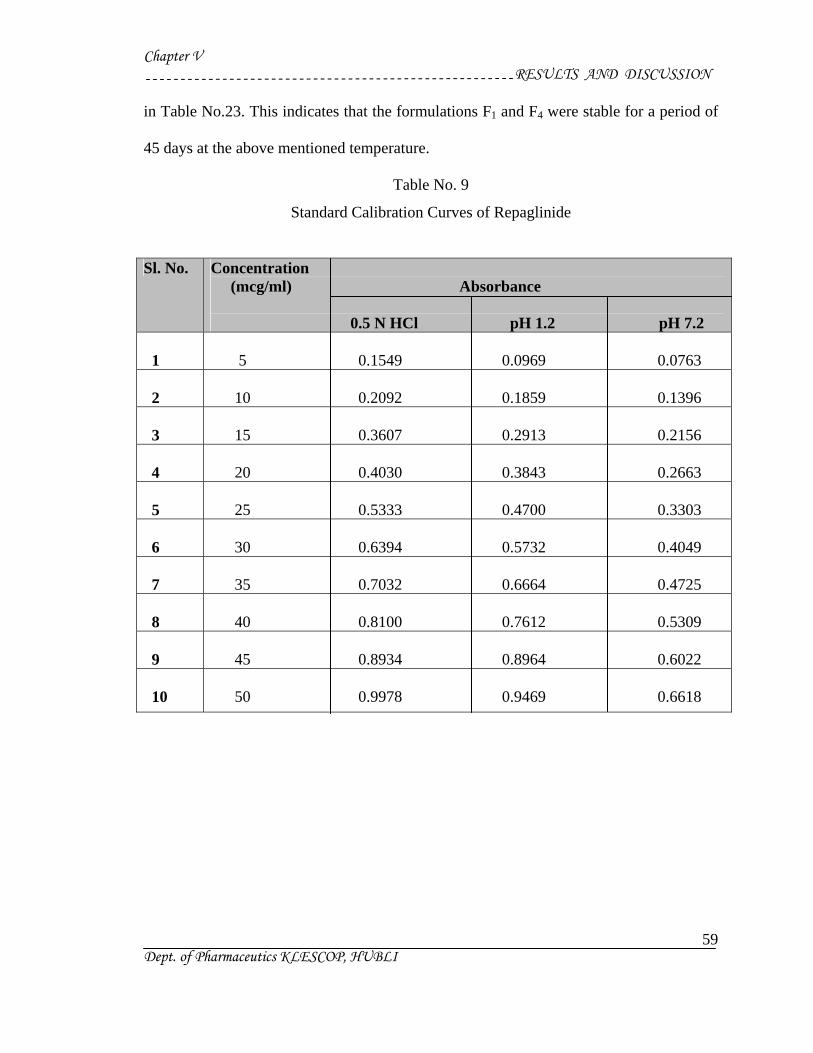
Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
59
in Table No.23. This indicates that the formulations F1 and F4 were stable for a period of
45 days at the above mentioned temperature.
Table No. 9
Standard Calibration Curves of Repaglinide
Sl. No. Concentration (mcg/ml) Absorbance 0.5 N HCl pH 1.2 pH 7.2
1
5 0.1549 0.0969 0.0763
2
10 0.2092 0.1859 0.1396
3
15 0.3607 0.2913 0.2156
4
20 0.4030 0.3843 0.2663
5
25 0.5333 0.4700 0.3303
6
30 0.6394 0.5732 0.4049
7
35 0.7032 0.6664 0.4725
8
40 0.8100 0.7612 0.5309
9
45 0.8934 0.8964 0.6022
10
50 0.9978 0.9469 0.6618
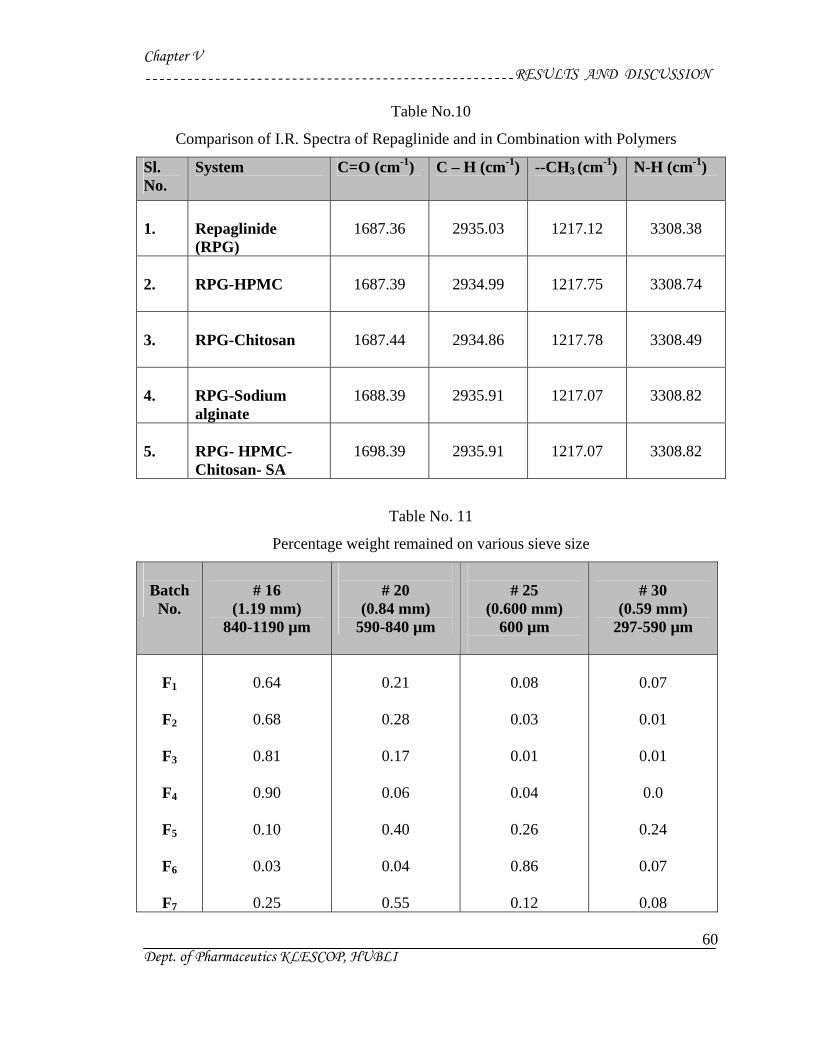
Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
60
Table No.10
Comparison of I.R. Spectra of Repaglinide and in Combination with Polymers
Sl. No.
System C=O (cm-1) C – H (cm-1) --CH3 (cm-1) N-H (cm-1)
1.
Repaglinide (RPG)
1687.36
2935.03
1217.12
3308.38
2.
RPG-HPMC
1687.39
2934.99
1217.75
3308.74
3.
RPG-Chitosan
1687.44
2934.86
1217.78
3308.49
4.
RPG-Sodium alginate
1688.39
2935.91
1217.07
3308.82
5.
RPG- HPMC- Chitosan- SA
1698.39
2935.91
1217.07
3308.82
Table No. 11
Percentage weight remained on various sieve size
Batch
No.
# 16
(1.19 mm) 840-1190 µm
# 20
(0.84 mm) 590-840 µm
# 25
(0.600 mm) 600 µm
# 30
(0.59 mm) 297-590 µm
F1
F2
F3
F4
F5
F6
F7
0.64
0.68
0.81
0.90
0.10
0.03
0.25
0.21
0.28
0.17
0.06
0.40
0.04
0.55
0.08
0.03
0.01
0.04
0.26
0.86
0.12
0.07
0.01
0.01
0.0
0.24
0.07
0.08
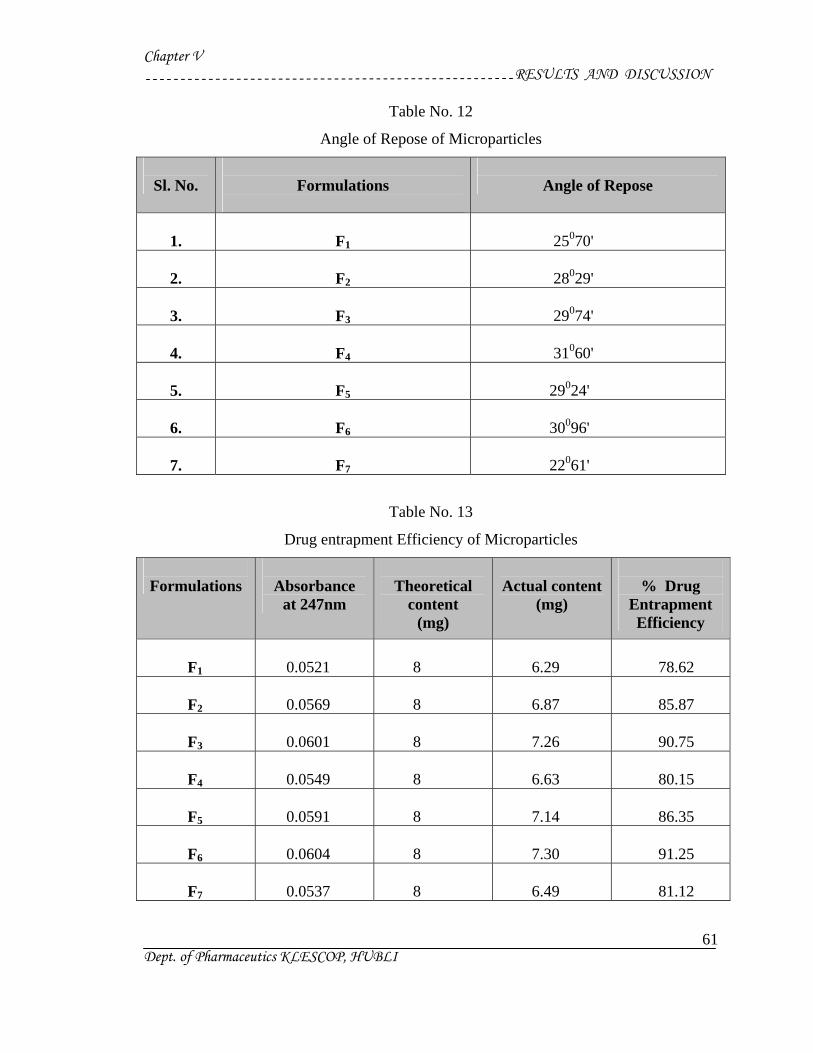
Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
61
Table No. 12
Angle of Repose of Microparticles
Sl. No.
Formulations
Angle of Repose
1.
F1
25070'
2.
F2
28029'
3.
F3
29074'
4.
F4
31060'
5.
F5
29024'
6.
F6
30096'
7.
F7
22061'
Table No. 13
Drug entrapment Efficiency of Microparticles
Formulations
Absorbance
at 247nm
Theoretical
content (mg)
Actual content
(mg)
% Drug
Entrapment Efficiency
F1
0.0521
8
6.29
78.62
F2
0.0569
8
6.87
85.87
F3
0.0601
8
7.26
90.75
F4
0.0549
8
6.63
80.15
F5
0.0591
8
7.14
86.35
F6
0.0604
8
7.30
91.25
F7
0.0537
8
6.49
81.12

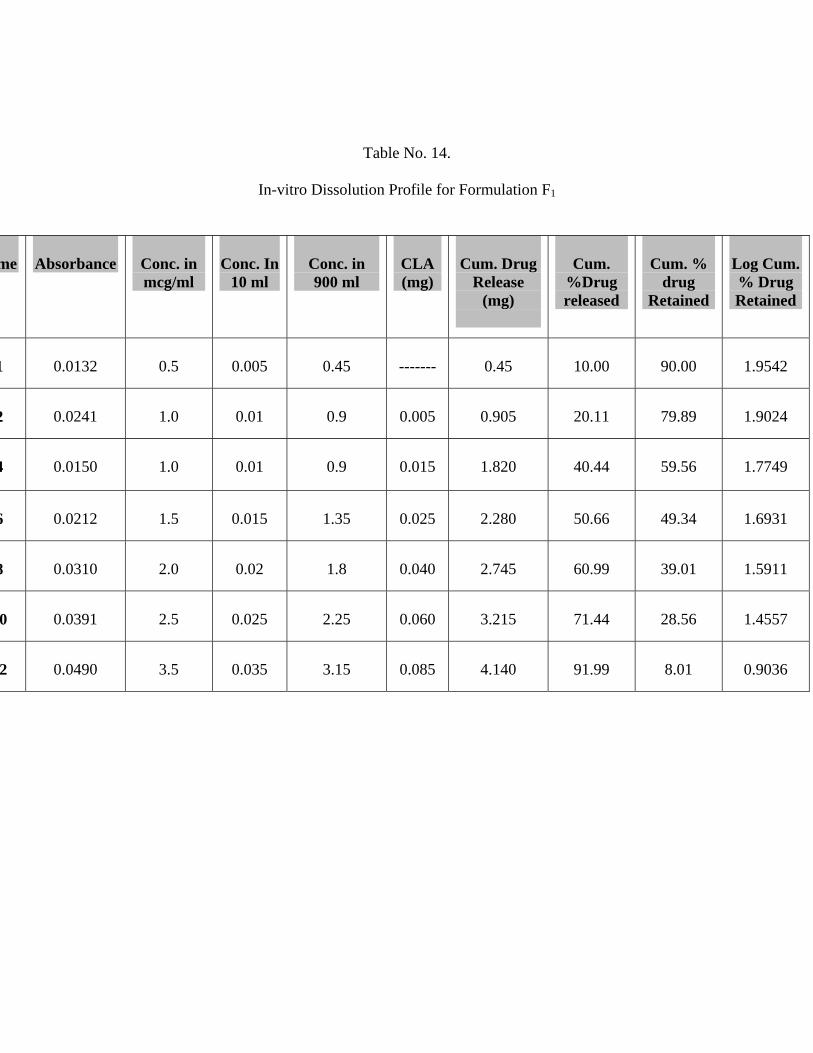
Table No. 14.
In-vitro Dissolution Profile for Formulation F1
me
Absorbance
Conc. in mcg/ml
Conc. In
10 ml
Conc. in 900 ml
CLA (mg)
Cum. Drug
Release (mg)
Cum.
