



1 23
Structural ChemistryComputational and ExperimentalStudies of Chemical and BiologicalSystems ISSN 1040-0400Volume 24Number 3 Struct Chem (2013) 24:967-980DOI 10.1007/s11224-012-0193-x
Structure–activity relationships of theantiviral D4T and seven 4′-substitutedderivatives using MP2 and DFT methods
M. Alcolea Palafox & N. Iza

1 23
Your article is protected by copyright and all
rights are held exclusively by Springer Science
+Business Media New York. This e-offprint is
for personal use only and shall not be self-
archived in electronic repositories. If you wish
to self-archive your article, please use the
accepted manuscript version for posting on
your own website. You may further deposit
the accepted manuscript version in any
repository, provided it is only made publicly
available 12 months after official publication
or later and provided acknowledgement is
given to the original source of publication
and a link is inserted to the published article
on Springer's website. The link must be
accompanied by the following text: "The final
publication is available at link.springer.com”.

ORIGINAL RESEARCH
Structure–activity relationships of the antiviral D4T and seven40-substituted derivatives using MP2 and DFT methods
M. Alcolea Palafox • N. Iza
Received: 20 December 2012 / Accepted: 22 December 2012 / Published online: 15 March 2013
� Springer Science+Business Media New York 2013
Abstract A comprehensive quantum chemical investiga-
tion of the conformational landscapes of the anti-HIV D4T
nucleoside analogue was carried out. Thus, all the possible
stable structures were determined with full optimization of
all the geometrical parameters. The whole conformational
parameters (v, b, c, P, mmax) were analyzed. The hydration
was simulated by explicit water molecules. The most stable
conformer was determined as C1 either in isolated state as
in aqueous solution, b = 63.8�, N-type. Conformer C3
(b = 165.08) is stable with 1–7 water molecules but with a
number larger than eight, C1 is the only stable form in a
close hydrated cluster. However, hydration of the natural
nucleoside thymidine with 13 water molecules stabilizes the
conformer C3 with b ca. 1808 due to the presence of the
C30-OH group. The first phosphorylation step in D4T was
simulated through the interaction with the ATP anion. This
simulation was performed for C1 and C3 conformers in
isolated state, showing that C1 changes to C3 by rotation of
C50-OH group until the value of b ca. 1808. Phosphorylation
of hydrated clusters is only possible with a number of water
molecules below of eight, which permits the C50-OH group
rotation to be accessible for the phosphate group. The
bonding of D4TTP to DNA viral through the reverse
transcriptase enzyme was also simulated. Seven 40-substituted
D4T derivatives were full optimized and analyzed on basis
of the activity reported on TK-1 enzyme and effective
concentration EC50, and several structure–activity rela-
tionships/tendencies were established. The two best cor-
relations correspond to those observed between the TK-1
phosphorylation activity of D4T derivatives and AZT, and
both the calculated exocyclic b angle and dipole moment.
Keywords Stavudine � Zerit � D4T � Structure–activity �Anti-HIV � Phosphorylation
Introduction
Significant progress has been made towards the chemother-
apy (and prophylaxis) of HIV infections [1–5], where
nucleoside analogues have an important role in the current
treatment of cancer and viral infections. Compounds con-
taining an unsaturated ribose ring and with lack of 20- and 30-OH groups, appear as the most effective alternative substrates
of the reverse transcriptase enzyme of the human immuno-
deficiency HIV virus. D4T (20,30-didehydro-20,30-dideoxyt-
hymidine, stavudine or Zerit) (Scheme 1) was one of the first
nucleosides synthesized nearly 40 years ago at the Michigan
Cancer Foundation [6], and Lin and Prusoff [7] at Yale
University discovered the capability of this molecule in
treating HIV/AIDS. Thus, D4T shows exceptional interest. It
was approved by the Food and Drug Administration (FDA)
for clinical use in the infection caused by virus HIV [8, 9]. It
belongs to the Nucleoside Reverse Transcriptase Inhibitors
(NRTIs) antiviral agents that have also wide range of others
biological activity as anti-tumour and antibiotic agents. These
nucleoside analogues are the inactive unphosphorylated form
(prodrugs). Their activation to the triphosphate form by cel-
lular kinases is required for drug potency. Currently, all
Dedicated to Professor Aldo Domenicano on the occasion of his 75th
birthday.
Electronic supplementary material The online version of thisarticle (doi:10.1007/s11224-012-0193-x) contains supplementarymaterial, which is available to authorized users.
M. A. Palafox (&) � N. Iza
Departamento de Quımica Fısica I, Facultad de Ciencias
Quımicas, Universidad Complutense, Ciudad Universitaria,
28040 Madrid, Spain
e-mail: [email protected]
123
Struct Chem (2013) 24:967–980
DOI 10.1007/s11224-012-0193-x
Author's personal copy

approved NRTIs with the exception of EFdA (40-ethynyl-
2-fluoro-20-deoxyadenosine) lack a 30-OH group. The other
20,30-didehydro-20,30-dideoxynucleosides, containing canon-
ical DNA and RNA bases are also considered as potential RT
inhibitors. It has been shown that there are not conformational
obstacles for incorporation of D4U and D4C into the double
helical A and B forms of DNA [10]. D4C has pronounced
antiretroviral activity [11]. However, D4U, neither its free
[12] nor phosphorylated [13] forms exhibit noticeable bio-
logical activity. This fact can be related to the presence of the
methyl group at the 5-position of the d4T nucleobase and
other modifications at this position of D4U can also transform
the compound into an efficacious RT inhibitors [14]. Intra-
molecular interactions govern conformational properties that
are a decisive factor for binding the active sites of enzymes.
On the other hand, the deoxyribose ring is the major source of
the conformational flexibility of DNA [15]. This is in accord
with the considerable reduction of the conformational
capacity on going from the natural nucleosides to their 20,30-didehydro-20,30-dideoxynucleosides analogues due to the
rigidity of C20=C30double bond in these derivatives.
Several studies have been performed aimed at establishing
the relationship between structure and physicochemical
properties/activity of these drugs. Thus, the anti-HIV-1
activity has been reported that depends upon ribose form
and differences in the ring puckering lead to noticeable
changes in the C50-OH group and in the thymine ring
positions [16]. Recently, relaxed force constants (RFC) and
vibrational root-mean-square (VRMS) deviations have
been used [17] to compare the flexibility of torsional angles
for 20-deoxyribonucleosides (2DRs). It has been found that
torsional angle c is the most rigid one (RFC 15–30 kcal
mol-1) and e, b and the pseudorotation phase angle P are
softer with low RFC values. The glycosidic torsional angle
v is characterized by intermediate values of RFC. The
phenomenon of non-planarity of the amino group in the
amines has also to be considered in nucleobases [18].
From our understanding an analysis of the different con-
formational possibilities for D4T would be interesting, and a
comparison of the results to the nucleoside natural thymidine
(dT) in order to see the facility to change of conformation, the
flexibility of the structure for the interpretation of drug–target
interactions. For this reason different authors have analyzed
the conformers of several nucleosides and nucleosides ana-
logues [19–24]. In previous works we have analyzed the
hydration effect on the two most stables conformers of D4T
[25], and their tautomeric forms [26]. In the present work a
full quantum chemical analysis of the conformational pref-
erences was carried out in an attempt to gain insights into
molecular features responsible for activity. We are also
interested in whether the different H-bondings in D4T make
significant contributions to its conformational behaviour. In
this sense, the importance of intramolecular H-bonds is
showed with 6-azaCyd molecule [27] where the presence of
N6 nitrogen atom leads to the formation of additional, as
compared to Cyd, H-bonds between the nucleoside base and
sugar atoms and allows the molecule to adopt the additional
conformations stabilized by these intramolecular H-bonds.
