Sheryl J. Hays, Bradley W. Caprathe, John L. Gilmore, Nilam Amin, Mark R. Emmerling, Walter Michael, Ravi Nadimpalli, Rathna Nath, Kadee J. Raser, Daniel Stafford, Desiree Watson, Kevin Wang, and Juan C. Jaen Departments of Chemistry and Neuroscience Therapeutics, Parke-Davis Pharmaceutical Research, Division of Warner-Lambert Company, 2800 Plymouth Road, Ann Arbor, Michigan 48105 JOURNAL OF MEDICINAL CHEMISTRY@ Reprinted from Volume 41, Number 7, Pages 1060-1067

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Sheryl J. Hays, Bradley W. Caprathe, John L. Gilmore,Nilam Amin, Mark R. Emmerling, Walter Michael,
Ravi Nadimpalli, Rathna Nath, Kadee J. Raser, Daniel Stafford,Desiree Watson, Kevin Wang, and Juan C. Jaen
Departments of Chemistry and Neuroscience Therapeutics,Parke-Davis Pharmaceutical Research, Division of Warner-Lambert
Company, 2800 Plymouth Road, Ann Arbor, Michigan 48105
JOURNAL OF
MEDICINAL
CHEMISTRY@Reprinted from
Volume 41, Number 7, Pages 1060-1067

1060 J. Med. Chem. 1998, 41, 1060-1067
2-Amino-4H-3,l-benzoxazin-4-ones as Inhibitors of Clr Serine Protease
Sheryl J. Hays,*.* Bradley W. Caprathe,* John L. Gilmore,* Nilam Amin,t Mark R. Emmerling,t Walter Michael,tRavi Nadimpalli,t Rathna Nath,t Kadee J. Raser,t Daniel Stafford,t Desiree Watson,t Kevin Wang,t andJuan C. Jaen*
Departments of Chemistry and Neuroscience Therapeutics, Parke-Davis Pharmaceutical Research, Division of Wamer-LambertCompany, 2800 Plymouth Road, Ann Arbor, Michigan 48105
Received June 13, 1997
A series of2-amino-4H-3,l-benzoxazin-4-ones have been synthesized and evaluated as inhibitorsof the complement enzyme Clr. Clr is a serine protease at the beginning of the complementcascade, and complement activation by f3-amyloid may represent a major contributing pathwayto the neuropathology of Alzheimer's disease. Compounds such as 7-chloro-2-[(2-iodophenyl)-amino]benz[d] [l,3]oxazin-4-one (32) and 7-methyl-2-[(2-iodophenyl)amino]benz[d] [l,3]oxazin-4-one (37) show improved potency compared to the reference compound FUT-l75. Many ofthese active compounds also possess increased selectivity for Clr compared to trypsin andenhanced hydrolytic stability relative to 2-(2-iodophenyl)-4H-3,l-benzoxazin-4-one (I).
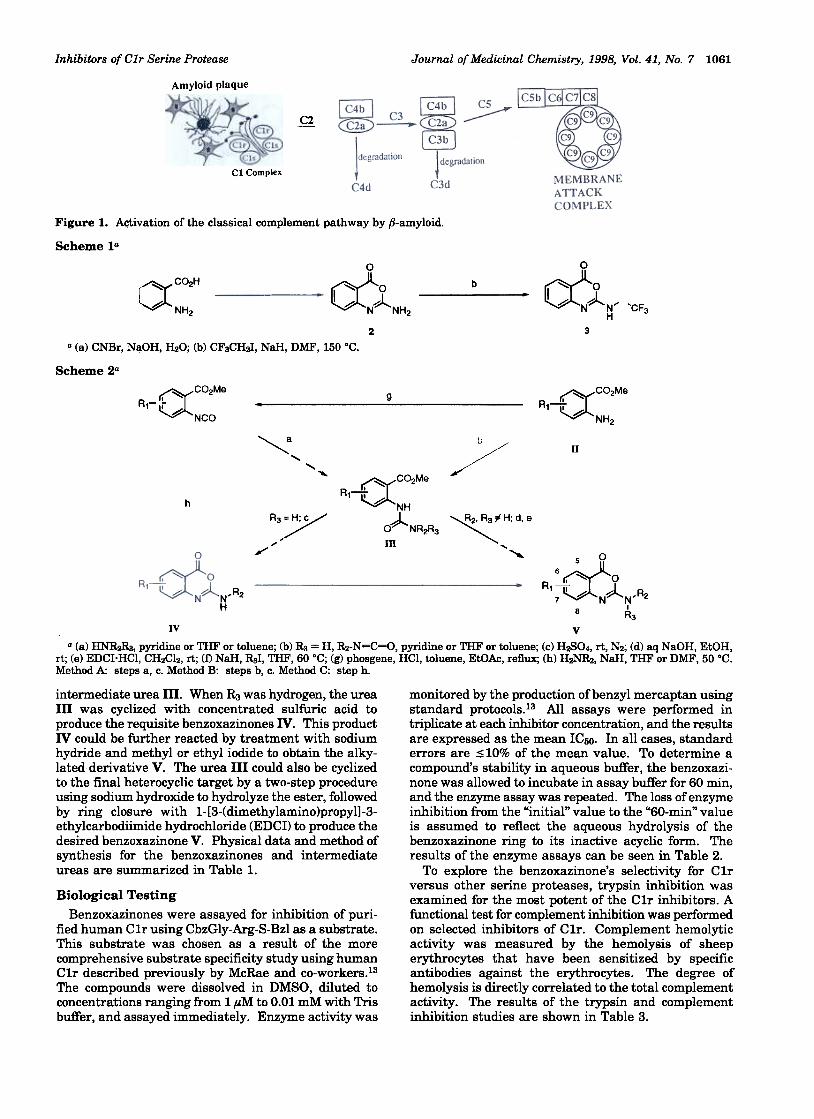
Introduction C1r is a trypsin-like serine protease that is part ofThe complement system is an important part of the the C1 complex (one C1q, two C1r, and two C1s
host's immune response to foreign antigens"1 The molecules) and is located at the beginning of theclassical complement pathway involves the sequential classical complement cascade" C1r is responsible for theactivation of nine major proteins or protein complexes proteolytic cleavage of C1s, the next protein in thedesignated as C1-C9 (Figure 1)" This activation re- pathway.9 By selecting a target that is early in thequires the interaction of the C1q subunit ofC1 with an cascade, it should be possible to prevent many of theantigen-antibody complex. Following activation, a amplification events that occur once the cascade is fullyhighly regulated cascade of events occurs in which actIvated.discrete interactions of soluble proteins catalyze sub- 6-Amidino-2-naphthyl-4-guanidinobenzoate (FUT-sequent steps. The final step in the pathway is the 175)10 and 2-(2-iodophenyl)-4H-3,1-benzoxazin-4-one (1)11formation ofa membrane attack complex which creates have been shown to act as inhibitors ofC1r" FUT-175a pore in any accessible cellular membrane causing has demonstrated little selectivity for C1r over otherlethal and sublethal damage. The complement cascade serine proteases, while compound 1 suffers from a lackcan additionally increase vascular permeability and of chemical stability. A series of 2-amino-benzo[d][3,1]-promote phagocytic recognition by chemotaxis. oxazin-4-ones have been targeted to overcome these
Alzheimer's disease (AD) is the most common degen- difficulties. The synthesis and C1r inhibition for thiserative dementia affecting primarily the elderly popula- series of compounds are reported here"tion. The disease is characterized by the decline ofmultiple cognitive functions and a progressive loss of d o
neurons in the central nervous system" Deposition of NH ~ O OfJ-amyloid peptide in the form of plaques is a classical Jl I A
~ ~feature of the disease, and these deposits have recently H2N ~ ~ I I ~ ~o Ibeen shown to activate the classical complement system ~ I A N6 ~(Figure 1).2,3 This activation by fJ-amyloid may repre- ~ I Asent a major contributing event to the neuropathology FUT-175 1of AD.4-6 These initial observations which suggest the HN NH2existence of an inflammatory component in the neuro-degeneration observed in AD have been extended to the Chemistryclinic. A small clinical study using the nonsteroidal 2 Am " b [d] [1 3] " 4 (2) h " dt ..
