Uniwersytet Warszawski Wydział Chemii Edyta Matysiak-Brynda Elektrograwimetryczna detekcja wybranych metaloprotein w roztworach i płynach ustrojowych Praca doktorska wykonana w Pracowni Teorii i Zastosowań Elektrod Zakładu Chemii Nieorganicznej i Analitycznej promotor: dr hab. Anna M. Nowicka Warszawa, 2017

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Uniwersytet Warszawski
Wydział Chemii
Edyta Matysiak-Brynda
Elektrograwimetryczna detekcja
wybranych metaloprotein
w roztworach i płynach ustrojowych
Praca doktorska wykonana
w Pracowni Teorii i Zastosowań Elektrod
Zakładu Chemii Nieorganicznej i Analitycznej
promotor: dr hab. Anna M. Nowicka
Warszawa, 2017


Pracę dedykuję Mężowi oraz Rodzicom
Za wiarę i ciągłe wsparcie w dążeniu do celu


Pragnę serdecznie podziękować:
Dr hab. Annie M. Nowickiej za opiekę naukową,
nieocenioną pomoc, cenne uwagi, poświęcony czas i wyrozumiałość.
Dr Agacie Kowalczyk za współpracę,
cenne uwagi i pomoc przy oprawie graficznej pracy.
Prof. dr hab. Mikołajowi Dontenowi,
za cenne uwagi dotyczące wpływu pola magnetycznego
na paramagnetyczne białka.
Koleżankom i Kolegom z Pracowni Teorii i Zastosowań Elektrod
za serdeczną atmosferę.


Niniejsza praca była częściowo finansowana z grantu Narodowego
Centrum Nauki, Preludium 9 Nr 2015/17/N/ST4/03933


Dorobek naukowy wypracowany na podstawie wyników badań
opisanych w nniejszej rozprawie doktorskiej stał się podstawą uzyskania
stypendium Ministra Nauki i Szkolnictwa Wyższego za wybitne
osiągnięcia w roku akademickim 2016/2017


Spis treści
11
Spis treści
- Część literaturowa -
1. WSTĘP .............................................................................................................. 17
2. CEL PRACY ..................................................................................................... 19
3. PODZIAŁ PŁYNÓW USTROJOWYCH, ICH SKŁAD
ORAZ FUNKCJE PEŁNIONE W ORGANIZMIE ...................................... 21
3.1. Skład i funkcje krwi ..................................................................................... 22
3.2. Równowaga kwasowo-zasadowa płynów ustrojowych .............................. 29
4. BIAŁKA WAŻNE SKŁADNIKI ORGANIZMU ...................................... 31
4.1. Definicja i budowa metaloprotein ............................................................... 33
4.2. Charakterystyka wybranych metaloprotein obecnych we krwi ................... 37
4.2.1. Hemoglobina, czerwony barwnik krwi wiążący
tlen i tlenek węgla(IV) ..................................................................... 37
4.2.2. Ceruloplazmina, nośnik jonów miedzi w organizmie ....................... 43
4.2.3. Transferyna, białko transportujące jony żelaza w organizmie .......... 49
4.2.4. Ferrytyna, białko magazynujące żelazo w organizmie ..................... 54
4.3. Metody oznaczania zawartości białek w płynach ustrojowych ................... 59
5. ELEKTROAKTYWNOŚĆ METALOPROTEIN ......................................... 65
5.1. Sposoby transportu elektronów pomiędzy metaloproteiną
a powierzchnią elektrody ............................................................................ 65
5.1.1. Bezpośredni transport elektronów .................................................... 67
5.1.2. Transport elektronów z wykorzystaniem mediatora ......................... 70
5.1.3. Sposoby unieruchamiania białek na wybranej powierzchni
i ich konsekwencje ........................................................................... 72
6. NANOMATERIAŁY TYPU „RDZEŃ − POWŁOKA” ............................... 79
6.1. Nanocząstki magnetyczne typu „rdzeń – powłoka” .................................... 80
6.1.1. Nanokapsuły węglowe zawierające żelazo ....................................... 81
6.2. Zastosowanie magnetycznych nanokapsuł węglowych
z metalicznym rdzeniem ............................................................................. 83

Spis treści
12
7. UDZIAŁ POLA MAGNETYCZNEGO W TRANSPORCIE
SUBSTANCJI ELEKTROAKTYWNEJ DO POWIERZCHNI
ELEKTRODY ................................................................................................... 87
8. TECHNIKI BADAWCZE STOSOWANE W PRACY................................. 91
8.1. Woltamperometria cykliczna i liniowa ........................................................ 91
8.2. Woltamperometria pulsowa róźnicowa ....................................................... 93
8.3. Elektrochemiczna mikrowaga kwarcowa .................................................... 95
8.4. Elektrochemiczna spektroskopia impedancyjna .......................................... 97
8.5. Skaningowy mikroskop elektrochemiczny .................................................. 99
8.6. Spektroskopia UV-vis ................................................................................ 100
- Część eksperymentalna -
9. ODCZYNNIKI CHEMICZNE I APARATURA ......................................... 103
9.1. Charakterystyka nanocząstek magnetycznych: składnika warstwy
receptorowej czujników ............................................................................ 106
9.1.1. Niezmodyfikowane nanokapsuły węglowe zawierające żelazo ..... 106
9.1.2. Nanokapsuły węglowe zawierające żelazo zmodyfikowane
grupami karboksylowymi ............................................................... 107
10. ELEKTROGRAWIMETRYCZNY SENSOR DO
BEZPOŚREDNIEJ DETEKCJI HEMOGLOBINY
W PRÓBKACH KRWI .................................................................................. 109
10.1. Optymalizacja warunków występowania paramagnetycznej formy
hemoglobiny ............................................................................................ 110
10.2. Procedura przygotowania warstwy receptorowej czujnika i jej
elektrochemiczna charakterystyka ........................................................... 112
10.3. Elektrochemiczna i grawimetryczna charakterystyka sensora do
bezpośredniej detekcji hemoglobiny ....................................................... 116
10.4. Funkcjonalność sensora do bezpośredniej detekcji Hb w próbkach
naturalnych .............................................................................................. 121
10.5. Podsumowanie ......................................................................................... 123

Spis treści
13
11. WPŁYW POLA MAGNETYCZNEGO NA AKTYWNOŚĆ
ELEKTROCHEMICZNĄ I KATALITYCZNĄ
CERULOPLAZMINY .................................................................................... 125
11.1. Analityczna charakterystyka czujnika .................................................... 125
11.1.1. Jednoczesna ilościowa analiza ceruloplazminy i hemoglobiny
we krwi szczurzej ......................................................................... 133
11.2. Wpływ pola magnetycznego na aktywność katalityczną
ceruloplazminy unieruchomionej na podłożu złotym
zmodyfikowanym ferromagnetykiem...................................................... 136
11.2.1. Wpływ ferromagnetycznego modyfikatora powierzchni na
elektroaktywność złota ................................................................ 136
11.2.2. Aktywność katalityczna ceruloplazminy wobec
jonów żelaza(II) ........................................................................... 138
11.3. Podsumowanie ......................................................................................... 146
12. ELEKTROCHEMICZNY IMMUNOSENSOR DO DETEKCJI
TRANSFERYNY ........................................................................................... 147
12.1. Procedura konstrukcji warstwy receptorowej immunosensora ............... 148
12.2. Analityczna charakterystyka immunosensora
do elektrochemicznej detekcji Tf ............................................................ 153
12.2.1. Wykorzystanie oporności przeniesienia ładunku
do ilościowego opisu zakresu pracy immunosensora .................. 153
12.2.2. Grawimetryczna charakterystyka immunosensora ...................... 155
12.2.3. Woltamperometryczna detekcja transferyny ............................... 158
12.2.4. Stabilność, selektywność oraz odtwarzalność warstwy
receptorowej immunosensora ...................................................... 161
12.2.5. Zastosowanie immunosensora w analizie próbek krwi ............... 164
12.3. Wpływ pola magnetycznego i ferromagnetycznego modyfikatora
powierzchni elektrody na integralność konformacyjną
i elektroaktywność transferyny ............................................................... 165
12.3.1. Procedura syntezy koniugatu transferyna-nanocząstka
magnetyczna ................................................................................ 166
12.3.2. Charakterystyka spektroskopowa koniugatu Fe@C-COOH/Tf .. 167
12.3.3. Analiza termograwimetryczna koniugatu Fe@C-COOH/Tf ....... 170

Spis treści
14
12.3.4. Charakterystyka struktur białkowych na powierzchni
nanokapsuł węglowych przy użyciu mikrowagi kwarcowej
z dyssypacją energii ..................................................................... 172
12.3.5. Elektroaktywność transferyny kowalencyjnie związanej
z nanokapsułami Fe@C-COOH Nps ........................................... 176
12.4. Podsumowanie ......................................................................................... 177
13. ELEKTROCHEMICZNA DETEKCJA FERRYTYNY
Z WYKORZYSTANIEM WYSOCE SELEKTYWNEGO
ODDZIAŁYWANIA ANTYGEN–PRZECIWCIAŁO ................................ 179
13.1. Procedura konstrukcji immunosensora do elektrochemicznej
detekcji ferrytyny ..................................................................................... 181
13.2. Określenie wpływu ułożenia Ab w warstwie receptorowej
na ilość unieruchamianej ferrytyny przy użyciu technik
LA-ICP-MS i QCM ................................................................................. 184
13.3. Elektrochemiczna charakterystyka etapów tworzenia
warstwy receptorowej immunosensora .................................................... 187
13.3.1. Wpływ pola magnetycznego na sygnał prądowy ferrytyny ........ 191
13.3.2. Woltamperometryczna charakterystyka immunosensora
do detekcji ferrytyny .................................................................... 194
13.4. Selektywność immunosensora do detekcji ferrytyny .............................. 198
13.5. Oznaczenie ferrytyny w próbkach krwi ................................................... 200
13.6. Podsumowanie ......................................................................................... 201
14. STRESZCZENIE ............................................................................................ 203
15. STRESZCZENIE W JĘZYKU ANGIELSKIM .......................................... 209
16. SPIS PRAC NAUKOWYCH OPUBLIKOWANYCH
W TRAKCIE STUDIÓW DOKTORANCKICH ......................................... 215
17. BIBLIOGRAFIA ............................................................................................ 217

Alfabetyczny spis skrótów i symboli stosowanych w pracy
15
Alfabetyczny spis skrótów stosowanych w pracy:
Ab: przeciwciało
AΔf: amplituda oscylacji drgań kryształu
kwarcu
apo-Tf: apo-transferyna
Au: złota elektroda dyskowa
CD: spektroskopia dichroizmu kołowego
CEINs: nanocząstki żelaza enkapsulowane
węglem
Cp: ceruloplazmina
CSH: chlorowodorek cysteaminy
CV: woltamperometria cykliczna
deoksyHb: deoksyhemoglobina
DET: bezpośredni transport elektronów
DPV: woltamperometria pulsowa różnicowa
ECF: płyn zewnątrzkomórkowy
EDC: chlorowodorek 1-etylo-3-(dimetylo-
aminopropylo)karbodiimidu
EIS: elektrochemiczna spektroskopia
impedancyjna
EQCM: elektrochemiczna mikrowaga
kwarcowa
Fab: fragment wiążący przeciwciało
FB: tryb sprzężenia zwrotnego
Fc: fragment krystalizowalny przeciwciała
Fe@C Nps: nanokapsuły węglowe zawie-
rające żelazo
Fe@C-COOH Nps: nanokapsuły węglowe
zawierające żelazo zmodyfikowane grupami
karboksylowymi
ferryloHb: ferrylohemoglobina
Ft: ferrytyna
FTIR: spektroskopia w podczerwieni z trans-
formacją Fouriera
GC: elektroda z węgla szklistego
α-GP: α-glikoproteina
H: łańcuch ciężki białka
Hb: hemoglobina
holo-Tf: holo-transferyna
HSA: albumina surowicy ludzkiej
ICF: płyn wewnątrzkomórkowy
L: łańcuch lekki białka
LA-ICP-MS: spektrometria mas z jonizacją
w plazmie indukcyjnie sprzężonej z ablacją
laserową
LOD: granica wykrywalności
LSV: woltamperometria z liniowo zmienia-
jącym się potencjałem
MET: transport elektronów z użyciem
mediatora
metHb: methemoglobina
M Nps: nanocząstki magnetyczne
NHE: standardowa elektroda wodorowa
NHS: N-hydroksysukcynimid
oksoferryloHb: oksoferrylohemoglobina
oksyHb: oksyhemoglobina
PB: bufor fosforanowy
PLT: płytki krwi
QCM-D: mikrowaga kwarcowa z dyssypacją
energii
RBC: krwinki czerwone
SAM: samoorganizująca się monowarstwa
SECM: skaningowa mikroskopia elektroche-
miczna
SEM: skaningowa mikroskopia elektronowa
SPIONs: superparamagnetyczne nanocząstki
tlenku żelaza, Fe3O4
SPR: powierzchniowy rezonans plazmonowy
Tf: transferyna
TGA: analiza termograwimetryczna
WBC: krwinki białe
Skróty symboli stosowanych we wzorach:
A: powierzchnia elektrody
B: indukcja magnetyczna
C: stężenie substancji
Cf: stała charakterystyczna dla danego
kryształu kwarcu
COx: stężenie substancji elektroaktywnej
w formie utlenionej
CRed: stężenie substancji elektroaktywnej
w formie zredukowanej
Cdl: pojemność warstwy podwójnej
D: dyssypacja energii
Di: współczynnik dyfuzji substancji
elektroaktywnej
DOx: współczynnik dyfuzji substancji
w formie utlenionej
DRed: współczynnik dyfuzji substancji
w formie zredukowanej
Eapp.: potencjał przykładany do elektrody
w trakcie pomiaru
Ef: potencjał formalny układu redoks
Ep: amplituda pulsu
Ep, a: potencjał piku anodowego
Ep, k: potencjał piku katodowego
F: stała Faraday’a (96 486 C·mol-1
)
f: częstotliwość drgań kryształu kwarcu
FG: siła gradientu pola magnetycznego zwaną
również siłą Kelvina
FL: siła Lorentza
g: współczynnik geometryczny
H: natężenie pola magnetycznego
Ip, a: prąd piku anodowego
Ip, k: prąd piku katodowego
Iss: prąd stacjonarny

Alfabetyczny spis skrótów i symboli stosowanych w pracy
16
Jdyfuz.: strumień substancji wywołany
transportem dyfuzyjnym
Jkonwek.: strumień substancji wywołany
konwekcją
Jmigr.: strumień substancji wywołany migracją
n: liczba elektronów uczestniczących
w reakcji elektrodowej/ numer nadtonu
częstotliwości rezonansowej
Rct: opór przeniesienia ładunku
t: czas
T: temperatura
Tdl: parametr pojemnościowy elementu stało-
fazowego
Q: ładunek
W: impedancja Warburga
W0.5: szerokość piku w połowie wysokości
v: szybkość przemiatania potencjałem
Zf: impedancja faradajowska
zi: ładunek i-tej substancji
Yb: średnia wartość sygnału ślepej próby
Symbole greckie:
Δ: zmiana
: gradient
: molowy współczynnik absorpcji
: potencjał elektryczny w roztworze
dl: parametr potęgowy elementu stało-
fazowego
Γ: stężenie powierzchniowe
σb: odchylenie standardowe dla ślepej próby

Wstęp
17
1. Wstęp
Białka, często nazywane prawdziwymi „cząsteczkami życia”, są
podstawowym elementem budulcowym każdej komórki ludzkiego ciała. Większość
białek występujących w organizmie ludzkim to białka wewnątrzkomórkowe, których
poziom ekspresji jest zróżnicowany i zależy od typu komórek lub tkanek, w których
one powstają. Zsyntezowane w odpowiednich komórkach białka wydzielane są do
osocza oraz płynu śródmiąższowego, gdzie zaczynają pełnić swoje funkcje. Osocze
zawiera zdecydowanie więcej białek niż płyn śródmiąższowy. Dodatkowo, białka
osocza, jako jedyne z jego składników, nie przedostają się do płynu
śródmiąższowego. Poziom białek w płynach ustrojowych może ulegać zmianie wraz
z wiekiem, a także w różnych stanach fizjologicznych i patologicznych. Zatem są
one doskonałym wskaźnikiem stanu zdrowia organizmu. Ważną grupę wśród białek
stanowią metaloproteiny, czyli białka złożone, zawierające w swojej strukturze
atomy lub jony metali. Metaloproteiny od lat nieustająco cieszą się bardzo dużym
zainteresowaniem wśród badaczy. Ich popularność wynika przede wszystkim
z funkcji, jakie pełnią w organizmie. Detekcja tych molekuł w roztworach i płynach
ustrojowych ma istotne znaczenie w diagnostyce wielu zakażeń, stanów zapalnych,
chorób nowotworowych, jak również innych wszelkiego typu zaburzeń homeostazy
organizmu. Precyzyjne oznaczanie metaloprotein we krwi umożliwia szybką
diagnostykę oraz ocenę stopnia zaawansowania choroby. Pomimo znacznej
intensyfikacji badań dotyczących analizy ilościowej metaloprotein wciąż poszukuje
się bardziej wydajnych, niezawodnych, szybkich oraz mało kosztownych systemów
analizy tego typu białek. Zastosowanie do tego celu czujników elektrochemicznych
wydaje się być bardzo konkurencyjne w stosunku do powszechnie stosowanych
metod klinicznych z powodu niskich kosztów eksploatacji, braku konieczności
stosowania toksycznych substancji, jak również krótkiego czasu analizy.
Jony metalu obecne w strukturze metaloprotein gwarantują ich
elektroaktywność. Niestety uzyskanie dobrze wykształconych, a przede wszystkim
miarodajnych sygnałów prądowych związanych z procesami redoks metaloprotein
nie jest proste. Metaloproteiny, podobnie jak inne białka, wykazują niezwykle silną
tendencję do spontanicznej adsorpcji, szczególnie na podłożu metalicznym. Ponadto
bezpośredni kontakt białka z powierzchnią metaliczną może doprowadzić do

Wstęp
18
uszkodzenia jego struktury (denaturacja), a w konsekwencji do utraty aktywności
katalitycznej, czy też elektrochemicznej białka. Produkty powstałe w procesie
denaturacji białka utrudniają lub wręcz uniemożliwiają wymianę elektronów
pomiędzy centrami elektroaktywnymi białka a powierzchnią elektrody. Dodatkowo
szybkiemu i efektywnemu transportowi elektronu pomiędzy białkiem a powierzchnią
elektrody nie sprzyja fakt, że centra elektroaktywane ulokowane są głęboko w ich
otoczce białkowej, co znacznie spowalnia szybkość przeniesienia elektronów.
Uniknięcie dezaktywacji elektrody, w wyniku adsorpcji łańcuchów
aminokwasowych białka na jej powierzchni, możliwe jest dzięki zastosowaniu
różnego typu modyfikatorów, których zadaniem jest skuteczne zabezpieczanie białka
przed bezpośrednim kontaktem z elektrodą. „Idealny modyfikator” powierzchni
elektrody powinien możliwie jak najmniej wpływać na stałą szybkości przeniesienia
elektronu oraz gwarantować najlepszą, z elektrochemicznego punktu widzenia,
orientację białka względem powierzchni elektrody.

Cel pracy
19
2. Cel pracy
Unikalne właściwości katalityczne metaloprotein są szeroko wykorzystywane
w medycynie, biotechnologii, kosmetologii, rolnictwie, czy też przemyśle [1,2].
W przypadku badań wykonywanych w laboratoriach chemicznych udział
metaloprotein ogranicza się głównie do katalizy wybranych procesów, jak np.:
redukcja tlenu do wody. W tym celu modyfikuje się powierzchnię elektrody
białkiem. Takie modyfikacje coraz powszechniej stosowane są w konstrukcji
elektrochemicznych bioczujników, a także elektrod w ogniwach przetwarzających
energię chemiczną w elektryczną. O przydatności tego typu układów decyduje
aktywność metaloproteiny zawartej w warstwie modyfikującej oraz jak najdłuższe
zachowanie tej aktywności. Cel ten można uzyskać w wyniku utworzenia gęstej
siatki połączeń elektrycznych w warstwie modyfikującej poprzez zastosowanie
dobrze przewodzących nanocząstek metali lub nanocząstek węglowych. Innym
sposobem jest kontrola orientacji cząsteczek białka na powierzchni elektrody
w wyniku zastosowania odpowiedniej warstwy pośredniczącej.
Głównym celem niniejszej rozprawy doktorskiej była konstrukcja
elektrochemicznych czujników do detekcji paramagnetycznych metaloprotein
obecnych we krwi, takich jak: hemoglobina, ceruloplazmina, transferyna i ferrytyna.
Kluczowym etapem badań była odpowiednia funkcjonalizacja powierzchni elektrody
pozwalająca na wzmocnienie sygnału prądowego procesu redoks wybranych
metaloprotein, z jednoczesnym zachowaniem ich właściwości elektrokatalitycznych.
W tym celu w roli modyfikatora powierzchni elektrody zastosowano
ferromagnetyczne nanokapsuły węglowe zawierające żelazo. Tworzone warstwy
sensorowe scharakteryzowano poprzez zastosowanie różnorodnych technik
pomiarowych, pozwalających na uzyskanie ilościowego i jakościowego opisu
badanych układów. Realizacja powyższego celu wymagała wykonania szeregu
czynności badawczych, dotyczących:
a) optymalizacji układu badawczego i warunków pomiarowych:
opracowania procedury uzyskiwania jednorodnej i stabilnej warstwy
ferromagnetyka na powierzchni elektrody,

Cel pracy
20
opracowania efektywnego i selektywnego sposobu unieruchamiania
metaloprotein na powierzchni elektrody złotej lub ich transportu do
powierzchni elektrody,
zbadania wpływu zastosowanego pola magnetycznego na intensywność
uzyskiwanych sygnałów prądowych metaloprotein,
b) pełnej charakterystyki analitycznej zaproponowanych czujników:
wyznaczenia zakresu pracy czujnika i jego granicy wykrywalności,
określenia jego selektywności i stabilności,
sprawdzenia przydatności zaproponowanych czujników w próbkach
rzeczywistych – próbkach krwi.

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
21
3. Podział płynów ustrojowych, ich skład oraz
funkcje pełnione w organizmie
Utrzymanie prawidłowych parametrów środowiska poza- i śródkomórkowego
jest podstawowym wymogiem życiowym każdego organizmu. Jest to możliwe dzięki
obecności wody w organizmie, która pełni rolę rozpuszczalnika, zarówno dla
substancji organicznych, jak i nieorganicznych. Jest ona niezbędna do prawidłowego
funkcjonowania wszystkich biologicznie czynnych makrocząsteczek, takich jak:
białka, czy też kwasy nukleinowe. Woda bierze również udział w procesach
metabolicznych zachodzących w komórkach, uczestniczy w regulacji temperatury
organizmu oraz jest ważnym ośrodkiem transportującym [3]. Stanowi ona około 60%
całkowitej masy ciała dorosłego człowieka [4] i zawarta jest w płynach ustrojowych
stanowiących środowisko wewnętrzne organizmu [5].
Płyny ustrojowe organizmu ludzkiego można podzielić na dwie grupy: płyn
wewnątrzkomórkowy, zwany również śródkomórkowym (ICF) oraz płyny
zewnątrzkomórkowe, tzw. pozakomórkowe (ECF). Płyn wewnątrzkomórkowy, czyli
cytoplazma komórkowa, zlokalizowany jest we wnętrzu komórek całego organizmu
i zawiera ok. 66% całkowitej zawartości wody w organizmie. Stanowi on środowisko
dla różnych procesów, takich jak: (i) wytwarzanie, magazynowanie
i wykorzystywanie energii, (ii) procesy samonaprawcze komórki oraz (iii) replikacja
[3]. Pozostała ilość wody (ok. 30%) wchodzi w skład płynów
zewnątrzkomórkowych, do których zalicza się [3]:
płyn śródmiąższowy, zwany tkankowym, wypełniający przestrzenie między
komórkami i tkankami;
płyn mózgowo-rdzeniowy zlokalizowany w komorach mózgu, kanałach
mózgu i rdzenia kręgowego oraz w przestrzeni podpajęczynówkowej;
osocze krwi: krew żylna, tętnicza i włośniczkowa;
chłonkę;
płyny znajdujące się w kościach, stawach oraz tkance łącznej zbitej;
płyn transkomórkowy: ślina, pot, mocz, śluz oraz soki trawienne.
Główną funkcją płynów zewnątrzkomórkowych jest dostarczanie do komórek
substancji odżywczych (np. glukozy, aminokwasów, kwasów tłuszczowych),
regulujących (np. hormonów) oraz podtrzymujących i koordynujących funkcje

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
22
komórki (np. tlen, różne jony, czy białka). Płyny pozakomórkowe odpowiedzialne są
również za usuwanie z organów tlenku węgla(IV), substancji odpadowych oraz
składników toksycznych i produktów detoksykacji. Płyn wewnątrzkomórkowy różni
się od płynów zewnątrzkomórkowych zawartością zarówno substancji organicznych,
jak i nieorganicznych.
Objętość, skład płynów ustrojowych oraz ich właściwości fizykochemiczne
uzależnione są od środowiska zewnętrznego organizmu. Dążenie organizmu do
utrzymywania odpowiedniej temperatury ciała, składu chemicznego płynów
ustrojowych, ciśnienia osmotycznego, czy też ciśnienia tętniczego krwi nosi nazwę
homeostazy. Należy podkreślić, że homeostaza to nie tylko zdolność organizmu do
utrzymywania stałości parametrów wewnętrznych, ale również zbiór procesów
samonaprawczych odpowiadających za podtrzymanie stałości różnych parametrów
fizjologicznych [3]. W utrzymaniu stałych parametrów środowiska uczestniczy
m.in.: przewód pokarmowy, a także wiele narządów wewnętrznych, np. nerki
i wątroba oraz niektóre układy, takie jak: krążenia, nerwowy, immunologiczny,
hormonalny oraz oddechowy. Zmiana nawet jednego z parametrów może prowadzić
do poważnych zaburzeń w funkcjonowaniu organizmu, a nawet do jego śmierci.
Przeciwdziałanie negatywnym skutkom zmiany właściwych parametrów środowiska
organizmu skutkuje uruchomieniem odpowiednich mechanizmów prowadzących do
zniwelowania działania czynnika zewnętrznego na organizm i powrotu do stanu
równowagi [6]. Uszkodzenie mechanizmu regulacji danego organizmu jest głównym
powodem powstania wielu chorób. Szacuje się, że organizm człowieka posiada
około tysiąca systemów kontroli i regulacji [7].
3.1. Skład i funkcje krwi
Jednym z ważniejszych zewnątrzkomórkowych płynów ustrojowych
organizmu, obok płynu mózgowo-rdzeniowego, płynu międzykomórkowego oraz
chłonki, jest krew, czyli rodzaj płynnej tkanki krążącej w naczyniach krwionośnych.
U dorosłego człowieka występuje średnio około 5 litrów krwi, co stanowi
w przybliżeniu 7% całkowitej masy ciała [8]. Dla normalnej temperatury ciała
(36.6 C) pH krwi wynosi 7.4. Płyn ten posiada właściwości buforowe, dzięki czemu
kwasy, które powstają w procesach związanych z przemianą materii nie wpływają
istotnie na pH krwi. Zmiany odczynu krwi mogą nastąpić jedynie w wyniku choroby

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
23
i wówczas prowadzą do zaburzeń wielu procesów fizjologicznych [9]. Krew pełni
wiele bardzo ważnych funkcji w organizmie człowieka, które zależne są od
substancji w niej obecnych. Główną rolą krwi jest transport tlenu, tlenku węgla(IV)
oraz substancji odgrywających istotną rolę w prawidłowym funkcjonowaniu
wszystkich komórek organizmu. Dzięki nieustającemu krążeniu uczestniczy ona
także w procesach oddychania. Cząsteczki tlenu przenoszone są z pęcherzyków
płucnych do tkanek, zaś cząsteczki tlenku węgla(IV) w kierunku odwrotnym,
z tkanek do płuc. Transport CO2 następuje z miejsca o wyższej prężności tego gazu
w cytoplazmie komórek do niższej wartości w pęcherzykach płucnych. Krew bierze
także udział w funkcjach odżywczych oraz odpornościowych organizmu. Reguluje
ona także temperaturę ciała, gospodarkę wodno-elektrolitową, równowagę kwasowo-
zasadową i hormonalną oraz utrzymuje stałość środowiska wewnętrznego [7]. Krew
pełni istotną rolę w mechanizmach obronnych organizmu poprzez niszczenie obcych
antygenów [10]. Z uwagi na fakt, że uczestniczy ona w wymianie różnych substancji
między tkankami oraz usuwa z nich zbędne produkty przemiany materii, jest ona
najbardziej podatna na wpływ czynników zewnętrznych.
Krew składa się z części płynnej – osocza oraz zawieszonych w nim
elementów morfotycznych: czerwonych i białych krwinek oraz płytek krwi. Skład
krwi schematycznie został przedstawiony na rysunku 1.
ELEMENTY MORFOTYCZNE
ZWIĄZKI
NIEORGANICZNEWODA
KRWINKI
BIAŁE
KRWINKI
CZERWONE
PŁYTKI
KRWI
KREW
OSOCZE
KWASY TŁUSZCZOWE
CHOLESTEROL
KWAS MOCZOWY
HORMONY
MOCZNIK
WITAMINY
BIAŁKA
GLUKOZA
ROZPUSZCZONE GAZY
JONY
SOLE MINERALNE
ZWIĄZKI
ORGANICZNE
GRANULOCYTY AGRANULOCYTY
EOZYNOFILE
NEUTROFILE
MONOCYTY
LIMFOCYTY (B I T)
NEUTROFILE
Rysunek 1. Schemat przedstawiający skład krwi. W oparciu o dane z [11].

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
24
Osocze stanowi około 50÷60% objętości krwi i charakteryzuje się
słomkowożółtym zabarwieniem. Główną substancją występującą w osoczu (około
92%) jest woda. Osocze zawiera także białka, lipidy, hormony, witaminy, sole
mineralne i glukozę [12]. Białka występujące w osoczu różnią się między sobą
zarówno składem, jak i strukturą, a także budową chemiczną [13]. Spośród
wszystkich białek można wyróżnić trzy główne grupy: albuminy, globuliny
i fibrynogen. Albuminy, białka globularne o masie cząsteczkowej około 66 kDa, są
jednymi z najmniejszych białek i stanowią 55% białek osocza krwi. Są one zdolne do
wiązania i transportu wielu różnych substancji, mogą przenosić niektóre jony metali,
np. Mg2+
, jak również transportować toksyczne metale ciężkie, hormony lub leki
[11]. Drugimi, co do zawartości w osoczu białkami (stanowiącymi około 38% jego
całkowitej objętości) są globuliny, które odpowiadają nie tylko za transport
substancji, ale uczestniczą również w procesach odpornościowych organizmu. Białka
te dzielą się na cztery podgrupy: α1-globuliny, α2-globuliny, β-globuliny
i γ-globuliny. Najlżejszymi białkami w grupie globulin są α-globuliny o masie
cząsteczkowej 92 kDa, natomiast największą masą cząsteczkową charakteryzują się
γ-globuliny (około 120 kDa). Ostatnim białkiem stanowiącym 7% całkowitej ilości
białek w osoczu jest fibrynogen. Cząsteczka tego białka składa się z trzech
łańcuchów polipeptydowych o masie cząsteczkowej 340 kDa. Stężenie fibrynogenu
w osoczu wynosi od około 2.5 do 3.0 mgmL-1
. Białko to pełni kluczową rolę
w procesie krzepnięcia krwi, gdyż przyczynia się do agregacji płytek krwi [14] oraz
w wyniku przekształcenia go przez trombinę w nierozpuszczalną fibrynę, prowadzi
do tworzenia skrzepów krwi, które są niezbędne do gojenia się ran [15].
Innymi ważnymi składnikami osocza są lipidy stanowiące około 0.6% jego
objętości, występujące w postaci substancji związanych z białkami – lipoprotein.
Poziom lipidów we krwi zależy od wieku i płci, i może ulegać zmianom pod
wpływem hormonów oraz również podczas chorób. Ludzkie osocze zawiera lipidy
w trzech podstawowych formach: estrów kwasów tłuszczowych i gliceryny,
fosfolipidów i steroidów [16]. Z uwagi na obecność w tej grupie cholesterolu, ilość
lipidów jest silnie powiązana z chorobami krążenia i chorobami serca. Lipidy
odgrywają istotną rolę w budowie błon komórkowych oraz pełnią rolę
rozpuszczalnika dla niektórych witamin, takich jak: witamina K, A, E i D [7].
Ponadto w osoczu wyróżnić można również mocznik, glukozę, kwas moczowy,
rozpuszczone gazy, witaminy oraz hormony. Pozostałą część osocza, około 1%,

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
25
stanowią elektrolity, do których należą kationy sodu, potasu czy wapnia oraz aniony,
zawierające atomy siarki, chloru czy fosforu [9]. Zarówno kationy, jak
i aniony nie są wytwarzane przez osocze, i pobierane są przez organizm z produktów
żywnościowych, a w przypadku zapotrzebowania organizmu są one uwalniane do
osocza.
Z kolei, do zawieszonych w osoczu elementów morfotycznych krwi należą:
krwinki czerwone (RBC), krwinki białe (WBC) i płytki krwi (PLT). Elementy te
stanowią około 45% całkowitej objętości krwi, a pozostałe 55% to osocze [17].
Krwinki czerwone, inaczej zwane erytrocytami, są okrągłymi dwuwklęsłymi
dyskami pozbawionymi kwasu nukleinowego. Ich obecność nadaje
charakterystyczny czerwony kolor krwi. Stosunek ilości czerwonych krwinek do
całkowitej objętości krwi nazywana jest hematokrytem (Hct) i jego ilość zależna jest
od płci, wieku, aktywności fizycznej oraz środowiska życia człowieka. W przypadku
mężczyzn ilość czerwonych krwinek to zazwyczaj 5 mlnmm-3
, natomiast dla kobiet
liczba ta jest nieco niższa, około 4.5 mlnmm-3
. Średnica erytrocytu wynosi
zazwyczaj około 8 µm, natomiast jego grubość różni się w części środkowej
i obwodowej, i wynosi ona odpowiednio około 2 oraz 2.5 µm [11]. Budowa
czerwonych krwinek jest ściśle związana z pełnioną przez nie funkcją. Brak
w strukturze erytrocytów jądra komórkowego powoduje ich większą elastyczność,
dzięki czemu mogą one łatwo przenikać przez ściany naczyń włosowatych
i efektywnie transportować tlen z płuc do odpowiednich komórek i tkanek,
a następnie wiązać tlenek węgla(IV) oraz przenosić go z komórek do płuc [18].
Krwinki czerwone pozostają z osoczem w równowadze osmotycznej, tworząc
roztwór izotoniczny, w którym ich kształt nie ulega zmianie. Jednak w sytuacji
wzrostu ciśnienia osmotycznego otaczającego je środowiska następuje zjawisko
hipertonii, przejawiające się zmniejszeniem rozmiaru erytrocytów na skutek utraty
wody. Natomiast w przypadku zjawiska hipotonii, związanego ze zmniejszeniem
ciśnienia osmotycznego, erytrocyty w wyniku chłonięcia wody zaczynają pęcznieć,
aż do momentu pęknięcia i uwolnienia ich zawartości, której głównym składnikiem
jest hemoglobina. Proces polegający na zniszczeniu struktury czerwonych krwinek,
prowadzący do uwolnienia hemoglobiny i innych ich składników nazywany jest
hemolizą [19]. Proces ten może przebiegać w warunkach in vivo, gdzie może być
powodowany różnymi schorzeniami, jak również w warunkach in vitro, gdzie
hemoliza może być wywołana wieloma różnymi czynnikami, w tym obróbką próbki

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
26
krwi [20]. Zmiany morfologiczne erytrocytów w roztworze hipertonicznym,
izotonicznym i hipotonicznym schematycznie przedstawia rysunek 2.
Rysunek 2. Schemat przedstawiający zmiany morfologii erytrocytów w zależności od rodzaju
roztworu. Rysunek zmodyfikowany w oparciu o [21].
Spośród elementów morfotycznych krwi dużo mniej liczną grupę,
w porównaniu do erytrocytów, stanowią krwinki białe, zwane leukocytami. Na 1 µl
krwi przypada od 4 500 do 10 000 leukocytów [22]. Leukocyty występują w wielu
różnych rozmiarach i kształtach, zawierają w swojej strukturze jądro komórkowe
oraz posiadają zdolność poruszania się ruchem pełzakowatym. Krwinki białe z uwagi
na budowę i pełnione funkcje można podzielić na granulocyty, które posiadają
ziarenka cytoplazmatyczne oraz agranulocyty – niezawierające tego typu struktur.
Istnieją trzy rodzaje granulocytów: neutrofile, eozynofile i bazofile [11]. Rolą
neutrofili, czyli granulocytów obojętnych jest wydzielanie oraz uwalnianie substancji
bakteriobójczych do otoczenia. Z kolei eozynofile (granulocyty kwasochłonne)
zabijają pasożyty oraz biorą udział w reakcjach alergicznych organizmu.
Granulocyty zasadochłonne, czyli bazofile, podobnie jak eozynofile uczestniczą
w procesach alergicznych, ale również usuwają tłuszcze z krwi i przeciwdziałają
krzepnięciu krwi. Z kolei granulocyty można podzielić na monocyty oraz limfocyty.
Monocyty stanowią najliczniejszą grupę spośród białych krwinek. Ich rola polega na
niszczeniu obcych bakterii oraz niszczeniu starych, uszkodzonych i martwych
komórek ciała. Limfocyty natomiast stanowią kluczowy element układu
odpornościowego organizmu. Wyróżnia się dwa rodzaje limfocytów: komórki T
i komórki B. Komórki T kierują działaniem układu odpornościowego, natomiast
limfocyty B biorą udział w wytwarzaniu przeciwciał [23].
Ostatnimi z elementów morfotycznych krwi, powstającymi w szpiku
kostnym, są płytki krwi, zwane trombocytami. Liczba trombocytów w 1 µl krwi
H2O
H2O H2OH2O
HIPERTONICZNY IZOTONICZNY HIPOTONICZNY
H2O

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
27
wynosi 15 0000 45 0000 [22]. Płytki krwi są fragmentami cytoplazmy, mają
okrągły kształt i nie posiadają jądra komórkowego, a ich średnica wynosi średnio
2 µm. Trombocyty uczestniczą w procesach krzepnięcia krwi poprzez tworzenie
skrzepów, które mają za zadanie zamykać uszkodzenia powstałe w ścianach naczyń
krwionośnych [11].
Środowisko wewnętrzne organizmu wraz z krwią stanowi wspomniana
wcześniej chłonka. Chłonka, inaczej zwana limfą, jest rodzajem płynu tkankowego
powstającego w wyniku przedostawania się płynnych składników osocza do obszaru
międzykomórkowego. Objętość limfy w organizmie może ulegać zmianie w wyniku
intensywnego wysiłku fizycznego. Skład chłonki początkowo jest identyczny, jak
skład osocza. Natomiast w wyniku przepływu limfy przez węzły chłonne jej skład
może ulec zmianie [24]. Z krwi przepływającej przez węzły chłonne przedostają się
do chłonki limfocyty, które mogą wytwarzać immunoglobuliny (γ-globuliny), inaczej
zwane przeciwciałami [7].
Przeciwciała (Ab) mogą występować w surowicy w formie wolnej lub
związanej z błoną limfocytów B [25]. Mogą one tworzyć kompleksy typu
antygenprzeciwciało z dużym wzajemnym powinowactwem i selektywnością.
Kompleksy takie tworzone są na zasadzie oddziaływań elektrostatycznych,
wodorowych, hydrofobowych oraz van der Waalsa. Każde przeciwciało zbudowane
jest z dwóch lekkich łańcuchów polipeptydowych (L) o masie cząsteczkowej około
23 kDa oraz dwóch ciężkich łańcuchów (H) o masie 50 ÷ 60 kDa [26]. Łańcuchy
lekkie połączone są z łańcuchami ciężkimi za pomocą wiązań niekowalencyjnych
oraz za pośrednictwem mostków disiarczkowych. Wiązania takie występują również
między dwoma łańcuchami ciężkimi. Zarówno łańcuchy lekkie, jak również
łańcuchy ciężkie posiadają dodatkowo wewnątrz-łańcuchowe mostki disiarczkowe,
dzięki czemu istnieje możliwość tworzenia domen, które zbudowane są ze 110
aminokwasów. Oba łańcuchy zawierają części zmienne, które posiadają miejsca
wiązania antygenu oraz części stałe, które określają dalszy los antygenu [27].
Antygen wiązany jest przez fragment Fab (ang. Fragment antigen binding)
przeciwciała, natomiast fragment Fc (ang. Fragment crystallizable) odpowiedzialny
jest za jego właściwości biologiczne. Schemat budowy przeciwciała został
przedstawiony na rysunku 3.

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
28
S S
S S
NH2
NH2H2N
H2N
COOHCOOH
SS
SS
SS
SS
SS
SS
SS
SS
S
S
S
S
S
S
S
S
LYS-NH2
LYS-NH2
LYS-NH2
LYS-NH2
LYS-NH2
LYS-NH2
LYS-NH2
LYS-NH2
DOMENA
STAŁA
ŁAŃCUCHLEKKI
DOMENA
ZMIENNA
ŁAŃCUCHCIĘŻKI
reszty lizynowerozmieszczone losowo
w cząsteczce przeciwciała
CZĘŚĆ
ZMIENNA
CZĘŚĆ
STAŁA
COOH COOH
Rysunek 3. Schemat budowy przeciwciała. Rysunek zmodyfikowany w oparciu o [28].
W zależności od wiązanej części antygenu, przeciwciała można podzielić na
monoklonalne oraz poliklonalne. Przeciwciała monoklonalne są wysoce specyficzne
i rozpoznają tylko określony fragment antygenu, który się z nimi wiąże. Natomiast
przeciwciała poliklonalne wiążą różne części antygenu i wykazują wobec niego
różne powinowactwo [29,30]. Dla tego typu przeciwciał trwałość utworzonego
z antygenem kompleksu w dużym stopniu zależy od pH roztworu [31]. W zależności
od właściwości fizycznych i chemicznych immunoglobuliny dzieli się na pięć klas:
IgA, IgD, IgE, IgG i IgM [28,32]. Przynależność immunoglobuliny do danej klasy,
jak również jej aktywność zależy od struktury ciężkiego łańcucha [33].
Immunoglobuliny IgG odgrywają istotną rolę w zastosowaniach analitycznych,
biotechnologicznych, farmaceutycznych oraz immunoterapii [34]. Są one najbardziej
rozpowszechnionym typem przeciwciała, który obecny jest we wszystkich płynach
ustrojowych. Specyficzne wiązanie przeciwciała z antygenem rozpoczyna ciąg
reakcji, których zadaniem jest obrona organizmu przed infekcjami bakteryjnymi
i wirusowymi. Na podstawie poziomu IgG w organizmie możliwa jest ocena
funkcjonowania układu odpornościowego [35]. Przeciwciała te odpowiadają za
odporność wobec różnych czynników przenikających do organizmu przez krew.
Z uwagi na obecność w tego typu przeciwciałach dwóch fragmentów Fab, mogą one
wiązać dwie cząsteczki antygenu, co może znacząco wpływać na efektywność
tworzenia kompleksu antygen−przeciwciało [28].

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
29
3.2. Równowaga kwasowo-zasadowa płynów ustrojowych
Utrzymanie stałego składu płynów ustrojowych jest gwarantem
prawidłowego funkcjonowania organizmu. Stan, w którym zachowany jest swoisty
stosunek kationów i anionów w płynach ustrojowych nazywany jest równowagą
kwasowo-zasadową [3]. Jest to jeden z najważniejszych mechanizmów regulacji
organizmu, warunkujący odpowiednie pH niezbędne do prawidłowego przebiegu
procesów życiowych. Stężenie jonów wodorowych w płynach ustrojowych zależy
bezpośrednio od dysocjacji kwasów i pośrednio od nasilenia procesów
katabolicznych w organizmie. Głównymi produktami przemian materii
w organizmach żywych są przede wszystkim kwasy lotne, np. kwas węglowy oraz
kwasy nielotne, np.: mlekowy, pirogronowy, moczowy, siarkowy oraz fosforowy.
W płynie wewnątrzkomórkowym stężenie jonów H+ wynosi ok. 100 nmoli·L
-1,
natomiast tylko w przypadku komórek o zredukowanym metabolizmie
(np. erytrocyty) jest ono niższe i wynosi ok. 65 nmoli·L-1
. W płynach
zewnątrzkomórkowych, tzw. pozakomórkowych stężenie jonów wodorowych
utrzymywane jest na poziomie 35 ÷ 45 nmoli·L-1
, co daje wartość pH w zakresie
7.35 ÷ 7.45. W warunkach fizjologicznych pH ulega tylko nieznacznym zmianom,
które są szybko niwelowne dzięki sprawnie działającym mechanizmom regulującym.
Stałość środowiska wewnętrznego, czyli homeostazę, zapewniają trzy czynniki:
układy buforowe krwi, układ oddechowy oraz czynność nerek.
Podstawowe mechanizmy homeostazy ustrojowej, odpowiedzialne za
utrzymanie równowagi kwasowo-zasadowej to roztwory buforowe [3]:
bufor wodorowęglanowy, obecny w płynach zewnątrzkomórkowych,
stanowiący 70% pojemności buforowej krwi;
CO2 + H2O H2CO3 HCO3
+ H+anhydraza węglanowa (1)
bufor hemoglobinowy, obecny w płynie wewnątrzkomórkowym, stanowiący
21% pojemności buforowej krwi;
22 COHHbO w pęcherzykach płucnych
2OHb w aktywnie oddychającej tkance (2)
H
2CO

Podział płynów ustrojowych, ich skład oraz funkcje pełnione w organizmie
30
Wiązanie CO2 przez cząsteczkę Hb zachodzi za pośrednictwem grup –NH2
histydyny.
bufor fosforanowy, obecny w płynie wewnątrzkomórkowym, stanowiący
6% pojemności buforowej krwi;
0.12pKHPOHPO
8.6pKHHPOPOH
0.2pKPOHHPOH
POHHHPO
3
2
1
a34
24
a2442
a4243
4224
(3)
Bufor fosforanowy jest najważniejszym pod względem ilościowym buforem
moczu, biorącym udział w wytwarzaniu kwaśności miareczkowej, która
określa wydalanie jonu H+ przez nerki w postaci NaH2PO4.
bufor białczanowy, obecny w płynie wewnątrzkomórkowym i w płynach
zewnątrzkomórkowych, stanowiący 3% pojemności buforowej krwi;
NaCOOHRHCOONaR (4)
Białka, jako związki wielkocząsteczkowe, nie mogą przenikać przez błony
półprzepuszczalne, ale dzięki temu, że występują w formie kationów lub
anionów wpływają na rozmieszczenie dyfundujących przez błony
półprzepuszczalne elektrolitów.

Białka ważne składniki organizmu
31
4. Białka ważne składniki organizmu
Białka stanowią większość związków organicznych występujących
w organizmie ludzkim. Odgrywają one istotną rolę w przebiegu wielu procesów
biologicznych w żywych komórkach. Zbudowane są z co najmniej 100
aminokwasów, które łącząc się ze sobą za pomocą wiązania peptydowego tworzą
łańcuchy polipeptydowe [36]. Białka można podzielić na dwie odrębne grupy: białka
proste i złożone, co schematycznie przedstawia rysunek 4.
CHROMOPROTEINY
DO ŁAŃCUCH BIAŁKOWEGO
DOŁĄCZONE SĄ BARWNIKI
(np.: HEMOGLOBINA ,
CYTOCHROM C)
FOSFOPROTEINY
DO ŁAŃCUCHABIAŁKOWEGO
DOŁĄCZONA JEST RESZTA
KWASU FOSFOROWEGO
(np.: KAZEINA, PEPSYNA)
BIAŁKA PROSTE
BIAŁKA FIBRYLARNE
KOLAGEN
KREATYNA
FIBRYNA
BIAŁKA GLOBULARNE
ALBUMINY
GLOBULINY
HISTONY
BIAŁKA
BIAŁKA ZŁOŻONE
GLIKOPROTEINY
DO ŁAŃCUCHABIAŁKOWEGO
DOŁĄCZONE SĄ CUKROWCE
(np.: SKŁADNIKI WYDZIELIN
ŚLUZOWATYCH)
LIPOPROTEINY
WIELKOCZĄSTECZKOWE
KOMPLEKSY BIAŁEK Z LIPIDAMI
(SKŁADNIKI BŁON)
METALOPROTEINY
DO ŁAŃCUCHABIAŁKOWEGO
DOŁĄCZONE SĄ METALE (np.:
TRANSFERYNA, FERRYTYNA,
LICZNE ENZYMY)
Rysunek 4. Podział białek w oparciu o dane z [3].
Białka proste zbudowane są tylko z aminokwasów, natomiast białka złożone
zawierają w swojej strukturze oprócz podstawowego łańcucha białkowego (białko
proste) także inne grupy, tzw. grupy prostetyczne. Do białek prostych zalicza się
białka fibrylarne oraz białka globularne. Podział ten związany jest z różnicą
w kształcie cząsteczek, co ma swoje przełożenie na ich rozpuszczalność w wodzie
[37]. Białka fibrylarne są nierozpuszczalne w wodzie, mają strukturę włókien, które
mogą być połączone między sobą wiązaniami wodorowymi. Białka te spełniają
głównie rolę budulcową tkanek. Z ich włókien zbudowana jest keratyna, kolagen, jak
również miozyna [3]. Z kolei białka globularne mają często pofałdowaną, zwartą

Białka ważne składniki organizmu
32
strukturę i są dobrze rozpuszczalne w roztworach wodnych. Odgrywają one istotną
rolę w regulacji wielu procesów zachodzących w organizmie. Do tej grupy należą
enzymy, hormony, przeciwciała, jak również białka uczestniczące w transporcie
różnych substancji [38].
Różnorodność białek związana jest z możliwością tworzenia różnych struktur
przestrzennych, które determinują funkcje poszczególnych białek w organizmie.
Wyróżnia się strukturę pierwszo-, drugo-, trzecio- oraz czwartorzędową białek [3],
co schematycznie ilustruje rysunek 5.
Rysunek 5. Organizacja struktury białek. Rysunek w oparciu o [3].
Struktura pierwszorzędowa określa miejsce występowania oraz rodzaj aminokwasów
tworzących łańcuch białkowy. Łańcuchy polipeptydowe mogą przybierać różne
struktury przestrzenne w wyniku oddziaływania łańcuchów bocznych aminokwasów
lub poprzez oddziaływanie łańcuchów polipeptydowych między sobą i utworzenie
wiązania wodorowego między atomem wodoru z grupy –NH– oraz atomem tlenu
z grupy karbonylowej [39]. Ułożenie łańcuchów polipeptydowych w przestrzeni
określa struktura drugorzędowa. Wśród struktur drugorzędowych najczęściej
występuje alfa helisa oraz beta kartka [40]. Struktura trzeciorzędowa białka odnosi
się do przestrzennego ułożenia wszystkich aminokwasów tworzących dane białko,
natomiast w przypadku białek zawierających więcej niż jeden łańcuch
polipeptydowy istnieje również struktura czwartorzędowa, która określa ich
oddziaływania i wzajemne położenie [41].
Arg
Met
Cys CysProTyrGlu Lys
Gln Cys AspProTyrHis Val
Arg Ala
Pro
ProTyrGlu Val
Lys
alfa helisa
beta kartka
BIAŁKO I-RZĘDOWE BIAŁKO II-RZĘDOWE
BIAŁKO IV-RZĘDOWEBIAŁKO III-RZĘDOWE

Białka ważne składniki organizmu
33
4.1. Definicja i budowa metaloprotein
Niezwykle istotną rolę w prawidłowym funkcjonowaniu organizmu
odgrywają metale wchodzące w skład białek. Nadmiar jonów metali może wpływać
niekorzystnie na tkanki i dlatego ich ilość musi być precyzyjnie regulowana.
Polipeptydy zawierające jeden lub więcej jonów metali w swojej strukturze stanowią
prawie połowę wszystkich białek występujących w przyrodzie [42] i określane są
mianem metaloprotein [43]. Metalami najczęściej występującymi w ich strukturze
są: Ca, Mg, Mn, Fe, Cu, Zn, Na oraz K [44]. Niektóre z nich, zwłaszcza Mg, Fe i Co
mogą być różnie kompleksowane, tworząc w ten sposób chlorofil, hem lub korynę
[3]. Białka zawierające Co i Ni dopiero stosunkowo niedawno zostały uznane za
niezbędne w niektórych żywych organizmach. Istnieją również przypadki, w których
struktura białka zawiera metale ciężkie, takie jak Cd, V i W. Pomimo, że metale te
nie uczestniczą w procesach metabolicznych, to jednak są one istotne w przypadku
niektórych gatunków, takich jak bakterie beztlenowe [45]. Atomy lub jony metali
występujące powszechnie w białkach odgrywają w nich różnorodne funkcje. Metale
występujące w białkach służą jako przenośnik elektronów, ułatwiając transport tlenu
oraz odgrywają kluczową rolę w reakcjach katalitycznych [45]. W przypadku
zaburzenia homeostazy organizmu poprzez działanie różnych czynników
zewnętrznych, takich jak bakterie, wirusy czy promieniowanie jonizujące dochodzi
do zmiany stężenia jonów metali we krwi [46]. Zmiany te powiązane są ze zmianą
ilości białek, które zawierają dane atomy metali w swojej strukturze. Białka takie
noszą nazwę białek ostrej fazy i zalicza się do nich między innymi ceruloplazminę,
transferynę, ferrytynę czy też białko CRP [47]. Wzrost lub zmniejszenie ich ilości we
krwi skutkuje uruchomieniem odpowiednich mechanizmów mających na celu
powrót organizmu do stanu równowagi. Stan zapalny organizmu związany jest
zarówno ze zmianą stężenia wymienionych białek, jak również wpływa na ilość
erytrocytów, a tym samym na zmianę stężenia hemoglobiny [48,49]. Dlatego też
kontrola białek zawierających jony metali ma istotne znaczenie w prawidłowym
funkcjonowaniu komórek. W niektórych proteinach metal jest częścią miejsca
aktywnego w procesach katalitycznych, u innych odgrywa on istotną rolę
w utrzymaniu struktury białka. Wiedza oraz poznanie struktury białek jest niezbędna
w zrozumieniu ich funkcji [50]. Różnorodność białek zawierających jony metali
schematycznie ilustruje rysunek 6.

Białka ważne składniki organizmu
34
METALOPROTEINY
ENZYMY
OKSYDOREDUKTAZY
OKSYDAZY, Fe, Cu
REDUKTAZY, Fe, Cu, Mo
NITROGENAZY, Fe, Mo, V
HYDROKSYLAZY, Fe, Cu, Mo
HYDROGENAZY, Fe, Ni
DYSMUTAZA PONADTLENKOWA, Fe, Cu, Mn
HYDROLAZY
FOSFATAZY, Mg, Zn, Cu
AMINOPEPTYDAZY, Mg, Zn
KARBOKSYPEPTYDAZY, Zn
IZOMERAZY I SYNTETAZY
WITAMINA B12, KOENZYM, Co
BIAŁKA
TRANSPORT
I MAGAZYNOWANIE
PRZENOŚNIKI ELEKTRONÓW
CYTOCHROMY, Fe
BIAŁKAŻELAZO-SIARKOWE
NIEBIESKIE MIEDZIO-PROTEINY, Cu
GOSPODARKA METALAMI
FERRYTYNA, Fe
TRANSFERYNA, Fe
CERULOPLAZMINA, Cu
GOSPODARKA TLENEM
MIOGLOBINA, Fe
HEMOGLOBINA, Fe
HEMOERYTRYNA, Fe
HEMOCYJANINA, Cu
INNE FUNKCJE
Rysunek 6. Podział metaloprotein w oparciu o dane z [3].
Metaloproteiny biorą udział w wielu rodzajach biologicznych procesów.
Białka przyspieszające zachodzenie wielu reakcji chemicznych w organizmie
nazywane są enzymami. W strukturze enzymów wyróżnić można część białkową,
tak zwane centrum aktywne lub inaczej centrum katalityczne oraz fragment
niepeptydowy, zwany grupą prostetyczną [38]. Centrum aktywne enzymu
zbudowane jest z aminokwasów, które uczestniczą w wiązaniu substratu
z wytworzeniem kompleksu enzym−substrat [51]. Część enzymów wykazuje
aktywność tylko w przypadku, gdy w ich składzie znajduje się niebiałkowa część lub
grupa funkcyjna, określana mianem kofaktora. Kofaktorem mogą być zarówno jony
metali, jak również cząsteczki organiczne. Organiczne związki uczestniczące
w transporcie wodoru, grup funkcyjnych oraz wymianie elektronów między
substratami [52], takie jak dinukleotyd nikotynoamidoadeninowy (NAD+), nazywa
się koenzymami. Część enzymu niezawierająca kofaktora, która zwykle jest
nieaktywna katalitycznie to apoenzym. Dopiero połączenie apoenzymu z częścią

Białka ważne składniki organizmu
35
niebiałkową, zwane holoenzymem nadaje takiemu białku aktywność katalityczną
[53]. Enzymy zawierające w swojej strukturze jony metali noszą nazwę
metaloenzymów i stanowią one około jedną trzecią wszystkich białek zawierających
jony metali [54].
Metaloproteiny, w tym metaloenzymy, do prawidłowego funkcjonowania
wymagają obecności atomów metali [55]. Miejsca, w których następuje wiązanie
jonów metali w białkach, można podzielić na pięć typów; podział ten opiera się na
funkcji, jaką pełni dany jon metalu:
strukturalna – udział w tworzeniu trzecio- i czwartorzędowych struktur
przestrzennych;
katalityczna – wiązanie substratu i jego aktywacja [56];
magazynująca – wychwytywanie, wiązanie i uwalnianie metali w formie
rozpuszczalnej;
transportowa elektronów;
wiązania cząsteczek tlenu [43].
Jony metali mogą uczestniczyć także w tworzeniu pofałdowanej struktury białek, czy
też zmianach konformacjnych [57]. Otoczenie chemiczne metali jest istotne dla
zrozumienia chemicznego zachowania białek. Jony metali mogą być połączone
z białkiem za pomocą wiązań kowalencyjnych lub koordynacyjnych, jak również
mogą one tworzyć kompleksy z białkami. Rolę ligandów w strukturze białka mogą
pełnić pojedyncze atomy lub grupy atomów. Ligandami grupowymi mogą być:
grupy imidazolowe histydyny (His) występujące w łańcuchu bocznym białka, grupy
karboksylowe pochodzące z kwasu asparginowego (Asp) i kwasu glutaminowego
(Glu), grupy wodorotlenowe seryny (Ser) i treoniny (Thr) oraz fenolany (analogi
tyrozyny (Tyr)), tiolany (cysteina (Cys)), jak również cząsteczki wody lub inne
cząsteczki niebiałkowe (węglany, wodorowęglany, itp.) obecne w miejscu wiązania
metalu. Stopień utlenienia metalu, liczba koordynacyjna, jak również charakter
ligandów mogą mieć wpływ na kąty i odległości pomiędzy atomami w cząsteczce
białka [58]. Z kolei atomy uczestniczące w koordynacji metalu przez białko to
głównie atomy tlenu z grupy karbonylowej łańcucha głównego, jak również tlen
z aminokwasowego łańcucha bocznego lub atomy azotu z histydyny, czy też atomy
siarki z cysteiny. W przypadku kompleksów chelatowych, w których ligand wiąże
się z jonem metalu za pośrednictwem więcej niż jednego wiązania koordynacyjnego,
pierścień chelatowy zawiera atom metalu i 3, 4 lub 5 innych atomów (C, N, O, S itp.)
Białka ważne składniki organizmu
36
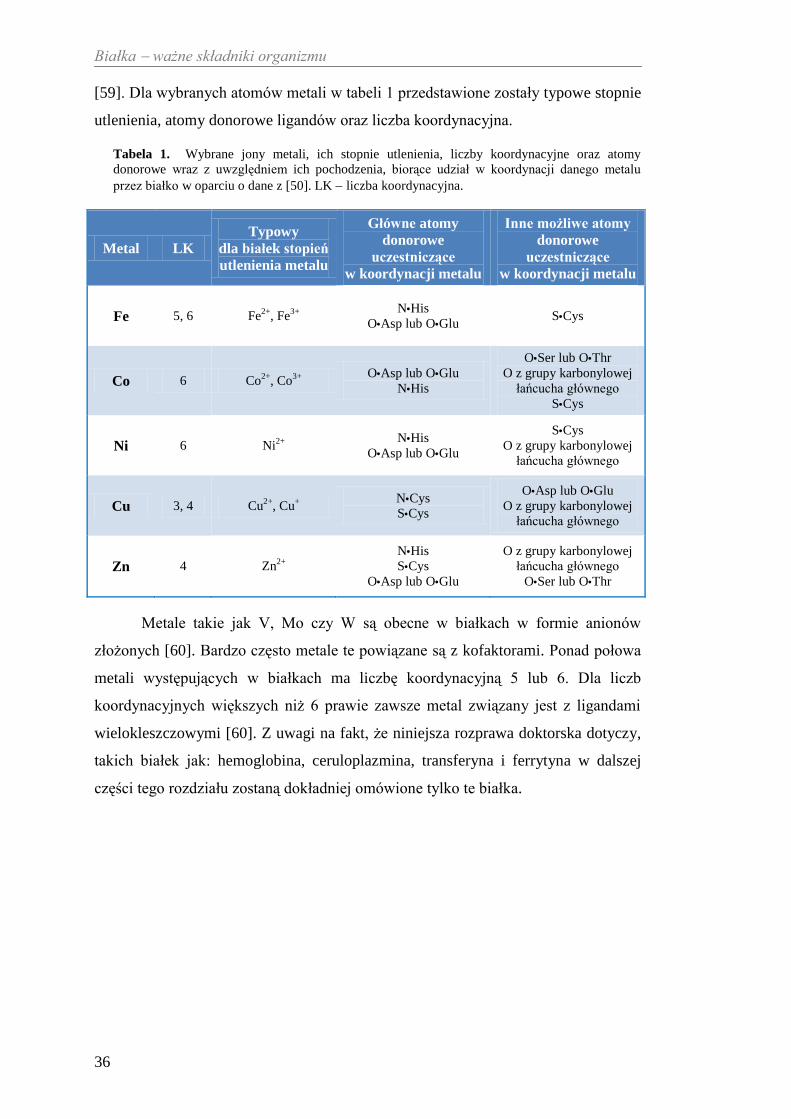
[59]. Dla wybranych atomów metali w tabeli 1 przedstawione zostały typowe stopnie
utlenienia, atomy donorowe ligandów oraz liczba koordynacyjna.
Tabela 1. Wybrane jony metali, ich stopnie utlenienia, liczby koordynacyjne oraz atomy
donorowe wraz z uwzględniem ich pochodzenia, biorące udział w koordynacji danego metalu
przez białko w oparciu o dane z [50]. LK liczba koordynacyjna.
Metal LK
Typowy
dla białek stopień
utlenienia metalu
Główne atomy
donorowe
uczestniczące
w koordynacji metalu
Inne możliwe atomy
donorowe
uczestniczące
w koordynacji metalu
Fe 5, 6 Fe2+
, Fe3+
NHis
OAsp lub OGlu SCys
Co 6 Co2+
, Co3+
OAsp lub OGlu
NHis
OSer lub OThr
O z grupy karbonylowej
łańcucha głównego
SCys
Ni 6 Ni2+
NHis
OAsp lub OGlu
SCys
O z grupy karbonylowej
łańcucha głównego
Cu 3, 4 Cu2+
, Cu+
NCys
SCys
OAsp lub OGlu
O z grupy karbonylowej
łańcucha głównego
Zn 4 Zn2+
NHis
SCys
OAsp lub OGlu
O z grupy karbonylowej
łańcucha głównego
OSer lub OThr
Metale takie jak V, Mo czy W są obecne w białkach w formie anionów
złożonych [60]. Bardzo często metale te powiązane są z kofaktorami. Ponad połowa
metali występujących w białkach ma liczbę koordynacyjną 5 lub 6. Dla liczb
koordynacyjnych większych niż 6 prawie zawsze metal związany jest z ligandami
wielokleszczowymi [60]. Z uwagi na fakt, że niniejsza rozprawa doktorska dotyczy,
takich białek jak: hemoglobina, ceruloplazmina, transferyna i ferrytyna w dalszej
części tego rozdziału zostaną dokładniej omówione tylko te białka.

Białka ważne składniki organizmu
37
4.2. Charakterystyka wybranych metaloprotein obecnych
we krwi
4.2.1. Hemoglobina, czerwony barwnik krwi wiążący tlen i tlenek
węgla(IV)
Hemoglobina (Hb) jest głównym białkiem odpowiedzialnym za przenoszenie
tlenu w układzie krwionośnym istot żywych [61]. Rozpuszczalność tlenu w wodzie
wynosi około 3·10-4
mol·L-1
, natomiast obecność hemoglobiny powoduje około
30-krotne zwiększenie jego rozpuszczalności we krwi [45]. Oprócz transportu tlenu
białko to uczestniczy także w przenoszeniu tlenku węgla(IV) z tkanek do płuc w celu
jego usunięcia z organizmu [62,3]. Udział hemoglobiny w przenoszeniu O2 i CO2
pomiędzy płucami a tkankami schematycznie przedstawia rysunek 7.
Rysunek 7. Rola hemoglobiny w organizmie.
Organizm ludzki zawiera około 750 g Hb, zlokalizowanej głównie w czerwonych
krwinkach [63]. Istnieje wiele przyczyn zaburzeń ilości tego białka we krwi, które
można podzielić na: (i) choroby genetycznie dziedziczone, jak np. anemia sierpowata
oraz (ii) schorzenia nabyte, jak methemoglobinemia polekowa, jak również
niedokrwistość spowodowana niedoborem żelaza w diecie [64]. Zatem zarówno
Fe2+
Fe2+
Fe2+
Fe2+
NHCOO-
NHCOO-
Fe2+
Fe2+
Fe2+
Fe2+
O2 O2
O2 O2
karbonylohemoglobina oksyhemoglobina
CO2 O2
CO2 O2
PŁUCA
TKANKA

Białka ważne składniki organizmu
38
zmiany funkcjonalne i strukturalne hemoglobiny związane są z wieloma chorobami
klinicznymi, takimi jak: białaczka, niedokrwistość czy choroby serca [65-67].
Dlatego też określenie ilości tego białka we krwi ma istotne znaczenie we wczesnym
wykrywaniu wielu chorób.
Hemoglobina zaliczana jest do kompleksów chelatowych, z uwagi na
obecność metalu wchodzącego w skład pierścienia tej cząsteczki [68]. Białko to jest
tetramerem, o masie cząsteczkowej wynoszącej ok. 67 kDa (67 000 g·mol-1
) [69],
który złożony jest z dwóch jednakowych łańcuchów polipeptydowych α oraz dwóch
identycznych łańcuchów β, nazwanych podjednostkami α i β. Łańcuch alfa złożony
jest ze 141 reszt aminokwasowych, gdzie N-koniec stanowi walina, zaś C-koniec
arginina. Z kolei łańcuch beta zawiera 146 reszt aminokwasowych, gdzie
N-końcowym aminokwasem jest walina, zaś C-końcowym histydyna. Każdy
z dwóch łańcuchów α pozostaje w kontakcie z dwoma łańcuchami β, natomiast
oddziaływania pomiędzy dwoma łańcuchami alfa lub beta są bardzo słabe [51,70].
Wszystkie cztery łańcuchy polipeptydowe zlokalizowane są w wierzchołkach
czworościanu [71]. Każdy z nich zawiera jedną grupę hemową, dzięki czemu
hemoglobina zdolna jest do wiązania czterech cząsteczek O2 [40,72]. Struktura
przestrzenna cząsteczki hemoglobiny przedstawiona została na rysunku 8.
Rysunek 8. Struktura przestrzenna cząsteczki hemoglobiny i hemu w oparciu o rysunek z [73].
Hem stanowi grupę prostetyczną hemoglobiny, ulokowany jest
w hydrofobowej kieszeni białka i jest to kompleks jonu żelaza z porfiryną, zwaną
protoporfiryną IX. To właśnie hem nadaje krwi charakterystyczny czerwony kolor;
w jego skład wchodzi cykliczny tetrapirol, w którym wszystkie cztery pierścienie
pirolowe połączone są za pomocą mostków α-metinowych, tworzących płaski
N
NN
N
CH3
O
OH
O
OH
CH3
CH3
Fe

Białka ważne składniki organizmu
39
cykliczny układ [3]. Żelazo utrzymywane jest w strukturze porfiryny głównie za
pośrednictwem oddziaływań hydrofobowych reszt aminokwasowych,
w szczególności izoleucyny, leucyny, fenyloalaniny i waliny, jak również za pomocą
jednego wiązania kowalencyjnego z resztą proksymalnej histydyny, oznaczanej jako
HisF8 [53]. Takie oznaczenie histydyny dotyczy jej umiejscowienia w łańcuchu
polipeptydowym, gdyż żelazo wiąże się z ósmą resztą tego aminokwasu w regionie F
α-helisy [53]. Atom żelaza zlokalizowany jest w centralnej części tego układu
i połączony jest on z czterema atomami azotu, piąte wiązanie koordynacyjne służy
do wiązania atomu żelaza z substancją białkową, natomiast szóste z cząsteczką tlenu.
Żelazo w hemoglobinie może występować w postaci jonu Fe2+
lub Fe3+
.
Natomiast tylko obecność żelaza na drugim stopniu utlenienia pozwala hemoglobinie
na transport tlenu w organizmie [74]. Podjednostki cząsteczki hemoglobiny
wykazują kooperatywność [75]; oznacza to, że każda z podjednostek ma inne
powinowactwo względem O2, co wynika z czwartorzędowej struktury Hb.
Przyłączenie jednej cząsteczki tlenu do jednej z grup hemowych powoduje, że
kolejne cząsteczki O2 coraz łatwiej przyłączają się do białka. Wzrost powinowactwa
pozostałych grup hemowych względem tlenu pozwala na wiązanie w płucach
maksymalnej ilości tlenu i uwalnianie go w tkankach. Utworzenie kompleksu
z tlenem prowadzi do zmian konformacyjnych cząsteczki hemoglobiny [3]. Zmiana
struktury przestrzennej hemoglobiny wynika z wsunięcia się atomu żelaza
w płaszczyznę pierścienia hemu w wyniku przyłączenia tlenu. W konsekwencji
oprócz żelaza wsunięciu ulega również histydyna proksymalna (bezpośrednio
związana z żelazem), co z kolei wymusza przemieszczenie się sąsiednich
aminokwasów łańcuchów globiny. W rezultacie następuje rozrywanie wiązań
poprzecznych występujących pomiędzy karboksylowymi końcami wszystkich
czterech podjednostek hemowych. Konformacja, w której hemoglobina jest tylko
częściowo utlenowana, określana jest jako stan T (z ang. taut – naprężony) [51].
Natomiast konformacja całkowicie utlenowanej hemoglobiny po wykonaniu przez
jedną z par podjednostki α/β obrotu o około 15° względem drugiej pary tej
podjednostki, w wyniku czego pary te zbliżają się do siebie i następuje wzrost
powinowactwa grup hemowych do O2 [3], określa się jako stan R (ang. relaxed –
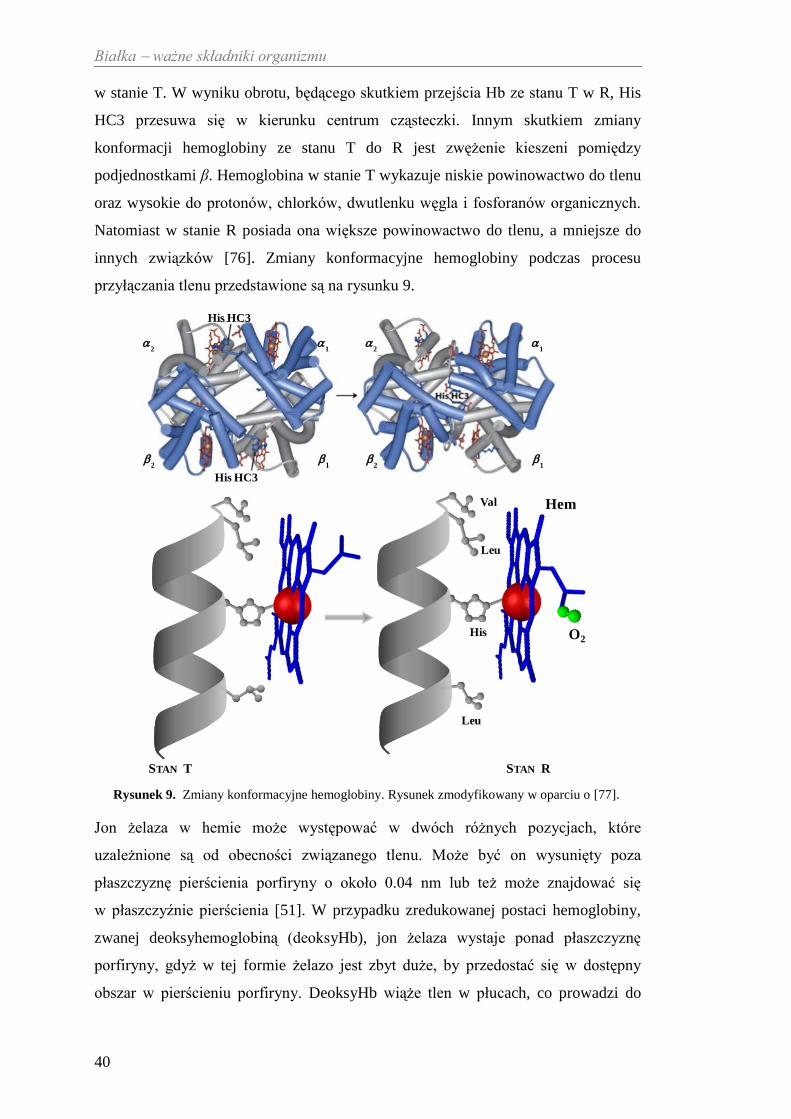
rozluźniony) [51]. Przejście ze stanu T do stanu R wpływa na położenie par
podjednostek α/β. Najbardziej zauważalna jest zmiana położenia His HC3 na
C-końcach podjednostek β, które są zaangażowane w tworzenie par jonowych
Białka ważne składniki organizmu
40
w stanie T. W wyniku obrotu, będącego skutkiem przejścia Hb ze stanu T w R, His
HC3 przesuwa się w kierunku centrum cząsteczki. Innym skutkiem zmiany
konformacji hemoglobiny ze stanu T do R jest zwężenie kieszeni pomiędzy
podjednostkami β. Hemoglobina w stanie T wykazuje niskie powinowactwo do tlenu
oraz wysokie do protonów, chlorków, dwutlenku węgla i fosforanów organicznych.
Natomiast w stanie R posiada ona większe powinowactwo do tlenu, a mniejsze do
innych związków [76]. Zmiany konformacyjne hemoglobiny podczas procesu
przyłączania tlenu przedstawione są na rysunku 9.
Rysunek 9. Zmiany konformacyjne hemoglobiny. Rysunek zmodyfikowany w oparciu o [77].
Jon żelaza w hemie może występować w dwóch różnych pozycjach, które
uzależnione są od obecności związanego tlenu. Może być on wysunięty poza
płaszczyznę pierścienia porfiryny o około 0.04 nm lub też może znajdować się
w płaszczyźnie pierścienia [51]. W przypadku zredukowanej postaci hemoglobiny,
zwanej deoksyhemoglobiną (deoksyHb), jon żelaza wystaje ponad płaszczyznę
porfiryny, gdyż w tej formie żelazo jest zbyt duże, by przedostać się w dostępny
obszar w pierścieniu porfiryny. DeoksyHb wiąże tlen w płucach, co prowadzi do
O2His
Val
Leu
Leu
Hem
STAN T STAN R
His HC3
His HC3
2
2
1
1
2
2
1
1

Białka ważne składniki organizmu
41
utworzenia utlenowanej formy hemoglobiny, tzw. oksyhemoglobiny (oksyHb),
oznaczanej również jako HbFe2+
lub HbFe(II)O2 [78]. Po związaniu cząsteczki tlenu
z kationem żelaza, następuje zmiana układu elektronów w obrębie tego atomu. Po
utlenowaniu średnica jonu żelaza zmniejsza się, co pozwala na jego przemieszczenie
w głąb płaszczyzny hemu. OksyHb jest stosunkowo stabilną formą hemoglobiny,
jednak powoli ulega ona samorzutnemu procesowi utlenienia, w wyniku którego
powstaje anion ponadtlenkowy O2¯ (reaktywna forma tlenu) oraz methemoglobina –
metHb [HbFe(III)], inna forma hemoglobiny zawierająca jon Fe3+
. Forma ta nie
wykazuje zdolności wiązania tlenu, natomiast może wiązać cząsteczki wody [79,80].
W wyniku utleniania oksyHb może powstać również nadtlenek wodoru (H2O2), który
może być toksyczny i dlatego jego nadmiar zostaje usunięty przez katalazę oraz
peroksydazę glutationową, dzięki czemu jego ilość w czerwonych krwinkach
pozostaje stała [81]. Utlenianie żelaza za pomocą nadtlenku wodoru
w oksyhemoglobinie oraz deoksyhemoglobinie może również prowadzić do
powstawania ferrylohemoglobiny (ferryloHb, HbFe(IV)=O) [64,82]. Schemat
tworzenia różnych form hemoglobiny przedstawia rysunek 10.
oksyhemoglobina
(oksyHb, HbFe(II)O2)
utlenowana
deoksyhemoglobina
(deoksyHb, HbFe(II))
nieutlenowana
methemoglobina
(metHb, HbFe(III))
forma utleniona
ferrylohemoglobina
(ferryloHb, HbFe(IV)=O)
forma utleniona
HEMOGLOBINA
H2O2
H2O2
H2O2
oksoferrylohemoglobina
(oksoferryloHb, HbFe(IV)=O)
forma utleniona
H2O2
H2O2
Rysunek 10. Formy hemoglobiny. Schemat wykonany na podstawie danych zawartych w [83].
W przypadku utleniania oksyhemoglobiny za pomocą nadtlenku wodoru powstaje
oksoferrylohemoglobina (oksoferryloHb, HbFe(IV)=O). Zarówno ferrylo-
hemoglobina, jak również oksoferryloHb są związkami silnie utleniającymi. Procesy
utleniania oksyHb, metHb, ferryloHb oraz oksoferryloHb za pomocą H2O2
przedstawiają poniższe równania [64]:

Białka ważne składniki organizmu
42
22222 OOHO)IV(HbFeOHO)II(HbFe (5)
OHO)IV(HbFeOH)III(HbFe 222 (6)
2222 OOH)III(HbFeOHO)IV(HbFe (7)
2222 OOH)III(HbFeOHO)IV(HbFe (8)
Hemoglobina uwolniona z czerwonych krwinek lub chemicznie zmodyfikowana jest
podatna na utlenianie pod wpływem nadtlenku wodoru. Jednak zbyt długie
oddziaływanie Hb z H2O2 może prowadzić nawet do zniszczenia struktury hemu
i uwolnienia żelaza [84].
W przypadku związania hemoglobiny z tlenem zmianie ulega charakter
wiązania pomiędzy atomem żelaza a resztą grupy hemowej i łańcucha
polipeptydowego. W deoskyhemoglobinie wiązanie to ma charakter jonowy,
natomiast w formie utlenowanej hemoglobiny występuje wiązanie kowalencyjne
[85,86]. Dla utlenionej formy hemoglobiny, methemoglobiny (metHb), wiązanie
pomiędzy jonem Fe3+
a grupą porfirynową i łańcuchem peptydowym, tak jak
w przypadku deoksyhemoglobiny ma charakter jonowy [87]. Przyłączenie tlenu do
deoksyHb powoduje zmiany w strukturze elektronowej cząsteczki, a tym samym
zmiany we właściwościach magnetycznych form Hb. Z uwagi na kowalencyjne
wiązanie w oksyhemoglobinie, cząsteczka ta jest diamagnetyczna. Dla form
hemoglobiny, w których żelazo jest związane z histydyną w łańcuchu
polipeptydowym za pomocą wiązania jonowego, występują niesparowane elektrony.
Konsekwencją obecności w deoksyhemoglobinie czterech niesparowanych
elektronów oraz pięciu w metHb, jest ich paramagnetyczność. Łańcuch peptydowy
hemoglobiny nie wpływa na właściwości paramagnetyczne tych cząsteczek,
ponieważ ma on właściwości diamagnetyczne [88]. Hem w stanie niezwiązanym
wykazuje dużo większe powinowactwo do tlenku węgla(II) niż do tlenu. Z uwagi na
dużo większą trwałość kompleksu CO−Hb, tlen nie jest w stanie wyprzeć tlenku
węgla(II) z kompleksu. Połączenie białka z CO zmienia strukturę cząsteczki Hb,
powstaje wówczas zawada przestrzenna i hemoglobina nie jest w stanie związać
tlenu oraz dostarczyć go do komórek [89]. Hemoglobina oprócz transportu tlenu
i tlenku węgla(IV) bierze także udział w wielu innych procesach. W pewnych
warunkach białko to może wykazywać aktywność katalazy, która obecna jest
w czerwonych krwinkach i uczestniczy w rozkładzie nadtlenku wodoru [90]. Może

Białka ważne składniki organizmu
43
ona także mieć właściwości utleniające, jak również katalizuje proces kondensacji
aminofenoli [91,92].
4.2.2. Ceruloplazmina, nośnik jonów miedzi w organizmie
Ceruloplazmina (Cp) należy do rodziny multimiedziowych oksydaz, do
których zalicza się również oksydazę askorbinianu oraz lakazę. Oksydazy miedziowe
to białka wykazujące aktywność enzymatyczną w reakcjach, w których utlenianie
substratów związane jest z czteroelektronową redukcją tlenu cząsteczkowego do
dwóch cząsteczek wody [93]. Zarówno z uwagi na strukturę, funkcje, jak i udział
w reakcjach katalitycznych najbardziej złożoną multimiedziową oksydazą jest
właśnie ceruloplazmina (oksydoreduktaza ditlenu, EC 1.16.3.1). Białko to kodowane
jest tylko przez gen CPN, znajdujący się na dłuższym ramieniu chromosomu
3q23-q24 [94]. Ludzka ceruloplazmina jest glikoproteiną o masie cząsteczkowej
około 132 kDa (~131 976 g·mol-1
). Po raz pierwszy została ona wyizolowana
z frakcji białek osocza (globulin) przez Holmberga w 1944 roku [95]. Cząsteczka ta
składa się z pojedynczego łańcucha polipeptydowego zbudowanego z 1046
aminokwasów i zawiera od 7 do 8% węglowodanów [96]. Ceruloplazmina
syntezowana jest głównie w wątrobie, jednak może być również produkowana przez
kilka innych organów wydzielniczych, takich jak: nerki, gruczoł piersiowy, łożysko
czy splot naczyniówkowy mózgu [97]. W wątrobie biosynteza tego białka zachodzi
w aparacie Golgiego hepatocytów, gdzie enzym ATP7B uczestniczący w transporcie
miedzi w organizmie powoduje przyłączenie 6 atomów miedzi do nieaktywnej formy
białka, apo-ceruloplazminy (apo-Cp), tworząc holo-ceruloplazminę (holo-Cp). Ta
forma ceruloplazminy jest aktywna chemicznie i wydzielana jest do krwioobiegu
[98]. Ceruloplazmina jest głównym białkiem transportującym miedź w organizmie,
wiąże ponad 95% całkowitej jej ilości w osoczu krwi [99].
Struktura ludzkiej Cp jest znacznie bardziej złożona w porównaniu do innych
multimiedziowych oksydaz. Na podstawie badań krystalografii rentgenowskiej
udowodniono, że cząsteczka ceruloplazminy ma zwartą strukturę (około 214 x 85 Å)
z 3 homologicznymi regionami, z których każdy składa się z dwóch części [100].
Cząsteczka tego białka zbudowana jest z sześciu β-beczułkowych domen i zawiera
unikalne centrum aktywne, które tworzy grupa prostetyczna złożona z trzech różnych
typów centrów miedziowych: T1, T2 oraz T3, zawierających w sumie sześć atomów
miedzi. Poszczególne centra miedziowe są odzwierciedleniem budowy miejsc
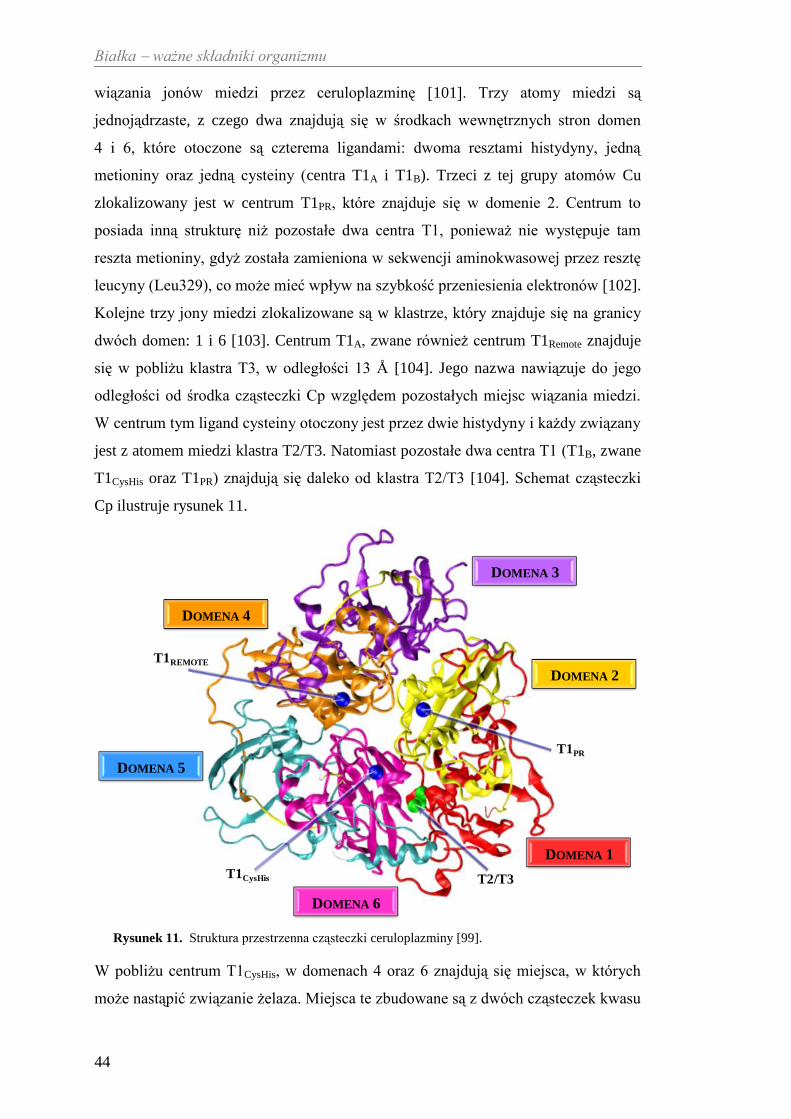
Białka ważne składniki organizmu
44
wiązania jonów miedzi przez ceruloplazminę [101]. Trzy atomy miedzi są
jednojądrzaste, z czego dwa znajdują się w środkach wewnętrznych stron domen
4 i 6, które otoczone są czterema ligandami: dwoma resztami histydyny, jedną
metioniny oraz jedną cysteiny (centra T1A i T1B). Trzeci z tej grupy atomów Cu
zlokalizowany jest w centrum T1PR, które znajduje się w domenie 2. Centrum to
posiada inną strukturę niż pozostałe dwa centra T1, ponieważ nie występuje tam
reszta metioniny, gdyż została zamieniona w sekwencji aminokwasowej przez resztę
leucyny (Leu329), co może mieć wpływ na szybkość przeniesienia elektronów [102].
Kolejne trzy jony miedzi zlokalizowane są w klastrze, który znajduje się na granicy
dwóch domen: 1 i 6 [103]. Centrum T1A, zwane również centrum T1Remote znajduje
się w pobliżu klastra T3, w odległości 13 Å [104]. Jego nazwa nawiązuje do jego
odległości od środka cząsteczki Cp względem pozostałych miejsc wiązania miedzi.
W centrum tym ligand cysteiny otoczony jest przez dwie histydyny i każdy związany
jest z atomem miedzi klastra T2/T3. Natomiast pozostałe dwa centra T1 (T1B, zwane
T1CysHis oraz T1PR) znajdują się daleko od klastra T2/T3 [104]. Schemat cząsteczki
Cp ilustruje rysunek 11.
Rysunek 11. Struktura przestrzenna cząsteczki ceruloplazminy [99].
W pobliżu centrum T1CysHis, w domenach 4 oraz 6 znajdują się miejsca, w których
może nastąpić związanie żelaza. Miejsca te zbudowane są z dwóch cząsteczek kwasu
T2/T3
T1REMOTE
T1CysHis
T1PR
DOMENA 2
DOMENA 3
DOMENA 1
DOMENA 6
DOMENA 5
DOMENA 4

Białka ważne składniki organizmu
45
glutaminowego, jednej histydyny oraz jednej cząsteczki kwasu asparaginowego.
Oprócz tych miejsc w domenie 2 obecne są również obszary podobne do ligandów
w domenie 4 i 6, które złożone są z jednej cząsteczki kwasu asparaginowego, jednej
tyrozyny oraz dwóch cząsteczek kwasu glutaminowego [105]. Klaster T2/T3,
zawierający trzy atomy miedzi, otoczony jest przez cztery pary reszt histydyny. Dwie
pary pochodzą z domeny 1 i oddzielone są od siebie resztą seryny, natomiast kolejne
pary związane są z domeną 6 i oddzielone są resztami cysteiny oraz fenyloalaniny.
Dodatkowo, dwa atomy miedzi wchodzące w skład centrum T2/T3 związane są
z trzema parami reszt histydyny, podczas gdy najdalej położony w domenie 6 atom
miedzi związany jest tylko z dwoma resztami histydyny, z tego powodu miejsce to
oznaczane jest jako typ II [106,107]. Klaster T2/T3 łączy N-końcową i C-końcową
domenę cząsteczki Cp, dzięki czemu stabilizuje strukturę ceruloplazminy
i powoduje, że ma ona globularny kształt [99,100,108]. Według danych
literaturowych centra T1 są akceptorami elektronów, natomiast klaster T2/T3 wiąże
tlen cząsteczkowy i redukuje go do wody. Istotną cechą centrów miedziowych typu
T1 jest to, że wykazują one optyczną absorpcję przy długości fali 610 nm. Natomiast
centrum T2 nie absorbuje światła, ale wykazuje właściwości paramagnetyczne.
Z kolei centrum miedziowe T3 jest diamagnetyczne i jest ono niezbędne do wiązania
oraz redukcji tlenu. Właściwości paramagnetyczne posiadają kolejne dwa centra T1:
T1Remote i T1CysHis [106,109,110].
Ceruloplazmina jest wielozadaniowym enzymem. Jedną z najważniejszych
fizjologicznych funkcji Cp jest transport jonów miedzi w organizmie. Miedź
niezwiązana z białkiem może wiązać się z wolnymi grupami tiolowymi
pochodzącymi z cysteiny i powodować ich utlenianie, co w konsekwencji może
prowadzić do utraty aktywności enzymów oraz powodować uszkodzenie różnych
białek strukturalnych [111]. Kolejną ważną funkcją ceruloplazminy jest jej
aktywność katalityczna w reakcji utleniania jonów Fe(II) do Fe(III). Z uwagi na ten
fakt, enzym ten nazywany jest również ferroksydazą [112]. Poprzez utlenianie jonów
żelaza(II) Cp reguluje metabolizm tego pierwiastka, umożliwia wychwytywanie
i wiązanie żelaza(III) przez transferynę, która jest głównym białkiem uczestniczącym
w transporcie tego metalu w organizmie [113-115]. Ograniczenie ilości jonów Fe(II)
powoduje, że Cp bierze udział w jednym z głównych systemów antyoksydacyjnych
(system Cp-Tf), który przeciwdziała stresowi oksydacyjnemu [116] wywołanemu
działaniem reaktywnych form tlenu na organizm [117]. Proces powstawania

Białka ważne składniki organizmu
46
szkodliwych dla zdrowia form tlenu katalizują jony metali przejściowych, w tym
jony żelaza(II) oraz miedzi(I). Nagromadzenie jonów Fe2+
(katalizatora
w wolnorodnikowej reakcji Fentona) prowadzi do powstania rodnika
hydroksylowego, w wyniku ich oddziaływania z nadtlenkiem wodoru [118,119].
Utlenienie jonów Cu+ oraz Fe
2+ do form Cu
2+ i Fe
3+ powoduje, że w tej postaci są
one znacznie mniej szkodliwe dla organizmu [120]. Zatem Cp jest skutecznym
przeciwutleniaczem, zapobiegającym oksydacyjnemu uszkodzeniu białek, lipidów
i DNA [121]. Oprócz utleniania jonów żelaza(II) Cp, jako enzym należący do
oksydoreduktaz, jest w stanie utleniać także rozległą grupę związków organicznych,
takich jak hormony, aminofenole czy katechole [122]. Białko to wykazuje również
właściwości utleniające względem miedzi(I) oraz tlenku azotu(II) [99].
Ceruloplazmina, jako białko ostrej fazy, może być wskaźnikiem
diagnostycznym wielu chorób. Prawidłowe stężenie ceruloplazminy w ludzkim
osoczu w przypadku kobiet mieści się w przedziale od 25 do 47 mgdL-1
,
a dla mężczyzn zakres ten wynosi od 25 do 37 mgdL-1
[123]. W przypadku
narządów wewnętrznych najwyższe stężenie miedzi jest w nerkach (7÷12 µg·g-1
),
nieco niższe w wątrobie (4÷6 µg·g-1
) i mózgu (3÷5 µg·g-1
). Natomiast w mięśniach
zawartość Cu to jedynie około 1 µg·g-1
. Stężenie miedzi w osoczu krwi i surowicy u
dorosłych osobników różnych ssaków jest dużo bardziej zmienne i można podzielić
je na trzy grupy, z których pierwszą stanowią myszy i psy (25÷40 mg·dL-1
), do
drugiej grupy zalicza się ludzi, szczury, krowy i owce (~100 mg·dL-1
), natomiast do
trzeciej grupy należą świnie (150÷200 mg·dL-1
). Stężenie miedzi w osoczu ulega
zmianie w odpowiedzi na specyficzne warunki otoczenia. Głównym sposobem
dostarczania miedzi do organizmu jest pożywienie (40÷70%). Miedź pochodząca
z pokarmów jest wchłaniana głównie w dwunastnicy i jelicie cienkim. Pierwszy etap
pobierania i rozkładu miedzi związany jest z transportem miedzi do krwi, gdyż
praktycznie cała miedź zostaje związana z białkami osocza, które następnie
wbudowywane są do hepatocytów. Większość miedzi ulega rozpuszczeniu
w wątrobie i jest wprowadzana do nieaktywnej formy Cp: apo-Cp. W kolejnym
etapie następuje wydzielanie do krwioobiegu holo-Cp. Nadmiar miedzi wydalany
jest przez wątrobę do żółci, gdzie pierwiastek ten jest usuwany z organizmu przez
defekację [124]. Schemat wchłaniania miedzi, jej cyrkulacji w organizmie oraz
wydalania przedstawia rysunek 12.