The variety of conformations made the 6-azaCyd radically
different from the Cyd molecule. Other study on DNA-
related molecules [28] using DFT calculations, Bader’s
quantum theory of atoms in molecules (AIM) and spectro-
scopic vibrational analysis reveals the strongest intramolec-
ular H-bonds with red-shifts over 40 cm-1 in the most cases.
In canonical 20-deoxy- and ribonucleosides, the O50H���N3H
H-bond in ribonucleoside guanosine was found to have the
maximum energy, 8.1 kcal mol-1. In relation to the weaker
H-bonds, MP2 and DFT quantum chemical calculations and
analysis AIM [29] conducts to five types of the CH���Ohydrogen bonds involving bases and sugar residues, and were
detected numerically from 1 to 3 per a conformer: C20H���O50, C10H���O2, C6H���O50, C8H���O50, and C6H���O40. The
energy values of H-bonds are in the range of 2.3–5.6
kcal mol-1, exceeding the kT value (0.62 kcal mol-1). The
Scheme 1 Molecular structure
and definition of the exocyclic
and endocyclic angles in D4T
nucleoside and in seven 40-substituted D4T derivatives
968 Struct Chem (2013) 24:967–980
123
Author's personal copy

calculation of the interactions in the dA-T nucleoside pair
gives evidence, that additional to the N6H���O4 and
N1���N3H canonical H-bonds between the bases adenine
and thymine, the third one C2H���O2 is formed, which,
though being rather weak (about 1 kcal mol-1), satisfies
the AIM criteria of H-bonding and may be classified as a
true H-bond. The nucleoside CH���O hydrogen bonds
appeared survive turns of bases against the sugar ring in
rather large ranges of the angle v values, pertinent to cer-
tain conformations. This fact points out to the source of the
DNA characteristic lability necessary for the conforma-
tional adaptation in processes of its functioning.
Another goal of present research is a simulation of the
solid state structure. Several crystallographic studies have
been reported on the polymorphism of D4T [30] yielding to
triclinic [31], monoclinic [32], and orthorhombic [33, 34]
crystals. We have simulated well the dimer form found in
these crystal unit cells.
Efficient phosphorylation depends largely on the spatial
structure of the nucleoside. Thus, an extensive conforma-
tional analysis identifying all minima on the potential
energy surface (PES) can be considered as a first step in the
design of nucleoside analogues as anti-HIV drugs. The first
step of this phosphorylation process was reproduced in the
present manuscript and a proton transfer mechanism in the
isolated state and in the hydrated form was observed.
The last goal was to obtain structure–activity relation-
ships. For this purpose a full optimization of the structural
parameters in seven 40-substituted D4T derivatives was
carried out and the data obtained were compared with the
biological activity reported for these derivatives. As the
formation of monophosphate D4TMP is the first step to
obtain the active metabolite D4TTP, the potential of these
derivatives to be phosphorylated by human TK-1 kinase
was studied. We have simulated the phosphorylation with
an ATP molecule in isolated state and in the presence of
different number of water molecules. In a previous work
[35, 36] we reported that C3 conformation with v =
-122.3�, b = 165�, c = 43.7� is the most adequate to be
phosphorylated at the C50-OH group. Thus, for the first
time structure–activity relations are described here in dT
nucleoside analogues and several conclusions are under-
lined that could be considered for further development of
new anti-HIV drugs.
Calculations
The computations were performed by using the MP2
ab initio method and by using Density Functional methods
(DFT), such as B3LYP and B971 hybrid functionals. These
methods appear implemented in the GAUSSIAN 03
program package [37]. An adequate compromise between
the desired chemical accuracy and the heavy demands put
on computer time and power is provided by DFT methods.
Moreover, these methods were used satisfactory in many
drug design studies [25, 38–40]. The B3LYP method was
selected because several studies have shown that the data
obtained at this level of theory are in good agreement with
those obtained by other more costly computational meth-
ods as MP2 calculations and it predicts the frequencies of
DNA bases better than the HF and MP2 methods [41–45].
Several basis sets were used starting from the 6-31G* to
the 6-311??G(3df,pd), but the 6-31G** basis set was
selected for all the calculations because it represents a
compromise between accuracy and computational cost.
Moreover, these methods and basis sets have been also
used in different nucleosides [10, 19, 46].
Under the TIGHT convergence criterion, the optimum
geometry was determined by minimizing the energy with
respect to all geometrical parameters without imposing
molecular symmetry constraints. The harmonic frequencies
were calculated at the same level of the respective opti-
mization process and by the analytic evaluation of the
second derivative of the energy with respect to the nuclear
displacement. In all the optimized conformers were per-
formed frequency calculations to assess that they corre-
spond to real minimum. All the optimized forms only
showed positive harmonic vibrations (true energy mini-
mum). Relative energies were determined by including
zero-point vibrational energies (ZPE). For the calculation
of ZPE, the wavenumbers were retainted unscaled. Atomic
charges were determined with the Natural NBO [47, 48]
procedure.
All quantum chemical calculations were carried out on
the alpha computer of the Centro de Calculo de la Uni-
versidad Complutense de Madrid, as well as on a HP
Integrity rx2600 server, with 2 Intel� Itanium� 2 proces-
sors at Computational Chemistry Laboratory from Uni-
versidad Nacional de Educacion a Distancia (Lab-QC,
UNED) [49].
The PES was determined in this molecule by rotation of
the torsional angles v (glycosidic bond), c (C40–C50 bond)
and b (C50–O50 bond). In a first study these dihedral angles
simultaneously held fixed at values varying between 0� and
360� in steps of 60� (Table S1 is a resume with the global
minimum) and of 20� in a more detailed second study
(Supplementary Material). During these optimizations all
other geometrical parameters were relaxed. The optimized
points were plotted by the SURFER program [50]. 216
optimized geometries were considered at the first step and
5,832 in the second one.
The ATP molecule was simulated in the completely
deprotonated anionic form with charge -4, and starting
the optimization with X-ray values for their torsional
angles.
Struct Chem (2013) 24:967–980 969
123
Author's personal copy

Results and discussion
Definition of the conformational angles
The atomic description of this molecule as well as the most
important endocyclic and exocyclic torsional angles are
defined in Scheme 1 according to the Saenger’s notation
[51]. The different conformations in D4T can be charac-
terized by the following four important structural parame-
ters: (i) the glycosylic torsional angle, v (O40–C10–N1–C2),
which determines the two orientations of the base relative
to the furanose ring, denoted as the anti and syn confor-
mations. (ii) The exocyclic torsional angle c (C30–C40–C50–O50) which describes the orientation of the 50-hydroxyl
group relative to the furanose ring. For minimizing non-
bonded interactions between their substituents this ring is
twisted out-of-plane. (iii) The exocyclic torsional angle b(C40–C50–O50–H50) describes the orientation of the
hydroxyl hydrogen H50 relative to the furanose ring. (iv)
The furanose pucker P defined in the bottom of Table 2,
which indicates north and south orientations.
An important structural characteristic of D4T is the
presence of the C20=C30 double bond, a feature that renders
the sugar ring nearly planar and imparts a high degree of
rigidity to the sugar moiety. However, there are many
conformational possibilities yet for the endocyclic and
exocyclic torsional angles of the molecule.