%Drug released
Cum. %
drug Retained
Log Cum. % Drug Retained
1
0.0132
0.5
0.005
0.45
-------
0.45
10.00
90.00
1.9542
2
0.0241
1.0
0.01
0.9
0.005
0.905
20.11
79.89
1.9024
4
0.0150
1.0
0.01
0.9
0.015
1.820
40.44
59.56
1.7749
6
0.0212
1.5
0.015
1.35
0.025
2.280
50.66
49.34
1.6931
8
0.0310
2.0
0.02
1.8
0.040
2.745
60.99
39.01
1.5911
0
0.0391
2.5
0.025
2.25
0.060
3.215
71.44
28.56
1.4557
2
0.0490
3.5
0.035
3.15
0.085
4.140
91.99
8.01
0.9036
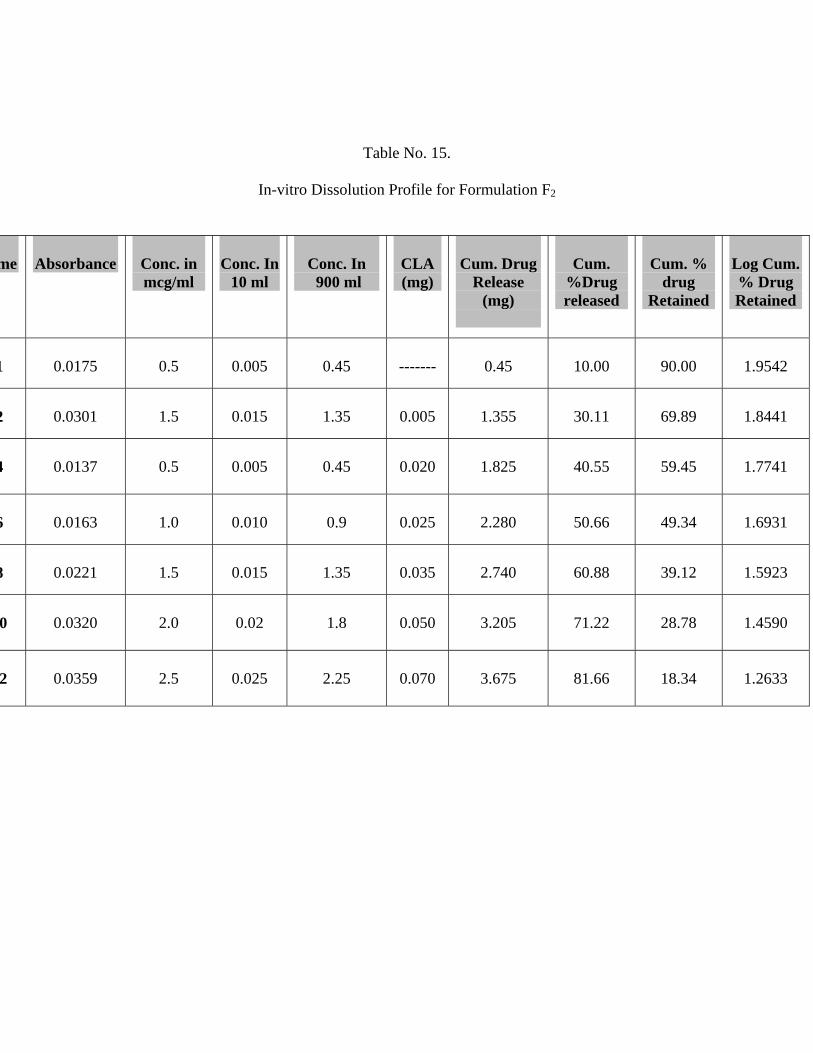
Table No. 15.
In-vitro Dissolution Profile for Formulation F2
me
Absorbance
Conc. in mcg/ml
Conc. In
10 ml
Conc. In 900 ml
CLA (mg)
Cum. Drug
Release (mg)
Cum.
%Drug released
Cum. %
drug Retained
Log Cum. % Drug Retained
1
0.0175
0.5
0.005
0.45
-------
0.45
10.00
90.00
1.9542
2
0.0301
1.5
0.015
1.35
0.005
1.355
30.11
69.89
1.8441
4
0.0137
0.5
0.005
0.45
0.020
1.825
40.55
59.45
1.7741
6
0.0163
1.0
0.010
0.9
0.025
2.280
50.66
49.34
1.6931
8
0.0221
1.5
0.015
1.35
0.035
2.740
60.88
39.12
1.5923
0
0.0320
2.0
0.02
1.8
0.050
3.205
71.22
28.78
1.4590
2
0.0359
2.5
0.025
2.25
0.070
3.675
81.66
18.34
1.2633
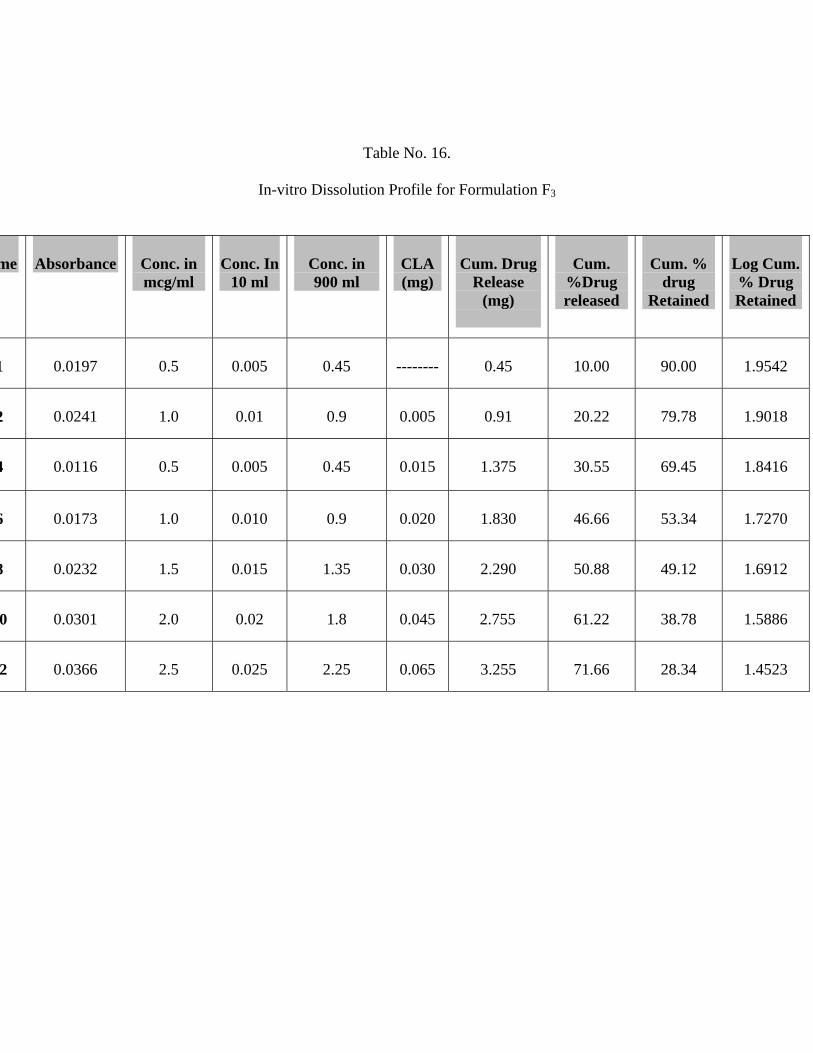
Table No. 16.
In-vitro Dissolution Profile for Formulation F3
me
Absorbance
Conc. in mcg/ml
Conc. In
10 ml
Conc. in 900 ml
CLA (mg)
Cum. Drug
Release (mg)
Cum.
%Drug released
Cum. %
drug Retained
Log Cum. % Drug Retained
1
0.0197
0.5
0.005
0.45
--------
0.45
10.00
90.00
1.9542
2
0.0241
1.0
0.01
0.9
0.005
0.91
20.22
79.78
1.9018
4
0.0116
0.5
0.005
0.45
0.015
1.375
30.55
69.45
1.8416
6
0.0173
1.0
0.010
0.9
0.020
1.830
46.66
53.34
1.7270
8
0.0232
1.5
0.015
1.35
0.030
2.290
50.88
49.12
1.6912
0
0.0301
2.0
0.02
1.8
0.045
2.755
61.22
38.78
1.5886
2
0.0366
2.5
0.025
2.25
0.065
3.255
71.66
28.34
1.4523
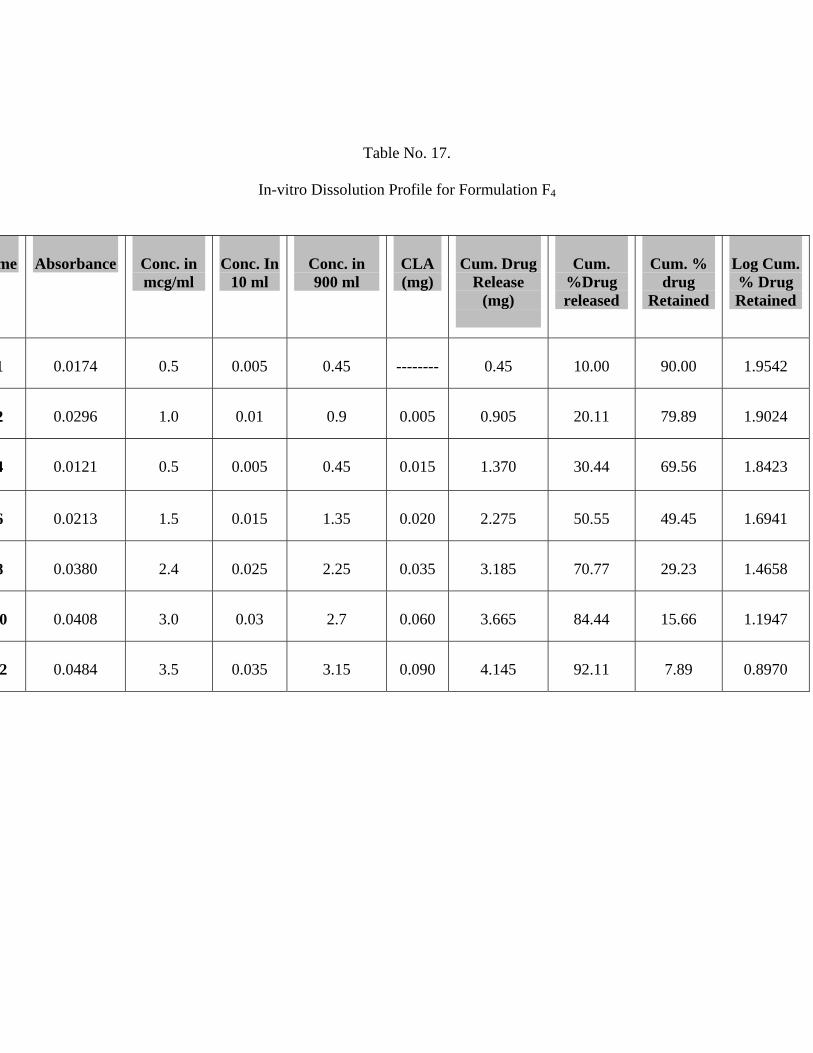
Table No. 17.
In-vitro Dissolution Profile for Formulation F4
me
Absorbance
Conc. in mcg/ml
Conc. In
10 ml
Conc. in 900 ml
CLA (mg)
Cum. Drug
Release (mg)
Cum.
%Drug released
Cum. %
drug Retained
Log Cum. % Drug Retained
1
0.0174
0.5
0.005
0.45
--------
0.45
10.00
90.00
1.9542
2
0.0296
1.0
0.01
0.9
0.005
0.905
20.11
79.89
1.9024
4
0.0121
0.5
0.005
0.45
0.015
1.370
30.44
69.56
1.8423
6
0.0213
1.5
0.015
1.35
0.020
2.275
50.55
49.45
1.6941
8
0.0380
2.4
0.025
2.25
0.035
3.185
70.77
29.23
1.4658
0
0.0408
3.0
0.03
2.7
0.060
3.665
84.44
15.66
1.1947
2
0.0484
3.5
0.035
3.15
0.090
4.145
92.11
7.89
0.8970
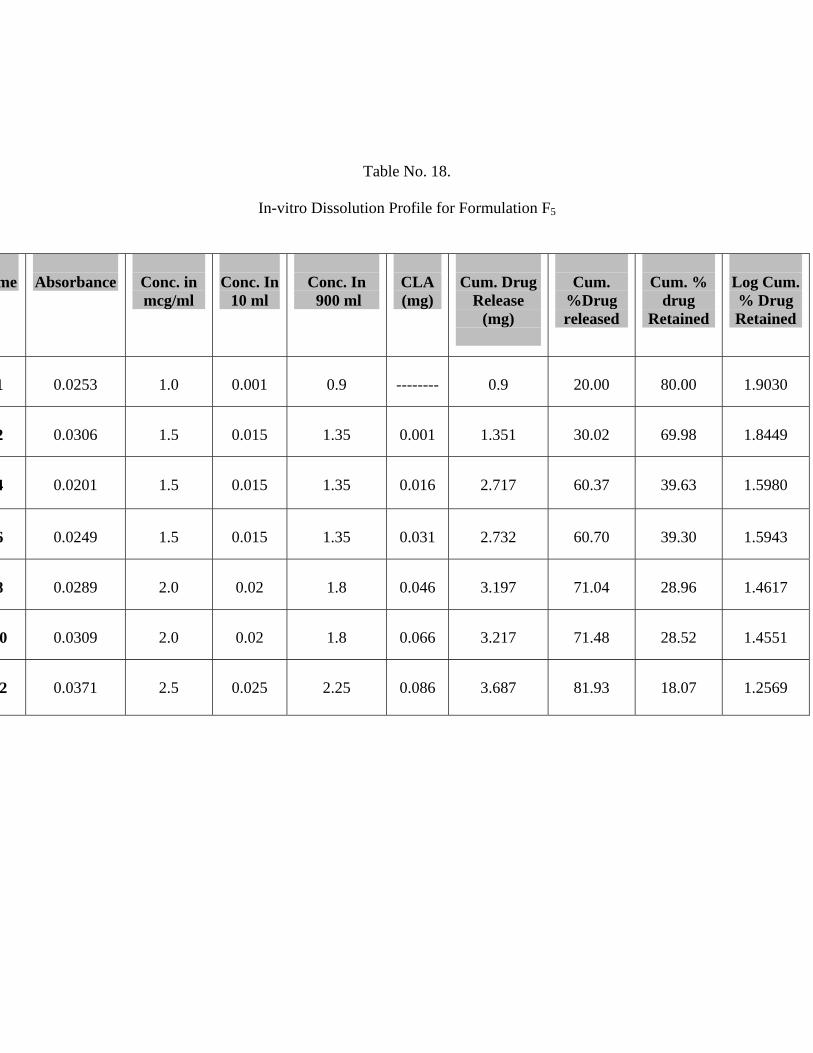
Table No. 18.
In-vitro Dissolution Profile for Formulation F5
me
Absorbance
Conc. in mcg/ml
Conc. In
10 ml
Conc. In 900 ml
CLA (mg)
Cum. Drug
Release (mg)
Cum.