Białka ważne składniki organizmu
47
MÓZG
KREW ŻÓŁĆ
CHOROBA
MENKESA
Cu ~ 6 mg
65 95%
miedzi w Cp
Cu ~ 2.5 mg/dzień
CHOROBA
WILSONA
CHOROBA
MENKESA
ORGANY
JE
LIT
OC
IEN
KIE
Cu ~ 0.5 2.5 mg/dzień
KAŁ
MIEDŹ Z POKARMU
PŁYNY WYDZIELNICZE
Cu ~ 2.0 mg/dzień
DWUNASTNICA
Cu ~ 4.5 mg/dzień
KRĄŻENIE WROTNE
Rysunek 12. Schemat procesu obiegu miedzi w organizmie [124].
Zmiany stężenia ceruloplazminy mają negatywny wpływ na zdrowie
człowieka. Spadek stężenia tego białka w osoczu krwi poniżej wartości 2 mgdL-1
związany jest z występowaniem choroby Menkesa lub choroby Wilsona [125].
Należy podkreślić, że ilość ceruloplazminy oraz całkowite stężenie miedzi maleją
także przy niedoborze miedzi w diecie [126]. Poziom Cp wzrasta w przypadku ciąży,
chorób nowotworowych, np. chłoniaka Hodgkina [127], jak również w stanie
zapalnym organizmu [128]. Białko to ma zatem duże znaczenie w diagnostyce
schorzeń, takich jak: białaczka, rak wątroby, gruźlica, zaburzenia neurologiczne
(np.: choroba Alzheimera, choroba Parkinsona) oraz udar [129]. Określenie stężenia
Cp w osoczu wymaga zastosowania bardzo czułych metod, z uwagi na fakt, że
w przypadku niektórych chorób jej stężenie jest zazwyczaj bardzo niskie.
Pacjenci z chorobą Menkesa (MNK, choroba krętych włosów) posiadają
zablokowany wychwyt miedzi, który spowodowany jest mutacją w genie ATP7A,
w wyniku niedoboru miedzi w układzie krążenia [123]. Choroba ta jest zaburzeniem
metabolizmu miedzi dziedziczonym recesywnie, prowadzącym do neurodegeneracji
organizmu [130]. Objawy tej choroby widoczne są już po urodzeniu i bardzo często
prowadzą do zgonu we wczesnych latach życia. Spadek aktywności białka
ATP7A-azy prowadzi do gromadzenia się miedzi w enterocytach, przez co następuje
obniżenie jej wchłaniania z pokarmu i kumulacja w nerkach [123]. Schorzenie to
występuje niezwykle rzadko, od 1/100 000 do 1/300 000 żywych urodzeń [131,132].

Białka ważne składniki organizmu
48
Nosicielami zmutowanego genu są kobiety, natomiast choroba ta pojawia się tylko
u chłopców. Typowymi objawami choroby Menkesa są kręte i szorstkie włosy oraz
zaburzenia neurologiczne, takie jak: brak kontroli głowy, zaburzenia wzroku,
napięcie mięśniowe kończyn oraz drgawki [123].
Niski poziom ilości Cp w osoczu krwi jest zwykle związany z chorobą
Wilsona (WD) [133], czyli dziedzicznym zaburzeniem metabolizmu miedzi
z poważnymi neurologicznymi objawami. Znana jest ona również pod nazwą
zwyrodnienia soczewkowo–wątrobowego. Nagromadzenie miedzi w różnych
narządach, głównie w wątrobie i ośrodkowym układzie nerwowym [134] może
prowadzić do marskości wątroby, zaburzeń neurologicznych oraz do innych
objawów powiązanych z zaburzeniem transportu miedzi [135]. Toksyczne
nagromadzenie miedzi w wątrobie jest związane z mutacją białka ATP7B, które
kontroluje równowagę miedzi w organizmie. Częstość występowania tego
zaburzenia wynosi od 1/30 000 do 1/ 100 000 [136]. U osób z tym zaburzeniem
często pojawia się marskość wątroby, przewlekłe zapalenie tego narządu oraz jego
niewydolność. Mogą wystąpić także objawy neurologiczne, w tym zaburzenia
ruchowe, drgawki, zmiany osobowości, depresja czy psychoza. Miedź może także
gromadzić się w innych tkankach, np. rogówce oka, powodując pojawienie się tzw.
pierścienia Kaysera-Fleischera, będącego objawem charakterystycznym tej choroby
[137].
Z kolei wzrost stężenia ceruloplazminy w osoczu krwi może być związany
głównie z chłoniakiem Hodgkina. Choroba ta nazywana jest również ziarnicą
złośliwą [127]. Chłoniak Hodgkina jest nowotworem układu limfatycznego, który
powoduje rozrost limfocytów B, które tworzą komórki nowotworowe Reed-
Sternberga (R-S). Nowotwór ten stanowi około 1% wszystkich występujących
nowotworów [138]. Przyczyną tej choroby są najprawdopodobniej uwarunkowania
genetyczne oraz różnego rodzaju infekcje. Rozpoznanie choroby jest niezwykle
trudne, ponieważ głównym jej objawem jest powiększenie węzłów chłonnych. Inne
objawy, takie jak gorączka, czy spadek masy ciała pojawiają się w znacznie
późniejszym okresie i związane są z zaawansowanym stadium nowotworu. Oprócz
układu chłonnego Chłoniak Hodgkina może zająć również wątrobę, płuca oraz szpik
kostny [139,140].

Białka ważne składniki organizmu
49
4.2.3. Transferyna, białko transportujące jony żelaza w organizmie
Transferyny to grupa białek uczestniczących w wiązaniu i transporcie żelaza
w organizmie. Do rodziny tej należą: laktoferyna (LF), owotransferyna (OTf),
melanotransferyna (MTf) oraz ludzka transferyna osocza (Tf). Laktoferyna jest
białkiem występującym głównie w mleku, łzach, białych krwinkach, żółci i soku
trzustkowym. Z kolei owotransferyna spotykana jest głównie w jajach kurzych,
a melanotransferyna jest białkiem membranowym obecnym w bardzo małej ilości na
powierzchni zdrowych komórek [141]. Najbardziej rozpowszechnionym białkiem
z tej grupy, występującym u wszystkich kręgowców, a także u krabów i owadów jest
transferyna [142].
U ssaków transferyna jest składnikiem krwi, limfy oraz płynów mózgowo-
rdzeniowego i owodniowego [143]. Białko to składa się z pojedynczego
glikoproteinowego łańcucha, zawierającego około 700 aminokwasów, o masie
cząsteczkowej bliskiej 80 kDa (~79 985 g·mol-1
) [144]. Łańcuch polipeptydowy Tf
tworzą dwie globularne podjednostki, nazywane podjednostkami N- oraz
C-końcowymi, wiążące w sumie dwa atomy żelaza. Strukturalnie podjednostki te są
do siebie bardzo podobne, masa każdej z nich wynosi około 40 kDa i połączone są ze
sobą za pomocą krótkiego peptydu. Strukturę przestrzenną cząsteczki transferyny
przedstawia rysunek 13.
Rysunek 13. Struktura przestrzenna cząsteczki transferyny oraz schemat budowy miejsc wiązania
jonów Fe(III) w domenach C- i N-końcowej. W nawiasach podano aminokwasy domeny
N-końcowej. Kolor niebieski symbolizuje atom azotu, zielony – atom tlenu, zaś szary – atom
węgla. Rysunek wykonano na podstawie danych literaturowych [143,145].
Każda z podjednostek Tf podzielona jest na dwie domeny o podobnym rozmiarze,
które zawierają fragmenty α-helisy oraz β-kartki [146]. Podjednostki te posiadają
Fe(III)
Fe(III)
domena C-końcowa
domena N-końcowa
Tyr188 (517)Tyr95 (426)
His249 (585) Asp63 (392)
CO32-
Fe3+

Białka ważne składniki organizmu
50
po jednym miejscu wiązania żelaza(III), składającego się z: kwasu
asparginowego, dwóch cząsteczek tyrozyny, jednej histydyny oraz anionu
węglanowego, nazywanego także anionem synergistycznym [147]. Miejsca te są
do siebie bardzo podobne zarówno pod względem budowy, jak i długości
wiązania metal-ligand, która zawiera się w przedziale od około 1.9 do 2.2 Å
[148]. Ligandy pochodzą z czterech różnych części transferyny. Jeden z ligandów
pochodzi z domeny 1 (Asp63), kolejny z domeny 2 (Tyr188), a ostatnie dwa
ligandy (Tyr95 i His249) z dwóch łańcuchów polipeptydowych, które występują
pomiędzy tymi domenami [147]. Jony Fe3+
wiązane są w domenie N-końcowej za
pośrednictwem Asp392, His585, Tyr426, Tyr517 oraz jonów CO32-
. Natomiast
wiązanie żelaza w domenie C-końcowej zachodzi poprzez atomy tlenu Asp63,
Tyr95, Tyr188, atom azotu His249 oraz dwa atomy tlenu pochodzące z jonu CO32-
[143]. Domeny mogą przemieszczać się względem siebie i prowadzić do zmiany
konformacji cząsteczki transferyny z zamkniętej, zawierającej jony Fe(III), do
otwartej – pozbawionej jonów żelaza [147]. Zmiany konformacyjne cząsteczki Tf
mogą mieć funkcjonalne znaczenie i odgrywają kluczową rolę w rozpoznawaniu
oraz wiązaniu receptora Tf. Schematycznie zmiany konformacyjne cząsteczki
transferyny ilustruje rysunek 14.
Fe(III)
KONFORMACJA OTWARTA KONFORMACJA ZAMKNIĘTA
Rysunek 14. Konformacja otwarta oraz konformacja zamknięta transferyny [149].
Silne wiązanie jonów żelaza(III) przez transferynę występuje tylko
w przypadku obecności anionu węglanowego, który odgrywa niezwykle istotną rolę
w tworzeniu miejsca wiązania żelaza. Brak tego jonu powoduje, że wiązanie żelaza
jest dużo słabsze [150]. W osoczu krwi tylko około 30% całkowitej ilości transferyny
występuje w postaci związanej z jonami żelaza(III), podczas gdy 70% pozostaje
w formie apo-transferyny (apo-Tf), która może również wiązać inne jony metali
w tych samych miejscach, co jony żelaza. Forma Tf zawierająca żelazo nosi nazwę
holo-transferyny (holo-Tf) [151,152] i może ona transportować jeden jon Fe3+

Białka ważne składniki organizmu
51
(Nt(Fe)Tf oraz Tf(Fe)Ct) lub też dwa jony tego metalu (Tf(Fe)2) [153]. Główna
funkcja transferyny polega na wiązaniu jonów żelaza(III), co ułatwia transport żelaza
z miejsc jego magazynowania do miejsc, w których następuje zapotrzebowanie na
ten metal [154]. Stężenie transferyny w osoczu utrzymuje się na stałym poziomie od
momentu urodzenia i wynosi około 0.2 ÷ 0.3 g·dL-1
[155]. Odpowiedni poziom tego
białka jest niezwykle ważny dla prawidłowego wzrostu komórek. Tf, podobnie jak
Cp, należy do białek ostrej fazy i spadek jej ilości w osoczu krwi poniżej 0.01 g·dL-1
może być związany z występowaniem zakażenia, opóźnienia wzrostu oraz
niedokrwistości [156].
Z uwagi na fakt, że żelazo może być zarówno akceptorem, jak i donorem
elektronów oraz dodatkowo może oddziaływać z różnymi cząsteczkami, jest ono
jednym z najbardziej rozpowszechnionych metali w organizmie człowieka,
pełniącym różnorodne funkcje. Metal ten może odwracalnie wiązać różne ligandy,
przez co uczestniczy w przemianach metabolicznych organizmu [157]. Żelazo
przenoszone przez transferynę trafia przede wszystkim do białek oddechowych, czyli
hemoglobiny znajdującej się w erytrocytach oraz mioglobiny, która obecna jest
w mięśniach. Metal ten jest niezbędny do pełnienia przez te białka ich funkcji
fizjologicznych [157]. Cześć żelaza trafia również do ferrytyny, czyli białka
magazynującego ten metal w organizmie [158]. Żelazo bierze także udział
w syntezie różnych związków, takich jak hormony czy kolagen. Może ono również
uczestniczyć w replikacji DNA i przemianie energii [159-161]. Obecność żelaza
w cząsteczce Tf sprawia, że białko to działa jako środek przeciwbakteryjny, gdyż
zapobiega dostarczaniu niezbędnych substancji odżywczych do komórek
bakteryjnych, jak również przeciwdziała powstawaniu toksycznych form tlenu
powstających w reakcji Fentona [162,163]. Transferyna może również pełnić rolę
wskaźnika informującego o przewlekłym nadużywaniu alkoholu [164]. Spożywanie
alkoholu ma wpływ na strukturę cząsteczki Tf [165] i prowadzi do zmniejszenia
ilości kwasu sialowego w cząsteczce transferyny, a powstała izoforma Tf nosi nazwę
transferyny desialowanej (CDT) [166]. Jednak zmiany te w strukturze białka są
odwracalne i w przypadku odstawienia alkoholu, po kilku dniach transferyna wraca
do wyjściowej formy [165]. Schematycznie funkcje transferyny i transport żelaza
w organizmie przedstawiono na rysunku 15.
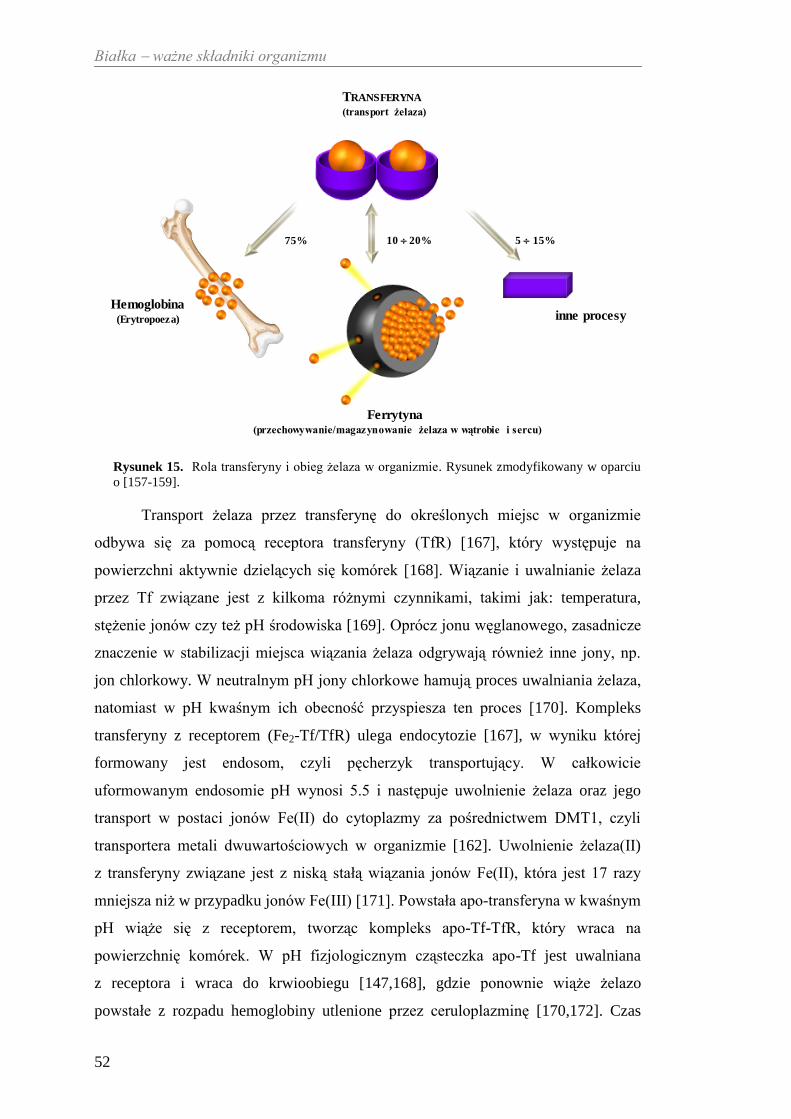
Białka ważne składniki organizmu
52
TRANSFERYNA
(transport żelaza)
Ferrytyna (przechowywanie/magazynowanie żelaza w wątrobie i sercu)
inne procesyHemoglobina
(Erytropoeza)
75% 10 20% 5 15%
Rysunek 15. Rola transferyny i obieg żelaza w organizmie. Rysunek zmodyfikowany w oparciu
o [157-159].
Transport żelaza przez transferynę do określonych miejsc w organizmie
odbywa się za pomocą receptora transferyny (TfR) [167], który występuje na
powierzchni aktywnie dzielących się komórek [168]. Wiązanie i uwalnianie żelaza
przez Tf związane jest z kilkoma różnymi czynnikami, takimi jak: temperatura,
stężenie jonów czy też pH środowiska [169]. Oprócz jonu węglanowego, zasadnicze
znaczenie w stabilizacji miejsca wiązania żelaza odgrywają również inne jony, np.
jon chlorkowy. W neutralnym pH jony chlorkowe hamują proces uwalniania żelaza,
natomiast w pH kwaśnym ich obecność przyspiesza ten proces [170]. Kompleks
transferyny z receptorem (Fe2-Tf/TfR) ulega endocytozie [167], w wyniku której
formowany jest endosom, czyli pęcherzyk transportujący. W całkowicie
uformowanym endosomie pH wynosi 5.5 i następuje uwolnienie żelaza oraz jego
transport w postaci jonów Fe(II) do cytoplazmy za pośrednictwem DMT1, czyli
transportera metali dwuwartościowych w organizmie [162]. Uwolnienie żelaza(II)
z transferyny związane jest z niską stałą wiązania jonów Fe(II), która jest 17 razy
mniejsza niż w przypadku jonów Fe(III) [171]. Powstała apo-transferyna w kwaśnym
pH wiąże się z receptorem, tworząc kompleks apo-Tf-TfR, który wraca na
powierzchnię komórek. W pH fizjologicznym cząsteczka apo-Tf jest uwalniana
z receptora i wraca do krwioobiegu [147,168], gdzie ponownie wiąże żelazo
powstałe z rozpadu hemoglobiny utlenione przez ceruloplazminę [170,172]. Czas
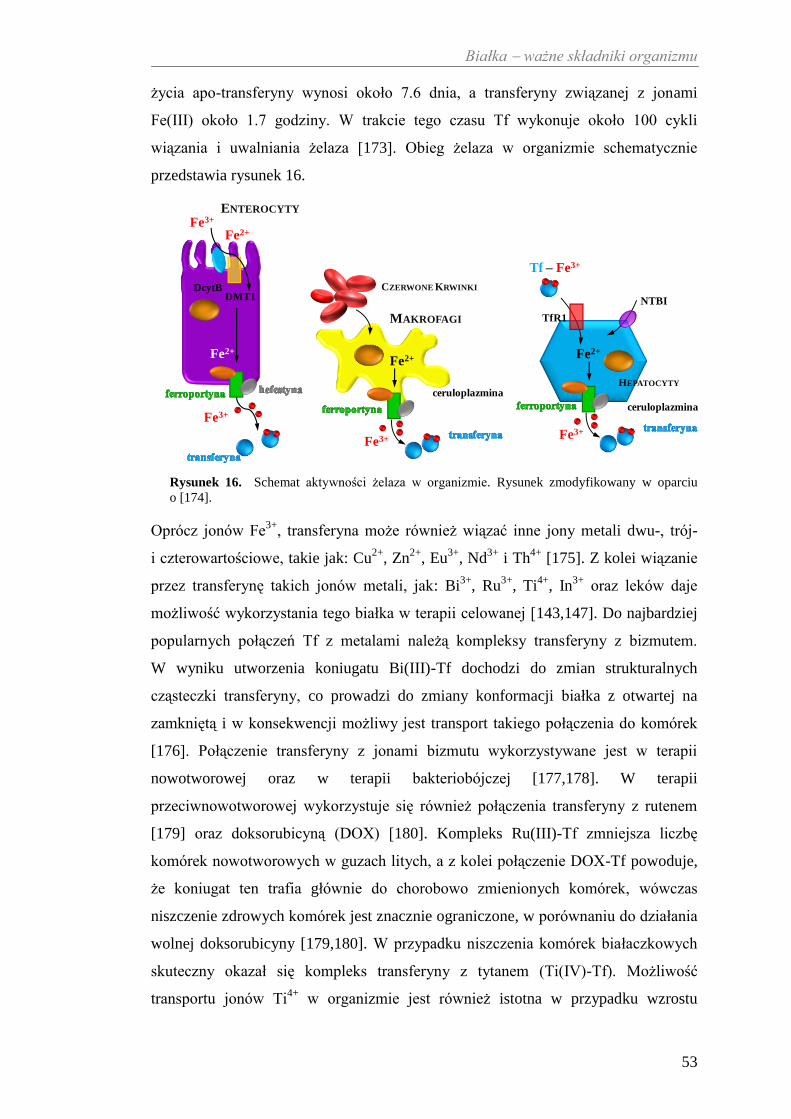
Białka ważne składniki organizmu
53
życia apo-transferyny wynosi około 7.6 dnia, a transferyny związanej z jonami
Fe(III) około 1.7 godziny. W trakcie tego czasu Tf wykonuje około 100 cykli
wiązania i uwalniania żelaza [173]. Obieg żelaza w organizmie schematycznie
przedstawia rysunek 16.
Rysunek 16. Schemat aktywności żelaza w organizmie. Rysunek zmodyfikowany w oparciu
o [174].
Oprócz jonów Fe3+
, transferyna może również wiązać inne jony metali dwu-, trój-
i czterowartościowe, takie jak: Cu2+
, Zn2+
, Eu3+
, Nd3+
i Th4+
[175]. Z kolei wiązanie
przez transferynę takich jonów metali, jak: Bi3+
, Ru3+
, Ti4+
, In3+
oraz leków daje
możliwość wykorzystania tego białka w terapii celowanej [143,147]. Do najbardziej
popularnych połączeń Tf z metalami należą kompleksy transferyny z bizmutem.
W wyniku utworzenia koniugatu Bi(III)-Tf dochodzi do zmian strukturalnych
cząsteczki transferyny, co prowadzi do zmiany konformacji białka z otwartej na
zamkniętą i w konsekwencji możliwy jest transport takiego połączenia do komórek
[176]. Połączenie transferyny z jonami bizmutu wykorzystywane jest w terapii
nowotworowej oraz w terapii bakteriobójczej [177,178]. W terapii
przeciwnowotworowej wykorzystuje się również połączenia transferyny z rutenem
[179] oraz doksorubicyną (DOX) [180]. Kompleks Ru(III)-Tf zmniejsza liczbę
komórek nowotworowych w guzach litych, a z kolei połączenie DOX-Tf powoduje,
że koniugat ten trafia głównie do chorobowo zmienionych komórek, wówczas
niszczenie zdrowych komórek jest znacznie ograniczone, w porównaniu do działania
wolnej doksorubicyny [179,180]. W przypadku niszczenia komórek białaczkowych
skuteczny okazał się kompleks transferyny z tytanem (Ti(IV)-Tf). Możliwość
transportu jonów Ti4+
w organizmie jest również istotna w przypadku wzrostu
Fe3+
Fe2+
DcytBDMT1
Fe2+
Fe3+
ENTEROCYTY
Fe2+
Fe3+
ceruloplazmina
CZERWONE KRWINKI
MAKROFAGI
HEPATOCYTY
Fe2+
Fe3+
Tf – Fe3+
ceruloplazmina
TfR1
NTBI

Białka ważne składniki organizmu
54
stężenia tych jonów z powodu obecności implantów tytanowych, środków
obrazujących opartych na tytanie oraz dodatków do żywności [181,182].
Dostarczanie przez transferynę leków do komórek nowotworowych możliwe jest
dzięki wykorzystaniu faktu, że na powierzchni chorych komórek występuje
zwiększona ilość receptorów Tf, co zwiększa również możliwość odpowiedniej
lokalizacji zmian nowotworowych [183].
4.2.4. Ferrytyna, białko magazynujące żelazo w organizmie
Ferrytyny to grupa białek zdolnych do wiązania, magazynowania
i uwalniania żelaza w miejscach, w których pojawia się zapotrzebowanie na ten
pierwiastek. Do rodziny tej zalicza się białka magazynujące żelazo (Ftn), ferrytyny
bakteryjne zawierające hem (Bfr) oraz białka wiążące DNA (Dps). Ferrytyny
uczestniczą w szlakach biochemicznych regulujących apoptozę, jak również
translację białek [184], co sprawia, że mają one istotne znaczenie w prawidłowym
funkcjonowaniu organizmów.
Ferrytyna (Ft) została po raz pierwszy odkryta i wyizolowana ze śledziony
konia przez Laufbergera w 1937 roku [185], jest ona obecna również u ludzi,
ssaków, roślin, grzybów i bakterii [186]. Ferrytyna odgrywa kluczową rolę
w utrzymaniu odpowiedniego stężenia żelaza wewnątrz komórek [187], poprzez jego
magazynowanie oraz kontrolowane uwalnianie [188]. Wiążąc wolne jony żelaza
zapobiega zarówno peroksydacji lipidów, zmianom w płynności błon komórkowych,
jak również rozrywaniu błony i śmierci komórek [189]. Białko to reguluje również
rozwój organizmu [190] oraz pełni rolę wskaźnika różnych procesów zapalnych
[191]. Dodatkowo ferrytyna zabezpiecza organizm przed stresem oksydacyjnym
poprzez ograniczenie nadmiaru wolnych jonów żelaza generujących reaktywne
formy tlenu w reakcji Fentona [192] oraz bierze udział w reakcjach obronnych
komórek [193].
Cząsteczka ferrytyny przypomina kształtem kulę zbudowaną z powłoki
białkowej, której wnętrze wypełniają związki żelaza, a jej masa cząsteczkowa
wynosi około 450 kDa (~449 920 g·mol-1
) [185]. Jedna cząsteczka ferrytyny może
związać ok. 4500 atomów Fe, przy czym zazwyczaj wiąże od 1000 do 2000 atomów
Fe [194]. Cząsteczka ferrytyny posiada powłokę białkową łączącą się z jądrem
żelaznym poprzez kanały wzdłuż 2, 3 i 4-krotnych osi symetrii [194]. Zewnętrzna
średnica cząsteczki ferrytyny wynosi około 12 nm, natomiast średnica wewnętrznej

Białka ważne składniki organizmu
55
kapsuły, w której tworzy się jądro rdzenia żelaznego, ma wielkość około 8 nm [195].
Schemat struktury cząsteczki ferrytyny przedstawiony został na rysunku 17.
Rysunek 17. Struktura przestrzenna cząsteczki ferrytyny [196].
Powłoka białkowa ferrytyny składa się z 24 podjednostek (łańcuchów) dwóch
typów: ciężkich (H) oraz lekkich (L) [197,198]. Rola łańcuchów H i L jest
zróżnicowana. Łańcuchy ciężkie, których masa cząsteczkowa wynosi około 24 kDa,
odpowiedzialne są za transport żelaza do wnętrza ferrytyny i na zewnątrz molekuły.
Biorą one udział w utlenianiu jonów Fe(II) do Fe(III) [194], dzięki obecności
w swojej strukturze ferroksydazy. Ferroksydaza zlokalizowana jest w czterech
spiralnych fragmentach łańcuchów polipeptydowych podjednostek H w odległości
7÷8 Å od wewnętrznej i 12÷13 Å od zewnętrznej granicy otoczki białkowej,
w regionie o charakterze silnie hydrofobowym [196]. Ludzka ferroksydaza składa się
z dwóch centrów wiązania żelaza (FeA i FeB), które są osłonięte przez następujące
aminokwasy: His65, Glu27, Glu107, Glu61 i Glu62, co pokazuje rysunek 18.
ZEWNĘTRZNA STRONA
OTOCZKI BIAŁKOWEJ
WEWNĘTRZNA STRONA
OTOCZKI BIAŁKOWEJ
OH
O
O
NH
O
Tyr - 34
Glu - 107
Gln - 141
Glu - 27
Glu - 62Glu - 61
O
O O O O+
H H
O
O
N+
N
H
His - 65FeB FeA
Rysunek 18. Położenie ferroksydazy w cząsteczce ferrytyny [195].
= 12 nm
= 8 nm

Białka ważne składniki organizmu
56
Dodatkowo łańcuchy H posiadają znaczną ilość reszt kwasu glutaminowego, które
łatwo reagują z reaktywnymi formami tlenu. Mutacje w aminokwasach Glu62 oraz
His65 mogą blokować wiązanie żelaza przez cząsteczkę ferrytyny. Z kolei łańcuch
lekki (L), o masie 19 kDa, z uwagi na obecność wielu grup karboksylowych nie
uczestniczy w utlenianiu żelaza [199], natomiast bierze udział w tworzeniu
wewnętrznej struktury ferrytyny, mineralnego rdzenia, który zbudowany jest głównie
z cząsteczek uwodnionego tlenku żelaza(III) [FeIII
10O14(OH)2] [200]. Dodatkowo
łańcuchy L stabilizują strukturę tego białka [194]. Struktura FeIII
10O14(OH)2 złożona
jest z pojedynczej fazy heksagonalnej i zawiera ona około 20% tetraedrycznie
skoordynowanego żelaza(III) oraz około 80% oktaedrycznie skoordynowanych
jonów Fe3+
[201].
Stężenie żelaza w cytoplazmie jest dużo większe niż innych metali
przejściowych. Otoczenie cytoplazmy o charakterze redukującym sprzyja obecności
żelaza na drugim stopniu utlenienia, gdzie jego stężenie wynosi od 10-7
do 10-18
M
[202]. Żelazo(II) wychwytywane jest najpierw przez apo-ferrytynę (cząsteczkę
niezawierającą w swojej strukturze jonów żelaza), która obecna jest głównie
w cytoplazmie [203]. Ujemnie naładowane kanały w ferrytynie przyciągają jony
Fe2+
, powodując tym samym wciąganie ich do wnętrza struktury ferrytyny [204],
gdzie następnie ulegają one utlenianiu do jonów Fe3+
i w tej postaci żelazo jest
przechowywane w cząsteczce Ft [205]. Reakcja utleniania jonów Fe(II) zachodzi
przy udziale tlenu i katalizowana jest przez podjednostkę ciężką ferrytyny, a ściślej
mówiąc przez ferroksydazę wchodzącą w skład łańcucha H. W reakcji tej wolne
miejsca ferroksydazy (P) oddziałują z jonami Fe(II), w wyniku czego tworzą się
odpowiednie tlenki żelaza oraz nadtlenek wodoru. Reakcję utleniania żelaza(II)
w cząsteczce ferrytyny ilustruje następujące równanie [206]:
H4OHFeOOH2P
)OH(OFePOFePPOH4OFe2
22
222
22222
2
(9)
W kolejnym etapie powstaje jądro rdzenia żelaznego i następuje jego wzrost
w wyniku oddziaływania z łańcuchem L. Powstawaniu rdzenia żelaznego
w ferrytynie mogą sprzyjać niektóre jony, jak np. jony fosforanowe [207].
W przypadku, kiedy rdzeń żelazny jest już dobrze uformowany i osiągnie
wystarczającą wielkość, a jony żelaza(II) dalej dostarczane są do wnętrza ferrytyny;
ich utlenianie może odbywać się bezpośrednio na powierzchni rdzenia żelaznego
za pośrednictwem tlenu, zgodnie z równaniem [194]:

Białka ważne składniki organizmu
57
H8FeOOH4OH6OFe4 222 (10)
Reakcja bezpośredniego utleniania żelaza jest reakcją dominującą. Schemat
mineralizacji żelaza w powłoce białkowej cząsteczki ferrytyny przedstawia rysunek
19.
Fe3+
Fe3+
Fe2+
Fe2+
Fe3+ łańcuch ciężki (H) łańcuch lekki (L)
UTLENIANIE Fe2+
PRZEZ FERROOKSYDAZĘ
W ŁAŃCUCHU CIĘŻKIM
TWORZENIE SIĘ
I WZROST MINERALNEGO
RDZENIA FERRYTYNY
UTLENIANIE Fe2+
NA POWIERZCHNI
MINERALNEGO RDZENIA
FERRYTYNY
WZROST MINERALNEGO
RDZENIA FERRYTYNY
Rysunek 19. Schemat mineralizacji żelaza w powłoce białkowej cząsteczki ferrytyny.
Prawidłowe funkcjonowanie cząsteczki Ft oparte jest na współdziałaniu dwóch
procesów pozwalających na szybkie i efektywne magazynowanie żelaza oraz jego
kontrolowane uwalnianie. Uwalnianie żelaza w postaci jonów Fe(II) z wnętrza
ferrytyny w warunkach fizjologicznych następuje w momencie, gdy stężenie żelaza
w cytoplazmie ulega znacznemu zmniejszeniu [208]. Ferrytyna oprócz jonów żelaza
może wiązać i magazynować również inne jony metali. Niektóre z tych jonów, takie
jak Zn(II), wiążąc się z ferrytyną hamują proces wiązania żelaza przez to białko
[209]. Z kolei jony miedzi(II), oddziałując z ferrytyną, katalizują wychwyt żelaza
przez cząsteczki tego białka. Ferrytyna może również oddziaływać z różnymi
biomolekułami, takimi jak: glutation (GSH), oksydaza ksantynowa i dysmutaza
ponadtlenkowa (SOD) [208].
Ferrytyna jest niezwykle stabilną cząsteczką, jest odporna na zmiany
temperatury aż do 70 °C i wartości pH w zakresie od 3 do 10. Przy pH poniżej 3
poszczególne podjednostki mogą odłączać się od reszty Ft, jednak zmiany te są
odwracalne, przy powrocie do pH większego od 3 cząsteczka wraca do wyjściowej
formy [210]. Ilość poszczególnych łańcuchów w organach zawierających ferrytynę
jest odmienna. W wątrobie i śledzionie dominują łańcuchy lekkie, a z kolei w mózgu
i sercu przeważa ilość łańcuchów ciężkich [211,212]. Łańcuchy L charakteryzują się

Białka ważne składniki organizmu
58
większą stabilnością niż łańcuchy H nawet w warunkach kwasowych i redukujących
[213].
Synteza podjednostek H i L ferrytyny oraz metabolizm żelaza regulowane są
przez dwa białka: IRP1 oraz IRP2 [214]. Białka te wiążą się do odpowiednich
sekwencji RNA, np. mRNA kodujących ferrytynę lub do receptorów transferyny.
Przy wzroście stężenia żelaza w cytoplazmie białko IRP1 tworzy klaster Fe-S.
Z kolei, gdy poziom żelaza jest niski białko to przyjmuje konformację otwartą
pozbawioną klastra Fe-S, która wiąże się z sekwencjami reagującymi na żelazo
(IRE) występującymi w mRNA ferrytyny i powodującymi blokowanie translacji
mRNA [214]. Obecność IRP2 w komórkach reguluje ich degradację spowodowaną
zbyt dużą ilością żelaza [215]. Regulacja stężenia żelaza przez białka IRP
w warunkach niedoboru oraz nadmiaru tego metalu w komórkach sprawia, że
organizm szybko powraca do stanu, w którym stężenie żelaza jest odpowiednie do
prawidłowego funkcjonowania komórek [216].
Zaburzenia funkcji ferrytyny mogą powodować typowe choroby powiązane
ze wzrostem lub zmniejszeniem ilości żelaza w organizmie. Poziom ferrytyny ma
istotne znaczenie w przypadku wystąpienia anemii, jak również może odgrywać
kluczową rolę w niektórych zaburzeniach neurologicznych, takich jak choroba
Parkinsona lub choroba Alzheimera [217]. Podstawowym elementem pozwalającym
na wprowadzenie skutecznego leczenia niedokrwistości jest ustalenie stężenia
ferrytyny w surowicy. Niedobór żelaza występuje przy stężeniu tego białka poniżej
2 µgdL-1
. Jednak przy stanach zapalnych nerek stężenie ferrytyny w osoczu wzrasta
i jest niezależne od ilości żelaza w surowicy. U osób z chorobą nerek niedobór żelaza
występuje, gdy ilość ferrytyny spada poniżej 10 µgdL-1
[218]. Przyjmuje się, że
prawidłowe stężenie żelaza występuje w przypadku, kiedy stężenie ferrytyny
w surowicy krwi mieści się w przedziale od 10 do 40 µgdL-1
[219]. Około 20%
całkowitej ilości żelaza zmagazynowane jest w ferrytynie. Synteza tego białka
wzrasta wraz ze wzrostem stężenia żelaza w organizmie [220]. Stężenie ferrytyny
w surowicy mężczyzn jest wyższe niż stężenie tego białka u kobiet [221]. Niższe
stężenie ferrytyny u kobiet prawdopodobnie związane jest z regularną utratą krwi
podczas miesiączki [222]. Wzrost stężenia żelaza, które zmagazynowane jest
w łańcuchach lekkich ferrytyny może prowadzić do peroksydacji lipidów, co
w konsekwencji powoduje zmiany miażdżycowe [223]. Wzrost stężenia ferrytyny
związany jest ze zwiększonym ryzykiem chorób sercowo-naczyniowych [224], np.

Białka ważne składniki organizmu
59
wzrost ilości tego białka u mężczyzn powoduje większe ryzyko zawału serca
w porównaniu do osób, u których stężenie ferrytyny było dużo niższe [225].
Wchłanianie żelaza, magazynowanie i przechowywanie przez ferrytynę może
stanowić informację przydatną w projektowaniu nowych środków terapeutycznych
opartych na metabolizmie żelaza [226]. Białko to, z uwagi na swoje unikalne
właściwości i możliwość zmian struktury w zależności od wartości pH może być
zastosowane, jako potencjalny nośnik leków przeciwnowotworowych, takich jak np.
cis-platyna [227]. Ferrytyna znalazła również zastosowanie w obrazowaniu metodą
rezonansu magnetycznego oraz w terapii radiofarmaceutykami [226]. Możliwości
zastosowania ferrytyny ilustruje rysunek 20.
Rysunek 20. Potencjalne możliwości zastosowania ferrytyny [228].
4.3. Metody oznaczania zawartości białek w płynach
ustrojowych
Z uwagi na fakt, że białka odgrywają niezwykle istotną rolę w każdym
organizmie, określenie zmian ich stężenia w płynach ustrojowych jest niezwykle
ważne. Istnieje wiele metod wykrywania i oznaczania stężenia białek (w tym
metaloprotein), które różnią się między sobą zakresem detekcji, czułością
i selektywnością. Do ilościowej analizy białek w płynach ustrojowych wykorzystuje
się najczęściej następujące techniki: immunonefelometrię, immunoturbidymetrię, test
ELISA, immunodyfuzję radialną, immunoelektroforezę, technikę Western Blot oraz
metody spektroskopowe. Zdecydowanie rzadziej używa się w tym celu technik
elektrochemicznych.
FERRYTYNA
MEDYCYNA URZĄDZENIAELERKTRONICZNE
MRI
SYSTEMY DOSTARCZANIA
LEKÓW
DIAGNOSTYKA BIOSENSORY
TRANZYSTOR POLOWY
URZĄDZENIA
PAMIĘCI

Białka ważne składniki organizmu
60
Metody immunoturbidymetryczne oraz immunonefelometryczne oparte są na
pomiarze zmian rozproszenia wiązki światła przechodzącej przez roztwór
zawierający badane białko oraz przeciwciało. W wyniku specyficznej reakcji białka
z przeciwciałem następuje utworzenie kompleksu antygen (białko)przeciwciało,
w wyniku czego roztwór ulega zmętnieniu, powodując większe rozproszenie wiązki
światła, niż w przypadku roztworów zawierających sam antygen lub tylko
przeciwciało [229]. Oznaczenie ilości białka opiera się na założeniu, że natężenie
światła rozproszonego jest wprost proporcjonalne do stężenia białka w analizowanej
próbce. Założenie to jest jednak prawdziwe wyłącznie w przypadku rozcieńczonych
roztworów, w których nie występuje możliwość całkowitego rozproszenia wiązki
światła przez duże stężenie cząsteczek znajdujących się blisko źródła światła, co
uniemożliwiałoby pomiar w głębi roztworu. Duże stężenie cząsteczek koloidu może
również powodować uzyskiwanie zaniżonych ilości białka [229]. W metodzie
immunoturbidymetrycznej w wyniku tworzenia się kompleksów
antygenprzeciwciało zwiększa się przenikalność wiązki światła przez roztwór, która
związana jest z ilością białka w próbce [230]. W technice tej detektor umieszczony
jest na wprost wiązki światła, podczas gdy w metodzie immunonefelometrycznej
detektor znajduje się pod kątem 90° w stosunku do wiązki światła.
W przeciwieństwie do immunoturbidymetrii detekcja w immunonefelometrii opiera
się na pomiarze światła rozproszonego [229]. Schematycznie zasadę pomiaru
w technice immunoturbidymetrycznej oraz immunonefelometrycznej ilustruje
rysunek 21.
Rysunek 21. Schemat układu pomiarowego do detekcji białek w metodzie immunoturbidy-
metrycznej i immunonefelometrycznej.
Test Elisa stosowany jest do wykrywania konkretnych białek w próbce
z użyciem przeciwciał mono- lub poliklonalnych, połączonych z enzymem. Odmianą
IMMUNOTURBIDYMETRIA IMMUNONEFELOMETRIA
ŹRÓDŁO ŚWIATŁAFOTODETEKTOR
FOTODETEKTOR
ŹRÓDŁO ŚWIATŁA
90

Białka ważne składniki organizmu
61
tej metody jest test podwójnego wiązania, zwany „Sandwich ELISA” lub też
„kanapkowa ELISA”. Test ten można wykonać metodą bezpośrednią lub pośrednią.
W metodzie bezpośredniej wykrywanie badanego białka odbywa się przy użyciu
jednego przeciwciała wyznakowanego enzymem. W metodzie pośredniej używa się
natomiast zarówno przeciwciał pierwszo-, jak i drugorzędowych. Znakuje się
wówczas przeciwciało drugorzędowe, które wiąże się z pierwszorzędowym
przeciwciałem wykrywającym dany antygen [231]. Zaletą zastosowania metody
pośredniej jest brak konieczności znakowania specyficznych przeciwciał względem
danego antygenu, dzięki czemu możliwe jest wykorzystanie różnych
pierwszorzędowych przeciwciał, co pozwala na szybszy pomiar i mniejsze koszty
analizy. Przeciwciała pierwszorzędowe związane z antygenem wykrywane są
następnie za pomocą zawsze tych samych znakowanych przeciwciał
drugorzędowych [232], co schematycznie ilustruje rysunek 22.
BEZPOŚREDNI TEST ELISA POŚREDNITEST ELISA
przeciwciało antygen znacznik
Rysunek 22. Schemat postępowania w teście Elisa wykorzystywanym do bezpośredniej
i pośredniej detekcji białek.
Kolejną techniką analityczną do oznaczania białek jest metoda
immunodyfuzji, której zasada działania opiera się na procesie tworzenia się
immunoprecypitatów w żelu agarowym lub agarozowym, w wyniku reakcji między
antygenem i specyficznym w stosunku do niego przeciwciałem. Istnieje kilka odmian
tej techniki, które różnią się między sobą ilością dyfundujących reagentów.
W metodzie immunodyfuzji jeden z reagentów może być związany z żelem,
natomiast drugi może dyfundować w żelu lub też oba reagenty mogą przemieszczać
się w żelu agarozowym [233]. Jedną z odmian immunodyfuzji jest technika

Białka ważne składniki organizmu
62
immunodyfuzji radialnej, zwana również testem Mancini, która służy do oznaczania
stężenia antygenu w próbce. Metoda ta opiera się na oddziaływaniu przeciwciała
umieszczonego w żelu agarozowym z roztworem zawierającym oznaczane białko
[234], w wyniku czego tworzy się nierozpuszczalny kompleks
antygenprzeciwciało, który widoczny jest w postaci tzw. pierścienia
precypitacyjnego [229]. Oznaczanie stężenia białka opiera się na pomiarach średnicy
utworzonego pierścienia precypitacyjnego, który jest wprost proporcjonalny do ilości
oznaczanego białka [235].
Z kolei technika immunoelektroforezy pozwala na określenie stężenia białek
w surowicy krwi i jest połączeniem techniki elektroforezy i immunodyfuzji [236].
Metoda ta opiera się na rozdziale białek na podstawie ich wielkości i ruchliwości
w żelu agarozowym pod wpływem pola elektrycznego. Mieszaninę specyficznych
dla białek przeciwciał umieszcza się w agarze i obserwuje się ich migrację
w kierunku określonych białek. W wyniku tworzenia kompleksu
antygenprzeciwciało powstają charakterystyczne linie związane z wytworzeniem
się osadu pochodzącego od utworzonego kompleksu [237].
Technika Western Blot, inaczej zwana również immunoblottingiem, jest
stosowana do identyfikacji poszczególnych białek ze złożonej mieszaniny [238].
Technika ta oparta jest na trzech elementach: rozdzieleniu białek według ich
wielkości, przeniesieniu ich na podłoże stałe oraz oznaczeniu białka docelowego za
pomocą odpowiedniego przeciwciała. W technice tej białka oddziela się od siebie
w oparciu o ich masę cząsteczkową [239]. Rozdzielanie białek prowadzi się za
pomocą elektroforezy w żelu, czyli pod wpływem pola elektrycznego. Białka o dużej
masie cząsteczkowej rozdzielane są na żelach charakteryzujących się niższą
gęstością, niż w przypadku białek o mniejszej masie cząsteczkowej. Ujemnie
naładowane białka pod wpływem pola elektrycznego wędrują w kierunku elektrody
o ładunku dodatnim. Początkowo odpowiednia ilość próbki rozcieńczana jest za
pomocą odpowiedniego buforu zawierającego glicerynę i następnie próbka jest
podgrzewana. W wyniku tych procesów następuje przecinanie mostków
disiarczkowych, co prowadzi do denaturacji drugorzędowej struktury białka. Ma to
na celu ograniczenie szybszej wędrówki białek o dużej masie cząsteczkowej,
charakteryzujących się bardziej zwartą strukturą w przeciwieństwie do lżejszych
białek, o rozluźnionej strukturze [239]. Po rozdzieleniu elektroforetycznym białka
przenoszone są z żelu na specjalną membranę pod wpływem prądu elektrycznego

Białka ważne składniki organizmu
63
[240], na której następuje proces migracji białek w kierunku dodatnio naładowanej
elektrody. Następnie białka ulegają zatrzymaniu na membranie znajdującej się za
żelem agarozowym [241]. Po przeniesieniu na membranę widoczny jest obraz
rozdzielonych w żelu białek. W celu zablokowania niespecyficznego wiązania
przeciwciała z membraną stosuje się środki blokujące. Z uwagi na cenę i dostępność
najczęściej stosowanym środkiem tego typu jest rozcieńczone, odtłuszczone mleko.
Po wysyceniu membrany stosuje się przeciwciało pierwszorzędowe, które wykazuje
powinowactwo do danego białka. Po tym etapie następuje odmycie nadmiaru
przeciwciała pierwszorzędowego, a w kolejnym etapie dodaje się przeciwciała
drugorzędowe, które mogą być znakowane radioaktywnie lub enzymem, np.
peroksydazą chrzanową, która wywołuje reakcję barwną [242]. Schemat działania
metody Western Blot przedstawiony został na rysunku 23.
-
+BUFORBUFOR
ŻEL
MEMBRANA
BIBUŁA
GĄBKA
BIBUŁA
GĄBKA
Rysunek 23. Schemat układu pomiarowego w metodzie Western Blot. Rysunek zmodyfikowany
w oparciu o [243].
Oznaczenia stężenia białek można dokonać również za pomocą metod
spektrofotometrycznych. Najbardziej popularną metodą spektroskopową jest pomiar
absorbancji w ultrafiolecie, przy długości fali 280 nm. Przy badanej długości fali
światło pochłaniane jest przez aminokwasy aromatyczne występujące w białkach.
W celu dokonania oznaczenia wykonuje się pomiary zarówno przy długości fali
280 nm, jak również przy ok. 260 nm. Stosunek absorbancji przy wspomnianych
długościach fali (A280 : A260) określany jest za pomocą litery F, której wartości są
stałe i stabelaryzowane. Stężenie białka uzyskuje się poprzez pomnożenie wartości F
i wartości absorbancji przy 280 nm [244]. W technice tej ilościowemu oznaczaniu
białek przeszkadzają obecne w próbce kwasy nukleinowe. Kolejną metodą
wykorzystywaną w oznaczaniu metaloprotein jest metoda Bradford’a, w której
następuje wiązanie białka z barwnikiem Coomassie Brillant Blue [245]. Po

Białka ważne składniki organizmu
64
związaniu się oznaczanego białka z barwnikiem zachodzi zmiana barwy roztworu,
a pomiar oparty jest na pomiarze absorbancji przy długości fali 595 nm. Natężenie
barwy roztworu jest wprost proporcjonalne do ilości białka w badanej próbce [245].
Większość opisanych powyżej metod niestety wykorzystuje często toksyczne
i rakotwórcze substancje, które są trudne do utylizacji. Charakteryzują się one
również dość niską czułością, są stosunkowo drogie oraz często wymagają
wcześniejszego przetworzenia próbki do pomiaru, co znacznie wydłuża czas analizy
[246]. Dobrą alternatywą dla powszechnie stosowanych technik w analizie klinicznej
białek są metody elektrochemiczne. Metody te można zastosować jedynie
w przypadku białek wykazujących elektroaktywność. Problematyka transportu
elektronów pomiędzy elektrodą a elektroaktywnym białkiem przedstawiona została
w rozdziale 5.

Elektroaktywność metaloprotein
65
5. Elektroaktywność metaloprotein
Transport elektronów w układach biologicznych jest jednym z najczęściej
badanych zjawisk w ciągu ostatnich 25 lat, obejmującym zagadnienia z pogranicza
biologii i elektrochemii [247]. Obecność jonów metali w strukturze metaloprotein
sprawia, że białka te są elektroaktywne, czyli mogą wymieniać elektrony z elektrodą
na drodze procesu elektroredukcji i/lub elektroutleniania. Poznanie procesu
transportu elektronów w tego typu układach oraz jego kontrola są bardzo ważne,
z uwagi na bardzo szeroki wachlarz zastosowań metaloprotein. Związki te biorą
udział w procesach metabolicznych, fotosyntezie, oddychaniu, jak również są one
ważnym elementem w bioogniwach paliwowych, urządzeniach elektronicznych,
biochipach, czy też w biosensorach [248]. Jednakże uzyskanie dobrze
wykształconych sygnałów prądowych pochodzących od metaloproteiny jest wciąż
dużym wyzwaniem dla elektrochemików. Białka nie ulegają łatwo reakcjom redoks
na powierzchni elektrody, ponieważ ich centra elektroaktywne są głęboko osadzone
w otoczce białkowej, co zdecydowanie utrudnia proces wymiany elektronów
pomiędzy elektrodą a białkiem [249]. Zatem ważną rolę w procesie elektroutleniania
lub elektroredukcji białka odgrywa jego orientacja względem powierzchni elektrody,
a ściślej mówiąc odległość centrum aktywnego od powierzchni materiału
przewodzącego. Niestety, bezpośrednia adsorpcja białka na powierzchni materiału
przewodzącego może doprowadzić do uszkodzenia jego wyższorzędowych struktur,
prowadząc jednocześnie do jego denaturacji, a tym samym do utraty aktywności
katalitycznej, czy też elektrochemicznej białka. Powstałe w tym procesie produkty
denaturacji białka utrudniają lub wręcz uniemożliwiają komunikację między
centrami elektroaktywnymi a powierzchnią elektrody [250].
5.1. Sposoby transportu elektronów pomiędzy
metaloproteiną a powierzchnią elektrody
Struktura metaloprotein, których znaczną część stanowią enzymy, generalnie
składa się z dwóch części: (i) miejsca biokatalitycznego, czyli apoenzymu służącego
do rozpoznania substratu oraz (ii) centrum elektrokatalitycznego, czyli grupy
prostetycznej, w której następuje przeniesienie elektronów. Z uwagi na budowę

Elektroaktywność metaloprotein
66
metaloprotein wyróżnia się dwa mechanizmy transportu elektronów pomiędzy takimi
związkami a powierzchnią elektrody: bezpośredni transport elektronów (DET; z ang.
direct electron transfer) oraz transport elektronów z udziałem mediatora (MET;
z ang. mediated electron transfer) [251]. Podczas bezpośredniej wymiany
elektronów z powierzchnią elektrody grupa prostetyczna lub centrum redoks
metaloproteiny ulega zmianom konformacyjnym. Natomiast w przypadku białek,
w których ten sam fragment struktury pełni zarówno rolę bio- i elektrokatalityczną
transport elektronów odbywa się zazwyczaj za pomocą rozpuszczonej w roztworze
substancji redoks, zwanej mediatorem [252]. W celu zwiększenia efektywności
katalitycznej metaloproteiny (głównie enzymu) względem danego substratu bardzo
często unieruchamia się kilka enzymów na wybranej powierzchni (np. niebieskie
oksydazy mulitimiedziowe: lakaza, ceruloplazmina, oksydaza askorbinianowa czy
oksydaza bilirubiny) katalizujących ten sam proces. Należy jednak pamiętać, że
bardzo często wiąże się to z koniecznością wprowadzenia do układu kilku różnych
mediatorów charakterystycznych dla danego enzymu, co zdecydowanie komplikuje
układ [252]. Schemat transportu elektronów w funkcji liczby enzymów
unieruchomionych na danej powierzchni ilustruje rysunek 24.
P
S
E1 E2 E3
E4
Ee- e- e- e- e-
M1 M2 M3
M4S
P
Rysunek 24. Schemat transportu elektronów w zależności od ilości unieruchomionych na
powierzchni elektrody enzymów. S oznacza substrat, P – produkt, En – enzym, Mn – mediator
charakterystyczny dla danego enzymu, e- – elektron [252].
Zgodnie z teorią Marcusa [253] kinetyka przeniesienia elektronu pomiędzy dwoma
substancjami redoks określona jest za pomocą siły napędowej (czyli różnicy
potencjałów), energii reorganizacji oraz odległości pomiędzy dwoma centrami
redoks. Oczywiste jest, że odległość pomiędzy grupą prostetyczną metaloproteiny
a powierzchnią elektrody jest raczej dość duża ze względu na występowanie otoczki
białkowej, co powoduje, że bezpośredni transport elektronów na drodze tunelowania
jest rzadko spotykany. Dodatkowo, trudno jest jednoznacznie wyznaczyć granicę
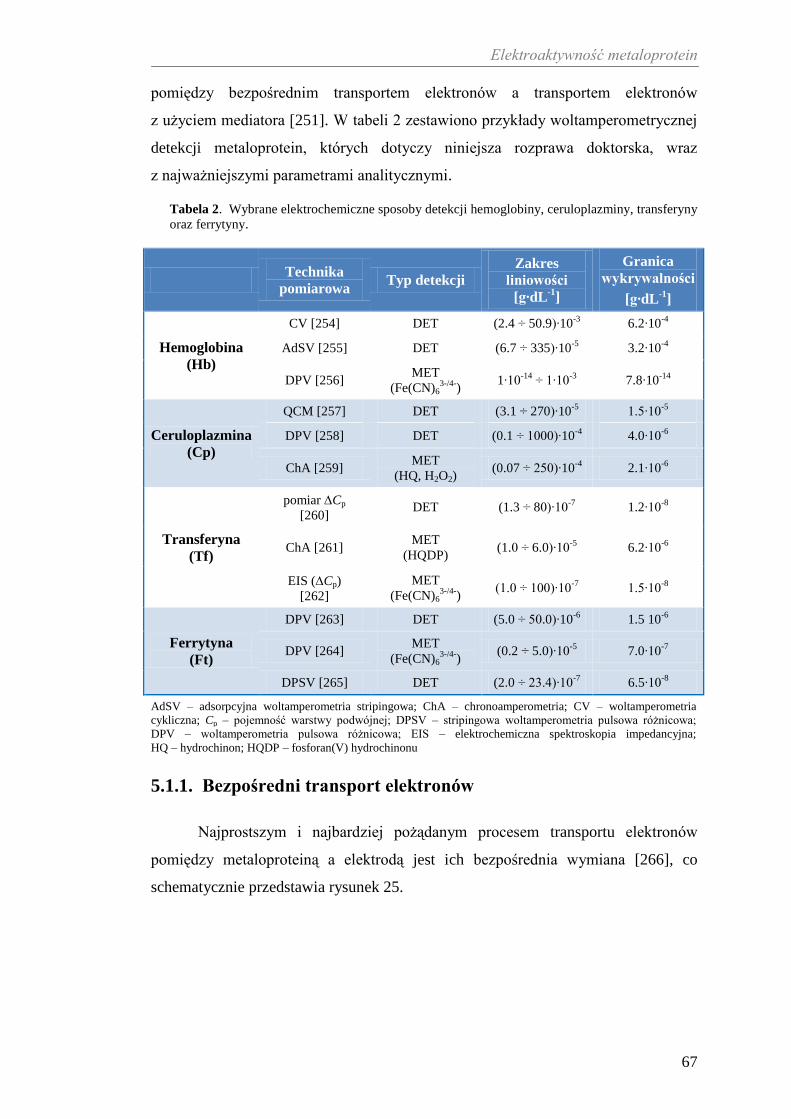
Elektroaktywność metaloprotein
67
pomiędzy bezpośrednim transportem elektronów a transportem elektronów
z użyciem mediatora [251]. W tabeli 2 zestawiono przykłady woltamperometrycznej
detekcji metaloprotein, których dotyczy niniejsza rozprawa doktorska, wraz
z najważniejszymi parametrami analitycznymi.
Tabela 2. Wybrane elektrochemiczne sposoby detekcji hemoglobiny, ceruloplazminy, transferyny
oraz ferrytyny.
Technika
pomiarowa Typ detekcji
Zakres
liniowości
[g·dL-1
]
Granica
wykrywalności
[g·dL-1
]
Hemoglobina
(Hb)
CV [254] DET (2.4 ÷ 50.9)·10-3
6.2·10-4
AdSV [255] DET (6.7 ÷ 335)·10-5
3.2·10-4
DPV [256] MET
(Fe(CN)63-/4-
) 1·10
-14 ÷ 1·10
-3 7.8·10
-14
Ceruloplazmina
(Cp)
QCM [257] DET (3.1 ÷ 270)·10-5
1.5·10-5
DPV [258] DET (0.1 ÷ 1000)·10-4
4.0·10-6
ChA [259] MET
(HQ, H2O2) (0.07 ÷ 250)·10
-4 2.1·10
-6
Transferyna
(Tf)
pomiar Cp
[260] DET (1.3 ÷ 80)·10
-7 1.2·10
-8
ChA [261] MET
(HQDP) (1.0 ÷ 6.0)·10
-5 6.2·10
-6
EIS (Cp)
[262]
MET
(Fe(CN)63-/4-
) (1.0 ÷ 100)·10
-7 1.5·10
-8
Ferrytyna
(Ft)
DPV [263] DET (5.0 ÷ 50.0)·10-6
1.5 10-6
DPV [264] MET
(Fe(CN)63-/4-
) (0.2 ÷ 5.0)·10
-5 7.0·10
-7
DPSV [265] DET (2.0 ÷ 23.4)·10-7
6.5·10-8
AdSV – adsorpcyjna woltamperometria stripingowa; ChA – chronoamperometria; CV – woltamperometria
cykliczna; Cp – pojemność warstwy podwójnej; DPSV – stripingowa woltamperometria pulsowa różnicowa;
DPV – woltamperometria pulsowa różnicowa; EIS – elektrochemiczna spektroskopia impedancyjna;
HQ – hydrochinon; HQDP – fosforan(V) hydrochinonu
5.1.1. Bezpośredni transport elektronów
Najprostszym i najbardziej pożądanym procesem transportu elektronów
pomiędzy metaloproteiną a elektrodą jest ich bezpośrednia wymiana [266], co
schematycznie przedstawia rysunek 25.

Elektroaktywność metaloprotein
68
Rysunek 25. Bezpośredni transport elektronów w oparciu o [267].
W celu uzyskania bezpośredniego transportu elektronów konieczne jest właściwe
unieruchomienie białka na powierzchni elektrody tak, aby odległość centrum
elektroaktywnego tego związku od powierzchni elektrody była możliwie jak
najmniejsza [268]. Zatem cząsteczki muszą posiadać odpowiednią orientację,
gwarantującą najmniejszą odległość pomiędzy grupą prostetyczną metaloproteiny
a materiałem przewodzącym [248]. Jak już wcześniej wspomniano, w przypadku
unieruchomienia metaloproteiny na niezmodyfikowanej elektrodzie, zwłaszcza
metalicznej, bardzo trudno jest osiągnąć bezpośredni transport elektronów, z uwagi
na niewłaściwą orientację białka oraz możliwy proces jego denaturacji [269], co
w konsekwencji blokuje wymianę elektronów pomiędzy centrum redoks białka
a elektrodą [270]. W celu uniknięcia denaturacji i/lub deaktywacji metaloproteiny
powierzchnię elektrody pokrywa się różnego typu modyfikatorami [271], które
wpływają zarówno na orientację unieruchamianej za ich pośrednictwem
metaloproteiny, jak również zabezpieczają elektrodę przed utratą jej właściwości
przewodzących [272]. Najczęściej w roli tego typu modyfikatorów powierzchni
elektrody, umożliwiających kontrolę orientacji metaloproteiny, wykorzystuje się
samoorganizujące się monowarstwy tiolowe zawierające odpowiednie grupy
funkcyjne (np. COOH; NH2) [273], polimery przewodzące [274], czy też
nanomateriały [275], takie jak: nanocząstki złota [276] oraz nanorurki węglowe
[277]. Schematycznie wpływ procesu unieruchomienia metaloproteiny na
efektywność transportu elektronów ilustruje rysunek 26.
SUBSTRAT PRODUKT
e-
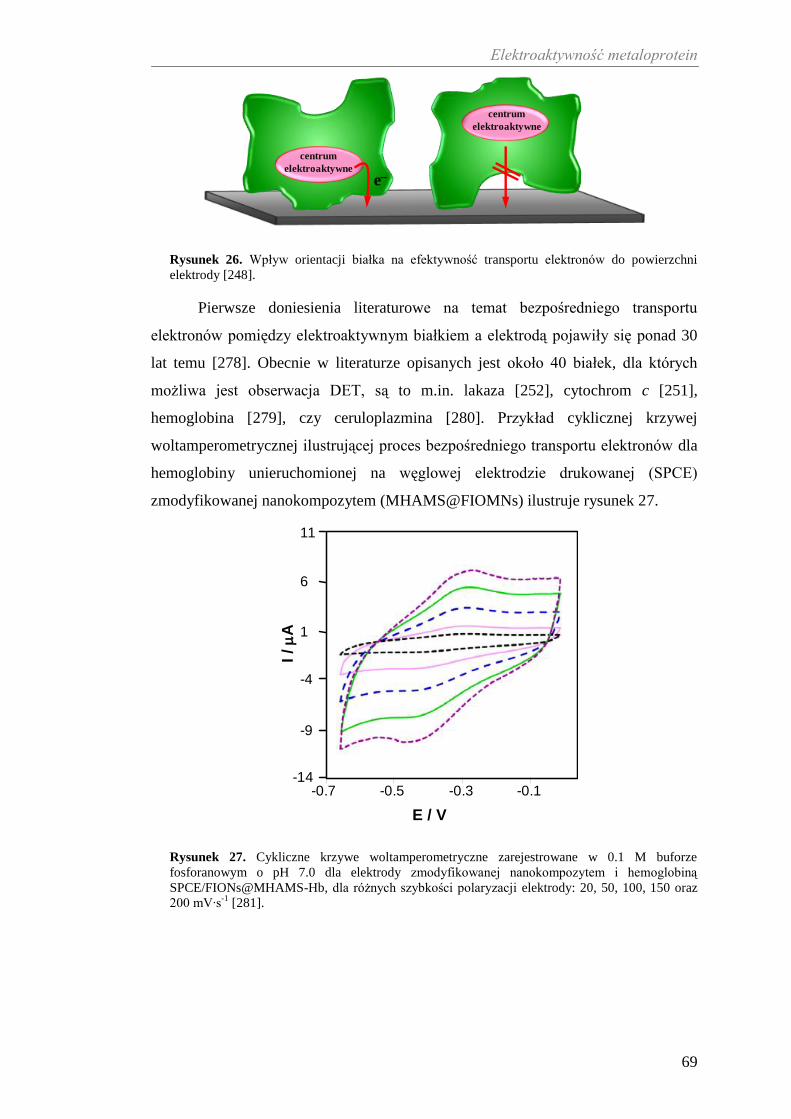
Elektroaktywność metaloprotein
69
centrum
elektroaktywne
e
centrum
elektroaktywne
Rysunek 26. Wpływ orientacji białka na efektywność transportu elektronów do powierzchni
elektrody [248].
Pierwsze doniesienia literaturowe na temat bezpośredniego transportu
elektronów pomiędzy elektroaktywnym białkiem a elektrodą pojawiły się ponad 30
lat temu [278]. Obecnie w literaturze opisanych jest około 40 białek, dla których
możliwa jest obserwacja DET, są to m.in. lakaza [252], cytochrom c [251],
hemoglobina [279], czy ceruloplazmina [280]. Przykład cyklicznej krzywej
woltamperometrycznej ilustrującej proces bezpośredniego transportu elektronów dla
hemoglobiny unieruchomionej na węglowej elektrodzie drukowanej (SPCE)
zmodyfikowanej nanokompozytem (MHAMS@FIOMNs) ilustruje rysunek 27.
11
6
1
-4
-9
-14-0.7 -0.5 -0.3 -0.1
I / m
A
E / V
Rysunek 27. Cykliczne krzywe woltamperometryczne zarejestrowane w 0.1 M buforze
fosforanowym o pH 7.0 dla elektrody zmodyfikowanej nanokompozytem i hemoglobiną
SPCE/FIONs@MHAMS-Hb, dla różnych szybkości polaryzacji elektrody: 20, 50, 100, 150 oraz
200 mV·s-1
[281].
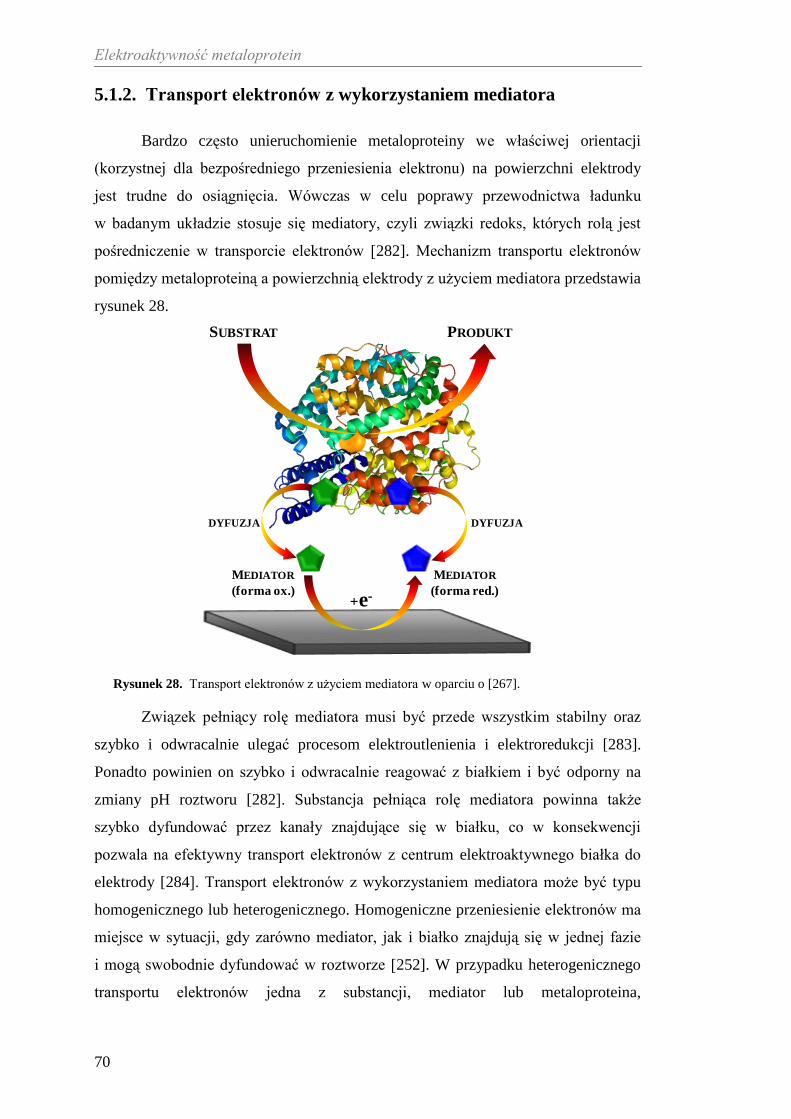
Elektroaktywność metaloprotein
70
SUBSTRAT PRODUKT
DYFUZJA DYFUZJA
+e-
MEDIATOR
(forma red.)
MEDIATOR
(forma ox.)
5.1.2. Transport elektronów z wykorzystaniem mediatora
Bardzo często unieruchomienie metaloproteiny we właściwej orientacji
(korzystnej dla bezpośredniego przeniesienia elektronu) na powierzchni elektrody
jest trudne do osiągnięcia. Wówczas w celu poprawy przewodnictwa ładunku
w badanym układzie stosuje się mediatory, czyli związki redoks, których rolą jest
pośredniczenie w transporcie elektronów [282]. Mechanizm transportu elektronów
pomiędzy metaloproteiną a powierzchnią elektrody z użyciem mediatora przedstawia
rysunek 28.
Rysunek 28. Transport elektronów z użyciem mediatora w oparciu o [267].
Związek pełniący rolę mediatora musi być przede wszystkim stabilny oraz
szybko i odwracalnie ulegać procesom elektroutlenienia i elektroredukcji [283].
Ponadto powinien on szybko i odwracalnie reagować z białkiem i być odporny na
zmiany pH roztworu [282]. Substancja pełniąca rolę mediatora powinna także
szybko dyfundować przez kanały znajdujące się w białku, co w konsekwencji
pozwala na efektywny transport elektronów z centrum elektroaktywnego białka do
elektrody [284]. Transport elektronów z wykorzystaniem mediatora może być typu
homogenicznego lub heterogenicznego. Homogeniczne przeniesienie elektronów ma
miejsce w sytuacji, gdy zarówno mediator, jak i białko znajdują się w jednej fazie
i mogą swobodnie dyfundować w roztworze [252]. W przypadku heterogenicznego
transportu elektronów jedna z substancji, mediator lub metaloproteina,

Elektroaktywność metaloprotein
71
unieruchomiona jest na powierzchni elektrody, natomiast druga znajduje się
w roztworze [282].
Niezbędnym warunkiem szybkiego transportu elektronów w MET jest
utrzymanie komunikacji pomiędzy mediatorem a białkiem. Jeśli mediator obecny
jest na powierzchni elektrody, nie zaś w roztworze, wówczas trwałość takiego
połączenia jest niezwykle istotna. Aby uzyskać trwałe połączenie mediatora
z powierzchnią materiału przewodzącego zazwyczaj wprowadza się go przy udziale
takich substancji, jak: pasta węglowa, związki koloidalne, materiały kompozytowe,
hydrożele, czy też polimery przewodzące [252]. Należy jednak podkreślić, że
w układach biologicznych pożądanym procesem transportu elektronów jest
bezpośrednia ich wymiana pomiędzy centrum elektroaktywnym metaloproteiny
a elektrodą. Brak mediatora w układzie zdecydowanie zwiększa selektywność
procesu wymiany elektronów, w wyniku zmniejszenia jego podatności na zakłócenia
oraz uproszczenia zapisu równania chemicznego, w porównaniu do sytuacji
z użyciem mediatora [278]. Z drugiej strony, obecność mediatora może potencjalnie
zwiększyć maksymalną szybkość transportu elektronów. Dodatkowo, dzięki
zastosowaniu mediatorów metaloproteina nie musi być w bezpośrednim kontakcie
z elektrodą, przez co zmniejsza się ryzyko procesu denaturacji białka.
W przypadku elektrochemicznej detekcji białek stosuje się wiele różnych
mediatorów redoks, różniących się liczbą przenoszonych elektronów oraz
wartościami potencjałów redoks. Przykładowo do oznaczania transferyny zazwyczaj
stosuje się takie mediatory, jak wiologen metylu [285], czy też barwniki cyjankowe
[286], natomiast dla ceruloplazminy mediatorami mogą być: hydroksyacetanilid,
hydroksybenzotriazol oraz kwas wiolurowy [287], Z kolei dla ferrytyny są to:
fenosafranina, indygokarmin, jak również błękit metylenowy [288]. Jeśli chodzi
o hemoglobinę często spotykanymi mediatorami są błękit metylenowy, metosiarczan
fenazyny oraz tionina [289], dzięki którym obserwuje się odwracalne sygnały
redoks, które zmieniają intensywność wraz z dodatkiem do roztworu oznaczanego
białka. Przykładowa krzywa zarejestrowana dla błękitu metylenowego, w obecności
hemoglobiny rozpuszczonej w roztworze przedstawiona została na rysunku 29.
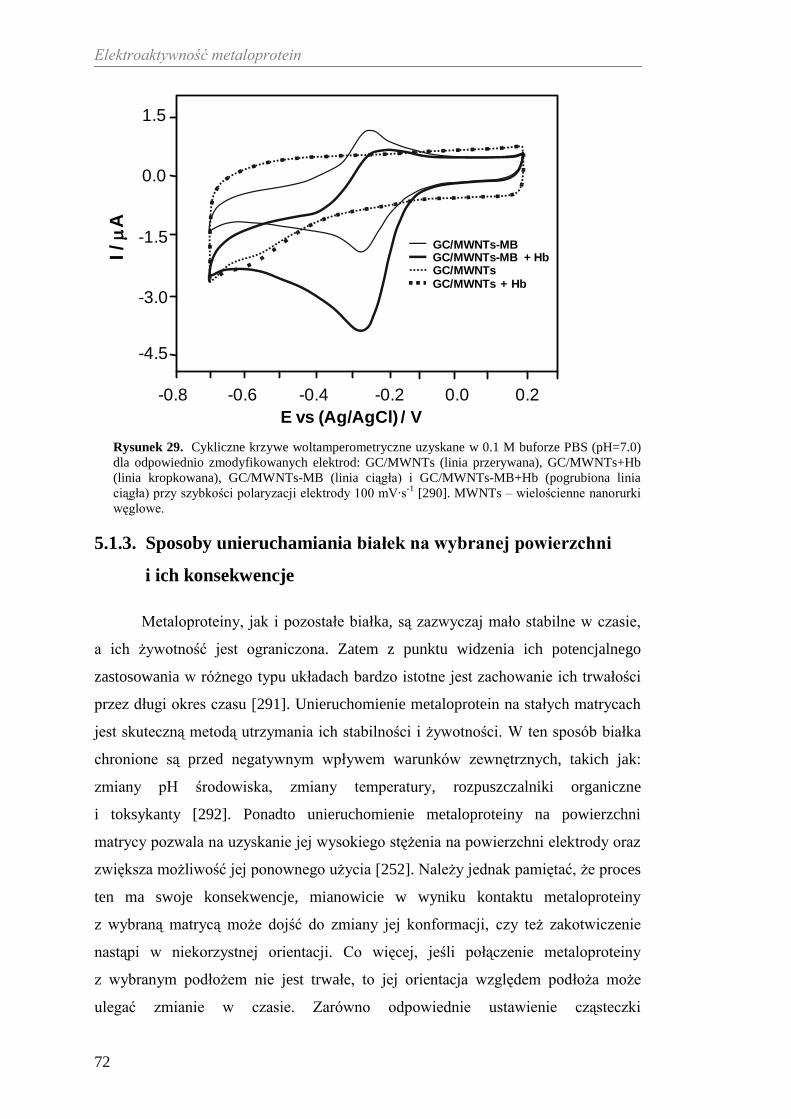
Elektroaktywność metaloprotein
72
Rysunek 29. Cykliczne krzywe woltamperometryczne uzyskane w 0.1 M buforze PBS (pH=7.0)
dla odpowiednio zmodyfikowanych elektrod: GC/MWNTs (linia przerywana), GC/MWNTs+Hb
(linia kropkowana), GC/MWNTs-MB (linia ciągła) i GC/MWNTs-MB+Hb (pogrubiona linia
ciągła) przy szybkości polaryzacji elektrody 100 mV·s-1
[290]. MWNTs – wielościenne nanorurki
węglowe.
5.1.3. Sposoby unieruchamiania białek na wybranej powierzchni
i ich konsekwencje
Metaloproteiny, jak i pozostałe białka, są zazwyczaj mało stabilne w czasie,
a ich żywotność jest ograniczona. Zatem z punktu widzenia ich potencjalnego
zastosowania w różnego typu układach bardzo istotne jest zachowanie ich trwałości
przez długi okres czasu [291]. Unieruchomienie metaloprotein na stałych matrycach
jest skuteczną metodą utrzymania ich stabilności i żywotności. W ten sposób białka
chronione są przed negatywnym wpływem warunków zewnętrznych, takich jak:
zmiany pH środowiska, zmiany temperatury, rozpuszczalniki organiczne
i toksykanty [292]. Ponadto unieruchomienie metaloproteiny na powierzchni
matrycy pozwala na uzyskanie jej wysokiego stężenia na powierzchni elektrody oraz
zwiększa możliwość jej ponownego użycia [252]. Należy jednak pamiętać, że proces
ten ma swoje konsekwencje, mianowicie w wyniku kontaktu metaloproteiny
z wybraną matrycą może dojść do zmiany jej konformacji, czy też zakotwiczenie
nastąpi w niekorzystnej orientacji. Co więcej, jeśli połączenie metaloproteiny
z wybranym podłożem nie jest trwałe, to jej orientacja względem podłoża może
ulegać zmianie w czasie. Zarówno odpowiednie ustawienie cząsteczki
1.5
0.0
-1.5
-3.0
-4.5
E vs (Ag/AgCl) / V
I / m
A
GC/MWNTs-MBGC/MWNTs-MB + Hb
GC/MWNTs + HbGC/MWNTs
-0.8 -0.6 -0.4 -0.2 0.0 0.2

Elektroaktywność metaloprotein
73
metaloproteiny względem wybranej powierzchni, jak i jej konformacja są gwarantem
zachowania jej naturalnych funkcji, a także w przypadku białek elektroaktywnych
decydują o mechanizmie transportu elektronów pomiędzy taką cząsteczką
a powierzchnią elektrody.
Ilość unieruchamianego białka, szybkość przebiegu tego procesu oraz
stabilność formowanej warstwy zależą od: fizycznej charakterystyki białek
(rozmiaru, elastyczności, ładunku), morfologii matrycy, na której następuje
osadzanie białka i środowiska, w którym zachodzi ten proces [293,294]. Proces
wiązania białka z wybraną matrycą następuje zasadniczo w trzech etapach, które
występują w różnej skali czasowej [295]. Pierwszym krokiem jest dyfuzja cząsteczek
metaloproteiny z głębi roztworu do powierzchni matrycy. Następnie białko ulega
związaniu z matrycą, a w końcowym etapie może nastąpić zmiana orientacji oraz
konformacji białka w czasie [296]. Ilość zmian konformacyjnych unieruchomionego
białka zależna jest od stabilności jego struktury. Białka posiadające wysoką
stabilność znacznie rzadziej ulegają zmianom konformacyjnym w porównaniu do
mniej stabilnych białek, u których zmiany konformacyjne mogą być znaczne i mogą
nawet prowadzić do utraty ich aktywności biologicznej. Istnieją jednak przypadki,
gdy zmiana konformacyjna wzmacnia funkcjonalność metaloprotein, jednak jest to
niezwykle rzadkie zjawisko. Na ilość unieruchamianego białka ma wpływ charakter
materiału, na którym następuje ten proces. W przypadku hydrofobowych matryc
występuje silniejsze związanie cząsteczek białka niż dla matryc o właściwościach
hydrofilowych, co wpływa na większą ilość zaadsorbowanych cząsteczek
metaloprotein na powierzchni elektrody [297,298]. Możliwe zmiany orientacji oraz
konformacji białka po jego unieruchomieniu na powierzchni elektrody
schematycznie ilustruje rysunek 30.
ZMIANA KONFORMACJI
czas
ZMIANA ORIENTACJI
Rysunek 30. Zmiany orientacji oraz konformacji białka w czasie [296].

