Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227 Contents lists available at ScienceDirect Journal of Molecular Catalysis A: Chemical journal homepage: www.elsevier.com/locate/molcata Catalytic oxidation of benzene over MnO x /TiO 2 catalysts and the mechanism study Junlin Zeng a,b , Xiaolong Liu a , Jian Wang a,c , Hanlei Lv a , Tingyu Zhu a,∗ a Beijing Research Centre of Process Pollution Control, National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China b Chemistry and Chemical Engineering, Guizhou University, Guiyang, Guizhou 550025, China c University of Chinese Academy of Sciences, Beijing 100049, China article info Article history: Received 5 June 2015 Received in revised form 16 July 2015 Accepted 18 July 2015 Available online 29 July 2015 Keywords: Catalytic oxidation Benzene Manganese Reaction mechanism abstract MO x /TiO 2 composites with various metal species (M = Mn, Ce, Co, Fe) were synthesized and employed as in the catalytic oxidation of benzene. Among those materials, MnO x /TiO 2 showed the highest catalytic efficiency. The catalytic activities of MnO x /TiO 2 with different MnO x contents were also studied. XRD, BET, SEM, TEM, and XPS characterizations were conducted to analyze the physiochemical properties of the MnO x /TiO 2 catalysts. Moreover, the reaction mechanism of benzene oxidation over MnO x /TiO 2 was studied through the in situ FTIR experiments. © 2015 Elsevier B.V. All rights reserved. 1. Introduction Volatile organic compounds (VOCs) emitted from industrial pro- cess and transportation activities are harmful to the atmosphere and human health [1–3]. Among the strategies explored for VOCs removal, catalytic oxidation is regarded as the most promising pathway to reduce VOCs emissions due to its advantages of high efficiency, low temperature, and high selectivity to harmless prod- ucts [4,5]. In recent years, numerous catalysts have been explored, includ- ing noble metals [6–8] and transition-metal oxides [9–19]. The noble metal catalysts such as Pd and Pt-based catalysts have attracted increasing attention for their excellent catalytic activities. However, the drawbacks of high costs and being likely to produce multi-chlorinated byproducts have greatly restricted their applica- tions. Non-noble metal catalysts (mainly Co, Mn, Ce and Fe) have also demonstrated a great catalytic performance in VOCs oxidations [20–22]. Moreover, the catalysts showed great advantages of lower costs and higher resistance to poison [23,24]. Within the non-noble metal catalysts developed, MnO x -based materials have gained increasing importance because of its high ∗ Corresponding author. E-mail address: [email protected] (T. Zhu). efficiency, which was ascribed to the reactive oxygen species of MnO x . Aguero et al. [25] prepared the MnO x /Al 2 O 3 and the material revealed as a well-defined catalyst in binary VOCs oxida- tion. For MnO x -containing composites, Wang et al. [26] reported CeO 2 –MnO x catalytic oxidation of benzene with high efficiency, and CeO 2 enhance the transfer ability of oxygen species. The meso- porous Mn–Ce composite exhibited higher catalytic activity in the total oxidation of VOCs [27]. Based on previous work, the Mn-containing materials are use- ful for developing a stable catalyst with low costs and high activity, and the mechanism study are helpful to optimize the MnO x -based catalytic system. However, the mechanism research such as the organic intermediates, the interaction between the Mn active cen- ter and organic species, and the reaction path, are far less explored. Herein, we present MnO x /TiO 2 catalyzed benzene oxidation and its mechanism studies. 2. Experimental 2.1. Catalyst preparation MO x /TiO 2 catalysts were prepared using impregnation method with the weight percentages of MO x and TiO 2 being 3 wt.% and 97 wt.%, respectively. In a typical preparation for CeO 2 /TiO 2 , Ce(NO 3 ) 2 ·6H 2 O (0.391 g, 0.9 mmol) was dissolved in 20 mL of http://dx.doi.org/10.1016/j.molcata.2015.07.024 1381-1169/© 2015 Elsevier B.V. All rights reserved.

Finandhita Hayu Pradana Jurnal-015
Feb 17, 2016
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227
Contents lists available at ScienceDirect
Journal of Molecular Catalysis A: Chemical
journa l homepage: www.e lsev ier .com/ locate /molcata
Catalytic oxidation of benzene over MnOx/TiO2 catalysts and themechanism study
Junlin Zenga,b, Xiaolong Liua, Jian Wanga,c, Hanlei Lva, Tingyu Zhua,∗
a Beijing Research Centre of Process Pollution Control, National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology,Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, Chinab Chemistry and Chemical Engineering, Guizhou University, Guiyang, Guizhou 550025, Chinac University of Chinese Academy of Sciences, Beijing 100049, China
a r t i c l e i n f o
Article history:Received 5 June 2015Received in revised form 16 July 2015Accepted 18 July 2015Available online 29 July 2015
Keywords:Catalytic oxidationBenzeneManganeseReaction mechanism
a b s t r a c t
MOx/TiO2 composites with various metal species (M = Mn, Ce, Co, Fe) were synthesized and employed asin the catalytic oxidation of benzene. Among those materials, MnOx/TiO2 showed the highest catalyticefficiency. The catalytic activities of MnOx/TiO2 with different MnOx contents were also studied. XRD,BET, SEM, TEM, and XPS characterizations were conducted to analyze the physiochemical properties ofthe MnOx/TiO2 catalysts. Moreover, the reaction mechanism of benzene oxidation over MnOx/TiO2 wasstudied through the in situ FTIR experiments.