nil d " d h " " di -mo enz , oxazm- -one was synt es1Zean 11 ammatory rug m omet acm m cated that fr thr " ti " d d b " d d"
d h ." "fi " om an anl c acl an cyanogen roml e as e-m omet acm SlgnI cantly slowed the progressIon of the "b d . 1 ( S h 1) 12
Tr t t f 2d " 7
Add " t " 1 t di " th .." nil scn e preVIous y see c eme .ea men 0Isease. 1 10na s u es Wl vanous antIl am- "th d . h d " d d t "fl th 1 " d "d fli d dd h fi ." ." " 8 Wl so lum y n e an n uoroe y 10 1 e a or e
matory rugs ave con rmed this mltIal observatIon. th alk 1 t d d t 3S " fi " nh "b " t " f h 1 e y a e pro uc .pecl c 1 1 1 Ion 0 t e comp ement system may Th th fth " " t fth b "
h " e syn eses 0 e maJon yo e enzoxazmones
represent anot er approach to modulate the neurom- h " S h 2 An . t 1 b t " t d.are s own m c eme .appropna e y su s 1 ute
flammatory component of AD. WhIle there are many th 1 th " I t I t t d "th "
t" 1 ti " me y an ranI a e was rea e W1 an Isocyana epotentIa targets or the preventIon of complement t d th III . d t t d " Id" " h " ...0 pro uce e urea m mo era e 0 goo YIe "
actIvatIon, we ave chosen to explore Clr inhIbItIon. Alt t " 1 th th 1 th " I t . t nema Ive y, e me y an ram a e Isocyana e
* Department of Chemistry. (purchased or prepared from the methyl anthranilate It Department of Neuroscience Therapeutics" and phosgene) was reacted with an amine to form the
SO022-2623(97)00394-4 CCC: $15"00 @ 1998 American Chemical SocietyPublished on Web 03/05/1998
Inhibitors of Clr Serine Protease Journal of Medicinal Chemistry, 1998, Vol. 41, No.71061
Amyloid plaque
C2
~,.
Cl Complex
Figure I. A<;tivation of the classical complement pathway by .8-amyloid.
Scheme la
o
., ,.Jl.o
~N~
.CO2H
::;cb
o
~o
~N) NH2
2
'N"'H
'CF3NH2
3
a (a) CNBr, NaOH, H20; (b) CF3CH2I, NaB, DMF, 150 °C.
Scheme 2a
RI- :t:::;(CO2MeNCO R1-CCcO2MeNH2
9
" II
~~
/, o:CO2Me RI-t
~ NH
,)...O NR2R3
III
h
~ i H; d, e
R3// ,
~
,/ "
R1-
'N'R2H
5 O
6.;,..)J..O
~~~I~N'R27 N I8 R3
IV Va (a) ~, pyridine or THF or toluene; (b) Rs = H, R2-N=C=O, pyridine or THF or toluene; (c) H2S04, rt, N2; (d) aq NaOH, EtOH,
rt; (e) EDCI.HC1, CH2C12, rt; (f) NaB, R3I, THF, 60 °C; (g) phosgene, HC1, toluene, EtOAc, reflux; (h) H~, NaB, THF or DMF, 50 °C.Method A: steps a, c. Method B: steps b, c. Method C: step h.
intermediate urea ill. When R3 was hydrogen, the ureaill was cyclized with concentrated sulfuric acid toproduce the requisite benzoxazinones IV. This productIV could be further reacted by treatment with sodiumhydride and methyl or ethyl iodide to obtain the alky-latedderivative V. The urea ill could also be cyclizedto the final heterocyclic target by a two-step procedureusing sodium hydroxide to hydrolyze the ester, followedby ring closure with 1-[3-(dimethylamino)proPYI]-3-ethylcarbodiimide hydrochloride (EDCI) to produce thedesired benzoxazinone V. Physical data and method ofsynthesis for the benzoxazinones and intermediateureas are summarized in Table 1.
monitored by the production of benzyl mercaptan usingstandard protocols.13 All assays were performed intriplicate at each inhibitor concentration, and the resultsare expressed as the mean IC50. In all cases, standarderrors are :$10% of the mean value. To determine acompound's stability in aqueous buffer, the benzoxazi-none was allowed to incubate in assay buffer for 60 min,and the enzyme assay was repeated. The loss of enzymeinhibition from the "initial" value to the "60-min" valueis assumed to reflect the aqueous hydrolysis of thebenzoxazinone ring to its inactive acyclic form. Theresults of the enzyme assays can be seen in Table 2.
To explore the benzoxazinone's selectivity for C1rversus other serine proteases, trypsin inhibition wasexamined for the most potent of the C1r inhibitors. Afunctional test for complement inhibition was performedon selected inhibitors of C1r. Complement hemolyticactivity was measured by the hemolysis of sheeperythrocytes that have been sensitized by specificantibodies against the erythrocytes. The degree ofhemolysis is directly correlated to the total complementactivity. The results of the trypsin and complementinhibition studies are shown in Table 3.
Biological TestingBenzoxazinones were assayed for inhibition of puri-
fied human C1r using CbzGly-Arg-S-Bzl as a substrate.This substrate was chosen as a result of the morecomprehensive substrate specificity study using humanC1r described previously by McRae and co-workers.13The compounds were dissolved in DMSO, diluted toconcentrations ranging from 1 p.M to 0.01 mM with Trisbuffer, and assayed immediately. Enzyme activity was
1062 Journal of Medicinal Chemistry, 1998, Vol. 41, No.7 Hays et al.
Table I. Synthetic Data
R1-0:CO 2Me
J:O NR2R3
Dl
s O
6 A)J... o
RI--!..~ J N,R27 N ,
8 R3
2-amino-4H-3,1-benzoxazin-4-one product (V)
Ra
H HCF3CH2 HMe H(CH3)2CH HC&H5 H2-I-C&H4 H3-I-C&H4 H4-I-C&H4 H2-I-C6fu Me2-I-C&H4 Et2-I-C6fuCH2 H2-I-C6fuCH2CH2 H2-F-C&H4 H2-CI-C&H4 H2-Br-C&H4 H2-Me-C&H4 H2-(OCF3)-C&H4 H2-(SMe)-C&H4 H2,6-diCI-C&H3 H2,5-diCI-C&H3 H2,6-diMe-C&H3 H3-thiophenylmethyl H2-(1,3,4-thiadiazolyl) H2-(1,3,4-triazolyl) H2-thiazolidinyl H2-thiazolyl H3-tetrahydrothiophenyl H2-I-C&H4 H2-I-C&H4 H2-I-C6fu H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-I-C&H4 H2-CI-C&H4 H2-CI-C&H4 H2,6-diCI-C&H3 H2-CI-C&H4 H2-CI-C&H4 H2,6-diCI-C&H3 H
Rl R2 method (yield)
a(92%)b (11%)A (80%)Ba (35%)CBb,d (86%)A (81%)A (67%)Abb (24%)A (92%)Ab (94%)A (99%)A (79%)A (91%)C (34%)A (60%)C (11%)A (55%)C (62%)B (84%)C (57%)A(31%)C (15%)C (24%)C (18%)C (71%)B (46%)B (51%)B (63%)B (77%)B (34%)B (55%)B (56%)C (4%)B (79%)B (40%)C (15%)B (59%)Bd (44%)B (72%)B (58%)B (37%)B (71%)B (63%)Bd (64%)B (43%)B (47%)Bd (53%)B (69%)B (56%)B (15%)
mp,OC
corresponding urea startingmaterial (ID)
no. mp, °C (yield)no.
