



1
Interaction Between Bradykinin Subtype 2 and Angiotensin II Type 2 Receptors
During Post-MI Left Ventricular Remodeling
David C. Isbell MD, Szilard Voros MD, Zequan Yang MD, Joseph M. DiMaria BA,
Stuart S. Berr PhD, Brent A. French PhD, Frederick H. Epstein PhD,
Sanford P. Bishop DVM PhD, Hongkun Wang PhD, Rene J. Roy, Brandon A. Kemp BA,
Hiroaki Matsubara MD, PhD, Robert M. Carey MD, Christopher M. Kramer MD
From the Cardiovascular Division, Departments of Medicine (DCI, SV, RMC, BAK,
CMK), Radiology (JMD, SSB, BAF, FHE, RJR, CMK), Biomedical Engineering (ZY,
SSB, BAF, FHE) and Public Health Sciences (HW), University of Virginia Health
System, Charlottesville, VA, Department of Pathology, University of Alabama,
Birmingham, AL (SPB), and Department of Cardiovascular Medicine, Kyoto Prefectural
University School of Medicine, Kyoto, Japan (HM)
Supported by AHA Mid-Atlantic Affiliate Grant-in-Aid 0455687U (CMK) and T32
HL07355 (SV).
Word count: 5,368
Brief Title: B2 and ang II type 2 receptors post-MI
Correspondence to: Christopher M. Kramer, MD University of Virginia Health SystemDepartments of Medicine and Radiology 1215 Lee Street Box 800170 Charlottesville, VA 22908E-mail: [email protected] Telephone: (434) 243-6060 Fax: (434) 982-1618
Page 1 of 34
Copyright Information
Articles in PresS. Am J Physiol Heart Circ Physiol (October 12, 2007). doi:10.1152/ajpheart.00997.2007
Copyright © 2007 by the American Physiological Society.

2
Background: AT2-receptor overexpression (AT2TG) attenuates left ventricular
remodeling in a mouse model of anterior myocardial infarction (MI). We hypothesized
that the beneficial effects of cardiac AT2TG are mediated via the bradykinin-2 receptor
(B2R).
Methods and Results: Fourteen transgenic mice overexpressing the AT2R (AT2TG), 10
mice with a B2R deletion (B2KO), 13 AT2TG with B2R deletion (AT2TG/B2KO), and 11
wild type (WT) mice were studied. All mice were on a C57BL/6 background. Mice were
studied by cardiac magnetic resonance imaging (CMR) at baseline and days 1, 7, and 28
post-MI induced by 1 hour of occlusion of the left anterior descending artery followed by
reperfusion. Short axis images from apex to base were used to compare ventricular
volumes and ejection fraction (EF). At baseline, end-diastolic volume index (EDVI) and
end-systolic volume index (ESVI) were lower and EF higher in AT2TG compared to the
other 3 strains. Infarct size was similar between groups. No differences were observed in
global remodeling parameters at day 28 between AT2TG and AT2TG/B2KO; however,
EDVI and ESVI were lower and EF higher in both transgenic groups than in WT or
B2KO. Both strains lacking B2R demonstrated increased collagen content and less
hypertrophy in adjacent noninfarcted regions at day 28.
Conclusion: Attenuation of post-infarct remodeling by overexpression of AT2R is not
directly mediated via a B2R pathway. However, B2R does appear to have a role in the
smaller cavity size and hyperdynamic function observed at baseline in AT2TG and in
limiting collagen deposition during post-infarct remodeling.
Page 2 of 34
Copyright Information

3
Key Words: magnetic resonance imaging, bradykinin, angiotensin, receptors,
remodeling
Page 3 of 34
Copyright Information

4
The renin-angiotensin system (RAS) plays an important role in post-infarction left
ventricular (LV) remodeling. Pharmocologic strategies devised to manipulate the RAS
can improve both LV function and clinical outcomes following infarction(30-32).
Angiotensin II (Ang II) is now recognized as a potent mediator of LV remodeling post-
MI, largely through promotion of vasoconstriction, cellular hypertrophy, and interstitial
fibrosis(26). Two major Ang II receptor subtypes have now been characterized, the Ang
II type 1 (AT1R) and Ang II type 2 (AT2R) receptors.
While the importance of AT1R mediated effects on the cardiovascular system are
well known(5), only recently has the role of the AT2R been recognized. AT2R, which is
expressed at low levels in normal myocytes, is upregulated post-MI and in other
pathologic states(6). AT2R promotes vasodilation and inhibits growth and
remodeling(34). The importance of this receptor is evident in models of AT2R deletion
in which there is an increased incidence of heart failure, myocardial rupture, and death
post-infarction(18). Furthermore, experimental evidence suggests that the clinical
benefits of AT1 inhibition post-MI, which have now been validated in a large,
randomized clinical trial(31), are mediated in part through the AT2R (20; 41). However,
the mechanisms through which an increase in cardiac AT2R expression might attenuate
post-MI remodeling are incompletely understood.
We have previously demonstrated in a murine model that cardiac overexpression
of the AT2R preserves left ventricular size and function during post- infarct myocardial
remodeling(46). While the nitric oxide (NO)/cGMP cascade mediates many of the anti-
remodeling benefits of AT2R overexpression (4; 43), the relative importance of
bradykinin and its subtype 2 receptor (B2R) has not been assessed. Previous studies in
Page 4 of 34
Copyright Information

5
mice with AT2-R overexpression in vascular smooth muscle demonstrate that AT2-R
blocks the Na+/H+ exchanger, causing intracellular acidosis, which increases
kininogenase activity and bradykinin production.(40) Cyclic GMP production was also
increased. The Ang II mediated pressor effect was abolished in these animals, but
reinstated by icatibant (HOE-140), a B2R blocker. This suggests that the AT2-R increases
bradykinin, which in turn stimulates the nitric oxide/cGMP system and promotes
vasodilatation in VSM.(40) AT2-R-mediated increases in cGMP and hypotensive effects
due to bradykinin and nitric oxide have been shown in the aorta of the spontaneously
hypertensive rat.(15) AT2-R effects in the kidney are also mediated by bradykinin.(7) The
effects of AT1-R blockade on blood pressure are affected by the AT2-R through release of
renal bradykinin, which then mediates NO production.(37)
The same pathways may apply to myocardial AT2-R signaling. We hypothesized
that in a murine B2R knockout model (B2KO), simultaneous cardiac overexpression of
the AT2R would fail to attenuate remodeling in the intact post-MI heart. To test this
hypothesis, we used CMR to image LV dimensions and global function serially after
reperfused MI in four strains of mice: 1) Wild Type (WT) 2) B2R knockout (B2KO) 3)
AT2R overexpression (AT2TG) and 4) AT2TG/ B2R knockout (AT2TG/B2KO).
Methods:
Mouse Model:
Animal protocols were performed in accordance with the Guide for the Care and
Use of Laboratory Animals (NIH publication No. 85-23, revised 1996) and were
approved by the University of Virginia Animal Care and Use Committee. C57BL/6 mice
Page 5 of 34
Copyright Information

