



ORIGINAL PAPER
Wood Modification Effects on Weathering of HDPE-Based WoodPlastic Composites
James S. Fabiyi Æ Armando G. McDonald ÆDavid McIlroy
Published online: 18 April 2009
� Springer Science+Business Media, LLC 2009
Abstract The effects of weathering on the constituents of
wood and polymer matrix behavior in wood plastic com-
posites (WPCs) were investigated. WPCs were produced
from pine, extractives-free pine, and pine holocellulose
fibers (60%) together with HDPE (40%). These composites
were subjected to xenon-arc accelerated and outside
weathering for a total of 1200 h and 120 days, respec-
tively. The color and chemical changes that occurred on the
surface of the WPCs were analyzed using a set of analytical
techniques. For pine and extractive-free pine filled com-
posites, the results showed that the total color change,
lightness, and oxidation increased, while the lignin content
decreased. In addition, the weight average molecular
weight (Mw) and number average molecular weight (Mn)
of extracted HDPE decreased with an increase in exposure
time of the composites. However, HDPE crystallinity
increased with longer exposure time. Lightness of holo-
cellulose-based WPC changed the least while the change in
its HDPE crystallinity was not significant compared to the
other composite types. Therefore, holocellulose-based
WPC may be preferred for applications where color sta-
bility is of high priority.
Keywords Wood plastic composite � Molecular weight �Crystallinity � Wood loss � Pine � Extractive free wood �Holocellulose
Introduction
Wood plastic composites (WPC) are polymeric materials
that are made up of wood, plastic, and other chemical
additives (lubricants, coupling agents, nucleating agents,
pigments, and UV stabilizers). Incorporation of wood fibers
into plastics improves certain properties of the resulting
composite material, such as the flexural and tensile stiff-
ness, relative to pure plastics [1]. Also, the dimensional
stability of these materials tends to be greater than that of
the traditional wood products, thereby rendering them
suitable for application in end-uses where stability is a
prerequisite attribute, especially in humid and water front
environments [1]. Due to its advantages over wood and
plastic lumber, WPCs are gaining entrance into the build-
ing and construction industries [2]. However, durability
issues especially degradation and structural failure sur-
rounding the use of WPCs are of concern because of
warranty claims [3].
Indeed, several attempts have been made to improve the
durability and stability of WPC [4–8]. However, photo-
degradation has continued to be a problem with these
products. The consequences of photodegradation of WPCs
are the loss of their aesthetic appearance and strength
properties [5–7]. In order to stabilize WPC against
weathering, many approaches have been employed. These
include the incorporation of chemical additives such as
UV-stabilizers, free radical scavengers, and pigments to
moderate the effects of weathering. However, little success
has been made even with the incorporation of additives
because WPC still undergo degradation and color change
upon weathering. Stark and Matuana [6] observed that the
addition of pigments to WPC had a greater influence on
reducing the effect of photodegradation on total color
change, lightness, and mechanical properties during
J. S. Fabiyi � A. G. McDonald (&)
Forest Products Department, University of Idaho, Moscow,
ID 83844-1132, USA
e-mail: [email protected]
D. McIlroy
Physics Department, University of Idaho, Moscow,
ID 83844-0903, USA
123
J Polym Environ (2009) 17:34–48
DOI 10.1007/s10924-009-0118-y

accelerated weathering of HDPE based WPC than the
addition of UV stabilizers.
In the course of studying the durability of WPC, it was
observed that wood lignin makes a significant contribution
to the color change during weathering [4–6, 9]. Unfortu-
nately, detailed investigation on the role of lignin on color
change of weathered WPC has not been considered. The
use of pure cellulose fibers over wood fibers in WPC offers
the benefit of higher thermal stability (up to 270 �C) in that
engineering thermoplastics (e.g., nylon) can be used to
obtain materials for structural applications [10]. Therefore,
cellulose fibers (commonly produced below 200 �C during
pulping) would be a suitable reinforcement/filler for HDPE
based WPC.
Apart from the effects of weathering on the wood
component of WPC, polymer matrix degradation in WPC
during weathering can result in changes such as molecular
weight distribution (MWD) and crystallinity. Chemical
changes that occur during weathering affect the overall
properties of the polymer including melt flow/viscosity,
molecular weight, and mechanical strength [11]. Also,
reduction in molecular weight (chain-scission) leads to
shorter polymer chains and lower mechanical properties.
Torik et al., [12] stated that crystallinity is one of the most
important factors in photostability of polyethylene. Chan-
ges in the degree of crystallinity have been shown to have
pronounced effects on the mechanical behavior and frac-
ture toughness of polymers [13]. Therefore, investigating
the effects of weathering on the MWD and crystallinity of
the plastic matrix is important for outdoor structural
applications of WPC.
This study was therefore aimed at examining the
behavior of wood constituents and polymer matrix during
weathering of WPC. MWD and percent crystallinity of
polymer matrix were investigated to establish the effects of
wood modification on contribution of modified wood to
WPC surface degradation during weathering. Color and
chemical changes of modified wood fiber based WPC were
investigated to establish the role of wood extractives and
lignin during weathering.
Materials and Methods
Materials Preparation
Wood fibers were extracted in accordance with a modified
TAPPI method (T204 om-88) [14] using acetone (98%) as
solvent. One-kilogram batches of pine wood fibers (60
mesh, American Wood Fibers) was dispersed in acetone
(30 L) with constant stirring for 24 h. Thereafter, the
extract was discarded and the extracted wood was further
washed (thrice) with acetone before air dried. Note that
only approximately 3.2% extractives based on initial
weight of wood were removed by this technique. The
remaining wood fibers after the extraction would be
henceforth referred to as extractive free wood in this paper.
Holocellulose was prepared following the method
developed by Wise et al. [15]. Air-dried extractives free
wood fiber (1 kg batch) was dispersed (under constant
stirring) in 32 L of deionised water containing 0.3 kg of
NaClO2 (99%) and 200 mL of acetic acid and heated to
70 �C for 1 h. After an hour, a further aliquot of 200 mL
acetic acid and NaClO2 (0.3 kg) was added and the reac-
tion proceeded for another 1 h. This was repeated four
more times to a total of 6 h. Finally, at the completion of
the sixth treatment, the reaction was allowed to cool to
room temperature, and the delignified wood fiber was
recovered by filtering through a polypropylene screen (100
mesh). The wood fiber was washed repeatedly (7–10 times)
with deionised water until the conductivity (Accumet
conductivity meter) of the solution was reduced to 1.5–
2.5 lS. It was finally rinsed with acetone to accelerate
drying. The resultant wood fiber (holocellulose containing
1–1.5% lignin) was then air-dried. This method yielded
67% holocellulose based on initial weight of wood. Finally,
untreated pine wood flour, extractive free wood, and ho-
locellulose were dried to below 0.5% moisture content
prior to being used for WPC production.
WPC Production
HDPE (Equistar petrothene, LB 0100-00, MFI = 0.3 g/
10 min, and density = 0.950 g/cm3) and the three types of
wood fibers already prepared were used for WPC produc-
tion. One formulation based on 60% of wood and 40% of
plastic was considered. Materials were compounded and
extruded on a 35 mm counter rotating conical twin-screw
extruder (Cincinnati Milacron) to a profiled dimension of
9.5 9 38 mm. The barrel and die temperatures were set
between 149 and 193 �C. The extruded profiles were then
knife milled to a thickness of 5.0 mm for the weathering
tests because many commercial WPC products are surface
finished.
Mechanical Properties of WPC Produced
from Modified Wood Fiber
Three point flexural tests (modulus of rupture (MOR),
modulus of elasticity (MOE)) were performed in accor-
dance with ASTM Standard D 790-00 [16]. Seven repli-
cates were performed for each WPC type (20 9
5.73 9 115 mm) on an Instron 5500R Universal test
machine equipped with a 454 kg load cell. Data was col-
lected and processed using Bluehill software (Instron).
J Polym Environ (2009) 17:34–48 35
123

Accelerated and Natural Weathering of WPC
WPC specimens (5 9 38 9 101 mm) were subjected to
accelerated weathering test, which was conducted in a
xenon-arc weatherometer (Q-Sun). The average irradiance
was 0.70 W/m2 at 340 nm wavelength with a chamber
temperature of 70 �C and water spray in accordance with
ASTM D 6662 [17], which is a very severe weathering
condition. The natural weathering test was conducted by
exposing the WPC specimens (5 9 38 9 610 mm) outside
in Moscow, Idaho, USA on a south-facing wall at an angle
of 45� (Moscow, ID) in accordance with ASTM D 1435
[18]. Average daylight in Moscow within the period of
exterior exposure was approximately 12–14 h/day for July
to August 2006 and 9–10 h/day for September to
November 2006. The sample condition was assessed from
0 to 1200 h and 0 to 120 days of exposure in xenon-arc and
outside weathering regimes, respectively.
