




Neuropharmacology 49 (2005) 465e476
www.elsevier.com/locate/neuropharm
Neuronal Kir3.1/Kir3.2a channels coupled to serotonin 1A andmuscarinic m2 receptors are differentially modulated by the
‘short’ RGS3 isoform
Cristina Jaen, Craig A. Doupnik*
Department of Physiology & Biophysics, University of South Florida College of Medicine, 12901 Bruce B. Downs Blvd. MDC8,
Tampa, FL 33612, USA
Received 21 December 2004; received in revised form 5 April 2005; accepted 7 April 2005
Abstract
‘Regulators of G protein signaling’ (RGS proteins) have profound effects on ion channels regulated by G protein-coupledreceptor (GPCR) signaling, including the G protein-gated inwardly rectifying KC (GIRK) channels that inhibit excitability ofneuronal, endocrine, and cardiac cells. Here we describe the effects of an alternatively spliced ‘short’ RGS3 isoform (RGS3s) in
comparison to RGS4, on the temporal and steady-state gating properties of neuronal GIRK channels (Kir3.1/Kir3.2a) activated byeither serotonin 1A (5-HT1A) receptors or muscarinic m2 receptors expressed in Chinese hamster ovary (CHO-K1) cells. RGS3s isabundantly expressed in brain and contains a unique short N-terminus via alternative splicing that distinguishes it from other RGS3
isoforms as well as other members of the B/R4 RGS gene subfamily. Our results indicate that RGS3s and RGS4 similarly affect thetemporal and steady-state gating properties of 5-HT1A receptor-coupled Kir3.1/Kir3.2a channels, but differentially modulatemuscarinic m2 receptor-coupled channels. RGS3s caused a significant w45% reduction in the maximal acetylcholine (ACh)-evoked
GIRK current amplitude and a marked shift in the steady-state ACh doseeresponse relation indicative of a reduction infunctionally coupled m2 receptor-GIRK channel complexes. Yet RGS3s still accelerated the m2 receptor-dependent GIRKactivation, deactivation, and acute desensitization time course consistent with the RGS-enhanced GAP activity that was alsoobserved with RGS4. Several mechanisms that may contribute to the receptor-dependent effects of RGS3s are discussed with
particular attention to the role of the distinct N-terminal domain. Our findings highlight the potential impact of selective RGS-GPCR interactions on neuronal GIRK channel function that may affect the properties of inhibitory postsynaptic potentialsactivated by different GPCR-GIRK channel complexes.
� 2005 Elsevier Ltd. All rights reserved.
Keywords: Inward rectifier potassium channels; G protein coupled receptors; Regulators of G protein signaling; Acetylcholine; Serotonin; Patch
clamp; Chinese hamster ovary cells
1. Introduction
Gbg-gated inwardly rectifying KC channels (GIRKs)are expressed predominantly in brain, heart, and
* Corresponding author. Tel.: C1 813 974 1507; fax: C1 813 974
3079.
E-mail address: [email protected] (C.A. Doupnik).
0028-3908/$ - see front matter � 2005 Elsevier Ltd. All rights reserved.
doi:10.1016/j.neuropharm.2005.04.010
endocrine tissue and suppress cell excitability duringneurotransmitter and hormone activation of pertussistoxin (PTX)-sensitive G protein-coupled receptors(GPCRs) (Stanfield et al., 2002). Consistent with this,gene knockout of neuronal GIRK channel subunitspromote spontaneous and pharmacologically inducedseizures and hyperactivity in mice (Signorini et al., 1997;Blednov et al., 2001). The recent discovery of neuronal

466 C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
GIRK channel involvement in drug-induced analgesiafurther highlight the physiological role of GPCR-activated GIRK channels and their modulators in thenervous system (Blednov et al., 2003).
The temporal gating properties of receptor-activatedGIRK currents are determined by the kinetic propertiesof the G protein cycle and dramatically accelerated by‘regulators of G protein signaling’ (RGSs) (Breitwieserand Szabo, 1988; Doupnik et al., 1997; Saitoh et al.,1997). RGS proteins speed up passage through theG-protein cycle by increasing the intrinsic GTPaseactivity of Ga subunits, thereby accelerating the re-association of the ‘inactive’ heterotrimeric Ga(GDP)bgcomplex (Ross and Wilkie, 2000). RGS proteins arecharacterized by a highly conserved ‘RGS domain’ ofw125 a.a. that confers direct binding to Ga subunitsand is flanked by less conserved N- and C-terminaldomains of variable length (Tesmer et al., 1997). Morethan 20 mammalian RGS genes have been identified todate and are classified into six subfamilies based onsequence homology within the RGS domain (Ross andWilkie, 2000; Hollinger and Hepler, 2002).
The specific RGS proteins that modulate endogenousneuronal GIRK channels are currently not known.Since GIRK channels can form stable signaling com-plexes with GPCRs (Lavine et al., 2002) and multipleRGS proteins are expressed within single GIRK-expressing neurons and atrial myocytes (Gold et al.,1997; Doupnik et al., 2001, 2004), different RGSproteins may assemble with different GPCR-coupledGIRK channels to provide distinct regulatory capabil-ities. The divergent amino terminal region of the B/R4subfamily of RGS proteins has been implicated in (1)mediating RGS selective coupling to GPCRs (Zenget al., 1998), (2) facilitating functional a2 adrenergicreceptor-GIRK channel coupling in rat sympatheticneurons (Jeong and Ikeda, 2001), and (3) promotingtranslocation of GPCReRGS complexes to the plasmamembrane (Saitoh et al., 2001; Roy et al., 2003). Thusthe divergent RGS amino terminus may provide a meansto confer selective RGS coupling to different GPCReeffector signaling complexes (Hollinger and Hepler,2002).
We report here the functional properties of analternatively spliced ‘short’ isoform of mouse RGS3(RGS3s) (Reif and Cyster, 2000) on neuronal GIRKchannels (Kir3.1/Kir3.2a) coupled to either serotonin1A (5-HT1A) receptors or muscarinic m2 receptors inChinese hamster ovary cells (CHO-K1). MammalianRGS3 isoforms are expressed in both brain and heart(Druey et al., 1996; Koelle and Horvitz, 1996), andalternatively spliced RGS3 transcripts generate at leastfour different protein isoforms having different aminoterminal domains that share a common RGS domain(Kehrl et al., 2002). The unique amino terminal regionof mouse RGS3s is 21 amino acids long and comparable
in size to the 33 amino acid N-terminus of RGS4. YetRGS3s lacks the amphipathic a-helix conserved inother members of the B/R4 subfamily that facilitatesmembrane association and is necessary for membranetranslocation (Chen et al., 1999; Bernstein et al., 2000;Heximer et al., 2001; Saitoh et al., 2001). We thereforequestioned whether the variant RGS3s isoform differ-entially affects GIRK channel gating properties com-pared to RGS4 (Doupnik et al., 1997). Our findingsdemonstrate RGS3s differentially modulates GPCR-GIRK channel complexes and suggest that differentRGS N-termini may influence the agonist sensitivity andmagnitude of GIRK channel activation in a GPCR-dependent manner.