6
were used as WT and served as the background for all other strains. The TG mouse
strain with cardiac overexpression of the AT2R was developed in the laboratory of H.
Matsubara, MD (Kansai Medical University, Osaka, Japan)(25). The presence of the
transgene was confirmed with methods as previously described (46). Bradykinin B2
receptor-deficient transgenic mice were generously provided by Dr. Samir El-Dahr at
Tulane University (9). After cross-breeding the AT2R transgenic mice with homozygous
B2R knockout mice, subsequent generations yielded mice with both cardiac
overexpression of the AT2R and systemic knockout of the B2R. Transgene and knockout
mutations were confirmed by Southern Blot analysis of genomic DNA from mouse tails.
All mice were male and 8-14 weeks of age at the start of the study period.
AT2-R protein levels
Plasma membranes from the mouse hearts were isolated using a triton solubilized
100,000 x G membrane preparation based upon the method of Nagamatsu et al.(29)
Protein concentrations were quantified using a bicinchoninic acid protein assay and
subjected to Western blot analysis. The membrane blots were incubated with AT2
receptor antibody and the 44 kDa band densities were measured by scanning
densitometry and were normalized with Ponceau staining.(35)
Surgery and Magnetic Resonance Imaging
Surgical procedures for the induction of reperfused myocardial infarction were as
reported previously(47). Murine CMR was performed as previously described (4; 41; 46).
A 4.7T MRI system (Varian 200/400 Inova) with Magnex gradients (80 G/cm maximum
Page 6 of 34
Copyright Information

7
strength) with a custom-built Litz radiofrequency coil (Doty Scientific, Columbia, SC)
was used. Contiguous short axis bright blood cine images of the left ventricle were
obtained with a 2D-FLASH gradient echo sequence (Figure 1) as previously
published(4; 41; 46). Infarct imaging was done on Day 1 post-MI as carefully validated
against pathologic measures of infarct size by our group(45).
Image Analysis
Short axis cine images were analyzed using ARGUS image analysis software
(Siemens Medical Solutions, Princeton, NJ). Epicardial and endocardial borders were
planimetered to determine end-diastolic volume (EDV), end-systolic volume (ESV), left
ventricular mass (LVM) and ejection fraction (EF). Volumes and mass were indexed to
body weight in grams (EDVI, ESVI, LVMI). Infarct size was measured as previously
described(41; 46) and expressed as % of LV mass.
Hemodynamic Measurements:
Invasive hemodynamic measurements were performed after the Day 28 CMR
examination. The LV pressure was obtained by direct puncturing of the LV via the left
5th interspace with a 27-gauge needle connected to PE-50 tubing. Blood pressure and LV
pressure as well as heart rate and developed pressure (dP/dt) were recorded by a MacLab
recording system..
Collagen Analysis:
Page 7 of 34
Copyright Information

8
We performed collagen analysis using paraffin embedded sections. Quantitative
morphometry was performed on 6 µm slices from infarct border (adjacent) and remote
myocardium which were stained with picric acid Sirius red. Volume percent collagen was
determined by measuring a minimum of 30 fields in 3 to 4 sections from each region.
Mean area was calculated for each region in each animal.
Myocyte Analysis:
Three to five tissue samples from each heart were sectioned at 5 micron thickness,
were stained with picric acid sirius red and examined with an Olympus AH2 research
microscope, using rhodamine epifluorescence. Using a 40X objective (660X on monitor),
cross sectional area was determined on a minimum of 60 myocytes from each animal in
adjacent and remote noninfarcted regions, selected from areas judged to be within 20
degrees of true cross section, using a Universal Imaging AT1 image analysis system
(Universal Imaging, West Chester, PA). Mean area was calculated for each animal.
Statistical Methods:
Infarct sizes between groups and noninvasive and invasive hemodynamic
parameters were compared by one-way analysis of variance (ANOVA). Regional %
collagen and regional myocyte size between groups were compared by two-way ANOVA
with Tukey subtesting. Volumetric parameters were compared between all 4 groups using
F-tests in repeated measures models, using day 0 data as a covariate. Analyses were
carried out using PROC MIXED in SAS 9.1 (SAS Institute Inc., Cary, NC). All values
are presented as mean ± SE; p<0.05 was considered significant.
Page 8 of 34
Copyright Information

9
Results:
Fourteen transgenic mice overexpressing the AT2R (AT2TG), 10 mice with a B2R
deletion (B2KO), 13 AT2TG with B2R deletion (AT2TG/B2KO), and 11 wild type (WT)
mice were studied. All mice were on a C57BL/6 background.
Baseline Parameters
Mean systolic blood pressure in conscious animals at baseline measured with
noninvasive tail-cuff apparatus was similar between groups. (Table 1) Resting HR at
baseline was 598 ± 55 bpm in WT, 522 ± 53 bpm in B2KO, 691 ± 27 bpm in AT2TG
(p<0.001 vs. B2KO and AT2TG / B2KO), and 518 ± 70 bpm in AT2TG /B2KO. These
differences were no longer apparent while the animals were under anesthesia during
baseline imaging (Table 1). Body weight was higher in the AT2TG compared to other
groups at baseline (Table 1). Baseline volumetric parameters were also different between
the four strains of mice (Figure 2). LV EDVI and ESVI were smaller and EF higher in
AT2TG.
AT2-R protein levels
By densitometry, AT2-R protein levels were 145 ± 27 densitometric units (D.U.)
in WT, 328 ± 23 D.U. in AT2TG (p<0.001 vs. WT), and 419 ± 41 D.U. in AT2TG /B2KO
(p<0.001 vs. WT). No difference was noted between AT2-R protein levels between
AT2TG and AT2TG /B2KO groups.
MRI parameters of post-infarct remodeling:
Page 9 of 34
Copyright Information

10
Infarct size on post-infarct day 1 imaging was similar between the four groups
(41± 5%, 43 ± 7% , 41 ± 10%, and 36 ± 6% in WT, B2KO, AT2TG, and AT2TG /B2KO;
p=NS). Systolic blood pressure was not different between groups during the course of the
study (Table 1). By day 7 and 28 post-MI, body weights were similar between groups
(Table 1). By day 28, all groups experienced a decline in LV systolic function and
increase in LV size when compared to baseline values (Table 1). However, both LV size
and function was better preserved in both transgenic strains compared to WT and B2KO
animals as evidenced by the lower ESVI, EDVI, and higher EF (Table 1 and Figures 1,
3, 4, and 5). Values shown in Figures 2-4 for days 1, 7 and 28 post-MI have been
adjusted for baseline differences in AT2TG.
Collagen analysis:
Morphometric analysis was performed in all mice in regions both adjacent and
remote to the infarct zone and in all groups collagen content was increased in adjacent
noninfarcted regions compared to remote (p<0.01) (Table 2). Both B2KO and AT2TG
/B2KO had greater collagen content in adjacent regions compared to WT. In remote
regions, no differences were found between the four strains of mice (Table 2).
Myocyte Hypertrophy
Myocyte hypertrophy occurred in all groups on Day 28 post-MI and was greater
in adjacent, noninfarcted regions than in remote (Table 3). For comparison purposes,
myocyte size in noninfarcted C57Bl/6 wild type controls is 164±31 µm2. In mice with
B2KO, less hypertrophy was noted in adjacent noninfarcted regions.
Page 10 of 34
Copyright Information