%Drug released
Cum. %
drug Retained
Log Cum. % Drug Retained
1
0.0253
1.0
0.001
0.9
--------
0.9
20.00
80.00
1.9030
2
0.0306
1.5
0.015
1.35
0.001
1.351
30.02
69.98
1.8449
4
0.0201
1.5
0.015
1.35
0.016
2.717
60.37
39.63
1.5980
6
0.0249
1.5
0.015
1.35
0.031
2.732
60.70
39.30
1.5943
8
0.0289
2.0
0.02
1.8
0.046
3.197
71.04
28.96
1.4617
0
0.0309
2.0
0.02
1.8
0.066
3.217
71.48
28.52
1.4551
2
0.0371
2.5
0.025
2.25
0.086
3.687
81.93
18.07
1.2569
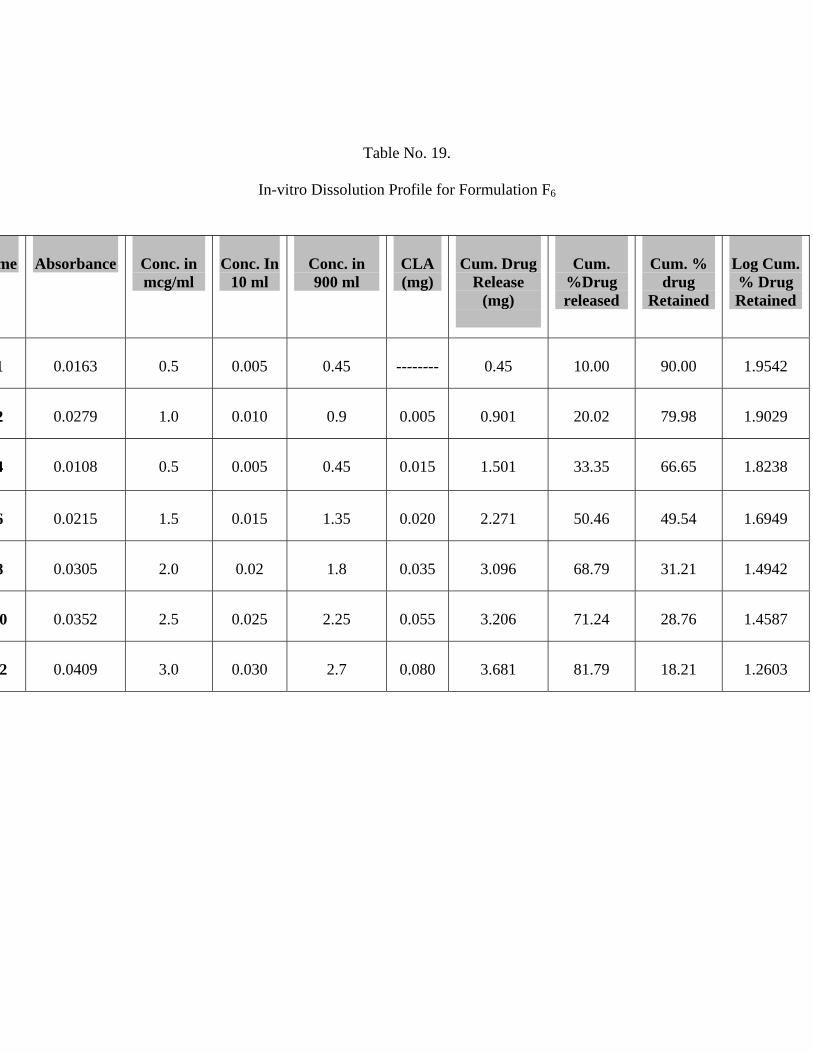
Table No. 19.
In-vitro Dissolution Profile for Formulation F6
me
Absorbance
Conc. in mcg/ml
Conc. In
10 ml
Conc. in 900 ml
CLA (mg)
Cum. Drug
Release (mg)
Cum.
%Drug released
Cum. %
drug Retained
Log Cum. % Drug Retained
1
0.0163
0.5
0.005
0.45
--------
0.45
10.00
90.00
1.9542
2
0.0279
1.0
0.010
0.9
0.005
0.901
20.02
79.98
1.9029
4
0.0108
0.5
0.005
0.45
0.015
1.501
33.35
66.65
1.8238
6
0.0215
1.5
0.015
1.35
0.020
2.271
50.46
49.54
1.6949
8
0.0305
2.0
0.02
1.8
0.035
3.096
68.79
31.21
1.4942
0
0.0352
2.5
0.025
2.25
0.055
3.206
71.24
28.76
1.4587
2
0.0409
3.0
0.030
2.7
0.080
3.681
81.79
18.21
1.2603
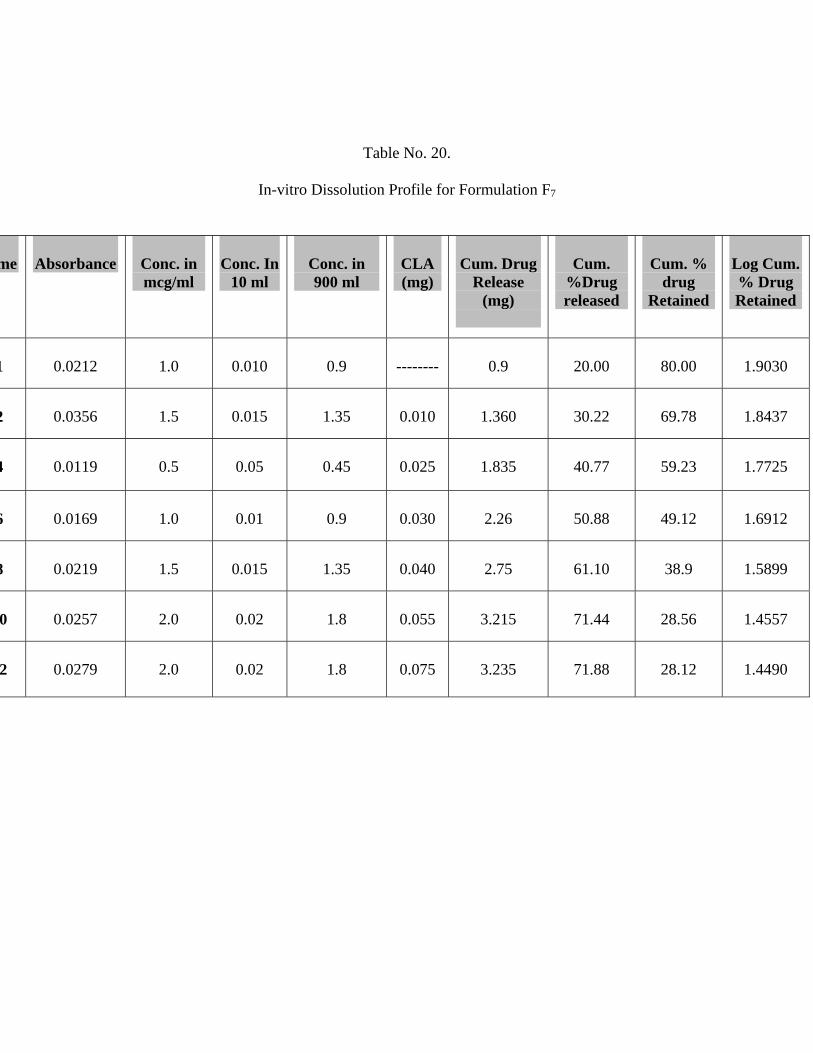
Table No. 20.
In-vitro Dissolution Profile for Formulation F7
me
Absorbance
Conc. in mcg/ml
Conc. In
10 ml
Conc. in 900 ml
CLA (mg)
Cum. Drug
Release (mg)
Cum.
%Drug released
Cum. %
drug Retained
Log Cum. % Drug Retained
1
0.0212
1.0
0.010
0.9
--------
0.9
20.00
80.00
1.9030
2
0.0356
1.5
0.015
1.35
0.010
1.360
30.22
69.78
1.8437
4
0.0119
0.5
0.05
0.45
0.025
1.835
40.77
59.23
1.7725
6
0.0169
1.0
0.01
0.9
0.030
2.26
50.88
49.12
1.6912
8
0.0219
1.5
0.015
1.35
0.040
2.75
61.10
38.9
1.5899
0
0.0257
2.0
0.02
1.8
0.055
3.215
71.44
28.56
1.4557
2
0.0279
2.0
0.02
1.8
0.075
3.235
71.88
28.12
1.4490
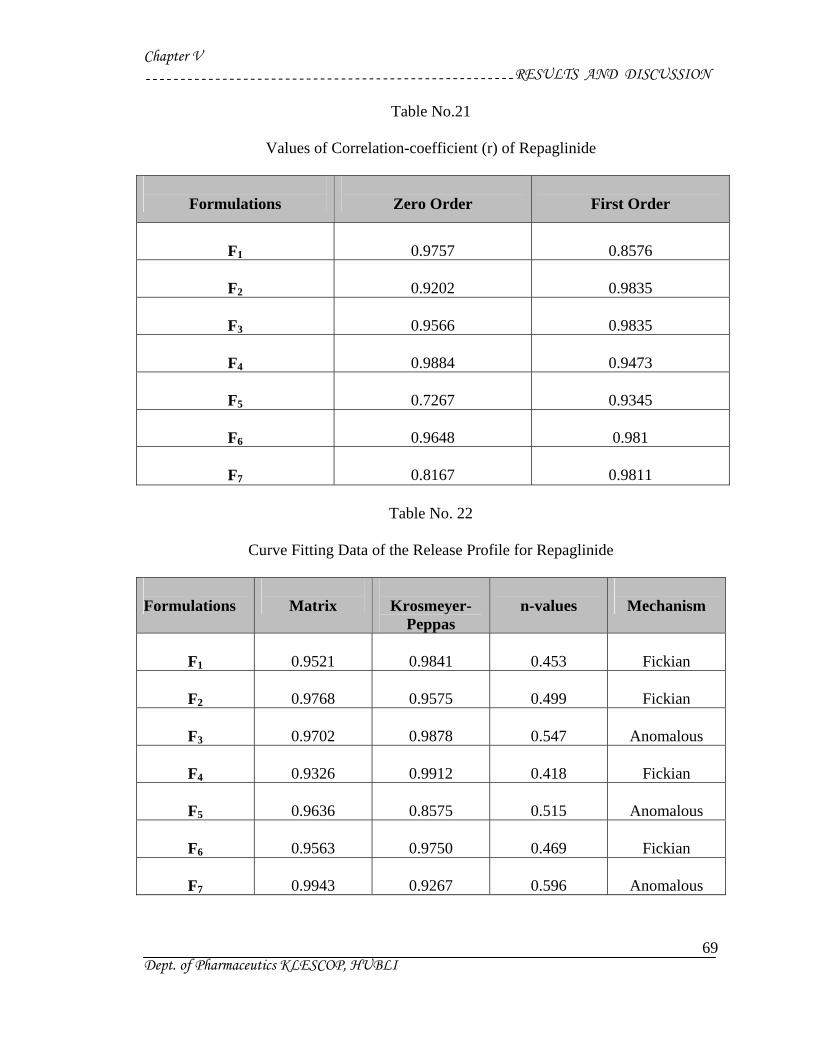
Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
69
Table No.21
Values of Correlation-coefficient (r) of Repaglinide
Formulations
Zero Order
First Order
F1
0.9757
0.8576
F2
0.9202
0.9835
F3
0.9566
0.9835
F4
0.9884
0.9473
F5
0.7267
0.9345
F6
0.9648
0.981
F7
0.8167
0.9811
Table No. 22
Curve Fitting Data of the Release Profile for Repaglinide Formulations
Matrix
Krosmeyer-
Peppas
n-values
Mechanism
F1
0.9521
0.9841
0.453
Fickian
F2
0.9768
0.9575
0.499
Fickian
F3
0.9702
0.9878
0.547
Anomalous
F4
0.9326
0.9912
0.418
Fickian
F5
0.9636
0.8575
0.515
Anomalous
F6
0.9563
0.9750
0.469
Fickian
F7
0.9943
0.9267
0.596
Anomalous
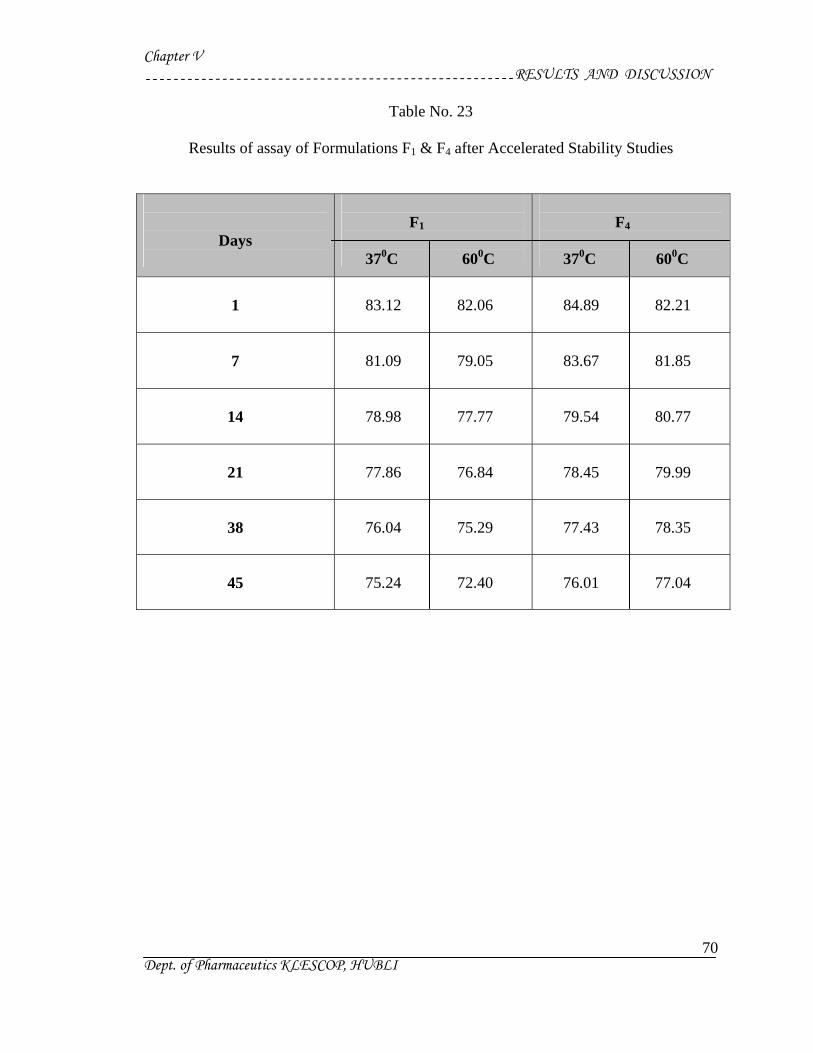
Chapter V RESULTS AND DISCUSSION
Dept. of Pharmaceutics KLESCOP, HUBLI
70
Table No. 23
Results of assay of Formulations F1 & F4 after Accelerated Stability Studies
Days
F1 370C 600C
F4 370C 600C
1
83.12 82.06
84.89 82.21
7
81.09 79.05
83.67 81.85
14
78.98 77.77
79.54 80.77
21
77.86 76.84
78.45 79.99
38
76.04 75.29
77.43 78.35
45
75.24 72.40
76.01 77.04
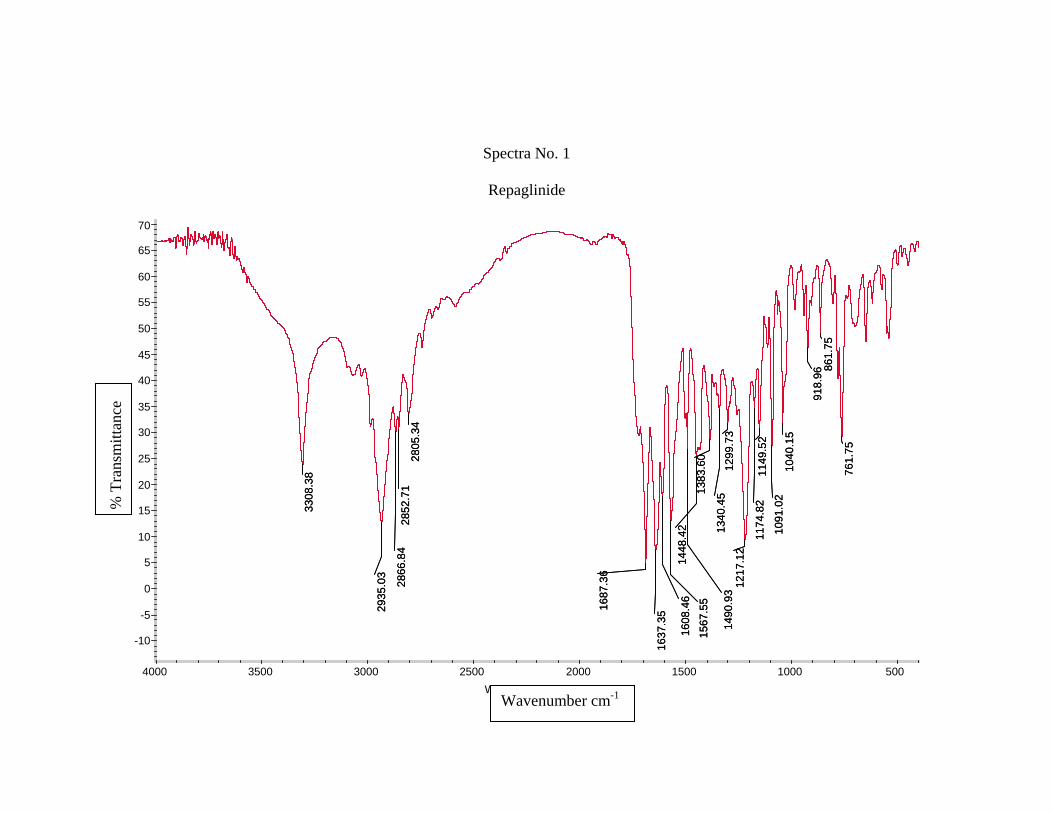
Spectra No. 1
Repaglinide
761.