PES maps in an isolated state
Optimization of the conformers with minimum energies
was carried out, aimed at finding the low-energy confor-
mational states of this molecule. Thus, 25 optimum stable
conformers were determined by rotation of the exocyclic v,
c, and b torsional angles. From all data collected (Sup-
plementary Material) only the 3D conformational map with
the conformers C1–C9 including the global minimum C1
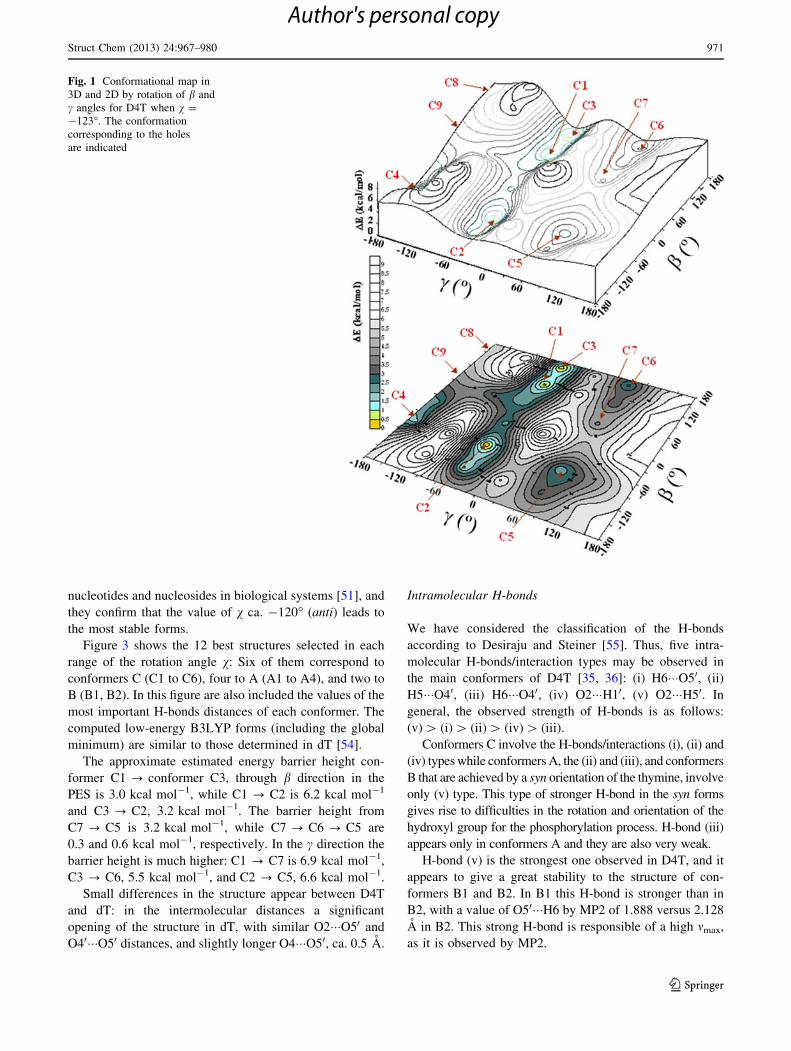
v = -103.68, b = 63.88, c = 60.68 was plotted, Fig. 1. In
this figure the holes correspond to the optimum conform-
ers, which are pointed on the map.
Results by X-ray [31, 32] and molecular mechanics
analysis of conformations [52, 53] have shown an
increased flexibility of the unsaturated thymidine ana-
logues as compared to the saturated compounds. From our
results, it is possible to see that the rotations around C50–O50 bond, b, is less restricted in D4T (energy barrier
height & 2.2 kcal mol-1) than in dT (energy barrier
height = 5.4 kcal mol-1), when v is in anti and c in gg
position. Energy barrier heights are defined here as the
energy difference between the global minimum and the
relevant highest energy point obtained in the PES map. It
can be noted a much lower barrier height in the b direction
than in the c angle. The rotation on the O50–H50 bond is
much easier than on C50–O50. The holes corresponding to
conformers C5, C7 and C6 appear in the conformational
map with v = -103�.
Conformers and energetics
In the isolated state
An extensive conformational study was carried out and the
relative calculated energies of the 25 optimum conformers
were obtained at the B3LYP, B971 and MP2 levels. In the
global minimum a detailed collection of the most impor-
tant conformational parameters, as well as the dipole
moments, is included in Table 1. The conformers were
classified in groups according to the three ranges of
rotation of v: conformers C (v: -119.1� ± 15.5� by MP2),
conformers A (v: -173.6� ± 5.0�) and conformers B
(v: 83.9� ± 18.1�), Fig. 2. The energy criterion by MP2
was followed for the numbering into each group. The C
group contains the most stable conformer at all, C1, and
the majority of more stable conformers. These conformers
C are found in the crystal. The A group conformers, also
found in the crystal, have high planarity. The B group
corresponds to expected non-active biological, syn forms.
Conformers A were difficult to be optimized by the dif-
ferent methods, and in many cases they moved to the
equivalent conformer C.
For each conformer two energy criteria were considered:
Gibbs energy DG, and the electronic energy DE ? ZPE
correction. In the different methods the stability order of the
five most stable conformers by the DE ? ZPE criterion is:
C1 [ C2 [ C3 [ B1 [ C4
by MP2=6-31G�� and B3LYP=6-31G��ð ÞC1 [ C4 [ C3 [ B1 [ C2
by B3LYP=6-311þþG 3df; pfð Þð ÞC1 [ C2 [ C3 [ C4 [ B1
by B971=6-31G��ð Þ
By Gibbs energy criterion is:
C1 [ C4 [ C3 [ C2 [ B1 by B3LYP=6-31G��ð ÞC1 [ C4 [ A1 [ C2 [ C3 by B971=6-31G��ð Þ
The global minimum at all the levels corresponds to the
anti-gg–gg form with respect to v, b and c torsional angles,
-103.6�, 63.8� and 60.6� values by MP2, respectively.
This conformer is denoted as C1 (Fig. 3) and it appears
stabilized by three intramolecular H-bonds/contacts. The
sugar puckering is unsymmetrical twisted, with major O40
atom in endo position and minor C40-exo, 0T4. These
values are close to the expected ones for the natural
970 Struct Chem (2013) 24:967–980
123
Author's personal copy
nucleotides and nucleosides in biological systems [51], and
they confirm that the value of v ca. -120� (anti) leads to
the most stable forms.