11
Invasive hemodynamics
Invasive hemodynamic measurements were performed after completion of day 28
post-infarct imaging in 6 mice per group. LV end-systolic pressure (LVESP) and LV end-
diastolic pressure (LVEDP) were similar among the four strains of mice (Table 4).
Positive dP/dt was higher and - dP/dt lower in B2KO and AT2TG/B2KO compared to WT
while a strong trend was noted between AT2TG and WT (p=0.06)(Table 4).
Discussion:
There were several findings of interest in this study: 1) Enhanced baseline
function in AT2TG is mediated, at least in part, by B2R; 2) Based on global volumetric
and functional parameters, AT2R-mediated attenuation of post-infarct LV remodeling is
largely independent of B2R; 3) In regions adjacent to infarction zones, B2R plays an
important role in limiting collagen expression but allowing cellular hypertrophy during
LV remodeling; and 4) AT2R overexpression offers protection against post-infarct LV
remodeling, and this protection is not dependent on the smaller cavity size and higher
ejection fraction at baseline in AT2TG.
This study sought to define the role of B2R in a model of post-MI LV remodeling.
In contrast to the low-level, continuous activation of the RAS in chronic CHF, there is
intense activation of RAS in the acute setting, which initially serves to maintain cardiac
output. RAS activation occurs rapidly in MI with Ang II levels peaking at approximately
3 days(27). Furthermore, RAS escape, through production of Ang II via pathways
Page 11 of 34
Copyright Information

12
independent of ACE, is less likely to occur in the acute rather than chronic setting.
Recently, the importance of bradykinin in mediating the benefits of AT1 blockade in a
chronic CHF model was demonstrated in rats (20; 21). The use of either a bradykinin
antagonist or kininogen deficient animals limited the benefits of AT1 receptor
antagonism. However, the study drugs were administered two months following
surgically induced MI, well after early LV remodeling had taken place(19).
In addition to its direct vasodilatory effects(17), bradykinin can influence cell
growth and division, inhibit collagen production, and influence myocardial energy
metabolism(2; 12). Two receptor subtypes for bradykinin have been identified, the B1R
and B2R. While the B2R mediates the majority of known biological effects of bradykinin,
the B1R is upregulated in pathological states and investigators have become increasingly
interested in its role in ischemic injury (3; 10).
Our group has previously demonstrated the importance of nitric oxide (NO) in
AT2R mediated attenuation of LV remodeling post-infarction(4). In AT2TG mice treated
with the NO synthase inhibitor N-nitro-L-arginine methyl ester, the beneficial effects of
AT2R overexpression post-infarct were largely abrogated. While bradykinin is important
in the AT2R mediated production of NO/cGMP in the aorta of spontaneously
hypertensive rats(15) and in the kidney(37), the receptor’s role in cardiac NO production
is less clear. AT2R activation does lead to the formation of bradykinin within the
myocardium through induction of an intracellular acidosis via blockade of the Na+/H+
exchanger. It is postulated that this acidic environment stimulates pH sensitive
kininogenases to cleave stored kininogens into kinins (40). Indeed, in a rat model of
hypertension(36), infusion of Ang II directly elicited bradykinin release, a response that
Page 12 of 34
Copyright Information

13
was AT2R-mediated. In one model of AT2R overexpression in vascular smooth muscle
cells, Ang II-mediated increases in aortic cGMP could be reversed by treatment with
either AT2R or B2R antagonists(40). However, the relative importance of the B2R in the
AT2R cascade remains incompletely understood. Recent evidence has shown that AT2R
may stimulate the release of cGMP and NO independent of B2R(1; 48). It is certainly
possible that AT2 signaling occurs downstream of the B2R.
Consistent with prior studies using this transgenic mouse model, cardiac AT2R
overexpression resulted in smaller cavity size and an increased ejection fraction
compared to WT(46). Our findings suggest that B2R may mediate much of these effects
of AT2TG on baseline parameters. Although no difference in mean SBP was noted
between the groups, noninvasive baseline HR in conscious animals was higher in the
AT2TG mice than in the other three strains. Frequency-dependent potentiation of cardiac
contractility could account for some of the enhanced baseline function observed in the
AT2TG animals, although some investigators have described a negative force-frequency
relationship in small rodent models(28). While under sedation for cardiac imaging no
difference in HR existed between the four groups, consistent with prior studies(46).
Heightened response to catecholamines during stress in AT2R overexpressed animals
mediated through B2R could account for these findings and is consistent with known
interactions between B2R and the sympathoadrenal system(13; 14).
At study completion, remodeling was attenuated in both TG strains to a similar
extent, and both TG strains remodeled to a lesser extent than either the WT or B2KO.
Even with adjustment for baseline differences, AT2TG and AT2TG/B2KO remodeled to a
similar degree suggesting that the B2R did not play an important role in global measures
Page 13 of 34
Copyright Information

14
of post-infarction LV remodeling. Furthermore, the fact that these transgenic strains
experienced similar remodeling despite differences in baseline cavity size and function
suggests that decreased wall tension (as a consequence of the smaller cavity at baseline)
is not the sole mechanism through which remodeling is attenuated in AT2TG animals.
Morphometric analysis demonstrates that B2R limited collagen deposition in
regions adjacent to the infarct zone but had no effect on collagen expression in remote
regions. Our findings are consistent with other studies demonstrating the role of B2R in
limiting fibrosis in noninfarcted segments post-MI(10; 21-23; 44). There may be
uncoupling of these 2 receptor systems for this particular endpoint. Despite regional
differences in collagen expression between strains, global parameters of post-infarct
remodeling did not differ between AT2TG/B2KO and AT2TG. Similarly, B2KO and WT
animals remodeled in a parallel fashion, despite increased collagen deposition in B2KO.
Interestingly, B2KO and AT2TG/B2KO strains demonstrated less myocyte hypertrophy in
infarct adjacent regions at day 28. The kinins are generally believed to exert anti-
hypertrophic effects in the myocardium but the relationship is a complex one. In one
study performed on isolated ventricular myocytes, bradykinin had direct hypertrophic
effects on the cells(33). However, in the presence of endothelial cells, bradykinin exerted
antihypertrophic effects.
One rationale for administration of both ACE inhibitor and Ang II receptor
blocker simultaneously in CHF is to augment levels of bradykinin. While AT2R
stimulation enhances production, ACE inhibition prolongs the kinins’ half-life. In
addition, ACE inhibition appears to potentiate angiotensin II mediated increases in
bradykinin(38). Our findings, when taken together with those of Liu et al.(21), suggest
Page 14 of 34
Copyright Information

15
that the role of bradykinin in AT1/AT2 manipulation may be less important in the acute
than in the chronic heart failure setting.
Although the B2R is known to be a potent stimulus for endothelial NO release, it
may only serve to potentiate AT2R-mediated NO production and not be an essential
regulator in the cascade. The B1 receptor, which is upregulated in the setting of
inflammation or injury, may have similar signal transduction pathways to the B2R and
promote the production of NO in certain tissues, which we know to be critical to AT2R
mediated attenuation of remodeling (24). Furthermore, the effects of the B1R may be
exaggerated in this mouse model as it is known to be upregulated in B2R deficient
mice(8) and may be responsible for cardioprotective effects(16). Further studies
investigating the importance of B1R mediated effects on remodeling and the interaction
between the two receptors are warranted.
Limitations:
One limitation of the present study is that no sham operated controls were studied
for each group. Renin-angiotensin system activation was not directly measured. A
subgroup of mice from each group was studied for hemodynamics and no hemodynamic
data was obtained in noninfarcted mice. Additionally, in mice genetically deficient in
B2R, some investigators have described progressive LV remodeling and functional
impairment while others have not(11; 44). The B2KO was not a cardiac specific knockout
and systemic effects of the deletion may have affected the results. The background
genetics of the mouse model appears to modulate the impact of B2KO with C57BL/6
mice being less susceptible to alterations in phenotype, perhaps because of a 10-fold
Page 15 of 34
Copyright Information