75
861.
7591
8.96
1040
.15
1091
.02
1149
.52
1174
.82
1217
.12
1299
.73
1340
.4513
83.6
0
1448
.42
1490
.93
1567
.55
1608
.46
1637
.3516
87.3
6
2805
.34
2852
.71
2866
.84
2935
.03
3308
.38 76
1.75
861.
7591
8.96
1040
.15
1091
.02
1149
.52
1174
.82
1217
.12
1299
.73
1340
.4513
83.6
0
1448
.42
1490
.93
1567
.55
1608
.46
1637
.3516
87.3
6
2805
.34
2852
.71
2866
.84
2935
.03
3308
.38
-10
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
%T
500 1000 1500 2000 2500 3000 3500 4000 Wavenumbers (cm-1)
% T
rans
mitt
ance
Wavenumber cm-1
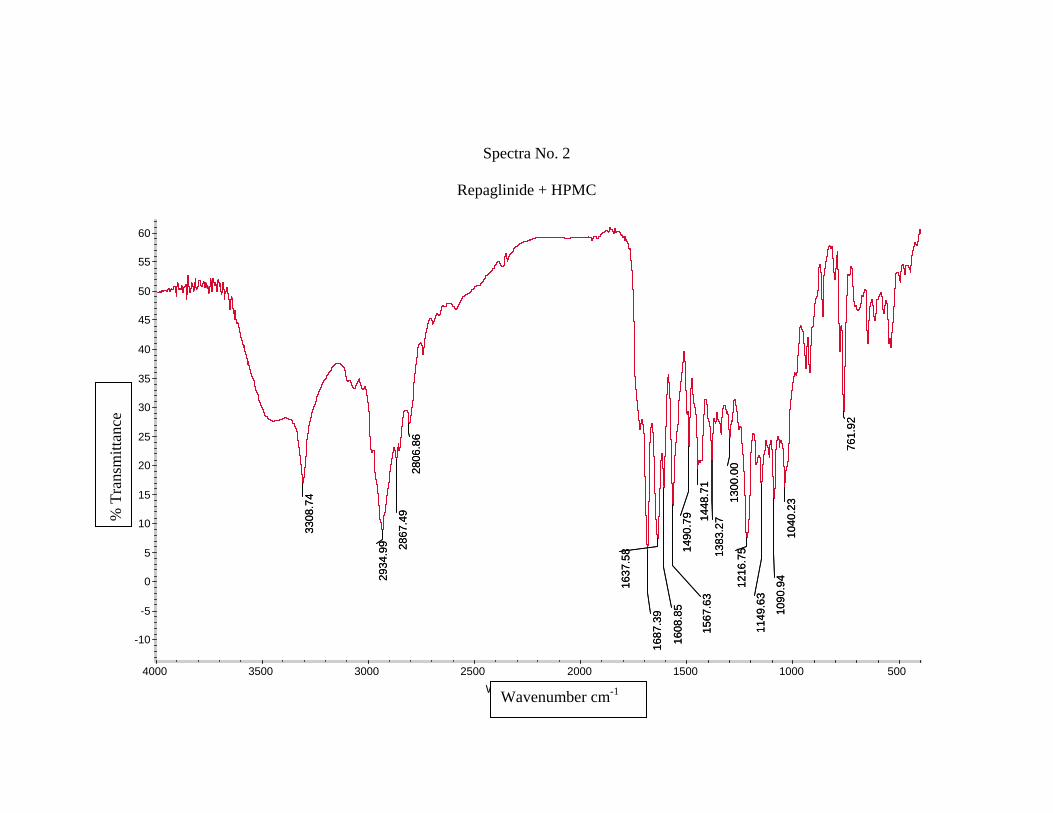
Spectra No. 2
Repaglinide + HPMC
761.
92
1040
.23
1090
.94
1149
.63
1216
.75
1300
.00
1383
.2714
48.7
114
90.7
9
1567
.63
1608
.85
1637
.58
1687
.39
2806
.86
2867
.49
2934
.99
3308
.74
761.
92
1040
.23
1090
.94
1149
.63
1216
.75
1300
.00
1383
.2714
48.7
114
90.7
9
1567
.63
1608
.85
1637
.58
1687
.39
2806
.86
2867
.49
2934
.99
3308
.74
-10
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
%T
500 1000 1500 2000 2500 3000 3500 4000 Wavenumbers (cm-1)
% T
rans
mitt
ance
Wavenumber cm-1
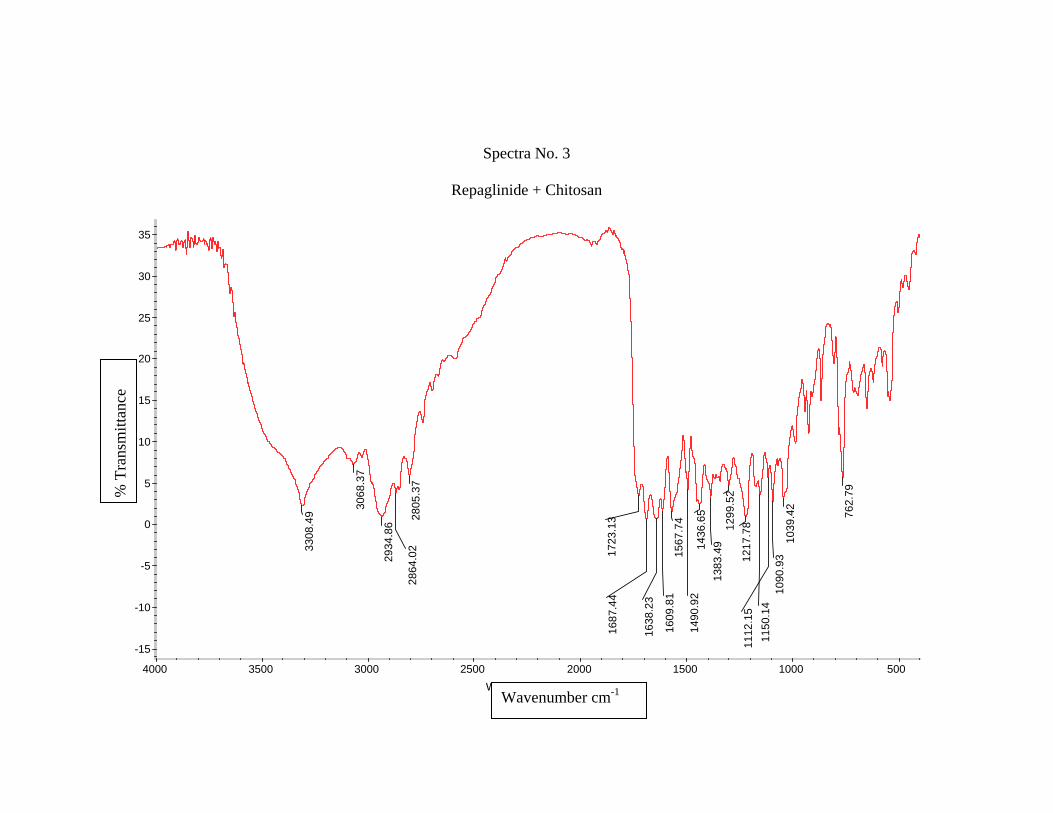
Spectra No. 3
Repaglinide + Chitosan
762.
79
1039
.42
1090
.93
1112
.15
1150
.14
1217
.7812
99.5
213
83.4
91436
.65
1490
.92
1567
.74
1609
.81
1638
.23
1687
.44
1723
.1328
05.3
728
64.0
2
2934
.86
3068
.37
3308
.49
-15
-10
-5
0
5
10
15
20
25
30
35
%T
500 1000 1500 2000 2500 3000 3500 4000 Wavenumbers (cm-1)
% T
rans
mitt
ance
Wavenumber cm-1
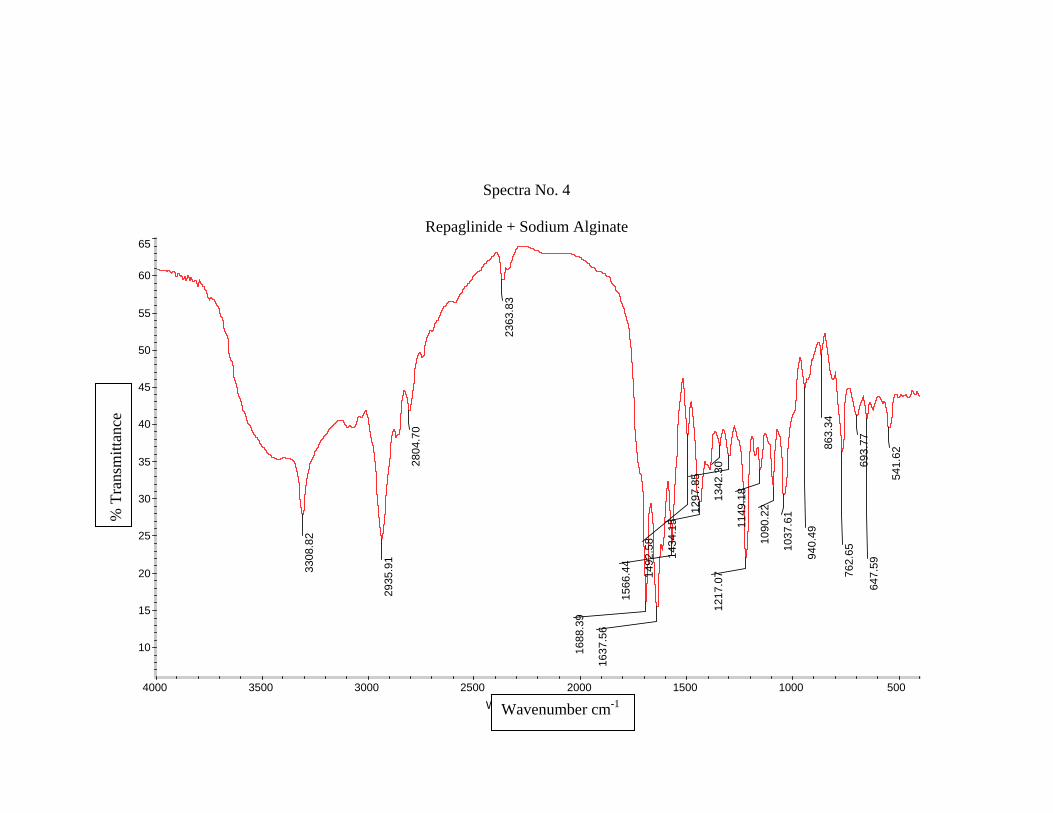
Spectra No. 4
Repaglinide + Sodium Alginate
541.
62
647.
5969
3.77
762.
65
863.
3494
0.49
1037
.61
1090
.22
1149
.18
1217
.07
1297
.85
1342
.30
1434
.15
1492
.58
1566
.44
1637
.56
1688
.39
2363
.83
2804
.70
2935
.91
3308
.82
10
15
20
25
30
35
40
45
50
55
60
65
%T
500 1000 1500 2000 2500 3000 3500 4000 Wa
% T
rans
mitt
ance
venumbers (cm-1)Wavenumber cm-1
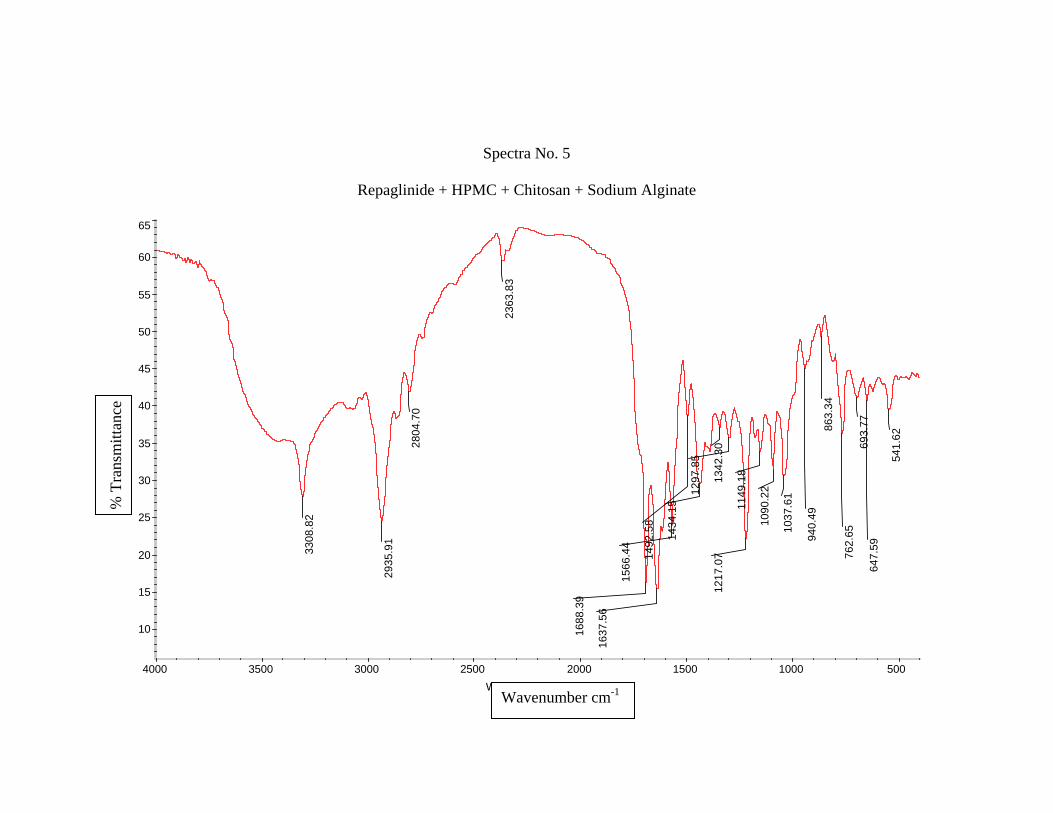
Spectra No. 5
Repaglinide + HPMC + Chitosan + Sodium Alginate
541.