Material Characterization after Weathering
Scanning Electron Microscopy Analysis
Scanning electron micrographs of WPC samples were
obtained on a LEO Gemini field emission SEM instrument.
Sections of unweathered and weathered (400 and 1200 h
xenon-arc weathered) WPCs were cut into approximately
8 9 8 mm pieces. The specimens were mounted on alu-
minum stubs using carbon tape. The weathered surfaces
were analyzed directly (without coating) at 1 kV at a
magnification of 709.
Color Measurement
A StellarNet EPP2000 UV-Vis spectrometer (190–
850 nm) using a krypton light source (SL1, Stellar Net)
and a diffuse reflection fiber optic probe was used to
measure color in accordance with the ASTM 2244 stan-
dard [19]. The spectrometer SpectraWiz software trans-
forms spectral data into CIELAB color coordinates based
on a D65 light source (L*, a* and b*) [20]. Color was
measured for five replicates per WPC sample at three
locations on each specimen. Relative lightness (DLrel) and
total color change (DEab) were calculated using the fol-
lowing equations:
DLrel ¼L�final � L�initial
L�initial
� 100 ð1Þ
DEab ¼ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
DL2 þ Da2 þ Db2p
ð2Þ
where, DL, Da, and Db represent the differences between
the initial and final values of L*, a*, and b*, respectively.
An increase in L means the sample is lightening.
Infrared Spectroscopy Analysis
Infrared measurements were performed with a Thermo-
Nicolet Avatar 370 FTIR spectrometer operating in the
attenuated total reflection (ATR) mode (SmartPerformer,
ZnSe crystal). Thin slices of about 50 lm of WPC (vacuum
dried) from three samples were analyzed on the exposed
side. Each spectrum was taken as an average of 64 scans at
a resolution of 4 cm-1. Special interest was focused on the
C=C double bond region (1630–1660 cm-1), carbonyl
region (1660–1800 cm-1), and the hydroxyl region (3200–
3500 cm-1) [21–23].
For the quantitative analysis, the spectra were normal-
ized and the absorbance of the peak of interest was
obtained. The concentrations of the carbonyl (C=O),
unsaturated carbon (C=C), and hydroxyl (OH) groups
(degradation products) were estimated based on the Lam-
bert–Beer equation (Eq. 3):
A ¼ ebc ð3Þ
where, A is the absorbance of the functional group band in
the infrared spectrum, c is the molar concentration of the
functional group mol/L (M), e is the molar absorptivity
(L/mol/cm), and b is the path length of the sample (that is,
the optical path of the infrared beam through the sample,
cm). A is obtained from the absorption band of interest in
the ATR spectrum. Values for the molar absorptivity, e used
in this study were taken from the work reported by Lacoste
et al., [24] from the spectra of model carboxylic acids
(1718 cm-1), esters (1744 cm-1), and C=C (1635 cm-1),
which were 350, 590 and 121 L/mol/cm, respectively. The
molar absorptivity of hydroperoxide from the secondary
alcohol was 660 L/mol/cm [23], this was used for the
investigation of weathering on wood. Optical path length b
was considered as the product of the effective path length,
de and the number of reflections in the internal reflection
element (IRE). The protocol of Fabiyi et al. [25] was
employed to determine the concentration of each functional
group was calculated as:
c ¼ A�
2dpe� �
ð4Þ
where de = 2dp and dp is the ATR depth of penetration.
X-ray Photoelectron Spectroscopy (XPS) Analysis
X-ray photoelectron spectroscopy (XPS) is a non-destruc-
tive surface analytical technique that provides information
on the oxidation or chemical bonding state of elements [26,
27]. It has been used for the characterization of wood fibers
[28] and the weathering of wood and WPC [27].
XPS measurements were performed on a limited number
of samples on both control and weathered WPC (made
from HDPE and untreated pine wood fibers) to determine
36 J Polym Environ (2009) 17:34–48
123

surface chemical composition. A custom built spectrometer
using two monochromators at 1253 and 1487 eV, with a
resolution of 50 meV (Courtesy of Dr. D. McIlroy Labo-
ratory, Dept of Physics, Univ. of Idaho) was employed. The
instrument has a high-resolution hemispherical analyzer.
Charging was minimized using a flood gun. Samples were
mounted on a stainless steel sample holder with carbon
tape. Care was taken to ensure that uncontaminated sample
with flat surface covered the carbon tape to avoid its
(carbon tape) exposure to X-ray beam.
Two types of spectra were obtained that include low
resolution spectra from 0 to 1100 eV binding energy to
determine elemental composition (oxygen and carbon
ratios) and high resolution spectra from 280 to 300 eV to
analyze carbon. For the quantitative analysis, the spectra
were baseline corrected, normalized, and curve fit using
IGOR Pro 5.05 software (WaveMetrics, Inc). The area of
each peak identified by curve fitting was mathematically
computed.
The following data processing restrictions were made in
accordance with procedure developed by McDonald et al.
[28]:
(i) The full widths at half height (FWHM), for the C1 to
4 peaks (285, 286.5, 288, and 289.5 eV) were kept
constant (1.6 eV).
(ii) The fitted peaks was made using Voigt.
(iii) The third restriction is that a peak C0 (C=C), at
283.5 eV was fitted when the residual of the band
indicating the presence of this peak (FWHM of 1.6
EV).
(iv) Therefore, with these restrictions in place, a peak
fitting routine was then used to minimize and
randomize the residual signal.
From the area of the curve fitted peaks, the percent
carbon, oxygen, and the ratios of oxygen to carbon (O/C)
present upon weathering were computed.
Pyrolysis Gas Chromatography-Mass Spectrometry
Wood derived compounds (lignin and polysaccharides)
were identified and quantified by GC-MS according to the
method described by Fabiyi et al., [25] and Schauwecker
et al., [29]. Two replicates specimens were used for each
treatment. Wood content in the weathered WPC samples
was quantified using pyrolysis-GC-MS by developing a
calibration curve from the total peak areas under the wood
derived peaks relative to the total peak areas under the
polyethylene derived peaks. Calibration curves were based
on tests of a series of WPC formulations of known wood
content (untreated and extractive free pine) ranging from 0
to 100% in HDPE-based WPC.
Polymer Matrix Characterization
The need to extract the polymer matrix (HDPE) prior to
analysis is important since one of the main interests was to
understand its behavior. The WPC surface (unweathered
and weathered) was scrapped with a razor blade and HDPE
was extracted. Approximately 500 mg of surface material
was weighed into a flat bottom flask and 1,2,4-trichloro-
benzene (TCB) (50 mL) was added. The contents were
then rapidly heated with a microwave oven (Sanyo,
600 W) for 2 to 3 min to minimize oxidation. The solu-
bilized HDPE was collected by separating wood fibers
from the heated contents using a cotton wool filter, then re-
filtered through filter paper (Whatman No.1) and dried
under a stream of nitrogen to obtain a film of HDPE.
Polymer Analysis by Gel Permeation Chromatography
The effect of weathering on the molecular weight and
MWD of plastic (HDPE) was determined by gel perme-
ation chromatography (GPC) on elution with TCB. Mw and
Mn for HDPE was conducted by Equistar Chemicals
(Cincinnati, OH) and ExxonMobil Chemicals (Baton
Rouge, LA) using high temperature GPC systems.
The HDPE from xenon-arc weathered extractive free
and exterior weathered pine composites were analyzed by
Equistar Chemicals. The HDPE were heated for 1 h at
175 �C in TCB (1–1.5 mg/mL) prior to injection (300 lL).
Analysis was performed on a Waters GPC2000 system and
separation was achieved using two mixed-bed columns at
145 �C and a TCB flow rate of 1 mL/min. The components
were detected using refractive index (RI), capillary vis-
cometer, and Precision Detector LS systems. The GPC
system was calibrated using a universal calibration curve
(PS standard, K = 0.0001387, a = 0.70) with a linear fit.
The extracted HDPE from unweathered and xenon-arc
weathered pine composites were also analyzed by Exxon-
Mobil Chemicals. Analysis was performed on GPC 220
system with an autosampler equipped with an RI and dif-
ferential viscosity detector (Viscotek). Separation was
carried out using three columns in series (10 lm mixed-
bed, 300 mm 9 7.6 mm from Polymer Laboratories). The
analysis was performed at 135 �C using TCB as the mobile
phase (1 mL/min). Calibration was conducted using a
narrow range PS standard. Molecular weight analysis was
performed using OmniSEC 4.1 software (Viscotek).
