




IDENTIFICATION AND CHARACTERISATION
OF A MADS-BOX GENE
FROM RAFFLESIA CANTLEYI SOLMS-LAUBACH
(RAFFLESIACEAE)
PHUA EK KIAN, EDWIN
(B.Sc., NUS)
A THESIS SUBMITTED FOR THE
DEGREE OF MASTER OF SCIENCE
DEPARTMENT OF BIOLOGICAL SCIENCES
NATIONAL UNIVERSITY OF SINGAPORE
2010

i
Acknowledgements
My deepest gratitude to my supervisor Associate Professor Hugh Tan Tiang Wah, and my co-supervisor Professor Prakash P. Kumar, for they have been most patient and understanding, and who believed in me and pushed me right to the end, despite a very long and tiring candidature.
I need to thank the Economic Planning Unit, Prime Minister’s Office, Government of Malaysia for permission to collect Rafflesia cantleyi buds from Pulau Tioman, Pahang, Peninsular Malaysia; and Associate Professor Lim Saw Hoon, formerly of the Malaysia University of Science and Technology for her help in this project.
I would like to thank Ang Kai Yang, Reuben Clements Gopalasamy, Norman
Lim T-Lon, and Alvin Lok for their expertise in the field and in help with the collection of the Rafflesia flowers.
I would also like to thank Dr. Rengasamy Ramamoorthy for his invaluable help
and expertise in the laboratory. Last, but not least, I thank all my colleagues and labmates from the Plant
Systematics Laboratory and the Plant Morphogenesis Laboratory, and friends in the Department of Biological Sciences for their support, advice, and help! I could not have done this without you!

ii
Table of Contents
Page
Acknowledgements i
Table of Contents ii
Summary iv
List of Abbreviations v
List of Tables vii
List of Figures viii
Chapter 1: General Introduction 1
Chapter 2: Literature Review
2.1. Rafflesia R.Br. 5
2.1.1. Floral morphology Rafflesia 5
2.1.2. Rafflesia evolution and systematics 6
2.1.3 Molecular studies in Rafflesia 8
2.1.4. Rafflesia cantleyi Solms-Laubach 8
2.2. MADS-box genes 8
2.2.1. Floral organ identity genes 12
2.2.2. Flowering time genes 15
2.3. Heterologous expression system for functional analysis of genes 17
Chapter 3: Material and Methods
3.1. Plant Materials 18
3.2. RNA and DNA isolation 18
3.3. Reverse transcription 20
3.4. PCR amplification 20
3.5. Cloning of PCR products 22
3.6. Plasmid DNA purification 23
3.7. DNA sequencing 24
3.8. Sequence analysis 24
3.9. Phylogenetic analysis 25
3.10. Rapid amplification of cDNA ends 25

iii
3.11. Preparation of ectopic expression construct 25
3.12. Transformation of Agrobacterium tumefaciens 26
3.13. Genetic transformation of Arabidopsis thaliana 28
3.14. Quantitative real-time PCR analysis 29
3.15. Genomic Southern blot analysis 29
Chapter 4: Results and Discussion
4.1. Collection of Rafflesia cantleyi flower buds 32
4.2. RNA isolation 32
4.3. DNA isolation 35
4.4. Cloning MADS-box genes by RT-PCR 35
4.5. Phylogenetic analysis 40
4.6. Functional characterisation of 35S::RcMADS1 in A. thaliana 41
4.6.1. Construction of ectopic expression plasmid 41
4.6.2. Transgenic Arabidopsis thaliana T1 phenotypes 43
4.6.3. 35S::RcMADS1 effects on flowering time 45
4.6.4. Floral morphology in 35S::RcMADS1 transgenic lines 47
4.7. Molecular characterisation of selected transgenic lines
4.7.1. Genomic Southern blot analysis 53
4.7.2. Quantitative real-time PCR analysis 53
Chapter 5: General Discussion and Future Work
5.1. RcMADS1 may be involved in regulation of flowering time 60
5.2. Temporal and spatial expression of RcMADS1 in Rafflesia cantleyi 61
5.3. Future work 62
5.4. Conclusions 62
References 64

iv
SUMMARY
Rafflesia is a distinctive genus of holoparasitic endophytes found only in the
Indo-Malayan region, with highly reduced vegetative morphology, and usually
manifest only as large, fleshy flowers on their host plants. Despite its distinct ecology
and morphology, very little is known about this taxon, including information on the
molecular processes of floral development. The MADS-box genes, which encode
transcription factors sharing a highly conserved MADS domain are known as the key
regulatory genes that mediate flower development. Therefore, in an attempt to learn
more about molecular floral development in Rafflesia, we cloned and characterised a
MADS-box cDNA, RcMADS1, from Rafflesia cantleyi.
Using RNA from flower buds of Rafflesia cantleyi, we performed RT-PCR with
degenerate primers specific for the MADS domain, followed by 5′-RACE. This
yielded a cDNA of 951 bp (named RcMADS1), encoding a polypeptide of 228 amino
acids. Sequence analysis of this polypeptide revealed about 57% similarity to AGL24
and SVP, two proteins from Arabidopsis thaliana that are involved in mediating
various flowering signals. Phylogenetic analysis showed RcMADS1 to be nested in the
StMADS11 clade, together with AGL24 and SVP. Ectopic expression of RcMADS1 in
Arabidopsis thaliana as a heterologous system produced several independent lines of
transgenic plants that showed alterations in flowering time and floral morphology in a
dose-dependent manner, similar to the phenotypes observed when AGL24 is
overexpressed. Our data suggest that RcMADS1 may be functionally similar to AGL24.

v
List of Abbreviations
Chemicals and Reagents
CTAB cetyltrimethylammonium bromide
DEPC diethyl pyrocarbonate
DTT dithiothreitol
EDTA ethylenediaminetetraacetic acid
HCl hydrochloric acid
LB lysogeny broth ( = Luria Bertani)
MgCl2 magnesium chloride
NaCl sodium chloride
NaOH sodium hydroxide
PVPP polyvinyl polypyrrolidone
SDS sodium dodecyl sulfate
SSC standard saline citrate
TBE Tris borate EDTA
TE Tris EDTA
Tris tris(hydroymethyl)aminomethane
Units and Measurements
bp base pairs
cm centimeter
cm2 square centimeter
g gram
g centrifugal force
h hour
l litre
M molar ( = moles per litre)
min minute
mg milligram
mJ millijoule
ml millilitre
mM millimolar

vi
mm
ng nanogram
pmol picomole
rpm revolutions per minute
s second
V volt
v/v volume per volume
w/v weight per volume
°C degree Celsius
µg microgram
µl microlitre
Others
BLAST Basic Local Alignment Search Tool
CaMV cauliflower mosaic virus
cDNA complementary deoxyribonucleic acid
DNA deoxyribonucleic acid
dNTP deoxynucleoside triphosphate
mRNA messenger ribonucleic acid
OD optical density
oligo(dT) oligodeoxythymidylic acid
PCR polymerase chain reaction
pH potential of hydrogen
RACE rapid amplification of cDNA ends
RNA ribonucleic acid
RT-PCR reverse transcription polymerase chain reaction
UTR untranslated region
UV ultraviolet

vii
List of Tables Page
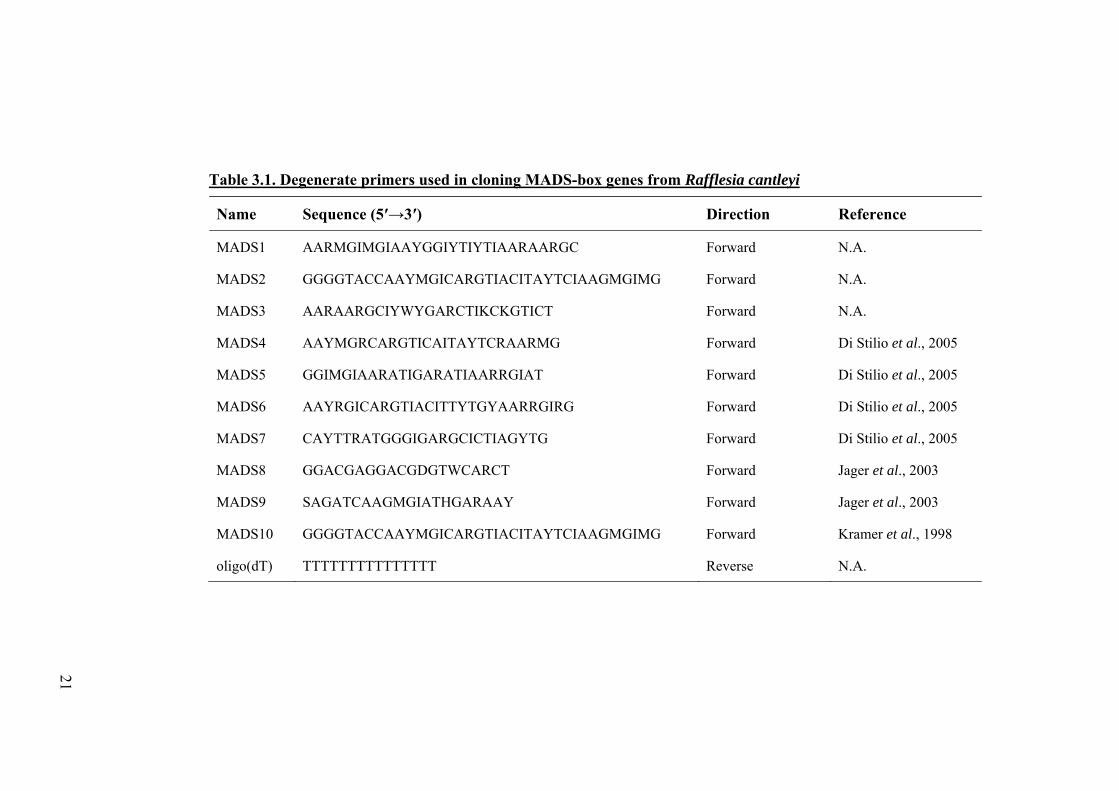
Table 3.1 Degenerate primers used in cloning MADS-box genes from Rafflesia cantleyi.
21
Table 3.2 Primer pairs used in quantitative real-time PCR. 30
Table 4.1 Phenotype analysis of T1 transgenic plants generated. 44
Table 4.2 Comparison of flowering times of 35S::RcMADS1 T2 lines.
46

viii
List of Figures Page
Figure 2.1 Schematic representation of the structure of plant MIKC-type MADS-box genes.
11
Figure 3.1 Schematic diagram of 35S::RcMADS1 ectopic expression construction.
27
Figure 4.1 Rafflesia cantleyi Solms-Laubach buds. 33
Figure 4.2 Gel electrophoresis of total RNA extracted from young Rafflesia cantleyi flower bud (~1 cm in diameter).
34
Figure 4.3 Cloning of MADS-box genes via degenerate PCR. 36
Figure 4.4 Structure of RcMADS1 cDNA. 38
Figure 4.5 Alignment of the derived amino acid sequences of RcMADS1 and other members of the StMADS11 clade.
39
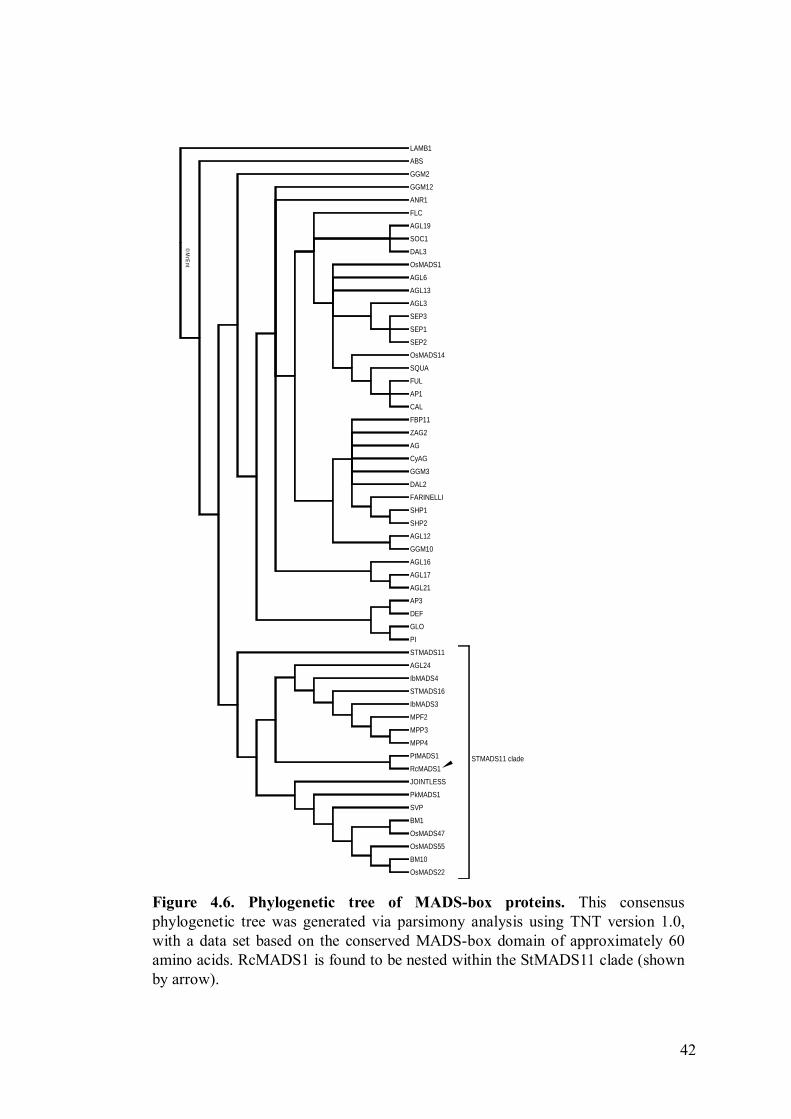
Figure 4.6 Phylogenetic tree of MADS-box proteins. 42
Figure 4.7 Phenotype of wild-type-looking transgenic line ETL01. 48
Figure 4.8 Phenotype of strong transgenic line ETL12. 49
Figure 4.9 Phenotype of strong transgenic line ETL14. 50
Figure 4.10 Altered floral morphology due to ectopic expression of 35S::RcMADS1 in Arabidopsis thaliana.
51
Figure 4.11 Secondary inflorescence development in 35S::RcMADS1 plants.
52
Figure 4.12 Development of profuse branching. 54

ix
Figure 4.13 Genomic Southern blot analysis. 55
Figure 4.14 Comparison of expression levels of RcMADS1 across transgenic lines.
56
Figure 4.15 Effect of RcMADS1 ectopic expression on FLC and FT. 58