16
lower plasma renin activity at baseline compared to 129/J mice(23; 42). In most studies
of C57BL/6 mice, the B2KO mutation ultimately results in significant cardiac effects but
this process is delayed. In our study on a C57BL/6 background, the B2KO and WT mice
were phenotypically similar at baseline suggesting that age-related dysfunction had yet to
occur. Also, the mice studied in this protocol were between 8-14 weeks in age, younger
than the age at which cardiomyopathies have been demonstrated to develop in
noninfarcted B2KO mice(11; 23; 39).
Conclusion
The B2R-receptor plays a role in the small cavity size and enhanced systolic
function at baseline in AT2R overexpressing mice. However, the B2R-receptor does not
mediate the attenuation of remodeling afforded by AT2R overexpression. This may have
implications for differential response to medical therapies for post-infarct LV remodeling
in the acute and chronic setting that involve the B2R.
Page 16 of 34
Copyright Information

17
Reference List
1. Abadir PM, Carey RM and Siragy HM. Angiotensin AT2 receptors directly
stimulate renal nitric oxide in bradykinin B2-receptor-null mice. Hypertension 42:
600-604, 2003.
2. Baxter GF and Ebrahim Z. Role of bradykinin in preconditioning and protection
of the ischaemic myocardium. Br J Pharmacol 135: 843-854, 2002.
3. Bouchard JF, Chouinard J and Lamontagne D. Role of kinins in the endothelial
protective effect of ischaemic preconditioning. Br J Pharmacol 123: 413-420, 1998.
4. Bove CM, Yang Z, Gilson WD, Epstein FH, French BA, Berr SS, Bishop SP,
Matsubara H, Carey RM and Kramer CM. Nitric oxide mediates benefits of
angiotensin II type 2 receptor overexpression during post-infarct remodeling.
Hypertension 43: 680-685, 2004.
5. Burnier M. Angiotensin II type 1 receptor blockers. Circulation 103: 904-912,
2001.
6. Busche S, Gallinat S, Bohle RM, Reinecke A, Seebeck J, Franke F, Fink L,
Zhu M, Sumners C and Unger T. Expression of angiotensin AT(1) and AT(2)
receptors in adult rat cardiomyocytes after myocardial infarction. A single-cell
reverse transcriptase-polymerase chain reaction study. Am J Pathol 157: 605-611,
2000.
Page 17 of 34
Copyright Information

18
7. Carey RM, Jin X, Wang Z and Siragy HM. Nitric oxide: a physiological
mediator of the type 2 (AT2) angiotensin receptor. Acta Physiol Scandi 168: 65-71,
2000.
8. Duka I, Kintsurashvili E, Gavras I, Johns C, Bresnahan M and Gavras H.
Vasoactive potential of the b(1) bradykinin receptor in normotension and
hypertension. Circ Res 88: 275-281, 2001.
9. El Dahr SS, Harrison-Bernard LM, Dipp S, Yosipiv IV and Meleg-Smith S.
Bradykinin B2 null mice are prone to renal dysplasia: gene-environment
interactions in kidney development. Physiol Genomics 3: 121-131, 2000.
10. Emanueli C, Bonaria SM, Stacca T, Pintus G, Kirchmair R, Isner JM, Pinna
A, Gaspa L, Regoli D, Cayla C, Pesquero JB, Bader M and Madeddu P.
Targeting kinin B(1) receptor for therapeutic neovascularization. Circulation 105:
360-366, 2002.
11. Emanueli C, Maestri R, Corradi D, Marchione R, Minasi A, Tozzi MG, Salis
MB, Straino S, Capogrossi MC, Olivetti G and Madeddu P. Dilated and failing
cardiomyopathy in bradykinin B(2) receptor knockout mice. Circulation 100: 2359-
2365, 1999.
12. Gallagher AM, Yu H and Printz MP. Bradykinin-induced reductions in collagen
gene expression involve prostacyclin. Hypertension 32: 84-88, 1998.
Page 18 of 34
Copyright Information

19
13. Gardiner SM, Kemp PA, Bennett T, Bose C, Foulkes R and Hughes B.
Involvement of beta 2-adrenoceptors in the regional haemodynamic responses to
bradykinin in conscious rats. Br J Pharmacol 105: 839-848, 1992.
14. Gavras I and Gavras H. Hypertension, vasoactive peptides and coagulation
factors. J Hypertens 22: 1091-1092, 2004.
15. Gohlke P, Pees C and Unger T. AT2 receptor stimulation increases aortic cyclic
GMP in SHRSP by a kinin-dependent mechanism. Hypertension 31: 349-355, 1998.
16. Griol-Charhbili V, Messadi-Laribi E, Bascands JL, Heudes D, Meneton P,
Giudicelli JF, Alhenc-Gelas F and Richer C. Role of tissue kallikrein in the
cardioprotective effects of ischemic and pharmacological preconditioning in
myocardial ischemia. FASEB J 04-3508fje, 2005.
17. Hatta E, Rubin LE, Seyedi N and Levi R. Bradykinin and cardioprotection: don't
set your heart on it. Pharmacol Res 35: 531-536, 1997.
18. Ichihara S, Senbonmatsu T, Price E Jr, Ichiki T, Gaffney FA and Inagami T.
Targeted deletion of angiotensin II type 2 receptor caused cardiac rupture after
acute myocardial infarction. Circulation 106: 2244-2249, 2002.
19. Liu YH, Yang XP, Nass O, Sabbah HN, Peterson E and Carretero OA. Chronic
heart failure induced by coronary artery ligation in Lewis inbred rats. Am J Physiol
272: H722-H727, 1997.
Page 19 of 34
Copyright Information

20
20. Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E and Carretero
OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1
receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type
2 receptors. J Clin Invest 99: 1926-1935, 1997.
21. Liu YH, Yang XP, Shesely EG, Sankey SS and Carretero OA. Role of
angiotensin II type 2 receptors and kinins in the cardioprotective effect of
angiotensin II type 1 receptor antagonists in rats with heart failure. J Am Coll
Cardiol 43: 1473-1480, 2004.
22. Madeddu P, Emanueli C, Maestri R, Salis MB, Minasi A, Capogrossi MC and
Olivetti G. Angiotensin II type 1 receptor blockade prevents cardiac remodeling in
bradykinin B(2) receptor knockout mice. Hypertension 35: 391-396, 2000.
23. Maestri R, Milia AF, Salis MB, Graiani G, Lagrasta C, Monica M, Corradi D,
Emanueli C and Madeddu P. Cardiac hypertrophy and microvascular deficit in
kinin B2 receptor knockout mice. Hypertension 41: 1151-1155, 2003.
24. Marceau F, Hess JF and Bachvarov DR. The B1 receptors for kinins. Pharmacol
Rev 50: 357-386, 1998.
25. Masaki H, Kurihara T, Yamaki A, Inomata N, Nozawa Y, Mori Y, Murasawa
S, Kizima K, Maruyama K, Horiuchi M, Dzau VJ, Takahashi H, Iwasaka T,
Inada M and Matsubara H. Cardiac-specific overexpression of angiotensin II
AT2 receptor causes attenuated response to AT1 receptor-mediated pressor and
chronotropic effects. J Clin Invest 101: 527-535, 1998.
Page 20 of 34
Copyright Information