Polymer Crystallinity using Differential Scanning
Calorimetric
Differential scanning calorimetric (DSC) was performed on
a TA2910 instrument to monitor the thermal behavior of
HDPE crystallization according to ASTM D 3418 [30].
J Polym Environ (2009) 17:34–48 37
123

The instrument was calibrated using indium. TCB-extrac-
ted HDPE (5–7 mg) was analyzed using a temperature
profile after 2 min equilibration at 50 �C, then ramped to
160 �C at a heating rate of 10 �C/min and held isother-
mally for 10 min, cooled to 50 �C at -10 �C/min. Two
replicate measurements were performed for each sample.
The degree of crystallinity was calculated from the
experimentally determined heat of fusion for the extracted
HDPE and the heat of fusion corresponding to a pure PE
crystal of 293.6 J/g [31, 32].
All the data was statistically analyzed using SPSS v11
software.
Results and Discussion
Flexural Properties of Modified Wood Based WPC
Flexural properties of modified wood based WPC are
presented in Table 1. The results showed that there were no
significant differences in the MOR, and MOE among the
composites made from pine fibers, extractive free pine
fibers, and pine holocellulose fibers. This was contrary to
the expectation because holocellulose based composites
supposed to be more hydrophilic (incompatible) than pine
fibers and extractive free pine fibers.
Characterization of WPC Weathered Surface
Visual Appearance
Figure 1 shows scanning electron micrographs of unweath-
ered, 400, and 1200 h xenon-arc weathered composites.
These composites degraded differently depending on wood
fiber types. Cracks were observed on the surface, which
might be as an effect of polymer chain-scission that normally
results in highly crystallized polymer zones. Cracking occurs
because of wetting and drying cycles between the surface
and interior sections. The extent of wood degradation and
erosion, leaving cracks and ‘‘pits’’ increased upon extended
weathering. These findings are in agreement with other
studies [33, 34]. Also, more wood fibers were found on the
surface of holocellulose based composites upon longer
exposure compared to pine based composites. After 400 h of
exposure in xenon-arc weatherometer, the surface layer of
the weathered WPC was eroded; thereby creating cavities on
the surface (Fig. 1b and e). Accelerated weathering exposure
for 1200 h resulted in the size and frequency of cavities
(Fig. 1c and f).
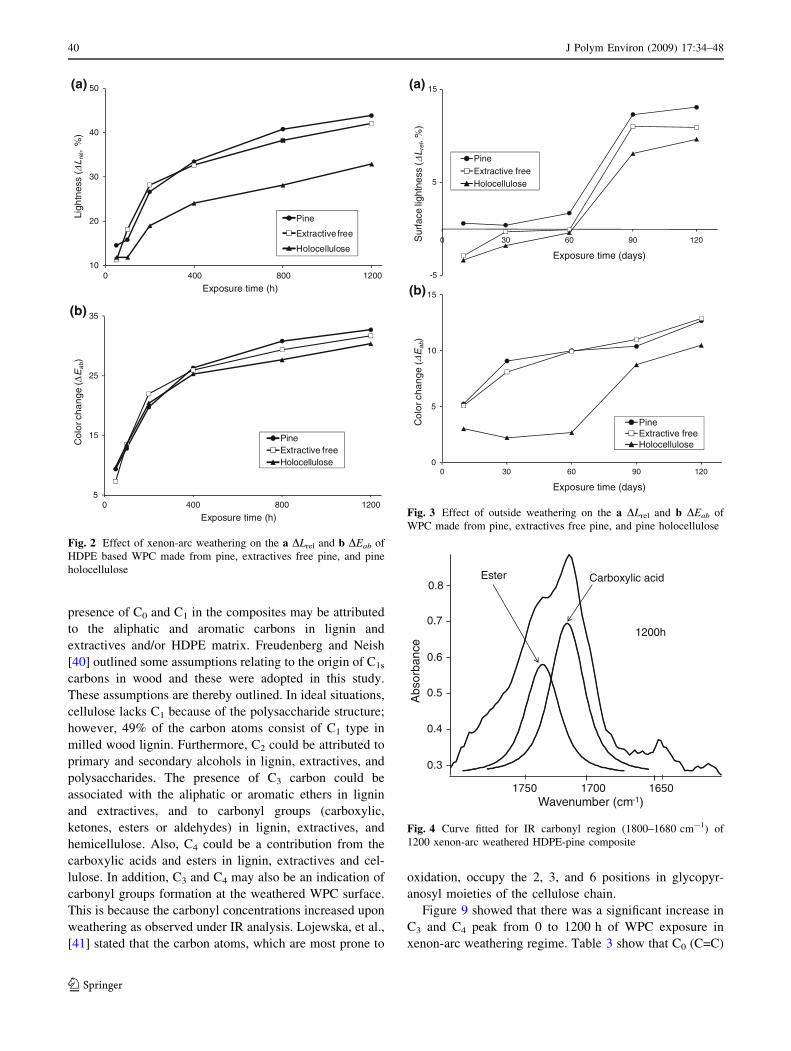
Color Changes
Relative lightness (DLrel) and total color change (DEab) of
xenon-arc and exterior weathered WPC made from
untreated, extractive free and holocellulose are shown in
Figs. 2 and 3. These results showed that WPC produced
from holocellulose fibers had the lowest DLrel and DEab for
both xenon-arc and outside weathering regimes. Stark [35]
conducted a study on the color change of HDPE/pine
composites in which HDPE (48.5 or 47%), pine flour
(50%), UV absorber (0.5 or 1%) and zinc ferrite pigment
(1 or 2%) were used. Surprisingly, HDPE/holocellulose
composites (after 1200 h xenon-arc exposure) performed
better in term of lightness than HDPE/pine composites with
UV absorber (0.5 or 1%) and zinc ferrite pigment (1 or 2%)
after 1000 h of xenon arc weathering.
There was no significant difference in DLrel for the WPC
produced from untreated and extractive free pine wood
(a = 0.05) subjected to both xenon arc and outside
weathering. This result is expected since the extractives
were not considered a significant contributor to weathering.
The DLrel and DEab for all the WPC increased upon
weathering until 1200 h and 120 days for xenon-arc and
outside weathering regimes, respectively (Figs. 2 and 3).
There was no significant difference between DEab of
holocellulose and untreated/extractive free composites.
This might be due to the contribution of combined effect of
the three color coordinates. The results obtained were
consistent with other weathering studies [4, 5].
Surface Chemical Characterization
IR Spectroscopy
IR spectrum of WPC showing the bands between carbonyl
group regions is presented in Fig. 4. The entire IR spectra
(not shown) conveyed some useful information on some
bands. For instance, the spectral structures between 1015–
1050 and 3500–3080 cm-1 regions which were assigned
to C–O and OH groups in wood (combination of cellulose,
hemicellulose and lignin) decreased upon weathering of the
WPC [36, 37]. It was shown that the lignin ether linkage
assigned band (1512–1508 cm-1) was not present in the
holocellulose composites. This implies that the lignin
content in the holocellulose was negligible. For pine and
Table 1 Flexural properties of modified wood fiber/HDPE
composites
WPC type MOR (MPa) MOE (GPa)
Pine 25.51 (2.5) 2.25 (3.2)
Extractive free pine 25.57 (5.0) 2.41 (7.4)
Holocellulose 25.03 (3.9) 2.54 (3.1)
Data presented are the average of seven replicated specimens while
those in bracket are the coefficients of variation
38 J Polym Environ (2009) 17:34–48
123

extractive free pine WPC, the band at 1508–1512 cm-1
was shown to decrease in intensity as a function of
weathering time [37]. This indicates that lignin degradation
occurred on the weathered WPC surface.
In this study, the extent of WPC oxidation was deter-
mined by its carbonyl functionality (1750–1690 cm-1).
The concentration of the carbonyl (carboxylic acids at
1715 cm-1 and esters at 1735 cm-1) increased upon WPC
weathering (Figs. 5 and 6). The increase in carbonyl con-
centration provided evidence that photodegradation
occurred with prolonged exposure time. It also means that
the composites were vulnerable to further degradation
because these carbonyl groups are photolabile [4]. The
concentration of the C=C groups at 1635 cm-1 present in
weathered WPC slightly increased upon weathering
(Fig. 7). This suggests that HDPE and wood oxidation
occurred resulting in an increase unsaturated functionality.
In addition, the hydroperoxide (from hydroxyl group at
3500–3080 cm-1) concentration decreased with longer
exposure time (Fig. 8). This indicates that wood content
decreased from the weathered surface since the band
between 3500 and 3080 cm-1 are usually assigned to pri-
mary alcohol. However, wood loss from holocellulose-
based WPC was minimal as compared to pine and
extractives free pine based composites.
