HAL Id: tel-03008820 https://tel.archives-ouvertes.fr/tel-03008820 Submitted on 17 Nov 2020 HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci- entific research documents, whether they are pub- lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers. L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés. Etude des mécanismes cellulaires et moléculaires des leucémies pédiatriques à mauvais pronostic présentant la fusion ETO2-GLIS2 Cecile Lopez To cite this version: Cecile Lopez. Etude des mécanismes cellulaires et moléculaires des leucémies pédiatriques à mauvais pronostic présentant la fusion ETO2-GLIS2. Hématologie. Université Paris Saclay (COmUE), 2018. Français. NNT : 2018SACLS413. tel-03008820

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

HAL Id: tel-03008820https://tel.archives-ouvertes.fr/tel-03008820
Submitted on 17 Nov 2020
HAL is a multi-disciplinary open accessarchive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come fromteaching and research institutions in France orabroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, estdestinée au dépôt et à la diffusion de documentsscientifiques de niveau recherche, publiés ou non,émanant des établissements d’enseignement et derecherche français ou étrangers, des laboratoirespublics ou privés.
Etude des mécanismes cellulaires et moléculaires desleucémies pédiatriques à mauvais pronostic présentant la
fusion ETO2-GLIS2Cecile Lopez
To cite this version:Cecile Lopez. Etude des mécanismes cellulaires et moléculaires des leucémies pédiatriques à mauvaispronostic présentant la fusion ETO2-GLIS2. Hématologie. Université Paris Saclay (COmUE), 2018.Français. �NNT : 2018SACLS413�. �tel-03008820�

Etude des mécanismes cellulaires et moléculaires des leucémies à mauvais pronostic présentant la
fusion ETO2-GLIS2
Thèse de doctorat de l'Université Paris-Saclay Préparée à l’université Paris-Saclay
École doctorale n°582 cancérologie : biologie, médecine, santé (CBMS) Spécialité de doctorat: Aspects moléculaires et cellulaires de la biologie
Thèse présentée et soutenue à Villejuif, le 16 novembre 2018, par
Cécile Lopez
Composition du Jury :
Mr Thierry Jaffredo Directeur de recherche, INSERM (U1156) Président
Mme Véronique Maguer-satta Directrice de recherche, INSERM (U1052) Rapporteur
Mr Eric Delabesse Professeur, Université de Toulouse Rapporteur
Mme Marie-laure Arcangeli Chargée de recherche, INSERM (UMR967) Examinateur
Mme Leïla Maouche-chretien Chargée de recherche, INSERM (U1163) Examinateur
Mr Thomas Mercher Directeur de recherche, INSERM (U1170) Directeur de thèse
Mr Olivier Bernard Directeur de recherche, INSERM (U1170) Co-directeur de thèse
NN
T :
20
18
SA
CLS
413

2

3
REMERCIEMENTS
Je tiens tout d’abord à remercier la Cancéropole Ile de France et la Fondation Recherche
Médicale d’avoir financé cette thèse. Merci à Véronique Maguer-satta et Eric Delabesse
d’avoir accepté d’évaluer ce travail, ainsi qu’à Marie-Laure Arcangeli, Leïla Maouche-
Chretien et Thierry Jaffredo pour faire partie de mon jury de thèse.
À Thomas, mon directeur de thèse, je tiens à te remercier pour ces 9 années passées dans
ton équipe. Merci pour les premières années en tant qu’ingénieur, j’ai adoré travailler avec
toi, Clarisse, Vincianne, Paola et le reste de l’équipe. Tu m’as fait découvrir le monde de
l’hématologie avec beaucoup de patience et de pédagogie. L’experte souris qui n’en avait
jamais touché une à bien grandit. Merci aussi de m’avoir poussé plus loin, et notamment
jusqu’à cette thèse. Tu m’as toujours chalengée et c’était très stimulant. J’ai adoré nos
discussions scientifiques aussi. Et surtout merci d’avoir composé avec mon caractère, hum,
doux et tellement facile à vivre …
À Olivier, mon deuxième directeur de thèse, merci de m’avoir permis de réaliser cette thèse
au sein de ton équipe et pour tes conseils.
À Jurg Schwaller, en premier lieu je tiens à te remercier pour le modèle de la fusion ETO2-
GLIS2 qui a été au cœur de ce travail et qui m’a permis de répondre à bon nombre de
questions. Merci aussi pour tous les échanges que nous avons pu avoir et qui m’ont toujours
beaucoup apporté. Et enfin, merci pour toutes les analyses d’histologies et de cytospins que
j’ai pu t’envoyer (et il y en a eu des colis fedex !!!). Ton expertise a été d’une aide précieuse !
La recherche est une grande aventure prenante, passionnante et pas toujours évidente.
Mais elle m’a permis de rencontrer des personnes incroyables au cours de ces 9 années :
mon boubou (alias Clarisse), Vinciane, Paola, Laurianne (mon cerveau externe), Aline,
Carrèle, Morgane, Elodie, Lucile, Claire, Enguerran, Damien, Dom et Olive merci pour tous
ces moments partagés à la paillasse et ailleurs.

4
Merci à tous les membres de l’équipe Erythro-Méga-Lympho. À Sébastien, je te remercie
pour ta disponibilité et toutes les discussions scientifiques que nous avons pu échanger
avant ton départ pour le pays des kangourous.
À Zak, un énorme merci ! J’ai adoré (et j’adore encore !) maniper avec toi. Tu es toujours
prête à aider tout le monde et c’est un vrai plaisir de t’avoir dans l’équipe. Merci pour ta
bonne humeur, toute l’aide que tu m’as apportée et tous les bons moments partagés. Merci
aussi pour tous les jeux de mot volontaire et involontaire qui ont mené à de cocasses
situations et à de nombreux fous rires. Tu es quelqu’un de génial. :)
À Anouch (et Robert), ma chouchou, Madame Prout-Prout, ça a été un vrai plaisir de
partager toutes ces pauses en ta compagnie, de te voir évoluer et grandir entre ton M2 et ta
quatrième année de thèse. Tu es un petit rayon de soleil, ne change pas ! Plein de bisous et
de bon courage pour la suite.
À Alex, Laetitia, Fabien, Julie et Muriel, merci pour tous ces moments passés avec vous. Les
réunions d’équipes et repas sont rarement mornes et silencieux en votre compagnie. Et un
énorme merci pour toute l’aide apportée : relecture, discussion, partage d’information, aide
de manip mais aussi pour les bons moments : gâteaux, apéro et fous rires.
Aux bio-informaticiens, Kadija et Esteve (prononcé « Steba », aller comprendre…), qui ont
énormément contribués à l’avancé des projets. Merci pour toute l’aide apportée. Kadija
merci pour ta disponibilité et ta pédagogie. La compréhension de la bioinformatique sous
ton aile a été précieuse !!!
Merci aux différents membres de l’unité U1170 avec une mention particulière pour véro.
Ma véro, tellement de chose à te dire que je ne sais par où commencer. J’ai adoré partager le
bureau avec toi pendant toutes ces années. Tu es toujours disponible et de bonne humeur.
Merci pour tous tes conseils et tous ces moments inoubliables. Je nous reverrais toujours
nous lançant « bille en tête » dans des analyses FACS alors qu’aucune de nous deux ne
savais l’utiliser (merci à William et Philippe de nous avoir sauvées du naufrage !) ou encore
en injectant des cellules à des souris qui n’étaient pas les nôtres… Vive l’isoflurane !
À Paule, merci pour tout ce que tu fais pour nous tous. Tu es la meilleure !!

5
A Yann, merci pour toutes ces heures passées avec toi devant l’ARIA. Grâce à toi j’ai appris à
compenser même avec de la Cherry, du PE (« P » « I » comme dirait ton amie M), du Pc7 et
du PercP5.5 ! Finger in the nose !!! À Valou, Madame Super Formatrice IVIS ! Merci !
Toujours disponible pour répondre aux milles questions ou faire des tests au pied levé (pas
toujours fructueux, la GFP ne se détecte pas dans les C57bl6 ! maintenant je le sais !). À
Karine, tu m’auras finalement appris à mettre une blouse pour maniper… Merci à vous trois
pour votre soutient, vos encouragements et toutes les discussions sur le perron de l’IGR (je
ne regarde plus les chats, les chiens et les canards de la même façon).
À toute l’équipe du PFEP, merci pour le bien être apporté aux petites pensionnaires et pour
votre aide.
À mes amis, (enfance, collège, lycée, FAC, colo…), merci à tous d’avoir été présent malgré
mon « hibernage » de ces 4 dernières années. J’ai une chance folle de vous avoir auprès de
moi et j’espère bien que l’on partagera encore longtemps les évènements importants
(mariage, naissance…) et les non moins importants (soirée crêpe annuelle, soirée jeux,
dégustation de vin…).
À ma famille, ma sœur, mon grand-père, ma mamie, mes tontons, tatas, cousines et pièces
rapportées, merci pour tous ces moments passés en famille. Nous avons la chance d’avoir
une famille unie et c’est toujours un plaisir de vous retrouver.
À mes neveux d’amour, j’ai eu la chance d’être tata deux fois pendant cette thèse et je ne
saurais vous dire à quel point cela a été une joie immense (Sébastien pourra en témoigner).
C’est un plaisir de vous voir grandir. Vous êtes mes rayons de soleil.
À mes parents, je tiens à vous remercier pour tout le soutien inconditionnel que vous
m’avez apporté. Cette belle aventure n’aurait pas pu avoir lieu sans vous deux. Vous m’avez
toujours épaulé et encouragé tout au long de ces 4 années (et bien plus encore) de la
meilleur de façon. Merci pour tout. Je vous aime énormément.

6
En guise de préface, voici une discussion que j’ai eue avec mon grand-père sur mon
orientation professionnelle…
Et maintenant laissons place à la science et aux « oncle Eugène » (oncogènes)…

7
TABLE DES MATIERES
INDEX DES FIGURES ET DES TABLEAUX ...................................................................................... 11
INTRODUCTION .................................................................................................................................... 14
I/ Hématopoïèse : ...................................................................................................................................... 15
A/ Ontogenèse du système hématopoïétique : .......................................................................... 15
1/ Les premières cellules hématopoïétiques dites « primitives » : .............................. 16
2/ Les cellules hématopoïétiques dites « définitives » transitoires : ........................... 17
3/ Les cellules souches hématopoïétiques HSC : ................................................................. 18
a/ Émergence des HSC : ............................................................................................................ 18
b/ Amplification des HSC : ....................................................................................................... 19
B/ Organisation de l’hématopoïèse adulte .................................................................................. 20
1/ Caractéristiques cellulaires des cellules souches hématopoïétiques adultes ..... 21
a/ Différenciation : ...................................................................................................................... 21
b/ Autorenouvellement : .......................................................................................................... 22
c/ Quiescence : .............................................................................................................................. 23
2/ Modèles de hiérarchie hématopoïétique ........................................................................... 23
a/ Modèle (pré)historique : ..................................................................................................... 24
b/ Vers des modèles de continuum : .................................................................................... 25
3/ Myélopoïèse : ............................................................................................................................... 28
a/ Monocyte et macrophage : ................................................................................................. 28
b/ Les cellules dendritiques : .................................................................................................. 29
c/ Granulocytes : .......................................................................................................................... 29

8
4/ Mégacaryopoïèse : ...................................................................................................................... 30
a/ Généralités :.............................................................................................................................. 30
b/ Origine des progéniteurs mégacaryocytaires ............................................................. 31
c/ Maturation des mégacaryocytes ...................................................................................... 32
d/ Différence fœtal et adulte ................................................................................................... 35
C/ Régulation de l’hématopoïèse : .................................................................................................. 36
1/ La niche hématopoïétique : .................................................................................................... 36
2/ Communication entre HSC et cellules de la niche : les voies de signalisation .... 38
a/ Contact cellule-cellule, exemple de la voie Notch : ................................................... 39
b/ Facteur soluble et récepteur, exemple de la TPO :.................................................... 39
c/ Exemple de la voie Sonic Hedgehog ................................................................................ 41
3/ Régulation transcriptionnelle : ............................................................................................. 41
a/ Facteur de transcription et complexe transcriptionnel .......................................... 42
b/ Organisation chromatinienne ........................................................................................... 46
II/ Hémopathies malignes ...................................................................................................................... 51
A / Les leucémies aiguës myéloïdes : ............................................................................................ 51
1/ Classification des LAM .............................................................................................................. 51
2/ Anomalies génétiques des LAM ............................................................................................ 52
a/ Types d’altérations génétiques ......................................................................................... 53
b/ Conséquences fonctionnelles des mutations .............................................................. 54
c/ Coopération oncogénique et évolution clonale .......................................................... 56
3/ Les cellules souches leucémiques ........................................................................................ 58
a/ Propriétés des LSC ................................................................................................................. 58

9
b/ Origine cellulaire .................................................................................................................... 59
4/ Différence entre leucémies pédiatriques et leucémies de l’adulte .......................... 60
a/ Différence de répartition des sous-types de LAM et génétique ........................... 61
b/ Origine in utero des leucémies pédiatriques ............................................................... 62
B/ Les LAM-M7 pédiatriques ............................................................................................................ 63
1/ Présentation clinique et traitements .................................................................................. 64
2/ Génétique des LAM-M7 pédiatriques ................................................................................. 65
a/ LAM-M7 associées aux trisomies 21 constitutionnelles ......................................... 66
b/ LAM-M7 de novo ..................................................................................................................... 67
3/ Modélisation fonctionnelle des LAM-M7 .......................................................................... 71
a/ LAM7 associées aux trisomies 21 constitutionnelles : ............................................ 71
b/ Fusion OTT-MAL : .................................................................................................................. 72
c/ Fusion NUP98-KDM5A :....................................................................................................... 73
d/ Fusions MLL ou HOX : .......................................................................................................... 73
3/ La fusion ETO2-GLIS2 ............................................................................................................... 74
RESULTATS ............................................................................................................................................ 78
CONTEXTE ET OBJECTIFS ................................................................................................... 79
ARTICLE 1 .................................................................................................................................. 86
ARTICLE 2 ............................................................................................................................... 112
DISCUSSION .......................................................................................................................................... 158
LISTE DES ABREVIATIONS .............................................................................................................. 170
ANNEXES ............................................................................................................................................... 174
Pediatric Acute Megakaryoblastic Leukemia : Multitasking Fusion Proteins
and Oncogenic Cooperations ........................................................................................... 174

10
Molecular pathways driven by ETO2-GLIS2 in aggressive pediatric leukemia
..................................................................................................................................................... 188
Leucémies à mégacaryoblastes de l’enfant : une affaire de complexes .......... 192
REFERENCES BIBLIOGRAPHIQUES .............................................................................................. 213

11
INDEX DES FIGURES ET DES TABLEAUX
Figure 1: Apparition spatiale et temporelle des cellules hématopoïétiques pendant le
développement murin et humain. ....................................................................................................... 16
Figure 2: Production des cellules hématopoïétiques au cours du développement
embryonnaire murin. ............................................................................................................................... 17
Figure 3 : Représentation schématique du mécanisme EHT lors de la formation des HSC. .. 19
Figure 4: Les stades de différenciation des HSC. ..................................................................................... 21
Figure 5: Modèles de l’hématopoïèse. ......................................................................................................... 25
Figure 6 : Évolution du modèle hématopoïétique. ................................................................................. 27
Figure 7 : La mégacaryopoïèse ....................................................................................................................... 31
Figure 8 : Processus d’endomitose lors de la mégacaryopoïèse. ...................................................... 33
Figure 9 : Processus de mégacaryopoïèse. ................................................................................................ 34
Figure 10 : Différenciation hématopoïétique fœtale et adulte. .......................................................... 35
Figure 11 : Organisation spatiale des cellules dans la moelle osseuse. .......................................... 37
Figure 12 : Voie de signalisation de la TPO. .............................................................................................. 40
Figure 13 : Activation de la transcription par les facteurs de transcriptions. ............................. 43
Figure 14 : Régulation de la transcription via les complexes transcriptionnels. ........................ 45
Figure 15 : Les différents niveaux de condensation de la chromatine. .......................................... 46
Figure 16 : Régulation de la transcription par des modifications épigénétiques. ...................... 47
Figure 17 : Le complexe de cohésines est un régulateur-clé de la transcription et de la
formation de boucle chromatinienne. ................................................................................................ 50

12
Figure 18 : Exemple d’évolution clonale de leucémie aiguë promyélocytaire (LAM-M3). ..... 57
Figure 19 : Répartition des sous-groupes de LAM en fonction de l’âge. ........................................ 61
Figure 20 : Modèle de transformation par la fusion TEL-AML1 chez des jumeaux. .................. 63
Figure 21 : Survie Globale (OS) d’une cohorte française de LAM pédiatriques. ......................... 65
Figure 22 : Altérations génétiques entre les LAM-M7 pédiatriques et adultes. .......................... 65
Figure 23 : Développement leucémique des DS-LAM-M7. .................................................................. 67
Figure 24 : Mutations additionnelles des LAM-M7. ............................................................................... 69
Figure 25 : Représentation schématique des gènes ETO2 et GLIS2 et des transcrits de fusion.
........................................................................................................................................................................... 75
Figure 26 : Influence de l’ontogenèse hématopoïétique sur le phénotype des leucémies. .. 167
Tableau 1 : Combinaison de marqueurs de surface pour identifier les progéniteurs
hématopoïétiques murins et humains. .............................................................................................. 26
Tableau 2 : Fréquence des altérations génétiques des LAM-M7 de novo. ..................................... 68
Tableau 3 : Modèles de LAM-M7 pédiatriques. ........................................................................................ 72

13

14
INTRODUCTION

15
I/ Hématopoïèse :
Le système sanguin est composé de cellules matures circulantes qui assurent certaines
fonctions indispensables au bon fonctionnement de l’organisme incluant le transport de
l’oxygène par les érythrocytes, la prévention des hémorragies par les plaquettes, ainsi que
la lutte contre les infections par les cellules myéloïdes (monocytes/macrophages et
neutrophiles) et les lymphocytes. Ces cellules matures ont une durée de vie généralement
courte, il est donc nécessaire de les renouveler tout au long de la vie. Chez l’Homme entre
1010 et 1012 cellules sanguines sont produites par jour par un processus très régulé connu
sous le nom d’hématopoïèse.
Les différents travaux de James Till et Ernest McCulloch ont permis la découverte d’une
cellule immature, appelée cellule souche hématopoïétique (HSC), présente dans la moelle
osseuse et ayant la capacité de former les cellules matures composant le sang (1). Depuis,
l’hématopoïèse est généralement décrite comme une hiérarchie avec au sommet les cellules
souches hématopoïétiques (HSC) capables de maintenir la production des différentes
cellules sanguines tout au long de la vie de l’individu.
A/ Ontogenèse du système hématopoïétique :
Les cellules hématopoïétiques apparaissent au cours de l’embryogenèse en plusieurs
vagues spatiales (différents organes comme le sac vitellin, le foie ou encore la moelle
osseuse) et temporelles (Figure 1). Par la suite, les HSC colonisent la moelle osseuse qui
représente le siège de l'hématopoïèse chez l'adulte. L’hématopoïèse embryonnaire a
principalement été étudiée dans des modèles animaux (murin, poisson zèbre…) à cause des
difficultés d'accès aux étapes précoces du développement humain. Les observations
obtenues dans le modèle murin seront principalement décrites ici.

16
Figure 1: Apparition spatiale et temporelle des cellules hématopoïétiques pendant le développement
murin et humain. Développement de l’hématopoïèse murine (A) et humaine (B). (Issue (2)).
1/ Les premières cellules hématopoïétiques dites « primitives » :
Les premières cellules sanguines sont détectées dans le sac vitellin au jour E7-7,5 du
développement murin (Figure 2), avant l’apparition de la circulation sanguine qui a lieu à
E8,5 (3). Les érythroblastes, les macrophages (3) et les mégacaryocytes (4) observés
descendraient d’un progéniteur commun aux cellules endothéliales, l’« hémangioblaste »
(5,6). Cette première vague hématopoïétique transitoire est également nommée
«primitive » Pour revue (7) car les cellules présentent des caractéristiques différentes des
cellules synthétisées lors de l’hématopoïèse adulte appelée « définitive ».
Ainsi, alors que les érythrocytes adultes sont énuclées, les érythroblastes primitifs
présentent un noyau et l’expression d’une hémoglobine fœtale (2) ; Pour revue (8).
Il est généralement admis que cette hématopoïèse primitive participe au développement de
l’embryon en oxygénant et en phagocytant les cellules durant la formation des tissus par les
érythrocytes et les macrophages, respectivement. Les mégacaryocytes assurent la
production de plaquettes qui participent à l'homéostasie du système vasculaire sanguin. De
plus, les macrophages participent à la protection l’embryon contre les infections.
Les cellules hématopoïétiques primitives sont amplifiées entre E7 et E8,25 et ne sont plus
détectées vers E9.

17
2/ Les cellules hématopoïétiques dites « définitives » transitoires :
Chez l’embryon, une deuxième vague de cellules sanguines transitoires dérivant de
progéniteurs érythro-myéloïde et lymphoïde est décrite avant l’émergence des cellules
souches hématopoïétiques (HSC) qui présentent une capacité de reconstitution
multipotente à long-terme.
À un stade de développement précoce vers E8,25, les cellules de la lignée myéloïde
dérivent d’un progéniteur bipotent EMP (Erythro-Myeloid Progeniteur) qui émerge dans le
sac vitellin (3). Ce progéniteur prolifère puis migre dans le foie fœtal vers E10,5 pour
produire les cellules matures érythrocytes, mégacaryocytes, et les différentes cellules
myéloïdes (macrophages, neutrophiles, basophiles, éosinophiles). Les cellules EMP
expriment les marqueurs de surface Kit, CD41 puis CD16/32 vers E9,5 (Figure 2, (9)).
Figure 2: Production des cellules hématopoïétiques au cours du développement embryonnaire murin. Les premières cellules hématopoïétiques composées d’érythrocytes, de mégacaryocytes et de macrophages apparaissent au stade E7-7,5 dans le sac vitellin. Par la suite les progéniteurs EMP (Erythro-myeloid Progenitors) et lymphoïdes (B-Lymphoid) se développent vers E8,25-8,5 dans le sac vitellin et la région de l’AGM (Aorte-Gonade-Mésonéphros) avant la formation des HSC au stade E10,5. (Issue de (9)).

18
La greffe de ces progéniteurs EMP dans des souris hôtes irradiées, confirme la capacité de
différenciation de ces progéniteurs vers les lignées myéloïdes et non vers les lignées
lymphoïdes. Ces progéniteurs ne semblent pas posséder de potentiel de reconstitution à
long terme dans ces analyses.
Une hypothèse récente suppose que les macrophages résidents des tissus de l’adulte
proviennent des progéniteurs EMP du sac vitellin (10).
La lignée lymphoïde est observée entre les stades E8,5 et E9,5 du développement
embryonnaire murin avec la présence des progéniteurs lymphoïdes dans le sac vitellin et la
région AGM (Aorte-Gonade-Mésonéphros) (11–13).
Un progéniteur pouvant se différencier vers les lignées myéloïdes (granulocytes et
monocytes) et lymphoïdes est aussi observé au stade E9,5 dans le sac vitellin (14).
La formation de ces cellules spécifiques du système immunitaire permet de protéger
l’embryon des pathogènes.
3/ Les cellules souches hématopoïétiques HSC :
Les HSC sont définies par leur capacité de reconstitution hématopoïétique à long terme de
toutes les cellules matures. Historiquement, les cellules de différents organes à différents
temps gestationnels ont donc été testées pour leur capacité de reconstitution lors d'une
greffe dans des animaux receveurs irradiés. Le développement d'anticorps spécifiquement
dirigés contre des protéines de surface, utilisées comme marqueurs cellulaires, a permis la
caractérisation phénotypique et fonctionnelle des HSC au cours du développement
embryonnaire ainsi que chez l’adulte.
a/ Émergence des HSC :
Les premières cellules souches hématopoïétiques sont détectées au stade E10,5 dans des
foyers de cellules hématopoïétiques attachés à l’endothélium de l’aorte de la région AGM
(Figure 3, (15,16)). Comme dans le cas de l'hémangioblaste, les premières HSC définitives

19
dérivent de cellules endothéliales embryonnaires présentant un potentiel hémogénique,
appelées les cellules endothéliales hémogéniques (HEC, hematopoietic endothelial cell).
Seulement certaines cellules endothéliales formant l’aorte sont des HEC qui présentent des
marqueurs endothéliaux (CD31) mais aussi hématopoïétiques (Sca1). Les HEC subissent
une transition morphologique et moléculaire, passant d’une forme endothéliale aplatie à
une cellule hématopoïétique ronde exprimant les marqueurs CD31+ Sca1+ Kit+ CD41+. Ce
mécanisme de transition d’une cellule endothéliale vers une cellule hématopoïétique est
appelé EHT (Endothelium to Hematopoietic Transition) (17,18), pour revue (7) et est un
processus conservé entre les espèces (19–21).
Plusieurs foyers de cellules hématopoïétiques sont observés à E10,5 au niveau de l’aorte
mais seulement quelques-uns semblent présenter des HSC fonctionnelles (22).
Figure 3 : Représentation schématique du mécanisme EHT lors de la formation des HSC.
Un sous-groupe de cellules endothéliales composant l’aorte, appelées cellules endothéliales hémogéniques (HE), peuvent subir un mécanisme de transdifférenciation pour former un foyer de cellules hématopoïétiques (IAHC) composé de cellules souches hématopoïétiques (HSC). (Issue de (7)).
Les HSC sont aussi détectées à E10,5 dans d’autres lieux comme le sac vitellin et le placenta
avant leur apparition dans la circulation sanguine supposant qu’un mécanisme similaire a
lieu dans différents territoires embryonnaires (23,24).
b/ Amplification des HSC :
Les HSC formées dans les différents sites colonisent ensuite le foie fœtal à partir de E11 et y
prolifèrent entre E12 et E16 (25).
Vers E15,5 les HSC migrent vers la rate, qui est un organe hématopoïétique important chez
la souris contrairement à l’Homme, pour s’y amplifier (26,27). A partir de E17,5 les HSC

20
sont aussi détectées dans la moelle osseuse où elles résideront tout le reste de la vie adulte
Pour revues (28,29).
Les HSC fœtales sont définies par leur capacité de prolifération importante (nécessaire pour
la mise en place du système hématopoïétique adulte), contrairement aux HSC adultes qui
assurent l’homéostasie. De même les HSC fœtales présentent un cycle cellulaire actif alors
que les HSC adultes sont majoritairement quiescentes (30). Ces caractéristiques permettent
une reconstitution de l’hématopoïèse plus rapide à partir des HSC fœtales lors de greffes
compétitives (31,32). Ces capacités de prolifération sont modifiées entre 3 et 4 semaines
après la naissance ou post greffe, une fois que le système hématopoïétique est mis en place,
pour adopter les caractéristiques des HSC adultes (32).
Les HSC peuvent être enrichies par cytométrie en flux sur la base de l’absence de certains
marqueurs de surface présents sur les cellules matures et de la présence d'une combinaison
d’autres marqueurs comme Sca1+ Kit+ CD45+ CD11b+ CD48- CD150+ Pour revue (33).
L’expression de certains marqueurs de surface comme CD11b et CD41, exprimés à la
surface des HSC fœtales va être modifiée durant l’acquisition des caractéristiques des HSC
adultes.
B/ Organisation de l’hématopoïèse adulte
La fonction de l’hématopoïèse adulte est la production en continu des cellules matures pour
assurer le maintien de l’homéostasie à la fois dans des conditions normales ou de stress (ex:
infections).
Historiquement, les cellules hématopoïétiques ont été classées en 4 groupes en fonction de
leur capacité d’autorenouvellement, de prolifération et de leur état de différenciation: 1-les
HSC, cellules immatures ayant une capacité d’autorenouvellement importante mais une
prolifération faible, 2-les progéniteurs hématopoïétiques (HPC) ayant une capacité
proliférative importante et pouvant se différencier vers plusieurs lignées différentes, 3-les
précurseurs, cellules immatures pouvant être identifiées sur leur morphologie et perdant
progressivement leurs capacités prolifératives pour donner les 4-cellules matures libérées
dans la circulation sanguine Pour revue (34). Le concept de cellule souche a développé la

21
vision de l’hématopoïèse en organisation hiérarchique où la HSC se différencierait en HPC
puis en précurseurs jusqu’aux cellules matures en une série d’étapes définies pour produire
la grande quantité de cellules nécessaire par jour. Cette organisation est classiquement
représentée sous la forme d’un « arbre hématopoïétique ».
1/ Caractéristiques cellulaires des cellules souches hématopoïétiques
adultes
Les HSC sont des cellules peu nombreuses (chez la souris, environ 3-4 HSC pour 105 cellules
de la moelle osseuse, pour revue (35)) localisées dans la moelle osseuse, et présentant des
caractéristiques particulières permettant leur maintien et leur différenciation (Figure 4).
L’analyse des HSC est généralement observée dans des souris transplantées pour vérifier
leur capacité d’autorenouvellement et de différenciation.
Figure 4: Les stades de différenciation des HSC. Les HSC quiescentes (phase G0 du cycle cellulaire) peuvent être activées pour entrer dans le cycle cellulaire (phase G1/S/G2/M) et permettre l'autorenouvellement et la différenciation. (Issue de (36)).
a/ Différenciation :
La différenciation cellulaire est le processus de formation d’une cellule spécialisée à partir
d’une cellule immature présentant le même matériel génétique.

22
Les HSC ont la capacité de se différencier vers toutes les cellules spécialisées du sang
incluant les érythrocytes, les mégacaryocytes, les cellules myéloïdes (monocytes,
macrophages, neutrophiles) et les lymphocytes. Les modèles de transplantation ainsi que
de nouveaux modèles d’étude utilisant des souris transgéniques ont contribué à la
compréhension du potentiel de différenciation des cellules souches hématopoïétiques.
Les mécanismes moléculaires contrôlant la différenciation seront plus amplement détaillés
par la suite.
b/ Autorenouvellement :
L’autorenouvellement, qui est la possibilité d’une cellule à se diviser en deux cellules filles
dont au moins une conserve les propriétés de la cellule mère, est une caractéristique des
cellules souches hématopoïétiques permettant de maintenir le pool des HSC constant. Deux
types de divisions peuvent être à l'origine de l'autorenouvellement. Lors d'une division
asymétrique, seulement une des 2 cellules filles conserve les caractéristiques de la cellule
mère alors que la seconde se différencie. Lors d’une division symétrique, les 2 cellules filles
obtenues peuvent présenter les mêmes caractéristiques que la cellule mère, conduisant
ainsi à une amplification des HSC. Dans le cas d’une division symétrique où les 2 cellules
filles obtenues s’engagent dans la différenciation, cela conduit à une diminution du nombre
absolu de HSC.
Plusieurs cellules présentent des capacités d’autorenouvellement à plus ou moins long
terme. Elles ont été classées en fonction de leur durée de reconstitution dans les souris
hôtes: <6 mois (Short Term-HSC, ST-HSC), >6mois (Intermédiaire) et >12 mois (Long Term
HSC, LT-HSC) Pour revues (37,38).
Une étude a pu mettre en évidence la présence d’une population LT-HSC conservant le
marqueur H2B-GFP in vivo sur une période de 22 mois. Cette population effectuerait 4
divisions symétriques pour amplifier le pool des LT-HSC avant d’entrer en phase de
quiescence (39).

23
c/ Quiescence :
Le cycle cellulaire est caractérisé par 4 phases : l’interphase G1, la phase de synthèse S,
l’interphase G2 puis la phase de mitose M. Les cellules peuvent sortir du cycle cellulaire et
entrer en phase G0 aussi appelée quiescence ou encore dormance Pour revue (36). En
utilisant l’expression du Ki67 (marqueur de prolifération) avec un intercalant de l’ADN
(Dapi), il a été montré que 70% des HSC sont en phase G0 du cycle cellulaire (40).
La quiescence est une caractéristique importante des HSC, qui permet de réguler leur
nombre et de les protéger de l’acquisition de mutation Pour revue (41,42). Il a été montré
que la résistance des HSC aux agents antiprolifératifs comme le 5 Fluorouracil (5FU) est due
à cet état quiescent Pour revue (43).
Le cycle cellulaire est un mécanisme fortement régulé, de même que l’entrée ou la sortie en
phase G0. Les cytokines sont des facteurs extrinsèques importants, dont certaines (SCF,
TPO…) ont été impliquées dans le maintien de la phase quiescente des HSC (44). De même,
plusieurs facteurs de transcription (PU.1, CEBPA) ont eux aussi été identifiés comme
régulateurs de la quiescence des HSC. Le facteur de transcription PU.1, en régulant
l’expression d’un certain nombre de régulateurs du cycle cellulaire (GFI1, E2F1, CDK1),
influence le statut du cycle cellulaire des HSC. Effectivement, le modèle murin hypomorphe
pour PU.1 présente une diminution de la quiescence des HSC. Le facteur CEBPA joue un rôle
important sur le changement de statut prolifératif à quiescent observé entre le HSC fœtales
et les HSC adultes, notamment par la régulation du facteur Mycn (45), pour revue (36).
Il a été montré que les HSC quiescentes peuvent être rapidement activées en réponse à
certains stimuli (inflammation, stimulation au G-CSF) puis retourner à leur état quiescent
une fois l’homéostasie rétablie (40,46).
2/ Modèles de hiérarchie hématopoïétique
Les étapes d'engagement précoce des HSC vers les différentes lignées sont généralement
étudiées au sein de la fraction de cellules négatives pour les marqueurs des lignées matures
tel que Ter119 (marquant les érythrocytes), B220 (les lymphocytes B), CD3 (les

24
lymphocytes T), Gr1 et CD11b (les myéloïdes), appelée fraction "lineage-negative" (LIN-).
L'utilisation de différents marqueurs de surface a permis de raffiner progressivement
l'identification de populations enrichies en cellules souches, en progéniteurs ou en
précurseurs. Il est apparu que le pool des HSC lui même est hétérogène sur le plan
fonctionnel et moléculaire Pour revues (47,48). L’utilisation de nouvelles technologies
comme le profil transcriptionnel à l’état unicellulaire, le profil chromatinien et le suivi de
différenciation à l’état homéostatique permet maintenant de suggérer que la différenciation
hématopoïétique est comparable à un continuum de progéniteurs hétérogènes ayant un
potentiel de différenciation flexible pour répondre au besoin de l’organisme (39,49,50).
a/ Modèle (pré)historique :
Les progéniteurs multipotents, présents dans la fraction LIN-Kit+Sca1+ (LSK) constituent
0,05% des cellules de la moelle. Ils sont enrichis en cellules souches et peuvent être divisés
en 3 sous-populations : Les LT-HSC, les ST-HSC et les progéniteurs multipotents (MPP). Le
système hématopoïétique est représenté comme une pyramide où les LT-HSC génèrent les
ST-HSC qui à leur tour vont donner les MPP (51,52). Le premier modèle d’hématopoïèse
décrit implique une première séparation entre les potentiels de différentiation lymphoïde
(Lymphocytes B, T et NK) et myéloïde (monocyte, granulocyte, macrophage, érythrocyte et
mégacaryocyte) avec la caractérisation des progéniteurs lymphoïdes communs (CLP)(53)
et des progéniteurs myéloïdes communs (CMP)(54). Dans ce modèle, les CMP et CLP
présents dans la fraction LIN-Sca1-Kit+ sont les premiers progéniteurs à présenter une
capacité non-multipotente de différenciation. Par la suite, les CMP se différencient en
progéniteurs granulocytaires et macrophagiques (GMP) formant les granulocytes, les
monocytes et les macrophages, et en progéniteurs érythro-mégacaryocytaires (MEP)
formant les érythrocytes et les progéniteurs mégacaryocytaires (MkP) conduisant aux
plaquettes (Figure 5A).

25
Figure 5: Modèles de l’hématopoïèse. A. Modèle hématopoïétique où la séparation des lignées myéloïdes et lymphoïdes s’effectue au niveau
des progéniteurs communs CLP et CMP. (53,54). B. Découverte du progéniteur LMPP présentant un potentiel myéloïde et lymphoïde sans capacité
érythroïde-mégacaryocytaire. (55–57).
b/ Vers des modèles de continuum :
Par la suite, l'utilisation de marqueurs additionnels et d'autres tests fonctionnels ont
conduit à remettre en question la proximité de certaines lignées. Par exemple, les cellules
érythroïdes, mégacaryocytaires et myéloïdes ne semblent pas aussi proches les unes des
autres. L’identification d’un progéniteur LMPP (55) présentant une capacité de
différenciation à la fois lymphoïde et myéloïde (en CLP et GMP sans passer par le
progéniteur CMP) avec une faible capacité de différenciation érythroïde et
mégacaryocytaire (56)(Figure 5B) suggère une séparation précoce entre les lignées
lymphoïdes/myéloïdes d'un côté et les lignées érythro/mégacaryocytaires d'un autre (57).
Dans ce modèle, il a été proposé que les MEP dérivent directement des ST-HSC.
Au cours de la dernière décennie, le compartiment des LSK a été divisé en sous-populations
sur la base de l’expression de marqueurs de surface : CD150, CD34, CD48 et CD135 (40)
associé à une caractérisation fonctionnelle in vivo et in vitro. A ce jour, une nomenclature
impliquant l'existence d'au moins 5 populations est largement utilisée pour réaliser les

26
analyses fonctionnelles. Ainsi, on peut identifier les HSC (LT-HSC), les MPP1 (ST-HSC), les
MPP2, les MPP3 et les MPP4 (aussi appelés LMPP, (55)) (Tableau 1, (58)).
Tableau 1 : Combinaison de marqueurs de surface pour identifier les progéniteurs hématopoïétiques
murins et humains.
Les HSC, MPP1-2 présentent une capacité de différenciation multipotente mais diffèrent sur
leur capacité d’autorenouvellement/reconstitution à long-terme in vivo et de prolifération.
Un biais de différenciation est observé pour les MPP3 vers la lignée myéloïde et pour les
MPP4 vers la lignée lymphoïde.
Au cours des dernières années, les analyses fonctionnelles de "lineage-tracing" in vivo
(49,59), de transplantation à l'état unicellulaire des HSC (60,61) ainsi que les
caractérisations moléculaires à l’état unicellulaire (ex: transcriptome, (50,61,62)) tendent à
montrer que l’engagement des HSC dans la différenciation vers une lignée se ferait de façon
précoce et graduelle et serait associée à un continuum de capacités d’autorenouvellement,
de multipotence et de différenciation Pour revues (47,48).
Ainsi, la notion de bipotentialité des progéniteurs LIN-Sca1+Kit+ au sein des compartiments
CMP, GMP et MEP a été remise en cause. En effet, les progéniteurs CMP semblent en fait être
Murin Humain
LT-HSC LSK+ CD150+CD34-CD48-CD135- LIN-Kit+CD34+CD38-CD90+CD45RA-
MPP1 LSK+ CD150+CD34+CD48-CD135-
LIN-Kit+CD34+CD38-CD90-CD45RA- MPP2 LSK+ CD150+CD34+CD48+CD135-
MPP3 LSK+ CD150-CD34+CD48+CD135-
MPP4 LSK+ CD150-CD34+CD48+CD135+
CLP LIN-Sca1low
Kit+CD135+IL7R+CD34+CD25+ LIN-Kitlow
CD34+CD38+CD90-CD45RA-
CMP LIN-Sca1-Kit+CD16/32low
CD34+ LIN-CD34+CD38+CD90-CD45RA-IL3Rlow
GMP LIN-Sca1-Kit+CD16/32+CD34+CD150- LIN-CD34+CD38+CD90-CD45RA-IL3+
MEP LIN-Sca1-Kit+CD16/32-CD34-CD150+ LIN-CD34+CD38+CD90-CD45RA-IL3-
27
une population hétérogène composée de progéniteurs déjà engagés vers une lignée (soit
érythro-mégacaryocytaire soit myéloïde ; (50,62,63)). De même, les MEP définis dans le
premier modèle présentent un programme transcriptionnel exclusivement érythroïde,
suggérant qu'ils sont en très grande majorité composés des progéniteurs érythroïdes et non
mégacaryocytaires (50). Ces différentes analyses ont suggéré que l’engagement vers une
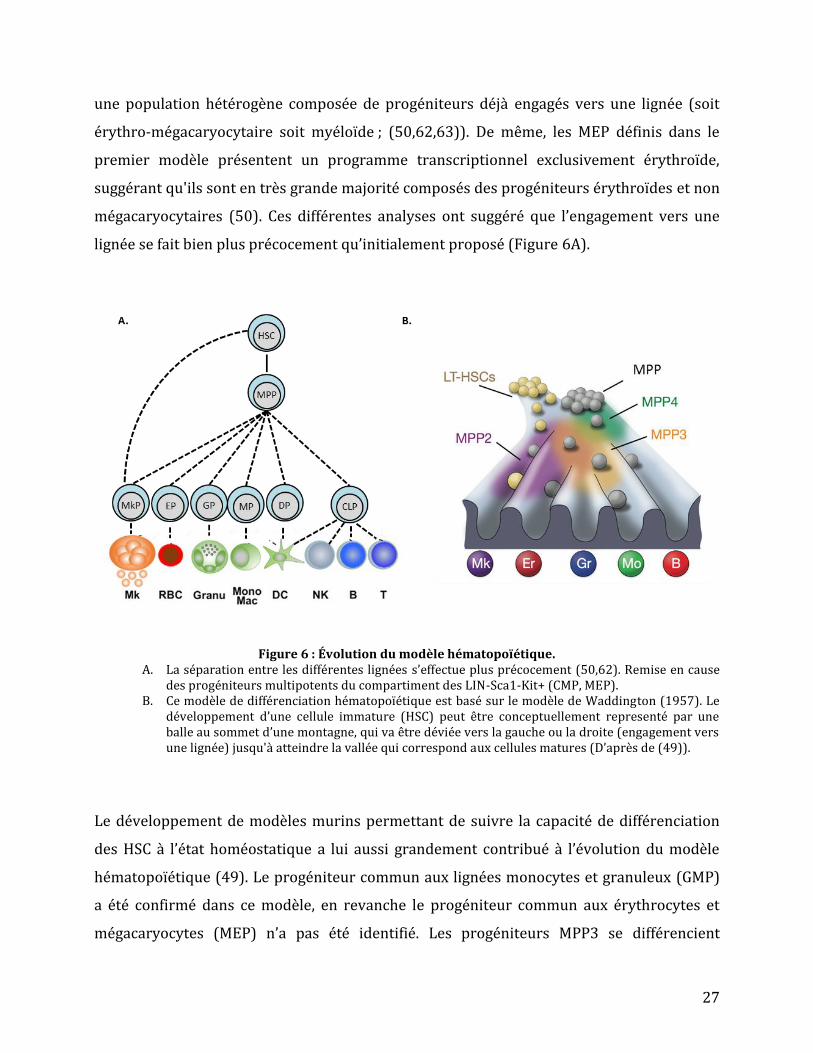
lignée se fait bien plus précocement qu’initialement proposé (Figure 6A).
Figure 6 : Évolution du modèle hématopoïétique.
A. La séparation entre les différentes lignées s’effectue plus précocement (50,62). Remise en cause des progéniteurs multipotents du compartiment des LIN-Sca1-Kit+ (CMP, MEP).
B. Ce modèle de différenciation hématopoïétique est basé sur le modèle de Waddington (1957). Le développement d’une cellule immature (HSC) peut être conceptuellement representé par une balle au sommet d’une montagne, qui va être déviée vers la gauche ou la droite (engagement vers une lignée) jusqu'à atteindre la vallée qui correspond aux cellules matures (D’après de (49)).
Le développement de modèles murins permettant de suivre la capacité de différenciation
des HSC à l’état homéostatique a lui aussi grandement contribué à l’évolution du modèle
hématopoïétique (49). Le progéniteur commun aux lignées monocytes et granuleux (GMP)
a été confirmé dans ce modèle, en revanche le progéniteur commun aux érythrocytes et
mégacaryocytes (MEP) n’a pas été identifié. Les progéniteurs MPP3 se différencient

28
principalement vers les lignées érythroïdes et myéloïdes alors que les MPP4 conservent une
différenciation lympho-érythro-myéloïde. Les MPP2 semblent produire majoritairement la
lignée mégacaryocytaire (63) et conserver une capacité multipotente restreinte. De plus,
dans ce modèle une partie des LT-HSC se différencierait directement en progéniteur
mégacaryocytaire sans passer par les différents intermédiaires du compartiment MPP
(Figure 6B).
3/ Myélopoïèse :
Les cellules myéloïdes sont composées des granulocytes, monocytes, macrophages et
cellules dendritiques jouant un rôle élémentaire dans l’immunité. Elles sont présentes dans
la circulation sanguine, le système lymphatique, certains tissus et sont rapidement
mobilisées par chimiotactisme sur un lieu endommagé ou infecté. A l’état non
inflammatoire, elles participent au développement et à la réparation tissulaire. Toutes ces
cellules dérivent d’un progéniteur commun (49,54). Une séparation entre les granulocytes
et les autres cellules est observée avec les progéniteurs monocyte/macrophage et cellules
dentritiques (MDP) et aux progéniteurs granulocytaires. Les progéniteurs MDP vont donner
lieu aux progéniteurs monocytaires communs (cMoPs) et aux précurseurs communs des
cellules dendritiques (CDP, (64)).
a/ Monocyte et macrophage :
La monopoïèse, régulée principalement par le facteur de transcription PU.1, et les cytokines
M-CSF et IL34, permet la formation des monocytes. Une forte expression de PU.1 va activer
les gènes spécifiques comme IRF8, KLF4, CD115 (récepteur au M-CSF) et inhiber les facteurs
de transcription spécifiques d’autres lignées comme GATA1, GATA2 ou encore CEBPA. Les
monocytes localisés dans la circulation sanguine jouent un rôle essentiel dans
l’inflammation ainsi que la régulation du nombre de certains pathogènes. Ils peuvent
infiltrer les tissus en traversant l’endothélium vasculaire et se différencier en macrophages.
Monocytes et macrophages sont de grosses cellules arrondies présentant un noyau excentré

29
et des vacuoles dans le cytoplasme. Leur rôle est de phagocyter les débris cellulaires ainsi
que les agents pathogènes. Ces cellules expriment les marqueurs Gr1+ F4/80+ (64).
La durée de vie d’un macrophage peut aller de plusieurs mois à plusieurs années.
b/ Les cellules dendritiques :
Les cellules dendritiques (DC) sont des cellules présentatrices d’antigènes permettant la
mise en place de l’immunité adaptative. Elles sont présentes dans les différents tissus
lymphoïdes tels la rate, les ganglions lymphatiques et la moelle osseuse.
La différenciation vers la lignée dendritique est dépendante des facteurs de transcription
PU.1, GFI1 et CBFB.
Les cellules dendritiques ont une durée de vie allant de 3 à 6 jours.
c/ Granulocytes :
Les granulocytes sont les cellules myéloïdes les plus représentées du système sanguin et
sont composées des neutrophiles, basophiles et éosinophiles. Elles dérivent du progéniteur
granulocyte/monocytaire commun (GMP) et expriment les marqueurs CD11b+ et Gr1+. La
granulopoïèse est dépendante des facteurs de transcription PU.1, CEBPA, GATA2 et des
cytokines GM-CSF et G-CSF.
Les neutrophiles, formés dans la moelle osseuse, sont attirés par les macrophages sur un
lieu endommagé ou infecté pour aider à l’élimination du pathogène par phagocytose.
Les éosinophiles sont principalement localisés dans les tissus et participent à l’homéostasie
tissulaire et immunitaire.
Les basophiles circulent dans le sang et jouent un rôle important dans la réponse
inflammatoire et les réactions allergiques.

30
4/ Mégacaryopoïèse :
a/ Généralités :
La mégacaryopoïèse est le processus de synthèse des mégacaryocytes (Mk) à partir de la
cellule souche hématopoïétique et qui se finit par la plaquettogenèse conduisant à la
libération des plaquettes sanguines dans le sang circulant.
Les plaquettes sanguines sont de petites cellules (2 à 5 microns) anucléées présentes dans
la circulation sanguine et qui proviennent de la fragmentation du cytoplasme des
mégacaryocytes, cellules rares et polyploïdes présentes dans la moelle osseuse. Les
plaquettes ont une durée de vie très courte (7 à 9 jours) et sont renouvelées chaque jour
(10.000 nouvelles plaquettes par jour chez l’Homme) pour maintenir leur nombre constant.
La diminution de leur nombre est appelée thrombopénie et engendre un risque élevé
d’hémorragie tandis qu’une augmentation est appelée thrombocytose, pouvant provoquer
des risques de thrombose (obstruction des vaisseaux sanguins). Elles jouent un rôle
primordial dans divers processus biologiques comme l’homéostasie, la cicatrisation,
l’angiogenèse, l’inflammation et l’immunité innée.
Lors d’un endommagement des vaisseaux sanguins, des cellules endothéliales induisent
l'expression du facteur Von Willebrand qui est reconnu par les plaquettes via un complexe
de glycoprotéines composé des molécules CD41, CD61 et CD42 présentes à leur surface.
Cela permet ainsi la formation d'un agrégat plaquettaire qui participe à la réparation
vasculaire indispensable pour l’homéostasie du système vasculaire.
Il a été démontré qu’en cas de thrombopénie, l’état inflammatoire peut engendrer des
hémorragies et des chocs septiques augmentant le risque de mortalité (65,66).
Il est donc primordial de maintenir un niveau constant de plaquettes à l’état homéostatique
et de pouvoir rapidement restaurer l’homéostasie plaquettaire en cas de thrombopénie.
La mégacaryopoïèse peut être schématiquement décrite en trois grandes étapes (Figure 7) :
1-engagement et prolifération des progéniteurs mégacaryocytaires
2-maturations nucléaire et cytoplasmique des mégacaryocytes

31
3-formation et libération des plaquettes dans la circulation sanguine.
Figure 7 : La mégacaryopoïèse La mégacaryopoïèse est composée de 3 étapes : L’engagement d’une cellule souche hématopoiètique vers les progéniteurs mégacaryocytaires (MkP), la maturation (nucléaire et cytoplasmique) jusqu’à la libération des plaquettes dans la circulation sanguine.
b/ Origine des progéniteurs mégacaryocytaires
Notre compréhension de l'origine des progéniteurs mégacaryocytaires a fortement évolué
depuis une 20aine d'années et reste l'objet d'intenses discussions. Dans le modèle
historique impliquant des étapes discrètes d'engagement des HSC dans la différenciation,
les progéniteurs mégacaryocytaires (MkP) dérivent des MEP qui eux-mêmes proviennent
des CMP (54). Quelques années après, une étude a suggéré que les MEP dérivent
directement des ST-HSC (56).
Par la suite, de nombreux facteurs communs au développement des HSC et des Mk ont été
mis en évidence et incluent des récepteurs membranaires (ex : MPL, CD150, CD41), des
voies de signalisation (ex : MPL, Notch) et des facteurs de transcriptions (ex : RUNX1, TAL1,

32
GATA2, GATA1) (56,57,67,68). Ces similarités moléculaires et phénotypiques ont conduit à
l’hypothèse d’un lien directe entre les HSC et la lignée mégacaryocytaire.
L’équipe de Sanjuan-Pla a observé qu’une partie des LT-HSC (60%) expriment le marqueur
Facteur Von Willebrand (vWF), une protéine impliquée dans l’agrégation des plaquettes et
présente sur les mégacaryocytes (69). Ces LT-HSC vWF positives présentent une capacité
de différenciation mégacaryocytaire, une capacité d’autorenouvellement à très long terme
ainsi que la capacité de former toutes les lignées cellulaires contrairement aux HSC vWF
négatives qui montrent un engagement préférentiel vers la lignée lymphoïde. La grande
majorité des LT-HSC du foie fœtal au stade E14,5 (LSK+CD48-CD150+) expriment aussi le
marqueur vWF. Il a été proposé que ces LT-HSC biaisées-Mk sont au sommet de la
hiérarchie hématopoïétique (69). En effet l’analyse de l’hématopoïèse à l’état
homéostatique a aussi confirmé qu’une partie des LT-HSC se différencient directement en
progéniteur mégacaryocytaire et que ces HSC « engagées » conservent une capacité
multipotente lors de transplantation dans des souris hôtes (49).
Une autre étude a montré qu’en cas de stress induit par des infections, certaines HSC
présentant un biais de différentiation vers la lignée mégacaryocytaire vont très rapidement
former des plaquettes sans passer par les différents intermédiaires MPP, CMP, MEP pour
conserver leur nombre constant. Cette voie permet une production rapide et importante de
plaquettes. Les SL-MkP contribuent faiblement à la mégacaryopoïèse à l’état homéostatique
et semblent utilisés principalement en cas de besoin urgent (70).
Ainsi, la mégacaryopoïèse pourrait être une voie de différenciation native des HSC (49) qui
pourrait avoir une origine dans le rôle essentiel de la mégacaryopoïèse au cours du
développement embryonnaire précoce.
c/ Maturation des mégacaryocytes
Les progéniteurs mégacaryocytaires (MkP) expriment des marqueurs immatures LIN-, Kit+,
CD34low, CD150+ et des marqueurs plus spécifiques de la lignée Mk comme le CD41 et le
CD61. Ces progéniteurs sont des cellules diploïdes (2n) qui prolifèrent dans la moelle
osseuse pour se renouveler de la même façon que les autres cellules hématopoïétiques. Les

33
MkP maturent en mégacaryoblastes qui vont devenir polyploïdes avec un contenu en ADN
pouvant aller de 4n à 128n (ploïdie modale: 16n chez l’Homme et la souris) sous la forme
d'un noyau polylobé.
La polyploïdie des Mk résulte de cycles d’endomitose associés à un défaut de karyokinèse
(pas de séparation des noyaux) et un défaut de cytokinèse (pas de séparation des 2 cellules
filles) (Figure 8).
Figure 8 : Processus d’endomitose lors de la mégacaryopoïèse. Le progéniteur mégacaryocytaire entre en mitose, il y a duplication du matériel génétique (passant de 2n à 4n), séparation des chromosomes aux 2 pôles de la cellule mais un défaut de karyokinèse (pas de séparation des noyaux) et un défaut de cytokinèse (pas de séparation des 2 cellules filles) entraînent la formation d’une cellule dont le matériel génétique est de 4n et dont le noyau forme 2 lobes. Cette étape de mitose incomplète peut se répéter jusqu'à former une cellule dont le matériel génétique atteint 128n. (Issue de (71))
Ainsi, dans les progéniteurs MkP qui entrent en endomitose, les chromosomes ségrégent
aux 2 pôles de la cellule mais la formation de la membrane nucléaire est incomplète
(observation de pont nucléoplasmique) ce qui engendre un noyau polylobé. La cytokinèse
est également incomplète aboutissant à la formation d’une cellule où le matériel génétique a

34
doublé. La survenue de plusieurs cycles consécutifs de ce type génère des cellules
polyploïdes de grande taille pouvant aller jusqu'à 100uM (72,73), pour revue (71).
Au cours de cette maturation des récepteurs spécifiques de la lignée mégacaryocytaire sont
exprimés à la surface des cellules comme la glycoprotéine CD42 (74), associée à une perte
des marqueurs immatures comme le CD34 et Kit.
Le processus d’endomitose est accompagné d’une maturation cytoplasmique où le
cytoplasme des cellules est déformé en de longues extensions appelées proplaquettes. Un
mégacaryoblaste peut former entre 10 et 20 proplaquettes qui vont se fragmenter pour
libérer les plaquettes dans la circulation sanguine en nombre suffisant pour le
renouvellement homéostatique (Figure 9). La mégacaryopoïèse est un processus pouvant
durer de 8 à 10 jours.
Figure 9 : Processus de mégacaryopoïèse.
La mégacaryopoïèse « classique » est caractérisée par la formation de proplaquettes et est dépendante de la thrombopoiétine (TPO). Ce mécanisme est mis en place à l’état homéostatique pour une production de plaquettes constante. La mégacaryopoïèse par rupture de la membrane est dépendant de l’IL-1a et est mis en place en période de stress. Ce dernier mécanisme produirait beaucoup plus de plaquettes pour répondre à une demande accrue. (Issue de (75)).
La thrombopoiétine (TPO) est une cytokine majeure de la mégacaryopoïèse, qui sera
présentée plus en détail par la suite. La mégacaryopoïèse est aussi dépendante de facteurs
de transcription, dont GATA1, impliqué dans la différentiation mégacaryocytaire et NFE2,
régulant la formation des proplaquettes Pour revue (76).

35
En cas de thrombopénie, un autre mécanisme est mis en place pour répondre au besoin
rapide de plaquettes.
Ce mécanisme qui semble indépendant de la TPO et de la formation des proplaquettes,
permet de produire un grand nombre de plaquettes fonctionnelles en peu de temps. Ce
mécanisme régulé par l’interleukine 1a (IL1a) permet de modifier la stabilité membranaire
du mégacaryoblaste et conduit à la formation de plaquettes, en nombre 20 fois supérieur
par rapport au mécanisme classique (Figure 8), (75).
Un équilibre entre TPO et IL1a semble donc contrôler la mégacaryopoïèse et permettre une
production en fonction d'un besoin de plaquettes à l'homéostasie ou en condition de stress.
d/ Différence fœtal et adulte
Les Mk sont détectés très tôt au cours du développement, au stade E7,5 chez la souris, et les
plaquettes sont observées dans la circulation sanguine vers E10,5. La mégacaryopoïèse
diffère au cours de l’ontogenèse. Dans le foie fœtal, le potentiel mégacaryocytaire est
observé à partir du compartiment des cellules souches et du compartiment des
progéniteurs alors que chez l’adulte il dérive principalement du compartiment souche
(Figure 10, (77)).
Figure 10 : Différenciation hématopoïétique fœtale et adulte.

36
De plus certaines caractéristiques cellulaires et moléculaires diffèrent. Les Mk fœtaux sont
de petite taille et ont une ploïdie moyenne moins élevée que la ploïdie des Mk adultes. Leur
taille et ploïdie vont augmenter au cours des différents stades du développement pour
atteindre les caractéristiques des Mk adultes vers l’âge de 4 ans chez l’Homme. La capacité
proliférative des Mk fœtaux est largement supérieure aux Mk adultes, mais ils forment 3
fois moins de proplaquettes et donc de plaquettes (78). Ces caractéristiques peuvent être
corrélées aux changements transcriptionnels observés au cours de l’ontogenèse
mégacaryocytaire sur le cycle cellulaire, l’expression de facteurs de transcription (NFE2,
GATA1) et de régulateurs mégacaryocytaires spécifiques (influençant la maturation
mégacaryocytaire) (79).
C/ Régulation de l’hématopoïèse :
Le nombre des cellules souches hématopoïétiques résulte d'un équilibre entre quiescence,
autorenouvellement et différentiation. L'équilibre entre ces propriétés varie en fonction du
stade de développement et du besoin en cellules matures et est contrôlé par des facteurs
extrinsèques (également appelés « niche hématopoïétique ») et intrinsèques.
1/ La niche hématopoïétique :
La niche hématopoïétique est composée de différentes cellules assurant des fonctions
régulatrices distinctes Pour revue (80).
Les avancées technologiques d’imagerie cellulaire ont permis de visualiser les HSC et leurs
cellules avoisinantes dans la moelle osseuse. L’implication des cellules dans la régulation
des HSC a ensuite été validée à l’aide de modèles d’inactivation conditionnelle permettant
de cibler spécifiquement certaines populations entourant les HSC.
Les cellules périvasculaires ou péricytes, sont des cellules encerclant les vaisseaux sanguins.
Il existe différents péricytes classées sur l’expression des marqueurs de surface NG2+ et
37
LepR. Un groupe de péricytes exprimant fortement la cytokine CXCL12, appelées cellules
CAR (CXCL12-abundant reticular) est fréquemment retrouvé près des cellules souches (81).
Les cellules de Schwann (82), les nerfs sympathiques, les ostéoclastes, les cellules endothéliales,
les macrophages, les neutrophiles, les lymphocytes T CD4+ et les mégacaryocytes font partie
des cellules fréquemment retrouvées près des HSC et ont été associées à leur régulation
(83), pour revue (80).
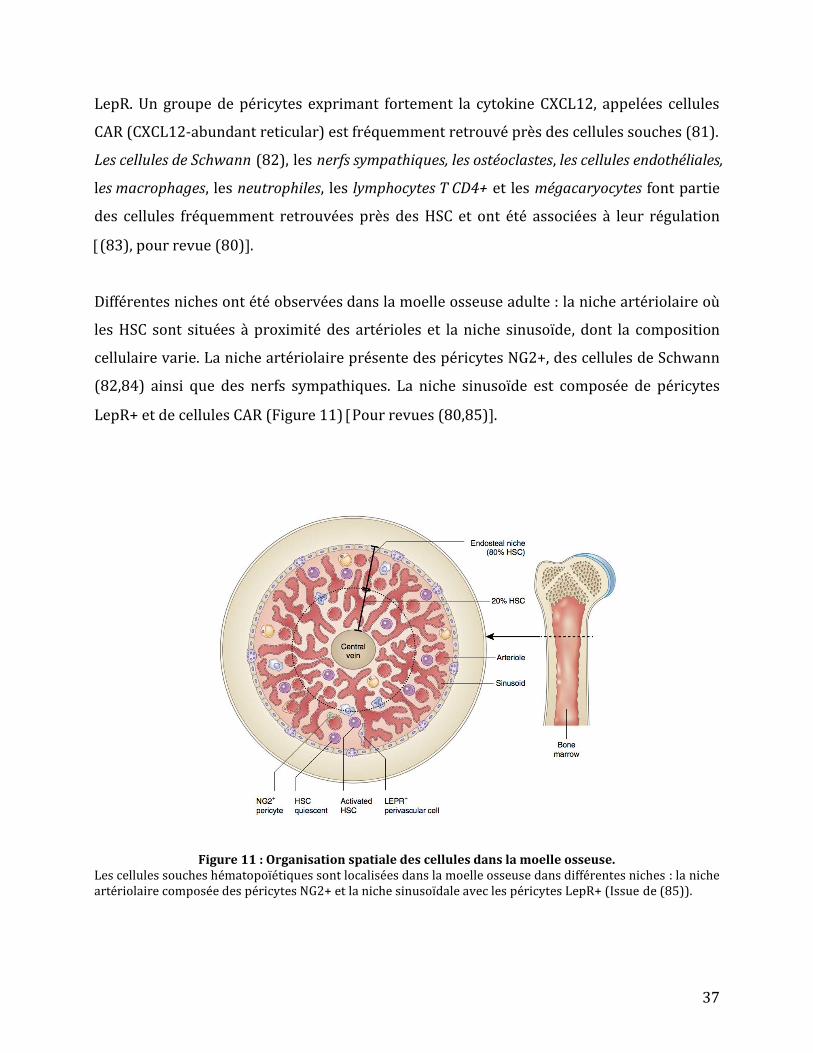
Différentes niches ont été observées dans la moelle osseuse adulte : la niche artériolaire où
les HSC sont situées à proximité des artérioles et la niche sinusoïde, dont la composition
cellulaire varie. La niche artériolaire présente des péricytes NG2+, des cellules de Schwann
(82,84) ainsi que des nerfs sympathiques. La niche sinusoïde est composée de péricytes
LepR+ et de cellules CAR (Figure 11) Pour revues (80,85).
Figure 11 : Organisation spatiale des cellules dans la moelle osseuse.
Les cellules souches hématopoïétiques sont localisées dans la moelle osseuse dans différentes niches : la niche artériolaire composée des péricytes NG2+ et la niche sinusoïdale avec les péricytes LepR+ (Issue de (85)).