1
CHAPTER 1
GENERAL INTRODUCTION
The parasitic plant genus Rafflesia is a distinctive flowering plant genus,
highly unusual in the plant kingdom owing to its highly reduced vegetative
morphology, prominent and large floral structures, and physiology. Rafflesia species
are holoparasitic endophytes — plants that grow completely embedded within their
host plants and completely dependent on them for nutrition. Unlike the majority of the
flowering plants, they lack leaves, stems, and roots, and only manifest as flowers for
sexual reproduction on host plants such as species of Tetrastigma (Kuijt, 1969).
Rafflesia has particularly distinctive, large fleshy flowers that can grow up to a metre
in diameter, producing the smell of rotting flesh which attracts carrion flies for
pollination (Meijer, 1997). Besides being recognised as the largest individual flower
among all extant angiosperms, Rafflesia flowers have some unusual structures, such
as a modified perianth (perigone) enclosed by a diaphragm; a central column with an
apical disk bearing long spike-like structures (processes), and the presence of ramenta,
which are fine hairs, on the interior surface of the perigone tube and diaphragm
(Meijer, 1997).
The genus Rafflesia is confined to the Indo-Malayan region (Meijer, 1997),
and has been little researched, with only a few studies (whether molecular or
ecological) published in the past 20 years (e.g., Beaman et al., 1988; Nickrent and
Starr, 1994; Barkman et al., 2004). There is a lack of extensive work on this genus
partly owing to its rarity and the inaccessibility of its habitats. Holoparasitic plants
like Rafflesia have many physiological and morphological adaptations as a result of
their evolution and have lost many plant structures such as leaves, stems and roots,

2
thus making phylogenetic relationships with non-parasitic plants difficult. Barkman et
al. (2004) sequenced the mitochondrial gene matR and produced a broad phylogenetic
tree that showed a placement of Rafflesia in the Malpighiales. Rafflesia was later
found to be nested in the Euphorbiaceae in a more restricted study of the Malpighiales
using more mitochondrial genes and a chloroplastic gene (Davis et al., 2007). It is
interesting to note that Rafflesia is considered to have evolved from a family with
very small flowers.
Because the flower is the only macroscopic structure of the plant that is visible,
and that the flowers are highly unusual, Rafflesia can be studied from the molecular
development perspective, which can help elucidate the evolutionary processes that
Rafflesia has undergone. Flowering is a complex process that involves the regulation
of various developmental programs by MADS-box genes, which encode transcription
factors containing a highly-conserved MADS-box which is part of the DNA-binding
domain (Becker and Theissen, 2003). Plant floral MADS-box genes also have three
other domains in addition to the MADS (M) domain: an intervening (I) domain; a
keratin-like coiled-coil (K) domain; and a C-terminal (C) domain. Together, these
genes have an MIKC structure which is specific to plants (Nam et al., 2003).
There are at least nine classes of MADS-box genes based on their function and
expression patterns (Nam et al., 2003): classes A, B, C, D, E, F, G, Bs (B-sister), and
T. Many of these MADS-box genes control flower formation and are known as floral
MADS-box genes (Nam et al., 2003). The ‘ABC’ model of flower formation was
originally proposed to explain how the genes (from classes A, B, and C) interact to
produce the different organs (Weigel and Meyerowitz, 1994), and this model is being
modified and updated as more information from continuing studies point to the
involvement of other gene classes in floral development: such as class E genes acting

3
synergistically with combinations of A, B, and C genes to produce petals, stamens and
carpels, as well as floral meristem formation (Honma and Goto, 2001); and class D
genes are required for ovule development (Favaro et al., 2003). Such genes are being
intensely studied using model organisms such as Arabidopsis thaliana (thale cress),
Antirrhinum majus (snapdragon), Zea mays (maize), and Oryza sativa (rice).
Changes in MADS-box genes are strongly correlated to the evolution of land
plant reproductive structures (Theissen et al., 2000). However, model organisms
represent only a small portion of the plant kingdom, and many more genes need to be
identified before a thorough understanding of the control and evolution of flower
development is achieved (Soltis et al., 2002, 2007). Work on many branches of plants
have begun to fill in the gaps, such as from bryophytes (e.g., Physcomitrella), ferns
(e.g., Ceratopteris), gymnosperms (e.g., Cycas, Gnetum, Ginkgo, Pinus) and basal
angiosperms (e.g., Michelia, Piper). The identification and characterisation of
MADS-box genes in Rafflesia could fill in some of these gaps in the genetic
architecture of floral development, leading to the objectives of this study:
1) To clone one or more MADS-box genes from Rafflesia cantleyi, a species of
Rafflesia from Pulau Tioman, Pahang, Malaysia, using a degerate PCR
approach;
2) To identify and analyse the cloned MADS-box gene(s) through sequencing and
phylogenetic analysis;
3) To characterise the function(s) of the cloned MADS-box gene(s) by studying
the effects of ectopic expression in Arabidopsis thaliana plants generated via
Agrobacterium-mediated transformation;

4
4) To characterise the molecular processes underlying the function(s) of cloned
MADS-box genes using quantitative real-time PCR and other appropriate
methods.

5
CHAPTER 2
LITERATURE REVIEW
2.1. Rafflesia R.Br.
The genus Rafflesia R.Br. is well-known as a parasitic plant genus, with the
largest known flowers in the world (Beaman et al., 1988). All species in Rafflesia are
holoparasitic endophytes of the vine Tetrastigma (Vitaceae): the vegetative parts of
the plant are wholly embedded inside the tissues of the host plants and completely
dependent on their host plants for nutrition. These plants have no visible leaves, stems
or roots; and only appear as flowers and fruits during sexual reproduction (Kuijt,
1969). The flowers are often large and can grow up to one metre in diameter, and
produce a smell of rotten flesh during anthesis to attract carrion flies for pollination
(Beaman et al. 1988). Besides these large flowers, and the unusual mode of animal-
aided pollination, Rafflesia flowers have unusual morphology.
2.1.1. Floral morphology of Rafflesia
Rafflesia flowers are unisexual, where the female flowers possess rudimentary
anthers (Meijer, 1997). The perianth is fused, forming a perigone partially closed by a
diaphragm at the apex (leaving an aperture). There are five perigone lobes (the
‘petals’) which are reddish and often with white warts. A central column widens into
a disk at the apex, which supports processes that are spike-like structures. The
processes are hypothesised to radiate heat to aid in dispersal of the odour (that
resembles decaying protein) as olfactory cues to attact carrion flies for pollination
(Beaman et al., 1988). Underneath the disk is a groove, known as the sulcus; in the

6
male flowers, the anthers are situated under the rim of the disk adjacent to the sulcus
(Meijer, 1997).
The floral structures of Rafflesia have been thought to be possibly homologous
to those in Passiflora (Kuijt, 1969; Barkman et al., 2004): the diaphragm of Rafflesia
being homologous to the annular corona of the Passifloraceae; the Rafflesiaceous
central column possibly homologous to the Passifloraceous androgynophore, and the
Rafflesiaceous perigone tube possibly homologous to the Passifloraceous hypanthium.
Phylogenetic data placing Passifloraceae as close relatives to Rafflesiaceae suggests a
shared origin for these floral structures (Barkman et al., 2004).
2.1.2. Rafflesia evolution and systematics
Because Rafflesia specimens are so rare and often found in remote habitats,
and the ecology of Rafflesia is dependent on its host plants, the exact number of
Rafflesia species is uncertain. Several species described in the 19th and early 20th
centuries are not completely known, owing to incomplete descriptions, or the lack of
type specimens (Meijer, 1997); these species include Rafflesia borneensis Koord.;
Rafflesia ciliata Koord.; Rafflesia titan Jack; Rafflesia tuan-mudae Becc.; and
Rafflesia witkampii Koord. In his treatment, Meijer (1997) accepted 13 species:
Rafflesia arnoldii R.Br., with two varieties: Rafflesia arnoldii var. arnoldii R.Br., and
Rafflesia arnoldii var. atjehensis (Koord.) Meijer; Rafflesia cantleyi Solms-Laubach;
Rafflesia gadutensis Meijer; Rafflesia hasseltii Suringar; Rafflesia keithii Meijer;
Rafflesia kerrii Meijer; Rafflesia manillana Teschemacher; Rafflesia micropylora
Meijer; Rafflesia patma Blume; Rafflesia pricei Meijer; Rafflesia rochussenii Teijsm.
& Binn.; Rafflesia schadenbergiana Göpp.; and Rafflesia tengku-adlinii Salleh &
Latiff. Since 2002, 10 or 11 new species have been discovered in the Philippines, as

7
well as two others from outside the Philippines, bringing the number of currently
recognised and described Rafflesia species to 27 (Barcelona et al., 2009). The new
Philippine species are: Rafflesia baletei Barcelona & Cajano; Rafflesia leonardi
Barcelona & Pelser; Rafflesia lobata R.Galang & Madulid; Rafflesia mira Fernando
& Ong; Rafflesia philippensis Blanco; and Rafflesia speciosa Barcelona & Fernando.
The other newly discovered species since the treatment by Meijer (1997) are Rafflesia
azlanii Latiff & M.Wong from Peninsular Malaysia; and Rafflesia bengkuluensis
Susatya, Arianto & Mat-Salleh from Sumatra, Indonesia.
Owing to the highly unusual morphology and evolution as endophytic
holoparasites, the taxonomy and phylogenetic affinities of Rafflesia were not clear.
Rafflesia had been grouped together with other parasitic plants (such as Apodanthus,
Pilostyles, Cytinus, Bdallophyton, and Mitrastema) in various taxonomic treatments
(Meijer, 1997). More recent phylogenetic studies using molecular data had more
precisely established the phylogenetic affinities of Rafflesiaceae sensu stricto
(comprising Rafflesia, Rhizanthes, and Sapria). Using data from the mitochondrial
gene matR from a wide analysis of 95 species of angiosperms and gymnosperms,
Barkman et al. (2004) placed Rafflesia and Rhizanthes within the order Malpighiales,
with sister families such as Passifloraceae, Salicaceae, and Violaceae. Rafflesiaceae
was more confidently placed within the Malpighiales as nested in Euphorbiaceae
using more data (five mitochondrial and one chloroplastic genes) from a focused
sampling of species from all families of Malpighiales (Davis et al., 2007). These
studies suggest a rapid evolution leading to highly specialised and unusual floral
morphology.

8
2.1.3. Molecular studies in Rafflesia
Apart from phylogenetic studies of Rafflesia (Nickrent et al., 1997; Barkman
et al., 2004; Davis et al., 2007) mentioned above, there have been no other published
studies of the molecular biology of Rafflesia, particularly the functional genomics and
developmental biology.
2.1.4. Rafflesia cantleyi Solms-Laubach
Rafflesia cantleyi Solms-Laubach is a species with relatively smaller flowers,
compared to some of the better-known and large-flowered species such as Rafflesia
arnoldii and Rafflesia keithii (Meijer, 1997). It is found in Malaysia, in the states of
Perak, Kelantan, Pahang, and Kedah. Up to 1984, this species was considered to be
identical with Rafflesia hasseltii by Meijer (1997) following identification by Ridley
and other botanists, but was later re-identified as Rafflesia cantleyi as conceived by
Solms-Laubach, owing to differences in the size and pattern of the warts on the
perigone lobes. Meijer (1997) views this species to be closely related to Rafflesia
hasseltii and that it seems to hybridise with it in the Malay Peninsula.
2.2. MADS-Box Genes
Many key processes in growth and development are regulated by transcription
factors, which are important proteins that bind to and affect the transcription of
various target genes. Transcription factors can be classified into gene families
according to the conserved DNA-binding domain present. In plants, the major
transcription factor gene families include the basic-region leucine zipper (bZIP),
MYB-related and MADS-box gene families (Pabo and Sauer, 1992; Martin and
Paz-Ares, 1997; Liu et al., 1999).