2. Experimental procedures
2.1. Heterologous expression in CHO-K1 cells
CHO-K1 cells (American Type Culture Collection,Manassas, VA) were cultured in a-modified Eagle’smedium containing 5% fetal bovine serum and 0.1 mg/mlstreptomycin, and maintained in a humidified 5% CO2
incubator at 37 �C. One day after low density platingin 35 mm dishes, cells were transfected with DNAeliposome complexes composed of lipofectamine (Invi-trogen, Carlsbad, CA) and a mixture of cDNAs clonedinto the mammalian expression vector pcDNA3.1(C)(Invitrogen). The total DNA (mg) to lipofectamine (mg)ratio was kept constant at 1:5 when pre-forming theDNAeliposome complexes. The amount of each DNAvector in the mixture per dish was as follows; 0.2 mg ratKir3.1 (GenBank accession # NM_031610), 0.2 mgmouse Kir3.2a (GenBank accession # NM_010606),0.2 mg GPCR either human muscarinic m2 receptor(GenBank accession # NM_000739) or human 5-HT1A
receptor (GenBank accession # NM_000524), with orwithout 1.0 mg RGS either mouse RGS3s (GenBankaccession # NM_134257) or rat RGS4 (GenBankaccession # NM_017214), with 0.1 mg enhanced greenfluorescent protein (GFP) cDNA (pGreenlantern-1,GIBCO) included as a reporter gene (Doupnik et al.,1997, 2004). The transfected cells were incubated over-night in serum-free OPTI-MEM media (Invitrogen).Twenty-four to thirty-six hours after transfection, singleGFP-positive cells were selected for electrophysiologicalrecordings. The RGS3s cDNA clone was generouslyprovided byDrs Karin Reif and Jason Cyster (Universityof California, San Francisco) (Reif and Cyster, 2000).All other cDNA clones were as described elsewhere(Doupnik et al., 1997, 2004). For pertussis toxin (PTX)pre-treatment experiments, transfected CHO-K1 cellswere incubated overnight (12e18 h) with 100 ng/ml PTX(P-7208, Sigma Chemical).

467C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
2.2. Electrophysiological recordings
Electrophysiological recordings were performed us-ing the whole-cell configuration of the patch clamptechnique (Hamill et al., 1981). GFP-positive cells wereidentified by epi-fluorescence microscopy using aninverted microscope (Nikon Diaphot with CF N PlanFluor Ph 20! objective) equipped with a mercury lampand GFP filter set (Endow GFP, Chroma TechnologyCorp.). Whole-cell recordings were performed witha patch clamp amplifier (Axopatch-1D, Axon Instru-ments) linked to a personal computer (Dell Dimension400R) via a digital acquisition system (Digidata 1200B,Axon Instruments). Current recordings were digitized at100 Hz and filtered at 50 Hz using the amplifier’sintegrated 4-pole Bessel-type filter. All experiments wereperformed at room temperature (22e25 �C).
Patch pipettes were prepared from borosilicate glasscapillaries (#BF150-86-10, Sutter Instruments) usinga programmable microelectrode puller (P-97, SutterInstruments). The patch pipettes had tip resistancesof 3e5 MU when filled with internal solution. Thecomposition of the internal pipette solution was (inmM): KCl 120, NaCl 10, MgCl2 5, EGTA 1, HEPES 5,ATP 5, GTP 0.2 (pH 7.2). Two minutes were allowedafter breaking into the cells to permit dialysis of theintracellular components. The initial external bathsolution composition was (in mM): NaCl 145, KCl 5,CaCl2 2, MgCl2 1; glucose 10, HEPES 5 (pH 7.4). Afterestablishing whole-cell recording, fast capacitive currenttransients were minimized by the amplifier’s analoguecompensation circuitry and the cell membrane potentialclamped to a holding potential of �100 mV.
Rapid application and washout of different agonistconcentrations was performed using a multi-barrel per-fusion system (SF-77B, Warner Instruments) (Doupniket al., 2004). Cells were initially superfused with anisotonic high KC external solution to shift the KC
equilibrium potential (EK) to �40 mV. The compositionof the ‘‘high KC solution’’ was (in mM): NaCl 125, KCl25, CaCl2 2, MgCl2 1; glucose 10, HEPES 5 (pH 7.4).Agonist-evoked inward GIRK currents were elicited bycomputer-controlled barrel movement that enabledrapid agonist application and washout via an adjacentbarrel containing ACh or 5-HT in high KC solution(Doupnik et al., 2004). Flow through the perfusionbarrels was gravity driven and the time constant forsolution exchange was w200 ms (Doupnik et al., 2004).Different concentrations of ACh or 5-HT were tested viaa manifold connecting multiple solution reservoirs to theagonist perfusion barrel. One minute was allowedbetween agonist concentration changes to accommodatedead space within the perfusion system. Voltage rampsfrom �100 to C50 mV (0.5 s) were applied before andduring agonist application to evaluate the voltage depen-dence of the agonist-evoked currents.
2.3. Electrophysiological data analysis
Time-dependent GIRK current kinetics were analyzedusing nonlinear curve fitting software that fit singleexponential functions to derive activation time constants(tact) and deactivation time constants (tdeact) (Clampfit8.0 software, Axon Instruments). Agonist doseeresponserelations were analyzed by fitting peak GIRK currentamplitudes with the Hill function, where the effectiveconcentration producing a 50% response (EC50) and Hillcoefficient value (nH) were derived from the best fit(Origin 6.0 software, OriginLab Corp.). For comparisonof GIRK current amplitudes across cells, agonist-evokedcurrents from each cell were normalized to the measuredcell membrane capacitance (Cm) determined duringcapacitive current compensation. The normalized currentamplitudes are expressed as GIRK current density( pA/pF ). Pairwise statistical analysis between experi-mental groups was performed by one-way ANOVAwhere p!0.05 was considered significant.