62
647.
5969
3.77
762.
65
863.
3494
0.49
1037
.61
1090
.22
1149
.18
1217
.07
1297
.85
1342
.30
1434
.15
1492
.58
1566
.44
1637
.56
1688
.39
2363
.83
2804
.70
2935
.91
3308
.82
10
15
20
25
30
35
40
45
50
55
60
65
%T
500 1000 1500 2000 2500 3000 3500 4000 Wavenumbers (cm-1)
% T
rans
mitt
ance
Wavenumber cm-1

Figure No. 1
SEM of Formulation F1 Under Low Magnification
Figure No. 2
SEM of Formulation F4 Under Low Magnification
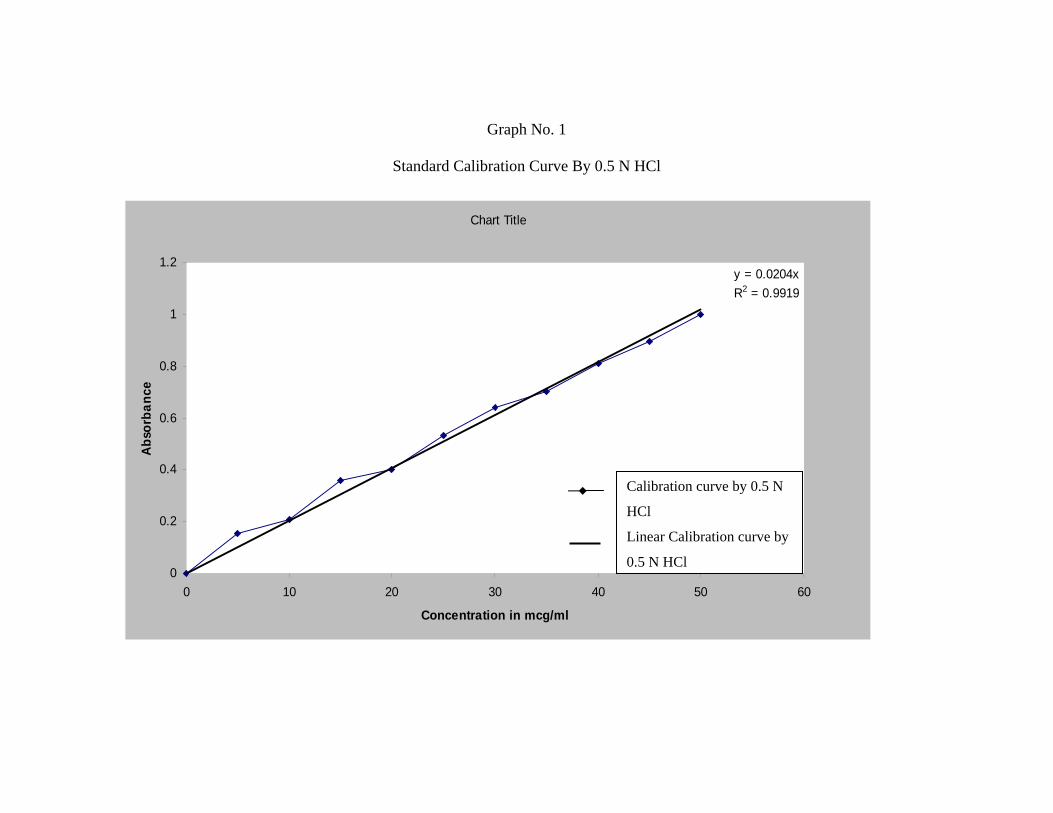
Graph No. 1
Standard Calibration Curve By 0.5 N HCl
Graph No. 2
Standard Cal y 1.2 pH
Chart Title
y = 0.0204xR2 = 0.9919
0
0.2
0.4
0.6
0.8
1
1.2
0 10 20
Concentrat
Abs
orba
nce
ibration Curve B
30 40 50 60
ion in mcg/ml
Calibration Curve by0.5N HCL
Linear (Calibration Curveby 0.5N HCL)
Calibration curve by 0.5 N
HCl
Linear Calibration curve by
0.5 N HCl
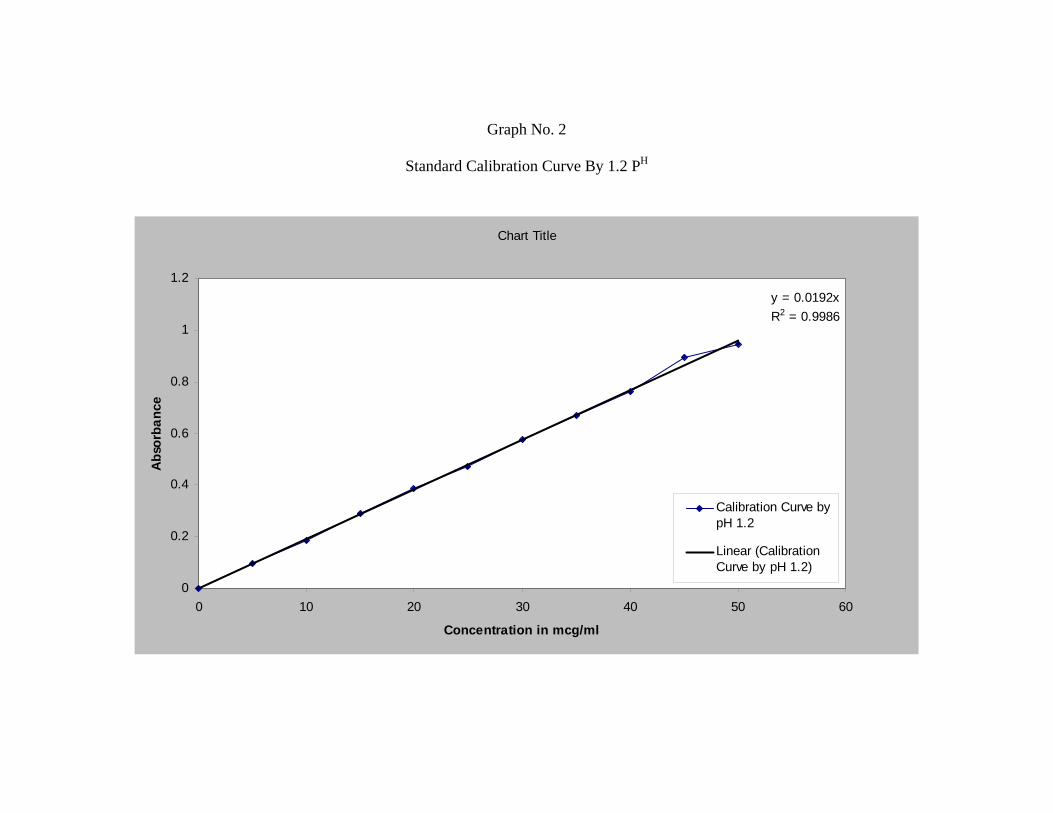
Graph No. 2
Standard Calibration Curve By 1.2 PH
Chart Title
y = 0.0192xR2 = 0.9986
0
0.2
0.4
0.6
0.8
1
1.2
0 10 20 30 40 50 60
Concentration in mcg/ml
Abs
orba
nce
Calibration Curve bypH 1.2
Linear (CalibrationCurve by pH 1.2)
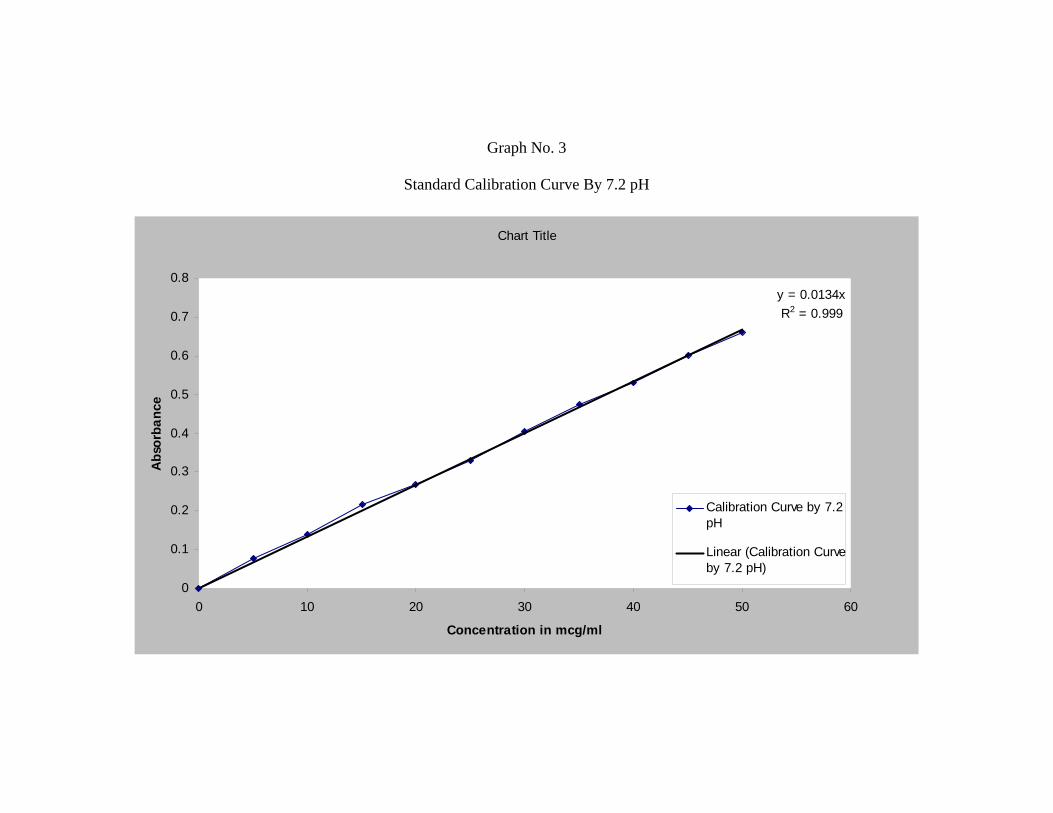
Graph No. 3
Standard Calibration Curve By 7.2 pH
Chart Title
y = 0.0134xR2 = 0.999
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0 10 20 30 40 50 60
Concentration in mcg/ml
Abs
orba
nce
Calibration Curve by 7.2pH
Linear (Calibration Curveby 7.2 pH)
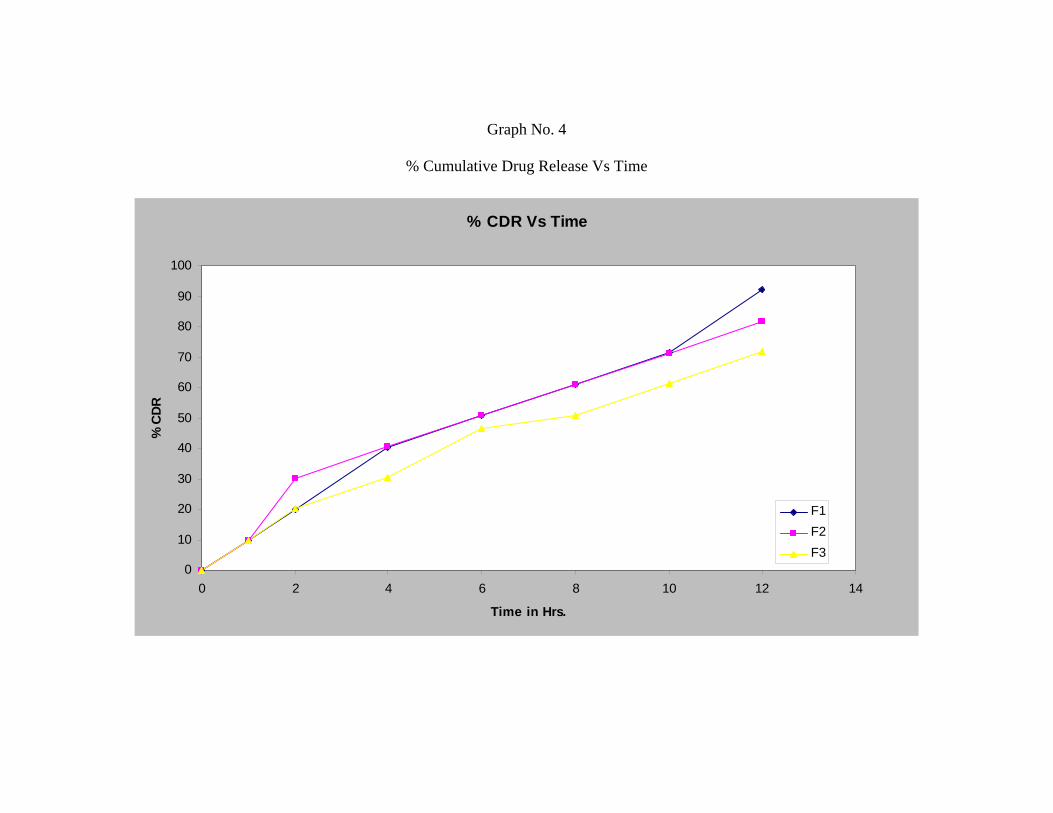
Graph No. 4
% Cumulative Drug Release Vs Time
% CDR Vs Time
0
10
20
30
40
50
60
70
80
90
100
0 2 4 6 8 10 12 14
Time in Hrs.
% C
DR
F1F2F3
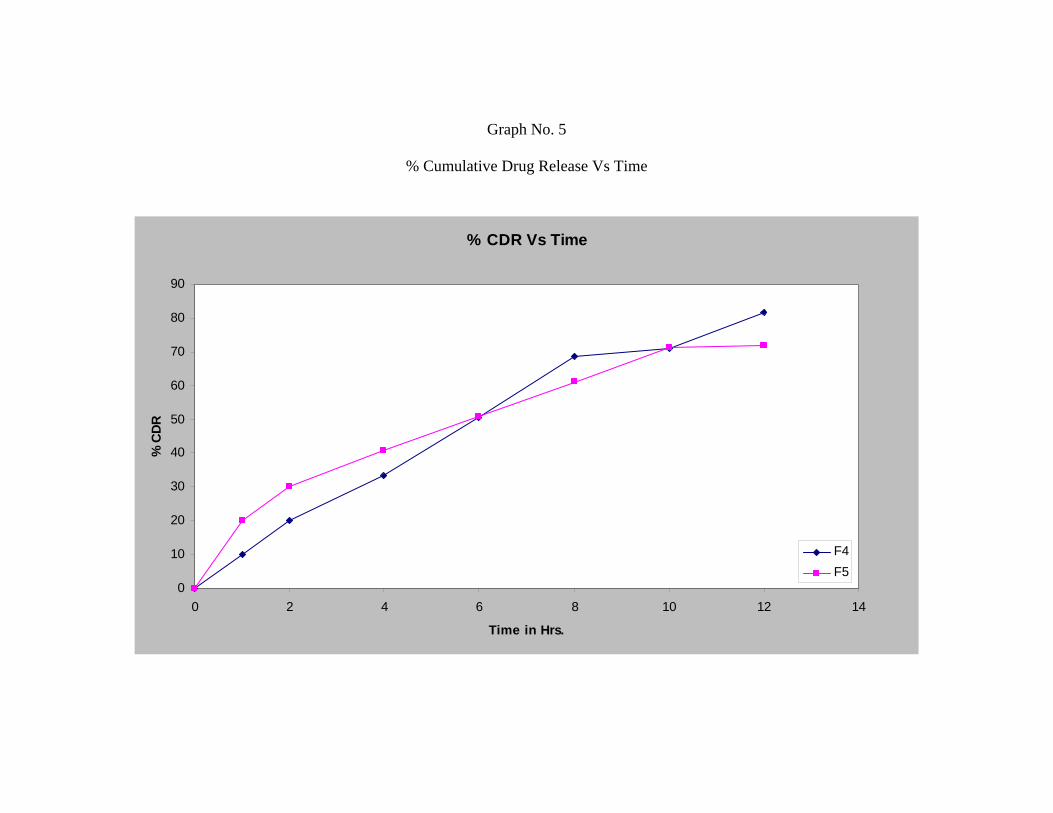
Graph No. 5
% Cumulative Drug Release Vs Time
% CDR Vs Time
0
10
20
30
40
50
60
70
80
90
0 2 4 6 8 10 12 14
Time in Hrs.