2
~
54555657585960
120-121 (63%)not taken (52%)141-142 (85%)162-164 (78%)167-168 (60%)190-192 (71%)144-148 (80%)
45~
91011121314151617181920212223242526272829303132333435363738394041424344454647484950515253
61626364656667
169-170 (60%)132-135 (86%)178-179 (56%)179-180 (25%)not taken (27%)201-202 (92%)140-142 (75%)
686970
205-206 (33%)219-222 (72%)237-239 (94%)
71 not taken (95%)
72737475767778
not taken (70%)183-185 (80%)226-227 (53%)187-188 (25%)212-214 (33%)220-221 (30%)185-187 (53%)
189-190 (64%)not taken (46%)
7980
8182838485868788899091929394
178-180 (70%)178-181 (87%)186-187 (29%)241-243 (19%)177-178 (30%)93-95 (84%)194-198 (79%)210-211 (36%)207-209 (27%)198-200 (82%)180-183 (40%)183-187 (43%)186-188 (72%)160-162 (74%)
HHH5-MeHHHHHHHHHHHHHHHHHHHHHHH7-NMe25-Cl6-Cl7-Cl6,7-diCl5,7-diCl5-Me6-Me7-Me6-0Me7-0Me6,7-diOMe7-NO27-F6,7-diF7-CF37-(CH3)2CH7-cyclohexyl6,7-CH=CH-CH=CH-7-Cl6,7-diCl7-Cl7-Me7-NO27-CF3
a Reference 12. b Procedure is described in the Experimental Section. c Reference 14. d Ethyl ester of the anthranilic acid
(IC5o = 16.7 .uM) are potent inhibitors of C1r asdescribed previously but are hydrolytically labile.llReplacement of the aryl substituent by primary oraliphatic amines has been used as a successful strategyto inhibit the serine protease human elastase andimprove benzoxazinone stability12 but was unsuccessfulfor inhibiting C1r (compounds 2-6, IC5o > 62.5 .uM).Additionally, benzyl- and phenethylamine derivatives12 and 13 were also inactive.
Results and DiscussionActive benzoxazinones have been shown to exhibit a
time-dependent inhibition of Clr. This is believed tooccur via a nucleophilic attack of the active site serineof the enzyme on the carbonyl of the heterocycle toproduce acyl enzyme intermediates. Substitution at the2-position of the benzoxazinone is a sensitive parameterthat affects enzyme activity as well as chemical stability .Directly bonded aryl derivatives such as compound 1

Journal of Medicinal Chemistry, 1998, Vol. 41, No.71063[nhibitors of Clr Serine Protease
Table 2. Clr Inhibition by 2-Amino-4H-3,l-benzoxazinones
O
R, -0:" J..N- R,N I
R3
Clr inhibition (lC5o, IlM)
FUT-1751234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253
HHHHHHHHMeEtHHHHHHHHHHHHHHHH
HHHHHHHHHHHHHHHHHHHHHHHHH
14.7
16.6
>62.58.0
15.238
>62.5
0.92.0
HCF3CH2Me(CH3)2CHCsH52-I-CsH43-I-CsH44-I-CsH42-I-CsH42-I-CsH42-I-CsH4CH22-I-CsH4CH2CH22-F-CsH42-CI-CsH42-Br-C6~2-Me-CsH42-0CF3-CsH42-SMe-CsH42,6-diCI-C6H32,5-diCI-CsH32,6-diMe-CsH33-thiophenylmethyl2-(1,3,4-thiadiazolyl)2-(1,3,4-triazolyl)2-thiazolidinyl2-thiazolyl3- tetrah ydrothiophen y 12-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-I-CsH42-CI-CsH42-CI-CsH42,6-diCI-CsH42-CI-CsH42-CI-CsH42,6-diCI-CsH4
HHH5-MeHHHHHHHHHHHHHHHHHHHHHHH7-N(CH3)25-Cl6-Cl7-Cl6.7-diCl5.7-diCl5-Me6-Me7-Me6-0Me7-0Me6,7-diOMe7-NO27-F6.7-diF7-CF37-(CH3)2CH7-cyclohexyl6.7-CH=CH-CH=CH-7.Cl6.7-diCl7-Cl7-Me7-NO27-CF3
>62.5>62.5
0.9>62.5
1.5
>62.5
47
7.8
2.8
>62.5
4.0
16.60.8
>62.50.90.9
>62.54.2
Appropriately substituted 2-anilinobenzoxazinoneshave demonstrated good activity as inhibitors of C1r.While no inhibition was observed for the unsubstitutedaniline 6, derivatives which contain at least one orthosubstituent (compounds 7, 15, 16, 18, 20, and 21) havedemonstrated improved potency relative to the referenceagent FUT-175 (IC5o = 12 /lM).ll The best anilinosubstituents included the o-iodo (7, IC5o = l.4/lM) and2,6-dichloro (20, IC5o = 0.9 /lM) analogues, while them- and p-iodoanilino derivatives (compounds 8 and 9)
were inactive. Several ortho substituents which weresmall such as fluoro or electronically more neutral suchas methyl and thiomethyl analogues ( compounds 14, 17,and 19, respectively) produced poor enzyme inhibition.Derivatives which replaced the ortho-substituted anilinegroup with a heterocycle (compounds 23-28) demon-strated no enzyme inhibition. Changing R3 from hy-drogen to methyl produced an equipotent derivative (10,IC5o = 1.4 !.tM), while the slightly larger ethyl replace-ment abolished activity (11, IC5o > 62.5 !.tM).

1064 Journal of Medicinal Chemistry, 1998, Vol. 41, No.7 Hays et al.
FUT-1751101621374142444853
0.01715.34.5>100>100>100>100>100>100>10087.1
a Represents percent of control at 200 JlM. b ND, not determined.
conformation strain than the larger iodo and bromosubstituents, thereby increasing the compounds' aque-ous stability.
Phenyl substitution of the benzoxazinone's benzenering also profoundly influences the compounds' hydro-lytic stability. The presence of a chloro, methyl, orfluoro group in the 7 -position greatly stabilizes theheterocyclic ring structure to aqueous addition (com-pounds 32, 37, and 42), while substitution at the6-position has a deleterious effect (compounds 31, 33,43, and 47). It is difficult to determine the influence ofsubstitution at the 5-position since only one analogueexhibited enzymatic inhibition. This result (30, IC5o =14.5 f.tM) suggests that this substitution pattern nega-tively influences hydrolytic stability.
In an effort to maximize C1r inhibition and to increaseinhibitor stability, a series of 2-o-chloroanilino deriva-tives with electron-withdrawing groups in the 7 -positionwere synthesized. Several of these compounds substi-tuted with either a 7 -chloro ( 48, IC5o = 0.8 f.tM; 50, IC5o= 0.4 f.tM), 7-methyl (51, IC5o = 1.4 f.tM), or 7-trifluo-romethyl (53, IC5o = 1.5 f.tM) are among the most stableand potent benzoxazinones tested for C1r activity. The6,7-dichloro derivative 49 and 7-nitro analogue 52 arepotent C1r inhibitors but are relatively unstable inaqueous media.