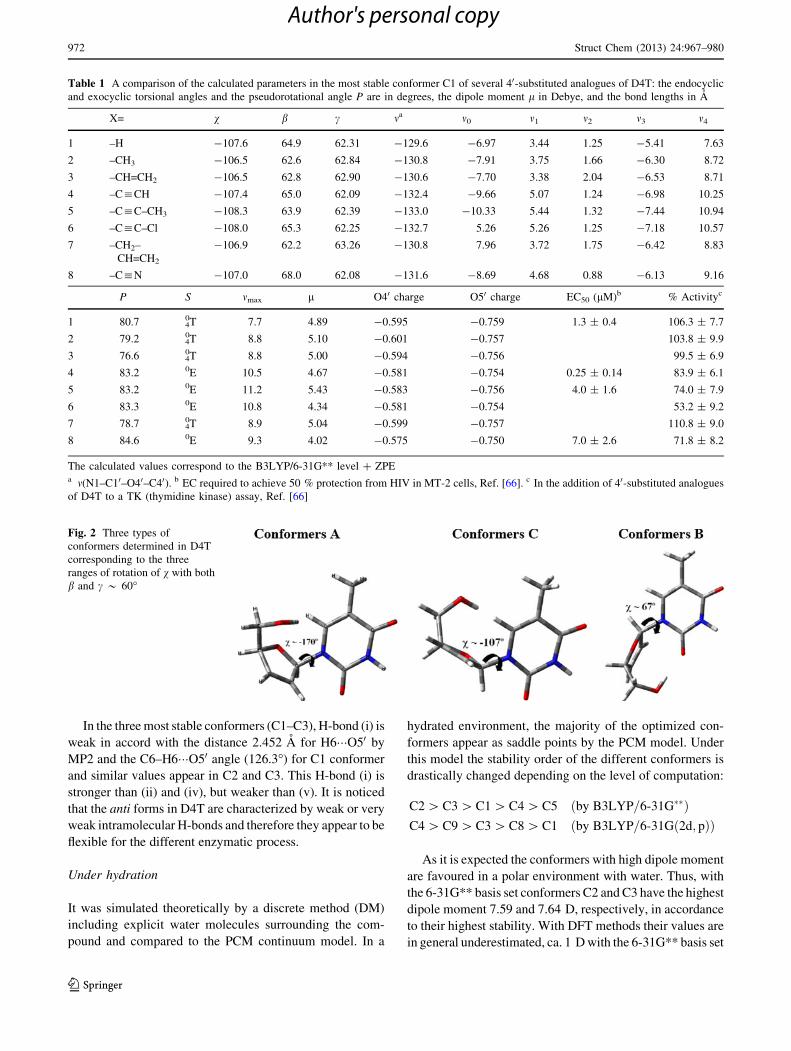
Figure 3 shows the 12 best structures selected in each
range of the rotation angle v: Six of them correspond to
conformers C (C1 to C6), four to A (A1 to A4), and two to
B (B1, B2). In this figure are also included the values of the
most important H-bonds distances of each conformer. The
computed low-energy B3LYP forms (including the global
minimum) are similar to those determined in dT [54].
The approximate estimated energy barrier height con-
former C1 ? conformer C3, through b direction in the
PES is 3.0 kcal mol-1, while C1 ? C2 is 6.2 kcal mol-1
and C3 ? C2, 3.2 kcal mol-1. The barrier height from
C7 ? C5 is 3.2 kcal mol-1, while C7 ? C6 ? C5 are
0.3 and 0.6 kcal mol-1, respectively. In the c direction the
barrier height is much higher: C1 ? C7 is 6.9 kcal mol-1,
C3 ? C6, 5.5 kcal mol-1, and C2 ? C5, 6.6 kcal mol-1.
Small differences in the structure appear between D4T
and dT: in the intermolecular distances a significant
opening of the structure in dT, with similar O2���O50 and
O40���O50 distances, and slightly longer O4���O50, ca. 0.5 A.
Intramolecular H-bonds
We have considered the classification of the H-bonds
according to Desiraju and Steiner [55]. Thus, five intra-
molecular H-bonds/interaction types may be observed in
the main conformers of D4T [35, 36]: (i) H6���O50, (ii)
H5���O40, (iii) H6���O40, (iv) O2���H10, (v) O2���H50. In
general, the observed strength of H-bonds is as follows:
(v) [ (i) [ (ii) [ (iv) [ (iii).
Conformers C involve the H-bonds/interactions (i), (ii) and
(iv) types while conformers A, the (ii) and (iii), and conformers
B that are achieved by a syn orientation of the thymine, involve
only (v) type. This type of stronger H-bond in the syn forms
gives rise to difficulties in the rotation and orientation of the
hydroxyl group for the phosphorylation process. H-bond (iii)
appears only in conformers A and they are also very weak.
H-bond (v) is the strongest one observed in D4T, and it
appears to give a great stability to the structure of con-
formers B1 and B2. In B1 this H-bond is stronger than in
B2, with a value of O50���H6 by MP2 of 1.888 versus 2.128
A in B2. This strong H-bond is responsible of a high mmax,
as it is observed by MP2.
Fig. 1 Conformational map in
3D and 2D by rotation of b and
c angles for D4T when v =
-123�. The conformation
corresponding to the holes
are indicated
Struct Chem (2013) 24:967–980 971
123
Author's personal copy
In the three most stable conformers (C1–C3), H-bond (i) is
weak in accord with the distance 2.452 A for H6���O50 by
MP2 and the C6–H6���O50 angle (126.3�) for C1 conformer
and similar values appear in C2 and C3. This H-bond (i) is
stronger than (ii) and (iv), but weaker than (v). It is noticed
that the anti forms in D4T are characterized by weak or very
weak intramolecular H-bonds and therefore they appear to be
flexible for the different enzymatic process.
Under hydration
It was simulated theoretically by a discrete method (DM)
including explicit water molecules surrounding the com-
pound and compared to the PCM continuum model. In a
hydrated environment, the majority of the optimized con-
formers appear as saddle points by the PCM model. Under
this model the stability order of the different conformers is
drastically changed depending on the level of computation:
C2 [ C3 [ C1 [ C4 [ C5 by B3LYP=6-31G��ð ÞC4 [ C9 [ C3 [ C8 [ C1 by B3LYP=6-31G 2d; pð Þð Þ
As it is expected the conformers with high dipole moment
are favoured in a polar environment with water. Thus, with
the 6-31G** basis set conformers C2 and C3 have the highest
dipole moment 7.59 and 7.64 D, respectively, in accordance
to their highest stability. With DFT methods their values are
in general underestimated, ca. 1 D with the 6-31G** basis set
Table 1 A comparison of the calculated parameters in the most stable conformer C1 of several 40-substituted analogues of D4T: the endocyclic
and exocyclic torsional angles and the pseudorotational angle P are in degrees, the dipole moment l in Debye, and the bond lengths in A
X= v b c ma m0 m1 m2 m3 m4
1 –H -107.6 64.9 62.31 -129.6 -6.97 3.44 1.25 -5.41 7.63
2 –CH3 -106.5 62.6 62.84 -130.8 -7.91 3.75 1.66 -6.30 8.72
3 –CH=CH2 -106.5 62.8 62.90 -130.6 -7.70 3.38 2.04 -6.53 8.71
4 –C:CH -107.4 65.0 62.09 -132.4 -9.66 5.07 1.24 -6.98 10.25
5 –C:C–CH3 -108.3 63.9 62.39 -133.0 -10.33 5.44 1.32 -7.44 10.94
6 –C:C–Cl -108.0 65.3 62.25 -132.7 5.26 5.26 1.25 -7.18 10.57
7 –CH2–
CH=CH2
-106.9 62.2 63.26 -130.8 7.96 3.72 1.75 -6.42 8.83
8 –C:N -107.0 68.0 62.08 -131.6 -8.69 4.68 0.88 -6.13 9.16
P S mmax l O40 charge O50 charge EC50 (lM)b % Activityc
1 80.7 40T 7.7 4.89 -0.595 -0.759 1.3 ± 0.4 106.3 ± 7.7
2 79.2 40T 8.8 5.10 -0.601 -0.757 103.8 ± 9.9
3 76.6 40T 8.8 5.00 -0.594 -0.756 99.5 ± 6.9
4 83.2 0E 10.5 4.67 -0.581 -0.754 0.25 ± 0.14 83.9 ± 6.1
5 83.2 0E 11.2 5.43 -0.583 -0.756 4.0 ± 1.6 74.0 ± 7.9
6 83.3 0E 10.8 4.34 -0.581 -0.754 53.2 ± 9.2
7 78.7 40T 8.9 5.04 -0.599 -0.757 110.8 ± 9.0
8 84.6 0E 9.3 4.02 -0.575 -0.750 7.0 ± 2.6 71.8 ± 8.2
The calculated values correspond to the B3LYP/6-31G** level ? ZPEa m(N1–C10–O40–C40). b EC required to achieve 50 % protection from HIV in MT-2 cells, Ref. [66]. c In the addition of 40-substituted analogues
of D4T to a TK (thymidine kinase) assay, Ref. [66]
Fig. 2 Three types of
conformers determined in D4T
corresponding to the three
ranges of rotation of v with both
b and c * 60�
972 Struct Chem (2013) 24:967–980
123
Author's personal copy
and ca. 0.4 D with the 6-311??G(3df,pd) basis set, as
compared to MP2.