9
MADS-box genes encode transcription factors involved in a variety of
important developmental and signal transduction processes in eukaryotes (Messenguy
and Dubois, 2003). The MADS-box encodes a DNA-binding domain comprising of
approximately 60 amino acids, the MADS domain, which is highly conserved across
plants, fungi, and animals (Theissen et al., 1996). “MADS” is an acronym for the four
DNA-binding proteins whose similarity led to the definition of this gene family
(Schwarz-Sommer et al., 1990): MINICHROMOSOME MAINTENANCE 1 (MCM1)
from Saccharomyces cerevisiae (yeast) (Passmore et al., 1989), AGAMOUS (AG)
from Arabidopsis thaliana (Yanofsky et al., 1990), DEFICIENS (DEF) from
Antirrhinum majus (Sommer et al., 1990), and SERUM RESPONSE FACTOR (SRF)
from Homo sapiens (Norman et al., 1988). This MADS domain folds into a structural
motif for DNA interaction consisting of an antiparellel coiled coil of α-helices that
lies flat on the DNA minor groove (Pellegrini et al., 1995).
All known MADS-domain proteins are transcription factors which regulate
target gene expression by binding to specific cis-acting DNA sequences, and have
diverse biological roles primarily in development or cell differentiation such as cell-
type determination and pheromone response in yeast; trachea development in insects;
muscle development in vertebrates and insects; and inflorescence and flower
development in angiosperms (Shore and Sharrocks, 1995). Besides development-
related processes, MADS-domain proteins in yeast have also been found to control
arginine metabolism (Messenguy and Dubois, 1993).
MADS-domain proteins are proposed to be classed into two main groups of
proteins comprising two lineages arising from an ancient duplication event: the Type I
lineage which includes SRF-like proteins and the Type II lineage which includes
MEF2-like (MYOCYTE-SPECIFIC ENHANCER FACTOR 2-like) proteins, both of

10
which are found in animals, fungi, and plants (Alvarez-Buylla et al., 2000). The two
classes of MADS-domain proteins are further classified into subfamilies on the basis
of sequence similarity of the C-terminal extensions (Theissen et al., 1996). In animals
and fungi, the Type I (SRF-like) proteins contain an SAM domain in the C-terminal
extension; this SAM domain (for SRF, ARG80, and MCM1) is based on the loose
similarity shared between SRF, ARG80 and MCM1 (Shore and Sharrocks, 1995).
Some plant MADS-box genes have been found to group with the animal and fungal
SRF-like genes to form the Type I lineage, although the C-terminal domain
extensions for these Type I plant MADS-domain proteins are not defined (Alvarez-
Buylla et al., 2000). In the Type II lineage, animal and fungal MEF2-like proteins
contain an MEF2 domain, originally described for vertebrates (Yu et al., 1992). In
plants, Type II proteins are of the MIKC structure characteristic of most known plant
MADS-box genes (Alvarez-Buylla et al., 2000). MIKC-type proteins are found only
in plants, and thus these proteins are thought to have evolved after plants have
diverged from animals (and fungi) (Kaufmann et al., 2005).
MIKC-type plant MADS-domain proteins characteristically have a modular
structure comprising of four domains: the MADS (M), intervening (I), keratin-like
(K), and C-terminal (C) domains (Figure 2.1) (Theissen et al., 1996, Alvarez-Buylla
et al, 2000). The MADS-domain, which is highly conserved across organisms/plants,
encodes a 60-amino-acid DNA-binding domain. This conserved domain binds DNA
at a consensus recognition sequence known as the CarG box [CC(A/T)6GG]
(Riechmann et al., 1996b). The K domain is a region approximately 70-amino-acid-
residues long, with a sequence similar to the coiled-coil of keratin, and found only in
plant Type II proteins (Theissen et al., 1996). This K domain is weakly conserved at
the primary sequence level but the predicted potential to form amphipathic helices

11
MADS I K C
DNA binding Dimerisation Multimerisation
Figure 2.1. Schematic representation of the structure of plant MIKC-typeMADS-box genes. From left (amino terminal): MADS domain, with DNAbinding and dimerisation functions; I and K domains, which are involved indimerisation; and C domain (at carboxyl terminal), which is variable in length, andpostulated to be involved in transactivation and formation of multimeric proteincomplexes. (Modified fromAlvarez-Buylla et al., 2000)

12
characterises this region. The weakly conserved I domain links the MADS domain to
the K domain, and is predicted to form an α-helix similar to the MEF2S and SAM
domains of non-plant MADS-domain proteins which are required for dimerisation
(Huang et al., 2000), and thus influences the specificity of DNA-binding dimer
formation (Riechmann et al., 1996a). The MADS+I domains have been found to be
sufficient for the formation of DNA-binding dimers, although some class B proteins
require part of the K domain as well (Huang et al., 1996; Riechmann et al., 1996a).
The C-terminal domain is the least conserved region and is variable in length.
However, there is differential conservation within subfamilies, and is particularly
conserved in the DEF subfamily (Kaufmann et al., 2005). This domain has been
postulated to function as a transactivation domain and contribute to the formation of
multimeric protein complexes (Cho et al., 1999; Egea-Cortines et al., 1999; Honma
and Goto, 2001).
The MADS-box gene family is particularly important in controlling various
aspects of plant development, such as floral transition, floral meristem identity, floral
organ specification, and fruit and ovule development (Ng and Yanofsky, 2001). Plant
MIKC-type MADS-box genes can be divided into at least nine classes based on their
function and expression patterns (Nam et al., 2003): classes A, B, C, D, E, F, G, Bs
(B-sister), and T. The classes of MADS-box genes which control flower formation are
known as floral MADS-box genes (Nam et al., 2003).
2.2.1. Floral organ identity genes
The best studied plant MADS-box transcription factors are those involved in
floral organ identity determination. In the ‘ABC’ genetic model of determination of
floral organ identity, combinatorial interactions between the three classes of floral

13
homeotic genes, A, B, and C, determine the identities of the four floral organs
(Haughn and Somerville, 1988; Coen and Meyerowitz, 1991; Weigel and Meyerowitz,
1994; Theissen, 2001). A typical flower consists of four different types of organs
arranged in four whorls. The first and outermost whorl usually comprises green, leaf-
like sepals. The second whorl is composed of usually showy, colourful petals. The
third whorl is the androecium, composed of the stamens, the reproductive organs that
produce pollen. The fourth and innermost whorl is the gynoecium, consisting of the
carpels, the reproductive organs that produce the ovules. The homeotic genes are
active in two adjacent whorls in the flower: Class A genes alone in the first whorl
specify sepals; both Class A and B genes in the second whorl specify petals; Class B
and C genes in the third whorl specify stamens; and Class C genes alone in the fourth
whorl specify carpels. In Arabidopsis thaliana, the Class A function is contributed by
two different genes, APETALA1 (AP1)and APETALA2 (AP2), the B function also by
two genes, APETALA3 (AP3) and PISTILLATA (PI), and the C function by just one
gene, AG. All these genes, with the exception of AP2, are members of the MADS-box
family.
The ‘classical ABC model’ has been later extended to include D and E functions,
yielding an ‘ABCDE model’ (Theissen, 2001; Krizek and Fletcher, 2005). The A, B,
and C functions are the same as in the earlier ABC model, but a D function specifying
ovules and an E function that is required for the specification of petal, stamen and
carpel identity have been added. First described in Petunia (Angenent et al., 1995;
Colombo et al., 1995), D-function genes act in concert with C-function genes to
specify ovule development. Homologous genes in Arabidopsis, SEEDSTICK (STK),
SHATTERPROOF1 (SHP1) and SHATTERPROOF2 (SHP2) were found to act
redundantly and regulate each other’s expression. A stk shp1 shp2 triple mutant has

14
arrested ovule development, but each of the genes is sufficient for ovule development
to proceed (Favaro et al., 2003).
Class E genes are a new class of floral homeotic genes required for the
specification of organ identity in the second, third, and fourth whorls (Jack, 2001). In
Arabidopsis thaliana, the first E-function genes characterised were the three
SEPATALLA genes (SEP1, 2, and 3). Loss of function of all three SEP genes caused
the transformation of the second to fourth whorls of the flower into sepals (Pelaz et al.,
2000). A fourth gene, SEP4, is required with the other three SEP genes to confer sepal
identity and also contributes to the development of the other three floral organs (Ditta
et al., 2004). A sep1 sep2 sep3 sep4 quadruple mutant shows a conversion of all four
floral organ types into reiterating whorls of leaf-like structures, instead of sepals as is
the case for the sep1 sep2 sep3 triple mutant.
Using yeast two-hybrid screening, analyses of protein-protein interaction have
shown that AG interacts with SEP1, SEP2, and SEP3, while AP1 interacts with SEP3
(Fan et al., 1997; Pelaz et al., 2001). Co-immunoprecipitation experiments suggest
that the AP3–PI heterodimer can interact directly with SEP3 and AP1, as well as with
SEP3 and AG to form ternary complexes in vitro (Honma and Goto, 2001). The
ability of MADS-box proteins to form multimeric complexes may therefore provide
the molecular basis for the combinatorial control of floral organ specification. In this
hypothesis, different MADS homo- or hetero-dimer combinations interact with
additional transcription factors, which then determine the functional specificity of the
complexes formed (Riechmann et al., 1996b). This led to the formulation of a ‘quartet
model’, which postulates that four different combinations of four different floral
homeotic proteins determine the identities of the four different floral organs (Theissen,
2001; Theissen and Saedler, 2001). Specifically, tetramers of AP1–AP3–PI–SEP,

15
AP3–PI–AG-SEP, and AG–AG–SEP–SEP would specify petals in the second whorl,
stamens in the third whorl, and carpels in the fourth whorl, respectively (Honma and
Goto, 2001; Theissen and Saedler, 2001). These protein quartets represent one model
of transactivation of genes for floral organ identity by MADS box protein complexes.
Each dimer of a MADS-box tetramer recognises and binds to a single CArG box
sequence; the C-terminal domains of the MADS-box proteins are involved in protein–
protein interaction to form the tetramer (Jack, 2001). However, for this model to work,
two closely linked CArG box sequences have to be present in the promoters of target
genes. Two other models were hypothesised. One model is where multimeric MADS-
box protein complexes bind to single CArG box sequences. Here, a single dimer binds
to the CArG box sequence while other proteins which bind to this dimer via protein–
protein interactions could provide either altered DNA-binding selection or affinity, or
a transcriptional activation domain to the multimeric complex (Jack, 2001). Another,
less likely, model is that dimers of MADS-box proteins cooperatively bind to adjacent
CArG box sequences where there is no protein–protein interaction. This is however
not well supported by existing data (Jack, 2001).
2.2.2. Flowering time genes
Besides floral organ identity genes, there are MADS-box genes involved in
flowering that have different functions, for example, in the control of flowering time,
such as SHORT VEGETATIVE PHASE (SVP) and AGAMOUS-LIKE 24 (AGL24).
SVP and AGL24 are members of the StMADS11 clade (Becker and Theissen, 2003)
which are involved in the contrasting functions of repression and promotion of
flowering, respectively. These genes have been categorised as Class T genes (Nam et
al., 2003).

16
SVP forms a repressor complex of flowering time along with FLOWERING
LOCUS C (FLC) (Liu et al., 2009a) which directly affects the expression of
SUPPESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1) and FLOWERING
LOCUS T (FT) (Liu et al., 2009b). SVP has been shown to repress transcription of
SOC1 in the shoot apex and leaves by binding directly to the SOC1 promoter (Li et al.,
2008). In contrast, AGL24 promotes the expression of SOC1 by binding to the SOC1
promoter. These observations clearly show that SVP and AGL24 are key integrators
of flowering signals, along with other floral transition signals (Liu et al., 2008).
Overexpression of SVP results in the loss of carpels as well as the conversion
of flowers into shoot-like structures with chimaeric characteristics of vegetative
shoots and flowers. Similarly, overexpression of AGL24 results in the transformation
of carpels into inflorescence-like structures, the sepals and petals into leaf-like
structures, and initiation of secondary inflorescences in the axils of sepals (Liu et al.,
2009a). Homologues of SVP and AGL24 have been isolated from a number of
dicotyledonous and monocotyledonous species, and when they were ectopically
expressed in Arabidopsis, phenotypes similar to those of 35S::SVP and 35S::AGL24,
respectively, have been observed. This shows that they are likely to have conserved
function in specifying floral meristem development (Liu et al., 2009a).
The coregulator of LEAFY (LFY), namely SEPALLATA3 (SEP3), is repressed
by SVP, AGL24 and SOC1 (Liu et al., 2009b). This is achieved by forming
complexes with two chromatin regulators: TERMINAL FLOWER 2/LIKE
HETEROCHROMATIN PROTEIN 1 (TFL2/LHP1) and SAP18. SVP interacts with
TFL2/LHP1 to modulate histone H3 methylation while AGL24 and SOC1 interacts
with SAP18 to modulate histone H3 acetylation in SEP3 chromatin.

17
2.2.3. Heterologous expression system for functional analysis of genes
Arabidopsis thaliana has been used for understanding functions of genes cloned
from species for which there is limited molecular and genetic information available.
The ease of genetic transformation coupled with the availability of numerous mutants
makes it a convenient heterologous system for such functional analyses. This is
particularly useful for plants that are not amenable to transformation, for plants that
lack mutants, and for plants with very long generation times. Examples include
Eucalyptus grandis (Brill and Watson, 2004) , Cycas edentata (Zhang et al., 2004),
and Paulownia kawakamii (Prakash and Kumar, 2002).
From the foregoing review of literature, it is clear that molecular regulation of
floral development in higher plants is understood in a fairly comprehensive manner.
However, there is a paucity of information on the developmental regulation of
parasitic plants. Despite having the world’s largest flowers, application of molecular
tools were rarely used in studying floral development in Rafflesia species. In view of
this, we initiated the current project of cloning MADS-box genes that might be
involved in regulating flower development in Rafflesia cantleyi. It is hoped that our
results will contribute to a better understanding of the development of highly
specialised flowers of Rafflesia, and parasitic plants in general.

18
CHAPTER 3
MATERIALS AND METHODS
3.1. Plant materials
Flower buds of various sizes of Rafflesia cantleyi Solms-Laubach were
collected from a few localities along a trail between Tekek and Juara in Pulau Tioman,
Pahang, Malaysia. Collection of Rafflesia cantleyi material in Peninsular Malaysia
required a permit (permit number: UPE40/200/19 SJ. 1200) from the Economic
Planning Unit, Prime Minister’s Department (Unit Perancang Ekonomi, Jabatan
Perdana Menteri), Putrajaya, Malaysia. The buds were surface-sterilised using a 10%
(v/v) Clorox® solution (1% sodium hypochlorite) for 5–10 min, followed by three
rinses with sterile water. Tissues were cut and weighed, then flash-frozen in liquid
nitrogen. All samples were stored at –80°C until further use.
Transgenic and mutant Arabidopsis thaliana plants used in the experiments
were of the same genetic background, Columbia ecotype. Arabidopsis thaliana seeds
were sown on soil (Flora Fleur) and stratified for 3–4 days at 4°C to break seed
dormancy and allow uniform germination, before being transferred to a growth
chamber. The plants were grown at 23 ± 2°C under long-day photoperiod conditions
(16 h of light / 8 h of darkness).
3.2. RNA and DNA isolation
Total RNA from the Rafflesia cantleyi flower buds was isolated using a
modified RNeasy® Plant Mini Kit (QIAGEN) method (Kim, 2004). The modification
involves an initial CTAB extraction (Doyle and Doyle, 1987). 100 mg fresh weight of
tissue was pulverised in liquid nitrogen and homogenised in 500 ml CTAB buffer

19
with 1 µl β-mercaptoethanol added by vigorously mixing using a vortex. The
homogenate was incubated at 60°C for 10 min before 500 µl of chloroform–isoamyl
alcohol (24:1) was added and then vigorously mixed. The homogenate was then
centrifuged at 14,000 g for 15 min to pellet the insoluble cell debris. 360–400 µl of
the aqueous phase was recovered and mixed with cold isopropanol (2/3 volume of the
recovered supernatant) and incubated at −20°C for 1 h or more to precipitate the RNA.
The preparation was then applied to an RNeasy® column and purification of the
preparation was done following the manufacturer’s instructions.
Total RNA from Arabidopsis thaliana plant tissues was isolated using the
RNeasy® Plant Mini Kit (QIAGEN) following manufacturer’s instructions.
Genomic DNA from Rafflesia cantleyi was isolated using a modified CTAB
method (Lodhi et al., 1994). 100 mg fresh weight of tissue was pulverised in liquid
nitrogen and homogenised in 500 ml CTAB buffer, with 1 µl β-mercaptoethanol and
PVPP (100 mg/g plant tissue) added, by vigorously mixing using a vortex. The
homogenate was incubated at 60°C for 25 min before 500 µl of chloroform–isoamyl
alcohol (24:1) was added and then vigorously mixed. The homogenate was then
centrifuged at 14,000 g for 15 min to pellet the insoluble cell debris. 360–400 µl of
the aqueous phase was recovered, and 1/2 volume of 5 M NaCl was added to the
supernatant. The resulting solution was then with cold isopropanol (2/3 volume of the
recovered supernatant) and incubated at −4°C for 1 h or more to precipitate the DNA.
The DNA was purified by repeated steps of centrifugation and washing with 76%
ethanol, and then stored in deionised water or TE buffer.