Elektroaktywność metaloprotein
74
Informacja na temat zmian strukturalnych metaloprotein w wyniku kontaktu
z wybranym podłożem jest niezwykle ważna z punktu widzenia ich potencjalnych
biotechnologicznych zastosowań. Zmiany konformacyjne białka zachodzące podczas
procesu jego unieruchamiania na powierzchni wybranej matrycy można badać za
pomocą wielu metod, do których zalicza się: spektroskopię Ramana, spektroskopię
w podczerwieni (IR), spektroskopię w podczerwieni z transformacją Fouriera
(FTIR), spektroskopię UV-Vis, dichroizm kołowy (CD), fluorescencję oraz
powierzchniowy rezonans plazmonowy (SPR) [299].
W zależności od charakteru matrycy, stabilności biocząsteczki i jej
właściwości stosuje się różne metody unieruchamiania białek, do których zalicza się:
adsorpcję fizyczną, wiązanie kowalencyjne, pułapkowanie, sieciowanie
i oddziaływanie na zasadzie powinowactwa [252,300]. Wybierając odpowiedni
sposób modyfikacji matrycy białkiem należy wziąć pod uwagę takie czynniki, jak:
utrzymanie aktywności białka, odporność na zmiany czynników fizykochemicznych,
czas życia białka, jego preferowaną orientację i konformację [292]. Modyfikacja
chemiczna powierzchni elektrody zdecydowanie zwiększa stabilność białka oraz
wprowadza możliwość kontroli jego orientacji i gęstości upakowania [301]. Sposoby
unieruchamiania białek na wybranej powierzchni schematycznie ilustruje rysunek
31.
ADSORPCJAFIZYCZNA WIĄZANIEKOWALENCYJNE POWINOWACTWO
PUŁAPKOWANIE SIECIOWANIE
Rysunek 31. Metody unieruchamiania białek na powierzchni elektrody [252].
W przypadku fizycznej adsorpcji wiązanie białka z daną powierzchnią jest
stosunkowo słabe i z reguły nie prowadzi do zmian konformacyjnych cząsteczki
metaloproteiny [302]. Dodatkowo, niemożliwe jest uzyskanie identycznej orientacji
wszystkich unieruchomionych cząsteczek, dlatego też bardzo często powierzchnię

Elektroaktywność metaloprotein
75
matrycy modyfikuje się różnego typu związkami ułatwiającymi kontrolę orientacji
unieruchamianego białka. Z uwagi na rozwiniętą powierzchnię właściwą oraz
wysoką chemiczną i termiczną stabilność, dużą popularnością cieszą się materiały na
bazie węgla, którymi może być sadza, grafit, włókna węglowe oraz różnego typu
nanostruktury węglowe [252]. Modyfikacja elektrody nanostrukturami, takimi jak
nanocząstki, nanowłókna, nanodziury, nanorurki czy też nanokompozyty, na ogół
prowadzi do wyższej gęstości prądu spowodowanej większym upakowaniem białka
na elektrodzie. Tego typu nanostruktury mogą przedłużać także żywotność białka,
zwiększać jego stabilność oraz aktywność. Umieszczenie nanostruktur w innych
materiałach (np.: polimery, żele) może prowadzić do poprawy ich właściwości
[303-305]. Z uwagi na niepowtarzalne właściwości chemiczne i fizyczne szeroko
badane są również nanocząstki metali. Struktury te zazwyczaj oparte są na złocie,
srebrze czy platynie [306,307] oraz na tlenkach metali, np. tlenku żelaza [308].
Popularność tego typu materiałów związana jest z możliwością łatwej kontroli ich
rozmiarów oraz powstawaniem dobrze zdefiniowanych struktur [309]. Szczególną
grupę nanstruktur stanowią nanocząstki magnetyczne, które charakteryzują się
wysoką zdolnością wiązania białek, w szczególności paramagnetycznych
metaloprotein. Dodatkowo, można je łatwo wyodrębnić z mieszaniny reakcyjnej za
pomocą magnesu [308].
Ostatnio dużo uwagi poświęca się cieczom jonowym, które posiadają wysoką
lepkość, hydrofobowość, strukturę jonową, charakteryzują się również
przewodnictwem jonowym i biozgodnością [310]. Ciecze jonowe składają się
z anionów lub kationów i możliwe są różne kombinacje kationów lub anionów
organicznych z nieorganicznymi [311]. Jedną z najważniejszych zalet tego typu
modyfikatorów, w przypadku zastosowania w elektrochemicznych układach, jest
posiadanie przez te struktury szerokiego okna potencjałowego [310].
Jako modyfikatory elektrod wykorzystuje się również materiały polimerowe,
np. nafion, chitozan, polipirol, polianilinę, alkohol winylowy i wiele innych [252].
Użycie biokompatybilnych polimerów gwarantuje transport elektronów pomiędzy
białkiem a elektrodą oraz zachowanie katalitycznej funkcji badanych białek [312].
Elektrody modyfikuje się także za pomocą kompozytów, dzięki czemu możliwe jest
poprawienie własności fizycznych lub chemicznych poszczególnych składników.
Zazwyczaj stosuje się układy zol-żel z nanocząstkami lub polimery z materiałami
węglowymi. Mieszanina dwóch lub więcej związków może wpływać na aktywność

Elektroaktywność metaloprotein
76
enzymatyczną białek, stabilność oraz szybkość transportu elektronów do elektrody
[313]. Struktury na bazie zol-żel są porowatymi matrycami polimerowymi
o zwiększonej powierzchni. Materiały te są biokompatybilne, dzięki czemu tworzą
stabilne środowisko dla prawidłowego funkcjonowania białek [314]. Stosuje się je do
pułapkowania białek wewnątrz ich struktury. Uważa się, że w celu efektywnej
adsorpcji metaloprotein wielkość porów powinna być podobna lub większa od
średnicy białek. Związek między wielkością porów i średnicą cząsteczki ma istotne
znaczenie, gdyż duży rozmiar porów zazwyczaj prowadzi do małej stabilności
białek. W stabilności układu ważną rolę odgrywa również ładunek wewnątrz porów.
W sytuacji, gdy ładunek ten jest przeciwny do ładunku białka, wówczas układ jest
stabilny, natomiast w przypadku ładunków jednoimiennych następuje odpychanie
między powierzchnią porów a białkiem. Kontrola ładunku możliwa jest poprzez
monitorowanie pH środowiska lub też poprzez modyfikację powierzchni porów
różnymi grupami funkcyjnymi, np. aminowymi lub karboksylowymi [315].
Kolejną metodą unieruchamiania metaloprotein jest kowalencyjne
przyłączenie białka do nośnika, np. polimeru syntetycznego, jak poliglikolu
etylenowego lub polimerów naturalnych – celulozy lub sacharozy [316]. Technika ta
pozwala na otrzymanie stabilnych układów, odpornych na podwyższoną temperaturę
[317]. Zazwyczaj kowalencyjne związanie metaloproteiny następuje poprzez grupę
karboksylową nośnika oraz grupę aminową białka, z użyciem karbodiimidu [316].
Zaletą zastosowania tego odczynnika jest jego dobra rozpuszczalność w wodzie,
dzięki czemu może być on bezpośrednio zastosowany w mieszaninie wodnej, bez
wstępnego rozpuszczania w związkach organicznych. W wyniku reakcji
karbodiimidu z grupami karboksylowymi wytwarza się reaktywny produkt pośredni,
którym jest acyloaminoester, który następnie reaguje z grupą aminową białka,
tworząc wiązanie amidowe [318]. Trwałe związanie białka z nośnikiem powoduje
usztywnienie konformacji białka, a tym samym zwiększa odporność na denaturację,
jednak sposób ten może prowadzić do nieodpowiedniej orientacji białka względem
elektrody i spowolnić proces transportu elektronów [316].
Atrakcyjnym podejściem wydaje się być również tworzenie
samoorganizujących się monowarstw (SAM) [319]. Warstwy takie są łatwe do
przygotowania, gdyż grupy -SH alkanotioli łatwo oddziałują z metaliczną
powierzchnią elektrody z wytworzeniem wiązania kowalencyjnego, a wolna grupa
funkcyjna znajdująca się na drugim końcu łańcucha węglowego jest zdolna do

Elektroaktywność metaloprotein
77
przyłączenia cząsteczek białka [320]. Warstwy takie charakteryzują się wysoką
organizacją i jednorodnością. Dodatkowo, przy użyciu samoorganizujących się
monowarstw możliwe jest kontrolowanie długości łańcucha hydrofobowego, co
pozwala na pożądane zmiany właściwości warstwy [321].
Kolejną metodą unieruchamiania białek jest ich pułapkowanie w sieci
polimerowej. Technika ta polega na zmieszaniu monomeru lub polimeru
z roztworem metaloproteiny w obecności czynnika sieciującego. Warunki
zachodzenia polimeryzacji mogą niestety prowadzić do denaturacji białka [316].
Alternatywą tej metody jest stosowanie polimerów pochodzenia naturalnego, np.
agarozy lub żelatyny [322], lub też hydrożeli [323]. Sieciowanie białek polega na
utworzeniu wielu wiązań kowalencyjnych w trzech wymiarach pomiędzy
cząsteczkami białka z użyciem substancji sieciującej, którą może być aldehyd
glutarowy. Zastosowanie tej substancji związane jest z jej dużą stabilnością, niską
ceną oraz łagodnymi warunkami procesu sieciowania, jak również wysoką
wydajnością [322]. Metoda ta ma jednak pewne ograniczenia, gdyż często może
prowadzić do utraty aktywności białka oraz dodatkowo problemem jest stosunkowo
mała powtarzalność wyników [316].
Ostatnią z często wykorzystywanych metod wiązania białek jest
wykorzystanie powinowactwa danej substancji do unieruchamianego białka
i związanie go w sposób trwały z substancją [324]. Proces ten w określonych
warunkach może być odwracalny. Za pomocą metody powinowactwa otrzymuje się
korzystną orientację białka niezbędną do procesu przeniesienia elektronu.
Substancjami kowalencyjnie wiążącymi metaloproteiny są zazwyczaj przeciwciała
mono- lub polikolonalne [325].


Nanomateriały typu „rdzeń – powłoka”
79
6. Nanomateriały typu „rdzeń – powłoka”
Nanotechnologia jest obecnie jednym z najszybciej rozwijających się
obszarów nauki, który łączy w sobie różne dziedziny, takie jak: elektronika, chemia,
fizyka, biologia czy inżynieria materiałowa [326]. Unikalne właściwości
nanomateriałów związane są głównie z ich niezwykle małymi rozmiarami,
mieszczącymi się zazwyczaj w przedziale od 1 do 100 nm [327,328]. Dzięki
niezwykle małemu rozmiarowi, nanostruktury posiadają zupełnie odmienne
właściwości, w porównaniu do ich odpowiedników w skali „makro”. Zmiany
właściwości fizykochemicznych nanomateriałów są konsekwencją dwóch zjawisk:
(i) zwiększonego stosunku liczby atomów/jonów powierzchniowych lub
przypowierzchniowych względem liczby atomów zlokalizowanych wewnątrz
nanomateriału oraz (ii) kwantowego ograniczenia elektronów w cząstkach rozmiaru
„nano”. Charakterystyczną cechą tego typu materiałów jest przede wszystkim duża
powierzchnia właściwa cząstek, wysoka aktywność oraz tendencja do procesu
aglomeracji [329]. Z uwagi na swój niezwykle mały rozmiar, nanomateriały cieszą
się dużą popularnością w nowatorskich rozwiązaniach technologicznych
obejmujących takie dziedziny, jak: biotechnologia, farmacja, elektronika czy linie
produkcyjne w przemyśle. Nanomateriały odgrywają również znaczącą rolę
w zwalczaniu zanieczyszczeń i zagrożeń środowiska, umożliwiając zaawansowane
oczyszczanie wody i innych mediów z jonów metali ciężkich: Hg2+
, Pb2+
, Cd2+
[330]. Najbardziej popularne nanomateriały to: fulereny [331], nanorurki węglowe
[332], nanodruty [333], nanocząstki złota [334], srebra [335], jak również
nanocząstki typu rdzeń – powłoka (z ang. core – shell), do których zalicza się m.in.
kropki kwantowe [336], czy coraz bardziej popularne nanocząstki magnetyczne
(M Nps) [337].
Najlepiej poznanym i najszerzej opisanym przedstawicielem nanocząstek
magnetycznych są superparamagnetyczne nanocząstki tlenku żelaza – Fe3O4
(SPIONs). Liczne doniesienia literaturowe opisują skuteczne metody syntezy
stabilnych, biokompatybilnych, monodyspersyjnych oraz kontrolowanych kształtem
nanostruktur tlenku żelaza [338]. Najpopularniejsze metody obejmują
współstrącanie, rozkład termiczny, syntezy hydrotermalne, mikroemulsje, czy
syntezy sonochemiczne. Ponadto omawiane nanocząstki można również wytwarzać
innymi sposobami, takimi jak: synteza elektrochemiczna [339], technika pirolizy

Nanomateriały typu „rdzeń – powłoka”
80
laserowej [340], synteza bakteryjna (zwłaszcza bakterie żelaza redukującego) [341].
Niestety nanocząstki oparte na tlenku żelaza bardzo trudno jest uchronić przed
procesem nieodwracalnego utleniania w środowisku komórek [342], co z kolei
znacznie wpływa na wzrost toksyczności tego typu układów. Ogromnego znaczenia
nabiera tutaj tzw. kapsułkowanie nanocząstek magnetycznych, które prowadzi do
uzyskania różnych pod wzglądem stabilności, heterogennych układów typu „rdzeń –
powłoka”.
6.1. Nanocząstki magnetyczne typu „rdzeń – powłoka”
Nanocząstki magnetyczne typu „rdzeń – powłoka” składają się z co najmniej
dwóch faz, różniących się między sobą strukturą i składem chemicznym. Schemat
budowy nanocząstki typu „rdzeń – powłoka” przedstawia rysunek 32.
Rysunek 32. Schemat budowy nanocząstki typu „rdzeń – powłoka”.
Rdzeń takiej nanocząstki stanowi zazwyczaj substancja pochodzenia
nieorganicznego pokryta jedną lub kilkoma powłokami o odmiennym składzie
chemicznym. W zależności od potrzeb, właściwości rdzenia można kontrolować
poprzez odpowiedni dobór substancji go budujących. Obecność magnetytu Fe3O4
nadaje takiej nanocząstce właściwości magnetyczne [343]. Z kolei zastosowanie
w roli rdzenia półprzewodnikowych kropek kwantowych, np.: ZnO, CdSe, CdS
nadaje tego typu materiałom właściwości luminescencyjne [344]. Powłoka może być
zarówno substancją prostą lub związkiem nieorganicznym, np.: C, TiO2 czy też SiO2,
jak i polimerem organicznym [345]. Taka powłoka powinna być obojętna lub
aktywna tylko w stosunku do określonych ugrupowań chemicznych lub może zostać
odpowiednio zmodyfikowana, w zależności od planowanych zastosowań.
Dodatkowo, powinna być mało czuła na zmiany pH i działanie czynników
agresywnych oraz nie powinna łatwo ulegać negatywnym wpływom środowiska
zewnętrznego. W grupie materiałów powłokowych pierwsze miejsce zdecydowanie
zajmują materiały węglowe [346].
OTOCZKA
RDZEŃ

Nanomateriały typu „rdzeń – powłoka”
81
6.1.1. Nanokapsuły węglowe zawierające żelazo
Nanokapsuły węglowe zawierające żelazo (Fe@C Nps), zwane również
nanocząstkami żelaza enkapsulowanymi węglem (CEINs), zbudowane są
z nieorganicznego rdzenia (zazwyczaj o sferycznym kształcie), którym jest żelazo,
kobalt, nikiel, czy też ich stopy oraz otaczającej rdzeń powłoki węglowej [346].
Struktury te zostały odkryte w 1993 roku podczas badań nad fulerenami, w wyniku
wyładowania w łuku węglowym [347]. Metaliczny rdzeń ma zazwyczaj średnicę od
10 do 100 nm, a powłoka węglowa składa się z 5 warstw grafenowych, o grubości od
5 do 15 nm [348,349]. Węgiel jest jednym z najlepszych materiałów do tworzenia
takich powłok, ze względu na to, że jest materiałem lekkim, ma niską wrażliwość
magnetyczną oraz posiada dużą odporność termiczną oraz chemiczną w stosunku do
wielu związków [350]. Dodatkowo, powłoka węglowa zabezpiecza nanocząstki
przed ich utlenianiem oraz procesem korozji, dzięki czemu zachowują one swoje
właściwości. Zastosowanie powłoki węglowej zabezpiecza nanocząstki również
przed procesem agregacji oraz sprawia, że są one biokompatybilne i stabilne w wielu
nieorganicznych oraz organicznych roztworach [348].
Synteza nanokapsuł węglowych zawierających nanocząstki metaliczne może
być prowadzona przy użyciu takich metod, jak: plazma łuku węglowego,
przepływowa plazma termiczna, synteza spaleniowa, a nawet kontrolowana reakcja
egzotermiczna oparta na termolizie prostych związków chemicznych. Zasada
działania tych metod opiera się na etapie wytworzenia gazu zawierającego pary
węgla i metalu, który ma ulec zakapsułkowaniu. Jednakże duża ilość parametrów
procesowych (ciśnienie, moc, stechiometria reagentów, źródło węgla, geometria
reaktora, etc.) utrudnia znalezienie warunków idealnych do wzrostu nanokapsuł
węglowych. Metoda plazmy łuku węglowego zapewnia stabilne środowisko
reakcyjne i polega na wytworzeniu łuku węglowego pomiędzy katodą i grafitową
anodą z domieszką proszku zawierającego żelazo. Wysoka temperatura prowadzi do
odparowania węgla i żelaza, a ich pary szybko kondensują, tworząc nanokapsuły
węglowe zawierające żelazo. Powstałe w ten sposób materiały umieszcza się we
wrzącym 4 M HCl na 24 h, w celu usunięcia z ich powierzchni niepokrytego węglem
żelaza [351]. Kolejny etap polega na kilkukrotnym przemywaniu uzyskanego
produktu wodą i etanolem. Zabiegi te mają na celu usunięcie wszelkich innych
pozostałości z powierzchni nanokapsuł. Oczyszczone w ten sposób nanocząstki

Nanomateriały typu „rdzeń – powłoka”
82
suszy się w atmosferze powietrza. Niestety jest to metoda periodyczna, co mocno
ogranicza możliwości powiększania skali procesu; w trakcie pojedynczego
eksperymentu można otrzymać zaledwie kilka gramów produktu. Drugą z często
stosowanych metod, pozwalających wyeliminować konieczność wymiany
zużywających się anod, jest metoda tworzenia Fe@C Nps w strumieniu plazmy
termicznej. W tym przypadku mieszaninę proszku metalicznego Fe14Nd2B oraz
proszku grafitowego wprowadza się do otworu znajdującego się w środku anody.
Następnie pod wpływem plazmy elektrody ulegają procesowi sublimacji. W celu
oddzielenia powstałych w tej metodzie produktów niepożądanych od nanokapsuł
węglowych zawierających żelazo, powstałe materiały umieszcza się we wrzącym
2 M HCl na 24 h, a następnie przemywa się je wodą destylowaną i etanolem oraz
suszy w atmosferze powietrza w temperaturze 350 K [352]. Metody wykorzystujące
plazmę jako środowisko reakcyjne mają dwie zasadnicze wady: wymagają
skomplikowanego i kosztownego sprzętu oraz są energochłonne.
Z ekonomicznego punktu widzenia najskuteczniejszą metodą syntezy
nanocząstek metalicznych zakapsułkowanych w węglu jest wysokoenergetyczna
termoliza opracowana przez dr hab. Bystrzejewskiego z WCh UW [349].
W metodzie tej proces syntezy Fe@C Nps prowadzi się w prostym reaktorze, który
nie wymaga ani systemów próżniowych, ani zaawansowanych zasilaczy. Siłą
napędową tego procesu jest reakcja pomiędzy dwoma prostymi i tanimi związkami
chemicznymi: azydkiem sodu i sześciochlorobenzenem [353]. Źródłem metalu mogą
być proste związki organiczne (np. ferrocen), czy nawet drobny proszek żelaza.
Termoliza jest procesem szybkim, reakcja zwykle nie trwa dłużej niż kilka sekund.
Schemat syntezy nanokapsuł węglowych podczas termolizy układu NaN3-C6Cl6-Fe
przedstawia rysunek 33.
Rysunek 33. Schemat syntezy nanokapsuł węglowych zawierających żelazo metodą
wysokoenergetycznej termolizy układu NaN3-C6Cl6-Fe.
+ 6 Na–N=N=N ++ –
(10 20 μm)Fe@C Nps
(30 80 nm)
2366 N9NaCl6C6NaN6ClC
Fe
a-C Nps(20 100 nm)

Nanomateriały typu „rdzeń – powłoka”
83
Powłoka węglowa nadaje nanokapsułom szczególne cechy, takie jak: dobra
aktywność elektrokatalityczna, jak również możliwość chemicznego
sfunkcjonalizowania powierzchni (np. COOH) [354], które może być wymagane
w procesach koniugacji niektórych cząsteczek, jak związki biologicznie ważne.
Modyfikacji powierzchni nanokapsuł węglowych za pomocą grup –COOH można
dokonać poprzez ich zanurzenie w 8 M HNO3 na czas 12 h. Następnie nanocząstki
przemywa się kilkukrotnie wodą, aż do momentu osiągnięcia neutralnego pH.
Utlenianie Fe@C Nps nie powoduje zmiany ich wielkości oraz kształtu, jak również
nie prowadzi do zmian właściwości chemicznych [355]. Otoczka węglowa oprócz
możliwości modyfikacji powierzchni nanocząstek poprawia również ich właściwości
magnetyczne. W przypadku SPIONs (superparamagnetycznych nanocząstek tlenku
żelaza) osiągalne maksymalne namagnesowanie wynosi pomiędzy 80 a 90 emug-1
,
natomiast dla nanokapsuł węglowych zawierających żelazo możliwe jest uzyskanie
nanomateriału z prawie dwukrotnie zwiększoną magnetyzacją (150 emug-1
) [356].
Dodatkowo, właściwości magnetyczne nanokapsuł zależą od ich rozmiarów [357].
W przypadku bardzo małych rozmiarów, gdy średnica pojedynczej nanokapsuły jest
mniejsza od średnicy pojedynczej domeny magnetycznej, nanocząstki takie mogą
posiadać właściwości superparamagnetyczne w temperaturze pokojowej [358]. Takie
nanokapsyły węglowe ulegają wysyceniu magnetycznemu w stosunkowo niewielkim
polu magnetycznym [349].
6.2. Zastosowanie magnetycznych nanokapsuł węglowych
z metalicznym rdzeniem
Właściwości magnetyczne nanocząstek mogą być wykorzystane w wielu
dziedzinach, np. do przechowywania danych [359], jako sorbenty [360], mobilne
katalizatory [361], w czujnikach [362] oraz do celów biomedycznych [363], w tym
również w obrazowaniu metodą rezonansu magnetycznego (MRI) [364].
Zastosowanie nanokapsuł węglowych, jako mobilnych sorbentów metali
ciężkich, takich jak: miedź, kobalt czy kadm ma zasadniczą zaletę w porównaniu do
standardowych sorbentów, z uwagi na fakt, że można je łatwo i szybko wyodrębnić
z roztworu przy użyciu magnesu. W celu wykorzystania nanokapsuł węglowych jako
sorbentów do wiązania metali należy je wcześniej zmodyfikować grupami
karboksylowymi poprzez poddanie ich procesowi utlenienia. Wiązanie metali

Nanomateriały typu „rdzeń – powłoka”
84
z sorbentem opiera się na elektrostatycznym oddziaływaniu zdeprotonowanych grup
funkcyjnych na powierzchni nanocząstek z kationami metali ciężkich [365].
Nanokapsuły węglowe z powodzeniem stosowane są również, jako
modyfikatory powierzchni elektrod, m.in. z węgla szklistego. Tego typu materiały
można wykorzystać do monitorowania efektów elektrokatalitycznych
paramagnetycznych enzymów (np. lakazy względem tlenu) w obecności
i nieobecności zewnętrznego źródła pola magnetycznego. Obserwuje się wówczas
wzrost uzyskiwanych sygnałów prądowych, który zależny jest od zastosowanej
wartości natężenia pola magnetycznego; im jest ono silniejsze, tym intensywność
rejestrowanych sygnałów prądowych jest większa. Ten wzrost jest wynikiem
zwiększonego transportu paramagnetycznej substancji elektroaktywnej do
powierzchni elektrody. Modyfikacja powierzchni elektrody z węgla szklistego
warstwą nanocząstek magnetycznych oraz unieruchomienie na takim podłożu lakazy,
pozwala na około dwukrotne zwiększenie prądu redukcji tlenu, w porównaniu do
elektrody niezmodyfikowanej nanocząstkami [366]. Zjawisko to może być
potencjalnie wykorzystane w przyszłości do konstrukcji bioogniw paliwowych. Efekt
silnego wzmocnienia intensywności woltamperometrycznych pików utleniania oraz
redukcji został również zaobserwowany dla paramagnetycznej pochodnej ferrocenu
(Fc-CH2COONa). Modyfikacja elektrody z węgla szklistego za pomocą nanokapsuł
węglowych oraz wprowadzenie do układu zewnętrznego źródła pola magnetycznego,
o stosunkowo małym natężeniu (około 3.4 mT), pozwoliło na znaczne
przyspieszenie procesu utlenienia oraz redukcji pochodnej ferrocenu, co skutkowało
wzrostem prądów pików o około 165%, w porównaniu do czystej elektrody z węgla
szklistego [366].
Fundamentalnie ważną zasadą zastosowania nanocząstek jest wielkość
i powierzchnia obszaru reakcji, które są do siebie odwrotnie proporcjonalne; wraz ze
zmniejszeniem się wielkości cząstek, powierzchnia tych nanostruktur wzrasta.
Z literatury wiadomo, że umieszczenie nanocząstek we krwi czy osoczu sprawia, że
oddziałują one z różnymi białkami [367]. W wyniku związania białka
z powierzchnią nanocząstki zmianie może ulec jej konformacja, co może wpływać
na oddziaływania pomiędzy poszczególnymi białkami [368]. Głównymi czynnikami
decydującymi o różnicy w wiązaniu białek z nanocząstkami, jest ich rozmiar oraz
ładunek powierzchniowy [369]. W celu selektywnego wiązania nanokapsuł
węglowych zawierających żelazo z białkami prowadzi się badania z wykorzystaniem

Nanomateriały typu „rdzeń – powłoka”
85
np. poliklonalnych przeciwciał. Modyfikacja nanokapsuł węglowych za pomocą
różnej długości łączników, zawierających grupy karboksylowe pozwala, po ich
aktywacji, na utworzenie wiązania kowalencyjnego z przeciwciałem, które następnie
oddziałuje z bydlęcymi i ludzkimi gamma globulinami. Koniugaty Fe@C Nps/Ab
mogą być wykorzystane do zastosowań biomedycznych, w tym w rezonansie
magnetycznym oraz terapii celowanej [370].
Niezwykłe fizyczne-, chemiczne- oraz biologiczne właściwości nanocząstek
magnetycznych otwierają również nowe możliwości projektowania różnorodnych
klinicznych aplikacji, takich jak, np. diagnostyka chorób czy terapia fotodynamiczna
[368]. Nanocząstki można również wykorzystać jako nośniki leków [371] oraz do
obrazowania diagnostycznego, czyli rezonansu magnetycznego z kontrastem, jako
środki fluorescencyjne lub jako struktury do wykrywania zmian patologicznych
w organizmie, czyli odróżnieniu guza od zdrowych tkanek [372]. Dodatkowo,
magnetyczne nanokapsuły o średnicy 10 100 nm mogą być potencjalnie
zastosowane w magnetofekcji, czyli dostarczaniu leków w obecności pola
magnetycznego [373]. Technika ta daje duże możliwości szybkiego i skutecznego
transportowania różnych leków bezpośrednio do chorych komórek pod wpływem
gradientu pola magnetycznego [348].


Udział pola magnetycznego w transporcie substancji elektroaktywnej…
87
7. Udział pola magnetycznego w transporcie
substancji elektroaktywnej do powierzchni
elektrody
W układzie elektrochemicznym wyróżnia się trzy zasadnicze rodzaje
transportu jonów oraz cząsteczek w roztworze do powierzchni elektrody, do których
zalicza się dyfuzję, migrację oraz konwekcję, co ilustruje poniższe równanie:
vCφCDRT
FzCDJJJJ r. iii
iiikonwek.migdyfuz.n (11)
gdzie: Jdyfuz. oznacza strumień substancji spowodowany transportem dyfuzyjnym,
Jmigr. dotyczy transportu migracyjnego, natomiast Jkonwek. to strumień substancji
wywołany konwekcją, Di – współczynnik dyfuzji i-tej substancji, Ci – stężenie i-tej
substancji w roztworze, F – stała Faradaya, R – stała gazowa, zi – ładunek i-tej
substancji, – potencjał elektryczny w roztworze, v – szybkość poruszania się
roztworu [374].
Transport dyfuzyjny substancji elektroaktywnej do powierzchni elektrody
towarzyszy wszystkim procesom elektrodowym, w konsekwencji następuje
wyrównanie stężeń depolaryzatora w roztworze, jak i potencjału chemicznego [375].
W przypadku naładowanych elektrycznie cząsteczek oraz w warunkach niedoboru
elektrolitu podstawowego, czyli dla niskiej mocy jonowej roztworu, proces
transportu może być wzmacniany lub osłabiany poprzez migrację. Proces migracji
ma miejsce w sytuacji, gdy transport depolaryzatora do powierzchni elektrody
wymuszony jest działaniem pola elektrycznego lub wynika on z różnicy potencjałów
[374]. Zjawisko migracji można wyeliminować poprzez dodanie do roztworu
nadmiaru elektrolitu podstawowego. Transport jonów lub cząsteczek może odbywać
się również pod wpływem gradientu gęstości, generowanego elektrochemicznie lub
termicznie lub pod wpływem zewnętrznych sił mechanicznych, czyli ruchu elektrody
lub roztworu. Zjawisko to nosi nazwę konwekcji [375].
Z literatury wiadomo, że pole magnetyczne może wpływać na procesy
zachodzące zarówno w pobliżu elektrody, jak również na jej powierzchni [376].
Zewnętrzne źródło pola magnetycznego może również mieć duży wpływ na
transport substancji [376]. W sytuacji, gdy w pobliżu elektrody umieszczony

Udział pola magnetycznego w transporcie substancji elektroaktywnej…
88
zostanie magnes, wówczas substancja może być transportowana do powierzchni
elektrody na drodze magnetycznego transportu migracyjnego. W przypadku
występowania w roztworze tego typu zjawisk, w układzie eksperymentalnym
pojawiają się dwa rodzaje sił magnetycznych: siła magnetohydrodynamiczna,
nazywana również siłą Lorentza oraz siła gradientu pola magnetycznego, zwana siłą
Kelvina. Siła Lorentza związana jest z istnieniem w pobliżu elektrody pola
magnetycznego (najczęściej o stałym natężeniu) i określana jest za pomocą
równania:
BJF L (12)
gdzie:
J to strumień obdarzonych ładunkiem elektrycznym molekuł wyrażany
w A·m-2
, natomiast
B to wektor indukcji magnetycznej wyrażony w teslach (T)
[377]. Z równania (12) wynika, że siła Lorentza działa wyłącznie na poruszające się
indywidua chemiczne obdarzone ładunkiem. Wkład tej siły w transport
depolaryzatora do powierzchni elektrody jest proporcjonalny do ładunku, jakim
obdarzona jest cząsteczka oraz jej prędkości, czyli zależy od natężenia prądu.
W zależności od wzajemnego usytuowania wektora strumienia
J i wektora indukcji
magnetycznej
B , siła ta może wpływać na kierunek poruszania się jonów, przez co
może przyspieszać lub hamować ich transport do powierzchni elektrody. Z uwagi na
fakt, że równanie (12) zawiera iloczyn wektorowy
BJ , największą wartość siły
Lorentza obserwuje się w sytuacji, gdy strumień substancji elektroaktywnej jest
prostopadły do wektora indukcji pola magnetycznego. Siła Lorentza zupełnie zanika
w przypadku, gdy substancja elektroaktywna nie jest obdarzona ładunkiem, jest
w stanie spoczynku lub wektory
J i
B są względem siebie równoległe [376]. Dużo
bardziej skomplikowany jest efekt związany z działaniem na procesy elektrodowe
pola magnetycznego o natężeniu gradientowym. W tym przypadku obserwuje się
efekty wywołane również oddziaływaniem pola z cząsteczkami obojętnymi
elektrycznie. Siłę gradientu pola magnetycznego (FG) opisuje równanie:
χμ
BB
μ
BχF
2
00
G
2 (13)

Udział pola magnetycznego w transporcie substancji elektroaktywnej…
89
gdzie: jest bezwymiarową podatnością magnetyczną, a 0m to przenikalność
magnetyczna próżni ( ]AN[104]mH[104 2717
0
m ) [377]. Pojawienie
się w układzie siły Kelvina najczęściej związane jest z obecnością gradientu wektora
indukcji magnetycznej, w mniejszym stopniu gradientu podatności magnetycznej
[378]. Gradient wektora indukcji magnetycznej (
B ) w dużej mierze zależy od
zastosowanych warunków eksperymentalnych i pojawia się w przypadku, gdy
w naczynku elektrochemicznym zewnętrzne pole magnetyczne jest niejednorodne
lub w sytuacji, gdy powierzchnia elektrody składa się z ferromagnetycznej substancji
lub jest nią zmodyfikowana [377]. Ten przypadek opisany jest pierwszym
składnikiem sumy, przedstawionym w równaniu (13) i najczęściej stanowi
dominujący wkład do wartości siły Kelvina. Drugi człon równania (13) opisującego
powstawanie siły Kelvina na skutek niejednorodności podatności magnetycznej ( )
ośrodka jest zwykle mniej znaczący i często zaniedbywalny. Istnienie gradientu
podatności magnetycznej ( ) jest konsekwencją występowania w roztworze
gradientu stężenia substancji paramagnetycznej ( C ) i określany jest on za pomocą
równania [377]:
Cm (14)
gdzie: m to podatność molowa w [m
3·mol
-1].
Przyczyną niejednorodności podatności magnetycznej roztworu może być np.
zachodząca w układzie reakcja redoks, w wyniku której może nastąpić zmiana
właściwości magnetycznych substancji w pobliżu powierzchni elektrody [379].
Zatem wartość siły Kelvina silnie powiązana jest z właściwościami magnetycznymi
molekuł, które powinny posiadać trwały moment magnetyczny, natomiast jest ona
w mniejszym stopniu zależna od orientacji pola magnetycznego względem
powierzchni elektrody [380].
Oprócz podatności molowej substancji istnieje także bezwymiarowa
podatność objętościowa oraz podatność masowa. Ogólnie podatność magnetyczną
definiuje się za pomocą równania:
B0
Mμ
H
Mχ (15)
gdzie: M oznacza namagnesowanie, H to natężenie pola, ǀ Bǀ określa wartość
indukcji pola magnetycznego, a 0m to przenikalność magnetyczna próżni. W

Udział pola magnetycznego w transporcie substancji elektroaktywnej…
90
zależności od wartości podatności magnetycznej substancje można podzielić na:
diamagnetyki, paramagnetyki oraz ferromagnetyki [381]. Substancje
diamagnetyczne, które są wypychane z pola magnetycznego, takie jak np. grafit,
posiadają ujemną podatność magnetyczną, gdyż indukowany przez zewnętrzne pole
magnetyczne diamagnetyczny moment magnetyczny zwrócony jest przeciwnie do
pola magnetycznego. Oddziaływania te są tak słabe, że nie mają praktycznego
wpływu na transport substancji w kierunku elektrody. W przypadku niektórych
jonów, takich jak: jony Fe2+
lub Co3+
, podatność magnetyczna zależna jest od
temperatury i wraz
z jej wzrostem przechodzi ona w podatność paramagnetyczną [381]. Właściwości
paramagnetyczne wykazują substancje, które posiadają niesparowane elektrony,
takie jak np.: tlen, wolne rodniki oraz metaloproteiny [382]. Substancje
paramagnetyczne są wciągane w obszar silniejszego pola i charakteryzują się
dodatnią podatnością magnetyczną nawet w przypadku braku zewnętrznego pola
magnetycznego. Natomiast po wprowadzeniu pola do układu, paramagnetyki
wykazują moment magnetyczny mający niezerową składową równoległą do
zewnętrznego pola [383].
Gradientowe pole magnetyczne wpływa nie tylko na transport substancji do
powierzchni elektrody, ale również decyduje o wydajności reakcji elektrodowych
oraz ich mechanizmach. Gradient podatności magnetycznej może wywoływać
konwekcję i powodować wciąganie paramagnetycznych substancji w kierunku pola
o większej indukcji magnetycznej lub wypychać z pola substancje o właściwościach
diamagnetycznych [384]. Transport paramagnetycznej substancji ulegającej
procesom redoks do powierzchni elektrody, spowodowany działaniem pola
magnetycznego, może być w pewnych warunkach silniejszy niż transport
zachodzący podczas konwekcji naturalnej [385].

Techniki badawcze stosowane w pracy
91
8. Techniki badawcze stosowane w pracy
W badaniach własnych, przedstawionych w niniejszej rozprawie doktorskiej,
do ilościowej i jakościowej charakterystyki zaproponowanych elektrochemicznych
czujników do detekcji wybranych metaloprotein w próbkach krwi zastosowano
szereg technik elektrochemicznych i spektroskopowych. Parametry analityczne, takie
jak: zakres pracy czujnika, jego granicę wykrywalności, powtarzalność
uzyskiwanych wyników, stabilność oraz selektywność wyznaczono na podstawie
eksperymentów wykonanych przy użyciu woltamperometrii z liniowo zmieniającym
się potencjałem (LSV), woltamperometrii cyklicznej (CV), woltamperometrii
pulsowej różnicowej (DPV), elektrochemicznej mikrowagi kwarcowej (EQCM) oraz
elektrochemicznej spektroskopii impedancyjnej (EIS). Informacji na temat jakości
i morfologii warstw analitycznie czynnych (tzw. warstw receptorowych)
konstruowanych czujników dostarczyły pomiary wykonane przy użyciu:
spektroskopii w podczerwieni z transformacją Fouriera (FTIR), spektroskopii
dichroizmu kołowego (CD), spektroskopii UV-vis, spektrometrii mas
z plazmą indukcyjnie sprzężoną z ablacją laserową (LA-ICP-MS), analizy
termograwimetrycznej (TGA), mikrowagi kwarcowej z dyssypacją energii
(QCM-D), powierzchniowego rezonansu plazmonowego (SPR) oraz skaningowego
mikroskopu elektronowego (SEM). Natomiast wpływ zastosowanych modyfikacji
oraz obecności zewnętrznego źródła pola magnetycznego na aktywność
elektrokatalityczną analizowanych metaloprotein zbadano za pomocą skaningowego
mikroskopu elektrochemicznego (SECM). W niniejszym rozdziale zostały
omówione tylko te techniki pomiarowe, które osobiście stosowałam
w prowadzonych badaniach.
8.1. Woltamperometria cykliczna i liniowa
Woltamperometria cykliczna (CV) jest techniką analityczną, w której
mierzona jest zależność pomiędzy natężeniem prądu (I) płynącego przez elektrodę
pracującą a potencjałem tej elektrody (E) [374]. W trakcie pomiaru do elektrody
pracującej przykładany jest zmieniający się liniowo w czasie potencjał od wartości,
przy której nie zachodzi reakcja elektrodowa (E1) do wartości, przy której ma
miejsce proces elektrodowy (E2), zgodnie z rysunkiem 34A. Jeśli kierunek

Techniki badawcze stosowane w pracy
92
przykładanego do elektrody potencjału zostanie zmieniony, wówczas produkty
powstałe w trakcie procesu elektrodowego ulegają procesowi odwrotnemu,
odpowiednio elektroredukcji lub elektroutlenianiu. Zależność I=f(E) uzyskiwana dla
zmian potencjału tylko w jednym kierunku nosi nazwę liniowej krzywej
woltamperometrycznej (LSV). Kształt uzyskiwanych sygnałów prądowych zależy od
rozmiaru elektrody. Dla elektrod o konwencjonalnych rozmiarach rejestrowana
zależność (I=f(E)) ma kształt piku (rysunek 34B), natomiast w przypadku
mikroelektrod otrzymuje się falę o kształcie sigmoidalnym, rysunek 34C.
E / V
I / A
C
t / s
E /
V
E1
E2
Cykl 1 Cykl 2 Cykl 3
A
E / V
I /
A
D
reakcja odwracalna
reakcja nieodwracalna
reakcja quasi-odwracalna
E / V
I /
A
B
Ip,a
Ip,k
Rysunek 34. Zależność przykładanego do elektrody pracującej potencjału w funkcji czasu (A),
cykliczne krzywe woltamperometryczne zarejestrowane na elektrodzie o konwencjonalnych
rozmiarach (B) i mikroelektrodzie (C); oraz cykliczne krzywe wltamperometryczne uzyskane dla
reakcji odwracalnej, quasi-odwracalnej i nieodwracalnej w warunkach dyfuzji liniowej [374].
Prąd piku w woltamperometrii z użyciem klasycznych (o konwencjonalnych
rozmiarach) elektrod dla procesów odwracalnych opisuje równanie Randlesa–Ńevčika
[374]:
i
2/1
i2/12/1
2/3446.0CvADn
TR
FI 1/23/2
p (16)
gdzie: Ip oznacza prąd piku [A], 0.446 – bezwymiarowy współczynnik liczbowy,
F – stała Faraday’a (96 485 C·mol-1
), R – stała gazowa (8.31 J·K-1
·mol-1
),
T – temperatura wyrażona w K, n – liczba elektronów uczestniczących w reakcji

Techniki badawcze stosowane w pracy
93
elektrodowej, A – powierzchnia elektrody [cm2], Di – współczynnik dyfuzji
substancji elektroaktywnej [cm2s
-1], v – szybkość przemiatania potencjałem [Vs
-1],
Ci – stężenie depolaryzatora w głębi roztworu [molcm-3
]. Dla temperatury
pokojowej (T = 298 K) równanie Randlesa–Ńevčika upraszcza się do postaci:
i
2/1
i
5
p 1069.2 CvADnI 1/23/2 (17)
W sytuacji, gdy warunki eksperymentalne nie ulegają zmianie, wartość prądu piku
jest proporcjonalna do stężenia substancji elektroaktywnej ulegającej procesom
redoks na powierzchni elektrody. W przypadku mikroelektrod, mierzony prąd
określany jest mianem prądu stacjonarnego (Iss; ang. steady-state current), który
opisuje poniższe równanie:
iiss CgnFrDI (18)
gdzie: g to współczynnik geometryczny (g = 4 dla dyskowej mikroelektrody),
a r – promień mikroelektrody.
Potencjały, przy których obserwowane są sygnały prądowe, związane
z zachodzącymi procesami elektrodowymi są charakterystyczne dla danego
badanego układu redoks, a ich położenie związane jest z potencjałem formalnym
tego układu. Potencjał formalny (Ef) określany jest za pomocą równania:
2
kp,ap,
f
EEE
(19)
gdzie: Ep,a to potencjał piku anodowego [V], a Ep,k – potencjał piku katodowego [V].
W przypadku odwracalnych reakcji redoks stosunek wartości Ip,a do Ip,k jest równy
jedności, natomiast różnicę potencjałów (Ep,a – Ep,k) dla T = 20 ºC określa się za
pomocą równania:
]mV[58
22.2k,pa,pnnF
RTEE (20)
W sytuacji jakichkolwiek odstępstw od odwracalnego przebiegu reakcji elektrodowej
wzrasta różnica potencjałów piku anodowego i katodowego, a także zmienia się
kształt krzywej woltamperometrycznej, zgodnie z rysunkiem 34D.
8.2. Woltamperometria pulsowa róznicowa
Technika woltamperometrii pulsowej różnicowej (DPV) jest niezwykle
użyteczna w analizie chemicznej ze względu na możliwość uzyskiwania niskiej
granicy oznaczalności substancji, poniżej 0.05 mM [386]. W metodzie tej, dzięki

Techniki badawcze stosowane w pracy
94
dwukrotnemu próbkowaniu prądu: tuż przed przyłożeniem pulsu oraz pod koniec
jego trwania następuje eliminacja prądu pojemnościowego [387]. Amplituda
przykładanych pulsów jest jednakowa podczas trwania całego eksperymentu. Zmianę
potencjału w woltamperometrii pulsowej różnicowej przedstawia rysunek 35.
t / s
E /
V
Eptp
Rysunek 35. Zmiany potencjału elektrody w czasie w metodzie woltamperometrii pulsowej
różnicowej w oparciu o rysunek z [386].
Woltamperogramy otrzymane za pomocą techniki DPV ilustrują zmianę prądu
ΔI [ΔI = I(t2) – I(t1)] względem przyłożonego potencjału. Rejestrowane sygnały
pradowe mają kształt pików, a wysokość prądu piku dla układu odwracalnego (Ip)
jest proporcjonalna do stężenia oznaczanej substancji i opisana jest poniższym
równaniem:
ζ
ζ
t
CnFADI
0.5
1
1iip (21)
gdzie:
RT
EnF p
2exp , t to czas trwania pulsu, natomiast ΔEp oznacza amplitudę
pulsu [387].
Technika woltamperometrii pulsowej różnicowej charakteryzuje się dobrą
rozdzielczością i pozwala na jednoczesny pomiar sygnałów różniących się względem
siebie o 50 mV. W metodzie tej elektronowość badanego procesu odwracalnego
wyznacza się na podstawie szerokości piku w połowie wysokości (W0.5), którą
określa równanie:
nF
RTW
52.35.0 (22)
Dzięki możliwości rejestracji sygnałów prądowych o kształcie pików oraz małemu
prądowi tła technika DPV jest niezwykle użyteczna w analizowaniu mieszanin [387].

Techniki badawcze stosowane w pracy
95
8.3. Elektrochemiczna mikrowaga kwarcowa
Mikrowaga kwarcowa (QCM) po raz pierwszy została wykorzystana do
pomiarów masy przez Sauerbrey’a w roku 1959 [388]. W porównaniu do
komercyjnych mikrowag analitycznych wykrywających substancje na poziomie
10-10
kg, technika QCM pozwalała na detekcję substancji nawet na poziomie 10-16
kg
[389]. Zastosowanie mikrowagi kwarcowej w pomiarach elektrochemicznych in situ
daje możliwość jednoczesnego rejestrowania parametrów elektrochemicznych
układu i zmian masy elektrody wywołanych procesem adsorpcji indywiduów
chemicznych na jej powierzchni. Wówczas stosuje się określenie elektrochemicznej
mikrowagi kwarcowej (EQCM). Metoda ta wykorzystywana jest do monitorowania
przebiegu procesu adsorpcji na powierzchni stałej.
Najważniejszym elementem każdej mikrowagi kwarcowej jest rezonator
kwarcowy, którego zasada działania oparta jest na zjawisku piezoelektrycznym
[388], a ściślej mówiąc na odwrotnym zjawisku piezoelektrycznym kryształu
kwarcu, polegającym na zmianie jego częstotliwości rezonansowej wywołanej
zmianą jego masy. Stosowany w przypadku elektrochemicznej mikrowagi
kwarcowej kryształ kwarcu pokryty jest z obu stron warstewką tlenku chromu bądź
tytanu, w celu zapewnienia dobrego przewodnictwa elektrycznego, na którą z obu
stron napylona jest cienka warstwa metalu, np. złota lub platyny [390]. Schemat
kryształu kwarcu stosowanego w pomiarach EQCM ilustruje rysunek 36.
ELEKTRODA ZŁOTA
KRYSZTAŁ KWARCU
KONTAKT
ELEKTRYCZNY
Rysunek 36. Schemat kryształu kwarcu. Rysunek oparty o [388].
Częstotliwość rezonansowa kryształu kwarcu zależy od całkowitej ilości
cząsteczek zaadsorbowanych na jego powierzchni. Jeśli zaadsorbowana na
powierzchni kryształu warstwa jest cienka, twarda, nie wykazuje właściwości

Techniki badawcze stosowane w pracy
96
wiskoelastycznych (fvis. 0) oraz jej masa jest dużo mniejsza od masy kryształu
(< 5 %), wówczas zmiana częstotliwości rezonansowej (Δf) kryształu kwarcu jest
proporcjonalna do zmian jego masy, zgodnie z równaniem Sauerbrey’a [391-394]:
2
02
ρμA
mnff
(23)
gdzie: Δf jest zmianą częstotliwości drgań kryształu kwarcu [Hz],
Δm − zmiana masy kryształu kwarcu [g], mq − współczynnik sprężystości
poprzecznej kryształu kwarcu (2.947·1011
g·cm-1
·s-2
), q – gęstość kwarcu
(2.648·1011
g·cm-3
), natomiast n − numer nadtonu częstotliwości rezonansowej.
Równanie Sauerbrey’a jest spełnione tylko dla cienkich, stabilnych i sztywnych
warstw jednorodnie rozmieszczonych na powierzchni kryształu kwarcu. Znak
ujemny w równaniu Sauerbrey’a wskazuje, że wzrost masy kryształu kwarcu
związany jest ze spadkiem jego częstotliwości rezonansowej. Równanie to często
przedstawiane jest w skróconej formie:
fC
mnf
(24)
gdzie: Cf jest stałą charakterystyczną dla danego kryształu kwarcu. Całkowitą zmianę
częstotliwości rezonansowej kryształu kwarcu, uwzgledniającą właściwości
wiskoelastyczne otoczenia, takie jak: gęstość ( lρ ) i lepkość cieczy ( lη ), opisuje
równanie [395]:
2/1
ll2/3
0
2
0.vis.mas.cał
2
μρ
ηρf
ρμA
mffff (25)
W przypadku pomiaru masy miękkich i sprężystych warstw niezwykle
pomocna jest mikrowaga kwarcowa z możliwością monitorowania dyssypacji energii
(ΔD), związanej z właściwościami wiskoelastycznymi zaadsorbowanej warstwy.
Wzrost wartości ΔD świadczy o utworzeniu na powierzchni kryształu kwarcu
miękkiej, elastycznej warstwy, podczas gdy niewielka zmiana dyssypacji energii
wskazuje na powstanie sztywnego filmu [396]. Wkład efektu wiskoelastycznego
w zmianę częstotliwości rezonansowej kryształu kwarcu może zostać pominięty, gdy
nDn/fn << 2.6·10-7
lub 4·10-7
Hz-1
, odpowiednio dla 6- i 5 MHz kryształu kwarcu
[397]. Metoda QCM-D cieszy się dużym powodzeniem w badaniach procesu
adsorpcji układów o znaczeniu biologicznym, gdyż pozwala określić orientację oraz
konformację zaadsorbowanych cząsteczek [398,399]. QCM-D umożliwia również

Techniki badawcze stosowane w pracy
97
badanie wpływu wielu czynników, takich jak: pH, siła jonowa oraz stężenie, na ilość
zaadsorbowanych molekuł. Należy podkreślić, że pomiar prowadzony jest w czasie
rzeczywistym.
8.4. Elektrochemiczna spektroskopia impedancyjna
W metodzie elektrochemicznej spektroskopii impedancyjnej (EIS) rejestruje
się liniową, elektryczną odpowiedź elektrody po przyłożeniu do niej potencjału
o małej amplitudzie, zmieniającego się sinusoidalnie w czasie i nie powodującego
zaburzeń badanej próbki. Na podstawie pomiarów EIS można uzyskać informacje na
temat kinetyki procesów elektrodowych, transportu zarówno ładunku pomiędzy
powierzchnią elektrody a substancją elektroaktywną, jak również masy
(substancji elektroaktywnej) do powierzchni elektrody [400]. Dodatkowo, metoda ta
pozwala na określenie stopnia porowatości utworzonej warstwy, jak i na ocenę
jakości warstwy utworzonej na powierzchni elektrody. Wadą tej techniki są bardzo
często problemy związane z właściwą interpretacją wartości uzyskanych parametrów
impedancyjnych [401].
Impedancja faradajowska jest wielkością zespoloną, złożoną z części
rzeczywistej (Z') oraz części urojonej (-Z'') [402], co przedstawia rysunek 37.
-Z"
Z'
ǀZǀ
oś u
rojo
na
oś rzeczywista
Rysunek 37. Wektor impedancji w przestrzeni zespolonej dla wybranej częstotliwości ω.
Impedancję faradajowską opisuje poniższe równanie matematyczne:
2s
2
s
0
f
1
ωCR
I
φZ
(26)
gdzie: ǀ Zfǀ to moduł impedancji, I0 − prąd wymiany w potencjale równowagowym
danego procesu elektrodowego, Δ − kąt przesunięcia fazowego, Rs − całkowita
oporność warstwy reakcyjnej, ω − częstość kątowa, natomiast Cs − całkowita
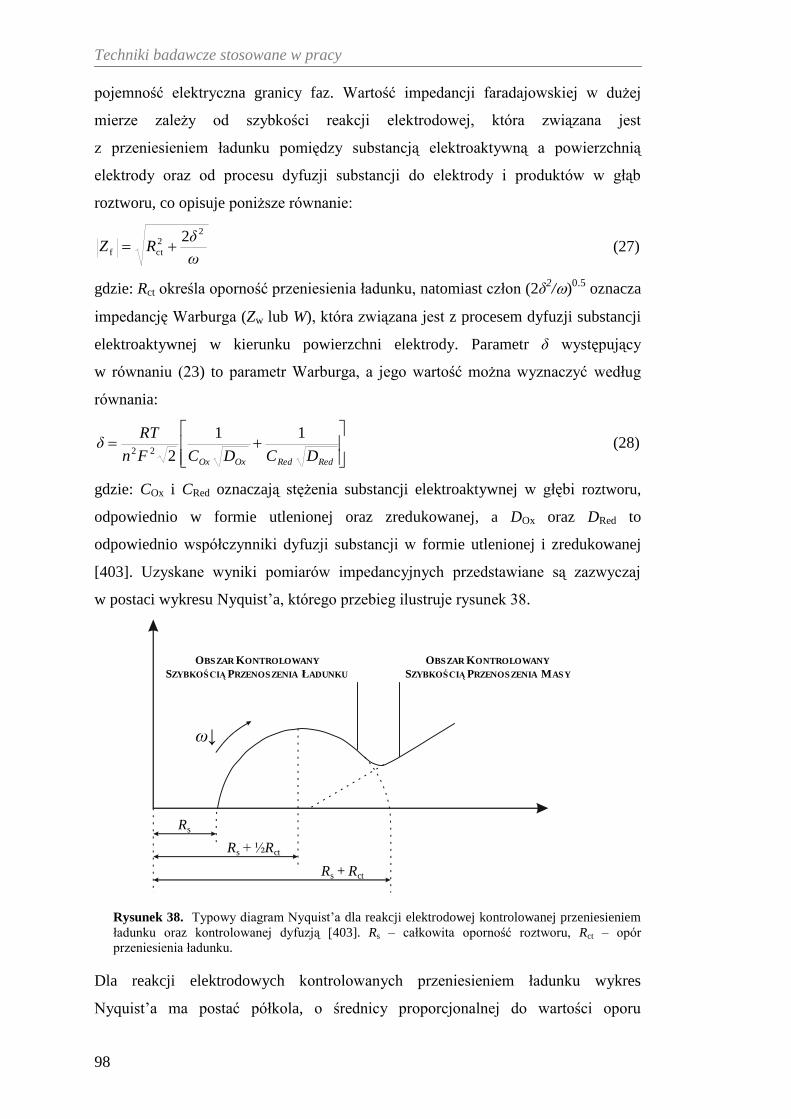
Techniki badawcze stosowane w pracy
98
pojemność elektryczna granicy faz. Wartość impedancji faradajowskiej w dużej
mierze zależy od szybkości reakcji elektrodowej, która związana jest
z przeniesieniem ładunku pomiędzy substancją elektroaktywną a powierzchnią
elektrody oraz od procesu dyfuzji substancji do elektrody i produktów w głąb
roztworu, co opisuje poniższe równanie:
ω
δRZ
22
ctf
2 (27)
gdzie: Rct określa oporność przeniesienia ładunku, natomiast człon (2δ2/)
0.5 oznacza
impedancję Warburga (Zw lub W), która związana jest z procesem dyfuzji substancji
elektroaktywnej w kierunku powierzchni elektrody. Parametr δ występujący
w równaniu (23) to parametr Warburga, a jego wartość można wyznaczyć według
równania:
RedRedOxOx DCDCFn
RTδ
11
222 (28)
gdzie: COx i CRed oznaczają stężenia substancji elektroaktywnej w głębi roztworu,
odpowiednio w formie utlenionej oraz zredukowanej, a DOx oraz DRed to
odpowiednio współczynniki dyfuzji substancji w formie utlenionej i zredukowanej
[403]. Uzyskane wyniki pomiarów impedancyjnych przedstawiane są zazwyczaj
w postaci wykresu Nyquist’a, którego przebieg ilustruje rysunek 38.
OBSZAR KONTROLOWANY
SZYBKOŚCIĄPRZENOSZENIA ŁADUNKU
OBSZAR KONTROLOWANY
SZYBKOŚCIĄPRZENOSZENIA MASY
Rs
Rs + Rct
Rs + ½Rct
ω↓
Rysunek 38. Typowy diagram Nyquist’a dla reakcji elektrodowej kontrolowanej przeniesieniem
ładunku oraz kontrolowanej dyfuzją [403]. Rs – całkowita oporność roztworu, Rct – opór
przeniesienia ładunku.
Dla reakcji elektrodowych kontrolowanych przeniesieniem ładunku wykres
Nyquist’a ma postać półkola, o średnicy proporcjonalnej do wartości oporu

Techniki badawcze stosowane w pracy
99
przeniesienia ładunku, natomiast w przypadku reakcji kontrolowanych dyfuzją
zależność ta ma charakter liniowy [404]. W celu dokonania ilościowej interpretacji
uzyskanych wyników eksperymentalnych, impedancję badanego układu opisuje się
za pomocą odpowiedniego obwodu zastępczego. W niniejszej rozprawie doktorskiej
w analizie wyników EIS stosowano obwód zastępczy, przedstawiony na rysunku 39.
RsTdl , dl
WRct
Rysunek 39. Obwód zastępczy opisujący badany układ pomiarowy. Rs – całkowita oporność
roztworu, Rct – opór przeniesienia ładunku, Tdl – parametr pojemnościowy elementu stało-
fazowego, dl – parametr potęgowy elementu stało-fazowego, W – impedancja Warburga.
8.5. Skaningowy mikroskop elektrochemiczny
W technice skaningowej mikroskopii elektrochemicznej (SECM)
wykorzystuje się mikroelektrody dyskowe najczęściej o średnicy od 0.6 do 25 µm
wykonane z platyny, złota lub węgla, które umożliwiają badanie profili stężeniowych
próbnika redoks w roztworze nad powierzchnią próbki [405]. W trakcie pomiaru
położenie mikroelektrody zmieniane jest zarówno w kierunku prostopadłym (z), jak
i w płaszczyźnie równoległej (x-y) do powierzchni próbki [406]. W ten sposób
uzyskuje się mapę elektroaktywnych miejsc badanej powierzchni [407]. W celu
wyeliminowania efektów związanych z dyfuzją z obszaru okołoelektrodowego,
stosunek średnicy mikroelektrody wraz z obudową do średnicy samej elektrody
powinien wynosić około 10. Tak dobrany rozmiar mikroelektrody czyni ją bardziej
czułą na procesy zachodzące na badanej powierzchni [407].
Obrazowanie za pomocą SECM prowadzi się zazwyczaj w trybie sprzężenia
zwrotnego (z ang. feedback mode, FB), w którym próbnik redoks umieszczony jest w
roztworze elektrolitu podstawowego. W sytuacji, gdy próbnik redoks jest w formie
zredukowanej, wówczas w wyniku przyłożenia wystarczająco dodatniego potencjału
na mikroelektrodzie następuje jego utlenienie. Szybkość zachodzącego procesu
kontrolowana jest przez szybkość dyfuzji sferycznej. Dla dostatecznie dużej
odległości mikroelektrody od badanej powierzchni dyfuzja zachodzi z obszaru
półnieskończonego i ustala się prąd stacjonarny (Iss), opisany równaniem (18).
W sytuacji, gdy mikroelektroda umieszczona jest w pobliżu powierzchni
nieprzewodzącej, rejestrowany prąd faradajowski maleje ze względu na utrudnioną

Techniki badawcze stosowane w pracy
100
dyfuzję próbnika redoks do mikroelektrody. Obserwowane zjawisko określane jest
mianem ujemnego sprzężenia zwrotnego. Jeśli natomiast mikroelektroda znajduje się
w pobliżu substratu przewodzącego następuje zwiększenie wartości rejestrowanego
prądu faradajowskiego, co określa się mianem dodatniego sprzężenia zwrotnego.
Obserwowane wzrosty prądu związane są z regeneracją próbnika redoks na
powierzchni mikroelektrody [408]. W metodzie SECM wyniki przedstawia się
w postaci krzywych zbliżania opisujących zmiany prądowe mikroelektrody w funkcji
jej odległości od powierzchni analizowanej próbki. Schemat działania skaningowego
mikroskopu elektrochemicznego w trybie sprzężenia zwrotnego ilustruje rysunek 40.
RedRed
RedRed
Red
e‒
Red
RedRed
Red
Red
Red
Red Ox
RedRed
RedRed
Red RedRed Ox
IZOLATOR PRZEWODNIK
e‒ e‒
A B C
e‒
Rysunek 40. Schemat działania skaningowego mikroskopu elektrochemicznego w trybie
sprzężenia zwrotnego dla różnych przypadków: półnieskończonego obszaru dyfuzji sferycznej
(A), umieszczenia w pobliżu mikroelektrody izolatora elektrycznego (B) oraz przewodnika (C),
w oparciu o [408].
8.6. Spektroskopia UV-vis
Spektroskopia w zakresie ultrafioletu (UV; 200 380 nm) oraz światła
widzialnego (vis; 380 780 nm) jest szeroko stosowaną techniką do ilościowej
analizy substancji absorbujących promieniowanie elektromagnetyczne z zakresu
charakterystycznego dla tej metody, jak również do analizy strukturalnej związków
organicznych. Absorpcja promieniowania elektromagnetycznego przez związki
związana jest z występowaniem w ich strukturze wiązań podwójnych, potrójnych
oraz sprzężonych układów wiązań. Te nienasycone ugrupowania określane są
mianem chromoforów [409]. W metodzie UV-vis rejestruje się zależność zmiany
absorbancji próbki (A) w funkcji długości fali (). Intensywność zaabsorbowanego
promieniowania zależy od: stężenia badanej substancji (C), grubości warstwy
absorbującej (l wyrażonej w cm) oraz molowego współczynnika absorpcji
(, wyrażonego w jednostce dm3·mol
-1·cm
-1), a jego miarą jest absorbancja lub

Techniki badawcze stosowane w pracy
101
transmitancja (T). Zależność tą w sposób matematyczny opisuje prawo Lamberta –
Beera, zgodnie z równaniem:
εlCI
I
TA 0log
1log (29)
gdzie: I0 i I to odpowiednio natężenie wiązki promieniowania padającego i po jego
przejściu przez ośrodek absorbujący. Molowy współczynnik absorpcji jest
wielkością stałą, charakterystyczną dla danej substancji, niezależną od jej stężenia,
natomiast zależną od długości fali i stosowanego rozpuszczalnika [410]. Obecność
w strukturze związku podstawników elektronodonorowych, takich jak np.: –OH,
–NH2 lub –OCH3 określanych jako auksochromy, powoduje przesunięcie położenia
pasma absorpcji w kierunku większych wartości długości fali oraz wzrost
intensywności pasma. Efekty te nazywane są odpowiednio tzw. przesunięciem
batochromowym oraz efektem hiperchromowym. Z kolei obecność nasyconych grup
elektronoakceptorowych, takich jak np.: –CHO, –NO, –NO2 powoduje przesunięcie
pasma absorpcji w kierunku mniejszych wartości długości fali (tzw. przesunięcie
hipsochromowe) oraz zmniejszenie intensywności pasma (tzw. efekt
hipochromowy).
Dla roztworów wieloskładnikowych, zgodnie z prawem addytywności,
absorbancja układu jest równa sumie absorbancji poszczególnych jego składników,
co opisuje poniższe równanie:
lCεCεCεAA nn2211
ni
1i
i ...
(30)
Prawo to jest spełnione tylko wtedy, gdy poszczególne składniki obecne w roztworze
nie oddziałują ze sobą i zależność A = f(C) ma charakter prostoliniowy [411].

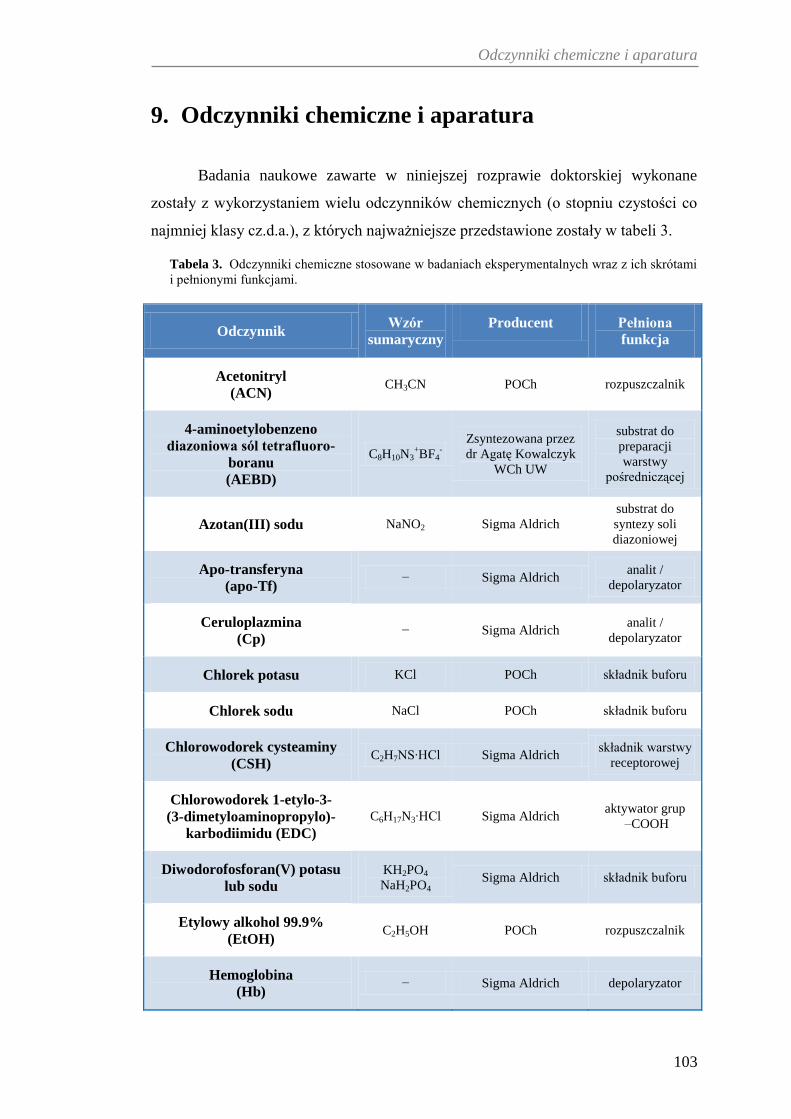
Odczynniki chemiczne i aparatura
103
9. Odczynniki chemiczne i aparatura
Badania naukowe zawarte w niniejszej rozprawie doktorskiej wykonane
zostały z wykorzystaniem wielu odczynników chemicznych (o stopniu czystości co
najmniej klasy cz.d.a.), z których najważniejsze przedstawione zostały w tabeli 3.
Tabela 3. Odczynniki chemiczne stosowane w badaniach eksperymentalnych wraz z ich skrótami
i pełnionymi funkcjami.
Odczynnik Wzór
sumaryczny
Producent Pełniona
funkcja
Acetonitryl
(ACN) CH3CN POCh rozpuszczalnik
4-aminoetylobenzeno
diazoniowa sól tetrafluoro-
boranu
(AEBD)
C8H10N3+BF4
-
Zsyntezowana przez
dr Agatę Kowalczyk
WCh UW
substrat do
preparacji
warstwy
pośredniczącej
Azotan(III) sodu NaNO2 Sigma Aldrich
substrat do
syntezy soli
diazoniowej
Apo-transferyna
(apo-Tf) − Sigma Aldrich
analit /
depolaryzator
Ceruloplazmina
(Cp) − Sigma Aldrich
analit /
depolaryzator
Chlorek potasu KCl POCh składnik buforu
Chlorek sodu NaCl POCh składnik buforu
Chlorowodorek cysteaminy
(CSH) C2H7NS·HCl Sigma Aldrich
składnik warstwy
receptorowej
Chlorowodorek 1-etylo-3-
(3-dimetyloaminopropylo)-
karbodiimidu (EDC)
C6H17N3·HCl Sigma Aldrich aktywator grup
–COOH
Diwodorofosforan(V) potasu
lub sodu
KH2PO4
NaH2PO4 Sigma Aldrich składnik buforu
Etylowy alkohol 99.9%
(EtOH) C2H5OH POCh rozpuszczalnik
Hemoglobina
(Hb) − Sigma Aldrich depolaryzator
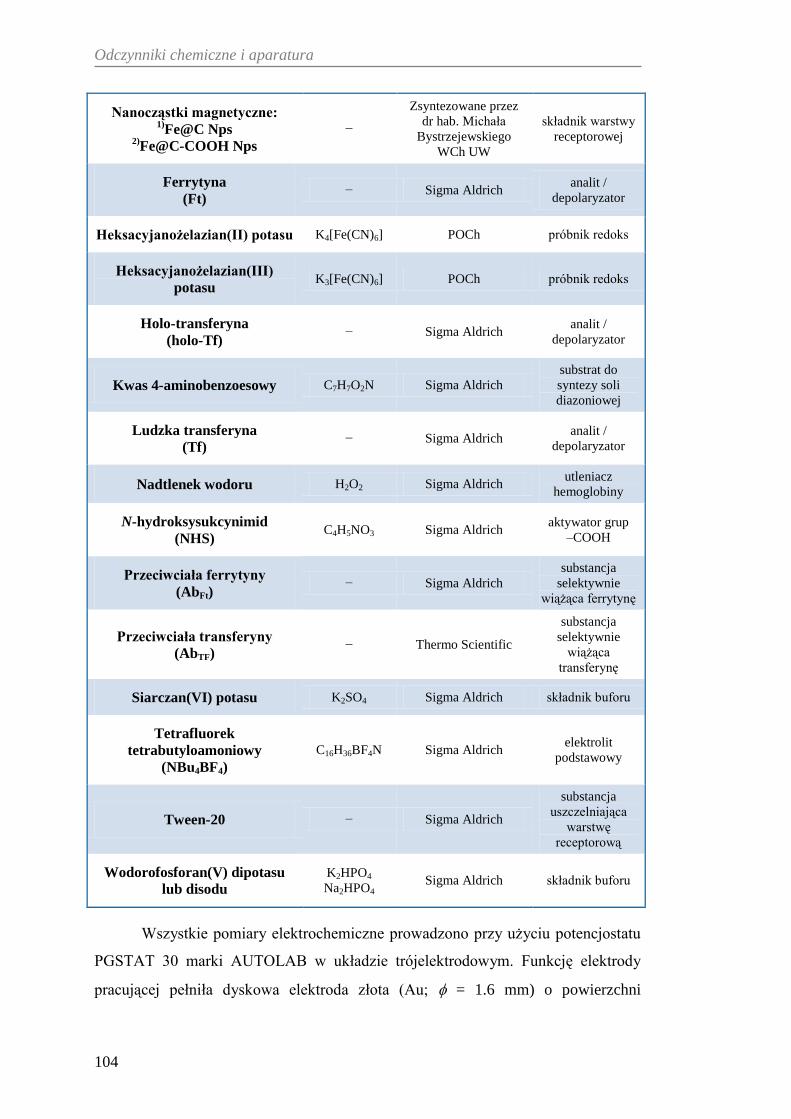
Odczynniki chemiczne i aparatura
104
Nanocząstki magnetyczne: 1)
Fe@C Nps 2)
Fe@C-COOH Nps
−
Zsyntezowane przez
dr hab. Michała
Bystrzejewskiego
WCh UW
składnik warstwy
receptorowej
Ferrytyna
(Ft) − Sigma Aldrich
analit /
depolaryzator
Heksacyjanożelazian(II) potasu K4[Fe(CN)6] POCh próbnik redoks
Heksacyjanożelazian(III)
potasu K3[Fe(CN)6] POCh próbnik redoks
Holo-transferyna
(holo-Tf) − Sigma Aldrich
analit /
depolaryzator
Kwas 4-aminobenzoesowy C7H7O2N Sigma Aldrich
substrat do
syntezy soli
diazoniowej
Ludzka transferyna
(Tf) − Sigma Aldrich
analit /
depolaryzator
Nadtlenek wodoru H2O2 Sigma Aldrich utleniacz
hemoglobiny
N-hydroksysukcynimid
(NHS) C4H5NO3 Sigma Aldrich
aktywator grup
–COOH
Przeciwciała ferrytyny
(AbFt) − Sigma Aldrich
substancja
selektywnie
wiążąca ferrytynę
Przeciwciała transferyny
(AbTF) − Thermo Scientific
substancja
selektywnie
wiążąca
transferynę
Siarczan(VI) potasu K2SO4 Sigma Aldrich składnik buforu
Tetrafluorek
tetrabutyloamoniowy
(NBu4BF4)
C16H36BF4N Sigma Aldrich elektrolit
podstawowy
Tween-20 − Sigma Aldrich
substancja
uszczelniająca
warstwę
receptorową
Wodorofosforan(V) dipotasu
lub disodu
K2HPO4
Na2HPO4 Sigma Aldrich składnik buforu
Wszystkie pomiary elektrochemiczne prowadzono przy użyciu potencjostatu
PGSTAT 30 marki AUTOLAB w układzie trójelektrodowym. Funkcję elektrody
pracującej pełniła dyskowa elektroda złota (Au; = 1.6 mm) o powierzchni

Techniki badawcze stosowane w pracy
105
0.0201 cm2 lub kryształ kwarcu z napyloną po obu stronach warstwą złota
(Au-EQCM) o powierzchni ok. 0.485 cm2. Jako elektrody odniesienia użyto
chlorosrebrową elektrodę z podwójnym płaszczem (Ag/AgCl/3 M KCl/0.1 M
KNO3), zaś elektrodę pomocniczą stanowiła blaszka platynowa lub złota
o powierzchni co najmniej 1 cm2.
W przypadku sensorów tworzonych na powierzchni wybranego materiału,
niezwykle ważny jest stan takiej powierzchni. Zatem konstrukcję poszczególnych
czujników zawsze poprzedzał etap właściwego przygotowania powierzchni materiału
przewodzącego. W zależności od typu elektrody pracującej zastosowano w tym celu
metody mechaniczne i/lub elektrochemiczne. Elektrody dyskowe najpierw
polerowano na kółku polerskim z użyciem tlenku glinu, o średnicy ziaren 1 μm,
poprzez wielokrotne wykonywanie ruchów ósemkowych. Następnie w celu usunięcia
pozostałości tlenku glinu z powierzchni elektrody, opłukiwano ją silnym-,
prostopadłym do powierzchni strumieniem wody destylowanej (o przewodnictwie
równym 0.056 mS·cm-1
z aparatu Hydrolab). Następnie tak przygotowaną elektrodę
poddawano elektrochemicznej procedurze czyszczenia. Pierwszy jej etap polegał na
zarejestrowaniu woltamperogramu w 0.1 M roztworze wodorotlenku sodu,
w zakresie potencjałowym 0 ÷ 1.8 ÷ 0 V z szybkością przemiatania potencjału 100
mV·s-1
. Kolejno rejestrowano woltamperogramy cykliczne w 0.1 M roztworze kwasu
siarkowego(VI), w zakresie potencjałów -0.3 ÷ 1.5 ÷ -0.3 V z szybkością polaryzacji
elektrody 50 mV·s-1
. Cykliczne krzywe rejestrowano do momentu uzyskania
stabilnego woltamperogramu charakterystycznego dla czystej elektrody złotej [412].
Końcowym etapem elektrochemicznej procedury czyszczenia elektrod złotych była
ich aktywacja w wyniku cyklizacji w 0.1 M roztworze kwasu chlorowego(VII),
w zakresie potencjałów -0.65 ÷ 0.95 ÷ -0.65 V, z szybkością polaryzacji elektrody
1 V·s-1
. Z uwagi na łatwość zniszczenia i uszkodzenia kryształu kwarcu, tego typu
elektrody poddawano tylko i włącznie elektrochemicznej procedurze czyszczenia,
analogicznej jak w przypadku elektrod dyskowych. Pomiary wykonywano
w temperaturze pokojowej, wynoszącej 21 °C, w roztworach odtlenionych za
pomocą argonu (czas odtleniania około 30 minut). W zależności od oznaczanego
białka stosowano różne bufory pomiarowe, które dobierane były tak, aby uzyskiwać
maksymalną aktywność danego białka. W badaniach zastosowano następujące
bufory pomiarowe:

Odczynniki chemiczne i aparatura
106
0.02 M bufor PBS: 0.02 M NaH2PO4, 0.02 M Na2HPO4, 2 mM KCl
i 150 mM NaCl o pH 6.0; w przypadku pomiarów dotyczących hemoglobiny,
0.02 M bufor PB: 0.02 M KH2PO4, 0.02 M K2HPO4 z dodatkiem 150 mM
K2SO4 o pH 7.4; w przypadku pomiarów dotyczących ceruloplazminy,
transferyny i ferrytyny.
9.1. Charakterystyka nanocząstek magnetycznych:
składnika warstwy receptorowej czujników
Podstawowym elementem warstw receptorowych opisanych w niniejszej
rozprawie czujników do detekcji wybranych metaloprotein w próbkach krwi były
magnetyczne nanokapsuły węglowe zawierające żelazo, które zostały zsyntezowane
przez dr hab. Michała Bystrzejewskiego z Wydziału Chemii Uniwersytetu
Warszawskiego. W badaniach wykorzystano niezmodyfikowane nanocząstki
magnetyczne (Fe@C Nps), jak i nanocząstki magnetyczne, których powierzchnia
zmodyfikowana została grupami karboksylowymi (Fe@C-COOH Nps).
9.1.1. Niezmodyfikowane nanokapsuły węglowe zawierające żelazo
Wykorzystane w eksperymentach nanokapsuły węglowe zawierające żelazo
charakteryzują się słabymi właściwościami ferromagnetycznymi. Magnetyczna pętla
histerezy Fe@C Nps została przedstawiona na rysunku 41. Analizując poniższy
histogram można zauważyć, że zastosowane nanocząstki osiągają maksymalny
moment magnetyczny (121 emu·g-1
) przy stosunkowo niskim polu magnetycznym,
o natężeniu ok. 8 kOe. W przeprowadzonych badaniach jako źródło pola
magnetycznego zastosowano magnesy neodymowe (Fe14Nd2B) o różnym kształcie,
których indukcja magnetyczna mieściła się w zakresie 40÷90 mT. Wiadomo, że
przenikalność magnetyczna wody jest praktycznie identyczna jak przenikalność
magnetyczna powietrza, dlatego też wartość indukcji pola magnetycznego
o natężeniu 10 Oe wynosi 1 mT. Zatem zastosowane w badaniach magnesy stałe
wytwarzały pole magnetyczne o natężeniu w zakresie wartości: 400÷900 Oe.
Analizując dane uzyskane z magnetycznej pętli histerezy nanokapsuł Fe@C można
stwierdzić, że w warunkach pomiarowych nanocząstki magnetyczne osiągają
moment magnetyczny rzędu 21÷36 emu·g-1
. Rozkład wielkości nanokapsuł Fe@C
Nps (dolny wykres wewnętrzny na rysunku 41) pokazuje, że ich średnica mieści się

Techniki badawcze stosowane w pracy
107
zakresie od 5 do 65 nm, a główną ich frakcję stanowią nanokapsuły o względnie
wąskim rozkładzie wielkości (5÷30 nm). Średnia wartość rozmiaru zastosowanych
nanokapsuł węglowych zawierających żelazo wynosiła ok. 20 nm.
pole magnetyczne / Oe
-10000 -5000 0 5000 10000
mag
ne
tyza
cja
/ e
mu
g
-1
-120
-80
-40
0
40
80
120
Hc / Oe
-500 0 500
Ms /
em
ug
-1
-40
-20
0
20
40
Ms = 121 emug
-1
/ nm
0 20 40 60 80 100
Zlicze
nia
0
50
100
150
200
śred. = 19 nm
Rysunek 41. Magnetyczna pętla histerezy (zmierzona w 25 °C) dla nanokapsuł Fe@C Nps.
Górny wykres wewnętrzny: powiększona magnetyzacja w obszarze -1000 ÷ 1000 Oe. Dolny
wykres wewnętrzny: rozkład wielkości nanokapsuł węglowych zawierających żelazo. Badania
wykonał dr hab. Michał Bystrzejewski.
9.1.2. Nanokapsuły węglowe zawierające żelazo zmodyfikowane
grupami karboksylowymi
Jedną z zalet nanokapsuł węglowych zawierających żelazo jest możliwość
modyfikacji ich powierzchni odpowiednimi grupami funkcyjnymi. W badaniach
zastosowano również nanocząstki magnetyczne zmodyfikowane grupami
karboksylowymi (Fe@C-COOH Nps). Rozkład wielkości tego typu nanokapsuł
mieścił się w zakresie od 5 do 140 nm, a główną ich frakcję stanowiły nanokapsuły
o średnicy 40 nm [413]. Maksymalna magnetyzacja nanocząstek wynosiła
ok. 54 emu·g-1
przy polu magnetycznym równym około 7 kOe. Nanokapsuły
węglowe Fe@C-COOH w warunkach pomiarowych, w których zastosowano pole
magnetyczne o indukcji magnetycznej wynoszącej 45÷90 mT, osiągały moment
magnetyczny od 8 do 26 emu·g-1
. Magnetyczną pętlę histerezy uzyskaną dla
Fe@C-COOH Nps przedstawia rysunek 42.