38
Ces deux niches joueraient un rôle différent sur la régulation des HSC. La niche artériolaire
serait associée à la maintenance des HSC, en régulant notamment leur statut cellulaire
(quiescentes) et leur mobilisation. En effet, la déplétion des péricytes NG2+ induit l’entrée
en cycle des HSC ainsi que leur mobilisation réduisant ainsi leur capacité de repopulation à
long terme (86).
Plus récemment, il a été montré que ces différentes niches étaient associées à différents
sous-types de HSC. La niche sinusoïdale, composée aussi de mégacaryocytes, serait la niche
préférentielle des cellules souches vWF+ (HSC qui présentent un biais vers la lignée
mégacaryocytaire et myéloïde et pouvant redonner toutes les cellules (69) alors que la
niche artériolaire régulerait les HSC vWF- (HSC associée aux lignées lymphoïdes)(87).
Les cellules de la niche hématopoïétique vont sécréter des facteurs solubles ou interagir
physiquement avec les cellules souches hématopoïétiques pour moduler l’expression des
gènes et influencer leur devenir.
2/ Communication entre HSC et cellules de la niche : les voies de
signalisation
De nombreuses voies de signalisation médient l'interaction des cellules hématopoïétiques
avec leur environnement. Les voies de signalisation sont fréquemment activées par
l’interaction entre deux protéines qui peuvent être un récepteur et son ligand. Ces ligands
peuvent être des molécules transmembranaires, impliquant que l'activation de la voie de
signalisation nécessite un contact entre 2 cellules comme pour la voie de signalisation
Notch. D'autres sont des molécules solubles comme les cytokines. Les voies de signalisation
impliquent généralement une succession d’intermédiaires et conduisent fréquemment à la
modulation de l’expression des gènes.

39
a/ Contact cellule-cellule, exemple de la voie Notch :
La signalisation de la voie Notch nécessite l’interaction entre 2 cellules via un récepteur et
son ligand, tous deux transmembranaires. Chez les mammifères il existe 4 récepteurs Notch,
NOTCH1 à NOTCH4 et 5 ligands, Jagged1, Jagged2, Delta-like1, Delta-like2, Delta-like4. La
partie extracellulaire du récepteur interagit avec son ligand, ce qui va entraîner une
succession de modifications et de clivages de la partie intracellulaire de NOTCH permettant
la libération du fragment NICD (Notch Intra Cellular Domain). Ce fragment va être
transporté dans le noyau et interagir avec un complexe protéique incluant RBPJ et MAML1
pour moduler l’expression de ces gènes cibles (HES1, GATA2…). En absence d’activation de
NOTCH, RBPJ interagit avec des co-represseurs comme SMRT, SHARP ou OTT1 pour inhiber
leur expression.
La voie Notch a été impliquée dans l’établissement et le maintien des HSC au cours de
l’ontogenèse hématopoïétique. Le ligand Jagged1, exprimé par les cellules endothéliales de
la moelle osseuse, participe à l’autorenouvellement des HSC adultes à l’état homéostatique
mais aussi à leur capacité régénérative (88), pour revue (89).
La génération des HSC au cours de l’embryogenèse (au stade E10,5 chez la souris) ainsi que
leur capacité multipotent est dépendante de la voie de signalisation Notch (90,91).
La voie Notch est aussi impliquée dans l’engagement hématopoïétique vers la lignée
lymphocytaire T Pour revue (92) et mégacaryocytaire (93).
b/ Facteur soluble et récepteur, exemple de la TPO :
Les cytokines sont des protéines produites par différentes cellules, pouvant agir sur
plusieurs cellules cibles dont la cellule sécrétrice (action autocrine), localement (action
paracrine) ou à distance (effet endocrine). Les cytokines peuvent avoir un effet redondant
(exercer la même action biologique), agir en synergie ou encore avoir des effets
antagonistes. Elles permettent la communication intercellulaire, sans contact entre les
cellules via la fixation à un récepteur.
La liaison des cytokines sur leurs récepteurs spécifiques va induire un signal intracellulaire.

40
Des études récentes ont montré qu’une grande variété de cytokines pouvait agir sur les
HSC tel SCF, TPO, CXCL12, CXCL4, TGFB, G-CSF…
La TPO est une cytokine largement impliquée dans la mégacaryopoïèse (engagement et
prolifération des progéniteurs mégacaryocytaires et influençant leur ploïdie) mais aussi
dans le maintien des HSC dans la moelle osseuse (94,95). Elle est synthétisée
principalement par le foie et se fixe sur son récepteur MPL présent sur les HSC, les
mégacaryocytes et les plaquettes. Le récepteur MPL ne possède pas d’activité kinase
intrinsèque mais s'associe avec des tyrosines kinases dont la protéine JAK2. La TPO en se
fixant à son récepteur MPL, entraîne sa dimérisation, un changement de conformation et le
rapprochement des protéines JAK qui vont s’activer par transphosphorylation et
phosphoryler les résidus tyrosines du récepteur. Cela conduit ensuite à l'induction de
plusieurs voies de signalisation incluant: JAK/STAT, PI3K/AKT et ERK/MAPK (Figure 12).
Figure 12 : Voie de signalisation de la TPO. La TPO en se fixant à son récepteur MPL entraîne sa dimérisation, un changement de conformation, le rapprochement des protéines JAK qui vont s’activer par transphosphorylation et phosphoryler les résidus tyrosines du récepteur. Plusieurs voies vont être activées : la voie JAK/STAT, PI3K/AKT et ERK/MAPK. (D’après (96)).
Exemple d’activation : la voie JAK/STAT. Il existe 4 membres de la famille JAK : JAK1 à 3 et
TYK2. Ces tyrosines kinases sont associées à la partie cytoplasmique des récepteurs

41
membranaires sans activité kinase. Les protéines de la famille STAT (STAT1 à 6 dont
STAT5a et STA5b) vont s’associer au récepteur phosphorylé et être phosphorylées par les
protéines JAK. Leur phosphorylation entraîne leur dimérisation puis leur migration dans le
noyau pour réguler l’expression de leurs gènes cibles (97).
La TPO induit l’activation de STAT3, STAT5a et STAT5b (98,99). STAT3 jouerait un rôle
dans l’expansion des progéniteurs mégacaryoblastiques (96), tandis que STAT5a et b
joueraient un rôle dans la formation des plaquettes, puisque les souris STAT5(a et b)-/-
présentent une thrombopénie (100).
c/ Exemple de la voie Sonic Hedgehog
Une autre voie activée par des molécules secrétées est la voie Sonic Hedgehog qui joue un
rôle au cours de l’embryogenèse en participant à la mise en place de différents tissus (tube
neural, poumons, peau…). Cette voie a aussi été impliquée dans la régulation des cellules
hématopoïétiques primitives via les protéines BMP dont BMP4 (101).
Il existe 3 ligands chez les vertébrés : Desert Hedgehog (DHH), Indian Hedgehog (IHH) et
Sonic Hedgehog (SHH), et deux récepteurs : PTCH1 et SMOH.
En absence de ligand, PTCH1 réprime l’activité de SMOH. Lorsque que SHH se lie à PTCH1,
cela induit l’internalisation du récepteur, la levée de la répression de SMOH et l’activation
des facteurs de transcription de la famille GLI. Les facteurs GLI migrent alors dans le noyau
pour réguler l’activité des gènes cibles de la voie (gènes HOX, BMP, BCL2, PDGFRA…).
In fine, cette voie participe à la prolifération et à la différenciation cellulaire. Elle est
principalement active au cours du développement embryonnaire et réprimée dans les
cellules adultes en dehors des progéniteurs immatures. Il a été montré que cette voie
participe au développement leucémique via une activation anormale Pour revue (102).
3/ Régulation transcriptionnelle :
La régulation transcriptionnelle de l’hématopoïèse implique plusieurs niveaux, incluant le
niveau d'expression des facteurs de transcription, la régulation de leur activité par des

42
mécanismes post-traductionnels, mais également leur accessibilité à l'ADN contrôlée par
l’organisation épigénétique de la chromatine.
a/ Facteur de transcription et complexe transcriptionnel
La découverte de l’activité ARN polymérase remonte aux débuts des années 1960, avec la
première synthèse in vitro d’un brin d’ARN complémentaire à la matrice ADN, ce qui a initié
la recherche de la régulation transcriptionnelle. La transcription des ARN messagers par
l’ARN polymérase PolII nécessite qu'elle soit guidée sur le locus à transcrire par des
facteurs de transcription liant spécifiquement l'ADN et nécessite également le recrutement
de facteurs généraux de la transcription Pour revue (103).
Les facteurs de transcription se fixent à des régions de l'ADN appelées promoteurs ou
"enhancers" Pour revue (104).
Les promoteurs sont les régions sur lesquelles la machinerie transcriptionnelle se fixe pour
initier la transcription du gène. Elles sont généralement situées juste en amont du gène et
permettent de définir le site d'initiation de la transcription ou Transcription Start Site (TSS).
Les enhancers sont des régions régulatrices de la transcription qui influencent l’activité
transcriptionnelle des promoteurs (Figure 13). Les enhancers peuvent être localisés à
plusieurs dizaines (voir centaines) de kilobases des gènes/promoteurs ciblés (cette
localisation varie en fonction du type cellulaire) et plusieurs enhancers peuvent réguler
l'expression d'un seul gène. Les enhancers présentent généralement des sites de liaison de
nombreux facteurs de transcription.
La différenciation des progéniteurs hématopoïétiques vers les différentes lignées est
associée à l’activation de programmes transcriptionnels régulés par des facteurs ou
combinaisons de facteurs de transcription spécifiques d’une lignée incluant, par exemple,
les facteurs GATA1, PU.1 et CEBPA Pour revue (29).

43
Figure 13 : Activation de la transcription par les facteurs de transcriptions. Les facteurs de transcription, en se fixant sur leur site de liaison (promoteur, enhancer), vont recruter la machinerie transcriptionnelle et moduler l’expression des gènes. (Issue de (105))
Le facteur de transcription GATA1 appartient à une famille de 6 membres (incluant GATA2
et GATA3 également exprimé dans le système hématopoïétique) qui possèdent un domaine
de liaison à l’ADN sur des séquences consensus de type (A/TGATAA/G) et un domaine de
transactivation permettant la transcription de ces gènes cibles. GATA1 est essentiel pour la
différenciation érythroïde, mégacaryocytaire et des éosinophiles. Pour la lignée
mégacaryocytaire, il a été montré qu’une absence de GATA1 était associée à une
prolifération des progéniteurs mégacaryocytaires dont la ploïdie est diminuée et présentant
un défaut de formation des proplaquettes. Au niveau transcriptionnel, les gènes clés
spécifiques de la lignée mégacaryocytaire comme NFE2, GP1bA (CD42b) et PF4 sont
fortement diminués en absence de GATA1 Pour revue (106).
Le facteur PU.1 (fréquemment appelé SPI1) est un facteur déterminant pour le
développement de différentes lignées myéloïdes et lymphoïdes. Son niveau d’expression
influence l’engagement vers une lignée par rapport à l’autre où une expression importante,
intermédiaire ou faible est associée aux macrophages, lymphocytes B ou les cellules
érythroïdes et mégacaryocytaires respectivement Pour revue (107). De même, il a été
enhancer'
enhancer'
enhancer'

44
montré qu’une faible expression de PU.1 dans les HSC adultes murines engendre une perte
de la quiescence des HSC et une augmentation de leur prolifération (108).
Le facteur CEBPA influence la différenciation myéloïde notamment granulocytaire et
macrophagique en régulant des gènes spécifiques de ces lignées et en bloquant la
prolifération cellulaire Pour revue (109). Une étude récente implique directement CEBPA
dans l’engagement de toutes les lignées myéloïdes (110). La perte de CEBPA dans les HSC
adultes est associée à une réactivation du programme transcriptionnel fœtal ainsi qu’à une
augmentation de leur prolifération (45) supposant un rôle de CEBPA dans la régulation de
l’état quiescent des HSC adultes.
Les facteurs de transcription peuvent agir de concert au sein de complexe (111,112). Un
complexe nommé « heptad » composé des facteurs SCL, GATA2, RUNX1, ERG, LYL1, LMO2 et
FLI-1 influence l’expression des gènes importants pour l’émergence, le maintien et la
prolifération des cellules souches hématopoïétiques. L’inactivation complète de Gata2
(Gata2-/-), Runx1 (Runx1-/-) et à l’état hétérozygote de ces 2 gènes (Gata2+/- Runx1+/-)
entraîne la mort des embryons murins (112–114) suggérant que GATA2 et RUNX1
contrôlent ensemble l’expression de certains gènes importants pour le bon développement
de l’embryon.
La formation des cellules souches hématopoïétiques se fait par un processus de transition
des cellules endothéliales (mécanisme EHT). Ce processus nécessite l’expression de certain
gènes (comme GPR56) régulée par le complexe « heptad » (18).
Une variation de la composition de ces complexes transcriptionnels a également été
impliquée dans la progression de la différenciation hématopoïétique, notamment
mégacaryocytaire. GATA1 et GATA2 sont des facteurs de transcription qui reconnaissent un
motif similaire sur l’ADN. Il a été montré que GATA1 remplaçait GATA2 dans un processus
appelé « GATA switch » au cours de la différenciation (115). Dans les progéniteurs
hématopoïétiques, le complexe « heptad », composé de GATA2, lie un certain nombre de
gènes spécifiques de la lignée mégacaryocytaire. L’expression de ces gènes est fortement
augmentée dans les cellules mégacaryocytaires où le remplacement de GATA2 par GATA1 a
eu lieu (Figure 14). C’est le cas pour le gène GP1bA (CD42b) et PF4 (116).
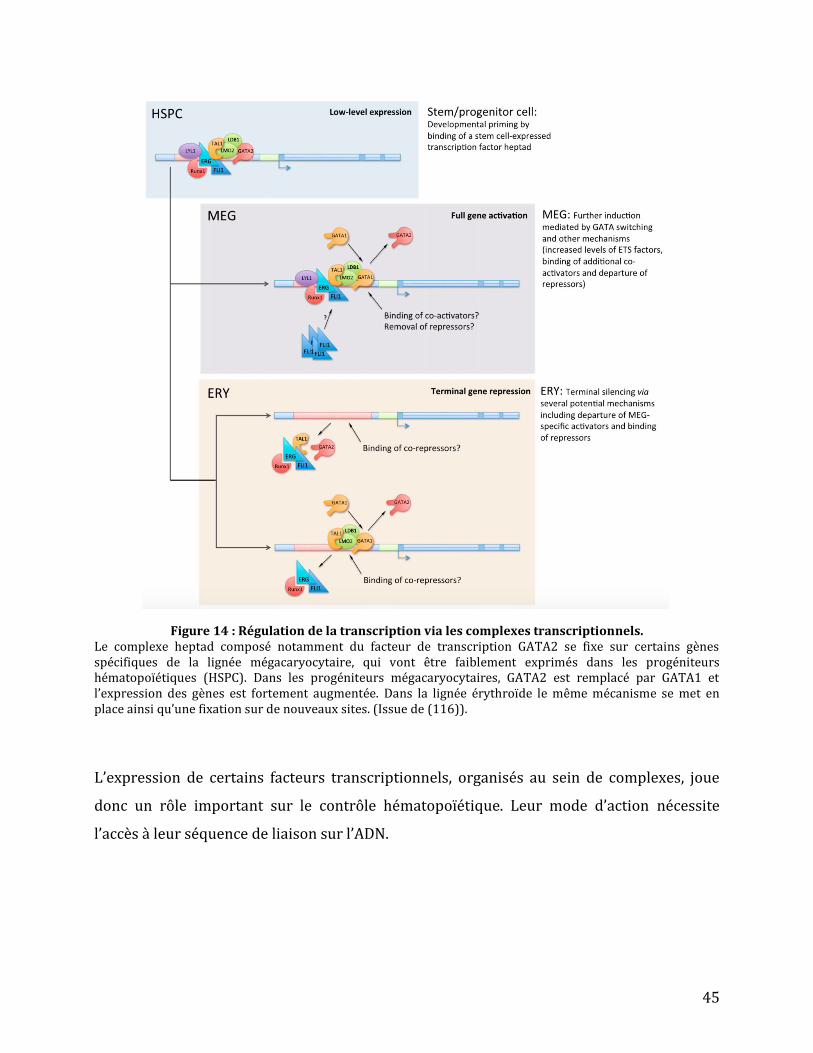
45
Figure 14 : Régulation de la transcription via les complexes transcriptionnels. Le complexe heptad composé notamment du facteur de transcription GATA2 se fixe sur certains gènes spécifiques de la lignée mégacaryocytaire, qui vont être faiblement exprimés dans les progéniteurs hématopoïétiques (HSPC). Dans les progéniteurs mégacaryocytaires, GATA2 est remplacé par GATA1 et l’expression des gènes est fortement augmentée. Dans la lignée érythroïde le même mécanisme se met en place ainsi qu’une fixation sur de nouveaux sites. (Issue de (116)).
L’expression de certains facteurs transcriptionnels, organisés au sein de complexes, joue
donc un rôle important sur le contrôle hématopoïétique. Leur mode d’action nécessite
l’accès à leur séquence de liaison sur l’ADN.

46
b/ Organisation chromatinienne
La molécule d’ADN, d’une taille d’environ 2 mètres, est compactée au sein de complexes
nucléoprotéiques formant ainsi la chromatine, dans le noyau des cellules dont la taille est
de quelques micromètres. L’ADN s’enroule autour d’un octamère d’histones (H3, H4, H2A et
H2B), structure appelée nucléosome, via des interactions électrostatiques entre l’ADN
chargé négativement et les histones chargées positivement. L’enchaînement régulier des
nucléosomes forme le nucléofilament qui se structure, à l’aide de l’histone H1, pour former
une fibre chromosomique (Figure 15).
Figure 15 : Les différents niveaux de condensation de la chromatine. La double hélice d’ADN s’enroule autour des nucléosomes qui vont à leur tour s’organiser en fibre chromosomique puis en chromosome. (D’après le manuscrit « Biologie moléculaire de Joelle Brodeur et martin toussaint, 2006).
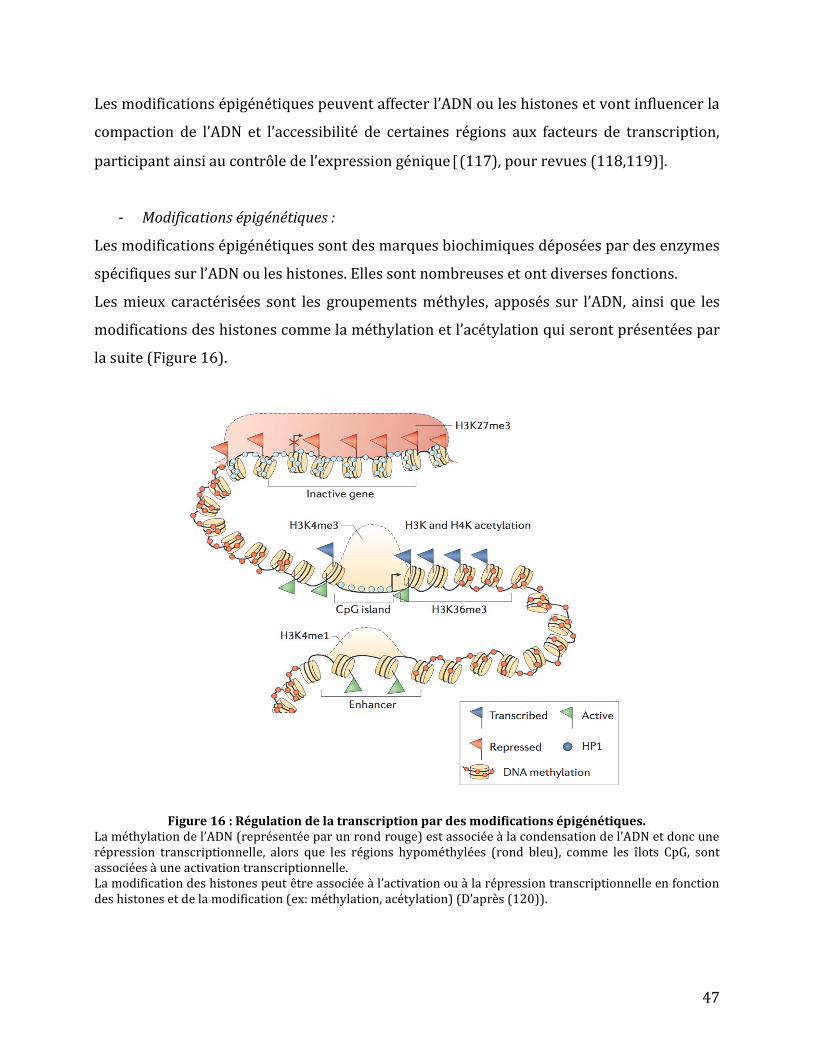
47
Les modifications épigénétiques peuvent affecter l’ADN ou les histones et vont influencer la
compaction de l’ADN et l’accessibilité de certaines régions aux facteurs de transcription,
participant ainsi au contrôle de l’expression génique (117), pour revues (118,119).
- Modifications épigénétiques :
Les modifications épigénétiques sont des marques biochimiques déposées par des enzymes
spécifiques sur l’ADN ou les histones. Elles sont nombreuses et ont diverses fonctions.
Les mieux caractérisées sont les groupements méthyles, apposés sur l’ADN, ainsi que les
modifications des histones comme la méthylation et l’acétylation qui seront présentées par
la suite (Figure 16).
Figure 16 : Régulation de la transcription par des modifications épigénétiques.
La méthylation de l’ADN (représentée par un rond rouge) est associée à la condensation de l’ADN et donc une répression transcriptionnelle, alors que les régions hypométhylées (rond bleu), comme les îlots CpG, sont associées à une activation transcriptionnelle. La modification des histones peut être associée à l’activation ou à la répression transcriptionnelle en fonction des histones et de la modification (ex: méthylation, acétylation) (D’après (120)).

48
La méthylation de l’ADN correspond à l’ajout d’un groupement méthyles –(CH3) sur un
résidu cytosine suivi d’une guanine, appelé dinucléotide CpG. Certaines régions promotrices
des gènes présentent un regroupement de dinucléotides CpG, appelé « îlots CpG ».
Cette modification épigénétique est régulée par des enzymes : les DNA méthyltransférases
(DNMT3a, DNMT3b, DNMT1) et est associée à un état condensé de la chromatine et donc à
une répression transcriptionnelle (Figure 16).
DNMT3a/b méthylent des sites de novo contrairement à DNMT1 qui permet la maintenance
des sites déjà méthylés pendant de la réplication. Le niveau de méthylation de l’ADN joue un
rôle dans la régulation de l’autorenouvellement des HSC ainsi que dans la différenciation,
notamment dans l’engagement vers une lignée. L’engagement myéloïde est associé à un
niveau de méthylation moins important que l’engagement vers les lignées lymphoïdes
(121). DNMT3a joue un rôle important dans la différenciation en réprimant les gènes
impliqués dans l’autorenouvellement des HSC comme RUNX1 et GATA3 (122).
La déméthylation de l’ADN s’effectue via des protéines dioxygénases comme les protéines
TET qui catalysent la conversion des 5-methylcytosines (5mC) en 5-
hydroxymethylcytosines (5hmC) Pour revue (123). Les protéines TET, dont TET2 et TET3,
travaillent en synergie avec les facteurs de transcription E2A et PU.1 pour déméthyler
l’ADN et permettre l’accessibilité de certains enhancers spécifiques de la lignée
lymphocytaire B. L’activité des protéines TET est nécessaire pour l’expression de IRF4, gène
cible de PU.1 (124).
Les modifications post-traductionnelles des histones comme la méthylation et l’acétylation
sont des phénomènes réversibles.
Le processus de méthylation des histones est dépendant de deux sortes d’enzymes: les
histones méthyltransférases et les histones déméthylases. Les lysines des histones peuvent
être mono-, bi- ou triméthylées par les histones méthyltransférases (K-methylases) et le
même résidu peut être modifié par différentes enzymes. C’est le cas de la lysine 9 de
l’histone H3 (H3K9) qui peut être modifiée par les protéines SUV39H1/2, G9a et SETD1B.
Certaines protéines n’agissent que sur un seul résidu, comme par exemple EZH2
responsable de la triméthylation de la lysine 27 de l’histone H3 (H3K27). Les histones

49
déméthylases permettent de déméthyler les histones comme la protéine JARID qui
déméthyle les lysines de l’histone H3.
L’acétylation des histones est le mécanisme de transfert d’un groupe acétyle sur certaines
lysines des histones par des acétyltransférases (HAT pour histone acétyltransférase). Cette
modification est associée à un changement de structure de la chromatine permettant ainsi
l’accessibilité des facteurs de transcription à certaines régions. La désacétylation est
effectuée par les histones désacétylases (HDAC), et est associée à une compaction de la
chromatine et une répression transcriptionnelle.
Les analyses d’immunoprécipitation de chromatine associée au séquençage (ChIPseq) ont
permis de corréler certaines marques (comme la marque H3K4me3) à l’activation de la
transcription et d'autres (comme la marque H3K27me3) à la répression de la transcription.
Parmi les marques généralement associées à l'activation transcriptionnelle, certaines (ex:
H3K4me3) sont associées préférentiellement aux promoteurs actifs et d'autres (ex:
H3K4me1 ou H3K27Ac) sont plus intenses au niveau des enhancers.
Les Super-Enhancers (SE) sont des enhancers de grande taille présentant une intensité
importante de marques H3K27Ac et H3K4me1 sur une large région chromosomique. Ces
régions sont généralement associées à la présence de nombreux facteurs de transcription et
du complexe co-activateur Mediator (dont Med12 et BRD4). Il a été montré que les SE sont
associés à des gènes clés spécifiques de l’identité cellulaire, ils sont donc très variables d'un
type cellulaire à l'autre (125). Dans les progéniteurs lymphoïdes B, les SE sont fortement
liés par le facteur PU.1 alors que dans les macrophages, les SE sont fixés par le facteur
CEBPA (126).
- Organisation chromatinienne :
Les cohésines forment un complexe multiprotéique jouant un rôle dans divers processus
biologiques comme la réplication (cohésion entre les chromatides sœurs), l’apoptose ou
encore la réparation de l’ADN (Figure 17, Pour revue (127)).
Elles participent aussi à la régulation hématopoïétique en modulant l’accessibilité de la
chromatine aux facteurs de transcription. L’utilisation de mutant des cohésines a montré

50
une fixation accrue de certains facteurs comme ERG, GATA2 et RUNX1 sur l’ADN, entraînant
une augmentation du programme transcriptionnel associé aux cellules souches et un
blocage de la différenciation dans des cellules primaires humaines (128).
Un autre rôle des cohésines a été proposé dans les interactions entre les promoteurs et les
enhancers en association avec la protéine CTCF. La formation de boucles permet le
rapprochement d’un promoteur avec un enhancer éloigné, ou au contraire une séparation
entre ces deux régions régulatrices, agissant ainsi sur la transcription (129).
Figure 17 : Le complexe de cohésines est un régulateur-clé de la transcription et de la formation de
boucle chromatinienne. Le complexe permet la cohésion entre les deux chromatides, interagit avec CTCF, forme des boucles chromatiniennes, recrute des facteurs de transcriptions et des complexes participant au contrôle de la transcription. (Issue de (127)).
L’ensemble de ces mécanismes permet une régulation fine de l’hématopoïèse. Leur
dérégulation peut engendrer un défaut de différentiation et/ou une production incontrôlée
de cellules, pouvant causer une hémopathie maligne.

51
II/ Hémopathies malignes
Le terme « hémopathie », composé des racines grecques « hémo » (sang) et « pathos » (ce
qu’on éprouve), désigne les maladies du sang et de ses composants. Les hémopathies
malignes sont diverses et les leucémies aiguës représentent généralement une forme
d’hémopathie agressive. Les leucémies aiguës myéloïdes (LAM) seront présentées plus en
détail ci-dessous.
A / Les leucémies aiguës myéloïdes :
1/ Classification des LAM
Les leucémies aiguës myéloïdes sont un groupe de leucémies cliniquement hétérogènes,
dues à la prolifération incontrôlée de blastes dans la moelle osseuse, pouvant engendrer
une diminution de l’hématopoïèse normale avec l’observation d’une anémie (diminution
des globules rouges), une neutropénie (diminution des globules blancs) ainsi qu’une
thrombopénie (diminution des plaquettes).
Depuis les années 1970, une classification internationale appelée FAB (pour Franco-
Américano-Britannique) est utilisée pour classer les leucémies, basée essentiellement sur la
morphologie des blastes. Dans les leucémies aiguës myéloïdes, huit sous-groupes sont
observés, notés de 0 à 7, classés historiquement sur la base de leurs statuts de
différenciation (130).
LAM0 : avec différenciation minime
LAM1 : sans maturation
LAM2 : avec maturation
LAM3 : LA promyélocytaire
LAM4 : LA myélomonocytaire
LAM5 : LA monoblastique/monocytaire
LAM6 : LA érythroïde
LAM7 : LA mégacaryoblastique

52
L’organisation mondiale de la santé (OMS, ou World Health Organization en anglais, WHO) a
progressivement intégré les données immunophénotypiques puis génétiques (c.f. infra)
pour affiner cette classification (131,132).
Il a ainsi été mis en évidence que certaines anomalies génétiques sont associées
spécifiquement à un sous-groupe FAB comme RUNX1-RUNX1T1 pour les LAM-M2, PML-
RARA pour les LAM-M3, ou encore OTT-MAL pour les LAM-M7. La recherche et la détection
de ces anomalies rendent possibles un diagnostic plus précis et l’utilisation d’un traitement
thérapeutique ciblé. Le sous-groupe des LAM-M3 présentant la mutation PML-RARA est
maintenant traité avec succès en utilisant l’acide rétinoïque (133).
2/ Anomalies génétiques des LAM
Les cellules cancéreuses sont caractérisées par des anomalies du matériel génétique (ADN)
porté par les chromosomes. Cette hypothèse découle des observations de David von
Hansemann et de Theodor Boveri sur la présence d’altérations des chromosomes dans les
cellules cancéreuses Pour revue (134).
L’observation des chromosomes et du caryotype par les approches de cytogénétique
(caryotype, fluorescent in situ hybridization: FISH) puis l'évolution des techniques de
séquençage (Sanger puis séquençage à haut débit) a ensuite permis d'identifier les
anomalies génétiques récurrentes.
Les mutations sont à la base de l’évolution des espèces. En effet, une mutation survenue
dans les cellules germinales et conférant un avantage sélectif, peut être transmise à la
descendance. Dans le cas des leucémies aiguës, les mutations apportent un avantage sélectif
aux progéniteurs hématopoïétiques (avantage prolifératif, d’autorenouvellement, de
résistance aux traitements) au détriment de la différenciation et de la production des
cellules matures et fonctionnelles, déséquilibrant ainsi l’homéostasie hématopoïétique. Les
mutations retrouvées dans les leucémies sont majoritairement somatiques (dans les
cellules hématopoïétiques). Elles sont parfois germinales (dans les cellules des gamètes)

53
comme dans le cas de pathologies familiales telles que le syndrome plaquettaire familial où
la mutation du gène RUNX1 est transmissible et prédispose au développement d’une LAM.
a/ Types d’altérations génétiques
La plupart des cellules humaines sont diploïdes (2n), c’est à dire que chaque chromosome
est représenté en deux exemplaires dans la cellule. Des anomalies de nombre peuvent être
observées avec la perte ou le gain d’un chromosome entier (ex: monosomie 7 ou trisomie
21) ou d'une région d'un chromosome (délétion ou duplication/amplification). Ces
anomalies peuvent entraîner des anomalies de dosage génique (surexpression ou sous-
expression) d’un ou plusieurs gènes. D’autres anomalies de structure fréquemment
retrouvées dans les leucémies sont les translocations chromosomiques (échange de
segments entre 2 chromosomes) et les inversions chromosomiques (échange au sein du
même chromosome).
Le développement des techniques de séquençage haut débit (NGS) a permis l’analyse de
plusieurs gènes simultanément dans un grand nombre d'échantillons tumoraux, et la
détection de mutations dites "ponctuelles" incluant les substitutions et les
insertions/délétions de petite taille, et dans certains cas, les altérations de structure
chromosomique.
Quand elles surviennent dans la séquence codante d'un gène, les mutations peuvent avoir
différentes conséquences incluant la substitution d’un nucléotide par un autre, conduisant
soit à une mutation silencieuse (mutation codant pour le même acide-aminé), une mutation
faux-sens (mutation modifiant l’acide aminé) ou une mutation non-sens (substitution par
un codon stop).

54
b/ Conséquences fonctionnelles des mutations
Les modifications génétiques qui contribuent fonctionnellement au développement
leucémique sont appelées mutations « driver » par opposition aux autres mutations dites
"passagères" Pour revue (135).
Un projet de séquençage à grande échelle des échantillons tumoraux dont le but est
l’identification des gènes directement impliqués dans la leucémogenèse a identifié une
centaine de gènes mutés dans les LAM (COSMIC,
https://cancer.sanger.ac.uk/census#cl_download). A l’état normal, ces gènes sont
impliqués dans divers processus biologiques comme les voies de signalisation (JAK2, FLT3),
la régulation transcriptionnelle (RUNX1, CEBPA), la régulation épigénétique (TET2,
DNMT3A), ou encore les cohésines (RAD21).
Les conséquences fonctionnelles de ces mutations sont variées et les plus fréquentes sont
décrites ci-dessous.
- Mutations activatrices :
Certaines mutations peuvent engendrer l’activation constitutive d’une protéine. C’est le cas
de la mutation faux-sens du gène JAK2 V617F où la substitution de la Guanine en position
1849 par la Thymine (G1849T) entraîne une modification de la séquence protéique. Cette
mutation est fréquemment retrouvée dans les hémopathies (136).
- Mutations inactivatrices :
L’inactivation de certains gènes par une grande variété de mutations (mutations
ponctuelles, délétion…) contribue à l’expansion des cellules tumorales Pour revue (137).
Ces gènes ont été nommés « gène suppresseur de tumeur » car à l’état normal ils
interviennent comme régulateurs négatifs de la prolifération cellulaire (ex : RB1, TP53). Le
premier gène caractérisé comme tel a été le gène RB1 dans les rétinoblastomes (tumeurs de
l’œil), où l’inactivation des 2 copies du gène est nécessaire pour le développement tumoral
(138). Dans certains cas, l’inactivation d’une des deux copies du gène peut
empêcher/limiter son rôle et favoriser l’apparition de cancers, il s’agit d’haplo-insuffisance.
C’est le cas du gène suppresseur de tumeur TET2 retrouvé muté dans 15% des hémopathies

55
myéloïdes mais où tous les patients ne présentent pas d’inactivation des 2 copies du gène
Pour revue (139).
- Création de gènes de fusion :
Les translocations ou les inversions chromosomiques peuvent engendrer des protéines de
fusion, protéine obtenue par la combinaison de 2 protéines différentes. C’est le cas de la
translocation t(8;21) associée aux leucémies LAM-M2 où les protéines AML1 (aussi appelée
RUNX1) et ETO sont fusionnées. La protéine AML1-ETO recrute des régulateurs
épigénétiques modifiant l’expression des gènes, dérégule les gènes cibles d’AML1 et
interfère dans le recrutement de cofacteurs essentiels pour la différentiation myéloïde
comme CEBPA et PU.1 Pour revue (140).
- Création de nouvelle fonction :
Dans ce type de mutation, la protéine issue de l’allèle muté fonctionne différemment de la
protéine physiologique. Par exemple les mutations des gènes IDH, fréquemment retrouvées
dans les LAM et autres cancers, modifient les propriétés enzymatiques de IDH1 ou IDH2.
Les protéines IDH1 et IDH2 sont des isocitrates déshydrogénases qui convertissent
l’isocitrate en Ketoglutarate. La protéine IDH1 mutée peut convertir l’Ketoglutarate
(KG) en 2-hydroxyglutarate, augmentant cette nouvelle molécule dans la cellule au
détriment de l’KG, réduisant ainsi l’activité des enzymes KG-dépendantes comme
certaines méthyltransférases (ex: TET2). Certaines mutations d'enzyme du métabolisme
pourraient donc in fine affecter la régulation transcriptionnelle et participer au
développement leucémique Pour revue (141).
- Modification du niveau d’expression :
Parmi les mutations décrites précédemment, plusieurs participent à l’altération du niveau
d’expression de gène dans les leucémies. Par exemple, l’amplification de certaines régions
ainsi que les anomalies du nombre de chromosomes comme la trisomie 21, conduisent à la
présence de trois copies de certains gènes. Le gène ERG, localisé sur le chromosome 21 est

56
fréquemment surexprimé dans les leucémies associées à un mauvais pronostic (142). Sa
surexpression dans des modèles murins conduit au développement de leucémies (143,144).
Certaines altérations modifient les régions régulatrices comme les promoteurs ou
enhancers, ce qui va avoir un impact direct sur l’expression des gènes associés. Certains
réarrangements chromosomiques, comme l’inversion du chromosome 3, sont associés à la
surexpression d’EVI1 et à l’inhibition de GATA2 dans certaines LAM. Cette dérégulation
transcriptionnelle est due au repositionnement d’un enhancer causé par la translocation
(145).
c/ Coopération oncogénique et évolution clonale
Dans certaines leucémies pédiatriques, le nombre de mutations est particulièrement faible
et suggère qu'une seule mutation driver serait nécessaire pour la transformation.
Cependant la plupart des LAM présentent plusieurs mutations « drivers » au sein d’un
même échantillon (146), pour revues (135,147).
La présence de combinaison récurrente de mutations et la modélisation de ces mutations in
vivo a ainsi conduit à la notion de coopération oncogénique. Par exemple, les mutations
activatrices du gène FLT3 (un intermédiaire de voie de signalisation), appelées FLT3-ITD,
font parties des mutations les plus récurrentes des LAM. Cependant, le modèle murin de
surexpression de Flt3 n’est pas suffisant à lui seul pour induire une leucémie aiguë.
Plusieurs coopérations ont été mises en évidence entre les mutations FLT3 et PML-RARA
pour le développement de leucémies promyélocytaires (LAM-M3) (148,149) ou une
surexpression de RUNX1, dans d'autres LAM (150).
La nécessité de l’acquisition de plusieurs mutations dans la même cellule pour le
développement tumoral est appelée « évolution clonale » Pour revue (146,151). Ce
processus peut conduire à l'apparition de différentes populations présentant diverses
combinaisons d'altérations génétiques, appelées clones. Cette hétérogénéité tumorale a des
conséquences sur l’initiation et la progression de la maladie ainsi que sur la survenue
d’éventuelles rechutes suite aux traitements.

57
Dans de nombreux cas de leucémies aiguës, l'hypothèse est que le premier évènement
mutationnel, appelé mutation initiatrice, confère un avantage compétitif aux progéniteurs
hématopoïétiques, conduisant à l'amplification d'un clone dit pré-leucémique, mais sans les
symptômes immédiats de leucémie.
Deux exemples illustrent cette hypothèse. Dans les leucémies de l'enfant, la fusion TEL1-
AML1 est retrouvée chez des jumeaux monozygotes qui peuvent développer des leucémies
avec des latences très différentes (c.f. infra). Dans les leucémies de l'adulte, les mutations
DNMT3A ou TET2 sont retrouvées chez des individus ne présentant pas de pathologie (152).
Par exemple, chez des individus présentant une LAM associant DNMT3A et NPM1 dans les
blastes leucémiques, certaines HSC présentent la mutation DNMT3A sans la mutation NPM1.
L'impact clinique de ces différences clonales pourrait être dans une réponse différente des
sous-clones aux traitements (Figure 18).
Figure 18 : Exemple d’évolution clonale de leucémie aiguë promyélocytaire (LAM-M3). Les progéniteurs hématopoïétiques acquièrent des mutations « passagères » de façon proportionnelle avec l’âge des patients (« X » représente les mutations « passagères »). La mutation initiatrice est associée à l’étape pré-leucémique qui précède l’acquisition des mutations additionnelles permettant la transformation. La présence de sous-clones ayant acquis des mutations additionnelles différentes peut générer une résistance aux traitements. (Réalisé d’après (146,151)).

58
L’association de données de séquençage et de clonalité des échantillons tumoraux à la
modélisation fonctionnelle permet ainsi de mieux comprendre la contribution de chaque
mutation (mutations initiatrices, coopératrices ou impliquées dans la progression
leucémique) sur le développement leucémique. La compréhension de cette évolution
clonale est donc un élément important d'un point de vue clinique pour évaluer les
conséquences des thérapies sur le clone fondateur ainsi que les sous-clones et limiter le
risque de rechute.
3/ Les cellules souches leucémiques
a/ Propriétés des LSC
L'observation que certains patients rechutent après traitement ainsi que les analyses de
greffe de cellules leucémiques humaines dans des souris immunodéficientes indiquent que
toutes les cellules leucémiques de patients n'ont pas les mêmes capacités et ont généré le
concept de cellules souches leucémiques "LSC".
L’existence de ces LSC a été démontrée dans des analyses de xénotransplantation dans les
années 1990. Plusieurs équipes ont montré que seule une fraction de cellules leucémiques
était capable de générer une leucémie chez ces souris. La capacité d’autorenouvellement est
observée dans des greffes secondaires, et est une propriété importante pour définir les LSC.
Ces cellules peuvent dériver des cellules souches (fraction CD34+CD38-) ou des
progéniteurs plus matures (fraction CD34-) ayant acquis certaines propriétés des HSC
(153,154).
Chez les patients la fréquence des LSC varie et peut être déterminante sur le pronostic des
leucémies. En effet, une forte fréquence de LSC au diagnostic est associée à un pronostic
défavorable (155,156).
Certaines LSC observées dans des modèles de xénotransplantation sont associées à un état
quiescent, ce qui pourrait expliquer leur résistance aux chimiothérapies classiques, qui
agissent sur des cellules prolifératives (157).

59
Le rôle du microenvironnement est aussi important pour les cellules leucémiques (158),
pour revue (159). Dans les modèles de xénotransplantation, il a été montré que les LSC
migraient très rapidement après leur injection dans des régions de la moelle osseuse
attribuées aux HSC normales et y résidaient tout au long du développement leucémique. La
diminution de l’hématopoïèse normale fréquemment observée durant la progression
leucémique, pourrait être due à une compétition entre HSC et LSC pour ce
microenvironnement (157).
Il est donc important d’étudier les cellules souches leucémiques dans l’espoir de trouver des
cibles thérapeutiques spécifiques. Les analyses transcriptionnelles (154,160) et
phénotypiques pourraient aboutir à de nouvelles stratégies. Par exemple, avec l’utilisation
d’anticorps ciblant les marqueurs de surface des LSC comme l’anti-CD33 ou l’anti-CD123.
b/ Origine cellulaire
Une partie de l'hétérogénéité génétique, phénotypique et clinique des leucémies pourrait
être due à des différences d’origine cellulaire, c’est-à-dire le type de cellule
hématopoïétique dans laquelle les mutations s'accumulent. Sur la base des expériences de
xénotransplantation des différentes sous-populations de cellules leucémiques humaines, les
leucémies pourraient dériver d'au moins deux types cellulaires : les cellules souches
hématopoïétiques ou les progéniteurs hématopoïétiques plus engagés dans la
différentiation.
L’hypothèse d’une cellule souche hématopoïétique normale comme origine de la leucémie
découle des caractéristiques similaires observées entre les HSC et les LSC, incluant une
capacité d’autorenouvellement et de maintien d'un état quiescent dans certaines conditions,
ainsi que l’expression de marqueurs de surface en commun (153).
L’hypothèse d’un progéniteur engagé comme cellule cible découle principalement des
différences phénotypiques observées chez les patients. Par exemple, l’association de la
fusion MLL-AF9 avec les LAM myélo-monoblastiques (LAM-M4) suggère l’acquisition de

60
cette altération dans un progéniteur granuleux-macrophagique (GMP) comme origine de la
maladie.
Comme les progéniteurs hématopoïétiques normaux ne possèdent pas de capacité
d’autorenouvellement à long-terme, cette hypothèse implique que les mutations drivers
restaurent/imposent une capacité d’autorenouvellement à long-terme. Certains oncogènes
comme MLL-AF9 ou MOZ-TIF2 semblent capables de cela. En effet, la transduction
rétrovirale de progéniteurs CMP ou GMP purifiés, par la fusion MOZ-TIF2, leurs confère des
propriétés d’autorenouvellement in vitro et permet le développement leucémique in vivo
(161). Pour d’autres oncogènes capables de transformer aussi bien des HSC que des
progéniteurs plus engagés, comme la fusion MLL-AF9, l’utilisation d’un modèle murin
inductible a montré une plus grande agressivité (latence et invasion tissulaire) et une
résistance aux traitements des leucémies générées in vivo à partir de HSC que celles
générées à partir de progéniteur GMP (162).
Cependant, tous les oncogènes ne sont pas capables d’apporter les capacités
d’autorenouvellement nécessaires à la transformation d’un progéniteur engagé comme
observé avec la mutation BCR-ABL1 (161).
4/ Différence entre leucémies pédiatriques et leucémies de l’adulte
Les cancers pédiatriques représentent 1% des nouveaux cas de cancer en France, dont la
fréquence est de 1 enfant sur 450 développant un cancer avant l’âge de 15 ans. Les
leucémies aiguës représentent le cancer pédiatrique le plus fréquent.
Les cancers pédiatriques diffèrent de ceux de l’adulte par leur évolution rapide et une plus
grande sensibilité aux chimiothérapies dans la plupart des cas. Ainsi, la survie à 5 ans des
leucémies de l’enfant de 1 à 15 ans est de 90,6% pour les LAL et de 66,5% pour les LAM
alors que chez l’adulte développant une LAM, la survie est de 30% entre 55 et 65 ans et
diminue à 3% pour les plus de 75 ans.
De plus, la fréquence des leucémies varie drastiquement entre ces deux groupes. Par
exemple, les LAL représentent 80% des leucémies aiguës (LA) pédiatriques et seulement
20% des LA de l’adulte. Au sein des LAM, la répartition des sous-types ainsi que les

61
altérations génétiques observées diffèrent aussi. Les différences entre les leucémies aiguës
myéloïdes pédiatriques et adultes seront présentées plus en détail par la suite.
a/ Différence de répartition des sous-types de LAM et génétique
La répartition des sous-types de LAM de la classification FAB est différente en fonction de
l’âge des patients (Figure 19) et particulièrement pour les patients très jeunes dont l’âge est
inférieur à 2 ans (163,164).
Figure 19 : Répartition des sous-groupes de LAM en fonction de l’âge.
La fréquence des sous-types de LAM varie entre les 3 groupes observés LAM inférieur à 2 ans (<2ans), LAM de 2 à 20 ans et les LAM de l’adulte (>21ans). (D’après (163,164)).
Les leucémies pédiatriques diffèrent également des leucémies observées chez l’adulte par
les altérations génétiques retrouvées. Les leucémies pédiatriques sont associées à peu de
mutations additionnelles, suggérant qu’un nombre faible d’évènements génétiques (1 ou 2)
serait suffisant pour induire une transformation leucémique (165–167). La fréquence des
altérations génétiques observées diffère aussi, comme la fréquence de la fusion MLL-AF9
qui décroît fortement avec l’âge allant de 19,6% chez les enfants de moins de 2 ans à moins
de 2% chez les adultes (168).
< 2 ans 2 à 20 ans
M0M1
M2M4
M5M6
M7
> 21 ans

62
Au vu du très jeune âge de certains patients au diagnostic, l’hypothèse d’une origine
prénatale de certaines leucémies pédiatriques est probable. Dans l’ensemble, ces analyses
suggèrent une influence du stade de développement sur le développement leucémique.
b/ Origine in utero des leucémies pédiatriques
L’analyse génétique de jumeaux monozygotes présentant tous les deux une leucémie a
permis d'affirmer l’origine in utero de certaines mutations observées dans les leucémies
pédiatriques.
La première observation de jumeaux monozygotes (qui possèdent le même matériel
génétique) présentant une leucémie similaire dans l’enfance a été décrite en 1882. Par la
suite, Greaves et ses collègues ont réalisé l’analyse génétique des cellules hématopoïétiques
à la naissance, prélevées lors des tests de Guthrie. Ce test mis en place dans les années
1960-1970, est un prélèvement sanguin effectué au niveau du talon du bébé quelques jours
seulement après sa naissance. Ainsi, en utilisant la séquence du point de fusion des
altérations TEL-AML1 retrouvées dans les échantillons leucémiques, il a pu être mis en
évidence que ce même point de fusion pouvait être détecté dans l'ADN extrait des cellules
présentes dans le test de Guthrie. Ces observations suggèrent qu'un évènement génétique
(fusion TEL-AML1), survenu in utero dans un progéniteur hématopoïétique, a donné lieu à
l'amplification d'un clone muté qui a "colonisé" l'autre jumeau, probablement par la
circulation sanguine, puis a conduit au développement d'une leucémie indépendamment
chez les deux jumeaux (Figure 20)(169,170).

63
Figure 20 : Modèle de transformation par la fusion TEL-AML1 chez des jumeaux.
La mutation initiatrice (TEL-AML1) survient dans un progéniteur hématopoïétique chez un des jumeaux avant la naissance. Ce clone est transmis au deuxième jumeau par la circulation sanguine in utéro. Des mutations coopératrices arrivent indépendamment chez les deux jumeaux conduisant au développement de leucémie dont la latence peut varier. (Réalisé d’après (169,170)).
La fusion MLL-AF4 a également été détectée à la naissance chez 3 jeunes enfants
diagnostiqués à 5, 6 et 24 mois pour une leucémie aiguë lymphoïde.
Ainsi, l'hypothèse actuelle est que la plupart des leucémies pédiatriques (LAM et LAL) ont
une origine prénatale (171). Cependant, les mécanismes cellulaires et moléculaires à
l'origine de la transformation spécifique chez l'enfant restent mal connus et les modèles
disponibles ne permettent généralement pas de reproduire précisément la transformation
par les oncogènes pédiatriques.
B/ Les LAM-M7 pédiatriques
La leucémie aiguë mégacaryoblastique a été décrite pour la première fois en 1931 par Von
Borost et incorporée dans la classification FAB en 1985, comme le sous-type LAM-M7 (172).

64
1/ Présentation clinique et traitements
Les LAM-M7 sont caractérisées par la présence de blastes anormaux dont la morphologie
généralement hétérogène peut rappeler celle des mégacaryoblastes normaux avec, dans
certains cas, des irrégularités cytoplasmiques ou des cellules multi-nucléées de grande taille.
Ces blastes peuvent exprimer les glycoprotéines plaquettaires comme le CD41, CD61 ou
encore le CD42.
Les LAM-M7 sont un sous-groupe de LAM plutôt rare dont la fréquence est plus importante
chez les enfants que chez les adultes (18% chez les moins de 2 ans, 2% de 2 à 21 ans et 1%
pour les plus de 21 ans, (163,164)). Les LAM-M7 pédiatriques peuvent survenir suite à une
prédisposition, telle la trisomie 21 constitutionnelle, également appelée syndrome de Down
(DS-LAM-M7), ou diagnostiquée de novo (sans prédisposition apparente). Chez l’adulte en
revanche, les LAM-M7 sont généralement des leucémies secondaires à un syndrome
myéloprolifératif ou myélodysplasique après chimiothérapie.
Le tableau clinique inclut fréquemment, particulièrement chez les enfants, une anémie, une
thrombopénie, une myélofibrose, une splénomégalie ainsi qu’une hépatomégalie (173). Il
n’existe pas de traitement spécifique pour les LAM-M7 qui repose sur une chimiothérapie
intensive classique en 3 phases (phase d’induction, de consolidation et intensification). La
phase d’induction utilise un mélange de molécules de la famille des anthracyclines (comme
la daunorubicine) associée à la cytarabine. Ces molécules ciblent principalement les cellules
en prolifération. Lorsque le taux de blastes dans la moelle osseuse est inférieur à 5% après
cette première phase, les patients sont dits en « rémission complète ». La phase de
consolidation dépend de la qualité de rémission après l’induction et utilise généralement de
fortes doses des mêmes molécules. La phase d’intensification peut comporter l’utilisation
de molécules anti-cancéreuses ou une transplantation de moelle osseuse (174).
Néanmoins, le risque élevé de rechute confère un pronostic défavorable aux LAM-M7
(Figure 21)(175,176).
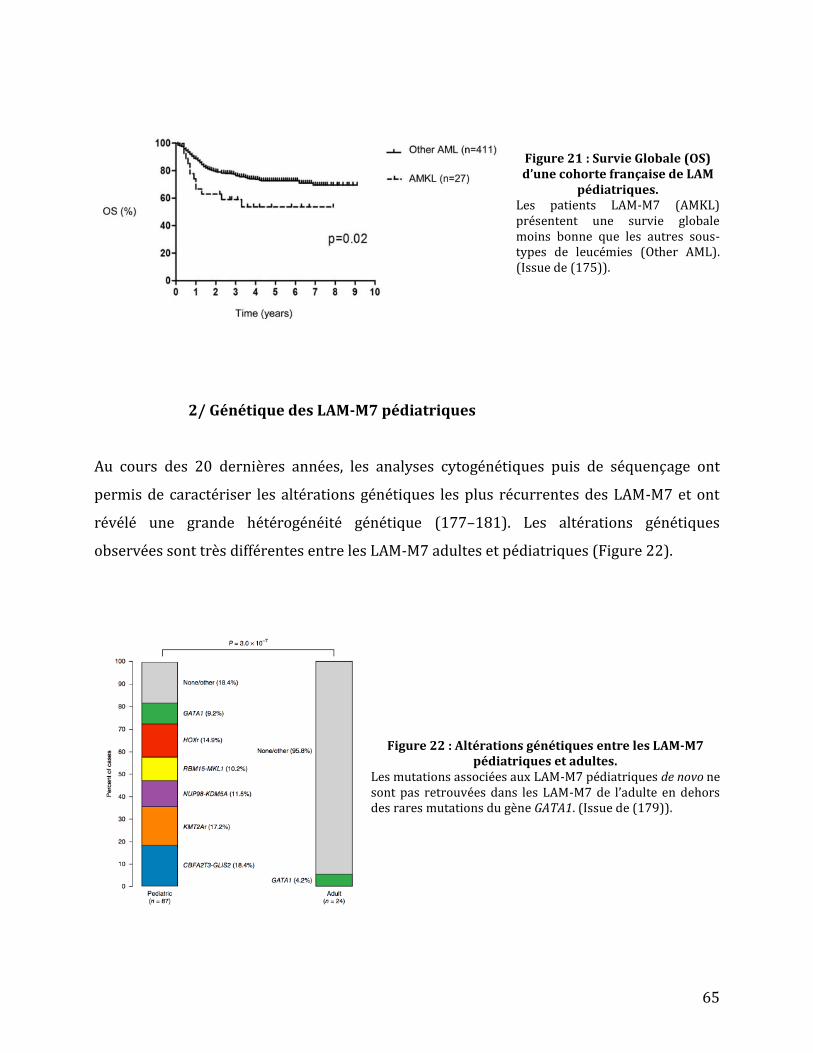
65
Figure 21 : Survie Globale (OS) d’une cohorte française de LAM
pédiatriques. Les patients LAM-M7 (AMKL) présentent une survie globale moins bonne que les autres sous-types de leucémies (Other AML). (Issue de (175)).
2/ Génétique des LAM-M7 pédiatriques
Au cours des 20 dernières années, les analyses cytogénétiques puis de séquençage ont
permis de caractériser les altérations génétiques les plus récurrentes des LAM-M7 et ont
révélé une grande hétérogénéité génétique (177–181). Les altérations génétiques
observées sont très différentes entre les LAM-M7 adultes et pédiatriques (Figure 22).
Figure 22 : Altérations génétiques entre les LAM-M7 pédiatriques et adultes.
Les mutations associées aux LAM-M7 pédiatriques de novo ne sont pas retrouvées dans les LAM-M7 de l’adulte en dehors des rares mutations du gène GATA1. (Issue de (179)).

66
Les altérations associées aux LAM-M7 pédiatriques, principalement de novo, sont détaillées
ci-dessous.
a/ LAM-M7 associées aux trisomies 21 constitutionnelles
Les enfants avec un syndrome de Down (trisomie 21 constitutionnelle) présentent un
risque 500 fois plus élevé de développer une LAM-M7 que le reste de la population Pour
revues (182,183). Un certain nombre de nouveau-nés (10%) développent une leucémie
transitoire appelée TMD (pour transient myeloid disorder), caractérisée par la présence de
mégacaryoblastes immatures. Dans la majorité des cas, cette leucémie transitoire régresse
spontanément dans les 3 mois. Cependant 20 à 30% des patients développent une LAM-M7
avant l’âge de 4 ans (182). Le développement leucémique résulte de l’acquisition d’une
succession d’évènements oncogéniques.
La leucémie transitoire TMD est associée à des mutations du gène GATA1, ayant lieu in utero,
engendrant la formation d’une protéine tronquée de l’extrémité N-terminale, appelée
GATA1s (184). Les patients développant une LAM-M7 présentent des mutations
additionnelles (moyenne de 5,8 mutations somatiques par patients) affectant des gènes
codant pour des intermédiaires de voie de signalisation (47%), des régulateurs
épigénétiques (65%) incluant les cohésines, CTCF et les protéines régulant les marques
épigénétiques (185).
Les DS-LAM-M7 résultent donc d’une succession d’altérations incluant: 1- une trisomie du
chromosome 21, 2- des mutations somatiques du gène GATA1 et 3- l’acquisition de
mutations additionnelles (Figure 23). On peut noter que la combinaison de ces trois types
d'altérations est retrouvée dans ~10% des LAM-M7 de novo à ceci près que la trisomie 21
est, dans ces cas-là, acquise de façon somatique et non constitutive.

67
Figure 23 : Développement leucémique des DS-LAM-M7.
L’acquisition de mutations du gène GATA1 in utero chez les enfants présentant une trisomie 21 constitutive (syndrome de Down) conduit au développement d’une leucémie transitoire (TMD) qui régresse de façon spontanée dans la plupart des cas. L’apparition de mutations additionnelles affectant des gènes codant pour des intermédiaires de voie de signalisation (Kinase), des régulateurs épigénétiques (PRC2) ainsi que les cohésines, conduit au développement d’une leucémie aiguë mégacaryoblastique appelée DS-LAM-M7. (Issue de (186)).
Dans l’ensemble, ces patients présentent une meilleure réponse à des doses faibles de
chimiothérapies et un meilleur pronostic que les LAM-M7 de novo (179), pour revue (182).
b/ LAM-M7 de novo
Dans le groupe de LAM7 de novo, la majorité des cas (75%) présente des altérations
chromosomiques conduisant à l’expression d’oncogène de fusion (Tableau 2).
- Oncogènes de fusion :
La première anomalie récurrente identifiée à la fin des années 1980 a été une translocation
entre le chromosome 1 et 22, visible en cytogénétique classique (188,189). L’âge très jeune
des patients et l’observation de cette translocation chez des jumeaux monozygotes
suggèrent une apparition in utero (190). Cette translocation code pour une protéine de
fusion OTT-MAL impliquant la quasi-totalité des deux protéines prédites (177).
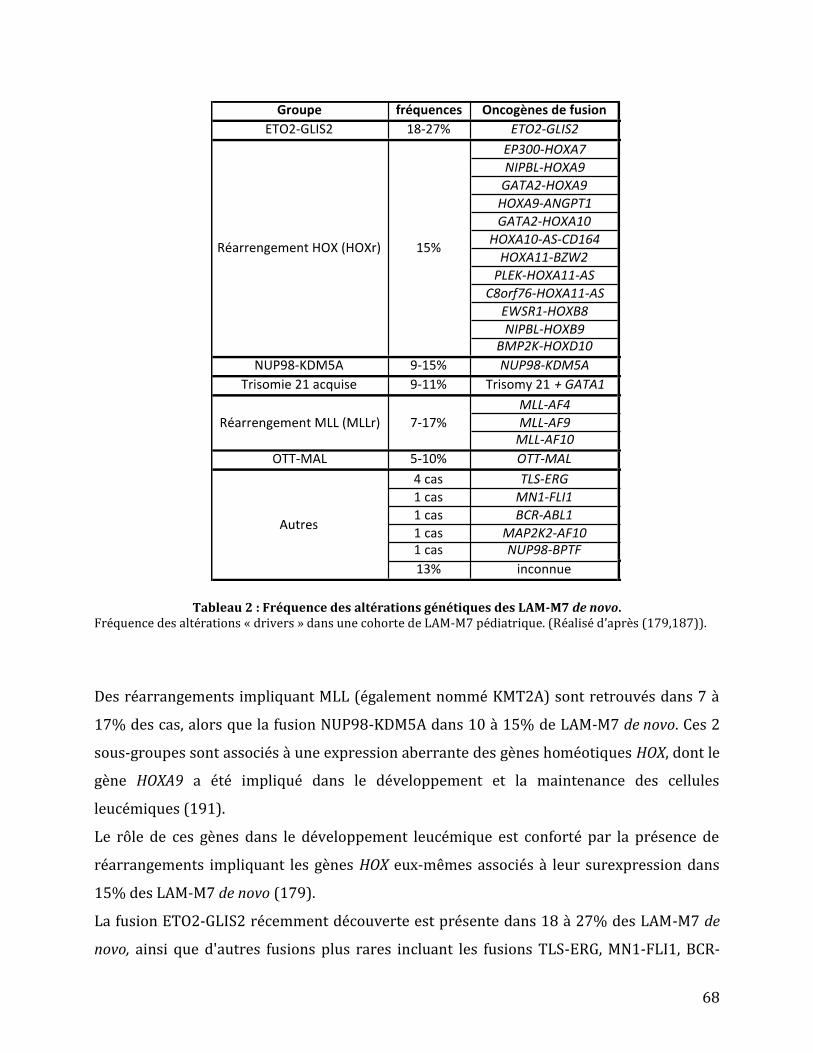
68
Tableau 2 : Fréquence des altérations génétiques des LAM-M7 de novo. Fréquence des altérations « drivers » dans une cohorte de LAM-M7 pédiatrique. (Réalisé d’après (179,187)).
Des réarrangements impliquant MLL (également nommé KMT2A) sont retrouvés dans 7 à
17% des cas, alors que la fusion NUP98-KDM5A dans 10 à 15% de LAM-M7 de novo. Ces 2
sous-groupes sont associés à une expression aberrante des gènes homéotiques HOX, dont le
gène HOXA9 a été impliqué dans le développement et la maintenance des cellules
leucémiques (191).
Le rôle de ces gènes dans le développement leucémique est conforté par la présence de
réarrangements impliquant les gènes HOX eux-mêmes associés à leur surexpression dans
15% des LAM-M7 de novo (179).
La fusion ETO2-GLIS2 récemment découverte est présente dans 18 à 27% des LAM-M7 de
novo, ainsi que d'autres fusions plus rares incluant les fusions TLS-ERG, MN1-FLI1, BCR-

69
ABL1, MAP2K2-AF10 (178–181,187). Finalement, 10 à 15% des patients ne présentent pas
de mutation causale identifiée à ce jour.
- Autres mutations :
En plus des oncogènes de fusion, certains sous-groupes de patients présentent des
mutations additionnelles fréquentes (Figure 24).
Figure 24 : Mutations additionnelles des LAM-M7. Représentation de la fréquence des mutations additionnelles observées dans les différents sous-groupes de LAM-M7.
La trisomie du chromosome 21 est retrouvée dans tous les sous-groupes moléculaires de
LAM-M7 avec une fréquence variable de 11% (sous-groupe "OTT-MAL") à 100% (sous-
groupe "Trisomie 21 acquise"). Plusieurs gènes du chromosome 21 ont été proposés pour
jouer un rôle dans le processus de leucémogenèse comme ERG, impliqué dans plusieurs
cancers (cancer de la prostate, sarcome d’Ewing, leucémies) et dont la surexpression est

70
associée à un mauvais pronostic. ERG est un facteur de transcription de la famille ETS ayant
un rôle important dans l’établissement de l’hématopoïèse, et nécessaire à la
mégacaryopoïèse. Son inactivation dans des lignées cellulaires de LAM-M7 (CMY, Meg-01,
tableau 3) impacte la prolifération cellulaire et l’expression de marqueurs
mégacaryocytaires (192), alors que sa surexpression dans des modèles murins induit des
leucémies aiguës lymphoblastiques ou des leucémies érythro-mégacaryoblastiques
(143,192). Ceci suggère que le dosage génique de ERG contribue à la leucémogenèse.
Les mutations GATA1 observées chez les patients DS-LAM-M7 sont aussi retrouvées dans
des sous-groupes de LAM-M7 de novo comme le groupe de trisomie 21 acquise (100%),
NUP98-KDM5A (22%) et les réarrangements MLL (7%). GATA1 interagit avec RB1 et E2F2
pour inhiber la prolifération et induire la différenciation terminale des progéniteurs
érythroïdes (193). De façon intéressante, des mutations inhibitrices de RB1 (délétions,
mutations ponctuelles) sont fréquemment retrouvées, notamment dans le groupe NUP98-
KDM5A (89%) et des réarrangements HOX (15%). Ces mutations sont mutuellement
exclusives avec les mutations GATA1, laissant supposer un rôle de ce complexe dans le
développement des LAM-M7.
Les mutations affectant des intermédiaires de voie de signalisation stimulent généralement
la survie et la prolifération des cellules. Les gènes JAK (JAK1, 2 et 3), STAT, RAS, MPL sont
mutés dans une proportion non négligeable (77% des réarrangements HOX, 50% des
réarrangements MLL, et des trisomies 21 acquises, 47% des DS-LAM-M7, 44% du groupe
« autres ») des différents groupes de LAM-M7.
Les membres du complexe des cohésines sont affectés par des mutations dans 13% des
LAM. L’haploinsuffisance des cohésines induit une augmentation de l’autorenouvellement
des progéniteurs, ainsi qu’une altération de l’accessibilité de la chromatine modifiant
l’expression génique Pour revue (194). Dans le groupe des LAM-M7 ces mutations sont
encore plus représentées que dans la plupart des LAM.