X-ray Photoelectron Spectroscopy (XPS)
The results obtained from XPS analysis (widescans) for the
unweathered and xenon-arc weathered HDPE/pine com-
posites showed only the presence of carbon and oxygen.
The peaks for C1s and O1s had chemical binding energies
of approximately 285–290 and 532 eV, respectively,
(Table 2) [38, 39]. High resolution scans of the C1s region
were analyzed after curve fitting and the spectra shown in
Fig. 9, while binding energies are given in Table 2. The
Fig. 1 Scanning electron
micrographs (709) of HDPE-
pine composites a unweathered,
b 400 h xenon-arc weathered,
c 1200 h xenon-arc weathered,
and HDPE-holocellulose
composites d unweathered,
e 400 h xenon-arc weathered,
f 1200 h xenon-arc weathered
J Polym Environ (2009) 17:34–48 39
123
presence of C0 and C1 in the composites may be attributed
to the aliphatic and aromatic carbons in lignin and
extractives and/or HDPE matrix. Freudenberg and Neish
[40] outlined some assumptions relating to the origin of C1s
carbons in wood and these were adopted in this study.
These assumptions are thereby outlined. In ideal situations,
cellulose lacks C1 because of the polysaccharide structure;
however, 49% of the carbon atoms consist of C1 type in
milled wood lignin. Furthermore, C2 could be attributed to
primary and secondary alcohols in lignin, extractives, and
polysaccharides. The presence of C3 carbon could be
associated with the aliphatic or aromatic ethers in lignin
and extractives, and to carbonyl groups (carboxylic,
ketones, esters or aldehydes) in lignin, extractives, and
hemicellulose. Also, C4 could be a contribution from the
carboxylic acids and esters in lignin, extractives and cel-
lulose. In addition, C3 and C4 may also be an indication of
carbonyl groups formation at the weathered WPC surface.
This is because the carbonyl concentrations increased upon
weathering as observed under IR analysis. Lojewska, et al.,
[41] stated that the carbon atoms, which are most prone to
oxidation, occupy the 2, 3, and 6 positions in glycopyr-
anosyl moieties of the cellulose chain.
Figure 9 showed that there was a significant increase in
C3 and C4 peak from 0 to 1200 h of WPC exposure in
xenon-arc weathering regime. Table 3 show that C0 (C=C)
10
20
30
40
50
12008004000
Ligh
tnes
s ( ∆
Lre
l, %
)
Exposure time (h)
Pine
Extractive free
Holocellulose
5
15
25
35
Col
or c
hang
e (∆
Eab
)
PineExtractive freeHolocellulose
12008004000
Exposure time (h)
(a)
(b)
Fig. 2 Effect of xenon-arc weathering on the a DLrel and b DEab of
HDPE based WPC made from pine, extractives free pine, and pine
holocellulose
-5
5
15
0 30 60 90 120Sur
face
ligh
tnes
s (
L rel, %
)
Exposure time (days)
PineExtractive freeHolocellulose
0
5
10
15
Col
or c
hang
e (
Eab
)PineExtractive freeHolocellulose
∆∆
0 30 60 90 120
Exposure time (days)
(a)
(b)
Fig. 3 Effect of outside weathering on the a DLrel and b DEab of
WPC made from pine, extractives free pine, and pine holocellulose
0.8
0.7
0.6
0.5
0.4
0.3
1750 1700 1650
Carboxylic acid
1200h
Abs
orba
nce
Wavenumber (cm-1)
Ester
Fig. 4 Curve fitted for IR carbonyl region (1800–1680 cm-1) of
1200 xenon-arc weathered HDPE-pine composite
40 J Polym Environ (2009) 17:34–48
123

and C1 (C–C or C–H) peak intensity decreased upon
weathering, indicating a decrease in C1 peak. This is con-
trary to the result obtained by IR data. The decreased C=C
groups from XPS data may be due to the contribution of the
decreased lignin content at the surface of weathered WPC.
On the other hand, C2, C3, and C4, which correspond to
C–OH, O–C–O or C=O, and O–C=O respectively,
increased upon WPC weathering. This suggests that the
concentrations of the oxidized carbons increase with longer
exposure time of composites to varying environmental
conditions. Note that C3 and C4 contributed mostly to the
increase in surface oxidation relative to C2 as they
increased from 8 to 21% (C3) and 7 to 17% and (C4)
(Table 3). Indeed, C2, C3 and C4 increased by 38, 167 and
153%, respectively, comparatively to the unweathered
composites. Relative atomic weight (%) for carbon and
oxygen were obtained from the integration of peak areas of
the different emissions corrected for intensity with an
appropriate sensitivity factor for each of the XPS spectra.
The carbon and oxygen compositions are presented in
Table 3. In order to substantiate that surface oxidation had
occurred, as suggested from the increased oxidized car-
bons, the degree of surface oxidation was calculated by
finding the ratios of oxygen to carbon atoms. In addition,
the relationship (Eq. 5) developed by Matuana and Kam-
dem [27] for the computation of the ratio of oxidized to
unoxidized carbon atoms was adopted.
Coxidized
Cunoxidized
¼ C2þ C3þ C4
C1ð5Þ
The ratios of oxygen to carbon and oxidized to
unoxidized carbon atoms increased upon weathering
(Fig. 10 and Table 3). This is an indication that oxidation
had occurred. This provides supporting evidence to the
increased carbonyl groups concentrations observed by IR
analysis. Matuana and Kamdem [27] suggested that the
0 400 800 12000.0
0.4
0.8
1.2
1.6
2.0
Pine Extractive free Holocellulose
Con
cent
ratio
n at
171
5 cm
-1 (
mol
/kg)
Exposure time (h)
0.4
0.8
Pine Extractive free Holocellulose
Con
cent
ratio
n at
173
5 cm
-1 (
mol
/kg)
0 400 800 1200Exposure time (h)
(a)
(b)
Fig. 5 Effect of xenon-arc accelerated weathering on the concentra-
tion of a carboxylic acid, 1715 cm-1 and b esters, 1735 cm-1 present
in composites made from pine, extractives free pine, and pine
holocellulose
0 30 60 90 1200.0
0.4
0.8
1.2
1.6
2.0
2.4
Pine Extractive free Holocellulose
Con
cent
ratio
n at
171
5 cm
-1 (
mol
/kg)
Exposure time (days)
0.0
0.4
0.8
1.2
1.6
Pine Extractive free Holocellulose
Con
cent
ratio
n at
173
5 cm
-1 (
mol
/kg)
0 30 60 90 120Exposure time (days)
(a)
(b)
Fig. 6 Effect of outside weathering on the concentration of a carbox-
ylic acid, 1715 cm-1 and b esters, 1735 cm-1 present in composites
made from pine, extractives free pine, and pine holocellulose
J Polym Environ (2009) 17:34–48 41
123

increased oxidation due to weathering might probably
have occurred from the oxidized carbon, such as C=O and
O–C=O. This might have originated from the carbonyl
functional groups of wood fiber.
WPC Material Composition and Characterization
Pyrolysis Gas Chromatography-Mass Spectrometry
The chromatograms of the pyrolyzed unweathered and
weathered composites produced from untreated and
extractive free pine wood fibers are shown in Figs. 11 and
12 respectively. Molecular fragments from both wood and
plastic were obtained from the pyrolyzed specimens. The
wood derived compounds in HDPE/pine composites are
given in Table 4 [9, 42]. The same wood derived peaks
were found in extractive free wood fibers composites with
some variation in quantity.