21
26. Matsubara H. Pathophysiological role of angiotensin II type 2 receptor in
cardiovascular and renal diseases. Circ Res 83: 1182-1191, 1998.
27. McMurray JJ, Pfeffer MA, Swedberg K and Dzau VJ. Which inhibitor of the
renin-angiotensin system should be used in chronic heart failure and acute
myocardial infarction? Circulation 110: 3281-3288, 2004.
28. Meyer M, Bluhm WF, He H, Post SR, Giordano FJ, Lew WY and Dillmann
WH. Phospholamban-to-SERCA2 ratio controls the force-frequency relationship.
Am J Physiol 276: H779-H785, 1999.
29. Nagamatsu S, Kornhauser JM, Burant CF, Seino S, Mayo KE and Bell GI.
Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the
brain facilitative glucose transporter isoform, and identification of sites of
expression by in situ hybridization. J Biol Chem 267: 467-472, 1992.
30. Pfeffer MA, Lamas GA, Vaughan DE, Parisi AF and Braunwald E. Effect of
captopril on progressive ventricular dilatation after anterior myocardial infarction.
N Engl J Med 319: 80-86, 1988.
31. Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP,
Solomon SD, Swedberg K, Van de WF, White H, Leimberger JD, Henis M,
Edwards S, Zelenkofske S, Sellers MA and Califf RM. Valsartan, captopril, or
both in myocardial infarction complicated by heart failure, left ventricular
dysfunction, or both. N Engl J Med 349: 1893-1906, 2003.
Page 21 of 34
Copyright Information

22
32. Pitt B, Williams G, Remme W, Martinez F, Lopez-Sendon J, Zannad F,
Neaton J, Roniker B, Hurley S, Burns D, Bittman R and Kleiman J. The
EPHESUS trial: eplerenone in patients with heart failure due to systolic dysfunction
complicating acute myocardial infarction. Eplerenone Post-AMI Heart Failure
Efficacy and Survival Study. Cardiovasc Drugs Ther 15: 79-87, 2001.
33. Ritchie RH, Marsh JD, Lancaster WD, Diglio CA and Schiebinger RJ.
Bradykinin blocks angiotensin II-induced hypertrophy in the presence of
endothelial cells. Hypertension 31: 39-44, 1998.
34. Sadoshima J and Izumo S. Molecular characterization of angiotensin II--induced
hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role
of the AT1 receptor subtype. Circ Res 73: 413-423, 1993.
35. Salomone LJ, Howell NL, McGrath HE, Kemp BA, Keller SR, Gildea JJ,
Felder RA and Carey RM. Intrarenal Dopamine D1-Like Receptor Stimulation
Induces Natriuresis via an Angiotensin Type-2 Receptor Mechanism. Hypertension
49: 155-161, 2007.
36. Siragy HM and Carey RM. Protective role of the angiotensin AT2 receptor in a
renal wrap hypertension model. Hypertension 33: 1237-1242, 1999.
37. Siragy HM, de Gasparo M and Carey RM. Angiotensin type 2 receptor mediates
valsartan-induced hypotension in conscious rats. Hypertension 35: 1074-1077,
2000.
Page 22 of 34
Copyright Information

23
38. Siragy HM, de Gasparo M, El Kersh M and Carey RM. Angiotensin-converting
enzyme inhibition potentiates angiotensin II type 1 receptor effects on renal
bradykinin and cGMP. Hypertension 38: 183-186, 2001.
39. Trabold F, Pons S, Hagege AA, Bloch-Faure M, Alhenc-Gelas F, Giudicelli JF,
Richer-Giudicelli C and Meneton P. Cardiovascular phenotypes of kinin B2
receptor- and tissue kallikrein-deficient mice. Hypertension 40: 90-95, 2002.
40. Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S,
Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y,
Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H
and Iwasaka T. Angiotensin II type 2 receptor overexpression activates the
vascular kinin system and causes vasodilation. J Clin Invest 104: 925-935, 1999.
41. Voros S, Yang Z, Bove CM, Gilson WD, Epstein FH, French BA, Berr SS,
Bishop SP, Conaway MR, Matsubara H, Carey RM and Kramer CM. The
interaction between AT1 and AT2 receptors during postinfarction left ventricular
remodeling. Am J Physiol Heart Circ Physiol 290: H1004-H1010, 2006.
42. Wang Q, Hummler E, Nussberger J, Clement S, Gabbiani G, Brunner HR and
Burnier M. Blood pressure, cardiac, and renal responses to salt and
deoxycorticosterone acetate in mice: role of Renin genes. J Am Soc Nephrol 13:
1509-1516, 2002.
43. Widdop RE, Jones ES, Hannan RE and Gaspari TA. Angiotensin AT2
receptors: cardiovascular hope or hype? Br J Pharmacol 140: 809-824, 2003.
Page 23 of 34
Copyright Information

24
44. Yang XP, Liu YH, Mehta D, Cavasin MA, Shesely E, Xu J, Liu F and
Carretero OA. Diminished cardioprotective response to inhibition of angiotensin-
converting enzyme and angiotensin II type 1 receptor in B(2) kinin receptor gene
knockout mice. Circ Res 88: 1072-1079, 2001.
45. Yang Z, Berr SS, Gilson WD, Toufektsian MC and French BA. Simultaneous
evaluation of infarct size and cardiac function in intact mice by contrast-enhanced
cardiac magnetic resonance imaging reveals contractile dysfunction in noninfarcted
regions early after myocardial infarction. Circulation 109: 1161-1167, 2004.
46. Yang Z, Bove CM, French BA, Epstein FH, Berr SS, DiMaria JM, Gibson JJ,
Carey RM and Kramer CM. Angiotensin II type 2 receptor overexpression
preserves left ventricular function after myocardial infarction. Circulation 106: 106-
111, 2002.
47. Yang Z, Zingarelli B and Szabo C. Crucial role of endogenous interleukin-10
production in myocardial ischemia/reperfusion injury. Circulation 101: 1019-1026,
2000.
48. Zhao Y, Biermann T, Luther C, Unger T, Culman J and Gohlke P.
Contribution of bradykinin and nitric oxide to AT2 receptor-mediated
differentiation in PC12 W cells. J Neurochem 85: 759-767, 2003.
Page 24 of 34
Copyright Information