71
3/ Modélisation fonctionnelle des LAM-M7
L’utilisation de xénogreffe de cellules de patients LAM-M7 dans des souris
immunodéficientes ainsi que les lignées cellulaires permettent de modéliser des LAM-M7
humaines in vivo et in vitro (178). Ces modèles permettent l’amplification des cellules
cancéreuses, l’étude moléculaire de ces cellules et permettent de tester in vivo et in vitro de
nouvelles stratégies thérapeutiques. L’efficacité des inhibiteurs de la kinase Aurora A a pu
être testée in vivo (195).
Les lignées cellulaires et les modèles de xénogreffe sont des modèles leucémiques déjà
établis. Pour comprendre l’impact d’une altération génétique sur les différents stades de
l’hématopoïèse, sur le progéniteur cible ou encore la nécessité de coopération pour le
développement leucémique, d’autres modèles d’études sont nécessaires comme les cellules
ES et iPS ou encore les modèles murins. Ces derniers seront plus amplement décrits par la
suite.
Les cellules ES et iPS (cellules somatiques reprogrammées) peuvent être utilisées pour
étudier les conséquences des certaines altérations génétiques sur l’ontogénie
hématopoïétique humaine (Tableau 3).
a/ LAM7 associées aux trisomies 21 constitutionnelles :
Différents modèles murins transgéniques modélisant la trisomie 21 ont été réalisés et
analysés mais aucun ne développe de leucémie (200–202). La coopération de la trisomie 21
avec les mutations GATA1 engendre des anomalies de l'hématopoïèse mais, là encore, sans
développement leucémique (201). Le premier modèle de DS-LAM-M7 a été obtenu par
l'expression de trois évènements génétiques (trisomie 21 + mutation GATA1s + mutation
MPLW515L), confirmant l’importance d'une coopération oncogénique pour le développement
in vivo d'une LAM7 correspondant à ce sous-groupe moléculaire (204).
Une autre équipe a montré l’implication du gène ERG, localisé sur le chromosome 21, dans
le développement des leucémies lymphoïdes et des leucémies érythro-mégacaryocytaires
dans des analyses de transplantation murine (143,192).
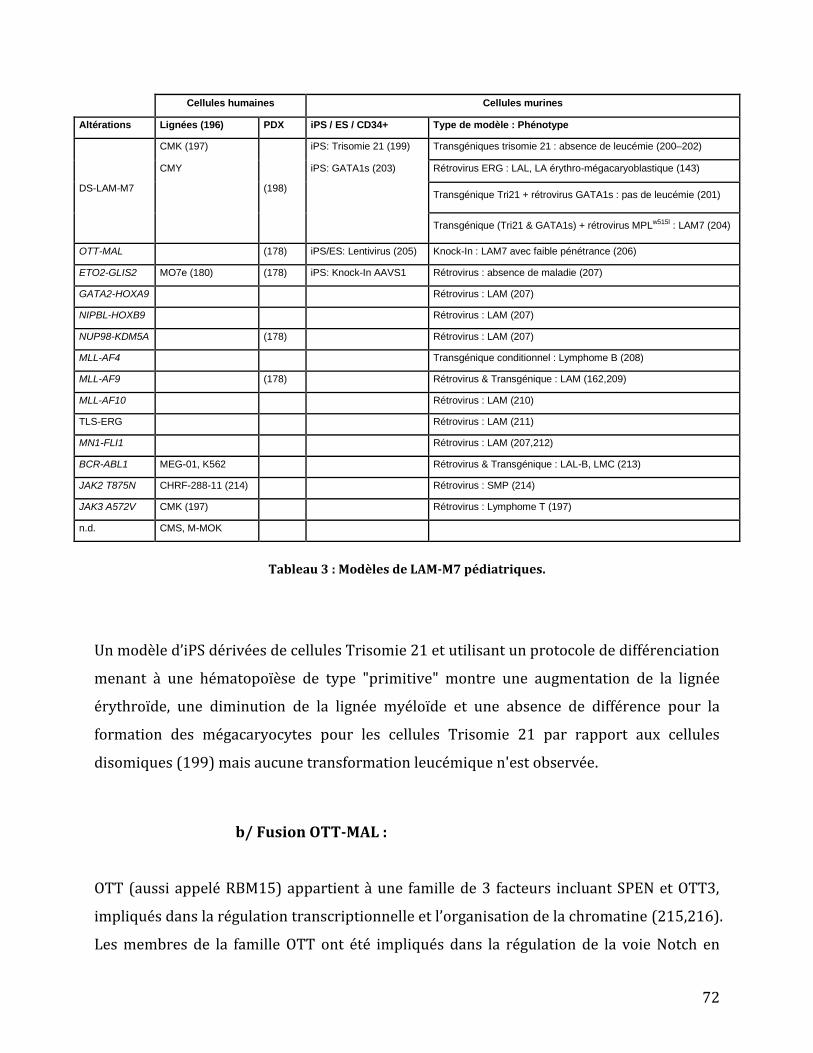
72
Cellules humaines Cellules murines
Altérations Lignées (196) PDX iPS / ES / CD34+ Type de modèle : Phénotype
DS-LAM-M7
CMK (197)
(198)
iPS: Trisomie 21 (199) Transgéniques trisomie 21 : absence de leucémie (200–202)
CMY iPS: GATA1s (203) Rétrovirus ERG : LAL, LA érythro-mégacaryoblastique (143)
Transgénique Tri21 + rétrovirus GATA1s : pas de leucémie (201)
Transgénique (Tri21 & GATA1s) + rétrovirus MPLw515l : LAM7 (204)
OTT-MAL (178) iPS/ES: Lentivirus (205) Knock-In : LAM7 avec faible pénétrance (206)
ETO2-GLIS2 MO7e (180) (178) iPS: Knock-In AAVS1 Rétrovirus : absence de maladie (207)
GATA2-HOXA9 Rétrovirus : LAM (207)
NIPBL-HOXB9 Rétrovirus : LAM (207)
NUP98-KDM5A (178)
Rétrovirus : LAM (207)
MLL-AF4 Transgénique conditionnel : Lymphome B (208)
MLL-AF9 (178) Rétrovirus & Transgénique : LAM (162,209)
MLL-AF10 Rétrovirus : LAM (210)
TLS-ERG Rétrovirus : LAM (211)
MN1-FLI1 Rétrovirus : LAM (207,212)
BCR-ABL1 MEG-01, K562 Rétrovirus & Transgénique : LAL-B, LMC (213)
JAK2 T875N CHRF-288-11 (214) Rétrovirus : SMP (214)
JAK3 A572V CMK (197) Rétrovirus : Lymphome T (197)
n.d. CMS, M-MOK
Tableau 3 : Modèles de LAM-M7 pédiatriques.
Un modèle d’iPS dérivées de cellules Trisomie 21 et utilisant un protocole de différenciation
menant à une hématopoïèse de type "primitive" montre une augmentation de la lignée
érythroïde, une diminution de la lignée myéloïde et une absence de différence pour la
formation des mégacaryocytes pour les cellules Trisomie 21 par rapport aux cellules
disomiques (199) mais aucune transformation leucémique n'est observée.
b/ Fusion OTT-MAL :
OTT (aussi appelé RBM15) appartient à une famille de 3 facteurs incluant SPEN et OTT3,
impliqués dans la régulation transcriptionnelle et l’organisation de la chromatine (215,216).
Les membres de la famille OTT ont été impliqués dans la régulation de la voie Notch en

73
s’associant au facteur RBPJ (217). MAL (aussi appelé MKL1) est un coactivateur
transcriptionnel de SRF (Serum response factor) capable de moduler ces gènes cibles. SRF
et MAL jouent un rôle dans la mégacaryopoïèse (218) dont l’inactivation de MAL dans un
modèle murin induit une thrombopénie avec une augmentation des progéniteurs
mégacaryocytaires dans la moelle osseuse (219).
La fusion OTT-MAL induit une LAM-M7 dans un modèle murin Knock-In avec une faible
pénétrance (206) en activant de façon anormale les gènes cibles de Notch régulés par RBPJ.
Cette dérégulation transcriptionnelle est retrouvée chez les patients OTT-MAL.
c/ Fusion NUP98-KDM5A :
NUP98 est une protéine du pore nucléaire, dont le rôle principal est le transport des
macromolécules entre le noyau et le cytoplasme. Le gène est fréquemment réarrangé dans
une grande variété de leucémies pédiatriques (3,8%) dont le pronostic est défavorable. Il
existe une trentaine de partenaires différents (220) dont KDM5A. KDM5A (aussi appelé
JARID1A) est une histone déméthylase, dont la surexpression est retrouvée dans plusieurs
cancers et qui a été impliquée dans le processus de métastase et de résistance aux
traitements (221). La fusion NUP98-KDM5A est une fusion récurrente des LAM-M7
pédiatriques (179,222). La surexpression de NUP98-KDM5A dans des progéniteurs murins
engendre une leucémie myéloïde (207).
Une fusion NUP98-BPTF a récemment été décrite chez un jeune enfant LAM-M7 (223).
d/ Fusions MLL ou HOX :
Le gène MLL code pour une histone méthyltransférase spécifique de la lysine 4 de l’histone
3 (H3K4), qui joue un rôle important dans la régulation transcriptionnelle, notamment des
gènes homéotiques HOX (dont HOXA7, HOXA9). Les gènes HOX sont impliqués dans la
régulation hématopoïétique, et sont fortement exprimés dans les progéniteurs précoces.
MLL est fusionné avec plus de 80 partenaires différents dans les leucémies humaines. Ces

74
fusions engendrent une dérégulation des gènes HOX, participant ainsi au développement
leucémique (224).
Trois partenaires de MLL sont retrouvés dans les LAM-M7 : AF4 (225), AF9 (178) et AF10
(226). Ces protéines font partie d’un complexe liant la polymérase II participant à l’étape
d’élongation de la transcription. Ces différentes fusions sont retrouvées dans d’autres sous-
types de leucémies, et leur expression dans différents modèles murins induit des leucémies
myéloïdes ou lymphoïdes sans phénotype mégacaryoblastique (162,208,210).
De même pour les fusions impliquant les gènes homéotiques HOX eux-mêmes, leur
expression engendre une LAM myéloïde chez la souris (207).
3/ La fusion ETO2-GLIS2
La fusion ETO2-GLIS2 est l’altération génétique la plus fréquemment retrouvée dans les
LAM-M7 de novo, et est associée à un pronostic particulièrement défavorable (178,180,227).
Elle résulte d’une inversion du chromosome 16 qui code pour une protéine de fusion
exprimant la grande majorité des gènes ETO2 et GLIS2. Plusieurs variants ont été observés,
impliquant les exons 10, 11 et 12 de ETO2 et les exons 1, 2 et 3 de GLIS2 (Figure 25).
La fusion ETO2-GLIS2 a été initialement découverte dans des LAM-M7 pédiatriques. Masetti
et coll. ont également identifié cette fusion dans d’autres sous-types de LAM M0 à M5 de la
classification FAB, excluant les groupes M3 et M6 (227), suggérant que cette fusion n’est pas
restreinte au sous-groupe des LAM-M7. Cette altération génétique est associée à un
pronostic particulièrement défavorable indépendamment du sous-type de LAM.

75
Figure 25 : Représentation schématique des gènes ETO2 et GLIS2 et des transcrits de fusion. Les gènes GLIS2 et ETO2 sont localisés sur le chromosome 16. Il existe plusieurs variants de la fusion ETO2-GLIS2 impliquant les exons 10, 11 et 12 de ETO2 et 1, 2 et 3 de GLIS2. La fusion la plus fréquemment retrouvée est la fusion "11-3" indiquée par une étoile rouge.
La fusion ETO2-GLIS2 implique deux régulateurs transcriptionnels.
Le gène ETO2, aussi nommé MTG16 ou CBFA2T3, code une protéine de la famille ETO qui est
exprimée dans toutes les cellules hématopoïétiques. ETO2 est un cofacteur transcriptionnel
qui interagit avec les facteurs de transcription du complexe Heptad (SCL, RUNX1, GATA…).
Une inactivation d'Eto2 chez la souris est associée à une perte de fonction/maintien des
HSC (228,229). ETO2 joue aussi un rôle clé dans la régulation de la mégacaryopoïèse (115)
en réprimant certains gènes impliqués dans la différenciation terminale (comme GATA1 et
PF4)(230).
GLIS2 est un facteur de transcription de la famille GLI impliqué dans la voie de signalisation
Hedgehog. Les patients présentant la fusion ETO2-GLIS2 montrent une signature
transcriptionnelle de surexpression des gènes cibles de cette voie (178). GLIS2 est
fortement exprimé dans le rein et serait impliqué dans le maintien et l’architecture du rein

76
(231). Des mutations de GLIS2 sont associées à une fibrose et une atrophie du rein appelée
néphronophtisie (232). Une étude récente d'inactivation par shRNA dans une approche de
criblage a impliqué GLIS2 dans la régulation de l’hématopoïèse, et plus spécifiquement
comme régulateur positif de la capacité de repopulation des progéniteurs hématopoïétiques
lors de greffe (233). Indépendamment, l'expression de GLIS2 aurait un effet défavorable sur
le processus de reprogrammation cellulaire (234).
Bien que l'expression de la fusion ETO2-GLIS2 conduise à une augmentation de
l’autorenouvellement des progéniteurs hématopoïétiques murin in vitro (180), aucun
modèle de leucémogenèse in vivo par expression de la fusion ETO2-GLIS2 n'a été rapporté à
ce jour.

77

78
RESULTATS

79
CONTEXTE ET OBJECTIFS
La fusion ETO2-GLIS2 a été décrite pour la première fois en 2012 par plusieurs laboratoires
(178,180). Au sein de notre équipe, l'identification et la caractérisation de cette fusion a été
réalisée par une approche combinée de xénogreffe de cellules de patients LAM-M7 et
d'analyse génétique par RNAseq (178). Avant le commencement de ma thèse, j'ai participé à
ce travail par la caractérisation de la récurrence de la fusion dans une cohorte de 31
patients LAM-M7 de novo pédiatriques en collaboration avec Jean-Pierre Bourquin (Zurich,
Suisse). L'identification des patients ETO2-GLIS2+ (26% des LAM7 pédiatriques de novo),
pour lesquels nous avions déjà des profils d'expression, a permis de mettre en évidence que
la fusion ETO2-GLIS2 est associée à une signature transcriptionnelle distincte des autres
LAM7 pédiatriques (235).
La fréquence élevée de la fusion ETO2-GLIS2 au sein des LAM7 pédiatriques de novo, son
association avec un mauvais pronostic (180) et la signature transcriptionnelle spécifique
constituaient des éléments moteurs pour l'étude des mécanismes de transformation par
cette fusion qui étaient inconnus.
Les données fonctionnelles disponibles, détaillées dans l'introduction générale, indiquaient
que ETO2 est un cofacteur transcriptionnel qui interagit avec les facteurs de transcription
du complexe Heptad, et est impliqué dans le maintien, la prolifération et le devenir des
progéniteurs hématopoïétiques. En revanche, aucune donnée n'était disponible pour GLIS2
et dehors de son appartenance à une famille de facteurs de transcription GLI/GLIS/ZIC
incluant les facteurs GLI impliqués dans la réponse transcriptionnelle à une stimulation de
la voie de signalisation Hedgehog. L'hypothèse était donc que la fusion ETO2-GLIS2
implique deux régulateurs transcriptionnels et entraîne une dérégulation de l'expression
génique contribuant à la transformation leucémique.
Notre objectif a donc été de caractériser la signature transcriptionnelle induite par
l'expression de la fusion pour identifier ses gènes cibles, de définir les contributions
relatives des facteurs ETO2 et GLIS2 et de mieux comprendre les mécanismes moléculaires

80
à la base de cette dérégulation transcriptionnelle. Les résultats obtenus ont été publiés dans
l'Article 1.
D'autre part, les capacités de transformation de la fusion ETO2-GLIS2 n'étaient pas connues.
En particulier, les bases cellulaires et/ou moléculaires de l'association spécifique de cette
fusion, tout comme d'autres oncogènes de fusion, avec les leucémies pédiatriques restent
inexpliquées.
Une autre observation importante, rapportée par l'équipe de F Locatelli, rapidement après
son identification, était que la fusion ETO2-GLIS2, initialement découverte chez des enfants
diagnostiqués pour une LAM-M7, est également retrouvée dans d’autres sous-types de LAM
pédiatriques (LAM-M0 à M5).
Afin d'obtenir des données expérimentales permettant de comprendre les déterminants de
la transformation et des phénotypes induits par ETO2-GLIS2, j'ai analysé d'autres
échantillons de patients et développé plusieurs approches de modélisation de l'expression
de la fusion ETO2-GLIS2 chez la souris. Les résultats de ces approches sont l'objet de
l'Article 2 qui est en cours de considération pour publication.
Article 1 :
Au cours de ce travail, réalisé en collaboration avec une autre étudiante en thèse (Cathy
Ignacimouttou) et une post-doctorante (Cécile Thirant) nous avons cherché à répondre à
plusieurs questions.
1-Quelles sont les conséquences de l'expression de la fusion sur la différenciation
mégacaryocytaire et l’autorenouvellement des progéniteurs hématopoïétiques ?
Pour cela, j'ai construit des vecteurs rétroviraux ETO2-GLIS2, ETO2 et GLIS2 qui ont été
utilisés pour transduire des progéniteurs hématopoïétiques murins et montrer que le
phénotype mégacaryocytaire est imposé par la partie GLIS2 de la fusion alors que
l’augmentation de l’autorenouvellement est due à une combinaison des fonctions ETO2 et
GLIS2.

81
2-Quels sont les gènes cibles de la fusion ?
Pour cela, nous avons surexprimé la fusion, ainsi que ETO2 et GLIS2 seuls, dans une lignée
humaine érythro-mégacaryocytaire ne présentant pas de fusion (HEL). L'expression de la
fusion dans ces cellules induit une forte dérégulation transcriptionnelle corrélée à la
signature retrouvée chez les patients. Certains facteurs impliqués dans le complexe avec
ETO2 sont fortement dérégulés dont GATA1 retrouvé diminué et ERG fortement augmenté.
Une dérégulation similaire a été retrouvée chez les patients ET02-GLIS2 par rapport aux
autres patients LAM7 pédiatriques.
3-Comment la fusion dérégule l'expression de ses gènes cibles ?
Pour cela, nous devions ensuite caractériser les sites de fixation de cette fusion, impliquant
deux régulateurs transcriptionnels, sur l'ADN. Une limitation importante était 1-l'absence
d'anticorps spécifique pour GLIS2 (plusieurs anticorps commerciaux ayant été testés sans
succès) et 2-le fait que, dans les cellules ETO2-GLIS2+, un anticorps ETO2 reconnaît à la fois
la fusion mais également ETO2 endogène, exprimé à partir de la deuxième copie d'ETO2
sauvage. Pour note, ce problème n'est pas présent pour GLIS2 dont l'expression est absente
de la différentiation hématopoïétique. J'ai donc réalisé une approche de modification ciblée
du génome de type CRISPR-Cas9 dans la lignée MO7e, dérivée de patient et exprimant de
façon endogène la fusion ETO2-GLIS2, afin d'introduire la GFP en fusion en C-terminal de
ETO2-GLIS2. Cette approche a permis de valider la localisation nucléaire de la fusion par
imagerie et la réalisation d’immunoprécipitation spécifique de la fusion avec un anticorps
anti-GFP pour des approches d'immunoprécipitation de chromatine (ChIPSeq).
La fusion ETO2-GLIS2 se fixe bien à l’ADN sur 1871 régions dans les MO7e. L’analyse des
motifs observés dans ces régions, révèle plusieurs motifs des partenaires d’ETO2 comme
ERG, GATA et RUNX ainsi que des motifs attribués à GLIS2. Ces résultats suggèrent la
fixation de la fusion sur l’ADN via la partie GLIS2 ainsi que par le complexe ETO2
notamment avec le partenaire ERG. En comparant les sites connus de fixation des
partenaires d’ETO2 dans les mégacaryocytes, aux sites de fixation de la fusion dans les
MO7e, nous avons pu voir que plus de la moitié des sites de la fusion sont des sites non
connus (de novo). En corrélant avec l’expression transcriptionnelle obtenue dans les HEL,

82
nous avons pu observer que les gènes associés des sites connus du complexe ETO2 sont
majoritairement inhibés alors que les gènes des nouveaux sites sont plutôt surexprimés en
présence de la fusion ETO2-GLIS2. Dans ces analyses, la fusion impose donc une fixation et
une régulation transcriptionnelle différente aux complexes transcriptionnels incluant ETO2.
Certaines marques épigénétiques sont fortement retrouvées associées aux sites de fixation
d’ETO2-GLIS2 comme la marque H3K27ac, associée aux enhancers et super-enhancers (SE).
Les SE sont des régions de fixation aux facteurs de transcription associés aux gènes clés
spécifiques de l’identité cellulaire. Dans les MO7e, 483 SE ont été observés, qui réguleraient
l’expression de 828 gènes putatifs dont ETO2, ERG et KIT. La fusion se lie à 1/3 des SE, et les
gènes associés sont majoritairement surexprimés (ex: ERG).
Le complexe ETO2 semble jouer un rôle important et particulièrement le facteur ERG,
surexprimé par la fusion et colocalisant fréquemment avec elle. L’inactivation d’ERG par
CRISPR-Cas9 dans les MO7e ou des cellules de patient ETO2-GLIS2 a validé son rôle dans le
maintien des cellules leucémiques et son implication dans la régulation de certains gènes
cibles de la fusion (ex: KIT).
4-Comment est régulée la stabilité de ces complexes protéiques ?
Nous sommes partis de l'observation que les protéines de la famille ETO, dont fait partie
ETO2, présentent un domaine NHR2 indispensable pour leur oligomérisation et pour le
recrutement de co-facteurs. Il a été montré, avec la fusion AML1-ETO, l’importance de ce
domaine sur l’autorenouvellement des cellules leucémiques, en utilisant un peptide nommé
NC128, contenant la séquence protéique du domaine NHR2 de ETO.
Nous avons montré que la fusion ETO2-GLIS2 peut également interagir avec elle-même et
avec ETO2. Étant donnée la grande homologie de séquence protéique entre ETO et ETO2 au
niveau du domaine NHR2, nous avons ensuite caractérisé les conséquences de l'expression
du peptide NC128 dans les cellules leucémiques ETO2-GLIS2. L’expression du NC128 dans
la lignée MO7e modifie la signature transcriptionnelle imposée par la fusion avec
notamment une diminution d’ERG et une surexpression de GATA1. De plus, le NC128 inhibe
la prolifération cellulaire en réduisant le cycle cellulaire des cellules et en augmentant
l’apoptose. L'expression du NC128 dans des cellules de patients ETO2-GLIS2 (transduction

83
lentivirale) inhibe également le développement leucémique dans des modèles de
xénotransplantation.
Article 2 :
Au cours de ce travail, j'ai cherché à répondre aux questions suivantes:
1-Quelle est la fréquence de la fusion ETO2-GLIS2 au sein des LAM pédiatriques ?
Afin de valider l’association de la fusion ETO2-GLIS2 avec les autres sous-types de LAM, j’ai
tout d'abord recherché sa récurrence dans une cohorte nationale de LAM pédiatriques en
collaboration avec les réseaux de la SFCE et les responsables du protocole ELAM02. La
fusion ETO2-GLIS2 a été retrouvée dans les sous-types de LAM-M0 à M5 en plus des LAM-
M7 avec une fréquence de 4,7%. De façon intéressante, nous avons pu observer que les
patients ETO2-GLIS2 LAM-M7 sont diagnostiqués très jeunes (moyenne d’âge de 1,7 ans)
par rapport aux autres sous-types (moyenne d’âge de 8,6 ans). Pour mieux caractériser les
cellules leucémiques, j’ai réalisé des xénogreffes des cellules de patients dans des souris
immunodéficientes (NSG) lorsque des cellules viables étaient disponibles. Pour un patient
diagnostiqué LAM-M2, j'ai observé un phénotype original. En effet, la majorité des blastes
leucémiques sont constitués d’une population immature (CD34+CD41-CD15-) mais une
petite fraction de cellules présente, d'une part, un phénotype mégacaryoblastique
(CD41+CD15-) et, d'autre part, un phénotype myéloïde (CD41-CD15+) suggérant une
capacité de différenciation soit myéloïde soit mégacaryocytaire. Cette hypothèse fut
confirmée par l'observation que seule la population immature est capable de développer
une leucémie dans des souris secondaires et régénère les 3 populations. Dans l'ensemble,
ces différentes observations suggèrent l’implication du stade de développement et/ou du
type de progéniteur cible sur le phénotype des leucémies ETO2-GLIS2.
2-L'expression de la fusion ETO2-GLIS2 induit-elle des leucémies dans un modèle
expérimental ?
Pour tester ces hypothèses expérimentalement, j’ai développé, en collaboration avec le
groupe de Jürg Schwaller, un modèle murin transgénique d’expression inductible de la

84
fusion ETO2-GLIS2 sous le contrôle de la doxycycline. L’induction de la fusion dans des
femelles induit des hémopathies létales avec une forte pénétrance (96,9%, 31/32 souris)
qui peuvent être classées en 3 groupes : phénotype immature (9/19), phénotype
mégacaryoblastique (expression du CD41, 4/19) ou phénotype myéloïde (expression des
marqueurs CD11b et Gr1, 6/19).
3-Quel est l'impact du stade de développement et du type de cellule hématopoïétique ciblé
pour la leucémogenèse et le phénotype des pathologies ?
Pour caractériser l'impact du stade de développement sur le phénotype, j'ai exprimé la
fusion dans des progéniteurs du foie fœtal ou de la moelle osseuse adulte. Le phénotype
mégacaryoblastique est fortement associé aux progéniteurs fœtaux et décroît au cours du
développement à l’inverse du phénotype myéloïde majoritaire dans les progéniteurs
adultes. Cette observation est valable pour d’autres oncogènes retrouvés dans différents
sous-types de LAM comme MLL-AF9 et NUP98-KDM5A, pour lesquels l'expression dans des
progéniteurs issus de foie fœtal E12.5 favorise le phénotype mégacaryoblastique.
Pour montrer que l’origine cellulaire influence également le phénotype, j'ai exprimé la
fusion dans différents types de HSC et progéniteurs purifiés. Les progéniteurs du foie fœtal
expriment exclusivement (HSC, MPP2) ou majoritairement (MPP3) un phénotype
mégacaryocytaire, alors que chez l’adulte seules les HSC présentent un potentiel
mégacaryoblastique exclusif. Les autres progéniteurs (MPP2, MPP3, MPP4) expriment de
façon graduelle les marqueurs myéloïdes. A l’état unicellulaire, j'ai montré que 48% des
progéniteurs MPP3 adultes exprimant la fusion possèdent une capacité d'engagement à la
fois de la différenciation myéloïde et mégacaryocytaire, comme observé chez le patient
LAM-M2. Ces résultats démontrent l’importance du type de progéniteur et du stade de
développement sur le phénotype des leucémies induit par un même oncogène.
Finalement, j’ai modélisé, à partir de HSC fœtales exprimant la fusion, le développement in
vivo d'une leucémie rapide (27 jours) et agressive présentant les caractéristiques des LAM-
M7 humaines. En revanche, les HSC adultes exprimant la fusion conduisent au
développement de leucémies avec une latence longue (>312 jours) et un phénotype
hétérogène. La possibilité d'éteindre l'expression de la fusion dans les cellules transformées

85
(par retrait du traitement à la doxycycline) a ensuite révélé, par greffe secondaire, qu'une
partie des cellules leucémiques ne présentant plus d'expression de la fusion conservent des
caractéristiques de reconstitution hématopoïétique à long-terme (>6 mois) attribuées
uniquement aux HSC normales.
4-Quelles sont les conséquences moléculaires de la fusion ETO2-GLIS2 dans les HSC ?
Pour cela, j'ai mené une analyse RNAseq à l’état unicellulaire des HSC en allant réaliser cette
technique, non disponible à l'institut, dans le laboratoire de Berthold Gottgens. Les analyses
bio-informatiques, réalisées en collaboration avec l'expertise de Esteve Noguera dans
l'équipe, ont montré une dérégulation transcriptionnelle importante de la fusion dans les
HSC dès 24h post-induction de la fusion. La signature obtenue corrèle avec celle des
patients EG LAM-M7 (ex: surexpression de Kit). Une prédiction de l’activité des facteurs de
transcription permet de suggérer que plusieurs facteurs sont régulés par la fusion EG,
incluant les facteurs impliqués dans la différenciation myéloïde (IRF8, GFI1b),
mégacaryocytaire (FLI1, IKZF1) ainsi que certains membres du complexe heptad (LDB1,
ERG, GATA2). Ces résultats sont compatibles avec l’observation d’un blocage précoce de la
différenciation des HSC vers la lignée mégacaryocytaire.
Fonctionnellement, j'ai pu mettre en évidence l’importance de certains facteurs de
transcription sur le phénotype myéloïde (rôle positif de CEBPA) ou mégacaryoblastique
(ex: ERG et GATA1) de la transformation par la fusion ETO2-GLIS2.

86
ARTICLE 1
ETO2-GLIS2 hijacks transcriptional complexes to drive cellular identity and self-renewal in pediatric acute megakaryoblastic
leukemia Cécile Thirant 1,2, #, Cathy Ignacimouttou 1,3, #, Cécile K. Lopez 1,4, #, M’Boyba Diop 2, Lou Le Mouël 2,4, Clarisse Thiollier 2,3, Aurélie Siret 1,2, Phillipe Dessen 1,2, Zakia Aid 1,2, Julie Rivière 1,2, Philippe Rameau 2, Céline Lefebvre2, Mehdi Khaled2, Guy Leverger 5, Paola Ballerini 5, Arnaud Petit 5, Hana Raslova1,2, Catherine L. Carmichael6, Benjamin T. Kile6, Eric Soler7, John D. Crispino8, Christian Wichmann9, Françoise Pflumio 7, Jürg Schwaller 10, William Vainchenker 1,2, Camille Lobry 1,2, Nathalie Droin 1,2,11, Olivier A. Bernard 1,2,4, Sébastien Malinge 1,2, Thomas Mercher 1,2,3,4, * #Contributed equally Cancer Cell. 2017. Mar 13;31(3):452-465. Commentaires: -Cancer Cell. 2017. ETO2-GLIS2: A Chimeric Transcription Factor Drives Leukemogenesis through a Neomorphic Transcription Network. Wheat JC, Steidl U. -Cancer Discovery. 2017. ETO2–GLIS2 Drives the Transcriptional Program Underlying AMKL. DOI: 10.1158/2159-8290

87

88

89

90
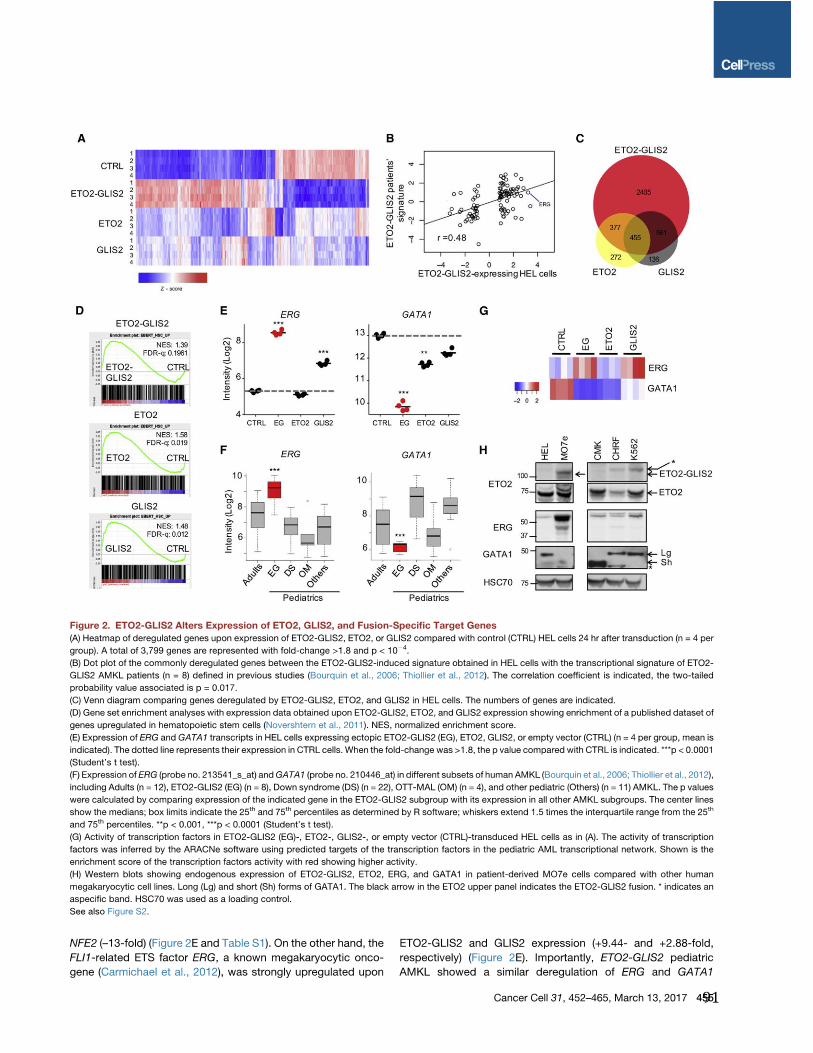
91

92
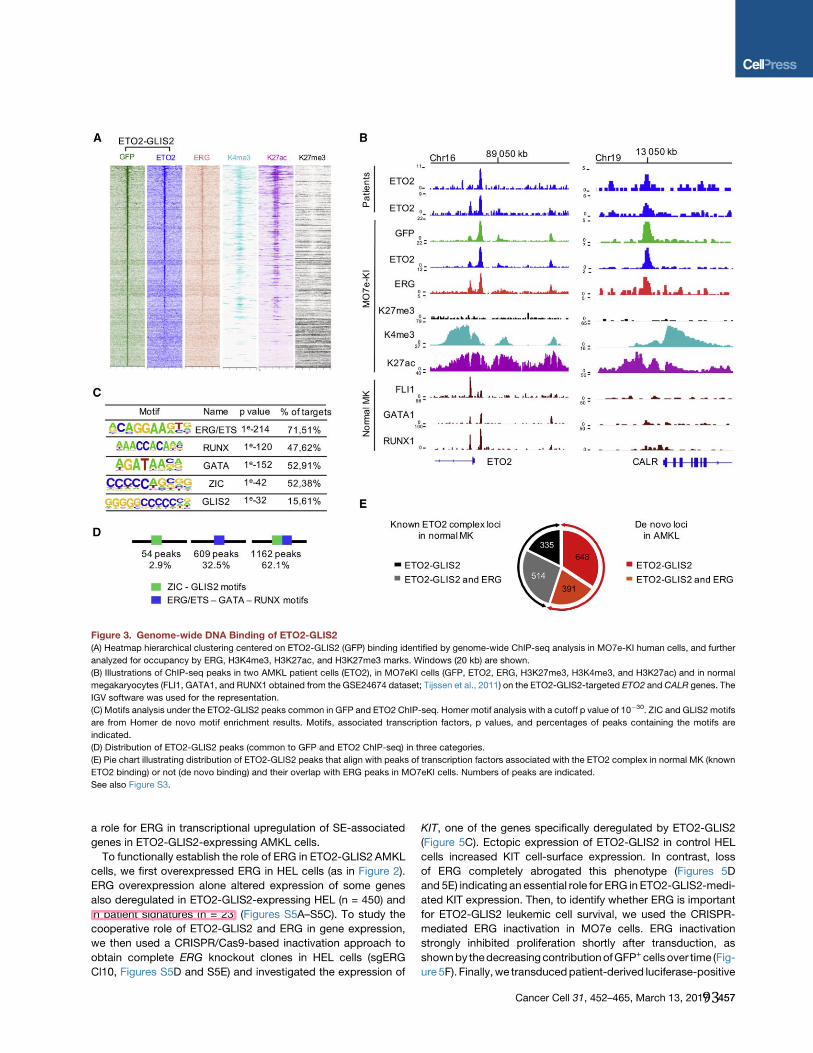
93
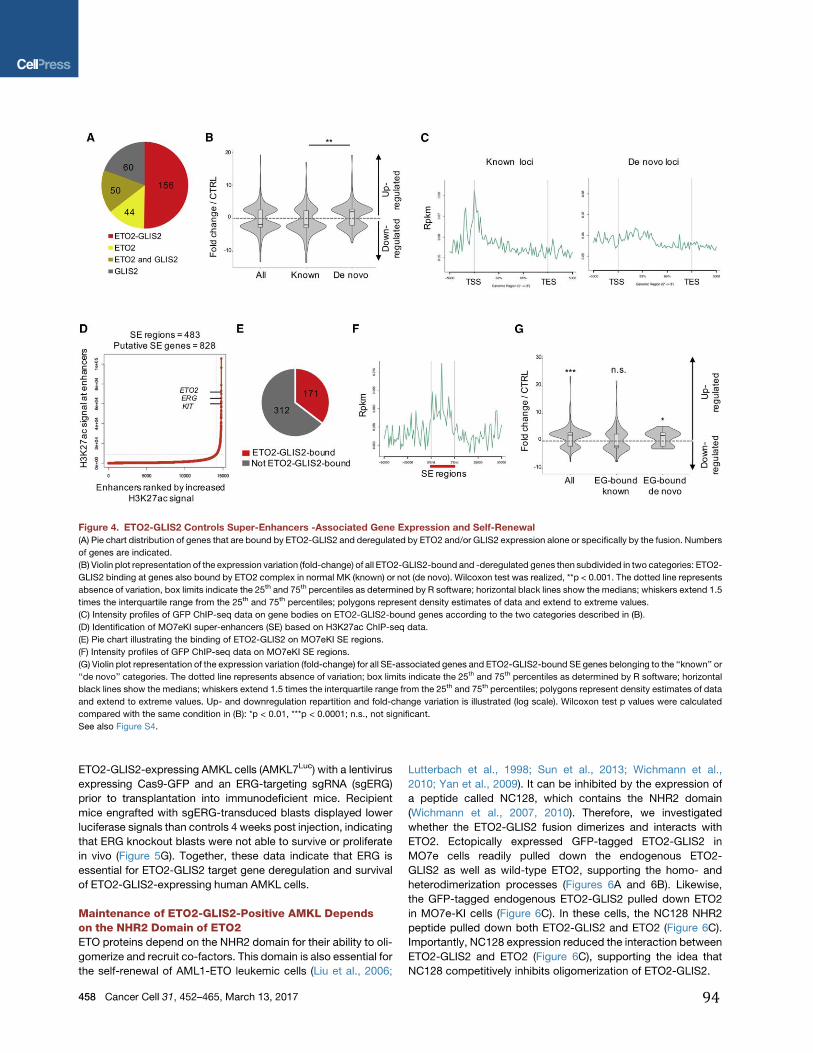
94
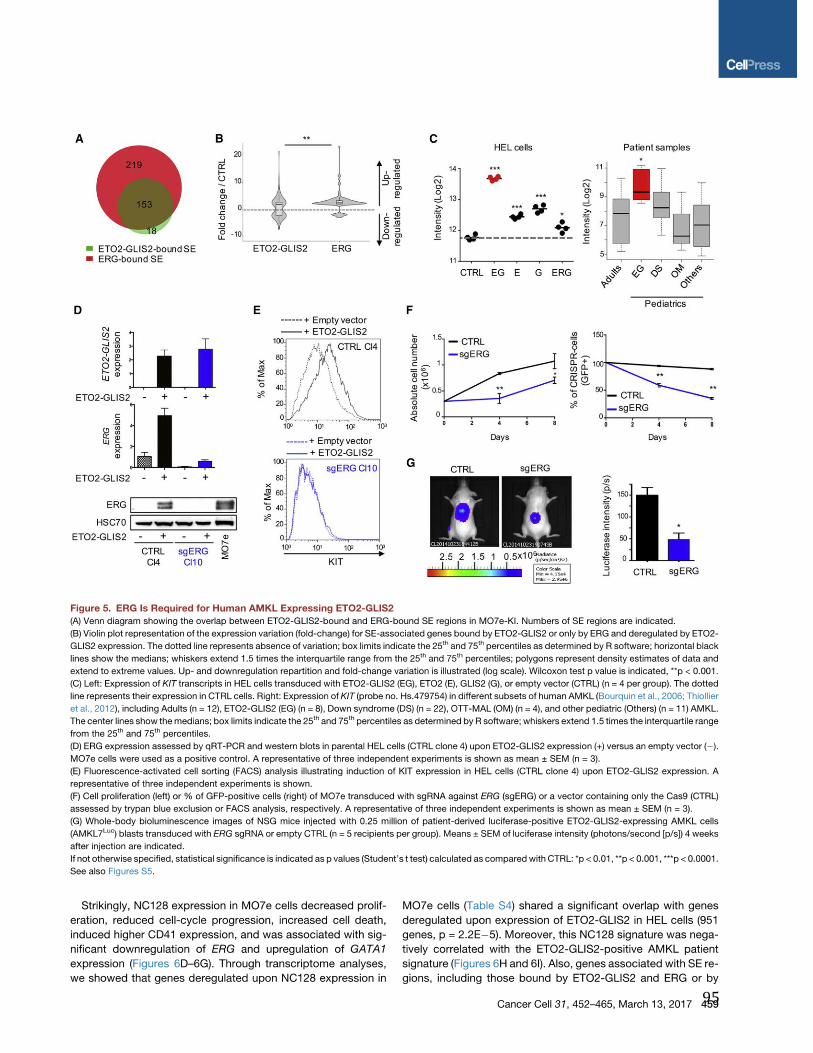
95

96
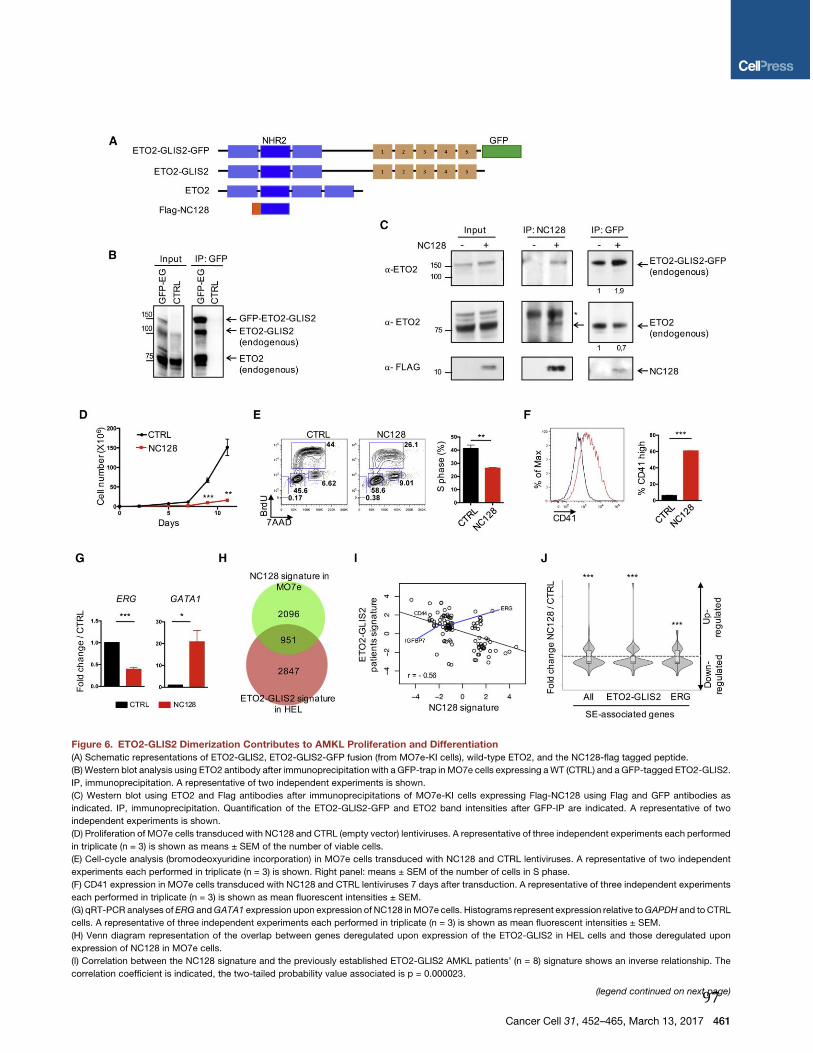
97
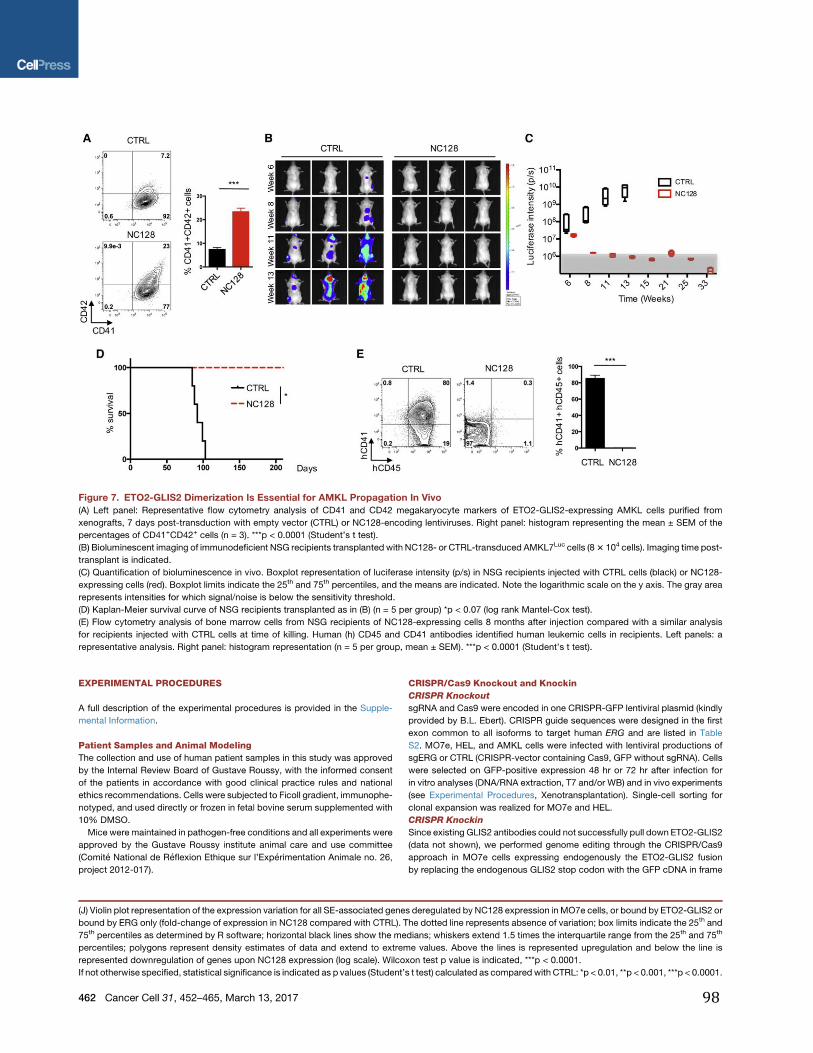
98

99

100

101

102
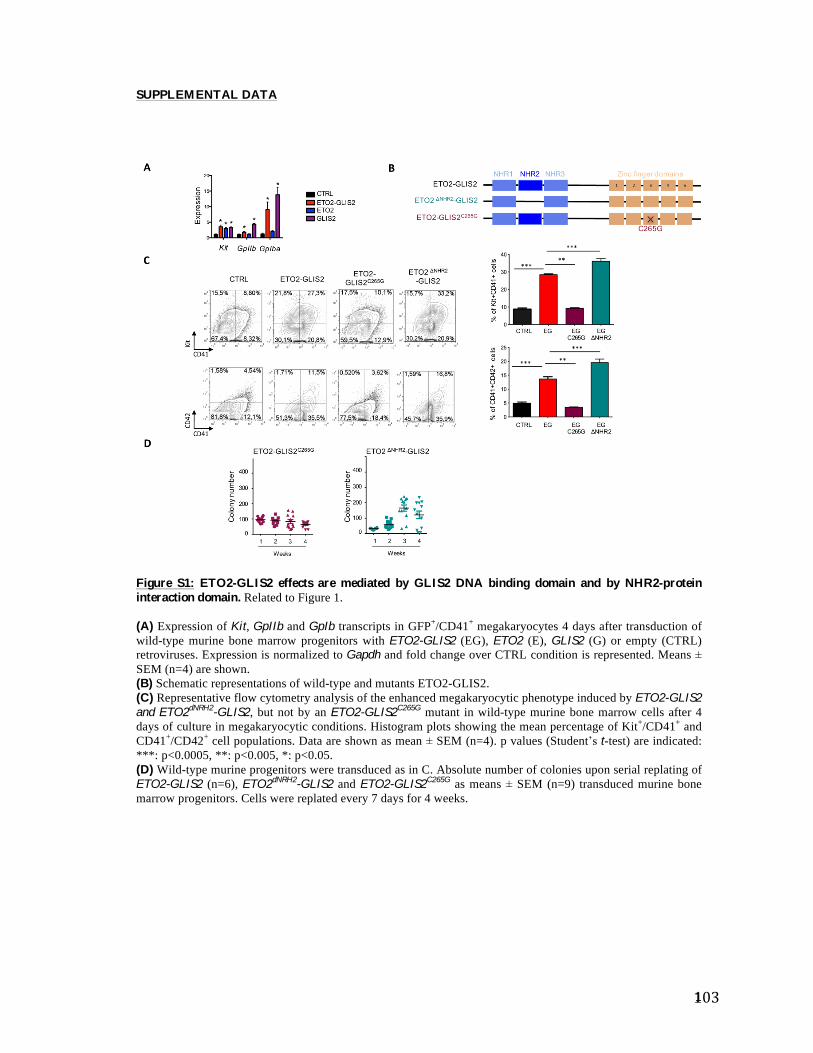
103
1
SUPPLEMENTAL DATA
Figure S1: ETO2-GLIS2 effects are mediated by GLIS2 DNA binding domain and by NHR2-protein
interaction domain. Related to Figure 1.
(A) Expression of Kit, GpIIb and GpIb transcripts in GFP
+/CD41
+ megakaryocytes 4 days after transduction of
wild-type murine bone marrow progenitors with ETO2-GLIS2 (EG), ETO2 (E), GLIS2 (G) or empty (CTRL)
retroviruses. Expression is normalized to Gapdh and fold change over CTRL condition is represented. Means ±
SEM (n=4) are shown.
(B) Schematic representations of wild-type and mutants ETO2-GLIS2.
(C) Representative flow cytometry analysis of the enhanced megakaryocytic phenotype induced by ETO2-GLIS2
and ETO2dNRH2-GLIS2, but not by an ETO2-GLIS2C265G mutant in wild-type murine bone marrow cells after 4
days of culture in megakaryocytic conditions. Histogram plots showing the mean percentage of Kit+/CD41
+ and
CD41+/CD42
+ cell populations. Data are shown as mean ± SEM (n=4). p values (Student’s t-test) are indicated:
***: p<0.0005, **: p<0.005, *: p<0.05.
(D) Wild-type murine progenitors were transduced as in C. Absolute number of colonies upon serial replating of
ETO2-GLIS2 (n=6), ETO2dNRH2-GLIS2 and ETO2-GLIS2C265G as means ± SEM (n=9) transduced murine bone
marrow progenitors. Cells were replated every 7 days for 4 weeks.
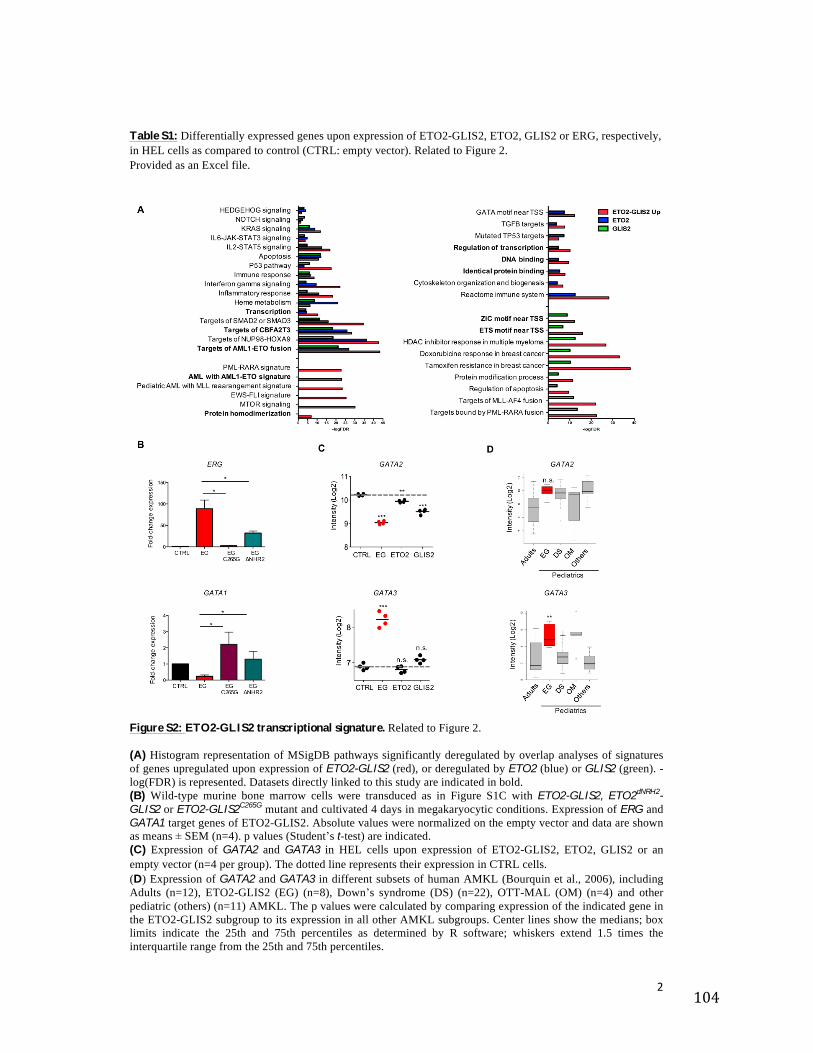
104
2
Table S1: Differentially expressed genes upon expression of ETO2-GLIS2, ETO2, GLIS2 or ERG, respectively,
in HEL cells as compared to control (CTRL: empty vector). Related to Figure 2.
Provided as an Excel file.
Figure S2: ETO2-GLIS2 transcriptional signature. Related to Figure 2.
(A) Histogram representation of MSigDB pathways significantly deregulated by overlap analyses of signatures
of genes upregulated upon expression of ETO2-GLIS2 (red), or deregulated by ETO2 (blue) or GLIS2 (green). -
log(FDR) is represented. Datasets directly linked to this study are indicated in bold. (B) Wild-type murine bone marrow cells were transduced as in Figure S1C with ETO2-GLIS2, ETO2dNRH2-
GLIS2 or ETO2-GLIS2C265G mutant and cultivated 4 days in megakaryocytic conditions. Expression of ERG and
GATA1 target genes of ETO2-GLIS2. Absolute values were normalized on the empty vector and data are shown
as means ± SEM (n=4). p values (Student’s t-test) are indicated.
(C) Expression of GATA2 and GATA3 in HEL cells upon expression of ETO2-GLIS2, ETO2, GLIS2 or an
empty vector (n=4 per group). The dotted line represents their expression in CTRL cells.
(D) Expression of GATA2 and GATA3 in different subsets of human AMKL (Bourquin et al., 2006), including
Adults (n=12), ETO2-GLIS2 (EG) (n=8), Down’s syndrome (DS) (n=22), OTT-MAL (OM) (n=4) and other
pediatric (others) (n=11) AMKL. The p values were calculated by comparing expression of the indicated gene in
the ETO2-GLIS2 subgroup to its expression in all other AMKL subgroups. Center lines show the medians; box
limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the
interquartile range from the 25th and 75th percentiles.
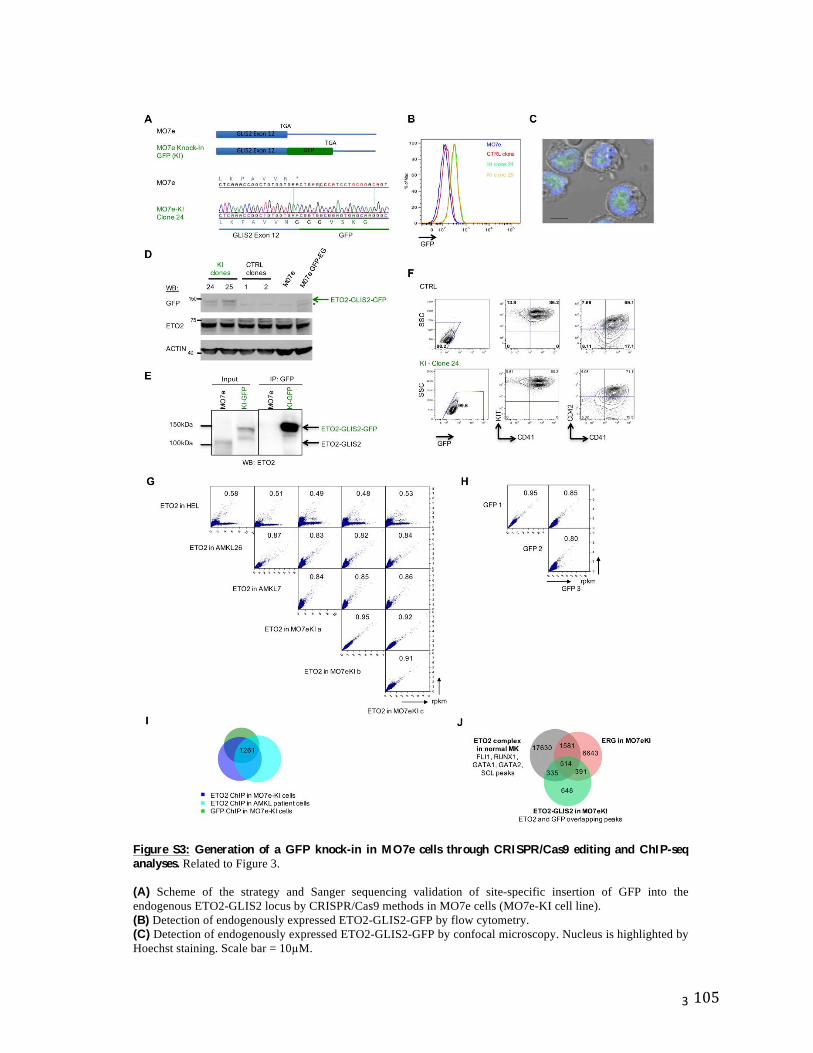
105
3
Figure S3: Generation of a GFP knock-in in MO7e cells through CRISPR/Cas9 editing and ChIP-seq analyses. Related to Figure 3.
(A) Scheme of the strategy and Sanger sequencing validation of site-specific insertion of GFP into the
endogenous ETO2-GLIS2 locus by CRISPR/Cas9 methods in MO7e cells (MO7e-KI cell line).
(B) Detection of endogenously expressed ETO2-GLIS2-GFP by flow cytometry.
(C) Detection of endogenously expressed ETO2-GLIS2-GFP by confocal microscopy. Nucleus is highlighted by
Hoechst staining. Scale bar = 10µM.