The benzoxazinones with the most potent C1r inhibi-tory activity and hydrolytic stability were examined fortrypsin inhibition. This serves as a quick determinationof the selectivity of the compound as an inhibitor ofC1rversus other serine proteases. The reference compoundsFUT-175 and 1 were better or equipotent inhibitors oftrypsin compared to C1r, and it was one of the goals ofthe project to improve the compounds' selectivity. Whilethe N-methylated analogue 10 shows no selectivity, allthe other derivatives demonstrated increased inhibitionof C1r when compared to trypsin (see Table 3).
These same compounds were also examined in afunctional assay using erythrocyte hemolysis as afunctional end point of complement inhibition. FUT -175 showed good activity in this model with an IC5o of16.6 f.tM. The benzoxazinones, however, were margin-ally active at 200 f.tM (compound lor 10) or showed noactivity in this model. Several possible explanationsexist for this lack of functional activity. Although theseaminobenzoxazinones showed good aqueous stability,one of the components of this assay is human serumand these compounds may be less stable to enzymaticdegradation. Alternatively, the inhibitors may be tightlybinding to plasma proteins. In conclusion, while theinitial goals of enhanced C1r inhibition, improved serineprotease selectivity, and increased aqueous stabilityover the 2-arylbenzoxazinone series were achieved, thecompounds proved to be inactive in the functionalhemolysis assay. Additional series of benzoxazinonesare under investigation to address this problem.
Substitution of the benzoxazinone ring also greatlyaffects enzyme activity. Chloro or methyl substitutionof the 5-position (30, 5-chloro, IC5o = 14.5 p,M; 34, 5,7-dichloro, IC5o > 62.5 p,M; 35, 5-methyl, IC5o > 62.5 p,M)reduced or abolished enzyme activity. This result wasin contrast with the 5-substituted benzoxazinones pre-pared as human elastase inhibitors where 5-alkylsubstitution was highly advantageous for enzyme inhi-bition.12 Compounds that are substituted in the 6-posi-tion with a neutral or electron-donating group (36,6-methyl, IC5o > 62.5 p,M; 38, 6-methoxy , IC5o = 55 p,M;40, 6,7 -dimethoxy , IC5o = 47 p,M) showed little or noactivity, while electron-withdrawing groups in thisposition (31, 6-chloro, IC5o = 4.0 p,M; 33, 6,7-dichloro,IC5o = 0.5 p,M; 43, 6,7-difluoro, IC5o = 7.0 p,M) main-tained or improved initial activity. Electron-donatinggroups in the 7-position of the benzoxazinone ringshowed no activity (29, 7-dimethylamino; 39, 7-meth-oxy), while neutral (37, 7-methyl, IC5o = 0.8 p,M) orelectron-withdrawing (32, 7-chloro, IC5o = 0.7 p,M; 41,7-nitro, IC5o = 0.8 p,M; 42, 7-fluoro, IC5o = 1.7 p,M; 44,7-trifluoromethyl, IC5o = O.4p,M) groups demonstratedimproved activity relative to the unsubstituted parent7. Neutral substituents in the 7-position that arerelatively bulky compared to the methyl group (45,7-isopropyl; 46, 7-cyclohexyl) demonstrated no appre-ciable enzyme inhibition.
Stability of the compounds was assessed using theenzyme assay as described above. The advantage of thistechnique is that it provides a fast and easy method todetermine a derivative's aqueous stability with thedisadvantage that a compound must be biologicallyactive for a "60-min" value to be obtained. Attempts todetermine the half-life of a compound in aqueous bufferusing ultraviolet spectroscopy were unsuccessful due tothe multiple chromophores in the molecule. Althoughaliphatic amino substitution has been reported toincrease benzoxazinone stability, anilino substitutiondoes not necessarily improve compound stability relativeto the 2-iodophenyl derivative 1. Comparison of 1 withthe 2-substituted iodoaniline derivative 7 shows acomparable decrease in enzyme inhibition after the 60-min incubation. Additionally, methyl substitution of theanilino nitrogen (compound 10) further destabilizes thebenzoxazinone ring structure. While these o-iodo de-rivatives do not improve stability, the o-chloro analoguesdo show increased hydrolytic stability with little or nobenzoxazinone breakdown after 60 min (compounds 15,20, and 21). The smaller chloro group may provide less
Experimental Section
Chemistry. All melting points were determined on aMELT-TEMP II capillary melting point apparatus and areuncorrected. Infrared (IR) spectra were determined as KBrpellets on a Mattson Cygnus 100 or a Nicolet MXl instrument.Proton magnetic resonance (NMR) was recorded on either aVarian XL-300 or a Broker AM250 spectrometer; chemicalshifts are reported in O units relative to internal tetrameth-

Journal of Medicinal Chemistry, 1998, Vol. 41, No.71065
of 1 M HCl in ether solution) in 500 mL of toluene was addeddropwise a 12.5% solution of phosgene in toluene (26 mL, 32.8mmol) at room temperature. The mixture was refluxed for 1h, cooled to room temperature, treated with another portionof phosgene in toluene solution (26 mL), and refluxed for anadditional 12 h. The cloudy solution was concentrated,suspended into 250 mL ofhexanes, and filtered through a padof Celite. The filtrate was concentrated and distilled (bp 50-60 °C, 0.50-0.25 mmHg) to give 5.0 9 (78%) of 2-iodophenylisocyanate as a colorless liquid: lH NMR (CDCI3) O 6.88-6.92(m, 1H), 7.13 (dd, 1H, J = 1.4, 8.0 Hz), 7.27-7.31 (m, 1H),7.79 (dd, 1H, J = 1.4, 8.0 Hz). Anal. Calcd for C7~INO: C,34.32; H, 1.65; N, 5.72. Found: C, 34.38; H, 1.68; N, 5.67.
A solution of 2-iodophenyl isocyanate (3.36 g, 13.7 mmol)and ethyl 2-aminobenzoate (5.1 mL, 34.5 mmol) in 250 mL ofTHF was stirred at room temperature for 24 h. The solutionwas concentrated and then partitioned between ethyl acetateand 10% HCl. The ethyl acetate extract was washed withbrine, dried (MgSO4), and concentrated to an oily white solid.The sample was suspended in ether and the insoluble whitesolid that resulted from filtration yielded 4.38 9 (78%) of ethyl2-[3-(2-iodophenyl)ureido]benzoate (57): mp 162-164 °C; lHNMR (CDCI3) O 1.41 (t, 3H, J = 7.2 Hz), 4.37 (q, 2H, J = 7.2Hz), 6.82-6.87 (m, 2H), 7.01-7.05 (m, 1H), 7.34-7.38 (m, 1H),7.51-7.55 (m, 1H), 7.80 (dd, 1H, J = 1.4,8.0 Hz), 7.99 (dd,1H, J = 1.7,8.2 Hz), 8.03 (dd, 1H, J = 1.7,8.0 Hz), 8.51 (dd,1H, J = 1.0, 8.4 Hz), 10.76 (s, 1H). Anal. Calcd for Cl6Hl5-IN203: C, 46.85; H, 3.69; N, 6.83. Found: C, 46.75; H, 3.62;N,6.56.