25
Figure Legends:
Figure 1: Representative end-diastolic (top) and end-systolic (bottom) short axis mid-
ventricular cine MRI images at day 28 post-MI from each of the 4 groups. Note that the
end-diastolic and end-systolic cavity areas are larger in the WT and B2KO groups than
AT2TG or AT2TG/B2KO
Figure 2: ESVI, EDVI, and EF (%) at baseline between the four groups. The center line
represents the median, the outer borders of the box the 25th and 75th percentiles, the lines
the 0th and 100th percentiles. Any outliers are represented by the dots. †p<0.03 vs. WT,
B2KO, and AT2TG/B2KO.
Figure 3: ESVI at baseline, days 1, 7, and 28 post-infarction in all 4 groups. Values have
been corrected for baseline differences among groups. *p<0.05 vs. WT; #p<0.05 vs.
B2KO.
Figure 4: EF (%) at baseline, days 1, 7, and 28 post-infarction in all 4 groups. Values
have been corrected for baseline differences among groups. *p<0.05 vs. WT; #p<0.05
vs. B2KO.
Figure 5: EDVI at baseline, days 1, 7, and 28 post-infarction in all 4 groups. Values have
been corrected for baseline differences among groups. *p<0.05 vs. WT; #p<0.05 vs.
B2KO.
Page 25 of 34
Copyright Information
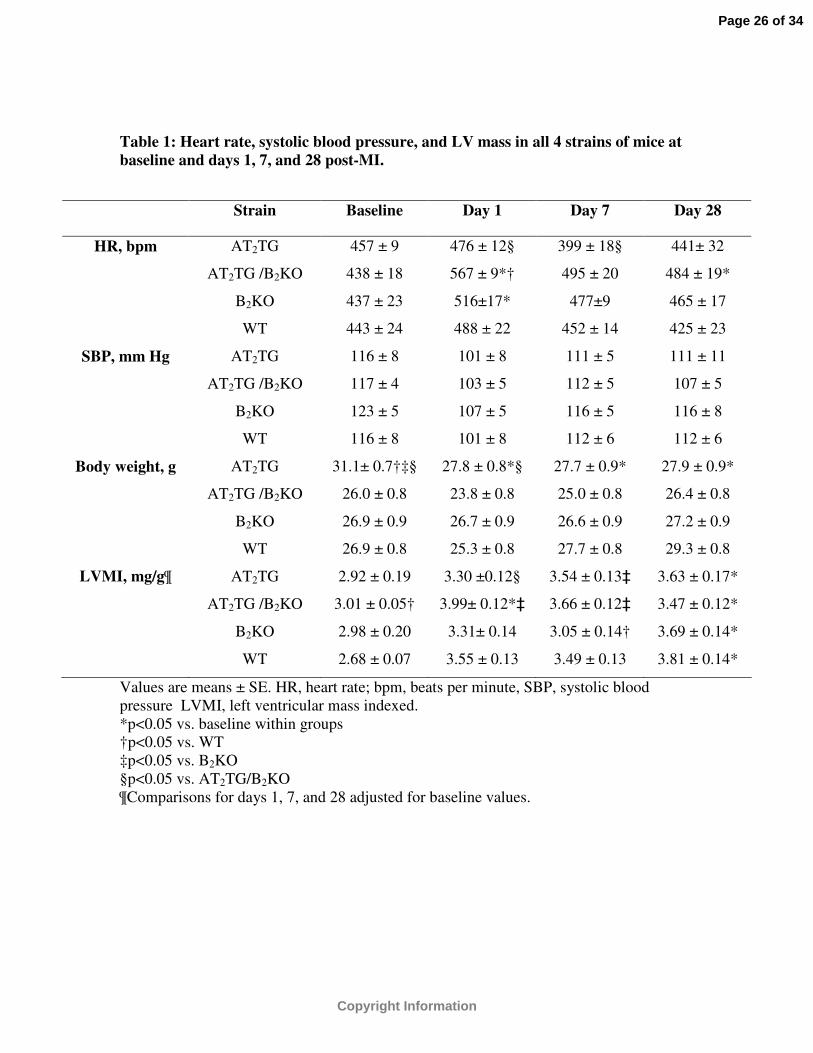
Table 1: Heart rate, systolic blood pressure, and LV mass in all 4 strains of mice at baseline and days 1, 7, and 28 post-MI.
Values are means ± SE. HR, heart rate; bpm, beats per minute, SBP, systolic blood pressure LVMI, left ventricular mass indexed.*p<0.05 vs. baseline within groups†p<0.05 vs. WT‡p<0.05 vs. B2KO§p<0.05 vs. AT2TG/B2KO¶Comparisons for days 1, 7, and 28 adjusted for baseline values.
Strain Baseline Day 1 Day 7 Day 28
HR, bpm AT2TG 457 ± 9 476 ± 12§ 399 ± 18§ 441± 32
AT2TG /B2KO 438 ± 18 567 ± 9*† 495 ± 20 484 ± 19*
B2KO 437 ± 23 516±17* 477±9 465 ± 17
WT 443 ± 24 488 ± 22 452 ± 14 425 ± 23
SBP, mm Hg AT2TG 116 ± 8 101 ± 8 111 ± 5 111 ± 11
AT2TG /B2KO 117 ± 4 103 ± 5 112 ± 5 107 ± 5
B2KO 123 ± 5 107 ± 5 116 ± 5 116 ± 8
WT 116 ± 8 101 ± 8 112 ± 6 112 ± 6
Body weight, g AT2TG 31.1± 0.7†‡§ 27.8 ± 0.8*§ 27.7 ± 0.9* 27.9 ± 0.9*
AT2TG /B2KO 26.0 ± 0.8 23.8 ± 0.8 25.0 ± 0.8 26.4 ± 0.8
B2KO 26.9 ± 0.9 26.7 ± 0.9 26.6 ± 0.9 27.2 ± 0.9
WT 26.9 ± 0.8 25.3 ± 0.8 27.7 ± 0.8 29.3 ± 0.8
LVMI, mg/g¶ AT2TG 2.92 ± 0.19 3.30 ±0.12§ 3.54 ± 0.13‡ 3.63 ± 0.17*
AT2TG /B2KO 3.01 ± 0.05† 3.99± 0.12*‡ 3.66 ± 0.12‡ 3.47 ± 0.12*
B2KO 2.98 ± 0.20 3.31± 0.14 3.05 ± 0.14† 3.69 ± 0.14*
WT 2.68 ± 0.07 3.55 ± 0.13 3.49 ± 0.13 3.81 ± 0.14*
Page 26 of 34
Copyright Information
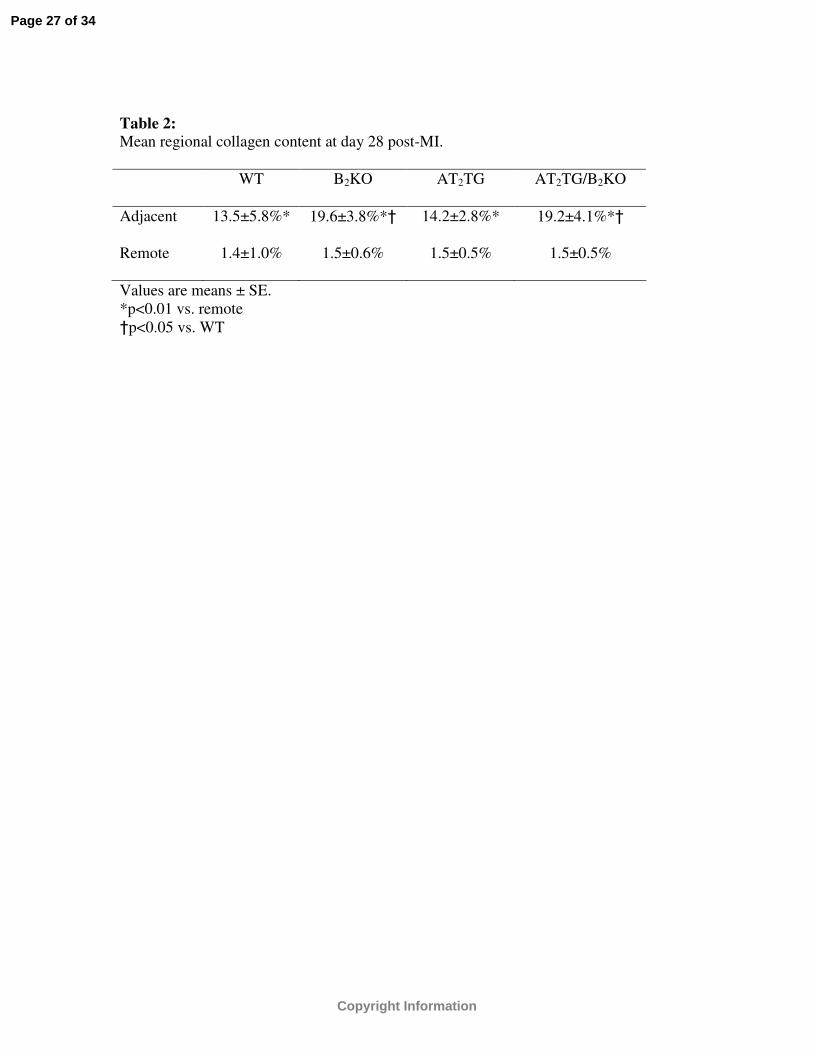
Table 2: Mean regional collagen content at day 28 post-MI.
WT B2KO AT2TG AT2TG/B2KO
Adjacent 13.5±5.8%* 19.6±3.8%*† 14.2±2.8%* 19.2±4.1%*†
Remote 1.4±1.0% 1.5±0.6% 1.5±0.5% 1.5±0.5%
Values are means ± SE. *p<0.01 vs. remote†p<0.05 vs. WT
Page 27 of 34
Copyright Information

