



Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
ORIGINAL ARTICLE
The prospects for peroxidase-based biorefining of petroleum fuels
MARCELA AYALA, JORGE VERDIN, & RAFAEL VAZQUEZ-DUHALT
Departamento de Ingenierıa Celular y Biocatalisis, Instituto de Biotecnologıa, Universidad Nacional Autonoma de Mexico,
UNAM. Apartado Postal 510-3 Cuernavaca, Mor. 62250 Mexico
AbstractPeroxidases have many potential uses for biotechnological processes. In this review, peroxidase-catalyzed reactionspotentially applicable to the petroleum industry are described. Although peroxidases are attractive catalysts fordesulfurization, aromatic oxidation and asphaltene transformation, there are important issues that must be overcomebefore any industrial application can be considered. The opportunities and challenges of enzymatic petroleum biorefiningare documented and discussed, with emphasis on the available tools to design a biocatalyst with appropriate performancefor the oil and other industries.
Keywords: Petroleum biorefining, peroxidase, biocatalysis, desulfurization, biocracking, oxidative inactivation
Introduction
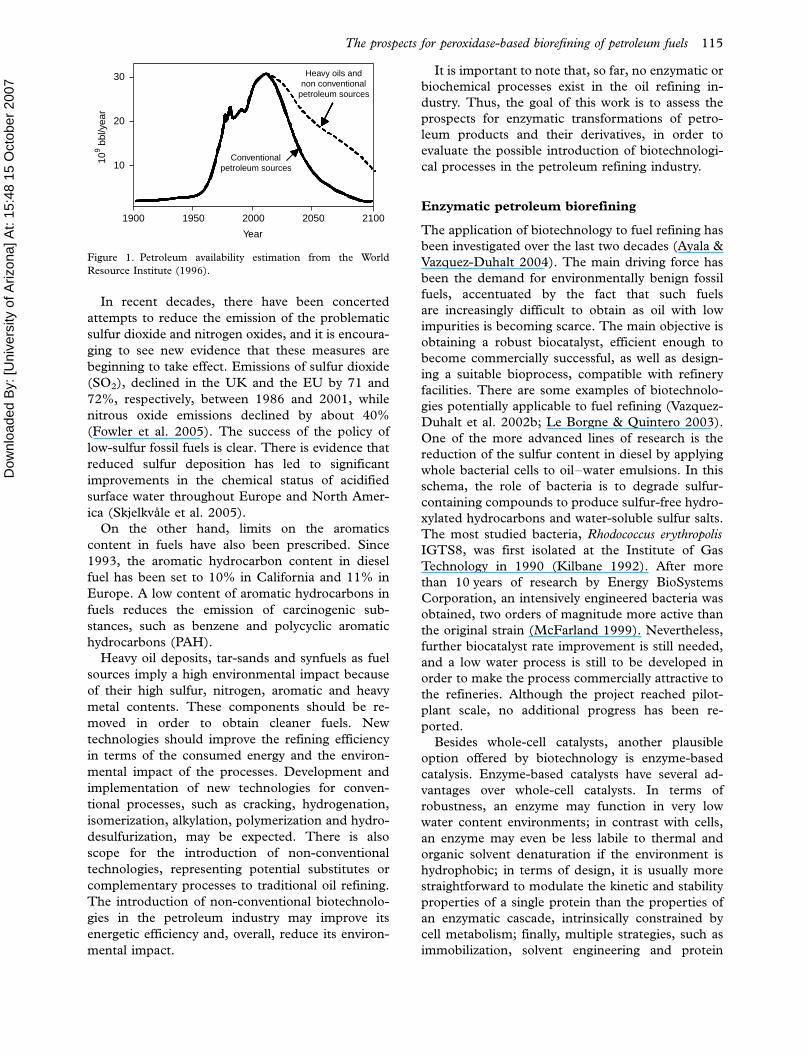
The exploitation of petroleum as a primary source
of both energy and raw material began a century
ago. Our society is highly dependent on oil for
energy, transportation and general industrial pro-
duction. Doubtless, history will describe our time
as the oil-based society. The world’s oil took
500 millions years to accumulate by nature, and it
will be consumed in only two centuries. The
production peak is estimated to occur sometime
between 2010 and 2020, and, by the end of this
century, oil resources will be drastically reduced
(Hall et al. 2003). When the world’s oil reserves
become scarce, more expensive fuel sources, such
as hard-to-extract oil deposits, tar sands, and
synfuels from coal, will come to the fore of
production (Figure 1).
The US Geological Survey recently estimated that
one trillion barrels have already been harvested, and
that about three trillion barrels of oil remain to be
recovered worldwide, half from proven reserves and
half from undeveloped or undiscovered sources
(Hall et al. 2003). On the other hand, the Institute
for the Analysis of Global Security (IAGS 2005)
estimated that since the shift from coal to oil,
the world has consumed over 875 billion barrels,
and less optimistically estimates that another
1000 billion barrels of proved and probable reserves
remain to be recovered. Finally, the Oil and Gas
Journal estimates that at the beginning of 2004,
worldwide reserves were 1.28 trillion barrels (Radler
2004). Thus, we can conclude from these estimates
that at least an equivalent amount of the total oil
consumed during the past 100 years is still to be
produced, but may require new methods of refining
during the next century.
Environment is a key issue for the petroleum
industry. Serious adverse environmental impact
arises from the emission of reactive hydrocarbons,
carbon monoxide, sulfur oxides and nitrogen oxides
when fossil fuels are burned. From the onset of the
Industrial Revolution in the eighteenth century up to
the mid-1970s, the emission of acidifying com-
pounds to the atmosphere increased steadily. These
pollutants are transported through the atmosphere
and deposited as ‘acid rain’, causing acidification of
soils and surface waters, with adverse effects on
terrestrial and freshwater biota. Increasing public
pressure to limit or reduce these emissions will
severely constrain the use of fossil fuels as a source
of energy supply. Unless this lost fossil fuel output is
replaced on a large scale by acceptable alternatives,
the cost, in terms of economic development fore-
gone, will be very high.
Correspondence: M. Ayala, Departamento de Ingenierıa Celular y Biocatalisis, Instituto de Biotecnologıa, Universidad Nacional
Autonoma de Mexico. A.P. 510-3. Cuernavaca, Morelos 62250, Mexico. Fax: 52 777 3172388. E-mail: [email protected]
Biocatalysis and Biotransformation, March�August 2007; 25(2�4): 114�129
ISSN 1024-2422 print/ISSN 1029-2446 online # 2007 Informa UK Ltd
DOI: 10.1080/10242420701379015
Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
In recent decades, there have been concerted
attempts to reduce the emission of the problematic
sulfur dioxide and nitrogen oxides, and it is encoura-
ging to see new evidence that these measures are
beginning to take effect. Emissions of sulfur dioxide
(SO2), declined in the UK and the EU by 71 and
72%, respectively, between 1986 and 2001, while
nitrous oxide emissions declined by about 40%
(Fowler et al. 2005). The success of the policy of
low-sulfur fossil fuels is clear. There is evidence that
reduced sulfur deposition has led to significant
improvements in the chemical status of acidified
surface water throughout Europe and North Amer-
ica (Skjelkvale et al. 2005).
On the other hand, limits on the aromatics
content in fuels have also been prescribed. Since
1993, the aromatic hydrocarbon content in diesel
fuel has been set to 10% in California and 11% in
Europe. A low content of aromatic hydrocarbons in
fuels reduces the emission of carcinogenic sub-
stances, such as benzene and polycyclic aromatic
hydrocarbons (PAH).
Heavy oil deposits, tar-sands and synfuels as fuel
sources imply a high environmental impact because
of their high sulfur, nitrogen, aromatic and heavy
metal contents. These components should be re-
moved in order to obtain cleaner fuels. New
technologies should improve the refining efficiency
in terms of the consumed energy and the environ-
mental impact of the processes. Development and
implementation of new technologies for conven-
tional processes, such as cracking, hydrogenation,
isomerization, alkylation, polymerization and hydro-
desulfurization, may be expected. There is also
scope for the introduction of non-conventional
technologies, representing potential substitutes or
complementary processes to traditional oil refining.
The introduction of non-conventional biotechnolo-
gies in the petroleum industry may improve its
energetic efficiency and, overall, reduce its environ-
mental impact.
It is important to note that, so far, no enzymatic or
biochemical processes exist in the oil refining in-
dustry. Thus, the goal of this work is to assess the
prospects for enzymatic transformations of petro-
leum products and their derivatives, in order to
evaluate the possible introduction of biotechnologi-
cal processes in the petroleum refining industry.
Enzymatic petroleum biorefining
The application of biotechnology to fuel refining has
been investigated over the last two decades (Ayala &
Vazquez-Duhalt 2004). The main driving force has
been the demand for environmentally benign fossil
fuels, accentuated by the fact that such fuels
are increasingly difficult to obtain as oil with low
impurities is becoming scarce. The main objective is
obtaining a robust biocatalyst, efficient enough to
become commercially successful, as well as design-
ing a suitable bioprocess, compatible with refinery
facilities. There are some examples of biotechnolo-
gies potentially applicable to fuel refining (Vazquez-
Duhalt et al. 2002b; Le Borgne & Quintero 2003).
One of the more advanced lines of research is the
reduction of the sulfur content in diesel by applying
whole bacterial cells to oil�water emulsions. In this
schema, the role of bacteria is to degrade sulfur-
containing compounds to produce sulfur-free hydro-
xylated hydrocarbons and water-soluble sulfur salts.
The most studied bacteria, Rhodococcus erythropolis
IGTS8, was first isolated at the Institute of Gas
Technology in 1990 (Kilbane 1992). After more
than 10 years of research by Energy BioSystems
Corporation, an intensively engineered bacteria was
obtained, two orders of magnitude more active than
the original strain (McFarland 1999). Nevertheless,
further biocatalyst rate improvement is still needed,
and a low water process is still to be developed in
order to make the process commercially attractive to
the refineries. Although the project reached pilot-
plant scale, no additional progress has been re-
ported.
Besides whole-cell catalysts, another plausible
option offered by biotechnology is enzyme-based
catalysis. Enzyme-based catalysts have several ad-
vantages over whole-cell catalysts. In terms of
robustness, an enzyme may function in very low
water content environments; in contrast with cells,
an enzyme may even be less labile to thermal and
organic solvent denaturation if the environment is
hydrophobic; in terms of design, it is usually more
straightforward to modulate the kinetic and stability
properties of a single protein than the properties of
an enzymatic cascade, intrinsically constrained by
cell metabolism; finally, multiple strategies, such as
immobilization, solvent engineering and protein
200019501900 2050 2100
Conventionalpetroleum sources
Heavy oils andnon conventional
petroleum sources
30
109 b
bl/y
ear
Year
20
10
Figure 1. Petroleum availability estimation from the World
Resource Institute (1996).
The prospects for peroxidase-based biorefining of petroleum fuels 115

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
engineering, may be combined in order to enhance
the desired characteristics of the enzyme.