106
4
(D) Expression of the ETO2-GLIS2-GFP fusion in individual clones by Western blot as compared to MO7e cells
and retrovirus-induced expression of a GFP-ETO2-GLIS2 (GFP-EG) construct in MO7e. Anti-GFP, anti-ETO2
and anti-ACTIN antibodies were used. * indicates an aspecific band.
(E) Immunoprecipitation using GFP-trap readily pull-down specifically ETO2-GLIS2-GFP but not untagged
ETO2-GLIS2.
(F) Multiparameter flow cytometer analysis of MO7e-KI clones indicating that they present a phenotype similar
to parental MO7e cells.
(G & H) Alignment of replicates of ETO2 (A) and GFP (B) ChIP-seq with Pearson’s coefficient.
(I ) Venn diagram showing the overlap between ETO2 ChIP-seq in MO7eKI cells, in AMKL patients cells and in
GFP ChIP-seq in MO7eKI cells.
(J) Venn diagram illustrating the overlap of ETO2-GLIS2 (common to GFP and ETO2 in MO7e) and ERG
peaks with ETO2 cofactors (SCL, RUNX1, FLI1, GATA1/2) peaks bound in normal megakaryocytes (MK)
previously published (GSE24674 dataset) (Tijssen et al., 2011).

107
5
Figure S4: Characterization of ETO2-GLIS2 super-enhancers genes. Related to Figure 4.
(A) Venn diagram showing the low overlap between SE-associated genes identified in MO7e and HEL cells.
(B) Investigate overlap analysis using MsigDB of 5 lists of genes (SE-associated genes identified in MO7e,
genes deregulated after ETO2-GLIS2, ETO2 and GLIS2 expression, or previously established ETO2-GLIS2
patient signature) showing a significant association with the WONG_ADULT_TISSUE_STEM_MODULE by
computing overlap of genes. FDR value (-logFDR) is represented.
(C) Venn diagram showing the overlap between the upregulated SE-associated genes upon ETO2-GLIS2, ETO2
and GLIS2 expression. Numbers of SE are indicated.
Table S2: Lists of the common super-enhancers regions of MO7e cells identified in the three replicates using the
H3K27Ac mark. Related to Figure 4.
Provided as an Excel file
Table S3: List of super-enhancers annotated genes in MO7e cells. Related to Figure 4.
Provided as an Excel file
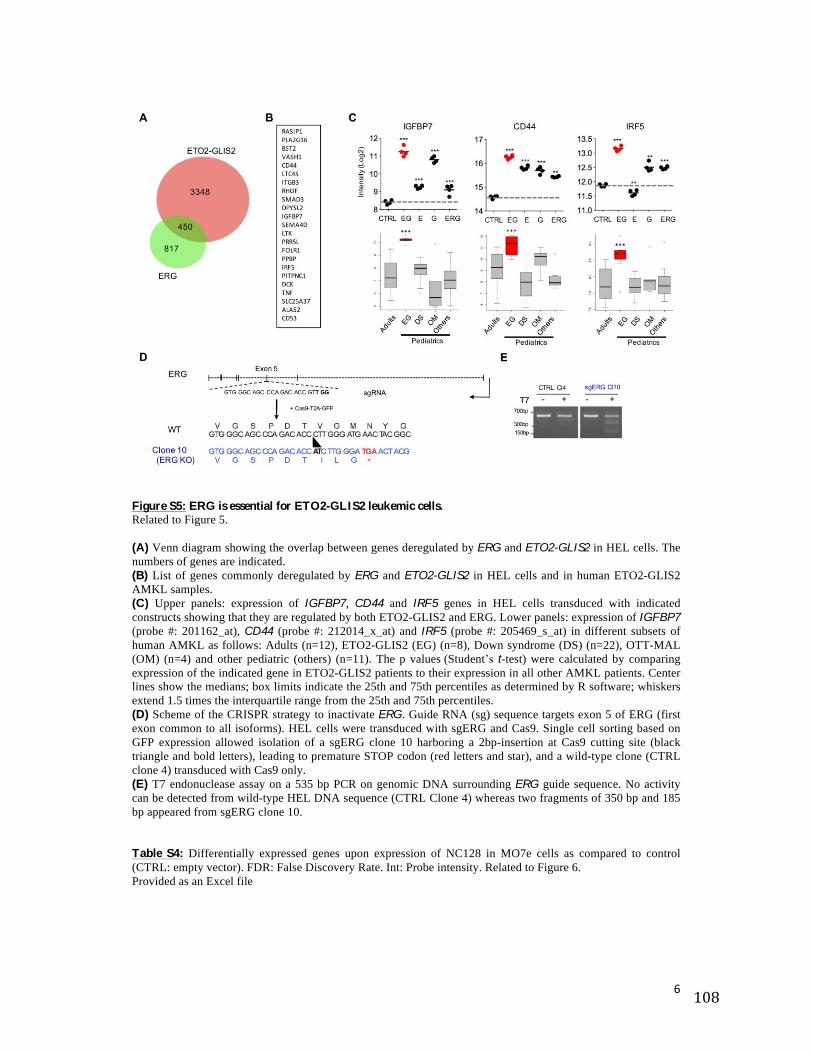
108
6
Figure S5: ERG is essential for ETO2-GLIS2 leukemic cells. Related to Figure 5.
(A) Venn diagram showing the overlap between genes deregulated by ERG and ETO2-GLIS2 in HEL cells. The
numbers of genes are indicated.
(B) List of genes commonly deregulated by ERG and ETO2-GLIS2 in HEL cells and in human ETO2-GLIS2
AMKL samples.
(C) Upper panels: expression of IGFBP7, CD44 and IRF5 genes in HEL cells transduced with indicated
constructs showing that they are regulated by both ETO2-GLIS2 and ERG. Lower panels: expression of IGFBP7
(probe #: 201162_at), CD44 (probe #: 212014_x_at) and IRF5 (probe #: 205469_s_at) in different subsets of
human AMKL as follows: Adults (n=12), ETO2-GLIS2 (EG) (n=8), Down syndrome (DS) (n=22), OTT-MAL
(OM) (n=4) and other pediatric (others) (n=11). The p values (Student’s t-test) were calculated by comparing
expression of the indicated gene in ETO2-GLIS2 patients to their expression in all other AMKL patients. Center
lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers
extend 1.5 times the interquartile range from the 25th and 75th percentiles.
(D) Scheme of the CRISPR strategy to inactivate ERG. Guide RNA (sg) sequence targets exon 5 of ERG (first
exon common to all isoforms). HEL cells were transduced with sgERG and Cas9. Single cell sorting based on
GFP expression allowed isolation of a sgERG clone 10 harboring a 2bp-insertion at Cas9 cutting site (black
triangle and bold letters), leading to premature STOP codon (red letters and star), and a wild-type clone (CTRL
clone 4) transduced with Cas9 only.
(E) T7 endonuclease assay on a 535 bp PCR on genomic DNA surrounding ERG guide sequence. No activity
can be detected from wild-type HEL DNA sequence (CTRL Clone 4) whereas two fragments of 350 bp and 185
bp appeared from sgERG clone 10. Table S4: Differentially expressed genes upon expression of NC128 in MO7e cells as compared to control
(CTRL: empty vector). FDR: False Discovery Rate. Int: Probe intensity. Related to Figure 6.
Provided as an Excel file

109
7
SUPPLEMENTAL EXPERIMENTAL PROCEDURES
Vectors, retroviral and lentiviral transduction For retroviral transduction experiments, we constructed murine stem cell virus (MSCV) backbone constructs
expressing GFP or mCherry and encoding human ETO2-GLIS2, ETO2, GLIS2 cDNA. The ETO2-GLIS2C265G
mutant was introduced using the Quickchange II XL site-directed mutagenesis Kit (Agilent technologies). To
this aim, a cysteine within the fourth zinc finger at a position equivalent to position 265 of wild-type GLIS2,
described as essential for GLIS2 DNA binding (Vasanth et al., 2011), was replaced by a glycine in the ETO2-
GLIS2 cDNA and validated by Sanger sequencing. Deletion of the NHR2 domain in ETO2 DNHR2-GLIS2 was
performed by fusion PCR. Retroviral supernatants production and transduction were described elsewhere
(Mercher et al., 2009). Briefly, 293T cells were transfected using Xtreme GENE transfection reagent according
to the manufacturer’s instructions (Roche). Retroviral particles were harvested 24h and 48h after transfection.
For retroviral transduction, murine bone marrow cells were spinoculated twice with retroviral particles in culture
media containing 5µg/mL Polybrene and 7.5mM HEPES buffer for 90 minutes at 2500 rpm, 33°C.
The lentiviral pSiEW-GFP and pSiEW-NC128-GFP vectors were described previously (Wichmann et al., 2010;
Wichmann et al., 2007). Lentiviral particles were produced in 293T cells which were co-transfected with
plasmids of interest, along with a packaging plasmid (pCMV 8.74) and a VSV envelope expression plasmid
(pMD2.G) using jetPRIME transfection reagent (Polyplus transfection, Ozyme). Supernatants were collected at
48h and concentrated by ultra-centrifugation.
Cell culture. Whole bone marrow cells were harvested from 8-10 weeks old mice, red cell lyzed for 5 minutes at 4°C and
washed twice in PBS 1X containing 2% FBS. Cells were then cultured overnight in RPMI1640 supplemented
with 10% FBS (Gibco), Penicillin (100U/mL)-Streptomycin (100µg/mL), 2mM L-Glutamine and containing
10ng/ml of recombinant murine Il-3, Il-6 and SCF (Peprotech). To promote megakaryocytic differentiation,
transduced bone marrow cells were cultured for 4 days in RPMI1640, Penicillin (100U/mL)-Streptomycin
(100µg/mL), 2mM L-Glutamine, 10% FBS supplemented with 50ng/ml murine(m)TPO and SCF. For serial
replating assays, transduced bone marrow cells were GFP sorted at 24h and plated at 5x104
cells/35mm2
dish in
methylcellulose M3434 (Stemcell Technologies), colonies were scored after 7 days and 1x104 cells were replated
every 7 days for at least 3 weeks to assess self-renewal capacities in vitro.
The human MO7e cell line was derived from a 6-month-old patient with AMKL (Avanzi et al., 1988). MO7e
cells were cultured in MEM-α supplemented with 20% FBS, Penicillin (100U/mL)-Streptomycin (100µg/mL)
and 2mM L-Glutamine (Gibco), and 5 ng/ml of human GM-CSF (PeproTech). MO7e-KI cells were derived from
MO7e cells through CRISPR/Cas9 mediated knock-in of the GFP at the endogeneous GLIS2 locus (Figure S2).
The human HEL 92.1.7 (HEL) cell line was derived from a 30-years-old patient with AML-M6 (Martin and
Papayannopoulou, 1982) and expresses megakaryocyte markers including CD41 and CD42 (Kieffer et al., 1986).
HEL cells do not express ETO2-GLIS2 but present the JAK2V617F
mutation also found in some cases of pediatric
AMKL (Gruber et al., 2012; Levine et al., 2005). Therefore, HEL cells represent a genetic and phenotypic
cellular context close to human AMKL cells. HEL cells are cultured in RPMI1640 supplemented with 10% FBS,
Penicillin (100U/mL)-Streptomycin (100µg/mL), 2mM L-Glutamine (Gibco). HEL cells were authenticated by
the presence of a homozygous JAK2V617F
mutation.
AMKL cells obtained directly from immunodeficient animals or following purification by flow cytometry were
cultured in RPMI1640 medium supplemented with 20% FBS, Penicillin (100U/mL)-Streptomycin (100µg/mL)
and 2mM L-Glutamine (Gibco), and 10 ng/ml each of human SCF, IL3, IL6, IL11, TPO, 10U/ml EPO and 5
ng/ml of GM-CSF (PeproTech). Cell proliferation was assessed by trypan blue exclusion.
Xenotransplantation. Xenograft of primary human AMKL patient samples was described previously (Thiollier et al., 2012). For
bioluminescent follow-up of human AMKL cells, human AMKL7 patient cells were transduced with the FUW-
Luc-mCherry-puro lentiviral construct (a kind gift from A. L. Kung, Dana Farber Cancer Institute, Boston),
sorted on mCherry expression and then used for further transduction with CRISPR vectors or NC128 constructs.
6-10 weeks-old female NSG mice (The Jackson Laboratory) intravenously injected with AMKL7Luc
blasts were
monitored every 4 weeks. For bioluminescence monitoring, mice received 150 mg/kg D-Luciferin (Beetle
luciferin, E1605; Promega, Charbonnières, France) intraperitoneally, were anesthetized with 3% isoflurane, and
imaged 1 to 5 min with an IVIS50 system (Perkin Elmer, Courtaboeuf, France). Bioluminescence intensity is
expressed as photons per second (p/s). Engraftment was monitored by BM sampling at 6-wk after injection and
performed under isoflurane anesthesia or by bioluminescence. For the in vivo NC128 assay, 7.104 AMKL7
luciferase leukemic blasts were engrafted with the empty vector or NC128 after sorted for GFP. To monitor the
development of the disease by bioluminescence, luciferase assay was performed monthly. For ERG sgRNA in
vivo assay, 25.104 AMKL7
Luc leukemic blasts transduced with ERG sgRNA or empty vector, sorted on GFP

110
8
expression were injected. Immunophenotyping of leukemic cells from diseased animals were performed by flow
cytometer.
Flow cytometry. Cells analyzed by flow cytometry were stained in PBS 1X supplemented with 2% FBS for 30 minutes at 4°C
with washing prior to FACS analysis. Antibodies were purchased from BD or eBioscience for human (h)
antibodies (hCD41: HIP8; hCD42: HIP1; hKIT: 104-D2) or BD, eBioscience or Emfret for murine (m)
antibodies (mCD41: MWReg30; mCD42: 1C2, Xia.G5; mKIT: 2B8). Whole BM and spleen cells from mice
were collected, subjected to red blood cell lysis, and stained for FACS analysis. Cells were analyzed on a
FACSCantoII or Fortessa with FACS Diva software (BD Biosciences). Apoptosis assays were performed
following manufacturer's recommendations (AnnexinV-APC Apoptosis Detection Kit, BD Pharmingen). Cell
cycle analyses were performed through bromodeoxyuridine (BrdU) incorporation and flow cytometer analysis
following the provider's instructions (APC BrdU Flow Kit, BD Biosciences). Briefly, cells were incubated in the
presence of BrdU for 1 hr, stained with anti-BrdU-APC antibody, prior to staining with 7-amino- actinomycin D
(7-AAD). For ploidy analyses, bone marrow cells were cultured in RPMI1640+10%FBS+50ng/ml of mTPO and
mSCF for 4 days prior to 2 hr incubation with Hoescht 33-342 (10µg/ml) and staining for 30 minutes with an
anti-mCD41-APC antibody. Analyses were gated on CD41+GFP
+ cells.
Western Blot and Immunoprecipitation. Total cells lysates were prepared as follows in 50mM Tris-HCl, pH 8, 150mM NaCl, 1% NP40, 1mM EDTA,
0,1% SDS, and 0,5% DOC supplemented with protease inhibitors (PMSF 1X, NAF 1X, Sodium Orthovanadate
1X, complete 1X). Western blotting was then performed using standard procedures using the following
antibodies from Abcam (ab) or Santa Cruz (sc): anti-CBFA2T3 (ab33072), anti-GFP (ab290), anti-ERG
(ab133264 and sc-271048), anti-GATA1 (sc-1233), anti-flag M2 (F1804), anti-HSC70 (sc-7298), anti-actin (AC-
40). Immunoprecipitation of GFP was performed with GFP-Trap agarose beads (Chromotek) with 1 hr
incubation.
DNA/RNA extraction, Real-time RT-PCR and Micro-array analyses. DNA and RNA were isolated using the RNeasy Mini/Micro or All prep DNA/RNA Micro Kit (Qiagen) and
quantified by NanoDrop (ThermoScientific). Reverse transcription was done with SuperScript II from
Invitrogen. Q-PCR was performed using SYBR Select master mix or Taqman Gene expression master mix
(Applied Biosystems) on 7500HT Fast Real-Time PCR System (Applied Biosystems) following manufacturer's
recommendations. Probes (Applied Biosystems) were used to detect ERG (Hs01554629_m1), Erg
(Mm01214246_m1), GATA1 (Hs01085823_m1), GAPDH (Hs02758991_g1) and Gapdh (Mm99999915_g1)
selected as housekeeping gene. For micro-array analyses, RNA were isolated from GFP+ cells purified by flow
cytometry 24 hr (HEL cells) or 2-7 days (MO7e) after transduction using RNeasy Micro Kit (Qiagen) with on-
column DNAse treatment. Hybridizations were performed for 3 to 4 independent biological replicates on whole-
human-genome 4x60K v2 oligonucleotide microarrays (design 039494; Agilent). The supervised analysis of the
microarray data was conducted with the Class comparison tool in BrB after multiple testing and 5.10-4
FDR.
Differentially expressed genes were identified using the following parameters: fold-change>1.8, p<10-4
(random-
variance t-test). Violin plots were obtained using BoxPlotR (http://boxplot.tyerslab.com).
Transcriptional network and transcription factor activity computation. The gene regulatory network was computed using the ARACNe algorithm (Margolin et al., 2006) on the
GSE35874 dataset, with the following parameters: mutual information was computed with adaptive partitioning
(p < 1e-9), and significant interactions were selected after 100 bootstraps (p < 7e-10).
The activity of a transcription factor (TF) in a sample was computed using the expression of its predicted targets
by ARACNe. Each set of target is separated in activated and repressed targets based on the sign of the
correlation between the expression of the TF and its predicted targets in the dataset. To infer the activity of each
TF in the HEL cells dataset, we first z-transformed the gene expression profiles to normalize the expression of
each gene across samples. We then computed the enrichment score (ES) for the set of activated and repressed
targets separately (ESpos and ESneg respectively) of each TF using this z-transformed matrix of expression, as
described in the Gene Set Enrichment Analysis (GSEA) original publication (Subramanian et al., 2005). The
final ES is computed as ESpos – ESneg, with higher positive values corresponding to higher activity of the TF in
a sample among the samples belonging to the HEL cells gene expression matrix.
Gene Set Enrichment Analysis (GSEA) analyses have been performed with the version 5.0 of Molecular
signature database (MisgDB) using weighted statistics (Subramanian et al., 2005).
Chromatin immunoprecipitation and sequencing. ChIP protocol was adapted from the MagnaChIP kit protocol (Millipore). Cells (AMKL and MO7e-KI) were
fixed with 1% formaldehyde, lysed with Cell Lysis and then Nuclear Lysis buffers respecting concentration of
111
9
20.106 cells per mL, and finally sonicated (30-min cycle on Covaris apparatus; KBioscience). Sheared chromatin
was immunoprecipitated overnight using the following antibodies: anti-CBFA2T3 (Abcam, ab33072), anti-ERG
(Santa Cruz, SC354), anti-H3K27ac (ActiveMotif, #39133), anti-H3K4me1 (Diagenode, #C15410037), anti-
H3K4me3 (Diagenode, #C15410003), anti-H3K27me3 (Cell Signaling, #9733S) and rabbit IgG (Upstate, #12-
370). 1/10 of the sheared chromatin was used as a reference (Input). Immune complex collection was realized
with Protein G Sepharose (Sigma-Aldrich, P3296), 1h30 at +4°C. For GFP-ChIP, GFP-Trap agarose beads were
used (Chromotek). Rinses were done according to MagnaChIP kit protocol with Low salt, High salt and LiCL
immune complex wash buffers. Finally, elution was realized according the IPure Kit protocol (Diagenode, Cat
No C03010012) following manufacturer’s instructions. Three independent ChIP-seq experiments were
conducted for GFP, ETO2, ERG, H3K27ac, H3K4me3 and two for H3K27me3.
Enriched DNA from ChIP and Input DNA fragments were end-repaired, extended with an 'A' base on the 3′end,
ligated with indexed paired-end adaptors (NEXTflex, Bioo Scientific) using the Bravo Platform (Agilent), size-
selected after 4 cycles of PCR with AMPure XP beads (Beckman Coulter) and amplified by PCR for 10 cycles
more. Fifty-cycle single-end sequencings were performed using Illumina HiSeq 2000 (Illumina, San Diego, CA).
Reads were aligned in toto to human genome hg19 with BWA aln (v0.7.5a) and peak calling assessed using
MACS 2.0 with a q-value cut-off of 10-4
for ERG, and 10-3
for GFP, ETO2 and all histone marks. Peak-calling
analyses for each ChIP-seq were realized independently and then we created one specific list of peaks for each
mark including common peaks in at least 2 replicates. Annotation and motif analyses have been done with
HOMER (v4.7.2), with a p value <10-30
. Integrative Genomics Viewer (IGV 2.3.34) was used for representation.
All regions bound by ETO2 cofactors in normal megakaryocytes presented Fig. 3C were obtained using
published ChIP-seq data (GSE24674 dataset)(Tijssen et al., 2011) by merging regions bound by GATA1,
GATA2, RUNX1, FLI1 and SCL. Figure 3D shows individual ChIP-seq for GATA1, FLI1 and RUNX1 from
this dataset. Intensity profiles of ChIPseq data were done using ngsplot version 2.61.
List of oligonucleotides List of CRISPR/Cas9 and PCR primer oligonucleotides used in this study.
LOCUS FORWARD REVERSE
CRISPR guides
GLIS2 caccGGTGAACTGAGCCCATCCTG aaacCAGGATGGGCTCAGTTCACC
ERG caccGTGGGCAGCCCAGACACCGT aaacACGGTGTCTGGGCTGCCCAC
PCR on genomic DNA
GLIS2 (5'arm) GAGGCTAGGCAGAGCTGGA CTCGACCAGGATGGGCAC
GLIS2 (3'arm) TCACTCTCGGCATGGACGAG CAACGTGTCATGGGTCCAGC
ERG GAGCTGGGGCTACATACTGC TGTCCTTTCTCCCCCAGTCT
Q-PCR
mmGAPDH TGCACCACCAACTGCTTAG GATGCAGGGATGATGTTC
mmKit GAGTTCCATAGACTCCAGCGTC AATGAGCAGCGGCGTGAACAGA
mmGpIIb ACAAGCCTATCTGCCACA CGACACGTTGAGGCTTAG
mmGpIba GACCGAGTCATAGCCTGGAA GCAGGAAGAGCAAGATGAGG
mmErg GTGCCCAAAACTGAAGACCAGC GAGCTGTCTGACAGGAGTTCGA
mmGata1 AGCACTGGCCTACTACAGAG AGGAATTCCCTCCATACTGT
HsGAPDH GAAGGTGAAGGTCGGAGT GAAGATGGTGATGGGATT
HsERG GCTGCTCAACCATCTCCTTC ACAGGAGCTCCAGGAGGAAC

112
ARTICLE 2 (En cours de soumission pour publication)
Pediatric myeloid fusion oncogenes control leukemogenesis
through rewiring of native hematopoiesis
Cécile K. Lopez1,2,3,4, Esteve Noguera1,2,4, Vaia Stavropoulou5, Zakia Aid1,2,4, Paola Ballerini6,
Chrystèle Bilhou-Nabera7, Hélène Lapillonne7, Cécile Thirant1,2,4, Alexandre Fagnan1,2,4,9, Sarah
J. Kinston11, M’Boyba Diop2, Yann Lecluse2, Erika Brunet12, Loelia Babin12, Jean-Luc Villeval1,2,
Antoine H.F.M. Peters15, Riccardo Masetti16, Franco Locatelli17, William Vainchenker1,2, Muriel
Gaudry1,2, Sébastien Malinge1,2,3,4, Isabelle Godin1,2, Olivier A. Bernard1,2,3,4, Berthold Göttgens11,
Arnaud Petit6, Françoise Pflumio10, Jürg Schwaller5,*, Thomas Mercher1,2,4,9,*

113
Pediatric myeloid fusion oncogenes control leukemogenesis
through rewiring of native hematopoiesis
Cécile K. Lopez1,2,3,4, Esteve Noguera1,2,4, Vaia Stavropoulou5, Zakia Aid1,2,4, Paola Ballerini6,
Chrystèle Bilhou-Nabera7,Hélène Lapillonne7, Cécile Thirant1,2,4, Alexandre Fagnan1,2,4,9, Sarah J.
Kinston11, M’Boyba Diop2, Yann Lecluse2, Erika Brunet12, Loélia Babin12, Jean-Luc Villeval1,2,
Antoine H.F.M. Peters14, William Vainchenker1,2, Muriel Gaudry1,2, Riccardo Masetti15, Franco
Locatelli16, Sébastien Malinge1,2,3,4, Claus Nerlov17, Isabelle Godin1,2, Olivier A. Bernard1,2,3,4,
Berthold Göttgens11, Arnaud Petit6, Françoise Pflumio10, Juerg Schwaller5,*, Thomas
Mercher1,2,4,9,*
1INSERM U1170, Gustave Roussy, 94800 Villejuif, France 2Gustave Roussy, 94800 Villejuif, France 3Université Paris-Saclay, 94800 Villejuif, France 4Equipe labellisée Ligue Nationale Contre le Cancer, 75013 Paris, France 5Department of Biomedicine, University Children’s Hospital Beider Basel (UKBB), 4056 Basel, Switzerland 6Hôpital Trousseau, AP-HP, 75012 Paris, France 7Sorbonne Université, CRSA – Unité INSERM, AP-HP, Hôpital Trousseau, F-75012 Paris, France 8Hôpital Saint Antoine, AP-HP, 75012 Paris, France 9Université Paris Diderot, 75013 Paris, France 10Unité Mixte de Recherche 967 Inserm-CEA/DRF/IBFJ/IRCM/LSHL- Université Paris-Diderot-Université Paris-Sud, Equipe labellisée Association Recherche contre le Cancer, 92265 Fontenay-aux-Roses Cedex, France 11Wellcome and MRC Cambridge Stem Cell Institute and the Cambridge Institute for Medical Research, University of Cambridge, Cambridge, UK. 12Genome Dynamics in the Immune System Laboratory, Institut Imagine, INSERM, UMR 1163, Université Paris Descartes, Sorbonne Paris Cité, Equipe Labellisée Ligue Contre le Cancer, 75015 Paris, France. 13INSERM U1037, Team 16, Center of Research of Cancerology of Toulouse, Hematology laboratory, IUCT-Oncopole, France 14Friedrich Miescher Institute for Biomedical Research (FMI), 4058 Basel, Switzerland; Faculty of Sciences, University of Basel, 4056 Basel, Switzerland. 15Department of Pediatrics, "Lalla Seràgnoli", Hematology-Oncology Unit, Sant'Orsola-Malpighi Hospital, University of Bologna, Via Massarenti 11, 40137, Bologna, Italy. 16Department of Pediatric Hematology-Oncology, IRCCS Ospedale Bambino Gesù, Rome and University of Pavia, Italy 17MRC Molecular Hematology Unit, MRC Weatherall Institute of Molecular Medicine, Radcliffe Department of Medicine, University of Oxford, Oxford OX3 9DS, UK
*Correspondence: Thomas Mercher Institut Gustave Roussy, INSERM U1170

114
39 rue Camille Desmoulins, 94800 Villejuif Phone: +33 1 42 11 44 83 E-mail: [email protected] or Juerg Schwaller University Children’s Hospital Basel (UKBB) Department of Biomedicine (DBM), University of Basel ZLF-Lab 202 Hebelstrasse 20 CH-4031 Basel, Switzerland Phone: +41 61 265 3504 E-mail: [email protected]
Keywords:
Leukemia, pediatric, hematopoietic stem cell, progenitor cell, cellular identity, self-renewal, transcription factor, megakaryocyte, HSC, patient-derived xenograft, PDX, childhood, transcription, AML-M7, AMKL, stemness.

115
Summary
Fusion oncogenes are frequently associated with high-risk pediatric cancers. How these genetic
alterations lead to age- and phenotype-specific cancers remains unclear. We report that
aggressive pediatric acute megakaryoblastic leukemia is associated with younger age at
diagnosis than other myeloid leukemia. Inducible transgenic murine models showed that
expression of pediatric leukemia-associated ETO2-GLIS2 and MLL-AF9 fusion oncogenes in
fetal hematopoiesis was required to induce a megakaryoblastic phenotype. Hematopoietic stem
cells, but not committed progenitor cells, presented massive rewiring of master transcription
factors activities at the single cell level. Switching off ETO2-GLIS2 expression in leukemic blasts
restored the long-term hematopoietic stem cell potential in vivo. These findings demonstrate that
age and phenotype specificities in pediatric myeloid leukemia are determined by ontogeny-
dependent differential susceptibility of hematopoietic stem and progenitor cells to transformation
by fusion oncogenes.

116
Increasing evidences support a correlation between age, driver oncogene and disease
phenotype in pediatric cancers1,2. The differences between adult and pediatric cancers may
stem from either the distinct properties of the driver oncogenes or inherent differences between
fetal and adult tissue. In acute leukemia, some fusions oncogenes are acquired in utero and are
diagnosed almost exclusively during childhood3,4,5 for which efficient leukemogenesis models are
lacking6. Childhood de novo acute megakaryoblastic leukemia (AMKL) is an aggressive subtype
of acute myeloid leukemia (AML) that harbors recurrent fusion oncogenes (e.g. ETO2-GLIS2,
OTT-MAL, NUP98-KDM5A)7–10. Intriguingly, several AMKL fusions are also detected in patients
with other AML subtypes8,11,12, although the determinants of the genotype/phenotype association
remain unknown. Similarly to other aggressive childhood cancers such as Ewing sarcoma13,
these fusions are rarely associated with additional recurrent mutations in patients9,14 and they
strongly impact the epigenome15,16, thereby suggesting that global transcriptional deregulation is
sufficient to drive the disease and a likely candidate for direct therapeutic targeting17,18.
Nevertheless, these cancers currently lack adequate animal models that precisely recapitulate
the characteristics of the disease observed in patients. Such models could provide mechanistic
insights to understand pediatric cancer specificity.
Several differences between fetal and adult normal hematopoiesis exist19,20. In adults,
hematopoietic stem cells (HSC) are at the top of a hierarchy and differentiate toward more
committed progenitors to generate the mature blood cell types. Although the precise trajectories
that govern normal hematopoiesis remain a matter of debate21–24, leukemogenic processes are
strongly influenced by the type of cell targeted by fusion oncogenes25. A recent study has
proposed that fetal liver (FL) hematopoiesis primarily involves multipotent progenitors, while
adult hematopoiesis is chiefly regulated by committed progenitors with more restricted lineage
potentials26. This view is largely supported by single cell transcriptome data showing that most
adult hematopoietic progenitors present signs of lineage commitment27–29,24,21. At the molecular
level, fetal and adult hematopoiesis present differential activity of LIN28B30–33, GATA factors-
regulated Polycomb complexes34 and CEBPA/MYCN transcription factors35.
To better understand the genotype/phenotype association in pediatric cancers, we investigated
how the fetal and adult cellular architectures22–24,36,27 impact the transformation and disease
phenotype driven by ETO2-GLIS2 (hereafter abbreviated EG). We developed an inducible
transgenic mouse model that efficiently phenocopies the human disease with striking differences

117
in latency, phenotype and molecular wiring, depending on the ontogenic stage at which the
driver fusion oncogene is activated.
AMKL is diagnosed at a younger age than AML in pediatric patients
We first screened a cohort of 276 pediatric leukemia patients for the presence of EG and
confirmed that EG occurred in AMKL patients and other AML (AML-M0 to AML-M5) subtypes12,
with an overall incidence of 4.7% (Fig. 1a,b, Supplementary Table 1). The overall survival was
poor for all patients with a trend towards a worse prognosis for those presenting with an AMKL
phenotype (Supplementary Fig. 1a). In addition to the known EG+ MO7e megakaryoblastic cell
line, we found that the CMS leukemia cell line was also EG+ and presented with a myeloid
phenotype (Supplementary Fig. 1b-e). Strikingly, EG+ AMKL patients were diagnosed at a
significantly younger age of (1.66±0.18 years [n=33]) compared with other EG+ AML patients
(8.53±1.73 years [n=15]; p<0.0001)(Fig. 1c). A similar trend was observed in AMKL patients
carrying either NUP98-KDM5A (Fig. 1d) or MLL fusions (Fig. 1e) as well as for all AMKL patients
regardless of the genetic subgroup (Fig. 1f).
We applied patient-derived xenografts (PDX)8 to expand EG+ AMKL and EG+ AML cells in
immunodeficient mice. Overall, 3/5 EG+ AMKL and 2/8 EG+ AML transplants successfully
engrafted (Supplementary Table 1). While AMKL xenografts generally expressed the
megakaryoblastic marker CD41+ but not the myeloid marker CD15- (as observed for patient
AMKL-26), AML blasts were CD15+CD41- (e.g. patient AML-17)(Fig. 1g). Interestingly, we
identified a six-year-old EG+ patient (AML-04) diagnosed as AML-M2, who presented with
immature blasts (CD34+CD15-CD41-) and two smaller cellular fractions (CD34+CD41+CD15- or
CD34+CD41-CD15+)(Fig. 1g). All cell populations presented with distinct morphologies (Fig.
1h,1i) and expressed EG (Supplementary Fig. 1f). Upon injection of the three flow-purified cell
populations into secondary recipients, only the immature CD34+CD41-CD15- cell population
successfully engrafted into recipients (Fig. 1j) and regenerated the three populations (Fig. 1k),
thereby indicating dual megakaryoblastic and myeloid differentiation.
Together, these results show that pediatric AMKL occurs at a significantly younger age than
other AML subtypes irrespective of the driver oncogene. Importantly, the dual
megakaryoblastic/myeloid phenotype in the EG+ patient indicates that AMKL-associated fusion
oncogenes can transform HSC or multipotent progenitors.

118
ETO2-GLIS2 efficiently induces leukemia in mice
To analyze the determinants of EG-driven leukemogenesis and the associated megakaryoblastic
and myeloid phenotypes, we established a doxycycline (Dox)-regulated transgenic mouse model
(Supplementary Fig. 2a). Hereby, EG expression was controlled by a reverse tetracycline-
controlled transactivator (rtTA) integrated into the Rosa26 gene locus. To assess the
consequences of EG expression, we continuously exposed double transgenic iEG:rtTA females
(hereafter abbreviated iEG) to Dox (Dox+, Fig. 2a). Although most hematological parameters
remained unchanged at six weeks post-induction (data not shown), iEG animals consistently
presented with an abnormal bone marrow (BM) Lin-CD150-Kit+CD41+ cell population
(Supplementary Fig. 2b) bearing a myeloid and megakaryoblastic clonogenic potential in
methylcellulose cultures (Supplementary Fig. 2c). Later, Dox-treated iEG animals developed
lethal hematological malignancies with a prevalence of 96.9% (31/32 mice, Supplementary Fig.
2d), which was classified into three groups based on the most abundant blast populations (Fig.
2b, Supplementary Fig. 2e). Approximately 50% of the mice (9/19, Group 1) presented with an
immature phenotype (Kit+CD41-) with a median latency of 152 days (Fig. 2c). Group 2 presented
with a megakaryoblastic phenotype (CD41+) with a median latency of 164 days (Fig. 2c), while
group 3 presented with a myeloid phenotype (CD11b+Gr1+) with a median latency of 311 days.
All diseased mice developed hepatosplenomegaly (Fig. 2d) and anemia (Fig. 2e). Group 2 was
associated with significantly higher white blood cell (WBC) counts, while group 3 was
thrombocytopenic (Fig. 2e). Histological analysis confirmed that BM and spleen from group 2
mice were infiltrated with immature von Willebrand factor (Vwf)+ megakaryoblasts (Fig. 2f,
Supplementary Fig. 2f). The cell-autonomous nature of the disease and its dependence on EG
expression was demonstrated by transplantation of BM cells into Dox+ wild-type (WT) recipients
that developed disease after a shorter mean latency (120+/- SD days), while Dox- recipients
never developed any disease (Fig. 2g-k, Supplementary Fig. 2f). Collectively, this inducible
transgenic model faithfully recapitulates EG+ leukemia development as observed in patients.
Developmental stage determines the phenotype of ETO2-GLIS2+ leukemia
To investigate the impact of the developmental stage, we induced iEG expression in Lin-
hematopoietic stem and progenitor cells (HSPC) obtained from fetal liver (FL) at embryonic day
(E) 12.5, newborn BM (1-2 weeks old) and adult BM (ABM, 8 weeks old) (Fig. 3a). iEG
expression increased the clonogenic activity over the course of four passages in all contexts (Fig.
3b). While iEG+ FL-derived cells homogeneously expressed CD41, iEG+ ABM-derived cells also
consistently showed a CD11b+Gr1+ myeloid phenotype (Fig. 3c, Supplementary Fig. 3a,b).

119
Newborn-derived iEG cells presented with a myelo/megakaryoblastic intermediate phenotype
between that of FL and ABM. Similar results were obtained upon retroviral expression of EG in
WT cells (Supplementary Fig. 3c). These data indicate that fetal hematopoiesis favors EG-
induced megakaryoblastic transformation in vitro.
To address the consequences of EG expression on fetal progenitors in vivo, we exposed
pregnant females with E13 embryos to Dox and analyzed the embryos 36 hours later. All EG-
expressing embryos showed gross morphological and hematological abnormalities (Fig. 3d),
with decreased erythroid and myeloid cells and increased megakaryoblastic progenitors (Fig. 3e).
To compare the capacity of FL- and ABM-derived iEG cells to develop disease, we transplanted
1x106 mononuclear iEG cells from E14.5 FL or ABM donors (carrying a ubiquitously-expressed
GFP reporter to track their progeny) donors into Dox+ WT recipients (Fig 3f). One month post-
engraftment, the recipients of FL-derived iEG cells showed a significantly higher percentage of
GFP+ cells in the BM than the recipients of ABM-derived cells (Fig. 3g), which was associated
with increased CD41+ cells and decreased myeloid cells (Fig. 3h). Importantly, iEG FL cell
recipients developed short latency leukemia (41.5 days, p=0.0008)(Fig. 3i) characterized by high
WBC, anemia, thrombocytopenia, splenomegaly, and megakaryoblast infiltration in several
organs (Fig. 3j-m and data not shown).
MLL-AF9 and NUP98-KDM5A are also found in both human AML and AMKL. However, current
murine models only displayed myeloid, but not megakaryoblastic, phenotype. To assess whether
the ontogenic stage affects the phenotype induced by these fusion oncogenes, we expressed
them in FL and ABM HSPC from WT mice. We found that the FL cells gave rise to an increased
CD41+ megakaryoblastic phenotype compared with ABM cells (Supplementary Fig. 3d,e). For
MLL-AF9, we also used a similar inducible MLL-AF9 transgenic model25 in which induction of
iMLL-AF9 in purified CD41+ FL cells resulted in long-term self-renewal and establishment of a
permanent cell line that presented striking maintenance of megakaryocytic potential
(Supplementary Fig. 3f). Taken together, these observations show that the developmental stage
determines the predominance of aggressive megakaryoblastic over myeloid leukemia induced
upon expression of pediatric fusion oncogenes.
ETO2-GLIS2-induced AMKL is an HSC-derived leukemia
To address the consequence of EG on distinct populations of the hematopoietic hierarchy, we
purified control (CTRL) and iEG long-term HSC (LT-HSC) and multipotent progenitors (MPP2,
MPP3 and MPP4)37 obtained from E14.5 FL or ABM (Supplementary Fig. 4a) and characterized
their clonogenic and phenotypic profile in Dox+ methylcellulose cultures. CTRL FL LT-HSC and

120
MPP generated both myeloid Gr1+ and megakaryoblastic CD41+ cells (Fig. 4a,b, Supplementary
Fig. 4b), thereby confirming that most normal mouse FL progenitors maintain a
megakaryoblastic potential as observed in humans26. iEG FL LT-HSC and MPP2 cells
exclusively generated CD41+ cells (Fig. 4b, Supplementary Fig. 4b), MPP3 cells yielded a large
majority of CD41+ cells and rare Gr1+ myeloid cells (Fig. 4b, Supplementary Fig. 4b), and MPP4
cells did not significantly form any colony (Supplementary Fig. 4c). iEG FL LT-HSC and MPP2/3
cells showed increased replating activity compared with CTRL (Fig. 4c). To define the
differentiation potential of iEG-transformed cells, iEG FL LT-HSC and MPP2/3 cells derived from
the fourth plating were cultured without Dox (Fig. 4a), which resulted in myeloid and
megakaryoblastic differentiation (Fig. 4d, Supplementary Fig. 4d).
CTRL ABM LT-HSC formed Gr1+ and CD41+ colonies (Fig. 4e,f), whereas MPP2/3/4 cells
generated almost exclusively Gr1+ cells (Fig. 4f, Supplementary Fig. 4b), thus indicating that the
megakaryoblastic potential is qualitatively and quantitatively lower in ABM than in FL
(Supplementary Fig. 4e). By contrast, iEG ABM LT-HSC gave rise almost exclusively to CD41+
cells, while MPP2/3/4 cells generated CD11b+Gr1+ myeloid cells with a higher proportion than
iEG FL MPP (Fig. 4b,f, Supplementary Fig. 4b). While iEG FL MPP4 cells did not generate
colonies, iEG ABM MPP4 cells showed increasing clonogenic activity upon serial replatings and
a myeloid morphology (Figure 4f-h).
We next investigated whether iEG endows single LT-HSC or MPP3 cells with dual
myeloid/megakaryoblastic phenotypic potential as observed in the AML-04 patient. The majority
(56%) of CTRL ABM LT-HSC-derived cells showed a dual phenotype, while others showed
either megakaryoblastic or myeloid phenotypes. Upon iEG induction, ABM LT-HSC formed
primarily megakaryoblastic colonies (89%) with only a few dual phenotype colonies (11%)(Fig.
4i,j, Supplementary Fig. 4f). While CTRL MPP3 cells exclusively generated myeloid cells under
these conditions, 48% of iEG MPP3 cells yielded a dual megakaryoblastic and myeloid profile,
thereby indicating that EG imposes megakaryoblastic as well as dual myeloid/megakaryocytic
potential on single MPP3 progenitors.
Collectively, these data show that differences between murine FL and ABM HSPC affect their
megakaryoblastic transformation potential. These findings also demonstrate that EG expression
in ABM LT-HSC results in an exclusively megakaryoblastic phenotype, while expression in
further committed progenitors is required for myeloid transformation.

121
ETO2-GLIS2 rewires the transcriptional network in LT-HSC
To characterise the molecular consequences of EG expression, we performed single cell
RNAseq (scRNAseq) using LT-HSC and MPP4 cells from CTRL and iEG mice and after a 24
hour Dox induction (Fig. 5a, Extended Tables 2-5). Compared with CTRL, iEG LT-HSC showed
151 differentially expressed genes (p<0.05, Supplementary Fig. 5a). Previously defined EG
signatures in AMKL patients were enriched in EG+ LT-HSC (Fig. 5b), thereby showing that iEG
expression in LT-HSC recapitulates the molecular consequences observed in EG+ AMKL
patients. By contrast, iEG MPP4 cells showed few differentially expressed genes and no
enrichment for EG+ AMKL patient signatures (Supplementary Fig. 5b-d, Table 5).
As scRNAseq is prone to so-called "drop-out" events for low-expression genes including
transcription factors (TF), we reasoned that computing the expression levels of the targets of a
given TF would provide a more robust assessment of the TF activity. Therefore, we used the
ARACNe and VIPER software programs38 to infer a network of TF targets and compute the
activity of each TF at the single-cell level (Supplementary Fig. 6a). Two networks were
computed from our iEG data to obtain TF targets in the context of EG expression ("EG network",
Extended Table 6) or published normal HSPC data39 to obtain TF targets in WT HSPC("normal
network", Extended Table 7). The capacity to correctly predict activity for several known TF was
confirmed using an independent normal HSPC dataset39 (Supplementary Fig. 6b). In our
scRNAseq data, iEG presented an expected higher expression level and activity in iEG+ cells
while ERG (a known EG target) showed significantly higher activity without difference in mRNA
expression (Fig. 5c, Supplementary Fig. 5c). Similarly, higher activity was inferred for many key
TF without significant differential mRNA expression (Fig. 5d), thus highlighting the value of TF
activity prediction in scRNAseq analysis. Regardless of the starting dataset, networks
consistently inferred higher activity levels for several human oncogenes or cofactors (e.g.
MECOM, HOXA10, MEIS1, ERG) and lower activity levels for factors of the heptad complex or
factors important for megakaryocyte maturation (e.g., LDB1, IKZF1, RBM15) (Fig. 5e,
Supplementary Fig. 7 and Tables 8-11)40–42.
Clustering of single cells using the t-SNE method revealed that TF activity profiles can be used
to better segregate populations than expression data and confirmed that iEG expression
impacted LT-HSC more than MPP4 cells (Fig. 5f,5g). Compared with WT LT-HSC or iEG MPP4
cells, iEG LT-HSC showed an enhanced stem cell signature (including higher expression and
activity of GATA2, MECOM, and higher expression of Esam), an higher expression of platelet-
biased HSC markers (e.g. Vwf and Pf4) without evidence of megakaryocytic maturation (lower

122
GATA1 activity) and a lower expression of myeloid (e.g. IRF8, CEBPA) and erythroid programs
(Fig.5f-j, Supplementary Fig. 5e,7b).
HSC lineage specification involves TF switches including a SPI1/CEBPA module directing
myeloid development43,44 and an ERG-GATA2/GATA1 balance45 regulating the megakaryocytic
fate, which is altered in human AMKL10. As these TF are also involved in hematopoietic
ontogeny34,35, we investigated TF expression and functional contribution to EG-induced
megakaryoblastic and myeloid phenotypes. Twenty-four hours after iEG induction, FL HSPC
expressed higher levels of Erg and Gata1 and lower levels of Cebpa and Spi1 compared with
ABM HSPC (Supplementary Fig. 8a). Similar results were obtained comparing FL and ABM
HSPC-derived cells from iMLL-AF9 (Supplementary Fig. 8b). We quantified these TF in iEG
ABM LT-HSC- and MPP4-transformed cells. Erg and Gata1 were more expressed in iEG LT-
HSC-derived cells than in iEG MPP4-derived cells, while Cebpa and Spi1 were highly expressed
in MPP4 cultures (Supplementary Fig. 8c). We then assessed whether increasing ERG or
GATA1 activity imposed a megakaryoblastic phenotype in iEG MPP4 cells and whether
increasing SPI1 or CEBPA activity biased iEG LT-HSC toward a myeloid phenotype
(Supplementary Fig. 8d-f). Retroviral ERG or GATA1 expression enhanced the megakaryocyte
phenotype in iEG MPP4 cells. In iEG LT-HSC, expression of CEBPA, but not SPI1, enhanced
the myeloid phenotype.
To corroborate these findings in human leukemia, we compared ERG, GATA1, SPI1 and
CEBPA expression levels in leukemic blasts from an AMKL patient (AMKL-26), in different
populations from the dual AML/AMKL phenotype patient (AML-04) and in an AML patient (AML-
02) (Supplementary Fig. 8g). AMKL blasts showed high ERG and GATA1 and low SPI1 and
CEBPA expression, while AML blasts presented an opposite pattern. Similar expression profiles
were observed in the human EG+ cell lines MO7e (AMKL) and CMS (AML) (Supplementary Fig.
1e). In the dual phenotype AML, the most immature CD34+CD41-CD15- blasts showed the
highest ERG expression compared to CD41+ and CD15+ blasts, while the CD41+ blasts showed
the highest GATA1 expression and the CD15+ blasts showed the highest SPI1 expression levels.
Notably, CD41+ blasts showed an unexpected combination of GATA1high and CEBPAhigh
expression.
Collectively, these data indicate that iEG expression in LT-HSC rapidly imposes conflicting TF
activities of multiple master regulators of myeloid and erythroid lineages, thereby supporting
early blockage of HSC differentiation in a megakaryoblastic state.

123
ETO2-GLIS2 reversibly transforms LT-HSC
As cellular and molecular observations indicate that EG hijacks of HSC properties, we assessed
in vivo disease development from purified populations and tracked the differentiation and long-
term hematopoietic potential of iEG-transformed cells after Dox removal.
GFP+ FL LT-HSC from E14.5 iEG embryos, or LT-HSC and MPP cells from iEG ABM were
transplanted into Dox-treated WT recipients (Fig. 6a). The iEG FL LT-HSC recipients developed
fully penetrant and rapid (median: 27 days) lethal leukemia (Fig. 6b). The iEG ABM LT-HSC
recipients developed disease after a strikingly longer latency (median: 312 days, p=0.017) (Fig.
6b, c). By contrast, iEG ABM MPP3 or MPP4 did not result in any disease (Fig. 6c). The FL iEG
LT-HSC recipients presented with a more severe disease than the ABM iEG LT-HSC mice as
assessed by greater degree of splenomegaly, higher white blood counts, higher percentage of
CD41+ cells and lower platelet counts, maturing myeloid populations and lymphoid cells in BM
and spleen (Fig. 6d-g). Histological analyses showed that the BM, spleen and liver were
infiltrated with GFP+ cells that were weakly vWF+. The iEG ABM LT-HSC recipients displayed
rare maturing vWF+ megakaryocytes that were all GFP-, which was not observed in the iEG FL
LT-HSC counterparts (Supplementary Fig. 9a, data not shown).
To investigate the differentiation potential of LT-HSC-derived iEG leukemic cells, we purified the
immature CD41+CD11b-Gr1- blasts from iEG FL LT-HSC diseased recipients and grew them in
cultures in absence of Dox. As compared with the immature blasts maintained with Dox, they
generated both maturing CD41+CD42+ megakaryocytes and CD11b+Gr1+ myeloid cells
(Supplementary Fig. 9b). This was associated with reduced expression of iEG and its known
target Erg, while Gata1 expression increased, consistent with broad differentiation capacities
(Supplementary Fig. 9c). To assess long-term hematopoietic potential, BM cells from the ABM
LT-HSC-engrafted diseased recipients were transferred into secondary Dox-treated recipients
(Fig. 6h). Two weeks post-transplant (=D0 time point), when recipients showed the first signs of
disease (Supplementary Fig. 9d,e), some recipients were maintained on Dox (Group A) while
Dox-treatment was ceased in group B. At three days after Dox-removal, the platelets counts
were back to normal in group B; 80% of platelets originated from iEG-transformed cells as
shown by GFP expression (Fig. 6i). GFP+ cells were also detected later in RBC and WBC (Fig. 6i,
Supplementary Fig. 9f). Analysis at six-months post Dox removal demonstrated that GFP+ cells
where still present in all analyzed hematopoietic compartments, including mature B and T cells
(Fig. 6j, Supplementary Fig. 9g). Together, these data show that EG expression in FL LT-HSC
results in a rapid and aggressive AMKL in vivo associated with maintenance of long-term HSC-
like differentiation potential once the driver oncogene is switched off.

124
DISCUSSION
In toto, our observations in patient samples and in inducible transgenic mouse models for
pediatric leukemia-associated fusion oncogenes, including ETO2-GLIS2 and MLL-AF9, provide
experimental evidence that aggressive pediatric AMKL originates in HSC and, more globally,
that age and phenotype-specific associations observed in human patients rely on changes in the
cellular architecture during hematopoietic ontogeny (Supplementary Fig. 10).
Our data show that expression of EG in HSC, but not in further committed progenitors, resulted
in AMKL development in vivo. The previous lack of in vivo transformation46,47,our unpublished data could
therefore be due to a poor targeting of HSC by retroviral transduction. This inducible model was
also instrumental in demonstrating that HSC-derived EG+ leukemic cells can maintain long-term
reconstitution and differentiation properties upon withdrawal of the driver oncogene. In adult
hematopoiesis, several cellular and gene expression studies have indicated the close proximity
between HSC and megakaryocytic states in normal and stress conditions22–24,29,48,49, thus
prompting the hypothesis that megakaryopoiesis may represent the native differentiation
pathway for HSC24. Our data indicate that EG strongly rewires master hematopoietic regulator
activities in HSC (e.g. heptad complex and HOX-related factors) toward a megakaryoblastic
state and that targeting HSC is required for pediatric AMKL pathogenesis. This hypothesis is
also supported by the role of other factors in both normal HSC and megakaryocyte biology and
their mutation in pediatric AMKL (e.g. OTT1/RBM15)50. Taken together, we propose a
conceptual framework for pediatric AMKL in which transcriptional constraints imposed by distinct
oncogenes represent molecular switches that block HSC as they engage the native
megakaryocytic differentiation pathway.
This study also demonstrates that the transforming potential of pediatric oncogenes is strongly
dependent on the ontogeny-related dynamics of the hematopoietic tissue architecture. Indeed,
EG expression in LT-HSC exclusively generated a megakaryoblastic phenotype, while the
development of a myeloid phenotype required targeting of adult MPP. Importantly, our finding
that phenotypic output of transformation by a pediatric oncogene relies on both the ontogenic
stage and the targeted cell type is consistent with recent findings that normal fetal hematopoiesis
primarily involves multipotent HSPC (endowed with megakaryocytic potential), while adult
hematopoiesis presents a higher ratio of committed progenitors (lacking megakaryocytic
potential)26. Therefore, we propose that the inherent differences between fetal and adult
hematopoiesis, including a higher proportion of multipotent versus myeloid-committed
progenitors at the fetal stage, underlies the association between AMKL and younger childhood

125
AML. As the prevalence of other megakaryocyte-related diseases decreases after birth (e.g.,
Trisomy 21 AMKL and thrombocytopenia absent radii syndrome)51–53, it is likely that the
importance of this ontogeny-related architectural change may extend to several other human
megakaryocyte/platelet-related diseases. Furthermore, multiple pediatric cancers are driven by
strong epigenetic fusion oncogenes in the context of a low mutational burden, including Ewing's
sarcoma and rhabdomyosarcoma, for which the cellular origin is poorly defined13, and
glioblastoma, for which oncogenic drivers have been recently shown to arise in stem cells54.
Therefore, a precise characterization of the changes in the cellular architecture of the relevant
tissues of origin should improve our understanding of the heterogeneous clinical presentations in
other pediatric cancers and facilitate the establishment of other disease models that may serve
as experimental platforms to develop more selective therapeutic strategies.
AUTHORS CONTRIBUTION
CKL designed, performed, analyzed experiments and drafted the manuscript.
EN and MD performed bioinformatics analyses.
VS established the iEG ES cell and mouse lines and initiated in vivo induction.
ZA and CT, performed analyses of murine models.
CBN performed FISH analyses.
YL performed flow cytometry analyses and cell sorting.
PB, HL, ED, RM, FL and AP provided patient samples and clinical information.
SJK and BG generated single RNAseq data.
EB, JLV, PDA, WV, MG, SM, IG, AHFMP, OAB, BG and FP provided major intellectual inputs
and reagents.
JS and TM conceived and supervised the project; designed, performed and analyzed
experiments and wrote the manuscript.
All authors revised and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We are grateful to Michael Kyba for the generous gift of the A2Lox-Cre ES cells and to Peter D.
Aplan and Olivier Delattre for critical reading of the manuscript. We thank Clarisse Thiollier,
Olivier Bluteau, Lou Le Mouël, Claude Preudhomme, Lila Diamanti, Nassera Abermil, Julie

126
Chaumeil, Camille Lobry and Sabine Juge for their expertise and helpful discussions. We thank
the Gustave Roussy institute facilities for mouse care (Patrick Gonin and Karine Ser-Leroux),
bioinformatics support (Philippe Dessen and Daniel Gautheret), and Paul Zanardo for excellent
administrative assistance.
This work was supported by the Association Laurette Fugain (ALF-2015/13 to TM), SIRIC-
SOCRATE (INCa-DGOS-INSERM 6043 to TM), Fondation pour la Recherche Médicale (to ZA:
FRM-ING20150532273, CKL), Fondation de France (FdF-00057925 to CT and TM), Lady Tata
Foundation (R14120LL to CT), Cancéropôle Ile de France (to CKL and Emergence 2015),
Gustave Roussy Genomic Core Facility – TA2016 (to CKL), Institut National Du Cancer (PLBIO-
2014-176 and PLBIO-2018-169 to TM), PAIR-Pédiatrie/CONECT-AML (COllaborative Network
for Children and Teenagers with Acute Myeloblastic Leukemia: INCa-ARC-LIGUE_11905 and
Association Laurette Fugain), Associazione Italiana Ricerca sul Cancro (to FL). AHFMP was
supported by the Novartis Research Foundation. SJK and BG were supported by a Bloodwise
Specialist Programme grant (12029) and infrastructure funding from the Wellcome to the
Wellcome & MRC Cambridge Stem Cell Institute. JS was supported by the Swiss National
Science Foundation (31003A_173224/1), the Swiss Cancer League (KFS-3487-08-2014), the
San Salvatore Foundation (Lugano, Switzerland), the Novartis Foundation for Biomedical
Research and the Gertrude von Meissner Foundation (Basel, Switzerland). TM is an Equipe
Labellisée LIGUE principal investigator.
The authors declare no competing financial interests.
DATA AVAILABILITY STATEMENT
Single cell RNAseq data were deposited into EBI - Array-Express under the accession number E-MTAB-7213.

127
REFERENCES
1. Gröbner, S. N. et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018).
2. Bolouri, H. et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 24, 103–112 (2018).
3. Greaves, M. In utero origins of childhood leukemia. Early Hum. Dev. 81, 123–129 (2005). 4. Hong, D. et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia.
Science 319, 336–339 (2008). 5. Slany, R. K. The molecular biology of mixed lineage leukemia. Hematologica 94, 984–993 (2009). 6. Sanjuan-Pla, A. et al. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia.
Blood 126, 2676–2685 (2015). 7. Creutzig, U. et al. Significance of age in acute myeloid leukemia patients younger than 30 years: a common
analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 112, 562–571 (2008).
8. Thiollier, C. et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J. Exp. Med. 209, 2017–2031 (2012).
9. de Rooij, J. D. E. et al. Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 49, 451–456 (2017).
10. Lopez, C. K., Malinge, S., Gaudry, M., Bernard, O. A. & Mercher, T. Pediatric Acute Megakaryoblastic Leukemia: Multitasking Fusion Proteins and Oncogenic Cooperations. Trends Cancer 3, 631–642 (2017).
11. Gough, S. M., Slape, C. I. & Aplan, P. D. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood 118, 6247–6257 (2011).
12. Masetti, R. et al. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121, 3469–3472 (2013).
13. Grünewald, T. G. P. et al. Ewing sarcoma. Nat. Rev. Dis. Primer 4, 5 (2018). 14. Bardini, M. et al. DNA copy-number abnormalities do not occur in infant ALL with t(4;11)/MLL-AF4.
Leukemia 24, 169–176 (2010). 15. Krivtsov, A. V. et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer
Cell 14, 355–368 (2008). 16. Thirant, C. et al. ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-
Renewal in Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell 31, 452–465 (2017). 17. Dawson, M. A. et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion
leukemia. Nature 478, 529–533 (2011). 18. Erb, M. A. et al. Transcription control by the ENL YEATS domain in acute leukemia. Nature 543, 270–274
(2017). 19. Babovic, S. & Eaves, C. J. Hierarchical organization of fetal and adult hematopoietic stem cells. Exp. Cell
Res. 329, 185–191 (2014). 20. Elagib, K. E., Brock, A. T. & Goldfarb, A. N. Megakaryocyte ontogeny: Clinical and molecular significance.
Exp. Hematol. 61, 1–9 (2018). 21. Laurenti, E. & Göttgens, B. From hematopoietic stem cells to complex differentiation landscapes. Nature
553, 418–426 (2018). 22. Sanjuan-Pla, A. et al. Platelet-biased stem cells reside at the apex of the hematopoietic stem-cell hierarchy.
Nature 502, 232–236 (2013). 23. Carrelha, J. et al. Hierarchically related lineage-restricted fates of multipotent hematopoietic stem cells.
Nature 554, 106–111 (2018). 24. Rodriguez-Fraticelli, A. E. et al. Clonal analysis of lineage fate in native hematopoiesis. Nature 553, 212–
216 (2018). 25. Stavropoulou, V. et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML
Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 30, 43–58 (2016). 26. Notta, F. et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny.
Science 351, aab2116 (2016). 27. Paul, F. et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163,
1663–1677 (2015). 28. Wilson, N. K. et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves
Heterogeneity within Stem Cell Populations. Cell Stem Cell 16, 712–724 (2015). 29. Velten, L. et al. Human hematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol.
19, 271–281 (2017).