3. Results
3.1. Properties of 5-HT1A and m2 receptor coupledGIRK currents reconstituted in CHO-K1 cells
Co-expression of neuronal Kir3.1/Kir3.2a channelsin CHO-K1 cells with either the 5-HT1A receptor or themuscarinic m2 receptor produced agonist-evoked cur-rents that were dose-dependent and displayed stronginward rectification (Fig. 1). To resolve the temporaland steady-state kinetic features of the receptor-activatedGIRK currents, 5-HT and ACh were rapidly appliedand washed out using concentrations ranging from 10�9
to 10�4 M. The reversal potentials for the 5-HT andACh-evoked currents were both near the experimentallypreset KC equilibrium potential (�40 mV) consistentwith KC-selective GIRK channels (Fig. 1B). Both the5-HT-activated GIRK currents (IK,5HT) and ACh-activated GIRK currents (IK,ACh) displayed a similaractivation and deactivation time course followingagonist washout (Fig. 1A). Notably, however, thesteady-state dose-dependence of 5-HT versus ACh-activated GIRK currents indicated a significantly higherpotency for 5-HT versus ACh (Fig. 1C). The EC50 valuefor 5-HT was 24G8 nM (nZ5) compared to the AChEC50 value of 820G162 nM (nZ10). This difference inEC50 values indicate either a higher number of 5-HT1A
receptors being expressed compared to m2 receptorsand/or a greater efficacy in 5-HT1A receptor versus m2receptor signaling. ThemaximalGIRKcurrent density atsaturating concentrations of receptor agonist (10 mM)was comparable indicating equivalent Kir3.1/Kir3.2achannel expression with the two GPCRs; maximal IK,5HT
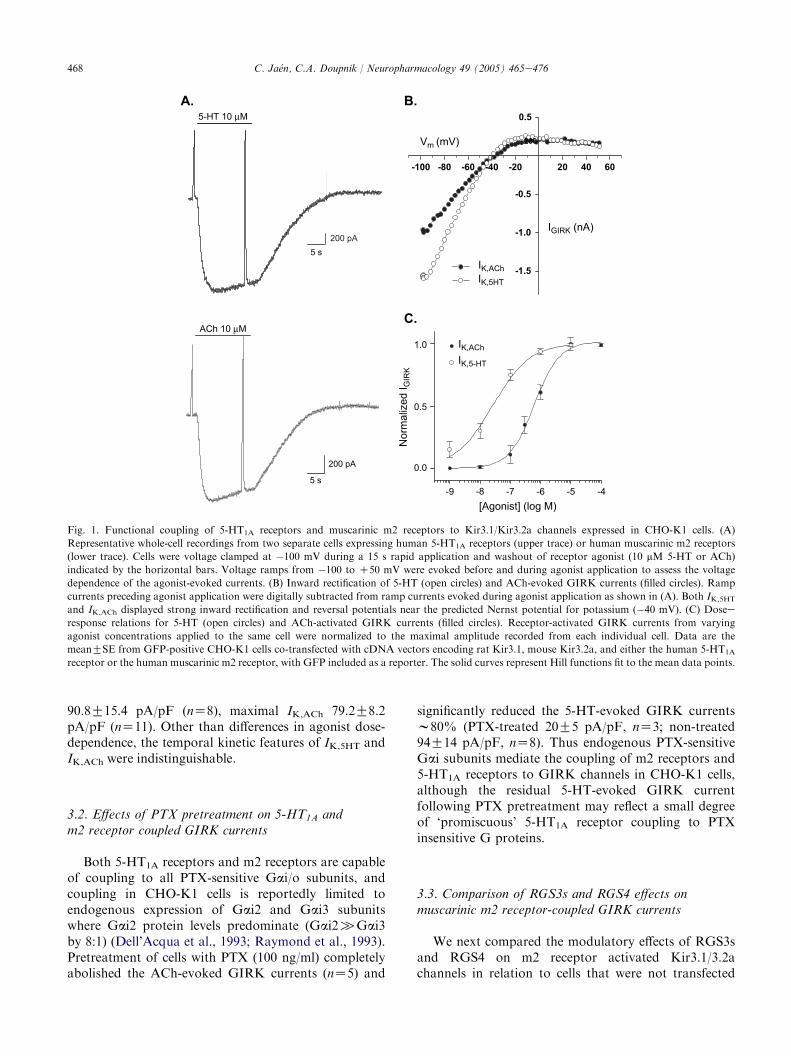
468 C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
A. B.
C.
-100 -80 -60 -40 -20 20 40 60
-1.0
200
0.5
IK,ACh
IGIRK (nA)
Vm (mV)
-0.5
-1.5
IK,5HT
IK,AChIK,5-HT
[Agonist] (log M)-9 -8 -7 -6 -5 -4
0.0
0.5
1.0
Nor
mal
ized
I GIR
K
200 pA
5 s
ACh 10 µM
5 s
5-HT 10 µM
Fig. 1. Functional coupling of 5-HT1A receptors and muscarinic m2 receptors to Kir3.1/Kir3.2a channels expressed in CHO-K1 cells. (A)
Representative whole-cell recordings from two separate cells expressing human 5-HT1A receptors (upper trace) or human muscarinic m2 receptors
(lower trace). Cells were voltage clamped at �100 mV during a 15 s rapid application and washout of receptor agonist (10 mM 5-HT or ACh)
indicated by the horizontal bars. Voltage ramps from �100 to C50 mV were evoked before and during agonist application to assess the voltage
dependence of the agonist-evoked currents. (B) Inward rectification of 5-HT (open circles) and ACh-evoked GIRK currents (filled circles). Ramp
currents preceding agonist application were digitally subtracted from ramp currents evoked during agonist application as shown in (A). Both IK,5HT
and IK,ACh displayed strong inward rectification and reversal potentials near the predicted Nernst potential for potassium (�40 mV). (C) Dosee
response relations for 5-HT (open circles) and ACh-activated GIRK currents (filled circles). Receptor-activated GIRK currents from varying
agonist concentrations applied to the same cell were normalized to the maximal amplitude recorded from each individual cell. Data are the
meanGSE from GFP-positive CHO-K1 cells co-transfected with cDNA vectors encoding rat Kir3.1, mouse Kir3.2a, and either the human 5-HT1A
receptor or the human muscarinic m2 receptor, with GFP included as a reporter. The solid curves represent Hill functions fit to the mean data points.
90.8G15.4 pA/pF (nZ8), maximal IK,ACh 79.2G8.2pA/pF (nZ11). Other than differences in agonist dose-dependence, the temporal kinetic features of IK,5HT andIK,ACh were indistinguishable.
3.2. Effects of PTX pretreatment on 5-HT1A andm2 receptor coupled GIRK currents
Both 5-HT1A receptors and m2 receptors are capableof coupling to all PTX-sensitive Gai/o subunits, andcoupling in CHO-K1 cells is reportedly limited toendogenous expression of Gai2 and Gai3 subunitswhere Gai2 protein levels predominate (Gai2[Gai3by 8:1) (Dell’Acqua et al., 1993; Raymond et al., 1993).Pretreatment of cells with PTX (100 ng/ml) completelyabolished the ACh-evoked GIRK currents (nZ5) and
significantly reduced the 5-HT-evoked GIRK currentsw80% (PTX-treated 20G5 pA/pF, nZ3; non-treated94G14 pA/pF, nZ8). Thus endogenous PTX-sensitiveGai subunits mediate the coupling of m2 receptors and5-HT1A receptors to GIRK channels in CHO-K1 cells,although the residual 5-HT-evoked GIRK currentfollowing PTX pretreatment may reflect a small degreeof ‘promiscuous’ 5-HT1A receptor coupling to PTXinsensitive G proteins.
3.3. Comparison of RGS3s and RGS4 effects onmuscarinic m2 receptor-coupled GIRK currents
We next compared the modulatory effects of RGS3sand RGS4 on m2 receptor activated Kir3.1/3.2achannels in relation to cells that were not transfected

469C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
with exogenous RGS (control). Shown in Fig. 2, theactivation and deactivation kinetics of ACh-evokedGIRK currents were accelerated by either RGS3s orRGS4 expression compared to the control cells. Kineticanalysis indicated RGS3s accelerated the GIRK de-activation time course somewhat greater than RGS4(RGS3s tdeactZ0.75G0.04 s, nZ8; RGS4 tdeactZ1.32G0.11 s, nZ5), although the effects of RGS3s onthe GIRK activation kinetics were equivalent to RGS4(Fig. 2C). The most striking difference between RGS3sand RGS4 was a significant reduction of GIRK currentamplitude (w45% decrease at 100 mM) and a 6-foldshift in the ACh doseeresponse curve associated withRGS3s expression (Fig. 2D,E). With RGS3s, the AChEC50 was 5.1G0.6 mM (nZ8) compared to 0.9G0.2 mM(nZ10) for control cells. By comparison, RGS4 did not
significantly affect the maximal GIRK current density asobserved previously in Xenopus oocytes (Doupnik et al.,1997), and caused a smaller shift in the ACh EC50 value(2.0G0.5 mM, nZ6) from the control group (Fig. 2D,E).Since the ACh doseeresponse curve with RGS3sexpression did not demonstrate saturation (Fig. 2D),GIRK current responses to 100 mM, 1 mM, and 10 mMACh were also compared in a separate set of cells(nZ9). These experiments confirmed that 100 mM AChwas indeed a saturating concentration, as maximalGIRK responses to 1 mM (95G2%) and 10 mM ACh(95G2%) were not significantly different than 100 mMACh (93G2%). Altogether these findings indicateRGS3s and RGS4 both accelerate GIRK channel gatingkinetics, but differentially affect steady-state m2 receptor-GIRK channel coupling properties.