© 2015 Elsevier B.V. All rights reserved.
1. Introduction
Volatile organic compounds (VOCs) emitted from industrial pro-cess and transportation activities are harmful to the atmosphereand human health [1–3]. Among the strategies explored for VOCsremoval, catalytic oxidation is regarded as the most promisingpathway to reduce VOCs emissions due to its advantages of highefficiency, low temperature, and high selectivity to harmless prod-ucts [4,5].
In recent years, numerous catalysts have been explored, includ-ing noble metals [6–8] and transition-metal oxides [9–19]. Thenoble metal catalysts such as Pd and Pt-based catalysts haveattracted increasing attention for their excellent catalytic activities.However, the drawbacks of high costs and being likely to producemulti-chlorinated byproducts have greatly restricted their applica-tions. Non-noble metal catalysts (mainly Co, Mn, Ce and Fe) havealso demonstrated a great catalytic performance in VOCs oxidations[20–22]. Moreover, the catalysts showed great advantages of lowercosts and higher resistance to poison [23,24].
Within the non-noble metal catalysts developed, MnOx-basedmaterials have gained increasing importance because of its high
∗ Corresponding author.E-mail address: [email protected] (T. Zhu).
efficiency, which was ascribed to the reactive oxygen speciesof MnOx. Aguero et al. [25] prepared the MnOx/Al2O3 and thematerial revealed as a well-defined catalyst in binary VOCs oxida-tion. For MnOx-containing composites, Wang et al. [26] reportedCeO2–MnOx catalytic oxidation of benzene with high efficiency,and CeO2 enhance the transfer ability of oxygen species. The meso-porous Mn–Ce composite exhibited higher catalytic activity in thetotal oxidation of VOCs [27].
Based on previous work, the Mn-containing materials are use-ful for developing a stable catalyst with low costs and high activity,and the mechanism study are helpful to optimize the MnOx-basedcatalytic system. However, the mechanism research such as theorganic intermediates, the interaction between the Mn active cen-ter and organic species, and the reaction path, are far less explored.Herein, we present MnOx/TiO2 catalyzed benzene oxidation and itsmechanism studies.
2. Experimental
2.1. Catalyst preparation
MOx/TiO2 catalysts were prepared using impregnation methodwith the weight percentages of MOx and TiO2 being 3 wt.%and 97 wt.%, respectively. In a typical preparation for CeO2/TiO2,Ce(NO3)2·6H2O (0.391 g, 0.9 mmol) was dissolved in 20 mL of
http://dx.doi.org/10.1016/j.molcata.2015.07.0241381-1169/© 2015 Elsevier B.V. All rights reserved.

222 J. Zeng et al. / Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227
100 150 200 250 300 350 400 450
0
20
40
60
80
100
(a)
Temperature/ oC
%noisrevnoC
MnOx(3%)/TiO
2
CeO2(3%)/TiO
2
Co3O
4(3%)/TiO
Fe2O
3(3%)/TiO
2
TiO2
100 150 200 250 300 350 400
0
20
40
60
80
100
%noisrevnoC
Temperature/ oC
MnOx(1%)/TiO
2
MnOx(3%)/TiO
2
MnOx(5%)/TiO
2
MnOx(10%)/TiO
2
MnOx(15%)/TiO
2
MnOx(20%)/TiO
2
(b)
Fig. 1. Benzene conversions as a function of reaction temperature over (a) MOx/TiO2
(M = Mn, Ce, Co, Fe) and (b) MnOx(y)/TiO2 (y = 1%, 3%, 5%, 10%, 15%, 20%).
deionized water, and the solution was then added into 80 mL ofaqueous suspension of TiO2 (5 g, 62.6 mmol). The mixture wasstirred for 2 h at 60 ◦C. The solvent was then removed with rotaryevaporation at 60 ◦C and kept for 2 h. The resulting sample wasdried at 110 ◦C for 10 h and calcined at 500 ◦C for 4 h, giving theexpected CeO2/TiO2 catalyst. Similarly, MnOx/TiO2, Co3O4/TiO2,Fe3O4/TiO2 catalysts were prepared adopting the same method.Additionally, MnOx(y)/TiO2 catalysts were prepared through thesame method, where y represents the weight percentage MnOx
(y = 1%, 3%, 5%, 10%, 15%, 20%).