Table 3: Mean myocyte cross-sectional area at day 28 post-MI.
WT B2KO AT2TG AT2TG/B2KO
Adjacent (µm2) 518±36* 403±15*† 536±45* 401±15*†§
Remote (µm2) 288±23 323±17 332±36 240±13‡
Values are means ± SE. *p<0.05 vs. remote†p<0.05 vs. WT‡p<0.05 vs. B2KO§p<0.05 vs. AT2TG
Page 28 of 34
Copyright Information
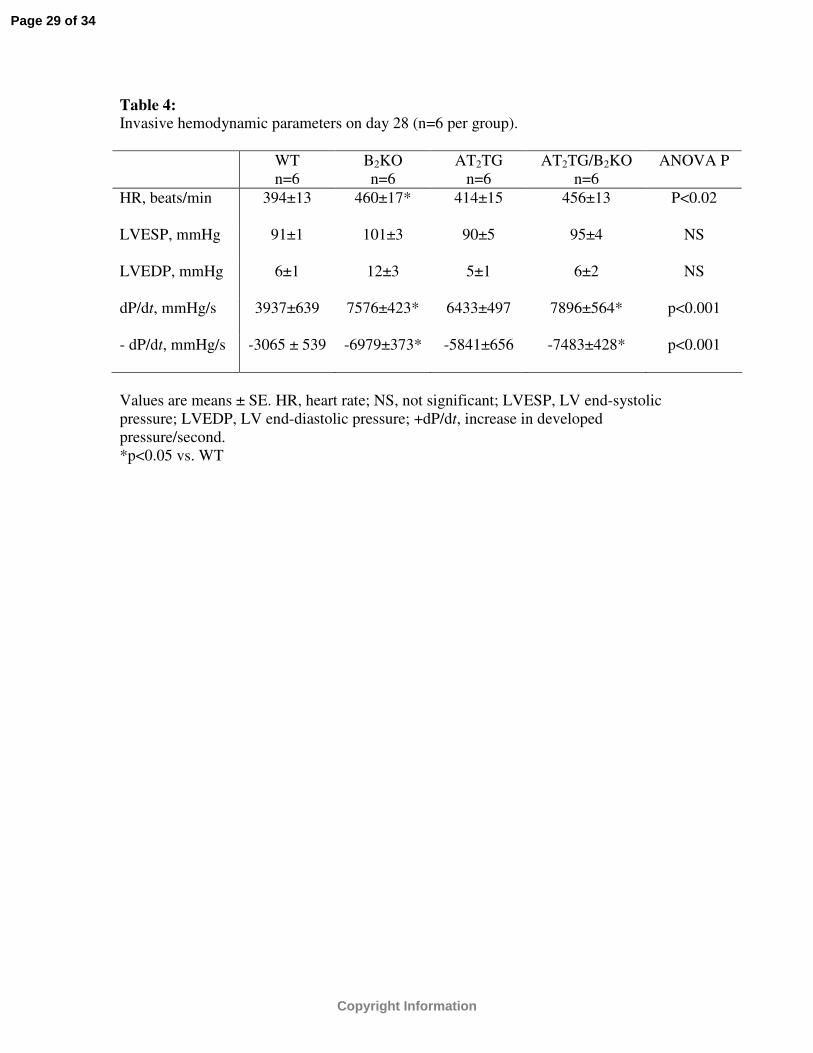
Table 4: Invasive hemodynamic parameters on day 28 (n=6 per group).
WTn=6
B2KOn=6
AT2TGn=6
AT2TG/B2KOn=6
ANOVA P
HR, beats/min 394±13 460±17* 414±15 456±13 P<0.02
LVESP, mmHg 91±1 101±3 90±5 95±4 NS
LVEDP, mmHg 6±1 12±3 5±1 6±2 NS
dP/dt, mmHg/s 3937±639 7576±423* 6433±497 7896±564* p<0.001
- dP/dt, mmHg/s -3065 ± 539 -6979±373* -5841±656 -7483±428* p<0.001
Values are means ± SE. HR, heart rate; NS, not significant; LVESP, LV end-systolic pressure; LVEDP, LV end-diastolic pressure; +dP/dt, increase in developed pressure/second.*p<0.05 vs. WT
Page 29 of 34
Copyright Information