Under DM model the first hydration shell of D4T was
simulated using 13 explicit water molecules [25] and 20
water molecules [35, 36] (Fig. 4), in the most stable con-
formers C1, C2 and C3 [56]. C1 and C2 are structurally
similar with b angle 60� and -60�, respectively. Thus,
their hydration clusters are basically the same. Conformer
C3 changes to C1 with addition of more than 8 water
molecules due to water molecules appear between the
thymine and furanose rings, therefore forcing the packing
of the structure and shielding close clusters. In these
clusters with more than eight water molecules, the orien-
tation of the b angle ca. 60� is not appropriate for the
phosphorylation by kinases. The hydration of other con-
formers was also carried out, and they follow similar trend
with a b value ca. 60�, which is neither appropriate for
phosphorylation. Therefore, the cluster with 20 water
Fig. 3 Geometry of the 12 best conformers in decreasing order of stability. The values of the strongest intramolecular H-bond distances, in A,
were determined at B3LY/6-31G**, B3LYP/6-311??G(3df,pd) and MP2/6-31G** (values in parentheses) levels
Struct Chem (2013) 24:967–980 973
123
Author's personal copy

molecules of Fig. 4 is the most stable one in a water
environment. We can conclude that in the presence of more
than 8 water molecules per nucleotide, the C1 conformer
will be the only stable one.
By contrast, the hydration of the natural nucleoside
thymidine lead to an orientation of the –O50H group
appropriate for phosphorylation with b ca. 180�, Fig. 5.
This is because the two hydroxyl groups –O50H and –O30Hcan be H-bonded through a water molecule. This fact can
explain the higher phosphorylation of the natural nucleo-
sides than their corresponding 30-deoxynucleosides
analogues.
In the solid state
By X-ray several kinds of D4T crystals have been observed
in the solid state: orthorhombic [33], rhombic [57, 58],
monoclinic [32, 33] and triclinic [31, 33]. In these studies,
as well as in other X-ray studies [59, 60] of related
nucleosides and uridine analogues [61, 62], it is shown that
the glycosyl torsion angle is in anti orientation and their
geometries are stabilized by the formation of self-associ-
ated species. We have well simulated [63] the dimer forms
of the X-ray data with C1 conformers or C1–A2. The
concordance of our calculated values with the experimental
X-ray data, Table 2, confirms the validity of our method-
ology performed on D4T.
Natural NBO atomic charges
It is observed that the largest negative charge in all the
conformers and levels is on the O50 hydroxyl oxygen. Its
value in conformer C1 is similar to that found in dT [25,
26], by MP2 -0.809 e- (where e- is the charge of an
electron). The second atom with large negative charge is
the N3 thymine nitrogen and O2 oxygen. The negative
charge on N3 is larger, ca. 0.2 e-, than on N1 atom. These
features are also in accordance to those calculated in dT
[25] and in AZT [54].
The highest positive charge is on C4 and C2 atoms in
agreement, respectively, to the high negative charge on the
O4 and O2 atoms. C2 has higher positive charge than C4
because of O2 has slight higher negative charge than O4.
The hydrogen atom with the highest positive value corre-
sponds to H50, i.e. it is the most reactive. Closely appears
H3 hydrogen.
First phosphorylation
The first phosphorylation of the nucleosides analogues by
the ATP kinase is a crucial step in the activity of these
prodrugs. The proportion of compound phosphorylated is
in general very small in the majority of the prodrugs. For
understanding this fact, we have simulated first in the
isolated state the interaction of ATP with conformers C1
and C3 of D4T, Fig. 6. It is observed that the interaction of
the phosphate group with the O50-H moiety of conformer
C1 produces a rotation of the b angle to a value ca. 180�,
i.e. conformer C1 changes to C3, and this conformer C3
appears with the b value appropriate for phosphorylation.
The interaction of ATP with the optimum hydrated
clusters of D4T was also simulated. The interaction in the
cluster of C1 conformer with 8 and 12 water molecules is
shown in Fig. 7. It is observed that this interaction of ATP
is not enough strong to rotate the b angle to a value
appropriate for phosphorylation, b ca. 180�. Moreover, the
phosphate groups little affect the values of the b and ctorsional angles. Thus, at this point, it is clear that the
number of water molecules surrounding the nucleoside in
the ATP kinase cavity should be lower than 8.
We study also the interaction of ATP with D4T in the
clusters with lower number than 8 water molecules. For
simplification, in this Fig. 8 is only shown the interaction
of C3 clusters with 6 and 7 water molecules. Similar trend
is observed in the interaction with C1 cluster, because the
phosphate group interaction is strong enough to rotate the
b angle toward the C3 form, which is appropriate for
phosphorylation. Successive steps in the simulation show
Fig. 4 Scheme followed in the
hydration of conformers C1 and
C3 of D4T up to the optimum
cluster with 20 water molecules
reproducing the first hydration
shell. H-bonds D4T-water and
water–water molecules are
denoted by dotted lines
974 Struct Chem (2013) 24:967–980
123
Author's personal copy

that the phosphate group with negative charge takes a
hydrogen atom from a neighbour water molecule. Then, the
new OH group formed from this water molecule takes
another H atom from the C50-OH group of D4T. Thus, the
O50 atom is negatively charged and ready to be bonded to a
P atom with positive charge in the ATP. This process
involves one water molecule as observed in the cluster with
6 H2O, or two water molecules in the cluster with 7 H2O.
This feature indicates that the hydrolysis process of ATP in
the phosphorylation, implicates a proton transfer between
ATP and D4T through the neighbour water molecules.
Thus, we can conclude that in the first phosphorylation
by the ATP kinase, the D4T cluster looses some of its water
molecules inside of the enzyme cavity facilitating the
rotation of C50-OH bond to get b ca. 1808, and leading to
interaction with the ATP molecule and the further phos-
phorylation. Therefore, the number of water molecules in
the cavity surrounding the nucleoside is predicted to be
lower than 8.