2.2. Catalyst characterization
The samples were characterized using the following equip-ments. X-ray diffraction (XRD) patterns were recorded by Cu K˛radiation on Netherlands PANalytical X ‘Pert PRO diffractometerfor phase identification in the samples. Specific surface areas (BET)were recorded by the US Quanta Chrome’s Autosorb-iQ automaticinstrument. Scanning electron microscope (SEM) images wereobtained using Hitachi S-4800. Transmission electron microscope(TEM) images and high-resolution transmission electron micro-scope (HRTEM) images were taken on JEOL JEM-2100 microscopeoperated at 120 kV. X-ray photoelectron spectroscopy (XPS) mea-surements were conducted on Thermo escalab 250Xi X-ray usingan Al K˛ source.
Fig. 2. XRD patterns of (a) MOx/TiO2 (M = Mn, Ce, Co, Fe) and (b) MnOx(y)/TiO2
(y = 1%, 3%, 5%, 10%, 15%, 20%).
Table 1The physical properties of MnOx(y)/TiO2.
Sample Specific surfacearea/(m2 g−1)
Particle size(nm)a
Particle size(nm)b
TiO2 51.3 25.0 25.0MnOx(1%)/TiO2 50.2 25.3 25.0MnOx(3%)/TiO2 49.7 25.5 25.0MnOx(5%)/TiO2 49.5 25.1 25.0MnOx(10%)/TiO2 49.6 25.6 25.0MnOx(15%)/TiO2 48.2 26.0 25.0MnOx(20%)/TiO2 48.4 25.9 25.0
a Data determined based on the XRD results according to the Scherrer equationusing the FWHM of the peak at 25.4◦ (2�).
b Data determined based on the TEM results.
2.3. Catalytic evaluation
Catalytic activities of the catalysts were evaluated in a contin-uous flow fixed-bed quartz microreactor (i.d. = 4 mm) from 100to 450 ◦C with 100 mg of catalyst (60–80 mesh). Benzene wasintroduced into reactant flow with its concentration of 500 ppm,and the O2 concentration is 20%. The total flow ratio of the reac-tant mixture was 100 mL/min with the weight hourly space velocity(WHSV) of 60,000 mL/(gh). Analysis of the reactants and productswere performed on-line with a gas chromatography–mass spec-trometry (GCMS-QP2010 Plus, Shimadzu).

J. Zeng et al. / Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227 223
Fig. 3. TEM images (a, d, g represent y = 5%, 10%, 15%, respectively), HRTEM images (b, e, h represent y = 5%, 10%, 15%, respectively) and EDX spectra (c, f, i represent y = 5%,10%, 15%, respectively) of the MnOx(y)/TiO2.
2.4. In situ FTIR experiments
FTIR spectra were collected with an FTIR spectrometer (Nicolet6700, Thermo) equipped with an MCT detector (cooled by liquidnitrogen) and a stainless steel IR cell (CaF2 windows). Primarily, theIR cell was flushed by flowing Ar (100 mL/min for 2 h) to ensure thatit was clean. Then the powder sample (about 45 mg) was pressedinto a supported disk and oxidized at 300 ◦C for 1 h in a 20% O2/Ar(100 mL/min) stream. Subsequently, the sample was cooled downto desired temperature and collected background in flowing 20%O2/Ar stream. Eventually, the IR spectra were collected at differ-ent times in flowing 20% O2/Ar and 50% benzene (100 mL/min)stream.
In order to examine the effect of the absorbed oxygen in catalyticoxidation of benzene, the powder sample was pressed into a sup-ported disk and desorbed at 300 ◦C for 1 h in an Ar (100 mL/min)stream. Then the sample was cooled down to desired tempera-ture and collected background in flowing Ar. Finally, the IR spectrawere collected at different times in flowing Ar and 50% benzene(100 mL/min) stream.
Table 2The XPS data of MnOx(y)/TiO2 catalysts.
Catalyst Mn2+
(%)Mn3+
(%)Mn4+
(%)Olatt
(%)Oads
(%)Osur
(%)Oads/Olatt
MnOx (1%)/TiO2 7 22.8 70.2 67.6 25.35 7.04 0.375MnOx (5%)/TiO2 6.6 64.1 29.3 70.76 20 9.23 0.283MnOx (15%)/TiO2 7 77.46 15.49 78.26 18.8 2.8 0.24MnOx (20%)/TiO2 1.66 83.3 15.01 85.09 14.58 0.92 0.17
3. Results and discussion
3.1. Catalytic performance
The catalytic oxidation of benzene over all catalysts was estab-lished within 100–450 ◦C. As shown in Fig. 1a, the catalytic activityincreased in sequence of TiO2, CeO2/TiO2, Fe2O3/TiO2, Co3O4/TiO2,and MnOx/TiO2, and T50 (50% benzene conversion rate correspond-ing to the reaction temperature) for the catalysts were 418 ◦C,417 ◦C, 332 ◦C, 320 ◦C, and 295 ◦C, respectively. MnOx/TiO2 showedthe lowest complete oxidation temperature of 350 ◦C, revealing

224 J. Zeng et al. / Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227
536 534 532 530 528
Olatt
MnOx(1%)/TiO
2
Osur
Oads
Binding energy(eV)
).u.a(ytisnetnIO1s
536 534 532 530 528
MnOx(5%)/TiO
2
Osur
Oads
Olatt
O1s
Inte
nsity
(a.u
.)