Semi-quantitative analysis was conducted based on the
calibration technique employed. The total percentage area
of all the wood derived compounds identified at the surface
0 400 800 12000
1
2
3
4C
once
ntra
tion
at 1
635
cm-1 (
mol
/kg)
Exposure time (h)
Pine Extractive free Holocellulose
0 30 60 90 1200
1
2
3
4
Con
cent
ratio
n at
163
5 cm
-1 (
mol
/kg)
Exposure time (days)
Pine Extractive free Holocellulose
(a)
(b)
Fig. 7 Effect of a xenon-arc and b outside weathering on the C=C
(1635 cm-1) concentration in HDPE-based WPC made from pine,
extractives free pine, and pine holocellulose
0 400 800 12000
1
2
3
4
5
6
7
Con
cent
ratio
n at
334
0 cm
-1 (
mol
/kg)
Exposure time (h)
Pine Extractive free Holocellulose
0 30 60 90 1200
2
4
6
8
Con
cent
ratio
n at
334
0 cm
-1 (
mol
/kg)
Exposure time (days)
Pine Extractive free Holocellulose
(a)
(b)
Fig. 8 Effect of a xenon-arc and b outside weathering on the
hydroxyl (3340 cm-1) concentration in HDPE-based WPC made
from pine, extractives free pine, and pine holocellulose
Table 2 Assignments of peaks with their corresponding binding
energy and bond type for XPS scan of the C1s and O1s regions of
xenon-arc weathered HDPE/pine composites
Element group Binding energy (eV) Bond type
C1s C0 282.5 C=C
C1 285.0 C–C or C–H
C2 286.5 C–OH
C3 288.0 O–C–O or C=O
C4 289.5 O–C=O
O1s O1 530.4
O2 532.3
O3 535.6
Source: Dorris and Gray [38]; Grigsby et al. [39]
42 J Polym Environ (2009) 17:34–48
123
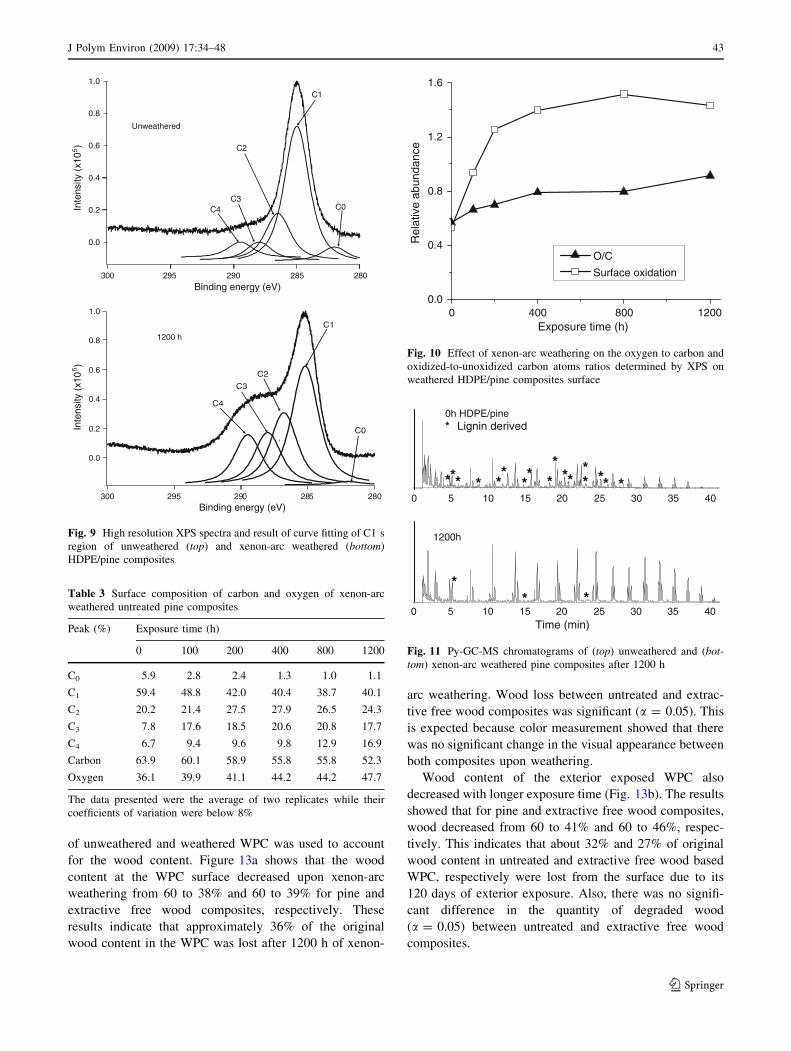
of unweathered and weathered WPC was used to account
for the wood content. Figure 13a shows that the wood
content at the WPC surface decreased upon xenon-arc
weathering from 60 to 38% and 60 to 39% for pine and
extractive free wood composites, respectively. These
results indicate that approximately 36% of the original
wood content in the WPC was lost after 1200 h of xenon-
arc weathering. Wood loss between untreated and extrac-
tive free wood composites was significant (a = 0.05). This
is expected because color measurement showed that there
was no significant change in the visual appearance between
both composites upon weathering.
Wood content of the exterior exposed WPC also
decreased with longer exposure time (Fig. 13b). The results
showed that for pine and extractive free wood composites,
wood decreased from 60 to 41% and 60 to 46%, respec-
tively. This indicates that about 32% and 27% of original
wood content in untreated and extractive free wood based
WPC, respectively were lost from the surface due to its
120 days of exterior exposure. Also, there was no signifi-
cant difference in the quantity of degraded wood
(a = 0.05) between untreated and extractive free wood
composites.
1.0
0.8
0.6
0.4
0.2
0.0
Inte
nsity
(x1
05)
300 295 290 285 280
Binding energy (eV)
C0
C1
C2
C3C4
Unweathered
1.0
0.8
0.6
0.4
0.2
0.0
300 295 290 285 280Binding energy (eV)
C0
C1
C2
C3
C4
1200 h
Inte
nsity
(x1
05)
Fig. 9 High resolution XPS spectra and result of curve fitting of C1 s
region of unweathered (top) and xenon-arc weathered (bottom)
HDPE/pine composites
Table 3 Surface composition of carbon and oxygen of xenon-arc
weathered untreated pine composites
Peak (%) Exposure time (h)
0 100 200 400 800 1200
C0 5.9 2.8 2.4 1.3 1.0 1.1
C1 59.4 48.8 42.0 40.4 38.7 40.1
C2 20.2 21.4 27.5 27.9 26.5 24.3
C3 7.8 17.6 18.5 20.6 20.8 17.7
C4 6.7 9.4 9.6 9.8 12.9 16.9
Carbon 63.9 60.1 58.9 55.8 55.8 52.3
Oxygen 36.1 39.9 41.1 44.2 44.2 47.7
The data presented were the average of two replicates while their
coefficients of variation were below 8%
0 400 800 12000.0
0.4
0.8
1.2
1.6
O/C
Surface oxidation
Rel
ativ
e ab
unda
nce
Exposure time (h)
Fig. 10 Effect of xenon-arc weathering on the oxygen to carbon and
oxidized-to-unoxidized carbon atoms ratios determined by XPS on
weathered HDPE/pine composites surface
0 5 10 15 20 25 30 35 40
*** * **
** *
**
**** ** *
0 5 10 15 20 25 30 35 40Time (min)
** *
1200h
0h HDPE/pine* Lignin derived
Fig. 11 Py-GC-MS chromatograms of (top) unweathered and (bot-tom) xenon-arc weathered pine composites after 1200 h
J Polym Environ (2009) 17:34–48 43
123
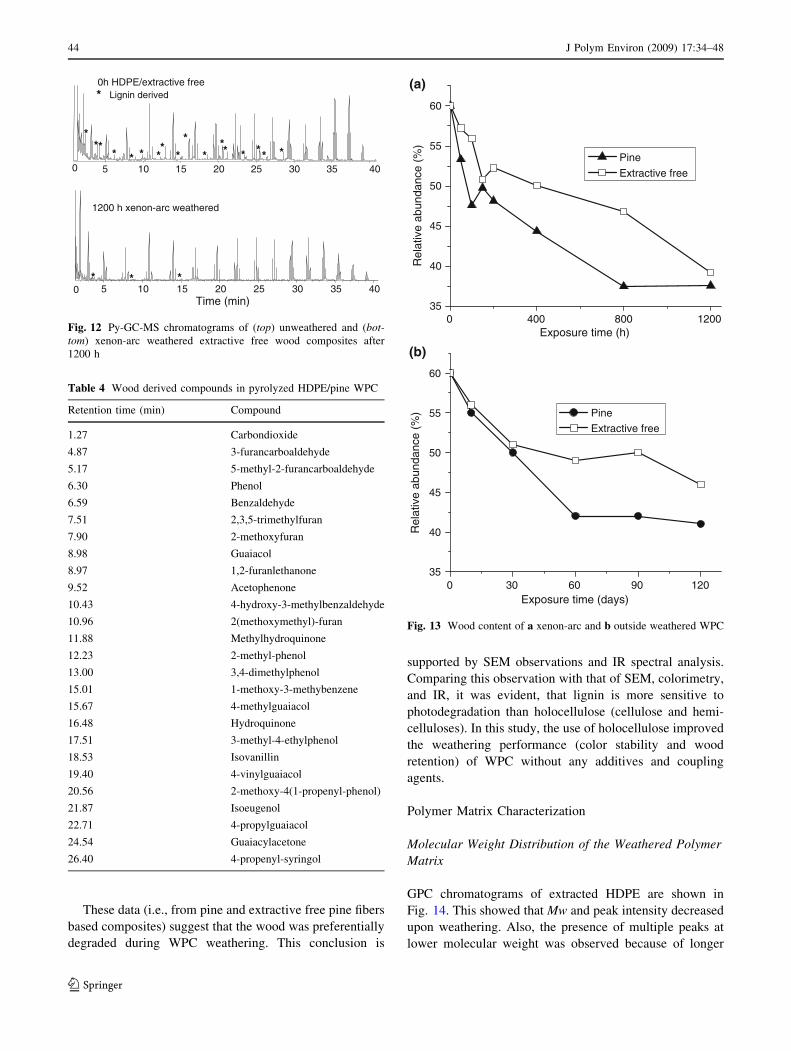
These data (i.e., from pine and extractive free pine fibers
based composites) suggest that the wood was preferentially
degraded during WPC weathering. This conclusion is
supported by SEM observations and IR spectral analysis.