Odczynniki chemiczne i aparatura
108
pole magnetyczne / Oe
-10000 -5000 0 5000 10000
ma
gn
ety
za
cja
/ e
mu
g
-1
-80
-40
0
40
80
Hc / Oe
-500 0 500M
s /
em
ug
-1-30
-20
-10
0
10
20
30
Ms = 54 emug
-1
/ nm
0 20 40 60 80 100 120 140
Zliczen
ia
0
5
10
15
20
25
śred.
= 40 nm
Rysunek 42. Magnetyczna pętla histerezy (zmierzona w 25 °C) dla nanokapsuł Fe@C-COOH.
Górny wykres wewnętrzny: powiększona magnetyzacja w obszarze -1000 ÷ 1000 Oe. Dolny
wykres wewnętrzny: rozkład wielkości nanokapsuł węglowych Fe@C-COOH. Badania wykonał
dr hab. Michał Bystrzejewski.
Nanocząstki te w swojej strukturze zawierają cztery fazy krystaliczne, takie jak:
grafit, bcc Fe, fcc Fe-C i Fe3C, a powierzchnia właściwa nanocząstki wynosiła
95 m2·g
-1. Z uwagi na obecność na ich powierzchni różnych grup kwasowych, takich
jak grupy karboksylowe, laktonowe czy fenolowe wykazują one właściwości
kwasowe, a przy wartości pH 4.5 osiagają punkt zerowego ładunku elektrycznego
[413].

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
109
10. Elektrograwimetryczny sensor do bezpośredniej
detekcji hemoglobiny w próbkach krwi
W analizie klinicznej zawartość hemoglobiny w próbkach krwi określa się
głównie przy użyciu metod spektrofotometrycznych [414], kolorymetrycznych lub za
pomocą procedur immunologicznych [415]. Niestety, niejednokrotnie metody te
wykorzystują toksyczne i często rakotwórcze substancje, jak np. odczynnik
Drabkina, składający się z K3[Fe(CN)6], NaHCO3 i KCN [416]. Dodatkowo,
charakteryzują się one niską wrażliwością na dyskretne zmiany ilości hemoglobiny
we krwi. Kolejną wadą powszechnie stosowanych metod do oznaczeń Hb w analizie
klinicznej jest ich czasochłonność, której główną przyczyną jest konieczność
wstępnej obróbki próbki krwi i relatywnie długi czas reakcji z odczynnikiem
charakterystycznym dla danej metody [417]. Elektrochemiczne czujniki do oznaczeń
hemoglobiny we krwi mogą być zatem dobrą alternatywą w stosunku do obecnie
stosowanych metod klinicznych, ze względu na krótki czas analizy, niski koszt oraz
brak konieczności stosowania toksycznych odczynników.
Opisane w literaturze procedury elektrochemicznej detekcji Hb zasadniczo
dotyczą dwóch typów układów: (i) elektroda pracująca/warstwa pośrednicząca/
Hbadsorpcja lub (ii) elektroda/mediator/Hbroztwór. Przy czym doniesienia literaturowe
dotyczące bezpośredniej elektrochemicznej detekcji Hb rozpuszczonej w roztworze
są znikome. Wynika to z faktu, że centra elektroaktywne Hb, czyli grupy hemowe, są
głęboko osadzone w otoczce białkowej i w konsekwencji przeniesienie elektronu jest
zbyt powolne. Jednym z zadań badawczych postawionych w celu niniejszego
projektu doktorskiego było opracowanie procedury woltamperometrycznego
oznaczania ilości tego białka w analizowanym roztworze, w tym również
w próbkach naturalnych, w szybki i prosty sposób, bez konieczności stosowania
mediatorów. Ideą zaproponowanej metody detekcji hemoglobiny z roztworu było
wykorzystanie magnetycznych właściwości białka i jego oddziaływania
z ferromagnetycznym składnikiem warstwy receptorowej w obecności zewnętrznego
źródła pola magnetycznego, dzięki czemu możliwa była bezpośrednia
elektrochemiczna detekcja Hb z roztworu.

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
110
10.1. Optymalizacja warunków występowania
paramagnetycznej formy hemoglobiny
Jak wspomniano w rozdziale 4.2.1. hemoglobina, w zależności od warunków,
może występować w roztworze w różnych formach różniących się między sobą
stopniem utlenienia żelaza, zawartością w swojej strukturze tlenu, stabilnością, czy
też właściwościami magnetycznymi. W celu uzyskania stabilnej, paramagnetycznej
pochodnej hemoglobiny (methemoglobiny), zdolnej do oddziaływania
z ferromagnetycznym modyfikatorem powierzchni elektrody oraz z zewnętrznym
źródłem pola magnetycznego, nieodtleniony roztwór hemoglobiny poddano
działaniu nadtlenku wodoru. Nadtlenek wodoru został zastosowany w 10-krotnym
nadmiarze w stosunku do stężenia Hb, np.: dla roztworu Hb o stężeniu 0.10 µM,
stężenie H2O2 w tej samej objętości próbki wynosiło 1 µM. Takie stężenie H2O2
pozwoliło na całkowite przeprowadzenie Hb w stabilną, paramagnetyczną formę
– methemoglobinę, bez zniszczenia grup hemowych [418]. Skuteczność działania
zastosowanej procedury sprawdzono wykonując pomiary przy użyciu techniki
UV-vis. Uzyskane widma przedstawiono na rysunku 43.
długość falinm
300 400 500 600
A
0.00
0.01
0.02
0.03
0.04
długość fali / nm
480 500 520 540 560 580 600 620 640
A
0.0000
0.0005
0.0010
0.0015
0.0020Hb
Hb + H2O
2 (1:10)
Hb+H2O
2 po 10 minutach
Rysunek 43. Widma UV-vis zarejestrowane dla 0.10 µM roztworu Hb przed i po dodaniu 1 µM
H2O2 w 0.02 M buforze PBS o pH 6.0. Rysunek wewnętrzny: powiększona część widma UV-Vis
w zakresie długości fali 460 ÷ 650 nm.
Widma UV-vis zarejestrowano dla roztworu hemoglobiny zarówno przed dodatkiem
nadtlenku wodoru, zaraz po jego dodaniu, jak i po 10 minutach oddziaływania

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
111
hemoglobiny z H2O2. Na uzyskanych widmach dobrze widoczne jest pasmo
absorpcji, charakterystyczne dla Hb, przy długości fali ok. 400 nm. Występowanie
tego pasma na widmie UV-vis potwierdza obecność nienaruszonych grup hemowych
w strukturze hemoglobiny [419]. Z literatury wiadomo, że położenie tego pasma jest
ściśle powiązane z formą hemoglobiny [420,421]. W tabeli 4 podano wartości
długości fali, przy których pojawia się pasmo absorpcji związane
z obecnością grup hemowych w strukturze Hb, w zależności od formy hemoglobiny
obecnej w roztworze. Należy podkreślić, że jego intensywność zależy nie tylko od
stężenia Hb, ale również od wartości molowego współczynnika absorpcji, wielkości
charakterystycznej dla danej formy hemoglobiny.
Tabela 4. Różne formy hemoglobiny występujące w roztworze i charakterystyczne dla nich
maksimum absorpcji przy danej długości fali w technice UV-vis [420,421].
Forma hemoglobiny Maksimum absorpcji
Deoksyhemoglobina
(deoksyHb) 430 nm
Oksyhemoglobina
(oksyHb) 415 nm
Methemoglobina
(metHb) 405 nm
Ferrylohemoglobina
(ferryloHb) 418 nm
Widmo UV-vis uzyskane dla roztworu hemoglobiny przed dodatkiem nadtlenku
wodoru (krzywa czarna na rysunku 43) pokazuje, że maksimum absorpcji pasma
pochodzącego od grup hemowych występuje przy długości fali ok. 412 nm.
Położenie tego pasma, zgodnie z danymi literaturowymi, wskazuje na obecność
w roztworze utlenowanej formy hemoglobiny, oksyhemoglobiny (oksyHb). Dodanie
do roztworu oksyHb 10-krotnego nadmiaru H2O2 spowodowało przesunięcie tego
pasma w kierunku mniejszych wartości długości fali, co potwierdza pojawienie się
w roztworze methemoglobiny (metHb). Zmiana intensywności tego pasma związana
jest ze zmianą formy Hb, jednakże jego obecność jest dowodem na brak destrukcji
grup hemowych w wyniku kontaktu Hb z taką ilością nadtlenku wodoru.
Dodatkowo, na widmach widoczne są zmiany intensywności pasm przy wartościach
długości fal: 540, 580 i 630 nm. Wzrost intensywności pasma przy długości fali 580
nm może wskazywać na utworzenie, w wyniku kontaktu hemoglobiny z nadtlenkiem

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
112
wodoru, mało stabilnej formy Hb, oksoferrylohemoglobiny (•HbFe(IV)=O) [420].
Rejestrując widma w funkcji czasu reakcji H2O2 z oksyHb widać również, że wzrost
intensywności absorbancji przy tej długości fali miał miejsce tylko w pierwszych
minutach reakcji H2O2 z oksyHb. W miarę wydłużania się czasu reakcji oksyHb
z nadtlenkiem wodoru następował spadek intensywności tego pasma, co wskazuje na
przejście mało stabilnej oksoferrylohemoglobiny w metHb. Badania spektroskopowe
pokazały, że pełne przejście oksyHb w metHb następuje w ciągu 10 minut
oddziaływania oksyHb z H2O2.
10.2. Procedura przygotowania warstwy receptorowej
czujnika i jej elektrochemiczna charakterystyka
Maksymalna elektroaktywność metaloprotein docierających do powierzchni
elektrody jest bezpośrednio związana z ich orientacją względem powierzchni
materiału przewodzącego. Zatem czynnikiem limitującym jest odległość centrów
redoks białka od powierzchni elektrody. Aby zapewnić maksymalną
elektroaktywność cząsteczek Hb docierających do przewodnika, w badaniach
wykorzystano pole magnetyczne wokół elektrody w trakcie przebiegu procesu
elektrodowego. W tym celu w roli modyfikatora powierzchni, umożliwiającego
kontrolę orientacji cząsteczek metHb, wykorzystano warstwę nanocząstek o silnych
właściwościach magnetycznych oddziałującą z paramagnetykiem (centrami
elektroaktywnymi białka). Schemat układu pomiarowego do bezpośredniej
woltamperometrycznej detekcji Hb w roztworze przedstawiony został na rysunku 44.
Hb(FeII)Hb(FeIII)
S
N
Au-EQCM/C/Fe@C Nps
e-
Rysunek 44. Schemat układu pomiarowego do detekcji Hb z roztworu.
Należy pamiętać, że jakość warstwy nanocząstek magnetycznych decyduje
o skuteczności działania takiej warstwy. Najbardziej popularną metodą tworzenia
warstwy nanocząstek magnetycznych na powierzchni elektrody jest ich adsorpcja
fizyczna. Jak powszechnie wiadomo, adsorpcja fizyczna nie prowadzi do
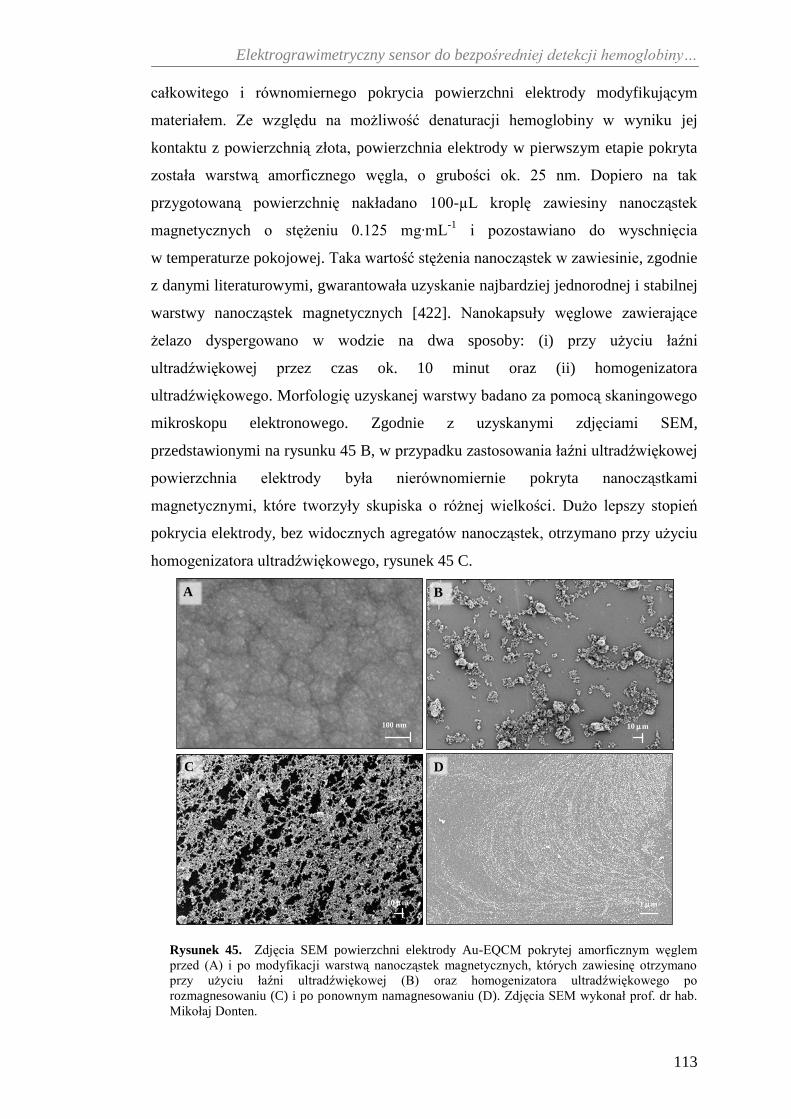
Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
113
całkowitego i równomiernego pokrycia powierzchni elektrody modyfikującym
materiałem. Ze względu na możliwość denaturacji hemoglobiny w wyniku jej
kontaktu z powierzchnią złota, powierzchnia elektrody w pierwszym etapie pokryta
została warstwą amorficznego węgla, o grubości ok. 25 nm. Dopiero na tak
przygotowaną powierzchnię nakładano 100-µL kroplę zawiesiny nanocząstek
magnetycznych o stężeniu 0.125 mg·mL-1
i pozostawiano do wyschnięcia
w temperaturze pokojowej. Taka wartość stężenia nanocząstek w zawiesinie, zgodnie
z danymi literaturowymi, gwarantowała uzyskanie najbardziej jednorodnej i stabilnej
warstwy nanocząstek magnetycznych [422]. Nanokapsuły węglowe zawierające
żelazo dyspergowano w wodzie na dwa sposoby: (i) przy użyciu łaźni
ultradźwiękowej przez czas ok. 10 minut oraz (ii) homogenizatora
ultradźwiękowego. Morfologię uzyskanej warstwy badano za pomocą skaningowego
mikroskopu elektronowego. Zgodnie z uzyskanymi zdjęciami SEM,
przedstawionymi na rysunku 45 B, w przypadku zastosowania łaźni ultradźwiękowej
powierzchnia elektrody była nierównomiernie pokryta nanocząstkami
magnetycznymi, które tworzyły skupiska o różnej wielkości. Dużo lepszy stopień
pokrycia elektrody, bez widocznych agregatów nanocząstek, otrzymano przy użyciu
homogenizatora ultradźwiękowego, rysunek 45 C.
100 nm
A
10 mm
B
10 mm
C
1 mm
D
Rysunek 45. Zdjęcia SEM powierzchni elektrody Au-EQCM pokrytej amorficznym węglem
przed (A) i po modyfikacji warstwą nanocząstek magnetycznych, których zawiesinę otrzymano
przy użyciu łaźni ultradźwiękowej (B) oraz homogenizatora ultradźwiękowego po
rozmagnesowaniu (C) i po ponownym namagnesowaniu (D). Zdjęcia SEM wykonał prof. dr hab.
Mikołaj Donten.

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
114
Na podstawie uzyskanych zdjęć SEM można stwierdzić, że stopień pokrycia
powierzchni elektrody silnie zależy od sposobu przygotowania wodnej zawiesiny
Fe@C Nps. Wykorzystanie w tym celu homogenizatora ultradźwiękowego
pozwoliło uzyskać warstwę Fe@C Nps na powierzchni elektrody o zdecydowanie
lepszych parametrach. Procedura przygotowywania takiej zawiesiny polegała na
zastosowaniu 1-sekundowych pulsów o mocy 80 W, przerywanych pulsami
„spoczynkowymi” przez 90 s. Oczywiście przed procesem rozpraszania nanocząstki
Fe@C poddano samoistnemu rozmagnesowaniu poprzez zastosowanie ciągłego,
intensywnego strumienia ultradźwiękowego, o mocy 50 W, z jednocześnie
zanikającym polem magnetycznym. Powstała w opisany sposób jednorodna
zawiesina nanocząstek magnetycznych wykazywała zmniejszoną skłonność do
aglomeracji, co w konsekwencji pozwoliło na uzyskanie cienkiej i stosunkowo
równomiernej warstwy ferromagnetycznego modyfikatora na powierzchni elektrody.
Ponowne namagnesowanie Fe@C Nps następowało w wyniku przyłożenia magnesu
do elektrody. Wprowadzenie zewnętrznego źródła pola magnetycznego skutkowało
reorganizacją warstwy nanokapsuł, które układały się wzdłuż linii sił pola
magnetycznego (rysunek 45 D).
Elektrochemicznej charakterystyki uzyskanej warstwy receptorowej
(Au-EQCM/C/Fe@C Nps) dokonano przy użyciu elektrochemicznej spektroskopii
impedancyjnej. Pomiary prowadzono w obecności próbnika redoks, którym była
równomolowa (5 mM) mieszanina heksacjanożelazianu(III) i (II) potasu w 0.02 M
buforze PBS o pH 6.0, przy potencjale formalnym układu redoks [Fe(CN)6]3-/4-
wynoszącym 0.243 V. Przykładowe wykresy Nyquist’a dla chemicznie
niezmodyfikowanego kryształu kwarcu (Au-EQCM) oraz kryształu pokrytego
warstwą amorficznego węgla (Au-EQCM/C) i następnie warstwą nanocząstek
magnetycznych (Au-EQCM/C/Fe@C Nps) przedstawiono na rysunku 46. Jak
wynika z uzyskanych widm EIS, pokrycie złota napylonego na kryształ kwarcu
warstwą amorficznego węgla zdecydowanie utrudnia wymianę elektronów, czego
dowodem jest wzrost półkola na widmie Nyquist’a. Porównując ze sobą widma EIS
uzyskane dla chemicznie niezmodyfikowanej elektrody złotej i elektrody wykonanej
z węgla szklistego można stwierdzić, że utworzona warstwa węgla była zwarta,
cienka i szczelnie pokrywała modyfikowaną powierzchnię.
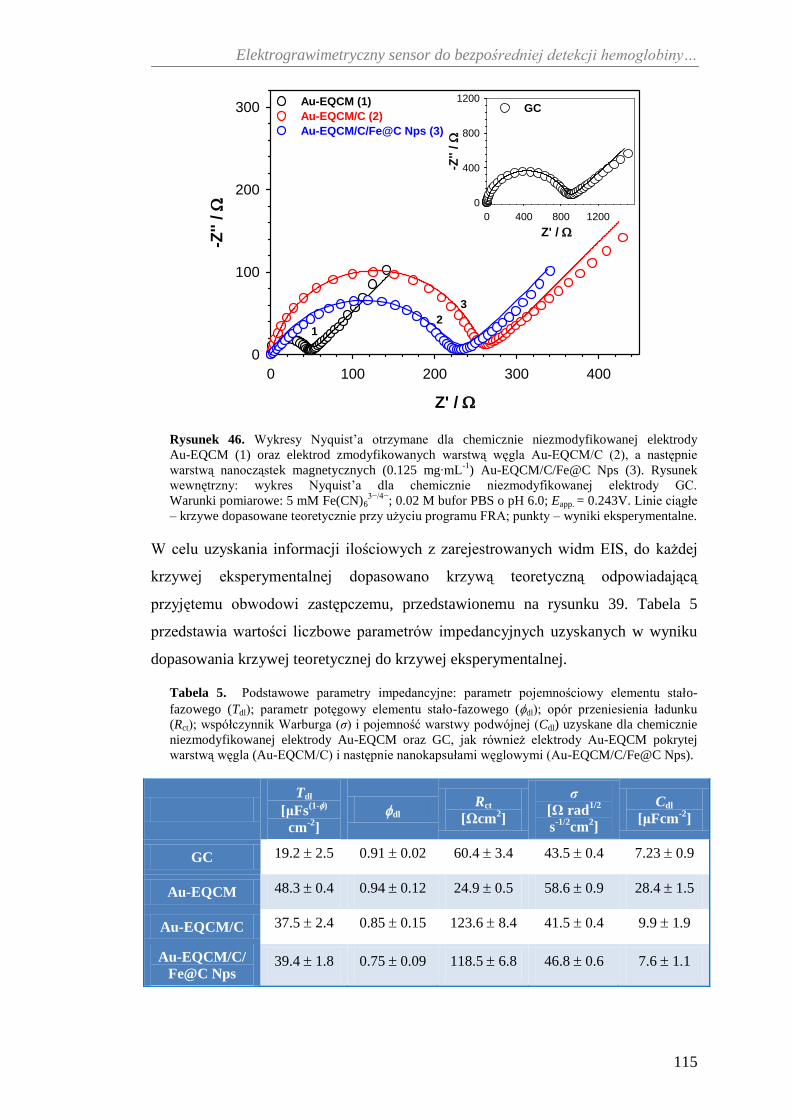
Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
115
Z' /
0 100 200 300 400
-Z'' /
0
100
200
300 Au-EQCM (1)
Au-EQCM/C (2)
Au-EQCM/C/Fe@C Nps (3)
Z' /
0 400 800 1200
-Z'' /
0
400
800
1200GC
12
3
Rysunek 46. Wykresy Nyquist’a otrzymane dla chemicznie niezmodyfikowanej elektrody
Au-EQCM (1) oraz elektrod zmodyfikowanych warstwą węgla Au-EQCM/C (2), a następnie
warstwą nanocząstek magnetycznych (0.125 mg·mL-1
) Au-EQCM/C/Fe@C Nps (3). Rysunek
wewnętrzny: wykres Nyquist’a dla chemicznie niezmodyfikowanej elektrody GC.
Warunki pomiarowe: 5 mM Fe(CN)63−/4−
; 0.02 M bufor PBS o pH 6.0; Eapp. = 0.243V. Linie ciągłe
– krzywe dopasowane teoretycznie przy użyciu programu FRA; punkty – wyniki eksperymentalne.
W celu uzyskania informacji ilościowych z zarejestrowanych widm EIS, do każdej
krzywej eksperymentalnej dopasowano krzywą teoretyczną odpowiadającą
przyjętemu obwodowi zastępczemu, przedstawionemu na rysunku 39. Tabela 5
przedstawia wartości liczbowe parametrów impedancyjnych uzyskanych w wyniku
dopasowania krzywej teoretycznej do krzywej eksperymentalnej.
Tabela 5. Podstawowe parametry impedancyjne: parametr pojemnościowy elementu stało-
fazowego (Tdl); parametr potęgowy elementu stało-fazowego (dl); opór przeniesienia ładunku
(Rct); współczynnik Warburga (σ) i pojemność warstwy podwójnej (Cdl) uzyskane dla chemicznie
niezmodyfikowanej elektrody Au-EQCM oraz GC, jak również elektrody Au-EQCM pokrytej
warstwą węgla (Au-EQCM/C) i następnie nanokapsułami węglowymi (Au-EQCM/C/Fe@C Nps).
Tdl
[μFs(1-)
cm-2
]
dl Rct
[Ωcm2]
σ
[Ω rad1/2
s-1/2
cm2]
Cdl
[μFcm-2
]
GC 19.2 2.5 0.91 0.02 60.4 3.4 43.5 0.4 7.23 0.9
Au-EQCM 48.3 0.4 0.94 0.12 24.9 0.5 58.6 0.9 28.4 1.5
Au-EQCM/C 37.5 2.4 0.85 0.15 123.6 8.4 41.5 0.4 9.9 1.9
Au-EQCM/C/
Fe@C Nps 39.4 1.8 0.75 0.09 118.5 6.8 46.8 0.6 7.6 1.1

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
116
Jednym z tych parametrów jest parametr Rct opisujący wartość oporności
przeniesienia ładunku. Wartość tego parametru, po modyfikacji elektrody warstwą
amorficznego węgla, początkowo znacząco wzrasta w stosunku do wartości
uzyskanej dla chemicznie niezmodyfikowanego kryształu kwarcu, a następnie
w wyniku dalszej modyfikacji nanokapsułami węglowymi nieznacznie spada.
Jednorodność i szczelność warstwy napylonego węgla potwierdza nieznaczna
zmiana wartości potęgowego elementu stało-fazowego (dl). Tylko dla idealnych
monokrystalicznych powierzchni elektrod jego wartość jest równa jedności [423].
Jak widać z uzyskanych danych przedstawionych w tabeli 5, wartość tego parametru
osiąga minimum dla elektrody Au-EQCM z napylonym węglem i nanokapsułami
węglowymi. Należy jednak podkreślić, że spadek wartości parametru dl stanowi
jedynie 20% jego początkowej wartości, uzyskanej dla chemicznie
niezmodyfikowanego kryształu kwarcu. Fakt ten wskazuje na równomierne pokrycie
elektrody zarówno warstwą węgla, jak również warstwą ferromagnetyka.
Dodatkowo, modyfikacja elektrody miała również niewielki wpływ na szybkość
transportu próbnika redoks, gdyż wartość parametru Warburga tylko nieznacznie
zmalała w porównaniu do wartości dla niezmodyfikowanej elektrody.
10.3. Elektrochemiczna i grawimetryczna charakterystyka
sensora do bezpośredniej detekcji hemoglobiny
W celu otrzymania informacji o wpływie pola magnetycznego
i ferromagnetycznego modyfikatora powierzchni elektrody na elektrochemiczny
sygnał paramagnetycznej formy hemoglobiny, przeprowadzono pomiary
woltamperometryczne w obecności i nieobecności zewnętrznego źródła pola
magnetycznego. Uzyskane wyniki, przedstawione na rysunku 47, stanowią
niezaprzeczalny dowód, że obecność pola magnetycznego nie tylko zwiększa
intensywność rejestrowanego sygnału prądowego, ale również powoduje jego
przesunięcie o ok. 120 mV w kierunku bardziej dodatnich wartości potencjału.
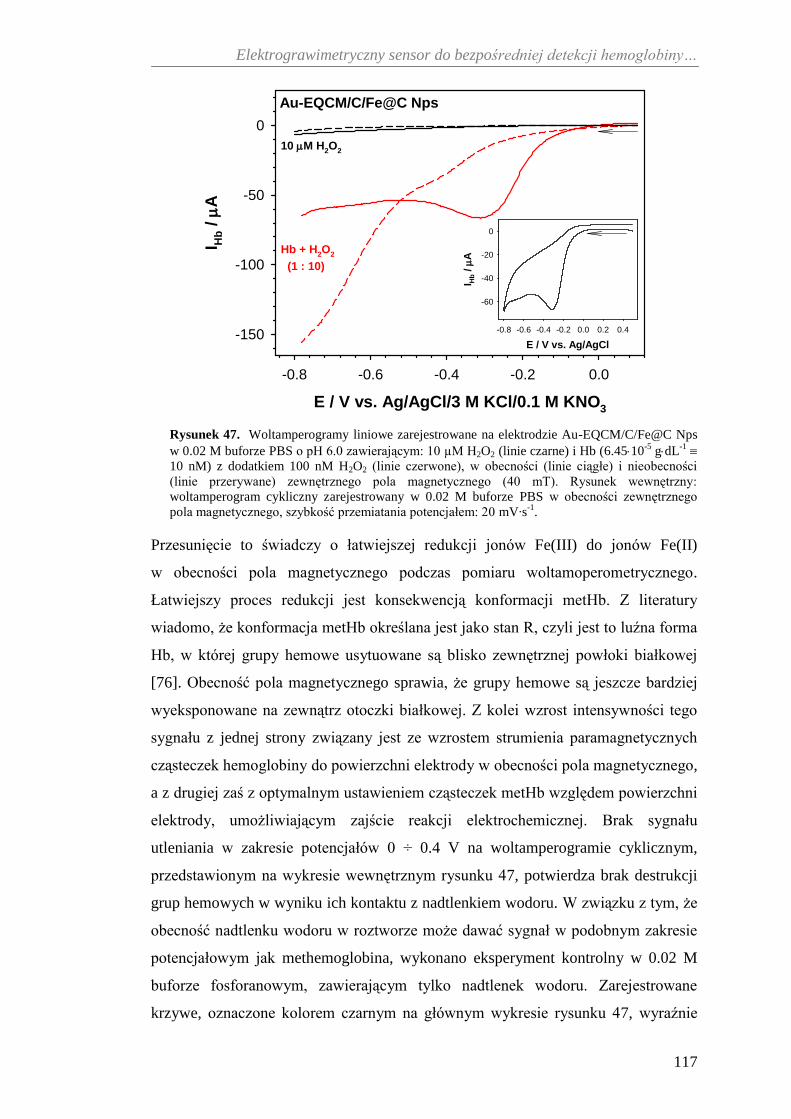
Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
117
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.8 -0.6 -0.4 -0.2 0.0
I Hb /
A
-150
-100
-50
0
Au-EQCM/C/Fe@C Nps
E / V vs. Ag/AgCl
-0.8 -0.6 -0.4 -0.2 0.0 0.2 0.4
I Hb /
A
-60
-40
-20
0
10 M H2O
2
Hb + H2O
2
(1 : 10)
Rysunek 47. Woltamperogramy liniowe zarejestrowane na elektrodzie Au-EQCM/C/Fe@C Nps
w 0.02 M buforze PBS o pH 6.0 zawierającym: 10 µM H2O2 (linie czarne) i Hb (6.4510-5
gdL-1
10 nM) z dodatkiem 100 nM H2O2 (linie czerwone), w obecności (linie ciągłe) i nieobecności
(linie przerywane) zewnętrznego pola magnetycznego (40 mT). Rysunek wewnętrzny:
woltamperogram cykliczny zarejestrowany w 0.02 M buforze PBS w obecności zewnętrznego
pola magnetycznego, szybkość przemiatania potencjałem: 20 mV·s-1
.
Przesunięcie to świadczy o łatwiejszej redukcji jonów Fe(III) do jonów Fe(II)
w obecności pola magnetycznego podczas pomiaru woltamoperometrycznego.
Łatwiejszy proces redukcji jest konsekwencją konformacji metHb. Z literatury
wiadomo, że konformacja metHb określana jest jako stan R, czyli jest to luźna forma
Hb, w której grupy hemowe usytuowane są blisko zewnętrznej powłoki białkowej
[76]. Obecność pola magnetycznego sprawia, że grupy hemowe są jeszcze bardziej
wyeksponowane na zewnątrz otoczki białkowej. Z kolei wzrost intensywności tego
sygnału z jednej strony związany jest ze wzrostem strumienia paramagnetycznych
cząsteczek hemoglobiny do powierzchni elektrody w obecności pola magnetycznego,
a z drugiej zaś z optymalnym ustawieniem cząsteczek metHb względem powierzchni
elektrody, umożliwiającym zajście reakcji elektrochemicznej. Brak sygnału
utleniania w zakresie potencjałów 0 ÷ 0.4 V na woltamperogramie cyklicznym,
przedstawionym na wykresie wewnętrznym rysunku 47, potwierdza brak destrukcji
grup hemowych w wyniku ich kontaktu z nadtlenkiem wodoru. W związku z tym, że
obecność nadtlenku wodoru w roztworze może dawać sygnał w podobnym zakresie
potencjałowym jak methemoglobina, wykonano eksperyment kontrolny w 0.02 M
buforze fosforanowym, zawierającym tylko nadtlenek wodoru. Zarejestrowane
krzywe, oznaczone kolorem czarnym na głównym wykresie rysunku 47, wyraźnie
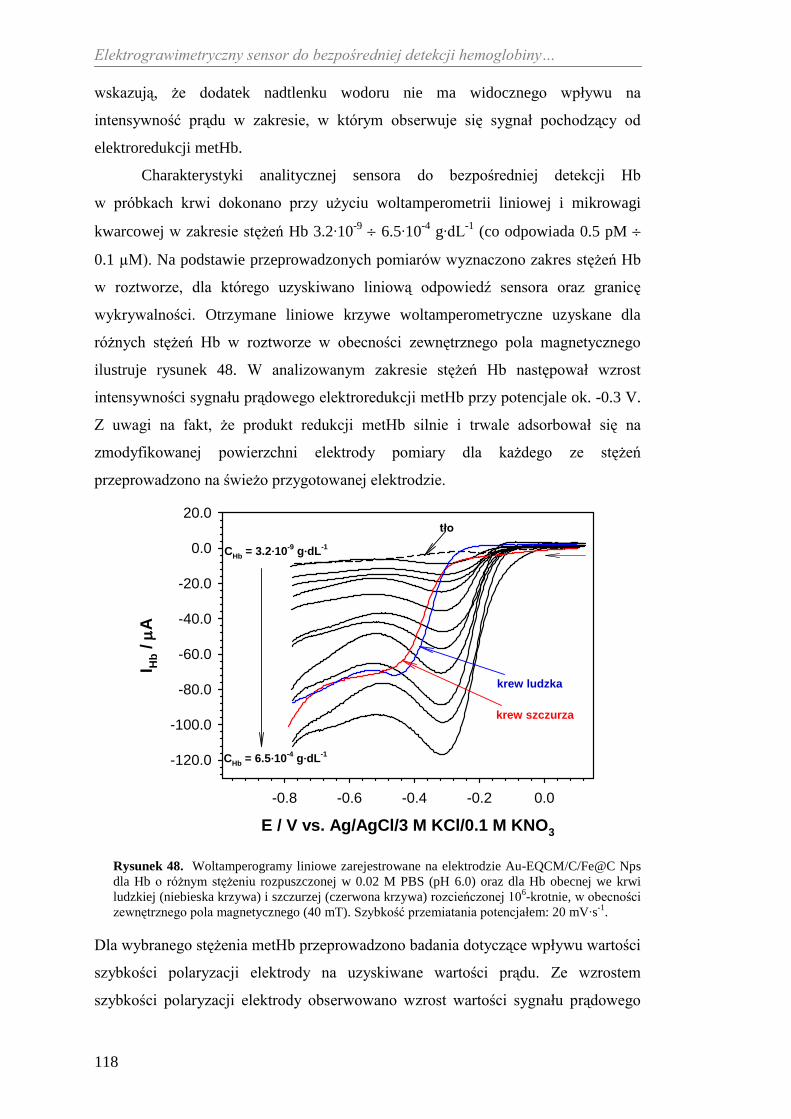
Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
118
wskazują, że dodatek nadtlenku wodoru nie ma widocznego wpływu na
intensywność prądu w zakresie, w którym obserwuje się sygnał pochodzący od
elektroredukcji metHb.
Charakterystyki analitycznej sensora do bezpośredniej detekcji Hb
w próbkach krwi dokonano przy użyciu woltamperometrii liniowej i mikrowagi
kwarcowej w zakresie stężeń Hb 3.2·10-9
6.5·10-4
g·dL-1
(co odpowiada 0.5 pM
0.1 µM). Na podstawie przeprowadzonych pomiarów wyznaczono zakres stężeń Hb
w roztworze, dla którego uzyskiwano liniową odpowiedź sensora oraz granicę
wykrywalności. Otrzymane liniowe krzywe woltamperometryczne uzyskane dla
różnych stężeń Hb w roztworze w obecności zewnętrznego pola magnetycznego
ilustruje rysunek 48. W analizowanym zakresie stężeń Hb następował wzrost
intensywności sygnału prądowego elektroredukcji metHb przy potencjale ok. -0.3 V.
Z uwagi na fakt, że produkt redukcji metHb silnie i trwale adsorbował się na
zmodyfikowanej powierzchni elektrody pomiary dla każdego ze stężeń
przeprowadzono na świeżo przygotowanej elektrodzie.
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.8 -0.6 -0.4 -0.2 0.0
I Hb /
A
-120.0
-100.0
-80.0
-60.0
-40.0
-20.0
0.0
20.0
krew szczurza
tło
krew ludzka
CHb
= 3.210-9 gdL
-1
CHb
= 6.510-4 gdL
-1
Rysunek 48. Woltamperogramy liniowe zarejestrowane na elektrodzie Au-EQCM/C/Fe@C Nps
dla Hb o różnym stężeniu rozpuszczonej w 0.02 M PBS (pH 6.0) oraz dla Hb obecnej we krwi
ludzkiej (niebieska krzywa) i szczurzej (czerwona krzywa) rozcieńczonej 106-krotnie, w obecności
zewnętrznego pola magnetycznego (40 mT). Szybkość przemiatania potencjałem: 20 mV·s-1
.
Dla wybranego stężenia metHb przeprowadzono badania dotyczące wpływu wartości
szybkości polaryzacji elektrody na uzyskiwane wartości prądu. Ze wzrostem
szybkości polaryzacji elektrody obserwowano wzrost wartości sygnału prądowego

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
119
i jego przesuniecie w kierunku bardziej ujemnych wartości potencjału. Wartość
prądu katodowego wzrastała liniowo, proporcjonalnie do pierwiastka kwadratowego
z szybkości polaryzacji, w zakresie od 5 do 500 mV·s-1
. Współczynnik korelacji
zależności I = f(v0.5
) wynosił 0.997. Uzyskane wyniki potwierdziły, że proces
redukcji metHb na zmodyfikowanej powierzchni elektrody
(Au-EQCM/C/Fe@C Nps), w przypadku niskich stężeń hemoglobiny w roztworze,
w obecności zewnętrznego pola magnetycznego, kontrolowany jest transportem
depolaryzatora do powierzchni elektrody. Porównanie masy hemoglobiny
zaadsorbowanej na powierzchni elektrody po procesie elektroredukcji (wyznaczonej
z pomiarów QCM) z wartością ładunku procesu elektrodowego pozwoliło
wyznaczyć liczbę elektronów biorących udział w procesie redukcji metHb.
Z uzyskanych danych wynika, że pojedyncza grupa hemowa podczas procesu
elektrodowego wymienia z powierzchnią przewodnika jeden elektron.
Zastosowanie elektrochemicznej mikrowagi kwarcowej pozwoliło na
jednoczesny pomiar zmian natężenia prądu w funkcji przykładanego potencjału, jak
i zmian częstotliwości drgań kryształu kwarcu w czasie przebiegu procesu
elektrodowego. Przykładowe krzywe ilustrujące zmiany częstotliwości drgań
kryształu kwarcu przedstawione zostały na rysunku 49.
czas / s
0 20 40 60 80 100 120 140
f
/ H
z
-120
-100
-80
-60
-40
-20
0 CHb
= 3.210-9 gdL
-1
krew szczurza
krew ludzka
CHb
= 6.510-4
gdL-1
Rysunek 49. Zmiany częstotliwości drgań kryształu kwarcu zarejestrowane na elektrodzie
Au-EQCM/C/Fe@C Nps w trakcie redukcji Hb rozpuszczonej w buforze PBS o pH 6.0 oraz Hb
obecnej we krwi ludzkiej (niebieska krzywa) i szczurzej (czerwona krzywa) rozcieńczonej
106-krotnie, w obecności zewnętrznego pola magnetycznego (40 mT).
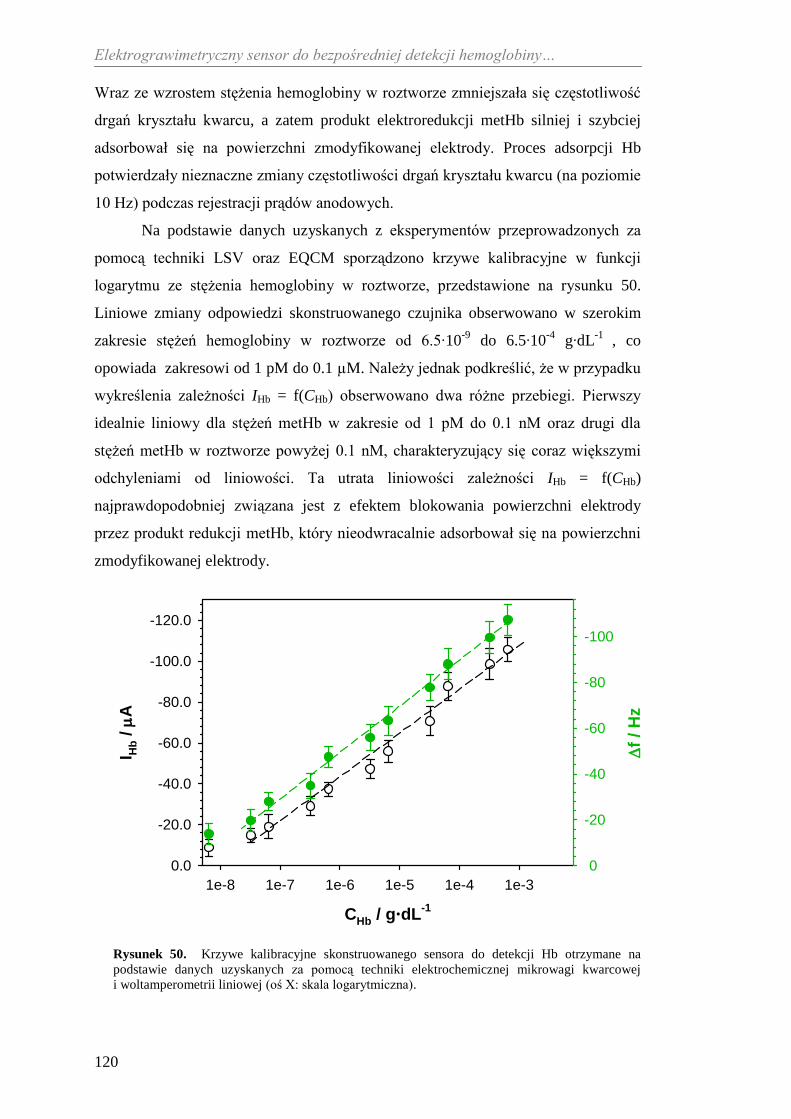
Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
120
Wraz ze wzrostem stężenia hemoglobiny w roztworze zmniejszała się częstotliwość
drgań kryształu kwarcu, a zatem produkt elektroredukcji metHb silniej i szybciej
adsorbował się na powierzchni zmodyfikowanej elektrody. Proces adsorpcji Hb
potwierdzały nieznaczne zmiany częstotliwości drgań kryształu kwarcu (na poziomie
10 Hz) podczas rejestracji prądów anodowych.
Na podstawie danych uzyskanych z eksperymentów przeprowadzonych za
pomocą techniki LSV oraz EQCM sporządzono krzywe kalibracyjne w funkcji
logarytmu ze stężenia hemoglobiny w roztworze, przedstawione na rysunku 50.
Liniowe zmiany odpowiedzi skonstruowanego czujnika obserwowano w szerokim
zakresie stężeń hemoglobiny w roztworze od 6.5·10-9
do 6.5·10-4
g·dL-1
, co
opowiada zakresowi od 1 pM do 0.1 µM. Należy jednak podkreślić, że w przypadku
wykreślenia zależności IHb = f(CHb) obserwowano dwa różne przebiegi. Pierwszy
idealnie liniowy dla stężeń metHb w zakresie od 1 pM do 0.1 nM oraz drugi dla
stężeń metHb w roztworze powyżej 0.1 nM, charakteryzujący się coraz większymi
odchyleniami od liniowości. Ta utrata liniowości zależności IHb = f(CHb)
najprawdopodobniej związana jest z efektem blokowania powierzchni elektrody
przez produkt redukcji metHb, który nieodwracalnie adsorbował się na powierzchni
zmodyfikowanej elektrody.
CHb / gdL-1
1e-8 1e-7 1e-6 1e-5 1e-4 1e-3
I Hb /
A
-120.0
-100.0
-80.0
-60.0
-40.0
-20.0
0.0
f
/ H
z-100
-80
-60
-40
-20
0
Rysunek 50. Krzywe kalibracyjne skonstruowanego sensora do detekcji Hb otrzymane na
podstawie danych uzyskanych za pomocą techniki elektrochemicznej mikrowagi kwarcowej
i woltamperometrii liniowej (oś X: skala logarytmiczna).

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
121
Granica wykrywalności hemoglobiny została wyznaczona z dolnego zakresu
prostoliniowości krzywej kalibracyjnej, zgodnie z równaniem:
bb 3LOD ζY (31)
gdzie: Yb oznacza średnią wartość sygnału ślepej próby (0.02 M PBS o pH 6.0),
natomiast b to odchylenie standardowe dla ślepej próby z 5 pomiarów. Wyznaczona
w ten sposób granica wykrywalności wynosiła 5·10-9
g·dL-1
(0.7 pM). W przypadku
wszystkich stężeń metHb z zakresu liniowego krzywej kalibracyjnej powtarzalność
wyników była bardzo dobra, a względne odchylenie standardowe wynosiło około
9.5% (n = 5). Sprawdzono również powtarzalność wyników uzyskanych dla tego
samego stężenia Hb, ale na sześciu różnych, niezależnie zmodyfikowanych
elektrodach i w tym przypadku względne odchylenie standardowe wynosiło około
5.2%.
10.4. Funkcjonalność sensora do bezpośredniej detekcji Hb
w próbkach naturalnych
Funkcjonalność zaproponowanego sensora do bezpośredniej
elektrochemicznej detekcji hemoglobiny w roztworze sprawdzono względem próbek
naturalnych, ludzkiej i szczurzej krwi. Próbki krwi otrzymano z Warszawskiego
Uniwersytetu Medycznego od prof. dr hab. I.P. Grudzińskiego. Hemoglobina jest
głównym składnikiem czerwonych krwinek (RBC, elementów morfotycznych krwi).
W pierwszej kolejności należało przeprowadzić hemolizę krwi w celu uwolnienia Hb
z RBC, w wyniku której następuje przejście hemoglobiny do osocza krwi na skutek
zniszczenia erytrocytów. Hemoliza może być wywołana różnego typu czynnikami,
np.: toksykantami, obniżeniem pH krwi lub też w wyniku kontaktu próbki krwi
z hipotonicznym roztworem (o niskiej zawartości jonów). W prowadzonych
badaniach hemolizę krwi przeprowadzono poprzez jej 106-krotne rozcieńczenie
0.02 M buforem PBS o pH 6.0 oraz późniejszą 20-minutową inkubację. Postęp
procesu hemolizy kontrolowano za pomocą skaningowego mikroskopu
elektronowego. Na podstawie uzyskanych zdjęć SEM, przedstawionych na rysunku
51, można stwierdzić, że czas 10 minut nie był wystarczający do całkowitego
uwolnienia hemoglobiny z czerwonych krwinek. Na zdjęciu SEM (rysunek 51A)
wciąż widoczne są czerwone krwinki. Optymalny czas hemolizy, pozwalający na
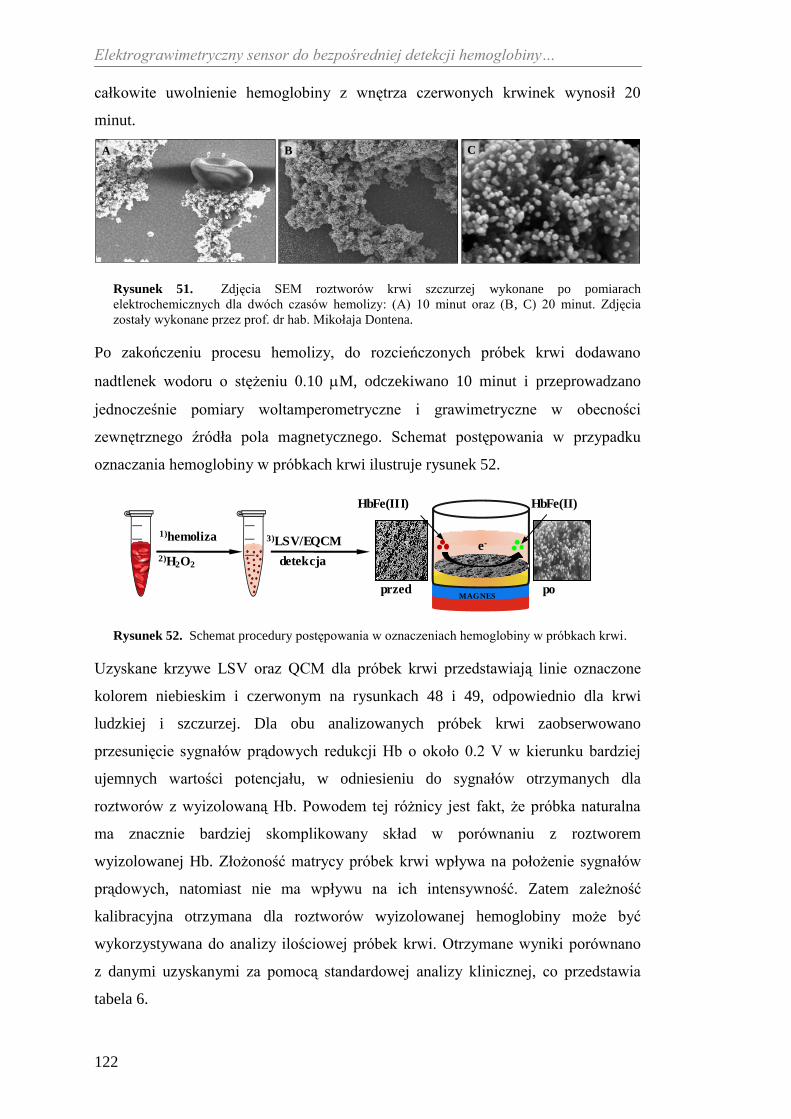
Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
122
całkowite uwolnienie hemoglobiny z wnętrza czerwonych krwinek wynosił 20
minut.
A B C
Rysunek 51. Zdjęcia SEM roztworów krwi szczurzej wykonane po pomiarach
elektrochemicznych dla dwóch czasów hemolizy: (A) 10 minut oraz (B, C) 20 minut. Zdjęcia
zostały wykonane przez prof. dr hab. Mikołaja Dontena.
Po zakończeniu procesu hemolizy, do rozcieńczonych próbek krwi dodawano
nadtlenek wodoru o stężeniu 0.10 mM, odczekiwano 10 minut i przeprowadzano
jednocześnie pomiary woltamperometryczne i grawimetryczne w obecności
zewnętrznego źródła pola magnetycznego. Schemat postępowania w przypadku
oznaczania hemoglobiny w próbkach krwi ilustruje rysunek 52.
e-1)hemoliza
2)H2O2
3)LSV/EQCM
detekcja
przed po
HbFe(III) HbFe(II)
MAGNES
Rysunek 52. Schemat procedury postępowania w oznaczeniach hemoglobiny w próbkach krwi.
Uzyskane krzywe LSV oraz QCM dla próbek krwi przedstawiają linie oznaczone
kolorem niebieskim i czerwonym na rysunkach 48 i 49, odpowiednio dla krwi
ludzkiej i szczurzej. Dla obu analizowanych próbek krwi zaobserwowano
przesunięcie sygnałów prądowych redukcji Hb o około 0.2 V w kierunku bardziej
ujemnych wartości potencjału, w odniesieniu do sygnałów otrzymanych dla
roztworów z wyizolowaną Hb. Powodem tej różnicy jest fakt, że próbka naturalna
ma znacznie bardziej skomplikowany skład w porównaniu z roztworem
wyizolowanej Hb. Złożoność matrycy próbek krwi wpływa na położenie sygnałów
prądowych, natomiast nie ma wpływu na ich intensywność. Zatem zależność
kalibracyjna otrzymana dla roztworów wyizolowanej hemoglobiny może być
wykorzystywana do analizy ilościowej próbek krwi. Otrzymane wyniki porównano
z danymi uzyskanymi za pomocą standardowej analizy klinicznej, co przedstawia
tabela 6.

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
123
Tabela 6. Porównanie danych zawartości Hb w próbkach naturalnych uzyskanych na podstawie
analizy klinicznej i proponowanego sensora.
Krew ludzka [g·dL-1
] Krew szczurza [g·dL-1
]
Analiza kliniczna 14.5 0.2 13.2 0.4
Bezpośrednia elektrochemiczna detekcja
LSV 13.9 0.8 13.1 0.6
EQCM 13.1 1.4 13.2 1.2
Analiza kliniczna zawartości hemoglobiny w próbkach krwi ludzkiej i szczurzej
przeprowadzona została na Warszawskim Uniwersytecie Medycznym przez grupę
prof. dr hab. I.P. Grudzińskiego. Wyniki zaprezentowane w tabeli 5 są wartością
średnią z trzech niezależnych pomiarów dla różnych próbek krwi. Oznaczenia
stężenia hemoglobiny w badanych próbkach krwi dokonano metodą
kolorymetryczną przy długości fali 546 nm, przy użyciu kompaktowego analizatora
hematologicznego BC-2800 VET (Mindray) i systemu ADVIA 120 Hematology
(Siemens) dla całej próbki krwi ludzkiej i szczurzej. Otrzymane wyniki pokazują, że
stężenia hemoglobiny w próbkach naturalnych wyznaczone za pomocą
zaproponowanego sensora oraz analizatora klinicznego są w bardzo dobrej
zgodności.
10.5. Podsumowanie
Opisany w niniejszym rozdziale sensor do bezpośredniej elektrochemicznej
detekcji hemoglobiny pozwolił z dużą dokładnością, w prosty i szybki sposób
oznaczyć zawartość Hb w skomplikowanych matrycach, takich jak krew ludzka
i szczurza. Jego zasada działania oparta była na oddziaływaniu paramagnetycznego
białka (hemoglobiny) z ferromagnetycznym składnikiem warstwy receptorowej
sensora w obecności zewnętrznego źródła pola magnetycznego. Zastosowanie
nanokapsuł węglowych zawierających żelazo oraz zewnętrznego źródła pola
magnetycznego zapewniło odpowiednią, do zajścia procesu elektrodowego,
orientację cząsteczek Hb względem powierzchni elektrody. Dużą zaletą
zaproponowanego sensora był brak konieczności specjalnego przygotowania próbek
krwi przed pomiarem. Ponadto przedstawione wyniki pokazują, że proponowany
sensor pozwolił na oznaczenie zawartości Hb w analizowanej próbce z dużą

Elektrograwimetryczny sensor do bezpośredniej detekcji hemoglobiny…
124
czułością, dobrą powtarzalnością i bardzo niską granicą wykrywalności, rzędu pM.
Należy podkreślić, że połączenie techniki LSV oraz QCM pozwoliło na uzyskanie
bardziej kompletnych informacji na temat przebiegu procesu detekcji. Z kolei analiza
zdjęć uzyskanych za pomocą skaningowego mikroskopu elektronowego wykazała,
że proces adsorpcji cząsteczek hemoglobiny ma miejsce głównie na
ferromagnetycznym modyfikatorze powierzchni elektrody. Opracowana procedura
może być z powodzeniem stosowana do analizy różnego typu paramagnetycznych
białek, zarówno w buforze, jak i w płynach ustrojowych.

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
125
11. Wpływ pola magnetycznego na aktywność
elektrochemiczną i katalityczną ceruloplazminy
Jednoczesna elektrochemiczna analiza kilku białek zawartych we krwi jest
niezwykle trudna do osiągnięcia, gdyż często produkty ich procesów elektrodowych,
jak również one same łatwo adsorbują się na powierzchni elektrody, blokując ją
i uniemożliwiając tym samym dalsze pomiary. Dodatkowo, analiza kilku białek przy
użyciu tego samego czujnika woltamperometrycznego jest możliwa tylko wtedy, gdy
sygnały prądowe pochodzące od procesów elektrodowych poszczególnych białek
różnią się położeniem na osi potencjału. Badania woltamperometryczne
hemoglobiny pokazały, że odpowiednie rozcieńczenie roztworu (CHb < 0.1 nM)
pozwala zminimalizować na tyle efekt blokowania powierzchni elektrody przez
produkt redukcji metHb, że możliwe jest wykonanie kolejnej analizy, bez
konieczności użycia świeżo przygotowanego czujnika. Zatem kolejny etap badań
dotyczył jednoczesnej analizy ilościowej ceruloplazminy i hemoglobiny w trakcie
pojedynczego pomiaru, bez konieczności specjalnego przygotowania analizowanej
próbki krwi. Taka analiza jest możliwa, z uwagi na fakt, że w skład ich struktury
wchodzą inne jony metali (hemoglobina zawiera jony Fe, zaś ceruloplazmina jony
Cu). Zatem, zgodnie z doniesieniami literaturowymi, sygnały prądowe Hb i Cp
pojawiają się w zupełnie innych miejscach na skali potencjałowej. Dodatkowo,
obecne w warstwie receptorowej (Au-EQCM/C/Fe@C Nps) magnetyczne
nanokapsuły węglowe w połączeniu z polem magnetycznym powinny zapewniać
maksymalną elektroaktywność cząsteczek białek docierających do powierzchni
czujnika.
11.1. Analityczna charakterystyka czujnika
Analogicznie, jak w przypadku czujnika do detekcji hemoglobiny, przed
przystąpieniem do badań próbek krwi, w pierwszej kolejności dokonano
charakterystyki analitycznej czujnika. Uściślając, wyznaczono: zakres pracy czujnika
oraz osiąganą przy jego użyciu granicę wykrywalności. Ta część badań została
wykonana na roztworach zawierających tylko ceruloplazminę w szerokim zakresie
stężeń tego białka od 2 do 500 mg·dL-1
. Ceruloplazmina, jak i inne białka, wykazuje
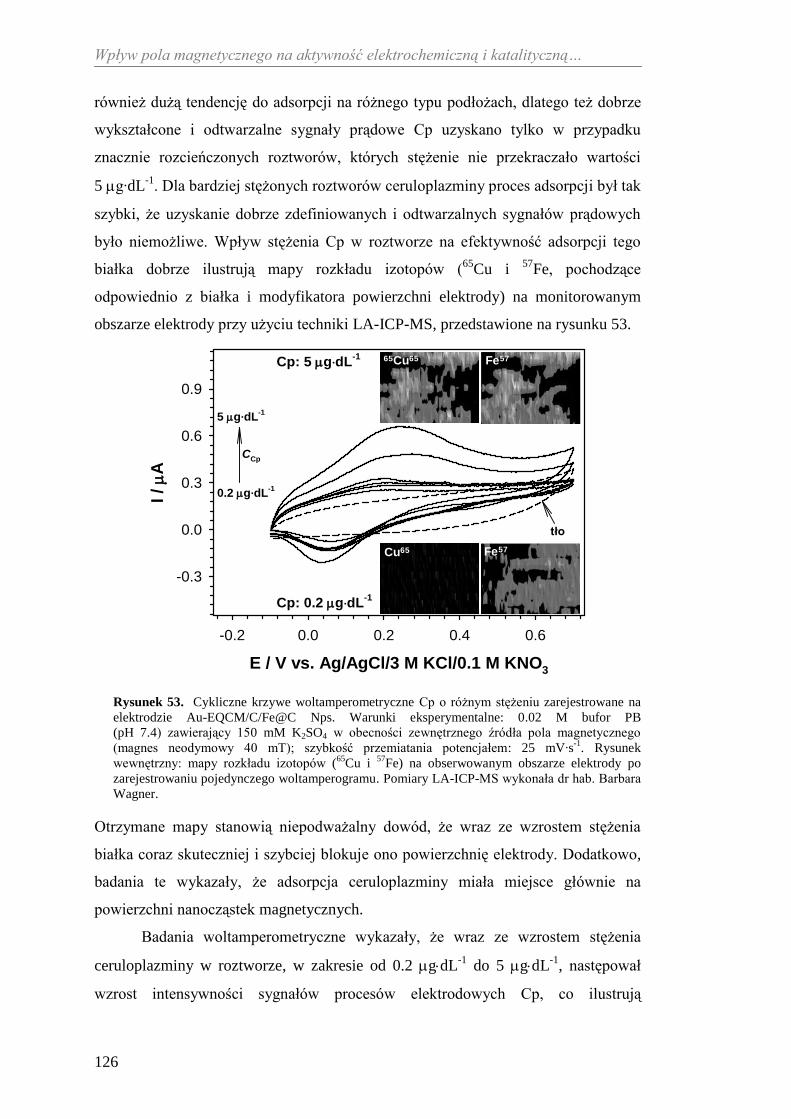
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
126
również dużą tendencję do adsorpcji na różnego typu podłożach, dlatego też dobrze
wykształcone i odtwarzalne sygnały prądowe Cp uzyskano tylko w przypadku
znacznie rozcieńczonych roztworów, których stężenie nie przekraczało wartości
5 mg·dL-1
. Dla bardziej stężonych roztworów ceruloplazminy proces adsorpcji był tak
szybki, że uzyskanie dobrze zdefiniowanych i odtwarzalnych sygnałów prądowych
było niemożliwe. Wpływ stężenia Cp w roztworze na efektywność adsorpcji tego
białka dobrze ilustrują mapy rozkładu izotopów (65
Cu i 57
Fe, pochodzące
odpowiednio z białka i modyfikatora powierzchni elektrody) na monitorowanym
obszarze elektrody przy użyciu techniki LA-ICP-MS, przedstawione na rysunku 53.
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.2 0.0 0.2 0.4 0.6
I /
A
-0.3
0.0
0.3
0.6
0.9
tło
5 gdL-1
0.2 gdL-1
CCp
Cp: 5 gdL-1
Cp: 0.2 gdL-1
65Cu65 Fe57
Cu65 Fe57
Rysunek 53. Cykliczne krzywe woltamperometryczne Cp o różnym stężeniu zarejestrowane na
elektrodzie Au-EQCM/C/Fe@C Nps. Warunki eksperymentalne: 0.02 M bufor PB
(pH 7.4) zawierający 150 mM K2SO4 w obecności zewnętrznego źródła pola magnetycznego
(magnes neodymowy 40 mT); szybkość przemiatania potencjałem: 25 mV·s-1
. Rysunek
wewnętrzny: mapy rozkładu izotopów (65
Cu i 57
Fe) na obserwowanym obszarze elektrody po
zarejestrowaniu pojedynczego woltamperogramu. Pomiary LA-ICP-MS wykonała dr hab. Barbara
Wagner.
Otrzymane mapy stanowią niepodważalny dowód, że wraz ze wzrostem stężenia
białka coraz skuteczniej i szybciej blokuje ono powierzchnię elektrody. Dodatkowo,
badania te wykazały, że adsorpcja ceruloplazminy miała miejsce głównie na
powierzchni nanocząstek magnetycznych.
Badania woltamperometryczne wykazały, że wraz ze wzrostem stężenia
ceruloplazminy w roztworze, w zakresie od 0.2 mgdL-1
do 5 mgdL-1
, następował
wzrost intensywności sygnałów procesów elektrodowych Cp, co ilustrują

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
127
odpowiednie krzywe woltamperometryczne na rysunku 53. Należy zaznaczyć, że
zmiany intensywności sygnałów prądowych w funkcji stężenia Cp są bardziej
widoczne w przypadku sygnałów anodowych, niż sygnałów katodowych.
Najprawdopodobniej, produkt utleniania Cp adsorbując się na powierzchni elektrody
w istotnym stopniu zaburzał przebieg reakcji katodowej. Ponadto kształt piku
anodowego (pik szeroki i rozmyty) wskazuje na złożoność tego procesu. Z literatury
wiadomo, że wszystkie centra miedziowe obecne w strukturze ceruloplazminy są
zdolne do wymiany elektronu z elektrodą, jednak szybkość tej wymiany jest ściśle
powiązana z ich otoczeniem. Ma to swoje odzwierciedlenie w wartościach
potencjałów redoks poszczególnych centrów, które znacznie się od siebie różnią.
Wartości potencjałów centrów redoks Cp przedstawione zostały w tabeli 7.
Tabela 7. Potencjały redoks poszczególnych centrów miedziowych ceruloplazminy wyznaczone
względem elektrody wodorowej (NHE) w buforze fosforanowym.
CENTRA MIEDZIOWE
T1CysHis T1Remote T1PR T2/T3
E v
s. N
HE
[mV
]
490 [424]
580 [109]
~1000 [106]
491/415 [109]
brak jonów Cl–
448 [109]
brak jonów Cl– 539/482 [109]
jony Cl– obecne w roztworze 434 [109]
jony Cl– obecne w roztworze
W celu uzyskania informacji, od którego z centrów T1 ceruloplazminy pochodzi
anodowy sygnał prądowy widoczny w zakresie potencjałów 0.08 ÷ 0.44 V,
wykonano pomiary z użyciem woltamperometrii fali prostokątnej.
Woltamperometria fali prostokątnej, z uwagi na sposób pomiaru prądu, jest
zdecydowanie bardziej czułą techniką niż woltamperometria cykliczna.
Zastosowanie tej techniki pozwoliło na rozdzielenie sygnałów prądowych
pochodzących od centrów T1 ceruloplazminy, co ilustruje rysunek 54. Sygnały
anodowe pojawiające się przy potencjałach 0.12 oraz 0.22 V względem elektrody
Ag/AgCl/3 M KCl/0.1 M KNO3 (0.40 i 0.50 V względem NHE), zgodnie z literaturą
odpowiadają sygnałom utleniania centrów miedziowych T1CysHis i T1Remote [109].
Sygnały te występują jednak przy bardziej ujemnych potencjałach w odniesieniu do
danych literaturowych przedstawionych w tabeli 7. Najprawdopodobniej, różnice
w położeniach tych sygnałów względem danych literaturowych wynikają ze zmian
konformacyjnych cząsteczki Cp zachodzących pod wpływem pola magnetycznego.
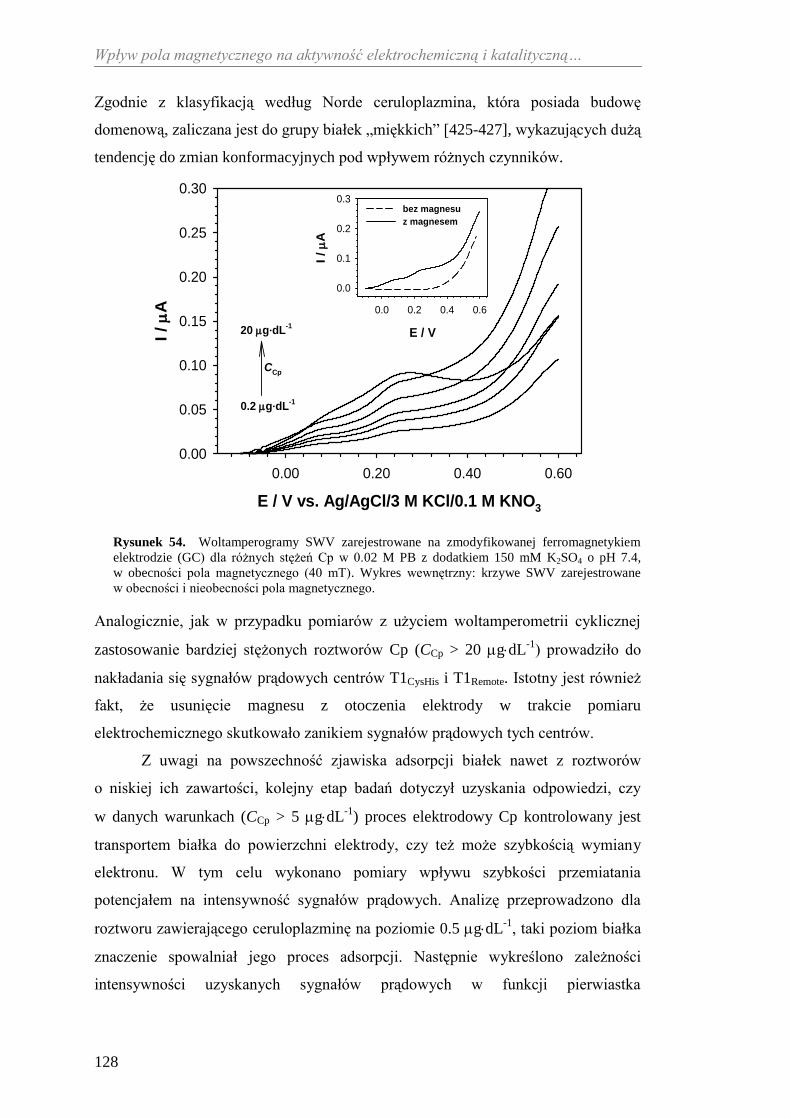
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
128
Zgodnie z klasyfikacją według Norde ceruloplazmina, która posiada budowę
domenową, zaliczana jest do grupy białek „miękkich” [425-427], wykazujących dużą
tendencję do zmian konformacyjnych pod wpływem różnych czynników.
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
0.00 0.20 0.40 0.60
I /
A
0.00
0.05
0.10
0.15
0.20
0.25
0.30
20 gdL-1
0.2 gdL-1
CCp
E / V
0.0 0.2 0.4 0.6I
/
A
0.0
0.1
0.2
0.3bez magnesu
z magnesem
Rysunek 54. Woltamperogramy SWV zarejestrowane na zmodyfikowanej ferromagnetykiem
elektrodzie (GC) dla różnych stężeń Cp w 0.02 M PB z dodatkiem 150 mM K2SO4 o pH 7.4,
w obecności pola magnetycznego (40 mT). Wykres wewnętrzny: krzywe SWV zarejestrowane
w obecności i nieobecności pola magnetycznego.
Analogicznie, jak w przypadku pomiarów z użyciem woltamperometrii cyklicznej
zastosowanie bardziej stężonych roztworów Cp (CCp > 20 mgdL-1
) prowadziło do
nakładania się sygnałów prądowych centrów T1CysHis i T1Remote. Istotny jest również
fakt, że usunięcie magnesu z otoczenia elektrody w trakcie pomiaru
elektrochemicznego skutkowało zanikiem sygnałów prądowych tych centrów.
Z uwagi na powszechność zjawiska adsorpcji białek nawet z roztworów
o niskiej ich zawartości, kolejny etap badań dotyczył uzyskania odpowiedzi, czy
w danych warunkach (CCp > 5 mgdL-1
) proces elektrodowy Cp kontrolowany jest
transportem białka do powierzchni elektrody, czy też może szybkością wymiany
elektronu. W tym celu wykonano pomiary wpływu szybkości przemiatania
potencjałem na intensywność sygnałów prądowych. Analizę przeprowadzono dla
roztworu zawierającego ceruloplazminę na poziomie 0.5 mgdL-1
, taki poziom białka
znaczenie spowalniał jego proces adsorpcji. Następnie wykreślono zależności
intensywności uzyskanych sygnałów prądowych w funkcji pierwiastka
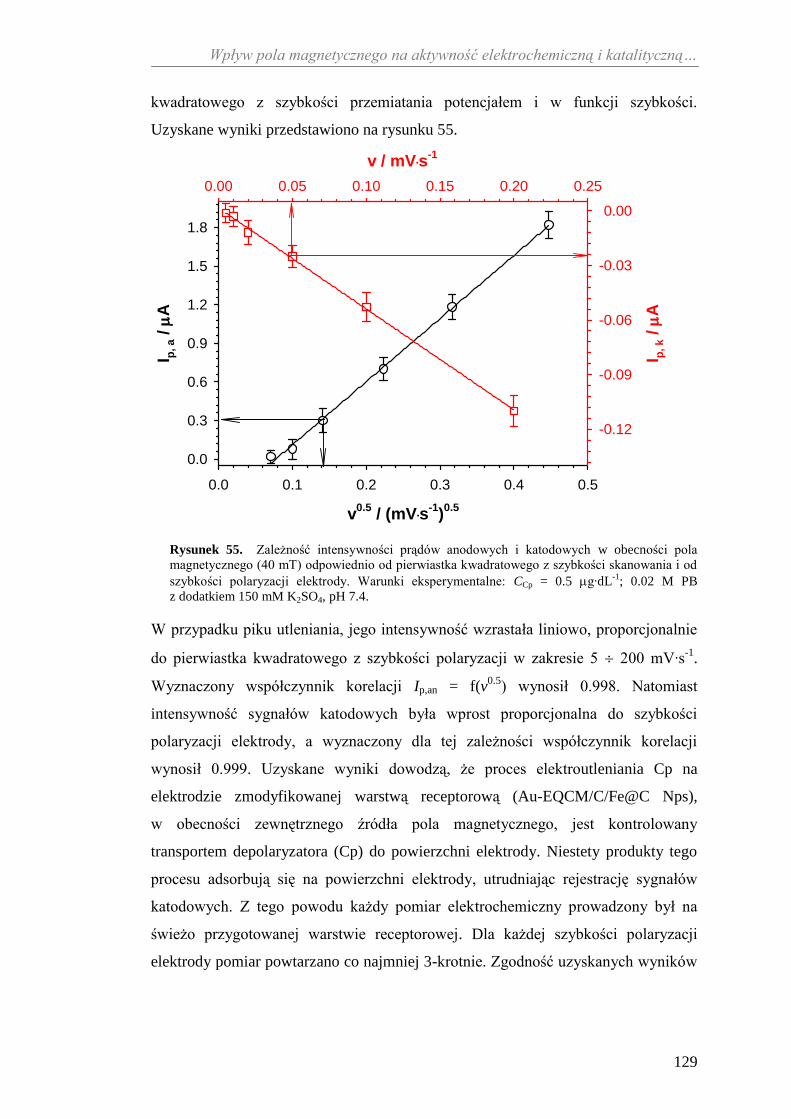
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
129
kwadratowego z szybkości przemiatania potencjałem i w funkcji szybkości.
Uzyskane wyniki przedstawiono na rysunku 55.
v0.5
/ (mVs-1
)0.5
0.0 0.1 0.2 0.3 0.4 0.5
I p, a /
A
0.0
0.3
0.6
0.9
1.2
1.5
1.8
v / mVs-1
0.00 0.05 0.10 0.15 0.20 0.25
I p, k /
A
-0.12
-0.09
-0.06
-0.03
0.00
Rysunek 55. Zależność intensywności prądów anodowych i katodowych w obecności pola
magnetycznego (40 mT) odpowiednio od pierwiastka kwadratowego z szybkości skanowania i od
szybkości polaryzacji elektrody. Warunki eksperymentalne: CCp = 0.5 mg·dL-1
; 0.02 M PB
z dodatkiem 150 mM K2SO4, pH 7.4.
W przypadku piku utleniania, jego intensywność wzrastała liniowo, proporcjonalnie
do pierwiastka kwadratowego z szybkości polaryzacji w zakresie 5 200 mV·s-1
.
Wyznaczony współczynnik korelacji Ip,an = f(v0.5
) wynosił 0.998. Natomiast
intensywność sygnałów katodowych była wprost proporcjonalna do szybkości
polaryzacji elektrody, a wyznaczony dla tej zależności współczynnik korelacji
wynosił 0.999. Uzyskane wyniki dowodzą, że proces elektroutleniania Cp na
elektrodzie zmodyfikowanej warstwą receptorową (Au-EQCM/C/Fe@C Nps),
w obecności zewnętrznego źródła pola magnetycznego, jest kontrolowany
transportem depolaryzatora (Cp) do powierzchni elektrody. Niestety produkty tego
procesu adsorbują się na powierzchni elektrody, utrudniając rejestrację sygnałów
katodowych. Z tego powodu każdy pomiar elektrochemiczny prowadzony był na
świeżo przygotowanej warstwie receptorowej. Dla każdej szybkości polaryzacji
elektrody pomiar powtarzano co najmniej 3-krotnie. Zgodność uzyskanych wyników
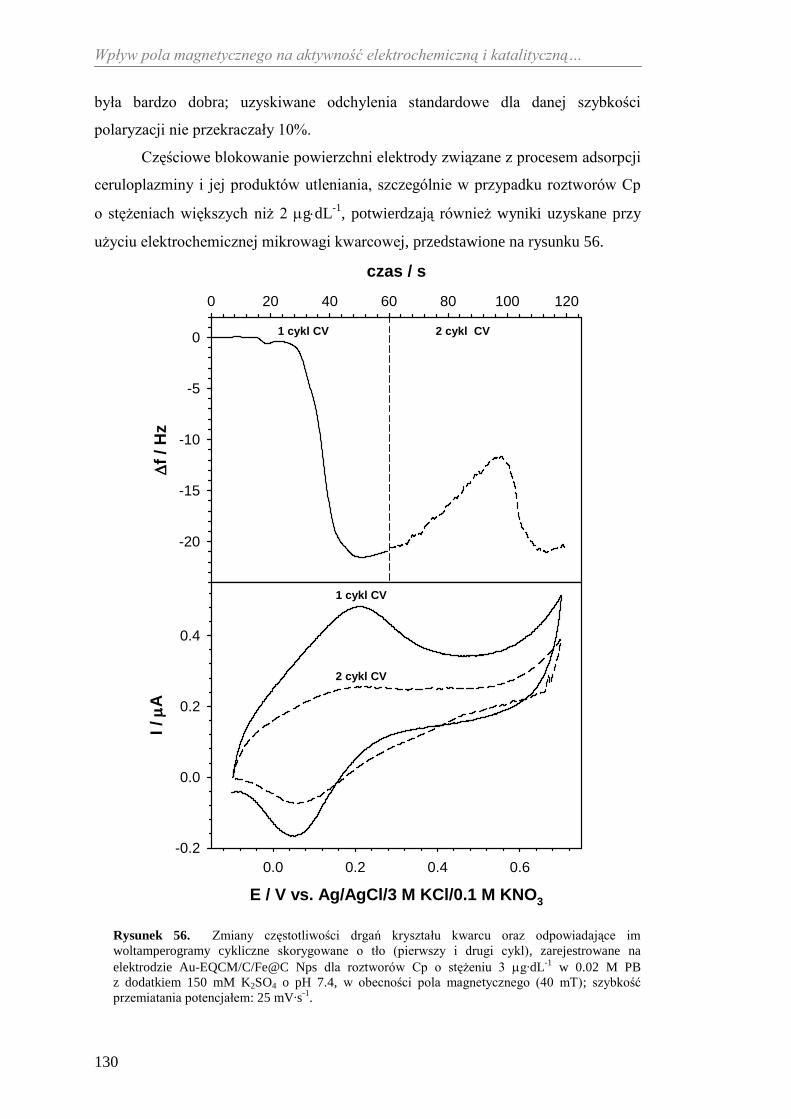
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
130
była bardzo dobra; uzyskiwane odchylenia standardowe dla danej szybkości
polaryzacji nie przekraczały 10%.
Częściowe blokowanie powierzchni elektrody związane z procesem adsorpcji
ceruloplazminy i jej produktów utleniania, szczególnie w przypadku roztworów Cp
o stężeniach większych niż 2 mgdL-1
, potwierdzają również wyniki uzyskane przy
użyciu elektrochemicznej mikrowagi kwarcowej, przedstawione na rysunku 56.
czas / s
0 20 40 60 80 100 120
f
/ H
z
-20
-15
-10
-5
01 cykl CV 2 cykl CV
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
0.0 0.2 0.4 0.6
I /
A
-0.2
0.0
0.2
0.4
1 cykl CV
2 cykl CV
Rysunek 56. Zmiany częstotliwości drgań kryształu kwarcu oraz odpowiadające im
woltamperogramy cykliczne skorygowane o tło (pierwszy i drugi cykl), zarejestrowane na
elektrodzie Au-EQCM/C/Fe@C Nps dla roztworów Cp o stężeniu 3 mg·dL-1
w 0.02 M PB
z dodatkiem 150 mM K2SO4 o pH 7.4, w obecności pola magnetycznego (40 mT); szybkość
przemiatania potencjałem: 25 mV·s-1
.

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
131
Pik utleniania Cp występuje jedynie w przypadku pierwszego cyklu
woltamperometrycznego, a w kolejnych cyklach zanika, ponieważ jak już
wspomiano, produkt utleniania Cp, jak i samo białko osadzone podczas pierwszego
cyklu efektywnie blokują powierzchnię elektrody. Obserwowane zmiany
częstotliwości drgań kryształu kwarcu podczas drugiego cyklu są znacznie mniejsze
w odniesieniu do pierwszego cyklu.
Zakres pracy czujnika do bezpośredniej elektrochemicznej detekcji
ceruloplazminy z roztworu określono na podstawie liniowego przedziału zmian
intensywności sygnału prądowego utleniania ceruloplazminy w funkcji jej stężenia
w roztworze, przedstawionego na rysunku 57.
CCp / gdL-1
0.0 1.0 2.0 3.0 4.0 5.0 6.0
I E=
0.2
2 V
/
A
0.0
0.1
0.2
0.3
0.4
0.5
0.6
Rysunek 57. Krzywa kalibracyjna wyznaczona na podstawie zmian intensywności sygnału
utleniania Cp w zależności od stężenia ceruloplazminy w roztworze. Warunki eksperymentalne:
0.02 M bufor PB (pH 7.4) zawierający 150 mM K2SO4 w obecności zewnętrznego źródła pola
magnetycznego (40 mT); szybkość przemiatania potencjałem: 25 mV·s-1
.
Otrzymana zależność charakteryzowała się liniową zmianą odpowiedzi prądowej
(Ip = (8.31 ± 0.15)CCp + (0.05 ± 0.004); r = 0.999) w zakresie stężeń od 0.2 mgdL-1
do ok. 5 mgdL-1
. Dla wszystkich stężeń Cp z zakresu liniowego krzywej
kalibracyjnej otrzymano dobrą powtarzalność wyników, a względne odchylenie
standardowe wynosiło około 8% (n = 5). Sprawdzono również powtarzalność
wyników uzyskanych dla tego samego stężenia ceruloplazminy, na dziesięciu
niezależnie zmodyfikowanych elektrodach, wówczas względne odchylenie
standardowe wynosiło około 4.7%.