A solution of ethyl 2-[3-(2-iodophenyl)ureido]benzoate (57;0.50 g, 1.22 mmol) in concentrated sulfuric acid (10 mL, 181.0mmol) was stirred at 50 °C for 1 h. The solution was cooledto room temperature and poured into ice-water. Afterneutralization with saturated NaHCO3 solution, the productwas collected by filtration. The solid was washed with waterand vacuum-dried to yield 0.38 9 (86%) of 7 as an off-whitesolid: mp 140-143 °C; FT-IR (KBr) 1763 cm-l; lH NMR(CDCI3) O 6.85-6.90 (m, 1H), 7.26-7.32 (m, 2H), 7.41-7.45(m, 1H), 7.68-7.73 (m, 1H), 7.82 (dd, 1H, J = 1.4,8.0 Hz),8.11 (dd, 1H, J = 1.6,7.8 Hz), 8.44 (dd, 1H, J = 1.6,8.1 Hz).Anal. Calcd for Cl4HgIN202: C, 46.18; H, 2.49; N, 7.69.Found: C, 46.13; H, 2.43; N, 7.51.
2-[ N -(2- Iodophenyl)-N -methylamino ]benz [ d] [ 1,3 ]oxazin -4-one (10). A solution of2-iodoaniline (6.0 g, 27.4 mmol) andethyl formate (11.1 mL, 137.4 mmol) in 200 mL ofTHF wasadded dropwise to a stirring suspension of sodium hydride (1.4g, 34.2 mmol, 60% dispersion in mineral oil washed twice with50 mL of hexanes) in 250 mL of THF at room temperature.The mixture was stirred for 24 h, cooled in an ice bath, andquenched by dropwise addition of water. The sample wasconcentrated (to remove the THF) and then partitionedbetween ethyl acetate and saturated KH2PO4 solution. Theethyl acetate extract was washed with brine, dried (MgSO4),filtered, and concentrated to give 6.3 9 (93%) ofN-(2-iodophen-yl)formamide as an off-white solid: mp 118-119 °C; lH NMR(DMSO-d6) O 6.93-7.02 (m, 1H), 7.33-7 .41 (m, 1H), 7. 78 (dd,1H, J = 1.1,8.1 Hz), 7.88 (dd, 1H, J = 1.1,7.8 Hz), 8.34 (s,1H), 9.54 (bs, 1H). Anal. Calcd for C7H6INO: C, 34.03; H,2.45; N, 5.67. Found: C, 34.16; H, 2.47; N, 5.67.
A suspension of sodium borohydride (3.6 g, 95.7 mmol) andN-(2-iodophenyl)formamide (6.3 g, 25.5 mmol) in 500 mL ofTHF was treated dropwise with boron trifluoride etherate (15.7mL, 127.6 mmol) at 0 °C. The mixture was allowed to warmto room temperature overnight. The mixture was cooled to 0°C and carefully quenched by dropwise addition ofwater (125mL). The THF was removed under vacuum, and the residuewas basified with concentrated ammonium hydroxide to a pHof 9-10 and then extracted with ethyl acetate. The organicextract was washed with brine, dried (MgSO4), filtered, andconcentrated to a tan oil. The residue was treated with HCland crystallized from ethanol to give 5.1 9 (74%) of N-methyl-2-iodoaniline hydrochloride as a white solid: mp 154-155 °Cdec; lH NMR (DMSO-d6) O 2.77 (s, 3H), 6.48-6.52 (m, 1H),6.70 (d, 1H, J= 8.0 Hz), 7.24-7.28 (m, 1H), 7.66 (dd, 1H, J =
Inhibitors of Clr Serine Protease
ylsilane. All mass spectra were obtained on a Finnigan 4500GCMS or a VG analytical 7070 ElF spectrometer. Elementalanalyses were performed on a CEC model 240 elementalanalyzer and are within 0.4% of the theoretical values unlessotherwise indicated. Medium-pressure liquid chromatographyutilized E. Merck silica gel, 230-400 mesh. All reactions wererun under N2, unless indicated otherwise. All analytical(C,H,N) and spectroscopic (lH NMR, IR, MS) data are inagreement with the proposed structures.
Method A. 2-[[2.(2-Iodophenyl)ethyl]amino]benz[d]-[1,3]oxazin-4-one (13). Thionyl chloride (13.9 mL, 190.6mmol) was added dropwise toa stirring solution of2-iodophen-ylacetic acid (5.0 g, 19.1 mmol) in 500 mL of methylenechloride. The solution was refluxed for 4 h, cooled, andconcentrated to a light-brown oil. Cold concentrated am-monium hydroxide (100 mL) was carefully added dropwise.Solid formed, and the mixture was filtered, washed with water,and vacuum-dried to yield 4.18 9 (84%) of 2-(2-iodophenyl)-acetamide as a tan solid: mp 180-182 °C; lH NMR (DMSO-d6) tJ 3.56 (s, 2H), 6.96-7.00 (m, 2H), 7.30-7.36 (m, 2H), 7,43(bs, IH), 7.82 (dd, IH, J = 0.8, 8.0 Hz). Anal. Calcd for C8H8-INO: C, 36.81; H, 3.09; N, 5.37. Found: C, 36.90; H, 3.09; N,5.22.
Boron trifluoride etherate (9.2 mL, 74.8 mmol) was addeddropwise to a stirring suspension of2-(2-iodophenyl)acetamide(3.9 g, 15.0 mmol) and sodium borohydride (2.1 g, 56.0 mmol)in 500 mL of THF maintained at 0 °C. The mixture wasallowed to warm to room temperature overnight. The mixturewas cooled to 0 °C, carefully quenched by dropwise additionof water (50 mL), concentrated to remove the THF, basifiedwith concentrated ammonium hydroxide to a pH of 9-10, andthen extracted into ethyl acetate. The ethyl acetate extractwas washed with brine, dried (MgSO4), filtered, and concen-trated to a hygroscopic white solid. The HCl salt was madeand crystallized from ethanol to give 3.1 9 (73%) of 2-(2-iodophenyl)ethylamine hydrochloride as a white solid: mp251-255 °C; lH NMR (DMSO-d6) tJ 2.96-3.04 (m, 4H), 7.01-7.05 (m, IH), 7.34-7.41 (m, 2H), 7.87 (dd, IH, J = 0.8,7.8Hz), 8.24 (bs, 3H). Anal. Calcd for C8HlolN.HCl: C,33.89;H, 3.91; N, 4.94. Found: C, 34.05; H, 3.83; N, 4.88.
A solution of2-carbomethoxyphenyl isocyanate (0.55 g, 3.10mmol), 2-(2-iodophenyl)ethylamine hydrochloride (0.80 g, 2.82mmol), and N ,N-diisopropylethylamine (0.75 mL, 4.30 mmol)in 100 mL of THF was stirred at room temperature for 24 h.The solution was concentrated and then partitioned betweenethyl acetate and 10% HCl. The ethyl acetate extract waswashed with brine, dried (MgSO4), and concentrated. Theresidue was chromatographed (silica gel, 20% ethyl acetatein hexanes) to yield 1.02 9 (86%) ofmethyl2-[3-[2-(2-iodophen-yl)ethyl]ureido]benzoate (62) as a white solid: mp 132-135°C; lH NMR (DMSO-d6) tJ 2.88 (t, 2H, J = 7.4 Hz), 3.28-3.30(m, 2H), 3.86 (s, 3H), 6.96-7.01 (m, 2H), 7.31-7 .38 (m, 2H),7.48-7.53 (m, IH), 7.66 (bs, IH), 7.84 (dd, IH, J = 0.8,7.8Hz), 7.89 (dd, IH, J = 1.6,8.1 Hz), 8.36 (dd, IH, J = 0.8,8.6Hz), 9.75 (bs, IH). Anal. Calcd for Cl7Hl7IN203: C, 48.13; H,4.04; N, 6.60. Found: C, 48.16; H, 4.09; N, 6.64.