128
30. Yuan, J., Nguyen, C. K., Liu, X., Kanellopoulou, C. & Muljo, S. A. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science 335, 1195–1200 (2012).
31. Copley, M. R. et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal hematopoietic stem cells. Nat. Cell Biol. 15, 916–925 (2013).
32. Bluteau, O. et al. Developmental changes in human megakaryopoiesis. J. Thromb. Haemost. JTH 11, 1730–1741 (2013).
33. Rowe, R. G. et al. Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J. Exp. Med. 213, 1497–1512 (2016).
34. Xu, J. et al. Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Mol. Cell 57, 304–316 (2015).
35. Ye, M. et al. C/EBPa controls acquisition and maintenance of adult hematopoietic stem cell quiescence. Nat. Cell Biol. 15, 385–394 (2013).
36. Pimkin, M. et al. Divergent functions of hematopoietic transcription factors in lineage priming and differentiation during erythro-megakaryopoiesis. Genome Res. 24, 1932–1944 (2014).
37. Cabezas-Wallscheid, N. et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522 (2014).
38. Alvarez, M. J. et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847 (2016).
39. Nestorowa, S. et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20-31 (2016).
40. Malinge, S. et al. Ikaros inhibits megakaryopoiesis through functional interaction with GATA-1 and NOTCH signaling. Blood 121, 2440–2451 (2013).
41. Saleque, S., Cameron, S. & Orkin, S. H. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 16, 301–306 (2002).
42. Wilson, N. K. et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7, 532–544 (2010).
43. Giladi, A. et al. Single-cell characterization of hematopoietic progenitors and their trajectories in homeostasis and perturbed hematopoiesis. Nat. Cell Biol. 20, 836–846 (2018).
44. Hoppe, P. S. et al. Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature 535, 299–302 (2016).
45. Bresnick, E. H., Lee, H.-Y., Fujiwara, T., Johnson, K. D. & Keles, S. GATA switches as developmental drivers. J. Biol. Chem. 285, 31087–31093 (2010).
46. Dang, J. et al. AMKL chimeric transcription factors are potent inducers of leukemia. Leukemia 31, 2228–2234 (2017).
47. Gruber, T. A. et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22, 683–697 (2012).
48. Takano, H., Ema, H., Sudo, K. & Nakauchi, H. Asymmetric division and lineage commitment at the level of hematopoietic stem cells: inference from differentiation in daughter cell and granddaughter cell pairs. J. Exp. Med. 199, 295–302 (2004).
49. Haas, S. et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 17, 422–434 (2015).
50. Niu, C. et al. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood 114, 2087–2096 (2009).
51. Roberts, I. & Izraeli, S. Hematopoietic development and leukemia in Down syndrome. Br. J. Hematol. 167, 587–599 (2014).
52. Albers, C. A., Newbury-Ecob, R., Ouwehand, W. H. & Ghevaert, C. New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr. Opin. Genet. Dev. 23, 316–323 (2013).
53. Liu, Z.-J. & Sola-Visner, M. Neonatal and adult megakaryopoiesis. Curr. Opin. Hematol. 18, 330–337 (2011).
54. Lee, J. H. et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 560, 243–247 (2018).
55. van Zutven, L. J. C. M. et al. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes. Chromosomes Cancer 45, 437–446 (2006).
56. Struski, S. et al. NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 31, 565–572 (2017).
57. Hara, Y. et al. Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes. Chromosomes Cancer 56, 394–404 (2017).
58. de Rooij, J. D. E. et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27, 2280–2288 (2013).
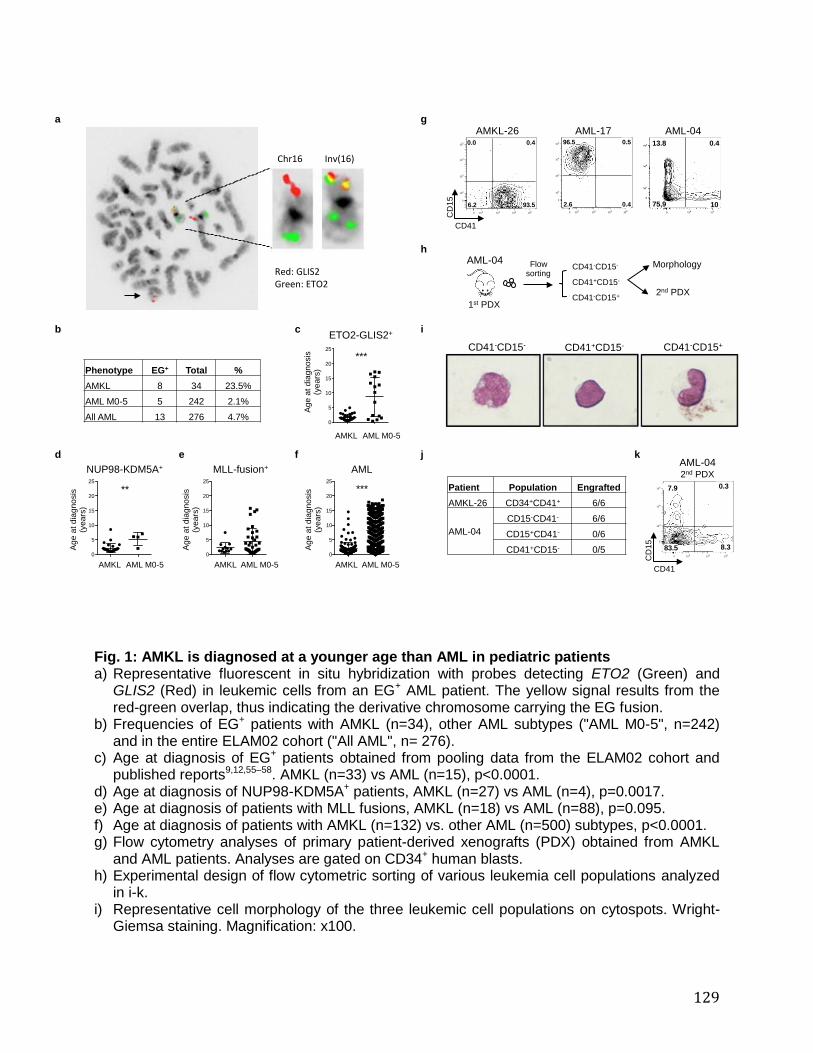
129
Fig. 1: AMKL is diagnosed at a younger age than AML in pediatric patients a) Representative fluorescent in situ hybridization with probes detecting ETO2 (Green) and
GLIS2 (Red) in leukemic cells from an EG+ AML patient. The yellow signal results from the red-green overlap, thus indicating the derivative chromosome carrying the EG fusion.
b) Frequencies of EG+ patients with AMKL (n=34), other AML subtypes ("AML M0-5", n=242) and in the entire ELAM02 cohort ("All AML", n= 276).
c) Age at diagnosis of EG+ patients obtained from pooling data from the ELAM02 cohort and published reports9,12,55–58. AMKL (n=33) vs AML (n=15), p<0.0001.
d) Age at diagnosis of NUP98-KDM5A+ patients, AMKL (n=27) vs AML (n=4), p=0.0017. e) Age at diagnosis of patients with MLL fusions, AMKL (n=18) vs AML (n=88), p=0.095. f) Age at diagnosis of patients with AMKL (n=132) vs. other AML (n=500) subtypes, p<0.0001. g) Flow cytometry analyses of primary patient-derived xenografts (PDX) obtained from AMKL
and AML patients. Analyses are gated on CD34+ human blasts. h) Experimental design of flow cytometric sorting of various leukemia cell populations analyzed
in i-k. i) Representative cell morphology of the three leukemic cell populations on cytospots. Wright-
Giemsa staining. Magnification: x100.
0
5
10
15
20
25
0
5
10
15
20
25
0
5
10
15
20
25
a
d e f
c
g
b
CD41-CD15+ CD41+CD15-
h
i
0 103 104 105
0
103
104
105 7.87 0.335
8.2983.5
7.9 0.3
8.3 83.5
ETO2-GLIS2+
Patient Population Engrafted
AMKL-26 CD34+CD41+ 6/6
AML-04
CD15-CD41- 6/6
CD15+CD41- 0/6
CD41+CD15- 0/5
CD41-CD15-
Flow sorting
CD41-CD15-
CD41+CD15-
CD41-CD15+
Morphology
2nd PDX
1st PDX
AML-04 AMKL-26 AML-17
CD
15
CD41
0 102 103 104 105
0
102
103
104
105 0.0 0.4
93.56.20 102 103 104 105
0
102
103
104
105 96.5 0.5
0.42.6
0.4 13.8
10 75,9
Chr16 Inv(16)
Ag
e a
t d
iag
nosis
(y
ea
rs)
MLL-fusion+
Ag
e a
t d
iag
no
sis
(y
ears
)
j k
AML
Ag
e a
t d
iag
no
sis
(y
ears
)
Red:GLIS2Green:ETO2
AML-04 2nd PDX
Phenotype EG+ Total %
AMKL 8 34 23.5%
AML M0-5 5 242 2.1%
All AML 13 276 4.7%
***
***
AMKL AML M0-5
AMKL AML M0-5 AMKL AML M0-5
0
5
10
15
20
25
Ag
e a
t d
iag
no
sis
(y
ears
)
NUP98-KDM5A+
**
AMKL AML M0-5
AML-04
CD
15
CD41

130
j) Secondary xenotransplantation of various cell populations from AML-04 indicates that only CD41-CD15- cells propagated the disease.
k) CD15 and CD41 expression on CD34+ cells obtained from recipients of 3x105 CD15-CD41- cells one-year post-transplant.
Statistical significance is indicated as p values (Student’s t test). *: p<0.05, **: p<0.01, ***: p<0.001.
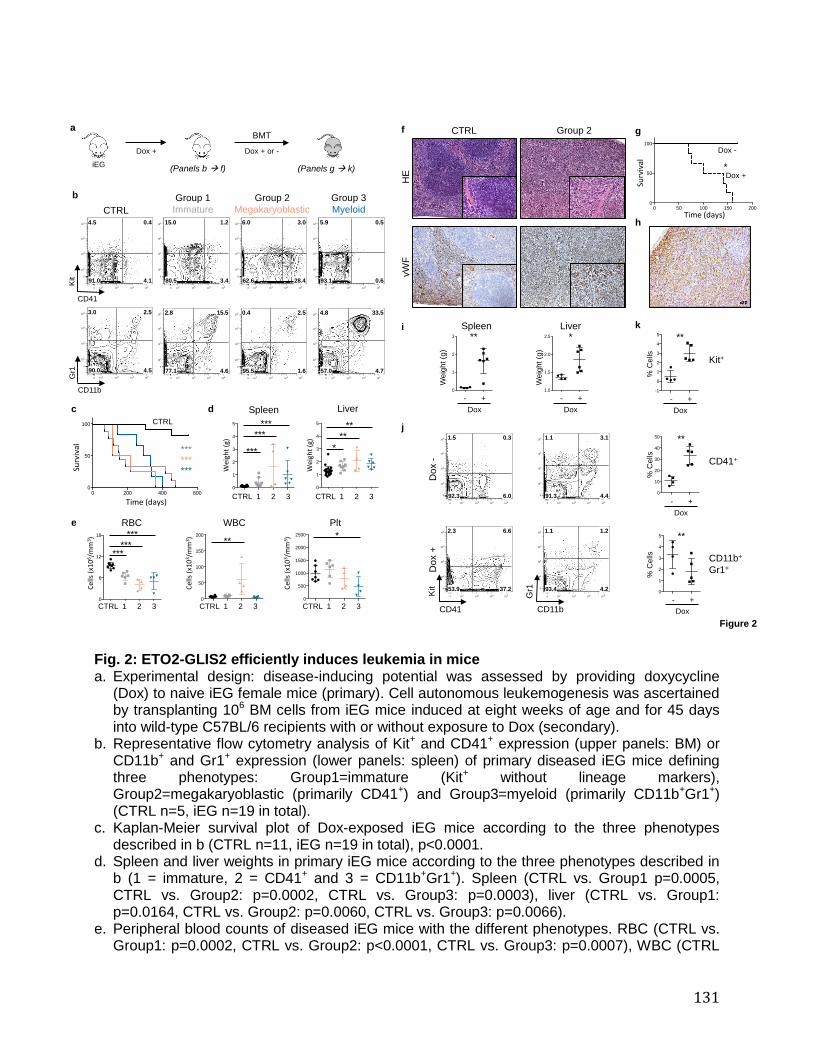
131
Fig. 2: ETO2-GLIS2 efficiently induces leukemia in mice a. Experimental design: disease-inducing potential was assessed by providing doxycycline
(Dox) to naive iEG female mice (primary). Cell autonomous leukemogenesis was ascertained by transplanting 106 BM cells from iEG mice induced at eight weeks of age and for 45 days into wild-type C57BL/6 recipients with or without exposure to Dox (secondary).
b. Representative flow cytometry analysis of Kit+ and CD41+ expression (upper panels: BM) or CD11b+ and Gr1+ expression (lower panels: spleen) of primary diseased iEG mice defining three phenotypes: Group1=immature (Kit+ without lineage markers), Group2=megakaryoblastic (primarily CD41+) and Group3=myeloid (primarily CD11b+Gr1+) (CTRL n=5, iEG n=19 in total).
c. Kaplan-Meier survival plot of Dox-exposed iEG mice according to the three phenotypes described in b (CTRL n=11, iEG n=19 in total), p<0.0001.
d. Spleen and liver weights in primary iEG mice according to the three phenotypes described in b (1 = immature, 2 = CD41+ and 3 = CD11b+Gr1+). Spleen (CTRL vs. Group1 p=0.0005, CTRL vs. Group2: p=0.0002, CTRL vs. Group3: p=0.0003), liver (CTRL vs. Group1: p=0.0164, CTRL vs. Group2: p=0.0060, CTRL vs. Group3: p=0.0066).
e. Peripheral blood counts of diseased iEG mice with the different phenotypes. RBC (CTRL vs. Group1: p=0.0002, CTRL vs. Group2: p<0.0001, CTRL vs. Group3: p=0.0007), WBC (CTRL
0
1
2
3
4
5
0
10
20
30
40
50
-1
0
1
2
3
4
5
1.0
1.5
2.0
2.5
0
1
2
3
0 50 100 150 2000
50
100
0
500
1000
1500
2000
2500
0
50
100
150
200
0
6
12
18
0
1
2
3
4
5
0
1
2
3
4
5
0 200 400 6000
50
100
CTRL
Group 1
Immature
b
a
iEG
Dox + Dox + or -
BMT
(Panels b à f) (Panels g à k)
c
e
f
Kit
CD41
Gr1
CD11b
Group 3
Myeloid
i
0 102 103 104 105
0
102
103
104
105 4.5 0.4
4.191.00 102 103 104 105
0
102
103
104
105 15.0 1.2
3.480.50 102 103 104 105
0
102
103
104
105 6.0 3.0
28.462.60 102 103 104 105
0
102
103
104
105 5.9 0.5
0.693.1
0 102 103 104 105
0
102
103
104
105 3.0 2.5
4.590.00 102 103 104 105
0
102
103
104
105 2.8 15.5
4.677.10 102 103 104 105
0
102
103
104
105 0.4 2.5
1.695.50 102 103 104 105
0
102
103
104
105 4.8 33.5
4.757.0
g
h
Figure 2
RBC WBC Plt
Spleen Liver
Time(days)
Cells(x103/m
m3)
Weight(g)
Time(days)
Survival
CTRL Group 2
HE
vW
F
Spleen Liver
We
igh
t (g
)
We
igh
t (g
)
j D
ox -
D
ox +
K
it
CD41
Gr1
CD11b
Kit+
CD41+
CD11b+
Gr1+
k
d
Group 2
Megakaryoblastic
*** ***
*** ***
CTRL 1 2 3
Weight(g)
*** ***
*
** **
CTRL 1 2 3
Survival
*** ***
***
CTRL 1 2 3
Cells(x106/m
m3)
**
CTRL 1 2 3
Cells(x103/m
m3) *
CTRL 1 2 3
CTRL
Dox -
Dox + *
- +
Dox
**
- +
Dox
*
% C
ells
**
**
**
% C
ells
%
Cells
0 102 103 104 105
0
102
103
104
105 1.5 0.3
6.092.30 102 103 104 105
0
102
103
104
105 1.1 3.1
4.491.3
0 102 103 104 105
0
102
103
104
105 2.3 6.6
37.253.90 102 103 104 105
0
102
103
104
105 1.1 1.2
4.293.4
- +
Dox
- +
Dox
- +
Dox

132
vs. Group1: p=0.1348, CTRL vs. Group2: p=0.0068, CTRL vs. Group3: p=0.1660), Platelet (Plt: CTRL vs. Group1: p=0.3663, CTRL vs. Group2: p=0.3902, CTRL vs. Group3: p=0.0440).
f. Spleen histopathology of a diseased iEG mouse from Group 2 showing infiltration of mostly von Willebrand factor (vWF)+ tumour cells. Upper panels: Hematoxylin-Eosin-Safran staining, lower panels: vWF staining (brown coloration).
g. Kaplan-Meier survival plot of secondary recipients with or without Dox-exposure (Dox -: n=4, Dox +: n=6). p=0.0110 (Log rank Mantel-Cox test).
h. Representative spleen section of a secondary recipient stained with vWF (brown coloration) shows significant organ infiltration.
i. Spleen and liver weights in secondary recipients with or without Dox-exposure (Dox -: n=4, Dox +: n=6). Spleen: p=0.0025, liver: p=0.0220.
j. Representative flow cytometry analysis of spleen of secondary recipients with or without Dox (Dox -: n=4, Dox +: n=5).
k. Quantification of flow cytometry analyses described in (j). Kit+: p=0.0026, CD41+: p=0.0015, CD11b+Gr1+: p=0.1002.
Statistical significance is indicated as p values (Student’s t test except when otherwise specified). *: p<0.05, **: p<0.01, ***: p<0.001.

133
Fig. 3: Developmental stage determines the phenotype of ETO2-GLIS2+ leukemia a. Experimental design to address the impact of EG on the clonogenic potential of fetal liver (FL)
or adult bone marrow (ABM) progenitors in methylcellulose shown in b and c. b. Number of colonies upon culture of 2500 fetal liver (FL) or Lin- BM cells at two weeks
(newborn) or two months post-partum (ABM) in Dox-containing methylcellulose conditions. Serial replating was performed every seven days. P1: first plate, P2: secondary plate, P3: tertiary plate, P4: quaternary plate.
c. Representative flow cytometry analysis of CD41+ and Gr1+ expression at P3 (upper panels) and percentages of CD41+ and Gr1+ cells at P1 to P3 (lower panels).
d. Pictures of E14.5 embryos (left and middle panels) and corresponding FL (right panels) obtained from pregnant females on Dox for 36 hours.
e. Flow cytometry analyses of FL cells obtained in d (CTRL n=8, iEG n=10). CD71+Ter119+: p<0.0001, CD11b+Gr1+: p=0.0380, CD41+: p<0.0001.
f. Experimental design: 1x106 ABM or FL cells from double transgenic iEG+Ubiquitin-GFP+ (GFP+) mice into wild type Dox-treated recipients. The progeny of engrafted cells is GFP+ cells.
g. Percentages of GFP+ cells in the BM of recipients at one month post-engraftment (CTRL n=6, FL n=6 and ABM n=6). CTRL vs. FL: p=0.0701, CTRL vs. ABM: p=0.0043.
0
25
50
0
10
20
30
40
50
0.0
0.5
1.0
0
300
600
900
1200
0
6
12
18
0
20
40
60
80
0 25 50 75 1000
25
50
75
100
0
25
50
75
100
0
25
50
75
100
0
25
50
75
100
0.0
1.5
3.0
0
1
2
3
4
0
25
50
75
100
0
20
40
60
80
100
0
100
200
300
400
0
100
200
300
400
0
100
200
300
400 P1P2
P3
P4
b
c
f
d
e
Gr1
CD41
h
l
Kit
CD41
Gr1
CD11b
CT
RL
FL
-le
uk
(Panels g à m)
0 102 103 104 105
0
102
103
104
105 42.7 6.8
41.69.00 102 103 104 105
0
102
103
104
105 21.1 1.5
56.321.10 102 103 104 105
0
102
103
104
105 0.0 0.5
82.117.4
0 102 103 104 105
0
102
103
104
105 2.5 0.1
0.696.80 103 104 105
0
102
103
104
105 1.6 44.2
4.050.1
0 102 103 104 105
0
102
103
104
105 8.7 1.5
19.969.80 103 104 105
0
102
103
104
105 4.9 24.0
2.468.8
BM
CD41+ CD11b+Gr1+ i
Dox +
Figure 3
iEG GFP+
FL12.5 Newborn BM
P1 P4
… iEG
Dox +
a
ABM
g
Plt RBC WBC k
Spleen
CD41+ CD11b+Gr1+
% c
ells
FL12.5
ABM
FL12.5 Newborn BM ABM
CT
RL
iEG
j
m
(Panels b, c)
CD11b+Gr1+ CD41+ CD71+Ter119+
Newborn BM
# o
f co
lon
y
CTRL iEG CTRL iEG CTRL iEG
% c
ells
P1 P2 P3
% c
ells
CTRL iEG
***
CTRL iEG CTRL iEG
*** *
BMT FL12.5
ABM
% G
FP
+
CTRL FL ABM
***
Cells(x103/m
m3)
CTRL FL ABM FL-leuk
Cells(x106/m
m3)
CTRL FL ABM FL-leuk
*** * **
Cells(x103/m
m3)
CTRL
FL
ABM ***
% c
ells
CTRL FL ABM
***
***
CTRL FL ABM
***
***
WT recipients
CTRL FL-leuk
CTRL FL-leuk CTRL FL-leuk
**
*** ** ***
CTRL FL ABM FL-leuk
*** ***
0
20
40
60
80
100
0
20
40
60
80
100
CD41+
Gr1+
Time(days)
Survival
***
We
igh
t (g
)
P1 P2 P3 P1 P2 P3
Leukemia

134
h. Flow cytometry quantification of CD41+ and CD11b+Gr1+ in GFP+ BM cells. CD41+ (CTRL vs. FL: p<0.0001, CTRL vs. ABM: p<0.0001, FL vs. ABM: p<0.0001), CD11b+Gr1+ (CTRL vs. FL: p=0.0002, CTRL vs. ABM: p=0.1493, FL vs. ABM: p<0.0001.
i. Kaplan-Meier survival plot of recipients of ABM or FL-derived iEG cells with Dox treatment. Log rank Mantel-Cox test: p=0.0008.
j. Peripheral blood counts of recipients of ABM (CTRL and iEG) and FL at one month post-engraftment and in recipients of FL at time of leukemia and final analysis (abbreviated FL-leuk). WBC (CTRL vs. ABM: p<0.0001, CTRL vs. FL-leuk: p=0.0009), RBC (CTRL vs. FL: p=0.0004, CTRL vs. ABM: p=0.0093, CTRL vs. FL-leuk: p<0.0001), Platelets (Plt: CTRL vs. FL: p=0.0007, CTRL vs. ABM: p=0.0163, CTRL vs. FL-leuk: p=0.0044).
k. Spleen weights in CTRL and diseased FL at time of sacrifice, p=0.0003. l. Representative flow cytometry analyses of BM cells from diseased FL recipients at time of
sacrifice. m. Quantification of CD41+ (p=0.0327) and CD11b+Gr1+ (p=0.0017) in GFP+ BM cells analyzed
in l. Statistical significance is indicated as p values (Student’s t test except when otherwise specified). *: p<0.05, **: p<0.01, ***: p<0.001.

135
Fig. 4: ETO2-GLIS2-induced AMKL is an HSC-derived leukemia a. Experimental design to address the clonogenic potential of HSC and MPP from CTRL and
iEG E14.5 embryos. Serial replating was performed every seven days for four weeks (P1 to P4).
b. Representative flow cytometry analyses of cells obtained from the first plate (P1). c. Number of colonies upon serial replating. Mean+/-SEM is indicated. d. Representative flow cytometry analyses of cells grown in methylcellulose cultures with and
without Dox. e. Experimental design to address the clonogenic potential of adult (8-10 weeks-old) HSC and
MPP from iEG and CTRL mice. f. Representative flow cytometry analyses of cells obtained at P1. g. Number of colonies upon serial replating. Mean+/-SEM is indicated. h. Wright-Giemsa staining of cytospots from iEG ABM HSC- and MPP4-derived methylcellulose
cultures at P3. Magnification: x100. i. Single HSC and MPP3 ABM cells from CTRL or iEG mice were evaluated for their potential to
develop MK and/or myeloid cells in 96-wells liquid cultures with Dox treatment. A
0
100
200
500
1000
0
100
200
500
1000
0
100
200
500
1000
0
100
200
500
1000
0
100
200
500
1000
0
100
200
500
1000
0
100
200
500
1000
0
50
100
Gr1+CD41+CD41+Gr1+
n=53 n=85
0
50
100n=50 n=47
0 102 103 104 105
0
102
103
104
105 44.1 7.8
10.038.20 102 103 104 105
0
102
103
104
105 6.4 5.8
17.270.60 102 103 104 105
0
102
103
104
105 14.5 4.6
16.264.7
HSC
# o
f co
lon
y
CTRL iEG
HSC
# o
f co
lon
y
0 102 103 104 105
0
102
103
104
105 15.0 0.3
36.148.6
c
f
CT
RL
iEG
HSC MPP2 MPP3 b
Gr1
CD41
HSC MPP2 MPP3 MPP4
Do
x +
D
ox -
0 102 103 104 105
0
102
103
104
105 0.5 0.5
94.15.00 102 103 104 105
0
102
103
104
105 1.3 0.9
82.615.20 102 103 104 105
0
102
103
104
105 15.0 4.3
46.833.9
0 102 103 104 105
0
102
103
104
105 11.0 0.3
26.162.6
0 102 103 104 105
0
102
103
104
105 0.1 0.2
94.25.4
0 102 103 104 105
0
102
103
104
105 84.8 0.8
3.610.80 102 103 104 105
0
102
103
104
105 69.1 0.3
1.429.2
0 102 103 104 105
0
102
103
104
105 22.8 0.3
63.113.7
0 102 103 104 105
0
102
103
104
105 88.9 0.6
0.310.4
0 102 103 104 105
0
102
103
104
105 33.8 0.6
57.28.20 102 103 104 105
0
102
103
104
105 13.6 0.5
79.76.2
0 102 103 104 105
0
102
103
104
105 2.8 5.1
88.04.0
0 102 103 104 105
0
102
103
104
105 58.2 7.3
32.61.9
0 102 103 104 105
0
102
103
104
105 4.6 2.0
91.32.0
0 102 103 104 105
0
102
103
104
105 34.4 4.5
50.910.2
0 102 103 104 105
0
102
103
104
105 31.2 16.0
46.76.0
0 102 103 104 105
0
102
103
104
105 40.8 12.6
44.02.6
HSC MPP2 MPP3
0 102 103 104 105
0
102
103
104
105 0.0 0.0
98.81.20 102 103 104 105
0
102
103
104
105 96.2 0.5
0.62.7
g
HSC
MPP
P1 P4 …
Dox +
Dox -
(Panels b, c) (Panel d)
Figure 4
d
Gr1
CD41
i
j
Single cell culture
HSC
MPP
…
(Panels i, j)
Dox + (Panels f à h)
MPP2
CTRL iEG
MPP3
CTRL iEG
MPP2
CTRL iEG
Gr1+ CD41+ CD41+Gr1+
% C
ells
CTRL iEG
% C
ells
CTRL iEG
HSC MPP3
MPP3
CTRL iEG
MPP4
CTRL iEG
FL12.5 iEG or CTRL
ABM iEG or CTRL Dox +
Dox +
CT
RL
iEG
H
SC
M
PP
4
h
CTRL iEG
Gr1
CD41
Gr1
CD41
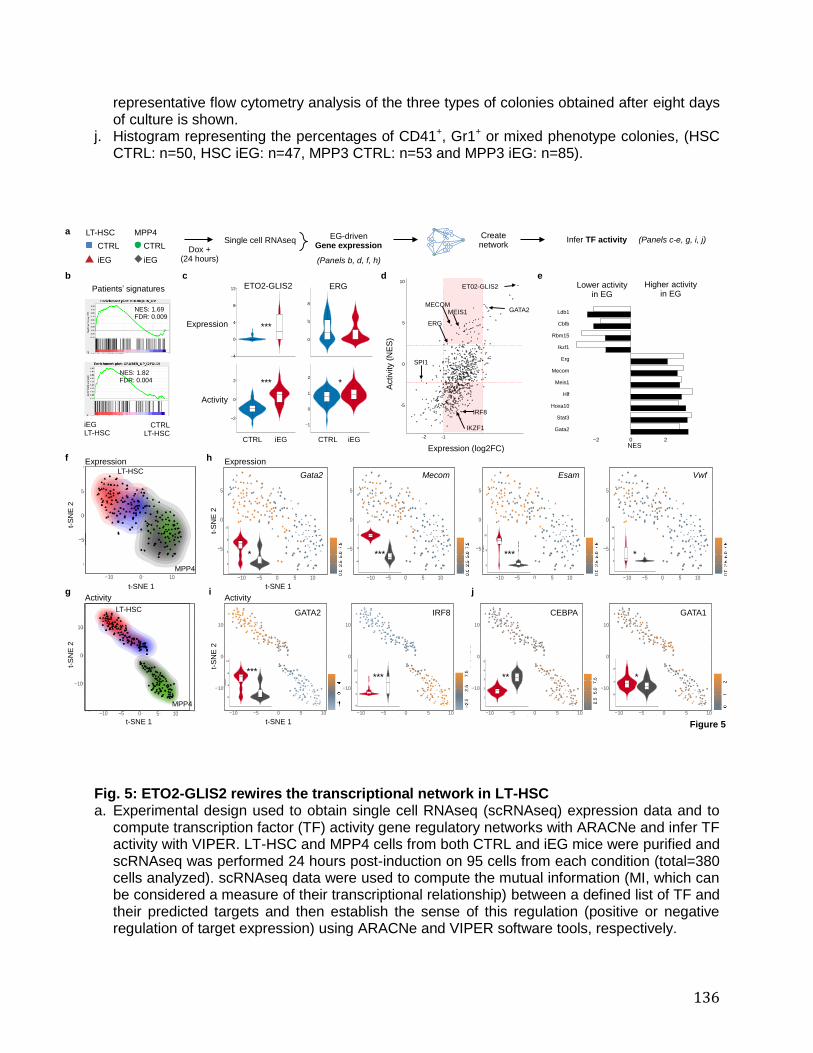
136
representative flow cytometry analysis of the three types of colonies obtained after eight days of culture is shown.
j. Histogram representing the percentages of CD41+, Gr1+ or mixed phenotype colonies, (HSC CTRL: n=50, HSC iEG: n=47, MPP3 CTRL: n=53 and MPP3 iEG: n=85).
Fig. 5: ETO2-GLIS2 rewires the transcriptional network in LT-HSC a. Experimental design used to obtain single cell RNAseq (scRNAseq) expression data and to
compute transcription factor (TF) activity gene regulatory networks with ARACNe and infer TF activity with VIPER. LT-HSC and MPP4 cells from both CTRL and iEG mice were purified and scRNAseq was performed 24 hours post-induction on 95 cells from each condition (total=380 cells analyzed). scRNAseq data were used to compute the mutual information (MI, which can be considered a measure of their transcriptional relationship) between a defined list of TF and their predicted targets and then establish the sense of this regulation (positive or negative regulation of target expression) using ARACNe and VIPER software tools, respectively.
CTRL
iEG
−10
0
10
−15 −10 −5 0 5 10 15t−SNE 1
t−S
NE
2
−10
−5
0
5
10
−10 0 10t−SNE 1
t−S
NE
2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Mecom
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
−10
0
10
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
−10
0
10
−10 −5 0 5 10t−SNE 1
t−S
NE
2
a Infer TF activity
Dox + (24 hours)
Single cell RNAseq
Figure 5
Create network
EG-driven Gene expression
b c
MPP4
LT-HSC
MPP4
LT-HSC
g i
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
−10
0
10
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
−10
0
10
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●
1
2
3
4
5
Ceb
pa
●●
●
0
1
2
Gata
1
●
●●●●●●●
●
●●
●●
0
4
8
12
Gata
2
●
●●
●
−5
0
5
10
Irf8
●
●●●●●●●
●
●●
●●
0
4
8
12
Gata
2
●
−4
0
4
Me
co
m
d e
f
j
*** *** ** *
GATA2 IRF8
NES: 1.69 FDR: 0.009
iEG LT-HSC
Patients’ signatures
NES: 1.82 FDR: 0.004
CTRL LT-HSC
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
2.55.07.510.0Type ●HSC EG − HSC EG + MPP4 EG −
●
●
●
●●
●
●
●●●
0
5
10
15
Vw
f
* * ***
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Esam
●
●
●
●
●●●●
●
●
●
0
5
10
Esa
m
***
h
●
●
−1
0
1
2
Erg
●●
0
4
8E
rg
●
●
●
−2
0
2
ET
O2−
GL
IS2
●
●
●
●
●
●
●
−4
0
4
8
12
ET
O2−
GL
IS2
CTRL iEG
***
Activity
***
ERG
CTRL iEG
*
Expression
ETO2-GLIS2
CTRL
(Panels b, d, f, h)
LT-HSC MPP4 (Panels c-e, g, i, j)
CEBPA GATA1
Gata2 Mecom Esam Vwf
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
−5
0
5
−10 −5 0 5 10t−SNE 1
t−S
NE
2
●HSC EG − HSC EG + MPP4 EG − MPP4 EG + Gata2
t-SNE 1
t-S
NE
2
t-SNE 1
t-S
NE
2
t-SNE 1
t-S
NE
2
t-SNE 1
t-S
NE
2
iEG
Activity
Expression
Activity
Expression
−5
0
5
10
−2 −1 0 1 2 3
Expression (log2 FC)
Activity (
t−sta
tistic)
Activity (
NE
S)
ET02-GLIS2
ERG
MECOM
SPI1
MEIS1
IKZF1
IRF8
Expression (log2FC)
GATA2
Gata2
Stat3
Hoxa10
Hlf
Meis1
Mecom
Erg
Ikzf1
Rbm15
Cbfb
Ldb1
−2 0 2
NES
Network EG Normal
Higher activity in EG
Lower activity in EG
0 1 2 3 -1 -2
-5
0
5
10

137
b. GSEA on CTRL and EG+ LT-HSC scRNAseq data using published EG+ patient-derived gene expression signatures16. Violin plot of EG expression in LT-HSC. The boxplots indicate median and SD.
c. Violin plots of EG and ERG expression and activity in LT-HSC. Log2 values of the normalized read counts are represented.
d. Scatter plot of differential expression (log2FC) against activity (Normalized Enrichment Score: NES) obtained by comparing for each gene iEG+ vs. CTRL LT-HSC. Horizontal lines represent the activity threshold of NES (FDR < 0.05 corresponding to a NES of +/- 2.29) and vertical lines represent a threshold two-fold change in expression in iEG+ vs. CTRL LT-HSC. Tinted squares indicate factors for which a significant differential activity is inferred without a significant differential expression in iEG vs. CTRL LT-HSC.
e. Master Regulator Analysis (MRA package from VIPER) consistently inferred similar differential activity for key factors upon EG expression regardless of the initial scRNAseq dataset used to compute the TF targets (empty bars: network computed using wild-type HSPC scRNAseq data, full bars: network computed using our scRNAseq data). Enrichment score (NES) of indicated TF activity in EG vs. CTRL LT-HSC is indicated. Positive NES indicates higher activity in iEG vs CTRL cells. Negative NES indicates lower activity in iEG vs CTRL cells.
f. 2D representation of the cells using t-SNE analysis of expression data. Density plots for each condition was overlaid (Red: LT-HSC iEG [triangles], blue: LT-HSC CTRL [squares], Grey: MPP4 iEG [diamonds], green: MPP4 CTRL [dots]).
g. 2D representation of the cells using t-SNE analysis of activity data inferred with the EG network.
h. Expression of key genes plotted in the t-SNE space shown in f. Log2 values of the normalized read counts are represented as a gradient (orange: higher expression, light grey: lower expression). Insert: violin plot representation of the expression of the same gene in iEG LT-HSC (red) vs. iEG MPP4 (grey) cells.
i. Activity of key factors using targets inferred in the EG network and plotted in the t-SNE space shown in g. NES scores are represented as a gradient (orange: higher activity, light grey: lower activity). Insert: violin plot representation of the activity of the same factor in iEG LT-HSC (red) vs. iEG MPP4 (grey) cells.
j. Activity of key factors using targets inferred in the normal network and plotted in the t-SNE space shown in g. NES scores are represented as a gradient (orange: higher activity, light grey: lower activity). Insert: violin plot representation of the activity of the same factor in iEG LT-HSC (red) vs. iEG MPP4 (grey) cells.
Statistical significance is indicated as p values (Student’s t test). *: p<0.05, **: p<0.01, ***: p<0.001.

138
Fig. 6: ETO2-GLIS2 reversibly transforms LT-HSC a. Experimental design: purified LT-HSC and MPP cells from wild type (WT) or iEG mice
expressing the Ubiquitin-GFP (GFP+) were transplanted into wild-type Dow-treated recipients. Note that GFP expression is not Dox-dependent and therefore engraftment of all cells can be followed using GFP regardless of the Dox treatment.
b. Kaplan-Meier survival curve of recipients of naive iEG HSC from ABM or FL, on Dox. Recipients were treated with Dox after transplantation. Log rank Mantel-Cox test iEG FL vs. ABM recipients: p=0.0171. *: p<0.05, **: p<0.01, ***: p<0.001
c. Percentages of GFP+ cells within nucleated blood cells of recipient mice. Analyses were performed at 2, 4, 6, 16, 28 weeks and 40 weeks. Mean+/-SEM (n=4) is shown.
d. Percentages of GFP+ cells in BM, spleen weights, white blood cell (WBC) counts and platelet counts (Plt) in CTRL and iEG ABM HSC recipients and iEG FL HSC recipients. Spleen (CTRL vs. FL: p<0.0001, CTRL vs. ABM: p=0.0039), WBC (CTRL vs. FL: p=0.0095, CTRL vs. ABM: p=0.0309), Plt (CTRL vs. FL: p=0.0079, CTRL vs. ABM: p=0.1536).
e. Representative flow cytometry analysis of diseased recipients at time of sacrifice.
0
30
60
90
0
10
20
30
40
50
0
20
40
60
0
20
40
60
80
0
10
20
30
40
0
25
50
75
100
0
25
50
75
100
0
2
4
6
8
10
0
500
1000
1500
0
20
40
60
80
100
0
10
20
30
40
50
0
20
40
60
80
100
0
20
40
60
80
100
0
200
400
600
800
1000
0
10
20
30
40
50
0.0
0.4
0.8
1.2
0
20
40
60
80
100
0
25
50
75
100
0
25
50
75
100
0
25
50
75
100
0 100 200 300 4000
50
100
0 102 103 104 105
0
102
103
104
105 1.6 1.0
12.385.2
Weig
ht
(g)
Kit
CD41
Gr1
CD11b
CTRL ABM
FL
d
e
f g
(Panels b à g)
0 102 103 104 105
0
102
103
104
105 1.5 1.1
36.261.2
0 102 103 104 105
0
102
103
104
105 0.9 15.3
10.972.9
0 102 103 104 105
0
102
103
104
105 18.4 3.3
5.073.2
0 102 103 104 105
0
102
103
104
105 11.9 32.3
4.151.7
0 102 103 104 105
0
102
103
104
105 3.9 0.5
1.793.9
0 102 103 104 105
0
102
103
104
105 3.5 44.4
3.049.2
CD41+ CD11b+Gr1+
Spleen
Figure 6
b
iEG GFP+
Dox +
1st HSC
MPP
HSC
MPP
a
Weeks
c
Weeks Weeks
HSC MPP3
Ce
lls (
10
3/m
m3)
% G
FP
+
Ce
lls (
10
6/m
m3)
% G
FP
+
Plt RBC
Surv
iva
l
Time (days)
HSC C
ells
(10
3/m
m3)
WBC
Cells
(10
3/m
m3)
Plt
% C
ells
% C
ells
B220+
% C
ells
T cells
% C
ells
h i
j
% C
ells
% C
ells
% C
ells
% C
ells
% C
ells
CD41+ CD11b+Gr1+ B220+CD19+
Thy
0 102 103 104 105
0
102
103
104
105 2.9 0.7
17.479.0
0 102 103 104 105
0
102
103
104
105 13.1 18.6
1.267.1
% G
FP
+
FL12.5
ABM
MPP4 CTRL
iEG Dox -
iEG Dox +
% G
FP
+
BM
*** **
** * **
#1: 30 days
*** ***
** ** *
*** **
ABM FL
*
B : Dox -
A : Dox +
#2: 315 days #3: 353 days
0 102 103 104 105
0
102
103
104
105 2.3 11.6
1.484.8
CD4+ CD8+ CD4+CD8+
GFP + GFP -
EG GFP+ from iEG
2nd
Group A : Dox +
Dox +
EG
EG
GFP- from recipient
Leukemia follow-up
(Panels i à j)
1st
BMT
2 weeks post-graft
Group B : Dox -
2 4 6 16 28 40 2 4 6 16 28 40 2 4 6 16 28 40 100 200 300 400
CTRL FL ABM CTRL FL ABM CTRL FL ABM CTRL FL ABM
CTRL FL ABM CTRL FL ABM CTRL FL ABM CTRL FL ABM
Days 0 3 7 Days 0 3 7
Days 0 3 7 Days 0 3 7
BM Spl Bld
0
5
10
15
20
% C
ells
CD71+Ter119+
BM Spl Bld BM Spl Bld LN
BM Spl Bld LN Thy BM Spl Bld LN Thy
0
5
10
15
% C
ells
BM Spl

139
f. Histogram of the percentages of CD41+ and CD11b+Gr1+ cells in the BM of primary diseased
recipients. CD41+ (CTRL vs. FL: p<0.0001, CTRL vs. ABM: p=0.0005), CD11b+Gr1+ (CTRL vs. FL: p=0.0016, CTRL vs. ABM: p=0.0717).
g. Histogram of the percentages of B220+, CD4+ and CD8+ cells in the spleen of primary diseased recipients. B220+ (CTRL vs. FL: p=0.012, CTRL vs. ABM: p=0.0102), T cells=CD4+ or CD8+ cells (CTRL vs. FL: p<0.0001, CTRL vs. ABM: p=0.0016).
h. Experimental design: 1x106 BM cells from the diseased primary recipients at time of analysis were transplanted into secondary sub-lethally irradiated Dox-treated recipients. After two weeks, Dox treatment was maintained for group A (GFP+ cells expressing iEG) and ceased in group B (GFP+ cells lacking iEG expression).
i. Plt and RBC counts (top panels) and respective percentages of GFP+ cells (lower panels) in secondary recipients of ABM LT-HSC-derived disease. D0: time at which Dox was maintained in group A and withdrawn in group B. Mean+/-SEM (n=6 / group) is shown.
j. Flow cytometry analyses in secondary recipients of iEG ABM LT-HSC-derived disease at six months after Dox withdrawal. Analyses were gated on GFP+ (iEG donor-derived) or GFP- (WT recipient) cells.
Statistical significance is indicated as p values (Student’s t test except when otherwise specified). *: p<0.05, **: p<0.01, ***: p<0.001.
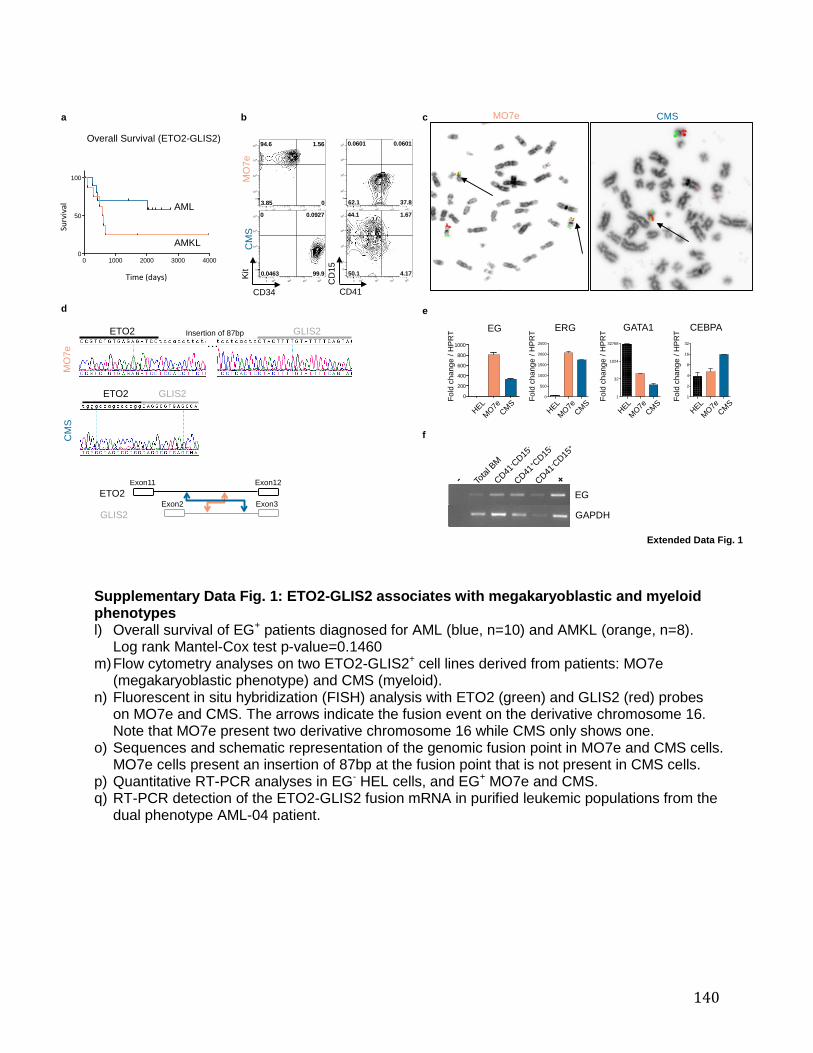
140
Supplementary Data Fig. 1: ETO2-GLIS2 associates with megakaryoblastic and myeloid phenotypes l) Overall survival of EG+ patients diagnosed for AML (blue, n=10) and AMKL (orange, n=8).
Log rank Mantel-Cox test p-value=0.1460 m) Flow cytometry analyses on two ETO2-GLIS2+ cell lines derived from patients: MO7e
(megakaryoblastic phenotype) and CMS (myeloid). n) Fluorescent in situ hybridization (FISH) analysis with ETO2 (green) and GLIS2 (red) probes
on MO7e and CMS. The arrows indicate the fusion event on the derivative chromosome 16. Note that MO7e present two derivative chromosome 16 while CMS only shows one.
o) Sequences and schematic representation of the genomic fusion point in MO7e and CMS cells. MO7e cells present an insertion of 87bp at the fusion point that is not present in CMS cells.
p) Quantitative RT-PCR analyses in EG- HEL cells, and EG+ MO7e and CMS. q) RT-PCR detection of the ETO2-GLIS2 fusion mRNA in purified leukemic populations from the
dual phenotype AML-04 patient.
0 1000 2000 3000 40000
50
100
0
200
400
600
800
1000
0
500
1000
1500
2000
2500
1
32
1024
32768
1
2
4
8
16
32
a
f
Extended Data Fig. 1
b
Overall Survival (ETO2-GLIS2)
AML
AMKL
- Tota
l BM
CD41
- CD15
-
+
EG
GAPDH
CMS MO7e c
Exon11 Exon12
Exon2 Exon3
ETO2
GLIS2
… Insertion of 87bp ETO2 GLIS2
CM
S
MO
7e
ETO2 GLIS2
0 102 103 104 105
0
102
103
104
105 0.0601 0.0601
37.862.1
0 102 103 104 105
0
102
103
104
105 44.1 1.67
4.1750.1
CD
15
CD41
MO
7e
C
MS
d e
EG
Fold
ch
ang
e / H
PR
T ERG GATA1 CEBPA
0 102 103 104 105
0
102
103
104
105 94.6 1.56
03.85
0 102 103 104 105
0
102
103
104
105 0 0.0927
99.90.0463Kit
CD34
Fold
ch
ang
e / H
PR
T
Fold
ch
ang
e / H
PR
T
Fold
ch
ang
e / H
PR
T
Time(days)
Survival
CD41
+ CD15
-
CD41
- CD15
+
HEL
M
O7e
CM
S
HEL
M
O7e
CM
S
HEL
M
O7e
CM
S
HEL
M
O7e
CM
S
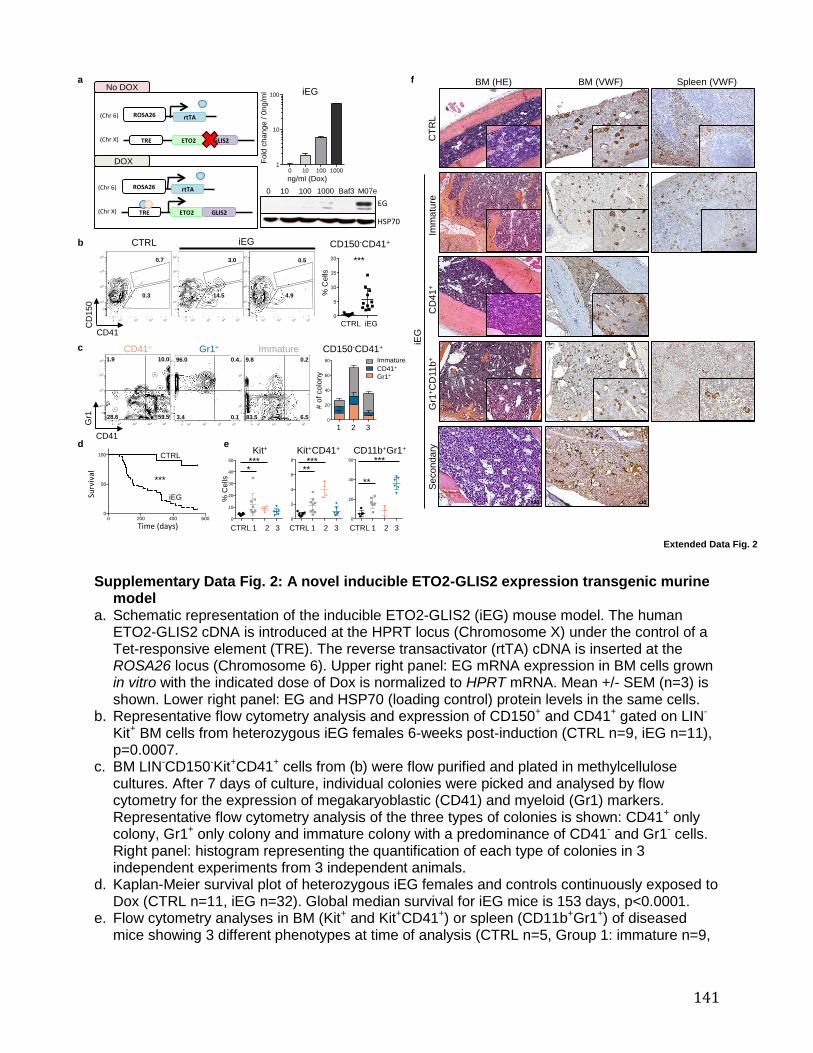
141
Supplementary Data Fig. 2: A novel inducible ETO2-GLIS2 expression transgenic murine
model a. Schematic representation of the inducible ETO2-GLIS2 (iEG) mouse model. The human
ETO2-GLIS2 cDNA is introduced at the HPRT locus (Chromosome X) under the control of a Tet-responsive element (TRE). The reverse transactivator (rtTA) cDNA is inserted at the ROSA26 locus (Chromosome 6). Upper right panel: EG mRNA expression in BM cells grown in vitro with the indicated dose of Dox is normalized to HPRT mRNA. Mean +/- SEM (n=3) is shown. Lower right panel: EG and HSP70 (loading control) protein levels in the same cells.
b. Representative flow cytometry analysis and expression of CD150+ and CD41+ gated on LIN-
Kit+ BM cells from heterozygous iEG females 6-weeks post-induction (CTRL n=9, iEG n=11), p=0.0007.
c. BM LIN-CD150-Kit+CD41+ cells from (b) were flow purified and plated in methylcellulose cultures. After 7 days of culture, individual colonies were picked and analysed by flow cytometry for the expression of megakaryoblastic (CD41) and myeloid (Gr1) markers. Representative flow cytometry analysis of the three types of colonies is shown: CD41+ only colony, Gr1+ only colony and immature colony with a predominance of CD41- and Gr1- cells. Right panel: histogram representing the quantification of each type of colonies in 3 independent experiments from 3 independent animals.
d. Kaplan-Meier survival plot of heterozygous iEG females and controls continuously exposed to Dox (CTRL n=11, iEG n=32). Global median survival for iEG mice is 153 days, p<0.0001.
e. Flow cytometry analyses in BM (Kit+ and Kit+CD41+) or spleen (CD11b+Gr1+) of diseased mice showing 3 different phenotypes at time of analysis (CTRL n=5, Group 1: immature n=9,
0
20
40
60
0
2
4
6
8
0
10
20
30
40
50
0 200 400 6000
50
100
0
20
40
60
80
0
5
10
15
20
0 10 100 10001
10
100
ROSA26 rtTA
TRE ETO2 GLIS2
(Chr6)
(ChrX)
ROSA26 rtTA
TRE ETO2 GLIS2
(Chr6)
(ChrX)
No DOX
DOX
a
b
c
CTRL iEG
CD41+ Gr1+ Immature
0 10 100 1000 Baf3 M07e
EG
HSP70
CD
150
CD41
Gr1
CD41 e
0 102 103 104 105
0
102
103
104
105
14.5
3.0
0 102 103 104 105
0
102
103
104
105
4.9
0.5
0 102 103 104 105
0
102
103
104
105
0.3
0.7
0 102 103 104 105
0
102
103
104
105 96.0 0.4
0.13.40 102 103 104 105
0
102
103
104
105 1.9 10.0
59.528.60 102 103 104 105
0
102
103
104
105 9.8 0.2
6.583.5
CD150-CD41+
CD150-CD41+
Kit+CD41+
iEG
Kit+ CD11b+Gr1+
Time(days)
Survival
% C
ells
f
C.
Spleen (VWF) BM (HE) BM (VWF)
CT
RL
Imm
atu
re
CD
41
+
Gr1
+C
D11
b+
S
eco
nd
ary
# o
f co
lony
% C
ells
iEG
* ***
CTRL 1 2 3 CTRL 1 2 3
** ***
CTRL 1 2 3
**
*** CTRL
iEG
***
Fold
cha
ng
e /
0n
g/m
l
ng/ml (Dox)
CTRL iEG
***
1 2 3
Immature
CD41+
Gr1+
d
Extended Data Fig. 2

142
Group 2: CD41+ n=4 and Group 3: CD11b+Gr1+ n=6). Kit+ (CTRL vs. Group 1: p=0.0308, CTRL vs. Group 2: p=0.0002, CTRL vs. Group 3: p=0.0562); Kit+CD41+ (CTRL vs. Group 1: p=0.0058, CTRL vs. Group 2: p<0.0001, CTRL vs. Group 3: p=01275); CD11b+Gr1+ (CTRL vs. Group 1: p=0.0034, CTRL vs. Group 2: p=0.4323, CTRL vs. Group 3: p<0.0001).
f. Histopathological analyses of BM and spleen sections of primary mice (CTRL and 3 groups of iEG) and secondary recipients.
Statistical significance is indicated as p values (Student’s t test with the exception of the survival curve where a log rank Mantel-Cox test is used). *: p<0.05, **: p<0.01, ***: p<0.001.
Supplementary Data Fig. 3: Fetal liver hematopoiesis favours the megakaryoblastic phenotype upon expression of various pediatric oncogenes a. Wright-Giemsa staining of cytospots from FL or ABM derived methylcellulose cultures at
passage (P)3. b. ABM cells from iEG animal were replated for 8 passages and the indicated populations were
flow purified to perform cytospots preparations that were then stained with Wright-Giemsa. c. Wild-type FL (E12.5), new-born BM and ABM progenitors were transduced with ETO2-GLIS2-
encoding retroviruses (rEG), flow sorted and replated for 3 rounds in methylcellulose cultures. Flow cytometry analysis at P3 for the in vitro analysis wild-type hematopoietic progenitors retrovirally transduced with ETO2-GLIS2. Compare to the phenotype obtained in Fig. 3c with the iEG model using the same conditions.
d. Wild-type FL (E12.5) and ABM progenitors were transduced with MLL-AF9 (rMA9) and NUP98-KDM5A (rNK)-encoding retroviruses, plated in methylcellulose for 7 days and analysed by flow cytometry for megakaryoblastic (CD41) and myeloid (Gr1) markers. A representative flow cytometry analysis at P1 is shown.
CD
41
+G
r1-
Gr1
+
b
c
Gr1
CD41
FL12.5 Newborn BM ABM
0 102 103 104 105
0
102
103
104
105 0.0 0.1
38.861.10 102 103 104 105
0
102
103
104
105 32.2 8.6
46.612.60 102 103 104 105
0
102
103
104
105 15.5 1.8
65.817.0
FL12
.5
AB
M
a
Gr1
CD41
FL12.5
A
BM
d rMA9 rNK
e
iMA9
FL
AB
M
Dox +
Sort CD41+
P1 P6
FL12.5
ABM
0
20
40
60
80
100
0
20
40
60
80
100
0
10
20
30
40
50
0
10
20
30
40
50
rMA9
f
% C
D4
1+
FL ABM
rNK
% C
D4
1+
FL ABM
FL ABM FL ABM
% G
r1+
% G
r1+
***
***
**
**
Wild-type FACS
Retroviral transduction rEG
P1 P3
FL12.5
ABM
Newborn BM
Extended Data Fig. 3
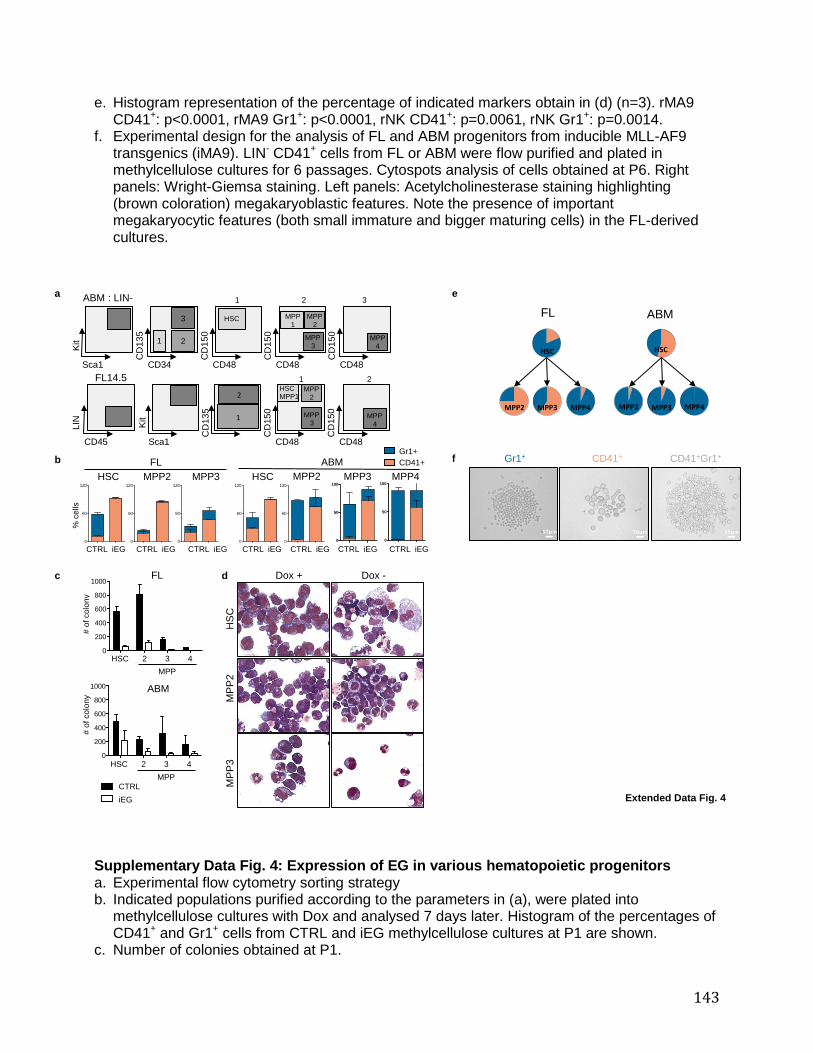
143
e. Histogram representation of the percentage of indicated markers obtain in (d) (n=3). rMA9 CD41+: p<0.0001, rMA9 Gr1+: p<0.0001, rNK CD41+: p=0.0061, rNK Gr1+: p=0.0014.
f. Experimental design for the analysis of FL and ABM progenitors from inducible MLL-AF9 transgenics (iMA9). LIN- CD41+ cells from FL or ABM were flow purified and plated in methylcellulose cultures for 6 passages. Cytospots analysis of cells obtained at P6. Right panels: Wright-Giemsa staining. Left panels: Acetylcholinesterase staining highlighting (brown coloration) megakaryoblastic features. Note the presence of important megakaryocytic features (both small immature and bigger maturing cells) in the FL-derived cultures.
Supplementary Data Fig. 4: Expression of EG in various hematopoietic progenitors a. Experimental flow cytometry sorting strategy b. Indicated populations purified according to the parameters in (a), were plated into
methylcellulose cultures with Dox and analysed 7 days later. Histogram of the percentages of CD41+ and Gr1+ cells from CTRL and iEG methylcellulose cultures at P1 are shown.
c. Number of colonies obtained at P1.
0
200
400
600
800
1000
0
50
100
0
50
100
0
60
120
0
60
120
0
60
120
0
60
120
0
60
120
c
a
HS
C
Dox + Dox -
MP
P2
M
PP
3
1 2
3
1 ABM : LIN-
Kit
Sca1
CD
13
5
CD34
HSC MPP 1
MPP 2
MPP 3
MPP 4
2 3
CD
15
0
CD48
CD
15
0
CD48
CD
15
0
CD48
1
2MPP
2
MPP 3
MPP 4
2 1 HSC MPP1
FL14.5
LIN
CD45
Kit
Sca1
CD
13
5
CD
15
0
CD48
CD
15
0
CD48
d
HSC MPP4 MPP2 MPP3 HSC MPP2 MPP3
b ABM FL
CTRL iEG
% c
ells
CTRL iEG CTRL iEG
Gr1+
CD41+
CTRL iEG CTRL iEG CTRL iEG CTRL iEG
# o
f co
lony
# o
f co
lon
y
HSC 2 3 4
MPP
HSC 2 3 4
MPP
HSC
MPP2 MPP3 MPP4
HSC
MPP2 MPP3 MPP4
FL ABM
e
f Gr1+ CD41+ CD41+Gr1+
Extended Data Fig. 4
CTRL
iEG
0
200
400
600
800
1000
ABM
FL
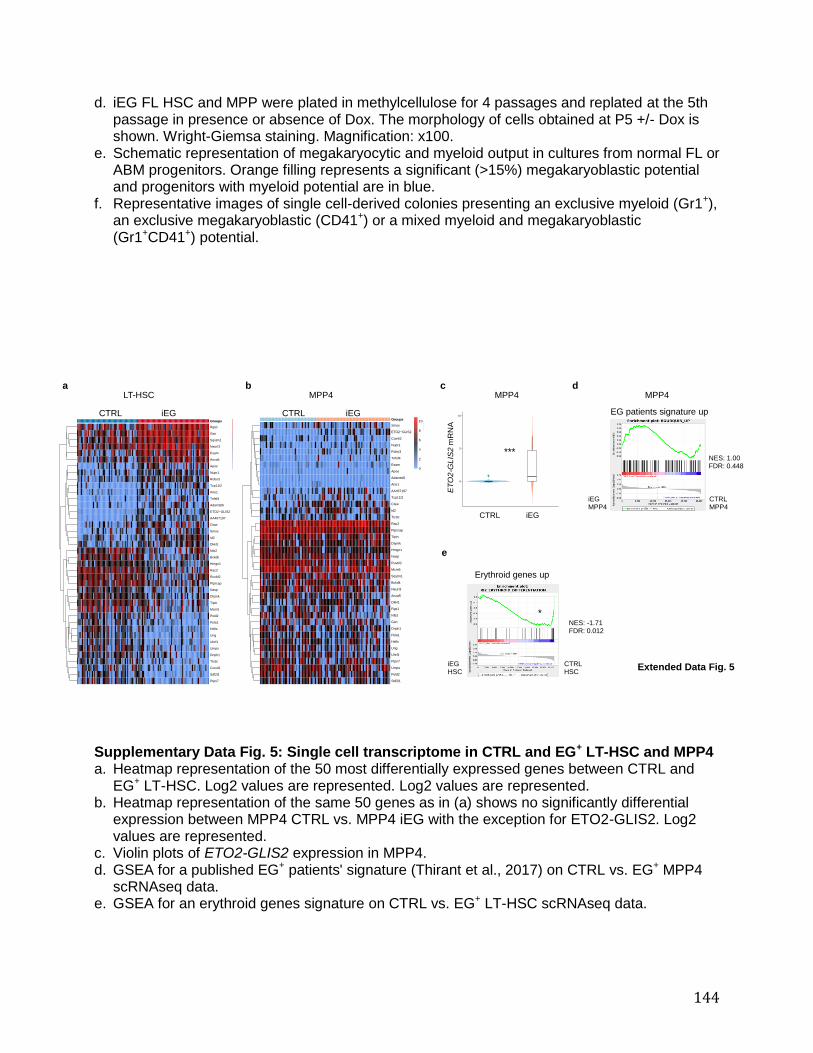
144
d. iEG FL HSC and MPP were plated in methylcellulose for 4 passages and replated at the 5th passage in presence or absence of Dox. The morphology of cells obtained at P5 +/- Dox is shown. Wright-Giemsa staining. Magnification: x100.
e. Schematic representation of megakaryocytic and myeloid output in cultures from normal FL or ABM progenitors. Orange filling represents a significant (>15%) megakaryoblastic potential and progenitors with myeloid potential are in blue.
f. Representative images of single cell-derived colonies presenting an exclusive myeloid (Gr1+), an exclusive megakaryoblastic (CD41+) or a mixed myeloid and megakaryoblastic (Gr1+CD41+) potential.
Supplementary Data Fig. 5: Single cell transcriptome in CTRL and EG+ LT-HSC and MPP4 a. Heatmap representation of the 50 most differentially expressed genes between CTRL and
EG+ LT-HSC. Log2 values are represented. Log2 values are represented. b. Heatmap representation of the same 50 genes as in (a) shows no significantly differential
expression between MPP4 CTRL vs. MPP4 iEG with the exception for ETO2-GLIS2. Log2 values are represented.
c. Violin plots of ETO2-GLIS2 expression in MPP4. d. GSEA for a published EG+ patients' signature (Thirant et al., 2017) on CTRL vs. EG+ MPP4
scRNAseq data. e. GSEA for an erythroid genes signature on CTRL vs. EG+ LT-HSC scRNAseq data.
a b
NES: 1.00 FDR: 0.448
EG patients signature up
iEG MPP4
CTRL MPP4
d c
Extended Data Fig. 5
iEG Smox
ETO2−GLIS2
Cox4i2
Nupr1
Robo3
Tnfsf4
Esam
Apoe
Adamtsl5
Ano1
AA467197
Tcp11l2
Ctsw
Id2
Ttc9c
Rac2
Ptprcap
Tipin
Dtymk
Hmgn1
Nasp
Ruvbl2
Mcm5
Sqstm1
Bckdk
Neurl3
Anxa5
Dkkl1
Rgs1
Nfe2
Gsn
Dnph1
Pola1
Hells
Ung
Uhrf1
Ptpn7
Umps
Pold2
Sdf2l1
Groups Groups
MPP4 EG −
MPP4 EG +
0
2
4
6
8
10
CTRL
MPP4
●●
0
5
10
CB
FA
2T
3−
GL
IS2
***
CTRL iEG
MPP4 MPP4
CTRL iEG
LT-HSC
Rgs1
Gsn
Sqstm1
Neurl3
Esam
Anxa5
Apoe
Nupr1
Robo3
Tcp11l2
Ano1
Tnfsf4
Adamtsl5
ETO2−GLIS2
AA467197
Ctsw
Smox
Id2
Dkkl1
Nfe2
Bckdk
Hmgn1
Rac2
Ruvbl2
Ptprcap
Nasp
Dtymk
Tipin
Mcm5
Pold2
Pola1
Hells
Ung
Uhrf1
Umps
Dnph1
Ttc9c
Cox4i2
Sdf2l1
Ptpn7
Groups Groups
HSC EG −
HSC EG +
0
2
4
6
8
10
ET
O2
-GL
IS2
mR
NA
e
iEG HSC
NES: -1.71 FDR: 0.012
CTRL HSC
Erythroid genes up
*
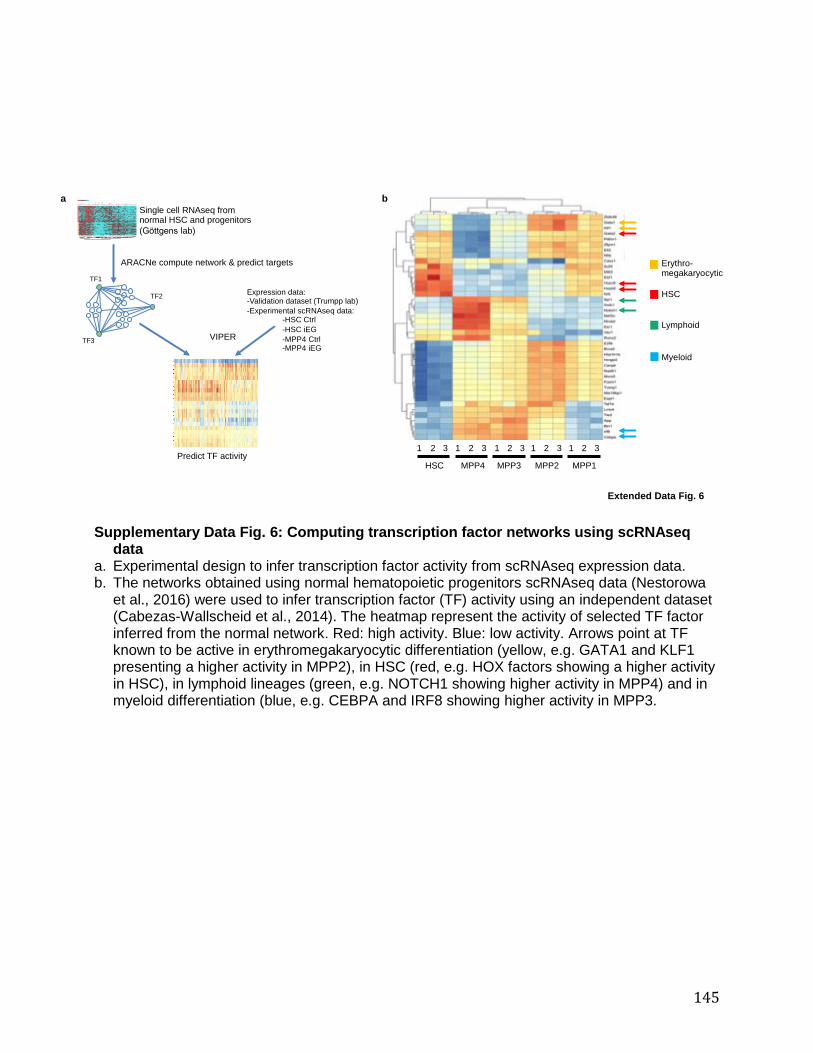
145
Supplementary Data Fig. 6: Computing transcription factor networks using scRNAseq
data a. Experimental design to infer transcription factor activity from scRNAseq expression data. b. The networks obtained using normal hematopoietic progenitors scRNAseq data (Nestorowa
et al., 2016) were used to infer transcription factor (TF) activity using an independent dataset (Cabezas-Wallscheid et al., 2014). The heatmap represent the activity of selected TF factor inferred from the normal network. Red: high activity. Blue: low activity. Arrows point at TF known to be active in erythromegakaryocytic differentiation (yellow, e.g. GATA1 and KLF1 presenting a higher activity in MPP2), in HSC (red, e.g. HOX factors showing a higher activity in HSC), in lymphoid lineages (green, e.g. NOTCH1 showing higher activity in MPP4) and in myeloid differentiation (blue, e.g. CEBPA and IRF8 showing higher activity in MPP3.
a Single cell RNAseq from normal HSC and progenitors
(Göttgens lab)
ARACNe compute network & predict targets
Expression data: -Validation dataset (Trumpp lab)
-Experimental scRNAseq data: -HSC Ctrl
-HSC iEG
-MPP4 Ctrl -MPP4 iEG
Predict TF activity HSC MPP4 MPP3 MPP2 MPP1
1 2 3 1 2 3 1 2 3 1 2 3 1 2 3
B b
Erythro- megakaryocytic
HSC
Lymphoid
Myeloid
VIPER
TF1
TF2
TF3
Nfe
2
Bckd
k
Ra
c2
Ptp
rca
p
Ru
vbl2
Fkbp
3
Rg
s1
Gs
n
Sq
stm
1
Ne
url
3
Gn
ai3
Esa
m
An
xa
5
Ap
oe
Ad
am
tsl5
Tnfs
f4
Bm
p4
AA
467
197
Du
pd
1
Tcp1
1l2
ET
O2−
GLIS
2
An
o1
Sm
ox
Id2
Dkkl1
Cts
w
Gm
665
2
Nu
pr1
Ro
bo
3
Na
sp
Dty
mk
Tip
in
Mcm
5
Pold
2
Hm
gn
1
Um
ps
Dn
ph
1
Ttc
9c
Msh
6
Dd
x27
Cox4
i2
Sd
f2l1
Ptp
n7
Cd
c45
Ga
tm
He
lls
Un
g
Uh
rf1
Pola
1
Zfp
36
7
Ty
pe
Typ
e HS
C E
G +
HS
C E
G −
0246810
Extended Data Fig. 6