20
3.3. Reverse transcription
Analysis of RNA was performed in a two-step reverse transcription–
polymerase chain reaction process (RT-PCR): cDNA was synthesised using poly(A)+
RNA primed with oligo(dT) in the first step; and PCR was performed using primers
specific for the gene of interest in the second step. First-strand cDNA was synthesised
using the SuperScript™ II RNase H–reverse transcriptase (Invitrogen), following
manufacturer’s instructions. 50 ng to 5 µg of RNA was mixed with 1 µl of
oligo(dT)12–18 (0.5 µg/µl) primer and 1 µl of 10 mM dNTP mix, and adjusted to a total
volume of 10 µl with DEPC-treated water. The RNA and primer mix was denatured
by incubation at 65°C for 5 min using a thermal cycler (Elmer Perkin) and
subsequently placed on ice for 1 min. A 9 µl reaction mixture containing 2 µl of 10×
RT buffer, 4 µl of 25 mM MgCl2 solution, 2 µl of 0.1 M DTT, and 1 µl of
RNaseOUT™ Recombinant RNase Inhibitor, was added to the reaction tube
containing the 10 µl mix of RNA and primer and the tube was incubated at 42°C for
2 min. 1 µl (50 units) of SuperScript™ II reverser transcriptase was then added, and
the 20 µl total reaction mixture was incubated at 42°C for 50 min for cDNA synthesis
to take place. The reaction was terminated by an incubation at 70°C for 15 min
followed by chilling on ice for 5–10 min. 1 µl of RNase H was then added and the
reaction mixture was incubated at 37°C for 20 min before being stored at −80°C.
3.4. PCR amplification
PCR amplification of MADS-box genes from Rafflesia cantleyi was
performed using degenerate primers and an oligo(dT)15 primer. These degenerate
primers were designed based on the conserved MADS box of MADS box genes. The
primers used were are listed in Table 3.1. PCR reactions were performed using
21
Table 3.1. Degenerate primers used in cloning MADS-box genes from Rafflesia cantleyi
Name Sequence (5′→3′) Direction Reference
MADS1 AARMGIMGIAAYGGIYTIYTIAARAARGC Forward N.A.
MADS2 GGGGTACCAAYMGICARGTIACITAYTCIAAGMGIMG Forward N.A.
MADS3 AARAARGCIYWYGARCTIKCKGTICT Forward N.A.
MADS4 AAYMGRCARGTICAITAYTCRAARMG Forward Di Stilio et al., 2005
MADS5 GGIMGIAARATIGARATIAARRGIAT Forward Di Stilio et al., 2005
MADS6 AAYRGICARGTIACITTYTGYAARRGIRG Forward Di Stilio et al., 2005
MADS7 CAYTTRATGGGIGARGCICTIAGYTG Forward Di Stilio et al., 2005
MADS8 GGACGAGGACGDGTWCARCT Forward Jager et al., 2003
MADS9 SAGATCAAGMGIATHGARAAY Forward Jager et al., 2003
MADS10 GGGGTACCAAYMGICARGTIACITAYTCIAAGMGIMG Forward Kramer et al., 1998
oligo(dT) TTTTTTTTTTTTTTT Reverse N.A.

22
step-up conditions with the following cycling parameters: an initial denaturation at
95°C for 1 min; 10 cycles of denaturation at 95°C for 30 s, annealing at 35°C for
1 min, and extension at 72°C for 1 min; 25 cycles of denaturation at 95°C for 30 s,
annealing at 40°C for 1 min, and extension at 72°C for 1 min; and a final extension at
72°C for 10 min. 1 µg of cDNA template was addeded to a reaction mixture
consisting of 0.4 µl DyNAzyme polymerase, 0.2 mM dNTP mix, 1× DyNAzyme PCR
buffer and 2 pmol each of forward and reverse primers. PCR reactions were visualised
by performing gel electrophoresis in a 1.2% agarose gel. Amplified fragments over
400 bp in size were selected for cloning and sequencing.
3.5. Cloning of PCR products
The PCR products were purified using the QIAquick® PCR purification kit (QIAGEN)
following manufacturer’s instructions. Five volumes of buffer PB were added to each
PCR sample and the mixture was applied to the QIAquick column and centrifuged at
14,000 g for 1 min. The flow-through was discarded and the column was washed with
0.75 ml of buffer PE diluted in ethanol. The column was centrifuged for 1 min to
remove residual ethanol. To elute the purified DNA, 30 µl of buffer EB (10 mM Tris
HCl, pH 8.5) was added to the column membrane and the column was allowed to
stand for 1 min before centrifugation for 1 min.
The purified PCR product was then cloned into the pGEM®-T Easy Vector
(Promega). The PCR product was added to a reaction mix containing 1× Rapid
Ligation buffer, 50 ng pGEM®-T Easy Vector and 3 units of T4 DNA ligase, and the
mixture was incubated overnight at 4°C to maximise ligation products. The resulting
recombinant plasmids were then introduced into competent Escherichia coli DH5α
cells.

23
Cell transformation was performed by adding 10 µl of ligation products to 100 µl of
competent cells. The mixture was incubated on ice for 30 min before a heat shock
treatment of the cells at 42°C for 90 s, followed by incubation on ice for 5 min. 1 ml
of LB medium was added to the mixture which was then incubated with shaking
at37°C for 1 h. The mixture was then centrifuged gently at 4,000 rpm for 4 min to
pellet the cells and the excess LB medium was removed. The cells were then plated
onto a LB agarose plate containing ampicillin (10 mg/l) and incubated at 37°C
overnight. Transformants were picked via blue/white colony selection and PCR was
performed to check for presence of the insert.
3.6. Plasmid DNA purification
Plasmid DNA was isolated from the Escherichia coli clones using the Wizard
SV Miniprep Kit (Promega) following manufacturer’s instructions. Clones were
picked from the agarose plates and grown overnight in 3 ml bacterial cultures using
LB medium containing ampicillin. Each bacterial culture was centrifuged at
3,700 rpm for 5 min to pellet the cells. The cells were then resuspended in 250 µl
resuspension buffer. The cells were lysed by addition of 250 µl of cell lysis solution
followed by 10 µl of alkaline protease, a step which did not exceed 5 min, as
recommended by the manufacturer, to prevent nicking of the plasmid DNA by the
alkaline protease. The cell lysis solution was neutralised by addition of 350 µl of
neutralisation solution. The cell debris was removed by centrifugation for 1 min at
14,000 rpm. The supernatant was then applied to the spin column, where the plasmid
DNA would bind to the silica membrane. The lysate was passed through the column
by centrifugation, and the column was rinsed with 750 µl of wash buffer. Residual

24
wash buffer was removed by an additional 2 min of centrifugation. The purified
plasmid DNA was eluted using 30 µl deionised water.
3.7. DNA sequencing
Selected clones were sequenced via an automated sequencing method using
ABI PRISM™ Big Dye™ Terminator Cycle Sequencing Ready Reaction Kit (Applied
Biosystems, USA). The sequencing reaction was prepared by mixing 150 ng of
double-stranded DNA with 1.6 pmol of forward or reverse primer and 2 µl of
Terminator Ready Reaction Mix and the final volume topped up to 5 µl with
nuclease-free water. The sequencing reaction was performed using a thermal cycler
(Elmer Perkin) for 25 cycles of denaturation at 96°C for 30 s, annealing at 52°C for
5 s, and extension at 60°C for 4 min. Sequence reactions were purified using the
CleanSEQ® kit (Agencourt) following manufacturer’s instructions with slight
modifications. 10 µl of CleanSEQ reagent containing magnetic beads and 31 µl of 85%
ethanol were added into each sequence reaction tube, with thorough mixing. The
tubes were then placed onto an Agencourt SPRIPlate, and incubated for 3 min, before
the supernatant was removed. The sequencing products were washed with 100 µl of
85% ethanol, then air-dried. The sequence products were eluted with 40 µl of sterile
water in each tube, and 12 µl of the elution was transferred out from each tube for
automated sequencing using the ABI PRISM™ 3100 DNA Sequencer (Applied
Biosystems, USA).
3.8. Sequence analysis
Sequences obtained after automated sequencing were collated and compared
with published sequences in the GenBank databases using the Basic Local Alignment

25
Search Tool (BLAST) program on the National Center for Biotechnology Information
(NCBI) website. The algorithms used were blastn (to search the nucleotide database
using a nucleotide query) and tblastx (to search the translated nucleotide database
using a translated nucleotide query).
3.9. Phylogenetic analysis
Multiple sequence alignments were carried out using the program CLUSTALW.
Since the ~60 amino acid MADS domain is highly conserved and alignment is
unproblematic, the data set for the phylogenetic analysis was based on this region.
This data set was subjected to a parsimony analysis in TNT version 1.0 (Tree
Analyses Using New Technology, Goloboff et al., 2000).
3.10. Rapid amplification of cDNA ends
5′-rapid amplification of cDNA ends (5′-RACE) was performend using BD
SMART™ RACE cDNA Amplification Kit (Clontech) following manufacturer’s
instructions. The 5′-region of the putative cDNA was amplified using UPM (forward
primer provided by manufacturer) and a gene-specific reverse primer (5′-ACA GCT
GCA GAC AAC AGT GG-3′).
3.11. Preparation of ectopic expression construct
The full open reading frame of the putative gene RcMADS1 from Rafflesia
cantleyi was amplified from the cDNA clone (obtained as above) using the following
primers containing restriction enzyme sites: RcM1-F-HindIII (5′-CCC AAG CTT
GGT CGT GCC GTA TTT GTT CT-3′, HindIII recognition site underlined) and
RcM1-R-XbaI (5′-TGC TCT AGA CCT CTC TCT CCG TCA GCT TG-3′, XbaI

26
recognition site underlined). The amplified fragments were digested for 2 h with
HindIII and XbaI restriction endonucleases to generate sticky ends, which were then
inserted between the CaMV 35S promoter and CaMV terminator in a sense direction
in the pGreen 0229 vector digested with the same restriction endonucleases (Figure
3.1). This ectopic expression construct was named 35S::RcMADS1.
3.12. Transformation of Agrobacterium tumefaciens
The 35S::RcMADS1 construct was introduced into Agrobacterium tumefaciens
strain GV3101 carrying the pSoup helper vector. A 2 ml culture LB medium
containing 20 mg/l gentamycin and 10 mg/l tetracycline was inoculated with GV3101
cells and incubated at 28°C in for 2 days. 100 µl of the bacterial culture was then
transferred to 50 ml of fresh LB medium and incubated at 28°C until an OD600 of 0.8–
1.0 was reached. This bacterial culture was transferred into a 50 ml polypropylene
tube and incubated on ice for 5 min, before being centrifuged at 3,700 rpm for 10 min
at 4°C, and the resulting pellet was resuspended in 5 ml of ice-cold water. The
resuspended cells were centrifuged again at 3,700 rpm for 10 min at 4°C and the
pellet was resuspended in 1 ml of ice-cold water and used immediately for
transformation.
1 µl of recombinant DNA was added to 100 µl of freshly prepared competent
cells and allowed to incubate on ice for 30 min. This mixture was transferred into an
electroporation cuvette (Eppendorf, 1 mm gap width, 100 µl volume) and
electroporation was carried out at 2,300 V (Eppendorf, Electroporator 2510). After
electroporation, 500 µl of LB medium was added and the mixture was incubated with
shaking at 28°C for 4 h before plating on an LB agar plate supplemented with
antibiotics (50 mg/l kanamycin, 20 mg/l gentamycin, and 10 mg/l tetracycline). The

27
bar
LB RB
Nospro 2× 35Spro
XbaI
CaMVterNoster RcMADS1
HindIII
Figure 3.1. Schematic diagram of 35S::RcMADS1 ectopic expressionconstruct. The RcMADS1 cDNA fragment was ligated into a pGreen 0229 vectorwith the following features: LB, left border; Noster, nopaline synthase terminator;bar, bialaphos encoding sequencing, conferring resistance to glufosinateammonium, a broad spectrum herbicide which is available as Basta®; Nospro,nopaline synthase promoter; 2× 35Spro, tandem copies of the cauliflower mosaicvirus (CaMV) 35S promoter; CaMVter, CaMV terminator; RB, right border.Arrows indicate directions of transcription. HindIII and XbaI are restriction sites.