Bonding of D4TTP to DNA viral
One of the goals of the present paper is the prediction of
new drugs with higher activity than D4T. For this purpose,
we have simulated how the bonding process of D4TTP to
DNA viral is in the cavity of the reverse transcriptase
enzyme, Fig. 9, based on the CHARMm25 force field [64,
65]. It looks clear in this figure that substituents in
Fig. 5 Hydration of the natural
nucleoside thymidine with 13
water molecules stabilizes the
conformer C3 due to C30-OH
group presence. The formation
of two hydrogen bonds between
C50-OH and C30-OH groups and
one water molecule permits a
value of b ca. 180�
Table 2 A comparison of the
calculated dimer forms in D4T
molecule with those reported in
the crystal
The exocyclic torsional angles
and the pseudorotational angle
P are in degrees. The calculated
values correspond to the
B3LYP/6-31G** level ? ZPEam(N1–C10–O40–C40)
Dimer Molecule v c ma P mmax
I A Calculated conformer C1 -108.70 62.04 -129.7 80.86 8.0
X-ray data, orthorhombic -102.1 50.5 -129.0
X-ray data, rhombic -102.0 52.0
B Calculated conformer C1 -107.75 62.80 -129.5 80.69 7.5
X-ray data, orthorhombic -117.2 62.0 -127.6
X-ray data, rhombic -159.9
G/H A Calculated conformer A2 -174.80 44.91 -127.0 90.43 5.3
X-ray data, triclinic -172.6 54.1 -125.6 90.4 4.8
X-ray data, monoclinic -174.1 53.8 -123.1
B Calculated conformer C1 -108.61 62.54 -130.0 81.03 8.3
X-ray data, triclinic -85.1 55.5 -128.8 103.6 6.2
X-ray data, monoclinic -118.0 60.6 -130.5
Fig. 6 First step in the
phosphorylation of conformers
C1 and C3 of D4T by one ATP
molecule in the isolated state. In
this interaction conformer C1
changes to conformer C3 where
C50-OH group with b ca. 180�has the adequate orientation to
loose the proton by the
phosphate moiety
Struct Chem (2013) 24:967–980 975
123
Author's personal copy
20-position should be very small for steric interaction with
Residue Y115. In 30-position the substituents can be
something larger than in 20, but not too much. In 40-position
substituents with three carbon atoms are admitted, and
thus, several authors [66, 67] have synthesized D4T
derivatives with different substituents in this position,
Scheme 1. We have studied them in the next section.
Relationships/tendencies observed
The possibility to establish general structure–activity rela-
tionships/tendencies in a series of anti-HIV D4T derivatives
dideoxynucleosides thymidine analogues is another objec-
tive of the present work. Unfortunately, only two papers
report the anti-HIV activity with more than three D4T
derivatives under the same experimental conditions [66, 67].
We have selected Dutschman et al. [66] data due to the bigger
derivative numbers and smaller size of substituents than
those by Argawal et al. [67]. These authors performed
experiments with dT, AZT, D4T and 40-substituted D4T
derivatives, with methyl, vinyl, ethynyl, methylethynyl,
chloroethynyl, allyl and cyano groups, Scheme 1, to evaluate
antiviral effect, cellular toxicity, interaction with TK-1 and
TP enzymes, and pharmacokinetic properties. To start this
study, a full optimization of all compounds was carried
out by B3LYP/6-31G** level ? ZPE to obtain the most
stable conformer. In all cases the global minimum corre-
sponds to the characteristic torsional angles of conformer C1,
as in the former compound D4T, Table 1 and Scheme 1.
Based in all these data, we have related two antiviral
activity parameters, the effective concentration EC50, and
Fig. 7 Interaction of ATP
molecule with the optimum
cluster of D4T in conformer
C1 ? 12H2O and ?8H2O. This
interaction is not enough strong
to rotate the b (C40–C50–O50–H50) exocyclic torsional angle.
H-bonds between D4T-water
and water–water molecules are
denoted by dotted lines
Fig. 8 First step in the phosphorylation of D4T in C3 cluster with 6H2O and 7H2O by one ATP molecule. A proton migration appears from O50-H to the phosphate moiety though one/two water molecules. H-bonds between D4T-water and water–water molecules are denoted by dotted lines
976 Struct Chem (2013) 24:967–980
123
Author's personal copy

the potential of these compounds to be phosphorylated,
named as % activity in a TK-1 kinase assay with [14C]dT
[66]. In this assay, the amount of conversion to [14C]dTMP
with or without addition of analogues in concentration
10-fold-higher than natural nucleoside was compared.
Thus, we have found several interesting structure–activity
relationships/tendencies with our calculated geometric
parameters and dipole moments, some of them are shown
in Fig. 10. The correlation is not so good because the
activity depends on other factors not considered here, but
the tendency is important. Thus, analyzing the different
parameters on these figures was noted the following:
(i) The different substituents on C40 of D4T structure
mainly produce a change in the NBO atomic charge on
O40. An increment in the negative charge on O40 is
linear related to an increase in the negative charge on
O50, a shortening of C40-C50 and a lengthening of C50-O50 and O50-H50. All these changes lead to other effects:
thus an increase in the negative charge on O40 is linear
related to a slight closing of the furanose pucker P and to
an increase in the negative charge on O2 and O4 of the
thymine ring, and as consequence a linear increment in
the dipole moment. Also it is observed a shortening in
the O50���O4 interatomic distance with the increase in
the negative charge on O40. All these features give to the
following conclusion: all the substituents on C40 that
produce an increase in the negative charge on O40,Fig. 10a, and as consequence on O2, O4 and the dipole
moment, increment the activity, Fig. 10e.
(ii) The D4T derivatives with high activity have in
general a low value of the furanose pucker P,
Fig. 10b, and of the exocyclic torsional angle b, but
high torsional angle c in the g? orientation, within
their corresponding ranges, Fig. 10c and d. In these
last figures we have considered important to include
the value of the most used anti-HIV drug AZT, which
follows the tendency observed.
(iii) An increase in the activity in thymidine derivatives is
linearly related to an increment in the dipole
moment, Fig. 10e.
(iv) An increment in the activity appears related to a
lower concentration required to achieve 50 % pro-
tection from HIV in MT-2 cells, EC50, Fig. 10f.
(v) In the few experimental data reported of EC50, a
tendency is suggested between b and c vs. EC50,
Fig. 10g.
(vi) Finally, a lower EC50 value is required in compounds
with a low value of furanose pucker P angle and
dipole moment, Fig. 10h.