% C
DR
F4F5
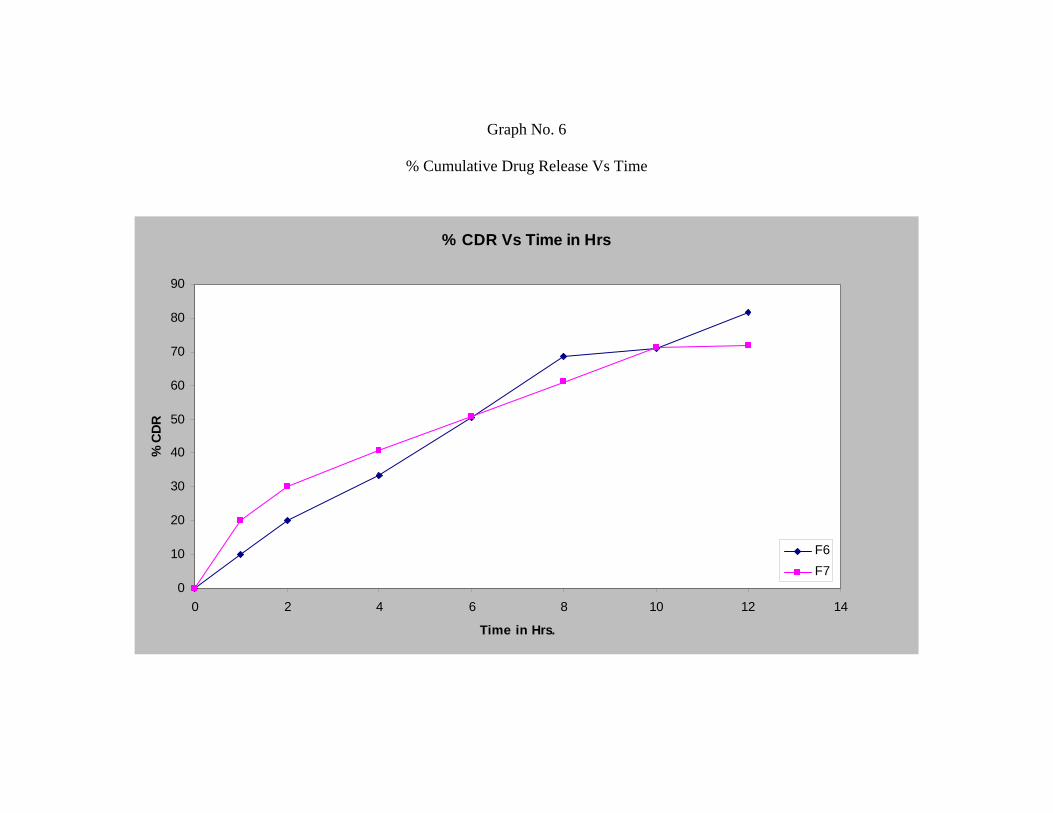
Graph No. 6
% Cumulative Drug Release Vs Time
% CDR Vs Time in Hrs
0
10
20
30
40
50
60
70
80
90
0 2 4 6 8 10 12 14
Time in Hrs.
% C
DR
F6F7
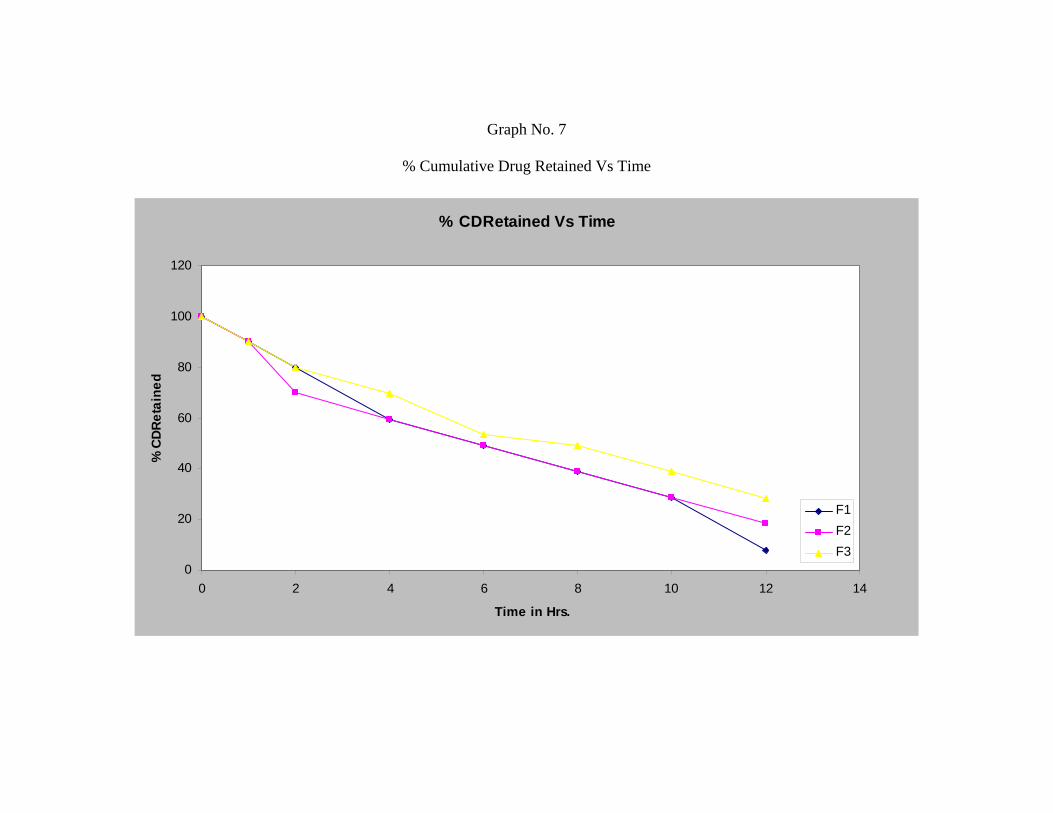
Graph No. 7
% Cumulative Drug Retained Vs Time
% CDRetained Vs Time
0
20
40
60
80
100
120
0 2 4 6 8 10 12 14
Time in Hrs.
% C
DRet
aine
d
F1F2F3
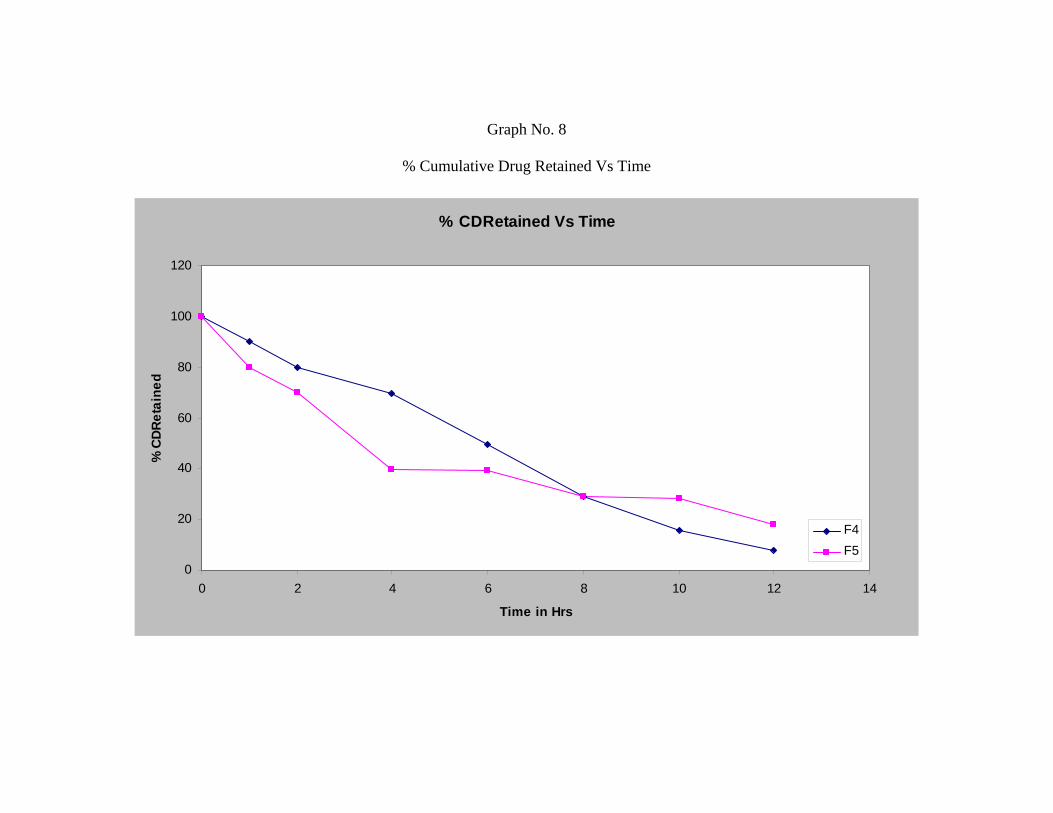
Graph No. 8
% Cumulative Drug Retained Vs Time
% CDRetained Vs Time
0
20
40
60
80
100
120
0 2 4 6 8 10 12 14
Time in Hrs
% C
DRet
aine
d
F4F5
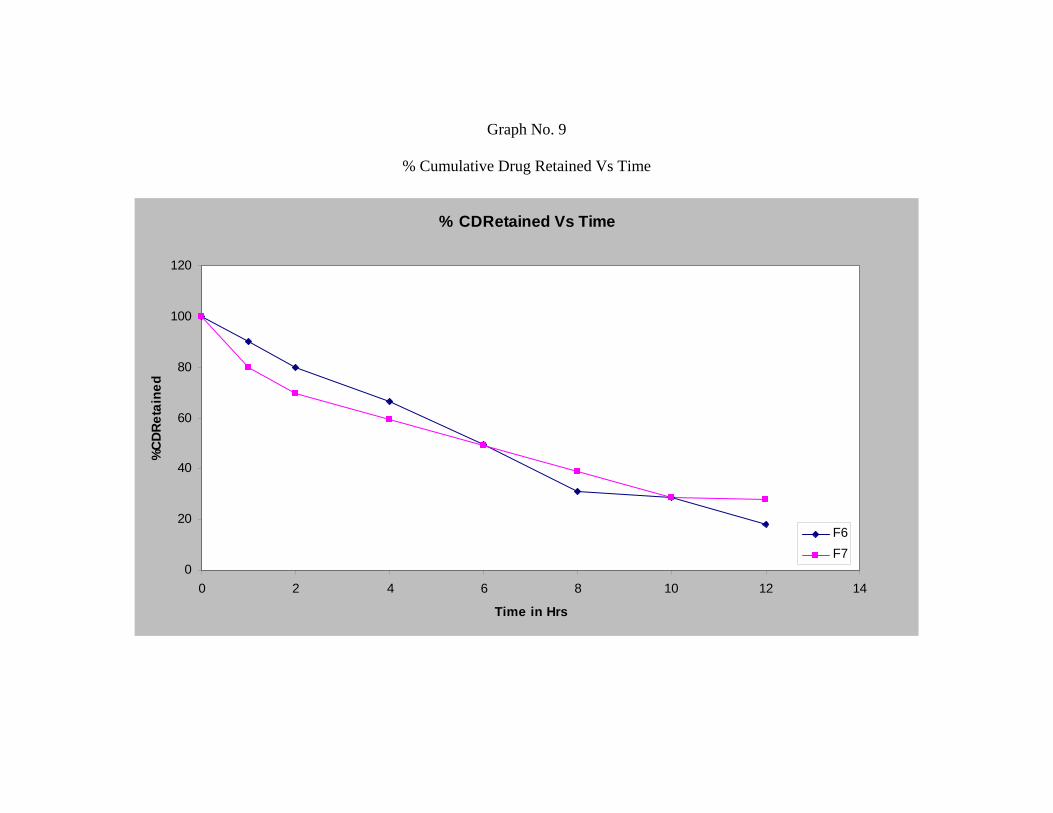
Graph No. 9
% Cumulative Drug Retained Vs Time
% CDRetained Vs Time
0
20
40
60
80
100
120
0 2 4 6 8 10 12 14
Time in Hrs
%C
DRet
aine
d
F6F7
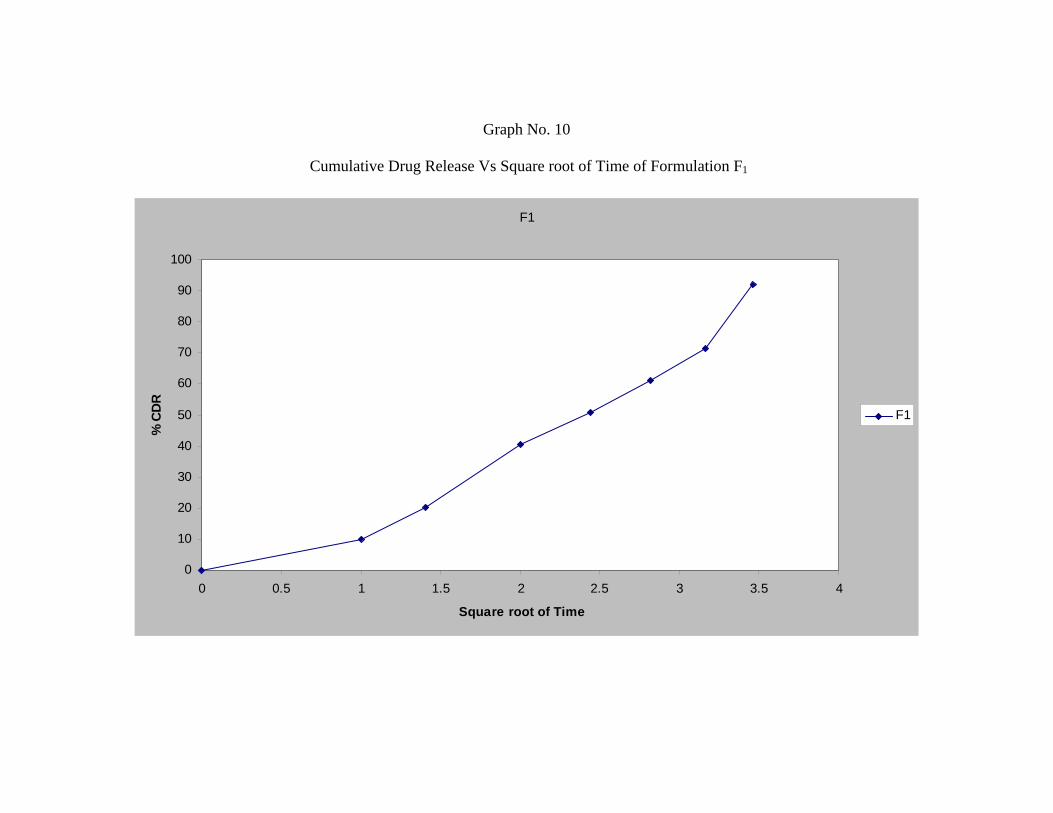
Graph No. 10
Cumulative Drug Release Vs Square root of Time of Formulation F1
F1
0
10
20
30
40
50
60
70
80
90
100
0 0.5 1 1.5 2 2.5 3 3.5 4
Square root of Time
% C
DR
F1
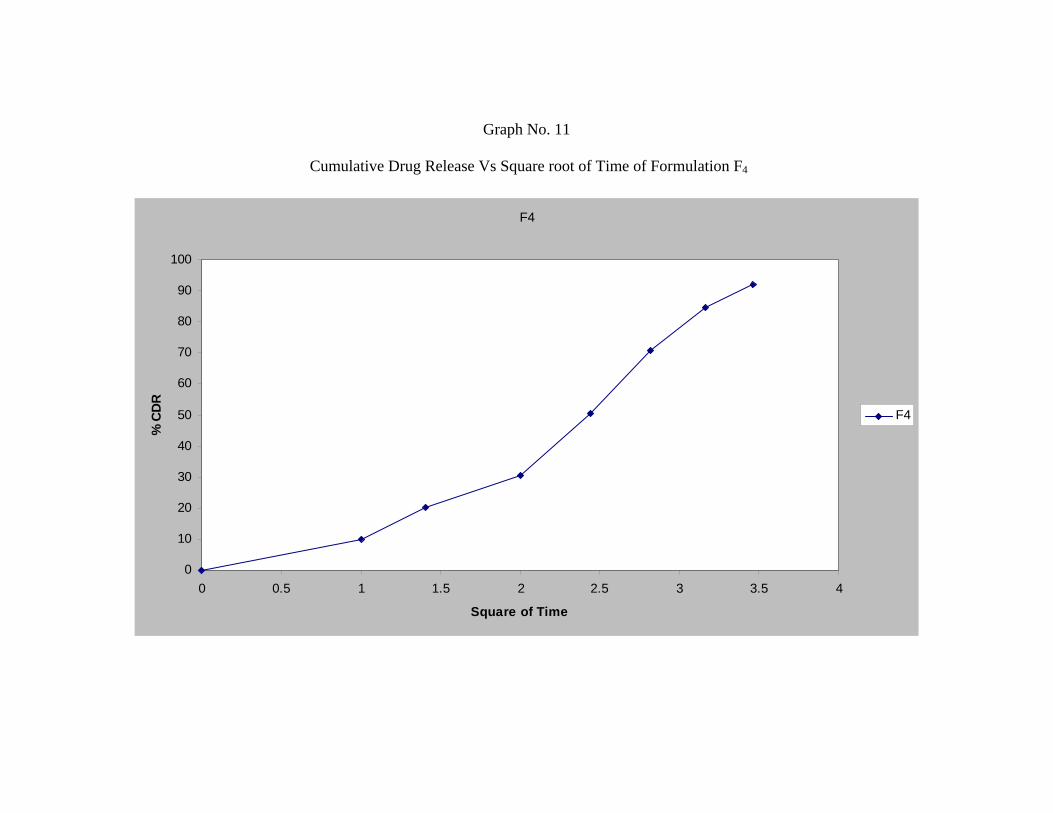