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
132
Granicę wykrywalności ceruloplazminy przy użyciu opisanego czujnika
wyznaczono dwoma różnymi sposobami. Według pierwszego sposobu,
najprostszego i najczęściej używanego, wartość LOD wyznaczono, analogicznie jak
w przypadku hemoglobiny, z dolnego zakresu prostoliniowości krzywej
kalibracyjnej, zgodnie z równaniem (31) i wynosił on 0.23 ± 0.06 mg·dL-1
. W drugim
podejściu, LOD obliczono za pomocą metody Hubaux-Vos [428]. Sposób
postępowania w metodzie Hubaux-Vos [429] opiera się na przewidywaniu
przedziału krzywej kalibracji, z której odrzuca się zarówno dodatnie, jak i ujemne
błędy pomiarowe, fałszujące uzyskiwany wynik. W metodzie tej wykorzystano dane
uzyskane z pięciu niezależnych pomiarów dla każdego stężenia obejmującego zakres
prostoliniowości krzywej kalibracyjnej. Wartość LOD oblicza się stosując metodę
najmniejszych kwadratów. Wyznaczona w ten sposób granica wykrywalności
wynosiła 1.14 ± 0.24 mg·dL-1
. Różnice pomiędzy wynikami otrzymanymi za pomocą
pierwszego i drugiego sposobu najprawdopodobniej związane są z tym, że
odchylenia standardowe dla poszczególnych wartości stężeń Cp były różne.
W diagnostyce klinicznej zawartość ceruloplazminy określa się na podstawie
analiz immunologicznych. W tabeli 8 porównano parametry pracy proponowanego
czujnika do bezpośredniej detekcji Cp z parametrami pracy popularnych
komercyjnych testów immunologicznych.
Tabela 8. Porównanie parametrów pracy komercyjnych testów immunochemicznych do
oznaczania ceruloplazminy we krwi [125] z danymi otrzymanymi przy użyciu czujnika opisanego
w niniejszym rozdziale.
TEST
IMMUNOLOGICZNY
ZAKRES
STĘŻEŃ
[mgdL-1
]
LIMIT
DETEKCJI
[mgdL-1
]
CZAS
ANALIZY
ELISA Kit PRO-20074F 0.19 ÷ 25 brak danych 4 h
ELISA Kit(Uscn E90909Hu96 0.94 ÷ 60 < 0.34 3 h 45 min
Abanova KA0470 0.125 ÷ 32 ~ 0.1 2 h 47 min
Mybiosource MBS701656 (1.5 ÷ 50)103 750 1 h 45 min
Opracowany sensor 0.2 ÷ 5 0.23
a
1.14b
30 min
awynik wyznaczony z dolnego zakresu krzywej kalibracyjnej, zgodnie z równaniem (31)
aLOD wyznaczony metodą Hubaux-Vos
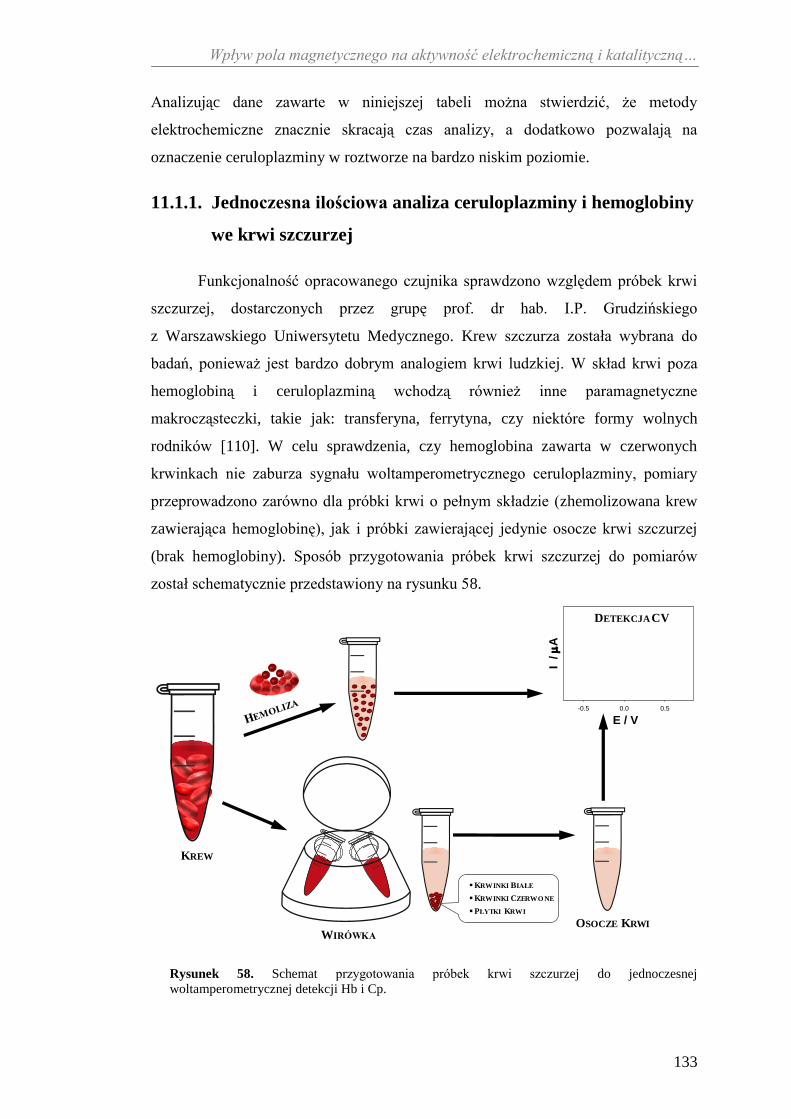
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
133
Analizując dane zawarte w niniejszej tabeli można stwierdzić, że metody
elektrochemiczne znacznie skracają czas analizy, a dodatkowo pozwalają na
oznaczenie ceruloplazminy w roztworze na bardzo niskim poziomie.
11.1.1. Jednoczesna ilościowa analiza ceruloplazminy i hemoglobiny
we krwi szczurzej
Funkcjonalność opracowanego czujnika sprawdzono względem próbek krwi
szczurzej, dostarczonych przez grupę prof. dr hab. I.P. Grudzińskiego
z Warszawskiego Uniwersytetu Medycznego. Krew szczurza została wybrana do
badań, ponieważ jest bardzo dobrym analogiem krwi ludzkiej. W skład krwi poza
hemoglobiną i ceruloplazminą wchodzą również inne paramagnetyczne
makrocząsteczki, takie jak: transferyna, ferrytyna, czy niektóre formy wolnych
rodników [110]. W celu sprawdzenia, czy hemoglobina zawarta w czerwonych
krwinkach nie zaburza sygnału woltamperometrycznego ceruloplazminy, pomiary
przeprowadzono zarówno dla próbki krwi o pełnym składzie (zhemolizowana krew
zawierająca hemoglobinę), jak i próbki zawierającej jedynie osocze krwi szczurzej
(brak hemoglobiny). Sposób przygotowania próbek krwi szczurzej do pomiarów
został schematycznie przedstawiony na rysunku 58.
KRWINKI BIAŁE
KRWINKI CZERWO NE
PŁYTKI KRWI
E / V -0.5 0.0 0.5
I /
A
KREW
OSOCZE KRWI
WIRÓWKA
DETEKCJACV
Rysunek 58. Schemat przygotowania próbek krwi szczurzej do jednoczesnej
woltamperometrycznej detekcji Hb i Cp.
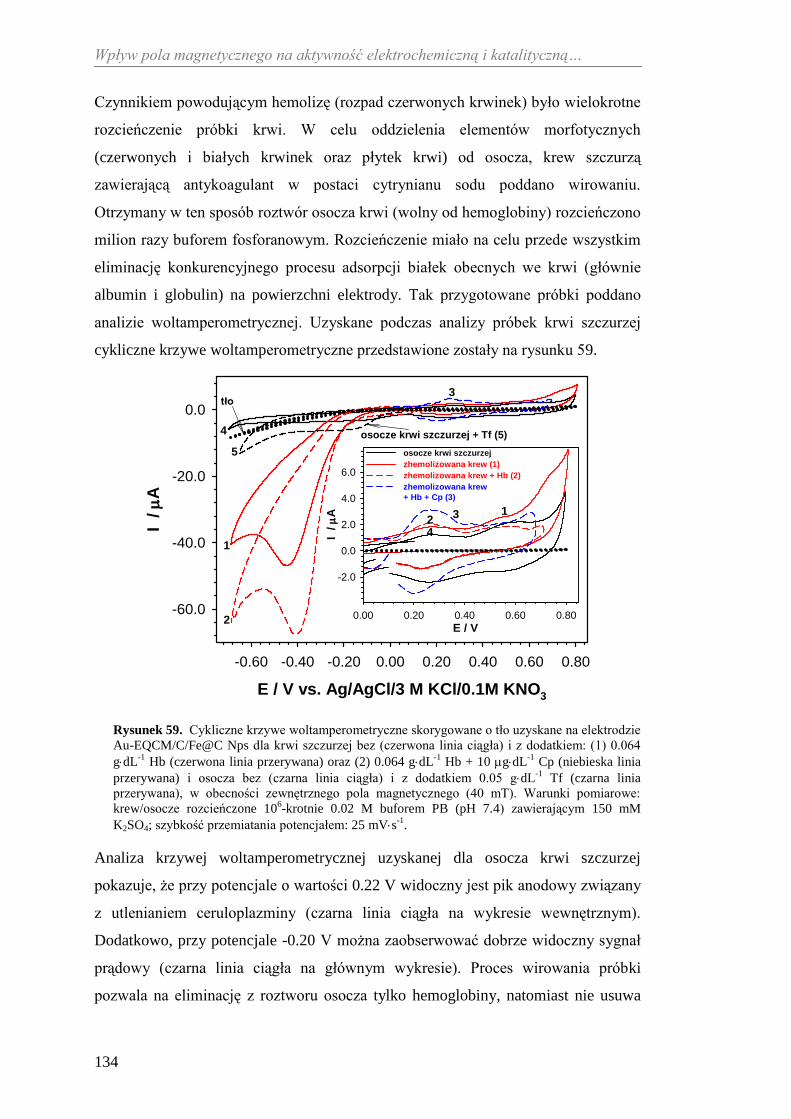
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
134
Czynnikiem powodującym hemolizę (rozpad czerwonych krwinek) było wielokrotne
rozcieńczenie próbki krwi. W celu oddzielenia elementów morfotycznych
(czerwonych i białych krwinek oraz płytek krwi) od osocza, krew szczurzą
zawierającą antykoagulant w postaci cytrynianu sodu poddano wirowaniu.
Otrzymany w ten sposób roztwór osocza krwi (wolny od hemoglobiny) rozcieńczono
milion razy buforem fosforanowym. Rozcieńczenie miało na celu przede wszystkim
eliminację konkurencyjnego procesu adsorpcji białek obecnych we krwi (głównie
albumin i globulin) na powierzchni elektrody. Tak przygotowane próbki poddano
analizie woltamperometrycznej. Uzyskane podczas analizy próbek krwi szczurzej
cykliczne krzywe woltamperometryczne przedstawione zostały na rysunku 59.
E / V vs. Ag/AgCl/3 M KCl/0.1M KNO3
-0.60 -0.40 -0.20 0.00 0.20 0.40 0.60 0.80
I /
A
-60.0
-40.0
-20.0
0.0tło
E / V 0.00 0.20 0.40 0.60 0.80
I /
A
-2.0
0.0
2.0
4.0
6.0
osocze krwi szczurzej
zhemolizowana krew (1)
zhemolizowana krew + Hb (2)
zhemolizowana krew
+ Hb + Cp (3)
osocze krwi szczurzej + Tf (5)
1
2
12
3
3
4
4
5
Rysunek 59. Cykliczne krzywe woltamperometryczne skorygowane o tło uzyskane na elektrodzie
Au-EQCM/C/Fe@C Nps dla krwi szczurzej bez (czerwona linia ciągła) i z dodatkiem: (1) 0.064
gdL-1
Hb (czerwona linia przerywana) oraz (2) 0.064 gdL-1
Hb + 10 mgdL-1
Cp (niebieska linia
przerywana) i osocza bez (czarna linia ciągła) i z dodatkiem 0.05 gdL-1
Tf (czarna linia
przerywana), w obecności zewnętrznego pola magnetycznego (40 mT). Warunki pomiarowe:
krew/osocze rozcieńczone 106-krotnie 0.02 M buforem PB (pH 7.4) zawierającym 150 mM
K2SO4; szybkość przemiatania potencjałem: 25 mVs-1
.
Analiza krzywej woltamperometrycznej uzyskanej dla osocza krwi szczurzej
pokazuje, że przy potencjale o wartości 0.22 V widoczny jest pik anodowy związany
z utlenianiem ceruloplazminy (czarna linia ciągła na wykresie wewnętrznym).
Dodatkowo, przy potencjale -0.20 V można zaobserwować dobrze widoczny sygnał
prądowy (czarna linia ciągła na głównym wykresie). Proces wirowania próbki
pozwala na eliminację z roztworu osocza tylko hemoglobiny, natomiast nie usuwa

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
135
innych paramagnetycznych substancji, takich jak: transferyna czy też ferrytyna.
W celu uzyskania informacji, od jakiej substancji paramagnetycznej pochodzi ten
sygnał prądowy, wykonano eksperyment kontrolny, w którym do roztworu osocza
dodano transferyny o stężeniu 0.05 g·dL-1
(czarna linia przerywana w głównym
wykresie). Otrzymana krzywa CV wyraźnie wskazuje, że sygnał przy -0.20 V
związany jest z obecnością w roztworze Tf. W przypadku analizy zhemolizowanej
próbki krwi (czerwona linia ciągła) sygnał redukcji Tf był niewidoczny, ponieważ
pokrywał się z sygnałem methemoglobiny pojawiającym się na skali potencjałowej
przy wartości ok -0.44 V. Aby uzyskać pewność, że obserwowany sygnał związany
jest z procesem elektroredukcji Hb wykonano pomiar kontrolny z dodatkiem
hemoglobiny do zhemolizowanej próbki krwi (czerwona linia przerywana). Po
wprowadzeniu znanej ilości Hb do próbki krwi, zaobserwowano nie tylko wzrost
intensywności sygnału katodowego po ujemnej stronie na skali potencjałowej, ale
również jego nieznaczne przesunięcie (ok. 15 mV) w kierunku bardziej dodatnich
wartości potencjałów. Dodatek Hb do próbki krwi miał wpływ tylko na sygnał
pochodzący od procesu elektrodowego tego białka, natomiast w żaden sposób nie
zaburzał sygnału elektroutleniania Cp (czerwona linia ciągła i przerwana na
wewnętrznym wykresie na rysunku 59). Dopiero dodatek ceruloplazminy
spowodował wzrost wartości prądu przy potencjale 0.22 V (niebieska linia
przerywana na wykresie wewnętrznym rysunku 59). Wyniki analizy zawartości Hb
i Cp w badanej próbce krwi szczurzej przy użyciu zaproponowanego czujnika
porównano z zakresami oznaczalności tych białek, uzyskanymi podczas typowej
analizy klinicznej wykonanej dla próbki krwi kobiety (K) i mężczyzny (M).
Odpowiednie wartości zostały podane w tabeli 9.
Tabela 9. Porównanie wyników jednoczesnej analizy ceruloplazminy i hemoglobiny w próbkach
krwi szczurzej przy użyciu zaproponowanego czujnika.
SKONSTRUOWANY
CZUJNIK
ANALIZA
KLINICZNA
ZAKRES
W LUDZKIEJ KRWI
Hb [g·dL-1
] 12.9 ± 1.8 14 ÷ 18 (M)
12 ÷ 16 (K) 11.5 ÷ 18
Cp [mg·dL-1
] 36.0 ± 2.0 30 ÷ 58 25.0 ÷ 47.0

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
136
Zgodność wyników była bardzo dobra, co przemawia za tym, że skonstruowany
czujnik może być z powodzeniem stosowany do bezpośredniego, jednoczesnego
oznaczania ceruloplazminy i hemoglobiny w próbkach krwi.
11.2. Wpływ pola magnetycznego na aktywność
katalityczną ceruloplazminy unieruchomionej na
podłożu złotym zmodyfikowanym ferromagnetykiem
Multimiedziowe ferroksydazy, których przedstawicielem jest ceruloplazmina,
ulegają ekspresji w wielu tkankach. Są one zdolne do utleniania szerokiej gamy
substratów. Obserwacje dotyczące skutków fenotypowych związanych z brakiem,
bądź uszkodzeniem genów kodujących multimiedziowe ferroksydazy pozwalają
przypuszczać, że najważniejszą rolą tych białek jest udział w metabolizmie żelaza
[112]. Główną funkcją ceruloplazminy w gospodarce żelaza w organizmie jest
utlenianie jonów Fe2+
do Fe3+
, co warunkuje wiązanie żelaza z transferyną (głównym
białkiem transportującym żelazo), jak i z ferrytyną (głównym białkiem
magazynującym żelazo). Unieruchomienie ceruloplazminy na powierzchni elektrody
we właściwej orientacji, odgrywa kluczową rolę nie tylko w transporcie elektronów
pomiędzy elektrodą a enzymem, ale również ma istotny wpływ na jego aktywność
katalityczną.
11.2.1. Wpływ ferromegnetycznego modyfikatora powierzchni
na elektroaktywność złota
Pierwszy etap badań dotyczył określenia wpływu ferromagnetycznego
modyfikatora powierzchni na elektrochemiczne parametry dyskowej elektrody złotej.
Pomiary przeprowadzono przy użyciu elektrochemicznej spektroskopii
impedancyjnej w obecności próbnika redoks w roztworze (równomolowa mieszanina
[Fe(CN)6]3-/4-
), przy potencjale formalnym zastosowanego układu redoks
wynoszącym 0.243 V, na niezmodyfikowanym chemicznie złotcie (Au) oraz
zmodyfikowanym nanokapsułami węglowymi zawierającymi żelazo
(Au/Fe@C Nps) oraz nanocząstkami węgla (Au/C Nps). Przykładowe wykresy
Nyquist’a (widma impedancyjne) otrzymane dla niezmodyfikowanej elektrody Au
oraz elektrod zmodyfikowanych odpowiednio Fe@C Nps i C Nps przedstawiono na
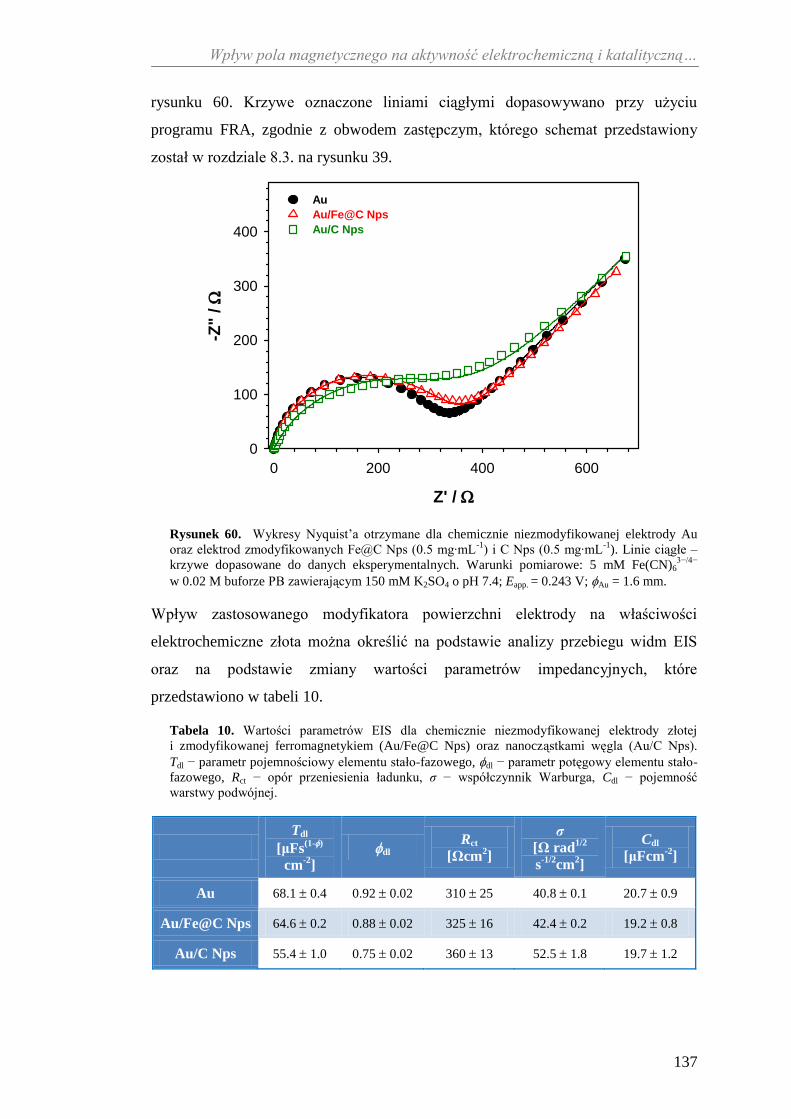
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
137
rysunku 60. Krzywe oznaczone liniami ciągłymi dopasowywano przy użyciu
programu FRA, zgodnie z obwodem zastępczym, którego schemat przedstawiony
został w rozdziale 8.3. na rysunku 39.
Z' /
0 200 400 600
-Z" /
0
100
200
300
400
Au
Au/Fe@C Nps
Au/C Nps
Rysunek 60. Wykresy Nyquist’a otrzymane dla chemicznie niezmodyfikowanej elektrody Au
oraz elektrod zmodyfikowanych Fe@C Nps (0.5 mg·mL-1
) i C Nps (0.5 mg·mL-1
). Linie ciągłe –
krzywe dopasowane do danych eksperymentalnych. Warunki pomiarowe: 5 mM Fe(CN)63−/4−
w 0.02 M buforze PB zawierającym 150 mM K2SO4 o pH 7.4; Eapp. = 0.243 V; Au = 1.6 mm.
Wpływ zastosowanego modyfikatora powierzchni elektrody na właściwości
elektrochemiczne złota można określić na podstawie analizy przebiegu widm EIS
oraz na podstawie zmiany wartości parametrów impedancyjnych, które
przedstawiono w tabeli 10.
Tabela 10. Wartości parametrów EIS dla chemicznie niezmodyfikowanej elektrody złotej
i zmodyfikowanej ferromagnetykiem (Au/Fe@C Nps) oraz nanocząstkami węgla (Au/C Nps).
Tdl − parametr pojemnościowy elementu stało-fazowego, dl − parametr potęgowy elementu stało-
fazowego, Rct − opór przeniesienia ładunku, σ − współczynnik Warburga, Cdl − pojemność
warstwy podwójnej.
Tdl
[μFs(1-)
cm-2
]
dl Rct
[Ωcm2]
σ
[Ω rad1/2
s-1/2
cm2]
Cdl
[μFcm-2
]
Au 68.1 0.4 0.92 0.02 310 25 40.8 0.1 20.7 0.9
Au/Fe@C Nps 64.6 0.2 0.88 0.02 325 16 42.4 0.2 19.2 0.8
Au/C Nps 55.4 1.0 0.75 0.02 360 13 52.5 1.8 19.7 1.2

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
138
To, czy modyfikator powierzchni elektrody w znaczący sposób wpływa na jej
elektroaktywność, najłatwiej zauważyć kontrolując zmiany wartości oporu
przeniesienia ładunku (Rct). Dane przedstawione w tabeli 10 pokazują, że
modyfikacja powierzchni złota nanokapsułami węglowymi zawierającymi żelazo,
czy też nanocząstkami węgla, nie wpływała na wartości parametru Rct. Ze wszystkich
wyznaczonych parametrów impedancyjnych tylko wartość potęgowego elementu
stało-fazowego ulega niewielkiemu zmniejszeniu, od 6 do 18%, w stosunku do
niezmodyfikowanej chemicznie elektrody złotej, odpowiednio w przypadku
nanocząstek zawierających żelazo i nanoczastek węgla. Małe odchylenia parametru
φdl w przypadku warstwy utworzonej przez Fe@C Nps wskazują, że nanocząstki te
dość równomiernie pokrywają powierzchnię elektrody. W sytuacji zastosowania
nanocząstek węgla parametr ten w większym stopniu różnił się od wartości
otrzymanej dla czystej elektrody złotej, dlatego też w dalszych badaniach stosowano
wyłącznie nanokapsuły węglowe zawierające żelazo. Na podstawie zmian wartości
parametru Warburga można wywnioskować, że modyfikacja powierzchni elektrody
warstwą Fe@C Nps nie utrudniała w żaden sposób transportu próbnika redoks do
powierzchni elektrody.
11.2.2. Aktywność katalityczna ceruloplazminy wobec
jonów żelaza(II)
W przypadku badań nad aktywnością katalityczną ceruloplazminy zarówno
proces tworzenia warstwy ferromagnetyka na powierzchni elektrody, jak również
sam proces unieruchamiania enzymu na tak zmodyfikowanej elektrodzie
prowadzone były w najprostszy sposób, czyli w wyniku nałożenia kropli
i pozostawienia jej do wyschnięcia. Etap modyfikacji podłoża cząsteczkami
ceruloplazminy prowadzony był w obecności pola magnetycznego. Wiadomo, że
modyfikacja podłoża na drodze adsorpcji fizycznej nie prowadzi do jego całkowitego
pokrycia. Dlatego też w celu uzyskania informacji na temat rozmieszczenia enzymu
na zmodyfikowanej powierzchni elektrody, wykonane zostały pomiary
z zastosowaniem techniki LA-ICP-MS, których wyniki ilustruje rysunek 61.
Analizując rozkład intensywności sygnałów uzyskanych dla izotopów miedzi oraz
żelaza można stwierdzić, że adsorpcja ceruloplazminy zachodzi głównie na
ferromagnetycznym modyfikatorze powierzchni elektrody. Na odsłoniętej
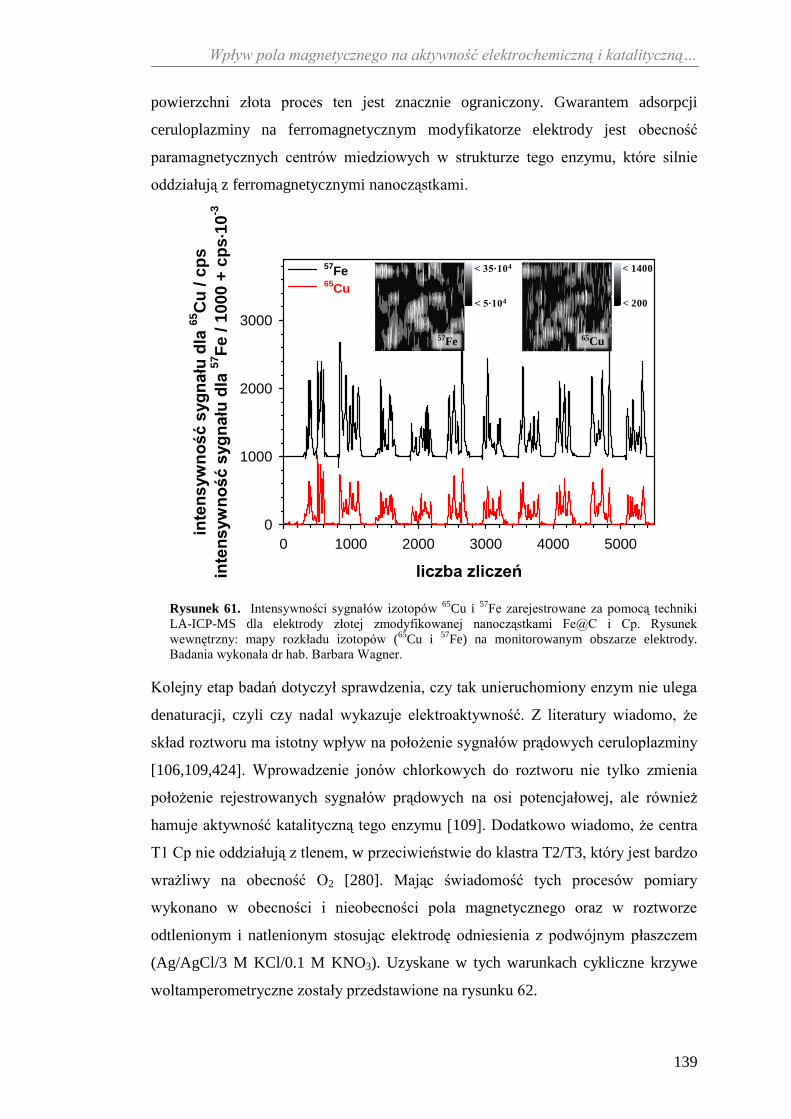
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
139
powierzchni złota proces ten jest znacznie ograniczony. Gwarantem adsorpcji
ceruloplazminy na ferromagnetycznym modyfikatorze elektrody jest obecność
paramagnetycznych centrów miedziowych w strukturze tego enzymu, które silnie
oddziałują z ferromagnetycznymi nanocząstkami.
liczba zliczeń
0 1000 2000 3000 4000 5000
inte
nsyw
no
ść s
yg
nału
dla
65C
u /
cp
s
inte
nsyw
no
ść s
yg
nału
dla
57F
e / 1
000 +
cp
s10
-3
0
1000
2000
3000
57Fe
65Cu
65Cu57Fe
< 35·104
< 5·104
< 1400
< 200
Rysunek 61. Intensywności sygnałów izotopów 65
Cu i 57
Fe zarejestrowane za pomocą techniki
LA-ICP-MS dla elektrody złotej zmodyfikowanej nanocząstkami Fe@C i Cp. Rysunek
wewnętrzny: mapy rozkładu izotopów (65
Cu i 57
Fe) na monitorowanym obszarze elektrody.
Badania wykonała dr hab. Barbara Wagner.
Kolejny etap badań dotyczył sprawdzenia, czy tak unieruchomiony enzym nie ulega
denaturacji, czyli czy nadal wykazuje elektroaktywność. Z literatury wiadomo, że
skład roztworu ma istotny wpływ na położenie sygnałów prądowych ceruloplazminy
[106,109,424]. Wprowadzenie jonów chlorkowych do roztworu nie tylko zmienia
położenie rejestrowanych sygnałów prądowych na osi potencjałowej, ale również
hamuje aktywność katalityczną tego enzymu [109]. Dodatkowo wiadomo, że centra
T1 Cp nie oddziałują z tlenem, w przeciwieństwie do klastra T2/T3, który jest bardzo
wrażliwy na obecność O2 [280]. Mając świadomość tych procesów pomiary
wykonano w obecności i nieobecności pola magnetycznego oraz w roztworze
odtlenionym i natlenionym stosując elektrodę odniesienia z podwójnym płaszczem
(Ag/AgCl/3 M KCl/0.1 M KNO3). Uzyskane w tych warunkach cykliczne krzywe
woltamperometryczne zostały przedstawione na rysunku 62.
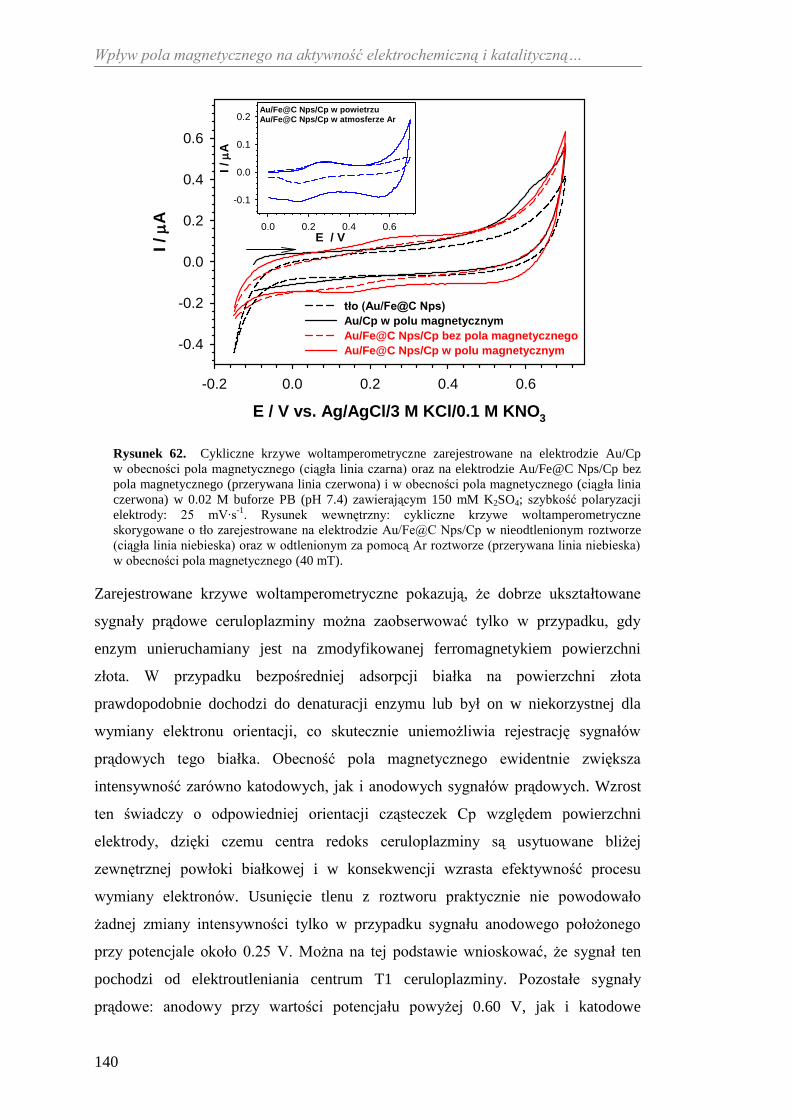
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
140
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.2 0.0 0.2 0.4 0.6
I /
A
-0.4
-0.2
0.0
0.2
0.4
0.6
tło (Au/Fe@C Nps)
Au/Cp w polu magnetycznym
Au/Fe@C Nps/Cp bez pola magnetycznego
Au/Fe@C Nps/Cp w polu magnetycznym
E / V0.0 0.2 0.4 0.6
I /
A
-0.1
0.0
0.1
0.2Au/Fe@C Nps/Cp w powietrzuAu/Fe@C Nps/Cp w atmosferze Ar
Rysunek 62. Cykliczne krzywe woltamperometryczne zarejestrowane na elektrodzie Au/Cp
w obecności pola magnetycznego (ciągła linia czarna) oraz na elektrodzie Au/Fe@C Nps/Cp bez
pola magnetycznego (przerywana linia czerwona) i w obecności pola magnetycznego (ciągła linia
czerwona) w 0.02 M buforze PB (pH 7.4) zawierającym 150 mM K2SO4; szybkość polaryzacji
elektrody: 25 mV·s-1
. Rysunek wewnętrzny: cykliczne krzywe woltamperometryczne
skorygowane o tło zarejestrowane na elektrodzie Au/Fe@C Nps/Cp w nieodtlenionym roztworze
(ciągła linia niebieska) oraz w odtlenionym za pomocą Ar roztworze (przerywana linia niebieska)
w obecności pola magnetycznego (40 mT).
Zarejestrowane krzywe woltamperometryczne pokazują, że dobrze ukształtowane
sygnały prądowe ceruloplazminy można zaobserwować tylko w przypadku, gdy
enzym unieruchamiany jest na zmodyfikowanej ferromagnetykiem powierzchni
złota. W przypadku bezpośredniej adsorpcji białka na powierzchni złota
prawdopodobnie dochodzi do denaturacji enzymu lub był on w niekorzystnej dla
wymiany elektronu orientacji, co skutecznie uniemożliwia rejestrację sygnałów
prądowych tego białka. Obecność pola magnetycznego ewidentnie zwiększa
intensywność zarówno katodowych, jak i anodowych sygnałów prądowych. Wzrost
ten świadczy o odpowiedniej orientacji cząsteczek Cp względem powierzchni
elektrody, dzięki czemu centra redoks ceruloplazminy są usytuowane bliżej
zewnętrznej powłoki białkowej i w konsekwencji wzrasta efektywność procesu
wymiany elektronów. Usunięcie tlenu z roztworu praktycznie nie powodowało
żadnej zmiany intensywności tylko w przypadku sygnału anodowego położonego
przy potencjale około 0.25 V. Można na tej podstawie wnioskować, że sygnał ten
pochodzi od elektroutleniania centrum T1 ceruloplazminy. Pozostałe sygnały
prądowe: anodowy przy wartości potencjału powyżej 0.60 V, jak i katodowe

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
141
położone przy ok. 0.56 V i 0.16 V wykazywały większą czułość na obecność tlenu
w roztworze. Obserwowany w warunkach tlenowych wzrost wartości rejestrowanego
prądu przy potencjale około 0.60 V najprawdopodobniej związany jest z utlenianiem
jonu miedzi(I) w centrum T1PR, który następnie ulega redukcji przy potencjale około
0.56 V. Znając stężenie Cp oraz wartość ładunku uzyskanego z krzywej
woltamperometrycznej (1.03·10-8
C) możliwe było wyznaczenie liczby elektronów
przenoszonych z elektrody do centrów T1CysHis i T1Remote w cząsteczce
ceruloplazminy. Dane literaturowe donoszą, że podczas tego procesu następuje
wymiana dwóch elektronów [280]. Z obliczeń natomiast wynika, że liczba ta jest
równa 2.4 ± 0.5. Przyjmując, że masa cząsteczkowa ceruloplazminy wynosi
132 kDa, można wyznaczyć stężenie powierzchniowe Cp unieruchomionej na
warstwie nanocząstek magnetycznych, zgodnie z równaniem:
nFA
QCp (32)
gdzie: ΓCp to stężenie powierzchniowe Cp, Q – ładunek sygnału utleniania z zakresu
potencjałowego 0.10.5 V , n – liczba elektronów wymienianych w procesie redoks
Cp, F – stała Faraday’a, zaś A – powierzchnia elektrody. Podstawiając uzyskane
wartości do równania (31) wyznaczono stężenie powierzchniowe białka, które
wynosiło 2.22 ± 0.6 pmol·cm-2
. Znając powierzchnię, jaką zajmuje pojedyncza
cząsteczka ceruloplazminy (7.5 x 4.0 6.6 nm) [430] można wyznaczyć teoretyczne
maksymalne stężenie powierzchniowe ceruloplazminy; wynosi ono
3.45÷5.5 pmol·cm-2
. Na podstawie wyznaczonych wartości stężeń
powierzchniowych Cp z danych eksperymentalnych i teoretycznych, można
powiedzieć, że utworzona warstwa enzymu na powierzchni nanocząstek
magnetycznych była dość gęsto upakowana.
Zdolność unieruchomionej ceruloplazminy na powierzchni nanocząstek
magnetycznych do katalitycznego utleniania jonów żelaza(II) do żelaza(III) zbadana
została przy wykorzystaniu skaningowego mikroskopu elektrochemicznego. Metoda
ta jest niezwykle użytecznym narzędziem wykorzystywanym do badań
enzymatycznie modyfikowanych elektrod, gdyż produkty i substraty reakcji
enzymatycznej wykrywane są z wysoką specyficznością. Źródłem jonów żelaza(II)
był 20 mM roztwór heksacyjanożelazianu(II) potasu. Obrazowanie SECM
przeprowadzono w trybie sprzężenia zwrotnego (ang. feedback mode, FB). Złota
elektroda dyskowa zmodyfikowana warstwą ferromagnetycznych nanocząstek oraz

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
142
ceruloplazminą umieszczona została wewnątrz kolistego magnesu o natężeniu pola
40 mT, w celu zwiększenia szybkości bezpośredniego przeniesienia elektronów
pomiędzy centrum T1 Cp a powierzchnią elektrody. Z uwagi na fakt, że tylko jony
miedzi(II) są zdolne do utleniania jonów żelaza(II) do jonów Fe3+
, w celu utrzymania
centrum miedziowego Cp na +II stopniu utlenienia, w trakcie badań do elektrody
przykładano stały dodatni potencjał wynoszący 0.25 V. Generowane w ten sposób
jony Fe(CN)63-
wykrywane były poprzez monitorowanie prądu redukcji tych jonów
przy potencjale 80 mV za pomocą mikroelektrody Pt o średnicy 10 µm. Schemat
ilustrujący zasadę pomiaru za pomocą skaningowego mikroskopu
elektrochemicznego przedstawia rysunek 63.
WE2: Pt ( = 10 µm)
MAGNES: Fe14Nd2B (40 mT)
x
yz
WE1:
Au/Fe@C Nps/Cp
(E = 250 mV)
ELEKTROLIT:
0.02 M PB (pH 7.4)
150 mM K2SO4
20 mM K4[Fe(CN)6]
Fe(CN)63-
ELEKTROLIT
Fe(CN)64-
WE1: Au ( = 2 mm)
WE2:
Pt (E = 80 mV)
Rysunek 63. Schemat układu pomiarowego aktywności elektrokatalitycznej Cp zaadsorbowanej
na Fe@C Nps w obecności zewnętrznego pola magnetycznego.
W celu uzyskania informacji, czy nanokapsuły Fe@C mogą również generować jony
Fe(CN)63-
zarejestrowane zostały krzywe zbliżania dla chemicznie
niezmodyfikowanej elektrody złotej oraz elektrody zmodyfikowanej warstwą
nanocząstek magnetycznych w obecności pola magnetycznego i 20 µM roztworu
K4[Fe(CN)6] w buforze fosforanowym, co ilustruje rysunek 64A. Uzyskane krzywe
zbliżania wyraźnie pokazują, że w przypadku modyfikacji powierzchni elektrody
warstwą nanocząstek magnetycznych jony Fe(CN)63-
praktycznie nie są generowane.
Dopiero wprowadzenie do układu ceruloplazminy miało zdecydowany wpływ na
ilość powstających jonów Fe(CN)63-
, gdyż następował gwałtowny wzrost wartości
prądu redukcji tych jonów, zgodnie z rysunkiem 64B. W sytuacji braku
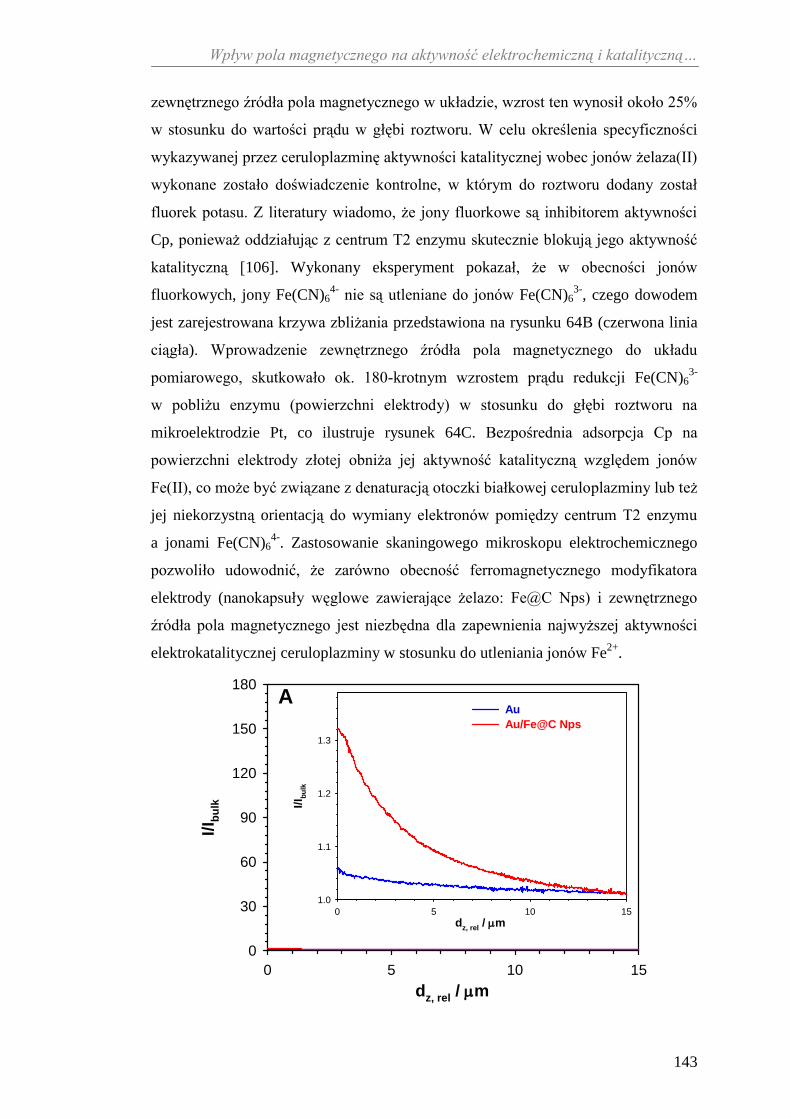
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
143
zewnętrznego źródła pola magnetycznego w układzie, wzrost ten wynosił około 25%
w stosunku do wartości prądu w głębi roztworu. W celu określenia specyficzności
wykazywanej przez ceruloplazminę aktywności katalitycznej wobec jonów żelaza(II)
wykonane zostało doświadczenie kontrolne, w którym do roztworu dodany został
fluorek potasu. Z literatury wiadomo, że jony fluorkowe są inhibitorem aktywności
Cp, ponieważ oddziałując z centrum T2 enzymu skutecznie blokują jego aktywność
katalityczną [106]. Wykonany eksperyment pokazał, że w obecności jonów
fluorkowych, jony Fe(CN)64-
nie są utleniane do jonów Fe(CN)63-
, czego dowodem
jest zarejestrowana krzywa zbliżania przedstawiona na rysunku 64B (czerwona linia
ciągła). Wprowadzenie zewnętrznego źródła pola magnetycznego do układu
pomiarowego, skutkowało ok. 180-krotnym wzrostem prądu redukcji Fe(CN)63-
w pobliżu enzymu (powierzchni elektrody) w stosunku do głębi roztworu na
mikroelektrodzie Pt, co ilustruje rysunek 64C. Bezpośrednia adsorpcja Cp na
powierzchni elektrody złotej obniża jej aktywność katalityczną względem jonów
Fe(II), co może być związane z denaturacją otoczki białkowej ceruloplazminy lub też
jej niekorzystną orientacją do wymiany elektronów pomiędzy centrum T2 enzymu
a jonami Fe(CN)64-
. Zastosowanie skaningowego mikroskopu elektrochemicznego
pozwoliło udowodnić, że zarówno obecność ferromagnetycznego modyfikatora
elektrody (nanokapsuły węglowe zawierające żelazo: Fe@C Nps) i zewnętrznego
źródła pola magnetycznego jest niezbędna dla zapewnienia najwyższej aktywności
elektrokatalitycznej ceruloplazminy w stosunku do utleniania jonów Fe2+
.
dz, rel / m
0 5 10 15
I/I b
ulk
0
30
60
90
120
150
180
dz, rel
/ m0 5 10 15
I/I b
ulk
1.0
1.1
1.2
1.3
Au
Au/Fe@C Nps
A
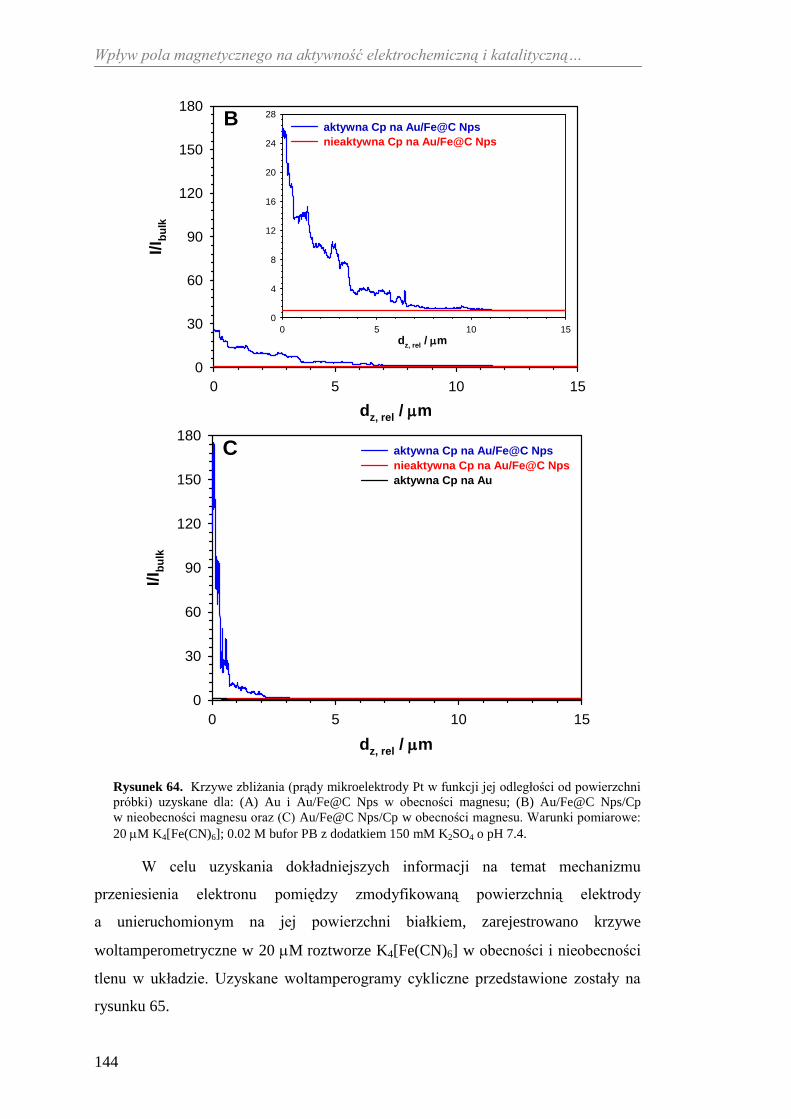
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
144
dz, rel / m
0 5 10 15
I/I b
ulk
0
30
60
90
120
150
180
dz, rel
/ m0 5 10 15
0
4
8
12
16
20
24
28
aktywna Cp na Au/Fe@C Nps
nieaktywna Cp na Au/Fe@C Nps
B
dz, rel / m
0 5 10 15
I/I b
ulk
0
30
60
90
120
150
180aktywna Cp na Au/Fe@C Nps
nieaktywna Cp na Au/Fe@C Nps
aktywna Cp na Au
C
Rysunek 64. Krzywe zbliżania (prądy mikroelektrody Pt w funkcji jej odległości od powierzchni
próbki) uzyskane dla: (A) Au i Au/Fe@C Nps w obecności magnesu; (B) Au/Fe@C Nps/Cp
w nieobecności magnesu oraz (C) Au/Fe@C Nps/Cp w obecności magnesu. Warunki pomiarowe:
20 mM K4[Fe(CN)6]; 0.02 M bufor PB z dodatkiem 150 mM K2SO4 o pH 7.4.
W celu uzyskania dokładniejszych informacji na temat mechanizmu
przeniesienia elektronu pomiędzy zmodyfikowaną powierzchnią elektrody
a unieruchomionym na jej powierzchni białkiem, zarejestrowano krzywe
woltamperometryczne w 20 mM roztworze K4[Fe(CN)6] w obecności i nieobecności
tlenu w układzie. Uzyskane woltamperogramy cykliczne przedstawione zostały na
rysunku 65.
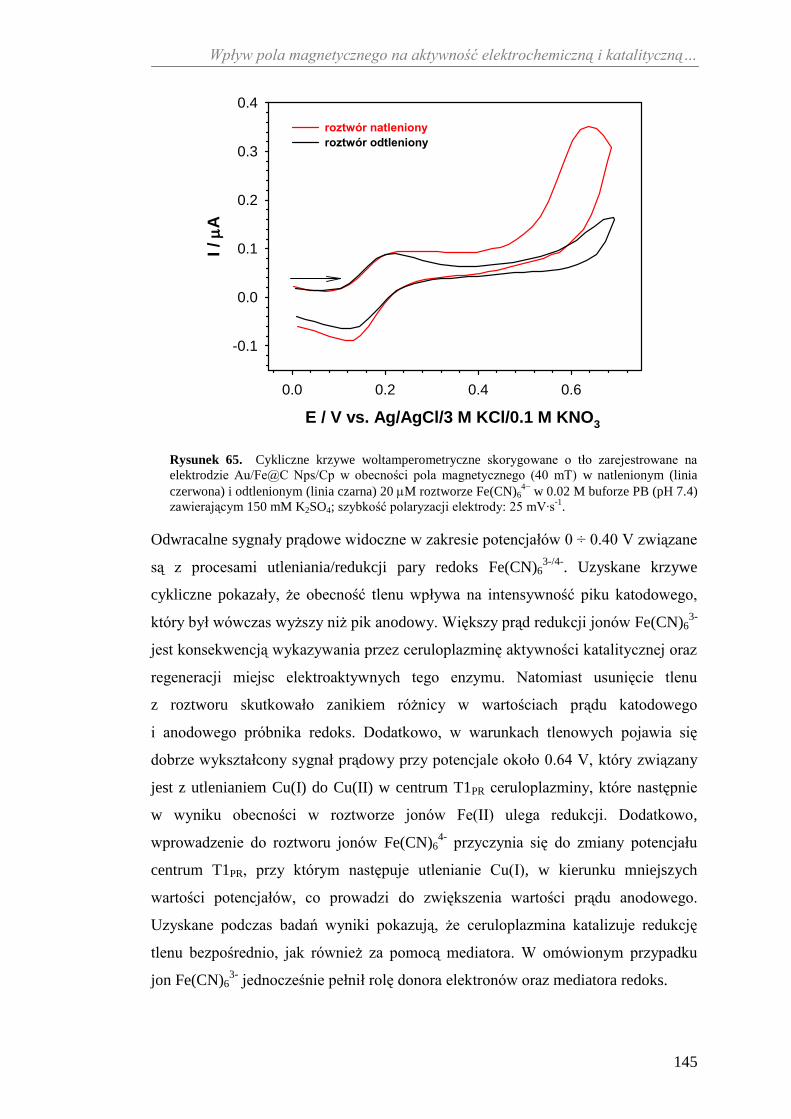
Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
145
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
0.0 0.2 0.4 0.6
I /
A
-0.1
0.0
0.1
0.2
0.3
0.4
roztwór natleniony
roztwór odtleniony
Rysunek 65. Cykliczne krzywe woltamperometryczne skorygowane o tło zarejestrowane na
elektrodzie Au/Fe@C Nps/Cp w obecności pola magnetycznego (40 mT) w natlenionym (linia
czerwona) i odtlenionym (linia czarna) 20 mM roztworze Fe(CN)64−
w 0.02 M buforze PB (pH 7.4)
zawierającym 150 mM K2SO4; szybkość polaryzacji elektrody: 25 mV·s-1
.
Odwracalne sygnały prądowe widoczne w zakresie potencjałów 0 ÷ 0.40 V związane
są z procesami utleniania/redukcji pary redoks Fe(CN)63-/4-
. Uzyskane krzywe
cykliczne pokazały, że obecność tlenu wpływa na intensywność piku katodowego,
który był wówczas wyższy niż pik anodowy. Większy prąd redukcji jonów Fe(CN)63-
jest konsekwencją wykazywania przez ceruloplazminę aktywności katalitycznej oraz
regeneracji miejsc elektroaktywnych tego enzymu. Natomiast usunięcie tlenu
z roztworu skutkowało zanikiem różnicy w wartościach prądu katodowego
i anodowego próbnika redoks. Dodatkowo, w warunkach tlenowych pojawia się
dobrze wykształcony sygnał prądowy przy potencjale około 0.64 V, który związany
jest z utlenianiem Cu(I) do Cu(II) w centrum T1PR ceruloplazminy, które następnie
w wyniku obecności w roztworze jonów Fe(II) ulega redukcji. Dodatkowo,
wprowadzenie do roztworu jonów Fe(CN)64-
przyczynia się do zmiany potencjału
centrum T1PR, przy którym następuje utlenianie Cu(I), w kierunku mniejszych
wartości potencjałów, co prowadzi do zwiększenia wartości prądu anodowego.
Uzyskane podczas badań wyniki pokazują, że ceruloplazmina katalizuje redukcję
tlenu bezpośrednio, jak również za pomocą mediatora. W omówionym przypadku
jon Fe(CN)63-
jednocześnie pełnił rolę donora elektronów oraz mediatora redoks.

Wpływ pola magnetycznego na aktywność elektrochemiczną i katalityczną…
146
11.3. Podsumowanie
Przeprowadzone badania udowodniły, że możliwa jest bezpośrednia
elektrochemiczna detekcja ceruloplazminy (Cp) i hemoglobiny (Hb) w trakcie
pojedynczego pomiaru, bez konieczności specjalnego przygotowania analizowanej
próbki krwi. Zaproponowana metoda oparta była na ferromagnetycznym
modyfikatorze elektrody (nanokapsułach węglowych zawierających żelazo) oraz
obecności zewnętrznego źródła pola magnetycznego. Nanocząstki ferromagnetyka
na powierzchni elektrody pełniły rolę bardzo specyficznego i selektywnego filtru,
który w ciągu pierwszych kilku minut przyśpieszał i zwiększał transport tylko
paramagnetycznych molekuł do powierzchni elektrody. Dodatkowo, wszystkie
paramagnetyczne cząsteczki docierające do powierzchni ferromagnetyka były
elektroaktywne. Co więcej, bezpośredni kontakt metaloproteiny z ferromagnetykiem
nie prowadził do jej deaktywacji. Opracowana metoda pozwala oznaczać
paramagnetyczne białka na poziomie granicy wykrywalności rzędu pM.
Dodatkowo, badania przeprowadzone przy użyciu skaningowego mikroskopu
elektrochemicznego (SECM) udowodniły, że zarówno obecność ferromagnetycznego
modyfikatora elektrody (nanokapsuły węglowe zawierające żelazo: Fe@C Nps), jak
i zewnętrznego źródła pola magnetycznego jest niezbędna w celu zapewnienia
najwyższej aktywności elektrokatalitycznej ceruloplazminy w stosunku do jonów
Fe2+
.

Elektrochemiczny immunosensor do detekcji transferyny
147
12. Elektrochemiczny immunosensor do detekcji
transferyny
Kolejną ważną z diagnostycznego punktu widzenia metaloproteiną obecną we
krwi jest transferyna. W klasycznej analizie medycznej ilościowego oznaczania tego
białka w osoczu krwi dokonuje się przy użyciu metod immunologicznych, których
zasada działania opiera się na bardzo selektywnym i specyficznym wiązaniu
antygenu (białka) przez charakterystyczne dla niego przeciwciało [431]. Niestety,
metody immunologiczne mają kilka znaczących wad. Największą z nich jest brak
możliwości zastosowania tych metod przy oznaczeniach białek na niskim poziomie,
ponadto są one bardzo często żmudne, czasochłonne oraz kosztowne [432].
Interesującą koncepcją i jednocześnie dobrą alternatywą dla klasycznych metod są
immunosensory z detekcją elektrochemiczną [433]. Ich prostota konstrukcji, bardzo
wysoka czułość oraz łatwość miniaturyzacji sprawiły, że coraz częściej sięga się po
tego typu narzędzia analityczne w nowoczesnej nanomedycynie [434,435].
Dodatkowo, immunosensory oparte na wykorzystaniu różnego typu nanomateriałów
charakteryzują się niższą granicą wykrywalności oraz szerszym zakresem
odpowiedzi liniowej w stosunku do tradycyjnych testów immunologicznych [436].
Z uwagi na fakt, że transferyna, podobnie jak hemoglobina, czy też ferrytyna,
zawiera w swoje strukturze jony żelaza, oznaczanie tego białka przy użyciu
elektrody zmodyfikowanej wyłącznie nanocząstkami magnetycznymi jest
niemożliwe. Jest to spowodowane silnymi interferencjami pochodzącymi od
hemoglobiny i ferrytyny. O ile hemoglobinę można łatwo oddzielić od osocza krwi
poprzez odwirowanie elementów morfotycznych, to jednak taki sposób
postępowania nie prowadzi do usunięcia z roztworu ferrytyny, która jest głównym
interferentem w analizie transferyny w osoczu krwi. Dlatego też, zastosowanie
wysoce selektywnego oddziaływania antygenprzeciwciało stanowiło podstawę
oznaczania transferyny we krwi.

Elektrochemiczny immunosensor do detekcji transferyny
148
12.1. Procedura konstrukcji warstwy receptorowej
immunosensora
Głównym elementem budującym warstwę receptorową immunosensora, poza
nanocząstkami magnetycznymi, były cząsteczki przeciwciała monoklonalnego typu
IgG charakterystyczne dla ludzkiej transferyny. Z literatury wiadomo, że orientacja
przeciwciała względem powierzchni bezpośrednio wynikająca z faktu, które grupy
funkcyjne przeciwciała biorą udział w procesie unieruchamiania, decyduje
o skuteczności jego działania [437]. W cząsteczce przeciwciała można wyróżnić
kilka potencjalnych grup, które mogą uczestniczyć w procesie jego unieruchamiania
na wybranej powierzchni. Najczęściej wykorzystuje się w tym celu grupy aminowe
zlokalizowane na końcach łańcuchów polipeptydowych w części zmiennej
przeciwciała, czy też grupy aminowe reszt lizyny obecne w części stałej Ab [438].
Zakotwiczenie przeciwciała na powierzchni elektrody za pośrednictwem grup
aminowych może prowadzić do pochylonego lub/i równoległego oraz pionowego
odwróconego ułożenia cząsteczek Ab względem jej powierzchni [437]. Z punktu
widzenia elektrochemii taki sposób ułożenia przeciwciała jest niezwykle korzystny,
gdyż zapewnia najkrótszy dystans pomiędzy centrum elektroaktywnym oznaczanego
białka a powierzchnią elektrody. Z kolei w sytuacji, gdy w procesie unieruchamiania
przeciwciała biorą udział grupy karboksylowe, znajdujące się na końcu łańcuchów
polipeptydowych domeny stałej [438], cząsteczki Ab są wówczas zorientowane
prostopadle względem powierzchni elektrody [437]. Taki sposób ułożenia
przeciwciała na wybranej powierzchni, zwany pionowym powoduje, że detekcja
elektrochemiczna białek jest znacznie utrudniona. W takim ustawieniu przeciwciało
stanowi największą barierę w transporcie elektronów. Możliwe sposoby ułożenia
przeciwciała na powierzchni elektrody ilustruje rysunek 66.

Elektrochemiczny immunosensor do detekcji transferyny
149
NH
2
SSSS
NH
2
H2 N
H2 N
CO
OH
HO
OC
SS
SS
SS
SS
S
S
S
S
S
S
S
S
S
S
S
S
S
S
LY
S-NH
2
H2 N
-LY
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
NH2
S S
S S
NH2H2N
H2N
COOHHOOC
SS
SS
SS S
S
SS
SS
S
S
S
S
S
S
LYS-NH2H2N-LYS S
S
SS
SS
PIONOWE ODWRÓCONEUŁOŻENIE
Ab vs Aelektrody
S S
S S
NH2
NH2H2N
H2N
COOHHOOC
SS
SS
SS
SS
SS
SS
SS
SS
S
S
S
S
S
S
S
S LYS-NH2H2N-LYS
POCHYLONEUŁOŻENIE
Ab vs Aelektrody
RÓWNOLEGŁE (ZWANE PŁASKIM)
UŁOŻENIE Ab vs Aelektrody
S SS S
NH2
H2NH
2N
COOH
HOOC
S
S
S
S
S
S
S
S
S S
S S
S S
S S
S
SS
S
S
S
S
S
LYS-NH2
H2N-LYS
PIONOWE UŁOŻENIE
Ab vs Aelektrody
WIĄZANIE PRZECIWCIAŁA Z POWIERZCHNIĄ PRZY UDZIALE JEGO GRUP COOH
WIĄZANIE PRZECIWCIAŁA Z POWIERZCHNIĄ PRZY UDZIALE JEGO GRUP NH2
Rysunek 66. Możliwe sposoby ułożenia cząsteczek przeciwciała w stosunku do powierzchni
elektrody. Rysunek wykonany w oparciu o dane literaturowe [437].
Orientacja przeciwciała względem powierzchni elektrody wpływa nie tylko na jego
efektywność działania, ale także na jego gęstość upakowania w warstwie. Znając
wymiary przeciwciała typu IgG (fragmentu Fab: 6.5×3.5 nm oraz fragmentu Fc:
5×3.5 nm) [439], przy założeniu maksymalnego upakowania Ab w płaszczyźnie,
można wyznaczyć teoretyczne maksymalne stężenie powierzchniowe przeciwciała,
które wynosi 7.30, 4.52 i 2.43 pmol·cm-2
, odpowiednio dla pionowego, pochylonego
oraz równoległego ułożenia Ab względem powierzchni elektrody.
W celu trwałego związania przeciwciała z powierzchnią złota, elektrodę
pokryto najpierw monowarstwą cysteaminy (CSH). Warstwa taka została utworzona
na drodze samoorganizacji, w wyniku umieszczenia elektrody złotej w 0.5 mM
etanolowym roztworze cysteaminy na czas 12 godzin. Po tym czasie elektrodę
delikatnie opłukiwano 96% alkoholem etylowym i osuszano bardzo delikatnym
strumieniem argonu. Kolejny etap polegał na związaniu z monowarstwą CSH
nanocząstek magnetycznych za pośrednictwem wiązania amidowego, utworzonego
pomiędzy grupami aminowymi cysteaminy i grupami karboksylowymi nanokapsuł
Fe@C-COOH. W celu ograniczenia procesu aglomeracji nanocząstek

Elektrochemiczny immunosensor do detekcji transferyny
150
magnetycznych oraz utworzenia równomiernej warstwy pod elektrodą umieszczony
został płaski magnes. Następnie, w celu wprowadzenia warstwy monoklonalnego
przeciwciała, elektrodę (Au-CSH/Fe@C-COOH Nps) zanurzono na 12 h do
20 mgmL-1
roztworu Ab w buforze fosforanowym z dodatkiem 0.2% v/v Tween-20.
Takie stężenie Ab gwarantowało związanie maksymalnej ilości przeciwciał
z powierzchnią nanocząstek magnetycznych. Niejonowy detergent, Tween-20, został
zastosowany w celu zablokowania powierzchni elektrody oraz wyeliminowania
niespecyficznych oddziaływań pomiędzy cząsteczkami przeciwciała a podłożem.
Aby usunąć z powierzchni elektrody nadmiar nieprzereagowanego przeciwciała,
elektrodę delikatnie opłukiwano buforem fosforanowym z dodatkiem Tweenu-20.
Przeciwciało unieruchamiane było na powierzchni nanocząstek magnetycznych
również za pośrednictwem wiązań amidowych utworzonych pomiędzy grupami
aminowymi Ab i grupami karboksylowymi nanocząstek. Tak zmodyfikowaną
elektrodę zanurzano do roztworu transferyny o różnym stężeniu w zakresie od 5·10-7
do 5·10-2
g·dL-1
(5·10-3
- 500 mg·mL-1
), na czas 60 minut w obecności zewnętrznego
źródła pola magnetycznego. Schemat ilustrujący poszczególne etapy konstrukcji
immunosensora do detekcji transferyny przedstawia rysunek 67.
czas / s
f
/ H
z
3) Ab
Au
4) Tf
CO
NH
C
O
NHCO
NH
CO
NH
2) zaktywowane
Fe@C-COOH Nps
1) CSH
E / V
I /
A
5) detekcja
Rysunek 67. Schemat ilustrujący etapy konstrukcji immunosensora do detekcji transferyny.
Związanie cząsteczek Tf z warstwą sensorową (Au-CSH/Fe@C-COOH
Nps/Ab) za pośrednictwem specyficznego oddziaływania antygenprzeciwciało
zostało potwierdzone za pomocą techniki powierzchniowego rezonansu
plazmonowego (SPR). Dzięki temu, że pomiary prowadzono w ciągłym przepływie
możliwe było zoptymalizowanie czasu oddziaływania transferyny
z charakterystycznym dla niej przeciwciałem. Typowe sensogramy SPR
zarejestrowane podczas formowania warstwy sensorowej oraz wiązania się do niej
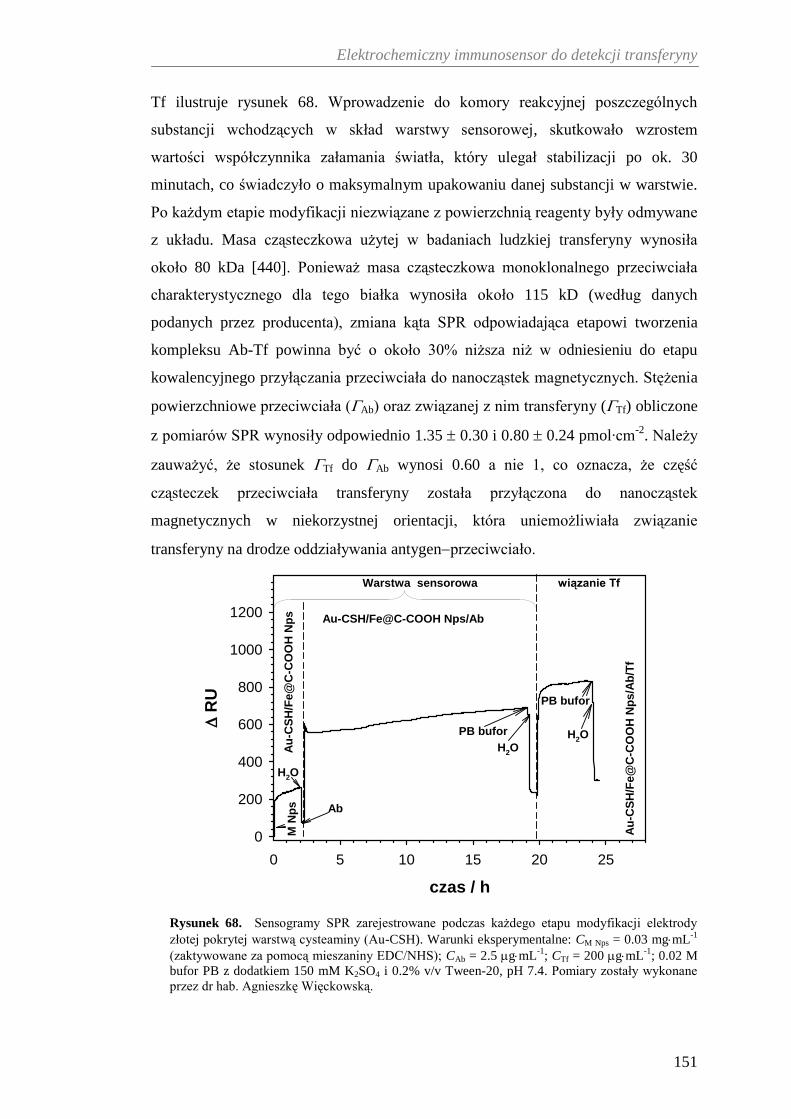
Elektrochemiczny immunosensor do detekcji transferyny
151
Tf ilustruje rysunek 68. Wprowadzenie do komory reakcyjnej poszczególnych
substancji wchodzących w skład warstwy sensorowej, skutkowało wzrostem
wartości współczynnika załamania światła, który ulegał stabilizacji po ok. 30
minutach, co świadczyło o maksymalnym upakowaniu danej substancji w warstwie.
Po każdym etapie modyfikacji niezwiązane z powierzchnią reagenty były odmywane
z układu. Masa cząsteczkowa użytej w badaniach ludzkiej transferyny wynosiła
około 80 kDa [440]. Ponieważ masa cząsteczkowa monoklonalnego przeciwciała
charakterystycznego dla tego białka wynosiła około 115 kD (według danych
podanych przez producenta), zmiana kąta SPR odpowiadająca etapowi tworzenia
kompleksu Ab-Tf powinna być o około 30% niższa niż w odniesieniu do etapu
kowalencyjnego przyłączania przeciwciała do nanocząstek magnetycznych. Stężenia
powierzchniowe przeciwciała (Ab) oraz związanej z nim transferyny (Tf) obliczone
z pomiarów SPR wynosiły odpowiednio 1.35 0.30 i 0.80 0.24 pmol·cm-2
. Należy
zauważyć, że stosunek Tf do Ab wynosi 0.60 a nie 1, co oznacza, że część
cząsteczek przeciwciała transferyny została przyłączona do nanocząstek
magnetycznych w niekorzystnej orientacji, która uniemożliwiała związanie
transferyny na drodze oddziaływania antygenprzeciwciało.
czas / h
0 5 10 15 20 25
R
U
0
200
400
600
800
1000
1200
M N
ps
H2O
Au
-CS
H/F
e@
C-C
OO
H N
ps Au-CSH/Fe@C-COOH Nps/Ab
Ab
H2O
PB bufor
Au
-CS
H/F
e@
C-C
OO
H N
ps
/Ab
/Tf
wiązanie Tf
H2O
PB bufor
Warstwa sensorowa
Rysunek 68. Sensogramy SPR zarejestrowane podczas każdego etapu modyfikacji elektrody
złotej pokrytej warstwą cysteaminy (Au-CSH). Warunki eksperymentalne: CM Nps = 0.03 mgmL-1
(zaktywowane za pomocą mieszaniny EDC/NHS); CAb = 2.5 mgmL-1
; CTf = 200 mgmL-1
; 0.02 M
bufor PB z dodatkiem 150 mM K2SO4 i 0.2% v/v Tween-20, pH 7.4. Pomiary zostały wykonane
przez dr hab. Agnieszkę Więckowską.

Elektrochemiczny immunosensor do detekcji transferyny
152
W celu potwierdzenia wysokiej specyficzności wiązania Tfprzeciwciało
przeprowadzono eksperyment kontrolny z użyciem przeciwciała
charakterystycznego dla innego białka. Wartości współczynników załamania światła
przed i po etapie oddziaływania Tf z warstwą sensorową zawierającą cząsteczki
przeciwciała charakterystycznego dla innego białka nie uległy zmianie, co świadczy
o tym, że etap odmywania usunął całkowicie cząsteczki Tf z powierzchni warstwy
receptorowej czujnika, ponieważ nie uległy one związaniu z warstwą przeciwciała.
Przeprowadzone eksperymenty pokazały, że opracowana procedura prowadzi do
związania transferyny z warstwą sensorową czujnika tylko na drodze wysokiego
powinowactwa białka do charakterystycznego dla niego przeciwciała, natomiast
wszelkie niespecyficzne oddziaływania praktycznie nie mają miejsca. Każdy etap
konstrukcji immunosensora do elektrochemicznej detekcji transferyny kontrolowany
był dodatkowo przy użyciu skaningowego mikroskopu elektronowego. Zmiany
morfologii powierzchni elektrody podczas kolejnych etapów jej modyfikacji
przedstawia rysunek 69.
200 nm
NIEZMODYFIKOWANE CHEMICZNIE Au
Au-CSH Au-CSH/Fe@C-COOH Nps
200 nm 200 nm
Au-CSH/Fe@C-COOH Nps/Ab Au-CSH/Fe@C-COOH Nps/Ab/Tf
200 nm 200 nm
Rysunek 69. Zdjęcia SEM powierzchni złota po kolejnych etapach jej modyfikacji. Zdjęcia
zostały wykonane przez prof. dr hab. Mikołaja Dontena.
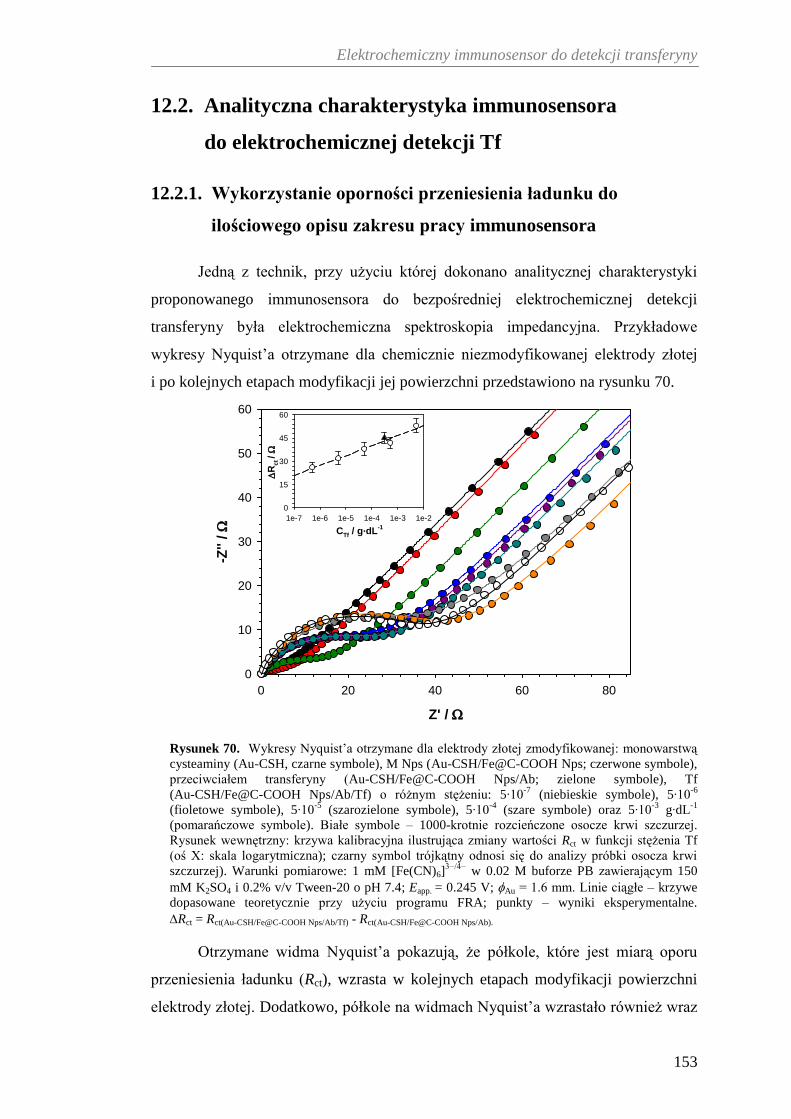
Elektrochemiczny immunosensor do detekcji transferyny
153
12.2. Analityczna charakterystyka immunosensora
do elektrochemicznej detekcji Tf
12.2.1. Wykorzystanie oporności przeniesienia ładunku do
ilościowego opisu zakresu pracy immunosensora
Jedną z technik, przy użyciu której dokonano analitycznej charakterystyki
proponowanego immunosensora do bezpośredniej elektrochemicznej detekcji
transferyny była elektrochemiczna spektroskopia impedancyjna. Przykładowe
wykresy Nyquist’a otrzymane dla chemicznie niezmodyfikowanej elektrody złotej
i po kolejnych etapach modyfikacji jej powierzchni przedstawiono na rysunku 70.
Z' /
0 20 40 60 80
-Z'' /
0
10
20
30
40
50
60
CTf
/ gdL-1
1e-7 1e-6 1e-5 1e-4 1e-3 1e-2
R
ct /
0
15
30
45
60
Rysunek 70. Wykresy Nyquist’a otrzymane dla elektrody złotej zmodyfikowanej: monowarstwą
cysteaminy (Au-CSH, czarne symbole), M Nps (Au-CSH/Fe@C-COOH Nps; czerwone symbole),
przeciwciałem transferyny (Au-CSH/Fe@C-COOH Nps/Ab; zielone symbole), Tf
(Au-CSH/Fe@C-COOH Nps/Ab/Tf) o różnym stężeniu: 5·10-7
(niebieskie symbole), 5·10-6
(fioletowe symbole), 5·10-5
(szarozielone symbole), 5·10-4
(szare symbole) oraz 5·10-3
g·dL-1
(pomarańczowe symbole). Białe symbole – 1000-krotnie rozcieńczone osocze krwi szczurzej.
Rysunek wewnętrzny: krzywa kalibracyjna ilustrująca zmiany wartości Rct w funkcji stężenia Tf
(oś X: skala logarytmiczna); czarny symbol trójkątny odnosi się do analizy próbki osocza krwi
szczurzej). Warunki pomiarowe: 1 mM [Fe(CN)6]3−/4−
w 0.02 M buforze PB zawierającym 150
mM K2SO4 i 0.2% v/v Tween-20 o pH 7.4; Eapp. = 0.245 V; Au = 1.6 mm. Linie ciągłe – krzywe
dopasowane teoretycznie przy użyciu programu FRA; punkty – wyniki eksperymentalne.
Rct = Rct(Au-CSH/Fe@C-COOH Nps/Ab/Tf) - Rct(Au-CSH/Fe@C-COOH Nps/Ab).
Otrzymane widma Nyquist’a pokazują, że półkole, które jest miarą oporu
przeniesienia ładunku (Rct), wzrasta w kolejnych etapach modyfikacji powierzchni
elektrody złotej. Dodatkowo, półkole na widmach Nyquist’a wzrastało również wraz
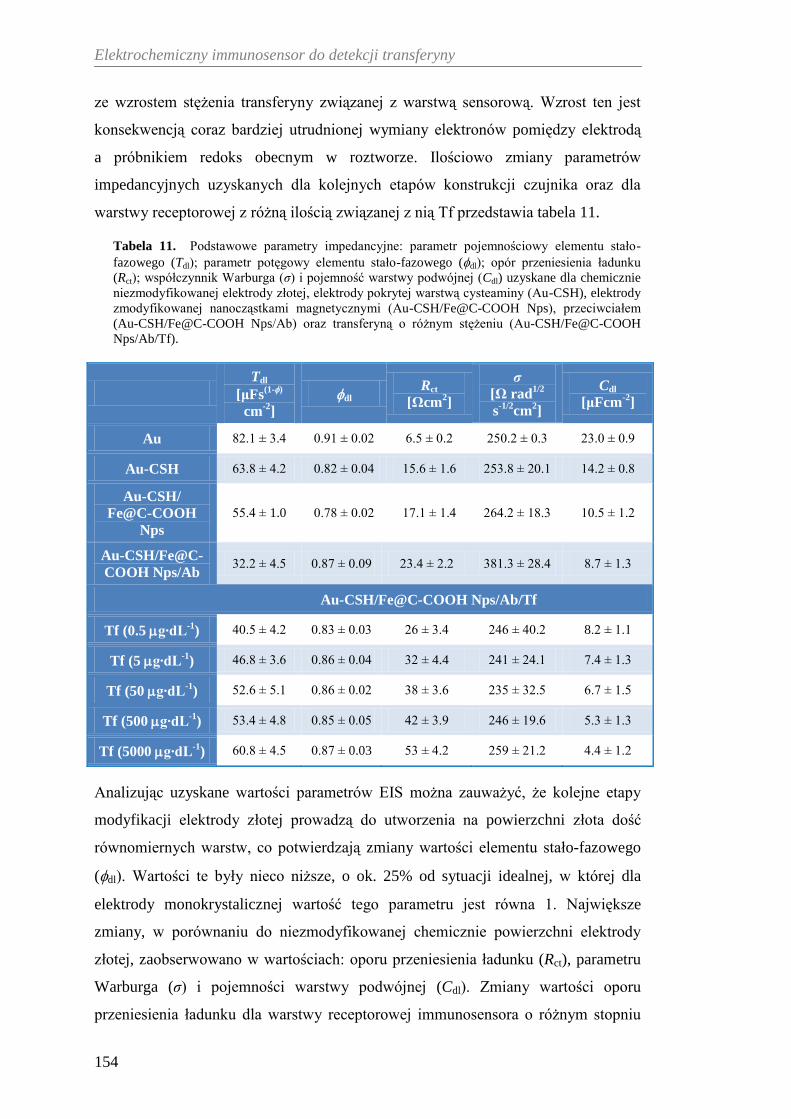
Elektrochemiczny immunosensor do detekcji transferyny
154
ze wzrostem stężenia transferyny związanej z warstwą sensorową. Wzrost ten jest
konsekwencją coraz bardziej utrudnionej wymiany elektronów pomiędzy elektrodą
a próbnikiem redoks obecnym w roztworze. Ilościowo zmiany parametrów
impedancyjnych uzyskanych dla kolejnych etapów konstrukcji czujnika oraz dla
warstwy receptorowej z różną ilością związanej z nią Tf przedstawia tabela 11.
Tabela 11. Podstawowe parametry impedancyjne: parametr pojemnościowy elementu stało-
fazowego (Tdl); parametr potęgowy elementu stało-fazowego (dl); opór przeniesienia ładunku
(Rct); współczynnik Warburga (σ) i pojemność warstwy podwójnej (Cdl) uzyskane dla chemicznie
niezmodyfikowanej elektrody złotej, elektrody pokrytej warstwą cysteaminy (Au-CSH), elektrody
zmodyfikowanej nanocząstkami magnetycznymi (Au-CSH/Fe@C-COOH Nps), przeciwciałem
(Au-CSH/Fe@C-COOH Nps/Ab) oraz transferyną o różnym stężeniu (Au-CSH/Fe@C-COOH
Nps/Ab/Tf).
Tdl
[μFs(1-)
cm-2
]
dl Rct
[Ωcm2]
σ
[Ω rad1/2
s-1/2
cm2]
Cdl
[μFcm-2
]
Au 82.1 ± 3.4 0.91 ± 0.02 6.5 ± 0.2 250.2 ± 0.3 23.0 ± 0.9
Au-CSH 63.8 ± 4.2 0.82 ± 0.04 15.6 ± 1.6 253.8 ± 20.1 14.2 ± 0.8
Au-CSH/
Fe@C-COOH
Nps
55.4 ± 1.0 0.78 ± 0.02 17.1 ± 1.4 264.2 ± 18.3 10.5 ± 1.2
Au-CSH/Fe@C-
COOH Nps/Ab 32.2 ± 4.5 0.87 ± 0.09 23.4 ± 2.2 381.3 ± 28.4 8.7 ± 1.3
Au-CSH/Fe@C-COOH Nps/Ab/Tf
Tf (0.5 mg·dL-1
) 40.5 ± 4.2 0.83 ± 0.03 26 ± 3.4 246 ± 40.2 8.2 ± 1.1
Tf (5 mg·dL-1
) 46.8 ± 3.6 0.86 ± 0.04 32 ± 4.4 241 ± 24.1 7.4 ± 1.3
Tf (50 mg·dL-1
) 52.6 ± 5.1 0.86 ± 0.02 38 ± 3.6 235 ± 32.5 6.7 ± 1.5
Tf (500 mg·dL-1
) 53.4 ± 4.8 0.85 ± 0.05 42 ± 3.9 246 ± 19.6 5.3 ± 1.3
Tf (5000 mg·dL-1
) 60.8 ± 4.5 0.87 ± 0.03 53 ± 4.2 259 ± 21.2 4.4 ± 1.2
Analizując uzyskane wartości parametrów EIS można zauważyć, że kolejne etapy
modyfikacji elektrody złotej prowadzą do utworzenia na powierzchni złota dość
równomiernych warstw, co potwierdzają zmiany wartości elementu stało-fazowego
(dl). Wartości te były nieco niższe, o ok. 25% od sytuacji idealnej, w której dla
elektrody monokrystalicznej wartość tego parametru jest równa 1. Największe
zmiany, w porównaniu do niezmodyfikowanej chemicznie powierzchni elektrody
złotej, zaobserwowano w wartościach: oporu przeniesienia ładunku (Rct), parametru
Warburga (σ) i pojemności warstwy podwójnej (Cdl). Zmiany wartości oporu
przeniesienia ładunku dla warstwy receptorowej immunosensora o różnym stopniu

Elektrochemiczny immunosensor do detekcji transferyny
155
wysycenia transferyną wykorzystano do konstrukcji krzywej kalibracyjnej
(Rct = Rct(Au-CSH/Fe@C-COOH Nps/Ab/Tf) - Rct(Au-CSH/Fe@C-COOH Nps/Ab) górny rysunek
wewnętrzny na wykresie 70). W szerokim zakresie stężeń transferyny od 5·10-7
do
5·10-2
g·dL-1
(5·10-3
÷ 500 mg·mL-1
) obserwowano liniową zależność wartości oporu
przeniesienia ładunku (Rct) w funkcji stężenia transferyny. Wyznaczona na podstawie
danych impedancyjnych, metodą Hubaux-Vos, granica wykrywalności wynosiła
15.0 2.4 ng·dL-1
.
12.2.2. Grawimetryczna charakterystyka immunosensora
Zastosowanie podczas pomiarów kryształu kwarcu z napyloną warstwą złota
(Au-EQCM) umożliwiło jednoczesną grawimetryczną i woltamperometryczną
charakterystykę zaproponowanego immunosensora do detekcji Tf. Pomiary
z użyciem elektrochemicznej mikrowagi kwarcowej przeprowadzono dla różnych
stężeń transferyny z zakresu od 5·10-7
do 5·10-2
g·dL-1
. Zależność zmian
częstotliwości drgań złotego kryształu kwarcu podczas procesu wiązania transferyny
z warstwą sensorową, w wyniku wysoce specyficznego oddziaływania
antygenprzeciwciało, przedstawia rysunek 71.
czas / s
0 5000 10000 15000
f
/ H
z
0.00510-4 gdL
-1
0.0510-4
gdL-1
0.510-4 gdL
-1
510-4 gdL
-1
5010-4 gdL
-1
-100 Hz
1000-krotnie rozcieńczone osocze
Rysunek 71. Zależności zmian częstotliwości drgań kryształu kwarcu podczas procesu
oddziaływania transferyny z warstwą sensorową w obecności zewnętrznego źródła pola
magnetycznego (90 mT). Czerwona krzywa ilustruje proces unieruchamiania transferyny
znajdującej się w 1000-krotnie rozcieńczonym osoczu krwi szczurzej. Warunki pomiarowe:
0.02 M bufor PB z dodatkiem 150 mM K2SO4 oraz 0.2 % v/v Tween-20, pH 7.4.