146
Supplementary Data Fig. 7: Transcription factor expression and activity in EG+ or CTRL
cells a. Far left panels: violin plot representation of mRNA expression of the indicated factor. Middle
left panels: predicted activity of the indicated factor using targets inferred in the EG network and represented as violin plots. Middle right panels: predicted activity of the indicated factor using targets inferred in the EG network plotted on the activity-based 2D representation of the cells. Far right panels: predicted activity of the indicated factor using targets inferred in the normal network and represented as violin plots.
b. Pf4 expression plotted on the expression-based 2D representation of the cells. Log2 values are represented.
Statistical significance is indicated as p values (Student’s t test). * indicates p<0.05, **: p<0.01, ***: p<0.001.
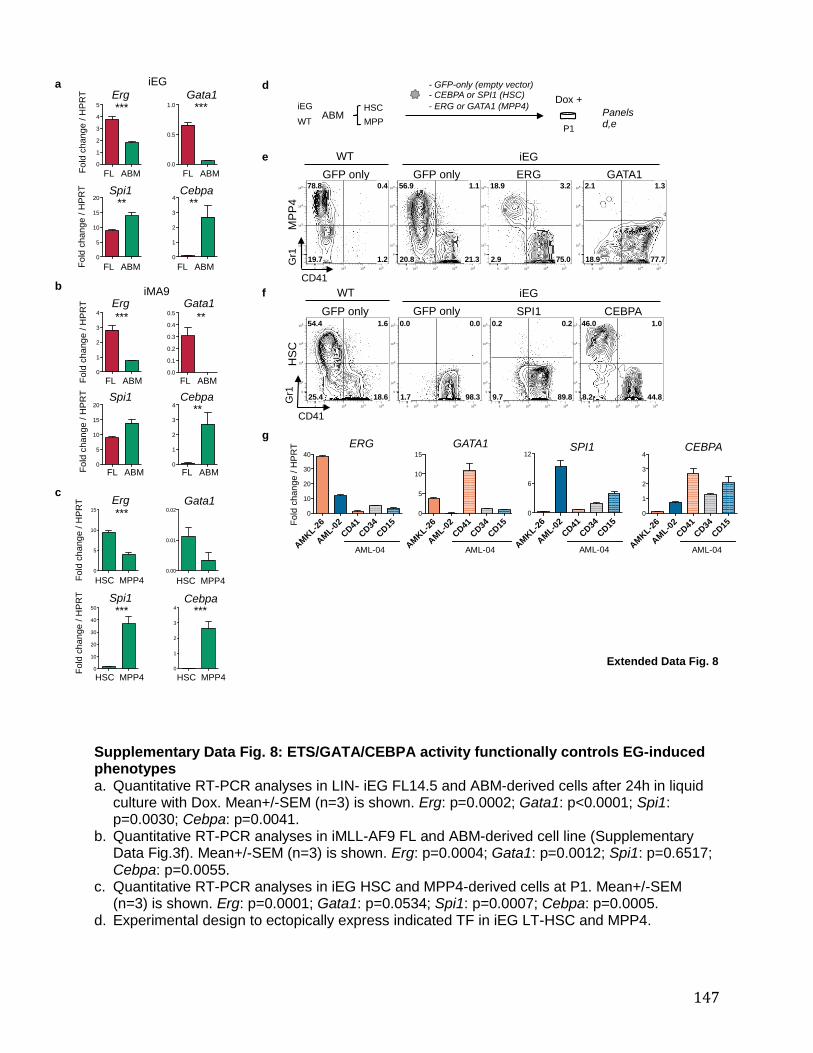
147
Supplementary Data Fig. 8: ETS/GATA/CEBPA activity functionally controls EG-induced phenotypes a. Quantitative RT-PCR analyses in LIN- iEG FL14.5 and ABM-derived cells after 24h in liquid
culture with Dox. Mean+/-SEM (n=3) is shown. Erg: p=0.0002; Gata1: p<0.0001; Spi1: p=0.0030; Cebpa: p=0.0041.
b. Quantitative RT-PCR analyses in iMLL-AF9 FL and ABM-derived cell line (Supplementary Data Fig.3f). Mean+/-SEM (n=3) is shown. Erg: p=0.0004; Gata1: p=0.0012; Spi1: p=0.6517; Cebpa: p=0.0055.
c. Quantitative RT-PCR analyses in iEG HSC and MPP4-derived cells at P1. Mean+/-SEM (n=3) is shown. Erg: p=0.0001; Gata1: p=0.0534; Spi1: p=0.0007; Cebpa: p=0.0005.
d. Experimental design to ectopically express indicated TF in iEG LT-HSC and MPP4.
AM
KL-2
6
AM
L-02
CD41
CD34
CD15
0
5
10
15
AML-04AM
KL-2
6
AM
L-02
CD41
CD34
CD15
0
10
20
30
40
AML-04
0.0
0.1
0.2
0.3
0.4
0.5
0
1
2
3
4
0
5
10
15
20
0
1
2
3
4
0
1
2
3
4
0
1
2
3
4
5
0
5
10
15
20
0.0
0.5
1.0
0
10
20
30
40
50
0
1
2
3
4
0
5
10
15
0.00
0.01
0.02
a
CEBPA
HS
C
MP
P4
SPI1
GATA1 ERG
d
GFP only
iEG
0 102 103 104 105
0
102
103
104
105 54.4 1.6
18.625.4
0 102 103 104 105
0
102
103
104
105 78.8 0.4
1.219.7
0 102 103 104 105
0
102
103
104
105 0.0 0.0
98.31.70 102 103 104 105
0
102
103
104
105 46.0 1.0
44.88.2
0 102 103 104 105
0
102
103
104
105 56.9 1.1
21.320.80 102 103 104 105
0
102
103
104
105 18.9 3.2
75.02.9
0 102 103 104 105
0
102
103
104
105 0.2 0.2
89.89.7
0 102 103 104 105
0
102
103
104
105 2.1 1.3
77.718.9
b WT
iEG WT
- GFP-only (empty vector) - CEBPA or SPI1 (HSC)
- ERG or GATA1 (MPP4)
P1
g GATA1
Fo
ld c
ha
ng
e /
HP
RT
ERG
f
Panels d,e
e
Fo
ld c
han
ge /
HP
RT
F
old
ch
an
ge
/ H
PR
T
HSC
MPP
Dox +
ABM iEG
GFP only GFP only
GFP only
Gr1
CD41 G
r1
CD41
WT
FL ABM
***
FL ABM
***
FL ABM FL ABM
** **
Erg Gata1
Spi1 Cebpa
Fo
ld c
han
ge
/ H
PR
T
Fold
ch
an
ge
/ H
PR
T
*** ***
***
HSC MPP4 HSC MPP4
HSC MPP4 HSC MPP4
Erg Gata1
Spi1 Cebpa
FL ABM
**
Gata1
***
Erg
FL ABM
FL ABM FL ABM
** Spi1 Cebpa
Fo
ld c
ha
ng
e /
HP
RT
F
old
ch
ang
e /
HP
RT
c
iEG
iMA9
AM
KL-2
6
AM
L-02
CD41
CD34
CD15
0
6
12
AML-04 AM
KL-2
6
AM
L-02
CD41
CD34
CD15
0
1
2
3
4
AML-04
SPI1 CEBPA
Extended Data Fig. 8

148
e. Representative flow cytometry analyses of cells obtained at P1 after transduction of WT and iEG MPP4 with ERG, GATA1 or GFP-only retroviruses. Analysis was gated on GFP+ cells.
f. Representative flow cytometry analyses of cells obtained at P1 after transduction of WT and iEG LT-HSC with SPI1, CEBPA or GFP-only retroviruses. Analysis was gated on GFP+ cells.
g. Quantitative RT-PCR analyses in blasts from patients with AMKL (AMKL-26), AML (AML-02) and in the different immature, megakaryoblastic and myeloid blast populations from the dual phenotype ETO2-GLIS2+ AML patient (AML-04).
Statistical significance is indicated as p values (Student’s t test). *: p<0.05, **: p<0.01, ***: p<0.001.
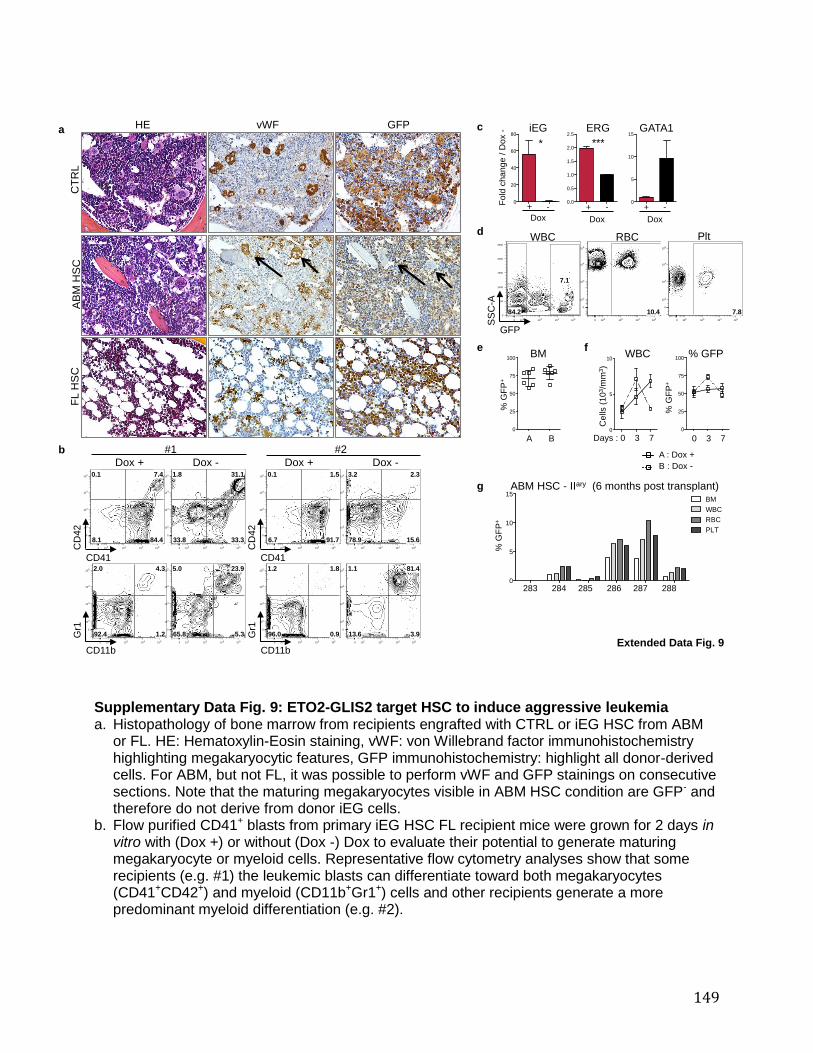
149
Supplementary Data Fig. 9: ETO2-GLIS2 target HSC to induce aggressive leukemia a. Histopathology of bone marrow from recipients engrafted with CTRL or iEG HSC from ABM
or FL. HE: Hematoxylin-Eosin staining, vWF: von Willebrand factor immunohistochemistry highlighting megakaryocytic features, GFP immunohistochemistry: highlight all donor-derived cells. For ABM, but not FL, it was possible to perform vWF and GFP stainings on consecutive sections. Note that the maturing megakaryocytes visible in ABM HSC condition are GFP- and therefore do not derive from donor iEG cells.
b. Flow purified CD41+ blasts from primary iEG HSC FL recipient mice were grown for 2 days in vitro with (Dox +) or without (Dox -) Dox to evaluate their potential to generate maturing megakaryocyte or myeloid cells. Representative flow cytometry analyses show that some recipients (e.g. #1) the leukemic blasts can differentiate toward both megakaryocytes (CD41+CD42+) and myeloid (CD11b+Gr1+) cells and other recipients generate a more predominant myeloid differentiation (e.g. #2).
0
25
50
75
100
0
5
10
0
25
50
75
100
0
5
10
15BM
WBC
RBC
PLT
0
5
10
15
0.0
0.5
1.0
1.5
2.0
2.5
0
20
40
60
80
d
c
b
a
CT
RL
HE vWF GFP
AB
M H
SC
BM
Ce
lls (
10
3/m
m3)
% G
FP
+
WBC % GFP
0 102 103 104 105
0
50K
100K
150K
200K
250K
7.1
84.20 102 103 104 105
0
102
103
104
105
7.80 102 103 104 105
0
102
103
104
105
10.4
SS
C-A
GFP
WBC RBC Plt
e
CD
42
CD41
Gr1
CD11b
H
Dox +
Dox -
0 102 103 104 105
0
102
103
104
105 0.1 7.4
84.48.1
0 102 103 104 105
0
102
103
104
105 2.0 4.3
1.292.4
0 102 103 104 105
0
102
103
104
105 1.8 31.1
33.333.8
0 102 103 104 105
0
102
103
104
105 5.0 23.9
5.365.8
0 102 103 104 105
0
102
103
104
105 0.1 1.5
91.76.7
0 102 103 104 105
0
102
103
104
105 1.2 1.8
0.996.0
0 102 103 104 105
0
102
103
104
105 3.2 2.3
15.678.9
0 102 103 104 105
0
102
103
104
105 1.1 81.4
3.913.6
CD
42
CD41
Gr1
CD11b
Dox +
Dox -
#1
#2
f
g ABM HSC - IIary (6 months post transplant)
FL H
SC
+ -
Dox
Fo
ld c
han
ge /
Do
x -
+ -
Dox
+ -
Dox
iEG ERG GATA1
*** *
A B
% G
FP
+
Days : 0 3 7 0 3 7
283 284 285 286 287 288
% G
FP
+
Extended Data Fig. 9
B : Dox -
A : Dox +

150
c. Quantitative RT-PCR analysis of ETO2-GLIS2 (iEG), Erg and Gata1 expression in cells obtained in (b). Expression was normalized to HPRT mRNA level. iEG: p=0.0121, Erg: p<0.0001, Gata1: p=0.0725.
d. Representative flow cytometry result of the percentage of GFP+ cells in white blood cells (WBC), erythrocytes (RBC) and platelets (Plt) of IIary ABM recipients.
e. Percentage of GFP in the BM of IIary recipients engrafted with cells from ABM LT-HSC-derived Iary recipients. Analysis was performed at 2-weeks post-graft in Dox-exposed IIary recipients. The same day, Dox was removed in Group B and maintained in Group A (Group A n=6, Group B n=6). These results indicate that the initial percentage of GFP is the same in the two groups.
f. Follow-up of the WBC and percentage of GFP in WBC upon removal of Dox treatment. D0 represents the time at which Dox was removed in Group B and maintained in Group A. Mean+/SEM (n=6 / group) is shown.
g. IIary recipients in Group B (Dox -) were analysed 6 months post-Dox removal to assess residual long-term HSC potential of iEG+ leukemic (GFP+) cells. Histograms indicate that within a recipient the percentages of GFP+ cells were similar in the BM, WBC, RBC and Plt indicating that GFP+ cells are endowed with LT-HSC potential.
Statistical significance is indicated as p values (Student’s t test). *: p<0.05, **: p<0.01, ***: p<0.001.
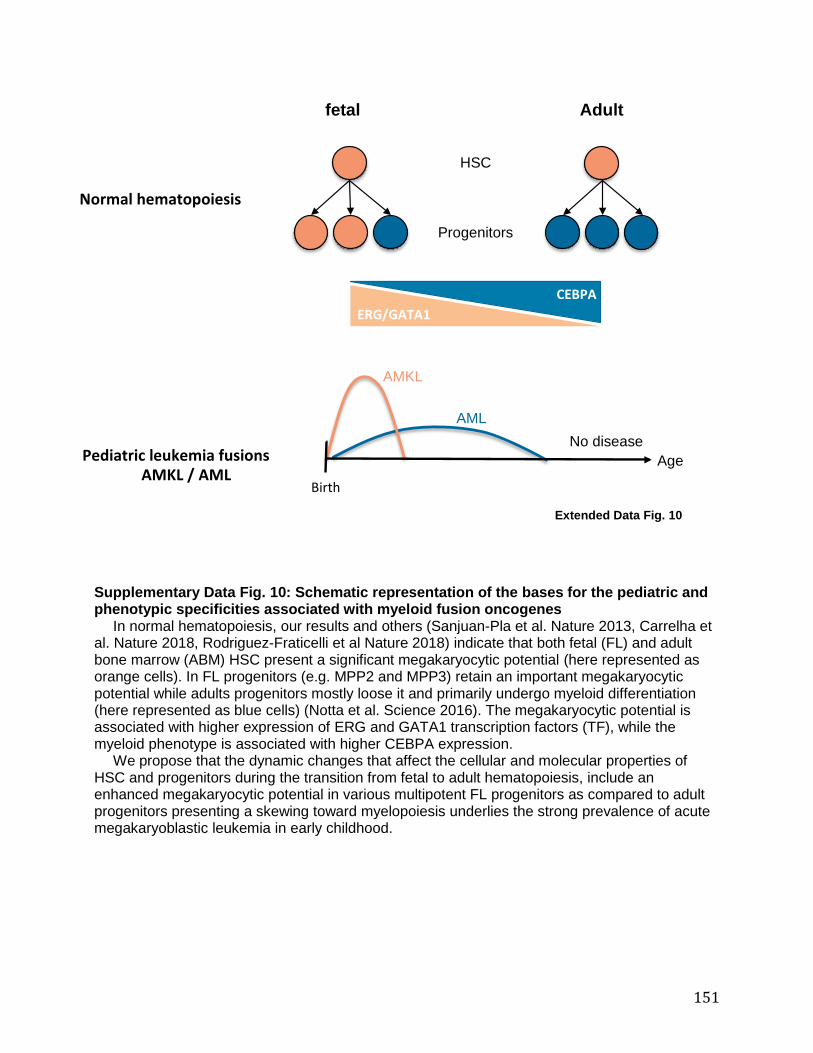
151
Supplementary Data Fig. 10: Schematic representation of the bases for the pediatric and phenotypic specificities associated with myeloid fusion oncogenes
In normal hematopoiesis, our results and others (Sanjuan-Pla et al. Nature 2013, Carrelha et al. Nature 2018, Rodriguez-Fraticelli et al Nature 2018) indicate that both fetal (FL) and adult bone marrow (ABM) HSC present a significant megakaryocytic potential (here represented as orange cells). In FL progenitors (e.g. MPP2 and MPP3) retain an important megakaryocytic potential while adults progenitors mostly loose it and primarily undergo myeloid differentiation (here represented as blue cells) (Notta et al. Science 2016). The megakaryocytic potential is associated with higher expression of ERG and GATA1 transcription factors (TF), while the myeloid phenotype is associated with higher CEBPA expression.
We propose that the dynamic changes that affect the cellular and molecular properties of HSC and progenitors during the transition from fetal to adult hematopoiesis, include an enhanced megakaryocytic potential in various multipotent FL progenitors as compared to adult progenitors presenting a skewing toward myelopoiesis underlies the strong prevalence of acute megakaryoblastic leukemia in early childhood.
Normalhematopoiesis
PediatricleukemiafusionsAMKL/AML
Extended Data Fig. 10
AMKL
AML
fetal Adult
ERG/GATA1
CEBPA
Birth
Age
No disease
HSC
Progenitors

152
METHODS
Reagents and resources Supplementary Table 13 contains a list of the relevant reagents. Patient-derived data and material Patient blood and BM samples were obtained with the informed consent of the patient or their family in accordance with national ethics law. Samples were subjected to Ficoll gradient, immunophenotyped, and used fresh or frozen in fetal bovine serum (FBS) supplemented with 10% DMSO for subsequent use. DNA and RNA were obtained from the mononuclear cell fractions using the manufacturer's recommendations (Qiagen). cDNA from 284 patients were available to detect the EG fusion via RT-PCR. The primer sequences are provided in Supplementary Table 14. Clinical data were obtained from the ELAM02 protocol. The present study focused on 385 of 438 children treated in the ELAM02 trial (Treating Patients with Childhood Acute Myeloid Leukemia with Interleukin-2; ClinicalTrials.gov NCT00149162). Patient selection was based on the availability of genomic DNA and RNA upon AML diagnosis. Children aged 0 to 18 years newly diagnosed with AML were enrolled between March 2005 and December 2011. Patients diagnosed with acute promyelocytic leukemia, therapy-related AML or Down syndromes were excluded from the ELAM02 trial. The study was approved by the Ethics Committee of Saint-Antoine Paris University Hospital (Assistance Publique-Hôpitaux de Paris) and by the Institutional Review Board of the French Regulatory Agency. The study was conducted in accordance with the declaration of Helsinki. Data concerning age at diagnosis was also collected from the ELAM02 protocol and de Rooij et al.9 including 87 AMKL patients. Patient-Derived Xenotransplantation (PDX) Patient-derived xenograft protocols have been described previously8. FISH The DNA probe set obtained from Empire genomics (CBFA2T3 and GLIS2 FISH probes, AmpliTech, Compiègne, France) hybridizes to chromosome 16p13.3 at CBFA2T3 locus (Red) and 16q24.3 at GLIS2 (green) in normal metaphase spreads or interphase nuclei. FISH analysis was performed following the manufacturer's protocol. Interphase nuclei and metaphases were scored on Imager Z2 fluorescence microscope (Zeiss, France) using Isis/Metafer/Relosys MetaSystems Software (MetaSystems, Altlussheim GmbH, Germany). In cells with the EG fusion, the overlap between the two probes generates a yellow signal indicative of the fusion oncogene. Cell lines The human AMKL MO7e cells59 (derived from a 6-month-old girl) were cultured in MEM-α supplemented with 20% FBS, Penicillin (100U/mL)-Streptomycin (100μg/mL), 2mM L-Glutamine (Gibco) and 5 ng/ml of human GM-CSF (PeproTech). The human CMS cells60 (derived from a 2-years-old girl) were cultured in RPMI1640 supplemented with 10% FBS, Penicillin (100U/mL)-Streptomycin (100μg/mL), and 2mM L-Glutamine (Gibco). The human AML-M6 HEL 92.1.7 (HEL) cells61 (derived from a 30-years-old patient) were cultured in RPMI1640 supplemented with 10% FBS, Penicillin (100U/mL)-Streptomycin (100μg/mL), and 2mM L-Glutamine (Gibco). All cell lines were karyotyped.

153
Mice To generate the iETO2-GLIS2:rtTA (iEG) model, the human EG cDNA was cloned into the p2Lox vector and electroporated into A2Lox-Cre embryonic stem (ES) cells, a generous gift from Michael Kyba62. ES cell clones were selected for the correct integration of EG at the Hprt locus on chromosome X and used to obtain germline-transmitting chimeras that were subsequently backcrossed into the C57BL/6 background. Genotyping of the reverse tetracycline-controlled transactivator (rtTA) and the EG transgene were genotyped using the primers indicated in Supplementary Table 14. To obtain GFP+-expressing iEG mice and track the iEG cells upon in vivo transplantations, we crossed iETO2-GLIS2:rtTA with UBI-GFP mice63. Note that GFP expression is not controlled by Dox and therefore engraftment of all CTRL and iEG donor cells can be followed using GFP regardless of the Dox treatment status or iEG expression. The transgenic mouse line for inducible expression of MLL-AF9 has been described previously25. The iETO2-GLIS2 and iMLL-AF9 transgenes were induced with doxycycline provided in impregnated food pellets (Dox food pellets, 545 mg/kg, Provider: SSNIF), which is predicted to correspond to an approximate dose of 2.2 mg/day. Mice were maintained at the Gustave Roussy preclinical facility and all experiments were approved by the French National Animal Care and Use Committee (CEEA 26, #2012-017). Bone Marrow Transplantation Transplantation of bone marrow (BM, 106) or sorted progenitor cells were performed through intravenous injection in lethally (9.5 Gy) or sub-lethally (5Gy) irradiated 8-week-old C57BL/6 recipient mice. Morphologic analysis of peripheral blood, BM, and spleen cells as well as histological analyses were performed according to standard procedures. Flow Cytometry Cells analyzed by flow cytometry were antibody stained in PBS 1X supplemented with 2% FBS for 30 minutes at 4°C with washing prior to FACS analysis. Whole BM and spleen cells from mice were collected, subjected to RBC lysis and stained for FACS analysis. To obtain hematopoietic stem and progenitor cells (HSPC), BM cells were depleted from the mature hematopoietic cells (Lin-) with the Becton Dickinson (BD) Mouse Hematopoietic Progenitor (stem) Cell enrichment Set (558451). To obtain adult BM (ABM) precise progenitors populations were further purified by FACS according to the following phenotypes: LT-HSC (HSC) were defined as LIN-Kit+Sca1+CD135-
CD34-CD48-CD150+, MPP2 cells were defined as LIN-Kit+Sca1+CD135-CD34+CD48+CD150+, MPP3 cells were defined as LIN-Kit+Sca1+CD135-CD34+CD48+CD150-, and MPP4 cells were defined as LIN-Kit+Sca1+CD135+CD34+CD48+CD150-. To obtain fetal liver HSPC, cells were depleted with Biotin-conjugated antibodies against CD3, Ter119, B220, Gr1, NK1.1, F4/80, CD19. To obtain FL HSC and progenitor populations, cells were purified by FACS according to the following phenotypes: LT-HSC were defined as LIN-Kit+Sca1+CD135-CD48-CD150+, while MPP2, MPP3 and MPP4 cells were defined with the same markers as ABM cells. Flow cytometry analyses were performed with an ARIAII, ARIAIII, CANTO-II or CANTO-X instruments (BD), and data were analyzed using FlowJo software (version Flowjo 9.3.2). Serial replating assays Total cells and/or progenitors from ABM or FL at E12.5 or E14.5 were isolated from iEG mice and plated in 3mL methylcellulose (Methocult M3434; StemCell Technologies, Canada), supplemented with IL3, IL6, SCF, EPO and 300ng/ml DOX. Colonies were scored under the microscope after five to seven days, harvested and replated (104 cells) for four rounds.

154
Single cell cultures Single cells were seeded in 96-well plates in 100ul RPMI supplemented with 10% FBS, cytokines (mIL3, mIL6, mIL11, mSCF, mTPO, mEPO, mGM-CSF, mFLT3l) and 100ng/ml Dox. Wells were scored for clonal growth after four days of culture and frequency of megakaryoblastic and myeloid cells was assessed by surface marker analysis using a previously described method64 after five to ten days. RNA extraction & quantitative RT-PCR analysis mRNA was isolated using an RNeasy Mini/Micro kit (Qiagen) and quantified using a NanoDrop (ThermoScientific). Reverse transcription was carried out with SuperScript II (Invitrogen). Q-PCR was performed with SYBR Select Master Mix or Taqman Gene Expression Master Mix (Applied Biosystems) using a 7500HT Fast Real-Time PCR System (Applied Biosystems) according to the manufacturer's recommendations. The primer sequences are provided in Supplementary Table 14. ETO2-GLIS2 genomic DNA fusion point DNA were isolated using an All Prep DNA/RNA Mini/Micro kit (Qiagen) and quantified using a NanoDrop (ThermoScientific). PCR was performed using a GeneAmp PCR System 9700. The primer sequences are provided in Supplementary Table 14. Immunoblotting Total cell lysates were prepared in lysate buffer containing 50 mM Tris-HCl (pH 8), 150 mM NaCl, 1% NP40, 1 mM EDTA, 0.1% SDS, 0.5% DOC, and protease inhibitors (PMSF 1X, NAF 1X, Sodium orthovanadate 1X, complete 1X). Western blotting was then performed using standard procedures using anti-CBFA2T3 (ETO2) and anti-HSC70 antibodies. Production of retroviral particles and transduction For retroviral transduction experiments, we constructed murine stem cell virus (MSCV) backbone constructs expressing GFP and encoding human ETO2-GLIS2, ERG, CEBPA, SPI1, GATA1 ORF. Briefly, 293T cells were transfected using Xtreme GENE transfection reagent according to the manufacturer’s instructions (Roche). Retroviral particles were harvested 24 and 48 hours after transfection. For retroviral transduction, murine progenitor cells were spinoculated twice with retroviral particles in culture media containing 5 μg/mL polybrene and 7.5 mM HEPES buffer for 90 minutes at 2500 rpm and 33°C. Single cell RNAseq (scRNAseq) Progenitor cells were isolated from iEG mice and cultured for 24 hours in RPMI supplemented with 10% FBS, cytokines (mIL3, mIL6, mSCF, mTPO, mFLT3l) and 100 ng/ml Dox. ScRNAseq analysis was performed as described previously28,65. Single cells were individually sorted in 96-well plates containing lysis buffer (0.2% Triton X-100 and 500U/ml Superase-In RNase Inhibitor). The Illumina Nextera XT DNA preparation kit was used to prepare libraries. Pooled libraries were sequenced using the Illumina Hisequation 4000 system (single-end 125 base pair reads). Statistical analyses Statistical analyses were performed using the GraphPad Prism software (version Prism 6-2) except otherwise mentioned. Bioinformatics analyses Reads quality control and alignment: Quality control of reads was performed using FastQC 0.11.7 and multiQC 1.5.dev0. The reads were aligned to the reference genome mm10

155
GRCm38.p4 81 with GSNAP (v 2015-09-29) with the following parameters: -A sam -B 5 -t 23 -n 1 -Q -N 1. Finally, gene expression was quantified using HTSeq 0.6.0 Counts pre-processing, quality controls and normalization: prior to data normalization, cells were filtered to remove low quality samples and to fulfil the following conditions: have at least 4000 detectable genes, have less than 20% of counts mapping to mitochondrial genes, have less than 50% of reads mapping to spike in controls and at least 25 counts that map to nuclear genes. Normalization was next performed by library size with the deconvolution of size factors method
described by Lun et al. using scran and scater R libraries66,67. Pre-clustering of cells and five
pool sizes from 20 to 100 (by an increment of 20) were used as normalization parameters. High variable genes (HVG) selection was performed as previously described68 for feature selection. Those HVG were used for t-SNE to explore possible underlying substructures in the data. Differential expression analysis and gene set enrichment analysis (GSEA): for detection of differences at the transcriptomic level between groups, differential expression analysis was performed with Wilcoxon test over the log2 counts values and p-value correction by Benjamini-Hochenberg (FDR cut-off at 0.05). GSEA was also performed to detect potential overexpressed
signatures in patients with default parameters except for –collapse false –permute phenotype69.
Gene Regulatory Network inference: ARACNe-AP software was used to infer a Gene Regulatory Network70 using iEG scRNAseq data to predicted a list of target genes of each TF in the context of cells expressing ETO2-GLIS2 (called "EG network", Supplementary Table 6). For that purpose, iEG cells with log2 counts < 1 for ETO2-GLIS2 were not taken into account. ARACNe was ran over the log2 normalized counts in bootstrap mode (100 iterations), with a p-value threshold of 1e-8 and a custom curated list of 2171 TFs. Similarly, we predicted the target genes of each TF in a normal hematopoietic context from published scRNAseq data39 obtained with the same protocol (called "normal network", Supplementary Table 7). Therefore, the activity of each TF was computed using two independent lists of target genes and consistent prediction of TF activity levels were obtained for key factors (Figure 5e). Transcription regulator activity prediction and Master Regulator Analysis: networks were used to infer activity (Normalized Enrichment Score, NES) of the transcriptomic regulator with R library viper38, as described in the package manual. NES were used to test differential activity by t-test and p-value correction by Benjamini-Hochenberg (FDR cut-off at 0.05). Master Regulator Analysis was also carried out as described in the manual, with calculation of bootstrapped null model and shadow correction (enrichment of a regulator driven by similar targets of another regulator) of top 25 regulators. Data availability Single cell RNAseq data were deposited into EBI - Array-Express under the accession number E-MTAB-7213.

156
References for the METHODS section 59. Avanzi, G. C. et al. Selective growth response to IL-3 of a human leukemic cell line with megakaryoblastic
features. Br. J. Hematol. 69, 359–366 (1988). 60. Sato, T. et al. HIV infection of megakaryocytic cell lines. Leuk. Lymphoma 36, 397–404 (2000). 61. Martin, P. & Papayannopoulou, T. HEL cells: a new human erythroleukemia cell line with spontaneous and
induced globin expression. Science 216, 1233–1235 (1982). 62. Iacovino, M. et al. Inducible cassette exchange: a rapid and efficient system enabling conditional gene
expression in embryonic stem and primary cells. Stem Cells Dayt. Ohio 29, 1580–1588 (2011). 63. Schaefer, B. C., Schaefer, M. L., Kappler, J. W., Marrack, P. & Kedl, R. M. Observation of antigen-
dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cell. Immunol. 214, 110–122 (2001). 64. Månsson, R. et al. Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem
cells and multipotent progenitors. Immunity 26, 407–419 (2007). 65. Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014). 66. Yip, S. H., Sham, P. C. & Wang, J. Evaluation of tools for highly variable gene discovery from single-cell
RNA-seq data. Brief. Bioinform. (2018). doi:10.1093/bib/bby011 67. Lun, A. T. L., Bach, K. & Marioni, J. C. Pooling across cells to normalize single-cell RNA sequencing data
with many zero counts. Genome Biol. 17, 75 (2016). 68. Brennecke, P. et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 10,
1093–1095 (2013). 69. Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting
genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 (2005).
70. Lachmann, A., Giorgi, F. M., Lopez, G. & Califano, A. ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinforma. Oxf. Engl. 32, 2233–2235 (2016).

157

158
DISCUSSION

159
Dans ce travail, j'ai développé un nouveau modèle expérimental d'expression inductible de
la fusion ETO2-GLIS2 chez la souris ainsi que des modèles de xénogreffes à partir
d'échantillons de patients, et contribué à décrire certaines conséquences moléculaires
imposées par cette fusion.
Dans l'ensemble, ces données expérimentales contribuent à:
1-Démontrer le rôle de dérégulateur épigénétique de la fusion ETO2-GLIS2.
2-Caractériser l'importance du contexte développemental, cellulaire et moléculaire pour la
transformation et le phénotype induit par la fusion ETO2-GLIS2.
3-Proposer de nouvelles perspectives thérapeutiques pour le traitement des LAM-M7
pédiatriques associées avec un mauvais pronostic.
Rôle de dérégulateur épigénétique de la fusion ETO2-GLIS2
L’épigénétique comprend l’ensemble des mécanismes moléculaires qui modulent
l’expression des gènes sans modifier la séquence d’ADN génomique. L’épigénome inclut des
modifications biochimiques de l’ADN et des histones, l’organisation structurale de la
chromatine, l’épissage alternatif et l’expression d’ARNs non codants. Nos observations
indiquent que la fusion ETO2-GLIS2 altère plusieurs de ces mécanismes épigénétiques.
Notamment, la fusion ETO2-GLIS2 1) implique deux facteurs transcriptionnels, 2) induit
une forte dérégulation de l'expression génique incluant des facteurs de transcription
impliqués dans la régulation hématopoïétique comme ERG et GATA1 (cf infra pour une
discussion détaillée du rôle des FT) et 3) est fréquemment retrouvée au niveau de marques
épigénétiques (ex: H3K27Ac) des super-enhancers (SE) qui contrôlent l’identité cellulaire et
est mutuellement exclusive avec la marque de répression transcriptionnelle H3K27me3.
Plusieurs questions restent en suspens pour comprendre comment les SE sont régulés par
la fusion. En particulier, est-ce que la fusion ETO2-GLIS2 joue un rôle pionnier et est
capable de créer de nouveaux SE dans des régions chromatiniennes inactives ou bien se
fixe-t-elle sur des SE ou enhancers déjà établis en interaction éventuelle avec des
partenaires protéiques présentant une activité acétyltransferase? Ces deux mécanismes
pourraient d'ailleurs coexister dans une cellule mais varier en fonction des loci.
Nous avons pu observer que la fusion se fixait à l’ADN via les partenaires d’ETO2, incluant
les facteurs de transcription ETS, GATA et RUNX ainsi que via GLIS2. Le facteur ERG

160
colocalise avec la fusion au niveau de certains SE et sa présence est associée à une
surexpression des gènes situés à proximité. De plus, des inactivations fonctionnelles
montrent le rôle essentiel de ERG sur la survie et le maintien des cellules leucémiques
ETO2-GLIS2.
Dans le cancer de la prostate, ERG a été associé à une forte expression de H3K27ac et
impliqué dans l’établissement de nouveaux SE entraînant une modification de
l’organisation chromatinienne et du profil transcriptionnel (236).
ERG pourrait donc être directement impliqué dans la génération des SE des LAM-M7 ETO2-
GLIS2. D'autre part, étant donné la différence entre les SE en fonction du contexte cellulaire,
il sera intéressant d'identifier les SE associés à la fusion dans un contexte myéloïde.
La fusion ETO2-GLIS2 semble également altérer l'épissage de certains transcrits. En effet,
chez ~40% des patients ETO2-GLIS2 LAM-M7, les cellules leucémiques expriment
également une autre fusion, DHH-RHEBL1, deux gènes contigus présentant la même
orientation sur le chromosome 12 (237). Cependant, aucune altération génomique n'a été
décrite à l'origine de cette fusion. L'hypothèse la plus probable est donc que cette fusion
DHH-RHEBL1 résulte d'un épissage aberrant survenant dans certaines cellules ETO2-GLIS2.
La formation de transcrit de fusion entre des gènes adjacents a été observée dans d’autres
cancers comme le cancer de la prostate ou le cancer du sein (238,239). Une hypothèse
concernant la formation de ces transcrits de fusion implique la diminution de la liaison de
CTCF sur les séquences « insulator » encadrant des gènes adjacents (238). CTCF est une
protéine ubiquitaire, composée de 11 domaines Zinc Finger (ZF), impliquée dans
l’organisation de la chromatine, la régulation génique, les interactions intra/inter
chromosomiques ainsi que l’épissage alternatif (240). L'expression du transcrit et de la
protéine CTCF n'est pas significativement diminuée dans les cellules ETO2-GLIS2 humaines
(nos données préliminaires). En revanche, GLIS2 présente des similitudes de motifs de
liaison à l’ADN avec CTCF. Ainsi, la formation du transcrit de fusion DHH-RHEBL1 pourrait
être due à la fixation de ETO2-GLIS2 sur certains sites CTCF engendrant une compétition
entre ces deux protéines, et une modification de l’organisation de la chromatine et de
l'épissage des transcrits DHH et RHEBL1. Une démonstration de ces hypothèses nécessitera
des analyses de capture de la conformation chromatinienne. D'autre part, il sera intéressant

161
de rechercher si la présence de cette fusion additionnelle constitue une étape épigénétique
vers le développement de leucémies plus agressives.
La réversibilité des conséquences de la fusion ETO2-GLIS2 a également pu être mise en
évidence par des approches moléculaires et cellulaires in vitro et in vivo.
Au niveau moléculaire, l’expression du peptide NC128 contenant la séquence protéique du
domaine NHR2 de ETO, inhibe l’oligomérisation de la fusion ETO2-GLIS2 et réverse
globalement l'expression des gènes associés aux SE.
Au niveau cellulaire, l’extinction de la fusion dans les blastes leucémiques murins, possible
grâce au système inductible DOX, permet de restaurer des capacités de reconstitution
hématopoïétique à long-terme ainsi qu’un potentiel de différenciation multi-lignées
attribués aux HSC normales. Ces résultats constituent une preuve de concept que l'effet
transformant de la fusion sur les HSC est réversible. Cette observation pourrait avoir des
conséquences cliniques car elle suggère qu’une inhibition spécifique de la fusion a le
potentiel de rétablir des capacités de différenciation normales et de reconstitution à long-
terme aux progéniteurs hématopoïétiques chez ces patients.
On peut noter que les fusions MLL, NUP98-KDM5A et OTT-MAL, retrouvées de façon
récurrente dans les LAM-M7, impliquent également des régulateurs transcriptionnels ayant
des conséquences épigénétiques (MLL-AF4 (241) ; OTT-MAL (242) ; MLL (243)). Il est donc
possible que certains mécanismes mis en œuvre par ETO2-GLIS2 soient communs à toutes
ces fusions.

162
Importance du contexte développemental, cellulaire et moléculaire pour la
transformation et le phénotype induit par la fusion ETO2-GLIS2.
Importance du stade de développement
Nous avons pu observer l’influence du stade de développement auquel la fusion est induite
sur le développement de leucémie in vivo chez la souris. En effet, la greffe de cellules de foie
fœtal murin à E14.5 (progéniteurs LIN- ou LT-HSC purifiés) exprimant ETO2-GLIS2 induit
des LAM-M7 agressives avec une latence faible (quelques semaines) dans des receveurs. En
revanche, une approche similaire avec des cellules de moelle osseuse adultes induit des
leucémies de façon peu efficace et seule la greffe de LT-HSC adulte conduit au
développement de leucémies avec une latence longue (plusieurs mois) et un phénotype
hétérogène.
Le stade développemental ciblé par les oncogènes pédiatriques est également relevant pour
d'autres leucémies. En effet, les LAL sont plus fréquentes chez les jeunes enfants que chez
les adultes. Certains oncogènes spécifiques des leucémies pédiatriques, dont l’origine
prénatale a été validée comme TEL-AML1 associée aux LAL-B ou MLL-AF4 dans les LAL-B,
semblent nécessiter un contexte particulier, apporté par les progéniteurs fœtaux, pour
induire une leucémie. L’analyse d’un modèle murin de la fusion MLL-AF4 suggère
l’influence d’une période particulière (E12-E14) du stade développemental ainsi que la
nécessité de cibler un progéniteur spécifique (LMPP fœtal) pour induire une pathologie
(208). Cependant, les pathologies rapportées avec ce modèle semblent se rapprocher plus
de lymphomes que de réelles leucémies aiguës suggérant que des analyses supplémentaires
seront requises pour développer un modèle fidèle de la pathologie humaine.
Dans le cas des autres oncogènes de fusion retrouvés dans les LAM-M7, nos résultats
indiquent que les propriétés des cellules fœtales ont un impact sur leur capacité à induire
une transformation présentant un phénotype mégacaryoblastique. En effet, l'expression de
MLL-AF9 et NUP98-KDM5A dans des progéniteurs fœtaux permet un phénotype
mégacaryoblastique alors que leur expression dans des progéniteurs adultes entraîne un
phénotype myéloïde. Ces résultats, obtenus in vitro, nécessitent d’être validés pour

163
confirmer la capacité de ces cellules à générer des LAM-M7 in vivo présentant les
caractéristiques retrouvées chez les patients.
Importance du type de cellule ciblée
De façon générale, la recherche de la cellule à l'origine des transformations leucémiques est
importante pour le développement de modèles récapitulant les leucémies et pour la
proposition de thérapies personnalisées. Il a été montré que certains oncogènes peuvent
transformer différents progéniteurs conduisant à des leucémies plus ou moins agressives et
sensibles aux traitements. Par exemple, la fusion MLL-AF9 peut transformer aussi bien des
cellules souches hématopoïétiques adultes que des progéniteurs GMP en leucémie
myélomonocytaire, mais son expression dans des HSC induit des leucémies plus agressives
(162).
Dans le cas des LAM-M7, au moins deux hypothèses existaient quant à leur origine cellulaire
au début de ce travail. La première était que les oncogènes arrivent dans un progéniteur
présentant une capacité de différenciation restreinte à la lignée mégacaryocytaire. La
deuxième était que les oncogènes surviennent dans les HSC ou progéniteurs multipotents et
que leurs conséquences fonctionnelles biaisent ces cellules vers un phénotype
mégacaryoblastique.
Nos résultats indiquent que les conséquences de fusion ETO2-GLIS2 sont drastiquement
différentes en fonction du progéniteur ciblé. En effet, l’expression de la fusion dans les HSC
issues de la moelle osseuse adulte engendre in vitro un phénotype exclusivement
mégacaryocytaire, tandis que les progéniteurs multipotents (MPP2 à 4) exprimant ETO2-
GLIS2 conservent aussi un phénotype myéloïde. L'implication d'un progéniteur multipotent
est également confirmé par l'existence d'un patient ETO2-GLIS2 présentant un double
phénotype et pour lequel les blastes immatures sont capables de régénérer des blastes
mégacaryoblastiques et myéloïdes dans des xénogreffes sériées.
Dans l'ensemble, ces résultats indiquent que le phénotype des leucémies induites par ETO2-
GLIS2 est dépendant du type de cellule hématopoïétique ciblé. Sur la base de ces résultats,
je propose que l'expression de la fusion dans une HSC soit nécessaire pour le
développement d'une leucémie à phénotype mégacaryoblastique (LAM-M7), et que le

164
développement d'une LAM d'un autre sous-type requiert l'expression de la fusion dans un
progéniteur plus engagé ayant perdu la capacité mégacaryocytaire.
Un lien direct entre HSC et différenciation mégacaryocytaire a été mis en évidence
récemment par plusieurs équipes. Ce modèle propose que les mégacaryocytes dérivent
directement des HSC sans passer par les progéniteurs intermédiaires, et que la
mégacaryopoïèse représente une différenciation par défaut des HSC (49). Nos résultats
soulignent que ce lien étroit entre HSC et le phénotype mégacaryoblastique pourrait
constituer le point commun entre les différents sous-groupes moléculaires de LAM-M7. En
effet, malgré l'hétérogénéité génétique des LAM-M7 pédiatriques, il est possible que les
dérégulations épigénétiques induites par les fusions des LAM-M7 aient pour point commun
d'induire le phénotype mégacaryoblastique par défaut des HSC. Dans cette hypothèse, les
LAM-M7 pourraient donc être considérées comme des leucémies de la cellule souche
hématopoïétique "primitive" plutôt que des leucémies d'un progéniteur mégacaryocytaire
engagé.
Contrôle moléculaire du phénotype
Plusieurs observations indiquent que la transformation et les phénotypes induits par la
fusion ETO2-GLIS2 impliquent une altération de l'activité de plusieurs facteurs de
transcription contrôlant l'engagement des HSC vers les différentes lignées
hématopoïétiques. En effet, les cellules humaines présentent une dérégulation de la balance
entre les facteurs ERG et GATA1. De plus, les analyses transcriptionnelles réalisées 24h post-
induction de la fusion montrent une dérégulation transcriptionnelle drastique dans les HSC
adultes murines (reproduisant la signature observée chez les patients LAM-M7 ETO2-
GLIS2), alors que les progéniteurs MPP4 sont peu affectés à ce stade. Dans l'ensemble, les
résultats obtenus dans les cellules murines et humaines indiquent que la fusion induit une
inhibition de l'activité de régulateurs majeurs de lignées comme CEBPA, PU.1 et IRF8
(myéloïde), KLF1 et NF-E2 (érythroïde), IKZF1 (lymphoïde) associée à une augmentation de
l'activité de facteurs importants pour le maintien des HSC incluant ERG, GATA2, MYCN. Ces
résultats sont compatibles avec un blocage de différenciation des HSC à un stade très
précoce. L'absence de différence significative d'expression de plusieurs facteurs myéloïdes,

165
tel que CEBPA ou IRF8 dans les MPP4, indique que la fusion ne dérégule pas les mêmes
cibles dans les deux contextes.
Dans l'ensemble, nos analyses d'expression et fonctionnelles indiquent que la fusion est
associée à une plus forte expression d’ERG et GATA1 dans les cellules leucémiques
présentant un phénotype mégacaryoblastique alors qu'une activité de CEBPA est plus
importante dans les cellules leucémiques présentant un phénotype myéloïde.
Pour le phénotype myéloïde, ces résultats sont en accord avec une étude récente impliquant
directement CEBPA dans l’engagement des HSC normales vers les différentes lignées
myéloïdes (110). CEBPA est un facteur de transcription qui régule les gènes spécifiques de
ces lignées et inhibe la prolifération cellulaire (109). Il a aussi été impliqué dans le
changement de statut prolifératif à quiescent observé entre le HSC fœtales et les HSC
adultes, notamment par la régulation du facteur Mycn (45,94). La perte de CEBPA dans les
HSC adultes est associée à une réactivation du programme transcriptionnel fœtal ainsi qu’à
une augmentation de leur prolifération (45). A l’état basal, CEBPA est moins exprimé dans
les HSC que les MPP4 (58). De façon intéressante, lorsque la fusion ETO2-GLIS2 est induite,
CEBPA ne semble pas exprimé dans les HSC alors que le niveau reste inchangé dans les
MPP4. Ceci suggère que la fusion ne parvient pas à inhiber CEBPA lorsqu’il est initialement
exprimé. Dans l'ensemble, l’acquisition d'une fusion ETO2-GLIS2 dans un progéniteur
exprimant fortement CEBPA serait en faveur du développement d’une LAM avec un
phénotype myéloïde.
En revanche, le facteur PU.1 ne semble pas déterminant pour le phénotype myéloïde et son
inhibition par la fusion pourrait donc principalement contribuer à une augmentation de
l'autorenouvellement des progéniteurs (108). Cette hypothèse nécessitera d'être testée
fonctionnellement.
Pour le phénotype mégacaryoblastique, nous proposons que la fusion ETO2-GLIS2 entraîne
une dérégulation de la balance entre le facteur ERG et GATA1 impliquée dans la progression
de la différenciation mégacaryocytaire (voir revue en annexe).
Dans les cellules murines, le facteur ERG semble exprimé de façon identique entre les HSC
et dans les progéniteurs engagés normaux tels que les MPP4 (58). L'expression de la fusion

166
ETO2-GLIS2 induit la surexpression de ERG seulement dans les cellules issues de HSC.
Cependant, la surexpression de ERG est commune à plusieurs cancers et entraîne différents
phénotypes dans des modèles murins. En effet, dans des progéniteurs murins issus de la
moelle osseuse adulte, la surexpression de ERG engendre exclusivement une leucémie aiguë
lymphoïde (LAL), alors que chez les receveurs de progéniteurs fœtaux surexprimant ERG,
30% développent une LAL et 70% développent des leucémies érythro-mégacaryoblastiques
(143). La surexpression de ERG seule ne peut expliquer le phénotype mégacaryoblastique
induit par la fusion ETO2-GLIS2.
Dans le cas de GATA1, la fusion ETO2-GLIS2 inhibe son expression de façon importante à la
fois dans les cellules humaines et murines. Cependant, il est important de noter que 1)
Gata1 est plus exprimé dans les HSC que dans les progéniteurs engagés normaux tels que
les MPP4 (58), 2) la fusion ETO2-GLIS2 inhibe l'expression de Gata1 dans les HSC et les
MPP4 mais que le niveau de Gata1 reste supérieur dans les HSC en comparaison des MPP4
et 3) dans les blastes de leucémies humaines, GATA1 est plus exprimé dans les
mégacaryoblastes que dans les blastes myéloïdes. Ainsi, mon hypothèse est que le
phénotype mégacaryoblastique des LAM-M7 induites par ETO2-GLIS2, nécessite une
réduction importante de l'activité GATA1 contribuant au blocage de différenciation, mais
requiert un niveau minimal supérieur à celui observé dans les cellules myéloïdes. Le niveau
d'expression de GATA1 et de CEBPA dans la cellule dans laquelle survient la fusion pourrait
donc être un déterminant important du phénotype mégacaryoblastique ou myéloïde
résultant.
De façon intéressante, les facteurs ERG et GATA1 sont fortement exprimés dans les
progéniteurs fœtaux par rapport aux adultes. On peut également noter que les patients
LAM-M7 présentent fréquemment une trisomie 21 acquise. Une hypothèse pour expliquer
cette association est que le gène ERG, localisé sur le chromosome 21, serait surexprimé de
façon encore plus élevé par la fusion dans un contexte trisomique, dérégulant de façon
encore plus importante la balance ERG/GATA1 et renforçant ainsi le blocage de
différenciation.
Les HSC fœtales présentent des caractéristiques cellulaires et moléculaires différentes des
HSC adultes. La modification de ces caractéristiques s’opère, chez la souris, dans les 3
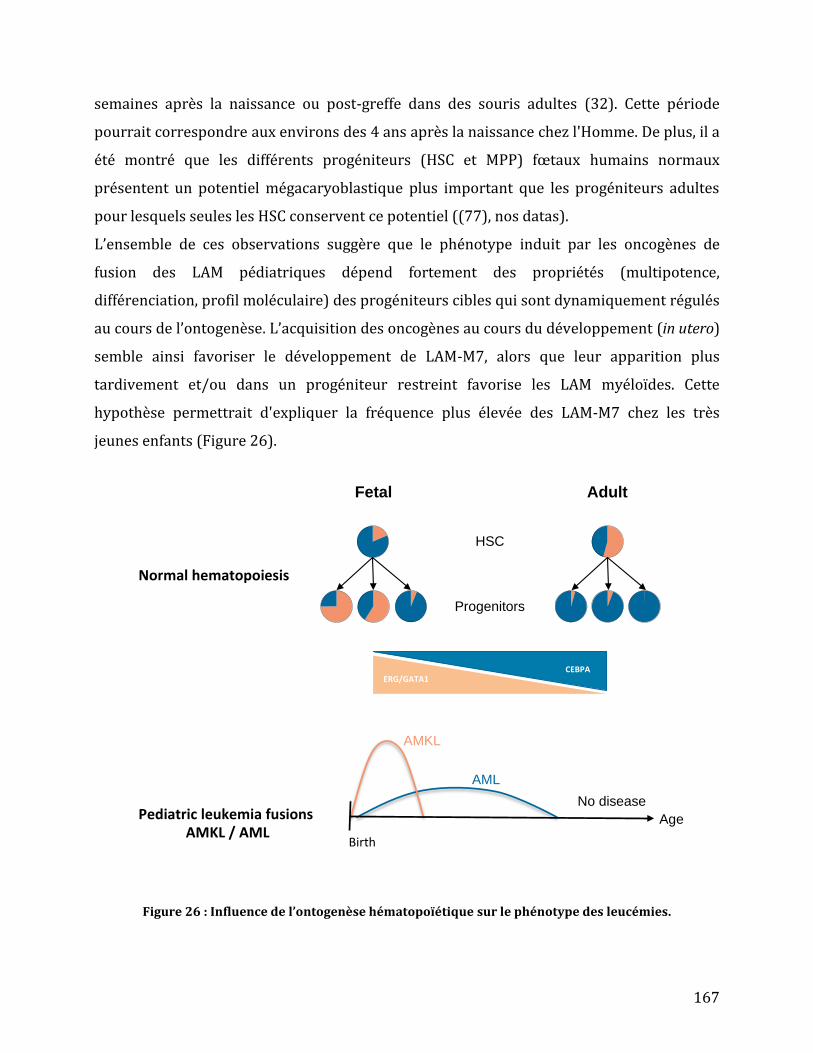
167
semaines après la naissance ou post-greffe dans des souris adultes (32). Cette période
pourrait correspondre aux environs des 4 ans après la naissance chez l'Homme. De plus, il a
été montré que les différents progéniteurs (HSC et MPP) fœtaux humains normaux
présentent un potentiel mégacaryoblastique plus important que les progéniteurs adultes
pour lesquels seules les HSC conservent ce potentiel ((77), nos datas).
L’ensemble de ces observations suggère que le phénotype induit par les oncogènes de
fusion des LAM pédiatriques dépend fortement des propriétés (multipotence,
différenciation, profil moléculaire) des progéniteurs cibles qui sont dynamiquement régulés
au cours de l’ontogenèse. L’acquisition des oncogènes au cours du développement (in utero)
semble ainsi favoriser le développement de LAM-M7, alors que leur apparition plus
tardivement et/ou dans un progéniteur restreint favorise les LAM myéloïdes. Cette
hypothèse permettrait d'expliquer la fréquence plus élevée des LAM-M7 chez les très
jeunes enfants (Figure 26).
Figure 26 : Influence de l’ontogenèse hématopoïétique sur le phénotype des leucémies.
Normalhematopoiesis
PediatricleukemiafusionsAMKL/AML
AMKL
AML
Fetal Adult
ERG/GATA1CEBPA
Birth
Age
No disease
HSC
Progenitors

168
Perspectives thérapeutiques pour les LAM-M7 et leucémies ETO2-GLIS2
La thérapie actuelle pour les LAM-M7 et leucémies ETO2-GLIS2 est une chimiothérapie
intensive non ciblée. De manière générale, le développement de nouveaux traitements pour
les leucémies s'est focalisé soit sur le ciblage de propriétés cellulaires générales
(prolifération, apoptose, différenciation) soit sur le ciblage des mécanismes moléculaires
spécifiques mis en œuvre par un oncogène.
Dans le cas des LAM-M7, l'équipe de John Crispino a proposé, il y a quelques années, une
nouvelle approche thérapeutique des hémopathies malignes affectant la lignée
mégacaryoblastique sur la base d'une propriété naturelle de cette lignée : la capacité à
former des cellules polyploïdes au cours de la différenciation terminale. Ainsi, une approche
de criblage de petites molécules pouvant induire une différenciation des blastes LAM-M7 en
induisant leur polyploïdisation a permis l'identification d'inhibiteurs de la kinase Aurora A,
tel que le dimethylfasudil (diMF) ou le MLN8237. Par la suite, l'utilisation de ces molécules
a montré qu’elles permettent l’induction de la ploïdie, la différenciation et l’apoptose de
leucémies mégacaryoblastiques murines et humaines in vitro indépendamment de
l’altération génétique observée. Ces molécules ont été testées avec succès in vivo sur des
modèles murins de xénogreffes de patient MLL-AF9 ou ETO2-GLIS2 (178,195). Ces données
suggèrent qu'une inhibition de la kinase Aurora A dans le contexte des LAM-M7 permet de
restaurer au moins en partie les capacités de différenciation des blastes de LAM-M7. Un
essai clinique testant cette approche est en cours pour les LAM-M7 et les myélofibroses de
l'adulte (clinicaltrials.gov: NCT02530619).
Pour obtenir une inhibition plus spécifique des mécanismes induits par la fusion ETO2-
GLIS2, nous avons au cours de ce travail utilisé un peptide contenant la séquence protéique
du domaine NHR2 de ETO, présentant une forte homologie avec ETO2. Ce peptide en
limitant l’oligomérisation de la fusion ETO2-GLIS2 inhibe la prolifération des cellules
leucémiques humaines in vitro et in vivo. Cette approche ciblant le complexe protéique
incluant les protéines ETO, développé pour la fusion AML1-ETO, démontre l’importance des
interactions protéiques au sein des complexes ETO/ETO2 pour le maintien des cellules
leucémiques de plusieurs sous-types de leucémies. Cependant, les limites probables de

169
cette approche pour un développement clinique incluent le mode d’administration aux
patients (ces peptides traversent difficilement la membrane des cellules) et l'observation
que la protéine ETO2 normale est essentielle au maintien des cellules souches
hématopoïétiques normales. Les perspectives de développement futures reposent donc
probablement sur l'identification d'un composant de ce complexe recruté plus
spécifiquement par ETO2-GLIS2 ou en commun avec d'autres oncogènes de fusions et dont
l'inhibition n'altère pas significativement l'hématopoïèse normale.
Nous avons pu voir que le facteur ERG était important pour le maintien et la prolifération
des cellules leucémiques ETO2-GLIS2 LAM-M7. ERG est impliqué dans de nombreux cancers,
associé à un mauvais pronostic. L’inhibition de ce facteur de transcription aurait un impact
sur les patients ETO2-GLIS2 mais aussi sur beaucoup d’autres cancers.
Les facteurs ETS sont fréquemment phosphorylés par les MAPK ce qui permet de réguler
leur activité. Il serait intéressant de regarder l’état de phosphorylation de ERG dans les
LAM-M7 en vue d’utiliser des inhibiteurs des MAPK. La phosphorylation d’ERG sur la serine
283 par ERK2, a été impliquée dans le processus de leucémogenèse de patients LAM et LAL-
T (244). De même, la phosphorylation d’ERG sur la serine 215 et la serine 96 induites par
ERK2 ont été impliquées dans la migration cellulaire des cancers de la prostate (245,246).
ERG présente un domaine de liaison à l’ADN qui peut être la cible de peptide, ce qui va
entraîner sa dégradation. Ces peptides ont été testés avec succès dans des modèles de
xénogreffe de cancer de la prostate (247). Une approche similaire pourrait être envisagée
pour la fusion ETO2-GLIS2.