Comparing this observation with that of SEM, colorimetry,
and IR, it was evident, that lignin is more sensitive to
photodegradation than holocellulose (cellulose and hemi-
celluloses). In this study, the use of holocellulose improved
the weathering performance (color stability and wood
retention) of WPC without any additives and coupling
agents.
Polymer Matrix Characterization
Molecular Weight Distribution of the Weathered Polymer
Matrix
GPC chromatograms of extracted HDPE are shown in
Fig. 14. This showed that Mw and peak intensity decreased
upon weathering. Also, the presence of multiple peaks at
lower molecular weight was observed because of longer
1200 h xenon-arc weathered
0h HDPE/extractive free* Lignin derived
5 10 15 20 25 30 35 40Time (min)
0
5 10 15 20 25 30 35 400
**
** *
* ** * ** *
**
**
**
*
*
Fig. 12 Py-GC-MS chromatograms of (top) unweathered and (bot-tom) xenon-arc weathered extractive free wood composites after
1200 h
Table 4 Wood derived compounds in pyrolyzed HDPE/pine WPC
Retention time (min) Compound
1.27 Carbondioxide
4.87 3-furancarboaldehyde
5.17 5-methyl-2-furancarboaldehyde
6.30 Phenol
6.59 Benzaldehyde
7.51 2,3,5-trimethylfuran
7.90 2-methoxyfuran
8.98 Guaiacol
8.97 1,2-furanlethanone
9.52 Acetophenone
10.43 4-hydroxy-3-methylbenzaldehyde
10.96 2(methoxymethyl)-furan
11.88 Methylhydroquinone
12.23 2-methyl-phenol
13.00 3,4-dimethylphenol
15.01 1-methoxy-3-methybenzene
15.67 4-methylguaiacol
16.48 Hydroquinone
17.51 3-methyl-4-ethylphenol
18.53 Isovanillin
19.40 4-vinylguaiacol
20.56 2-methoxy-4(1-propenyl-phenol)
21.87 Isoeugenol
22.71 4-propylguaiacol
24.54 Guaiacylacetone
26.40 4-propenyl-syringol
0 400 800 120035
40
45
50
55
60
Pine
Extractive free
Rel
ativ
e ab
unda
nce
(%)
Exposure time (h)
0 30 60 90 12035
40
45
50
55
60
Rel
ativ
e ab
unda
nce
(%)
Exposure time (days)
Pine Extractive free
(a)
(b)
Fig. 13 Wood content of a xenon-arc and b outside weathered WPC
44 J Polym Environ (2009) 17:34–48
123
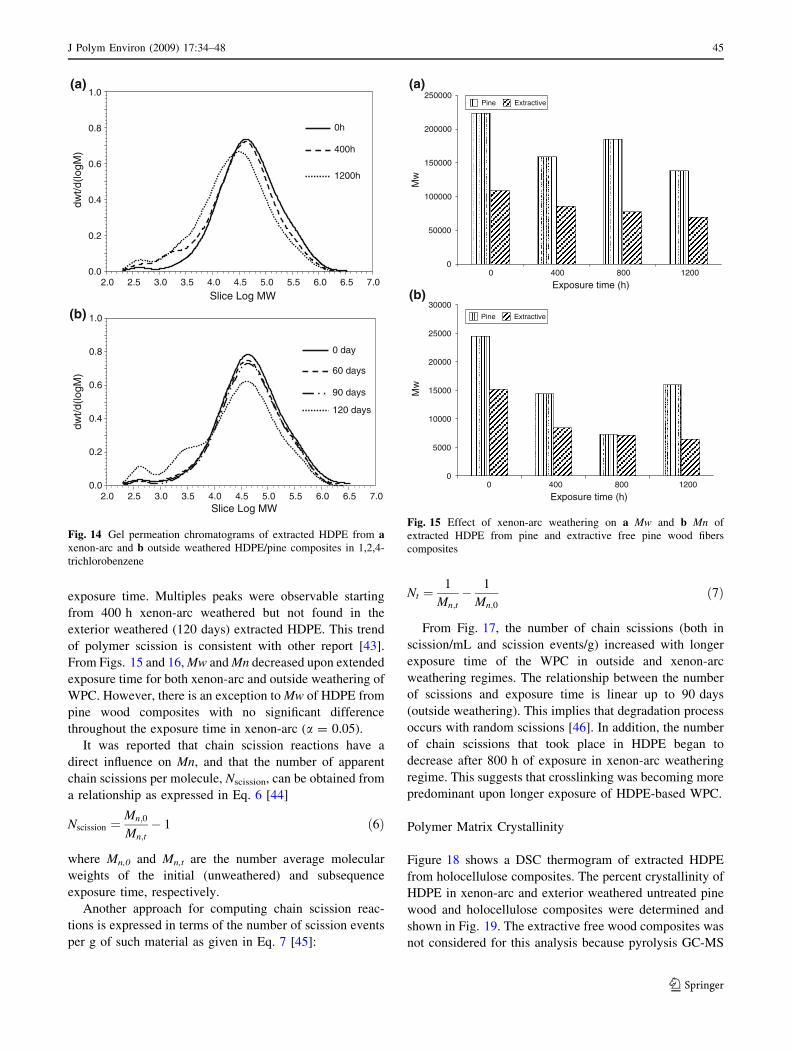
exposure time. Multiples peaks were observable starting
from 400 h xenon-arc weathered but not found in the
exterior weathered (120 days) extracted HDPE. This trend
of polymer scission is consistent with other report [43].
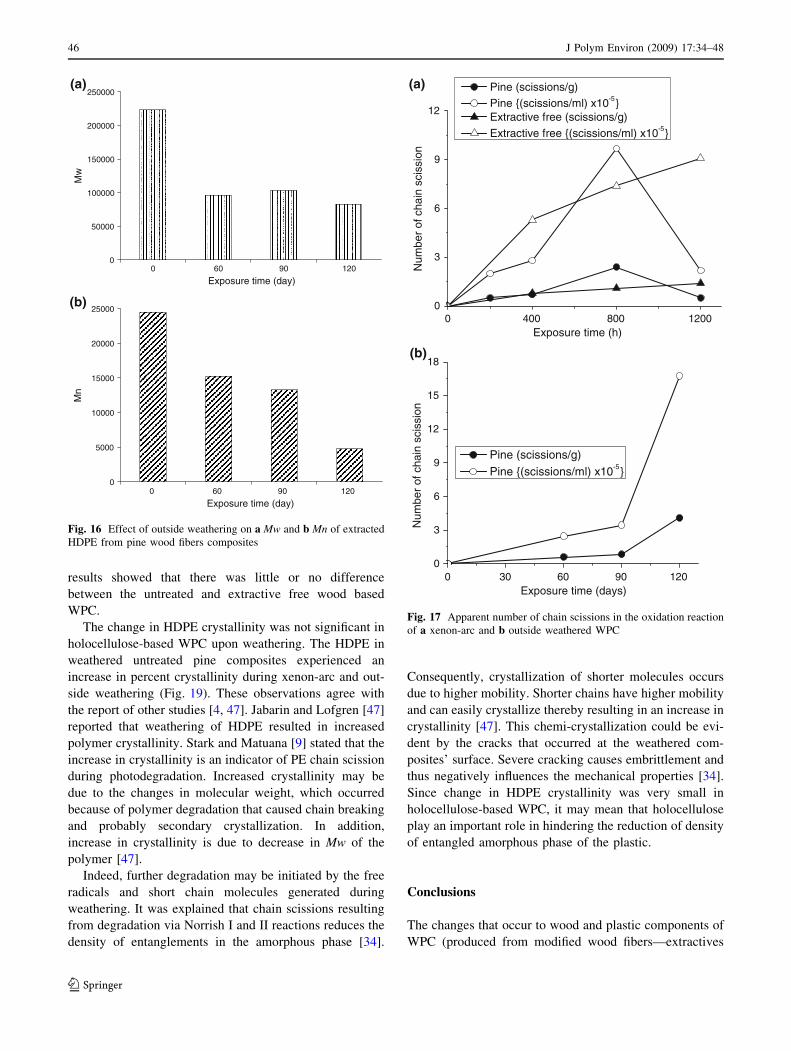
From Figs. 15 and 16, Mw and Mn decreased upon extended
exposure time for both xenon-arc and outside weathering of
WPC. However, there is an exception to Mw of HDPE from
pine wood composites with no significant difference
throughout the exposure time in xenon-arc (a = 0.05).