A.
C.
B.
E.
D.
-9 -8 -7 -6 -5 -40
20
40
60
80
100
120
RGS4 (n=6)RGS3s (n=8)Control (n=11)
[ACh] (log M)
I K,A
Ch
(pA/
pF)
Control RGS4RGS3s
τ dea
ct (s
)τ d
eact
(s)
0
10
20
* *
(9)
(8) (5)
-6 -5 -4[ACh] (log M)
0
1.0
2.0
(5)
(5) (5) (4)
(8)(8)
***
[ACh] (log M)-6 -5 -4
τ act
(s)
0
1.0
2.0
3.0
(10)
(10) (10)(6) (6) (6)(7) (9) (9)
* *
**
[ACh] (log M)
0
0.5
1.0
-9 -8 -7 -6 -5 -4
Nor
mal
ized
I K,A
Ch
RGS4 (n=6)RGS3s (n=8)Control (n=10)
ACh
Control
RGS3s
RGS4
200pA-5 5 s
3x-7
-4
-6-7
-8
*
Fig. 2. Comparative effects of RGS3s versus RGS4 on muscarinic m2 receptor-coupled Kir3.1/Kir3.2a channels expressed in CHO-K1 cells. (A)
Representative ACh-activated GIRK currents elicited from three separate expression conditions; either without exogenous RGS expression (control
traces), with exogenous RGS4 expression (RGS4 traces), or with exogenous RGS3s expression (RGS3s traces). GIRK currents evoked by a range of
ACh concentrations for each cell are superimposed for comparison after baseline adjustment of the holding current immediately preceding each ACh
application. ACh applications were 15 s in duration and separated by aw1 min washout period. (B) Deactivation kinetics of RGS-accelerated GIRK
currents. Upper panel: deactivation time constants (tdeact) derived from control (filled bar), RGS3s (grey bar), and RGS4 (open bar) groups following
10 mM ACh-evoked GIRK currents. Lower panel: comparison of tdeact values following three different ACh concentrations with either RGS3s (grey
bars) or RGS4 (open bars) expression. Data are the meanGSE where * indicates P!0.05. (C) Activation kinetics of RGS-accelerated GIRK
currents. Comparison of activation time constants (tact) derived from control (filled bar), RGS3s (grey bar), and RGS4 (open bar) groups with
increasing ACh concentrations. (D) ACh doseeresponse relations for control (filled squares), RGS3s (grey triangles), and RGS4 (open circles)
groups. GIRK currents were normalized to cell membrane capacitance and expressed as a current density ( pA/pF ) for group comparisons. (E)
Normalized ACh doseeresponse curves from data presented in (D). GIRK current amplitudes were normalized to the maximal amplitude recorded
from each cell (100 mM ACh) and fit with a Hill function to derive EC50 values and Hill coefficients.

470 C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
3.4. Comparison of RGS3s and RGS4 effects onserotonin 1A receptor-coupled GIRK currents
We next examined the effects of RGS3s and RGS4 on5-HT1A-coupled GIRK currents to determine whetherthe different effects of RGS3s and RGS4 observedwith m2 receptor coupling were similarly conferred with5-HT1A receptors. Shown in Fig. 3, co-expression ofeither RGS3s or RGS4 significantly accelerated the ac-tivation and deactivation time course of 5-HT-activatedGIRK currents. Kinetic analysis of both the GIRKactivation and deactivation time course indicated theaccelerating effects of RGS3s and RGS4 were notsignificantly different from each other (Fig. 3B,C).Interestingly, neither RGS3s nor RGS4 affected themaximal GIRK current density although both appearedto have subtle effects that were not statisticallysignificant (Fig. 3D). Similar to m2 receptor coupling,
RGS3s significantly shifted the 5-HT doseeresponsecurve yet RGS4 did not. For RGS3s, the 5-HT EC50 was128G36 nM (nZ5) compared to 30G9 nM (nZ4) forthe control cells and 48G6 nM (nZ9) with RGS4expression. Thus RGS3s, in contrast to RGS4, displaysGPCR dependence in that it dramatically reducessteady-state m2 receptor-activated GIRK currents butnot 5-HT1A receptor-coupled currents.
3.5. Effects of RGS3s and RGS4 on basal GIRKchannel activity
We also analyzed the effects of RGS3s and RGS4 onreceptor-independent basal GIRK current amplitudes asreflected in the holding currents in 25 mM KC solution.Previous reports indicate RGS3 (original 519 a.a.isoform) and RGS4 cause a significant increase in basal
RGS4 (n=10)RGS3s (n=5)Control (n=8)
[5-HT] (log M)
[5-HT] (log M)
RGS4RGS3sControl
-9 -8 -7 -6 -5
(6)
(5) (10)* *
(5) (10) (10) (10)(5) (3)
[5-HT] (log M)
[5-HT] (log M)
00.20.40.60.81.01.2
(9)
(11)(11)(11) (3)(5)(5)
(9)(9)
* *
** *
A.
C.
B.
E.
D.
-5-6
-8
5-HT
200pA
5 s
-4
-7
-9
Control
RGS3s
RGS4
0
10
20
0
1.0
2.0
τ dea
ct (s
)τ d
eact
(s)
τ act
(s)
Control RGS4RGS3s
-6 -5 -4
-6 -5 -4
I K,5
HT
(pA/
pF)
Nor
mal
ized
I K,5
HT
0
20
40
60
80
100
120
0
0.5
1.0
-9 -8 -7 -6 -5
Fig. 3. Comparative effects of RGS3s versus RGS4 on serotonin 1A (5-HT1A) receptor-coupled Kir3.1/Kir3.2a channels expressed in CHO-K1 cells.
(A) Representative 5-HT-activated GIRK currents elicited from three separate cell conditions, either without exogenous RGS expression (control
traces), with exogenous RGS4 expression (RGS4 traces), or with exogenous RGS3s expression (RGS3s traces). GIRK currents evoked by a range of
5-HT concentrations for each cell are superimposed for comparison after baseline adjustment of the holding current immediately preceding each
agonist application. 5-HT applications were 15 s in duration and separated by a w1 min washout period. (B) Deactivation kinetics of RGS-
accelerated GIRK currents. Upper panel: deactivation time constants (tdeact) derived from control (filled bar), RGS3s (grey bar), and RGS4 (open
bar) groups following 10 mM 5-HT-evoked GIRK currents. Lower panel: comparison of tdeact values following three different 5-HT concentrations
with either RGS3s (grey bars) or RGS4 (open bars) expression. Data are the meanGSE where * indicates P!0.05. (C) Activation kinetics of RGS-
accelerated GIRK currents. Comparison of activation time constants (tact) derived from control (filled bar), RGS3s (grey bar), and RGS4 (open bar)
groups with increasing 5-HT concentrations. (D) 5-HT doseeresponse relations for control (filled squares), RGS3s (grey triangles), and RGS4 (open
circles) groups. GIRK currents were normalized to cell membrane capacitance and expressed as a current density ( pA/pF ) for group comparisons.
(E) Normalized 5-HT doseeresponse curves from data presented in (D). GIRK current amplitudes were normalized to the maximal amplitude
recorded from each cell (10 mM 5-HT) and fit with a Hill function to derive EC50 values and Hill coefficients.