Although sulfur content is one of the main
concerns of refineries around the world in terms of
fuel quality and environmental impact, other issues,
such as nitrogen content, aromatic and other heavy
molecule content, are also being actively studied.
These issues will acquire significance as heavier
oils become the common refinery feed, and refi-
neries will have to address the problem in order
to comply with environmental regulations. In the
next section, we analyze the potential of heme-
peroxidases, for the production of clean fossil fuels.
Heme-peroxidases as multifunctional catalysts
Peroxidases are enzymes able to catalyze oxido-
reduction reactions, using peroxide as electron
acceptor. Iron-peroxidases contain a prosthetic
heme group that functions as the active redox site.
Welinder (1992) proposed a classification of heme-
peroxidases according to their structural properties.
Three superfamilies of peroxidases have been de-
scribed: plant peroxidases, animal peroxidases, and
catalases. Due to their versatility, the enzymes
belonging to the plant superfamily are probably the
best candidates for industrial applications. The plant
peroxidase superfamily comprises three classes:
prokaryotic (class I), fungal (class II) and plant
(class III) peroxidases. All of these enzymes are
monomeric proteins with a non-covalently attached
ferriprotoporphyrin IX as prosthetic group, which is
co-ordinated to the enzyme via the imidazole side
chain of a His residue in the proximal pocket. On
the opposite, distal face of the heme, His and
Arg catalytic residues participate during the per-
oxide cleavage. Unlike prokaryotic peroxidases from
class I, fungal and plant peroxidases are glycosylated
(0�25%) and contain two calcium ions and
four disulfide bridges in their structure. Some
exceptions to this general classification can be
found, such as the heme-chloroperoxidase (CPO)
from Caldariomyces fumago (Sundaramoorthy et al.
1995). Representative members of this super-
family are cytochrome c peroxidase (CcP; class I),
lignin and manganese peroxidase (LiP and MnP;
class II) and horseradish peroxidase (HRP; class III).
The former and the latter have been extensively
studied and represent the best models for heme-
peroxidases. A comprehensive review on these en-
zymes is provided elsewhere (Erman & Vitello 2002;
Veitch 2004).
During catalysis, the oxidation of substrate mole-
cules results from the interaction with peroxide-
activated enzyme species (Figure 2). The cycle
begins when the two oxidative equivalents from the
peroxide are transferred to the enzyme to form
Compound I, a ferryl species (FeIV�O) coupled to
a free radical, located either on the porphyrin ring
or a nearby residue (Svistunenko 2005). Depending
on the substrate and the characteristics of the
enzyme’s active site, the two-electron reduction of
peroxide is coupled either to the one-electron (A,
Figure 2) or two-electron (B, Figure 2) oxidation of
substrate molecules. In pathway A, one-electron is
transferred from the substrate to the protein to form
Compound II, through interaction with the d-meso
heme edge (Ator & Ortiz de Montellano 1987;
DePillis et al. 1990; Gilfoyle et al. 1996) or through
long-range pathways, usually involving solvent-
exposed residues (Blodig et al. 2001; Perez-Boada
et al. 2005). The second enzyme intermediate,
Compound II, accepts a second electron to return
to the ground ferric state. In some enzymes, access
to the ferryl oxygen in Compound I results in two-
electron oxidation of substrates (pathway B); this
Figure 2. Peroxidase cycle showing classical peroxidase pathway (A) and peroxygenase pathway (B).
116 M. Ayala et al.

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
peroxygenase activity might proceed in an enantio-
selective way (van Deurzen et al. 1997). The redox
potential, and hence the ability, of the enzyme
intermediates to oxidize certain substrates depends
on the heme environment, such as the residues
forming the heme cavity, as well as the topology of
the active site.
Peroxidases do not possess a highly substrate-
specific active site, where binding of a narrow set of
molecules take place, as in the classical key-lock
mechanism usually described for enzymes. On the
contrary, as most substrates interact with the rela-
tively exposed d-meso heme edge and with solvent-
exposed residues, peroxidases usually display low
specificity, and, thus, are able to catalyze the
oxidation of a variety of molecules (Smith & Veitch
1998). Table I lists a number of different reactions
catalyzed by peroxidases with different substrates.
Thus, the most attractive features for the potential
development of peroxidase-based industrial biocata-
lysts, are the strong oxidant character and low
specificity of these enzymes.
Peroxidase-catalyzed reactions applicable
for petroleum refining
Oxidation of aromatic compounds
The content of aromatic and PAHs in refined fuels
may be as high as 30%. Aromatic content is
undesirable, as incomplete combustion of these
molecules leads to the formation of particulate
matter, recognized as a health problem. Moreover,
some of the PAH species are highly recalcitrant and
mutagenic, so their release into the environment is
detrimental. The selective oxidation of aromatic
molecules could either facilitate their removal or
reduce their environmental impact.
Class II peroxidases are able to catalyze the
oxidation of PAH to produce quinones or hydro-
xylated species, in aqueous systems containing mis-
cible organic solvents (Vazquez-Duhalt et al. 1994;
Bogan et al. 1996; Wang et al. 2003). The products
have been found to be more water-soluble and
significantly less mutagenic than the parental com-
pounds, or not mutagenic at all. An inverse correla-
tion between the peroxidase specific activity and
the ionization energy of the substrates has been
observed. This correlation suggests that the mech-
anism involves the formation of free radical species
via oxidative dehydrogenation, followed by reaction
with oxygen or water to yield the final product. The
maximum substrate ionization energy varies depend-
ing on the enzyme, as shown in Table II. Some
significant issues, such as the ability of peroxidases to
catalyze this reaction in non-aqueous systems, as well
their efficiency in transforming polyaromatic com-
plex mixtures, remain to be studied.
Desulfurization
Current refining processes are able to reduce the
sulfur content of diesel fuels to B50 ppm (Song
2003). However, this technology is rather expensive
as it is based on metal catalysts that are prone to
inactivation in the presence of common fuel impu-
rities (such as nitrogen-containing compounds) and
it consumes hydrogen for sulfur elimination. The
technology is less efficient with chemically complex
species, such as large or sterically hindered com-
pounds. Thus, a complementary technology may
be useful to polish fuels that are not efficiently
and economically desulfurized by conventional
technologies.
Peroxidases have been shown to catalyze the
oxidation of sulfur-containing molecules. This reac-
tion proceeds through the peroxygenase pathway
(see Figure 2, pathway B). Particularly, CPO has
the highest sulfoxidation activity reported to date
for a heme-peroxidase (van Deurzen et al. 1997).
This enzyme has been described as an unusual
Table I. Examples of the diversity of reactions catalyzed by
peroxidases.
Activity Reaction catalyzed Substrates
Peroxidase Oxidative
dehydrogenation
Phenols, anilines, indoles
N- and O-
demethylation
Methoxylated aromatics,
n-alkylated anilines,
carbazoles
Oxidative
dehalogenation
Halogenated phenols
Peroxygenase N- and S-
oxidation
Aromatic sulfides, anilines
C�H bond
oxidation
Benzyl, allyl and propargyl
derivatives, indoles
Epoxidation Aromatic and aliphatic
olefins
Catalase Peroxide
dismutation
Hydrogen peroxide,
organic peroxides
Haloperoxidase Oxidative
halogenation
Alicyclic ketones, PAHs,
phenols, anilines, indoles,
flavonoids
Table II. Ionization energy limit for the peroxidase-catalyzed
oxidation of polycyclic aromatic compounds.
Peroxidase
Ionization energy
limit (eV) Reference
Chloroperoxidase 8.18 Ayala et al. (2000)
Manganese peroxidase 8.03 Bogan et al. (1996)
Lignin peroxidase 7.96 Vazquez-Duhalt et al.
(1994)
Versatile peroxidase 7.42 Wang et al. (2003)
Horseradish peroxidase 7.35 Cavalieri et al. (1983)
The prospects for peroxidase-based biorefining of petroleum fuels 117
Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
heme-peroxidase as it resembles enzymes from the
cytochrome P450 family, where substrates may
access the iron of the heme group (Sundaramoorthy
et al. 1995). The transfer of oxygen from the ferryl
iron of Compound I to certain substrates is catalyzed
by CPO, and it has been enhanced in some HRP
mutants that lack aromatic residues blocking access
to the iron (Newmyer & Ortiz de Montellano 1995).
Sulfoxidation has been shown to proceed in an
enantioselective way, which suggests more specific
binding of the substrate when the enzyme catalyzes
peroxygenation reactions instead of classical perox-
idatic reactions. Apparently, there is no correlation
between the ionization energy of the substrates and
the specific activity of the enzyme, which suggests
that the sulfoxidation reaction might also be influ-
enced by other substrate properties (Ayala et al.
2000).
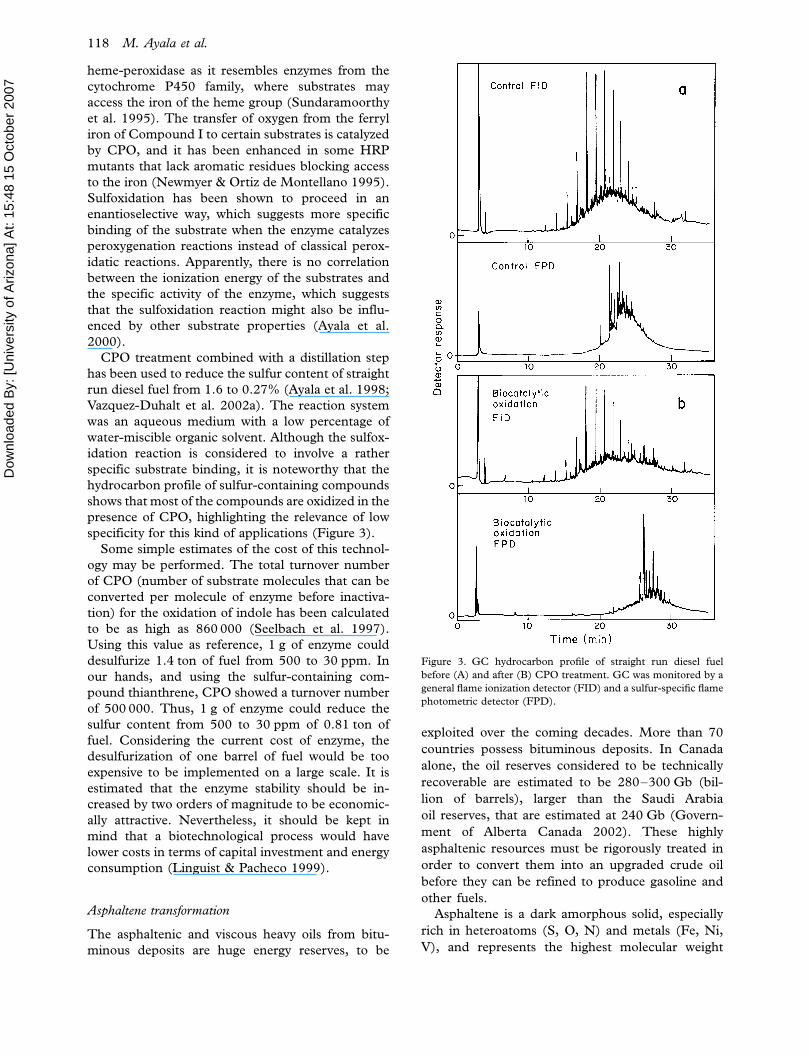
CPO treatment combined with a distillation step
has been used to reduce the sulfur content of straight
run diesel fuel from 1.6 to 0.27% (Ayala et al. 1998;
Vazquez-Duhalt et al. 2002a). The reaction system
was an aqueous medium with a low percentage of
water-miscible organic solvent. Although the sulfox-
idation reaction is considered to involve a rather
specific substrate binding, it is noteworthy that the
hydrocarbon profile of sulfur-containing compounds
shows that most of the compounds are oxidized in the
presence of CPO, highlighting the relevance of low
specificity for this kind of applications (Figure 3).