170
LISTE DES ABREVIATIONS
A
ABL1 v-abl abelson murine leukemia viral oncogene homolog 1 ABM Adult Bone Marrow ADN Acide désoxyribonucléique AF9 ALL1 fused gene on chromosome 9 AGM Aorte-Gonade-Mesonephros AKT v-akt murine thymoma viral oncogen homolog ARN Acide ribonucléique
B BCR B-cell receptor BMP Bone Morphogenic Protein BRD4 Bromodomain containing
C CAR CXCL12-abundant reticular CDP Common Dendritic Progenitor CEBP CCAAT/enhancer binding protein
ChIPseq Séquençage de l'immunoprécipitation de la chromatine CLP Common Lymphoid Progenitor cMoP Common Monocyte Progenitor CMP Common Myeloid Progenitor CTCF CCCTC-binding factor CXCL Chemiokine C-X-C motif ligand CXCR Chemiokine C-X-C motif receptor
D DC Dendritic cell
DHH Desert Hedgehog
E EHT Endothelial to Hematopoietic Transition EMP Erythro-Myeloid Progenitor ERG V-ETS avian erythoblastosis virus E26 Oncogene homolog ETO2 (CBFA2T3) CBFA2/RUNX1 translocation partner 3
F FAB Franco-Américano-Britanique
FACS Fluorescence-activated cell sorting FISH Fluorescent in situ hybridization FL Fetal liver

171
FLI1 Friend leukemia integration 1 FLT3 Fms-like tyrosine kinase 3 FLT3-ITD Internal tandem duplication of FLT3 FT Facteur de transcription
G GATA Globin transcription factor G-CSF Granulocte Colony Stimulating Factor GFP Green fluorescent protein
GLIS2 Kruppel-like Zinc finger protein GM-CSF Granulocyte Monocyte Colony Stimulating Factor GMP Granulocyte-Monocyte Progenitor GpIb Glycoprotéine Ib (CD42)
H H3 Histone 3 HE Hemogenic Endothelial HEC Hematopoietic Endothelial Cell HOX Homeobox gene
HPC Hematopoietic Progenitor Cell HSC Hematopoiétique Stem Cell
I iPS induce pluripotent stem cell IL1a Interleukine 1a
L LA Leucémie Aiguë
LAM Leucémie Aiguë Myéloide
LIN- Lineage negative LMPP Lymphoid Multipotent Progenitor LSC Leukemic Stem Cell LSK Cellules LIN- Kit+ Sca1+ LT-HSC Long Term-Hematopoietic Stem Cell
M MAL (MKL1) Myelin and lymphocyte protein M-CSF Monocyte Colony Stimulating Factor MDP Monocyte and Dendritic Progenitor
MEP Megakaryocyte Erythrocyte Progenitor Mk Megacaryocyte MkP Megakaryocyte Progenitor

172
MPL Myeloproliferative Leukemia virus oncogene MPP Multipotent Progenitor MSCV Murine stem cell virus
O OMS Organisation mondiale de la santé OTT (RBM15) RNA binding motif protein 15
P PML Promyelocytic leukemia
PU.1 Purine-rich box 1
R RARA Retinoidc acid receptor alpha
RBPJ Recombination signal binding protein for immunoglobulin kappa j region
RUNX1 Runt-related transcription factor 1
S SCF Stem Cell Factor
SE Super-Enhancer SHH Sonic Hedgehog SL-MkP Stem cell like Megakaryocyte Progenitor ST-HSC Short Term-Hematopoietic Stem Cell
T TMD Transcient Myeloproliferative Disorder TPO Thrombopoïétine
V
vWF Von Willebrand Factor
Z ZF Zinc finger

173

174
ANNEXES
Pediatric Acute Megakaryoblastic Leukemia : Multitasking Fusion Proteins and Oncogenic Cooperations
Trends in Cancer
Review

175

176

177
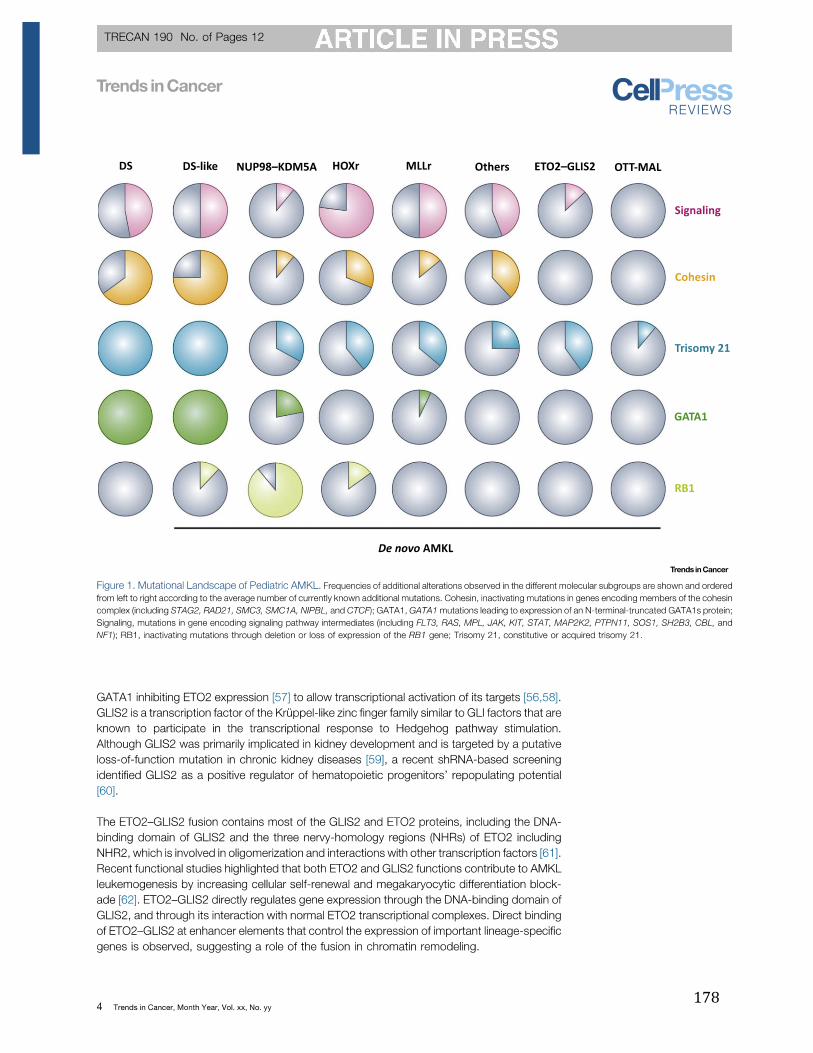
178
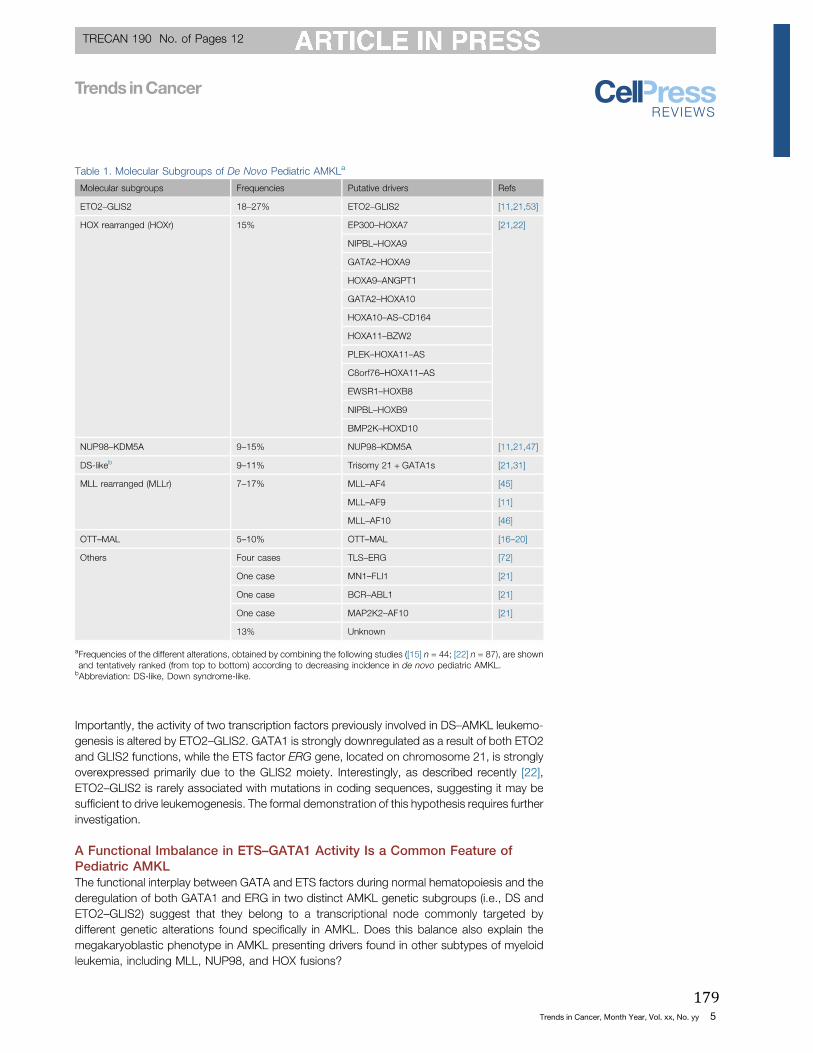
179

180

181
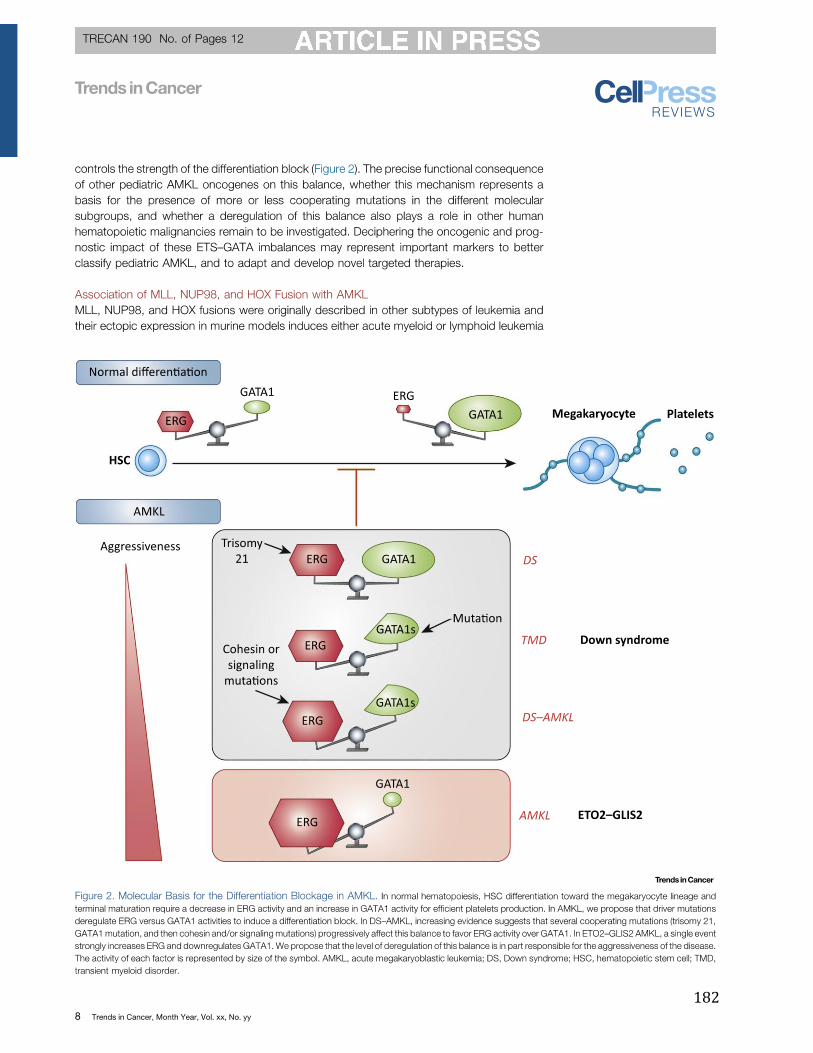
182

183

184

185

186

187

188
Molecular pathways driven by ETO2-GLIS2 in aggressive pediatric leukemia
Molecular and cellular oncology Review

189

190

191

192
Leucémies à mégacaryoblastes de l’enfant : une affaire de complexes
Medecine / Sciences Review
(En cours de publication)

193
Leucémies à mégacaryoblastes de l'enfant : une affaire de complexes
Cécile K Lopez, Thomas Mercher
Inserm U1170, Institut Gustave Roussy, Pavillon recherche 2 - 39 rue Camille Desmoulins,
94800 Villejuif
Résumé
Les leucémies aiguës mégacaryoblastiques de l'enfant (LAM7) sont généralement associées
à un mauvais pronostic et à l'expression d'oncogènes de fusion impliquant des régulateurs
transcriptionnels. Des résultats récents indiquent que la fusion ETO2-GLIS2 dérégule
l'activité de régions régulatrices de l'expression génique, appelées "enhancers", et altère
l'expression des facteurs GATA et ETS essentiels au développement des cellules souches
hématopoïétiques. Une dérégulation de l'équilibre GATA/ETS est également retrouvée dans
d'autres sous-groupes de LAM7. Cette revue porte sur les bases transcriptionnelles de la
transformation dans les LAM7 de l'enfant et les perspectives thérapeutiques.

194
Hématopoïèse normale et leucémies
L’hématopoïèse normale est le processus qui permet le développement de toutes les
cellules du sang incluant les lymphocytes, les érythrocytes ou globules rouges et les
plaquettes sanguines. Toutes ces cellules se développent à partir de cellules dites cellules
souches hématopoïétiques (HSC) localisées chez l’adulte dans la moelle des os [1]. Les HSC
sont capables de se maintenir (par des propriétés de quiescence et d'autorenouvellement)
et peuvent également proliférer sous la forme de progéniteurs qui se différencient pour
donner les milliards de cellules matures qui entrent dans le sang chaque jour pour mener
leur fonction. Pour maintenir un nombre constant de cellules, ce processus est régulé
finement par des signaux de l’environnement des cellules (ex: cellules à proximité ou des
facteurs de croissance solubles) qui peuvent stimuler des voies de signalisation et par des
facteurs propres aux cellules (ex: l’organisation de l’ADN et l’expression de facteurs de
transcription) qui peuvent réguler l’expression d’autres gènes importants pour la
prolifération ou la maturation. Certains facteurs de transcription contrôlent le
développement de lignées hématopoïétiques spécifiques et d'autres sont requis pour tous
les progéniteurs hématopoïétiques. Ces facteurs de transcription semblent fréquemment
regroupés au sein de complexes contenant plusieurs régulateurs transcriptionnels et dont
la composition contrôle l'identité et le niveau d'expression des gènes cibles [2]. En effet, les
facteurs peuvent être associés à des modifications de la chromatine, comme celles des
histones ou de l'ADN, conduisant à une activation ou une répression de l'expression des
gènes alentours. Certaines de ces modifications, telle que l'acétylation de la lysine 27 de
l'histone 3 (H3K27ac) peuvent s'étendre sur de larges régions (plusieurs 100aine de kb)
appelées enhancer ou super-enhancer [3]. Ces régions sont liées par de nombreux facteurs
de transcription et facteurs généraux de la machinerie transcriptionnelle (ex: facteurs BRD
et complexe Mediator) et sont associées à une transcription importante de gènes. La
localisation de ces régions enhancers a été associée à l’identité cellulaire et une hypothèse
actuelle est que l'activité de ces régions contrôle en partie l'équilibre entre auto-
renouvellement/prolifération des cellules souches et progéniteurs et progression vers la
différenciation terminale [4].

195
Parmi les facteurs de transcription important pour l'hématopoïèse, les facteurs de la famille
GATA lient l'ADN sur un motif consensus de type 5'-(A/T)GATAA(G/A/C)-3' et sont
fréquemment associés à des complexes multiprotéiques composés d'au moins 7 facteurs de
transcription (nommé heptad), incluant également TAL1, LYL1, LMO2, RUNX1, ERG et FLI1.
Ces complexes sont actifs dans les cellules souches et progéniteurs hématopoïétiques
adultes mais également dès l'apparition des premières cellules souches hématopoïétiques
au cours du développement [5]. Les facteurs GATA sont également essentiels pour le
développement des globules rouges et des mégacaryocytes qui sont à l'origine des
plaquettes sanguines [6]. Un changement d'activité des facteurs GATA est observé au cours
de la différenciation avec une activité GATA2 importante dans les cellules souches
hématopoïétiques et progéniteurs alors que l'activité GATA1 est plus importante dans les
cellules engagées dans la différenciation érythroïde ou mégacaryocytaire. Ce processus
d'échange des facteurs GATA au cours de la différenciation est appelé GATA-switch [7]. Des
approches de localisation de ces facteurs sur la chromatine (immunoprécipitation de
chromatine couplée au séquençage haut-débit : ChIPseq) confirment que GATA1 remplace
GATA2 au niveau de plusieurs gènes importants pour la différenciation mégacaryocytaire
[8] et que ce GATA-switch constitue une base moléculaire importante du remodelage des
enhancers au cours du développement érythroïde [9]. Une interaction fonctionnelle entre
les facteurs GATA et d'autres membres des complexes transcriptionnels, tels que les
facteurs ETS liant des motifs 5′-GGA(A/T)-3′ [2] est également importante pour la
différenciation mégacaryocytaire. En effet, le facteur ETS FLI1, dont l'expression est
augmentée au cours de la maturation, régule conjointement avec GATA l'expression de
gènes cibles mégacaryocytaires, tels que ITGA2b [10]. Ainsi, il a été proposé que cette
interaction fonctionnelle GATA/ETS joue un rôle important dans les HSC et dans leur
engagement vers la lignée mégacaryocytaire alors qu'une interaction fonctionnelle entre
GATA et TAL1 serait plus importante pour la différenciation érythroïde [8,9,11,12]. Au sein
de ces complexes, les cofacteurs de la famille ETO/ETO2 interagissent avec les facteurs de
transcription et des régulateurs transcriptionnels [13] et un changement de stœchiométrie
de ETO2 dans des complexes TAL1 participe également à la transition entre prolifération et
différenciation terminale au cours de l'érythropoïèse [14].

196
Les leucémies aiguës sont des altérations de l’hématopoïèse caractérisées par une
accumulation de progéniteurs associée à une altération de la différenciation terminale et
une insuffisance du système hématopoïétique. Les leucémies sont généralement classées en
fonction de la lignée hématopoïétique préférentiellement atteinte et présentent des
incidences variables en fonction de l’âge. Les leucémies aiguës myéloïdes (LAM) sont
caractérisées par la présence d'altérations génétiques dans les cellules souches et
progéniteurs hématopoïétiques [15]. Ces mutations affectent plusieurs fonctions
biologiques incluant la régulation de la transcription (ex: facteurs de transcription), de la
méthylation (ex: TET2, DNMT3A), de la chromatine (ex: CTCF), de l'épissage (ex: SRSF2), de
la signalisation (ex: JAK2) et du cycle cellulaire (ex: TP53). Les mutations affectant les
facteurs du complexe heptad (ex: TAL1, RUNX1) et ETO/ETO2 sont récurrentes et
généralement mutuellement exclusives dans les leucémies aiguës humaines, suggérant
qu'elles ciblent un mécanisme conservé de la biologie des cellules souches
hématopoïétiques. L'étude des leucémies représente donc une opportunité d'identifier et de
caractériser la fonction des régulateurs transcriptionnels importants contrôlant
l'hématopoïèse et la biologie des cellules souches humaines.
Génétique des leucémies mégacaryoblastiques
Les leucémies aiguës à mégacaryoblastes (LAM7) qui affectent la lignée mégacaryocytaire,
représentent un sous-type de leucémie rare mais diagnostiqué plus fréquemment chez
l’enfant que chez l'adulte [16]. Elles peuvent survenir suite à une prédisposition, telle que
les LAM7 associées à une trisomie 21 constitutionnelle (nommées ici DS-LAM7) et à un
pronostic favorable, ou diagnostiquées de novo (sans prédisposition apparente) et
généralement associées à un mauvais pronostic [17].
Les DS-LAM7 sont un groupe relativement homogène de patients et leur développement
représente un mécanisme de progression tumorale en 3 étapes, chacune associée à
l'acquisition d'altérations génétiques se cumulant et incluant: 1-une trisomie constitutive
du chromosome 21 associée à un prédisposition aux leucémies, 2-des mutations
somatiques du gène GATA1 entrainant une troncation de la partie Nt de la protéine (mutant

197
GATA1short ou GATA1s) associées à un syndrome myéloïde transitoire (SMT) diagnostiqué
à la naissance et 3-des mutations additionnelles dans des intermédiaires de voie de
signalisation (47%, ex: MPL ou JAK) et/ou des régulateurs épigénétiques (65%, ex: EZH2,
CTCF, RAD21) qui sont associées à l'étape de leucémie aiguë (DS-LAM7) diagnostiquée
généralement après l’âge de 3 ans (Pour revue voir [18]).
Dans le groupe de LAM7 de novo, les analyses cytogénétiques et génomiques globales ont
montré, dans la majorité des cas, la présence d'altérations chromosomiques conduisant à
l’expression d’oncogène de fusion (Figure 1A) [19,20]. La première anomalie récurrente
identifiée a été une translocation entre le chromosome 1 et 22, visible en cytogénétique
classique, et codant pour la fusion OTT-MAL incluant la quasi-totalité des deux protéines
[21]. Des réarrangements impliquant MLL (également nommé KMT2A) sont retrouvés dans
7-17% des cas, alors que la fusion NUP98-KDM5a dans 10-15% de LAM7 de novo. Ces 2
sous-groupes sont associés à une expression aberrante des gènes homéotiques HOX, dont le
gène HOXA9 qui a été impliqué dans le développement et la maintenance de certaines
leucémies [22]. Le rôle de ces gènes dans le développement leucémique est également
souligné par la présence de réarrangements impliquant les gènes HOX eux-mêmes associés
à leur surexpression dans 15% des LAM7 de novo [20]. La fusion ETO2-GLIS2 récemment
découverte est présente dans 18 à 27% des LAM7 de novo et résulte d’une inversion du
chromosome 16 qui code pour une protéine de fusion exprimant la quasi-totalité des
protéines ETO2 et GLIS2 normales. Ces fusions se réalisent entre les exons 10 à 12 de ETO2
et les exons 1 à 3 de GLIS2 [19]. D'autres fusions plus rares sont également observées
incluant TLS-ERG, MN1-FLI1, BCR-ABL1, MAP2K2-AF10. Un sous-groupe (nommé DS-like)
présente également les mêmes types de mutations que celles observées pour les DS-LAM7 à
ceci près que la trisomie 21 est acquise et non constitutive. D'autre part, l'importance d'une
coopération oncogénique pour la transformation est soulignée par la présence dans
plusieurs sous-groupes de mutations additionnelles incluant une trisomie 21 acquise, des
mutations GATA1 et RB1 ou des mutations des cohésines et d'intermédiaires de
signalisation (Figure 1B). Finalement, 10-15% des patients ne présentent pas de mutation
causale identifiée à ce jour.

198
Mécanismes moléculaires de la fusion ETO2-GLIS2
La fusion ETO2-GLIS2 est l’altération génétique la plus fréquemment retrouvée dans les
LAM7 de novo, et est associée à un pronostic particulièrement défavorable.
Le gène ETO2 code une protéine de la famille ETO qui est exprimée dans toutes les cellules
hématopoïétiques. ETO2 est un cofacteur transcriptionnel qui interagit avec les facteurs de
transcription du complexe heptad. Une inactivation d'ETO2 chez la souris est associée à une
absence de maintien des HSC [23]. ETO2 joue également un rôle d'inhibition de la
mégacaryopoïèse [8] en réprimant certains gènes impliqués dans la différenciation
terminale (comme GATA1 et PF4, [24]). GLIS2 est un facteur de transcription de la famille
GLI impliqué dans la voie de signalisation Hedgehog et les patients présentant ETO2-GLIS2
montrent une signature transcriptionnelle de surexpression de gènes cibles de cette voie
par rapport aux autres patients [19]. De récentes études suggèrent un rôle positif de GLIS2
dans la capacité de reconstitution des HSC [25]. La fusion implique donc deux régulateurs
transcriptionnels.
Récemment, nous avons caractérisé les contributions relatives de ETO2 et GLIS2 à la
transformation par ETO2-GLIS2 et observé que la fusion augmente l’autorenouvellement
des progéniteurs hématopoïétiques à la fois par la partie ETO2 et la partie GLIS2 [26]. De
plus, la partie GLIS2 est responsable du phénotype mégacaryocytaire. Ces résultats
indiquent que la fusion ETO2-GLIS2 est capable seule d'augmenter l'autorenouvellement
des progéniteurs présentant un phénotype mégacaryocytaire. Au niveau moléculaire, la
fusion ETO2-GLIS2 dérégule l'expression de nombreux de gènes, incluant ceux codant pour
les protéines du complexe heptad. Ainsi, l’expression de GATA1 est fortement diminuée
alors que celle de ERG est fortement augmentée. Cette dérégulation est également observée
dans les signatures transcriptionnelles des blastes de patients ETO2-GLIS2. L'identification
des régions de l’ADN sur lesquelles se fixe la fusion a ensuite montré qu'elle est localisée sur
des enhancers. L'analyse des motifs associés à ces régions suggère que cette liaison se fait
soit par l'intermédiaire des facteurs de transcription ERG, RUNX et GATA interagissant avec
ETO2 soit directement via la partie GLIS2.
Comme l’activité des régions enhancer est importante pour les cellules cancéreuses [27],
nous avons cherché comment la stabilité des complexes est régulée. Tout d'abord, nous

199
avons montré que le facteur ERG colocalise fréquemment avec la fusion et est associé à une
expression forte des gènes proches des enhancers. L’inactivation fonctionnelle de ERG par
ciblage du génome de type CRISPR/cas9 dans des cellules exprimant la fusion a finalement
montré que ERG joue un rôle essentiel dans la régulation transcriptionnelle de certains
gènes cibles de la fusion comme KIT et dans la survie des blastes leucémiques humains
ETO2-GLIS2.
Mécanismes communs aux leucémies à mégacaryoblastes
La différenciation mégacaryocytaire terminale implique un changement dynamique de la
composition/activité des complexes de facteurs de transcription avec l'acquisition d'une
forte activité GATA1 et d'une faible activité ERG dans les mégacaryocytes matures (Figure
2). Les résultats obtenus avec ETO2-GLIS2 indiquent que la fusion peut imposer seule, une
forte activité ERG et une faible activité de GATA1 qui participe au blocage de la
différenciation. Globalement, l'analyse des modèles de LAM7 obtenus jusqu'à présent
(Tableau 1) suggère que la modification de l'activité des facteurs ETS (ex : ERG) et de
GATA1 est un mécanisme commun aux LAM7 pédiatriques.
En effet, le gène ERG, localisé sur le chromosome 21, est sujet à une augmentation du
dosage génique dans les cellules trisomie 21 et sa surexpression peut transformer la lignée
érythroïde ou mégacaryocytaire [28]. La trisomie 21 est retrouvée dans tous les sous-
groupes de LAM7 avec une fréquence de 11 à 100% (Figure 1B). La présence de fusions
TLS-ERG et MN1-FLI1 (Figure 1, groupe "Autres") montre également l'implication directe
des facteurs ETS ERG et FLI1 présentant une forte homologie de séquence et de fonction
[29]. Dans le cas de MN1-FLI1, les capacités d'autorenouvellement des progéniteurs murins
in vitro et le phénotype mégacaryocytaire sont contrôlés par la partie FLI1 [30]. D’autres
mutations fréquemment retrouvées dans les différents sous-groupes de LAM7 participent à
modifier l’activité de ERG. Par exemple, les cohésines contrôlent l’organisation
chromatinienne et leur inactivation fonctionnelle favorise l’accessibilité de ERG à certaines
régions de l'ADN [31]. D'autre part, certaines mutations d'intermédiaires de voie de
signalisation (ex: MPL et JAK2) activent la voie MAPK qui régule également l’activité de ERG
[32]. Finalement, dans les DS- et DS-like LAM7, l'activité GATA1 est ciblé par les mutations

200
GATA1s qui contribuent à la transformation mégacaryoblastique et coopère avec une
surexpression de ERG [33].
Dans le cas des fusions MLL, NUP98 et HOX, les bases de leur association fréquente avec les
LAM7 de l'enfant restent à définir. En effet, ces fusions ont été initialement décrites dans
d'autres sous-types de leucémie et leur expression ectopique dans des modèles murins
induit soit une leucémie myéloïde aiguë, soit une leucémie lymphoïde dépourvue de
caractéristiques mégacaryocytaires typiques [34]. Leur association avec les LAM7 pourrait
résulter d'un rôle spécifique des partenaires de ces fusions au cours du développement des
mégacaryocytes comme pour HOXA10 [35]. Cependant, l'importance d'une dérégulation de
l'activité GATA1 et ERG par les mutations additionnelles retrouvées chez ces patients
pourrait également expliquer l'association avec un phénotype mégacaryocytaire. En effet,
les patients LAM7 avec NUP98-KDM5A ou des fusions HOX présentent une association
remarquable avec des mutations inactivatrices de RB1 et des mutations activatrices de MPL,
respectivement (Figure 1B). Comme GATA1 interagit avec RB1 et E2F2 pour réguler la
prolifération des progéniteurs érythro-mégacaryocytaires [36], il sera important de définir
si le rôle des mutations inactivatrices de RB1 dans la leucémogenèse des LAM7 est en
rapport avec l'activité GATA1. L'activité accrue des facteurs mégacaryocytaires comme
GATA ou ETS par une activation de la signalisation MPL pourrait également être à la base de
l'association entre les fusions HOX et les mutations MPL dans les LAM7. Alternativement, le
phénotype mégacaryoblastique dans les sous-groupes MLL / NUP98 / HOX pourrait
également résulter de la survenue de ces fusions dans des progéniteurs mégacaryocytaires
restreints.
Ciblage thérapeutique des complexes transcriptionnels : perspectives et difficultés
Les LAM7 de novo sont essentiellement caractérisées par la présence d’oncogène de fusion
impliquant des partenaires de complexe transcriptionnel (Figure 3A). Ces fusions
constituent des marqueurs moléculaires maintenant utilisés pour établir le diagnostic de
façon plus précise et suivre l’évolution de la maladie résiduelle au cours du traitement.
Dans le cas de la fusion ETO2-GLIS2 qui est associée à une mauvaise réponse aux

201
chimiothérapies, cela permet également d’orienter la stratégie thérapeutique vers une
greffe de moelle.
D'autre part, l'existence de fusions spécifiques affectant des complexes transcriptionnels
anormaux suggère de nouvelles stratégies thérapeutiques potentielles. Dans le cas des
fusions MLL, des inhibiteurs des partenaires du complexe comme DOT1L, BRD ou ENL sont
ainsi en cours de tests précliniques pour évaluer leur efficacité et spécificité.
Les membres de la famille ETO présentent la capacité de former des tétramères grâce à la
présence du domaine nervy homology region (NHR)2. L'expression d’un peptide identique
au domaine NHR2 d’ETO interfère dans la formation des tétramères et induit une inhibition
de la prolifération des cellules leucémiques présentant la fusion AML1-ETO [37],
représentant le sous-type cytogénétique le plus fréquent des LAM. En partant de
l’observation que le domaine NHR2 est fortement conservé entre les protéines de la famille
ETO, nous avons observé que la fusion ETO2-GLIS2 est capable de dimériser et d'interagir
avec la protéine ETO2 sauvage. L’expression du peptide correspondant au domaine NHR2
dans les cellules de patient leucémique ETO2-GLIS2 réduit l'interaction entre la fusion et
ETO2 sauvage (Figure 3B), inhibe la prolifération et augmente la différenciation des blastes
leucémiques in vitro et in vivo [26]. Ces résultats suggèrent que l’expression d'un petit
peptide NHR2 et l'interférence avec les complexes transcriptionnels anormaux impliquant
une protéine de fusion pourraient représenter une stratégie d'inhibition dans les LAM7.
Cependant, il sera nécessaire de définir si la stabilité des peptides ainsi que leur ciblage
attendu des complexes ETO endogènes représentent une stratégie thérapeutique
acceptable chez les patients ou si le développement d'autres méthodes d'interférence
transcriptionnelle avec ces complexes sera requis.
Dans l'ensemble, l'étude de la fusion ETO2-GLIS2 souligne le rôle d'une dérégulation de
complexes transcriptionnels multi-protéiques et d'une activité GLIS2 aberrante dans la
transformation des LAM7 de l'enfant. L'implication de GLIS2 dans le phénotype d'autres
leucémies (ex: MOZ-TIF2) [38] suggère un rôle plus large des facteurs GLIS dans
l'hématopoïèse normale et pathologie. D'autre part, il reste également à définir les bases
cellulaires et moléculaires de l'association spécifique de certaines fusions avec les
leucémies de l'enfant.

202
Remerciements
Ce travail est soutenu par l'Institut National du Cancer (PLBIO-2014-176), l'Association
Laurette Fugain (ALF-2015/13), la Fédération Enfants et Santé et la Société Française de
Lutte Contre les Cancers et les Leucémies de l'Enfant et l'Adolescent (SFCE: Projet
CAMELIAT), le SIRIC-SOCRATE (INCa-DGOS-INSERM 6043), la Fondation pour la Recherche
Médicale (FRM-ING20150532273), Cancéropôle Ile de France (à CL et Emergence 2015) et
le PAIR-Pédiatrie 2017 (Cancéropôle-INCA: projet CONECT-AML). L'équipe de TM est
labellisée Ligue contre le cancer.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet
article.
Summary
Pediatric de novo acute megakaryoblastic leukemia: an affair of complexes
Pediatric acute megakaryoblastic leukemia (AMKL) are generally associated with poor
prognosis and the expression of fusion oncogenes involving transcriptional regulators.
Recent results indicate that the ETO2-GLIS2 fusion, associated with 25-30% of pediatric
AMKL binds and alters the activity of regulatory regions of gene expression, called
"enhancers", resulting in the deregulation of GATA and ETS factors essential for the
development of hematopoietic stem cells. An imbalance in GATA/ETS factor activity is also
found in other AMKL subgroups. This review addresses the transcriptional bases of
transformation in pediatric AMKL and therapeutic perspectives.

203
Bibliographie
1. Stik G, Petit L, Charbord P, et al. Vésicules extracellulaires stromales et régulation des
cellules souches et progéniteurs hématopoïétiques. Med Sci Paris 2018 ; 34 : 114–116.
2. Pimkin M, Kossenkov AV, Mishra T, et al. Divergent functions of hematopoietic
transcription factors in lineage priming and differentiation during erythro-megakaryopoiesis.
Genome Res. 2014 ; 24 : 1932–1944.
3. Pott S, Lieb JD. What are super-enhancers? Nat. Genet. 2015 ; 47 : 8–12.
4. Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish
super-enhancers at key cell identity genes. Cell 2013 ; 153 : 307–319.
5. Wilson NK, Foster SD, Wang X, et al. Combinatorial transcriptional control in blood
stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell
Stem Cell 2010 ; 7 : 532–544.
6. Debili N, Vainchenker W. De macro à micro : l’histoire de la plaquette. Med Sci Paris 2008 ;
24 : 467–469.
7. Elagib KE, Racke FK, Mogass M, et al. RUNX1 and GATA-1 coexpression and cooperation
in megakaryocytic differentiation. Blood 2003 ; 101 : 4333–4341.
8. Doré LC, Chlon TM, Brown CD, et al. Chromatin occupancy analysis reveals genome-wide
GATA factor switching during hematopoiesis. Blood 2012 ; 119 : 3724–3733.
9. Huang J, Liu X, Li D, et al. Dynamic control of enhancer repertoires drives lineage and
stage-specific transcription during hematopoiesis. Dev. Cell 2016 ; 36 : 9–23.
10. Lemarchandel V, Ghysdael J, Mignotte V, et al. GATA and Ets cis-acting sequences mediate
megakaryocyte-specific expression. Mol. Cell. Biol. 1993 ; 13 : 668–676.
11. Fujiwara T, O’Geen H, Keles S, et al. Discovering hematopoietic mechanisms through
genome-wide analysis of GATA factor chromatin occupancy. Mol. Cell 2009 ; 36 : 667–681.
12. Tripic T, Deng W, Cheng Y, et al. SCL and associated proteins distinguish active from
repressive GATA transcription factor complexes. Blood 2009 ; 113 : 2191–2201.
13. Wei Y, Liu S, Lausen J, et al. A TAF4-homology domain from the corepressor ETO is a
docking platform for positive and negative regulators of transcription. Nat. Struct. Mol. Biol.
2007 ; 14 : 653–661.
14. Goardon N, Lambert JA, Rodriguez P, et al. ETO2 coordinates cellular proliferation and
differentiation during erythropoiesis. EMBO J. 2006 ; 25 : 357–366.

204
15. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in
acute myeloid leukemia. N. Engl. J. Med. 2016 ; 374 : 2209–2221.
16. Creutzig U, Büchner T, Sauerland MC, et al. Significance of age in acute myeloid leukemia
patients younger than 30 years: a common analysis of the pediatric trials AML-BFM 93/98
and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 2008 ; 112 : 562–571.
17. Teyssier A-C, Lapillonne H, Pasquet M, et al. Acute megakaryoblastic leukemia (excluding
Down syndrome) remains an acute myeloid subgroup with inferior outcome in the French
ELAM02 trial. Pediatr. Hematol. Oncol. 2017 ; 34 : 425–427.
18. Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br. J.
Haematol. 2014 ; 167 : 587–599.
19. Thiollier C, Lopez CK, Gerby B, et al. Characterization of novel genomic alterations and
therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J. Exp.
Med. 2012 ; 209 : 2017–2031.
20. Rooij JDE de, Branstetter C, Ma J, et al. Pediatric non-Down syndrome acute
megakaryoblastic leukemia is characterized by distinct genomic subsets with varying
outcomes. Nat. Genet. 2017 ; 49 : 451–456.
21. Bernard OA, Mercher T. Activation de la voie Notch par OTT-MAL dans les leucémies
aiguës mégacaryoblastiques. 2009 ; Med Sci (Paris) : 676–678.
22. Faber J, Krivtsov AV, Stubbs MC, et al. HOXA9 is required for survival in human MLL-
rearranged acute leukemias. Blood 2009 ; 113 : 2375–2385.
23. Fischer MA, Moreno-Miralles I, Hunt A, et al. Myeloid translocation gene 16 is required for
maintenance of haematopoietic stem cell quiescence. EMBO J. 2012 ; 31 : 1494–1505.
24. Hamlett I, Draper J, Strouboulis J, et al. Characterization of megakaryocyte GATA1-
interacting proteins: the corepressor ETO2 and GATA1 interact to regulate terminal
megakaryocyte maturation. Blood 2008 ; 112 : 2738–2749.
25. Holmfeldt P, Ganuza M, Marathe H, et al. Functional screen identifies regulators of murine
hematopoietic stem cell repopulation. J. Exp. Med. 2016 ; 213 : 433–449.
26. Thirant C, Ignacimouttou C, Lopez CK, et al. ETO2-GLIS2 hijacks transcriptional
complexes to drive cellular identity and self-renewal in pediatric acute megakaryoblastic
leukemia. Cancer Cell 2017 ; 31 : 452–465.

205
27. Gröschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer
rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014 ;
157 : 369–381.
28. Carmichael CL, Metcalf D, Henley KJ, et al. Hematopoietic overexpression of the
transcription factor Erg induces lymphoid and erythro-megakaryocytic leukemia. Proc. Natl.
Acad. Sci. U. S. A. 2012 ; 109 : 15437–15442.
29. Kruse EA, Loughran SJ, Baldwin TM, et al. Dual requirement for the ETS transcription
factors Fli-1 and Erg in hematopoietic stem cells and the megakaryocyte lineage. Proc. Natl.
Acad. Sci. U. S. A. 2009 ; 106 : 13814–13819.
30. Wenge DV, Felipe-Fumero E, Angenendt L, et al. MN1-Fli1 oncofusion transforms murine
hematopoietic progenitor cells into acute megakaryoblastic leukemia cells. Oncogenesis 2015
; 4 : e179.
31. Mazumdar C, Shen Y, Xavy S, et al. Leukemia-associated cohesin mutants dominantly
enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell
Stem Cell 2015 ; 17 : 675–688.
32. Huang Y, Thoms J a. I, Tursky ML, et al. MAPK/ERK2 phosphorylates ERG at serine 283 in
leukemic cells and promotes stem cell signatures and cell proliferation. Leukemia 2016 ; 30 :
1552–1561.
33. Stankiewicz MJ, Crispino JD. ETS2 and ERG promote megakaryopoiesis and synergize with
alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood 2009 ; 113 :
3337–3347.
34. Dang J, Nance S, Ma J, et al. AMKL chimeric transcription factors are potent inducers of
leukemia. Leukemia 2017 ; 31 : 2228–2234.
35. Magnusson M, Brun ACM, Miyake N, et al. HOXA10 is a critical regulator for
hematopoietic stem cells and erythroid/megakaryocyte development. Blood 2007 ; 109 :
3687–3696.
36. Kadri Z, Shimizu R, Ohneda O, et al. Direct binding of pRb/E2F-2 to GATA-1 regulates
maturation and terminal cell division during erythropoiesis. PLoS Biol. 2009 ; 7 : e1000123.
37. Wichmann C, Becker Y, Chen-Wichmann L, et al. Dimer-tetramer transition controls
RUNX1/ETO leukemogenic activity. Blood 2010 ; 116 : 603–613.

206
38. Shima H, Takamatsu-Ichihara E, Shino M, et al. Ring1A and Ring1B inhibit expression of
Glis2 to maintain murine MOZ-TIF2 AML stem cells. Blood 2018 ; 131 : 1833–1845.
39. Hara Y, Shiba N, Ohki K, et al. Prognostic impact of specific molecular profiles in pediatric
acute megakaryoblastic leukemia in non-Down syndrome. Genes. Chromosomes Cancer
2017 ; 56 : 394–404.
40. Drexler HG. Guide to Leukemia-Lymphoma Cell Lines. Braunschweig, Germany : German
Collection of Microorganisms and Cell Cultures, 2005 : p.
41. Walters DK, Mercher T, Gu T-L, et al. Activating alleles of JAK3 in acute megakaryoblastic
leukemia. Cancer Cell 2006 ; 10 : 65–75.
42. Saida S, Watanabe K, Sato-Otsubo A, et al. Clonal selection in xenografted TAM
recapitulates the evolutionary process of myeloid leukemia in Down syndrome. Blood 2013 ;
121 : 4377–4387.
43. Chou ST, Byrska-Bishop M, Tober JM, et al. Trisomy 21-associated defects in human
primitive hematopoiesis revealed through induced pluripotent stem cells. Proc. Natl. Acad.
Sci. U. S. A. 2012 ; 109 : 17573–17578.
44. Alford KA, Slender A, Vanes L, et al. Perturbed hematopoiesis in the Tc1 mouse model of
Down syndrome. Blood 2010 ; 115 : 2928–2937.
45. Byrska-Bishop M, VanDorn D, Campbell AE, et al. Pluripotent stem cells reveal erythroid-
specific activities of the GATA1 N-terminus. J. Clin. Invest. 2015 ; 125 : 993–1005.
46. Malinge S, Bliss-Moreau M, Kirsammer G, et al. Increased dosage of the chromosome 21
ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down
syndrome. J. Clin. Invest. 2012 ; 122 : 948–962.
47. Ayllón V, Vogel-González M, González-Pozas F, et al. New hPSC-based human models to
study pediatric Acute Megakaryoblastic Leukemia harboring the fusion oncogene RBM15-
MKL1. Stem Cell Res. 2017 ; 19 : 1–5.
48. Gruber TA, Larson Gedman A, Zhang J, et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-
GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic
leukemia. Cancer Cell 2012 ; 22 : 683–697.
49. Cardin S, Laramee L, MacRae T, et al. Modeling of pediatric acute megakaryoblastic
leukemia using cord blood stem/progenitor cells. Blood 2016 ; 128 : 1535–1535.
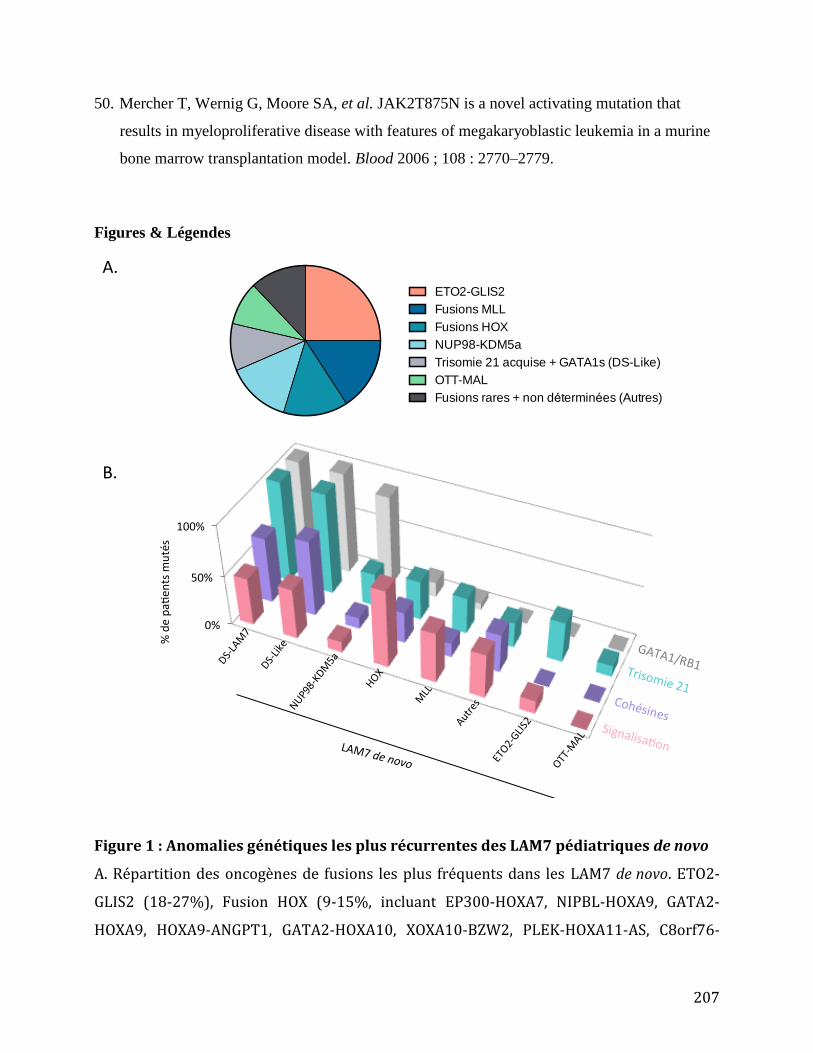
207
50. Mercher T, Wernig G, Moore SA, et al. JAK2T875N is a novel activating mutation that
results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine
bone marrow transplantation model. Blood 2006 ; 108 : 2770–2779.
Figures & Légendes
Figure 1 : Anomalies génétiques les plus récurrentes des LAM7 pédiatriques de novo
A. Répartition des oncogènes de fusions les plus fréquents dans les LAM7 de novo. ETO2-
GLIS2 (18-27%), Fusion HOX (9-15%, incluant EP300-HOXA7, NIPBL-HOXA9, GATA2-
HOXA9, HOXA9-ANGPT1, GATA2-HOXA10, XOXA10-BZW2, PLEK-HOXA11-AS, C8orf76-
A.
B.
0%
50%
100%
GATA1/RB1Trisomie21Cohésines
Signalisa on
%depa
entsm
utés
LAM7denovo
DS-Like
DS-LAM
7
NUP98-KDM
5a
HOX
MLL
Autres
ETO2-GLIS2
OTT-MAL
ETO2-GLIS2
Fusions MLL
Fusions HOX
NUP98-KDM5a
Trisomie 21 acquise + GATA1s (DS-Like)
OTT-MAL
Fusions rares + non déterminées (Autres)
LAM7 de novo

208
HOXA11-AS, EWSR1-HOXB8, NIPBL-HOXB9, BMP2K-HOXD10), Fusion MLL (7-17%,
incluant MLL-AF4, MLL-AF9, MLL-AF10), NUP98-KDM5a (9-15%), "Autres": fusions rares
(incluant TLS-ERG, MN1-FL1, BCR-ABL1, MAP2K2-AF10) et autres mutations non
déterminées (au total 13%), Trisomie 21 + GATA1s: "DS-like" (9-11%), OTT-MAL (5-10%).
La fréquence indiquée est sur la base des publications suivantes ((248): n=44, (249): n=87).
B. Fréquences des mutations additionnelles observées dans les différents sous-groupes de
LAM7 pédiatriques. GATA1/RB1: mutations conduisant à l’expression de la protéine GATA1
tronquée (GATA1s) ou mutations inactivatrices de RB1, Trisomie 21: trisomie acquise ou
constitutive, Cohésines: mutations inactivatrices de gènes codant pour des membres du
complexe des cohésines (incluant STAG2, RAD21, SMC3, SMC1A, NIPBL et CTCF),
Signalisation: mutations dans des gènes codant pour des intermédiaires de voie de
signalisation (incluant FLT3, RAS, MPL, JAK, KIT, STAT, MAP2K2, PTPN11, SOS1, SH2B3, CBL
et NF1).
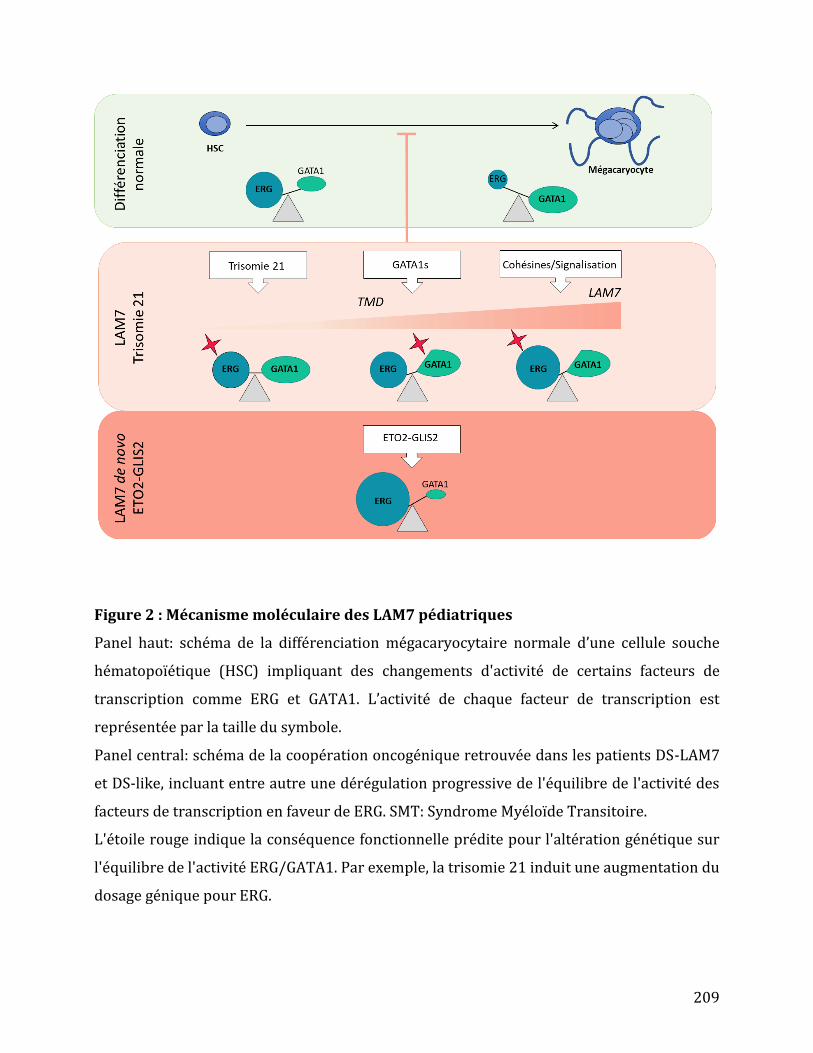
209
Figure 2 : Mécanisme moléculaire des LAM7 pédiatriques
Panel haut: schéma de la différenciation mégacaryocytaire normale d’une cellule souche
hématopoïétique (HSC) impliquant des changements d'activité de certains facteurs de
transcription comme ERG et GATA1. L’activité de chaque facteur de transcription est
représentée par la taille du symbole.
Panel central: schéma de la coopération oncogénique retrouvée dans les patients DS-LAM7
et DS-like, incluant entre autre une dérégulation progressive de l'équilibre de l'activité des
facteurs de transcription en faveur de ERG. SMT: Syndrome Myéloïde Transitoire.
L'étoile rouge indique la conséquence fonctionnelle prédite pour l'altération génétique sur
l'équilibre de l'activité ERG/GATA1. Par exemple, la trisomie 21 induit une augmentation du
dosage génique pour ERG.
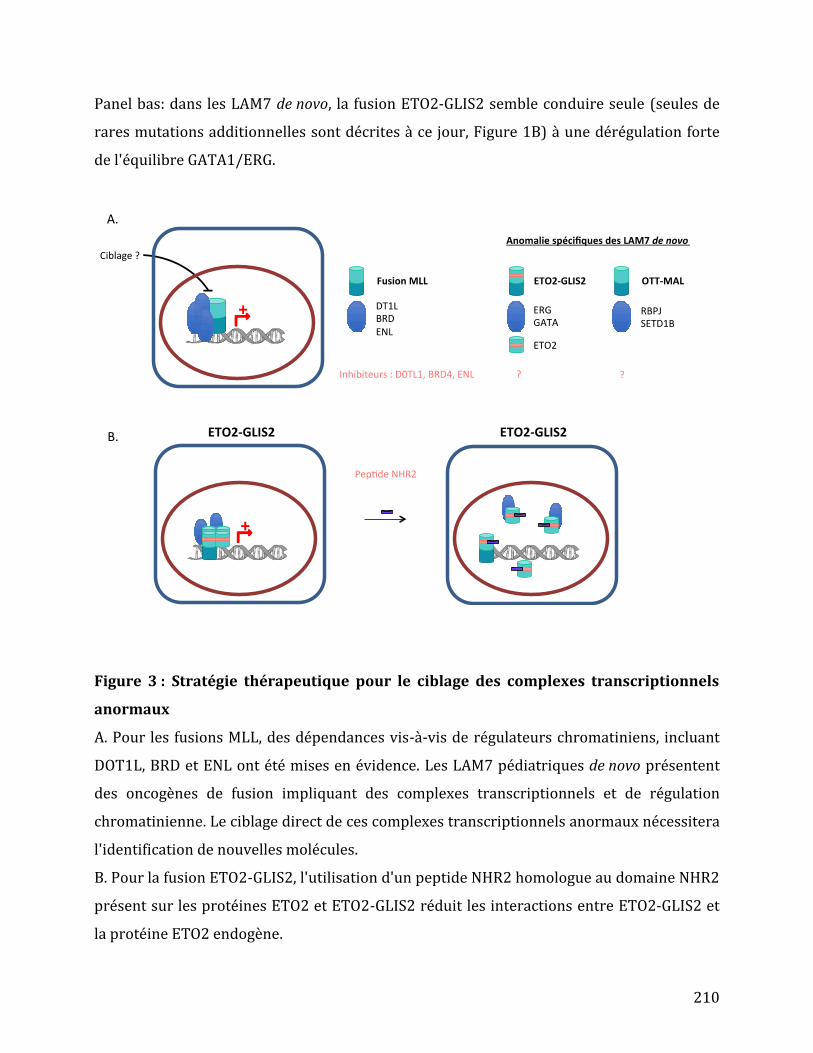
210
Panel bas: dans les LAM7 de novo, la fusion ETO2-GLIS2 semble conduire seule (seules de
rares mutations additionnelles sont décrites à ce jour, Figure 1B) à une dérégulation forte
de l'équilibre GATA1/ERG.
Figure 3 : Stratégie thérapeutique pour le ciblage des complexes transcriptionnels
anormaux
A. Pour les fusions MLL, des dépendances vis-à-vis de régulateurs chromatiniens, incluant
DOT1L, BRD et ENL ont été mises en évidence. Les LAM7 pédiatriques de novo présentent
des oncogènes de fusion impliquant des complexes transcriptionnels et de régulation
chromatinienne. Le ciblage direct de ces complexes transcriptionnels anormaux nécessitera
l'identification de nouvelles molécules.
B. Pour la fusion ETO2-GLIS2, l'utilisation d'un peptide NHR2 homologue au domaine NHR2
présent sur les protéines ETO2 et ETO2-GLIS2 réduit les interactions entre ETO2-GLIS2 et
la protéine ETO2 endogène.
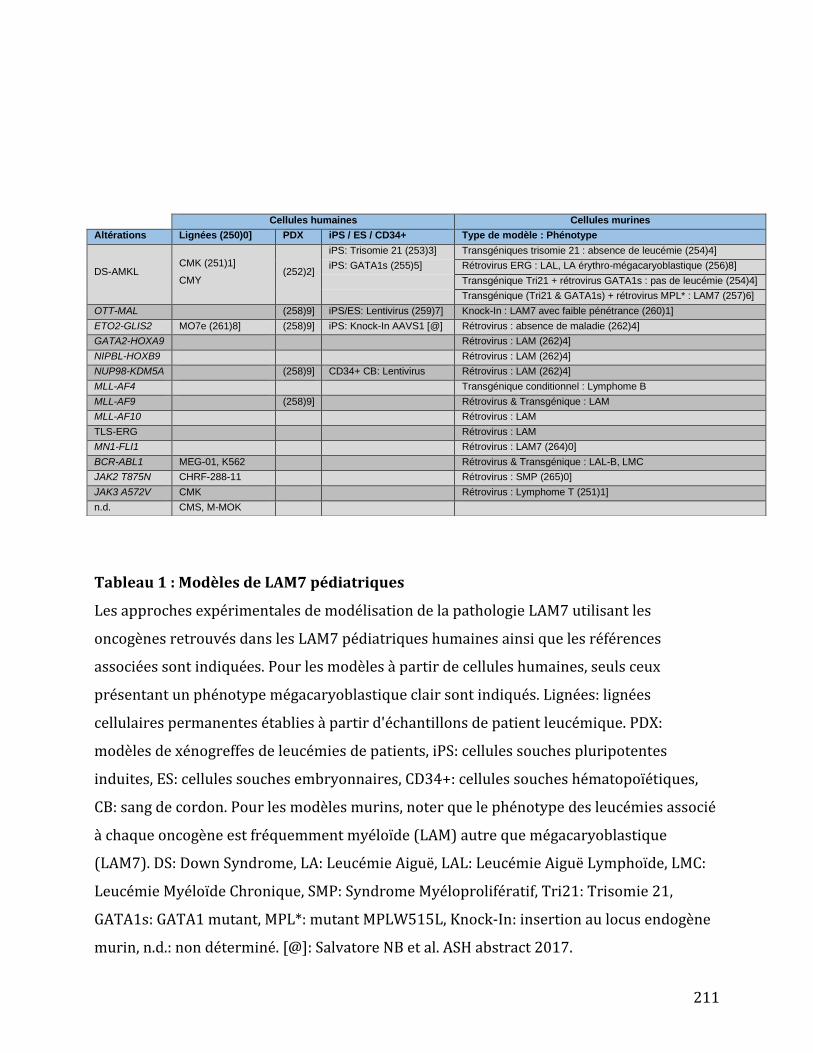
211
Tableau 1 : Modèles de LAM7 pédiatriques
Les approches expérimentales de modélisation de la pathologie LAM7 utilisant les
oncogènes retrouvés dans les LAM7 pédiatriques humaines ainsi que les références
associées sont indiquées. Pour les modèles à partir de cellules humaines, seuls ceux
présentant un phénotype mégacaryoblastique clair sont indiqués. Lignées: lignées
cellulaires permanentes établies à partir d'échantillons de patient leucémique. PDX:
modèles de xénogreffes de leucémies de patients, iPS: cellules souches pluripotentes
induites, ES: cellules souches embryonnaires, CD34+: cellules souches hématopoïétiques,
CB: sang de cordon. Pour les modèles murins, noter que le phénotype des leucémies associé
à chaque oncogène est fréquemment myéloïde (LAM) autre que mégacaryoblastique
(LAM7). DS: Down Syndrome, LA: Leucémie Aiguë, LAL: Leucémie Aiguë Lymphoïde, LMC:
Leucémie Myéloïde Chronique, SMP: Syndrome Myéloprolifératif, Tri21: Trisomie 21,
GATA1s: GATA1 mutant, MPL*: mutant MPLW515L, Knock-In: insertion au locus endogène
murin, n.d.: non déterminé. [@]: Salvatore NB et al. ASH abstract 2017.
Cellules humaines Cellules murines
Altérations Lignées (250)0] PDX iPS / ES / CD34+ Type de modèle : Phénotype
DS-AMKL CMK (251)1]
CMY (252)2]
iPS: Trisomie 21 (253)3] Transgéniques trisomie 21 : absence de leucémie (254)4]
iPS: GATA1s (255)5] Rétrovirus ERG : LAL, LA érythro-mégacaryoblastique (256)8]
Transgénique Tri21 + rétrovirus GATA1s : pas de leucémie (254)4]
Transgénique (Tri21 & GATA1s) + rétrovirus MPL* : LAM7 (257)6]
OTT-MAL (258)9] iPS/ES: Lentivirus (259)7] Knock-In : LAM7 avec faible pénétrance (260)1]
ETO2-GLIS2 MO7e (261)8] (258)9] iPS: Knock-In AAVS1 [@] Rétrovirus : absence de maladie (262)4]
GATA2-HOXA9 Rétrovirus : LAM (262)4]
NIPBL-HOXB9 Rétrovirus : LAM (262)4]
NUP98-KDM5A (258)9] CD34+ CB: Lentivirus
(263)9]
Rétrovirus : LAM (262)4]
MLL-AF4 Transgénique conditionnel : Lymphome B
MLL-AF9 (258)9] Rétrovirus & Transgénique : LAM
MLL-AF10 Rétrovirus : LAM
TLS-ERG Rétrovirus : LAM
MN1-FLI1 Rétrovirus : LAM7 (264)0]
BCR-ABL1 MEG-01, K562 Rétrovirus & Transgénique : LAL-B, LMC
JAK2 T875N CHRF-288-11
(265)0]
Rétrovirus : SMP (265)0]
JAK3 A572V CMK Rétrovirus : Lymphome T (251)1]
n.d. CMS, M-MOK
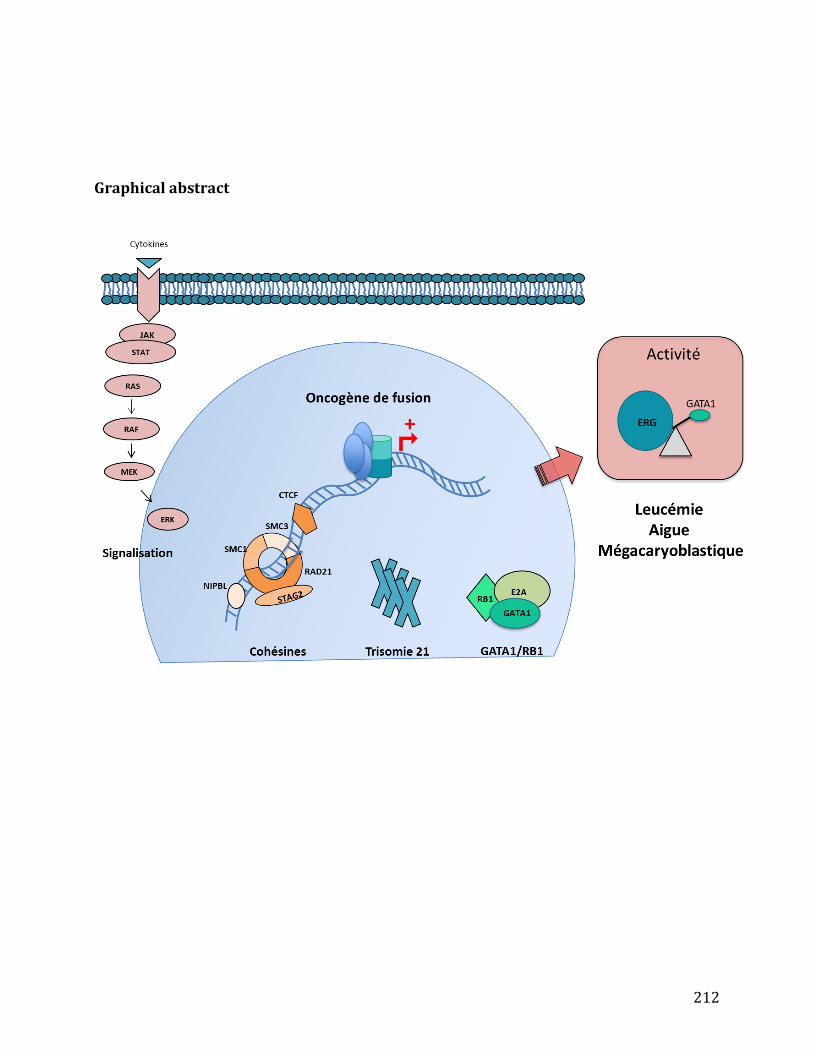
212
Graphical abstract