Elektrochemiczny immunosensor do detekcji transferyny
156
Zarejestrowane krzywe charakteryzują się dobrze widocznymi oscylacjami, których
amplituda (AΔf) silnie zależała od zastosowanego stężenia transferyny. Dodatkowo,
oscylacje te widoczne były tylko podczas procesu unieruchamiania transferyny
w warstwie sensorowej. Wyjaśnienie obserwowanych oscylacji nie jest proste.
W początkowym etapie wiązania Tf z warstwą receptorową immunosensora
widoczny jest spadek częstotliwości drgań kryształu kwarcu. Najprawdopodobniej
w wyniku obecności pola magnetycznego ma miejsce zmiana: (i) konformacji
paramagnetycznej cząsteczki transferyny i (ii) gęstości jej upakowania w warstwie.
To z kolei mogło skutkować utratą otoczki hydratacyjnej i zmianą
wiskoelastyczności warstwy Tf. W konsekwencji obserwowany był wzrost
częstotliwości drgań kryształu kwarcu. Następnie warstwa białka ulegała ponownie
uwodnieniu i znów obserwowany był spadek częstotliwości drgań kryształu kwarcu.
Wzrost stopnia wysycenia warstwy receptorowej cząsteczkami transferyny
skutkował znacznie wolniejszymi zmianami częstotliwości drgań kryształu kwarcu,
gdyż tworzona warstwa była bardziej upakowana. Z uwagi na fakt, że roztwór
transferyny był odtleniany podczas pomiaru, obserwowane oscylacje można
przypisać jedynie oddziaływaniu pola magnetycznego z paramagnetyczną
transferyną.
W celu uzyskania informacji na temat wpływu natężenia pola magnetycznego
na wartości amplitudy oscylacji częstotliwości drgań kryształu kwarcu
przeprowadzono eksperyment, w którym zmieniano odległość magnesu względem
powierzchni elektrody. Zarejestrowane krzywe ilustrujące zmiany częstotliwości
drgań kryształu kwarcu ilustruje rysunek 72. Na podstawie uzyskanych krzywych
można stwierdzić, że obecność pola magnetycznego oraz jego natężenie silnie
wpływają na kształt pojawiających się oscylacji drgań kryształu kwarcu, jak również
na ich amplitudę. Usunięcie magnesu skutkowało całkowitym zanikiem oscylacji.
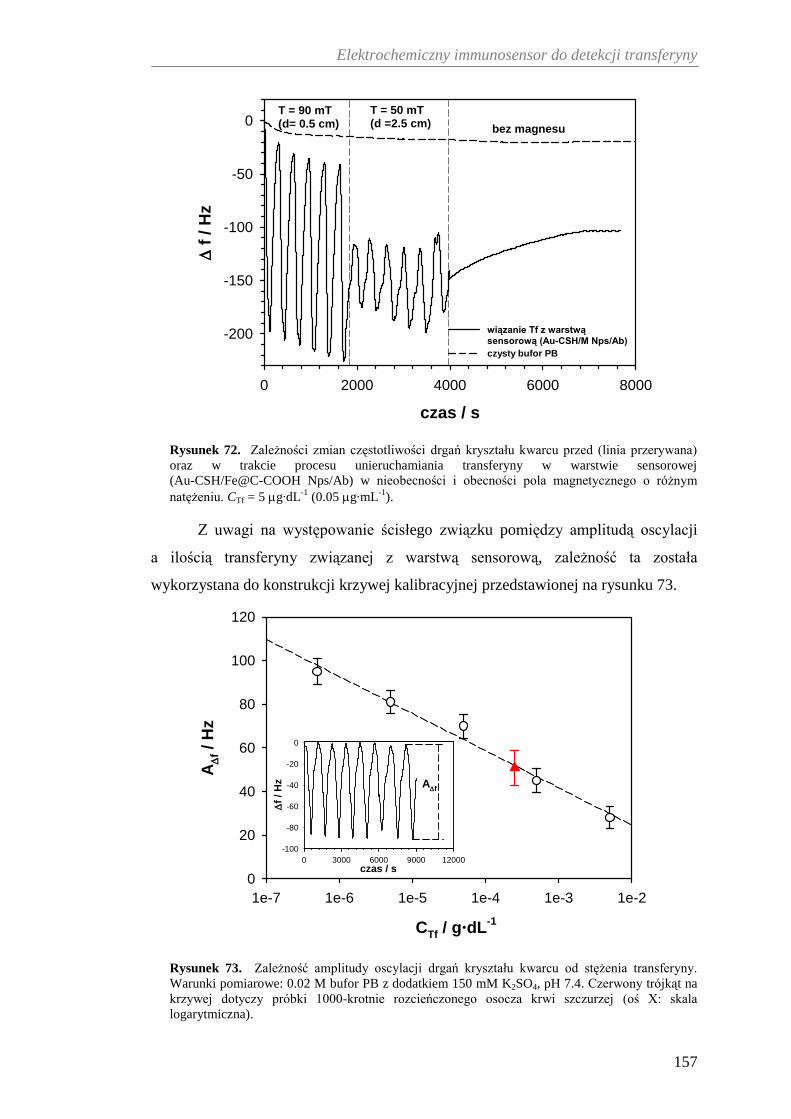
Elektrochemiczny immunosensor do detekcji transferyny
157
0.005 g/ml
0.05 g/ml
0.5 g/ml
5 g/ml
50 g/ml
czas/s vs f/Hz
czas / s
0 2000 4000 6000 8000
f
/ H
z
-200
-150
-100
-50
0
wiązanie Tf z warstwą
sensorową (Au-CSH/M Nps/Ab)
czysty bufor PB
T = 90 mT(d= 0.5 cm)
T = 50 mT(d =2.5 cm)
bez magnesu
Rysunek 72. Zależności zmian częstotliwości drgań kryształu kwarcu przed (linia przerywana)
oraz w trakcie procesu unieruchamiania transferyny w warstwie sensorowej
(Au-CSH/Fe@C-COOH Nps/Ab) w nieobecności i obecności pola magnetycznego o różnym
natężeniu. CTf = 5 mg·dL-1
(0.05 mg·mL-1
).
Z uwagi na występowanie ścisłego związku pomiędzy amplitudą oscylacji
a ilością transferyny związanej z warstwą sensorową, zależność ta została
wykorzystana do konstrukcji krzywej kalibracyjnej przedstawionej na rysunku 73.
CTf / gdL
-1
1e-7 1e-6 1e-5 1e-4 1e-3 1e-2
A
f /
Hz
0
20
40
60
80
100
120
czas / s0 3000 6000 9000 12000
f
/ H
z
-100
-80
-60
-40
-20
0
Af
Rysunek 73. Zależność amplitudy oscylacji drgań kryształu kwarcu od stężenia transferyny.
Warunki pomiarowe: 0.02 M bufor PB z dodatkiem 150 mM K2SO4, pH 7.4. Czerwony trójkąt na
krzywej dotyczy próbki 1000-krotnie rozcieńczonego osocza krwi szczurzej (oś X: skala
logarytmiczna).

Elektrochemiczny immunosensor do detekcji transferyny
158
Uzyskana w ten sposób krzywa kalibracyjna charakteryzowała się liniowością
w zakresie stężeń transferyny od 5·10-7
(0.005 mg·mL-1
) do 5·10-2
g·dL-1
(500 mg·mL-1
). Granica wykrywalności wyznaczona metodą Hubaux–Vos wynosiła
12.0 1.8 ng·dL-1
.
12.2.3. Woltamperometryczna detekcja transferyny
Biorąc pod uwagę fakt, że transferyna zawiera w swojej strukturze jony
Fe(III), które są odpowiedzialne za jej elektroaktywność, kolejny etap badań
dotyczył sprawdzenia możliwości woltamperometrycznej detekcji transferyny.
Rejestracja dobrze wykształconych sygnałów prądowych transferyny możliwa jest
tylko w przypadku osiągnięcia odpowiedniej jej orientacji względem powierzchni
elektrody, która umożliwia bezpośredni transport elektronów pomiędzy centrum
redoks białka a elektrodą. Ze względu na przynależność transferyny do grupy
paramagnetycznych białek zastosowanie zewnętrznego pola magnetycznego
w połączeniu z ferromagnetycznym modyfikatorem powierzchni elektrody
umożliwia osiągnięcie wymaganej orientacji transferyny. Z kolei zastosowana
procedura wiązania przeciwciała z powierzchnią elektrody zapewniała odpowiednie
ułożenie przeciwciała w warstwie sensorowej, które pozwalało na bezpośredni
transport elektronów pomiędzy centrum elektroaktywnym transferyny a elektrodą.
Pomiary woltamperometryczne przeprowadzone zostały w obecności oraz
nieobecności zewnętrznego źródła pola magnetycznego w układzie. W celu
potwierdzenia, że uzyskany sygnał prądowy pochodzi od transferyny związanej
z warstwą sensorową immunosensora jedynie na drodze jej oddziaływania
z przeciwciałem, wykonano eksperyment kontrolny, w którym niezmodyfikowana
chemicznie elektroda złota, elektroda zmodyfikowana warstwą cysteaminy
(Au-CSH), następnie nanocząstkami magnetycznymi (Au-CSH/Fe@C-COOH Nps)
i ostatecznie przeciwciałem (Au-CSH/Fe@C-COOH Nps/Ab) została zanurzona na
60 minut do roztworu transferyny w obecności pola magnetycznego. Po tym czasie
elektroda była opłukiwana delikatnym strumieniem buforu PB z dodatkiem 0.15 M
K2SO4 i 0.2% v/v Tweenu-20, a następnie przeprowadzany był pomiar przy użyciu
woltamperometrii liniowej w odtlenionym 0.02 M buforze fosforanowym
z dodatkiem 0.15 M K2SO4. Uzyskane krzywe woltamperometryczne przedstawia
rysunek 74.
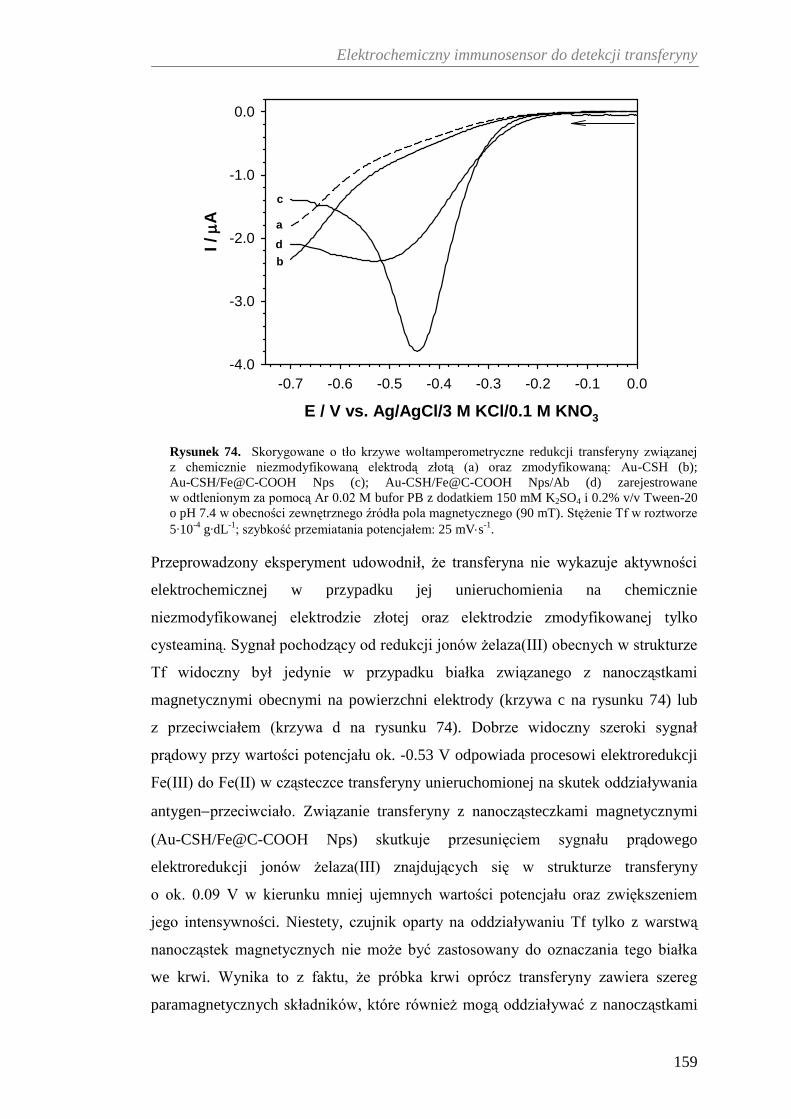
Elektrochemiczny immunosensor do detekcji transferyny
159
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.7 -0.6 -0.5 -0.4 -0.3 -0.2 -0.1 0.0
I /
A
-4.0
-3.0
-2.0
-1.0
0.0
a
b
d
c
Rysunek 74. Skorygowane o tło krzywe woltamperometryczne redukcji transferyny związanej
z chemicznie niezmodyfikowaną elektrodą złotą (a) oraz zmodyfikowaną: Au-CSH (b);
Au-CSH/Fe@C-COOH Nps (c); Au-CSH/Fe@C-COOH Nps/Ab (d) zarejestrowane
w odtlenionym za pomocą Ar 0.02 M bufor PB z dodatkiem 150 mM K2SO4 i 0.2% v/v Tween-20
o pH 7.4 w obecności zewnętrznego źródła pola magnetycznego (90 mT). Stężenie Tf w roztworze
5·10-4
g·dL-1
; szybkość przemiatania potencjałem: 25 mVs-1
.
Przeprowadzony eksperyment udowodnił, że transferyna nie wykazuje aktywności
elektrochemicznej w przypadku jej unieruchomienia na chemicznie
niezmodyfikowanej elektrodzie złotej oraz elektrodzie zmodyfikowanej tylko
cysteaminą. Sygnał pochodzący od redukcji jonów żelaza(III) obecnych w strukturze
Tf widoczny był jedynie w przypadku białka związanego z nanocząstkami
magnetycznymi obecnymi na powierzchni elektrody (krzywa c na rysunku 74) lub
z przeciwciałem (krzywa d na rysunku 74). Dobrze widoczny szeroki sygnał
prądowy przy wartości potencjału ok. -0.53 V odpowiada procesowi elektroredukcji
Fe(III) do Fe(II) w cząsteczce transferyny unieruchomionej na skutek oddziaływania
antygenprzeciwciało. Związanie transferyny z nanocząsteczkami magnetycznymi
(Au-CSH/Fe@C-COOH Nps) skutkuje przesunięciem sygnału prądowego
elektroredukcji jonów żelaza(III) znajdujących się w strukturze transferyny
o ok. 0.09 V w kierunku mniej ujemnych wartości potencjału oraz zwiększeniem
jego intensywności. Niestety, czujnik oparty na oddziaływaniu Tf tylko z warstwą
nanocząstek magnetycznych nie może być zastosowany do oznaczania tego białka
we krwi. Wynika to z faktu, że próbka krwi oprócz transferyny zawiera szereg
paramagnetycznych składników, które również mogą oddziaływać z nanocząstkami
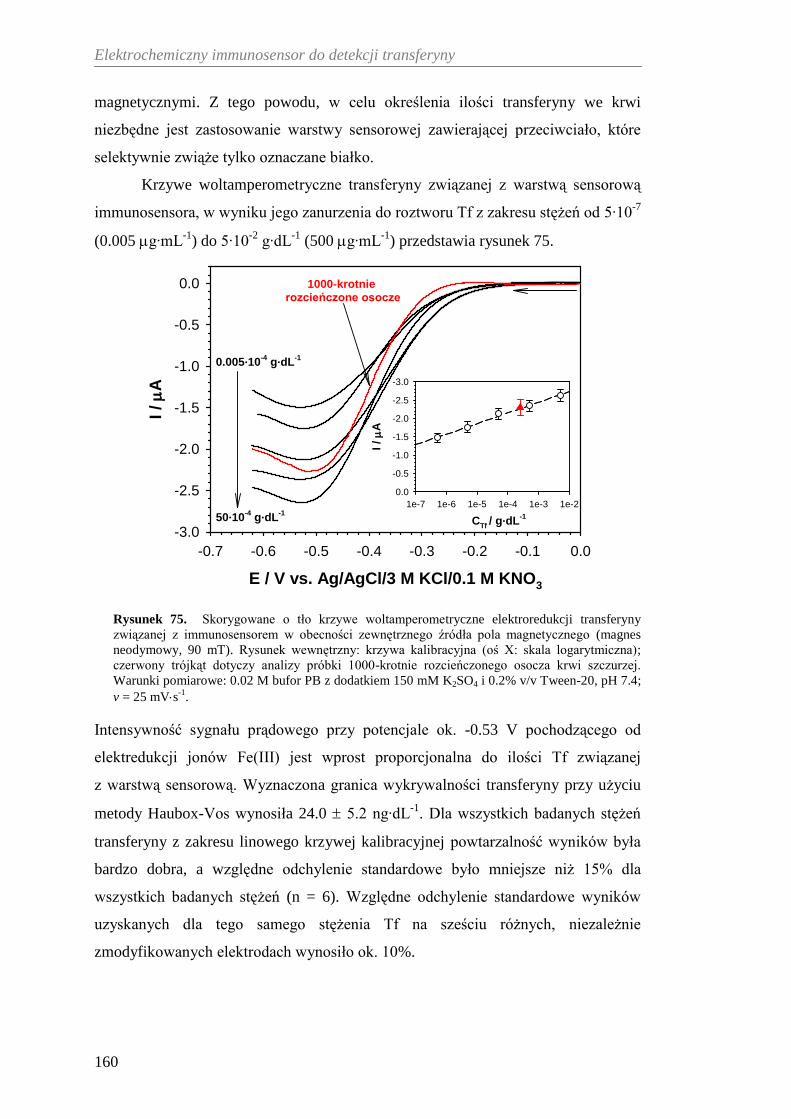
Elektrochemiczny immunosensor do detekcji transferyny
160
magnetycznymi. Z tego powodu, w celu określenia ilości transferyny we krwi
niezbędne jest zastosowanie warstwy sensorowej zawierającej przeciwciało, które
selektywnie zwiąże tylko oznaczane białko.
Krzywe woltamperometryczne transferyny związanej z warstwą sensorową
immunosensora, w wyniku jego zanurzenia do roztworu Tf z zakresu stężeń od 5·10-7
(0.005 mg·mL-1
) do 5·10-2
g·dL-1
(500 mg·mL-1
) przedstawia rysunek 75.
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.7 -0.6 -0.5 -0.4 -0.3 -0.2 -0.1 0.0
I /
A
-3.0
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
0.00510-4
gdL-1
5010-4 gdL
-1
1000-krotnie rozcieńczone osocze
CTf
/ gdL-1
1e-7 1e-6 1e-5 1e-4 1e-3 1e-2
I /
A-3.0
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
Rysunek 75. Skorygowane o tło krzywe woltamperometryczne elektroredukcji transferyny
związanej z immunosensorem w obecności zewnętrznego źródła pola magnetycznego (magnes
neodymowy, 90 mT). Rysunek wewnętrzny: krzywa kalibracyjna (oś X: skala logarytmiczna);
czerwony trójkąt dotyczy analizy próbki 1000-krotnie rozcieńczonego osocza krwi szczurzej.
Warunki pomiarowe: 0.02 M bufor PB z dodatkiem 150 mM K2SO4 i 0.2% v/v Tween-20, pH 7.4;
v = 25 mVs-1
.
Intensywność sygnału prądowego przy potencjale ok. -0.53 V pochodzącego od
elektredukcji jonów Fe(III) jest wprost proporcjonalna do ilości Tf związanej
z warstwą sensorową. Wyznaczona granica wykrywalności transferyny przy użyciu
metody Haubox-Vos wynosiła 24.0 5.2 ng·dL-1
. Dla wszystkich badanych stężeń
transferyny z zakresu linowego krzywej kalibracyjnej powtarzalność wyników była
bardzo dobra, a względne odchylenie standardowe było mniejsze niż 15% dla
wszystkich badanych stężeń (n = 6). Względne odchylenie standardowe wyników
uzyskanych dla tego samego stężenia Tf na sześciu różnych, niezależnie
zmodyfikowanych elektrodach wynosiło ok. 10%.
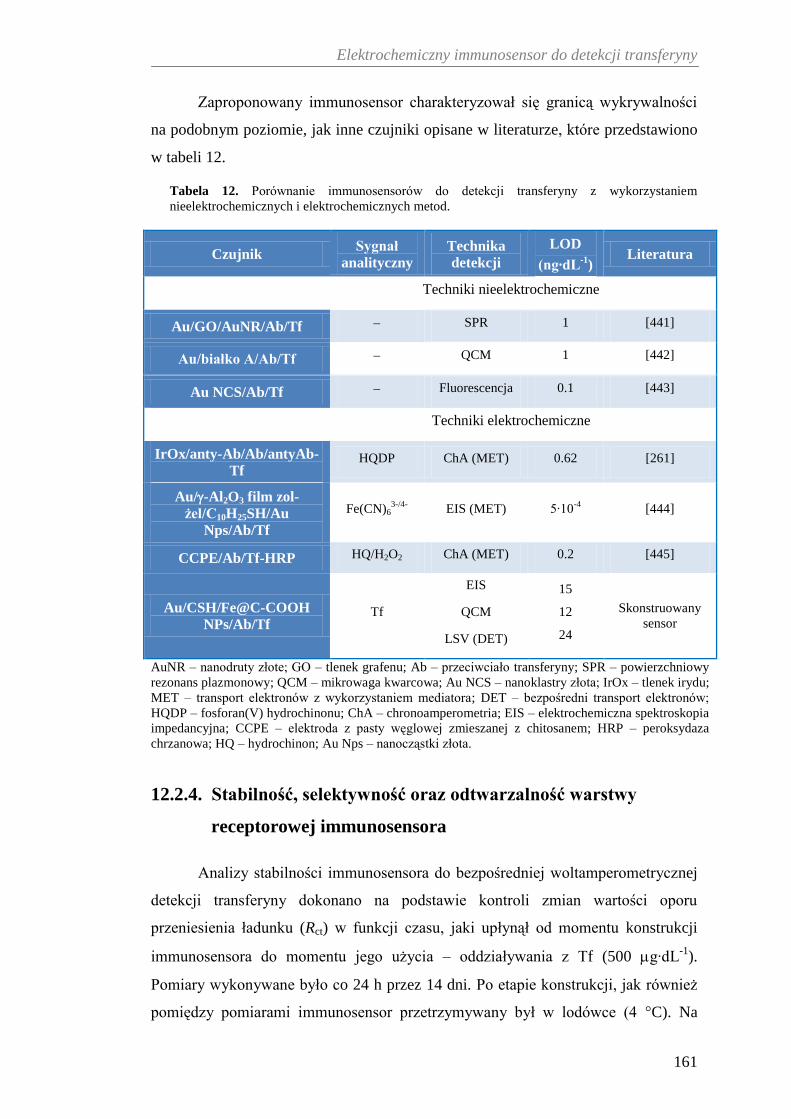
Elektrochemiczny immunosensor do detekcji transferyny
161
Zaproponowany immunosensor charakteryzował się granicą wykrywalności
na podobnym poziomie, jak inne czujniki opisane w literaturze, które przedstawiono
w tabeli 12.
Tabela 12. Porównanie immunosensorów do detekcji transferyny z wykorzystaniem
nieelektrochemicznych i elektrochemicznych metod.
Czujnik Sygnał
analityczny
Technika
detekcji
LOD
(ng·dL-1
) Literatura
Techniki nieelektrochemiczne
Au/GO/AuNR/Ab/Tf SPR 1 [441]
Au/białko A/Ab/Tf QCM 1 [442]
Au NCS/Ab/Tf Fluorescencja 0.1 [443]
Techniki elektrochemiczne
IrOx/anty-Ab/Ab/antyAb-
Tf HQDP ChA (MET) 0.62 [261]
Au/-Al2O3 film zol-
żel/C10H25SH/Au
Nps/Ab/Tf
Fe(CN)63-/4-
EIS (MET) 5·10-4
[444]
CCPE/Ab/Tf-HRP HQ/H2O2 ChA (MET) 0.2 [445]
Au/CSH/Fe@C-COOH
NPs/Ab/Tf Tf
EIS
QCM
LSV (DET)
15
12
24
Skonstruowany
sensor
AuNR – nanodruty złote; GO – tlenek grafenu; Ab – przeciwciało transferyny; SPR – powierzchniowy
rezonans plazmonowy; QCM – mikrowaga kwarcowa; Au NCS – nanoklastry złota; IrOx – tlenek irydu;
MET – transport elektronów z wykorzystaniem mediatora; DET – bezpośredni transport elektronów;
HQDP – fosforan(V) hydrochinonu; ChA – chronoamperometria; EIS – elektrochemiczna spektroskopia
impedancyjna; CCPE – elektroda z pasty węglowej zmieszanej z chitosanem; HRP – peroksydaza
chrzanowa; HQ – hydrochinon; Au Nps – nanocząstki złota.
12.2.4. Stabilność, selektywność oraz odtwarzalność warstwy
receptorowej immunosensora
Analizy stabilności immunosensora do bezpośredniej woltamperometrycznej
detekcji transferyny dokonano na podstawie kontroli zmian wartości oporu
przeniesienia ładunku (Rct) w funkcji czasu, jaki upłynął od momentu konstrukcji
immunosensora do momentu jego użycia – oddziaływania z Tf (500 mg·dL-1
).
Pomiary wykonywane było co 24 h przez 14 dni. Po etapie konstrukcji, jak również
pomiędzy pomiarami immunosensor przetrzymywany był w lodówce (4 °C). Na

Elektrochemiczny immunosensor do detekcji transferyny
162
podstawie uzyskanych wyników można stwierdzić, że skonstruowany immunosensor
do detekcji Tf charakteryzował się bardzo dobrą stabilnością, a zastosowana
procedura unieruchomienia przeciwciała pozwala na zachowanie jego specyficznej
aktywności względem transferyny. Różnice w wartościach oporu przeniesienia
ładunku w ciągu pierwszych 5 dni nie były większe niż 7%. Przez kolejne 8 dni
obserwowano coraz większe spadki wartości Rct, aż do ok. 35%, w odniesieniu do
wartości początkowej. Po czasie dłużyszm niż 13 dni uzyskiwane wartości oporu
przeniesienia ładunku były mniejsze niż 50% wartości początkowej.
Selektywność narzędzia analitycznego określana jest jako pewnego rodzaju
"przedział ufności", w którym może być ono stosowane do oznaczania substancji
poszukiwanej w obecności innych składników próbki bez występowania
interferencji, czyli bez wpływu składników matrycy na sygnał analitu. Selektywność
zaproponowanego immunosensora sprawdzono w obecności potencjalnych
substancji występujących we krwi, które mogą zakłócać oznaczanie transferyny.
W badaniach wykorzystano hemoglobinę (Hb), albuminę surowicy ludzkiej (HSA),
-glikoproteinę z ludzkiego osocza (-GP) i apo-transferynę (apo-Tf).
Eksperymenty zostały przeprowadzone dla dwóch różnych stężeń transferyny:
0.5·10-4
oraz 50·10-4
g·dL-1
, dzięki czemu uzyskano różne wysycenie warstwy
sensorowej cząsteczkami białka. Miało to na celu sprawdzenie, czy ilość transferyny
wpływa na oddziaływanie substancji zakłócających z immunosensorem oraz czy
substancje te mogą tworzyć bardziej stabilne kompleksy z przeciwciałem niż Tf.
W tym celu immunosensor (Au-CSH/Fe@C-COOH Nps/Ab) zanurzany był na czas
2 h do buforu PB zawierającego Tf oraz interferent, oba o tym samym stężeniu. Po
tym czasie elektroda była opłukiwana delikatnym strumieniem buforu PB
z dodatkiem 150 mM K2SO4 i 0.2% v/v Tween-20, a następnie przeprowadzany był
pomiar przy użyciu woltamperometrii liniowej w czystym odtlenionym 0.02 M
buforze fosforanowym z dodatkiem 0.15 M K2SO4. Wpływ substancji zakłócających
na sygnał elektroredukcji Tf (ITf + interferent / ITf) przedstawiony został na rysunku 76.
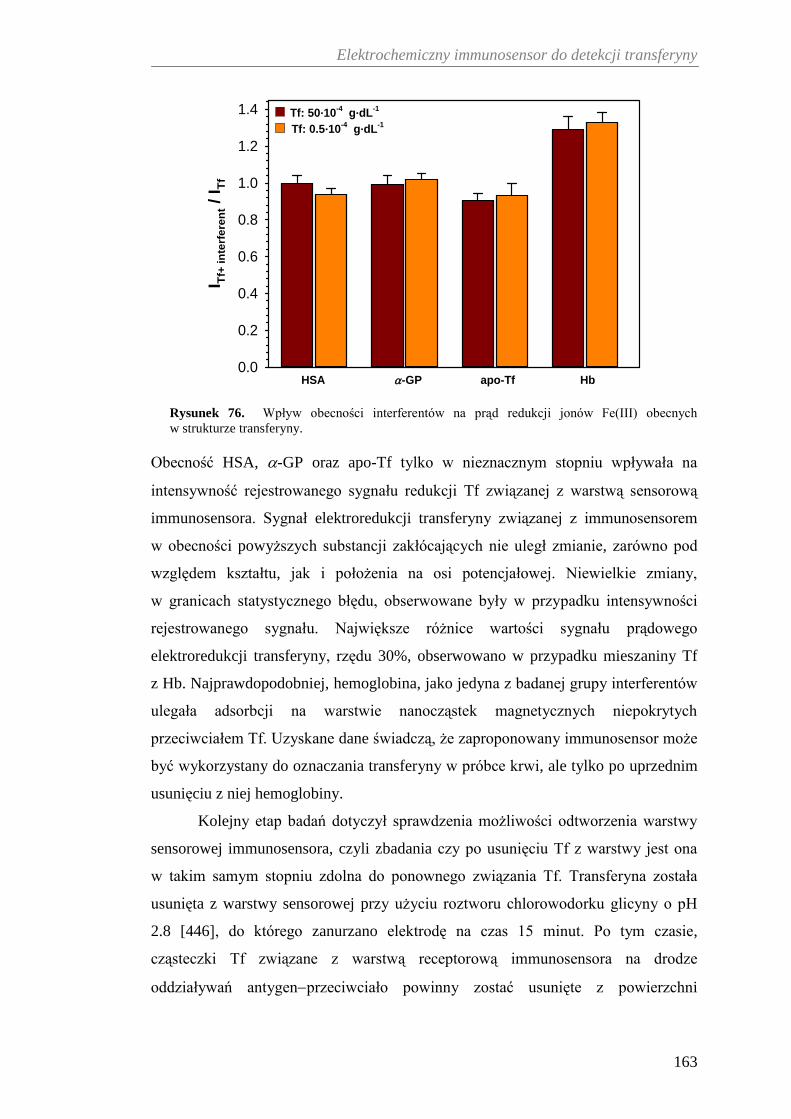
Elektrochemiczny immunosensor do detekcji transferyny
163
I Tf+
in
terf
ere
nt /
I Tf
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4 Tf: 5010-4 gdL
-1
Tf: 0.510-4
gdL-1
-GPHSA apo-Tf Hb
Rysunek 76. Wpływ obecności interferentów na prąd redukcji jonów Fe(III) obecnych
w strukturze transferyny.
Obecność HSA, -GP oraz apo-Tf tylko w nieznacznym stopniu wpływała na
intensywność rejestrowanego sygnału redukcji Tf związanej z warstwą sensorową
immunosensora. Sygnał elektroredukcji transferyny związanej z immunosensorem
w obecności powyższych substancji zakłócających nie uległ zmianie, zarówno pod
względem kształtu, jak i położenia na osi potencjałowej. Niewielkie zmiany,
w granicach statystycznego błędu, obserwowane były w przypadku intensywności
rejestrowanego sygnału. Największe różnice wartości sygnału prądowego
elektroredukcji transferyny, rzędu 30%, obserwowano w przypadku mieszaniny Tf
z Hb. Najprawdopodobniej, hemoglobina, jako jedyna z badanej grupy interferentów
ulegała adsorbcji na warstwie nanocząstek magnetycznych niepokrytych
przeciwciałem Tf. Uzyskane dane świadczą, że zaproponowany immunosensor może
być wykorzystany do oznaczania transferyny w próbce krwi, ale tylko po uprzednim
usunięciu z niej hemoglobiny.
Kolejny etap badań dotyczył sprawdzenia możliwości odtworzenia warstwy
sensorowej immunosensora, czyli zbadania czy po usunięciu Tf z warstwy jest ona
w takim samym stopniu zdolna do ponownego związania Tf. Transferyna została
usunięta z warstwy sensorowej przy użyciu roztworu chlorowodorku glicyny o pH
2.8 [446], do którego zanurzano elektrodę na czas 15 minut. Po tym czasie,
cząsteczki Tf związane z warstwą receptorową immunosensora na drodze
oddziaływań antygenprzeciwciało powinny zostać usunięte z powierzchni

Elektrochemiczny immunosensor do detekcji transferyny
164
immunosensora. Kontroli wydajności tego procesu dokonano na podstawie zmian
wartości oporu przeniesienia ładunku świeżo przygotowanej warstwy sensorowej
(Au-CSH/Fe@C-COOH Nps/Ab) i po etapie jej regeneracji w roztworze
chlorowodorku glicyny. Wykresy Nyquist’a zarejestrowane dla dwóch kolejnych
etapów regeneracji warstwy receptorowej immunosensora przedstawia rysunek 77.
Z' /
0 20 40 60 80 100 120 140
-Z'' /
0
20
40
60
80
100 świeżo przygotowana warstwa sensorowa
I regeneracja warstwy sensorowej
II regeneracja warstwy sensorowej
Rysunek 77. Wykresy Nyquist’a uzyskane dla pomiarów po oddziaływaniu transferyny
ze świeżo przygotowanym immunosensorem Au-CSH/Fe@C-COOH Nps/Ab i po jego
regeneracji. Warunki eksperymentalne: CTf = 0.05 µg·mL-1
; 0.02 M bufor PB z dodatkiem
150 mM K2SO4 i 0.2% v/v Tween-20 o pH 7.4, zawierający 1 mM [Fe(CN)6]3−/4−
; Eapp = 0.245 V.
Wykonane dwa testy regeneracyjne wykazały, że wydajność procesu
odtwarzania warstwy receptorowej immunosensora jest bardzo wysoka i wynosiła
około 90% w porównaniu do świeżo przygotowanego immunosensora do detekcji Tf.
W kolejnych cyklach regeneracji wartość Rct wzrosła (o około 25%) w odniesieniu
do wartości Rct zarejestrowanej dla świeżo przygotowanego czujnika. Oznacza to, że
skuteczność usuwania Tf z warstwy receptorowej uległa zmniejszeniu. Uzyskane
dane pozwalają stwierdzić, że skonstruowany immunosensor może być używany
trzykrotnie bez znacznej utraty czułości.
12.2.5. Zastosowanie immunosensora w analizie próbek krwi
Funkcjonalność skonstruowanego immunosensora do woltamperometrycznej
detekcji Tf została sprawdzona na dwóch niezależnych próbkach krwi szczurzej.
Z przeprowadzonych eksperymentów (rysunek 74) wynika, że tylko hemoglobina

Elektrochemiczny immunosensor do detekcji transferyny
165
(obecna w czerwonych krwinkach) może zakłócić określenie ilości transferyny
w analizowanej próbce krwi. Biorąc pod uwagę powyższy fakt, przed pomiarami
próbkę krwi, stabilizowaną jonami cytrynianu sodu, poddawano wirowaniu w celu
oddzielenia elementów morfotycznych od osocza. Następnie do osocza
rozcieńczonego 1000-krotnie buforem fosforanowym zanurzano immunosensor na
czas 60 minut. Po upływie tego czasu elektrodę delikatnie opłukiwano strumieniem
wody destylowanej, a następnie przeprowadzano pomiar w czystym odtlenionym
0.02 M buforze PB z dodatkiem 150 mM K2SO4 określoną metodą analityczną.
Stężenie transferyny w badanych próbkach krwi zostało wyznaczone na podstawie
pomiarów z użyciem elektrochemicznej spektroskopii impedancyjnej oraz
elektrochemicznej mikrowagi kwarcowej i wynosiło: 0.45 0.09 (EIS) i 0.42 0.08
(QCM) i 0.31 0.1 g·dL-1
(LSV) (uwzględniając 1000-krotne rozcieńczenie próbki
krwi). Poziom transferyny we krwi szczurzej, zgodnie z literaturą, mieści się
w zakresie od 0.356 do 0.550 g·dL-1
[447]. Zatem uzyskane wyniki zawartości Tf we
krwi przy użyciu zaproponowanego immunosensora dowodzą, że metoda ta może
z powodzeniem być wykorzystywana w oznaczeniach tego białka również we krwi
ludzkiej.
12.3. Wpływ pola magnetycznego i ferromagnetycznego
modyfikatora powierzchni elektrody na integralność
konformacyjną i elektroaktywność transferyny
Spośród wszystkich paramagnetycznych białek obecnych we krwi transferyna
jest najbardziej podatna na zmiany konformacyjne, prowadzące nawet do uwolnienia
jonów Fe(III) z otoczki białkowej [448-450]. Cząsteczka transferyny wiążąc
i uwalniając jony żelaza ulega cyklicznym zmianom konformacyjnym, dzięki czemu
możliwe jest jej rozpoznanie przez receptory znajdujące się w błonach
plazmatycznych [143]. Z uwagi na fakt, że komórki nowotworowe posiadają na
powierzchni swoich błon zwiększoną liczbę receptorów transferyny, w porównaniu
do komórek prawidłowych, obecność tego białka na powierzchni nośnika leku
przeciwnowotworowego może ułatwiać jego transport do jądra komórkowego [180].
Z literatury wiadomo, że kontakt Tf z powierzchnią superparamagnetycznych
nanocząstek tlenku żelaza prowadzi do uwalniania żelaza ze struktury Tf [451].

Elektrochemiczny immunosensor do detekcji transferyny
166
Proces ten jest tym efektywniejszy, im mniejsza jest wartość stosunku stężenia
transferyny względem stężenia nanocząstek magnetycznych. Powstaje zatem pytanie,
jaki wpływ na konformację transferyny, a tym samym na jej elektroaktywność, mają
nanokapsuły węglowe zawierające żelazo w połączeniu z polem magnetycznym?
12.3.1. Procedura syntezy koniugatu transferyna-nanocząstka
magnetyczna
W badaniach związanych z tym zagadnieniem zastosowano nanokapsuły
węglowe zawierające żelazo, których powierzchnię zmodyfikowano grupami
karboksylowymi. Transferyna unieruchamiana była na powierzchni takiej
nanocząstki za pośrednictwem wiązania amidowego utworzonego pomiędzy grupami
aminowymi białka i grupami karboksylowymi nanocząstek magnetycznych. Synteza
koniugatu nanocząstka magnetyczna – transferyna (Fe@C-COOH/Tf) przebiegała
w trzech prostych etapach. Pierwszy etap związany był z aktywacją grup
karboksylowych znajdujących się na powierzchni nanocząstek magnetycznych za
pomocą mieszaniny EDC/NHS. Następnie do zaktywowanej mieszaniny nanocząstek
dodawana była transferyna, a proces sprzęgania Tf z Fe@C-COOH Nps przebiegał
w obecności i nieobecności pola magnetycznego przez 1 h. W ostatnim etapie
syntezy nieprzereagowana transferyna odmywana była od koniugatu za pomocą
buforu fosforanowego w obecności magnesu. Schemat syntezy koniugatu
Fe@C-COOH/Tf ilustruje rysunek 78.
Kapsuły węglowe zawierające żelazo
sfunkcjonalizowane grupami –COOH
(Fe@C-COOH Nps)
AKTYWACJA
GRUP COOH (1 h)
TF
(1 h)
WYTRZĄSANIE
(1 h)
WIELOKROTNE
PRZEMYWANIE
MAGNES
Rysunek 78. Schemat syntezy koniugatu Fe@C-COOH/Tf.

Elektrochemiczny immunosensor do detekcji transferyny
167
12.3.2. Charakterystyka spektroskopowa koniugatu
Fe@C-COOH/Tf
Z uwagi na fakt, że białka wykazują silną tendencję do procesu adsorpcji,
pierwszy etap badań związany był z potwierdzeniem sposobu unieruchomienia Tf na
powierzchni nanocząstek magnetycznych. W tym celu zsyntezowano dwa koniugaty
Fe@C-COOH/Tf. W jednym z nich Tf unieruchomiona została na powierzchni
nanocząstki magnetycznej za pośrednictwem wiązania amidowego pomiędzy
grupami –NH2 obecnymi w jej otoczce białkowej i grupami –COOH nanokapsuł.
Drugi koniugat otrzymano w wyniku fizycznej adsorpcji Tf na powierzchni
Fe@C-COOH Nps (etap aktywacji grup karboksylowych przy użyciu mieszaniny
EDC/NHS został pominięty). Dodatkowo, syntezę koniugatów przeprowadzono
w nieobecności i obecności magnesu. Otrzymane koniugaty Fe@C-COOH/Tf
zostały scharakteryzowane przy użyciu spektroskopii w podczerwieni
z transformacją Fouriera (FTIR). Pomiary wykonano zarówno dla koniugatów, jak
i czystych składników, czyli kapsuł węglowych oraz transferyny. Uzyskane widma
FTIR przedstawione zostały na rysunku 79. Widmo FTIR czystych nanokapsuł
węglowych charakteryzuje się obecnością trzech głównych pasm położonych przy
następujących wartościach liczb falowych: (1) ok. 1700 cm-1
odpowiadających
drganiom rozciągającym –C=O grupy karboksylowej, (2) ok. 1580 i 1530 cm-1
związanych z drganiami rozciągającymi C=C w heksagonalnej sieci grafitu oraz
(3) ok. 1200 cm-1
odpowiadających drganiom wiązania CO. Z kolei widmo FTIR
transferyny, jak i każdego białka, posiada dwa dobrze widoczne, charakterystyczne
pasma przy ok. 1530 (amid II) i 1650 cm-1
(amid I) odpowiadające drganiom
rozciągającym pomiędzy atomami −N−H i −C=O [452]. Utworzenie trwałego
wiązania kowalencyjnego (wiązania amidowego) pomiędzy nanocząstką
magnetyczną a cząsteczką Tf skutkowało nie tylko spadkiem intensywności pasm
amid I i amid II, ale również przesunięciem pasma amid I w kierunku mniejszych
wartości liczb falowych o około 20 cm-1
. Położenie tego pasma w przypadku
koniugatu Fe@C-COOH/Tf otrzymanego w wyniku fizycznej adsorpcji białka nie
uległo zmianie. Spektroskopia w podczerwieni z transformacją Fouriera
potwierdziła, że cząsteczki transferyny unieruchamiane są na powierzchni
nanocząstek magnetycznych głównie za pośrednictwem wiązań amidowych, pomimo
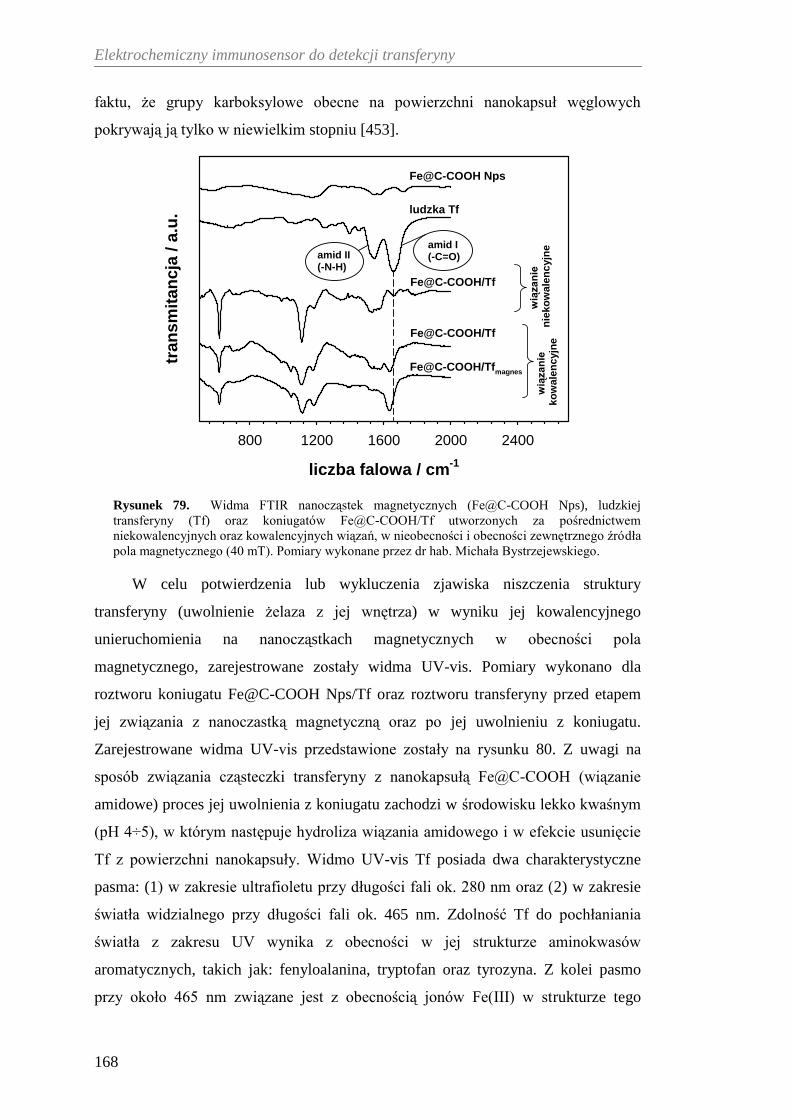
Elektrochemiczny immunosensor do detekcji transferyny
168
faktu, że grupy karboksylowe obecne na powierzchni nanokapsuł węglowych
pokrywają ją tylko w niewielkim stopniu [453].
liczba falowa / cm-1
800 1200 1600 2000 2400
tra
ns
mit
an
cja
/ a
.u.
Fe@C-COOH/Tf
Fe@C-COOH/Tf
Fe@C-COOH/Tfmagnes
wią
za
nie
n
ieko
wa
len
cyjn
ew
iąza
nie
k
ow
ale
nc
yjn
e
ludzka Tf
Fe@C-COOH Nps
amid II(-N-H)
amid I(-C=O)
Rysunek 79. Widma FTIR nanocząstek magnetycznych (Fe@C-COOH Nps), ludzkiej
transferyny (Tf) oraz koniugatów Fe@C-COOH/Tf utworzonych za pośrednictwem
niekowalencyjnych oraz kowalencyjnych wiązań, w nieobecności i obecności zewnętrznego źródła
pola magnetycznego (40 mT). Pomiary wykonane przez dr hab. Michała Bystrzejewskiego.
W celu potwierdzenia lub wykluczenia zjawiska niszczenia struktury
transferyny (uwolnienie żelaza z jej wnętrza) w wyniku jej kowalencyjnego
unieruchomienia na nanocząstkach magnetycznych w obecności pola
magnetycznego, zarejestrowane zostały widma UV-vis. Pomiary wykonano dla
roztworu koniugatu Fe@C-COOH Nps/Tf oraz roztworu transferyny przed etapem
jej związania z nanoczastką magnetyczną oraz po jej uwolnieniu z koniugatu.
Zarejestrowane widma UV-vis przedstawione zostały na rysunku 80. Z uwagi na
sposób związania cząsteczki transferyny z nanokapsułą Fe@C-COOH (wiązanie
amidowe) proces jej uwolnienia z koniugatu zachodzi w środowisku lekko kwaśnym
(pH 4÷5), w którym następuje hydroliza wiązania amidowego i w efekcie usunięcie
Tf z powierzchni nanokapsuły. Widmo UV-vis Tf posiada dwa charakterystyczne
pasma: (1) w zakresie ultrafioletu przy długości fali ok. 280 nm oraz (2) w zakresie
światła widzialnego przy długości fali ok. 465 nm. Zdolność Tf do pochłaniania
światła z zakresu UV wynika z obecności w jej strukturze aminokwasów
aromatycznych, takich jak: fenyloalanina, tryptofan oraz tyrozyna. Z kolei pasmo
przy około 465 nm związane jest z obecnością jonów Fe(III) w strukturze tego
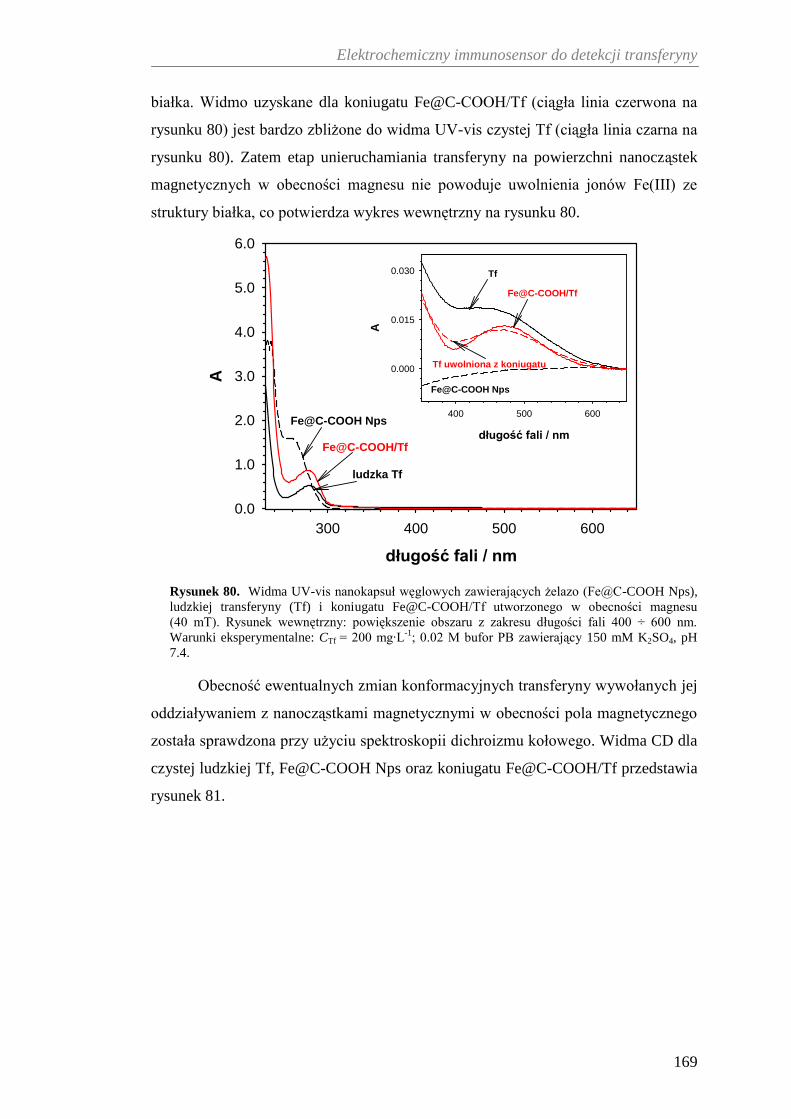
Elektrochemiczny immunosensor do detekcji transferyny
169
białka. Widmo uzyskane dla koniugatu Fe@C-COOH/Tf (ciągła linia czerwona na
rysunku 80) jest bardzo zbliżone do widma UV-vis czystej Tf (ciągła linia czarna na
rysunku 80). Zatem etap unieruchamiania transferyny na powierzchni nanocząstek
magnetycznych w obecności magnesu nie powoduje uwolnienia jonów Fe(III) ze
struktury białka, co potwierdza wykres wewnętrzny na rysunku 80.
długość fali / nm
300 400 500 600
A
0.0
1.0
2.0
3.0
4.0
5.0
6.0
długość fali / nm
400 500 600
A0.000
0.015
0.030
ludzka Tf
Fe@C-COOH/Tf
Fe@C-COOH Nps
Tf
Fe@C-COOH/Tf
Tf uwolniona z koniugatu
Fe@C-COOH Nps
Rysunek 80. Widma UV-vis nanokapsuł węglowych zawierających żelazo (Fe@C-COOH Nps),
ludzkiej transferyny (Tf) i koniugatu Fe@C-COOH/Tf utworzonego w obecności magnesu
(40 mT). Rysunek wewnętrzny: powiększenie obszaru z zakresu długości fali 400 ÷ 600 nm.
Warunki eksperymentalne: CTf = 200 mg·L-1
; 0.02 M bufor PB zawierający 150 mM K2SO4, pH
7.4.
Obecność ewentualnych zmian konformacyjnych transferyny wywołanych jej
oddziaływaniem z nanocząstkami magnetycznymi w obecności pola magnetycznego
została sprawdzona przy użyciu spektroskopii dichroizmu kołowego. Widma CD dla
czystej ludzkiej Tf, Fe@C-COOH Nps oraz koniugatu Fe@C-COOH/Tf przedstawia
rysunek 81.
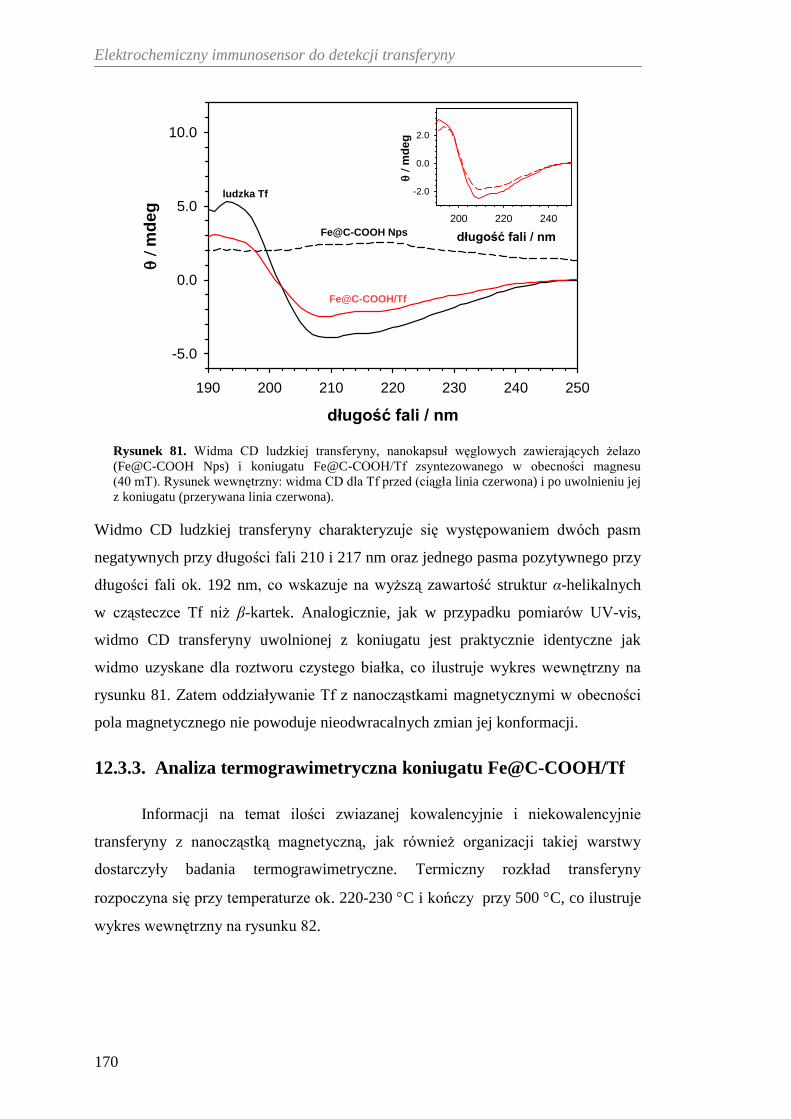
Elektrochemiczny immunosensor do detekcji transferyny
170
długość fali / nm
190 200 210 220 230 240 250
m
deg
-5.0
0.0
5.0
10.0
ludzka Tf
Fe@C-COOH/Tf
Fe@C-COOH Nps długość fali / nm
200 220 240
m
deg
-2.0
0.0
2.0
Rysunek 81. Widma CD ludzkiej transferyny, nanokapsuł węglowych zawierających żelazo
(Fe@C-COOH Nps) i koniugatu Fe@C-COOH/Tf zsyntezowanego w obecności magnesu
(40 mT). Rysunek wewnętrzny: widma CD dla Tf przed (ciągła linia czerwona) i po uwolnieniu jej
z koniugatu (przerywana linia czerwona).
Widmo CD ludzkiej transferyny charakteryzuje się występowaniem dwóch pasm
negatywnych przy długości fali 210 i 217 nm oraz jednego pasma pozytywnego przy
długości fali ok. 192 nm, co wskazuje na wyższą zawartość struktur α-helikalnych
w cząsteczce Tf niż β-kartek. Analogicznie, jak w przypadku pomiarów UV-vis,
widmo CD transferyny uwolnionej z koniugatu jest praktycznie identyczne jak
widmo uzyskane dla roztworu czystego białka, co ilustruje wykres wewnętrzny na
rysunku 81. Zatem oddziaływanie Tf z nanocząstkami magnetycznymi w obecności
pola magnetycznego nie powoduje nieodwracalnych zmian jej konformacji.
12.3.3. Analiza termograwimetryczna koniugatu Fe@C-COOH/Tf
Informacji na temat ilości zwiazanej kowalencyjnie i niekowalencyjnie
transferyny z nanocząstką magnetyczną, jak również organizacji takiej warstwy
dostarczyły badania termograwimetryczne. Termiczny rozkład transferyny
rozpoczyna się przy temperaturze ok. 220-230 C i kończy przy 500 C, co ilustruje
wykres wewnętrzny na rysunku 82.
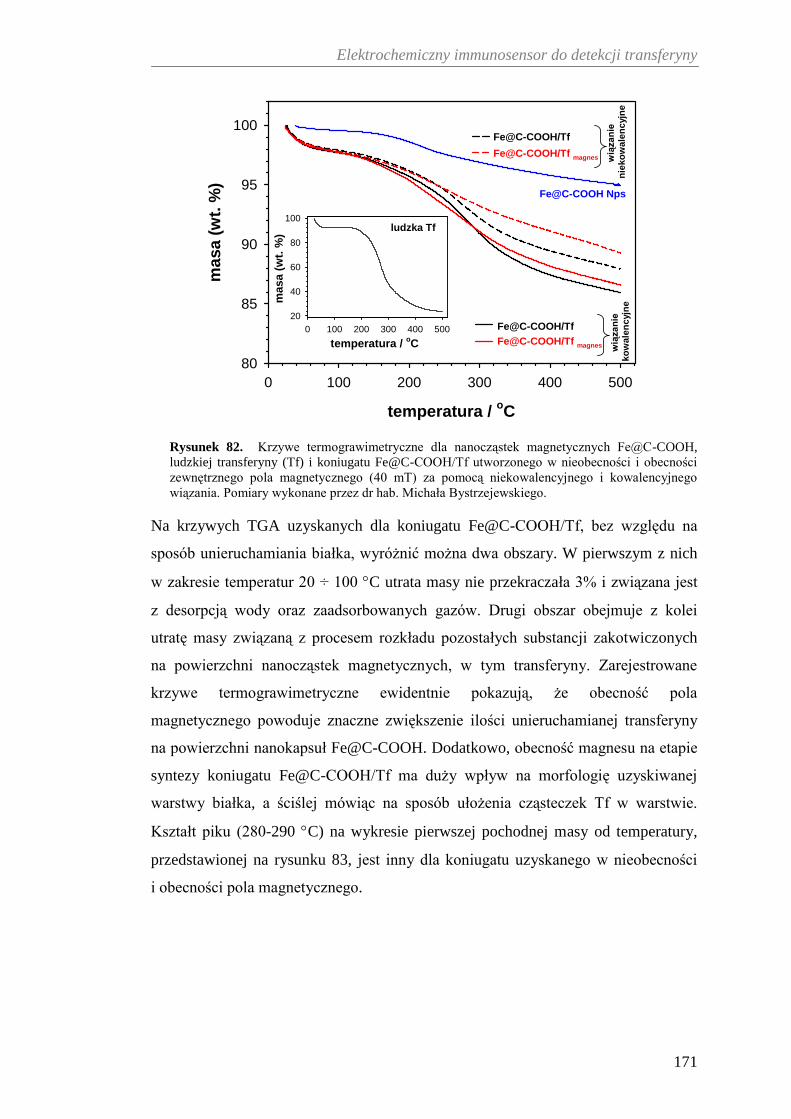
Elektrochemiczny immunosensor do detekcji transferyny
171
temperatura / oC
0 100 200 300 400 500
masa
(w
t. %
)
80
85
90
95
100
temperatura / oC
0 100 200 300 400 500
ma
sa (
wt.
%)
20
40
60
80
100ludzka Tf
Fe@C-COOH Nps
Fe@C-COOH/Tf
Fe@C-COOH/Tf magnes wią
za
nie
nie
ko
wale
ncyjn
ew
iąza
nie
ko
wale
ncyjn
e
Fe@C-COOH/Tf
Fe@C-COOH/Tf magnes
Rysunek 82. Krzywe termograwimetryczne dla nanocząstek magnetycznych Fe@C-COOH,
ludzkiej transferyny (Tf) i koniugatu Fe@C-COOH/Tf utworzonego w nieobecności i obecności
zewnętrznego pola magnetycznego (40 mT) za pomocą niekowalencyjnego i kowalencyjnego
wiązania. Pomiary wykonane przez dr hab. Michała Bystrzejewskiego.
Na krzywych TGA uzyskanych dla koniugatu Fe@C-COOH/Tf, bez względu na
sposób unieruchamiania białka, wyróżnić można dwa obszary. W pierwszym z nich
w zakresie temperatur 20 ÷ 100 C utrata masy nie przekraczała 3% i związana jest
z desorpcją wody oraz zaadsorbowanych gazów. Drugi obszar obejmuje z kolei
utratę masy związaną z procesem rozkładu pozostałych substancji zakotwiczonych
na powierzchni nanocząstek magnetycznych, w tym transferyny. Zarejestrowane
krzywe termograwimetryczne ewidentnie pokazują, że obecność pola
magnetycznego powoduje znaczne zwiększenie ilości unieruchamianej transferyny
na powierzchni nanokapsuł Fe@C-COOH. Dodatkowo, obecność magnesu na etapie
syntezy koniugatu Fe@C-COOH/Tf ma duży wpływ na morfologię uzyskiwanej
warstwy białka, a ściślej mówiąc na sposób ułożenia cząsteczek Tf w warstwie.
Kształt piku (280-290 C) na wykresie pierwszej pochodnej masy od temperatury,
przedstawionej na rysunku 83, jest inny dla koniugatu uzyskanego w nieobecności
i obecności pola magnetycznego.

Elektrochemiczny immunosensor do detekcji transferyny
172
temperatura / oC
100 200 300 400
dm
/dT
/ %
(oC
)-1
0.00
0.02
0.04
0.06
0.08
0.10
0.12
temperatura / oC
100 200 300 400 500d
m/d
T /
%(o
C)-1
0.00
0.02
0.04
Fe@C-COOH Nps
ludzka Tf
Fe@C-COOH/Tf
Fe@C-COOH/Tf magnes
Fe@C-COOH/Tf
Fe@C-COOH/Tf magnes
wią
za
nie
nie
ko
wa
len
cyjn
ew
iąza
nie
ko
wa
len
cyjn
e
Rysunek 83. Pochodne (dm/dT) dla nanocząstek Fe@C-COOH, ludzkiej transferyny (Tf)
i koniugatu Fe@C-COOH/Tf utworzonego w nieobecności i obecności zewnętrznego pola
magnetycznego (40 mT) za pomocą niekowalencyjnego i kowalencyjnego wiązania. Pomiary
wykonane przez dr hab. Michała Bystrzejewskiego.
12.3.4. Charakterystyka struktur białkowych na powierzchni
nanokapsuł węglowych przy użyciu mikrowagi kwarcowej
z dyssypacją energii
W celu uzyskania pełniejszych informacji na temat sposobu upakowania
transferyny w warstwie wykonane zostały eksperymenty z użyciem mikrowagi
kwarcowej z dyssypacją energii. Podczas pomiarów zmian częstotliwości drgań
kryształu kwarcu oraz zmian współczynnika rozproszenia energii (D) zastosowano
różne konfiguracje pola magnetycznego w stosunku do powierzchni elektrody, co
schematycznie przedstawia rysunek 84. W celu uzyskania stabilnej warstwy
nanocząstek magnetycznych powierzchnia złotego kryształu kwarcu została pokryta
monowarstwą cysteaminy. Następnie, w wyniku utworzenia wiązań amidowych
pomiędzy zaktywowanymi grupami karboksylowymi nanokapsuł węglowych
Fe@C-COOH a grupami aminowymi cysteaminy, monowarstwa CSH została
pokryta warstwą nanocząstek magnetycznych. W celu zwiększenia szybkości
opadania nanocząstek magnetycznych, ograniczenia ich aglomeracji na powierzchni
i utworzenia równomiernej warstwy pod elektrodą umieszczony został płaski
magnes. Tak zmodyfikowaną elektrodę (Au-CSH/Fe@C-COOH) umieszczano

Elektrochemiczny immunosensor do detekcji transferyny
173
w naczynku QCM-D. Pomiaru dokonano w nieobecności i obecności magnesu
w różnych jego konfiguracjach względem powierzchni elektrody.
1) CSH 3) UNIERUCHAMIANIE
TF
2) ZAKTYWOWANE FE@C-COOH
2a) Z MAGNESEM
2b) BEZ MAGNESU
Au
o0:AvsB
o90:AvsB
Rysunek 84. Schemat przygotowania elektrody do pomiarów z użyciem mikrowagi kwarcowej
z dyssypacją energii.
Otrzymane zależności zmian częstotliwości drgań kryształu kwarcu oraz zmian
współczynnika rozproszenia energii w funkcji czasu podczas etapu unieruchamiania
transferyny na powierzchni nanocząstek magnetycznych, w nieobecności i obecności
magnesu przedstawia rysunek 85. Po uzyskaniu stabilizacji częstotliwości drgań
kryształu kwarcu pokrytego monowarstwą cysteaminy oraz nanoczastkami
magnetycznymi (Au-CSH/Fe@C-COOH) w czystym 0.02 M buforze HEPES
z dodatkiem 150 mM Na2SO4, do układu wprowadzony został roztwór transferyny
o stężeniu 200 mg·L-1
. Wprowadzenie transferyny powodowało gwałtowny, znaczny
spadek częstotliwości drgań kryształu kwarcu. Obserwowany spadek był
konsekwencją utworzenia kowalencyjnego wiązania pomiędzy cząsteczkami
transferyny i grupami –COOH nanokapsuł węglowych. Po upływie około 20 minut
częstotliwość drgań kryształu kwarcu osiągnęła minimalną wartość i nie ulegała już
dalszym zmianom, co oznaczało, że maksymalna ilość cząsteczek Tf została
unieruchomiona na warstwie nanocząstek magnetycznych. Wprowadzenie magnesu
w trakcie wiązania Tf powodowało większe zmiany częstotliwości drgań kryształu
kwarcu, co bezpośrednio wiąże się z większą ilością cząsteczek Tf oddziałujących
z nanokapsułami. W celu usunięcia transferyny unieruchomionej na drodze fizycznej
adsorpcji z warstwy nanocząstek, do układu wprowadzony został czysty 0.02 M
bufor HEPES (bez dodatku Na2SO4). Na uzyskanych krzywych fn = f(t) praktycznie
nie obserwowano zmian częstotliwości drgań kryształu kwarcu, dzięki czemu można
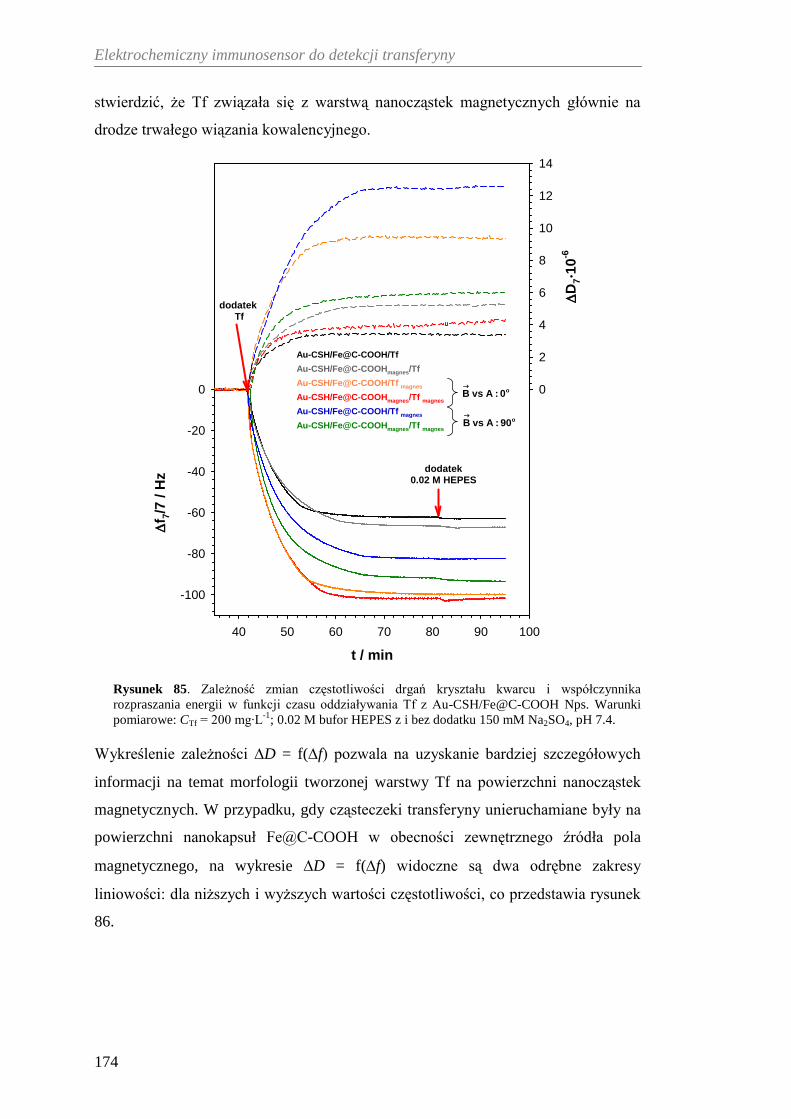
Elektrochemiczny immunosensor do detekcji transferyny
174
stwierdzić, że Tf związała się z warstwą nanocząstek magnetycznych głównie na
drodze trwałego wiązania kowalencyjnego.
D
710
-6
0
2
4
6
8
10
12
14
t / min
40 50 60 70 80 90 100
f 7
/7 /
Hz
-100
-80
-60
-40
-20
0
Au-CSH/Fe@C-COOH/Tf
Au-CSH/Fe@C-COOHmagnes/Tf
Au-CSH/Fe@C-COOH/Tf magnes
Au-CSH/Fe@C-COOHmagnes/Tf magnes
Au-CSH/Fe@C-COOH/Tf magnes
Au-CSH/Fe@C-COOHmagnes/Tf magnes
o0:AvsB
o90:AvsB
dodatek Tf
dodatek0.02 M HEPES
Rysunek 85. Zależność zmian częstotliwości drgań kryształu kwarcu i współczynnika
rozpraszania energii w funkcji czasu oddziaływania Tf z Au-CSH/Fe@C-COOH Nps. Warunki
pomiarowe: CTf = 200 mg·L-1
; 0.02 M bufor HEPES z i bez dodatku 150 mM Na2SO4, pH 7.4.
Wykreślenie zależności D = f(f) pozwala na uzyskanie bardziej szczegółowych
informacji na temat morfologii tworzonej warstwy Tf na powierzchni nanocząstek
magnetycznych. W przypadku, gdy cząsteczeki transferyny unieruchamiane były na
powierzchni nanokapsuł Fe@C-COOH w obecności zewnętrznego źródła pola
magnetycznego, na wykresie D = f(f) widoczne są dwa odrębne zakresy
liniowości: dla niższych i wyższych wartości częstotliwości, co przedstawia rysunek
86.

Elektrochemiczny immunosensor do detekcji transferyny
175
f7/7 /Hz
-100 -80 -60 -40 -20 0
D
710
-6
0
2
4
6
8
10
12
14
Au-CSH/Fe@C-COOH/Tf
Au-CSH/Fe@C-COOHmagnes/Tf
Au-CSH/Fe@C-COOH/Tf magnes
Au-CSH/Fe@C-COOHmagnes/Tf magnes
Au-CSH/Fe@C-COOH/Tf magnes
Au-CSH/Fe@C-COOHmagnes/Tf magnes
o0:AvsB
o90:AvsB
Rysunek 86. Zależność zmian ΔD w funkcji Δf. Warunki pomiarowe: CTf = 200 mg·L-1
; 0.02 M
bufor HEPES z i bez dodatku 150 mM Na2SO4, pH 7.4.
Pierwszy obszar, poniżej -20 Hz, dotyczy etapu tworzenia monowarstwy Tf
i charakteryzuje się on małą wartością stosunku D/f wynoszącym ok.
8.3·10-9
Hz-1
. Tak mała wartość D/f wskazuje na dość ścisłe upakowanie
cząsteczek transferyny w warstwie. Jony Fe(III) obecne w strukturze białka posiadają
właściwości paramagnetyczne [454], zatem są przyciągane przez nanokapsuły
Fe@C-COOH – źródło pola magnetycznego na powierzchni kryształu kwarcu.
Z kolei reszty wodorowęglanowe i węglanowe, również obecne w strukturze Tf,
wykazują właściwości diamagnetyczne [455], są zatem wypychane na zewnątrz
przez pole magnetyczne. Skutkuje to utworzeniem na drodze oddziaływań
elektrostatycznych kolejnej warstwy, grubszej i zdecydowanie luźniejszej (D/f
~(3÷10)·10-6
Hz-1
). Zjawisko to występuje niezwykle rzadko w przypadku białek
oraz stałego pH. Podobne zachowanie się cząsteczek adsorbowanego białka na
powierzchni elektrody zaobserwował Höök i jego współpracownicy w trakcie
procesu adsorpcji hemoglobiny w funkcji pH [456].

Elektrochemiczny immunosensor do detekcji transferyny
176
12.3.5. Elektroaktywność transferyny kowalencyjnie związanej
z nanokapsułami Fe@C-COOH Nps
W badaniach sprawdzono również, czy transferyna unieruchomiona na
powierzchni nanocząstek magnetycznych wykazuje zdolność do bezpośredniej
wymiany elektronów z powierzchnią elektrody. W tym celu przeprowadzono
pomiary z użyciem techniki woltamperometrii cyklicznej. Z uwagi na położenie
jonów Fe(III) głęboko w otoczce białkowej Tf, kluczową kwestią w zachowaniu
elektroaktywności tego białka jest jego odpowiednia orientacja względem
powierzchni elektrody. W przypadku paramagnetycznej cząsteczki, jaką jest Tf, pole
magnetyczne pozwala uzyskać najbardziej korzystną orientację dla tego typu procesu
wymiany elektronów. Schemat przygotowania elektrody do pomiaru oraz orientację
pola magnetycznego względem powierzchni elektrody ilustruje rysunek 87.
1) CSH
Au
4) POMIAR
o0:AvsB
o60:AvsB
2) ZAKTYWOWANE FE@C-COOH
2a) Z MAGNESEM
2b) BEZ MAGNESU
3) UNIERUCHAMIANIE TF
3a) Z MAGNESEM
3b) BEZ MAGNESU
Rysunek 87. Schemat przygotowania elektrody do pomiarów woltamperometrycznych.
Przykładowe cykliczne krzywe woltamperometryczne uzyskane dla koniugatu
Fe@C-COOH/Tf w nieobecności i obecności pola magnetycznego zostały
przedstawione na rysunku 88. Dobrze ukształtowane sygnały elektroredukcji Fe(III)
do Fe(II) obserwowane były tylko w przypadku warstwy Tf utworzonej w obecności
pola magnetycznego. Dodatkowo, intensywność tych sygnałów prądowych zależała
od orientacji pola magnetycznego w stosunku do powierzchni elektrody. Wyższe
sygnały redukcji Tf obserwowano w sytuacji, gdy kąt pomiędzy powierzchnią
elektrody a polem magnetycznym wynosił 60. Wzrost inetnsywności sygnałów
prądowych wraz ze zmianą orientacji linii sił pola magnetycznego względem
powierzchni elektrody związany jest z wpływem sił generowanych przez pole
magnetyczne na stabilność warstwy białka na powierzchni nanocząstki
magnetycznej.
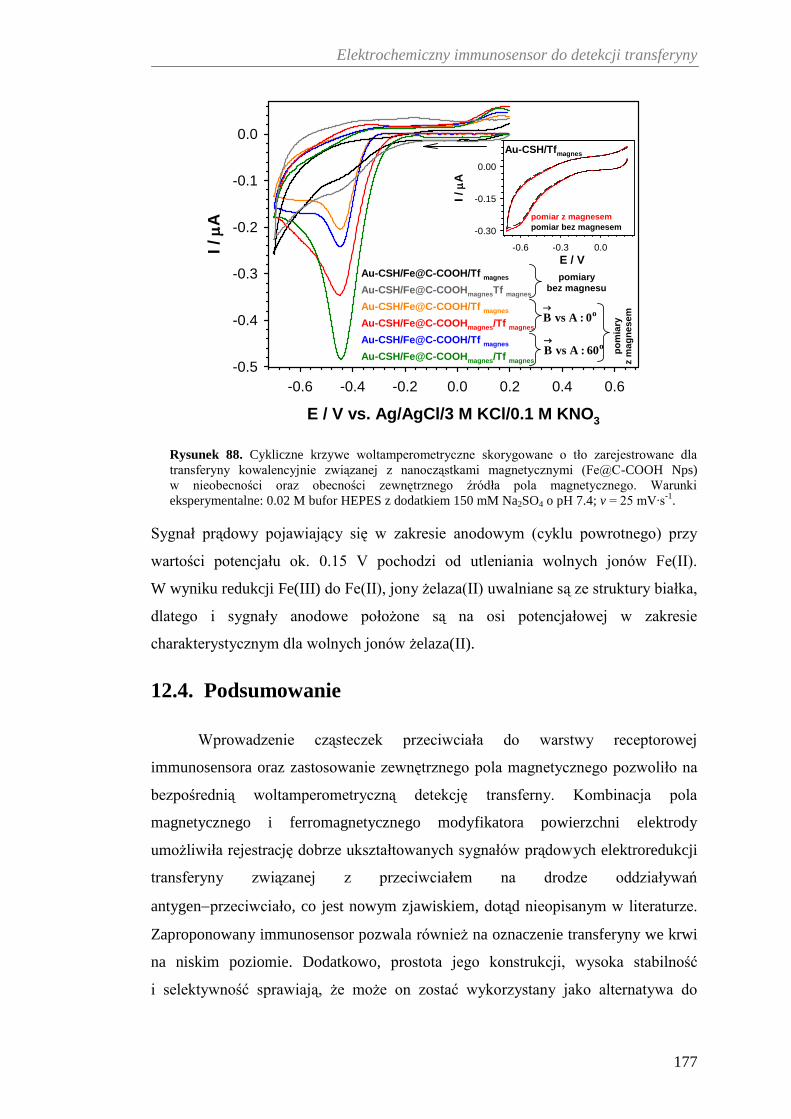
Elektrochemiczny immunosensor do detekcji transferyny
177
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.6 -0.4 -0.2 0.0 0.2 0.4 0.6
I /
A
-0.5
-0.4
-0.3
-0.2
-0.1
0.0
Au-CSH/Fe@C-COOH/Tf magnes
Au-CSH/Fe@C-COOHmagnesTf magnes
Au-CSH/Fe@C-COOH/Tf magnes
Au-CSH/Fe@C-COOHmagnes/Tf magnes
Au-CSH/Fe@C-COOH/Tf magnes
Au-CSH/Fe@C-COOHmagnes/Tf magnes
E / V -0.6 -0.3 0.0
I /
A
-0.30
-0.15
0.00
pomiary
bez magnesu
po
mia
ry
z m
ag
ne
se
m
Au-CSH/Tfmagnes
pomiar z magnesem
pomiar bez magnesem
o0:AvsB
o60:AvsB
Rysunek 88. Cykliczne krzywe woltamperometryczne skorygowane o tło zarejestrowane dla
transferyny kowalencyjnie związanej z nanocząstkami magnetycznymi (Fe@C-COOH Nps)
w nieobecności oraz obecności zewnętrznego źródła pola magnetycznego. Warunki
eksperymentalne: 0.02 M bufor HEPES z dodatkiem 150 mM Na2SO4 o pH 7.4; v = 25 mV·s-1
.
Sygnał prądowy pojawiający się w zakresie anodowym (cyklu powrotnego) przy
wartości potencjału ok. 0.15 V pochodzi od utleniania wolnych jonów Fe(II).
W wyniku redukcji Fe(III) do Fe(II), jony żelaza(II) uwalniane są ze struktury białka,
dlatego i sygnały anodowe położone są na osi potencjałowej w zakresie
charakterystycznym dla wolnych jonów żelaza(II).
12.4. Podsumowanie
Wprowadzenie cząsteczek przeciwciała do warstwy receptorowej
immunosensora oraz zastosowanie zewnętrznego pola magnetycznego pozwoliło na
bezpośrednią woltamperometryczną detekcję transferny. Kombinacja pola
magnetycznego i ferromagnetycznego modyfikatora powierzchni elektrody
umożliwiła rejestrację dobrze ukształtowanych sygnałów prądowych elektroredukcji
transferyny związanej z przeciwciałem na drodze oddziaływań
antygenprzeciwciało, co jest nowym zjawiskiem, dotąd nieopisanym w literaturze.
Zaproponowany immunosensor pozwala również na oznaczenie transferyny we krwi
na niskim poziomie. Dodatkowo, prostota jego konstrukcji, wysoka stabilność
i selektywność sprawiają, że może on zostać wykorzystany jako alternatywa do

Elektrochemiczny immunosensor do detekcji transferyny
178
metod rutynowo stosowanych w analizie klinicznej do oznaczeń Tf w osoczu krwi.
Dużą zaletą immunosensora jest również możliwość jego kilkukrotnego użycia
w wyniku regeneracji warstwy sensorowej w roztworze chlorowodorku glicyny, co
znacznie obniża koszty analizy. Zaproponowany sensor otwiera nowe możliwości
w badaniach biologicznych białek i wykrywania ich na poziomie rzędu około
15 ngdL-1
.
Przeprowadzone badania udowodniły, że zarówno obecność
ferromagnetycznego modyfikatora elektrody (nanokapsuły węglowe zawierające
żelazo: Fe@C-COOH Nps), jak i zewnętrznego źródła pola magnetycznego jest
niezbędna do zapewnienia najwyższej aktywności elektrochemicznej transferyny
kowalencyjnie związanej z podłożem. Wprowadzenie zewnętrznego źródła pola
magnetycznego do układu elektrochemicznego prowadzi do powstania silnego
gradientu pola magnetycznego w pobliżu powierzchni elektrody zmodyfikowanej
ferromagnetykiem, co wpływa na strukturę transferyny. Paramagnetyczne centra Tf,
jony Fe(III), są wciągane przez pole magnetyczne i w konsekwencji zmieniają swoje
położenie, natomiast diamagnetyczne składniki otoczki białkowej Tf, głównie
wodorowęglany, są wypychane przez pole na zewnątrz. W związku z tym, w wyniku
oddziaływań elektrostatycznych, tworzy się kolejna warstwa Tf na powierzchni
nanocząstek. Jest to bardzo rzadkie zjawisko w przypadku białek w warunkach
stałego pH. Ponadto pomiary UV-vis wykazały, że związanie Tf z nanocząstkami nie
powoduje uwolnienia żelaza ze struktury białka.

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
179
13. Elektrochemiczna detekcja ferrytyny
z wykorzystaniem wysoce selektywnego
oddziaływania antygen–przeciwciało
Białka ostrej fazy stanowią grupę białek surowicy krwi syntezowanych przez
wątrobę, których stężenie we krwi zmienia się w wyniku odpowiedzi na stan zapalny
organizmu. Są one zatem niezwykle ważnym wskaźnikiem diagnostycznym
pozwalającym na wczesne rozpoznanie zmian zachodzących w organizmie. Na
podstawie ich poziomu w płynach ustrojowych, głównie we krwi, potwierdza lub
wyklucza się takie schorzenia, jak: choroby nowotworowe, procesy zapalne oraz
rozprzestrzeniające się infekcje. Podstawową funkcją tych białek jest przywrócenie
zaburzonej homeostazy organizmu poprzez hamowanie proteinaz, regulowanie
procesów krzepnięcia, wiązanie i neutralizację patogenów oraz transportowanie
metabolitów. Do tego typu białek zaliczana jest m.in. ferrytyna, która podobnie jak
hemoglobina i transferyna, zawiera w swojej strukturze jony żelaza odpowiedzialne
za jej aktywność elektrochemiczną. Fakt ten narzuca konieczność wykorzystania
wysoce selektywnego i specyficznego oddziaływania antygen–przeciwciało podczas
oznaczania poziomu tego białka w osoczu krwi. Ponadto żelazo obecne w strukturze
ferrytyny nadaje jej właściwości paramagnetyczne, umożliwiając tym samym
wykorzystanie pola magnetycznego podczas etapu jej detekcji.
Analogicznie, jak w przypadku hemoglobiny, transferyny, czy też
ceruloplazminy, głównym elementem warstwy receptorej czujnika były nanocząstki
magnetyczne, a ścisłej mówiąc nanokapsuły węglowe zawierające żelazo.
Najbardziej popularną metodą tworzenia warstwy nanocząstek magnetycznych na
powierzchni elektrody jest ich adsorpcja fizyczna. Zjawiskiem niepożądanym
w trakcie tworzenia takiej warstwy jest silna tendencja nanocząstek magnetycznych
do agregacji. Ponadto warstwa utworzona na drodze adsorpcji jest podatna na
działanie pola magnetycznego, nanocząstki w obecności magnesu zmieniają swoje
położenie, ustawiając się zgodnie z liniami sił pola magnetycznego [457].
W konsekwencji taka warstwa jest całkowicie niejednorodna, mało stabilna i podatna
na uszkodzenia mechaniczne. W celu utworzenia jednorodnej i stabilnej powłoki,

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
180
nanocząstki magnetyczne pokryte zostały warstwą fenylową uzyskaną na drodze
elektroredukcji odpowiedniej soli diazoniowej.
Zgodnie z literaturą, modyfikacja powierzchni elektrody warstwą fenylową
w wyniku jednoelektronowej elektroredukcji danej soli diazoniowej [458] zachodzi
z wytworzeniem formy przejściowej, którą jest rodnik [459]. Teoretycznie sól
diazoniowa może tworzyć dwie formy rodnikowe; wytworzenie rodnika może
nastąpić na atomie węgla pierścienia aromatycznego lub na atomie azotu grupy
diazoniowej. Forma rodnikowa tworzy z podłożem niezwykle trwałe i stabilne
wiązanie kowalencyjne, zgodnie z rysunkiem 89.
+
+e-
R
+
N+
N R
NN R
lub
NN R
R- N2
N
R
N
Rysunek 89. Schemat tworzenia warstwy fenylowej na powierzchni elektrody w wyniku
jednoelektronowej redukcji soli diazoniowej [460].
Proces elektroredukcji soli diazoniowej może być prowadzony na dwa sposoby:
w warunkach ex situ i in situ. W pierwszym przypadku w rozpuszczalniku
aprotycznym, np. acetonitrylu, elektroredukcji ulegają tylko wcześniej zsyntezowane
sole diazoniowe [461462]. Z kolei w drugim podejściu, warunki in situ, sól
diazoniowa syntezowana jest bezpośrednio w 0.5 M roztworze kwasu solnego,
zawierającym pierwszorzędową aminę aromatyczną i azotan(III) sodu [460,463,464],
w którym jednocześnie zachodzi proces elektroredukcji, co schematycznie ilustruje
rysunek 90.
R
R
NH2
NO2+0.5 M HCl
++ e-
Rysunek 90. Schemat tworzenia warstwy fenylowej na powierzchni elektrody w warunkach
in situ w wyniku jednoelektronowej redukcji soli diazoniowej, uzyskanej z pierwszorzędowej
aminy aromatycznej oraz azotanu(III) sodu [460].