Some simple estimates of the cost of this technol-
ogy may be performed. The total turnover number
of CPO (number of substrate molecules that can be
converted per molecule of enzyme before inactiva-
tion) for the oxidation of indole has been calculated
to be as high as 860 000 (Seelbach et al. 1997).
Using this value as reference, 1 g of enzyme could
desulfurize 1.4 ton of fuel from 500 to 30 ppm. In
our hands, and using the sulfur-containing com-
pound thianthrene, CPO showed a turnover number
of 500 000. Thus, 1 g of enzyme could reduce the
sulfur content from 500 to 30 ppm of 0.81 ton of
fuel. Considering the current cost of enzyme, the
desulfurization of one barrel of fuel would be too
expensive to be implemented on a large scale. It is
estimated that the enzyme stability should be in-
creased by two orders of magnitude to be economic-
ally attractive. Nevertheless, it should be kept in
mind that a biotechnological process would have
lower costs in terms of capital investment and energy
consumption (Linguist & Pacheco 1999).
Asphaltene transformation
The asphaltenic and viscous heavy oils from bitu-
minous deposits are huge energy reserves, to be
exploited over the coming decades. More than 70
countries possess bituminous deposits. In Canada
alone, the oil reserves considered to be technically
recoverable are estimated to be 280�300 Gb (bil-
lion of barrels), larger than the Saudi Arabia
oil reserves, that are estimated at 240 Gb (Govern-
ment of Alberta Canada 2002). These highly
asphaltenic resources must be rigorously treated in
order to convert them into an upgraded crude oil
before they can be refined to produce gasoline and
other fuels.
Asphaltene is a dark amorphous solid, especially
rich in heteroatoms (S, O, N) and metals (Fe, Ni,
V), and represents the highest molecular weight
Figure 3. GC hydrocarbon profile of straight run diesel fuel
before (A) and after (B) CPO treatment. GC was monitored by a
general flame ionization detector (FID) and a sulfur-specific flame
photometric detector (FPD).
118 M. Ayala et al.

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
fraction of petroleum (Strausz et al. 1992). The
presence of a high concentration of asphaltenes is
the origin of many problems associated with either
recovery, separation or processing of heavy oils and
bitumen. This fraction is thought to be largely
responsible for other adverse oil properties, such as
high viscosity and the propensity to form emulsions,
polymers and coke. Considerable research has been
performed to elucidate the chemical nature of
asphaltenes, and their molecular structure has been
an enigma for several decades (Strausz et al. 1992;
Groenzin & Mullins 2000). There are indications
that asphaltenes are chemically diverse and, in
general, contain condensed aromatic cores substi-
tuted with alkyl and alicyclic moieties. Nitrogen,
oxygen and sulfur heteroatoms are present as non-
and heterocyclic groups. A significant amount of
porphyrins (petroporphyrins) can be found contain-
ing mainly nickel and vanadium. The proposed
chemical nature of asphaltene molecules has evolved
from a very complex, high molecular weight species,
comprising several functional groups in the same
molecule, to a collection of lower molecular weight
species, each bearing different functionalities. To
illustrate this evolution, the structures of asphaltene
molecules, proposed by Strausz et al. (1992)
and Groenzin and Mullins (2000), are shown in
Figure 4.
So far, there is no clear evidence that asphaltenes
are degraded by microbial activity. Thus, the asphal-
tenic fraction is recognized as the most recalcitrant
fraction of oil. Although microorganisms have been
found associated with bitumen containing high
amounts of asphaltenes (Wyndham & Costerton
1981), no changes in asphaltene content could be
found after bioconversion of heavy oils; furthermore,
the asphaltenic fraction did not support bacterial
growth (Premuzic et al. 1999; Thouand et al. 1999).
Nevertheless, the first clear experimental evidence
that enzymes are able to modify asphaltene mole-
cules was reported 13 years ago (Fedorak et al.
1993). CPO from the fungus C. fumago was able to
transform petroporphyrins and asphaltenes prefer-
ably in systems containing organic solvent (Fedorak
et al. 1993; Mogollon et al. 1998). Mass transfer
limitations are expected in aqueous reactions for
highly hydrophobic materials, such as asphaltenes
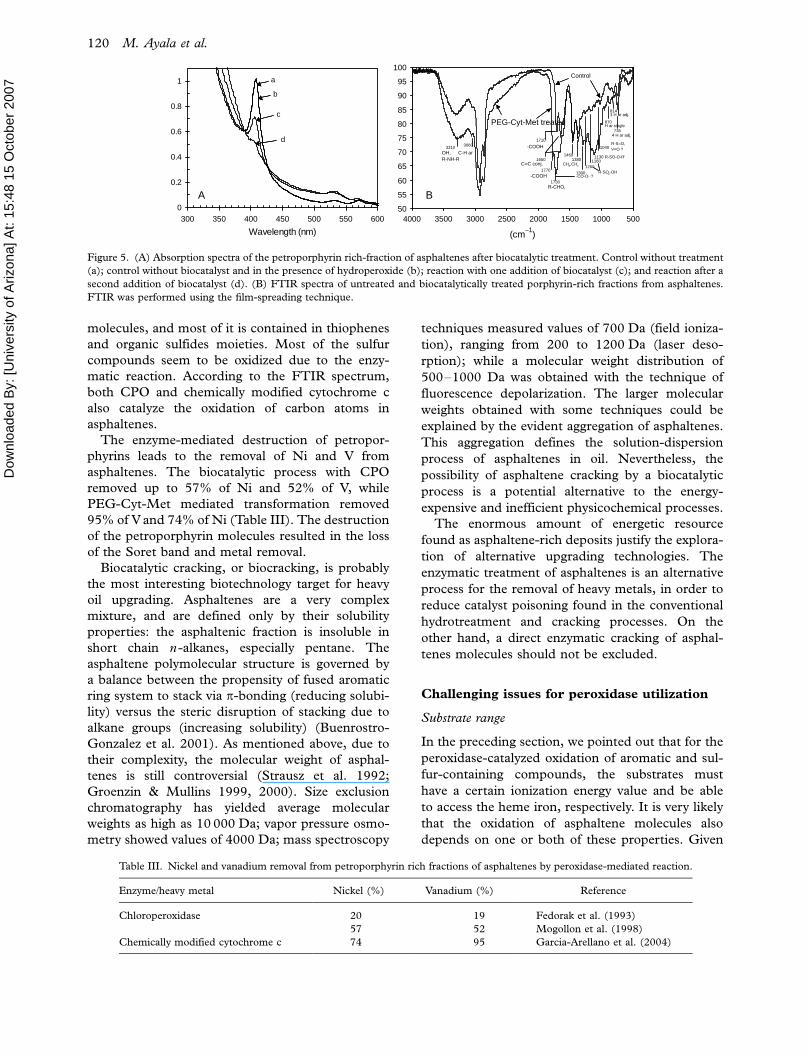
and petroporphyrins. CPO catalyzed the oxidation
of a petroporphyrin rich-fraction of asphaltenes in a
ternary solvent system and in the presence of
hydrogen peroxide, producing notable spectral
changes (Figure 5A).
Cytochrome c is a heme-protein that has perox-
idase-like activity in vitro. As CPO, a doubly
modified cytochrome c (PEG-Cyt-Met) was also
able to catalyze the oxidation of a petroporphyrin
rich-fraction of asphaltenes in a ternary solvent
system and in the presence of tert-butyl hydroper-
oxide (Garcia-Arellano et al. 2004). Both CPO and
PEG-Cyt-Met reactions produced spectral changes
in this petroporphyrin rich-fraction, as shown in
Figure 5(A) (Fedorak et al. 1993). Fourier trans-
form infrared spectroscopy (FTIR) analysis, be-
fore and after biocatalytic treatment, showed
significant differences, as illustrated in Figure 5(B)
(Garcia-Arellano et al. 2004). An increased pro-
portion of oxygen-containing groups, such as hy-
droxyl (3310 cm�1), carboxyl (1300, 1770 and
1710 cm�1), aldehydes (1730 cm�1), sulfoxides
(1040 cm�1), sulfones (1130 cm�1), and sulfonates
(1160 and 1260 cm�1) was detected. Sulfur is, after
carbon, the most important element in asphaltene
Figure 4. Proposed asphaltene molecules from (A) Strausz et al. (1992) and (B) Groenzin and Mullins (2000).
The prospects for peroxidase-based biorefining of petroleum fuels 119
Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
molecules, and most of it is contained in thiophenes
and organic sulfides moieties. Most of the sulfur
compounds seem to be oxidized due to the enzy-
matic reaction. According to the FTIR spectrum,
both CPO and chemically modified cytochrome c
also catalyze the oxidation of carbon atoms in
asphaltenes.
The enzyme-mediated destruction of petropor-
phyrins leads to the removal of Ni and V from
asphaltenes. The biocatalytic process with CPO
removed up to 57% of Ni and 52% of V, while
PEG-Cyt-Met mediated transformation removed
95% of V and 74% of Ni (Table III). The destruction
of the petroporphyrin molecules resulted in the loss
of the Soret band and metal removal.
Biocatalytic cracking, or biocracking, is probably
the most interesting biotechnology target for heavy
oil upgrading. Asphaltenes are a very complex
mixture, and are defined only by their solubility
properties: the asphaltenic fraction is insoluble in
short chain n-alkanes, especially pentane. The
asphaltene polymolecular structure is governed by
a balance between the propensity of fused aromatic
ring system to stack via p-bonding (reducing solubi-
lity) versus the steric disruption of stacking due to
alkane groups (increasing solubility) (Buenrostro-
Gonzalez et al. 2001). As mentioned above, due to
their complexity, the molecular weight of asphal-
tenes is still controversial (Strausz et al. 1992;
Groenzin & Mullins 1999, 2000). Size exclusion
chromatography has yielded average molecular
weights as high as 10 000 Da; vapor pressure osmo-
metry showed values of 4000 Da; mass spectroscopy
techniques measured values of 700 Da (field ioniza-
tion), ranging from 200 to 1200 Da (laser deso-
rption); while a molecular weight distribution of
500�1000 Da was obtained with the technique of
fluorescence depolarization. The larger molecular
weights obtained with some techniques could be
explained by the evident aggregation of asphaltenes.