213
REFERENCES BIBLIOGRAPHIQUES
1. Till JE, McCulloch EA. Hemopoietic stem cell differentiation. Biochim Biophys Acta.
26 nov 1980;605(4):431‑59.
2. Baron MH, Isern J, Fraser ST. The embryonic origins of erythropoiesis in mammals.
Blood. 24 mai 2012;119(21):4828‑37.
3. Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and
myeloid progenitors in the yolk sac and embryo proper of the mouse. Dev Camb Engl. nov
1999;126(22):5073‑84.
4. Tober J, Koniski A, McGrath KE, Vemishetti R, Emerson R, de Mesy-Bentley KKL, et al.
The megakaryocyte lineage originates from hemangioblast precursors and is an integral
component both of primitive and of definitive hematopoiesis. Blood. 15 févr
2007;109(4):1433‑41.
5. Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for
hematopoietic and endothelial cells. Dev Camb Engl. févr 1998;125(4):725‑32.
6. Huber TL, Kouskoff V, Fehling HJ, Palis J, Keller G. Haemangioblast commitment is
initiated in the primitive streak of the mouse embryo. Nature. 2 déc
2004;432(7017):625‑30.
7. Kauts M-L, Vink CS, Dzierzak E. Hematopoietic (stem) cell development - how
divergent are the roads taken? FEBS Lett. 2016;590(22):3975‑86.
8. Godin I, Cumano A. The hare and the tortoise: an embryonic haematopoietic race.
Nat Rev Immunol. août 2002;2(8):593‑604.
9. McGrath KE, Frame JM, Fegan KH, Bowen JR, Conway SJ, Catherman SC, et al. Distinct
Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood
Cells in the Mammalian Embryo. Cell Rep. 30 juin 2015;11(12):1892‑904.
10. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-
resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature.
26 févr 2015;518(7540):547‑51.
11. Yokota T, Huang J, Tavian M, Nagai Y, Hirose J, Zúñiga-Pflücker J-C, et al. Tracing the
first waves of lymphopoiesis in mice. Dev Camb Engl. mai 2006;133(10):2041‑51.

214
12. Yoshimoto M, Porayette P, Glosson NL, Conway SJ, Carlesso N, Cardoso AA, et al.
Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before
HSC emergence. Blood. 14 juin 2012;119(24):5706‑14.
13. Kobayashi M, Shelley WC, Seo W, Vemula S, Lin Y, Liu Y, et al. Functional B-1
progenitor cells are present in the hematopoietic stem cell-deficient embryo and depend on
Cbfβ for their development. Proc Natl Acad Sci U S A. 19 août 2014;111(33):12151‑6.
14. Böiers C, Carrelha J, Lutteropp M, Luc S, Green JCA, Azzoni E, et al. Lymphomyeloid
contribution of an immune-restricted progenitor emerging prior to definitive
hematopoietic stem cells. Cell Stem Cell. 7 nov 2013;13(5):535‑48.
15. Müller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of
hematopoietic stem cell activity in the mouse embryo. Immunity. juill 1994;1(4):291‑301.
16. Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the
AGM region. Cell. 20 sept 1996;86(6):897‑906.
17. Boisset J-C, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo
imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 4
mars 2010;464(7285):116‑20.
18. Solaimani Kartalaei P, Yamada-Inagawa T, Vink CS, de Pater E, van der Linden R,
Marks-Bluth J, et al. Whole-transcriptome analysis of endothelial to hematopoietic stem cell
transition reveals a requirement for Gpr56 in HSC generation. J Exp Med. 12 janv
2015;212(1):93‑106.
19. Jaffredo T, Gautier R, Eichmann A, Dieterlen-Lièvre F. Intraaortic hemopoietic cells
are derived from endothelial cells during ontogeny. Dev Camb Engl. nov
1998;125(22):4575‑83.
20. Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DYR, Traver D. Haematopoietic stem
cells derive directly from aortic endothelium during development. Nature. 4 mars
2010;464(7285):108‑11.
21. Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel
type of cell transition. Nature. 4 mars 2010;464(7285):112‑5.
22. Kumaravelu P, Hook L, Morrison AM, Ure J, Zhao S, Zuyev S, et al. Quantitative
developmental anatomy of definitive haematopoietic stem cells/long-term repopulating

215
units (HSC/RUs): role of the aorta-gonad-mesonephros (AGM) region and the yolk sac in
colonisation of the mouse embryonic liver. Dev Camb Engl. nov 2002;129(21):4891‑9.
23. Gekas C, Dieterlen-Lièvre F, Orkin SH, Mikkola HKA. The placenta is a niche for
hematopoietic stem cells. Dev Cell. mars 2005;8(3):365‑75.
24. Ottersbach K, Dzierzak E. The murine placenta contains hematopoietic stem cells
within the vascular labyrinth region. Dev Cell. mars 2005;8(3):377‑87.
25. Ema H, Nakauchi H. Expansion of hematopoietic stem cells in the developing liver of
a mouse embryo. Blood. 1 avr 2000;95(7):2284‑8.
26. Morita Y, Iseki A, Okamura S, Suzuki S, Nakauchi H, Ema H. Functional
characterization of hematopoietic stem cells in the spleen. Exp Hematol. mars
2011;39(3):351‑359.e3.
27. Christensen JL, Wright DE, Wagers AJ, Weissman IL. Circulation and chemotaxis of
fetal hematopoietic stem cells. PLoS Biol. mars 2004;2(3):E75.
28. Babovic S, Eaves CJ. Hierarchical organization of fetal and adult hematopoietic stem
cells. Exp Cell Res. 10 déc 2014;329(2):185‑91.
29. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 22
févr 2008;132(4):631‑44.
30. Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ.
Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific
engraftment defect. J Clin Invest. oct 2006;116(10):2808‑16.
31. Pawliuk R, Eaves C, Humphries RK. Evidence of both ontogeny and transplant dose-
regulated expansion of hematopoietic stem cells in vivo. Blood. 15 oct 1996;88(8):2852‑8.
32. Bowie MB, Kent DG, Dykstra B, McKnight KD, McCaffrey L, Hoodless PA, et al.
Identification of a new intrinsically timed developmental checkpoint that reprograms key
hematopoietic stem cell properties. Proc Natl Acad Sci U S A. 3 avr 2007;104(14):5878‑82.
33. Crisan M, Dzierzak E. The many faces of hematopoietic stem cell heterogeneity. Dev
Camb Engl. 15 2016;143(24):4571‑81.
34. Mayani H. The regulation of hematopoietic stem cell populations. F1000Research.
2016;5.
35. Purton LE, Scadden DT. Limiting factors in murine hematopoietic stem cell assays.

216
Cell Stem Cell. 13 sept 2007;1(3):263‑70.
36. Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of
quiescence in hematopoietic stem cells. Dev Camb Engl. déc 2014;141(24):4656‑66.
37. Ema H, Morita Y, Suda T. Heterogeneity and hierarchy of hematopoietic stem cells.
Exp Hematol. févr 2014;42(2):74‑82.e2.
38. Wang Z, Ema H. Mechanisms of self-renewal in hematopoietic stem cells. Int J
Hematol. mai 2016;103(5):498‑509.
39. Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic Stem Cells
Count and Remember Self-Renewal Divisions. Cell. 17 2016;167(5):1296‑1309.e10.
40. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al.
Hematopoietic stem cells reversibly switch from dormancy to self-renewal during
homeostasis and repair. Cell. 12 déc 2008;135(6):1118‑29.
41. Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into
cell-cycle regulation. Nat Rev Genet. févr 2008;9(2):115‑28.
42. Wang JCY, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. sept
2005;15(9):494‑501.
43. Arai F, Suda T. Maintenance of quiescent hematopoietic stem cells in the osteoblastic
niche. Ann N Y Acad Sci. juin 2007;1106:41‑53.
44. Gibbs BF, Yasinska IM, Oniku AE, Sumbayev VV. Effects of stem cell factor on
hypoxia-inducible factor 1 alpha accumulation in human acute myeloid leukaemia and
LAD2 mast cells. PloS One. 2011;6(7):e22502.
45. Ye M, Zhang H, Amabile G, Yang H, Staber PB, Zhang P, et al. C/EBPa controls
acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol. avr
2013;15(4):385‑94.
46. Qiu J, Papatsenko D, Niu X, Schaniel C, Moore K. Divisional history and hematopoietic
stem cell function during homeostasis. Stem Cell Rep. 8 avr 2014;2(4):473‑90.
47. Laurenti E, Göttgens B. From haematopoietic stem cells to complex differentiation
landscapes. Nature. 24 2018;553(7689):418‑26.
48. Haas S, Trumpp A, Milsom MD. Causes and Consequences of Hematopoietic Stem Cell
Heterogeneity. Cell Stem Cell. 3 mai 2018;22(5):627‑38.

217
49. Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, et al.
Clonal analysis of lineage fate in native haematopoiesis. Nature. 11 2018;553(7687):212‑6.
50. Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Transcriptional
Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 17 déc
2015;163(7):1663‑77.
51. Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, Weissman IL. Identification of a
lineage of multipotent hematopoietic progenitors. Dev Camb Engl. mai
1997;124(10):1929‑39.
52. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem
cells. Nature. 1 nov 2001;414(6859):105‑11.
53. Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid
progenitors in mouse bone marrow. Cell. 28 nov 1997;91(5):661‑72.
54. Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid
progenitor that gives rise to all myeloid lineages. Nature. 9 mars 2000;404(6774):193‑7.
55. Adolfsson J, Borge OJ, Bryder D, Theilgaard-Mönch K, Astrand-Grundström I, Sitnicka
E, et al. Upregulation of Flt3 expression within the bone marrow Lin(-)Sca1(+)c-kit(+) stem
cell compartment is accompanied by loss of self-renewal capacity. Immunity. oct
2001;15(4):659‑69.
56. Adolfsson J, Månsson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, et al.
Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential
a revised road map for adult blood lineage commitment. Cell. 22 avr 2005;121(2):295‑306.
57. Månsson R, Hultquist A, Luc S, Yang L, Anderson K, Kharazi S, et al. Molecular
evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and
multipotent progenitors. Immunity. avr 2007;26(4):407‑19.
58. Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, et al.
Identification of regulatory networks in HSCs and their immediate progeny via integrated
proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2 oct
2014;15(4):507‑22.
59. Sawai CM, Babovic S, Upadhaya S, Knapp DJHF, Lavin Y, Lau CM, et al. Hematopoietic
Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity.

218
20 2016;45(3):597‑609.
60. Carrelha J, Meng Y, Kettyle LM, Luis TC, Norfo R, Alcolea V, et al. Hierarchically
related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. 01
2018;554(7690):106‑11.
61. Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human
haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. avr
2017;19(4):271‑81.
62. Perié L, Duffy KR, Kok L, de Boer RJ, Schumacher TN. The Branching Point in Erythro-
Myeloid Differentiation. Cell. 17 déc 2015;163(7):1655‑62.
63. Hoppe PS, Schwarzfischer M, Loeffler D, Kokkaliaris KD, Hilsenbeck O, Moritz N, et al.
Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios.
Nature. 14 2016;535(7611):299‑302.
64. De Kleer I, Willems F, Lambrecht B, Goriely S. Ontogeny of myeloid cells. Front
Immunol. 2014;5:423.
65. Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O’Donnell E, Zhao B-Q, et al.
Inflammation induces hemorrhage in thrombocytopenia. Blood. 15 mai
2008;111(10):4958‑64.
66. Xiang B, Zhang G, Guo L, Li X-A, Morris AJ, Daugherty A, et al. Platelets protect from
septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1
signalling pathway. Nat Commun. 2013;4:2657.
67. Huang H, Yu M, Akie TE, Moran TB, Woo AJ, Tu N, et al. Differentiation-dependent
interactions between RUNX-1 and FLI-1 during megakaryocyte development. Mol Cell Biol.
août 2009;29(15):4103‑15.
68. Huang H, Cantor AB. Common features of megakaryocytes and hematopoietic stem
cells: what’s the connection? J Cell Biochem. 1 août 2009;107(5):857‑64.
69. Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, et al. Platelet-biased
stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 10 oct
2013;502(7470):232‑6.
70. Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, et al.
Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-

219
like Megakaryocyte Progenitors. Cell Stem Cell. 1 oct 2015;17(4):422‑34.
71. Mazzi S, Lordier L, Debili N, Raslova H, Vainchenker W. Megakaryocyte and
polyploidization. Exp Hematol. 2018;57:1‑13.
72. Zimmet J, Ravid K. Polyploidy: occurrence in nature, mechanisms, and significance
for the megakaryocyte-platelet system. Exp Hematol. janv 2000;28(1):3‑16.
73. Lordier L, Pan J, Naim V, Jalil A, Badirou I, Rameau P, et al. Presence of a defect in
karyokinesis during megakaryocyte endomitosis. Cell Cycle Georget Tex. 1 déc
2012;11(23):4385‑9.
74. Ruggeri ZM, De Marco L, Gatti L, Bader R, Montgomery RR. Platelets have more than
one binding site for von Willebrand factor. J Clin Invest. juill 1983;72(1):1‑12.
75. Nishimura S, Nagasaki M, Kunishima S, Sawaguchi A, Sakata A, Sakaguchi H, et al. IL-
1α induces thrombopoiesis through megakaryocyte rupture in response to acute platelet
needs. J Cell Biol. 11 mai 2015;209(3):453‑66.
76. Chang Y, Bluteau D, Debili N, Vainchenker W. From hematopoietic stem cells to
platelets. J Thromb Haemost JTH. juill 2007;5 Suppl 1:318‑27.
77. Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, et al. Distinct routes of
lineage development reshape the human blood hierarchy across ontogeny. Science. 8 janv
2016;351(6269):aab2116.
78. Elagib KE, Brock AT, Goldfarb AN. Megakaryocyte ontogeny: Clinical and molecular
significance. Exp Hematol. mai 2018;61:1‑9.
79. Bluteau O, Langlois T, Rivera-Munoz P, Favale F, Rameau P, Meurice G, et al.
Developmental changes in human megakaryopoiesis. J Thromb Haemost JTH. sept
2013;11(9):1730‑41.
80. Birbrair A, Frenette PS. Niche heterogeneity in the bone marrow. Ann N Y Acad Sci.
2016;1370(1):82‑96.
81. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem
cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches.
Immunity. déc 2006;25(6):977‑88.
82. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al.
Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone

220
marrow niche. Cell. 23 nov 2011;147(5):1146‑58.
83. Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes
maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic
stem cells. Nat Med. nov 2014;20(11):1321‑6.
84. Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential
cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol.
2017;19(3):214‑23.
85. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during
homeostasis and regeneration. Nat Med. août 2014;20(8):833‑46.
86. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar
niches maintain haematopoietic stem cell quiescence. Nature. 31 oct
2013;502(7473):637‑43.
87. Pinho S, Marchand T, Yang E, Wei Q, Nerlov C, Frenette PS. Lineage-Biased
Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev Cell. 12
2018;44(5):634‑641.e4.
88. Poulos MG, Guo P, Kofler NM, Pinho S, Gutkin MC, Tikhonova A, et al. Endothelial
Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 12 sept
2013;4(5):1022‑34.
89. Gao X, Xu C, Asada N, Frenette PS. The hematopoietic stem cell niche: from embryo to
adult. Dev Camb Engl. 22 2018;145(2).
90. Souilhol C, Lendinez JG, Rybtsov S, Murphy F, Wilson H, Hills D, et al. Developing
HSCs become Notch independent by the end of maturation in the AGM region. Blood. 22
2016;128(12):1567‑77.
91. Saito K, Nobuhisa I, Harada K, Takahashi S, Anani M, Lickert H, et al. Maintenance of
hematopoietic stem and progenitor cells in fetal intra-aortic hematopoietic clusters by the
Sox17-Notch1-Hes1 axis. Exp Cell Res. 1 avr 2018;365(1):145‑55.
92. Radtke F, Wilson A, Mancini SJC, MacDonald HR. Notch regulation of lymphocyte
development and function. Nat Immunol. mars 2004;5(3):247‑53.
93. Mercher T, Cornejo MG, Sears C, Kindler T, Moore SA, Maillard I, et al. Notch signaling
specifies megakaryocyte development from hematopoietic stem cells. Cell Stem Cell. 11 sept

221
2008;3(3):314‑26.
94. Nakamura-Ishizu A, Takubo K, Kobayashi H, Suzuki-Inoue K, Suda T. Correction:
CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the
bone marrow. J Exp Med. 14 déc 2015;212(13):2323.
95. Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes
regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. nov
2014;20(11):1315‑20.
96. Kirito K, Kaushansky K. Transcriptional regulation of megakaryopoiesis:
thrombopoietin signaling and nuclear factors. Curr Opin Hematol. mai 2006;13(3):151‑6.
97. Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol
Cell Biol. sept 2002;3(9):651‑62.
98. Drachman JG, Sabath DF, Fox NE, Kaushansky K. Thrombopoietin signal transduction
in purified murine megakaryocytes. Blood. 15 janv 1997;89(2):483‑92.
99. Schulze H, Ballmaier M, Welte K, Germeshausen M. Thrombopoietin induces the
generation of distinct Stat1, Stat3, Stat5a and Stat5b homo- and heterodimeric complexes
with different kinetics in human platelets. Exp Hematol. mars 2000;28(3):294‑304.
100. Snow JW, Abraham N, Ma MC, Abbey NW, Herndier B, Goldsmith MA. STAT5
promotes multilineage hematolymphoid development in vivo through effects on early
hematopoietic progenitor cells. Blood. 1 janv 2002;99(1):95‑101.
101. Bhardwaj G, Murdoch B, Wu D, Baker DP, Williams KP, Chadwick K, et al. Sonic
hedgehog induces the proliferation of primitive human hematopoietic cells via BMP
regulation. Nat Immunol. févr 2001;2(2):172‑80.
102. Watson S, Serrate C, Vignot S. [Sonic Hedgehog signaling pathway: from embryology
to molecular targeted therapies]. Bull Cancer (Paris). déc 2010;97(12):1477‑83.
103. Sentenac A. [The Nobel goes to gene transcription]. Med Sci MS. nov
2006;22(11):995‑8.
104. Lelli KM, Slattery M, Mann RS. Disentangling the many layers of eukaryotic
transcriptional regulation. Annu Rev Genet. 2012;46:43‑68.
105. Tijssen MR, Cvejic A, Joshi A, Hannah RL, Ferreira R, Forrai A, et al. Genome-wide
analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes

222
identifies hematopoietic regulators. Dev Cell. 17 mai 2011;20(5):597‑609.
106. Crispino JD. GATA1 in normal and malignant hematopoiesis. Semin Cell Dev Biol. févr
2005;16(1):137‑47.
107. Carotta S, Wu L, Nutt SL. Surprising new roles for PU.1 in the adaptive immune
response. Immunol Rev. nov 2010;238(1):63‑75.
108. Staber PB, Zhang P, Ye M, Welner RS, Nombela-Arrieta C, Bach C, et al. Sustained PU.1
levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells.
Mol Cell. 7 mars 2013;49(5):934‑46.
109. Pulikkan JA, Tenen DG, Behre G. C/EBPα deregulation as a paradigm for
leukemogenesis. Leukemia. 19 juill 2017;
110. Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, et al. Single-cell
characterization of haematopoietic progenitors and their trajectories in homeostasis and
perturbed haematopoiesis. Nat Cell Biol. juill 2018;20(7):836‑46.
111. Soler E, Andrieu-Soler C, de Boer E, Bryne JC, Thongjuea S, Stadhouders R, et al. The
genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation.
Genes Dev. 1 févr 2010;24(3):277‑89.
112. Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, et al. Combinatorial
transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major
transcriptional regulators. Cell Stem Cell. 8 oct 2010;7(4):532‑44.
113. Tsai FY, Keller G, Kuo FC, Weiss M, Chen J, Rosenblatt M, et al. An early
haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 15 sept
1994;371(6494):221‑6.
114. Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of
multiple chromosomal translocations in human leukemia, is essential for normal fetal liver
hematopoiesis. Cell. 26 janv 1996;84(2):321‑30.
115. Doré LC, Chlon TM, Brown CD, White KP, Crispino JD. Chromatin occupancy analysis
reveals genome-wide GATA factor switching during hematopoiesis. Blood. 19 avr
2012;119(16):3724‑33.
116. Pimkin M, Kossenkov AV, Mishra T, Morrissey CS, Wu W, Keller CA, et al. Divergent
functions of hematopoietic transcription factors in lineage priming and differentiation

223
during erythro-megakaryopoiesis. Genome Res. déc 2014;24(12):1932‑44.
117. Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, et al.
Immunogenetics. Chromatin state dynamics during blood formation. Science. 22 août
2014;345(6199):943‑9.
118. Sharma S, Gurudutta G. Epigenetic Regulation of Hematopoietic Stem Cells. Int J Stem
Cells. 30 mai 2016;9(1):36‑43.
119. Antoniani C, Romano O, Miccio A. Concise Review: Epigenetic Regulation of
Hematopoiesis: Biological Insights and Therapeutic Applications. Stem Cells Transl Med.
déc 2017;6(12):2106‑14.
120. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and
translational implications. Nat Rev Cancer. 23 sept 2011;11(10):726‑34.
121. Ji H, Ehrlich LIR, Seita J, Murakami P, Doi A, Lindau P, et al. Comprehensive
methylome map of lineage commitment from haematopoietic progenitors. Nature. 16 sept
2010;467(7313):338‑42.
122. Trowbridge JJ, Orkin SH. Dnmt3a silences hematopoietic stem cell self-renewal. Nat
Genet. 27 déc 2011;44(1):13‑4.
123. Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA
demethylation and transcription. Nat Rev Mol Cell Biol. juin 2013;14(6):341‑56.
124. Lio C-W, Zhang J, González-Avalos E, Hogan PG, Chang X, Rao A. Tet2 and Tet3
cooperate with B-lineage transcription factors to regulate DNA modification and chromatin
accessibility. eLife. 21 2016;5.
125. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers
in the control of cell identity and disease. Cell. 7 nov 2013;155(4):934‑47.
126. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master
transcription factors and mediator establish super-enhancers at key cell identity genes. Cell.
11 avr 2013;153(2):307‑19.
127. Panigrahi AK, Pati D. Higher-order orchestration of hematopoiesis: is cohesin a new
player? Exp Hematol. déc 2012;40(12):967‑73.
128. Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, et al. Leukemia-Associated
Cohesin Mutants Dominantly Enforce Stem Cell Programs and Impair Human

224
Hematopoietic Progenitor Differentiation. Cell Stem Cell. 3 déc 2015;17(6):675‑88.
129. Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, et al.
Cohesin Loss Eliminates All Loop Domains. Cell. 5 oct 2017;171(2):305‑320.e24.
130. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al.
Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-
operative group. Br J Haematol. août 1976;33(4):451‑8.
131. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The
2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms
and acute leukemia: rationale and important changes. Blood. 30 juill 2009;114(5):937‑51.
132. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016
revision to the World Health Organization classification of myeloid neoplasms and acute
leukemia. Blood. 19 2016;127(20):2391‑405.
133. Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, et al.
Management of acute promyelocytic leukemia: recommendations from an expert panel on
behalf of the European LeukemiaNet. Blood. 26 févr 2009;113(9):1875‑91.
134. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. avr
2009;458(7239):719‑24.
135. Pon JR, Marra MA. Driver and passenger mutations in cancer. Annu Rev Pathol.
2015;10:25‑50.
136. Vainchenker W, Constantinescu SN. A unique activating mutation in JAK2 (V617F) is
at the origin of polycythemia vera and allows a new classification of myeloproliferative
diseases. Hematol Am Soc Hematol Educ Program. 2005;195‑200.
137. Weinberg RA. Tumor suppressor genes. Science. 22 nov 1991;254(5035):1138‑46.
138. Valverde JR, Alonso J, Palacios I, Pestaña A. RB1 gene mutation up-date, a meta-
analysis based on 932 reported mutations available in a searchable database. BMC Genet. 4
nov 2005;6:53.
139. Bernard OA, Delhommeau F, Fontenay M, Vainchenker W. [Mutations in TET2 in
myeloid cancers]. Med Sci MS. oct 2009;25(10):785‑8.
140. Petrie K, Zelent A. AML1/ETO, a promiscuous fusion oncoprotein. Blood. 15 mai
2007;109(10):4109‑10.

225
141. Llopis L, Nibourel O, Boissel N, Huchette P, Renneville A, Abdelhamid E, et al.
Implication des mutations des gènes IDH1 et IDH2 dans les leucémies aiguës myéloïdes.
Hématologie. 1 mai 2011;17(2):132‑44.
142. Metzeler KH, Dufour A, Benthaus T, Hummel M, Sauerland M-C, Heinecke A, et al.
ERG expression is an independent prognostic factor and allows refined risk stratification in
cytogenetically normal acute myeloid leukemia: a comprehensive analysis of ERG, MN1, and
BAALC transcript levels using oligonucleotide microarrays. J Clin Oncol Off J Am Soc Clin
Oncol. 20 oct 2009;27(30):5031‑8.
143. Carmichael CL, Metcalf D, Henley KJ, Kruse EA, Di Rago L, Mifsud S, et al.
Hematopoietic overexpression of the transcription factor Erg induces lymphoid and
erythro-megakaryocytic leukemia. Proc Natl Acad Sci U S A. 18 sept
2012;109(38):15437‑42.
144. Goldberg L, Tijssen MR, Birger Y, Hannah RL, Kinston SJ, Schütte J, et al. Genome-
scale expression and transcription factor binding profiles reveal therapeutic targets in
transgenic ERG myeloid leukemia. Blood. 10 oct 2013;122(15):2694‑703.
145. Gröschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, et
al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2
deregulation in leukemia. Cell. 10 avr 2014;157(2):369‑81.
146. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and
evolution of mutations in acute myeloid leukemia. Cell. 20 juill 2012;150(2):264‑78.
147. Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat
Rev Cancer. juill 2002;2(7):502‑13.
148. Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, Lin L, et al. Sequencing a mouse
acute promyelocytic leukemia genome reveals genetic events relevant for disease
progression. J Clin Invest. avr 2011;121(4):1445‑55.
149. Sohal J, Phan VT, Chan PV, Davis EM, Patel B, Kelly LM, et al. A model of APL with
FLT3 mutation is responsive to retinoic acid and a receptor tyrosine kinase inhibitor,
SU11657. Blood. 15 avr 2003;101(8):3188‑97.
150. Behrens K, Maul K, Tekin N, Kriebitzsch N, Indenbirken D, Prassolov V, et al. RUNX1
cooperates with FLT3-ITD to induce leukemia. J Exp Med. 06 2017;214(3):737‑52.

226
151. Ferrando AA, López-Otín C. Clonal evolution in leukemia. Nat Med. oct
2017;23(10):1135‑45.
152. Busque L, Buscarlet M, Mollica L, Levine RL. Concise Review: Age-Related Clonal
Hematopoiesis: Stem Cells Tempting the Devil. Stem Cells Dayt Ohio. 8 juin 2018;
153. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that
originates from a primitive hematopoietic cell. Nat Med. juill 1997;3(7):730‑7.
154. Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene
stemness score for rapid determination of risk in acute leukaemia. Nature. 15
2016;540(7633):433‑7.
155. van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, et al. High
stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual
disease and poor survival. Clin Cancer Res Off J Am Assoc Cancer Res. 15 sept
2005;11(18):6520‑7.
156. Witte K-E, Ahlers J, Schäfer I, André M, Kerst G, Scheel-Walter H-G, et al. High
proportion of leukemic stem cells at diagnosis is correlated with unfavorable prognosis in
childhood acute myeloid leukemia. Pediatr Hematol Oncol. mars 2011;28(2):91‑9.
157. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-
resistant human AML stem cells home to and engraft within the bone-marrow endosteal
region. Nat Biotechnol. nov 2007;25(11):1315‑21.
158. Korn C, Méndez-Ferrer S. Myeloid malignancies and the microenvironment. Blood.
16 2017;129(7):811‑22.
159. Shafat MS, Gnaneswaran B, Bowles KM, Rushworth SA. The bone marrow
microenvironment - Home of the leukemic blasts. Blood Rev. sept 2017;31(5):277‑86.
160. Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al.
Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia
stem cells. Sci Transl Med. 3 févr 2010;2(17):17ra9.
161. Huntly BJP, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2,
but not BCR-ABL, confers properties of leukemic stem cells to committed murine
hematopoietic progenitors. Cancer Cell. déc 2004;6(6):587‑96.
162. Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, et al. MLL-AF9

227
Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-
Related Genes Linked to Poor Outcome. Cancer Cell. 11 juill 2016;30(1):43‑58.
163. Creutzig U, Büchner T, Sauerland MC, Zimmermann M, Reinhardt D, Döhner H, et al.
Significance of age in acute myeloid leukemia patients younger than 30 years: a common
analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and
AMLSG HD93/98A. Cancer. 1 févr 2008;112(3):562‑71.
164. Appelbaum FR, Gundacker H, Head DR, Slovak ML, Willman CL, Godwin JE, et al. Age
and acute myeloid leukemia. Blood. 1 mai 2006;107(9):3481‑5.
165. Basilico S, Göttgens B. Dysregulation of haematopoietic stem cell regulatory
programs in acute myeloid leukaemia. J Mol Med Berl Ger. juill 2017;95(7):719‑27.
166. Biondi A, Cimino G, Pieters R, Pui CH. Biological and therapeutic aspects of infant
leukemia. Blood. 1 juill 2000;96(1):24‑33.
167. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall
AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N
Engl J Med. 30 2013;368(22):2059‑74.
168. Creutzig U, Zimmermann M, Reinhardt D, Rasche M, von Neuhoff C, Alpermann T, et
al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from
childhood to adult age groups. Cancer. 15 déc 2016;122(24):3821‑30.
169. Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural
history. Blood. 1 oct 2003;102(7):2321‑33.
170. Greaves M. In utero origins of childhood leukaemia. Early Hum Dev. 1 janv
2005;81(1):123‑9.
171. Wiemels JL, Xiao Z, Buffler PA, Maia AT, Ma X, Dicks BM, et al. In utero origin of
t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood. 15 mai
2002;99(10):3801‑5.
172. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Criteria
for the diagnosis of acute leukemia of megakaryocyte lineage (M7). A report of the French-
American-British Cooperative Group. Ann Intern Med. sept 1985;103(3):460‑2.
173. Dastugue N, Lafage-Pochitaloff M, Pagès M-P, Radford I, Bastard C, Talmant P, et al.
Cytogenetic profile of childhood and adult megakaryoblastic leukemia (M7): a study of the

228
Groupe Français de Cytogénétique Hématologique (GFCH). Blood. 15 juill
2002;100(2):618‑26.
174. Zwaan CM, Kolb EA, Reinhardt D, Abrahamsson J, Adachi S, Aplenc R, et al.
Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J Clin Oncol Off
J Am Soc Clin Oncol. 20 sept 2015;33(27):2949‑62.
175. Teyssier A-C, Lapillonne H, Pasquet M, Ballerini P, Baruchel A, Ducassou S, et al.
Acute megakaryoblastic leukemia (excluding Down syndrome) remains an acute myeloid
subgroup with inferior outcome in the French ELAM02 trial. Pediatr Hematol Oncol. nov
2017;34(8):425‑7.
176. Ishiyama K, Yamaguchi T, Eto T, Ohashi K, Uchida N, Kanamori H, et al. Acute
megakaryoblastic leukemia, unlike acute erythroid leukemia, predicts an unfavorable
outcome after allogeneic HSCT. Leuk Res. 2016;47:47‑53.
177. Mercher T, Coniat MB, Monni R, Mauchauffe M, Nguyen Khac F, Gressin L, et al.
Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22)
translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A. 8 mai
2001;98(10):5776‑9.
178. Thiollier C, Lopez CK, Gerby B, Ignacimouttou C, Poglio S, Duffourd Y, et al.
Characterization of novel genomic alterations and therapeutic approaches using acute
megakaryoblastic leukemia xenograft models. J Exp Med. 22 oct 2012;209(11):2017‑31.
179. de Rooij JDE, Branstetter C, Ma J, Li Y, Walsh MP, Cheng J, et al. Pediatric non-Down
syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets
with varying outcomes. Nat Genet. 23 janv 2017;
180. Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An
Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype
of pediatric acute megakaryoblastic leukemia. Cancer Cell. 13 nov 2012;22(5):683‑97.
181. Inaba H, Zhou Y, Abla O, Adachi S, Auvrignon A, Beverloo HB, et al. Heterogeneous
cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a
retrospective international study. Blood. 24 sept 2015;126(13):1575‑84.
182. Khan I, Malinge S, Crispino J. Myeloid leukemia in Down syndrome. Crit Rev Oncog.
2011;16(1‑2):25‑36.

229
183. Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and
mechanisms of leukemogenesis in Down syndrome. Blood. 19 mars 2009;113(12):2619‑28.
184. Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, et al. Acquired
mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. sept
2002;32(1):148‑52.
185. Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, et al. The
landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet.
nov 2013;45(11):1293‑9.
186. Gruber TA, Downing JR. The biology of pediatric acute megakaryoblastic leukemia.
Blood. 20 août 2015;126(8):943‑9.
187. Hara Y, Shiba N, Ohki K, Tabuchi K, Yamato G, Park M-J, et al. Prognostic impact of
specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down
syndrome. Genes Chromosomes Cancer. mai 2017;56(5):394‑404.
188. Sait SN, Brecher ML, Green DM, Sandberg AA. Translocation t(1;22) in congenital
acute megakaryocytic leukemia. Cancer Genet Cytogenet. sept 1988;34(2):277‑80.
189. Baruchel A, Daniel MT, Schaison G, Berger R. Nonrandom t(1;22)(p12-p13;q13) in
acute megakaryocytic malignant proliferation. Cancer Genet Cytogenet. 15 juill
1991;54(2):239‑43.
190. Ng KC, Tan AM, Chong YY, Lau LC, Lou J. Congenital acute megakaryoblastic leukemia
(M7) with chromosomal t(1;22)(p13;q13) translocation in a set of identical twins. J Pediatr
Hematol Oncol. oct 1999;21(5):428‑30.
191. Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, et al.
HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 12 mars
2009;113(11):2375‑85.
192. Salek-Ardakani S, Smooha G, de Boer J, Sebire NJ, Morrow M, Rainis L, et al. ERG is a
megakaryocytic oncogene. Cancer Res. 1 juin 2009;69(11):4665‑73.
193. Kadri Z, Shimizu R, Ohneda O, Maouche-Chretien L, Gisselbrecht S, Yamamoto M, et al.
Direct binding of pRb/E2F-2 to GATA-1 regulates maturation and terminal cell division
during erythropoiesis. PLoS Biol. 9 juin 2009;7(6):e1000123.
194. Fisher JB, Peterson J, Reimer M, Stelloh C, Pulakanti K, Gerbec ZJ, et al. The cohesin

230
subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic
repression of Hoxa7 and Hoxa9. Leukemia. mars 2017;31(3):712‑9.
195. Wen Q, Goldenson B, Silver SJ, Schenone M, Dancik V, Huang Z, et al. Identification of
regulators of polyploidization presents therapeutic targets for treatment of AMKL. Cell. 3
août 2012;150(3):575‑89.
196. Drexler HG. Guide to leukemia-lymphoma cell lines. Braunschweig, Germany:
German collection of microorganisms and cell cultures; 2005.
197. Walters DK, Mercher T, Gu T-L, O’Hare T, Tyner JW, Loriaux M, et al. Activating
alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. juill 2006;10(1):65‑75.
198. Saida S, Watanabe K, Sato-Otsubo A, Terui K, Yoshida K, Okuno Y, et al. Clonal
selection in xenografted TAM recapitulates the evolutionary process of myeloid leukemia in
Down syndrome. Blood. 23 mai 2013;121(21):4377‑87.
199. Chou ST, Byrska-Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB, et al. Trisomy
21-associated defects in human primitive hematopoiesis revealed through induced
pluripotent stem cells. Proc Natl Acad Sci U S A. 23 oct 2012;109(43):17573‑8.
200. Kirsammer G, Jilani S, Liu H, Davis E, Gurbuxani S, Le Beau MM, et al. Highly
penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome.
Blood. 15 janv 2008;111(2):767‑75.
201. Alford KA, Slender A, Vanes L, Li Z, Fisher EMC, Nizetic D, et al. Perturbed
hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 8 avr
2010;115(14):2928‑37.
202. Carmichael CL, Majewski IJ, Alexander WS, Metcalf D, Hilton DJ, Hewitt CA, et al.
Hematopoietic defects in the Ts1Cje mouse model of Down syndrome. Blood. 26 févr
2009;113(9):1929‑37.
203. Byrska-Bishop M, VanDorn D, Campbell AE, Betensky M, Arca PR, Yao Y, et al.
Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. J Clin
Invest. 2 mars 2015;125(3):993‑1005.
204. Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al.
Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic
leukemia in a murine model of Down syndrome. J Clin Invest. mars 2012;122(3):948‑62.

231
205. Ayllón V, Vogel-González M, González-Pozas F, Domingo-Reinés J, Montes R, Morales-
Cacho L, et al. New hPSC-based human models to study pediatric Acute Megakaryoblastic
Leukemia harboring the fusion oncogene RBM15-MKL1. Stem Cell Res. 2017;19:1‑5.
206. Mercher T, Raffel GD, Moore SA, Cornejo MG, Baudry-Bluteau D, Cagnard N, et al. The
OTT-MAL fusion oncogene activates RBPJ-mediated transcription and induces acute
megakaryoblastic leukemia in a knockin mouse model. J Clin Invest. avr
2009;119(4):852‑64.
207. Dang J, Nance S, Ma J, Cheng J, Walsh MP, Vogel P, et al. AMKL chimeric transcription
factors are potent inducers of leukemia. Leukemia. 10 mars 2017;
208. Barrett NA, Malouf C, Kapeni C, Bacon WA, Giotopoulos G, Jacobsen SEW, et al. Mll-
AF4 Confers Enhanced Self-Renewal and Lymphoid Potential during a Restricted Window
in Development. Cell Rep. 26 2016;16(4):1039‑54.
209. Somervaille TCP, Cleary ML. Identification and characterization of leukemia stem
cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. oct 2006;10(4):257‑68.
210. DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. The AF10
leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10.
Blood. 15 mai 2002;99(10):3780‑5.
211. Pan J, Zou J, Wu DY, Roberson RS, Hennings LJ, Ma X, et al. TLS-ERG leukemia fusion
protein deregulates cyclin-dependent kinase 1 and blocks terminal differentiation of
myeloid progenitor cells. Mol Cancer Res MCR. mai 2008;6(5):862‑72.
212. Wenge DV, Felipe-Fumero E, Angenendt L, Schliemann C, Schmidt E, Schmidt LH, et
al. MN1-Fli1 oncofusion transforms murine hematopoietic progenitor cells into acute
megakaryoblastic leukemia cells. Oncogenesis. 21 déc 2015;4:e179.
213. Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell
leukaemia induced by BCR-ABL1. Nat Genet. janv 2000;24(1):57‑60.
214. Mercher T, Wernig G, Moore SA, Levine RL, Gu T-L, Fröhling S, et al. JAK2T875N is a
novel activating mutation that results in myeloproliferative disease with features of
megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 15 oct
2006;108(8):2770‑9.
215. Xiao N, Laha S, Das SP, Morlock K, Jesneck JL, Raffel GD. Ott1 (Rbm15) regulates

232
thrombopoietin response in hematopoietic stem cells through alternative splicing of c-Mpl.
Blood. 5 févr 2015;125(6):941‑8.
216. Oswald F, Winkler M, Cao Y, Astrahantseff K, Bourteele S, Knöchel W, et al. RBP-
Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol Cell Biol.
déc 2005;25(23):10379‑90.
217. Ma X, Renda MJ, Wang L, Cheng E-C, Niu C, Morris SW, et al. Rbm15 modulates Notch-
induced transcriptional activation and affects myeloid differentiation. Mol Cell Biol. avr
2007;27(8):3056‑64.
218. Tijssen MR, Ghevaert C. Transcription factors in late megakaryopoiesis and related
platelet disorders. J Thromb Haemost JTH. avr 2013;11(4):593‑604.
219. Ragu C, Boukour S, Elain G, Wagner-Ballon O, Raslova H, Debili N, et al. The serum
response factor (SRF)/megakaryocytic acute leukemia (MAL) network participates in
megakaryocyte development. Leukemia. juin 2010;24(6):1227‑30.
220. Struski S, Lagarde S, Bories P, Puiseux C, Prade N, Cuccuini W, et al. NUP98 is
rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group
with a poor prognosis. Leukemia. mars 2017;31(3):565‑72.
221. Gale M, Sayegh J, Cao J, Norcia M, Gareiss P, Hoyer D, et al. Screen-identified selective
inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance.
Oncotarget. 28 juin 2016;7(26):39931‑44.
222. van Zutven LJCM, Onen E, Velthuizen SCJM, van Drunen E, von Bergh ARM, van den
Heuvel-Eibrink MM, et al. Identification of NUP98 abnormalities in acute leukemia: JARID1A
(12p13) as a new partner gene. Genes Chromosomes Cancer. mai 2006;45(5):437‑46.
223. Roussy M, Bilodeau M, Jouan L, Tibout P, Laramée L, Lemyre E, et al. NUP98-BPTF
gene fusion identified in primary refractory acute megakaryoblastic leukemia of infancy.
Genes Chromosomes Cancer. 2018;57(6):311‑9.
224. Hess JL. MLL, Hox genes, and leukemia: the plot thickens. Blood. 15 avr
2004;103(8):2870‑1.
225. Takita J, Motomura A, Koh K, Ida K, Taki T, Hayashi Y, et al. Acute megakaryoblastic
leukemia in a child with the MLL-AF4 fusion gene. Eur J Haematol. août 2009;83(2):149‑53.
226. Borkhardt A, Haas OA, Strobl W, Repp R, Mann G, Gadner H, et al. A novel type of

233
MLL/AF10 fusion transcript in a child with acute megakaryocytic leukemia (AML-M7).
Leukemia. oct 1995;9(10):1796‑7.
227. Masetti R, Pigazzi M, Togni M, Astolfi A, Indio V, Manara E, et al. CBFA2T3-GLIS2
fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not
restricted to FAB M7 subtype. Blood. 25 avr 2013;121(17):3469‑72.
228. Chyla BJ, Moreno-Miralles I, Steapleton MA, Thompson MA, Bhaskara S, Engel M, et al.
Deletion of Mtg16, a target of t(16;21), alters hematopoietic progenitor cell proliferation
and lineage allocation. Mol Cell Biol. oct 2008;28(20):6234‑47.
229. Fischer MA, Moreno-Miralles I, Hunt A, Chyla BJ, Hiebert SW. Myeloid translocation
gene 16 is required for maintenance of haematopoietic stem cell quiescence. EMBO J. 21
mars 2012;31(6):1494‑505.
230. Hamlett I, Draper J, Strouboulis J, Iborra F, Porcher C, Vyas P. Characterization of
megakaryocyte GATA1-interacting proteins: the corepressor ETO2 and GATA1 interact to
regulate terminal megakaryocyte maturation. Blood. 1 oct 2008;112(7):2738‑49.
231. Kim Y-S, Kang HS, Herbert R, Beak JY, Collins JB, Grissom SF, et al. Kruppel-like zinc
finger protein Glis2 is essential for the maintenance of normal renal functions. Mol Cell Biol.
avr 2008;28(7):2358‑67.
232. Vasanth S, ZeRuth G, Kang HS, Jetten AM. Identification of nuclear localization, DNA
binding, and transactivating mechanisms of Kruppel-like zinc finger protein Gli-similar 2
(Glis2). J Biol Chem. 11 févr 2011;286(6):4749‑59.
233. Holmfeldt P, Ganuza M, Marathe H, He B, Hall T, Kang G, et al. Functional screen
identifies regulators of murine hematopoietic stem cell repopulation. J Exp Med. 7 mars
2016;213(3):433‑49.
234. Lee S-Y, Noh HB, Kim H-T, Lee K-I, Hwang D-Y. Glis family proteins are differentially
implicated in the cellular reprogramming of human somatic cells. Oncotarget. 29 sept
2017;8(44):77041‑9.
235. Bourquin J-P, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, et al.
Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene
expression profiling. Proc Natl Acad Sci U S A. 28 févr 2006;103(9):3339‑44.
236. Kron KJ, Murison A, Zhou S, Huang V, Yamaguchi TN, Shiah Y-J, et al. TMPRSS2-ERG

234
fusion co-opts master transcription factors and activates NOTCH signaling in primary
prostate cancer. Nat Genet. sept 2017;49(9):1336‑45.
237. Masetti R, Togni M, Astolfi A, Pigazzi M, Manara E, Indio V, et al. DHH-RHEBL1 fusion
transcript: a novel recurrent feature in the new landscape of pediatric CBFA2T3-GLIS2-
positive acute myeloid leukemia. Oncotarget. oct 2013;4(10):1712‑20.
238. Qin F, Song Z, Babiceanu M, Song Y, Facemire L, Singh R, et al. Discovery of CTCF-
sensitive Cis-spliced fusion RNAs between adjacent genes in human prostate cells. PLoS
Genet. févr 2015;11(2):e1005001.
239. Miglio U, Berrino E, Panero M, Ferrero G, Coscujuela L, Miano V, et al. THE
EXPRESSION OF LINE1-MET CHIMERIC TRANSCRIPT IDENTIFIES A SUBGROUP OF
AGGRESSIVE BREAST CANCERS. Int J Cancer. 25 août 2018;
240. Marshall AD, Bailey CG, Rasko JEJ. CTCF and BORIS in genome regulation and cancer.
Curr Opin Genet Dev. févr 2014;24:8‑15.
241. Godfrey L, Kerry J, Thorne R, Repapi E, Davies JOJ, Tapia M, et al. MLL-AF4 binds
directly to a BCL-2 specific enhancer and modulates H3K27 acetylation. Exp Hematol.
2017;47:64‑75.
242. Lee J-H, Skalnik DG. Rbm15-Mkl1 interacts with the Setd1b histone H3-Lys4
methyltransferase via a SPOC domain that is required for cytokine-independent
proliferation. PloS One. 2012;7(8):e42965.
243. Dafflon C, Craig VJ, Méreau H, Gräsel J, Schacher Engstler B, Hoffman G, et al.
Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia.
Leukemia. 2017;31(6):1269‑77.
244. Huang Y, Thoms J a. I, Tursky ML, Knezevic K, Beck D, Chandrakanthan V, et al.
MAPK/ERK2 phosphorylates ERG at serine 283 in leukemic cells and promotes stem cell
signatures and cell proliferation. Leukemia. juill 2016;30(7):1552‑61.
245. Selvaraj N, Kedage V, Hollenhorst PC. Comparison of MAPK specificity across the ETS
transcription factor family identifies a high-affinity ERK interaction required for ERG
function in prostate cells. Cell Commun Signal CCS. 19 févr 2015;13:12.
246. Kedage V, Strittmatter BG, Dausinas PB, Hollenhorst PC. Phosphorylation of the
oncogenic transcription factor ERG in prostate cells dissociates polycomb repressive

235
complex 2, allowing target gene activation. J Biol Chem. 20 2017;292(42):17225‑35.
247. Wang X, Qiao Y, Asangani IA, Ateeq B, Poliakov A, Cieślik M, et al. Development of
Peptidomimetic Inhibitors of the ERG Gene Fusion Product in Prostate Cancer. Cancer Cell.
12 2017;31(6):844‑7.
248. Hara Y, Shiba N, Ohki K, Tabuchi K, Yamato G, Park M-J, et al. Prognostic impact of
specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down
syndrome. Genes Chromosomes Cancer. 2017;56(5):394‑404.
249. de Rooij JDE, Branstetter C, Ma J, Li Y, Walsh MP, Cheng J, et al. Pediatric non-Down
syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets
with varying outcomes. Nat Genet. mars 2017;49(3):451‑6.
250. Drexler HG. Guide to Leukemia-Lymphoma Cell Lines. Braunschweig, Germany:
German Collection of Microorganisms and Cell Cultures; 2005.
251. Walters DK, Mercher T, Gu T-L, O’Hare T, Tyner JW, Loriaux M, et al. Activating
alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. juill 2006;10(1):65‑75.
252. Saida S, Watanabe K, Sato-Otsubo A, Terui K, Yoshida K, Okuno Y, et al. Clonal
selection in xenografted TAM recapitulates the evolutionary process of myeloid leukemia in
Down syndrome. Blood. 23 mai 2013;121(21):4377‑87.
253. Chou ST, Byrska-Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB, et al. Trisomy
21-associated defects in human primitive hematopoiesis revealed through induced
pluripotent stem cells. Proc Natl Acad Sci U S A. 23 oct 2012;109(43):17573‑8.
254. Alford KA, Slender A, Vanes L, Li Z, Fisher EMC, Nizetic D, et al. Perturbed
hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 8 avr
2010;115(14):2928‑37.
255. Byrska-Bishop M, VanDorn D, Campbell AE, Betensky M, Arca PR, Yao Y, et al.
Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. J Clin
Invest. 2 mars 2015;125(3):993‑1005.
256. Carmichael CL, Metcalf D, Henley KJ, Kruse EA, Di Rago L, Mifsud S, et al.
Hematopoietic overexpression of the transcription factor Erg induces lymphoid and
erythro-megakaryocytic leukemia. Proc Natl Acad Sci U S A. 18 sept
2012;109(38):15437‑42.

236
257. Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al.
Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic
leukemia in a murine model of Down syndrome. J Clin Invest. mars 2012;122(3):948‑62.
258. Thiollier C, Lopez CK, Gerby B, Ignacimouttou C, Poglio S, Duffourd Y, et al.
Characterization of novel genomic alterations and therapeutic approaches using acute
megakaryoblastic leukemia xenograft models. J Exp Med. 22 oct 2012;209(11):2017‑31.
259. Ayllón V, Vogel-González M, González-Pozas F, Domingo-Reinés J, Montes R, Morales-
Cacho L, et al. New hPSC-based human models to study pediatric Acute Megakaryoblastic
Leukemia harboring the fusion oncogene RBM15-MKL1. Stem Cell Res. 2017;19:1‑5.
260. Bernard OA, Mercher T. Activation de la voie Notch par OTT-MAL dans les leucémies
aiguës mégacaryoblastiques. 2009;Med Sci (Paris)(25):676–678.
261. Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An
Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype
of pediatric acute megakaryoblastic leukemia. Cancer Cell. 13 nov 2012;22(5):683‑97.
262. Dang J, Nance S, Ma J, Cheng J, Walsh MP, Vogel P, et al. AMKL chimeric transcription
factors are potent inducers of leukemia. Leukemia. oct 2017;31(10):2228‑34.
263. Cardin S, Laramee L, MacRae T, Chagraoui J, Sauvageau G, Humphries RK, et al.
Modeling of Pediatric Acute Megakaryoblastic Leukemia Using Cord Blood Stem/Progenitor
Cells. Blood. 2 déc 2016;128(22):1535‑1535.
264. Wenge DV, Felipe-Fumero E, Angenendt L, Schliemann C, Schmidt E, Schmidt LH, et
al. MN1-Fli1 oncofusion transforms murine hematopoietic progenitor cells into acute
megakaryoblastic leukemia cells. Oncogenesis. 21 déc 2015;4:e179.
265. Mercher T, Wernig G, Moore SA, Levine RL, Gu T-L, Fröhling S, et al. JAK2T875N is a
novel activating mutation that results in myeloproliferative disease with features of
megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 15 oct
2006;108(8):2770‑9.

237

Etude des mécanismes cellulaires et moléculaires des leucémies à mauvais pronostic présentant
la fusion ETO2-GLIS2
Mots clés : LAM-M7, pédiatrique, facteur de transcription, contexte développemental, contexte cellulaire
Résumé : La fusion ETO2-GLIS2, récemment
découverte dans les leucémies aiguës
mégacaryoblastiques (LAM-M7) et d’autres sous-
types de leucémies aiguës myéloïdes (LAM), est
associée à un mauvais pronostic.
L’objectif de ce travail a été d’étudier les
mécanismes mis en jeu par ETO2-GLIS2 dans les
cellules leucémiques ainsi que son association
spécifique avec les leucémies pédiatriques. Par des
analyses moléculaires, nous avons montré que
ETO2-GLIS2 se fixe à l’ADN via la partie GLIS2
ainsi que via ETO2 et ses partenaires (incluant
GATA, ETS, RUNX) entrainant ainsi une forte
dérégulation de l’expression de facteurs de
transcription incluant ERG et GATA1.
J’ai développé un modèle murin inductible de la
fusion ETO2-GLIS2 qui reproduit efficacement
les différentes hémopathies observées chez
l’Homme. Ce modèle a permis d’observer
l’influence du stade développemental (hématopoïèse
fœtale vs. adulte) et du type cellulaire (cellule souche
hématopoïétique vs. Progéniteur multipotent) sur le
phénotype et l’agressivité des leucémies. De plus, la
dérégulation transcriptionnelle imposée par ETO2-
GLIS2 est différente en fonction du contexte
cellulaire. Dans l’ensemble ces résultats indiquent
que la fusion ETO2-GLIS2 est suffisante pour
induire une leucémie dont le phénotype et
l’agressivité sont dépendants du contexte cellulaire
dans lequel l’oncogène est introduit. Ils indiquent
également que les changements cellulaires et
moléculaires au cours du développement sont à
l’origine de la forte prévalence des LAM-M7 chez
les enfants.
Molecular and cellular mechanisms of ETO2-GLIS2 pediatric leukemia
Keywords : AMKL, pediatric, transcriptional factor, developmental stage, cellular context
Abstract : The ETO2-GLIS2 fusion, recently
discovered in acute megakaryoblastic leukemia
(AMKL) and other subtypes of acute myeloid
leukemia (AML), is associated with poor prognosis.
The aim of this work was to study the mechanisms
involved in ETO2-GLIS2 leukemic cells and its
specific association with pediatric leukemia.
Molecular analyses have shown that ETO2-GLIS2
binds to DNA via the GLIS2 moiety as well as via
ETO2 and its partners (including GATA, ETS,
RUNX), leading to a strong dysregulation of the
expression of transcription factors including ERG
and GATA1.
I have developed an inducible murine model of
ETO2-GLIS2 fusion that efficiently reproduces the
different hematopoietic malignancies
observed in humans. We were able to observe the
influence of developmental stage (fetal vs. Adulte
hematopoiesis) and cell type (hematopoietic stem
cell vs. Multipotant progenitor) on the phenotype
and aggressiveness of leukemia. In addition, the
transcriptional dysregulation imposed by ETO2-
GLIS2 was different according to the cellular
context.
Overall, these results indicate that ETO2-GLIS2
fusion is sufficient to induce leukemia whose
phenotype and aggressiveness are dependent on the
cellular context in which the oncogene is expressed.
They also indicate that cellular and molecular
changes during development are responsible for the
high prevalence of AMKL in children.
Related Documents