471C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
GIRK current activity when expressed in either CHO orHEK293 cells by apparently increasing the availabilityof free Gbg subunits via RGS sequestration of Gasubunits (Bunemann and Hosey, 1998). This finding is incontrast to observations in the oocyte expression system,where RGS3 and RGS4 reduce IK,basal amplitudes byapparently shifting the equilibrium of Ga subunitstowards their GDP-bound state due to RGS-enhancedGTP hydrolysis and effectively sequestering free Gbgdimers that cause basal GIRK channel activity (Doupniket al., 1997). In the CHO-K1 experiments reportedhere, expression of Kir3.1/Kir3.2a channels significantlyincreased the IK,basal amplitude compared to non-transfected CHO-K1 cells (Table 1), thus demonstratinga significant level of receptor-independent ‘basal’ GIRKchannel activity in the absence of exogenous RGSexpression. Comparison of IK,basal amplitudes from thecontrol groups (RGS�) with co-expression of eitherRGS3s or RGS4 did not reveal a significant differencewith either m2 receptor or 5-HT1A receptor expression(Table 1). Thus the effects reported by Bunemann andHosey (1998) may result from significantly higher RGSprotein levels produced with their transfection methods,since expression conditions that elevate RGS4 levels inoocytes has also been reported to increase basal GIRKchannel activity (Keren-Raifman et al., 2001).
3.6. Effects of RGS3s and RGS4 on acutedesensitization of GIRK currents
In the absence of RGS co-expression, GIRK currentsmodestly desensitize during the short 15 s agonistapplication period (!10% of their amplitude). Co-expression of RGS4 causes a significant increase in therate of ‘acute’ desensitization which is attributable to theaccelerated rate of signal termination during sustainedreceptor activation (Doupnik et al., 1997; Chuang et al.,1998). Shown in Fig. 4, comparisons of the extent ofacute desensitization with RGS3s versus RGS4 expres-sion during the 15 s agonist application period indicateequivalent effects on both IK,5HT and IK,ACh. Thesefindings are consistent with the rate of acute GIRKcurrent desensitization being closely correlated with GaGTPase activity and best reflected in the GIRKdeactivation rates (Chuang et al., 1998; Leaney et al.,
2004). As shown earlier for RGS3s and RGS4 (Figs. 2and 3), both RGS proteins accelerate GIRK deactiva-tion rates to a similar degree.
4. Discussion
The goal of this study was to evaluate the modulatoryeffects of a recently identified ‘short’ RGS3 isoform onneuronal GIRK channels activated by different GPCRsin a mammalian cell expression system (CHO-K1 cells).The RGS3s mRNA transcript is abundant in mousebrain and heart (Reif and Cyster, 2000) and thereforemay modulate GPCR regulation of neuronal and cardiaccell excitability. We assessed the effects of RGS3s incomparison to the closely related and previously studiedRGS4 protein (Doupnik et al., 1997; Zhang et al., 2002)that is co-expressed with RGS3 in individual neuronsand atrial cardiomyocytes (Doupnik et al., 2001, 2004).The major finding of our experiments is that RGS3smodulates GIRK channels in a GPCR-dependentmanner, whereas RGS4 modulated GIRK channelssimilarly for both of the GPCRs studied. RGS3ssignificantly reduced GIRK current amplitudes withm2 receptor coupling and shifted the steady-state agonistdoseeresponse relations, whereas RGS4 affected m2receptor-activated GIRK currents similar to that ob-served with 5-HT1A receptors. These results indicateRGS3s has distinct interactions with muscarinic m2versus 5-HT1A receptor complexes, whereas RGS4interacts similarly with both GPCR-GIRK channelcomplexes. There are several possible RGS-affectedcellular processes that may contribute toward themodulatory differences that we have identified and arebriefly discussed below.
4.1. GTPase accelerating activity of RGS proteins
The GTPase accelerating activity of RGS proteins ismediated by direct interactions between the RGSdomain and the Ga subunit (Ross and Wilkie, 2000),and differences in RGS modulation of GIRK channelscan reflect differences in RGS-Ga subunit selectivity(Doupnik et al., 1997; Zhang et al., 2002). Although
Table 1
Effects of RGS3s and RGS4 on basal GIRK channel activity in CHO-K1 cells
IK,basal ( pA/pF )a
Control PTX-treated CRGS3s CRGS4
Non-transfected CHO-K1 cells �10G4 (nZ6) e e e
Muscarinic m2 receptorCKir3.1/Kir3.2a �130G17 (nZ10) �91G16 (nZ5) �162G26 (nZ8) �135G23 (nZ6)
Serotonin 1A receptorCKir3.1/Kir3.2a �84G23 (nZ11) �106G21 (nZ3) �120G31 (nZ5) �89G10 (nZ11)
a Data are resting membrane currents in 25 mM KC at a holding potential of �100 mV divided by the cell membrane capacitance.

472 C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
A.
B.
(11)
(5) (9)
**
0
5
10
15
20
25
30
Control RGS3s RGS4
GIR
K D
esen
sitiz
atio
n (%
)
(12)
(8)
(6)
*
*
0
5
10
15
20
25
30
Control RGS3s RGS4
GIR
K D
esen
sitiz
atio
n (%
)
5 s
ACh (10µM)
5 s
5-HT (10µM)
Fig. 4. Acute GIRK current desensitization associated with different GPCR-RGS coupling conditions. (A) Comparative effects of RGS3s (red trace)
and RGS4 (blue trace) on acute desensitization of 5-HT1A receptor-activated GIRK currents without exogenous RGS expression (control, black
trace). Peak amplitudes of the superimposed recordings were normalized for kinetic comparisons. Right panel: the percent desensitization was
quantified by measuring the percent decline in the peak GIRK current amplitude measured at the end of the 15 s application period, as denoted by
the application ‘‘window’’ (dotted box in left panel). Data are the meanGSE where * indicates a P!0.05 for comparisons between the control and
RGS groups. (B) Comparative effects of RGS3s (red trace) and RGS4 (blue trace) on acute desensitization of muscarinic m2 receptor-activated
GIRK currents without exogenous RGS expression (control, black trace). Right panel: quantification of acute GIRK desensitization was determined
as described in (A).

473C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
RGS3 and RGS4 both interact with Gai/o and Gaq/11subunits, RGS3 displays a higher affinity for Ga11versus Gai3 (Neill et al., 1997; Dulin et al., 1999) andRGS4 shows preferential interactions with Gai/o sub-units versus Gaq (Berman et al., 1996). So for theGai-coupled receptors examined in our CHO-K1experiments, these preferred RGS-Ga associations wouldgenerally favor greater accelerated GIRK deactivationrates with RGS4 compared to RGS3s. Yet to thecontrary, these kinetic differences were not observed andin fact RGS3s accelerated the GIRK deactivation ratesomewhat greater than RGS4. Thus differences inRGS3s versus RGS4 affinity for Gai subunits are notapparent in the accelerated GIRK channel gating pro-perties that reflect RGS-enhanced GTPase acceleratingactivity and seem unlikely to explain our findings.