This aggregation defines the solution-dispersion
process of asphaltenes in oil. Nevertheless, the
possibility of asphaltene cracking by a biocatalytic
process is a potential alternative to the energy-
expensive and inefficient physicochemical processes.
The enormous amount of energetic resource
found as asphaltene-rich deposits justify the explora-
tion of alternative upgrading technologies. The
enzymatic treatment of asphaltenes is an alternative
process for the removal of heavy metals, in order to
reduce catalyst poisoning found in the conventional
hydrotreatment and cracking processes. On the
other hand, a direct enzymatic cracking of asphal-
tenes molecules should not be excluded.
Challenging issues for peroxidase utilization
Substrate range
In the preceding section, we pointed out that for the
peroxidase-catalyzed oxidation of aromatic and sul-
fur-containing compounds, the substrates must
have a certain ionization energy value and be able
to access the heme iron, respectively. It is very likely
that the oxidation of asphaltene molecules also
depends on one or both of these properties. Given
50
55
60
65
70
75
80
85
90
95
100
5001000150020002500300035004000
(cm–1)
OH, R-NH-R
3310C-H ar
3060
1730R-CHO,
1770 -COOH
-COOH1710
C=C conj.1650
CH3,CH 2
14601380
1300
12601160
1130
1040R-S=O, V=O ?
7354 H ar adj.
8143 H ar adj.
870H ar single
R-SO2-OH
R-SO-O-R'
-CO-O- ?
Control
PEG-Cyt-Met treated
0
0.2
0.4
0.6
0.8
1
300 350 400 450 500 550 600
Wavelength (nm)
d
a
b
c
A B
Figure 5. (A) Absorption spectra of the petroporphyrin rich-fraction of asphaltenes after biocatalytic treatment. Control without treatment
(a); control without biocatalyst and in the presence of hydroperoxide (b); reaction with one addition of biocatalyst (c); and reaction after a
second addition of biocatalyst (d). (B) FTIR spectra of untreated and biocatalytically treated porphyrin-rich fractions from asphaltenes.
FTIR was performed using the film-spreading technique.
Table III. Nickel and vanadium removal from petroporphyrin rich fractions of asphaltenes by peroxidase-mediated reaction.
Enzyme/heavy metal Nickel (%) Vanadium (%) Reference
Chloroperoxidase 20 19 Fedorak et al. (1993)
57 52 Mogollon et al. (1998)
Chemically modified cytochrome c 74 95 Garcia-Arellano et al. (2004)
120 M. Ayala et al.

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
the complexity of the refinery streams, it would be
desirable that the enzymatic catalyst could recognize
a broad spectrum of substrates. In order to further
broaden the substrate range for peroxidases, we can
envisage two strategies.
Modulation of the redox potential of the enzyme. It is
known that the heme environment influences the
electronic states of Compound I and Compound II,
which ultimately determine the enzyme’s reactivity.
Table IV lists the experimental redox potential data
of the species involved in peroxidase catalysis. The
redox potential of the Fe�3/Fe�2 couple has been
interpreted to reflect the redox potential of Com-
pound I and Compound II (Millis et al. 1989).
Thus, one of the more oxidant enzymes in Table IV
would be manganese peroxidase, which catalyzes the
oxidation of Mn(II) to Mn(III) (Em�1500 mV).
There are a number of residues that are conserved
throughout the peroxidase plant superfamily which
surround the heme group, and form the distal and
proximal cavities. Following HRP numbering, Arg
38, Phe 41, His 42 and Asn 70 are present in the
distal cavity, while His 170, Phe 221 and Asp 247
comprise the proximal side. Distal Arg and His are
catalytic residues involved in peroxide cleavage,
while proximal His pentaco-ordinates the heme
group. Exceptions are found, such as CcP, that
bears Trp instead of Phe residues in both proximal
and distal sides; and ascorbate peroxidase (APX)
and Coprinus cinereus peroxidase (CiP), that bear a
Trp and Leu residue in the proximal side, respec-
tively, instead of a Phe residue.
Experimental evidence and theoretical studies
support the generalization that an anionic environ-
ment and a nearby electron-donating residue on the
proximal side, favor the transfer of the free radical
originally formed in the porphyrin ring. This is the
case for CcP and some F221W and F221Y HRP
mutants mimicking CcP, where proximal Trp or
Tyr residues provide an oxidizable site for the
formation of a protein-based free radical (Miller
et al. 1995; Morimoto et al. 1998; Wirstam et al.
1999). The redox potential of peroxidases depends
on the location of the free radical, as observed for the
F221W HRP mutant, which showed a more positive
redox potential of �178 mV for the Fe�3/Fe�2
couple, closer to that of CcP (Morimoto et al. 1998).
Thus, the introduction of cationic sites and removal
of the electron-donating residues, as well as a
diminished polarity of the heme proximal environ-
ment may favor the retention of the porphyrin-based
radical, as happens for HRP, and as theoretically and
experimentally determined for CcP and for APX
(Bonagura et al. 1996; Jensen et al. 1998; Wirstam
et al. 1999; de Visser et al. 2003; Barrows et al.
2004). Similar studies with other peroxidases
bearing protein-based radicals, such as LiP (Blodig
et al. 2001), versatile peroxidase (Perez-Boada et al.
2005), and turnip peroxidase (Ivancich et al. 2001),
would further test this hypothesis.
Two structural factors, on the proximal side, have
been proposed to determine the redox potential of
peroxidases. The first is related to the bond length
between the heme iron and the protein, which
depends on the relative position of the proximal
His. A weaker and longer bond between the His
residue and the heme iron would make the latter
more electron deficient, and, thus, destabilize the
higher oxidation states; this would lead to a more
positive redox potential for the Fe�3/Fe�2 couple,
and, thus, an increased oxidative capability of the
enzyme. Such a correlation has been observed for
CcP, LiP and MnP, but not for HRP (Banci 1997;
Table IV. Redox potential of different electronic states of peroxidases from the plant superfamily.
Peroxidase pH Redox couple Redox potential (mV) Reference
Horseradish peroxidase 7 Fe�3/Fe�2 �278* Yamada et al. (1975)
Compound I/Compound II 880 Farhangrazi et al. (1995)
898 Hayashi and Yamazaki (1979)
Compound II/Fe�3 870 Farhangrazi et al. (1995)
900 Hayashi and Yamazaki (1979)
Turnip peroxidase 8 Fe�3/Fe�2 �233 to 160* Ricard et al. (1972)
Cytochrome c peroxidase 6.1 Fe�3/Fe�2 �194* Conroy et al. (1978)
Compound I/Compound II 740* Mondal et al. (1996)
Compound II/Fe�3 1080 Purcell and Erman (1976)
C. cinereus peroxidase 7 Fe�3/Fe�2 �183* Battistuzzi et al. (2006)
Compound I/Compound II 915 Farhangrazi et al. (1994)
Compound II/Fe�3 982 Farhangrazi et al. (1994)
Chloroperoxidase 6.9 Fe�3/Fe�2 �140* Makino et al. (1976)
Lignin peroxidase 7 Fe�3/Fe�2 �120 to �140* Millis et al. (1989)
Manganese peroxidase 7 Fe�3/Fe�2 �88 to �93* Millis et al. (1989)
*Voltammetric data.
The prospects for peroxidase-based biorefining of petroleum fuels 121

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
Choinowski et al. 1999). The second structural
factor is related to the hydrogen bonding of the
proximal His. It has been suggested that strong
hydrogen bonding to neighboring residues increases
its basicity. The stronger imidazolate character
would stabilize the higher oxidations states, which
would result in a reduced redox potential value of
the Fe�3/Fe�2 couple (Poulos & Kraut 1981; Banci
et al. 1991). In this sense, the most important
residue is the proximal Asp, which forms a hydrogen
bond with the proximal His. Its substitution with
residues that modify the hydrogen bond network
would have an effect on the redox potential of the
Fe�3/Fe�2 couple. However, mixed results have
been observed. In CcP, substitution with a Glu
residue led to an increase of 70 mV in the redox
potential (Goodin & McRee 1993), whereas in MnP,
the same substitution resulted in a decreased redox
potential (Santucci et al. 2000). Moreover, substitu-
tion with the poor hydrogen-bonding Asn in CiP
decreased the redox potential of the enzyme, which
is in clear contradiction with the hypothesis men-
tioned above (Ciaccio et al. 2003). Clearly, the
modulation of the redox potential in peroxidases is
multifactorial and there are probably other contri-
buting factors.
One of these may be the two calcium-binding sites
in class II and III peroxidases. For instance, Verdin
et al. (2006) measured the redox potential of the Fe�3/
Fe�2 couple of calcium-depleted versatile peroxidase,
and noted a drastic reduction of 200 mV at catalytic
pH. Moreover, Howes et al. (2005) have shown that
the T171S HRP mutant retained the functional and
structural properties of the native enzyme. However, in
the mutant, the redox potential of the Fe�3/Fe�2
couple was increased by about 100 mV. Apparently,
the Thr 171 regulates the hydrogen bonding between
the structural calcium ion and the proximal His. It
would be interesting to test the ability of this mutant to
oxidize a broader range of substrates, with higher
redox potential values.
It has been noted that the environment in the distal
side mainly directs the peroxide activation process,
whereas the environment in the proximal side reg-
ulates the stability of enzyme intermediates and the
electron transfer process. However, Smulevich et al.
(1991) proved that changes in the distal side also
exert long-distance perturbations in the proximal
side. The substitution of His 181, Arg 48 and Trp
51 in the distal cavity of CcP disrupted the hydrogen
bond between the proximal Asp and His. Nagano
et al. (1996) substituted the distal Asn 70 in HRP for
Val or Asp, and noted that although the redox
potential of the Compound I/Compound II couple
was similar to the native enzyme, in the mutants the
Compound II was very unstable and the redox
potential of the Compound II/ferric enzyme couple
was about 100 mV higher. Finally, modification of the
heme itself also provides a means to manipulate the
redox potential of the enzyme. The role of heme
propionates has been discussed as modulators of the
nature and stability of Compound I (Barrows &
Poulos 2005; Guallar & Olsen 2006). For instance,
He et al. (1996) showed that introduction of electron-
withdrawing groups in the pyrrole ring of HRP
increased, by 100 mV, the redox potential of the
Compound I/Compound II couple.
Unfortunately, the redox potential data are not
available for most of the constructed peroxidase
mutants. Although the kinetic characterization pro-
vides some information on the substitution effects, it
does not reflect the effect on redox potential values.
Thus, better voltammetric or indirect methods to
estimate the redox potential values of the higher
oxidation states (Compound I and Compound II)
would greatly benefit experimental data interpreta-
tion, and the elucidation of other factors modulating
the oxidizing strength of peroxidases.
Topology of the active site. A second strategy to
broaden the substrate range of peroxidases has to
do with the topology found in the heme cavity.
Enhanced access to the heme iron or enlarged heme
cavity would improve the kinetics of the peroxygen-
ase activity, as demonstrated for plant peroxidases.