28
plate was incubated at 28°C for 2 days, and transformants were selected via PCR and
confirmed by sequencing.
3.13. Genetic transformation of Arabidopsis thaliana
Transformation of Arabidopsis thaliana plants was carried out using the floral
dip method (Clough and Bent, 1998). Healthy Arabidopsis thaliana plants were
grown on soil under long-day photoperiod conditions (16 h of light / 8 h of darkness),
until flowering.
An Agrobacterium tumefaciens transformant selected via PCR and confirmed
by sequencing to carry the ectopic expression construct 35S::RcMADS1 was
inoculated into a 3 ml culture of LB medium containing 50 mg/l kanamycin and the
culture was incubated at 28°C for 2 days. 25 µl of the bacterial culture was then
transferred to a fresh 25 ml culture and incubated at 28°C overnight. This bacterial
culture was centrifuged at 3,700 rpm for 10 min and the resulting pellet was
resuspended in a 5% sucrose solution. Before commencement of the floral dipping,
Silwet L-77 was added to the Agrobacterium tumefaciens cell suspension to a final
concentration of 0.03% (v/v).
Arabidopsis thaliana inflorescences were immersed in the bacterial cell
suspension for 5–10 s with gentle agitation. If possible, the rosette portions of the
plants were immersed in the bacterial cell suspension as well, to maximise transgenic
seed production. After dipping, the plants were covered with plastic bags for 16–24 h,
to maintain high humidity. The plants were then allowed to grow under long-day
conditions until siliques had developed. The seeds were harvested, germinated and the
resulting seedlings were screened for herbicide resistance.

29
35S::RcMADS1 plants were grown under long-day conditions and sprayed
with 250 mg/l Basta® solution (Finale, AgrEvo, California, USA) 3 days and 10 days
after germination. After 2 weeks, the surviving seedlings were selected as putative
transgenic plants and grown for the next generation prior to phenotypic
characterisation.
3.14. Quantitative real-time PCR analysis
Real-time PCR experiments were carried out using the Power SYBR® Green
PCR Master Mix (Applied Biosystems, USA) on the ABI Prism 7000 Sequence
Detection System (Applied Biosystems, USA). PCR was performed in 20 µl reactions
containing 1 µl of the diluted first strand cDNA samples, 4 pmol of primers, and 10 µl
of the SYBR Green PCR mix. The PCR thermocycling profile used was as follows:
1 cycle of 50°C for 2 min; 1 cycle of 95°C for 10 s; 40 cycles of 95°C for 15 s, and
60°C for 1 min. The gene-specific primer pairs used are listed in Table 3.2.
Analysis of the results was carried out using the ABI Prism 7000 Sequence
Detection System software.
3.15. Genomic Southern blot analysis
Genomic DNA samples from Rafflesia cantleyi was prepared as described in the
previous section (Section 3.2). The quality and quantity of genomic DNA were
analysed by spectrophotometer (NanoDrop, Thermo Fisher Scientific, USA).
A 1% agarose gel (w/v) containing 0.5× TBE buffer (45 mM Tris-boric acid,
1 mM EDTA, pH 8.0) was prepared. Rafflesia cantleyi genomic DNA was digested
by the appropriate restriction endonuclease, and electrophoresis of the digested DNA
was conducted using an agarose gel in 0.5× TBE buffer until the bromophenol blue

30
Table 3.2. Primer pairs used in quantitative real-time PCR
Target Forward Primer (5′→3′) Reverse Primer (5′→3′)
RcMADS1 5′- CCAAGCCAGCCATCTCTTGA-3′ 5′- GCTCAGTCGCACCCGATT-3′
AP1 5′-CATGGGTGGTCTGTATCAAGAAGAT-3′ 5′-CATGCGGCGAAGCAGCCAAGGTT-3′
AGL24 5′-GAGGCTTTGGAGACAGAGTCGGTGA-3′ 5′-AGATGGAAGCCCAAGCTTCAGGGAA-3′
CO 5′-TCAGGGACTCACTACAACGACAATGG-3′ 5′-TTGGGTGTGAAGCTGTTGTGACACAT-3′
FLC 5′-CCAAACGTCGCAACGGTCTC-3′ 5′-GTCCAGCAGGTGACATCTCC-3′
FT 5′-CTTGGCAGGCAAACAGTGTATGCAC-3′ 5′-GCCACTCTCCCTCTGACAATTGTAGA-3′
LFY 5′-ATCGCTTGTCGTCATGGCTG-3′ 5′-GCAACCGCATTGTTCCGCTC-3′
SEP3 5′-AGACTAAGGTTAGCTGATGGGTA-3′ 5′-ATGATGACGACCGTAGTGATC-3′
SOC1 5′-AGCTGCAGAAAACGAGAAGCTCTCTG-3′ 5′-GGGCTACTCTCTTCATCACCTCTTCC-3′
SVP 5′-CAAGGACTTGACATTGAAGAGCTTCA-3′ 5′-CTGATCTCACTCATAATCTTGTCAC-3′
TUB2 5′-GAGAATGCTGATGAGTGCATGG-3′ 5′-AGAGTTGAGTTGACCAGGGAACC-3′

31
dye had indicated the sample had been separated for a sufficient distance. The gel was
processed sequentially with depurination (in 250 mM HCl for 10 min), denaturation
(in 1.5 M NaCl, 0.5 M NaOH for 30 min) and neutralization (in 1.5 M NaCl, 0.5 M
Tris-HCl pH 7.5, for 30 min). Genomic DNA was then blotted onto a positively
charged nylon membrane by capillary blocking overnight. DNA was fixed to the
membrane by UV crosslinking for 20 s at 120 mJ/cm2.
Blots were hybridised with denatured probes in hybridisation buffer (1% SDS
(w/v), 1 M NaCl, 10% dextran sulfate (w/v) and 100 µg/ml denatured salmon sperm
DNA) at 65°C overnight. After hybrisation, the blots were washed twice with 2× SSC
(0.3 M NaCl and 0.03 M sodium citrate, pH 7.0) and 0.1% SDS (w/v) at 65°C for
15 min, once with 1× SSC and 0.1% SDS (w/v) at 65°C for 30 min, and then 0.1×
SSC and 0.1% SDS (w/v) at 65°C for 5 min. The blots were then exposed to X-ray
film.

32
CHAPTER 4
RESULTS AND DISCUSSION
4.1. Collection of Rafflesia cantleyi flower buds
Flower buds of Rafflesia cantleyi were collected from Pulau Tioman, Malaysia
(see Methods section for details). The size ranged from 1.0 cm to 11.4 cm in diameter
(Figure 4.1). Our attempts to extract RNA from buds of various sizes yielded mixed
results. The quality of RNA was generally poor in the extracts from the larger buds.
However, we succeeded in optimising nucleic acid extraction from buds of 1.0 cm to
2.95 cm in diameter. These were used for PCR cloning of MADS-box genes.
4.2. RNA isolation
Preliminary attempts at RNA extraction include the use of protocols such as
TRIzol®, RNeasy® and CTAB. Tissues turned brown and highly viscous upon
homogenisation, due to rapid polyphenol production, leading to poor quality RNA
with all the methods tried. RNA recovery was negligible using TRIzol® and RNeasy®
protocols, whereas it was low for the CTAB protocol, where the yield was less than
20 ng/µl. Optimisation of the CTAB protocol included the combination of the initial
steps of the CTAB protocol with an additional series of purification steps using an
RNeasy® column. This modification yield more appreciable amounts of total RNA,
from 83.8 ng/µl to 614.2 ng/µl, as well as fairly good quality total RNA (A260/A280
ratio of about 1.5 to 1.9) (Figure 4.2). Young buds yielded better quality RNA than
older buds.

33
A
1 cm
DB
C
Figure 4.1. Rafflesia cantleyi Solms-Laubach buds. (A) Buds of a range of sizescollected from Pulau Tioman, Malaysia, from the largest on the left to the smalleston the right. Not all buds were viable for DNA and RNA extraction; some wereaborted during development. (B) A bud of approximately 2.5 cm developing on aTetrastigma sp. vine in the forest in Pulau Tioman, Malaysia. (C) A longitudinalsection of a young bud (approximately 2 cm in diameter), showing generallyundifferentiated tissue. The layers of tissue on the top would develop into petalsand bracts, and while the layers of tissue in the centre would develop into thecolumn. Browning due to polyphenol formation was rapid after the tissue wasexposed to air; when cut, the bud is pale yellow and white in colour. (D) Alongitudinal section of a bud close to anthesis (approximately 8 cm in diameter).

34
Figure 4.2. Gel electrophoresis of total RNA extracted from young Rafflesiacantleyi flower bud (~1 cm in diameter). The quality of RNA appeared to begood, due to the presence of the 28S and 18S rRNA bands.
1 2 3

35
4.3. DNA isolation
Extraction of genomic DNA was achieved by using the CTAB method.
Although the yield was appreciable and the quality of DNA was good, problems were
encountered later during genomic DNA blot analysis. Digestion by restriction
endonucleases was poor, indicating that polyphenolic compounds produced by the
Rafflesia tissue may not have been thoroughly removed during extraction and
genomic DNA was contaminated by such compounds, preventing cleaving of the
genomic DNA.
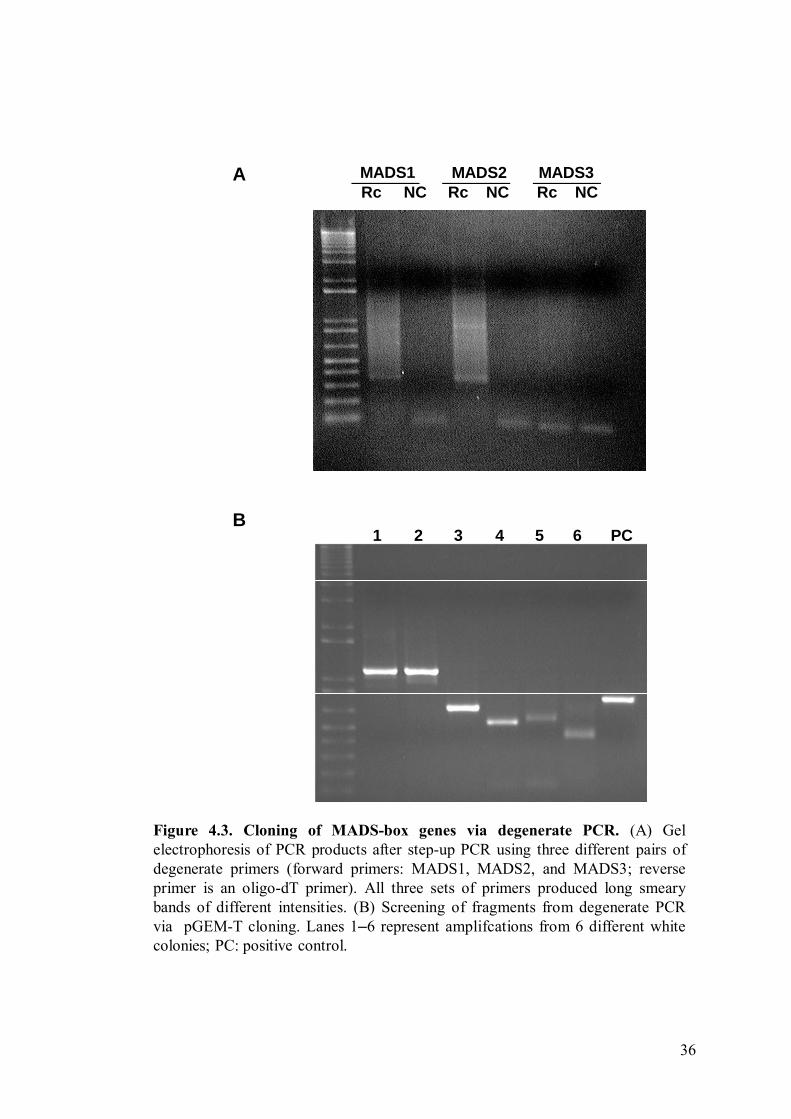
4.4. Cloning MADS-box genes by RT-PCR
The general approach to cloning the MADS-box genes from Rafflesia cantleyi
is based on reverse-transcribing mRNA from the total RNA extracted from R. cantleyi
flower buds and performing PCR using degenerate primers designed from consensus
sequences of known MADS-box genes from various angiosperms species, as little
nuclear gene data is available for Rafflesia. Using a step-up PCR strategy, long faint
smeary bands were obtained after amplification with degenerate primers. As there
were no particularly distinct bands (Figure 4.3A), these PCR products were purified
and cloned into pGEM®-T, and the recombinant plasmids were used for another round
of PCR amplification using SP6 and T7 primers, where distinct bands of 200 to 2000
bp were obtained. Plasmids containing fragments of 400 bp and longer were used for
DNA sequencing (Figure 4.3B). One fragment of 750 bp, amplified from RNA from a
young flower bud (~1 cm in diameter) using the primer MADS2, was found to be
highly similar to MADS-box genes, with scores up to 148 bits using a tblastx search.
The MADS-box genes with high similarity to this sequence include SVP, AGL24,
36
Figure 4.3. Cloning of MADS-box genes via degenerate PCR. (A) Gelelectrophoresis of PCR products after step-up PCR using three different pairs ofdegenerate primers (forward primers: MADS1, MADS2, and MADS3; reverseprimer is an oligo-dT primer). All three sets of primers produced long smearybands of different intensities. (B) Screening of fragments from degenerate PCRvia pGEM-T cloning. Lanes 1–6 represent amplifcations from 6 different whitecolonies; PC: positive control.
MADS1 MADS2 MADS3Rc NC Rc NC Rc NC
A
1 2 3 4 5 6 PCB

37
STMADS11, and IbMADS4, which are all members of the STMADS11 clade of
MADS-box genes.
A sequence-specific primer was designed from this fragment sequence for use
in Rapid Amplification of cDNA Ends (RACE) in sequencing the 5′ region of the
mRNA of this putative MADS-box gene. 5′-RACE PCR using this new primer
yielded an 800 bp fragment. A gene-specific primer meant for cloning the full-length
cDNA was designed based on the 5′ untranslated region (UTR) from this fragment.
We obtained a 951 base-pair-long sequence when we used the 5′ primer and the
oligo(dT) primer. This cDNA sequence contains a 687 bp open reading frame (ORF)
before a stop codon encoding a polypeptide of 228 amino acids, as well as 5′ and 3′
UTRs (Figure 4.4). The conserved MADS domain (69 amino acids) was identified
using the NCBI conserved domain search, while comparison with known MADS-box
genes allowed the identification of the K domain (79 amino acids).
Results from a tblastx search using the online BLAST program revealed high
similarities in protein sequence alignment to MADS-box proteins such as MADS1
from Populus tomentosa, MPF2 and MPF4 from Physalis pubescens, IbMADS4 from
Ipomoea batatas, StMADS11 and StMADS16 from Solanum tuberosum, and SVP
and AGL24 from Arabidopsis thaliana, all from the StMADS11 clade of MADS-
domain proteins. An alignment of some of these proteins from the StMADS11 with
RcMADS1 showed that the MADS domain and K domains are highly conserved
amongst these members of the StMADS11 clade., with the I domain somewhat highly
conserved as well (Figure 4.5).
RcMADS1 showed 57.6% and 56.7% amino acid similarity respectively to
AGL24 and SVP from Arabidopsis thaliana. At the nucleotide level, the RcMADS1
cDNA has a 64.6% and 61.2% similarity to AGL24 and SVP respectively.