Graph No. 11
Cumulative Drug Release Vs Square root of Time of Formulation F4
F4
0
10
20
30
40
50
60
70
80
90
100
0 0.5 1 1.5 2 2.5 3 3.5 4
Square of Time
% C
DR
F4
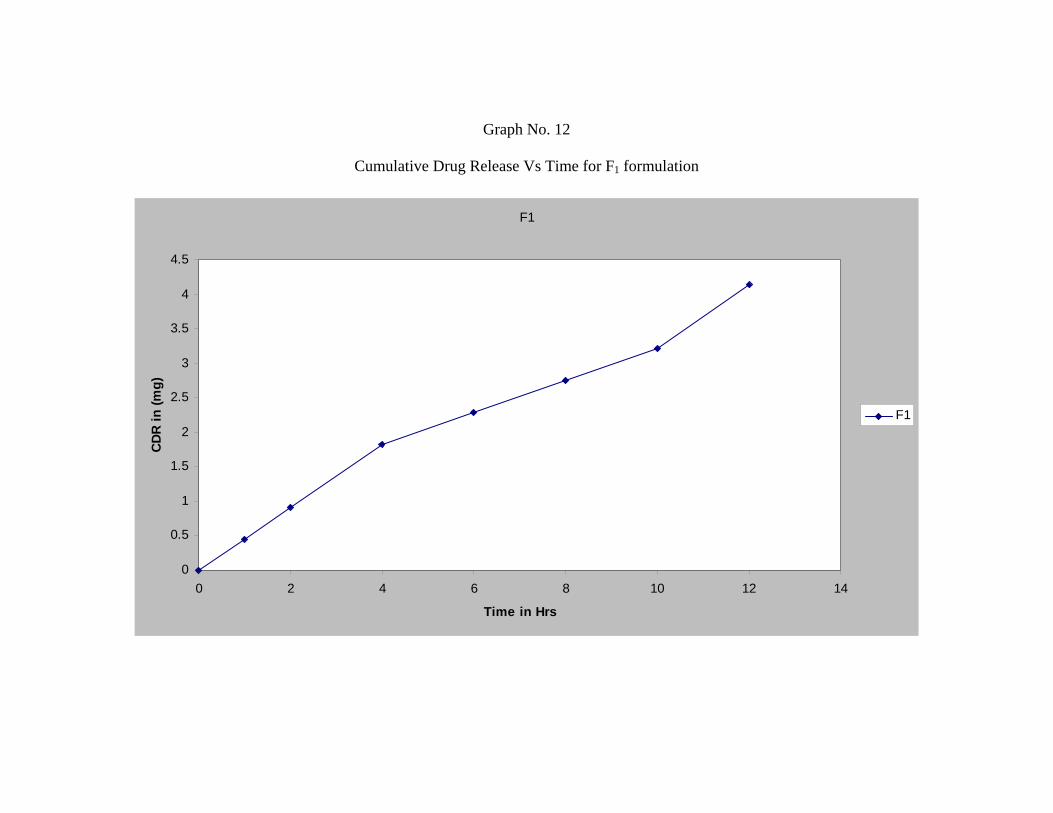
Graph No. 12
Cumulative Drug Release Vs Time for F1 formulation
F1
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
0 2 4 6 8 10 12 14
Time in Hrs
CDR
in (m
g)
F1
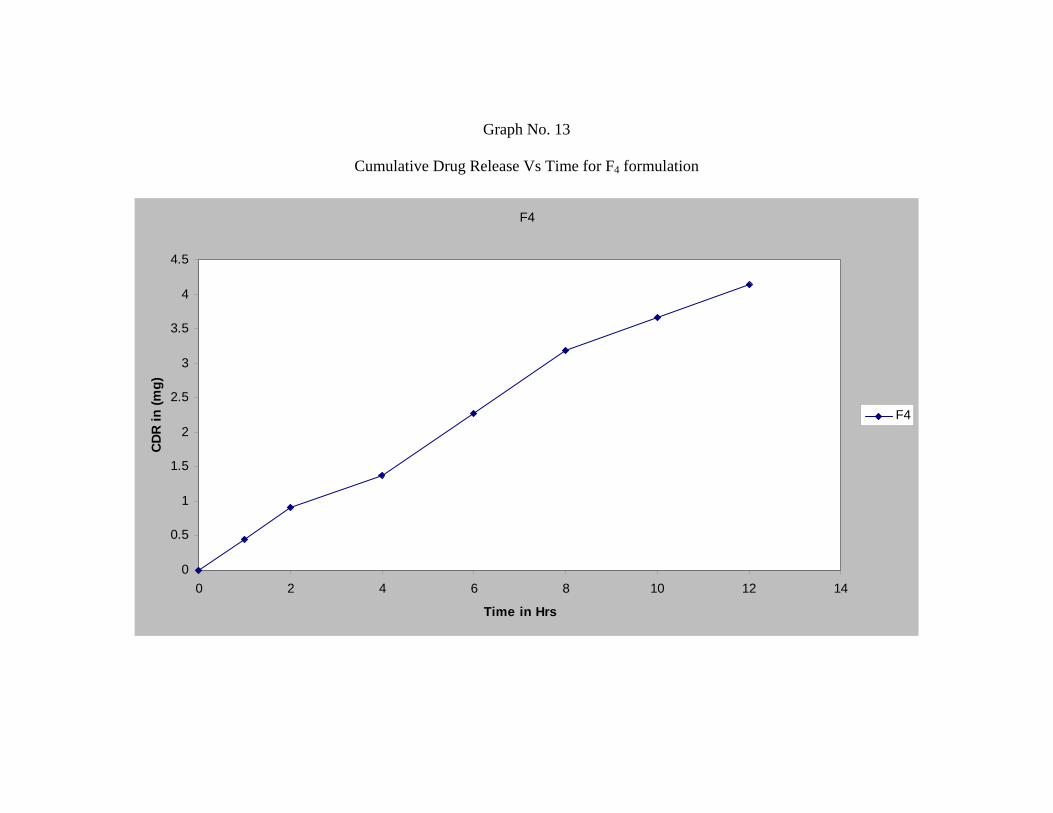
Graph No. 13
Cumulative Drug Release Vs Time for F4 formulation
F4
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
0 2 4 6 8 10 12 14
Time in Hrs
CDR
in (m
g)
F4
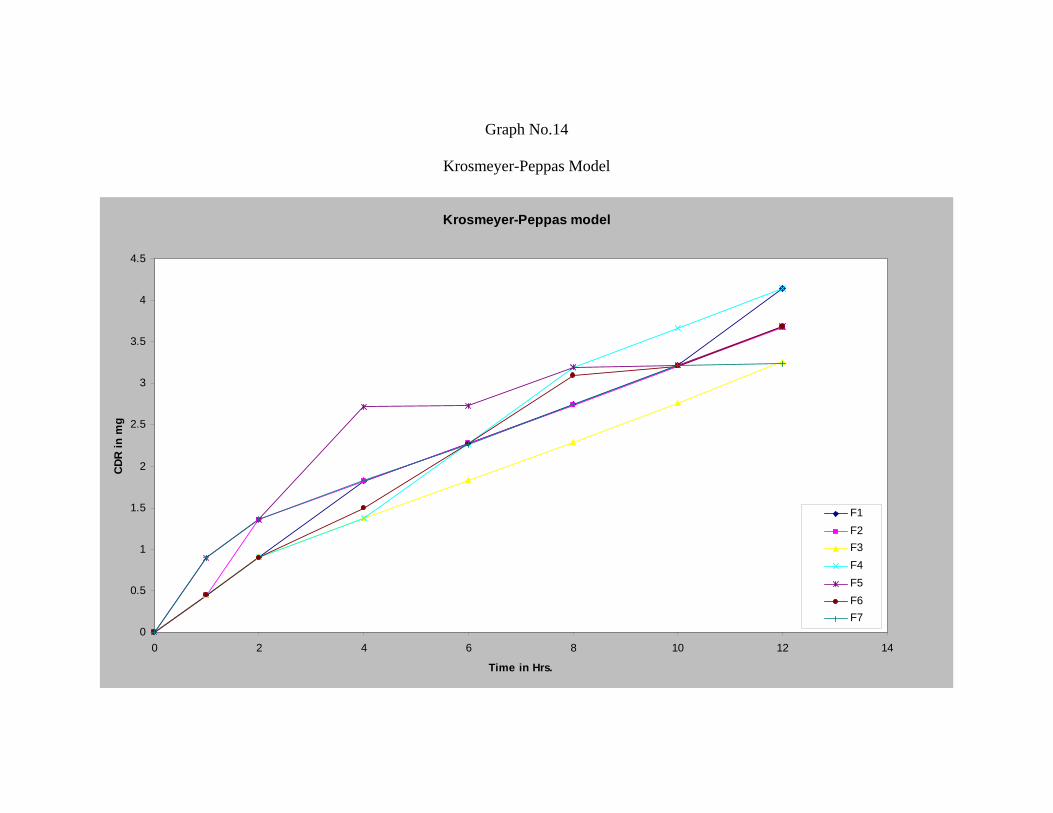
Graph No.14
Krosmeyer-Peppas Model
Krosmeyer-Peppas model
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
0 2 4 6 8 10 12 14
Time in Hrs.
CDR
in m
g
F1F2F3F4F5F6F7
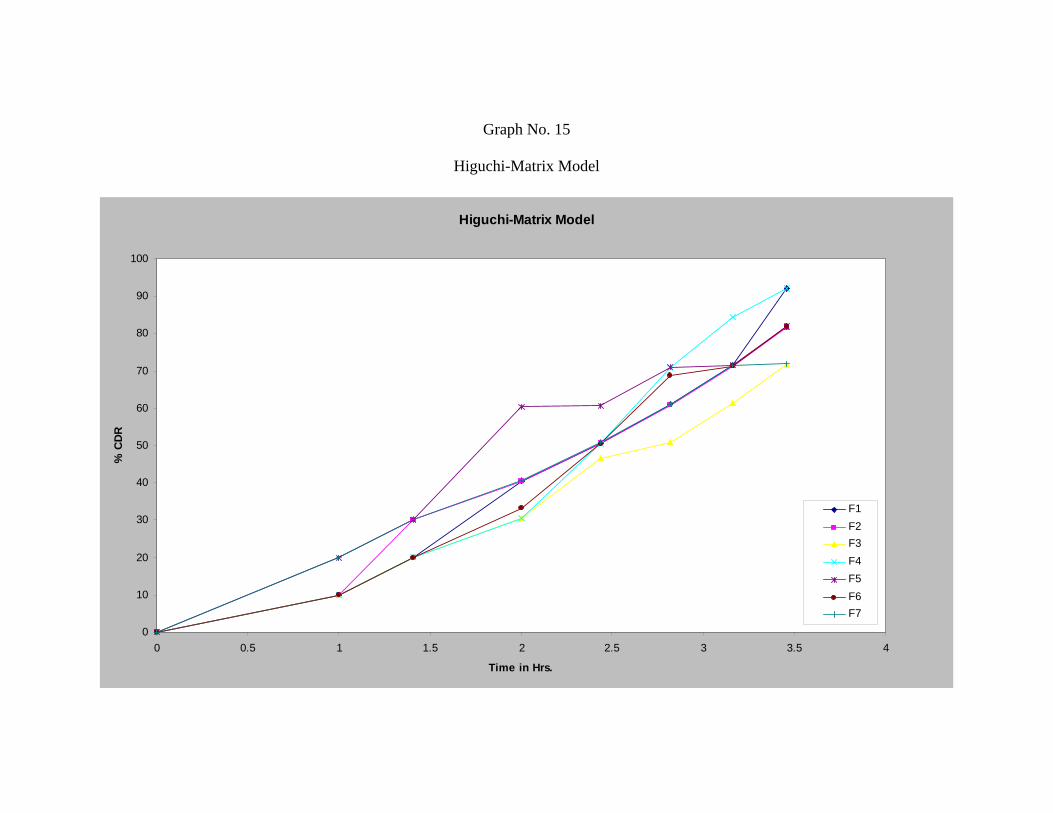
Graph No. 15
Higuchi-Matrix Model
Higuchi-Matrix Model
0
10
20
30
40
50
60
70
80
90
100
0 0.5 1 1.5 2 2.5 3 3.5 4
Time in Hrs.