Binding energy(eV)
536 534 532 530 528
Osur
Oads
Olatt
O1s
MnOx(20%)/TiO
2
Binding energy(eV)
Inte
nsity
(a.u
.)
536 534 532 530 528
MnOx(15%)/TiO
2
Osur
Oads
).u.a(ytisnetnI
Binding energy(eV)
O1s
Olatt
a
c
b
d
Fig. 4. XPS spectra of O 1s for MnOx(y)/TiO2 of (a), (b), (c), (d) (y = 1%, 5%, 15%, 20%).
that the Mn-based catalyst exhibit the highest catalytic activ-ity. Accordingly, a series of MnOx/TiO2 catalysts with differentMnOx contents (1–20 wt.%) were prepared. The samples were thenemployed in the catalytic oxidation of benzene and the resultswere shown in Fig. 1b. It can be seen that the catalytic activ-ity rose as the increase of the MnOx weight percentage withinthe range of (1–10 wt.%). MnOx(10%)/TiO2 contributed the lowestcomplete oxidation temperature of 325 ◦C. However, the samplesof MnOx(10%)/TiO2, MnOx(15%)/TiO2 and MnOx(20%)/TiO2 showedsimilar catalytic performance, revealing that further increase of theMnOx amount led to no higher efficiency. Noteworthy, no byprod-ucts were observed in our experiments. In order to evaluate thecatalytic stability of the samples, on-stream benzene oxidationexperiments were performed at 325 ◦C for 12 h. The results areshown in Fig. S1. The samples of MnOx(10%)/TiO2, MnOx(15%)/TiO2,and MnOx(20%)/TiO2 gave the complete oxidation, whereas theconversions for MnOx(3%)/TiO2 and MnOx(5%)/TiO2 were 83% and92%, respectively and it was consistent with Fig. 1b. Therefore, itwas disclosed that the MnOx(y)/TiO2 have extremely stability.
3.2. Characterization results and analysis
From the XRD patterns of the as-prepared MOx/TiO2 samplesin Fig. 2a, it can be seen that these materials exhibited 25.4◦,48.1◦, 53.6◦, 55.2◦ (2�) peaks due to anatase TiO2 (PDF 21-1272),and 27.5◦, 36.2◦, 54.4◦ (2�) peaks attributable to rutile TiO2 (PDF
21-1276). Any MnOx, CeO2, Co3O4, and Fe2O3 phases were notobserved, possibly because that little MOx was loaded leading towell-dispersion of the small crystallite particles [28–30]. The XRDpatterns of MnOx(y)/TiO2 catalysts were shown in Fig. 2b. Forthe samples with MnOx contents above 5%, the diffraction peaksat 28.8◦, 42.9◦, 56.7◦ (2�) ascribed to MnO2 (PDF 24-0735) wereobserved, and the peak intensity was gradually strengthened asthe increasing content of MnOx. When the MnOx content reachedto 20%, the diffraction peaks at 33.0◦, 55.3◦, 65.9◦ (2�) characteris-tic of Mn2O3 (PDF 41-1442) appeared. Therefore, MnOx(20%)/TiO2sample contains at least two valence states of Mn (Mn4+, Mn3+). Forother catalysts, multiple valence states might also exist, whereasthe XRD diffraction signals could not be detected possibly due totheir less multi-valence contents or the detection limitation.
The N2 adsorption–desorption isotherms and BJH pore size dis-tribution analysis were performed to evaluate the specific surfacearea and the particle size of the MnOx(y)/TiO2 catalysts. As shownin Table 1, with the increased of MnOx contents, the specific surfacearea showed a very slight decreasing tendency. The particles sizedetermined based on XRD and TEM results were also illustratedwith an average value of 25 nm, showing no significant difference.