38
1 GGGTTAGGTTTCCATATGCCCCACGGGCCACATATTCTTGAACCTGTCTAGCCTTCCCCA 60
61 ATTTATTGCCCGAAGCTGTACGTTCATATTTCGAAACTAAAGATTCGGTCGTGCCGTATT 120
121 TGTTCTGTTTCGGAGCAGATAATATAATGGCTCGAGAAAAGATCAAGATCAAAAAGATCG 1801 M A R E K I K I K K I 11181 ACAACATCACTGCAAGGCAAGTCACCTTCTCTAAGAGGAGACGAGGGCTCTTCAAGAAAG 24012 D N I T A R Q V T F S K R R R G L F K K 31241 CAGAAGAGCTATCAGTTCTTTGTGATGCTCATGTGGCTGTCATTATCTTTTCTGCCACAG 30032 A E E L S V L C D A H V A V I I F S A T 51301 GGAAGCTCTTCGACTATTCCAGCTCCAGCATGAAGGACATACTCTCGAGGTATGATGATC 36052 G K L F D Y S S S S M K D I L S R Y D D 71361 TGCATTTCAATAACAAAGAGAAGCCAAGCCAGCCATCTCTTGAACTGCAGCTAGAAATTA 42072 L H F N N K E K P S Q P S L E L Q L E I 91421 GCAATCGGGTGCGACTGAGCAAAGAAGTTGCAGACAAGACTCGCCAACTAAGGCAAATGA 48092 S N R V R L S K E V A D K T R Q L R Q M 111481 GAGGAGAAGATCTGAACGAATTAAATGTGGAGGAACTGCAGCAACTGGAGAACTTGCTTG 540112 R G E D L N E L N V E E L Q Q L E N L L 131541 AGGTGGGCCTCCAGCGCGTTACTGATGCCAAGGGCAAGCGCATCACAAATGAGATATCTG 600132 E V G L Q R V T D A K G K R I T N E I S 151601 AACTCGAAAGGAAGGGAGCGCAGCTGATGGAAGAAAATAAGCAACTAAAGCAAAAAATGG 660152 E L E R K G A Q L M E E N K Q L K Q K M 171661 TGATGATGTGCAGTGGAACAAGACCTGCCATCCTGGAGTCTGATATCACAACCCATGAAG 720172 V M M C S G T R P A I L E S D I T T H E 191721 AGGGCATGTCCTCTGATTCTGCCACTGTTGTCTGCAGCTGTAGCAATGGCCCCCCCGTGG 780192 E G M S S D S A T V V C S C S N G P P V 211781 AAGATGATATCTCCGATACGTATCTTAAATTGGGATTGTCCTTCTCAAGCTGACGGAGAG 840212 E D D I S D T Y L K L G L S F S S * 229841 AGAGGTGCAGGATTCTTCAATGGCGGTGCAATGAATGAACACATACAAATCTTTACACG 899
Figure 4.4. Structure of RcMADS1 cDNA. The upper row indicates thenucleotide sequence, and the lower row the deduced amino acid sequence. Thetermination (TGA) codon is shown by an asterisk (*). The MADS-box and Kdomains are shown in bold type.

39
80RcM1 79MPF2 79MPP3 79AGL24 79SVP 79STMADS16 79STMADS11
MAREKIKIKKIDNITARQVTFSKRRRGLFKKAEELSVLCDAHVAVIIFSATGKLFDYSSSSMKDILSRYDDLHFNNKEKPMAREKIKIKKIDNITARQVTFSKRRRGLFKKAEELSVLCDADVALIIFSSTGKLFDFSSSSMKDILGKYK.LQSANLDKVMAREKIKIKKIDNITARQVTFSKRRRGLFKKAEELSILCDADVALIIFSSTGKLFDFSSSSMKDILGKYK.LQSANLDKVMAREKIRIKKIDNITARQVTFSKRRRGIFKKADELSVLCDADVALIIFSATGKLFEFSSSRMRDILGRYS.LHASNINKLMAREKIQIRKIDNATARQVTFSKRRRGLFKKAEELSVLCDADVALIIFSSTGKLFEFCSSSMKEVLERHN.LQSKNLEKLMAREKIKIKKIDNITARQVTFSKRRRGLFKKAEELSVLCDADVALIIFSATGKLFDFASTSMKDILGKYK.LQSASLEKVMVRQKIQIKKIDNLTARQVTFSKRRRGLFKKAQELSTLCDADIGLIVFSATGKLFEYSSSSMMQLIEKHK.MQSERDSMD
MMMMMMM
AAAAAA
RRRRRRR
EEEEEEQ
KKKKKKK
IIIIIII
KKKR
K
IIIIIII
KKKKRKK
KKKKKKK
IIIIIII
DDDDDDD
NNNNNNN
IIII
IL
TTTTTTT
AAAAAAA
RRRRRRR
QQQQQQQ
VVVVVVV
TTTTTTT
FFFFFFF
SSSSSSS
KKKKKKK
RRRRRRR
RRRRRRR
RRRRRRR
GGGGGGG
LLLILLL
FFFFFFF
KKKKKKK
KKKKKKK
AAAAAAA
EEEDEEQ
EEEEEEE
LLLLLLL
SSSSSSS
VVIVVV
LLLLLLL
CCCCCCC
DDDDDDD
AAAAAAA
DDDDDD
VVVVVVI
AAAAAAG
VLLLLLL
IIIIIII
IIIIIIV
FFFFFFF
SSSSSSS
A
A
AA
TTTTTTT
GGGGGGG
KKKKKKK
LLLLLLL
FFFFFFF
DDDEEDE
YFFFFFY
SSSS
S
SSSSSSS
SSSSS
S
SSS
SSS
MMMMMMM
KKKRKK
DDDDEDQ
IIIIVIL
LLLLLLI
SGGGEGE
RKKRRKK
YYYY
Y
KK
KK
LLLLLLM
HQQHQQQ
SS
SSS
NNNNN
LLILL
KKKKKK
157RcM1 156MPF2 156MPP3 157AGL24 157SVP 156STMADS16 159STMADS11
..SQPSLELQL.EISNRVRLSKEVADKTRQLRQMRGEDLNELNVEELQQLENLLEVGLQRVTDAKGKRITNEISELERKG
..DQPSLDLQL.ENSLNVRLRKQVADKTRELRQMKGEELEGLSLEELQQIEKRLEAGFNRVLEIKGTRIMDEIANLQRKG
..DQPFLDLQL.ENSLNVRLRKQVADKTRELRQMKGEELEGLSLEELQQIEKRLEAGFNRVLEIKGTRIMDEIANLQRKGM.DPPSTHLRL.ENCNLSRLSKEVEDKTKQLRKLRGEDLDGLNLEELQRLEKLLESGLSRVSEKKGECVMSQIFSLEKRG..DQPSLELQLVENSDHARMSKEIADKSHRLRQMRGEELQGLDIEELQQLEKALETGLTRVIETKSDKIMSEISELQKKG..DEPSLDLQL.ENSLNMRLSKQVADKTRELRQMRGEELEGLSLEELQQIEKRLEAGFNRVLEIKGTRIMDEITNLQRKGNPEQLHSSNLLSEKKTHAMLSRDFVEKNRELRQLHGEELQGLGLDDLMKLEKLVEGGISRVLRIKGDKFMKEISSLKKKE
DDDDDE
QQQ
QEQ
PPPPPP
SS
SSS
LLL
LL
LLLLLL
QQQ
LLLLLLL
EEEEEEE
NNNNN
SSS
SS
RRRRRR
LLLLMLL
S
SSSS
KKKKKKR
VVVVIV
AAA
AA
DDDDDDE
KKKKKKK
TTTT
T
RRRKHRR
QEEQ
EE
LLLLLLL
RRRRRRR
QQQ
QQQ
MMMLMML
RKKRRRH
GGGGGGG
EEEEEEE
DEEDEEE
LLLLLLL
EGGGGGG
LLLLLLL
VLLLILL
EEEEEED
EEEEEED
LLLLLLL
QQQQQQ
QQQ
LIILLIL
EEEEEEE
KKKKKK
LLLLLLV
EEEEEEE
GGGGGGG
RRRRRRR
VVVVVVV
LL
ILL
DEEEEE
II
II
KKKKKKK
GGGGSGG
RRR
KRK
IIIVIIF
MMMMMM
EEEQEEE
IIIIIII
LLLLLLL
EQQEQQ
RRRKKRK
KKKRKKK
GGGGGGE
228RcM1 236MPF2 236MPP3 220AGL24 226SVP 222STMADS16 208STMADS11
AQLMEENKQLKQKMVMMCSGTRPAILESDITTHEEGMSSDSATV...VCSCSNGPPVEDDISDTYLKLGLSFSS......AELMEENKKLKQKMEMMKLGKFPLLTDMDCMVIEEGQSSDSIITTNNVCSSNSGPPPEDDSSNASLKLGCNNGLAAVDDDAELMEENKKLKQKMEMMKLGKLPLLTDMDCMVMEEGQSSDSIITTNNVCSSNTGPPPEDDSSNASLKLGCNNGLAPVDDDSELVDENKRLRDKLETLERAKLTTLKEALETESV..........TTNVSSYDSGTPLEDDSDT.SLKLGLPSWE......MQLMDENKRLRQQGTQLTEENERLGMQICNNVHAHGGAE...........SENAAVYEEGQSSESITNAGNSTGAPVDSEAELMEENKQLKHKMEIMKKGKFPLLTD...MVMEEGQSSESIITTNN........PDQDDSSNASLKLG...GTTAVEDEAQLQEENSQLKQQSQARLNE...............................EGQNVIEQGHSADSITNNRSLVNSHQDYN
AAA
AA
QEEEQEQ
LLLLLLL
MMMVMM
EEEDDEE
EEEEEEE
NNNNNNN
KKKKKK
LLLLLLL
KKKRRKK
QQQDQHQ
KKKK
K
MMML
M
EEE
EQ
MMMLLM
GGGAEGE
KKK
K
PPP
P
LL
LL
ILLL
L
VV
VV
EEE
E
EEE
E
GGG
GG
SSS
S
SSS
S
SSS
S
TTT
T
NNN
N
VVVV
SSSS
GGGGA
PPPP
P
EEEEEQE
DDDDEDQ
DDDDGDG
SSS
S
SSS
SSS
SSSSSS
LLLLILI
KKKK
K
LLLL
L
GGGGAG
VV
VV
DD
DED
228RcM1 249MPF2 249MPP3 220AGL24 239SVP 235STMADS16 221STMADS11
.............CSITSLKLGLPLSCSITSLKLGLPLS.............SSDTSLRLGLPYGCSITSLKLGLPFSDSDTSLKLCLAFP
SS
SSS
TT
TTT
SS
SSS
LL
LLL
KK
RKK
LL
LLL
GG
GG
LL
LLL
PP
PPA
MADS domain I domain
K domain
C-terminal
Figure 4.5. Alignment of the derived amino acid sequences of RcMADS1 andother members of the StMADS11 clade. The MADS, I and K domains are allrelatively conserved across the various proteins. Identical residues are coloureddark blue, conserved residues Key to sequences included: RcM1 = RcMADS1from Rafflesia cantleyi; MPF2 and MPF3 from Physalis pubescens; AGL24 andSVP from Arabidopsis thaliana; and StMADS16 and StMADS11 from Solanumtuberosum.

40
As this cDNA had been amplified from a young and small flower bud, perhaps
just past the floral transition stage and which had not quite started developing distinct
floral organs (see Figure 4.1C), it seems highly probable that RcMADS1 would have a
function in promoting or repressing flowering. Also, with relatively high amino acid
sequence similarity to AGL24 and SVP, two proteins known to promote and repress
flowering in Arabidopsis thaliana respectively, RcMADS1 could be a functional
homologue of AGL24 or SVP in Rafflesia cantleyi.
4.5. Phylogenetic analysis
Phylogenetic analysis of the conserved MADS-box domain using proteins
from the StMADS11 clade as well as representative members of other MADS-box
protein clades showed that RcMADS1 is nested with the StMADS11 clade (Figure
4.6), and it appears to be more related to AGL24 than SVP based on the subclades
they are nested in. RcMADS1 is grouped with PtMADS1, a protein from Populus
tomentosa, and both these two proteins are sister to a clade containing AGL24 and
StMADS16. Together, RcMADS1 and AGL24 form a sister clade to a clade
containing SVP and JOINTLESS (from Solanum lycopersicum). The StMADS11
clade comprises of proteins with diverse functions from a wide variety of plants from
both gymnosperms and angiosperms (Becker and Theissen, 2003). The presence of
homologues from gymnosperms seems to be evidence that StMADS11-like proteins
have ancestral functions in controlling aspects of vegetative development, and the
present functions of StMADS11 proteins in angiosperms are newly evolved after the
split from gymnosperms (Becker and Theissen, 2003).
This phylogenetic analysis shows that RcMADS1 is more closely related to
AGL24 than to SVP, despite the higher alignment similarity between RcMADS1 and

41
SVP as shown by a BLAST search. Since AGL24 and SVP have contrasting functions
in regulating flowering time, it appears likely that RcMADS1 would have a function
more similar to AGL24 than to SVP.
4.6. Functional characterisation of 35S::RcMADS1 in Arabidopsis thaliana
As genetic transformation of Rafflesia is not possible, due to the inherent
difficulty of cultivating the endophytic and holoparasitic Rafflesia, the functions of
identified genes from Rafflesia will have to be studied using some other plant model
organism such as Arabidopsis thaliana. Although this would be a heterologous plant
system involving genes foreign to the model organism, there are benefits: many
mutants are available for complementation studies, and there is a lot of published
information on ectopic expression of putative homologues in Arabidopsis thaliana. In
the case of RcMADS1, there are many studies already published for AGL24 and SVP,
both StMADS11-like genes. Therefore, in order to elucidate the function of the gene,
we used an Arabidopsis thaliana transgenic plant system to study the expression of
the gene in comparison with AGL24 and SVP.
Note: RcMADS1 cDNA was recently (in January 2010) independently cloned
and sequenced by Dr. Rengasamy Ramamoorthy in our laboratory. The independent
sequence verification showed a 100% similarity to the original clone.
4.6.1. Construction of ectopic expression plasmid
A plasmid for ectopic expression of RcMADS1 was constructed using a
pGreen 0229 vector containing a BAR gene conferring resistance to Basta® (a
herbicide containing glufosinate ammonium) (Figure 4.7). Arabidopsis thaliana plants
transformed with the pGreen vector would survive Basta® application. The
42
LAMB1
ABS
GGM2
GGM12
ANR1
FLC
AGL19
SOC1
DAL3
OsMADS1
AGL6
AGL13
AGL3
SEP3
SEP1
SEP2
OsMADS14
SQUA
FUL
AP1
CAL
FBP11
ZAG2
AG
CyAG
GGM3
DAL2
FARINELLI
SHP1
SHP2
AGL12
GGM10
AGL16
AGL17
AGL21
AP3
DEF
GLO
PI
STMADS11
AGL24
IbMADS4
STMADS16
IbMADS3
MPF2
MPP3
MPP4
PtMADS1
RcMADS1
JOINTLESS
PkMADS1
SVP
BM1
OsMADS47
OsMADS55
BM10
OsMADS22
STMADS11 clade
©M
rEnt
Figure 4.6. Phylogenetic tree of MADS-box proteins. This consensusphylogenetic tree was generated via parsimony analysis using TNT version 1.0,with a data set based on the conserved MADS-box domain of approximately 60amino acids. RcMADS1 is found to be nested within the StMADS11 clade (shownby arrow).