We can conclude that the % activity, is related to
high values of the negative charge on O4’ into the
(-0.575 to -0.601) range, dipole moment (4.02-5.43),
c angle (62.088-63.268) and low values of pseudorotation
angle P (76.588-84.608) and b angle (62.248-68.008).Two good correlations are observed for % activity of
D4T derivatives and AZT with b angle and dipole
moment. On the other hand, correlations for antiviral
activity as EC50 from only four derivatives suggest that
Fig. 9 Bonding of D4TTP and
their derivatives under study to
DNA viral through the reverse
transcriptase enzyme
Struct Chem (2013) 24:967–980 977
123
Author's personal copy
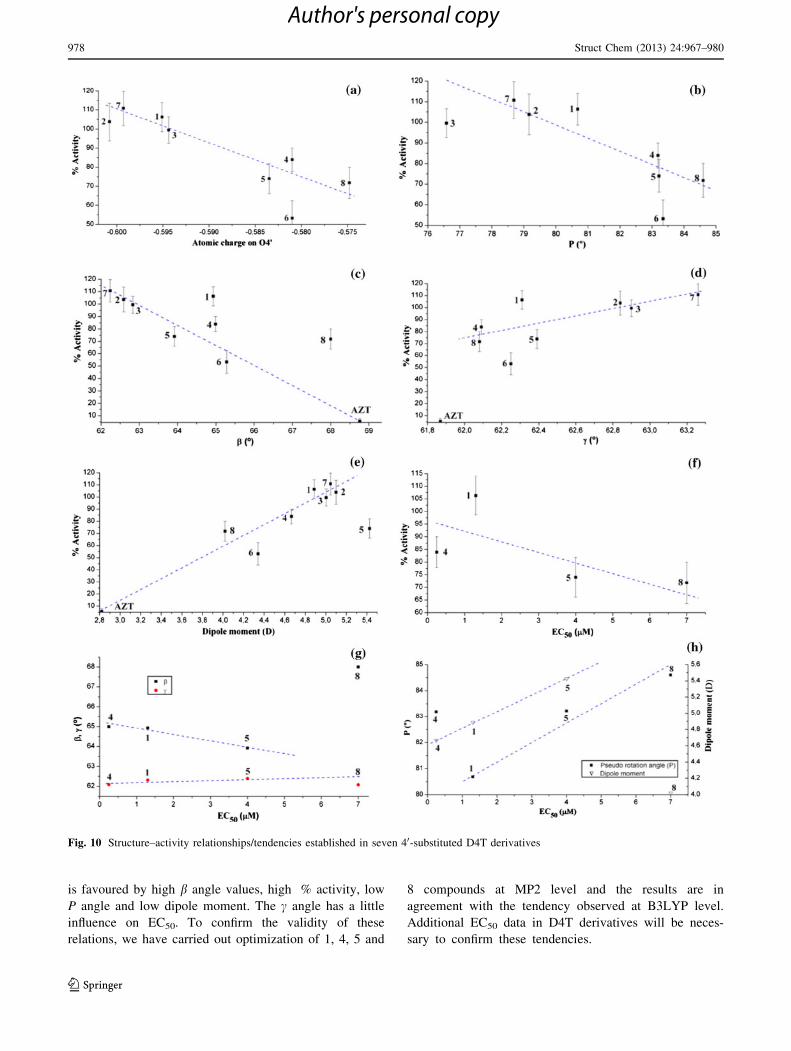
is favoured by high b angle values, high % activity, low
P angle and low dipole moment. The c angle has a little
influence on EC50. To confirm the validity of these
relations, we have carried out optimization of 1, 4, 5 and
8 compounds at MP2 level and the results are in
agreement with the tendency observed at B3LYP level.
Additional EC50 data in D4T derivatives will be neces-
sary to confirm these tendencies.
Fig. 10 Structure–activity relationships/tendencies established in seven 40-substituted D4T derivatives
978 Struct Chem (2013) 24:967–980
123
Author's personal copy

Summary and Conclusions
The conformational landscape of the HIV-1 reverse tras-
criptase inhibitor, the nucleoside analogue D4T was explored
using a comprehensive set of modern computational tech-
niques and using four levels of calculation in the isolated
state and two levels in the hydrated form. Comparisons of our
simulated dimer forms with the crystal structure data provide
support for the quality of our quantum chemical calculations.
Thus, the geometries and values presented here appear to
improve the results to date. In the present work the most
important findings are the following:
(1) In general conformers type C are more stable than
conformers type A and B. The calculated five first
optimum conformers are the same by all the methods
and levels. Conformer C1 corresponds to the global
minimum at all the levels of computation, either in
isolated state as in aqueous solution (v = -103.68,b = 63.88, c = 60.68, N-type). In the NBO charges
small differences were observed with dT in AZT.
(2) The first hydration shell was simulated with explicit
water molecules up to 20. The cluster with conformer
C1 was the most stable one. Conformer C3 (v =
-122.38, b = 165.08, c = 43.78), is stable with 1–8
water molecules but with a number of water mole-
cules larger eight, C1 is the only stable form with a
close hydrated cluster.
(3) Hydration of the natural nucleoside thymidine with
13 water molecules stabilizes the conformer C3 with
b ca. 1808 due to the presence of the group C30-OH.
The formation of a molecular pincers between C50-OH and C30-OH groups through two hydrogen bonds
with one water molecule permits a value of b ca. 1808and is stabilized by a four hydrogen bonded water
molecules net.
(4) The first phosphorylation step in D4T was simulated
through the interaction with the ATP anion and for C1
and C3 conformers. The simulation was carried out
under the isolated state consideration as well as under
hydration environment with different amounts of
explicit water molecules. In the isolated state this
interaction is enough strong to change C1 to C3 form
by rotation of C50-OH group until the value of b ca.
1808. This conformer C3 has the value of the b angle
appropriate for the phosphorylation.
(5) Phosphorylation of the D4T hydrated clusters is only
possible with a number of water molecules below of
eight, which permits the C50-OH group rotation to be
accessible for phosphate group. For a successful
phosphorylation, the ATP kinase enzyme must
remove the majority of the water molecules around
the nucleoside and therefore stabilizing conformer
C3, which is the stable form with less than 8 water
molecules.
(6) Hydrolysis process of ATP in the phosphorylation,
implicates a proton transfer between ATP and D4T
through the neighbour water molecules.
(7) Bonding of D4TTP and their derivatives to DNA viral
through the reverse transcriptase enzyme provides
further support for the fact that the biological activity
of 20,30-didehydro-20,30-dideoxy analogues is directly
related to the lack of an OH group at the C30-position.
The study shows also that substituents in 20-position
should be very small for steric interaction with
Residue Y115, substituents in 30-position should not
be large, while substituents in 40-position can be
large.
(8) For the first time structure–activity relationships/
tendencies were established in seven 40-substituted
D4T derivatives and AZT. All the substitutents on C40
in D4T that produce an increase in the negative
charge on O40 (and as consequence on O2, O4 and the
dipole moment) increment the TK-1 % enzymatic
activity.
The first phosphorylation is a crucial step in the activity
of anti-HIV drugs. Good comprehension of the mechanism
that has been proposed for this phosphorylation and all the
parameters investigated here could be very useful for
developing drugs with high anti-HIV activity and low
toxicity. Any information that could shed light on this
problem is important.
Acknowledgments The authors wish to thank to the MCI (Minis-
terio de Ciencia e Innovacion) through CTQ2010-18564 (subprogram
BQU), and to Dra. M. de la Fuente for her great help in the present
work.