The SEM images of MnOx(1%)/TiO2 and MnOx(20%)/TiO2 arelisted as Fig. S2. As shown in Fig.S2a, the sample presents a uni-form size and round structure, and the particles are well dispersed.The morphology of MnOx(1%)/TiO2 showed no apparent differencefrom that of MnOx(20%)/TiO2 shown in Fig.S2b. Hence, it could be
J. Zeng et al. / Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227 225
0.15 0.20 0.25 0.30 0.35 0.400
5
10
15
20
T=225 oC
(F
OT
x10
6-S
-1)
Oads
/Olatt
molar ratio (mol/mol)
T=275 oC
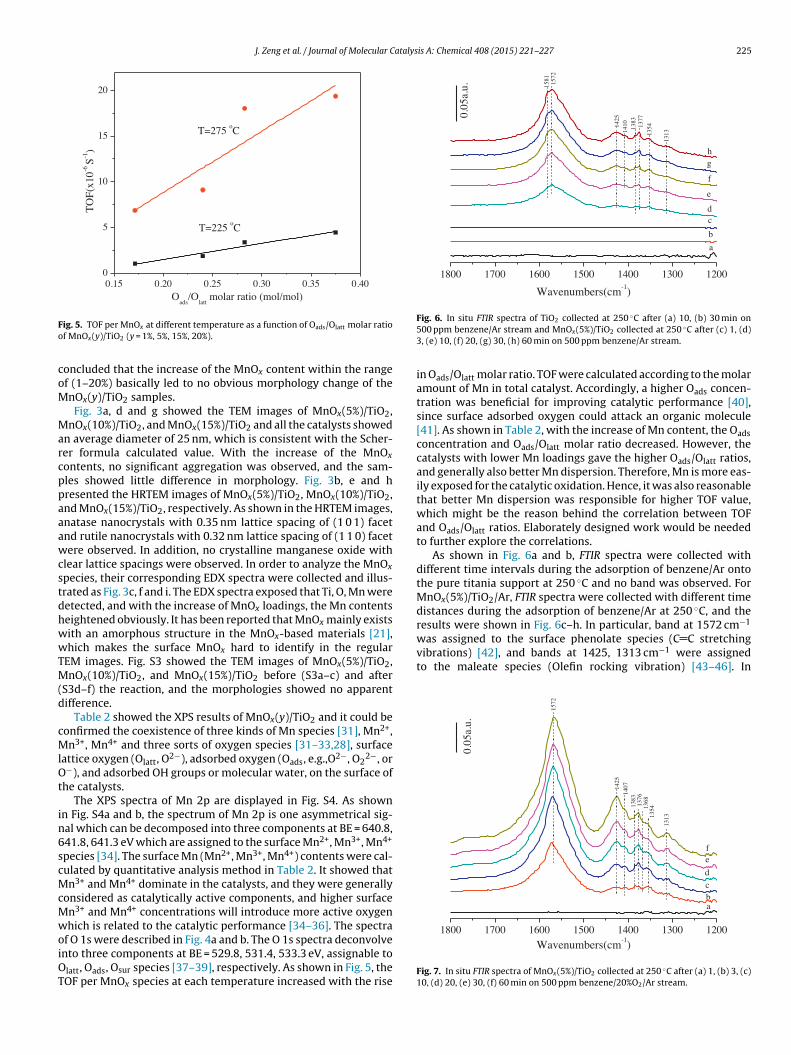
Fig. 5. TOF per MnOx at different temperature as a function of Oads/Olatt molar ratioof MnOx(y)/TiO2 (y = 1%, 5%, 15%, 20%).
concluded that the increase of the MnOx content within the rangeof (1–20%) basically led to no obvious morphology change of theMnOx(y)/TiO2 samples.
Fig. 3a, d and g showed the TEM images of MnOx(5%)/TiO2,MnOx(10%)/TiO2, and MnOx(15%)/TiO2 and all the catalysts showedan average diameter of 25 nm, which is consistent with the Scher-rer formula calculated value. With the increase of the MnOx
contents, no significant aggregation was observed, and the sam-ples showed little difference in morphology. Fig. 3b, e and hpresented the HRTEM images of MnOx(5%)/TiO2, MnOx(10%)/TiO2,and MnOx(15%)/TiO2, respectively. As shown in the HRTEM images,anatase nanocrystals with 0.35 nm lattice spacing of (1 0 1) facetand rutile nanocrystals with 0.32 nm lattice spacing of (1 1 0) facetwere observed. In addition, no crystalline manganese oxide withclear lattice spacings were observed. In order to analyze the MnOx
species, their corresponding EDX spectra were collected and illus-trated as Fig. 3c, f and i. The EDX spectra exposed that Ti, O, Mn weredetected, and with the increase of MnOx loadings, the Mn contentsheightened obviously. It has been reported that MnOx mainly existswith an amorphous structure in the MnOx-based materials [21],which makes the surface MnOx hard to identify in the regularTEM images. Fig. S3 showed the TEM images of MnOx(5%)/TiO2,MnOx(10%)/TiO2, and MnOx(15%)/TiO2 before (S3a–c) and after(S3d–f) the reaction, and the morphologies showed no apparentdifference.
Table 2 showed the XPS results of MnOx(y)/TiO2 and it could beconfirmed the coexistence of three kinds of Mn species [31], Mn2+,Mn3+, Mn4+ and three sorts of oxygen species [31–33,28], surfacelattice oxygen (Olatt, O2−), adsorbed oxygen (Oads, e.g.,O2−, O2
2−, orO−), and adsorbed OH groups or molecular water, on the surface ofthe catalysts.