Figure 1 200x101mm (300 x 300 DPI)
Page 30 of 34
Copyright Information
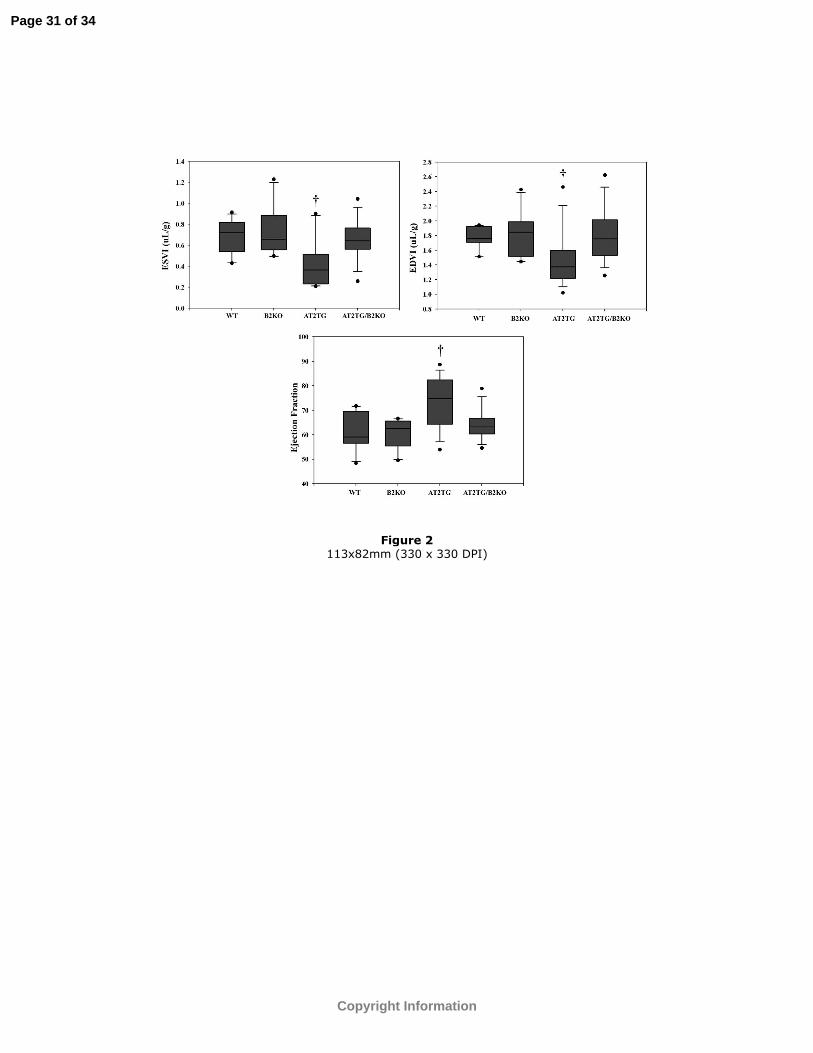
Figure 2 113x82mm (330 x 330 DPI)
Page 31 of 34
Copyright Information
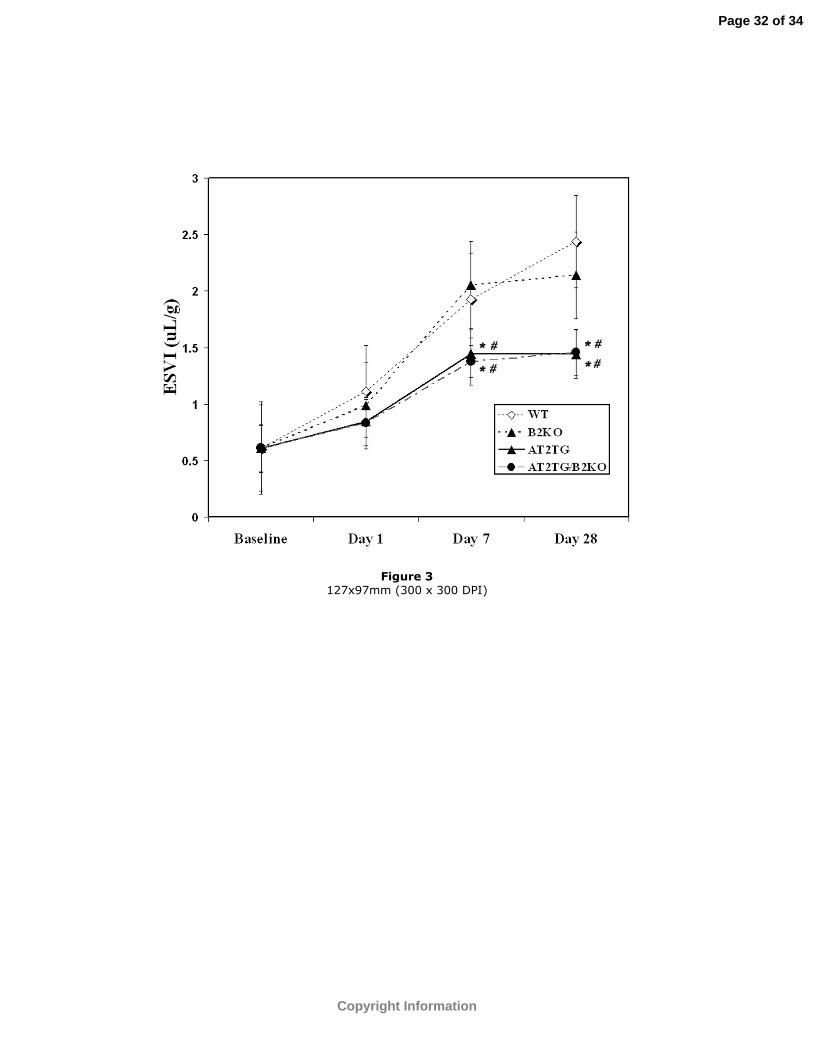
Figure 3 127x97mm (300 x 300 DPI)
Page 32 of 34
Copyright Information
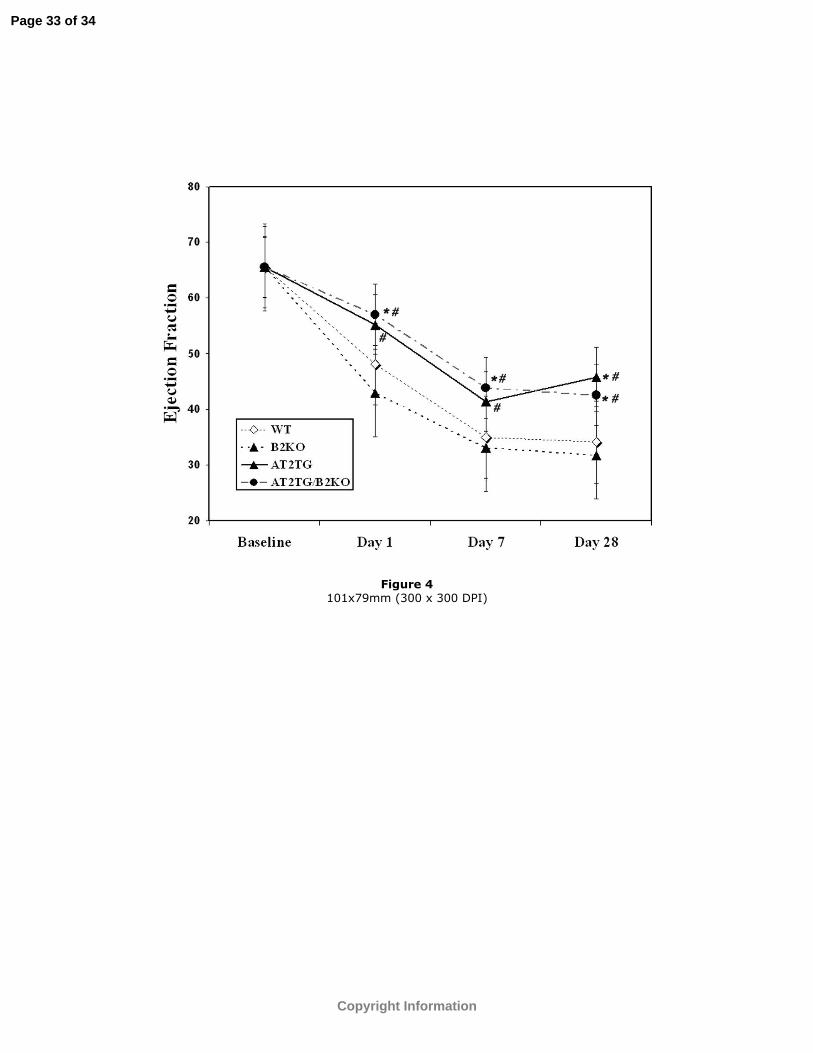
Figure 4 101x79mm (300 x 300 DPI)
Page 33 of 34
Copyright Information

Figure 5 101x79mm (300 x 300 DPI)
Page 34 of 34
Copyright Information






