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
181
Złożoność procesów, jakim mogą ulegać poszczególne składniki roztworów soli
diazoniowych wymusiły na badaczach optymalizację warunków tworzenia warstwy
fenylowej. Wiadomo, że rodniki powstające podczas procesu elektroredukcji mogą
ulegać reakcjom ubocznym (rysunek 91) zarówno w roztworze, jak i na powierzchni,
mogą m.in. oddziaływać z cząsteczkami rozpuszczalnika, przyłączać się do
pierścienia aromatycznego drugiej cząsteczki, czyli ulegać reakcji dimeryzacji,
a nawet oligomeryzacji, tworząc strukturę wielowarstwową [465,466].
RR
R
R
R
R
CH2CN+
CH3CN
dimeryzacja
tworzenie
wielowarstwy
tworzenie
monowarstwy
reakcja z
rozpuszczalnikiem
R
R
R
Rysunek 91. Możliwe reakcje rodnika arylowego zachodzące na powierzchni elektrody oraz
w roztworze acetonitrylu [466].
Badania przeprowadzone w grupie dr hab. Nowickiej wykazały, że przyłożenie do
elektrody potencjału o wartości woltamperometrycznego piku redukcji stosowanej
soli diazoniowej oraz zastosowanie równomolowego stosunku substratów pozwala
uzyskać warstwę najbardziej zbliżoną do monowarstwy, charakteryzującą się
największą jednorodnością rozmieszczenia grup fenylowych [467-469].
13.1. Procedura konstrukcji immunosensora
do elektrochemicznej detekcji ferrytyny
W przypadku czujników z elektrochemiczną detekcją ważne jest, aby
warstwa modyfikująca powierzchnię elektrody była odpowiednio cienka, jednorodna
i dobrze przewodząca. Powyższe parametry idealnie spełniają warstwy fenylowe
utworzone na drodze elektroredukcji odpowiednich soli diazonowych. W badaniach
zastosowano dwa typy soli diazoniowych różniących się między sobą grupami
funkcyjnymi: jedna sól posiadała grupę karboksylową (bezpośrednio związaną
z pierścieniem aromatycznym), druga zaś grupę aminową (połączoną z pierścieniem

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
182
za pomocą łącznika etylowego). Zróżnicowanie grup funkcyjnych na powierzchni
elektrody pozwoliło uzyskać różną orientację zastosowanego w warstwie
receptorowej przeciwciała. Z literatury wiadomo, że sposób ułożenia cząsteczek
przeciwciała w warstwie wpływa na wydajność oddziaływania antygenprzeciwciało
[437], co z kolei przekłada się na wydajność procesu elektrodowego ferrytyny
związanej z przeciwciałem.
Procedura konstrukcji immunosensora do elektrochemicznej detekcji
ferrytyny składała się z 5 etapów. W pierwszym, powierzchnię elektrody złotej
pokryto warstwą nanocząstek magnetycznych (Fe@C Nps). W tym celu na elektrodę
nałożono 6-µl kroplę zawiesiny nanocząstek i pozostawiono ją do wyschnięcia
w temperaturze pokojowej. Następnie tak uzyskaną warstwę nanocząstek
magnetycznych pokryto filmem fenylowym utworzonym w wyniku elektroredukcji
soli diazoniowej zawierającej odpowiednio grupę karboksylową i grupę aminową.
Elektroredukcja wybranej soli została przeprowadzona chronoamperometrycznie
przy stałej wartości potencjału, równej wartości woltamperometrycznego piku
redukcji odpowiedniej soli diazoniowej. W celu określenia wartości potencjałów
redukcji soli diazoniowych, w pierwszej kolejności zarejestrowano
woltamperogramy cykliczne zastosowanych soli, które przedstawione zostały na
rysunku 92. Proces elektroredukcji soli diazoniowej zawierającej grupę aminową
(+N2-C6H4(CH2)2NH2), zsyntezowanej przez dr Agatę Kowalczyk, prowadzono
w acetonitrylu. Zaś sól zawierająca grupę karboksylową (+N2-C6H4COOH)
zsyntezowana została w środowisku 0.5 M kwasu solnego. Na podstawie analizy
krzywych woltamperometrycznych, dla soli diazoniowej z grupą NH2 wybrano
wartość potencjału elektroredukcji -0.65 V, zaś w przypadku soli diazoniowej
z grupą COOH 0.22 V. Kolejny etap konstrukcji immunosensora związany był
z aktywacją grup –COOH oraz –NH2. W tym celu elektrody złote zmodyfikowane
nanocząstkami Fe@C i filmem karboksyfenylowym umieszczano na 1 godzinę
w wodnym roztworze zawierającym 40 mM EDC i 10 mM NHS, natomiast
elektrody pokryte filmem aminoetylofenylowym w roztworze 40 mM EDC na 0.5 h
[470].

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
183
E / V vs. Ag/AgCl/3 M KCl
-1.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2 0.4
prą
d z
no
rma
lizo
wa
ny (
I /
I p)
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
0.2
+N
2-C
6H
4(CH
2)2NH
2
+N
2-C
6H
4COOH
Rysunek 92. Znormalizowane względem wartości prądu piku cykliczne krzywe
woltamperometryczne uzyskane w wyniku jednoelektronowej elektroredukcji soli +N2-C6H4COOH w 0.5 M HCl (niebieska linia ciągła) oraz soli
+N2-C6H4(CH2)2NH2 (czarna linia
ciągła) w ACN z dodatkiem 0.1 M NBu4BF4. Warunki eksperymentalne: Csoli diaz. = 5 mM;
CNaNO2 = 5 mM (warunki in situ); szybkość przemiatania potencjałem 100 mV·s
-1.
Następnie na tak przygotowaną elektrodę nakładano 20-µl kroplę roztworu
przeciwciała (5 mgmL-1
) sporządzonego w 0.02 M buforze PB zawierającym
150 mM K2SO4 z dodatkiem 0.2% v/v Tween-20 na 2.5 h. Podczas etapu
modyfikacji podłoża cząsteczkami przeciwciała elektrody przechowywano
w lodówce, w temperaturze 4 C. Związanie przeciwciała z filmem fenylowym
następowało w wyniku utworzenia wiązania amidowego pomiędzy odpowiednimi
grupami funkcyjnymi pierścienia aromatycznego a grupami –COOH lub –NH2
przeciwciała. W przypadku filmu karboksyfenylowego proces kowalencyjnego
unieruchamiania cząsteczek przeciwciała odbywał się za pomocą grup aminowych
Ab, co prowadziło do pochylonego lub/i równoległego, a nawet pionowego
odwróconego ułożenia cząsteczek Ab względem danej powierzchni (rysunek 66).
Z kolei w przypadku filmu aminoetylofenylowego w procesie unieruchamiania
przeciwciała brały udział grupy karboksylowe obecne w domenie stałej przeciwciała,
prowadząc do pionowego ułożenia Ab względem powierzchni elektrody. Po
utworzeniu warstwy receptorowej (Au/Fe@C Nps/-C6H4R/Ab) elektrody delikatnie
i uważnie opłukiwano buforem PB, w celu usunięcia nadmiaru nieprzereagowanego
przeciwciała. Następnie zanurzano je do roztworu ferrytyny na czas 90 minut
w obecności zewnętrznego źródła pola magnetycznego (magnes neodymowy, 40

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
184
mT) w temperaturze pokojowej . Poszczególne etapy konstrukcji immunosensora do
detekcji ferrytyny ilustruje rysunek 93.
Magnes
Magnes
1ADSORPCJA
Fe@C Nps
5ZWIĄZANIE Ft Z Ab
(1.5 h)
Magnes
Rysunek 93. Schemat konstrukcji immunosensora do elektrochemicznej detekcji ferrytyny.
13.2. Określenie wpływu ułożenia Ab w warstwie
receptorowej na ilość unieruchamianej ferrytyny
przy użyciu technik LA-ICP-MS i QCM
Sposób ułożenia przeciwciała w warstwie receptorowej decyduje
o skuteczności oddziaływania antygen–przeciwciało. Zatem pierwszy etap badań
dotyczył sprawdzenia, czy wraz ze wzrostem stężenia ferrytyny w roztworze wzrasta
również ilość związanych cząsteczek tego białka z warstwą receptorową
immunosensora. W tym celu przeprowadzone zostały badania z wykorzystaniem
techniki LA-ICP-MS. Z uwagi na fakt, że zarówno nanocząstki magnetyczne, jak
i ferrytyna zawierają w swojej strukturze żelazo, w przypadku pomiarów
LA-ICP-MS elektrody zmodyfikowano tylko i wyłącznie odpowiednim filmem
fenylowym i ferrytyną o określonym stężeniu. Taka modyfikacja elektrody dawała
pewność, że rejestrowane sygnały izotpu 57
Fe pochodzą tylko i wyłącznie od żelaza
obecnego w strukturze ferrytyny. Uzyskane wyniki, przedstawione na wykresie
wewnętrznym rysunku 94, ewidentnie pokazują, że wprowadzenie cząsteczek Ab do
warstwy receptorowej za pośrednictwem grup karboksylowych Ab (HOOC-Ab)
prowadzi do ok. 3-krotnego wzrostu ilości ferrytyny związanej z immunosensorem
na drodze oddziaływań antygen–przeciwciało w porównaniu do wiązania Ft za
pośrednictem grup aminowych Ab (H2N-Ab), co jest zgodne z danymi opisanymi
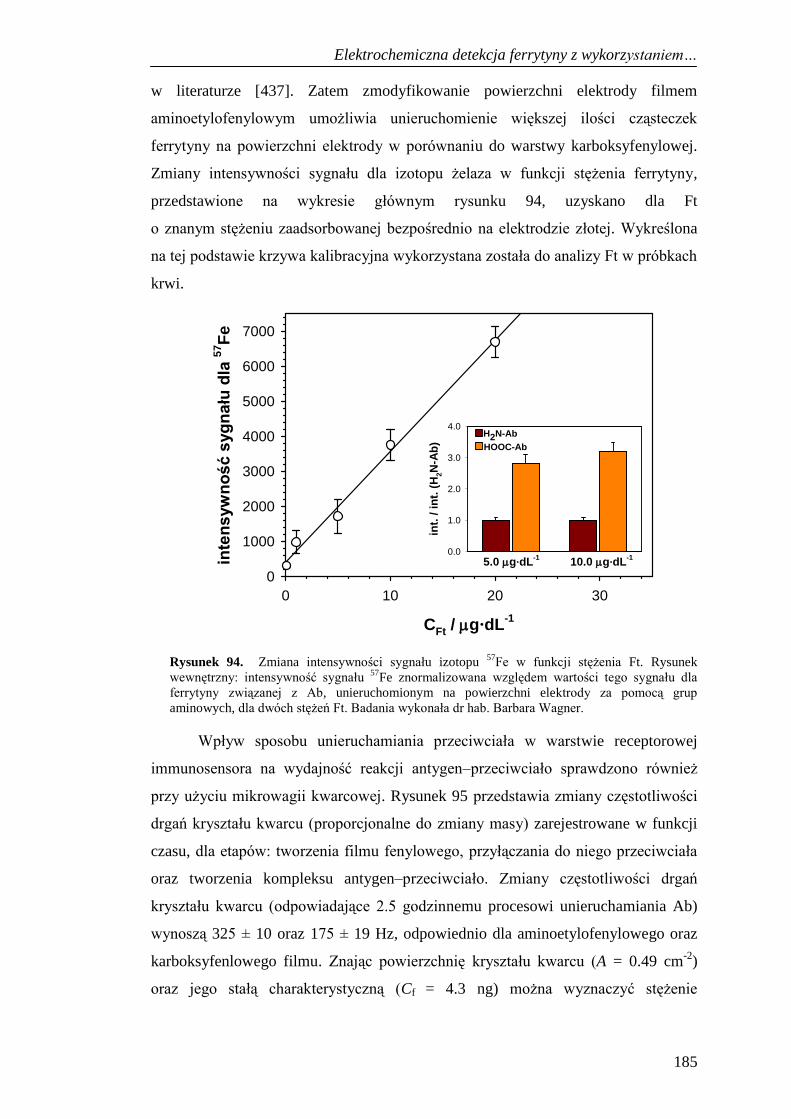
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
185
w literaturze [437]. Zatem zmodyfikowanie powierzchni elektrody filmem
aminoetylofenylowym umożliwia unieruchomienie większej ilości cząsteczek
ferrytyny na powierzchni elektrody w porównaniu do warstwy karboksyfenylowej.
Zmiany intensywności sygnału dla izotopu żelaza w funkcji stężenia ferrytyny,
przedstawione na wykresie głównym rysunku 94, uzyskano dla Ft
o znanym stężeniu zaadsorbowanej bezpośrednio na elektrodzie złotej. Wykreślona
na tej podstawie krzywa kalibracyjna wykorzystana została do analizy Ft w próbkach
krwi.
CFt / gdL-1
0 10 20 30
inte
nsyw
no
ść s
yg
nału
dla
57F
e
0
1000
2000
3000
4000
5000
6000
7000
int.
/ i
nt.
(H
2N
-Ab
)
0.0
1.0
2.0
3.0
4.0H2N-Ab
HOOC-Ab
5.0 gdL-1
10.0 gdL-1
Rysunek 94. Zmiana intensywności sygnału izotopu 57
Fe w funkcji stężenia Ft. Rysunek
wewnętrzny: intensywność sygnału 57
Fe znormalizowana względem wartości tego sygnału dla
ferrytyny związanej z Ab, unieruchomionym na powierzchni elektrody za pomocą grup
aminowych, dla dwóch stężeń Ft. Badania wykonała dr hab. Barbara Wagner.
Wpływ sposobu unieruchamiania przeciwciała w warstwie receptorowej
immunosensora na wydajność reakcji antygen–przeciwciało sprawdzono również
przy użyciu mikrowagii kwarcowej. Rysunek 95 przedstawia zmiany częstotliwości
drgań kryształu kwarcu (proporcjonalne do zmiany masy) zarejestrowane w funkcji
czasu, dla etapów: tworzenia filmu fenylowego, przyłączania do niego przeciwciała
oraz tworzenia kompleksu antygen–przeciwciało. Zmiany częstotliwości drgań
kryształu kwarcu (odpowiadające 2.5 godzinnemu procesowi unieruchamiania Ab)
wynoszą 325 ± 10 oraz 175 ± 19 Hz, odpowiednio dla aminoetylofenylowego oraz
karboksyfenlowego filmu. Znając powierzchnię kryształu kwarcu (A = 0.49 cm-2
)
oraz jego stałą charakterystyczną (Cf = 4.3 ng) można wyznaczyć stężenie
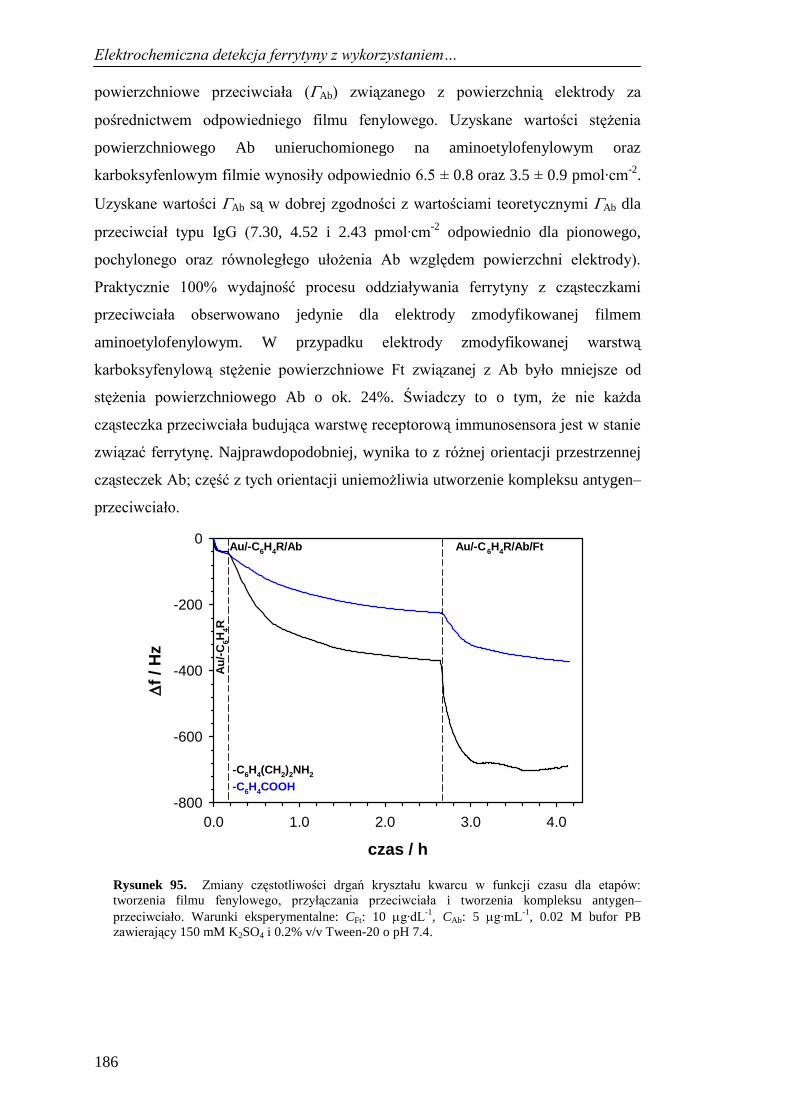
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
186
powierzchniowe przeciwciała (Ab) związanego z powierzchnią elektrody za
pośrednictwem odpowiedniego filmu fenylowego. Uzyskane wartości stężenia
powierzchniowego Ab unieruchomionego na aminoetylofenylowym oraz
karboksyfenlowym filmie wynosiły odpowiednio 6.5 ± 0.8 oraz 3.5 ± 0.9 pmol·cm-2
.
Uzyskane wartości Ab są w dobrej zgodności z wartościami teoretycznymi Ab dla
przeciwciał typu IgG (7.30, 4.52 i 2.43 pmol·cm-2
odpowiednio dla pionowego,
pochylonego oraz równoległego ułożenia Ab względem powierzchni elektrody).
Praktycznie 100% wydajność procesu oddziaływania ferrytyny z cząsteczkami
przeciwciała obserwowano jedynie dla elektrody zmodyfikowanej filmem
aminoetylofenylowym. W przypadku elektrody zmodyfikowanej warstwą
karboksyfenylową stężenie powierzchniowe Ft związanej z Ab było mniejsze od
stężenia powierzchniowego Ab o ok. 24%. Świadczy to o tym, że nie każda
cząsteczka przeciwciała budująca warstwę receptorową immunosensora jest w stanie
związać ferrytynę. Najprawdopodobniej, wynika to z różnej orientacji przestrzennej
cząsteczek Ab; część z tych orientacji uniemożliwia utworzenie kompleksu antygen–
przeciwciało.
czas / h
0.0 1.0 2.0 3.0 4.0
f
/ H
z
-800
-600
-400
-200
0Au/-C
6H
4R/Ab Au/-C
6H
4R/Ab/Ft
Au
/-C
6H
4R
-C6H
4(CH
2)2NH
2
-C6H
4COOH
Rysunek 95. Zmiany częstotliwości drgań kryształu kwarcu w funkcji czasu dla etapów:
tworzenia filmu fenylowego, przyłączania przeciwciała i tworzenia kompleksu antygen–
przeciwciało. Warunki eksperymentalne: CFt: 10 mg·dL-1
, CAb: 5 mg·mL-1
, 0.02 M bufor PB
zawierający 150 mM K2SO4 i 0.2% v/v Tween-20 o pH 7.4.
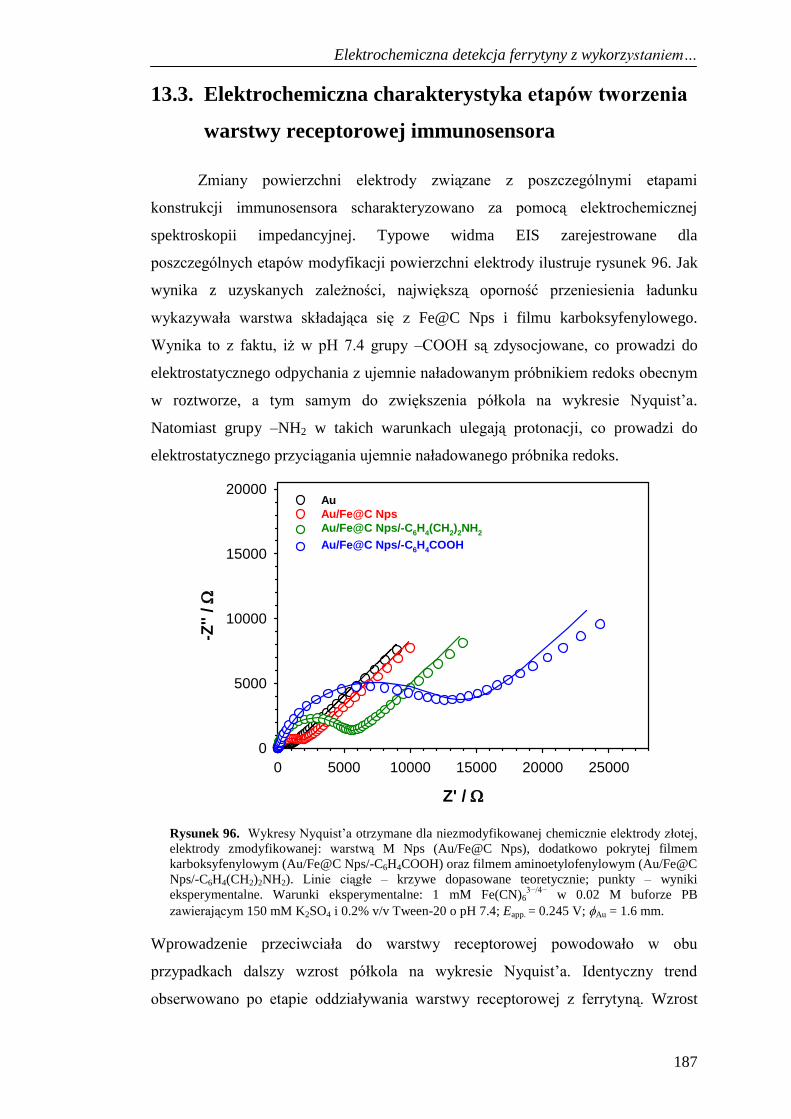
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
187
13.3. Elektrochemiczna charakterystyka etapów tworzenia
warstwy receptorowej immunosensora
Zmiany powierzchni elektrody związane z poszczególnymi etapami
konstrukcji immunosensora scharakteryzowano za pomocą elektrochemicznej
spektroskopii impedancyjnej. Typowe widma EIS zarejestrowane dla
poszczególnych etapów modyfikacji powierzchni elektrody ilustruje rysunek 96. Jak
wynika z uzyskanych zależności, największą oporność przeniesienia ładunku
wykazywała warstwa składająca się z Fe@C Nps i filmu karboksyfenylowego.
Wynika to z faktu, iż w pH 7.4 grupy –COOH są zdysocjowane, co prowadzi do
elektrostatycznego odpychania z ujemnie naładowanym próbnikiem redoks obecnym
w roztworze, a tym samym do zwiększenia półkola na wykresie Nyquist’a.
Natomiast grupy –NH2 w takich warunkach ulegają protonacji, co prowadzi do
elektrostatycznego przyciągania ujemnie naładowanego próbnika redoks.
Z' /
0 5000 10000 15000 20000 25000
-Z'' /
0
5000
10000
15000
20000Au
Au/Fe@C Nps
Au/Fe@C Nps/-C6H
4(CH
2)2NH
2
Au/Fe@C Nps/-C6H
4COOH
Rysunek 96. Wykresy Nyquist’a otrzymane dla niezmodyfikowanej chemicznie elektrody złotej,
elektrody zmodyfikowanej: warstwą M Nps (Au/Fe@C Nps), dodatkowo pokrytej filmem
karboksyfenylowym (Au/Fe@C Nps/-C6H4COOH) oraz filmem aminoetylofenylowym (Au/Fe@C
Nps/-C6H4(CH2)2NH2). Linie ciągłe – krzywe dopasowane teoretycznie; punkty – wyniki
eksperymentalne. Warunki eksperymentalne: 1 mM Fe(CN)63−/4−
w 0.02 M buforze PB
zawierającym 150 mM K2SO4 i 0.2% v/v Tween-20 o pH 7.4; Eapp. = 0.245 V; Au = 1.6 mm.
Wprowadzenie przeciwciała do warstwy receptorowej powodowało w obu
przypadkach dalszy wzrost półkola na wykresie Nyquist’a. Identyczny trend
obserwowano po etapie oddziaływania warstwy receptorowej z ferrytyną. Wzrost
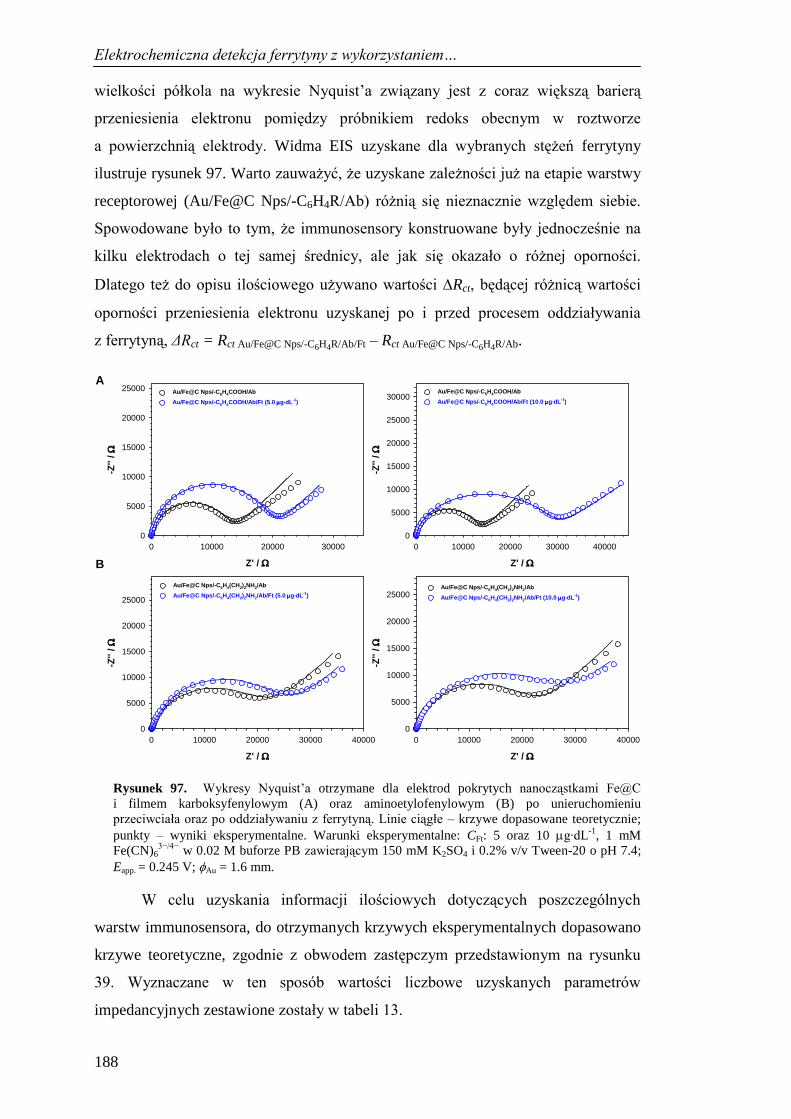
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
188
wielkości półkola na wykresie Nyquist’a związany jest z coraz większą barierą
przeniesienia elektronu pomiędzy próbnikiem redoks obecnym w roztworze
a powierzchnią elektrody. Widma EIS uzyskane dla wybranych stężeń ferrytyny
ilustruje rysunek 97. Warto zauważyć, że uzyskane zależności już na etapie warstwy
receptorowej (Au/Fe@C Nps/-C6H4R/Ab) różnią się nieznacznie względem siebie.
Spowodowane było to tym, że immunosensory konstruowane były jednocześnie na
kilku elektrodach o tej samej średnicy, ale jak się okazało o różnej oporności.
Dlatego też do opisu ilościowego używano wartości Rct, będącej różnicą wartości
oporności przeniesienia elektronu uzyskanej po i przed procesem oddziaływania
z ferrytyną, ΔRct = Rct Au/Fe@C Nps/-C6H4R/Ab/Ft – Rct Au/Fe@C Nps/-C6H4R/Ab.
Z' /
0 10000 20000 30000
-Z'' /
0
5000
10000
15000
20000
25000 Au/Fe@C Nps/-C6H4COOH/Ab
Au/Fe@C Nps/-C6H4COOH/Ab/Ft (5.0 gdL-1
)
Z' /
0 10000 20000 30000 40000
-Z'' /
0
5000
10000
15000
20000
25000
30000Au/Fe@C Nps/-C6H4COOH/Ab
Au/Fe@C Nps/-C6H4COOH/Ab/Ft (10.0 gdL-1
)
Z' /
0 10000 20000 30000 40000
-Z'' /
0
5000
10000
15000
20000
25000
Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab
Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab/Ft (5.0 gdL-1
)
Ab
Ft
Z' /
0 10000 20000 30000 40000
-Z'' /
0
5000
10000
15000
20000
25000Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab
Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab/Ft (10.0 gdL-1
)
A
B
Rysunek 97. Wykresy Nyquist’a otrzymane dla elektrod pokrytych nanocząstkami Fe@C
i filmem karboksyfenylowym (A) oraz aminoetylofenylowym (B) po unieruchomieniu
przeciwciała oraz po oddziaływaniu z ferrytyną. Linie ciągłe – krzywe dopasowane teoretycznie;
punkty – wyniki eksperymentalne. Warunki eksperymentalne: CFt: 5 oraz 10 mg·dL-1
, 1 mM
Fe(CN)63−/4−
w 0.02 M buforze PB zawierającym 150 mM K2SO4 i 0.2% v/v Tween-20 o pH 7.4;
Eapp. = 0.245 V; Au = 1.6 mm.
W celu uzyskania informacji ilościowych dotyczących poszczególnych
warstw immunosensora, do otrzymanych krzywych eksperymentalnych dopasowano
krzywe teoretyczne, zgodnie z obwodem zastępczym przedstawionym na rysunku
39. Wyznaczane w ten sposób wartości liczbowe uzyskanych parametrów
impedancyjnych zestawione zostały w tabeli 13.
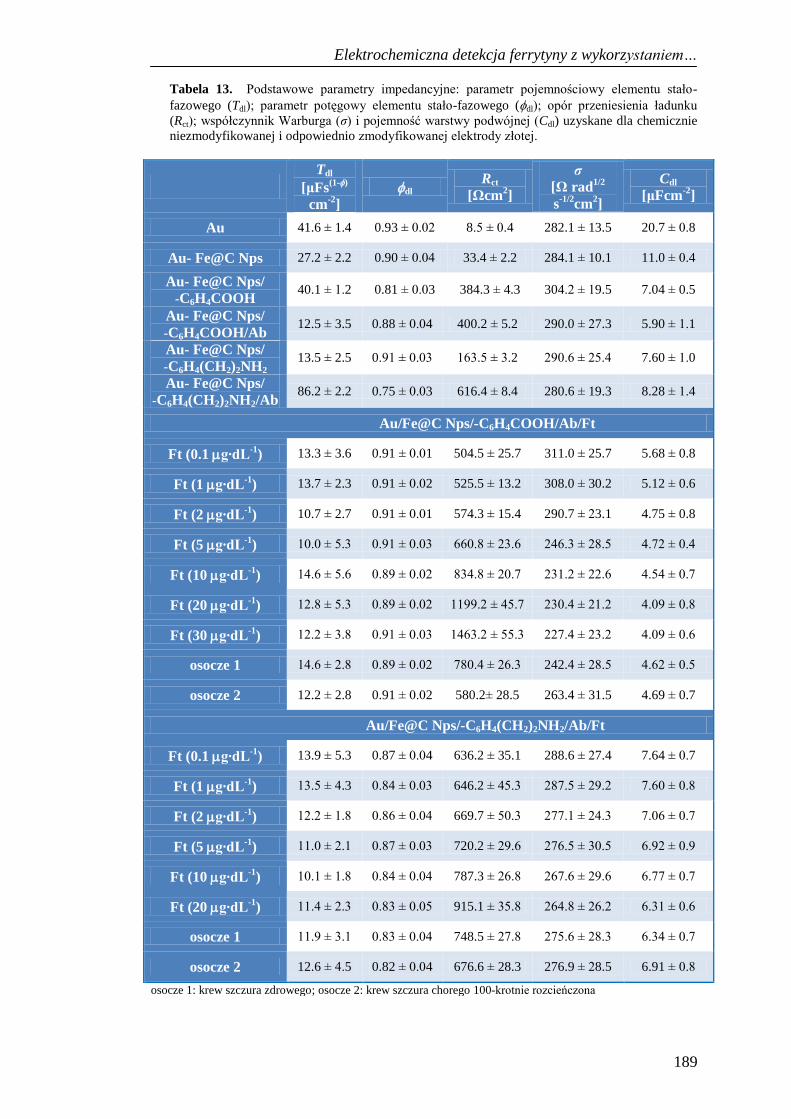
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
189
Tabela 13. Podstawowe parametry impedancyjne: parametr pojemnościowy elementu stało-
fazowego (Tdl); parametr potęgowy elementu stało-fazowego (dl); opór przeniesienia ładunku
(Rct); współczynnik Warburga (σ) i pojemność warstwy podwójnej (Cdl) uzyskane dla chemicznie
niezmodyfikowanej i odpowiednio zmodyfikowanej elektrody złotej.
Tdl
[μFs(1-)
cm-2
]
dl Rct
[Ωcm2]
σ
[Ω rad1/2
s-1/2
cm2]
Cdl
[μFcm-2
]
Au 41.6 ± 1.4 0.93 ± 0.02 8.5 ± 0.4 282.1 ± 13.5 20.7 ± 0.8
Au- Fe@C Nps 27.2 ± 2.2 0.90 ± 0.04 33.4 ± 2.2 284.1 ± 10.1 11.0 ± 0.4
Au- Fe@C Nps/
-C6H4COOH 40.1 ± 1.2 0.81 ± 0.03 384.3 ± 4.3 304.2 ± 19.5 7.04 ± 0.5
Au- Fe@C Nps/
-C6H4COOH/Ab 12.5 ± 3.5 0.88 ± 0.04 400.2 ± 5.2 290.0 ± 27.3 5.90 ± 1.1
Au- Fe@C Nps/
-C6H4(CH2)2NH2 13.5 ± 2.5 0.91 ± 0.03 163.5 ± 3.2 290.6 ± 25.4 7.60 ± 1.0
Au- Fe@C Nps/
-C6H4(CH2)2NH2/Ab 86.2 ± 2.2 0.75 ± 0.03 616.4 ± 8.4 280.6 ± 19.3 8.28 ± 1.4
Au/Fe@C Nps/-C6H4COOH/Ab/Ft
Ft (0.1 mg·dL-1
) 13.3 ± 3.6 0.91 ± 0.01 504.5 ± 25.7 311.0 ± 25.7 5.68 ± 0.8
Ft (1 mg·dL-1
) 13.7 ± 2.3 0.91 ± 0.02 525.5 ± 13.2 308.0 ± 30.2 5.12 ± 0.6
Ft (2 mg·dL-1
) 10.7 ± 2.7 0.91 ± 0.01 574.3 ± 15.4 290.7 ± 23.1 4.75 ± 0.8
Ft (5 mg·dL-1
) 10.0 ± 5.3 0.91 ± 0.03 660.8 ± 23.6 246.3 ± 28.5 4.72 ± 0.4
Ft (10 mg·dL-1
) 14.6 ± 5.6 0.89 ± 0.02 834.8 ± 20.7 231.2 ± 22.6 4.54 ± 0.7
Ft (20 mg·dL-1
) 12.8 ± 5.3 0.89 ± 0.02 1199.2 ± 45.7 230.4 ± 21.2 4.09 ± 0.8
Ft (30 mg·dL-1
) 12.2 ± 3.8 0.91 ± 0.03 1463.2 ± 55.3 227.4 ± 23.2 4.09 ± 0.6
osocze 1 14.6 ± 2.8 0.89 ± 0.02 780.4 ± 26.3 242.4 ± 28.5 4.62 ± 0.5
osocze 2 12.2 ± 2.8 0.91 ± 0.02 580.2± 28.5 263.4 ± 31.5 4.69 ± 0.7
Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab/Ft
Ft (0.1 mg·dL-1
) 13.9 ± 5.3 0.87 ± 0.04 636.2 ± 35.1 288.6 ± 27.4 7.64 ± 0.7
Ft (1 mg·dL-1
) 13.5 ± 4.3 0.84 ± 0.03 646.2 ± 45.3 287.5 ± 29.2 7.60 ± 0.8
Ft (2 mg·dL-1
) 12.2 ± 1.8 0.86 ± 0.04 669.7 ± 50.3 277.1 ± 24.3 7.06 ± 0.7
Ft (5 mg·dL-1
) 11.0 ± 2.1 0.87 ± 0.03 720.2 ± 29.6 276.5 ± 30.5 6.92 ± 0.9
Ft (10 mg·dL-1
) 10.1 ± 1.8 0.84 ± 0.04 787.3 ± 26.8 267.6 ± 29.6 6.77 ± 0.7
Ft (20 mg·dL-1
) 11.4 ± 2.3 0.83 ± 0.05 915.1 ± 35.8 264.8 ± 26.2 6.31 ± 0.6
osocze 1 11.9 ± 3.1 0.83 ± 0.04 748.5 ± 27.8 275.6 ± 28.3 6.34 ± 0.7
osocze 2 12.6 ± 4.5 0.82 ± 0.04 676.6 ± 28.3 276.9 ± 28.5 6.91 ± 0.8
osocze 1: krew szczura zdrowego; osocze 2: krew szczura chorego 100-krotnie rozcieńczona

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
190
Analizując uzyskane wartości parametrów EIS można zauważyć, że warstwy
tworzone podczas kolejnych etapów modyfikacji elektrody złotej charakteryzowały
się równomiernością i jednorodnością. Potwierdzają to niewielkie zmiany wartości
elementu stało-fazowego, które różnią się nie więcej niż 10% od wartości otrzymanej
dla chemicznie niezmodyfikowanej elektrody złotej. Znaczne zmiany,
w porównaniu do chemicznie niezmodyfikowanej powierzchni elektrody złotej,
obserwowano dla takich parametrów, jak: opór przeniesienia ładunku, parametr
Warburga oraz pojemność warstwy podwójnej. Wartości parametru Rct wzrastały
wraz z kolejnymi etapami modyfikacji powierzchni elektrody, bez względu na typ
użytej soli diazoniowej. Dodatkowo, parametr ten był bardzo czuły na ilość ferrytyny
oddziałującej z charakterystycznym dla niej przeciwciałem. Jeśli chodzi o pozostałe
parametry, takie jak parametr Warburga, czy też pojemność warstwy podwójnej,
intensywność zmian ich wartości była zdecydowanie mniejsza i mniej czuła na
zmiany stężenia ferrytyny. Na podstawie zmian parametru Warburga, ok. 20%
w odniesieniu do chemicznie niezmodyfikowanej elektrody złotej, można stwierdzić,
że transport próbnika redoks ulegał nieistotnym zmianom w wyniku modyfikacji
powierzchni elektrody. Porównując z kolei wartości pojemności warstwy podwójnej
wyraźnie widać, że wraz z kolejną modyfikacją wartość tego parametru zmniejszała
się, stabilizując się na poziomie 4÷5 mF·cm-2
. Taka wartość Cdl świadczy o znacznym
uwodnieniu tworzonych warstw.
Charakterystyki analitycznej zaproponowanego immunosensora dokonano na
podstawie kontroli zmian oporności przeniesienia ładunku w funkcji stężenia
ferrytyny. Uzyskane w ten sposób krzywe kalibracyjne ilustruje rysunek 98.
W przypadku modyfikacji elektrody filmem karboksyfenylowym liniową zależność
zmian wartości oporu przeniesienia ładunku w funkcji stężenia ferrytyny
obserwowano w zakresie 0.1 30 mg·dL-1
. Natomiast w sytuacji wykorzystania
filmu aminoetylofenylowego do stabilizacji warstwy nanocząstek magnetycznych,
liniowa zależność Rct = f(CFt) dotyczyła zakresu stężeń ferrytyny od 0.1 do
20 mg·dL-1
. Wyznaczona na podstawie uzyskanych zależności granica
wykrywalności ferrytyny wynosiła 0.40 0.04 oraz 0.20 0.02 µg·dL-1
,
odpowiednio dla filmu karboksyfenylowego i aminoetylofenylowego.

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
191
CFt / gdL-1
0 5 10 15 20 25 30 35
R
ct /
0
10000
20000
30000
40000
50000
CFt / gdL-1
0 5 10 15 20 25
R
ct /
0
2000
4000
6000
8000
10000
12000
A
B
Rysunek 98. Krzywe kalibracyjne ilustrujące zmianę wartości Rct w funkcji stężenia ferrytyny,
uzyskane dla elektrody zmodyfikowanej A: warstwą karboksyfenylową oraz B: warstwą
aminoetylofenylową. Czerwone symbole: osocze krwi szczura zdrowego; zielone symbole: osocze
krwi szczura. Rct = Rct(Au/Fe@C Nps/-C6H4R/Ab/Ft) - Rct(Au/Fe@C Nps/-C6H4R/Ab).
13.3.1. Wpływ pola magnetycznego na sygnał prądowy ferrytyny
Ze względu na fakt, że ferrytyna zawiera w swojej strukturze
paramagnetyczne jony żelaza(III), które jednocześnie odpowiadają za jej aktywność
elektrochemiczną, kolejny etap badań dotyczył sprawdzenia możliwości
wykorzystania woltamperometrii w detekcji ferrytyny. Obserwacja dobrze
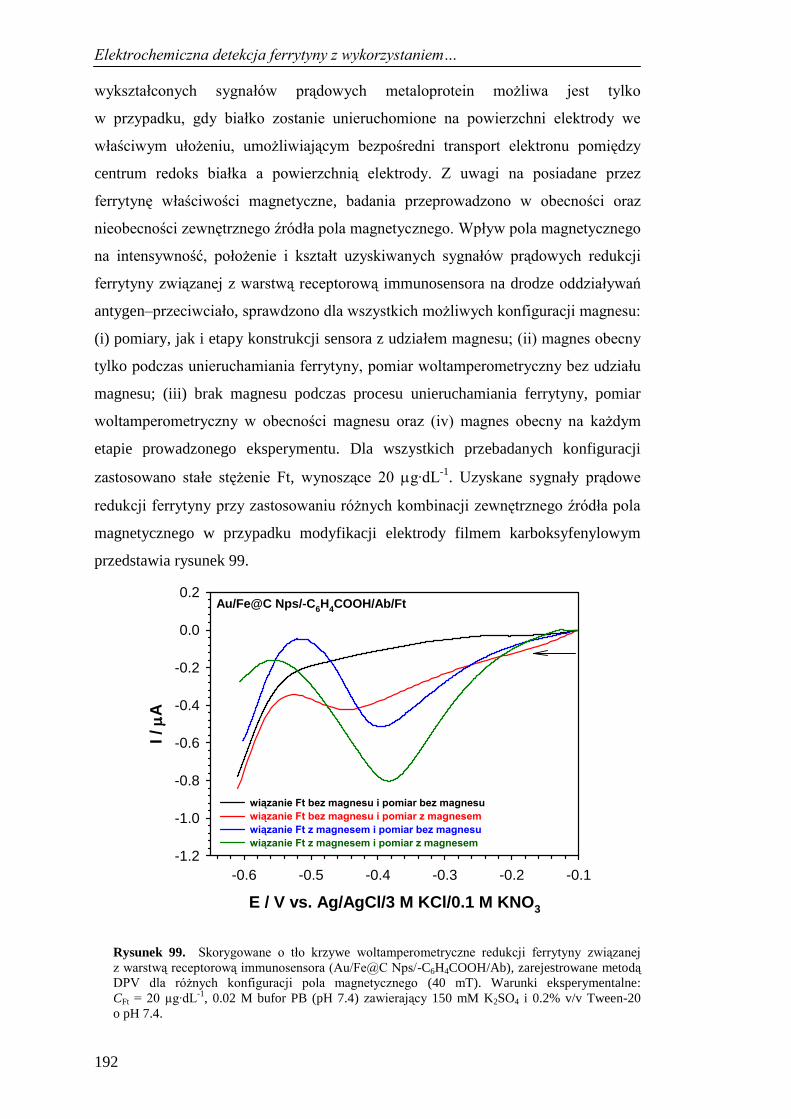
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
192
wykształconych sygnałów prądowych metaloprotein możliwa jest tylko
w przypadku, gdy białko zostanie unieruchomione na powierzchni elektrody we
właściwym ułożeniu, umożliwiającym bezpośredni transport elektronu pomiędzy
centrum redoks białka a powierzchnią elektrody. Z uwagi na posiadane przez
ferrytynę właściwości magnetyczne, badania przeprowadzono w obecności oraz
nieobecności zewnętrznego źródła pola magnetycznego. Wpływ pola magnetycznego
na intensywność, położenie i kształt uzyskiwanych sygnałów prądowych redukcji
ferrytyny związanej z warstwą receptorową immunosensora na drodze oddziaływań
antygen–przeciwciało, sprawdzono dla wszystkich możliwych konfiguracji magnesu:
(i) pomiary, jak i etapy konstrukcji sensora z udziałem magnesu; (ii) magnes obecny
tylko podczas unieruchamiania ferrytyny, pomiar woltamperometryczny bez udziału
magnesu; (iii) brak magnesu podczas procesu unieruchamiania ferrytyny, pomiar
woltamperometryczny w obecności magnesu oraz (iv) magnes obecny na każdym
etapie prowadzonego eksperymentu. Dla wszystkich przebadanych konfiguracji
zastosowano stałe stężenie Ft, wynoszące 20 mg·dL-1
. Uzyskane sygnały prądowe
redukcji ferrytyny przy zastosowaniu różnych kombinacji zewnętrznego źródła pola
magnetycznego w przypadku modyfikacji elektrody filmem karboksyfenylowym
przedstawia rysunek 99.
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.6 -0.5 -0.4 -0.3 -0.2 -0.1
I /
A
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
0.2
wiązanie Ft bez magnesu i pomiar bez magnesu
wiązanie Ft bez magnesu i pomiar z magnesem
wiązanie Ft z magnesem i pomiar bez magnesu
wiązanie Ft z magnesem i pomiar z magnesem
Au/Fe@C Nps/-C6H
4COOH/Ab/Ft
Rysunek 99. Skorygowane o tło krzywe woltamperometryczne redukcji ferrytyny związanej
z warstwą receptorową immunosensora (Au/Fe@C Nps/-C6H4COOH/Ab), zarejestrowane metodą
DPV dla różnych konfiguracji pola magnetycznego (40 mT). Warunki eksperymentalne:
CFt = 20 µg·dL-1
, 0.02 M bufor PB (pH 7.4) zawierający 150 mM K2SO4 i 0.2% v/v Tween-20
o pH 7.4.
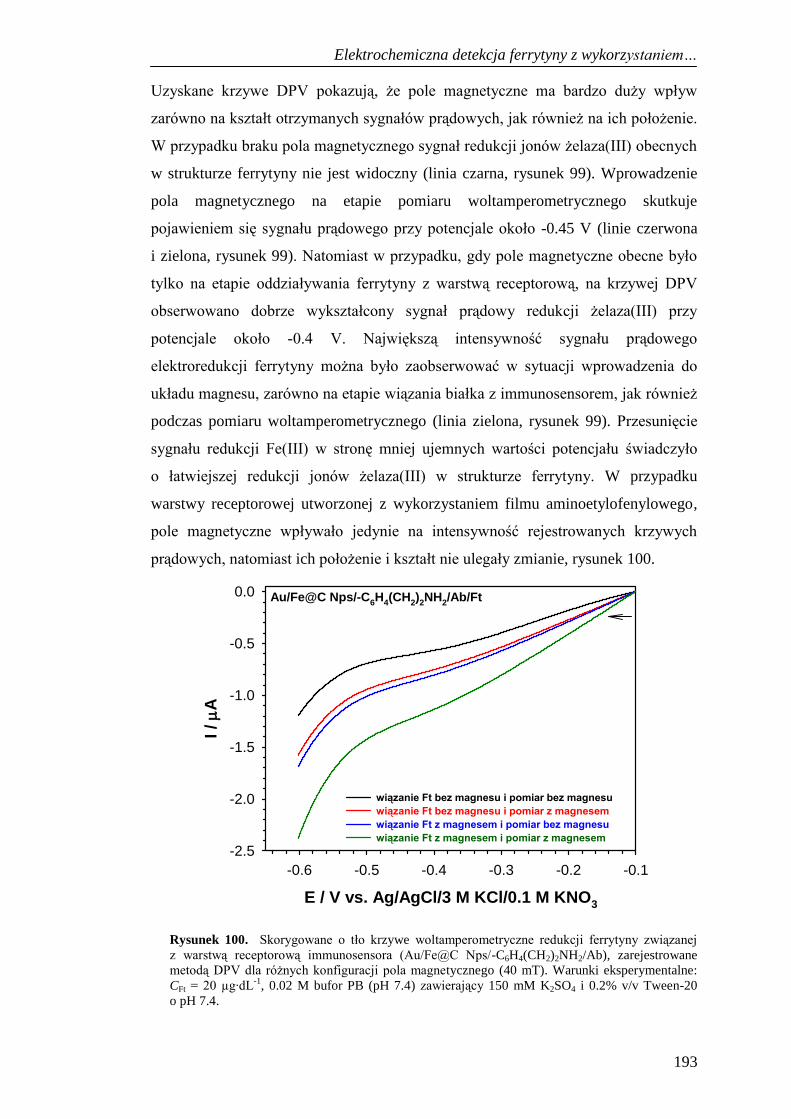
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
193
Uzyskane krzywe DPV pokazują, że pole magnetyczne ma bardzo duży wpływ
zarówno na kształt otrzymanych sygnałów prądowych, jak również na ich położenie.
W przypadku braku pola magnetycznego sygnał redukcji jonów żelaza(III) obecnych
w strukturze ferrytyny nie jest widoczny (linia czarna, rysunek 99). Wprowadzenie
pola magnetycznego na etapie pomiaru woltamperometrycznego skutkuje
pojawieniem się sygnału prądowego przy potencjale około -0.45 V (linie czerwona
i zielona, rysunek 99). Natomiast w przypadku, gdy pole magnetyczne obecne było
tylko na etapie oddziaływania ferrytyny z warstwą receptorową, na krzywej DPV
obserwowano dobrze wykształcony sygnał prądowy redukcji żelaza(III) przy
potencjale około -0.4 V. Największą intensywność sygnału prądowego
elektroredukcji ferrytyny można było zaobserwować w sytuacji wprowadzenia do
układu magnesu, zarówno na etapie wiązania białka z immunosensorem, jak również
podczas pomiaru woltamperometrycznego (linia zielona, rysunek 99). Przesunięcie
sygnału redukcji Fe(III) w stronę mniej ujemnych wartości potencjału świadczyło
o łatwiejszej redukcji jonów żelaza(III) w strukturze ferrytyny. W przypadku
warstwy receptorowej utworzonej z wykorzystaniem filmu aminoetylofenylowego,
pole magnetyczne wpływało jedynie na intensywność rejestrowanych krzywych
prądowych, natomiast ich położenie i kształt nie ulegały zmianie, rysunek 100.
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.6 -0.5 -0.4 -0.3 -0.2 -0.1
I /
A
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
wiązanie Ft bez magnesu i pomiar bez magnesu
wiązanie Ft bez magnesu i pomiar z magnesem
wiązanie Ft z magnesem i pomiar bez magnesu
wiązanie Ft z magnesem i pomiar z magnesem
Au/Fe@C Nps/-C6H
4(CH
2)2NH
2/Ab/Ft
Rysunek 100. Skorygowane o tło krzywe woltamperometryczne redukcji ferrytyny związanej
z warstwą receptorową immunosensora (Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab), zarejestrowane
metodą DPV dla różnych konfiguracji pola magnetycznego (40 mT). Warunki eksperymentalne:
CFt = 20 µg·dL-1
, 0.02 M bufor PB (pH 7.4) zawierający 150 mM K2SO4 i 0.2% v/v Tween-20
o pH 7.4.

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
194
Warstwa aminoetylofenylowa pozwala uzyskać pionowe ułożenie przeciwciała
w warstwie receptorowej. Zgodnie z doniesieniami literaturowymi, stężenie
powierzchniowe przeciwciał w warstwie, przy ich pionowym ustawieniu, jest
największe z możliwych. Co więcej, taka orientacja Ab sprawia, że wszystkie
cząsteczki przeciwciała są w stanie oddziaływać z antgenem, czyli z ferrytyną.
Niestety wraz ze wzrostem wydajności reakcji antygen–przeciwciało spada jej
użyteczność w stosunku do immunosensorów z woltamperometryczną detekcją.
Łańcuchy polipeptydowe są bardzo słabymi nośnikami ładunku. Przeprowadzone
pomiary wyraźnie pokazują, że intensywność sygnałów prądowych ferrytyny silnie
zależy od obecności pola magnetycznego. Najprawdopodobniej, podczas procesu
oddziaływania Ft z przeciwciałem, pole magnetyczne nie tylko wymusza na
cząsteczkach ferrytyny orientację zapewniającą ich maksymalną elektroaktywność,
ale również dodatkowo skraca odległość pomiędzy centrum elektroaktywnym białka
a powierzchnią elektrody. Ta zmiana odległości jest konsekwencją silnego
przyciągania pomiędzy paramagnetycznym białkiem i ferromagnetycznym
modyfikatorem powierzchni elektrody.
13.3.2. Woltamperometryczna charakterystyka immunosensora
do detekcji ferrytyny
Charakterystykę analityczną skonstruowanego immunosensora do
bezpośredniej woltamperometrycznej detekcji ferrytyny wykonano prowadząc
pomiary w obecności pola magnetycznego o wartości 40 mT, generowanego przez
magnes neodymowy (Fe14Nd2B) umieszczony w pobliżu elektrody. W tym celu
immunosensor zanurzano do roztworu Ft o odpowiednim stężeniu, z zakresu
0.1÷30 mg·dL-1
w obecności magnesu na czas 1.5 h. Następnie elektrodę
(Au/Fe@C Nps/-C6H4R/Ab/Ft), po opłukaniu wodą destylowaną, umieszczano
w czystym, odtlenionym 0.02 M buforze PB o pH 7.4 z dodatkiem 0.15 M K2SO4
i 0.2% v/v Tween-20 i wykonywano pomiar woltamperometryczny. Uzyskane
woltamperogramy redukcji ferrytyny związanej z warstwą receptorową, utworzoną
w wyniku modyfikacji elektrody filmem karboksyfenylowym, ilustruje rysunek 101.
Intensywność sygnału prądowego, pojawiającego się przy potencjale ok. -0.4 V
związanego z elektredukcją jonów Fe(III) obecnych w strukturze ferrytyny, była
wprost proporcjonalna do ilości ferrytyny związanej z warstwą receptorową
w zakresie stężeń Ft 0.1 ÷ 30 µg·dL-1
.
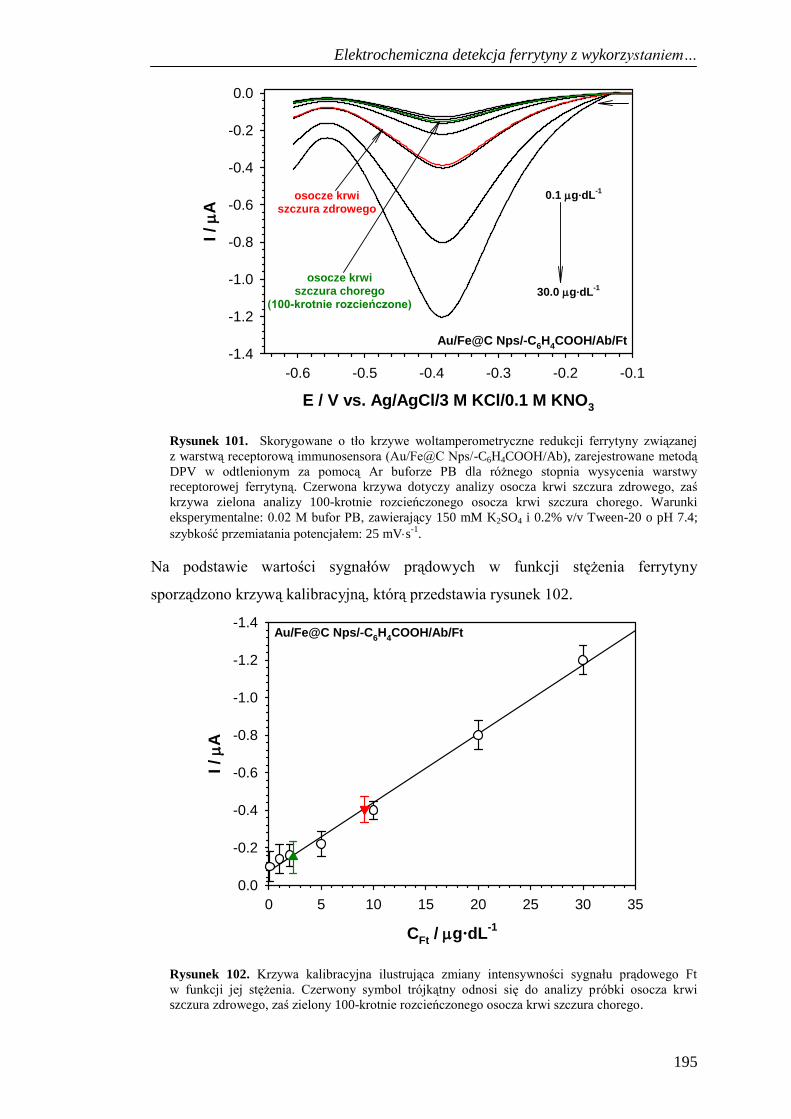
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
195
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.6 -0.5 -0.4 -0.3 -0.2 -0.1
I /
A
-1.4
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
0.1 gdL-1
30.0 gdL-1
osocze krwi szczura zdrowego
Au/Fe@C Nps/-C6H
4COOH/Ab/Ft
osocze krwi szczura chorego
(100-krotnie rozcieńczone)
Rysunek 101. Skorygowane o tło krzywe woltamperometryczne redukcji ferrytyny związanej
z warstwą receptorową immunosensora (Au/Fe@C Nps/-C6H4COOH/Ab), zarejestrowane metodą
DPV w odtlenionym za pomocą Ar buforze PB dla różnego stopnia wysycenia warstwy
receptorowej ferrytyną. Czerwona krzywa dotyczy analizy osocza krwi szczura zdrowego, zaś
krzywa zielona analizy 100-krotnie rozcieńczonego osocza krwi szczura chorego. Warunki
eksperymentalne: 0.02 M bufor PB, zawierający 150 mM K2SO4 i 0.2% v/v Tween-20 o pH 7.4;
szybkość przemiatania potencjałem: 25 mVs-1
.
Na podstawie wartości sygnałów prądowych w funkcji stężenia ferrytyny
sporządzono krzywą kalibracyjną, którą przedstawia rysunek 102.
CFt / gdL-1
0 5 10 15 20 25 30 35
I /
A
-1.4
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
Au/Fe@C Nps/-C6H
4COOH/Ab/Ft
Rysunek 102. Krzywa kalibracyjna ilustrująca zmiany intensywności sygnału prądowego Ft
w funkcji jej stężenia. Czerwony symbol trójkątny odnosi się do analizy próbki osocza krwi
szczura zdrowego, zaś zielony 100-krotnie rozcieńczonego osocza krwi szczura chorego.

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
196
Najmniejsze stężenie ferrytyny rozpuszczonej w roztworze, możliwe do
precyzyjnego oznaczenia zaproponowaną metodą z wykorzystaniem filmu
karboksyfenylowego, wyznaczono z dolnego zakresu prostoliniowego krzywej
kalibracyjnej, zgodnie z równaniem 31. Uzyskana w ten sposób granica
wykrywalności wynosiła 0.13 0.04 µg·dL-1
. Ponadto dla wszystkich badanych
stężeń ferrytyny z zakresu liniowego krzywej kalibracyjnej powtarzalność wyników
była dobra, a względne odchylenie standardowe (n = 5) było mniejsze niż 10%.
Z kolei względne odchylenie standardowe wyników otrzymanych dla tego samego
stężenia ferrytyny przy użyciu sześciu różnych, oddzielnie zmodyfikowanych
elektrod wynosiło ok. 14%.
Unieruchomienie przeciwciała w warstwie receptorowej za pośrednictwem
filmu aminoetylofenylowego niestety nie sprzyjało dobrze wykształconym sygnałom
prądowym redukcji ferrytyny. Związanie przeciwciała z grupami aminowymi filmu
fenylowego następuje przy udziale grup karboksylowych zlokalizowanych
w części stałej Ab. Skutkuje to pionowym ułożeniem cząsteczek Ab względem
powierzchni elektrody. Takie ustawienie cząsteczek przeciwciała z jednej strony
zdecydowanie wzmacnia wydajność oddziaływania Ab z charakterystycznym dla
niego antygenem, z drugiej zaś drastycznie spowalnia transport elektronów
pomiędzy Ft a powierzchnią elektrody. Pomimo tych trudności, wraz ze wzrostem
stężenia ferrytyny w roztworze, do którego zanurzano immunosensor, wzrastała
intensywność rejestrowanego sygnału prądowego, co ilustrują krzywe
woltamperometryczne uzyskane metodą DPV, przedstawione na rysunku 103.
Zakres pracy immunosensora z wykorzystaniem filmu aminoetylofenylowego do
bezpośredniej elektrochemicznej detekcji ferrytyny wyznaczono na podstawie
liniowej odpowiedzi sygnału analitycznego (intensywność sygnału redukcji Ft)
względem stężenia tego białka w roztworze, do którego na 1.5 h zanurzano
immunosensor. W badaniach wykorzystano roztwory Ft o różnym stężeniu,
z zakresu 0.1 ÷ 30 mg·dL-1
. W zakresie stężeń 0.1 ÷ 20 mg·dL-1
obserwowano liniowy
wzrost wysokości sygnału elektroredukcji ferrytyny związanej z warstwą
receptorową immunosensora na drodze oddziaływania antygen–przeciwciało. Dla
stężeń Ft większych niż 20 mg·dL-1
nie obserwowano już dalszych zmian
intensywności piku redukcji. Z kolei w przypadku stężeń ferrytyny poniżej
0.1 mg·dL-1
uzyskiwane sygnały prądowe Ft były bardzo nieodtwarzalne.
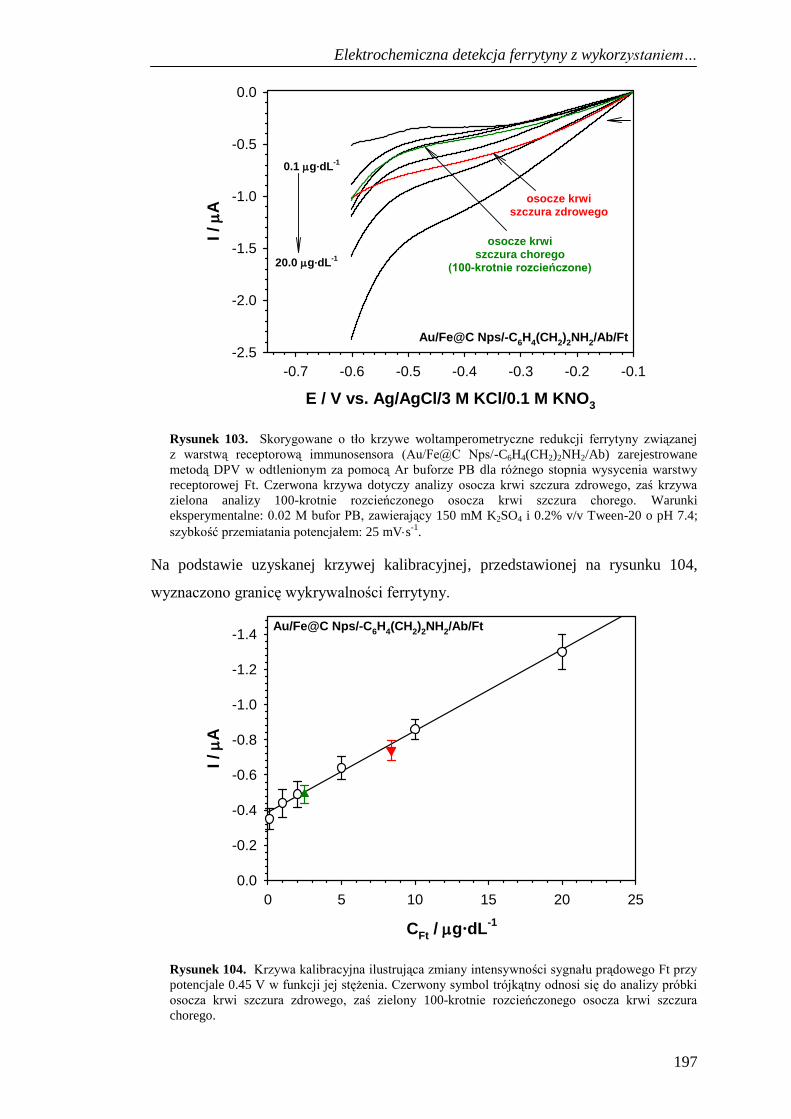
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
197
E / V vs. Ag/AgCl/3 M KCl/0.1 M KNO3
-0.7 -0.6 -0.5 -0.4 -0.3 -0.2 -0.1
I /
A
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
osocze krwi szczura zdrowego
0.1 gdL-1
20.0 gdL-1
Au/Fe@C Nps/-C6H
4(CH
2)2NH
2/Ab/Ft
osocze krwi szczura chorego
(100-krotnie rozcieńczone)
Rysunek 103. Skorygowane o tło krzywe woltamperometryczne redukcji ferrytyny związanej
z warstwą receptorową immunosensora (Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab) zarejestrowane
metodą DPV w odtlenionym za pomocą Ar buforze PB dla różnego stopnia wysycenia warstwy
receptorowej Ft. Czerwona krzywa dotyczy analizy osocza krwi szczura zdrowego, zaś krzywa
zielona analizy 100-krotnie rozcieńczonego osocza krwi szczura chorego. Warunki
eksperymentalne: 0.02 M bufor PB, zawierający 150 mM K2SO4 i 0.2% v/v Tween-20 o pH 7.4;
szybkość przemiatania potencjałem: 25 mVs-1
.
Na podstawie uzyskanej krzywej kalibracyjnej, przedstawionej na rysunku 104,
wyznaczono granicę wykrywalności ferrytyny.
CFt / gdL-1
0 5 10 15 20 25
I /
A
-1.4
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
Au/Fe@C Nps/-C6H
4(CH
2)2NH
2/Ab/Ft
Rysunek 104. Krzywa kalibracyjna ilustrująca zmiany intensywności sygnału prądowego Ft przy
potencjale 0.45 V w funkcji jej stężenia. Czerwony symbol trójkątny odnosi się do analizy próbki
osocza krwi szczura zdrowego, zaś zielony 100-krotnie rozcieńczonego osocza krwi szczura
chorego.

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
198
Wyznaczona, zgodnie z równaniem 31, granica wykrywalności ferrytyny związanej
z warstwą receptorową immunosensora, utworzonego na bazie nanocząstek
magnetycznych i filmu aminoetylofenylowego oraz monoklonalnego przeciwciała,
wynosiła 0.16 0.02 mg·dL-1
. Podobnie, jak w przypadku zastosowania filmu
karboksyfenylowego do utworzenia warstwy receptorowej immunosensora,
powtarzalność wyników dla badanych stężeń ferrytyny z zakresu liniowego krzywej
kalibracyjnej była bardzo dobra. Względne odchylenie (n = 5) było mniejsze niż
11%. Z kolei, względne odchylenie standardowe wyników uzyskanych dla tego
samego stężenia ferrytyny na sześciu różnych elektrodach wynosiło ok. 13%.
13.4. Selektywność immunosensora do detekcji ferrytyny
Selektywność zaproponowanego czujnika do bezpośredniej
woltamperometrycznej detekcji ferrytyny sprawdzono wobec innych
elektroaktywnych białek występujących w próbkach krwi, które zawierają w swojej
strukturze jony żelaza(III), czyli hemoglobiny oraz transferyny. Badania
przeprowadzono dla mieszaniny zawierającej ferrytynę i dany interferent
w równomolowym stosunku. Pomiary wykonano dla dwóch różnych stężeń
ferrytyny: 1 oraz 20 mg·dL-1
, dzięki czemu uzyskano różne wysycenie warstwy
receptorowej cząsteczkami białka. Podczas tego eksperymentu skonstruowane
immunosensory zanurzane były w roztworze zawierającym ferrytynę oraz badany
interferent o tym samym stężeniu, na czas 1.5 h w obecności zewnętrznego źródła
pola magnetycznego. Po upływie tego czasu, w celu usunięcia nieprzereagowanych
substancji, elektrodę opłukiwano buforem PB. Pomiaru woltamperometrycznego
dokonywano w czystym odtlenionym buforze PB z dodatkiem 0.15 M K2SO4 oraz
0.2% v/v Tween-20. Wpływ badanych interferentów na sygnał elektroredukcji
ferrytyny (IFt + interferent / IFt) ilustruje rysunek 105.
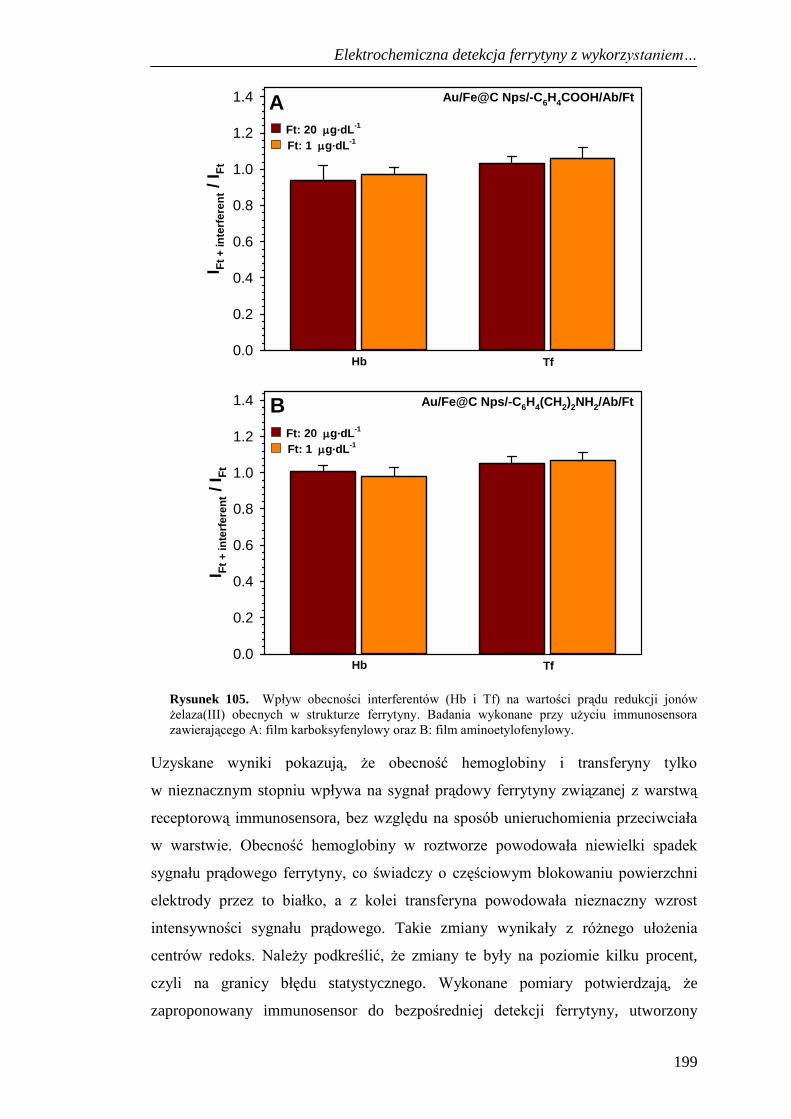
Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
199
I Ft
+ in
terf
ere
nt /
I Ft
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
Ft: 20 gdL-1
Ft: 1 gdL-1
TfHb
I Ft
+ in
terf
ere
nt /
I Ft
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
Ft: 20 gdL-1
Ft: 1 gdL-1
TfHb
Au/Fe@C Nps/-C6H
4(CH
2)2NH
2/Ab/Ft
Au/Fe@C Nps/-C6H
4COOH/Ab/FtA
B
Rysunek 105. Wpływ obecności interferentów (Hb i Tf) na wartości prądu redukcji jonów
żelaza(III) obecnych w strukturze ferrytyny. Badania wykonane przy użyciu immunosensora
zawierającego A: film karboksyfenylowy oraz B: film aminoetylofenylowy.
Uzyskane wyniki pokazują, że obecność hemoglobiny i transferyny tylko
w nieznacznym stopniu wpływa na sygnał prądowy ferrytyny związanej z warstwą
receptorową immunosensora, bez względu na sposób unieruchomienia przeciwciała
w warstwie. Obecność hemoglobiny w roztworze powodowała niewielki spadek
sygnału prądowego ferrytyny, co świadczy o częściowym blokowaniu powierzchni
elektrody przez to białko, a z kolei transferyna powodowała nieznaczny wzrost
intensywności sygnału prądowego. Takie zmiany wynikały z różnego ułożenia
centrów redoks. Należy podkreślić, że zmiany te były na poziomie kilku procent,
czyli na granicy błędu statystycznego. Wykonane pomiary potwierdzają, że
zaproponowany immunosensor do bezpośredniej detekcji ferrytyny, utworzony

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
200
zarówno na bazie filmu karboksyfenylowego i aminoetylofenylowego,
charakteryzował się wysoką selektywnością.
13.5. Oznaczenie ferrytyny w próbkach krwi
Funkcjonalność skonstruowanego immunosensora do detekcji ferrytyny
sprawdzono wobec próbki naturalnej: krwi szczura zdrowego oraz krwi szczura
chorego o podniesionym poziomie ferrytyny. W pierwszym etapie krew szczurzą,
która zawierała antykoagulant w postaci cytrynianu sodu, poddano wirowaniu.
Proces wirowania pozwolił na oddzielenie elementów morfotycznych krwi od
osocza. W kolejnym etapie immunosensor zanurzany był do roztworu osocza na czas
1.5 h w obecności pola magnetycznego. Następnie elektrodę delikatnie opłukiwano
strumieniem wody destylowanej. Pomiary prowadzono w czystym odtlenionym
0.02 M buforze fosforanowym z dodatkiem 150 mM K2SO4 przy użyciu
elektrochemicznej spektroskopii impedancyjnej, woltamperometrii pulsowej
różnicowej oraz techniki LA-ICP-MS. Wyznaczone na podstawie krzywych
kalibracyjnych stężenia ferrytyny w badanych próbkach krwi szczurzej związanej
z karboksyfenylową oraz aminoetylofenylową warstwą sensorową przedstawia tabela
14.
Tabela 14. Porównanie zawartości ferrytyny we krwi szczura zdrowego i chorego.
EIS DPV LA-ICP-MS
Au/Fe@C Nps/-C6H4COOH/Ab/Ft
Osocze krwi szczura
zdrowego [mg·dL-1
] 8.1 ± 0.2 7.9 ± 0.2 8.4 ± 0.8
Osocze krwi szczura
chorego [mg·dL-1
] 220.8 ± 16.2 230.5 ± 21.5 223.0 ± 30.1
Au/Fe@C Nps/-C6H4(CH2)2NH2/Ab/Ft
Osocze krwi szczura
zdrowego [mg·dL-1
] 8.3 ± 0.2 8.4 ± 0.3 8.1 ± 0.7
Osocze krwi szczura
chorego [mg·dL-1
] 253.3 ± 19.5 248.5 ± 20.4 240.4 ± 45
Osocze krwi szczura chorego rozcieńczano 100-krotnie
Z literatury wiadomo, że zakres stężeniowy ferrytyny we krwi u zdrowej osoby
wynosi 1 40 µg·dL-1
[219]. Z kolei wysoka zawartość ferrytyny (41 390 µg·dL-1
)
[471] we krwi związana jest z nadmiarem żelaza w organizmie wywołanym

Elektrochemiczna detekcja ferrytyny z wykorzystaniem…
201
schorzeniami typu: stan zapalny, zakażenia, czy choroby nowotworowe. Analiza
krwi szczura zdrowego i chorego przy użyciu zaproponowanego narzędzia
analitycznego dowodzi, że może być ono z powodzeniem stosowane do oznaczenia
ilości tego białka we krwi.
13.6. Podsumowanie
Modyfikacja powierzchni elektrody odpowiednio sfunkcjonalizowanym
filmem fenylowym pozwoliła na kontrolę orientacji unieruchamianych za jego
pośrednictwem cząsteczek przeciwciała i w konsekwencji na efektywne oznaczanie
Ft. Dodatkowo, zastosowanie nanocząstek magnetycznych i wprowadzenie do
układu zewnętrznego źródła pola magnetycznego pozwoliło na osiągnięcie właściwej
orientacji ferrytyny, umożliwiającej jej bezpośrednią woltamperometryczną detekcję.
Wykorzystanie filmu karboksyfenylowego, jako składnika warstwy receptorowej
czujnika, umożliwiło wprowadzenie cząsteczek przeciwciała do warstwy sensorowej
poprzez jego grupy aminowe, co gwarantowało najkrótszy dystans pomiędzy
centrum redoks białka a powierzchnią elektrody, a tym samym obserwację dobrze
wykształconych sygnałów prądowych ferrytyny. Z kolei związanie przeciwciała za
pomocą grup karboksylowych obecnych w jego strukturze z grupami aminowymi
filmu aminoetylofenylowego sprawiało, że cząsteczki przeciwciała znajdowały się
wówczas w położeniu pionowym w stosunku do powierzchni elektrody [437]. Takie
ułożenie Ab powodowało, że sygnały prądowe ferrytyny nie były dobrze
wykształcone, ponieważ odległość pomiędzy centrum redoks białka a powierzchnią
elektrody była zbyt duża. Zatem w przypadku oznaczania ferrytyny bardziej
korzystne jest zastosowanie filmów karboksyfenylowych do unieruchamiania
przeciwciała specyficznie oddziałującego z oznaczanym białkiem. Zaproponowany
immunosensor pozwala na oznaczenia ferrytyny na poziomie około 0.1 mg·dL-1
.
Dodatkowo, za pomocą skonstruowanego czujnika możliwe jest selektywne
oznaczanie ferrytyny w próbce krwi. Czujnik ten może zatem stanowić alternatywę
do obecnych metod stosowanych w analizie klinicznej.


Streszczenie
203
14. Streszczenie
W ostatnich latach nastąpił duży wzrost zainteresowania wykorzystaniem
metaloprotein w różnych dziedzinach życia, takich jak: medycyna, biotechnologia,
czy przemysł. Jednak największe znaczenie białka te odgrywają w badaniu zmian
zachodzących w organizmie ludzkim. Oznaczenie zawartości metaloprotein we krwi
pozwala na szybkie rozpoznanie wielu schorzeń, jak również wczesne wykrycie
nowotworów. Z uwagi na coraz większą liczbę zachorowań na choroby
nowotworowe poszukuje się szybkich, tanich i skutecznych narzędzi analitycznych
umożliwiających diagnostykę tego typu chorób. Wykorzystanie elektrochemii do
oznaczania zawartości białek we krwi wydaje się być konkurencyjne w stosunku do
metod stosowanych w analizie klinicznej, ze względu na brak konieczności
stosowania toksycznych substancji, łatwość przygotowania próbek do pomiarów, jak
również relatywnie krótki czas wykonywania analizy. Dzięki zastosowaniu technik
elektrochemicznych możliwe jest również badanie wpływu oddziaływania różnych
substancji na właściwości metaloprotein i ich aktywność.
Niniejsza rozprawa doktorska dotyczy konstrukcji czujników do oznaczania
zawartości paramagnetycznych metaloprotein obecnych we krwi. Praca ma
klasyczny układ składający się z dwóch części: literaturowej i eksperymentalnej.
Część literaturowa obejmuje osiem rozdziałów. Dwa pierwsze rozdziały zawierają
uzasadnienie podjętej tematyki badawczej oraz sprecyzowane cele badawcze.
W części literaturowej scharakteryzowano płyny ustrojowe ze szczególnym
uwzględnieniem krwi oraz budowę i funkcje analizowanych metaloprotein:
hemoglobiny, ceruloplazminy, transferyny oraz ferrytyny. Z uwagi na fakt, że
niniejsza praca dotyczy elektrochemicznych czujników do oznaczania
paramagnetycznych metaloprotein we krwi, część literaturowa zawiera również
rozdział poświęcony ferromagnetycznym modyfikatorom powierzchni, sposbom
wymiany elektronów pomiędzy białkiem a powierzchnią elektrody oraz wpływowi
pola magnetycznego na transport depolaryzatora do powierzchni elektrody.
Teoretyczna część pracy zawiera także opis stosowanych w pomiarach technik
badawczych.
Badania opisane w niniejszej pracy doktorskiej dotyczą konstrukcji
elektrochemicznych czujników do detekcji paramagnetycznych metaloprotein we
krwi w oparciu o nanocząstki magnetyczne (nanokapsuły węglowe zawierające

Streszczenie
204
żelazo) i zewnętrzne źródło pola magnetycznego. Elektrochemiczna detekcja białek
jest wciąż dużym wyzwaniem dla badaczy, gdyż ich centra elektroaktywne są
głęboko osadzone w otoczce białkowej, co zdecydowanie utrudnia wymianę
elektronów pomiędzy tymi związkami a powierzchnią przewodnika. Ponadto białka
w kontakcie z powierzchnią elektrody, w szczególności z powierzchnią metaliczną,
ulegają denaturacji, a produkty tego procesu efektywnie blokują powierzchnię
przewodnika. Dlatego, ważnym czynnikiem ułatwiającym bezpośrednią wymianę
elektronów jest odpowiednia orientacja metaloproteiny względem powierzchni
elektrody. Odpowiednio modyfikując powierzchnię elektrody można „wymusić” na
cząsteczce takiego związku korzystną z elektrochemicznego punktu widzenia
orientację. W przypadku paramagnetycznych metaloprotein idealnymi kandydatami
są ferromagnetyczne nanokapsuły węglowe zawierające żelazo, które w połączeniu
z polem magnetycznym oddziałując z paramagnetycznymi- elektroaktywnymi
centrami umożliwiają wymianę elektronów pomiędzy białkiem a elektrodą.
Ważnym etapem konstrukcji czujników do detekcji wybranych metaloprotein
było opracowanie procedury tworzenia jednorodnej i stabilnej warstwy nanocząstek
magnetycznych na powierzchni elektrody. Przeprowadzone badania pokazały, że
utworzeniu warstwy o takich parametrach sprzyja zastosowanie homogenizatora
ultradźwiękowego podczas przygotowywania zawiesiny nanocząstek. Skuteczność
współdziałania nanocząstek Fe@C (modyfikatora powierzchni) i zewnętrznego
źródła pola magnetycznego w pierwszej kolejności sprawdzono wobec hemoglobiny,
białka o największej zawartości we krwi. Nanocząstki magnetyczne obecne na
powierzchni elektrody pełniły rolę nanomagnesów, które z jednej strony wzmacniały
transport paramagnetycznej Hb do powierzchni elektrody, z drugiej zaś zapewniały
korzystną dla bezpośredniej wymiany elektronów jej orientację. Dzięki czemu
rejestracja dobrze wykształconych sygnałów elektroredukcji Hb była możliwa. Dużą
zaletą zaproponowanej metody oznaczania hemoglobiny we krwi był brak
konieczności specjalnego przygotowania próbek. Hemoglobina obecna jest
w czerwonych krwinkach, dlatego też przed procesem jej oznaczania
przeprowadzono hemolizę krwi poprzez odpowiednie jej rozcieńczenie. Uzyskane
zdjęcia SEM pokazały, że czas hemolizy ma duży wpływ na proces uwolnienia Hb
z wnętrza czerwonych krwinek, a całkowite uwolnienie tego białka następuje po
ok. 20 minutach. Skonstruowany czujnik pozwolił na oznaczenie hemoglobiny
w próbce krwi ludzkiej oraz szczurzej z dużą dokładnością, dobrą powtarzalnością

Streszczenie
205
oraz niską granicą wykrywalności, rzędu pM. Badania dotyczące oznaczania
hemoglobiny w próbkach krwi przy wykorzystaniu czujnika z warstwą nanocząstek
magnetycznych zostały opisane w publikacji naukowej w czasopiśmie Biosensors
and Bioelectronics 64 (2015) 554.
Jednym ze sposobów ograniczenia kosztów analizy ilościowej białek jest
oznaczanie kilku białek podczas jednego pomiaru. Jednoczesna elektrochemiczna
detekcja kilku białek obecnych we krwi jest trudna do przeprowadzenia, gdyż
produkty zachodzących procesów elektrodowych mogą adsorbować się na
powierzchni elektrody, uniemożliwiając dalszą detekcję. W niniejszej rozprawie
doktorskiej wykazano, że jednoczesne oznaczanie białek jest możliwe przy użyciu
tego samego czujnika, wówczas gdy sygnały prądowe badanych białek pojawiają się
w innym zakresie na skali potencjałowej. Przeprowadzone badania udowodniły, że
możliwa jest bezpośrednia elektrochemiczna detekcja ceruloplazminy i hemoglobiny
w trakcie pojedynczego pomiaru, bez konieczności specjalnego przygotowania
analizowanej próbki krwi. Nanocząstki ferromagnetyka na powierzchni elektrody
pełniły rolę specyficznego i selektywnego filtru, który przyśpieszał oraz wzmacniał
transport tylko paramagnetycznych cząsteczek do powierzchni elektrody.
Dodatkowo, bezpośredni kontakt metaloproteiny z powierzchnią zastosowanego
ferromagnetyka nie prowadził do jej deaktywacji. Badania związane z bezpośrednią,
jednoczesną detekcją ceruloplazminy i hemoglobiny w próbce krwi szczurzej
opublikowane zostały w czasopiśmie Sensors and Actuators B: Chemical 221 (2015)
1461.
Ceruloplazmina jest niezwykle ważnym białkiem w osoczu krwi,
transportującym miedź, ale również katalizującym utlenianie żelaza Fe(II) do Fe(III),
co umożliwia wiązanie tego pierwiastka z transferyną i transport w osoczu krwi.
Właściwości katalityczne Cp są związane z jej unikalnym centrum aktywnym,
zwanym grupą prostetyczną, która złożona jest z 3 typów centrów miedziowych.
Właściwe unieruchomienie cząsteczek ceruloplazminy na powierzchni elektrody ma
niezwykle istotny wpływ na jej aktywność katalityczną. Przeprowadzone badania
z wykorzystaniem skaningowego mikroskopu elektrochemicznego udowodniły, że
zarówno obecność ferromagnetycznego modyfikatora elektrody, czyli nanokapsuł
węglowych zawierających żelazo, jak również zewnętrznego źródła pola
magnetycznego gwarantowały maksymalną aktywność katalityczną cerulolazminy
względem jonów Fe(II). Dodatkowo, wyniki uzyskane za pomocą metody

Streszczenie
206
LA-ICP-MS pokazały, że adsorpcja ceruloplazminy zachodziła przede wszystkim na
ferromagnetycznym modyfikatorze powierzchni elektrody. Uzyskane wyniki zostały
opisane w publikacji naukowej: Langmuir 31 (2015) 8176.
Kolejną ważną paramagnetyczną metaloproteiną obecną we krwi jest
transferyna. Z uwagi na fakt, że białko to zawiera w swojej strukturze, podobnie jak
Hb i Ft, jony żelaza oznaczanie tej metaloproteiny przy użyciu elektrody
zmodyfikowanej wyłącznie nanocząstkami magnetycznymi było niemożliwe. O ile
hemoglobinę można łatwo usunąć z oznaczanego roztworu poprzez odwirowanie
elementów morfotycznych krwi, o tyle ferrytyna wciąż pozostaje w roztworze i jest
głównym białkiem przeszkadzającym w analizie transferyny w osoczu krwi. Zatem
w celu dokonania oznaczenia transferyny niezbędne jest zmodyfikowanie
powierzchni elektrody przeciwciałem, które selektywnie wiąże oznaczane białko.
Głównym elementem budującym warstwę receptorową immunosensora było
przeciwciało monoklonalne typu IgG, które charakteryzuje się dużą zdolnością do
swoistego rozpoznawania antygenów. Wprowadzenie cząsteczek przeciwciała do
warstwy receptorowej immunosensora oraz zastosowanie zewnętrznego pola
magnetycznego pozwoliło na bezpośrednią woltamperometryczną detekcję
transferyny. Dzięki wykorzystaniu kombinacji pola magnetycznego
i ferromagnetycznego modyfikatora powierzchni elektrody możliwa była rejestracja
dobrze ukształtowanych sygnałów prądowych pochodzących od redukcji jonów
Fe(III) obecnych w strukturze transferyny. Wykonane badania pokazały również, że
skonstruowany immunosensor można zastosować do wykrywania dyskretnych zmian
ilości tego białka we krwi, co jest niezwykle istotne w diagnostyce wielu schorzeń.
Dużą zaletą zaproponowanego immunosensora jest prostota jego wykonania, duża
selektywność, jak również możliwość kilkukrotnego jego wykorzystania, co
znacznie obniża koszty przeprowadzenia analizy. Użyteczność skonstruowanego
immunosensora do detekcji transferyny w próbkach krwi została przedstawiona
w publikacji naukowej w czasopiśmie Sensors and Actuators B: Chemical 249
(2017) 105.
Transferyna, zaliczana do białek „miękkich” według klasyfikacji Norde, jest
podatna na zmiany konformacyjne w wyniku kontaktu z wybraną powierzchnią.
W organizmie ulega ona cyklicznym zmianom konformacyjnym podczas procesu
wiązania i uwalniania jonów Fe(III), umożliwiając tym samym rozpoznanie tego
białka przez receptory zlokalizowane na powierzchni błon plazmatycznych komórek.