The XPS spectra of Mn 2p are displayed in Fig. S4. As shownin Fig. S4a and b, the spectrum of Mn 2p is one asymmetrical sig-nal which can be decomposed into three components at BE = 640.8,641.8, 641.3 eV which are assigned to the surface Mn2+, Mn3+, Mn4+
species [34]. The surface Mn (Mn2+, Mn3+, Mn4+) contents were cal-culated by quantitative analysis method in Table 2. It showed thatMn3+ and Mn4+ dominate in the catalysts, and they were generallyconsidered as catalytically active components, and higher surfaceMn3+ and Mn4+ concentrations will introduce more active oxygenwhich is related to the catalytic performance [34–36]. The spectraof O 1s were described in Fig. 4a and b. The O 1s spectra deconvolveinto three components at BE = 529.8, 531.4, 533.3 eV, assignable toOlatt, Oads, Osur species [37–39], respectively. As shown in Fig. 5, theTOF per MnOx species at each temperature increased with the rise
1800 1700 1600 1500 1400 1300 1200
1410
.u.a50.0
b
d c
f
e
h g
a
131313
541377
1383
1425
1581 1572
Wavenumbers(cm-1)
Fig. 6. In situ FTIR spectra of TiO2 collected at 250 ◦C after (a) 10, (b) 30 min on500 ppm benzene/Ar stream and MnOx(5%)/TiO2 collected at 250 ◦C after (c) 1, (d)3, (e) 10, (f) 20, (g) 30, (h) 60 min on 500 ppm benzene/Ar stream.
in Oads/Olatt molar ratio. TOF were calculated according to the molaramount of Mn in total catalyst. Accordingly, a higher Oads concen-tration was beneficial for improving catalytic performance [40],since surface adsorbed oxygen could attack an organic molecule[41]. As shown in Table 2, with the increase of Mn content, the Oadsconcentration and Oads/Olatt molar ratio decreased. However, thecatalysts with lower Mn loadings gave the higher Oads/Olatt ratios,and generally also better Mn dispersion. Therefore, Mn is more eas-ily exposed for the catalytic oxidation. Hence, it was also reasonablethat better Mn dispersion was responsible for higher TOF value,which might be the reason behind the correlation between TOFand Oads/Olatt ratios. Elaborately designed work would be neededto further explore the correlations.
As shown in Fig. 6a and b, FTIR spectra were collected withdifferent time intervals during the adsorption of benzene/Ar ontothe pure titania support at 250 ◦C and no band was observed. ForMnOx(5%)/TiO2/Ar, FTIR spectra were collected with different timedistances during the adsorption of benzene/Ar at 250 ◦C, and theresults were shown in Fig. 6c–h. In particular, band at 1572 cm−1
was assigned to the surface phenolate species (C C stretchingvibrations) [42], and bands at 1425, 1313 cm−1 were assignedto the maleate species (Olefin rocking vibration) [43–46]. In
1800 1700 1600 1500 1400 1300 1200
.u.a50.0
1368
131313
54
1376
1383
140714
25
1572
b
d
fe
c
Wavenumbers(cm-1)
a
Fig. 7. In situ FTIR spectra of MnOx(5%)/TiO2 collected at 250 ◦C after (a) 1, (b) 3, (c)10, (d) 20, (e) 30, (f) 60 min on 500 ppm benzene/20%O2/Ar stream.

226 J. Zeng et al. / Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227
Fig. 8. Proposed mechanism for the catalytic oxidation of benzene over MnOx/TiO2 catalysts.
addition, bands at 1383 (CH in plane bending), 1377 (CH2 stretch-ing vibration), 1354 (CH3 stretching vibration) cm−1 were assignedto the acetate species [47,48] and band at 1410 cm−1 was assignedto o-benzoquinone [45], respectively. As the reaction proceeding, alow intensity band at 1581 cm−1 was emerged that can be deemedthe unsaturated species (C C stretching vibrations) [49,50], and inthis research it could be phenolate other than benzene. If the bandat 1581 cm−1 was ascribed to benzene, it should be observed inthe Fig. 6a and b. Consequently, it could be concluded that ben-zene decomposed over MnOx(5%)/TiO2 at 250 ◦C under Ar, andphenolate, o-benzoquinone, maleate, and acetate species wereaccordingly produced. In combination with the XPS analysis, it wasbelieved that the Oads species (active oxygen) play a major role inthe oxidation process.
As shown in Fig. 7a–f, MnOx(5%)/TiO2 FTIR spectra were col-lected during the adsorption of benzene/20%O2/Ar at 250 ◦C. Incomparison to Fig. 6, similar bands at 1572, 1425, 1383, 1377,1354, 1313 cm−1 due to different organic species were foundin Fig. 7. Meanwhile, band at 1410 cm−1 shifted to 1407 cm−1,band at 1368 cm−1 was emerged and band at 1581 cm−1 disap-peared. Hence, it could be proposed that benzene was oxidizedinto phenolate, o-benzoquinone, maleate and acetate species overMnOx(5%)/TiO2 at 250 ◦C in the presence of an O2/Ar mixture. By thecomparison of Figs. 6 and 7, it could be concluded that the oxidationof benzene proceeds in the same path under oxygen-rich or oxygen-free conditions. It was believed that benzene reacted with the activeoxygen species of the catalyst, other than the gas oxygen. Whilethe active oxygen was consumed, the gas oxygen was adsorbedon the catalyst and transformed into active oxygen species,accomplishing the catalytic oxidation process on the catalystsurface.