References
1. Hammond E, McKinnon E, Nolan D (2010) Clin Infect Dis
51(5):591
2. Strydom S, Liebenberg W, Yu L, de Villiers M (2009) Int J
Pharm 379(1):72
3. De Clercq E (2004) Med Chem Res 13(6–7):439
4. De Clercq E (2002) Biochim Biophys Acta 1587:258
5. De Clercq E (2002) Med Res Rev 22(6):531
6. Horwitz JP, Chua J, Rooge MAD, Noel R, Klundt IL (1966)
J Org Chem 31:205
7. Lin TS, Prusoff WH (1987) US Patent 4710492
8. Seaton RA, Fox R, Bodasing N, Peters SE, Gourlay Y (2003)
Aids 17:445
9. Naeger LK, Margot NA, Miller MD (2002) Antimicrob Agents
Chemother 46:2179
10. Ponomareva AG, Yurenko YP, Zhurakivsky RO, Van Mourik T,
Hovorun DM (2012) Phys Chem Chem Phys 14:6787
11. Chu CK, Schinazi RF, Arnold BH, Cannon DL, Doboszewski B,
Bhadti VB, Gu ZP (1988) Biochem Pharmacol 37:3543
Struct Chem (2013) 24:967–980 979
123
Author's personal copy

12. Balzarini J, Kang GJ, Dalal M, Herdewijn P, Declerq E, Broder S,
Johns DG (1987) Mol Pharmacol 32:162
13. Mehellou Y, Balzarini J, McGuigan C (2009) Org Biomol Chem
7:2548
14. Hunter R, Muhanji CI, Hale I, Bailey CM, Basavapathruni A,
Anderson KS (2007) Bioorg Med Chem Lett 17:2614
15. Foloppe N, MacKerell AD (1999) Biophys J 76:3206
16. Yates PC, Kirby SV (1993) Struct Chem 4:299
17. Nikolaienko TY, Bulavin LA, Hovorun DM (2012) Phys Chem
Chem Phys 14(44):15554
18. Nikolaienko TY, Bulavin LA, Hovorun DM (2011) J Biomol
Struct Dyn 29(3):563
19. Yurenko YP, Zhurakivsky RO, Ghomi M (2007) J Phys Chem B
111:9655
20. Yurenko YP, Zhurakivsky RO, Ghomi M, Samijlenko SP,
Hovorun DM (2007) J Phys Chem B 111:6263
21. Yurenko YP, Zhurakivsky RO, Ghomi M, Samijlenko SP,
Hovorun DM (2008) J Phys Chem B 112:1240
22. Choi Y, George C, Comin MJ, Barchi JJ Jr, Kim HS, Jacobson
KA, Balzarini J, Mitsuya H, Boyer PL, Hughes SH, Marquez VE
(2003) J Med Chem 46:3292
23. Saran A, Ojha RP (1993) J Mol Struct (Theochem) 284:223
24. Saran A, Ojha RP (1993) J Mol Struct (Theochem) 286:247
25. Alcolea Palafox M, Iza N, De la Fuente M, Navarro R (2009)
J Phys Chem B 113(8):2458
26. Alcolea Palafox M, Iza N (2010) Phys Chem Chem Phys
12(4):881
27. Mishchuk Ya R, Potyagaylo AL, Hovorun DM (2000) J Mol
Struct 552:283
28. Nikolaienko TY, Bulavin LA, Hovorun DM (2012) Phys Chem
Chem Phys 28:7441
29. Yurenko YP, Zhurakivsky RO, Samijlenko SP, Hovorum DM
(2011) J Biomol Struct Dyn 29(1):51
30. Lu J, Rohani S (2009) Org Process Res Dev 13(6):1262
31. Gurskaya GV, Bochkarev AV, Zhdanov AS, Dyatkina NB,
Kraevskii AA (1991) Mol Biol 25(2):401
32. Harte WE, Starret JE Jr, Martin JC Jr, Mansuri MM (1991)
Biochem Biophys Res Commun 175(1):298
33. Mirmehrabi M, Rohani S, Jennings MC (2005) Acta Crystallogr
Sect C 61:0695
34. Mirmehrabi M, Rohani S, Keshava Murthy KS, Radatus B (2006)
Cryst Growth Des 1:141
35. Alcolea Palafox M, Iza N (2012) J Mol Struct 1028:181
36. Alcolea Palafox M, Iza N, de la Fuente M, Navarro R (2005) In:
11th European Conference on the Spectroscopy of biological
molecules, 188NI Aschaffenburg, Alemania
37. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA,
Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant
JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B,
Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada
M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nak-
ajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE,
Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R,
Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C,
Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P,
Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain
MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K,
Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cio-
slowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Ko-
maromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY,
Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen
W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03,
Revision B.04. Gaussian, Inc., Pittsburgh, PA
38. Hoffmann M, Rychlewski J (2002) Rev Mod Quantum Chem
2:1767
39. Alcolea Palafox M, Posada-Moreno P, Villarino-Marın AL,
Martinez-Rincon C, Ortuno-Soriano I, Zaragoza-Garcıa I (2011)
J Comput Aided Mol Des 25:145
40. Tamara Molina A, Alcolea Palafox M (2011) Chem Phys 387:11
41. Alcolea Palafox M, Nielsen OF, Lang K, Garg P, Rastogi VK
(2004) Asian Chem Lett 8:81
42. Alcolea Palafox M, Rastogi VK (2002) Spectrochim Acta A
58:411
43. Alcolea Palafox M (1998) Recent research developments in
physical chemistry. Transworld Research Network, Trivandrum,
India, 2:213
44. Alcolea Palafox M (2000) Int J Quantum Chem 77:661
45. Alcolea Palafox M, Iza N, Gil M (2002) J Mol Struct (Theochem)
585:69
46. Arissawa M, Taft CA, Felcman J (2003) Int J Quantum Chem
93:422
47. Carpenter JE, Weinhold F (1988) J Mol Struct (Theochem)
169:41
48. Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899
49. Laboratorio de Quımica Computacional (Lab-QC)—UNED.
http://www.uned.es/lab-qc, 2004
50. SURFER program. Version 7 (1999) Golden Software, Inc.
51. Saenger W (1984) Principles in nucleic acid structure. Springer,
New York
52. Van Roey P, Taylor EW, Chu CK, Schinazi RF (1993) J Am
Chem Soc 115:5365
53. Rao SN (1998) Nucleosides Nucleotides 17:791
54. Merchante DGG, Tamara A, Alcolea Palafox M, Iza N (2010) In:
Twenty-second austin symposium on molecular structure, Austin,
Texas, USA, S4, pp 67
55. Desiraju GR, Steiner T (1999) The weak hydrogen bond. Oxford
University Press, New York
56. de la Fuente M, Navarro R, Alcolea Palafox M, Iza N, (2007) In:
12th European Conference on the Spectroscopy of biological
molecules, pp. 263, 169, Paris
57. Gurskaya GV, Tsapkina EN (1989) In: Abstracts of the twelfth
European Crystallographic Meeting, Moscow, vol 2, p 380
58. Tsapkina EN (1989) The molecular and crystalline structures of
nucleic analogs of nucleic acid biosynthesis substrates (in Rus-
sian). Dissertation for Doctorate I Chemical Sciences, Institute of
Molecular Biology, Moscow
59. Shishkin OV, Pelmenschikov A, Hovorun DM, Leszczynski J
(2000) J Mol Struct 526:329
60. Hocquet A, Leulliot N, Ghomi M (2000) J Phys Chem B
104:4560
61. Van Roey P, Salerno JM, Duax WL, Chu CK, Ahn MK, Schinazi
RF (1988) J Am Chem Soc 110:2277
62. Allen FH, Bellard S, Brice MD, Cartwright BA, Doubleday A,
Higgs H, Hummelink T, Hummelink-Peters BG, Kennard O,
Motherwell WDS, Rodgers JR, Watson DG (1979) Acta Crys-
tallogr Sect B 35:2331
63. Alcolea Palafox M, Iza N (2009) In: 13th European Conference
on the Spectroscopy of biological molecules (ECSBM) Palermo,
Italia, pp PB-121
64. Singh K, Marchand B, Kirby KA, Michailidis E, Sarafianos SG
(2010) Viruses 2:606
65. Mu L, Sarafianos SG, Nicklaus MC, Russ P, Siddiqui MA, Ford
H Jr, Mitsuya H, Le R, Kodama E, Meier C, Knispel T, Anderson
L, Barchi JJ Jr, Marquez VE (2000) Biochemistry 39:11205
66. Dutschman GE, Grill SP, Gullen EA, Haraguchi K, Takeda S,
Tanaka H, Baba M, Cheng Y-C (2004) Antimicrob Agents
Chemother 48(5):1640
67. Agarwal HK, Loethan K, Mandal D, Doncel GF, Parang K (2011)
Bioorg Med Chem Lett 21:1917
980 Struct Chem (2013) 24:967–980
123
Author's personal copy






