Concentrated sulfuric acid (3.0 mL, 54.3 mmol) was addedto 62 (0.60 g, 1.41 mmol), and the solid slowly dissolved. Thesample was stirred at room temperature for 1.5 h and thencarefully poured into a vigorously stirred mixture of sodiumbicarbonate (14.0 g, 166.6 mmol, ca. 3 equiv vs H2S04 used)in water (250 mL) and ethyl acetate (250 mL). The ethylacetate extract was washed with brine, dried (MgSO4), filtered,and concentrated to afford 0.52 g (94%) of the title compoundas a white solid: mp 151-153 °C; FT-IR (KBr) 3298,1746,1632, 1476 cm-l; lH NMR (CDCls) tJ 3.09-3.13 (m, 2H), 3.68-3.73 (m, 2H), 4.97 (bs, IH), 6.91-6.95 (m, IH), 7.16-7.24 (m,IH), 7.24-7.32 (m, 3H), 7.62-7.66 (m, IH), 7.83 (d, IH, J =8,0 Hz), 8.02 (dd, IH, J = 1.5, 7.8 Hz). Anal. Calcd for Cl~l3-IN2O2: C, 49.00; H, 3.34; N, 7.14. Found: C, 48.93; H, 3.25;N, 7.07.
Method B. 2.[(2.Iodophenyl)amino]benz[d][1,3]oxazin-4-one (7). To a suspension of 2-iodoaniline hydrochloride(prepared from 5.7 g, 26.0 mmol, of 2-iodoaniline and 33 mL

1066 Journal of Medicinal Chemistry, 1998, Vol. 41, No.7
1.4, 7.7 Hz), 7.80 (bs, 2H). Anal. Calcd for C7HslN .HCl: C,31.20; H, 3.37; N, 5.20. Found: C, 30.90; H, 3.20; N, 5.17.
A solution of2-carbomethoxyphenyl isocyanate (0.93 g, 5.25mmol), N-methyl-2-iodoaniline hydrochloride (1.56 g, 5.79mmol), and N,N-diisopropylethylamine (1.1 mL, 6.32 mmol)in 100 mL of toluene was refluxed for 36 h. The solution wasconcentrated and then partitioned between chloroform andsaturated KH2PO4 solution. The chloroform extract waswashed with brine, dried (MgSO4), and concentrated. Theresidue was chromatographed (silica gel, CH2CI2) to yield 1.189 (55%) ofmethyI2-[3-(2-iodophenyl)-3-methylureidoJbenzoateas a light-yellow solid: mp 144-148 °C; lH NMR (CDCI3) <53.29 (s, 3H), 3.67 (s, 3H), 6.91-6.95 (m, IH), 7.12-7.16 (m,IH), 7.41 (dd, IH, J = 1.6,7.8 Hz), 7.46-7.52 (m, 2H), 7.87(dd, IH, J = 1.7, 8.0 Hz), 7.99 (dd, IH, J = 1.4, 8.0 Hz), 8.60(dd, IH, J = 1.0, 8.7 Hz), 9.91 (bs, IH). Anal. Calcd forC1~l5IN203: C, 46.85; H, 3.69; N, 6.83. Found: C, 47.15; H,3.77; N, 6.86.
A mixture of methyl 2-[3-(2-iodophenyl)-3-methylureidoJ-benzoate (0.50 g, 1.22 mmol) in 20 mL of ethanol and 0.1 NNaOH (17 mL, 1.70 mmol) was refluxed for 30 min. Thesolution was concentrated to remove the ethanol, acidified with10% HCl to pH 1-2, and then extracted into ethyl acetate.The organic extract was washed with brine, dried (MgSO4),filtered, and concentrated to give 2-[3-(2-iodophenyl)-3-methyl-ureido]benzoic acid as a light-yellow solid. The solid wasdissolved into 150 mL of chloroform (ethanol-free) and treatedwith 1-[3-(dimethylamino)propyIJ-3-ethylcarbodiimide hydro-chloride (0.26 g, 1.36 mmol). The solution was stirred at roomtemperature fQr 2 h and washed with saturated NaHCO3solution. The organic extract was dried (MgSO4) and concen-trated to give a colorless oil, which upon crystallization fromether-hexanes afforded 0.42 9 (91%) of2-[N-(2-iodophenyl)-N-methylamino]benz[d] [1,3Joxazin-4-one as a light-yellowsolid: mp 104-108 °C dec; FT-IR (KBr) 1749, 1620,1603 cm-l;lH NMR (CDC~) <5 3.44 (s, 3H), 7.08-7.12 (m, IH), 7.17-7.24(m, IH), 7.31-7 .46 (m, 3H), 7.62-7 .65 (m, IH), 7.93 (dd, IH,J = 1.2, 8.0 Hz), 8.02 (dd, IH, J = 1.2, 8.0 Hz). Anal. Calcdfor Cl5HlJN2O2: C, 47.64; H, 2.93; N, 7.41. Found: C,47.50;H, 3.00; N, 7.37.
MethOd C. 2-[[(3-Thiopheneyl}methyl]amino]benz[d].[1,3]oxazin-4-one (23). 3-Thiophenecarboxaldehyde (10.0 g,89 mmol), hydroxylamine hydrochloride (18.8 g, 270 mmol),and sodium acetate (22.1 g, 270 mmol) were combined inethanol (175 mL) and water (175 mL) and refluxed overnight.The reaction mixture was cooled, and most of the ethanol wasremoved in vacuo. The sample was extracted into ethyl acetate(200 mL). The organic layer was dried (MgSO4), and thevolatiles were removed under reduced pressure. Water (400mL) was added to the residue, and a crystalline product wasobtained. The product was filtered and recrystallized fromethyl acetate-hexanes to afford 3.0 g (27%) of the oxime of3-thiophenecarboxaldehyde. This oxime (1.03 g, 8.1 mmol) wasdissolved in methanol saturated with ammonia (100 mL) andhydrogenated with Raney nickel (1.0 g). The reaction mixturewas filtered, and the volatiles were removed under reducedpressure. A 1 N solution of HCl in ether (150 mL) was addedto the crude residue, and a precipitate formed. The solid wasfiltered and washed well with ether to afford 0.582 g (49%) of(3-thienyl)methylamine hydrochloride as a solid: lH NMR(DMSO-d6) <5 3.95-4.05 (s, 2H), 7.2-7 .3 (m, IH), 7.5-7 .7 (m,2H), 8.3-8.7 (bs, IH).
To a suspension of (3-thienyl}methylamine hydrochloride(0.57 g, 3.80 mmol) in 10 mL of DMF was added sodiumhydride (0.34 g of 60% dispersion in mineral oil, 8.50 mmol)at room temperature. After bubbling ceased, 2-carbomethox-yphenyl isocyanate (0.34 g, 8.4 mmol) was added. The samplewas heated at 75 °C for 5 days, cooled to room temperature,and poured into water. A precipitate formed which wasrecrystallized from methanol-ethyl acetate to give 0.56 g(57%) of 2-[ [(3-thiopheneyl)methyl]aminoJbenz[d] [1,3Joxazin-4-one (23) as a solid: mp 251-254 °C; lH NMR (DMSO-d6) <55.07 (s, 2H), 7.09-7.11 (dd,lH, J = 1.2,4.8 Hz), 7.18-7.23(m, 2H), 7.37-7.38 (m, IH), 7.45-7.47 (m, IH), 7.63-7.68 (m,
Hays et al.
1H), 7.93-7.96 (dd, 1H, J = 1.4, 8.4 Hz), 11.5 (s, 1H). Anal.Calcd for ClsHloN202S: C, 60.45; H, 3.90; N, 10.85. Found:C, 60.24; H, 3.92; N, 10.72.