Based on the results of in situ FTIR experiments, a reaction mech-anism was proposed and shown in Fig. 8. As one can see, benzene (A)first reacts with the active Mn center, giving phenolate (B) specieswith two conjugated structures (C). The oxygen-containing group isgenerally considered as an electron-donating and ortho-para pos-itioned director. Accordingly, the phenolate species can be easilyoxidized into o-benzoquinone (D) and p-benzoquinone (E). How-ever, p-benzoquinone (E) was not observed possibly due to its highreactivity. The quinone species were then further oxidized. Withthe catalyst promoting and the attack of the active oxygen species,a ring opening occurred, affording the small molecule interme-diates, such as maleate (F) and acetate (G) species, which wereindeed observed in the in situ experiments. Furthermore, oxida-tion of maleate (F) and acetate (G) species gave the final inorganicproducts of CO2, CO, and H2O.
4. Conclusion
MOx/TiO2 (M = Mn, Ce, Co, Fe) samples with certain contentswere prepared and employed as the catalysts for benzene oxida-tion, and MnOx/TiO2 showed the highest catalytic efficiency. Asthe MnOx content increased, the catalytic activity also showed anincreasing tendency. However, the catalytic activity increased lit-tle when the MnOx content was above 5%. XPS characterizationsrevealed that Oads species (active oxygen) play a major role in theoxidation process. Many organic intermediates were observed inthe in situ FTIR experiments. Accordingly, a reaction mechanismfor the catalytic oxidation of benzene over MnOx/TiO2 was pro-posed, which is meaningful to the future development of the VOCsremoval catalysts.
Acknowledgements
This work was supported by Project of the Natural Science Foun-dation of China (No. 21177129), and the Strategic Priority ResearchProgram of the Chinese Academy of Sciences (No. XDB05050100).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.molcata.2015.07.024
References
[1] G. Liu, M. Zheng, P. Lv, W. Liu, C. Wang, B. Zhang, K. Xiao, Environ. Sci. Technol.44 (2010) 8156–8161.
[2] G. Liu, M. Zheng, Environ. Sci. Technol. 47 (2013) 8093–8094.[3] B. Du, M. Zheng, Y. Huang, A. Liu, H. Tian, L. Li, N. Li, T. Ba, Y. Li, S. Dong, W. Liu,
G. Su, Environ. Sci. Technol. 44 (2010) 5818–5823.[4] B. Solsona, E. Aylón, R. Murillo, A.M. Mastral, A. Monzonís, S. Agouram, T.E.
Davies, S.H. Taylor, T. Garcia, J. Hazard. Mater. 187 (2011) 544–552.[5] M. Chen, L. Fan, L. Qi, X. Luo, R. Zhou, X. Zheng, Catal. Commun. 10 (2009)
838–841.[6] F. Regali, L.F. Liotta, A.M. Venezia, V. Montes, M. Boutonnet, S. Järås, Catal. Today
223 (2014) 87–96.[7] F. Regali, L.F. Liotta, A.M. Venezia, M. Boutonnet, S. Järås, Appl. Catal. A: Gen.
469 (2014) 328–339.[8] L.F. Liotta, Catalysts 4 (2014) 299–304.[9] F. Yu, Z. Qu, X. Zhang, Q. Fu, Y. Wang, J. Energy. Chem. 22 (2013) 845–852.
[10] W. Li, H. Gong, Acta Phys.-Chim. Sin. 26 (2010) 885–894.[11] W. Li, J. Wang, H. Gong, Catal. Today 148 (2009) 81–87.[12] G. Zhou, B. Zhong, W. Wang, X. Guan, B. Huang, D. Ye, H. Wu, Catal. Today 175
(2011) 157–163.[13] Y. Su, B. Fan, L. Wang, Y. Liu, B. Huang, M. Fu, L. Chen, D. Ye, Catal. Today 201
(2013) 115–121.[14] M. Lu, R. Huang, J. Wu, M. Fu, L. Chen, D. Ye, Catal. Today B 242 (2015) 274–286.

J. Zeng et al. / Journal of Molecular Catalysis A: Chemical 408 (2015) 221–227 227
[15] L. Tian, D. Ye, H. Liang, Catal. Today 78 (2003) 159–170.[16] H. Liang, Y. Hong, C. Zhu, S. Li, Y. Chen, Z. Liu, D. Ye, Catal. Today 201 (2013)
98–102.[17] H. Huang, D. Ye, X. Guan, Catal. Today 139 (2008) 43–48.[18] B. Huang, R. Huang, D. Jin, D. Ye, Catal. Today 126 (2007) 279–283.[19] Y. Guo, X. Liao, J. He, W. Ou, D. Ye, Catal. Today 153 (2010) 176–183.[20] S. Lin, G. Su, M. Zheng, D. Ji, M. Jia, Y. Liu, Appl. Catal. B: Environ. 123 (2012)
440–447.[21] Y. Ma, Y. Li, M. Mao, J. Hou, M. Zeng, X. Zhao, J. Mater. A: Chem. 3 (2015)
5509–5516.[22] A.M. Venezia, V. La Parola, L.F. Liotta, G. Pantaleo, M. Lualdi, M. Boutonnet, S.