4.2. RGS membrane association
Several members of the B/R4 subfamily, includingRGS4, enhance membrane binding through a mecha-nism requiring their short N-terminal domain. TheseRGS proteins (RGS2, RGS4, RGS5, RGS8, RGS16,RGS18) possess N-terminal palmitoylated cysteineresidues and a conserved basic amphipathic a-helix thatconfers membrane association and orientation thatenhances their GTPase activating activity (Chen et al.,1999; Bernstein et al., 2000; Heximer et al., 2001; Saitohet al., 2001; Tu et al., 2001). The RGS4 N-terminusalso contains a ubiquitin degradation signal (Davydovand Varshavsky, 2000). The RGS3s N-terminal domainnotably lacks these features. Yet for both RGS4 andRGS8, deleting the N-terminal domain does notsignificantly affect RGS-accelerated activation anddeactivation kinetics for GPCR-activated GIRK chan-nels expressed in Xenopus oocytes, indicating RGSdomain-Ga interactions are sufficient for these kineticeffects (Inanobe et al., 2001; Saitoh et al., 2001).However, deleting the RGS8 N-terminus does reduceacute desensitization during dopamine D2 receptorGIRK channel activation (Saitoh et al., 2001) which isattributable to RGS-enhanced GTPase activity(Chuang et al., 1998). Remarkably, a ‘short’ RGS8splice variant (RGS8s) differing only by the first 7e9N-terminal residues shows diminished effects on GIRKactivation and deactivation kinetics and altered selec-tivity for Gq-coupled receptor signaling (Saitoh et al.,2002). Furthermore, overexpression of the RGS8 N-terminal domain (1e5 a.a.) in rat sympathetic neuronsdramatically accelerates a2-adrenergic receptor activa-tion of heterologously expressed GIRK channels,supporting an important role of the RGS8 N-terminusin facilitating receptor-GIRK channel coupling (Jeongand Ikeda, 2001). In our comparison of RGS3s andRGS4 on GIRK kinetics reported here, despite their
divergent N-terminal sequences, both displayed similaraccelerating effects on GIRK activation and deactiva-tion kinetics and equivalent effects on acute desensiti-zation of receptor-activated GIRK currents. Thereforedifferences in RGS3s and RGS4 N-terminal domainsdo not confer obvious kinetic differences in receptor-dependent GIRK channel gating, despite their differ-ential effects on receptor-dependent steady-state gatingproperties.
4.3. RGS-mediated translocation of GPCRs
RGS4 is predominantly a cytosolic protein recruitedto membranes by interactions with G protein subunits(Druey et al., 1998). RGS-specific translocation from thecytosol to the plasma membrane involves directinteractions with the GPCR complex and is determinedin part by the relative affinity of the RGS-Ga subunitinteraction (Roy et al., 2003; Masuho et al., 2004). Itremains unclear whether cytosolic RGS proteins canincorporate into mature GPCReGIRK channel com-plexes already located at the plasma membrane, orwhether they co-assemble within the GPCReGIRKchannel complexes synthesized and assembled within theendoplasmic reticulum and Golgi apparatus (Lavineet al., 2002). Current evidence, however, clearly indicateRGS4 facilitates trafficking and recruitment ofG proteins (Chuang et al., 1998) and m2 receptoreGai2 complexes (Roy et al., 2003) from intracellularpools to the plasma membrane, and thereby increasesthe density of functional receptors at the plasmamembrane. For RGS8, deletion of the N-terminaldomain (DN-RGS8) prevents G protein-induced sub-cellular translocation of DN-RGS8 to the plasmamembrane (Saitoh et al., 2001). Similarly, translocationof the original RGS3 isoform from the cytosol to theplasma membrane also occurs but in an agonist- andCa2C-dependent manner (Dulin et al., 1999). The Ca2C-dependent translocation of RGS3 was recently shown tobe mediated by Ca2C binding to an EF-hand motiflocated in the N-terminus of RGS3, which is not presentin the shorter N-terminus of RGS3s (Tosetti et al.,2003). Together, these findings suggest RGS4 maytranslocate m2 receptor/Gai complexes to the plasmamembrane more effectively than RGS3s, due either toa lower RGS3s-Gai affinity and/or a reduced efficacy ofthe RGS3s N-terminal domain in the translocationprocess. The consequence in either case would be a lowercell surface concentration of receptors with RGS3sexpression, which is consistent with the reduced GIRKcurrent responses and rightward shift in the ACh doseeresponse curve observed with RGS3s expression. Whatis puzzling with this working hypothesis is why theRGS3s effect on steady-state receptor-dependent GIRKactivation properties are most prominent for m2receptors and less so for 5-HT1A receptor complexes.

474 C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
4.4. Direct RGS-GPCR interactions
Similar to our observations reported here, differentB/R4 RGS proteins exhibit GPCR-selective modulationof Gq/11-coupled (Xu et al., 1999; Wang et al., 2002)and other Gi/o-coupled signaling pathways (Ghavamiet al., 2004). Moreover, the N-terminal domains ofRGS3, RGS4, and RGS8 have been shown to affect theselective regulation among Gq/11-coupled receptors(Chatterjee et al., 1997; Zeng et al., 1998; Saitoh et al.,2002). Recently, the N-terminus of RGS2 was shown tohave direct and selective interactions with the 3rdintracellular (i3) loop of Gq/11-coupled muscarinicreceptors in vitro (Bernstein et al., 2004). Full-lengthRGS4 also interacts with the i3 loop of Gq/11-coupledmuscarinic m1 and m5 receptors, but similar to RGS2,does not interact with the i3 loops of Gi/o-coupled m2or m4 receptors (Bernstein et al., 2004). Thus directinteraction of RGS4 with the m2 receptor remains to beresolved, yet apparently does not involve interactionswith the i3 loop. Given the divergent nature of theRGS3s N-terminal domain compared to RGS4, differ-ential interactions of the RGS3s N-terminus withGPCRs seems plausible and could thereby affect theefficacy of receptor translocation to the plasma mem-brane and/or G protein activation in a GPCR-selectivemanner.
The intrinsic G protein coupling properties ofdifferent GPCRs may also impact RGS interactionswithin the signaling complex. In the absence of over-expressed RGS proteins in CHO-K1 cells, GIRKchannels activated by m2 receptors and 5-HT1A
receptors displayed significantly different agonist poten-cies with 5-HT being w30-fold more potent than ACh(cf. Fig. 1). Differences in receptor expression, cellsurface density, and receptor translocation (i.e.5-HT1A>m2 receptors) are all possible contributors tothis observed difference as discussed above. The 5-HT1A
receptor-coupled GIRK channels also displayed a PTX-insensitive component that may reflect promiscuousG protein coupling or coupling of residual Gai subunits(not ADP-ribosylated) due to a higher coupling affinity.Reconstitution experiments comparing GPCR-Gaibinding affinities recently found agonist-bound 5-HT1A
receptors to have a 12-fold higher affinity forGai1(GDP)bg compared to agonist-bound m2 receptors(Slessareva et al., 2003), indicating agonist-activated5-HT1A receptors have an intrinsically higher efficacyfor Gai(GDP)bg coupling compared to muscarinic m2receptors. GPCR differences in intrinsic G proteincoupling (i.e. precoupling) and the influence of associ-ated RGS proteins are important considerations forfuture mechanistic investigations (Shea and Linderman,1997). From our results described here, RGS3s andRGS4 could produce equivalent modulatory effectson 5-HT1A receptor-coupled GIRK channels due to
a higher degree of G protein precoupling compared tom2 receptors (Zhang et al., 2002).