% C
DR
F1F2F3F4F5F6F7

Chapter VI SUMMARY
Dept. of Pharmaceutics KLESCOP HUBLI
71
SUMMARY
• The goal of any drug delivery system is to provide a therapeutic amount of drug
to the proper site in the body and also to achieve and maintain the desired drug
concentration.
• Microencapsulation is a process whereby small discrete solid particles or small
liquid droplets are surrounded or enclosed, by an intact shell.
• Repaglinide is an antidiabetic agent used in the treatment of type II diabetes
mellitus. The plasma half life of Repaglinide is about one hour.
• An attempt is made to microencapsulate repaglinide by Ionotropic Gelation
Technique with a view to prevent the gastric side effects and to achieve an oral
controlled release of the drug.
• The literature survey discussed reveals the research work done to develop
microparticulates drug delivery system containing anti-diabetic, anti-hypertensive
and NSAID’s drugs by ionotropic gelation technique.
• Polymers used in this technique are Hydroxy Propyl Methyl Cellulose and
Chitosan, calcium chloride as a crosslinking agent, and sodium alginate as a
viscosity builder.
• In the present study seven formulations were formulated by using Hydroxy Propyl
Methyl Cellulose and Chitosan in various proportions.
• All the formulations were subjected for evaluation. Results of preformulation
studies, granulometric study, angle of repose, entrapment efficiency, in-vitro
dissolution study have shown satisfactory results.

Chapter VI SUMMARY
Dept. of Pharmaceutics KLESCOP HUBLI
72
• The in-vitro release study of formulations F1 to F7 shows a retarded release with
increase percentage of Hydroxy Propyl Methyl Cellulose and Chitosan.
• On the basis of release data and graphical analysis formulation F1 and F4 showed
a good controlled release profile with maximum entrapment efficiency.
• The release kinetics of formulations F1 and F4 shows a good correlation of
Krosmeyer-Peppas with a good ‘n’ values. According to ‘n’ values obtained F1
and F4 follows Fickian diffusion as release mechanism.
• The surface study of F1 and F4 viewed through SEM shows an uniform matrix
formulation with dense nature and low porosity.
• The formulation were subjected to accelerated stability study. Both the
formulation were found to be stable as there was no change in the drug content.

Chapter VII CONCLUSION
Dept. of Pharmaceutics KLESCOP HUBLI
73
CONCLUSION
From the above experimental results it can be concluded that :-
• Oral controlled release of Repaglinide can be achieved by ionotropic gelation
technique using HPMC and Chitosan as a polymer.
• The IR spectras revealed that, there was no interaction between polymers and
drug. All the polymers used were compatible with the drug.
• Prepared microparticles exhibited Krosmeyer-Peppas kinetics/Higuchi model and
the release profile was by Fickian and Anomalous.
• From the study it is evident that a promising controlled release microparticulate
drug delivery of repaglinide can be developed. Further in-vivo investigation is
required to establish efficacy of these formulations.
• The study also indicated that the amount of drug release decreases with an
increase in the polymer concentration.
• Microparticles formulated with a combination of HPMC and Chitosan prolonged
the release but gave unsatisfactory kinetic results with low correlation coefficient
values.

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
73

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
74
References :
1. Tripathi K.D., 'Essentials of Medical Pharmacology', 5th Edition, Jaypee Brothers
Medical Publications (P) Ltd., New Delhi, 2003; 167-184.
2. Yie W. Chien, "Concepts and System Design for Rate-controlled Drug Delivery",
Chapter 1 in Novel Drug Delivery System', 2nd Edition, Marcel Dekker, Inc, New
York, 1992; l-42.
3. Yie W. Chien, 'Rate-controlled Drug Delivery Systems'. Ind. J. Pharm. Sci.,
1988; Mar-April: 63-65.
4. Thomas Wai-Yip Lee and Joseph R. Robinson, "Controlled / Release
Drug-Delivery Systems", Chapter 47 in 'Remington's Pharmaceutical Sciences',
20th Edition, Mack Publishing Company, Volume-I, 2000; 903-929.
5. D. M. Brahmankar, Sunil B. Jaiswal., Biopharmaceutics and Pharmacokinetics a
Treatise, First edition, Vallabh Prakashan Pitampura, Delhi-2001; 337-341.
6. Herbert A. Lieberman, Leon Lachman, and Joseph B. Schwartz Pharmaceutical
dosage forms IInd Eidition Volume-I, 7.
7. Edith M. and Mark R.K., "Microencapsulation" in 'Encyclopedia of Controlled
Release', John Wiley and Sons, Inc. London, Volume-2, 1998; 493-510.
8. Chowdary K.P.R. and Sri Ram Murthy A., "Microencapsulation in Pharmacy". Indian
Drugs, 1998; 25(10) : 389-402.
9. Simon Bonita, "A survey of Microencapsulation process" chapter 1 in
'Microencapsulation, Method and Industrial Application' 2nd Edition, Marcel Dekker,
Inc. New York, 198; 2-5.

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
75
10. Manekar N.C. and Joshi, S.B., “Microencapsulation Technique". The Eastern
Pharmacist, 1998; 47-49.
11. Patrick B. Deasy, "Microencapsulation and Related Drug Processes", Chapter 1 in
'Drugs and the Pharmaceutical Sciences', 2nd Edition, James Swarbrick, Marcel
Dekker Inc, New York, Volume 20, 1984; l-13.
12. Vyas and Khar, “Targeted and Controlled Drug Delivery Novel Carrier System” First
Edition, C B S Publishers and Distributers, New Delhi, 2002; 419-424.
13. Rajesh, K.S., Khanrah, A. and Biswanath Sa, "Release of Ketoprofen from Alginate
Microparticles Containing Film forming Polymer". J. Sci. Ind. Res., 2003, 62(10);
987.
14. Chowdary K. P. R. and N. Koteswara rao, , “Ethyl cellulose microspheres of
glipizide: characterization in-vitro and in-vivo evaluation.” Ind. J. Pharm. Sci., 2004;
66 (4), 412-416.
15. Arul B. and R. Kothai, “Formulation and evaluation of chitosan microspheres
containing isoniazid”, Ind. J. Pharm. Sci., 2003; 66 (5), 640.
16. Stephen N. Devis and Granner K. Daryl, Alfred Goodman Gilman’s The
pharmacological basis of therapeutics, 10th Edn., McGraw-Hill, Newyork, 2001;
1686-1705.
17. Andhimathi M. G., and T. K. Ravi, “Determination of Repaglinide in pharmaceutical
formulation by HPLC with UV spectroscopy” Analytical Sci., 2003; (19), 1675-1676.
18. Bhalla H. l. and Kusha A. V., “Controlled release indomethacin”. Indian Drugs,
1988; 26(4) : 154-157.

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
76
19. Chowdary K.P.R. and Suresh Babu K.V.V., "Studies on Micro encapsulation by
Calcium Alginate". The Eastern Pharmacist, 1989; 2 : 125-126.
20. Pandey S., Singh U.V. and Udupa N., “Implantable Flurbiprofen for Treating
Inflammation Associated with Arthritis". Indian Drug, 1994; 31(6) : 254-257.
21. Kakkar A. P., "Characterization of Ibuprofen Loaded Microcapsules Prepared
by Ionotropic Gelation". Ind. J. Pharm. Sci., 1995; 57(2) : 56-60.
22. M. Guzman, J. Moleceres F. Garcia and M. R. Aberturas, “Preparation,
characterization and in vitro drug release of poly-e-caprolactone and hydroxyl
propyl methyl cellulose phathalate ketoprofen loaded microspheres”, J. of
Microencapsulation, 1996; 13(1): 25-39.
23. M. Cuna, M. l. Lorenzo-Lamosa, J. L. Vila-Jato, D. Torres, and M. J. Alonso,
“pH-Dependent Cellulosic Microsphers Containing Cefuroxime Axetil : Stability
and InVitro Release Behavior. Drug Develop. Ind. Pharm. 1997; 23(3): 259-265.
24. Lim-Ly and Wan-LS, "Propranolol Binding in Calcium Alginate Beads". Drug
Develop. Ind. Pharm. 1997; 23(10) : 973-980.
25. Lim-Ly et. al. "Chitosan Microspheres Prepared by Emulsification of Ionotropic
Gelation". Drug Develop. Ind. Pharm., 1997; 23(10) : 981-985.
26. Pilley V. and C. M. Dangor, “Ionotropic Gelation : encapsulation of indomethacin in
calcium alginate gel discs.” J. of microencapsulation : 1998; 15(2):215-26.
27. Manna A., Ghosh I., Goswami N., Ghosh L.K. and Gupta B.K., "Design and
Evaluation of an Oral Controlled Release Microparticulate Drug Delivery
System of Nimesulide by Ionotropic Gelation Technique and Statistical
Optimization by Factorial Analysis". J. Sci. Ind. Res., 1999; 58(9) : 717-722.

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
77
28. Alf Lamprecht, Ulrich Schafer, and Claus-Michael Lehr, “Structural Analysis of
Microparticles by Confocal Laser Scanning Microscopy”. AAPS Pharm. Sci. Tech.,
2001; 1 (3). (http://www.aapspharmsci.com)
29. Patil V.B. and Varsha B. Pokharkar, "Preparation and Evaluation of Sustained
Release Nimesulide Microspheres Prepared from Sodium alginate". Ind. J. Pharm.
Sci., 2001; 63(1) : 15-19.
30. Choi B. Y. and H. J. Park., “Preparation of alginate beads for floating drug delivery
system: effect of carbon dioxide gas-forming agents” Int. J. Pharm., 2002; 239
(1-2) : 81-91.
31. Chowdary, K. P. R. and Srinivasa Rao Y., "Preparation and Evaluation of
Mucoadhesive Microcapsules of Indomethacin". Ind. J. Pharm. Sci., 2003; 65(1) :
49-52.
32. Jelena Filipovie-Grcie, Beatrice Perissutti, Mariarosa Moneghini, Dario Voinovich,
Anita Martinac, Ivan Jalsenjak, J. Pharmacy and Pharmacology (2003); 55 (7), 921.
33. Nikhil O. Dhoot and Margaret A. Wheatley, “Microencapsulated Liposomes
in Controlled Drug Delivery : Strategies to Modulate Drug Release and
Eliminate the Burst Effect". J. Pharm. Sci., 2003; 92(3) : 679-689.
34. Arpita Bhattacharya, Parna Maitra and Mukherjee A., "Alginate-based Nanocapsular
Antineoplastic Drug Delivery System by Pneumatic Nebulization". Indian J. Pharm.
Sci., 2003; 65(5) : 477-481.
35. Pralhad T. Tayade, and Ranjendrakumar D. Kale, “Encapsulation of water-
Insoluble drug by a cross-linking Technique : Effect of Process and Formulation

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
78
Variables on Encapsulation Efficiency, Particle Size, and In Vitro Dissolution
Rate”. AAPS Pharm. Sci., 2004; 6(1) (http://www.aapspharmsci.org).
36. Mingshi Yang, Fude acaui, Bengang You, Jian You, Liang Wang, Liqiang Zhang,
Yoshiaki Kawashima, “A novel pH-dependent gradient-release delivery system for
nitrendipin”. J. Control. Release 98 : (2004); 219-229pp.
37. Govindrajan G., and K. Muttusamy, “Formulation and evaluation of floating hollow
microspheres of lansoprazole”, Ind. J. Pharm. Sci., 2004; 66 (4), 486.
38. Jamagondi L. N. and V. B. Patil, “Studies on Microencapsulation of metformin
hydrochloride using ethyl cellulose” Ind. J. Pharm. Sci., 2004; 66 (4), 494.
39. Dandagi P. M. and F. V. Manvi, “Microencapsulation of Verapamil Hydrochloride
by ionotropic gelation technique” Ind. J. Pharm. Sci., 2004; 66 (5), 631-635.
40. NovoNordisk. Prandin package insert [online]. NovoNordisk from
http://www.novonordiskus.com
41. www.drugs.com
42. Lin L.Y., et al, Hydroxy propyl methyl cellulose in 'Hand Book of Pharmaceutical
Excipients', Published by American Pharmaceutical Association and the
Pharmaceutical Society of Great Britain, 1986; 138-140.
43. 'Hydroxy propyl methyl cellulose', Indian Pharmacopoeia, Ministry of Health and
Welfare, Government of India, New Delhi, Volume II : 1996; 382-383.
44. Dusel R. et al., Sodium Alginate in 'Hand Book of Pharmaceutical Excipients',
Published by American Pharmaceutical Association and the Pharmaceutical Society
of Great Britain, 1986; 257-258.

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
79
45. 'Sodium Alginate', Indian Pharmacopoeia, Ministry of Health and Welfare,
Government of India, New Delhi, Volume II : 1996; 680.
46. Lin L.Y. et al, Chitosan in 'Hand Book of Pharmaceutical Excipients',
Published by American Pharmaceutical Association and the Pharmaceutical
Society of Great Britain, 1986 : 138-140.
47. Indian Pharmacopoeia Ministry of Health and Family Welfare, Government
of India, New Delhi, 1996; Volume-I : A-144.
48. Jain S. K., Agrawal G.P., “Spectrophotometric Determination of Repaglinide in
Tablet Dosage Forms” Ind. J. Pharm. Sci., 2005; 249.
49. Subrahmanyam C.V.S., Text Book of Physical Pharmaceutics', 2nd Edition, Dehi,
Vallabh Prakashan, 2000; 222-224.
50. Subrahmanyam C.V.S., Text Book of Physical Pharmaceutics', 2nd Edition, Dehli,
Vallabh Prakashan, 2000; 85.
51. The United States Pharmacopoeia, XXIV-NF XIX : Asian Edition, USP
Convention Inc., 2000; 1739-1742.
52. Sharma P. and Hamsa V., "Formation and Evaluation of Buccal Mucoadhesive
Patches of Terbutaline Sulphate". S.T.D. Pharm. Sci., 2000; 11(4) : 275-281.
53. Iannuccelli V., et al., "Air Compartment Multiple-unit System for Prolonged Gastric
Residence. Part I Formulation Study". Int. J. Pharm. 1996; 174 : 47-54.
54. Kumar V., Damien B. and Potdar A.R., "Designing of Stability Programme". The
Eastern Pharmacist, 1992; 8 : 29-32.

Chapter VIII References
Dept. of Pharmaceutics KLESCOP, HUBLI
80
55. Banker G.S. and Anderson N.R., "Kinetic Principles and Stability Testing",
in the Theory and Practice of Industrial Pharmacy', by Lachman, et al., 3rd edition,
1991; 760-769.
56. Indian Pharmacopoeia Ministry of Health and Family Welfare, Government
of India, New Delhi, 1996; Volume-I : 328.
57. Indian Pharmacopoeia Ministry of Health and Family Welfare, Government
of India, New Delhi, 1996; Volume-I : 5-43.
.
Related Documents