43
completed construct was sequenced to verify that the RcMADS1 gene was correctly
inserted into the vector.
4.6.2. Transgenic Arabidopsis thaliana T1 phenotypes
Transgenic Arabidopsis thaliana plants were obtained using the floral dip
method (see Section 3.13) (Clough and Bent, 1998). Seeds produced after floral
dipping were sown and germinated, and seedlings were subjected to 205 mg/l Basta®
selection. For the T1 generation, over 30 transgenic plants were generated after Basta®
screening (Table 4.1), with three distinctive phenotypes observed.
Seven of the T1 plants had a ‘strong’ phenotype where some homeotic
conversion of floral organs were observed: sepals and petals were converted to leaf-
like structures bearing conspicuous trichomes. Additionally, the development of the
carpel was abnormal. Instead of developing into a silique after self-fertilisation, the
gynophore elongated and the carpel itself usually split and developed an inflorescence.
Most of the time, siliques were not formed, and the plants did not produce viable seed.
Six plants had a ‘weak’ phenotype where full homeotic conversion of floral
organs did not occur, unlike in the ‘strong’ phenotype, but there were some alterations
to the development of the floral organs. The sepals became more leaf-like. Both
sepals and petals were persistent, not abscising when the flower grew older. Similar to
those in the ‘strong’ phenotype, the carpels did not develop normally into siliques
most of the time, and viable seed was not produced. This phenotype is similar to what
was observed for the attenuated phenotype with only bract-like sepals in 35S::AGL24
hemizygotes (Yu et al., 2004).
The remaining 18 plants generated after Basta® screening of the T1 seeds had a
‘normal’ phenotype where the phenotype was similar to that of wild-type plants.

44
Table 4.1. Phenotype analysis of T1 transgenic plants generated
Phenotype Morphology Number of plants generated
Mean number of rosette leaves formed before bolting
Normal Flowers look like wild type. 18 11.7 ± 1.7
Weak Sepals become leaf-like, persistent, not abscising when silique matures.
6 10.8 ± 2.6
Strong Sepals and petals become leaf-like, with presence of trichomes; abnormal carpel development; iterative inflorescence formation
7 11.7 ± 1.7

45
Most of the plants showing altered phenotype (especially with severely
deformed carpels) did not produce viable seeds. Hence, seeds from only 17
independent transgenic lines harbouring 35S::RcMADS1 were recovered.
Due to the ‘strong’ phenotype exhibited by some of the T1 plants which bear
some similarities to AGL24 overexpression in Arabidopsis thaliana plants (Yu et al.,
2002), some lines were chosen for further analysis in the T2 generation and beyond.
4.6.3. 35S::RcMADS1 effects on flowering time
Some transgenic Arabidopsis lines harbouring 35S::RcMADS1 were selected
for an analysis of the effect of the transgene on flowering time (Table 4.2). Lines
ETL01, ETL04, ETL05, and ETL09 had earlier exhibited no significant changes in
flower morphology in the T1 generation, while ETL06 has shown some mild changes
in floral morphology (i.e. the ‘weak’ phenotype), and ETL12 and ETL14 are
transgenic lines with total conversion of floral organs into leaf-like structures (i.e. the
‘strong’ phenotype). The T2 plants show a correlation in terms of flowering time.
Lines ETL01, ETL04, ETL05, and ETL09 had similar flowering time to wild-type
plants, with 10–11 rosette leaves before bolting. ETL06 (a ‘weak’ phenotype line)
segregated into a normal-looking phenotype, and a ‘weak’ phenotype with mild floral
organ homeosis, and the flowering time was lowered to a mean of 8.2 ± 1.2 and
5.5 ± 0.7 rosette leaves, respectively, before bolting. ETL12 (a ‘strong’ phenotype
line) segregated into a normal-looking phenotype, and a ‘strong’ phenotype exhibiting
total strong floral organ homeosis, and the flowering time was a mean of 10.4 ± 1.9
and 7.4 ± 1.4 rosette leaves before bolting. ETL14 (another ‘strong’ phenotype line)
only produced ‘strong’ phenotype plants in the T2 generation, and the flowering time
was 6.5 ± 0.9 leaves before bolting. These results suggest that an earlier flowering
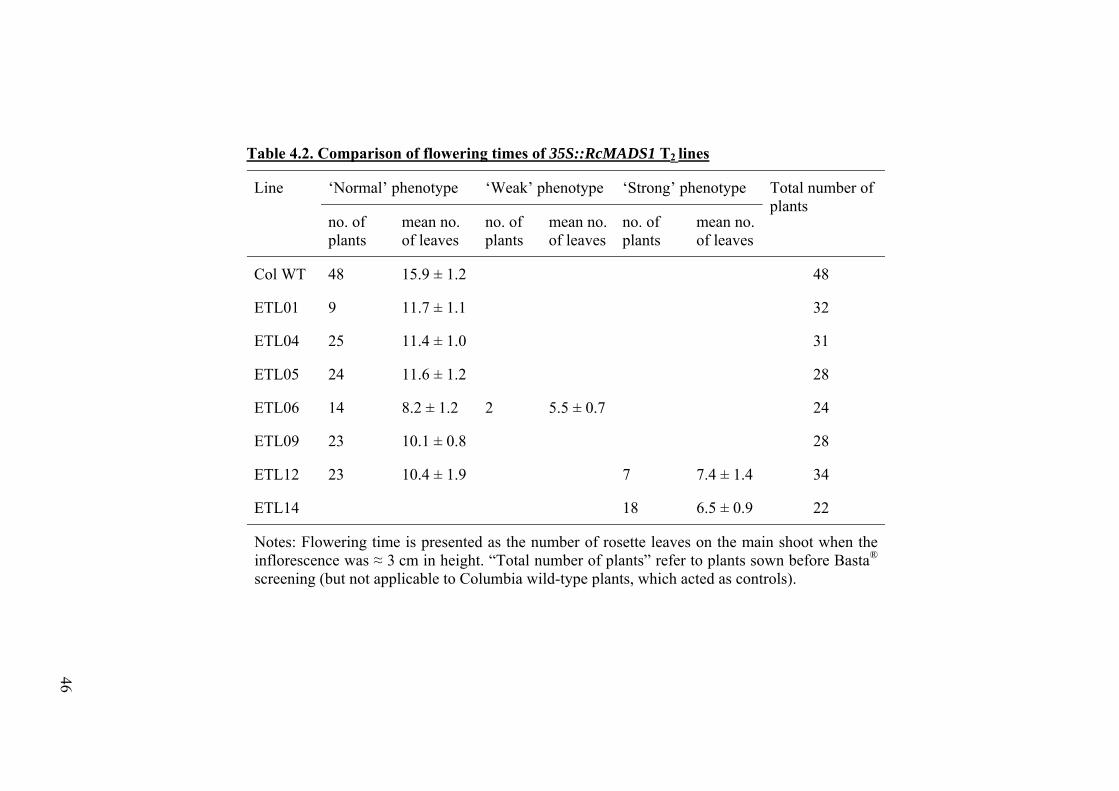
46
Table 4.2. Comparison of flowering times of 35S::RcMADS1 T2 lines
Line ‘Normal’ phenotype ‘Weak’ phenotype ‘Strong’ phenotype Total number of plants
no. of plants
mean no. of leaves
no. of plants
mean no. of leaves
no. of plants
mean no. of leaves
Col WT 48 15.9 ± 1.2 48
ETL01 9 11.7 ± 1.1 32
ETL04 25 11.4 ± 1.0 31
ETL05 24 11.6 ± 1.2 28
ETL06 14 8.2 ± 1.2 2 5.5 ± 0.7 24
ETL09 23 10.1 ± 0.8 28
ETL12 23 10.4 ± 1.9 7 7.4 ± 1.4 34
ETL14 18 6.5 ± 0.9 22
Notes: Flowering time is presented as the number of rosette leaves on the main shoot when the inflorescence was ≈ 3 cm in height. “Total number of plants” refer to plants sown before Basta® screening (but not applicable to Columbia wild-type plants, which acted as controls).

47
time was associated with floral organ homeosis. Lines that had a transgene insert
(conferring resistance to Basta®) but no discernible difference in phenotype had
flowering times similar to that of wild-type plants. Lines that showed observable
changes in floral morphology had distinctly earlier flowering times (as seen in ETL06,
ETL12, and ETL14).
4.6.4. Floral morphology in 35S::RcMADS1 transgenic lines
The morphology of the transgenic lines that showed the ‘strong’ phenotype
were further studied in the T2 and T3 generations, alongside two lines that expressed
the ‘strong’ phenotype only in the T2 generation and beyond. In the T1 generation, the
lines ETL10 and ETL17 did not express any discernible changes in floral morphology,
but expressed floral homeosis only in the T2 generation.
As a comparison, ETL01, a wild-type-looking line was also studied (Figure
4.7). The habit, floral morphology, and silique development of ETL01 all closely
resembled that of wild-type plants.
In the ‘strong’ phenotype, changes in floral morphology were drastic in the
lines observed, namely ETL10, ETL12 (Figure 4.8), ETL14 (Figure 4.9), and ETL17.
Both sepals and petals were converted into leaf-like structures, with the second whorl
losing the petalloid appearance, and developed conspicuous trichomes typically seen
in leaves (Figure 4.10). Secondary inflorescences developed from the axils of the
whorls. The carpel developed abnormally, with an elongation of the gynophore, and
silique formation was aborted. Secondary inflorescences were observed developing
from the aborted silique (Figure 4.11). In some plants of ETL14, the development of
the inflorescence proceeded directly from the fourth whorl, and bypassed the
intermediate step of carpel formation and development (see Figure 4.8B).

48
A B
D E
C
F
Figure 4.7. Phenotype of wild-type-looking transgenic line ETL01. (A)Flower, showing typical whorls, resembling wild-type flower. (B) Habit. (C)Comparison of the habits of wild-type plant (left) and ETL01 transgenic plant(right). (D) and (E) Branch of plant showing an inflorescence with wild-type-looking flowers. (F) Comparison of the inflorescences and flowers of wild-typeplant (left) and ETL01 transgenic plant (right).