2-[ (2,2,2- Trifluoroethyl)amino ]benz[d] [ 1,3]oxazin-4-one (3). 2-Aminobenz[d] [1,3Joxazin-4-one (2; 1.0 g, 6.2 mmol),trifluoroethyl iodide (2.6 g, 12.4 mmol), and sodium hydride(0.32 9 of a 60% oil dispersion, 8.0 mmol) were combined withDMF (25 mL) in a rocking autoclave and heated to 150 DC for4 h. The reaction mixture was cooled and partitioned betweenCH2C12 (200 mL) and water (2 x 200 mL). The organic layerwas dried (MgSO4), and the volatiles were removed underreduced pressure. The crude residue was chromatographed(silica gel, 25% ethyl acetate in hexanes). The product wasthen recrystallized from ethyl acetate-hexanes to afford 170mg (11 %) of2-[(2,2,2-trifluoroethyl)aminoJbenz[d] [1,3Joxazin-4-one as a solid: mp 244-245 DC; FT-IR (KBr) 3075, 2946,1728, 1669, 1453 cm-l; lH NMR (DMSO-d6) 6 4.68-4.78 (q,2H, J = 9.2, 18.4 Hz), 7.20-7.28 (m, 2H), 7.69-7.75 (m, 1H),7.95-7.98 (d, 1H, J = 7.9 Hz), 11.70 (bs, 1H). Anal. CalcdforCloH7FsN2O2: C, 49.19; H, 2.89; N, 11.47. Found: C,49.11;H, 3.07; N, 11.50.
2-[N- Ethyl-N -(2-iodophenyl)amino ]benz[d] [ 1,3 ]oxazin-4-one (11). 2-[(2-Iodophenyl)aminoJbenz[dJ [1,3Joxazin-4-one(7; 0.308 g, 0.85 mmol) was dissolved in THF (5 mL), and NaH(0.040 9 of a 60% oil dispersion, 1.0 mmol) was added. Thereaction mixture was stirred for 30 min, and ethyl iodide (0.312g, 2.0 mol) was added. The reaction mixture was heated to60 DC and stirred for 24 h. The reaction mixture waspartitioned between water (200 mL) and ethyl acetate (200mL). The ethyl acetate layer was dried (MgSO4), and thevolatiles were removed under reduced pressure. The cruderesidue was chromatographed (silica gel, 30% ethyl acetatein petroleum ether). This residue was then recrystallized fromether-hexanes to afford 80 mg (25%) of the desired product(11) as a white solid: mp 129-130 DC; FT-IR (KBr) 3422,2980,1713, 1664, 1609, 1481 cm-l; lH NMR (CDCls) 6 1.39-1.43 (t,3H, J= 7.0 Hz), 4.17-4.35 (m, 2H), 7.14-7.19(m, 1H), 7.28-7.34 (m, 2H), 7.48-7.53 (m, 1H), 7.72-7.77 (m, 1H), 7.96-7.98 (dd, 1H, J = 1.4, 8.2 Hz), 8.28-8.31 (dd, 1H, J = 1.4, 8.0Hz). Anal. Calcd for Cl6HlsIN202: C, 49.00; H, 3.34; N, 7.14.Found: C, 49.24; H, 3.52; N, 7.03.
Preparation of Starting Materials. Methyl 2-Amino-4-isopropylbenzoate (Starting Material for 45 and 86).Nitric acid (63 g, 1 mol) was added to a solution of 4-isopro-pylbromobenzene (50 g, 0.25 mol) dropwise at 0 DC. Thereaction mixture was stirred for 3 h, poured onto ice, andextracted with ether (2 x 500 mL). The organic layer wasdried (MgSO4), and the volatiles were removed under reducedpressure. The crude residue was distilled (1 mmHg, 105-129DC) to afford 19.5 9 (32%) of 2-nitro-4-isopropylbromoben-zene: FT-IR (LF) 2966, 1535, 1358 cm-l; lH NMR (CDCls) 61.26-1.28 (d, 6H, J = 7.0 Hz), 2.93-3.00 (m, 1H), 7.28-7.31(dd, 1H, J = 2.2, 8.2 Hz), 7.62-7 .64 (d, 1H, J = 8.4 Hz), 7.68-7.69 (d, 1H, J = 2.2 Hz). Anal. Calcd for C9HloBrNO2: C,44.29; H, 4.13; N, 5.74. Found: C, 43.91; H, 4.10; N, 5.48.
2-Nitro-4-isopropylbromobenzene (19.5 g, 80 mmol) andcopper(I) cyanide (14.3 g, 160 mmol) were refluxed in DMF(250 mL) for 24 h. The reaction mixture was poured into water(500 mL) and extracted with ether (3 x 500 mL). Thecombined organic layers were filtered through Celite andwashed with water (3 x 500 mL). The organic layer was dried(MgSO4), and the volatiles were removed under reducedpressure. The crude residue was chromatographed (silica gel,CH2C12-petroluem ether, 1:1) to afford 4.3 9 (28%) of2-nitro-4-isopropylbenzonitrile: FT-IR (KBr) 2969,1539, 1344cm-l; lH NMR (CDCls) 6 1.32-1.34 (d, 6H, J = 7.0 Hz), 3.08-3.13 (m, 1H), 7.65-7 .68 (dd, 1H, J = 1.7, 8.0 Hz), 7.82-7 .84(d, 1H, J = 8.0 Hz), 8.18-8.19 (d, 1H, J = 1.7 Hz). Anal. Calcdfor CloHloN202: C, 63.15; H, 5.30; N, 14.73. Found: C,63.13;H, 5.22; N, 14.90.
2-Nitro-4-isopropylbenzonitrile (4.3 g, 22.6 mmol) was re-fluxed overnight in a mixture ofwater-acetic acid- H2S04 (2:1:1, 60 mL). The reaction mixture was poured onto ice water(200 mL), basified with 1 N NaOH, and extracted with ether

Inhibitors of Clr Serine Protease
(250 mL). The aqueous layer was separated, acidifed withconcentrated HC1, and extracted with ether (250 mL). Theorganic layer was dried (MgSO4), and the volatiles wereremoved under reduced pressure to afford 4.8 9 (68%) of2-nitro-4-isopropylbenzoic acid as a solid: FT-IR (KBr) 2968,2663, 1703, 1535, 1356 cm-l; IH NMR (DMSO-d6) 6 1.22-1.25(d, 6H, J = 7.0 Hz), 3.02-3.10 (m, IH), 7.65-7.68 (dd, IH, J= 1.4, 8.0 Hz), 7.79-7.83 (m, 2H). Anal. Calcd for C1oHll-NO4: C, 57.41; H, 5.30; N, 6.70. Found: C, 57.78; H, 5.46; N,6.84.
2-Nitro-4-isopropylbenzoic acid (3.09 g, 14.8 mmol) washydrogenated with Raney nickel in water (100 mL) and NH4-OH (10 mL). The reaction mixture was filtered and acidifiedwith concentrated HC1. A precipitate formed and was collectedby filtration. The solid was heated in MeOH and filtered againto afford 1.2 g (45%) of 2-amino-4-isopropylbenzoic acidhydrochloride, which was carried on without further purifica-tion: IH NMR (DMSO-d6) 6 1.05-1.20 (d, 6H, J = 7.0 Hz),2.6-2.8 (m, IH), 6.3-6.5 (m, 2H), 7.6-7.7 (m, 3H).