Fungal and plant peroxidases have an aromatic
residue (either Phe or Trp) adjacent to the distal
His. Apparently, the removal of this aromatic residue
unblocks the access to the ferryl oxygen, and, thus,
enhances peroxygenase activity. In a study by
Newmyer and Ortiz de Montellano (1995), the
sulfoxidation activity of HRP was increased 100-
fold by substituting the bulky Phe 41 by Ala.
Moreover, the mutants catalyzed styrene epoxida-
tion, an activity absent in the native enzyme.
Similarly, Celik et al. (2001) removed the distal
Trp in APX, increasing the sulfoxidation activity of
the mutant from 10- to 100-fold, as well as the
enantioselectivity of the reaction.
To increase heme cavity size, Tanaka et al. (1997)
constructed a H42E HRP mutant, inspired by the
unusual distal Glu residue found in CPO. They
confirmed that a Glu residue performs as an efficient
acid�base catalyst. Although the redox potential of
the Fe�3/Fe�2 couple remained practically un-
changed, the enlarged heme cavity led to a 50- to
1000-fold increase in the peroxygenase activity of
mutant HRP. Furthermore, during crystallographic
studies of recombinant soybean peroxidase (SBP),
Henriksen et al. (2001) observed that the Ile 74
caused displacement of the distal Phe 41 and His 42,
thus rendering a more open active site compared to
122 M. Ayala et al.

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
HRP, which bears an Ala residue in the analogous
position. Indeed, the sulfoxidation of thioanisole
was faster when catalyzed with SBP than with
HRP (Dai & Klibanov 2000). However, the hypoth-
esis that this enlarged heme cavity enhances perox-
ygenase activity by increasing the transformation
rates, has not been explored with fungal peroxidases.
Activity in organic media
One of the characteristics of an enzyme-based
process for the oil industry would be the ability to
function in organic media, with low water content.
Several features must be addressed in this regard.
Enzyme stabilization may be achieved by means
of chemical modification and immobilization, as
already demonstrated for several heme-proteins
(Takahashi et al. 1984; Miland et al. 1996; Ayala
et al. 2002; Garcia-Arellano et al. 2002; Wang et al.
2003; Bruns & Tiller 2005; Song et al. 2005; Liu
et al. 2006). The preparation of the enzyme also
influences the activity (Lee & Dordick 2002; Cao
et al. 2003); in the case of peroxidases, some studies
are available on the influence of the enzyme micro-
environment and biocatalyst preparation (Dai &
Klibanov 1999; Kimura et al. 2004; Trevisan et al.
2004; Bruns & Tiller 2005; Park & Clark 2006). In
this regard, it is known that the hydration level in
protein molecules dramatically influences enzymatic
activity in organic solvents (Yang et al. 2004).
Although hydration level is difficult to estimate,
some studies have shown good correlation between
the thermodynamic water activity of the system and
the enzyme’s activity, rendering aw as a reliable
indicator of hydration water. The optimum value
of aw depends on the enzyme and the reaction
system. Few studies are available for peroxidases
(Dai & Klibanov 1999; van de Velde et al. 2001a;
Michizoe et al. 2003), therefore further research is
still needed in order to overcome this drawback.
However, the most limiting factor for the use of
peroxidases in organic solvents is substrate desolva-
tion (Ryu & Dordick 1992). In organic systems, the
ground state stabilization of hydrophobic substrates
reduces their availability to the enzyme, which is
reflected by increased affinity constants and lower
catalytic activity. Some strategies, such as manipula-
tion of the microenvironment and solvent engineer-
ing, may be applied to alleviate this constraint.
Thermodynamic approaches to estimate substrate
desolvation in certain systems have been described
(Ryu & Dordick 1992; Schmitke et al. 1996). An
application for the oil industry will certainly require
more sophisticated modeling in order to lessen the
impact of substrate desolvation on the enzymatic
reaction efficiency. However, to date, no studies are
available regarding the protein or solvent engineer-
ing of peroxidases in order to favor substrate binding
in hydrophobic media.
Oxidative inactivation
Oxidative inactivation is the most troublesome issue
hampering the commercial application of heme
peroxidases (van de Velde et al. 2001b). It occurs
when the enzyme reacts with excess H2O2 or in the
absence of reducing substrate (Jenzer et al. 1986;
Arnao et al. 1990; Weinryb 1996). The oxidative
inactivation mechanism for peroxidases has been
recently reviewed (Valderrama et al. 2002). Oxida-
tively inactivated peroxidases characteristically show
heme degradation, although the oxidation of redox
active amino acids has also been observed (Jenzer
et al. 1986; Wariishi & Gold 1990; Villegas et al.
2000; Pfister et al. 2001). Normally, peroxidases
react with H2O2 to form Compound I, which is
sequentially reduced by an exogenous substrate, first
to Compound II and, afterwards, to native ferric
enzyme, as shown in Figure 2 (Renganathan &
Gold 1986). Under inactivation conditions, Com-
pound II reacts with H2O2 to yield Compound III,
a Fe3��O2� complex (Wariishi & Gold 1990). The
decay of this intermediate produces superoxide
radicals that may react with an additional H2O2
molecule to produce hydroxyl radicals (Jenzer et al.
1986; Wariishi & Gold 1990; Chen & Schopfer
1999). These very reactive species attack the por-
phyrin ring leading to irreversible peroxidase inacti-
vation (Jenzer et al. 1986; Wariishi & Gold 1990).
Besides Compound III, Compound I also acts as an
oxidizing agent during the inactivation process. In
the absence of an exogenous substrate, Compound I
decays into Compound II by substracting electrons
from the apoprotein (Jenzer et al. 1986; Hiner et al.
2001; Pfister et al. 2001). The oxidation of Trp and
Tyr residues coupled to Compound I reduction has
been observed in several peroxidases (Choinowski
et al. 1999; Hiner et al. 2001; Pfister et al. 2001;
Perez-Boada et al. 2005; Pogni et al. 2005). In yeast
CcP, Tyr oxidation leads to protein cross-linking
(Pfister et al. 2001), while Trp oxidation in LiP and
APX produces hydroxylated adducts (Choinowski et
al. 1999; Hiner et al. 2001). To date, the deleterious
effect of Compound I reduction during oxidative
inactivation has not been clearly elucidated, but
could be expected to be important in peroxidases
that contain functionally compromised oxidizable
amino acids, such as LiP and versatile peroxidase
(Choinowski et al. 1999; Perez-Boada et al. 2005;
Pogni et al. 2005).
An alternative inactivation mechanism, mainly
supported by the formation of the inactive compound
The prospects for peroxidase-based biorefining of petroleum fuels 123

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
P670 at the level of Compound I, has been proposed
(Arnao et al. 1990; Hiner et al. 2002). In this model,
Compound I reacts with H2O2 to form the complex
Compound I�H2O2, from which three catalytic
pathways originate: catalase activity pathway, Com-
pound III-forming pathway, and the inactivation
pathway, that, afterwards, leads to compound
P670. Both catalase activity and Compound III-
forming pathways are proposed to be peroxidase
protection mechanisms that avoid the accumula-
tion of Compound I and, hence, irreversible inactiva-
tion. Unlike the Compound III-centered inactivation
model, this mechanism rules out any deleterious role
for such species.
Several strategies have been implemented in order
to circumvent oxidative inactivation. They include
the fine regulation (feed-on-demand) of hydrogen
peroxide concentration during catalyzed reactions
(Seelbach et al. 1997), as well as the construction of
random and site-directed peroxidase mutants with
improved oxidative stability (see Table V). Substitu-
tion of Met and Tyr residues for less oxidizable
amino acids has been the rationale for producing
site-directed peroxidase mutants with improved
oxidative stability. MnP was stabilized 6.3-fold after
substituting methionine residues in positions 67, 237
and 273 with leucine residues (Miyasaki & Takaha-
shi 2001). MnP was also stabilized by engineering
the H2O2-binding pocket (Miyasaki-Imamura et al.
2003). In this case, Ala 79, Asn 81 and Ile 83
residues were saturation mutated in order to con-
struct a library from which a variant (A79S, N81L,
I83L) 4.4-times more stable than the wild-type MnP
was selected. The most successful stabilization case,
so far, is Coprinus cinereus peroxidase (CiP). This
enzyme was submitted to a combination of site-
directed mutagenesis, random mutagenesis and in
vivo shuffling, followed by screening for higher
thermal and oxidative stability. This strategy yielded
a multiple mutant (I49S, V53A, T121A, M166F,
E239G, M242I and Y272F) 100-times more stable
than wild-type CiP in the presence of hydrogen
peroxide (Cherry et al. 1999). Except for the
obvious and modest role of mutations M166F,
M242I and Y272F, the molecular characterization
of stabilized CiP does not clearly explain the
important contribution of mutations I49S, V53A,
T121A and E239G to oxidative stability (Houborg
et al. 2003). Rationalization of the molecular causes
of CiP oxidative stabilization would contribute to
understanding the oxidative inactivation process.
In this sense, iso-1-cytochrome c has been ration-
ally stabilized against peroxide inactivation following
a redox-inspired approach (Valderrama et al. 2006).
Although not an enzyme, the in vitro peroxidase
activity of cytochrome c is well characterized
(Vazquez-Duhalt 1999). By analyzing simulta-
neous events during oxidative inactivation (i.e.
cross-linking, heme bleaching, activity loss, iron
co-ordination), it was possible to alter them inde-
pendently by protein engineering, and, thus, mod-
ulate the electron transfer pathways (Valderrama &
Vazquez-Duhalt 2005). As a result, a cytochrome c
variant with a 15-fold improved total turnover was
obtained. This approach serves as an example of the
strategies that could produce more stable peroxidase
variants.
Molecular tools for the generation of
tailor-made heme peroxidases
Before the arrival of recombinant DNA technolo-
gies, covalent chemical modification was the only
molecular tool available to modify and/or enhance
peroxidase properties. Increased thermal stability
and tolerance to non-aqueous media have been
the main goals pursued with this approach. Inter-
and intra-crosslinking of o-amino lysine groups
with bifunctional reagents, such as glutaraldehyde
and ethylene-glycol-bis(N-hydroxysuccinimidylsuc-
cinate) have been successfully utilized to obtain
more thermostable peroxidases, in both soluble
and crystalline forms (O’Brien et al. 2001; Ayala
et al. 2002). Modification of heme proteins with
polyethylene glycol has improved not only thermo-
stability, but also tolerance to organic solvents
(Garcia-Arellano et al. 2002; Wang et al. 2002).
Indeed, cytochrome c, a non-enzymatic heme pro-
tein with in vitro peroxidase activity, shows a wider
substrate range after double chemical modification
with polyethylene glycol and methyl ester (Tinoco &
Vazquez-Duhalt 1998). Chemical modification, a
non-site-specific tool, often leads to activity reduc-
tion due to the reaction of the modifying agent with
residues close to, or inside the catalytic site. More-
over, the yield of heterogeneous and irreproducible
Table V. Site-directed and random heme peroxidase mutants with improved H2O2 stability.