Järås, Catal. Today 197 (2012) 18–23.[23] J. Deng, L. Zhang, H. Dai, H. He, C.T. Au, J. Mol. Catal. A: Chem. 299 (2009) 60–67.[24] T. Ataloglou, J. Vakros, K. Bourikas, C. Fountzoula, C. Kordulis, A. Lycourghiotis,
Appl. Catal B: Environ. 57 (2005) 299–312.[25] F.N. Aguero, B.P. Barbero, L. Gambaro, L.E. Cadús, Appl. Catal. B: Environ. 91
(2009) 108–112.[26] Z. Wang, G. Shen, J. Li, H. Liu, Q. Wang, Y. Chen, Appl. Catal. B: Environ. 138
(2013) 253–259.[27] W. Tang, X. Wu, G. Liu, S. Li, D. Li, W. Li, Y. Chen, J. Rare Earths 33 (2015) 62–69.[28] Q. Dai, S. Bai, Z. Wang, X. Wang, G. Lu, Appl. Catal. B: Environ. 126 (2012) 64–75.[29] L.F. Liotta, A.M. Venezia, G. Deganello, A. Longo, A. Martorana, Z. Schay, L. Guczi,
Catal. Today 66 (2001) 271–276.[30] L.F. Liotta, G. Pantaleo, A. Macaluso, G. Di Carlo, G. Deganello, Appl. Catal. A:
Gen. 245 (2003) 167–177.[31] W. Tang, W. Li, D. Li, G. Liu, X. Wu, Y. Chen, Catal. Lett. 144 (2014) 1900–1910.
[32] Q. Dai, S. Bai, J. Wang, M. Li, X. Wang, G. Lu, Appl. Catal. B: Environ. 142–143(2013) 222–233.
[33] H. Huang, Q. Dai, X. Wang, Appl. Catal. B: Environ. 158–159 (2014) 96–105.[34] S. Azalim, R. Brahmi, M. Agunaou, A. Beaurain, J.M. Giraudon, J.F. Lamonier,
Chem. Eng. J. 223 (2013) 536–546.[35] J. Trawczynski, B. Bielak, W. Mista, Appl. Catal. B: Environ. 55 (2005) 277–285.[36] D. Döbber, D. Kießling, W. Schmitz, G. Wendt, Appl. Catal. B: Environ. 52 (2004)
135–143.[37] A.F. Carley, M.W. Roberts, A.K. Santra, J. Phys. B: Chem. 101 (1997) 9978–9983.[38] J. Niu, J. Deng, W. Liu, L. Zhang, G. Wang, H. Dai, H. He, X. Zi, Catal. Today 126
(2007) 420–429.[39] L.F. Liotta, F. Puleo, V. La Parola, S.G. Leonardi, N. Donato, D. Aloisio, G. Neri,
Electroanalysis 27 (2015) 684–692.[40] S. Rousseau, S. Loridant, P. Delichere, A. Boreave, J.P. Deloume, P. Vernoux, Appl.
Catal. B: Environ. 88 (2009) 438–447.[41] S. Ponce, M.A. Pena, J.L.G. Fierro, Appl. Catal. B: Environ. 24 (2000) 193–205.[42] J. Bandara, J.A. Mielczarski, J. Kiwi, Appl. Catal. B: Environ. 34 (2001) 307–320.[43] Z. Sarbak, React. Kinet. Catal. Lett. 84 (2005) 263–270.[44] G. Busca, G. Ramis, V. Lorenzelli, J. Mol. Catal. 50 (1989) 231–240.[45] J. Lichtenberger, M.D. Amiridis, J. Catal. 223 (2004) 296–308.[46] C.E. Hetrick, J. Lichtenberger, M.D. Amiridis, Appl. Catal. B: Environ. 77 (2008)
255–263.[47] X. Li, X. Zou, Z. Qu, Q. Zhao, L. Wang, Chemosphere 83 (2011) 674–679.[48] S. Krishnamoorthy, J.A. Rivas, M.D. Amiridis, J. Catal. 193 (2000) 264–272.[49] J. Wang, X. Wang, X. Liu, T. Zhu, Y. Guo, H. Qi, Catal. Today 241 (2015) 92–99.[50] S. Krishnamoorthy, M.D. Amiridis, Catal. Today 51 (1999) 203–214.
Related Documents




![Artikel Ilmiah [Agasi Dwi Pradana - C1B008067]](https://static.cupdf.com/doc/110x72/54a1c991ac7959ce688b4889/artikel-ilmiah-agasi-dwi-pradana-c1b008067.jpg)