2-Amino-4-isopropylbenzoic acid hydrochloride was refluxedin H2SO4 (20 mL) and methanol (80 mL) overnight. Thereaction mixture was poured onto ice water (400 mL) andextracted with methylene chloride (200 mL). The organic layerwas washed with saturated NaHCO3 (200 mL) and dried(MgSO4), and the volatiles were removed under reducedpressure to afford 0.805 g (62%) of methyl 2-amiDo-4-isopro-pylbenzoate as a solid: mp 40-41"C; Fr-IR (KBr) 3472,3367,2960, 1691, 1622, 1437, 1307 cm-l; IH NMR (CDC13) 6 1.1-1.34 (d, 6H, J = 7.0 Hz), 2.7-2.9 (m, IH), 3.85 (s, 3H), 6.5-6.6 (m, 2H), 7.7-7.87 (d, IH). Anal. Calcd for C11H1sNO2: C,72.07; H, 8.21; N, 6.00. Found: C, 71.87; H, 8.04; N, 5.97.
Biological Methods. Inhibition of Clr (Initial): Com-pounds to be tested were dissolved in DMSO at concentrationsranging from 1 mM to 0.01 mM, and a 10-/lL aliquot of eachconcentration was deposited into a well of a 96-well microtiterplate; 50 /lL ofCbz-Gly-Arg-S-Bzl (1.5 mM in DMSO/H2O, 3:7)and 50 /lL of 5,5'-dithiobis(2-nitrobenzoic acid) (DTNB) solution(0.75 mM in 150 mM Tris buffer) were dispensed in the wellsof the microtiter plate, followed by the immediate addition of50 /lL of the Clr enzyme solution (20 /lg/mL purified humanClr in 10 mM Tris buffer (pH 7.4». After a 30-s incubation,the hydrolyzed substrate was read in the kinetic mode usinga Molecular Devices Thermomax microplate reader set to awavelength of 405 nm. The assay background was followedby monitoring wells that contain substrate, buffer, and DTNBsolutions but no enzyme. All assays were performed intriplicate at each inhibitor concentration tested. The resultsare expressed in IC5o values.
Inhibition of Clr (60 lOin): A 50-/lL aliquot of 150 mMTris buffer (pH 7.5) was added to wells containing 10 /lL ofinhibitor in DMSO. The mixture sat at room temperature for60 miD; then 50 /lL of substrate solution, 50 /lL of DTNB, and50 /lL of enzyme solution were added to the wells to start thereaction. The plate was read as described above.
Inhibition of Trypsin: A microplate caseinolysis assaywas carried out as previously described.15 Briefly, 0.5 mg/mLcasein (sodium salt; Sigma Co.), 20 mM dithiothreitol, and 50mM Tris-HCl (pH7.4) were mixed in the presence ofvariousconcentrations of an inhibitor. After 1.25 /lg of trypsin (bovinepancreas; Sigma Co.) was added, the microtiter plate (250 /lL)was incubated for 60 miD at 25 "C. The samples wereprocessed for colorimetric development and read on a Molec-ular Devices Thermomax microplate reader at an absorbance
Journal of Medicinal Chemistry, 1998, Vol. 41, No.71067
of 595 nm. Based on a plot of percent inhibition of trypsinagainst log [inhibitor], an IC5o was generated using Sigma plot.Data are the average of two to four determinations with dataranges within 20% of the average values.
Hemolysis Assay: The hemolysis assay was performedusing a DiaMedix EZ Complement kit. Compounds (5 IlL) tobe tested were added to a test tube containing a standardizedsuspension of sheep erythrocytes sensitized with antibodiesto sheep erythrocytes. The mixture was incubated for 60 minat 25 °C and then centrifuged. The absorbance of the super-nate was read at 405 nm. The results are expressed as percentof control at 200 IlM.
References
(1) For reviews see: Tomlinson, S. Complement defense mecha-nisms. Curr. Opin. Immunol. 1993, 5, 83-89. (b) Benjamini, E.;Leskowitz, S. Immunology: A Short Course; Wiley-Liss: NewYork, 1991; pp 135-150.
(2) Rogers, J.; Cooper, N. R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S. D.; Civin, W. H.; Brachova, L.; Bradt, B.; Ward,P .; Lieberburg, I. Complement activation by f3-arnyloid in Alzhe-imer's disease. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 10016-10020.
(3) Jiang, H.; Burdick, D.; Glabe, C. G.; Cotman, C. W.; Tenner, A.f3-Amyloid activates complement by binding to a specific regionof the collagen-like domain of the C1q A chain. J. Immunol.1994, 152, 5050-5059.
(4) Kalaria, R. N. The immunopathology of Alzheimer's disease andsome related disorders. Brain Pathol. 1993, 3, 333-347.
(5) McGeer, P. L.; McGeer, E. G. The inflammatory response systemof brain: Implications for therapy of Alzheimer and otherneurodegenerative diseases. Brain Res. Rev. 1995, 21, 195-218.
(6) Eikelenboom, P.; Zban, S.-S.; van Gaol, W. A.; Allsop, D.Inflammatory mechanisms in Alzheimer's disease. Trends Phar-macol. Sci. 1994, 15, 447-450.
(7) Rogers, J.; Kirby, L. C.; Hempelman, S. R.; Berry, D. L.; McGeer,P. L.; Kaszniak, A. W.; Zalinski, J.; Cofield, M.; Mansukhani,L.; Willson, P.; Kogan, F. Clinical trial of indomethacin inAlzheimer's disease. Neurology 1993, 43, 1609-1611.
(8) McGeer, P. L.; Schulzer, M.; McGeer, E. G. Arthritis andantiinflammatory agents as possible protective factors for Alzhe-imer's disease: A review of 17 epidemiologic studies. Neurology1996, 47, 425-432.
(9) Fothergill, J.; Kemp, G.; Paton, N.; Carter, P.; Gray, P. Thestructures of human C1r and C1s and their relationship to otherserine proteases. Behring Inst. Mitt. 1989, 84, 72-79.
(10) Aoyama, T.; Ino, Y.; Ozeki, M.; Oda, M.; Sato, T.; Koshiyarna,Y.; Suzuki, S.; Fujita, M. Pharmacological studies ofFUT-175,nafarnstat mesilate I. Inhibition of protease activity in in vitroand in vivo experiments. Jpn. J. Pharmacol. 1984, 35, 203-227.
(11) Gilmore, J. L.; Hays, S. J.; Caprathe, B. W.; Lee, C.; E=erling,M. R.; Michael, W.; Jaen, J. C. Synthesis and evaluation of2-aryl-4H-3,1-benzoxazin-4-ones as C1r serine protease inhibi-tors. Bioorg. Med. Chem. Lett. 1996, 6, 679-682.
(12) Krantz, A.; Spencer, R. W.: Tarn, T. F.; Liak, T. J.; Copp, L. J.;Thomas, E. M.; Tafferty, S. P. Design and synthesis of 4H-3,1-benzoxazin-4-ones as potent alternate substrate inhibitors ofhuman leukocyte elastase. J. Med. Chem. 1990, 33, 464-479.
(13) McRae, B. J.; Lin, T. Y.; Powers, J. C. Mapping the substratebinding site of human C1r and C1s with peptide thioesters.Development of new sensitive substrates. J. Biol. Chem. 1981,256, 12362-12366.
(14) Papadopoulos, E. P.; Torres, C. D. Convenient preparation ofN-substituted 2- arnino-4H-3,1-benzoxazin-4-ones and 3-substi-tuted 2, 4(lH,3H)-quinazoline-diones. J. Heterocycl. Chem. 1982,19, 269-272.
(15) Buroker-Kilgore, M.; Wang, K K W. A Coomassie brilliant blueG-250-based colorimetric assay for measuring activity of calpainand other proteases. Anal. Biochem. 1993, 208, 389-392.
JM970394D
Related Documents