Enzyme Improving factor Mutations Reference
Manganese peroxidase 6.3 M67L, M237L, M273L Miyasaki and Takahashi (2001)
4.4 A79S, N81L, I83L Miyasaki-Imamura et al. (2003)
C. cinereus peroxidase 100 I49S, V53A, T121A, M166F, E239G,
M242I, Y272F
Cherry et al. (1999)
124 M. Ayala et al.

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
chemically modified mixtures is an important draw-
back. Nevertheless, when structural information is
available, chemical modification combined with site-
directed mutagenesis is a powerful tool suited for
introducing non-natural side chains in specific sites
(DeSantis & Jones 1999). Introduction of non-
codified functionalities into proteins, by this mean
or by modifying the protein synthesis machinery to
achieve the in vivo incorporation of non-natural
amino acids, could provide novel approaches to
enhance peroxidase properties.
Site-directed mutagenesis, in addition to an in-
creasing knowledge of the structure�function rela-
tionships in peroxidases, has yielded enzymes with
improved stability and broader substrate range with-
out the drawbacks of chemical modification. Intro-
duction of disulfide bonds to improve thermal
stability, as well as the substitution of bulky amino
acids around the active site to allow substrates access
to the heme edge, have become common strategies
(Reading & Aust 2000; Celik et al. 2001). In spite of
those successes, engineering of peroxidases for im-
proving oxidative stability remains very challenging.
A general strategy consisting of the substitution of
Met and/or Tyr residues for less oxidizable amino
acids has been applied to several peroxidases
(Cherry et al. 1999; Miyasaki & Takahashi 2001;
Morawski et al. 2001). However, this strategy gave
modest results in MnP (Miyasaki & Takahashi 2001)
and it was completely ineffective for HRP (Morawski
et al. 2001).
Directed evolution has emerged as an alternative
to rational protein engineering. It has been shown
very useful when structural data is limited and/or
when the protein-engineering problem has a diffuse
solution. Low levels of peroxidase heterologous
expression is representative of such kind of problem.
When homologous expression is not possible or
convenient, heme peroxidases have been heterolo-
gously expressed in Aspergillus sp. (Steward et al.
1996; Conesa et al. 2001), tobacco plants (Pelle-
grineschi et al. 1995), recombinant baculovirus
(Johnson & Li 1991; Pease et al. 1991) and
Escherichia coli followed by in vitro folding of purified
inclusion bodies (Smith et al. 1990; Whitwam et al.
1995; Doyle & Smith 1996), always with very low
yields. By using directed evolution techniques, it was
possible to express functional HRP in E. coli without
further in vitro folding (Lin et al. 1999). In addition
to heterologous expression and thermal and oxida-
tive stabilization cases, directed evolution has also
been utilized to produce specificity changes, such as
that of CcP evolved for increased activity against
guaiacol (Iffland et al. 2000).
Although genetic tools have displaced traditional
covalent chemical modification, the latter has
recently reappeared, enhancing the capabilities of
site-directed mutagenesis. Heme peroxidases are so
complex that the application of only one of the
molecular tools available today is unlikely to give rise
to more commercially suited enzymes. Successful
cases clearly show that complex problems may only
be attacked with powerful eclectic strategies.
Conclusions
The introduction of new technologies to the refining
industry will ensure the supply of clean fuels during
the next decades. The systems described here may
serve as models for future process development,
either enzyme-based or enzyme-inspired. Peroxi-
dases are enzymes with great potential, but with
important disadvantages, impeding industrial appli-
cation. Some of the structural features affecting the
redox properties, substrate recognition and oxidative
stability of peroxidases are already identified. Ther-
modynamic analysis of substrate�enzyme binding in
hydrophobic media combined with protein and
medium engineering are essential to the successful
development of a peroxidase-based process. The
synergistic combination of theoretical and molecular
tools will certainly provide a better understanding of
peroxidase functioning and better grounds to design
the desired thermodynamic and kinetic properties
into the enzyme.
Acknowledgements
This work was supported by CONACYT 2004-
C01-42 grant.
References
Arnao MB, Acosta M, del Rio JA, Varon R, Garcia-Canovas F.
1990. A kinetic study on the suicide inactivation of peroxidase
by hydrogen peroxide. Biochim Biophys Acta 1041:43�47.
Ator MA, Ortiz de Montellano PR. 1987. Protein control of
prosthetic heme reactivity. Reaction of substrates with the heme
edge of horseradish peroxidase. J Biol Chem 262:1542�1551.
Ayala M, Vazquez-Duhalt R. 2004. Enzymatic catalysis on
petroleum products. In: Vazquez-Duhalt R, Quintero-Ramirez
R, editors. Petroleum biotechnology: Developments and per-
spectives Vol. 151. Amsterdam: Elsevier. p 67�111.
Ayala M, Tinoco R, Hernandez V, Bremauntz P, Vazquez-Duhalt
R. 1998. Biocatalytic oxidation of fuel as an alternative to
biodesulfurization. Fuel Process Technol 57:101�111.
Ayala M, Robledo NR, Lopez-Munguia A, Vazquez-Duhalt R.
2000. Substrate specificity and ionization potential in chlor-
operoxidase-catalyzed oxidation of diesel fuel. Environ Sci
Technol 34:2804�2809.
Ayala M, Horjales E, Pickard MA, Vazquez-Duhalt R. 2002.
Cross-linked crystals of chloroperoxidase. Biochem Biophys
Res Commun 295:828�831.
Banci L. 1997. Structural properties of peroxidases. J Biotechnol
53:253�263.
The prospects for peroxidase-based biorefining of petroleum fuels 125

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
Banci L, Bertini I, Turano P, Tien M, Kirk TK. 1991. Proton
NMR investigation into the basis for the relatively high redox
potential of lignin peroxidase. Biochemistry 88:6956�6960.
Barrows TP, Poulos TL. 2005. Role of electrostatics and salt
bridges in stabilizing the Compound I radical in ascorbate
peroxidase. Biochemistry 44:14062�14068.
Barrows TP, Bhaskar B, Poulos TL. 2004. Electrostatic control of
the tryptophan radical in cytochrome c peroxidase. Biochem-
istry 43:8826�8834.
Battistuzzi G, Bellei M, De Rienzo F, Sola M. 2006. Redox
properties of the Fe�2/Fe�3 couple in Arthromyces ramosus
class II peroxidase and its cyanide adduct. J Biol Inorg Chem
11:586�592.
Blodig W, Smith AT, Doyle WA, Piontek K. 2001. Crystal
structures of pristine and oxidatively processed lignin perox-
idase expressed in Escherichia coli and of the W171F variant
that eliminates the redox active tryptophan 171. Implications
for the reaction mechanism. J Mol Biol 305:851�861.
Bogan BW, Lamar RT, Hammel KE. 1996. Fluorene oxidation in
vivo by Phanerochaete chrysosporium and in vitro during
manganese peroxidase-dependent lipid peroxidation. Appl
Environ Microbiol 62:1788�1792.
Bonagura CA, Sundaramoorthy M, Pappa HS, Patterson WR,
Poulos TL. 1996. An engineered cation site in cytochrome c
peroxidase alters the reactivity of the redox active tryptophan.
Biochemistry 35:6107�6115.
Bruns N, Tiller JC. 2005. Amphiphilic network as nanoreactor for
enzymes in organic solvents. Nano Lett 5:45�48.
Buenrostro-Gonzalez E, Groenzin H, Lira-Galeana C, Mullins
OC. 2001. The overriding chemical principles that define
asphaltenes. Energy Fuels 15:972�978.
Cao LQ, van Langen L, Sheldon RA. 2003. Immobilised
enzymes: Carrier-bound or carrier-free? Curr Opin Biotechnol
14:387�394.
Cavalieri EL, Rogan EG, Roth RW, Saugier RK, Hakam A. 1983.
The relationship between ionization potential and horseradish
peroxidase/hydrogen peroxide-catalyzed binding of aromatic
hydrocarbons to DNA. Chem Biol Interact 47:87�109.
Celik A, Cullis PM, Sutcliffe MJ, Sangar R, Raven EL. 2001.
Engineering the active site of ascorbate peroxidase. Eur J
Biochem 268:78�85.
Chen S, Schopfer P. 1999. Hydroxyl-radical production in
physiological reactions. Eur J Biochem 260:726�735.
Cherry JR, Lamsa MH, Schneider P, Vind J, Svendsen A, Jones A,
Pedersen AH. 1999. Directed evolution of a fungal peroxidase.
Nat Biotechnol 17:379�384.
Choinowski T, Blodig W, Winterhalter KH, Piontek K. 1999. The
crystal structure of lignin peroxidase at 1.70 A resolution
reveals a hydroxy group on the Cb of tryptophan 171: A novel
radical site formed during the redox cycle. J Mol Biol 286:
809�827.
Ciaccio C, Rosati A, De Sanctis G, Sinibaldi F, Marini S,
Santucci R, Ascenzi P, Welinder KG, Coletta M. 2003.
Relationships of ligand binding, redox properties, and proto-
nation in Coprinus cinereus peroxidase. J Biol Chem
278:18730�18737.
Conesa A, van de Velde F, van Rantwijk F, Sheldon RA, van den
Hondel CAMJJ, Punt PJ. 2001. Expression of the Caldario-
myces fumago chloroperoxidase in Aspergillus niger and char-
acterization of the recombinant enzyme. J Biol Chem
276:17635�17640.
Conroy CW, Tyma P, Daum PH, Erman JE. 1978. Oxidation�reduction potential measurements of cytochrome c peroxidase
and pH dependent spectral transitions in the ferrous enzyme.
Biochim Biophys Acta 537:62�69.
Dai L, Klibanov AM. 1999. Striking activation of oxidative
enzymes suspended in nonaqueous media. Proc Natl Acad
Sci USA 96:9475�9478.
Dai L, Klibanov AM. 2000. Peroxidase-catalyzed asymmetric
sulfoxidation in organic solvents versus in water. Biotechnol
Bioeng 70:353�357.
de Visser SP, Shaik S, Sharma PK, Kumar D, Thiel W. 2003.
Active species of horseradish peroxidase HRP and cytochrome
P450: Two electronic chameleons. J Am Chem Soc
125:15779�15788.
DePillis GD, Wariishi H, Gold MH, Ortiz de Montellano PR.
1990. Inactivation of lignin peroxidase by phenylhydrazine and
sodium azide. Arch Biochem Biophys 280:217�223.
DeSantis G, Jones JB. 1999. Chemical modification of enzymes
for enhanced functionality. Curr Opin Biotechnol 10:324�330.
Doyle WA, Smith AT. 1996. Expression of lignin peroxidase H8 in
Escherichia coli : Folding and activation of the recombinant
enzyme with Ca2� and haem. Biochem J 315:15�19.
Erman JE, Vitello LB. 2002. Yeast cytochrome c peroxidase:
Mechanistic studies via protein engineering. Biochim Biophys
Acta 1597:193�220.