It was reported that chain scission reactions have a
direct influence on Mn, and that the number of apparent
chain scissions per molecule, Nscission, can be obtained from
a relationship as expressed in Eq. 6 [44]
Nscission ¼Mn;0
Mn;t� 1 ð6Þ
where Mn,0 and Mn,t are the number average molecular
weights of the initial (unweathered) and subsequence
exposure time, respectively.
Another approach for computing chain scission reac-
tions is expressed in terms of the number of scission events
per g of such material as given in Eq. 7 [45]:
Nt ¼1
Mn;t� 1
Mn;0ð7Þ
From Fig. 17, the number of chain scissions (both in
scission/mL and scission events/g) increased with longer
exposure time of the WPC in outside and xenon-arc
weathering regimes. The relationship between the number
of scissions and exposure time is linear up to 90 days
(outside weathering). This implies that degradation process
occurs with random scissions [46]. In addition, the number
of chain scissions that took place in HDPE began to
decrease after 800 h of exposure in xenon-arc weathering
regime. This suggests that crosslinking was becoming more
predominant upon longer exposure of HDPE-based WPC.
Polymer Matrix Crystallinity
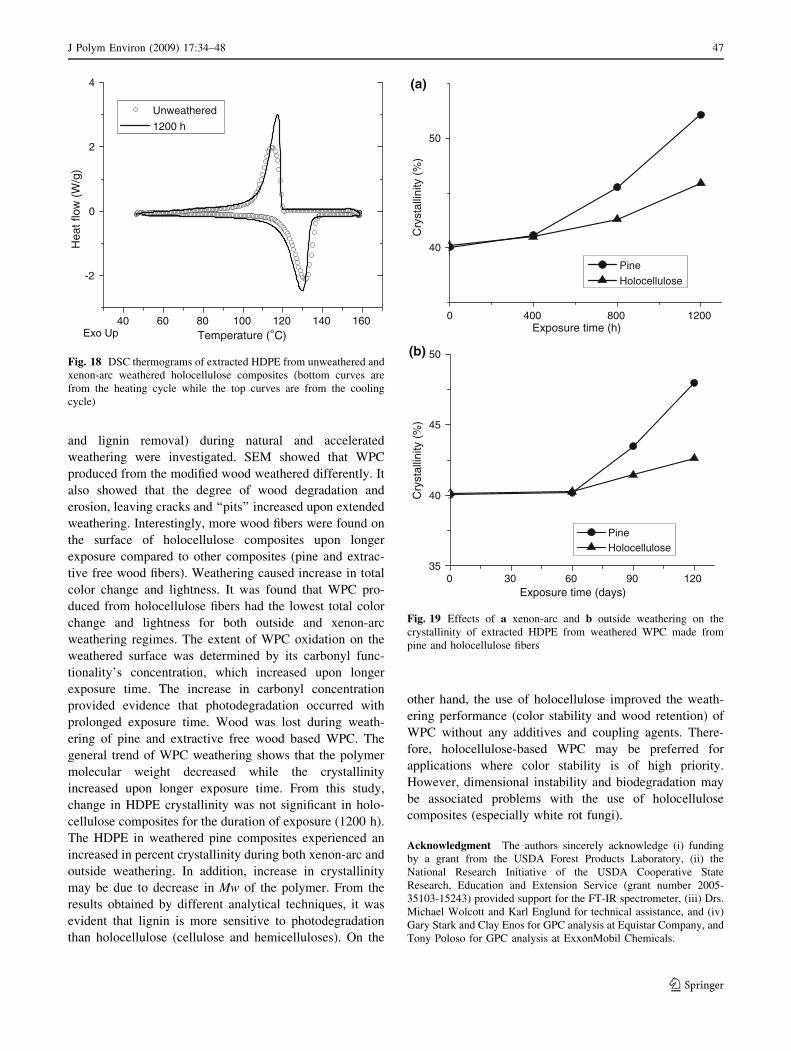
Figure 18 shows a DSC thermogram of extracted HDPE
from holocellulose composites. The percent crystallinity of
HDPE in xenon-arc and exterior weathered untreated pine
wood and holocellulose composites were determined and
shown in Fig. 19. The extractive free wood composites was
not considered for this analysis because pyrolysis GC-MS
0.0
0.2
0.4
0.6
0.8
1.0
Slice Log MW2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0
0h
400h
1200h
dwt/d
(logM
)dw
t/d(lo
gM)
0.0
0.2
0.4
0.6
0.8
1.0
Slice Log MW2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0
0 day
60 days
90 days
120 days
(a)
(b)
Fig. 14 Gel permeation chromatograms of extracted HDPE from axenon-arc and b outside weathered HDPE/pine composites in 1,2,4-
trichlorobenzene
0
50000
100000
150000
200000
250000
0 400 800 1200
Exposure time (h)
Mw
0
5000
10000
15000
20000
25000
30000
Mw
Pine Extractive
Pine Extractive
0 400 800 1200
Exposure time (h)
(a)
(b)
Fig. 15 Effect of xenon-arc weathering on a Mw and b Mn of
extracted HDPE from pine and extractive free pine wood fibers
composites
J Polym Environ (2009) 17:34–48 45
123
results showed that there was little or no difference
between the untreated and extractive free wood based
WPC.
The change in HDPE crystallinity was not significant in
holocellulose-based WPC upon weathering. The HDPE in
weathered untreated pine composites experienced an
increase in percent crystallinity during xenon-arc and out-
side weathering (Fig. 19). These observations agree with
the report of other studies [4, 47]. Jabarin and Lofgren [47]
reported that weathering of HDPE resulted in increased
polymer crystallinity. Stark and Matuana [9] stated that the
increase in crystallinity is an indicator of PE chain scission
during photodegradation. Increased crystallinity may be
due to the changes in molecular weight, which occurred
because of polymer degradation that caused chain breaking
and probably secondary crystallization. In addition,
increase in crystallinity is due to decrease in Mw of the
polymer [47].
Indeed, further degradation may be initiated by the free
radicals and short chain molecules generated during
weathering. It was explained that chain scissions resulting
from degradation via Norrish I and II reactions reduces the
density of entanglements in the amorphous phase [34].
Consequently, crystallization of shorter molecules occurs
due to higher mobility. Shorter chains have higher mobility
and can easily crystallize thereby resulting in an increase in
crystallinity [47]. This chemi-crystallization could be evi-
dent by the cracks that occurred at the weathered com-
posites’ surface. Severe cracking causes embrittlement and
thus negatively influences the mechanical properties [34].
Since change in HDPE crystallinity was very small in
holocellulose-based WPC, it may mean that holocellulose
play an important role in hindering the reduction of density
of entangled amorphous phase of the plastic.
Conclusions
The changes that occur to wood and plastic components of
WPC (produced from modified wood fibers—extractives
0
50000
100000
150000
200000
250000
0 60 90 120
Exposure time (day)
Mw
0
5000
10000
15000
20000
25000
Mn
0 60 90 120
Exposure time (day)
(a)
(b)
Fig. 16 Effect of outside weathering on a Mw and b Mn of extracted
HDPE from pine wood fibers composites
0 400 800 12000
3
6
9
12
Num
ber
of c
hain
sci
ssio
n
Exposure time (h)
Pine (scissions/g) Pine {(scissions/ml) x10-5} Extractive free (scissions/g) Extractive free {(scissions/ml) x10-5}
0 30 60 90 1200
3
6
9
12
15
18
Num
ber
of c
hain
sci
ssio
n
Exposure time (days)
Pine (scissions/g) Pine {(scissions/ml) x10-5}
(a)
(b)
Fig. 17 Apparent number of chain scissions in the oxidation reaction
of a xenon-arc and b outside weathered WPC
46 J Polym Environ (2009) 17:34–48
123
and lignin removal) during natural and accelerated
weathering were investigated. SEM showed that WPC
produced from the modified wood weathered differently. It
also showed that the degree of wood degradation and
erosion, leaving cracks and ‘‘pits’’ increased upon extended
weathering. Interestingly, more wood fibers were found on
the surface of holocellulose composites upon longer
exposure compared to other composites (pine and extrac-
tive free wood fibers). Weathering caused increase in total
color change and lightness. It was found that WPC pro-
duced from holocellulose fibers had the lowest total color
change and lightness for both outside and xenon-arc
weathering regimes. The extent of WPC oxidation on the
weathered surface was determined by its carbonyl func-
tionality’s concentration, which increased upon longer
exposure time. The increase in carbonyl concentration
provided evidence that photodegradation occurred with
prolonged exposure time. Wood was lost during weath-
ering of pine and extractive free wood based WPC. The
general trend of WPC weathering shows that the polymer
molecular weight decreased while the crystallinity
increased upon longer exposure time. From this study,
change in HDPE crystallinity was not significant in holo-
cellulose composites for the duration of exposure (1200 h).