5. Summary and future directions
In this study we compared the functional propertiesof the RGS3s isoform and RGS4 due to their expressionin brain and heart (Reif and Cyster, 2000; Kehrl et al.,2002) and in native GIRK-expressing neurons and atrialmyocytes (Doupnik et al., 2001, 2004). The GPCR-dependent effects of RGS3s we observed on neuronalGIRK channel function raise new questions regardingRGS-dependent modulation of GPCReGIRK channelcomplexes. We provisionally interpret the selectiveRGS3s effect on GPCReGIRK channels as eithera reduction in cell surface m2 receptoreGIRK channelcomplexes (Lavine et al., 2002) and/or a reduction in theefficacy of m2 receptoreGai proteineGIRK channelcoupling (Slessareva et al., 2003). In either case, thefunctional effect indicates selective interactions ofRGS3s with different GPCReGIRK signaling com-plexes. Selective interaction of RGS proteins withvarious GPCReeffector signaling complexes are ofimportance as both RGS3 and RGS4 mRNA levelsare transcriptionally regulated in the nervous systemduring pathophysiologic conditions, and may representimportant compensatory responses to elevated oraltered stimuli (Costigan et al., 2003). Our findingshighlight the impact of selective RGSeGPCR inter-actions on neuronal GIRK channel activity that candifferentially affect cellular responses to differing stimuliand their integrated output.
Acknowledgements
The authors thank Drs Karin Reif and Jason Cyster(University of California, San Francisco) for generouslyproviding the mouse RGS3s cDNA. This work wasfunded in part by a grant from the American HeartAssociation, Florida and Puerto Rico Affiliate.
References
Berman, D.M., Kozasa, T., Gilman, A.G., 1996. The GTPase-
activating protein RGS4 stabilizes the transition state for
nucleotide hydrolysis. Journal of Biological Chemistry 271,
27209e27212.
Bernstein, L.S., Grillo, A.A., Loranger, S.S., Linder, M.E., 2000.
RGS4 binds to membranes through an amphipathic alpha-helix.
Journal of Biological Chemistry 275, 18520e18526.
Bernstein, L.S., Ramineni, S., Hague, C., Cladman, W., Chidiac, P.,
Levey, A.I., Hepler, J.R., 2004. RGS2 binds directly and selectively

475C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
to the M1 muscarinic acetylcholine receptor third intracellular loop
to modulate Gq/11a signaling. Journal of Biological Chemistry
279, 21248e21256.
Blednov, Y.A., Stoffel, M., Chang, S.R., Harris, R.A., 2001. GIRK2
deficient mice. Evidence for hyperactivity and reduced anxiety.
Physiology and Behavior 74, 109e117.
Blednov, Y.A., Stoffel, M., Alva, H., Harris, R.A., 2003. A pervasive
mechanism for analgesia: activation of GIRK2 channels. Proceed-
ings of the National Academy of Sciences USA 100, 277e282.
Breitwieser, G.E., Szabo, G., 1988. Mechanism of muscarinic receptor-
induced KC channel activation as revealed by hydrolysis-resistant
GTP analogues. Journal of General Physiology 91, 469e493.Bunemann, M., Hosey, M.M., 1998. Regulators of G protein signaling
(RGS) proteins constitutively activate Gbg-gated potassium
channels. Journal of Biological Chemistry 273, 31186e31190.
Chatterjee, T.K., Eapen, A.K., Fisher, R.A., 1997. A truncated form
of RGS3 negatively regulates G protein-coupled receptor stimula-
tion of adenylyl cyclase and phosphoinositide phospholipase C.
Journal of Biological Chemistry 272, 15481e15487.Chen, C., Seow, K.T., Guo, K., Yaw, L.P., Lin, S.C., 1999. The
membrane association domain of RGS16 contains unique amphi-
pathic features that are conserved in RGS4 and RGS5. Journal of
Biological Chemistry 274, 19799e19806.
Chuang, H.H., Yu, M., Jan, Y.N., Jan, L.Y., 1998. Evidence that the
nucleotide exchange and hydrolysis cycle of G proteins causes
acute desensitization of G-protein gated inward rectifier KC
channels. Proceedings of the National Academy of Sciences USA
95, 11727e11732.
Costigan, M., Samad, T.A., Allchorne, A., Lanoue, C., Tate, S.,
Woolf, C.J., 2003. High basal expression and injury-induced down
regulation of two regulator of G-protein signaling transcripts,
RGS3 and RGS4 in primary sensory neurons. Molecular and
Cellular Neuroscience 24, 106e116.
Davydov, I.V., Varshavsky, A., 2000. RGS4 is arginylated and
degraded by the N-end rule pathway in vitro. Journal of Biological
Chemistry 275, 22931e22941.
Dell’Acqua, M.L., Carroll, R.C., Peralta, E.G., 1993. Transfected m2
muscarinic acetylcholine receptors couple to Gai2 and Gai3 in
Chinese hamster ovary cells. Activation and desensitization of the
phospholipase C signaling pathway. Journal of Biological Chem-
istry 268, 5676e5685.
Doupnik, C.A., Davidson, N., Lester, H.A., Kofuji, P., 1997. RGS
proteins reconstitute the rapid gating kinetics of Gbg-activated
inwardly rectifying KC channels. Proceedings of the National
Academy of Sciences USA 94, 10461e10466.
Doupnik, C.A., Xu, T., Shinaman, J.M., 2001. Profile of RGS
expression in single rat atrial myocytes. Biochimica et Biophysica
Acta 1522, 97e107.
Doupnik, C.A., Jaen, C., Zhang, Q., 2004. Measuring the modulatory
effects ofRGSproteins onGIRKchannels.Methods in Enzymology
389, 131e154.
Druey, K.M., Blumer, K.J., Kang, V.H., Kehrl, J.H., 1996. Inhibition
of G-protein-mediated MAP kinase activation by a new mamma-
lian gene family. Nature 379, 742e746.
Druey, K.M., Sullivan, B.M., Brown, D., Fischer, E.R., Watson, N.,
Blumer, K.J., Gerfen, C.R., Scheschonka, A., Kehrl, J.H., 1998.
Expression of GTPase-deficient Gia2 results in translocation of
cytoplasmic RGS4 to the plasma membrane. Journal of Biological
Chemistry 273, 18405e18410.
Dulin, N.O., Sorokin, A., Reed, E., Elliott, S., Kehrl, J.H., Dunn, M.J.,
1999. RGS3 inhibits G protein-mediated signaling via translocation
to the membrane and binding to Ga11. Molecular and Cell Biology
19, 714e723.
Ghavami, A., Hunt, R.A., Olsen, M.A., Zhang, J., Smith, D.L.,
Kalgaonkar, S., Rahman, Z., Young, K.H., 2004. Differential
effects of regulator of G protein signaling (RGS) proteins on
serotonin 5-HT1A, 5-HT2A, and dopamine D2 receptor-mediated
signaling and adenylyl cyclase activity. Cellular Signaling 16,
711e721.
Gold, S.J., Ni, Y.G., Dohlman, H.G., Nestler, E.J., 1997. Regulators
of G-protein signaling (RGS) proteins: region-specific expression
of nine subtypes in rat brain. Journal of Neuroscience 17,
8024e8037.
Hamill, O.P., Marty, A., Neher, E., Sakmann, B., Sigworth, F.J., 1981.
Improved patch-clamp techniques for high-resolution current
recording from cells and cell-free membrane patches. Pflugers
Archiv: European Journal of Physiology 391, 85e100.
Heximer, S.P., Lim, H., Bernard, J.L., Blumer, K.J., 2001. Mecha-
nisms governing subcellular localization and function of human
RGS2. Journal of Biological Chemistry 276, 14195e14203.
Hollinger, S., Hepler, J.R., 2002. Cellular regulation of RGS proteins:
modulators and integrators of G protein signaling. Pharmacology
Reviews 54, 527e559.Inanobe, A., Fujita, S., Makino, Y., Matsushita, K., Ishii, M.,
Chachin, M., Kurachi, Y., 2001. Interaction between the RGS
domain of RGS4 with G protein alpha subunits mediates the
voltage-dependent relaxation of the G protein-gated potassium
channel. Journal of Physiology (London) 535, 133e143.