Farhangrazi ZS, Copeland BR, Nakayama T, Amachi T, Yamazaki
I, Powers LS. 1994. Oxidation�reduction properties of com-
pounds I and II of Arthromyces ramosus peroxidase. Biochem-
istry 33:5647�5652.
Farhangrazi ZS, Fosset ME, Powers LS, Ellis WR. 1995. Variable-
temperature spectroelectrochemical study of horseradish per-
oxidase. Biochemistry 34:2866�2871.
Fedorak PM, Semple KM, Vazquez-Duhalt R, Westlake DWS.
1993. Chloroperoxidase mediated modifications of petropor-
phyrins and asphaltenes. Enzyme Microb Technol 15:429�437.
Fowler D, Smith RI, Mullera JBA, Hayman G, Vincent KJ. 2005.
Changes in the atmospheric deposition of acidifying com-
pounds in the UK between 1986 and 2001. Environ Pollut
137:15�25.
Garcia-Arellano H, Valderrama B, Saab-Rincon G, Vazquez-
Duhalt R. 2002. High temperature biocatalysis by chemically
modified cytochrome c. Bioconj Chem 13:1336�1344.
Garcia-Arellano H, Buenrostro-Gonzalez E, Vazquez-Duhalt R.
2004. Biocatalytic transformation of petroporphyrins by che-
mically modified cytochrome c. Biotechnol Bioeng 85:790�798.
Gilfoyle D, Rodriguez-Lopez JN, Smith AT. 1996. Probing the
aromatic-donor-binding site of horseradish peroxidase using
site-directed mutagenesis and the suicide substrate phenylhy-
drazine. Eur J Biochem 236:714�722.
Goodin DB, McRee DE. 1993. The Asp�His�Fe triad of
cytochrome c peroxidase controls the reduction potential,
electronic structure, and coupling of the tryptophan free radical
to the heme. Biochemistry 32:3313�3324.
Government of Alberta, Canada [Internet] Department of
Energy, Canada. 2002. Available from: http://www.energy.
gov.ab.ca
Groenzin H, Mullins OC. 1999. Asphaltene molecular size and
structure. J Phys Chem A 103:11237�11245.
Groenzin H, Mullins OC. 2000. Molecular size and structure of
asphaltenes from various sources. Energy Fuels 14:677�684.
Guallar V, Olsen B. 2006. The role of the heme propionates in
heme biochemistry. J Inorg Biochem 100:755�760.
Hall C, Tharakan P, Hallock J, Cleveland C, Jefferson M. 2003.
Hydrocarbons and the evolution of human culture. Nature
426:318�322.
Hayashi Y, Yamazaki I. 1979. The oxidation�reduction potentials
of Compound I/Compound II and Compound II/ferric couples
of horseradish peroxidases A2 and C. J Biol Chem 254:101�106.
126 M. Ayala et al.

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
He B, Sinclair R, Copeland BR, Makino R, Powers LS, Yamazaki
I. 1996. The structure�function relationship and reduction
potentials of high oxidation states of myoglobin and peroxidase.
Biochemistry 35:2413�2420.
Henriksen A, Mirza O, Indiani C, Teilum K, Smulevich J,
Welinder KG, Gajhede M. 2001. Structure of soybean seed
coat peroxidase: A plant peroxidase with unusual stability and
haem�apoprotein interactions. Protein Sci 10:108�115.
Hiner A, Martinez JI, Arnao MB, Acosta M, Turner DD, Raven
EL, Rodriguez-Lopez JN. 2001. Detection of a tryptophan
radical in the reaction of ascorbate peroxidase with hydrogen
peroxide. Eur J Biochem 268:3091�3098.
Hiner A, Hernandez-Ruiz J, Rodriguez-Lopez JN, Garcia-Cano-
vas F, Brisset NC, Smith AT, Arnao MB, Acosta M. 2002.
Reactions of the class II peroxidases, lignin peroxidase and
Arthromyces ramosus peroxidase, with hydrogen peroxide. J Biol
Chem 277:26879�26885.
Houborg K, Harris P, Poulsen JC, Schneider P, Svendsen A,
Larsen S. 2003. The structure of a mutant enzyme of Coprinus
cinereus peroxidase provides an understanding of its increased
thermostability. Acta Crystallogr D59:997�1003.
Howes BD, Brissett NC, Doyle WA, Smith AT, Smulevich G.
2005. Spectroscopic and kinetic properties of the horseradish
peroxidase mutant T171S: Evidence for selective effects on the
reduced state of the enzyme. FEBS J 272:5514�5521.
Iffland A, Tafelmeyer P, Saudan C, Johnsson K. 2000. Directed
molecular evolution of cytochrome c peroxidase. Biochemistry
39:10790�10798.
Institute for the Analysis of Global Security [Internet] Washington
DC, US. 2005. Available from: http://www.iags.org
Ivancich A, Mazza G, Desbois A. 2001. Comparative electron
paramagnetic resonance study of radical intermediates in
turnip peroxidase isozymes. Biochemistry 40:6860�6866.
Jensen GM, Bunte SW, Warshel A, Goodin DB. 1998. Energetics
of cation radical formation at the proximal active site trypto-
phan of cytochrome c peroxidase and ascorbate peroxidase. J
Phys Chem B 102:8221�8228.
Jenzer H, Jones W, Kohler H. 1986. On the molecular mechanism
of lactoperoxidase-catalyzed H2O2 metabolism and irreversible
enzyme inactivation. J Biol Chem 261:15550�15556.
Johnson TM, Li JKK. 1991. Heterologous expression and
characterization of an active lignin peroxidase from Phaner-
ochaete chrysosporium using recombinant baculovirus. Arch
Biochem Biophys 291:371�378.
Kilbane JJ II. 1992. Mutant microorganisms useful for cleavage of
organic C�S bonds. US Patent 5,104,801.
Kimura M, Michizoe J, Oakazaki S, Furusaki S, Goto M, Tanaka
H, Wariishi H. 2004. Activation of lignin peroxidase in organic
media by reversed micelles. Biotechnol Bioeng 88:495�501.
Le Borgne S, Quintero R. 2003. Biotechnological processes for
the refining of petroleum. Fuel Process Technol 81:155�169.
Lee MY, Dordick JS. 2002. Enzyme activation for nonaqueous
media. Curr Opin Biotechnol 13:376�384.
Lin Z, Thorsen T, Arnold FH. 1999. Functional expression of
horseradish peroxidase in E. coli by directed evolution.
Biotechnol Prog 15:467�471.
Linguist L, Pacheco M. 1999. Enzyme-based diesel desulfuriza-
tion process offers energy, CO2 advantages. Oil G J 22:46�48.
Liu J, Wang T, Huang M, Song H, Weng L, Ji L. 2006. Increased
thermal and organic solvent tolerance of modified horseradish
peroxidase. Prot Eng Des Selec 19:169�173.
Makino R, Chiang R, Hager LP. 1976. Oxidation-reduction
potential measurements on chloroperoxidase and its com-
plexes. Biochemistry 15:4748�4754.
McFarland BL. 1999. Biodesulfurization. Curr Opin Microbiol
2:257�264.
Michizoe J, Uchimura Y, Maruyama T, Kamiya N, Goto M.
2003. Control of water content by reverse micellar solutions for
peroxidase catalysis in a water-immiscible organic solvent. J
Biosci Bioeng 95:425�427.
Miland E, Smyth MR, O’Fagain C. 1996. Modification of
horseradish peroxidase with bifunctional hydroxysuccinimide
esters: Effects on molecular stability. Enzyme Microb Technol
19:242�249.
Miller VP, Goodin DB, Friedman AE, Hartmann C, Ortiz de
Montellano PR. 1995. Horseradish peroxidase Phe1720Tyr
mutant. Sequential formation of Compound I with a porphyrin
radical cation and a protein radical. J Biol Chem 270:181413�181419.
Millis CD, Cai D, Stankovich MT, Tien M. 1989. Oxidation-
reduction potentials and ionization states of extracellular
peroxidases from the lignin-degrading fungus Phanerochaete
chrysosporium . Biochemistry 28:8484�8489.
Miyasaki C, Takahashi H. 2001. Engineering of the H2O2-binding
pocket region of a recombinant manganese peroxidase to be
resistant to H2O2. FEBS Lett 509:111�114.
Miyasaki-Imamura C, Oohira K, Kitagawa R, Nakano H, Yamane
T, Takahashi H. 2003. Improvement of H2O2 stability of
manganese peroxidase by combinatorial mutagenesis and
high-throughput screening using in vitro expression with
protein disulfide isomerase. Prot Eng 16:423�428.
Mogollon L, Rodriguez R, Larrota W, Ortiz C, Torres R. 1998.
Biocatalytic removal of nickel and vanadium from petropor-
phyrins and asphaltenes. Appl Biochem Biotechnol 70�72:765�777.
Mondal MS, Fuller HA, Armstrong FA. 1996. Direct measure-
ment of the reduction potential of catalytically active cyto-
chrome c peroxidase Compound I: Voltammetric detection of a
reversible, cooperative two-electron transfer reaction. J Am
Chem Soc 118:263�264.
Morawski B, Quan S, Arnold FH. 2001. Functional expression
and stabilization of horseradish peroxidase by directed evolu-
tion in Saccharomyces cerevisiae. Biotechnol Bioeng 76:99�107.
Morimoto A, Tanaka M, Takahashi S, Ishimori K, Hori H,
Morishima I. 1998. Detection of a tryptophan radical as an
intermediate species in the reaction of horseradish peroxidase
mutant Phe-2210Trp and hydrogen peroxide. J Biol Chem
273:14753�14760.
Nagano S, Tanaka M, Ishimori K, Watanabe Y, Morishima I.
1996. Catalytic roles of the distal site asparagine�histidine
couple in peroxidases. Biochemistry 35:14251�14258.
Newmyer SL, Ortiz de Montellano PR. 1995. Horseradish
peroxidase His-420Ala, His-420Val, and Phe-410Ala mu-
tants. Histidine catalysis and control of substrate access to the
heme iron. J Biol Chem 270:19430�19438.
O’Brien AM, O’Fagain C, Nielsen PF, Welinder KG. 2001.
Location of crosslinks in chemically stabilized horseradish
peroxidase: Implications for design of crosslinks. Biotechnol
Bioeng 76:277�284.
Park JB, Clark DS. 2006. New reaction system for hydrocarbon
oxidation by chloroperoxidase. Biotechnol Bioeng 94:189�192.
Pease EA, Aust SD, Tien M. 1991. Heterologous expression of
active manganese peroxidase from Phanerochaete chrysosporium
using the baculovirus expression system. Biochem Biophys Res
Commun 179:897�903.
Pellegrineschi A, Kis M, Dix I, Kavanagh TA, Dix PJ. 1995.
Expression of horseradish peroxidase in transgenic tobacco.
Biochem Soc Trans 23:247�250.