The HDPE in weathered pine composites experienced an
increased in percent crystallinity during both xenon-arc and
outside weathering. In addition, increase in crystallinity
may be due to decrease in Mw of the polymer. From the
results obtained by different analytical techniques, it was
evident that lignin is more sensitive to photodegradation
than holocellulose (cellulose and hemicelluloses). On the
other hand, the use of holocellulose improved the weath-
ering performance (color stability and wood retention) of
WPC without any additives and coupling agents. There-
fore, holocellulose-based WPC may be preferred for
applications where color stability is of high priority.
However, dimensional instability and biodegradation may
be associated problems with the use of holocellulose
composites (especially white rot fungi).
Acknowledgment The authors sincerely acknowledge (i) funding
by a grant from the USDA Forest Products Laboratory, (ii) the
National Research Initiative of the USDA Cooperative State
Research, Education and Extension Service (grant number 2005-
35103-15243) provided support for the FT-IR spectrometer, (iii) Drs.
Michael Wolcott and Karl Englund for technical assistance, and (iv)
Gary Stark and Clay Enos for GPC analysis at Equistar Company, and
Tony Poloso for GPC analysis at ExxonMobil Chemicals.
40 60 80 100 120 140 160
-2
0
2
4
Unweathered
1200 h
Hea
t flo
w (
W/g
)
Temperature (°C)Exo Up
Fig. 18 DSC thermograms of extracted HDPE from unweathered and
xenon-arc weathered holocellulose composites (bottom curves are
from the heating cycle while the top curves are from the cooling
cycle)
0 400 800 1200
40
50
Cry
stal
linity
(%
)
Exposure time (h)
Pine
Holocellulose
0 30 60 90 12035
40
45
50
Pine Holocellulose
Cry
stal
linity
(%
)
Exposure time (days)
(a)
(b)
Fig. 19 Effects of a xenon-arc and b outside weathering on the
crystallinity of extracted HDPE from weathered WPC made from
pine and holocellulose fibers
J Polym Environ (2009) 17:34–48 47
123

References
1. Clemons C (2002) For Prod J 52:10–18
2. Smith PM, Wolcott MP (2005) In: Proceedings of the 8th inter-
national conference on woodfiber-plastic composites, Madison,
WI, pp 335–343, May 23–25
3. Klyosov A (2006) In: Proceeding progress in wood and bio-fibre
plastic composites international conference, Toronto
4. Stark NM, Matuana LM (2004) Polym Degrad Stab 86:1–9
5. Stark NM, Matuana LM, Clemons CM (2004) J Appl Polym Sci
93:1021–1030
6. Stark NM, Matuana LM (2006) Polym Degrad Stab 91:3048–
3056
7. Muasher M, Sain M (2006) Polym Degrad Stab 91:1156–1165
8. Stark NM, Matuana LM (2007) Polym Degrad Stab 92:1883–
1890
9. Fabiyi JS, McDonald AG, Wolcott MP (2005) In: Proceeding of
the 8th international conference on woodfiber-plastic composites,
Madison, WI, pp 191–196, May 23–25
10. Sears KD, Jacobson R, Caulfield DF, Underwood J (2001) In:
Proceedings of the 6th International Confrence on Wood-plastic
composites, Madison, WI, pp 27–34
11. Philip M, Attwood J, Hulme A, Williams G, Shipton P (2004)
The waste and resources action programme, p 113
12. Torik AA, Shirakawa H, Nagaya S (1990) J Appl Polym Sci
40:1637–1646
13. Isasi JR, Mandelkern L, Galante MAJ, Alamo RG (1999) J Polym
Sci Part B: Polym Phys 37:323–334
14. Technical Association of Pulp and Paper Industry (TAPPI T 204
om-88) (1996) Atlanta, Georgia, pp 1–3
15. Wise LE, Murphy M, D’ Addireco AA (1946) Paper Trade J
122:35–43
16. American Society Testing and Materials (ASTM D 790-00)
(2001) Annual book of ASTM standards. Conshohocken, PA,
08(01):145–153
17. American Society Testing and Materials (ASTM D 6662) (2003)
Annual book of ASTM standards. Conshohocken, PA,
08(03):1035–1049
18. American Society Testing and Materials (ASTM D 1435) (2003)
ASTM annual book. Conshohocken, PA, 08(03):308–312
19. American Society Testing and Materials (ASTM D 2244) (2001)
Annual book of ASTM standards. Conshohocken, PA, 06(01):
240–249
20. CIE (1986) CIE publication number 15.2, 2nd edn. Vienna,
Austria, p 74
21. Tidjani A (2000) Polym Degrad Stab 68:465–469
22. Mayo DW (2003) In: Mayo DW, Miller FA, Hannah RW (eds)
Course Notes on the interpretation of infrared and Raman spectra.
John, USA, pp 179–204
23. Lacoste J, Carlsson DJ (1992) J Polym Sci Part A Polym Chem
30:493–500
24. Lacoste J, Carlsson DJ, Falicki S, Wiles DM (1991) Polym
Degrad Stab 34:309–329
25. Fabiyi JS, McDonald AG, Wolcott MP, Griffiths PJ (2008) Polym
Degrad Stab (in press)
26. Kamdem DP, Rield B, Adnot A, Kaliaguine S (1991) J Appl
Polym Sci 43:1901–1912
27. Matuana LM, Kamdem DP (2002) Polym Eng Sci 42:1657–1666
28. McDonald AG, Clare AB, Dawson D (1999) In: Proceeding
of the 53rd appita annual confrence, Rotorua, New Zealand 1,
pp 51–56
29. Schauwecker C, Morrell JJ, McDonald AG, Fabiyi JS (2006) For
Prod J 56:123–129
30. American Society Testing and Materials (ASTM D 3418) (1999)
Philadelphia, PA, 8(03):337–341
31. Kodjie SL, Li L, Li B, Cai W, Li CY, Keating M (2006) J
Macromol Sci-Part B: Phys 45:231–245
32. Supaphol P, Spruiell JE (2002) J Appl Polym Sci 86:1009–1022
33. Selden R, Nystrom B, Langstrom R (2004) Polym Comp 25:543–
553
34. Rabello MS, White JR (1997) Polym 38:6379–6387
35. Stark NC (2003) PhD thesis, School of Forest Research and
Environment Science, Michigan Technical University, p 217
36. Faix O (1992) In: Lin SY, Dence CW (eds) Methods in Lignin
Chemistry. Springer-Verlag, Berlin, pp 83–132
37. Mononen K, Jaaskelainen AS (2005) Holzforschung 59:381–388
38. Dorris GM, Gray DG (1978) Cellul Chem Tech 12:721–734
39. Grigsby W, McDonald AG, Thumm A, Loxton C (2004) Holz
Roh ahs und Werkstoff 62:358–364
40. Freudenberg K, Neish AC (1968) Constitution and biosynthesis
of lignin. Springer Verlag, Berlin
41. Lojewska J, Miskowiec P, Lojewski T, Proniewicz LM (2005)
Polym Degrad Stab 88:512–520
42. Meier D, Faix O (1992) In: Lin SY, Dence CW (eds) Springer-
Verlag, Berlin Heidelberg, pp 177–199
43. Robertson JE (2001) PhD Dissertation, Virginia Polytechnic
Institute and State University, p 185
44. Chiantore O, Tripodi S, Sarmoria C, Valle AE (2001) Polym
42:3981–3987
45. Hoekstra HD, Spoormaker JL, Breent J, Audouin L, Verdu J
(1995) Polym Degrad Stab 49:251–262
46. MacCallum JR (1965) Makromol Chem 83:129–136
47. Jabarin SA, Lofgren EA (1994) J Appl Polym Sci 53:411–423
48 J Polym Environ (2009) 17:34–48
123






