Jeong, S.W., Ikeda, S.R., 2001. Differential regulation of G protein-
gated inwardly rectifying K(C) channel kinetics by distinct
domains of RGS8. Journal of Physiology (London) 535, 335e347.
Kehrl, J.H., Srikumar, D., Harrison, K., Wilson, G.L., Shi, C.S., 2002.
Additional 5# exons in the RGS3 locus generate multiple mRNA
transcripts, one of which accounts for the origin of human PDZ-
RGS3. Genomics 79, 860e868.
Keren-Raifman, T., Bera, A.K., Zveig, D., Peleg, S., Witherow, D.S.,
Slepak, V.Z., Dascal, N., 2001. Expression levels of RGS7 and
RGS4 proteins determine the mode of regulation of the G protein-
activated K(C) channel and control regulation of RGS7 by G b 5.
FEBS Letters 492, 20e28.
Koelle, M.R., Horvitz, H.R., 1996. EGL-10 regulates G protein
signaling in the C. elegans nervous system and shares a conserved
domain with many mammalian proteins. Cell 84, 115e125.
Lavine, N., Ethier, N., Oak, J.N., Pei, L., Liu, F., Trieu, P.,
Rebois, R.V., Bouvier, M., Hebert, T.E., Van Tol, H.H., 2002.
G protein-coupled receptors form stable complexes with inwardly
rectifying potassium channels and adenylyl cyclase. Journal of
Biological Chemistry 277, 46010e46019.
Leaney, J.L., Benians, A., Brown, S., Nobles, M., Kelly, D., Tinker, A.,
2004. Rapid desensitization of G protein-gated inwardly rectifying
K(C) currents is determined by G protein cycle. American Journal
of Physiology: Cell Physiology 287, C182eC191.Masuho, I., Itoh, M., Itoh, H., Saitoh, O., 2004. The mechanism of
membrane-translocationof regulatorofG-protein signaling (RGS) 8
induced by Ga expression. Journal of Neurochemistry 88, 161e168.
Neill, J.D., Duck, L.W., Sellers, J.C., Musgrove, L.C.,
Scheschonka, A., Druey, K.M., Kehrl, J.H., 1997. Potential role
for a regulator of G protein signaling (RGS3) in gonadotropin-
releasing hormone (GnRH) stimulated desensitization. Endocri-
nology 138, 843e846.Raymond, J.R., Olsen, C.L., Gettys, T.W., 1993. Cell-specific physical
and functional coupling of human 5-HT1A receptors to inhibitory
G protein a-subunits and lack of coupling to Gsa. Biochemistry 32,
11064e11073.
Reif, K., Cyster, J.G., 2000. RGS molecule expression in murine
B lymphocytes and ability to down-regulate chemotaxis to lymphoid
chemokines. Journal of Immunology 164, 4720e4729.Ross, E.M., Wilkie, T.M., 2000. GTPase-activating proteins for
heterotrimeric G proteins: regulators of G protein signaling
(RGS) and RGS-like proteins. Annual Review of Biochemistry
69, 795e827.
Roy, A.A., Lemberg,K.E., Chidiac, P., 2003.Recruitment of RGS2 and
RGS4 to the plasma membrane by G proteins and receptors reflects
functional interactions. Molecular Pharmacology 64, 587e593.

476 C. Jaen, C.A. Doupnik / Neuropharmacology 49 (2005) 465e476
Saitoh, O., Kubo, Y., Miyatani, Y., Asano, T., Nakata, H., 1997.
RGS8 accelerates G-protein-mediated modulation of KC currents.
Nature 390, 525e529.
Saitoh, O., Masuho, I., Terakawa, I., Nomoto, S., Asano, T.,
Kubo, Y., 2001. Regulator of G protein signaling 8 (RGS8)
requires its NH2 terminus for subcellular localization and acute
desensitization of G protein-gated KC channels. Journal of
Biological Chemistry 276, 5052e5058.Saitoh, O., Murata, Y., Odagiri, M., Itoh, M., Itoh, H., Misaka, T.,
Kubo, Y., 2002. Alternative splicing of RGS8 gene determines
inhibitory function of receptor type-specific Gq signaling.
Proceedings of the National Academy of Sciences USA 99,
10138e10143.
Shea, L., Linderman, J.J., 1997. Mechanistic model of G-protein signal
transduction. Determinants of efficacy and effect of precoupled
receptors. Biochemical Pharmacology 53, 519e530.
Signorini, S., Liao, Y.J., Duncan, S.A., Jan, L.Y., Stoffel, M., 1997.
Normal cerebellar development but susceptibility to seizures in
mice lacking G protein-coupled, inwardly rectifying KC channel
GIRK2. Proceedings of the National Academy of Sciences USA
94, 923e927.
Slessareva, J.E., Ma, H., Depree, K.M., Flood, L.A., Bae, H.,
Cabrera-Vera, T.M., Hamm, H.E., Graber, S.G., 2003. Closely
related G-protein-coupled receptors use multiple and distinct
domains on G-protein a-subunits for selective coupling. Journal
of Biological Chemistry 278, 50530e50536.
Stanfield, P.R., Nakajima, S., Nakajima, Y., 2002. Constitutively
active and G-protein coupled inward rectifier KC channels: Kir2.0
and Kir3.0. Reviews in Physiology, Biochemistry, and Pharmacol-
ogy 145, 47e179.
Tesmer, J.J., Berman, D.M., Gilman, A.G., Sprang, S.R., 1997.
Structure of RGS4 bound to AlF4-activated Gia1: stabilization of
the transition state for GTP hydrolysis. Cell 89, 251e261.
Tosetti, P., Pathak, N., Jacob, M.H., Dunlap, K., 2003. RGS3
mediates a calcium-dependent termination of G protein signaling in
sensory neurons. Proceedings of the National Academy of Sciences
USA 100, 7337e7342.
Tu, Y., Woodson, J., Ross, E.M., 2001. Binding of regulator of
G protein signaling (RGS) proteins to phospholipid bilayers.
Contribution of location and/or orientation to GTPase-activating
protein activity. Journal of Biological Chemistry 276, 20160e
20166.
Wang, Q., Liu, M., Mullah, B., Siderovski, D.P., Neubig, R.R., 2002.
Receptor-selective effects of endogenous RGS3 and RGS5 to
regulate mitogen-activated protein kinase activation in rat vascular
smooth muscle cells. Journal of Biological Chemistry 277, 24949e
24958.
Xu, X., Zeng, W., Popov, S., Berman, D.M., Davignon, I., Yu, K.,
Yowe, D., Offermanns, S., Muallem, S., Wilkie, T.M., 1999. RGS
proteins determine signaling specificity of Gq-coupled receptors.
Journal of Biological Chemistry 274, 3549e3556.
Zeng, W., Xu, X., Popov, S., Mukhopadhyay, S., Chidiac, P.,
Swistok, J., Danho, W., Yagaloff, K.A., Fisher, S.L., Ross, E.M.,
Muallem, S., Wilkie, T.M., 1998. The N-terminal domain of RGS4
confers receptor-selective inhibition of G protein signaling. Journal
of Biological Chemistry 273, 34687e34690.
Zhang, Q., Pacheco, M.A., Doupnik, C.A., 2002. Gating properties of
GIRK channels activated by Gao- and Gai-coupled muscarinic m2
receptors in Xenopus oocytes: the role of receptor precoupling in
RGS modulation. Journal of Physiology (London) 545, 355e373.





