Perez-Boada M, Ruiz-Duenas FJ, Pogni R, Basosi R, Choinowski
T, Martinez MJ, Piontek K, Martinez AT. 2005. Versatile
peroxidase oxidation of high redox potential aromatic
compounds: Site-directed mutagenesis, spectroscopic and
The prospects for peroxidase-based biorefining of petroleum fuels 127

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
crystallographic investigation of three long-range electron
transfer pathways. J Mol Biol 354:385�402.
Pfister TD, Gengenbach AJ, Syn S, Lu Y. 2001. The role of redox-
active amino acids on Compound I stability, substrate oxida-
tion, and protection cross linking in yeast cytochrome c
peroxidase. Biochemistry 40:14942�14951.
Pogni R, Baratto MC, Giansanti S, Teutloff C, Verdin J,
Valderrama B, Lendzian F, Lubitz W, Vazquez-Duhalt R,
Basosi R. 2005. Tryptophan-based radical in the catalytic
mechanism of versatile peroxidase from Bjerkandera adusta .
Biochemistry 44:4267�4274.
Poulos TL, Kraut J. 1981. The stereochemistry of peroxidase
catalysis. J Biol Chem 255:8199�8205.
Premuzic E, Lin MS, Bohenek M, Zhou WM. 1999. Bioconver-
sion reactions in asphaltenes and heavy crude oils. Energy Fuels
13:297�304.
Purcell WL, Erman JE. 1976. Cytochrome c peroxidase catalyzed
oxidations of substitution inert iron II complexes. J Am Chem
Soc 98:7033�7037.
Radler M. 2004. Crude oil production climbs as reserves post
modest rise. Oil G J 102:18�20.
Reading NS, Aust SD. 2000. Engineering a disulfide bond in
recombinant manganese peroxidase results in increased ther-
mostability. Biotechnol Prog 16:326�333.
Renganathan V, Gold MH. 1986. Spectral characterization of the
oxidized states of lignin peroxidase, an extracellular heme
enzyme from the white rot basidiomycete Phanerochaete chry-
sosporium . Biochemistry 25:1626�1631.
Ricard J, Mazza G, William RJP. 1972. Oxidation�reduction
potentials and ionization states of two turnip peroxidases. Eur J
Biochem 28:566�578.
Ryu K, Dordick JS. 1992. How do organic solvents affect
peroxidase structure and function? Biochemistry 31:2588�2598.
Santucci R, Bongiovanni C, Marini S, Del Conte R, Tien M,
Banci L, Coletta M. 2000. Redox equilibria of manganese
peroxidase from Phanerochaete chrysosporium : Functional role
of residues on the proximal side of the haem pocket. Biochem J
349:85�90.
Schmitke JL, Wescott CR, Klibanov AM. 1996. The mechanistic
dissection of the plunge in enzymatic activity upon transition
from water to anhydrous solvents. J Am Chem Soc 118:3360�3365.
Seelbach K, van Deurzen MPJ, van Rantwijk F, Sheldon RA.
1997. Improvement of the total turnover number and space�time yield for chloroperoxidase catalyzed oxidation. Biotechnol
Bioeng 55:283�288.
Skjelkvale BL, Stoddard JL, Jeffries DS, Tørseth K, Høgasen T,
Bowman J, Mannio J, Monteith DT, Mosello R, Rogora M, et
al. 2005. Regional scale evidence for improvements in surface
water chemistry 1990�2001. Environ Pollut 137:165�176.
Smith AT, Veitch NC. 1998. Substrate binding and catalysis in
heme peroxidases. Curr Opin Chem Biol 2:269�278.
Smith AT, Santama N, Dacey S, Edwards M, Bray RC, Thorneley
RNF, Burke JF. 1990. Expression of a synthetic gene for
horseradish peroxidase c in Escherichia coli and folding and
activation of the recombinant enzyme with Ca2� and heme. J
Biol Chem 265:13335�13343.
Smulevich G, Miller MA, Kraut J, Spiro TG. 1991. Conforma-
tional change and histidine control of heme chemistry in
cytochrome c peroxidase: Resonance Raman evidence from
Leu-52 and Gly-181 mutants of cytochrome c peroxidase.
Biochemistry 30:9546�9558.
Song C. 2003. An overview of new approaches to deep desulfur-
ization for ultra-clean gasoline, diesel fuel and jet fuel. Catal
Today 86:211�263.
Song H, Yaob J, Liu J, Zhou S, Xiong Y, Ji L. 2005. Effects of
phthalic anhydride modification on horseradish peroxidase
stability and structure. Enzyme Microb Technol 36:605�611.
Steward P, Whitwam RE, Kersten PJ, Cullen D, Tien M. 1996.
Efficient expression of a Phanerochaete chrysosporium manga-
nese peroxidase gene in Aspergillus oryzae . Appl Environ
Microbiol 62:860�864.
Strausz OP, Mojelsky TW, Lown EM. 1992. The molecular
structure of asphaltenes: An unfolding story. Fuel 71:1355�1363.
Sundaramoorthy M, Terner J, Poulos TL. 1995. The crystal
structure of chloroperoxidase: A heme peroxidase�cytochrome
P450 functional hybrid. Structure 3:1367�77.
Svistunenko DA. 2005. Reaction of haem containing proteins and
enzymes with hydroperoxides: The radical view. Biochim
Biophys Acta 1707:127�155.
Takahashi K, Nishimura H, Yoshimoto T, Saito Y, Inada Y. 1984.
A chemical modification to make horseradish peroxidase
soluble and active in benzene. Biochem Biophys Res Commun
121:261�265.
Tanaka M, Ishimori K, Mukai M, Kitagawa T, Morishima I.
1997. Catalytic activities and structural properties of horse-
radish peroxidase distal His42 of Glu or Gln mutant.
Biochemistry 36:9889�9898.
Thouand G, Bauda P, Oudot J, Kirsh G, Sutton C, Vidalie JF.
1999. Laboratory evaluation of crude oil biodegradation with
commercial or natural microbial inocula. Can J Microbiol
45:106�115.
Tinoco R, Vazquez-Duhalt R. 1998. Chemical modification of
cytochrome c improves their catalytic properties in oxidation of
polycyclic aromatic hydrocarbons. Enzyme Microb Technol
22:8�12.
Trevisan V, Signoretto M, Colonna S, Pironti V, Strukul G. 2004.
Microencapsulated chloroperoxidase as a recyclable catalyst for
the enantioselective oxidation of sulfides with hydrogen perox-
ide. Angew Chem Int Ed Engl 43:4097�4099.
Valderrama B, Vazquez-Duhalt R. 2005. Electron-balance during
the oxidative self-inactivation of cytochrome c. J Mol Catal B
Enzym 35:41�44.
Valderrama B, Ayala M, Vazquez-Duhalt R. 2002. Suicide
inactivation of peroxidases and the challenge of engineering
more robust enzymes. Chem Biol 9:555�565.
Valderrama B, Garcia-Arellano H, Giansanti S, Baratto MC,
Pogni R, Vazquez-Duhalt R. 2006. Oxidative stabilization of
iso-1-cytochrome c by redox-inspired protein engineering.
FASEB J 20:1233�1235.
van de Velde F, Bakker M, van Rantwijk F, Sheldon RA. 2001a.
Chloroperoxidase-catalyzed enantioselective oxidations in hy-
drophobic organic media. Biotechnol Bioeng 72:523�529.
van de Velde F, van Rantwijk F, Sheldon RA. 2001b. Improving
the catalytic performance of peroxidases in organic synthesis.
Trends Biotechnol 19:73�80.
van Deurzen MPJ, van Rantwijk F, Sheldon RA. 1997. Selective
oxidations catalyzed by peroxidases. Tetrahedron 53:13183�13220.
Vazquez-Duhalt R. 1999. Cytochrome c as a biocatalyst. J Mol
Catal B Enzym 7:241�249.
Vazquez-Duhalt R, Westlake DWS, Fedorak PM. 1994. Lignin
peroxidase oxidation of aromatic compounds in systems con-
taining organic solvents. Appl Environ Microbiol 60:459�466.
Vazquez-Duhalt R, Bremauntz MP, Barzana E, Tinoco R. 2002a.
Enzymatic oxidation process for desulfurization of fossil fuels.
US Patent 6,461,859.
Vazquez-Duhalt R, Torres E, Valderrama B, Le Borgne S. 2002b.
Will biochemical catalysis impact the petroleum refining
industry? Energy Fuels 16:1239�1250.
128 M. Ayala et al.

Dow
nloa
ded
By:
[Uni
vers
ity o
f Ariz
ona]
At:
15:4
8 15
Oct
ober
200
7
Veitch NC. 2004. Horseradish peroxidase: A modern view of a
classic enzyme. Phytochemistry 65:249�259.
Verdin J, Pogni R, Baeza A, Baratto MC, Basosi R, Vazquez-
Duhalt R. 2006. Mechanism of versatile peroxidase inactivation
by Ca2� depletion. Biophys Chem 121:163�170.
Villegas JA, Mauk AG, Vazquez-Duhalt R. 2000. A cytochrome c
variant resistant to heme degradation by hydrogen peroxide.
Chem Biol 7:237�244.
Wang Y, Vazquez-Duhalt R, Pickard MA. 2002. Purification,
characterization, and chemical modification of manganese
peroxidase from Bjerkandera adusta UAMH 8258. Curr Micro-
biol 45:77�87.
Wang Y, Vazquez-Duhalt R, Pickard MA. 2003. Manganese-
lignin peroxidase hybrid from Bjerkandera adusta oxidizes
polycyclic aromatic hydrocarbons more actively in the absence
of manganese. Can J Microbiol 49:675�682.
Wariishi H, Gold MH. 1990. Lignin peroxidase compound III,
mechanism of formation and decomposition. J Biol Chem
265:2070�2077.
Weinryb I. 1996. The behavior of horseradish peroxidase at high
hydrogen peroxide concentrations. Biochemistry 5:2003�2008.
Welinder KG. 1992. Superfamily of plant, fungal and bacterial
peroxidases. Curr Opin Struct Biol 2:388�393.
Whitwam RE, Gazarian IG, Tien M. 1995. Expression of fungal
Mn peroxidase in E. coli and refolding to yield active enzyme.
Biochem Biophys Res Commun 216:1013�1017.
Wirstam M, Blomberg MRA, Siegbahn PEM. 1999. Reaction
mechanism of compound I formation in heme peroxidases: A
density functional theory study. J Am Chem Soc 121:10178�10185.
Wyndham RC, Costerton JW. 1981. In vitro microbial degrada-
tion of bituminous hydrocarbons and in situ colonization of
bitumen surfaces within the Athabasca oil sands deposit. Appl
Environ Microbiol 41:791�800.
Yamada H, Makino R, Yamazaki I. 1975. Effects of 2,4-
substituents of deutero-heme upon redox potentials of horse-
radish peroxidases. Arch Biochem Biophys 169:344�353.
Yang L, Dordick JS, Garde S. 2004. Hydration of enzyme in
nonaqueous media is consistent with solvent dependence of its
activity. Biophys J 87:812�821.
The prospects for peroxidase-based biorefining of petroleum fuels 129






