Streszczenie
207
Dodatkowo, ilość receptorów Tf na powierzchni komórek nowotworowych jest
zdecydowanie większa w porównaniu do komórek zdrowych. Z tego względu
transferyna może pełnić funkcję nośnika leku w celowanej terapii
przeciwnowotworowej. Wykonane badania pokazały, że zastosowanie nanocząstek
magnetycznych (potencjalne nośniki leków przeciwnowotworowych) zawierających
na powierzchni grupy karboksylowe (Fe@C-COOH Nps) w obecności pola
magnetycznego prowadzi do kowalencyjnego unieruchomienia tego białka w jego
elektroaktywnej formie. Wprowadzenie zewnętrznego źródła pola magnetycznego
podczas procesu unieruchamiania Tf na powierzchni Fe@C-COOH Nps wpływało
na strukturę tworzonej warstwy transferyny. Paramagnetyczne centra Tf (jony
Fe(III)) były wciągane przez pole magnetyczne i w konsekwencji zmieniały swoje
położenie, natomiast diamagnetyczne składniki otoczki białkowej Tf (głównie
wodorowęglany) były wypychane przez pole na zewnątrz. W związku z tym,
w wyniku oddziaływań elektrostatycznych, tworzyła się kolejna warstwa Tf na
powierzchni nanocząstek, co potwierdziły badania z wykorzystaniem mikrowagi
kwarcowej z dyssypacją energii. Jest to bardzo rzadkie zjawisko w przypadku białek
w warunkach stałego pH. Ponadto pomiary z użyciem techniki UV-vis i CD
udowodniły, że zmiany konformacyjne cząsteczki transferyny zachodzące pod
wpływem pola magnetycznego nie uszkadzały jej struktury. Transferyna związana
z powierzchnią nanocząstki magnetycznej wciąż wykazywała wysoką aktywność
elektrochemiczną. Wyniki dotyczące wpływu sposobu związania transferyny
z nanocząstkami magnetycznymi na jej elektroaktywność oraz zmiany
konformacyjne opisane zostały w czasopiśmie Acta Biomaterialia 45 (2016) 367.
Ostatnią metaloproteiną poddaną analizie elektrochemicznej była ferrytyna
obecna, podobnie jak Cp i Tf, w osoczu krwi. Białko to, podobnie jak hemoglobina
i transferyna, zawiera jony żelaza, co uniemożliwia jego elektrochemiczne
oznaczanie we krwi w obecności tych białek. W celu zapewniania wysokiej
skuteczności działania zaproponowanego czujnika jego warstwę receptorową
wzbogacono o cząsteczki przeciwciała selektywnie oddziałującego tylko z Ft.
Konstrukcja immunosensora opierała się na wykorzystaniu warstwy nanocząstek
magnetycznych pokrytych filmem karboksyfenylowym lub aminoetylofenylowym
otrzymanym w wyniku elektroredukcji odpowiednej soli diazoniowej. Modyfikacja
powierzchni elektrody filmem fenylowym pozwoliła na kontrolę ułożenia
unieruchamianych cząsteczek przeciwciała, a w konsekwencji na efektywność ich

Streszczenie
208
oddziaływania z ferrytyną. Zastosowanie nanocząstek magnetycznych
i zewnętrznego źródła pola magnetycznego podczas reakcji antygenprzeciwciało
pozwoliło na osiągnięcie właściwej dla wymiany elektronu orientacji ferrytyny,
umożliwiającej jej bezpośrednią woltamperometryczną detekcję. Przeprowadzone
badania pokazały, że film karboksyfenylowy jest zdecydowanie korzystniejszy
w elektrochemicznej detekcji Ft w porównaniu do warstwy aminoetylofenylowej,
z uwagi na jakość uzyskiwanych sygnałów prądowych. Zaproponowany
immunosensor pozwala na selektywne oznaczanie ferrytyny w próbce krwi. Wyniki
uzyskane podczas tego etapu badań będą stanowiły podstawę publikacji naukowej.

Streszczenie w języku angielskim
209
15. Streszczenie w języku angielskim
In recent years there has been a great increase in interest of using
metalloproteins in various fields of life, such as medicine, biotechnology or industry.
However, the most important role of metalloproteins occurs in the study of changes
in the human body. The determination of the content of metalloproteins in blood
allows for a quick diagnosis of many diseases as well as early detection of cancer.
Due to the increasing number of cancer occurrences fast, cheap and effective
analytical tools for diagnosing such diseases are sought. The electrochemical
detection of these proteins seems to be competitive compared to clinical analysis due
to the low costs, no need to use the toxic substances, easiness of sample preparation
and short analysis time. The use of electrochemical techniques also allows to study
the effects of different substances on properties of metalloproteins and their activity.
This PhD thesis relates to the construction of sensors for determination of
paramagnetic metalloproteins present in the blood. Traditionally, this thesis consists
of two parts: literature and the experimental one. The literature section of this thesis
consists of eight chapters. The first two chapters describe the justification of the
research topic and the aim of this work. The next section describes the body fluids,
particularly blood. The structure and functions of the selected metalloproteins such
as hemoglobin, ceruloplasmin, transferrin and ferritin are also described. Due to the
fact that this thesis regards electrochemical sensors for determination of
paramagnetic metalloproteins in the blood, the literature part contains a chapter
devoted to ferromagnetic modifier of the surface, the way of electron exchange
between a protein and electrode surface as well as the influence of the magnetic field
on the transport of the substances to electrode surface. The theoretical part of this
thesis contains the description of the experimental techniques.
The research described in this thesis concerns the construction of
electrochemical sensors for detection of paramagnetic metalloproteins in the blood.
The sensors are based on magnetic nanoparticles (carbon encapsulated ion
nanoparticles) and the source of the external magnetic field. Detection of the
electrochemical protein is still a big challenge for researchers due to the fact that
protein redox centers are deeply embedded in the protein shell and, as a consequence,
the electron transfer rate is too slow. Moreover, the direct adsorption of large protein

Streszczenie w języku angielskim
210
onto the metallic electrode surface may lead to its denaturation, which in turn
efficiently blocks the electron communication between the redox centers and the
electrode. Therefore, an important factor allowing to direct electron exchange is the
appropriate orientation of the proteins on the electrode surface. The proper
modification of the electrode surface can lead to a favorable orientation of proteins
necessary for an electrochemical reaction. In the case of paramagnetic
metalloproteins, the ideal candidates for electrode modifier are ferromagnetic carbon
encapsulated iron nanoparticles (Fe@C Nps). The nanoparticles in combination with
the magnetic field, interact with the paramagnetic electroactive centers and allow for
an exchange of electrons between the protein and the electrode. An important step in
the construction of the sensors for the detection of selected metalloproteins was to
develop a procedure for creating a homogeneous and stable nanoparticle layer on the
electrode surface. The studies have shown that the formation of a layer with such
parameters is possible when using a homogenizer to prepare a nanoparticle
suspension.
The effectiveness of the interaction of Fe@C Nps (surface modifier) and the
external magnetic field was first tested on hemoglobin, a protein with the highest
content in blood. The magnetic nanoparticles have strengthened the transport of
paramagnetic Hb to the electrode surface causing the hemoglobin to be in a favorable
orientation for direct electron transfer. Due to the presence of nanoparticles on the
electrode surface the registration of well visible Hb signals was possible. The great
advantage of the proposed method for hemoglobin determination in the blood is the
lack of special preparation of the samples. Due to the fact that hemoglobin is present
in red blood cells, blood hemolysis was performed prior to Hb determination by
appropriate dilution. The obtained SEM images showed that hemolysis time has
a major impact on the release of Hb from the interior of red blood cells. The
complete release of Hb from red blood cells takes about 20 minutes. The constructed
sensor allowed the determination of hemoglobin in human and rat blood samples
with high accuracy, good repeatability and low detection limit in the pM range.
Studies on the determination of hemoglobin in blood samples using a sensor with
magnetic nanoparticles layer have been presented in the paper published in
Biosensors and Bioelectronics 64 (2015) 554.
The determination of several proteins in one measurement allows the
reduction the costs of analysis. The electrochemical detection of several proteins

Streszczenie w języku angielskim
211
present in the blood is difficult to perform because the products of the redox reaction
may adsorb on the electrode surface, preventing further detection. This thesis has
shown that the determination of proteins is possible using the same sensor when the
current signals of the investigated proteins appear in a different ranges on the
potential scale. The research has shown that direct electrochemical detection of
ceruloplasmin and hemoglobin is possible during a single measurement without any
special preparation of a blood sample. The construction of the proposed sensor was
based on the use of carbon nanocapsulated iron nanoparticles as electrode surface
modifier and on the presence of the source of external magnetic field. Ferromagnetic
nanoparticles on the electrode surface acted as a specific and selective filter that
stimulated and amplified the transport of only paramagnetic molecules to the
electrode surface. In addition, the direct contact of the metalloproteins with the
nanoparticles surface did not lead to its deactivation. The studies related to direct
detection of ceruloplasmin and hemoglobin in rat blood samples are presented in
Sensors and Actuators B: Chemical 221 (2015) 1461.
Ceruloplasmin is an extremely important protein in the blood plasma. The
ceruloplasmin transports copper and also catalyzes Fe(II) to Fe(III), which makes it
possible to bind Fe(III) with transferrin which transports it into the blood plasma.
Catalytic properties of Cp are associated with its unique active center called
a prosthetic group, which is composed of 3 types of copper centers. Proper
immobilization of ceruloplasmin molecules on the electrode surface is extremely
important for its catalytic activity. Studies conducted using a scanning
electrochemical microscopy (SECM) demonstrated that both the presence of
ferromagnetic nanoparticles (Fe@C Nps) as well as the source of external magnetic
field guarantee the maximum catalytic activity of ceruloplasmin towards Fe(II) ions.
In addition, the results obtained by LA-ICP-MS technique showed that adsorption of
ceruloplasmin occurred mainly on the surface of ferromagnetic electrode modifier.
The obtained results have been described in a scientific paper: Langmuir 31 (2015)
8176.
Another important paramagnetic metalloprotein present in the blood is
transferrin. Due to the fact that this protein contains in its structure the same iron ions
as Hb and Ft, the determination of this metalloprotein using an electrode modified
only with magnetic nanoparticles was impossible. Hemoglobin can be easily
removed from the solution by centrifugation of blood but this method does not

Streszczenie w języku angielskim
212
remove ferritin, which is the major protein that interfere with the transferrin analysis
in blood plasma. To determine the amount of transferrin it is necessary to modify the
electrode surface with an antibody that selectively binds the assayed protein. The
main component of building the receptor layer of immunosensor was the monoclonal
antibody type IgG which is characterized by its high ability to specifically recognize
the antigens. The introduction of antibody molecules onto the receptor layer and the
use of an external magnetic field allowed the direct electrochemical detection of
transferrin. Using a combination of a magnetic field and a ferromagnetic modifier of
electrode surface, it was possible to record well visible current signals derived from
the reduction of Fe(III) ions present in the transferrin structure. Studies have also
shown that a constructed immunosensor can be used to detect discrete changes in the
amount of this protein in the blood, which is extremely important in the diagnosis of
many diseases. The proposed immunosensor has many advantages such as its
simplicity, high selectivity, the ability to use it several times and also costs of
analysis. The utility of the constructed immunosensor for transferrin detection in
blood samples is presented in the scientific paper in Sensors and Actuators B:
Chemical 249 (2017) 105.
Transferrin, classified by Norde's as "soft" proteins, is susceptible to
conformational changes during a contact with a selected surface. In the human body
transferrin undergoes cyclic conformational changes during the binding and release
of Fe(III) ions. These changes thereby enable the recognition of this protein by
receptor localized on the surface of plasma membrane cells. In addition, the amount
of Tf receptors on the surface of tumor cells is considerably higher in comparison to
healthy cells. For this reason, transferrin may be used in targeted anti-cancer therapy.
The studies have shown that the use of magnetic nanoparticles (potential drug
carrier) with carboxylic groups (Fe@C-COOH Nps) in the presence of the magnetic
field leads to covalent immobilization of this protein in its electroactive form. The
introduction of the source of external magnetic field during the process of Tf
immobilization on F@C-COOH Nps affected the structure of a created transferrin
layer. The Tf undergoes conformation changes, in which the paramagnetic Fe(III)
ions (electroactive centers) are not deeply hidden in the protein shell in the presence
of the magnetic field, whilst the diamagnetic components of Tf molecule (mainly
carbohydrate) are pushed away towards the solution. Consequently, as a result of
electrostatic interactions the adlayer is formed. This is a very rare phenomenon in the

Streszczenie w języku angielskim
213
case of proteins in constant pH. In addition, the measurements using UV-vis and CD
techniques have proved that the conformational changes of the transferrin molecule
occurring under the influence of the magnetic field did not damage the structure of
transferrin. Transferrin linked to the surface of the magnetic nanoparticle still
exhibited electrochemical activity. The results on the effect of the method of binding
transferrin to magnetic nanoparticles on its electroactivity and conformational
changes have been described in the scientific paper in Acta Biomaterialia 45 (2016)
367.
The last metalloprotein submitted to electrochemical analysis was ferritin.
This protein, like hemoglobin and transferrin, also contains iron ions, what prevents
its electrochemical detection in the blood in the presence of Hb and Tf. In order to
ensure high performance of the proposed sensor, its receptor layer was enriched with
antibody molecules selectively interacting only with Ft. The construction of
immunosensor was based on the use of a magnetic layer coated with carboxyphenyl
or aminoethylphenyl film obtained by one-electron reduction of a diazonium salt.
Modification of the electrode surface with a phenyl film allowed the control of the
placement of immobilized antibody molecules and, consequently, their effectiveness
of interaction with ferritin. The use of magnetic nanoparticles and the source of
external magnetic field during the antigen-antibody interaction allowed to achieve
appropriate orientation of ferritin, enabling its direct voltammetric detection. Studies
have shown that the carboxyphenyl film is better at electrochemical Ft detection
compared to the aminoethylphenyl layer. The proposed immunosensor allows for
a selective detection of ferritin in a blood sample. The results obtained during this
stage of research will be the basis for a scientific publication.


Spis prac naukowych opublikowanych w trakcie studiów doktoranckich
215
16. Spis prac naukowych opublikowanych w trakcie
studiów doktoranckich
Publikacje i monografie bezpośrednio związane z tematem rozprawy doktorskiej:
1. E. Matysiak, M. Donten, A. Kowlaczyk, M. Bystrzejewski, I.P. Grudzinski,
A.M. Nowicka; „A novel type of electrochemical sensor based on
ferromagnetic carbon-encapsulated iron nanoparticles for direct determination
of hemoglobin in blood samples” Biosensors and Bioelectronics 64, 2015,
554-559.
2. E. Matysiak, B. Wagner, M. Bystrzejewski, I.P. Grudzinski, A.M. Nowicka;
„Direct voltammetric detection of ceruloplasmin in blood in presence of other
paramagnetic species” Sensors and Actuators, B: Chemical 221, 2015,
1461-1468.
3. E. Matysiak, A.J.R. Botz, J. Clausmeyer, B. Wagner, W. Schuhmann,
Z. Stojek, A.M. Nowicka; „Assembling paramagnetic ceruloplasmin at
electrode surfaces covered with ferromagnetic nanoparticles. Scanning
electrochemical microscopy in the presence of a magnetic field” Langmuir
31, 2015, 8176-8183.
4. A. Kowlaczyk, E. Matysiak-Brynda, M. Bystrzejewski, D.S. Sutherland,
Z Stojek, A.M. Nowicka; „Conformational control of human transferrin
covalently anchored to carbon-coated iron nanoparticles in presence of
a magnetic field” Acta Biomaterialia 45, 2016, 367-374.
5. E. Matysiak-Brynda, M. Bystrzejewski, A. Wieckowska, I.P. Grudzinski,
A.M. Nowicka; „Novel ultrasensitive immunosensor based on magnetic
particles for direct detection of transferrin in blood” Sensors and Actuators,
B: Chemical 249, 2017, 105-113.
6. E. Matysiak, A. Kowalczyk, M. Donten, A.M. Nowicka; „Aspekty
oddziaływań magnetycznych w elektrochemicznej charakterystyce substancji
istotnych biologicznie”, w: „Postępy Elektroanalizy”, B. Baś,
M. Jakubowska, W.W. Kubiak (Eds.), 2016, rodział 21, str. 235-249,
Wydawnictwo Naukowe AKAPIT, ISBN: 978-83-63663-78-0.

Spis prac naukowych opublikowanych w trakcie studiów doktoranckich
216
7. E. Matysiak-Brynda, A. Kowalczyk, M. Bystrzejewski, D.S. Sutherland,
Z. Stojek, A.M. Nowicka; „Kontrolowane unieruchamianie transferyny na
powierzchni nanocząstki magnetycznej w obecności pola magnetycznego
sposobem na wzrost jej elektroaktywności”, w: „Nowe Strategie w Analizie
Elektrochemicznej”, B. Baś, M. Jakubowska, W.W. Kubiak (Eds.), 2017,
rodział 1, str. 15-28, Wydawnictwo Naukowe AKAPIT, ISBN: 978-83-
63663-90-2
Inne publikacje i rozdziały w książkach:
1. E. Matysiak, S. Sek, Z. Stojek, A.M. Nowicka; „Accuracy of determination
of mass of DNA films on gold electrodes” Sensors and Actuators, B:
Chemical 210, 2015, 624-632.
2. E. Matysiak, A.M. Nowicka, B. Wagner, M. Donten; „Space-oriented
immobilization of fully active laccase on PPy-ferromagnetic nanoparticles
composite layer” Electrochimica Acta 191, 2016, 586-593.
3. A.M. Nowicka, A. Kowalczyk, E. Matysiak, M. Hepel; „DNA damage by
highly oxidizing environmental pollutants”, w: „Oxidative Stress:
Diagnostics, Prevention, and Therapy Volume 2”, 2015, rozdział 12, str. 279-
299, ACS Symposium Series, ISBN: 9780841231009.
Publikacje wysłane do recenzji:
1. E. Matysiak-Brynda, P. Bujak, E. Augustin, A. Kowalczyk, Z. Mazerska,
A. Pron, A.M. Nowicka; „Stable nanoconjugate of transferrin with alloyed
quaternary nanocrystals Ag-In-Zn-S as biological entity for tumor
recognition”

Bibliografia
217
17. Bibliografia
[1] A.A. Khan, M.A. Alzohairy; Res. J. Biol. Sci. 5, 2010, 565.
[2] P. Bajpai; Biotechnol. Prog. 15, 1999, 147.
[3] R.K. Murray, D.K. Granner, P.A. Mayes, V.W. Rodwell; „Biochemia Harpera”,
Wydawnictwo Lekarskie PZWL, Warszawa, 2006.
[4] B.R. Waterhouse, A.D. Farmery; Anaesth. Intens. Care Med. 3, 2012, 603.
[5] G.B. Forbes; „Human Body Composition: Growth, Aging, Nutrition, and Activity”, Springer-
Verlag, New York, 1987.
[6] E. Ziółko; „Podstawy fizjologii człowieka”, Oficyna Wydawnicza PWSZ w Nysie,
Nysa, 2006.
[7] W.Z. Traczyk, A. Trzebski; „Fizjologia człowieka z elementami fizjologii stosowanej
i klinicznej”, Wydawnictwo Lekarskie PZWL, Warszawa, 2009.
[8] M. Tafil-Klawe, J.J. Klawe; „Wykłady z fizjologii człowieka”, Wydawnictwo Lekarskie PZWL,
Warszawa, 2009.
[9] A. Michajlik, W. Ramotowski; „Anatomia i fizjologia człowieka”, Wydawnictwo Lekarskie
PZWL, Warszawa, 2009.
[10] G. Zhao, M. Viehrig, M. Pumera; Lab Chip 13, 2013, 1930.
[11] W. Sawicki, J. Malejczuk; „Histologia”, Wydawnictwo Lekarskie PZWL, Warszawa, 2012.
[12] M. Kersaudy-Kerhoas, E. Sollier; Lab Chip 13, 2013, 3323.
[13] S.H.D.P. Lacerda, J.J. Park, C. Meuse, D. Pristinski, M.L. Becker, A. Karim, J.F. Douglas;
ACS Nano 4, 2010, 365.
[14] M.-J. Wang, W.-B. Tsai; „Biomaterials in Blood-contacting Devices: Complications and
Solutions”, Nova Science Publishers, New York, 2010.
[15] Kenry, K.P. Loh, Ch.T. Lim; Nanoscale 8, 2016, 9425.
[16] A.C. Frazer; Discussions Faraday Soc. 6, 1949, 81.
[17] C.D. Hillyer, L.E. Silberstein, P.M. Ness, K.C. Anderson, J.D. Roback; „Blood Banking and
Transfusion Medicine: Basic Principles & Practice”, Churchill Livingstone, Philadelphia,
2006.
[18] A. Blann; „Routine Blood Results Explained”, M&K Publishing, Bath, 2013.
[19] G. Lippi, M. Plebani, S. Di Somma, G. Cervellin; Crit. Rev. Clin. Lab. Sci. 48, 2011, 143.
[20] G. Lippi, G. Cervellin, E.J. Favaloro, M. Plebani; „In Vitro and In Vivo Hemolysis:
An Unresolved Dispute in Laboratory Medicin”, De Gruyter, Berlin, 2012.
[21] https://en.wikipedia.org/wiki/Tonicity
[22] N.W. Tietz; „Fundamentals of clinical chemistry”, Elsevier, St. Louis, 2008.
[23] X. Chen; „On-chip pretreatment of whole blood by using MEMS technology”, eBooks,
Bentham Science Publishers, 2012.
[24] Ch.H. Sloop, L. Dory, P.S. Poheim; J. Lipid Res. 28, 1987, 225.
[25] W. Ptak, M. Ptak, M. Szczepanik; „Podstawy immunologii”, Wydawnictwo Lekarskie PZWL,
Warszawa, 2008.
[26] R. Nezlin; „The immunoglobulins: Structure and Function”, Academic Press, San Diego, 1998.
[27] R. Toughiri, X. Wu, D. Ruiz, F. Huang, J.W. Crissman, M. Dickey, K. Froning, E.M. Conner,
T.P. Cujec, S.J. Demarest; MAbs 8, 2016, 1276.
[28] P.M. Lydyard, M.W. Fanger, A. Whelan; „Immunologia. Krótkie wykłady”, Wydawnictwo
Naukowe PWN, Warszawa, 2012.
[29] M. Zieliński; Biotechnologia 2, 2009, 113.
[30] Ł. Sędek, B. Mazur; Postępy Biologii Komórki 24, 2008, 17.
[31] T. Boenisch; Appl. Immunohistochem. 7, 1999, 300.
[32] M. Basu, S.S. Lenka, M. Paichha, B. Swain, B. Patel, R. Banerjee, P. Jayasankar, S. Das,
M. Samanta; Vet. Immunol. Immunopathol. 179, 2016, 77.
[33] T. Honjo, F.W. Alt; „Immunoglobulin Genes”, Academic Press, London, 1995.
[34] Y.H. Chow, Y.J. Yap, P.L. Show, J.Ch. Juan, M.S. Anuar, E.-P. Ng, Ch.-W. Ooi, T.Ch. Ling;
J. Biosci. Bioeng. 122, 2016, 613.
[35] A.H. Loo, A. Bonanni, A. Ambrosi, H.L. Poh, M. Pumera; Nanoscale 4, 2012, 921.
[36] S. Doonan; „Białka i peptydy”, Wydawnictwo Naukowe PWN, Warszawa, 2008.

Bibliografia
218
[37] A.V. Finkelstein, O. Ptitsyn; „Protein Physics: A course of Lectures”, Academic Press,
Amsterdam, 2002.
[38] R.T. Morrison, R.N. Boyd; „Chemia organiczna. T.2”, Wydawnictwo Naukowe PWN,
Warszawa, 2010.
[39] A. Kołodziejczyk; „Naturalne związki organiczne”, Wydawnictwo Naukowe PWN, Warszawa,
2004.
[40] J. Kączkowski; „Podstawy Biochemii”, Wydawnictwo WNT, Warszawa, 2012.
[41] D.B. Hames, N.M. Hooper; „Biochemia. Krótkie wykłady”, Wydawnictwo Naukowe PWN,
Warszawa, 2012.
[42] Y. Lu, N. Yeung, N. Sieracki, N.M. Marshall; Nature 460, 2009, 855.
[43] R.H. Holm, P. Kennepohl, E.I. Solomon; Chem. Rev. 96, 1996, 2239.
[44] M.M. Harding; Acta Cryst. D60, 2004, 849.
[45] R.W. Hay; „Chemia bionieorganiczna”, Wydawnictwo Naukowe PWN, Warszawa, 1990.
[46] R.F. Boyer, B.E. Schori; Biochem. Biophys. Res. Commun. 116, 1983, 244.
[47] A. Koj, A. Kurdowska, D. Magielska-Zero, H. Rokita, J.D. Sipe, J.M. Dayer, S. Demczuk,
J. Gauldie; Biochem. Int. 14, 1987, 553.
[48] B. Mariańska; Diagn. Lab. 38, 2002, 339.
[49] G. Winsauer, R. de Martin; Thromb. Haemost. 97, 2007, 364.
[50] M.M. Harding, M.W. Nowicki, M.D. Walkinshaw; Crystallogr. Rev. 16, 2010, 247.
[51] L. Stryer; „Biochemia”, Wydawnictwo Naukowe PWN, Warszawa, 2003.
[52] A.F. Wagner, K.A. Folkers; „Vitamins and coenzymes”, Interscience Publishers, New York,
1975.
[53] R.M. Roat-Malone; „Chemia bionieorganiczna”, Wydawnictwo Naukowe PWN, Warszawa,
2010.
[54] C.G. Fraga; Mol. Aspects Med. 26, 2005, 235.
[55] H. Sun, Z.-F. Chai; Annual. Rep. Prog. Chem., Sect. A 106, 2010, 20.
[56] A. Rodacka, J. Gerszon, M. Puchala, G. Bartosz; Radiat. Phys. Chem. 128, 2016, 112.
[57] K. Hemavathi, M. Kalaivani, A. Udayakumar, G. Sowmiya, J. Jeyakanthan, K. Sekar; J. Appl.
Cryst. 43, 2010, 196.
[58] M. Harding; Acta Cryst. D55, 1999, 1432.
[59] M.M. Harding; Acta Cryst. D56, 2000, 857.
[60] K. Hsin, Y. Sheng, M.H. Harding, P. Taylor, M.D. Walkinshaw; J. Appl. Cryst. 41, 2008, 963.
[61] Y. Yuan, V. Simplaceanu, J.A. Lukin, C. Ho; J. Mol. Biol. 321, 2002, 863.
[62] P. Norouzi, B. Larijani, F. Faridbod, M. R. Ganjali; Int. J. Electrochem. Sci. 5, 2010, 1550.
[63] E. Nagababu, J.M. Rifkind; Antioxid. Redox Signal 6, 2004, 967.
[64] T. Kanias, J.P. Acker; FEBS Journal 277, 2010, 343.
[65] X.Y. Bao, Z.W. Zhu, N.Q. Li, J.G. Chen; Talanta 54, 2001, 591.
[66] E.J. Parker-Williams; Medicine 37, 2009, 137.
[67] I.S. Anand; J. Am. Coll. Cardiol. 52, 2008, 501.
[68] P. Karlson; „Zarys biochemii”, Wydawnictwo Naukowe PWN, Warszawa, 1987.
[69] M. Weissbluth; „Molecular Biology: Biochemistry and Biophysics”, Springer, New York,
1974.
[70] F.W. Scheller, N. Bistolas, S. Liu, M. Jänchen, M. Katterle, U. Wollenberger; Adv. Colloid
Interface Sci. 116, 2005, 111.
[71] M.K. Chan; J. Porphyr. Phthaloc. 4, 2000, 358.
[72] T. Yonetani, M. Laberge; BBA – Proteins Proteom. 1784, 2008, 1146.
[73] https://www.google.pl/search?q=hemoglobina.com
[74] G.M. Tush, R.J. Kuhn; Ann. Pharmacother. 30, 1996, 1251.
[75] W.A. Eaton, E.R. Henry, J. Hofrichter, A. Mozzarelli; Nat. Struct. Biol. 6, 1999, 352.
[76] D.M. Harmening, V.C. Hughes, C. Del Toro; „Clinical Hematology and Fundamentals of
Hemostasis”, FA Davis, Philadelphia, 2002.
[77] D.L. Nelson, M.M. Cox; „Lehninger Principles of Biochemistry“, W.H. Freeman and
Company, New York, 2008.
[78] E. Nagababu, J.M. Rifkind; Biochemistry 39, 2000, 12503.
[79] J.W. Wallace, J.C. Maxwell; Biochem. Biophys. Res. Commun. 57, 1974, 1104.
[80] J. Umbreit; Am. J. Hematol. 82, 2007, 134.
[81] C. Giulivi, P. Hochstein, K.J. Davies; Free Radical Biol. Med. 16, 1994, 123.
[82] G.N. La Mar, J.S. de Ropp, L. Latos-Grazynski, A.L. Balch, R.B. Johnson, K.M. Smith,
D.W. Parish, R.J. Cheng; J. Am. Chem. Soc. 105, 1983, 782.

Bibliografia
219
[83] E. Zapora, I. Jarocka; Postepy Hig. Med. Dosw. 67, 2013, 214.
[84] Y. Jia, P.W. Buehler, R.A. Boykins, R.M. Venable, A.I. Alayash; J. Biol. Chem. 282, 2007,
4894.
[85] L. Pauling, C. Coryell; Proc. Natl. Acad. Sci. USA 22, 1936, 159.
[86] L. Pauling, C. Coryell; Proc. Natl. Acad. Sci. USA 22, 1936, 210.
[87] C. Coryell, F. Stitt, L. Pauling; J. Am. Chem. Soc. 59, 1937, 633.
[88] J.P. Savicky, G. Lang, M. Ikeda-Sato; Proc. Natl. Acad. Sci. USA 81, 1984, 5417.
[89] V.L. Davidson, D.B. Sittmano; „Biochemia”, Urban & Partner, Wrocław, 2002.
[90] N. Masuoka, H. Kodama, T. Abe, D.H. Wang, T. Nakano; Biochim. Biophys. Acta 1637, 2003,
46.
[91] A. Kapralov, I.I. Vlasova, W. Feng, A. Maeda, K. Walson, V.A. Tyurin, Z. Huang,
R.K. Anjea, J. Carcillo, H. Bayir, V.E. Kagan; J. Biol. Chem. 284, 2009, 30395.
[92] W. Tao, Y. Liu, D. Pan, L. Nie, S. Yao; Bioelectrochemistry 65, 2004, 51.
[93] G. Vaschenko, T. Ross, A. MacGillivray; Nutrients 5, 2013, 2289.
[94] P. Hahn, G. Yong, J. Beard, J. Dunaief; NeuroReport 17, 2006, 1803.
[95] C.G. Holmberg; Acta Physiol. Scand. 8, 1944, 227.
[96] N.Takahashi, T.L. Ortel, F.W. Putnam; Proc. Natl. Acad. Sci. USA 81, 1984, 390.
[97] S.A. Donley, B.J. Ilagan, H. Rim, M.C. Linder; Am. J. Physiol. Endocrinol. Metab. 283, 2002,
E667.
[98] N.E. Hellman, J.D. Gitlin; Annu. Rev. Nutr. 22, 2002, 439.
[99] D. Wierzbicka, G. Gromadzka; Postepy Hig. Med. Dosw. 68, 2014, 912.
[100] P. Bielli, L. Calabrese; Cell. Mol. Life Sci. 59, 2002, 1413.
[101] H. Chen, Z.K. Attieh, B.A. Syed, Y.M. Kuo, V. Stevens, B.K. Fuqua, H.S. Andersen,
C.E. Naylor, W.R. Evans, L. Gambling, R. Danzeisen, M.B. Bacouri-Haidar, J. Usta,
C.D. Vulpe, H.J. McArdle; J. Nutr. 140, 2010, 1728.
[102] T.L. Ortel, N. Takahashi, F.W. Putnam; Proc. Natl. Acad. Sci. USA 81, 1984, 4761.
[103] A. Messerschmidt, R. Ladenstein, R. Huber, M. Bolognesi, L. Avigliano, R. Petruzzelli,
A. Rossi, A. Finazzi-Agro; J. Mol. Biol. 224, 1992, 179.
[104] A. Messerschmidt, A. Rossi, R. Ladenstein, R. Huber, M. Bolognesi, G. Gatti, A. Marchesini,
R. Petruzzelli, A. Finazzi-Agro; J. Mol. Biol. 206, 1989, 513.
[105] S.M. Lee, Z.K. Attieh, H.S. Son, H. Chen, M.B. Bacouri-Haidar, C.D. Vulpe; Biochem.
Biophys. Res. Commun. 421, 2012, 449.
[106] T.E. Machonkin, H.H. Zhang, B. Hedman, K.O. Hodgson, E.I. Solomon; Biochemistry
37, 1998, 9570.
[107] I. Bento, C. Peixoto, V.N. Zaitsev, P.F. Lindley; Acta Crystallogr. D Biol. Crystallogr.
63, 2007, 240.
[108] E. Sedlak, P. Wittung-Stafshede; Biochemistry 46, 2007, 9638.
[109] T.E. Machonkin, E.I. Solomon; J. Am. Chem. Soc. 122, 2000, 12547.
[110] T. Kubiak, R. Krzyminiewski, B. Dobosz; Curr. Top. Biophys 36, 2013, 7.
[111] P. Dusek, J. Jankovic, W. Le; Neurobiol. Dis. 46, 2012, 1.
[112] X. He, P. Hahn, J. Iacovelli, R. Wong, C. King, R. Bhisitkul, M. Massro-Giordano,
J.L. Dunaief; Prog. Retin Eye Res. 26, 2007, 649.
[113] C. Eid, M. Hémadi, N.T. Ha-Duong, J.M. El Hage Chahine; Biochim. Biophys. Acta 1840,
2014, 1771.
[114] S. Osaki, D.A. Johnson; J. Biol. Chem. 244, 1969, 5757.
[115] D.M. De Silva, C.C. Askwith, J. Kaplan; Physiol. Rev. 76, 1996, 31.
[116] J. Healy, K. Tipton; J. Neural Transm. 114, 2007, 777.
[117] A. Michalak, J. Krzeszowiak, I. Markiewicz-Górka; Postepy Hig. Med. Dosw. 68, 2014, 1483.
[118] J.M. Gutteridge; Clin. Chem. 41, 1995, 1819.
[119] P. Bielli, L. Calabrese; Cell. Mol. Life Sci. 59, 2002, 1413.
[120] A. Augustyniak, E. Skrzydlewska; Postepy Hig. Med. Dosw. 58, 2004, 194.
[121] J.M. Gutteridge; Ann. Neurol. 32, 1992, S16.
[122] E. Frieden, H.S. Hsieh; Adv. Enzymol. Ramb. 44, 1976, 187.
[123] I. Kochanowska, E. Hampel-Osipowicz, P. Waloszczyk; Neurologia Dziecięca 17, 2008, 63.
[124] P.V. van den Berghe, L.W. Klomp; Nutr. Rev. 67, 2009, 658.
[125] I. Ojeda, M. Moreno-Guzmán, A. González-Cortés, P. Yáñez-Sedeño, J.M. Pingarrón;
Electroanal. 25, 2013, 2166.
[126] M.C. Linder; Metallomics 8, 2016, 887.

Bibliografia
220
[127] T.C. Larkin, T.A. Fingleton, M. Mccusker, M. Cassidy, G. Gunter, C.A. Lepp, E. Gamboa;
Clin. Lab. 50, 2004, 193.
[128] M. Nowak, T. Wielkoszyński, B. Marek, B. Kos-Kudła, E. Świętochowska, L. Siemińska,
J. Karpe, D. Kajdaniuk, J. Głogowska-Szeląg, K. Nowak; Clin. Exp. Med. 10, 2010, 185.
[129] M. Siotto, P. Pasqualetti, M. Marano, R. Squitti; J. Neural Transm. 121, 2014, 1281.
[130] R.N. Rosenberg, S.B. Prusiner, S. DiMauro, R. Barchi; „Kliniczne kompendium do
molekularnych i genetycznych podstaw chorób neurologicznych”, D.W. Publishing Co.,
Szczecin, 1999.
[131] T. Tonnesen, W.J. Kleijer, N. Horn; Hum. Genet. 86, 1991, 408.
[132] Y.H. Gu, H. Kodama, K. Shiga, S. Nakata, Y. Yanagawa, H. Ozawa; J. Inherit. Metab. Dis. 28,
2005, 473.
[133] I.H. Scheinberg, I. Sternlieb; Annu. Rev. Med. 16, 1965, 119.
[134] I.H. Scheimberg, D. Gitlin; Science 116, 1952, 484.
[135] G. Crisponi, V.M. Nurchi, D. Fanni, C. Gerosa, S. Nemolato, G. Faa; Coord. Chem. Rev. 254,
2010, 876.
[136] A. Ala, A.P. Walker, K. Ashkan, J.S. Dooley, M.L. Schilsky; Lancet. 369, 2007, 397.
[137] J.M. Walshe; Lancet. 270, 1956, 25.
[138] J.M. Zaucha; Hematologia 2, 2011, 15.
[139] P. Knekt, A. Aromaal, J. Maatelal, A. Rissanen, M. Hakama, R.-K. Aaran, T. Nikkari,
T. Hakulinen, R. Peto, L. Teppo; Br. J. Cancer 65, 1992, 292.
[140] T. Wróbel; Acta Haematologica Polonica 42, 2011, 375.
[141] T.M. Rose, G.D. Plowman, D.B. Teplow, W.J. Dreyer, K.E. Hellström, J.P. Brown; Proc. Natl.
Acad. Sci. USA 83, 1986, 1261.
[142] R.P. Palmour, H.E. Sutton; Biochemistry 10, 1971, 4026.
[143] D. Łubgan, A. Marczak, L. Distel, Z. Jóźwiak; Postępy Biochemii 52, 2005, 72.
[144] E.N. Baker; Adv. Inorg. Chem. 41, 1994, 389.
[145] https://en.wikipedia.org/wiki/Transferrin
[146] G.V. Louie; Curr. Opin. Struct. Biol. 3, 1993, 401.
[147] H. Sun, H. Li, P.J. Sadler; Chem. Rev. 99, 1999, 2817.
[148] C.L. Day, B.F. Anderson, J.W. Tweedie, E.N. Baker; J. Mol. Biol. 232, 1993, 1084.
[149] R.F. Fischetti, D.J. Rodi, D.B. Gore, L. Makowski; Chemistry & Biology 11, 2004, 1431.
[150] M.R. Schlabach, G.W. Bates; J. Biol. Chem. 250, 1975, 2182.
[151] R.T.A. MacGillivray, E. Mendez, S.K. Sinha, M.R. Sutton, J. Lineback-Zins, K. Brew; Proc.
Natl. Acad. Sci. USA 79, 1982, 2504.
[152] O. Mazuryk, K. Kurpiewska, K. Lewiński, G. Stochel, M. Brindell; J. Inorg. Biochem.
116, 2012, 11.
[153] D. Furmanowicz, I. Pietrzak, K. Wiktorowicz; Przegląd Lekarski 64, 2007, 483.
[154] C.J. Parker Siburt, E.M. Lin, S.J. Brandt, A.D. Tinoco, A.M. Valentine, A.L. Crumbliss;
J. Inorg. Biochem. 104, 2010, 1006.
[155] A. Van Campenhout, C.M. Campenhout, A.R. Lagrou, B. Manuel-y-Keenoy; Free Radic. Res.
37, 2003, 1069.
[156] A. Hayashi, Y. Wada, T. Suzuki, A. Shimizu; Am. J. Hum. Genet. 53, 1993, 201.
[157] J. Artym, M. Zimecki; Kosmos 3, 2014, 345.
[158] P. Lipiński, Z. Jarząbek, S. Broniek, T. Zagulski; Int. J. Exp. Pathol. 72, 1991, 623.
[159] P. Aisen, C. Enns, M. Wessling-Resnick; Int. J. Biochem. Cell. Biol. 33, 2001, 940.
[160] J.L. Beard; J. Nutr. 131, 2001, 568S.
[161] T. Kubiak; Kosmos 62, 2013, 501.
[162] N.C. Andrews; N. Engl. J. Med. 341, 1999, 1986.
[163] M.D. Garrick, L.M. Garrick; Biochim. Biophys. Acta 1790, 2009, 309.
[164] B. Cylwik, L. Chrostek, M. Szmitkowski; Postepy Hig. Med. Dosw. 60, 2006, 101.
[165] H. Stibler, K.G. Kjellin; J. Neurol. Sci. 30, 1976, 269.
[166] H. Stibler; Clin. Chem. 37, 1991, 2029.
[167] P. Aisen, M. Wessling-Resnick, E.A. Leibold; Curr. Opin. Chem. Biol. 3, 1999, 200.
[168] M. Hémadi, P.H. Kahn, G. Miguel, J.M. El Hage Chahine; Biochemistry 43, 2004, 1736.
[169] Q.-Y. He, A.B. Mason, V. Nguyen, R.T.A. MacGillivray, R.C. Woodworth; Biochem. J. 350,
2000, 909.
[170] P.T. Gomme, K.B. McCann; Drug Discov. Today 10, 2005, 267.
[171] W.R. Harris; J. Inorg. Biochem. 27, 1986, 41.
[172] M. Shen, J. Wang, M. Yang, G. Li; Electrochem. Commun. 13, 2011, 114.

Bibliografia
221
[173] P. Aisen; Metal Ions Biol. Sys. 35, 1998, 585.
[174] R. Mariani, P. Trombini, M. Pozii, A. Piperno; Mediterr. J. Hematol. Infect. Dis. 1, 2009,
e2009006.
[175] M. Ching-Ming Chung; Biochemical Education 12, 1984, 146.
[176] H. Sun, H. Li, A.B. Mason, R.C. Woodworth, P.J. Sadler; J. Biol. Chem. 276, 2001, 8829.
[177] A. Ando, I. Ando, T. Hiraki, K. Hisada; Int. J. Rad. Appl. Instrum. B 15, 1988, 133.
[178] P. Domenica, J. Reich, W. Madonia, B.A. Cunha; J. Antimicrob. Chemother. 38, 1996, 1031.
[179] A. Ando, I. Ando, T. Hiraki, K. Hisada; Int. J. Rad. Appl. Instrum. B 15, 1988, 133.
[180] H. Li, Z.M. Qian; Med. Res. Rev. 22, 2002, 225.
[181] H. Li, H. Sun, Z.M. Qian; Trends Pharmacol. Sci. 23, 2002, 206.
[182] N.J. Hallab, A. Skipor, J.J. Jacobs; J. Biomed. Mater. Res. 65A, 2003, 311.
[183] J. Walley, P.J. Halbrooks, C. Vonrhein, M.A. Rould, S.J. Everse, A.B. Mason; J. Biol. Chem.
281, 2006, 24934.
[184] V. Thiruselvam, P. Sivaraman, T. Kumarevel, M.N. Ponnuswamy; Biochem. Biophys. Res.
Commun. 453, 2014, 636.
[185] P. Arosio, R. Ingrassia, P. Cavadini; Biochim. Biophys. Acta 1790, 2009, 589.
[186] E.C. Theil; Annu. Rev. Biochem. 56, 1987, 289.
[187] K. Orino, L. Lehman, Y. Tsuji, H. Ayaki, S.V. Torti, F.M. Torti; Biochem. J. 357, 2001, 241.
[188] G.J. Andreson, D.M. Frazer; Semin. Liver Dis. 25, 2005, 420.
[189] E.J. Lesnefsky; Adv. Exp. Med. Biol. 366, 1994, 129.
[190] C.W. Levenson, C.A. Fitch; Brain Res. Dev. Brain Res. 119, 2000, 105.
[191] S.V. Torti, E.L. Kwak, S.C. Miller, L.L. Miller, G.M. Ringold, K.B. Myambo, A.P. Young,
F.M. Torti; J. Biol. Chem. 263, 1988, 12638.
[192] K.B. Storey; J. Med. Biol. Res. 29, 1996, 1715.
[193] G. Beck, T.W. Ellis, G.S. Habicht, S.F. Schluter, J.J. Marchalonis; Dev. Comp. Immunol.
26, 2002, 11.
[194] P.M. Harrison, P. Arosio; Biochim. Biophys. Acta 1275, 1996, 161.
[195] F. Bou-Abdallah, G. Zhao, G. Biasiotto, M. Poli, P. Arosio, N.D. Chasteen; J. Am. Chem. Soc.
130, 2008, 17801.
[196] D.M. Lawson, P.J. Artymiuk, S.J. Yewdall, J.M. Smith, J.C. Livigstone, A. Treffry,
A. Luzzago, S. Levi, P. Arosio, G. Cesareni, C.D. Thomas, W.V. Shaw, P.M. Harrison; Nature
349, 1991, 541.
[197] R.R. Crixhton; Angew. Chem. Int. Ed. Engl. 12, 1973, 57.
[198] S.H. Banyard, D.K. Stammers, P.M. Harrison; Nature 271, 1978, 282.
[199] P. Rucker, F.M. Torti, S.V. Tprti; J. Biol. Chem. 271, 1996, 33352.
[200] N.D. Chasteen, P.M. Harrison; J. Struct. Biol. 126, 1999, 182.
[201] F.M. Michel, L. Ehm, S.M. Antao, P.L. Lee, P.J. Chupas, G. Liu, D.R. Strongin,
M.A.A. Schoonen, B.L. Phillips, J.B. Parise; Science 316, 2007, 1726.
[202] R.C. Hider, X. Kong; Dalton Trans. 42, 2013, 3220.
[203] E.C. Theil, R.K. Behera, T. Tosha; Coord. Chem. Rev. 257, 2013, 579.
[204] T. Douglas, D.R. Ripoll; Protein Sci. 7, 1998, 1083.
[205] X. Liu, E.C. Theil; Acc. Chem. Res. 38, 2005, 167.
[206] G. Jutz, P. van Rijn, B. Santos Miranda, A. Böker; Chem. Rev. 115, 2015, 1653.
[207] J.L. Johnson, M. Cannon, R.K. Watt, R.B. Frankel, G.D. Watt; Biochemistry 38, 1999, 6706.
[208] F. Carmona, Ò. Palacios, N. Gálvez, R. Cuesta, S. Atrian, M. Capdevila, J.M. Domínguez-
Vera; Coord. Chem. Rev. 257, 2013, 2752.
[209] N.E. Le Brun, A.M. Keech, M.R. Mauk, A.G. Mauk, S.C. Andrews, A.J. Thomson,
G.R. Moore; FEBS Lett. 397, 1996, 159.
[210] S.P. Martsev, A.P. Vlasov, P. Arosio, Protein Eng. 11, 1998, 377.
[211] M. Wagstaff, M. Worwood, A. Jacobs; Biochem. J. 173, 1978, 969.
[212] A. Friedman, P. Arrosio, D. Finazzi, D. Koziorowski, J. Galazka-Friedman; Park. Rel. Disord.
17, 2011, 423.
[213] K. Caban-Hernandez, J.F. Gaudier, A.M. Espino; Mol. Biochem. Parasitol. 182, 2012, 54.
[214] J. Zähringer, B.S. Baliga, H.N. Munro; Proc. Natl. Acad. Sci. USA 73, 1976, 857.
[215] K. Iwai, S.K. Drake, N.B. Wehr, A.M. Weissman, T. LaVaute, N. Minato, R.D. Klausner, R.L.
Levine, T.A. Rouault; Proc. Natl. Acad. Sci. USA 95, 1998, 4924.
[216] P. Lipiński, R.R. Starzyński; Postepy Hig. Med. Dosw. 60, 2006, 322.
[217] D. Berg, G. Beceker, P. Riederer, O. Rieb; Neurotox. Res. 4, 2002, 637.
[218] A. Grzegorzewska; Pol. Arch. Med. Wewn. 117, 2007, 172.

Bibliografia
222
[219] A. Szczeklik; Choroby wewnętrzne, Tom II, Wydawnictwo Medycyna Praktyczna, Kraków,
2005.
[220] T. Podolecki, J. Wasilewski, L. Poloński; Choroby Serca i Naczyń 6, 2009, 180.
[221] J. McCord; Circulation 83, 1991, 1112.
[222] J.L. Sullivan; Lancet 1, 1981, 1293.
[223] J.L. Sullivan; Circulation 86, 1992, 1036.
[224] S. Kiechl, J. Willeit, G. Egger, W. Poewe, F. Oberhollenzer; Circulation 96, 1997, 3300.
[225] J.T. Salonen, K. Nyyssönen, H. Korpela, J. Tuomilehto, R. Seppänen, R. Salonen; Circulation
86, 1992, 803.
[226] M. Truffi, L. Fiandra, L. Sorrentino, M. Monieri, F. Corsi, S. Mazzucchelli; Pharmacol. Res.
107, 2016, 57.
[227] J.M. Dominguez-Vera; J. Inorg. Biochem. 75, 2004, 3145.
[228] G. ker; Chem. Rev. 115, 2015, 1653.
[229] I. Kętnik-Prastowska; „Immunochemia w biologii medycznej, metody laboratoryjne”,
Wydawnictwo Naukowe PWN, Warszawa, 2009.
[230] J. Nowak, M. Boncler, C. Watała; Diagn. Lab. 50, 2014, 53.
[231] E. Turlej; Postepy Hig. Med. Dosw. 63, 2009, 213.
[232] T.W. Kragstrup, T. Vorup-Jensen, B. Deleuran, M. Hvid; SpringerPlus 2, 2013, 263.
[233] A.J. Crowle; „Immunodiffusion”, Academic Press, New York, 1973.
[234] B.H. Berne; Clin. Chem. 20, 1974, 61.
[235] G. Mancini, A.O. Carbonara, J.F. Heremans; Immunochemistry 2, 1965, 235.
[236] H.C.M. Clark, T. Freeman; Clin. Sci. 35, 1968, 403.
[237] G. Csako; Methods Mol. Biol. 869, 2012, 339.
[238] E.M. Southern; J. Mol. Biol. 98, 1975, 503.
[239] T. Mahmood, P. Yang; N. Am. J. Med. Sci. 4, 2002, 429.
[240] B.T. Kurien, R.H. Scofield; Methods 38, 2006, 283.
[241] J. Towbin, T. Staehlin, J. Gordon; Proc. Natl. Acad. Sci. USA 76, 1979, 4350.
[242] J. Opiela; Wiadomości Zootechniczne 1, 2006, 11.
[243] http://metlab.pl/Western_blotting-_teoria_p41.html
[244] O. Warburg, W. Christian; Biochem. Z 310, 1941, 384.
[245] M.M. Bradford; Anal. Biochem. 72, 1976, 248.
[246] K. Takahata, S. Igarashi; Clin. Chim. Acta 283, 1999, 129.
[247] J. Wang; Electroanal. 13, 2001, 983.
[248] R.S. Freire, C.A. Pessoa, L.D. Mello, L.T. Kubota; J. Braz. Chem. Soc. 14, 2003, 230.
[249] J. Xu, W. Li, Q. Yin, H. Zhong, Y. Zhu, L. Jin; J. Colloid. Interf. Sci. 315, 2007, 170.
[250] T. Ruzgas, E. Csöregi, J. Emnéns, L. Gorton, G. Marko-Varga; Anal. Chim. Acta 330, 1996,
123.
[251] K. Habermüller, M. Mosbach, W. Schuhman; Fresenius J. Anal. Chem. 366, 2000, 560.
[252] X. Dominguez-Benetton, S. Srikanth, Y. Satyawali, K. Vanbroekhoven, D. Pant; J. Microbial.
Biochem. Technol. 5, 2013, S6.
[253] R.A. Marcus; Angew. Chem. Int. Ed. 32, 1993, 1111.
[254] J. Yang, F. Pang, R. Zhang, Y. Xu, P. He, Y. Fang; Electroanal. 20, 2008, 2134.
[255] E. Alvarezde Eulate, L. Serls, D.W.M. Arrigan; Anal. Bioanal. Chem. 405, 2013, 3801.
[256] Y. Sun, H. Du, Y. Lan, W. Wang, Y. Liang, C. Feng, M. Yang; Biosens. Bioelectron. 77, 2016,
894.
[257] H. Wang, D. Li, Z. Wu, G. Shen, R. Yu; Talanta 62, 2004, 199.
[258] I. Ojeda, M. Moreno-Guzmán, A. González-Cortés, P. Yáñez-Sedeño, J.M. Pingarrón;
Electroanal. 25, 2013, 2166.
[259] B. Garcinuño, I. Ojeda, M. Moreno-Guzmán, A. González-Cortés, P. Yáñez-Sedeño,
J.M. Pingarrón; Talanta 118, 2014, 61.
[260] S.-Q. Hi, Z.-Y. Wu, Y.-M. Zhou, Z.-X. Cao, G.-L. Shen, R.-Q. Yu; Anal. Chim. Acta 458,
2002, 297.
[261] M.S. Wilson, R.D. Rauh; Biosens. Bioelectron. 19, 2004, 693.
[262] L. Yang, W. Wei, X. Gao, J. Xia, H. Tao; Talanta 68, 2005, 40.
[263] X. Zhang, S. Wang, M. Hu, Y. Xiao; Biosens. Bioelectron. 21, 2006, 2180.
[264] S.-F. Wang, Y.-M. Tan; Anal. Bioanal. Chem. 387, 2007, 703.
[265] S. Patra, E. Roy, R. Madhuri, P.K. Sharma; Biosens. Bioelectron. 63, 2015, 301.
[266] A.L. Ghindilis, P. Atanasov, E. Wilkins; Electroanal. 9, 1997, 661.
[267] D. Leech, P. Kavanagh, W. Schuhmann; Electrochim. Acta 84, 2012, 223.

Bibliografia
223
[268] M. Zhao, Y. Gao, J. Sun, F. Gao; Anal. Chem. 87, 2015, 2615.
[269] J.A. Cracknell, K.A. Vincent, F.A. Armstrong; Chem. Rev. 108, 2008, 2439.
[270] J.-F. Rusling; Acc. Chem. Res. 31, 1998, 363.
[271] W.H. Scouten, J.H.T. Luong, R.S. Brown; Trends Biotechnol. 13, 1995, 178.
[272] E. Stellwagen; Nature 275, 1978, 73.
[273] T. Lötzbeyer, W. Schuhmann, E. Katz, J. Falter, H.-L. Schmidt; J. Electroanal. Chem. 377,
1994, 291.
[274] Z.Y. Wang, S.N. Liu, P. Wu, C.X. Cai; Anal. Chem. 81, 2009, 1638.
[275] D. Ivnitski, K. Artyushkova, R.A. Rincón, P. Atanassov, H.R. Luckarift, G.R. Johnson; Small
4, 2008, 357.
[276] S.X. Zhang, N. Wang, H.J. Yu, Y.M. Niu, C.Q. Sun; Bioelectrochemistry 67, 2005, 15.
[277] C.X. Cai, J. Chen; Anal. Biochem. 332, 2004, 75.
[278] E.E. Ferapontova, S. Shleev, T. Ruzgas, L. Stoica, A. Christenson, J. Tkac, A.I. Yaropolov,
L. Gorton; Prospectives in Bioanalysis 1, 2005, 517.
[279] Z.-Q. Zhai, J. Wu, W. Sun, K. Jiao; J. Chin. Chem. 56, 2009, 561.
[280] S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A.I. Yaropolov, J.W. Whittaker, L. Gorton;
Biosens. Bioelectron. 20, 2005, 2517.
[281] H. Bagheri, E. Ranjbari, M. Amiri-Aref, A. Hajian, Y. H. Ardakani, S. Amidi; Biosens.
Bioelectron. 85, 2016, 814.
[282] A. Chaubey, B.D. Malhotra; Biosens. Bioelectron. 17, 2002, 441.
[283] K. Karnicka, B. Kowalewska, K. Miecznikowski, P.J. Kulesza; Biotechnologia 4, 2007, 25.
[284] K. Markowska, A.M. Grudniak, K.I. Wolska; Post. Mikrobiol. 52, 2013, 29.
[285] M. Tominaga; Electrochem. Commun. 4, 2002, 968.
[286] X. Zhang, L. Chen, Q. Yang, Q. Li, X. Sun, H. Chen, G. Yang, Y. Tang; Luminescence 30,
2015, 1176.
[287] J. Polak, A. Jarosz-Wilkołazka; Biotechnologia 4, 2007, 82.
[288] J. Tatur, W.R. Hagen, H.A. Heering; Dalton Trans. 21, 2009, 2837.
[289] J. Ye, R.P. Baldwin; Anal. Chem. 60, 1988, 2263.
[290] S. Pakapongpana, R. Palangsuntikula, W. Surareungchai; Electrochim. Acta 56, 2011, 6831.
[291] C.F. Meunier, X.-Y. Yang, J.C. Rooke, B.-L. Su; ChemCatChem 3, 2011, 476.
[292] P. Jochems, Y. Statyawali, L. Diels, W. Dejonghe; Green Chem. 13, 2011, 1609.
[293] A.W.P. Vermeer, W. Norde; J. Colloid Interf. Sci. 225, 2000, 394.
[294] W. Norde, C.E. Giacomelli; J. Biotechnol. 79, 2000, 259.
[295] T.E. Benavidez, D. Torrente, M. Marucho, C.D. Garcia; Langmuir 31, 2015, 2455.
[296] D.G. Castner, B.D. Ratner; Surf. Sci. 500, 2002, 28.
[297] M. Malmsten; J. Colloid Interf. Sci. 207, 1998, 186.
[298] K. Nakanishi, T. Sakiyama, K. Imamura; J. Biosci. Bioeng. 91, 2001, 233.
[299] F. Secundo; Chem. Soc. Rev. 42, 2013, 6250.
[300] W.H. Scouten, J.H.T. Luong, R.S. Brown; Trends. Biotechnol. 13, 1995, 178.
[301] J. Wang; Electroanal. 3, 1991, 255.
[302] W. Norde, C.E. Giacomelli; J. Biotechnol. 79, 2000, 259.
[303] Y. Xiao, F. Patolsky, E. Katz, J.F. Hainfeld, I. Willner; Science 299, 2003, 1877.
[304] E. Katz, I. Willner; Angew. Chem. Int. Ed. Engl. 43, 2004, 6042.
[305] S. Liu, Z. Dai, H. Chen, H, Ju; Biosens. Bioelectron. 19, 2004, 963.
[306] Z.S. Beiranvand, A.R. Abbasi, S. Dehdashtian, Z. Karimi, A. Azadbakht; Anal. Biochem. 518,
2017, 35.
[307] Z. Li, L. Sheng, A. Meng, C. Xie, K. Zhao; Microchim. Acta 183, 2016, 1625.
[308] A. Dyal, K. Loos, M. Noto, S.W. Chang, C. Spagnoli, K.V. Shafi, A.Ulman, M. Cowman, R.A.
Gross; J. Am. Chem. Soc. 125, 2003, 1684.
[309] S.A. Ansari, Q. Husain; Biotechnol. Adv. 30, 2012, 512.
[310] M. Opallo, A. Lesniewski; J. Electroanal. Chem. 656, 2011, 2.
[311] C.M. DiCarlo, D.L. Compton, K.O. Evans, J.A. Laszlo; Bioelectrochemistry 68, 2006, 134.
[312] S.J. Updike, G.P. Hicks; Nature 214, 1967, 986.
[313] X.Y. Yang, A. Léonard, A. Lemaire, G. Tian, B.L. Su; Chem. Commun. 47, 2011, 2763.
[314] N. Nassif, J. Livage; Chem. Soc. Rev. 40, 2011, 849.
[315] J. Kim, H. Jia, P. Wang; Biotechnol. Adv. 24, 2006, 296.
[316] J. Bryjak; Wiadomości chemiczne 58, 2004, 691.
[317] X.-P. Dai, Z.-F. Yang, R.G. Luo, K.K. Sirkan; J. Membr. Sci. 171, 2000, 183.
[318] L. Nobs, F. Buchegger, R. Gurny, E. Allémann; J. Pharm. Sci. 93, 2004, 1980.

Bibliografia
224
[319] J.B. Jia, B.Q. Wang, A.G. Wu, G.J. Cheng, Z. Li, S.J. Dong; Anal. Chem. 74, 2002, 2217.
[320] A. Ulman; Adv. Mater. 2, 1990, 573.
[321] D. Mandler, I. Turyan; Electroanal. 8, 1996, 207.
[322] S.F. D’Souza; Curr. Sci. 77, 1999, 69.
[323] I.Y. Galaev, B. Mattiasson; Enzyme Microb. Technol. 15, 1993, 354.
[324] J. Turkova; J. Chromotogr. B 722, 1999, 11.
[325] M. Saalemuddin; Adv. Biochem. Eng. 64, 1999, 203.
[326] S.K. Sahoo, V. Labhasetwar; Drugs Deliv. Today 8, 2003, 1112.
[327] E. Oberdörster; Environ. Health Prospect. 112, 2004, 1058.
[328] A.M. Świdwińska-Gajewska; Medycyna Pracy 58, 2007, 253.
[329] J. Rzeszutek, M. Matysiak, M. Czajka, K. Sawicki, P. Rachubik, M. Kruszewski, L. Kapka-
Skrzypczak; Hygeia Public Health 49, 2014, 449.
[330] I. Najarula; „Nanotechnology: Recent Trends, Emerging Issues and Future Directions”, Nova
Science Publishers, New York, 2014.
[331] D. Zhang, H. Li, H. Wang, L. Li; Int. J. Quantum Chem. 116, 2016, 1846.
[332] R.M. Allaf, I.V. Rivera, I.N. Ivanov; Int. J. Polym. Mater. Polym. Biomater. 66, 2017, 183.
[333] D. Han, X. Zhang, Z. Hua, S. Yang; App. Surf. Sci. 390, 2016, 760.
[334] J.S. Kim, E. Kuk, K.N. Yu, J.-H. Kim, S.J. Park, H.J. Lee, S.H. Kim, Y.K. Park, Y.H. Park,
C.-Y. Hwang, Y.-K. Kim, Y.-S. Lee, D.H. Jeong, M.-H. Cho; Nanomed. Nanotechnol. Biol.
Med. 3, 2007, 95.
[335] X. Huang, P.K. Jain, I.H. El-Sayed, M.A. El-Sayed; Lasers Med. Sci. 23, 2008, 217.
[336] L. Zhang, S.J. Xiao, L.L. Zheng, Y.F. Li, C.Z. Huang; J. Mater. Chem. B 2, 2014, 8558.
[337] S.E. McNeil; J. Leukoc. Biol. 78, 2005, 585.
[338] J. Aswathy, M. Suresh; ChemPlusChem 79, 2014, 1382.
[339] L. Cabrera, S. Gutierrez, N. Menendezb, M.P. Morales, P. Herrasti; Electrochim. Acta 53,
2008, 3436.
[340] O. Bomatı´-Miguel, L. Mazeina, A. Navrotsky, S. Veintemillas-Verdaguer; Chem. Mater.
20, 2008, 591.
[341] Y. Roh, H. Vali, T.J. Phelps, J.W. Moon; J. Nanosci. Nanotechnol. 11, 2006, 517.
[342] E.E. Carpenter, S. Calvin, R.H. Stroud, V. G. Harris; Chem. Mater. 15, 2003, 3245.
[343] J. Wang, S. Zheng, Y. Shao, J. Liu, Z. Xu, D. Zhu; J. Colloid Interface Sci. 349, 2010, 293.
[344] H. Hsu, Y.Y. Lin, S. Huang, K.W. Lem, D.H. Nguyen, D.S. Lee; Nanotechnology 24, 2013,
475102.
[345] R.A. Ramli, W.A. Laftah, S. Hashim; RSC Adv. 3, 2013, 15543.
[346] M. Zhu, G. Diao; Nanoscale 3, 2013, 2748.
[347] R.S. Ruoff, D.C. Lorents, B. Chan, R. Malhotra, S. Subramoney; Science 259, 1993, 346.
[348] M. Bystrzejewski, A. Huczko, H. Lange; Sens. Actuators, B 109, 2005, 81.
[349] J. Borysiuk, A. Grabias, J. Szczytko, M. Bystrzejewski, A. Twardowski, H. Lange; Carbon
46, 2008, 1693.
[350] M. Bystrzejewski; J. Solid State Chem. 184, 2011, 1492.
[351] M. Bystrzejewski, K. Pyrzyńska; Colloids Surf. A: Physicochem. Eng. Asp. 377, 2011, 402.
[352] M. Bystrzejewski, H. Lange, A. Huczko; Fuller. Nanotube. Car. N. 15, 2007, 167.
[353] M. Bystrzejewski, A. Huczko, M. Soszyński, H. Lange; Fuller. Nanotube. Car. N. 17, 2009,
600.
[354] N. Aguiló-Aguayo, L. Maurizi, S. Galmarini, M.G. Ollivier-Beuzelin, G. Coullerez, E. Bertran,
H. Hofmann; Dalton Trans. 43, 2014, 13764.
[355] K. Pyrzyńska, M. Bystrzejewski; Colloids Surf. A: Physicochem. Eng. Asp. 362, 2010, 102.
[356] A.M. Nowicka, A. Kowalczyk, M. Bystrzejewski, M. Donten, M.L. Donten, Z. Stojek,
Electrochim. Acta 126, 2014, 115.
[357] X.M. Lin, A.C.S. Samia; J. Magn. Magn. Mater. 305, 2006, 100.
[358] V. Panchal, M. Neergat, U. Bhandarkar; J. Nanopart. Res. 13, 2011, 3825.
[359] G. Reiss, A. Hutten; Nat. Mater. 10, 2005, 725.
[360] M. Bystrzejewski, K. Pyrzyńska; Mater. Chem. Phys. 141, 2013, 454.
[361] U. Laska, C.G. Frost, G.J. Price, P.K. Plucinski; J. Catal. 268, 2009, 318.
[362] J.S. Beveridge, J.R. Stephens, M.E. Williams; Annu. Rev. Anal. Chem. 4, 2011, 251.
[363] M. Bystrzejewski, S. Cudziło, A. Huczko, H. Lange, G. Soucy, G. Cota-Sanchez,
W. Kaszuwara; Biomol. Eng. 24, 2007, 555.
[364] H. Song, X. Chen; Chem. Phys. Lett. 374, 2003, 400.
[365] M. Bystrzejewski, K. Pyrzyńska, A. Huczko, H. Lange; Carbon 47, 2009, 1201.

Bibliografia
225
[366] A.M. Nowicka, A. Kowalczyk, M. Bystrzejewski, M. Donten, Z. Stojek; Electrochem.
Commun. 20, 2012, 4.
[367] M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervell, K.A. Dawson; Proc. Natl. Acad. Sci.
USA 105, 2008, 14265.
[368] M.I. Setyawati, C.Y. Tay, D. Docter, R.H. Stauber, D.T. Leong; Chem. Soc. Rev. 44, 2015,
8174.
[369] A. Gessner, A. Lieske, B. Paulke, R. Müller; Eur. J. Pharm. Biopharm. 54, 2002, 165.
[370] M. Poplawska, M. Bystrzejewski, I.P. Grudziński, M.A. Cywińska, J. Ostapko,
A. Cieszanowski; Carbon 74, 2014, 180.
[371] K.K.L. Phua, H.F. Staats, K.W. Leong, S.K. Nair; Sci. Rep. 4, 2014, 5128.
[372] S. Aula, S. Lakkireddy, K. Jamil, A. Kapley, A.V.N. Swamy, H.R. Lakkireddy; RSC Adv.
5, 2015, 47830.
[373] A.M. Nowicka, A. Kowalczyk, A. Jarzebinska, M. Donten, P. Krysinski, Z. Stojek,
E. Augustin, Z. Mazerska; Biomacromol. 14, 2013, 828.
[374] Z. Galus; „Fundamentals of Electrochemical Analysis”, Wydawnictwo Naukowe PWN,
Warszawa, 1994.
[375] A. Lasia; „Advanced Electrochemistry. Interfaces, thermodynamics, and electrochemical
techniques”, Département de chimie, Université de Sherbrooke, 2014.
[376] A. Bund, S. Kohler, H.H. Kuhnlein, W. Plieth; Electrochim. Acta 49, 2003, 147.
[377] A. Bund, H. H. Kuehnlein; J. Pys. Chem. 109, 2005, 19845.
[378] N. Leventis, N. Chandrasekaran, A. Dass, X. Gao; ECS Trans. 13, 2008, 25.
[379] S.R. Ragsdale, K.M. Grant, H.S. White; J. Am. Chem. Soc. 120, 1998, 13461.
[380] K.M. Grant, J.W. Hemmert, H.S. White; Electrochem. Commun. 1, 1999, 319.
[381] A. Szewczyk, A. Wiśniewski, R. Puźniak, H. Szymczak; „Magnetyzm i nadprzewodnictwo”,
Wydawnictwo Naukowe PWN, Warszawa, 2012.
[382] E. Katz, O. Lioubashevski, I. Willner; J. Am. Chem. Soc. 127, 2005, 3979.
[383] H. Stöcker; „Nowoczesne kompendium fizyki”, Wydawnictwo Naukowe PWN, Warszawa,
2012.
[384] M. Waskaas, Y.I. Kharkats; J. Electroanal. Chem. 502, 2001, 51.
[385] G. Mutschke, A. Bund; Electrochem. Commun. 10, 2008, 597.
[386] F. Scholtz; „Electroanalytical Methods”, Springer, Berlin, 2010.
[387] J. Wang; „Analytical Electrochemistry”, Wiley – VCH, New York, 2000. [388] V.M. Mecea; Sens. Actuators A 128, 2006, 270.
[389] V.M. Mecea; Anal. Lett. 38, 2005, 753.
[390] C.K. O'Sullivan, G.G. Guilbault; Biosens. Bioelectron. 14, 1999, 663.
[391] G. Sauerbrey; Z. Phys. 155, 1959, 206.
[392] M.V. Voinova, M. Jonson, B. Kasemo; Biosens. Bioelectron. 17, 2002, 835.
[393] K.A. Marx; Biomacromolecules 4, 2003, 1099.
[394] I. Reviakine, D. Johannsmann, R.P. Richter; Anal. Chem. 83, 2011, 8838.
[395] K.K. Kanazawa, J.G. Gordon; Anal. Chem. 57, 1985, 1770.
[396] R. Konradi, M. Textor, E. Reimhult; Biosensors 2, 2012, 341.
[397] F. Höök, A. Ray, B. Norden, B. Kasemo; Langmuir 17, 2001, 8305.
[398] J. Malmström, H. Agheli, P. Kingshott, D.S. Sutherland; Langmuir 23, 2007, 9760.
[399] S.H. Kristensen, G.A. Pedersen, L.N. Nejsum, D.S. Sutherland; J. Phys. Chem. B 117, 2013,
10376.
[400] C. Gabrielli; „Identification of electrochemical processes by frequency response analysis”,
Schlumberger Instrumentation Group, Farnborough, 1980.
[401] A. Królikowski; „Metody elektrochemiczne w badaniach korozyjnych”, Politechnika
Wrocławska, ZETiK, Karpacz, 1991.
[402] A. Lasia; „Electrochemical Impedance Spectroscopy and Its Applications, Modern Aspects of
Electrochemistry”, B.E. Conway, J. Bockris, R.E. White, Kluwer Academic/Plenum
Publishers, New York, 1999.
[403] H. Scholl, T. Błaszczyk, P. Knyczmonik; „Elektrochemia. Zarys teorii i praktyki”,
Wydawnictwo Uniwersytetu Łódzkiego, Łódź, 1998.
[404] R.N. Vyas, K. Li, B. Wang; J. Phys. Chem. B 114, 2010, 15818.
[405] A.J. Bard, F-R.F. Fan, J. Kwak, O. Lev; Anal. Chem. 61, 1989, 132.
[406] A.L. Barker, J.V. Macpherson, C.J. Slevin, P.R. Unwin; J. Phys. Chem. B 102, 1998, 1586.
[407] M.A. Edwards, S. Martin, A.L. Whitworth, J.V. Macpherson, P.R. Unwin; Physiol. Meas.
27, 2006, R63.

Bibliografia
226
[408] G. Wittstock, M. Burchardt, S.E. Pust,Y. Shen, C. Zhao; Angew. Chem. Int. Ed. 46, 2007,
1584.
[409] F. Jaroszyk; „Biofizyka. Podręcznik dla studentów”, Wydawnictwo lekarskie PZWL,
Warszawa, 2013.
[410] L. Komorowski, A. Olszewski; „Chemia fizyczna. Tom 4”, Wydawnictwo Naukowe PWN,
Warszawa, 2013.
[411] K. Pigoń, Z. Ruziewicz; „Chemia fizyczna. Tom 2”, Wydawnictwo Naukowe PWN, Warszawa,
2007.
[412] H.O. Finklea, S. Avery, M. Lynch, T. Furtsch; Langmuir 3, 1987, 409.
[413] I.P. Grudzinski, M. Bystrzejewski, M.A. Cywinska, A. Cieszanowski, A. Ostrowska;
J. Nanopart. Res. 15, 2013, 1835.
[414] A. Zwart; Clin. Chem. 39, 1993, 1570.
[415] F. Darain, P. Yager, K.L. Gan, S.C. Tjin; Biosens. Bioelectron. 24, 2009, 1744.
[416] http://dl.cmuj.krakow.pl
[417] K. Takahata, S. Igarashi, Clin. Chim. Acta 283, 1999, 129.
[418] A. Paulus, S. Gijsbertus, H. Rossius, M. Dijk, S. de Vries; J. Biol. Chem. 287, 2012, 8830.
[419] D. Li, X. Zhang, Y. Long, X. Sun; Life Sci. J. 3, 2006, 52.
[420] C. Giulivi, K.J.A. Davies; J. Biol. Chem. 265, 1990, 19453.
[421] L. Gebicka, E. Banasiak; Acta Biochim. Pol. 56, 2009, 509.
[422] A.M. Nowicka, A. Kowalczyk, M. Bystrzejewski, M. Donten, Z. Stojek; Electrochem.
Commun. 20, 2012, 4.
[423] G.J. Brug, A.L.G. Van Den Eden, M. Sluyters-Rehbach, J.H. Sluyters; J. Electroanal. Chem.
176, 1984, 275.
[424] J. Deinum, T. Vänngård; Biochim. Biophys. Acta 310, 1973, 321.
[425] W. Norde, F. MacRitchie, G. Nowicka, J. Lyklema; J. Colloid Interface Sci. 112, 1986, 447.
[426] W. Norde; Colloids Surf. B 61, 2008, 1.
[427] W. Norde, J. Lyklema; Adv. Colloid Interface Sci. 179, 2012, 5.
[428] L.A. Currie; Anal. Chim. Acta 391, 1999, 127.
[429] A. Hubaux, G. Vos; Anal. Chem. 42, 1970, 849.
[430] K. Haberska, C. Vaz-Domínguez, A.L. De Lacey, M. Dagys, C.T. Reimann, S. Shleev;
Bioelectrochemistry 76, 2009, 34.
[431] A.M. Yates, S.J. Elvin, D.E. Williamson; J. Immunoass. Immunochem. 20, 1999, 31.
[432] R. Akter, B. Jeong, J.-S. Choi, Md.A. Rahman; Biosens. Bioelectron. 80, 2016, 123.
[433] D. Dan, X. Xiaoxing, W. Shengfu, Z. Aidong; Talanta 71, 2007, 1257. [434] S.Q. Hu, Z.Y. Wu, Y.M. Zhou, Z.X. Cao, G.L. Shen, R.Q. Yu; Anal. Chim. Acta 458, 2002,
297.
[435] T. Yin, W. Wei, L. Yang, X. Gao, Y. Gao; Sens. Actuators, B 117, 2006, 286.
[436] E. Arkan, R. Saber, Z. Karimi, M. Shamsipur; Anal. Chim. Acta 874, 2015, 66.
[437] M. Stobiecka, A. Chalupa, B. Dworakowska; Biosens. Bioelectron. 84, 2016, 37.
[438] M.C. Garnett; Adv. Drug Deliv. Rev. 53, 2001, 171.
[439] E. Brynda; „Methods for attachment of antibodies onto optical biosensors”, w: „Optical
Chemical Sensors”, F. Baldini (Eds), Springer, New York, 2006.
[440] P. Aisen, A. Leibman, J. Zweier; J. Biol. Chem. 253, 1978, 1930.
[441] J. Zhang, Y. Sun, B. Xu, H. Zhang, Y. Gao, H. Zhang, D. Song; Biosens. Bioelectron. 45,
2013, 230.
[442] E. Prusak-Sochaczewski, J.H.T. Luong; J. Anal. Chem. 23, 1990, 183.
[443] X. Le Guevel, N. Daum, M. Schneider; Nanotechnology 22, 2011, 275103.
[444] T. Yin, W. Wei, L. Yang, X. Gao, Y. Gao; Sens. Actuators, B 117, 2006, 286.
[445] C.-X. Lei, J. Wu, H. Wang, G.-L. Shen, R.-Q. Yu; Talanta 63, 2004, 469.
[446] Z. Liu, S. Huang, D.C. Jiang, B.H. Liu, J.L. Kong; Anal. Lett. 37, 2004, 2283.
[447] M.C. Powanda, F.B. Abeles, K.A. Bostian, J.P. Fowler, E.C. Hauer; Biochem. J. 178, 1979,
633.
[448] Y. Cheng, O. Zak, P. Aisen, S.C. Harrison, T. Walz; Cell 116, 2004, 565.
[449] J. Wall, P.J. Halbrooks, C. Vonrhein, M.A. Rould, S.J. Everse, A.B. Mason, S.K. Buchanan;
J. Biol. Chem. 281, 2006, 24934.
[450] A. Bhattacharya, S. Chatterjee, V. Khorwal, T.K. Mukherjee; Phys. Chem. Chem. Phys. 18,
2016, 5148.
[451] M. Mahmoudi, M.A. Shokrgozar, S. Sardari, M.K. Moghadam, H. Vali, S. Laurent, P. Stroeve;
Nanoscale 3, 2011, 1127.

Bibliografia
227
[452] J. Banker; Biochim. Biophys. Acta 1120, 1992, 123.
[453] I.P. Grudzinski, M. Bystrzejewski, M.A. Cywinska, A. Kosmider, M. Poplawska,
A. Cieszanowski, A. Ostrowska; J. Nanopart. Res. 15, 2013, 1835.
[454] J. Dubach, B.J. Gaffney, K. More, G.R. Eaton, S.S. Eaton; Biophys. J. 59, 1991, 1091.
[455] O.P. Ivakhnenko, D.K. Potter; Phys. Chem. Earth 29, 2004, 899D.
[456] F. Höök, M. Rodahl, P. Brzezinski, B. Kasemo; Langmuir 14, 1998, 729.
[457] E. Matysiak, A.M. Nowicka, B. Wagner, M. Donten; Electrochim. Acta 191, 2016, 586.
[458] A.J. Kariuki-Downard; Electroanal. 12, 2000, 1085.
[459] M. Damar, R. Hitami, J. Pison, J.M. Saveant; J. Am. Chem. Soc. 114, 1992, 5883.
[460] J. Lyskawa, D. Belanger; Chem. Mater. 18, 2006, 4755.
[461] J. Pinson, F. Podvorica; Chem. Soc. Rev. 34, 2005, 249.
[462] H. Uetsuka, D. Shin, N. Tokuda, K. Saeki, C.E. Nobel; Langmuir 23, 2007, 3466.
[463] A. Adenier, E. Cabet-Deliry, A. Chausse, S. Griveau, F. Mercier, J. Pinson, C. Vautrin-Ul;
Chem. Mater. 17, 2005, 491.
[464] J. Agullo, S. Canesi, F. Schaper, M. Morin, D. Belanger; Langmuir 28, 2012, 4889.
[465] P. Allongue, M. Delawar, B. Debat, O. Fageboume, R. Hitmi, J. Pinson, J.M. Saveat; J. Am.
Chem. Soc. 119, 1997, 201.
[466] A. Berisha, C. Combellas, F. Kanoufi, J. Pinson, S. Ustaze, F.J. Podvorica; Chem. Mater. 22,
2010, 2962.
[467] A.M. Nowicka, M. Fau, A. Kowalczyk, M. Strawski, Z. Stojek; Electrochim. Acta 126, 2014,
11.
[468] M. Fau, A. Kowalczyk, P. Olejnik, A.M. Nowicka; Anal. Chem. 83, 2011, 9281.
[469] A. Kowalczyk, M. Fau, A.M. Nowicka, Z. Stojek; Electroanal. 24, 2012, 2053.
[470] G.T. Hermanson; „Bioconjugate Techniques”, Academic Press is an imprint of Elsevier,
Amsterdam, 2013.
[471] D.H. Parry, M. Worwood, A. Jacobs; Br. Med. J. 1, 1975, 245.
Related Documents