49
A B
D E
C
F
Figure 4.8. Phenotype of strong transgenic line ETL12. (A) Flower, showingconversion of sepals and petals into leaf-like structures. (B) Habit. (C)Comparison of the habits of wild-type plant (left) and ETL12 transgenic plant(right). ETL12 plants display more profuse branching. (D) Branch of ETL12transgenic plant. (E) Comparison of branches of wild-type plant (left) and ETL12transgenic plant (right). (F) Asecondary inflorescence developing from an abortedcarpel.
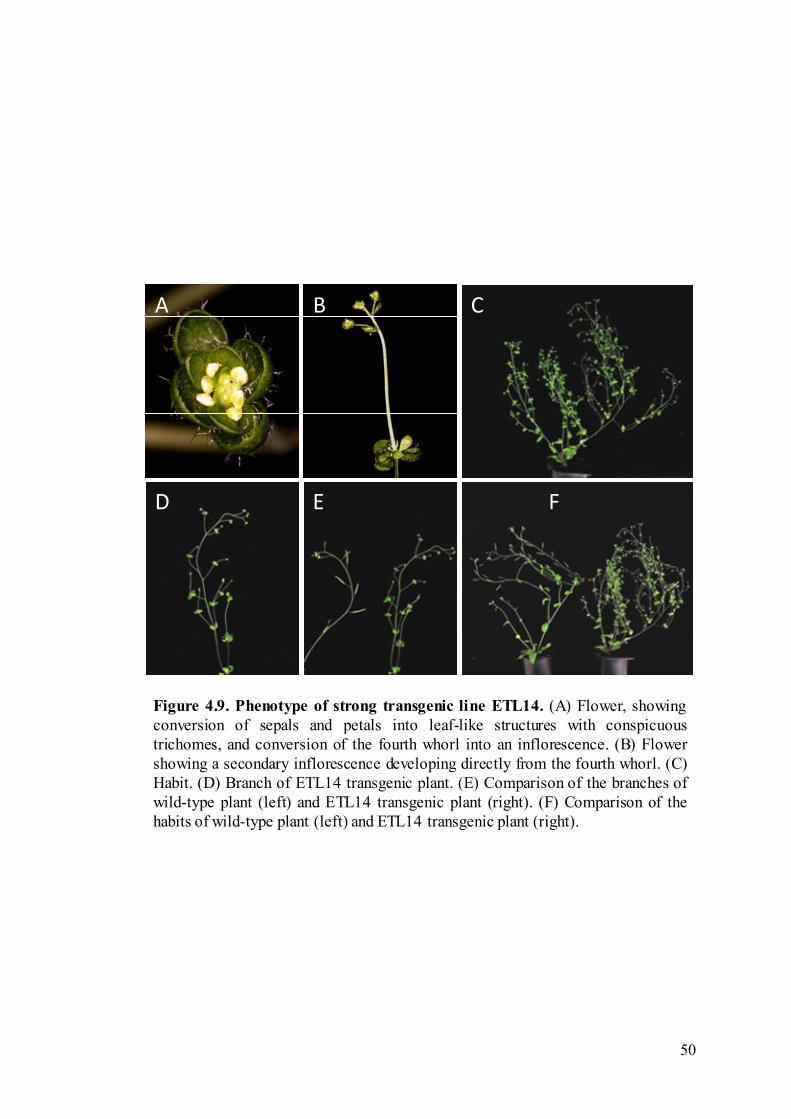
50
A B C
D E F
Figure 4.9. Phenotype of strong transgenic line ETL14. (A) Flower, showingconversion of sepals and petals into leaf-like structures with conspicuoustrichomes, and conversion of the fourth whorl into an inflorescence. (B) Flowershowing a secondary inflorescence developing directly from the fourth whorl. (C)Habit. (D) Branch of ETL14 transgenic plant. (E) Comparison of the branches ofwild-type plant (left) and ETL14 transgenic plant (right). (F) Comparison of thehabits of wild-type plant (left) and ETL14 transgenic plant (right).
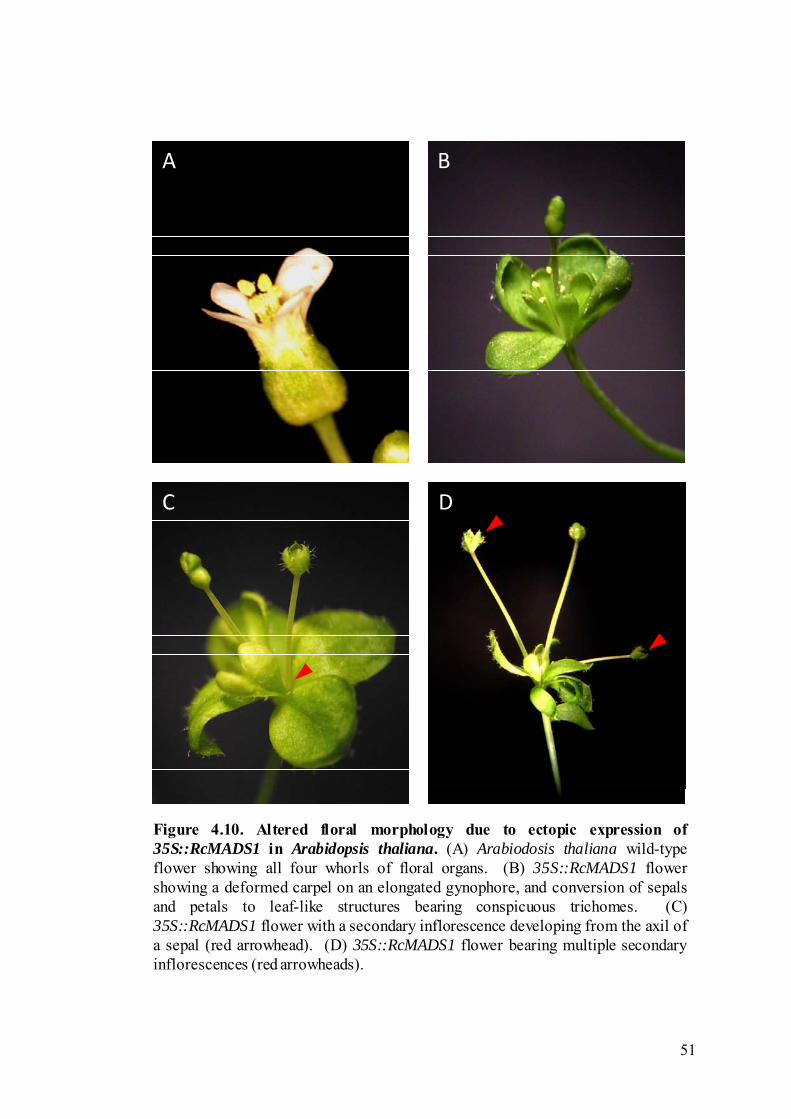
51
A B
C D
Figure 4.10. Altered floral morphology due to ectopic expression of35S::RcMADS1 in Arabidopsis thaliana. (A) Arabiodosis thaliana wild-typeflower showing all four whorls of floral organs. (B) 35S::RcMADS1 flowershowing a deformed carpel on an elongated gynophore, and conversion of sepalsand petals to leaf-like structures bearing conspicuous trichomes. (C)35S::RcMADS1 flower with a secondary inflorescence developing from the axil ofa sepal (red arrowhead). (D) 35S::RcMADS1 flower bearing multiple secondaryinflorescences (red arrowheads).
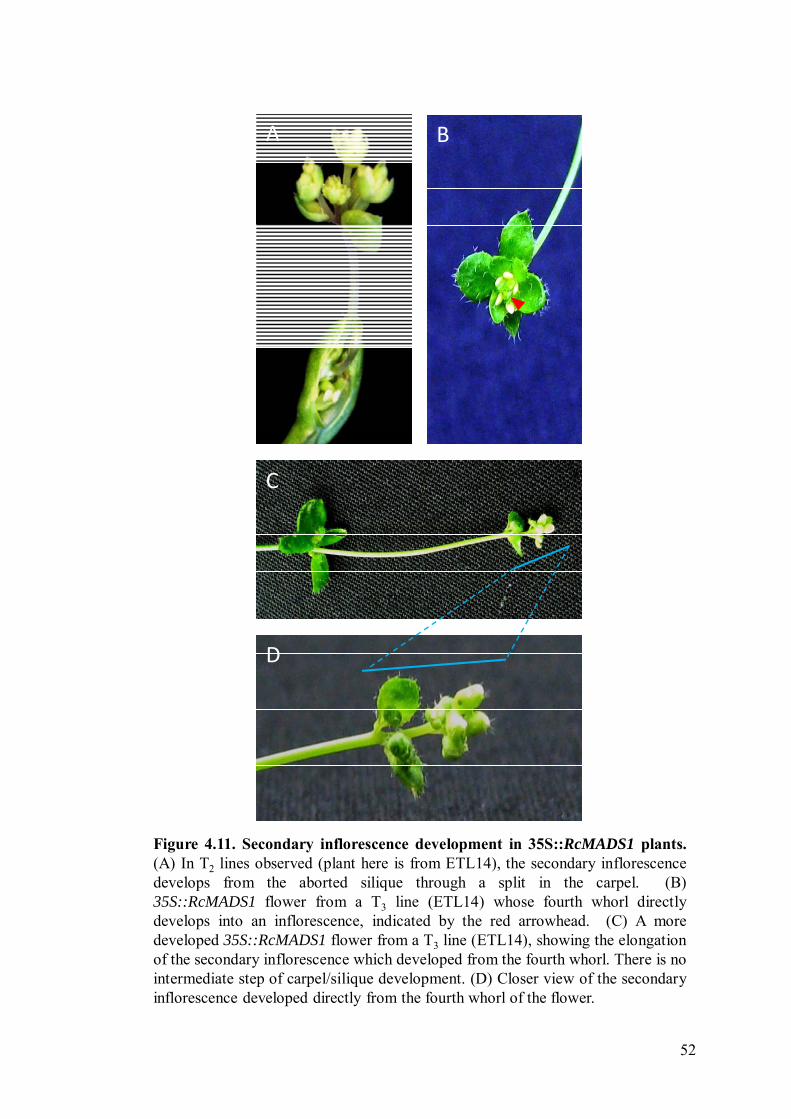
52
A
D
C
B
Figure 4.11. Secondary inflorescence development in 35S::RcMADS1 plants.(A) In T2 lines observed (plant here is from ETL14), the secondary inflorescencedevelops from the aborted silique through a split in the carpel. (B)35S::RcMADS1 flower from a T3 line (ETL14) whose fourth whorl directlydevelops into an inflorescence, indicated by the red arrowhead. (C) A moredeveloped 35S::RcMADS1 flower from a T3 line (ETL14), showing the elongationof the secondary inflorescence which developed from the fourth whorl. There is nointermediate step of carpel/silique development. (D) Closer view of the secondaryinflorescence developed directly from the fourth whorl of the flower.

53
Due to the iterative inflorescence development, transgenic lines harbouring the
35S::RcMADS1 displayed rather profuse branching habits (Figure 4.12). Secondary
inflorescences would develop from the axils of the fourth whorl or from
carpels/gynophores, which would in turn give rise to further inflorescences
developing from the abnormal flowers on the secondary inflorescences.
4.7. Molecular characterisation of selected transgenic lines
4.7.1. Genomic Southern blot analysis
All T2 transgenic Arabidopsis thaliana lines analysed in the genomic Southern
blot were found to have two insertions of the 35S::RcMADS1 transgene (Figure 4.13).
The use of two different restriction endonucleases in the analysis revealed that the
insertions were in different loci and that the transgenic lines were independently
generated. This indicates that the changes in flowering time and morphology observed
were due to the effect of the transgene and not likely to be due to a direct disruption of
native gene function by the insertions.
4.7.2. Quantitative real-time PCR analysis
To show that the transgene RcMADS1 is expressed in the various transgenic
lines, we performed quantitative real-time PCR and compared its expression across
the different transgenic lines (Figure 4.14). Five transgenic lines (ETL01, ETL10,
ETL12, ETL14, and ETL17) were selected. ETL01 and ETL10 are transgenic lines
that are wild-type-looking, with no discernible changes in floral morphology or in
flowering time. ETL12, ETL14, and ETL17 are transgenic lines showing a ‘strong’
phenotype, with severe altered floral morphology and early flowering. All lines
examined showed increases in expression levels of RcMADS1, indicating successful

54
A
B C
Figure 4.12. Development of profuse branching. (A) Wild-typeArabidopsis thaliana on the left, 35S::RcMADS1 transgenic line on the right. Notethe more profuse branching in the transgenic plant. (B) General habit of wild-typeArabidopsis thaliana (left) and 35S::RcMADS1 transgenic line (right). (C) Branchfrom a 35S::RcMADS1 transgenic plant, showing development of secondaryinflorescences from an aborted carpel (indicated by red arrowhead), andsubsequent abnormal development of the carpels in the flowers on the secondaryinflorescences.
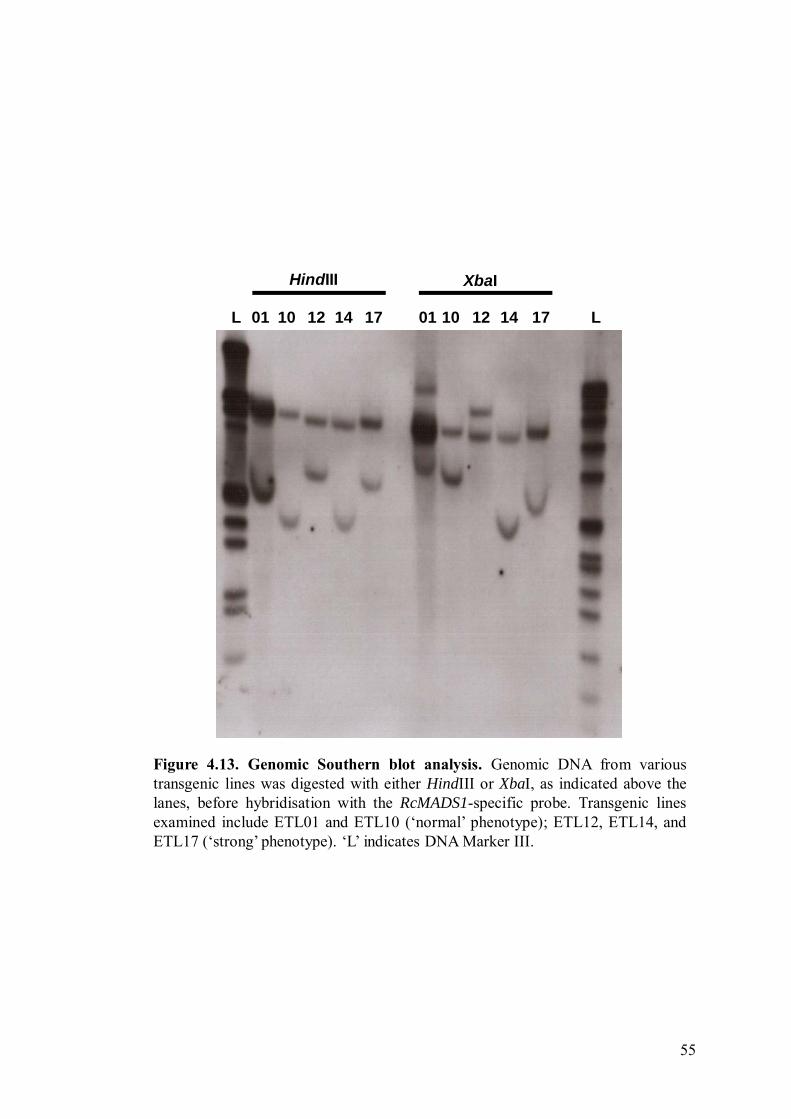
55
1401 10L L12 17 01 10 12 14 17
HindIII XbaI
Figure 4.13. Genomic Southern blot analysis. Genomic DNA from varioustransgenic lines was digested with either HindIII or XbaI, as indicated above thelanes, before hybridisation with the RcMADS1-specific probe. Transgenic linesexamined include ETL01 and ETL10 (‘normal’ phenotype); ETL12, ETL14, andETL17 (‘strong’ phenotype). ‘L’ indicates DNA Marker III.
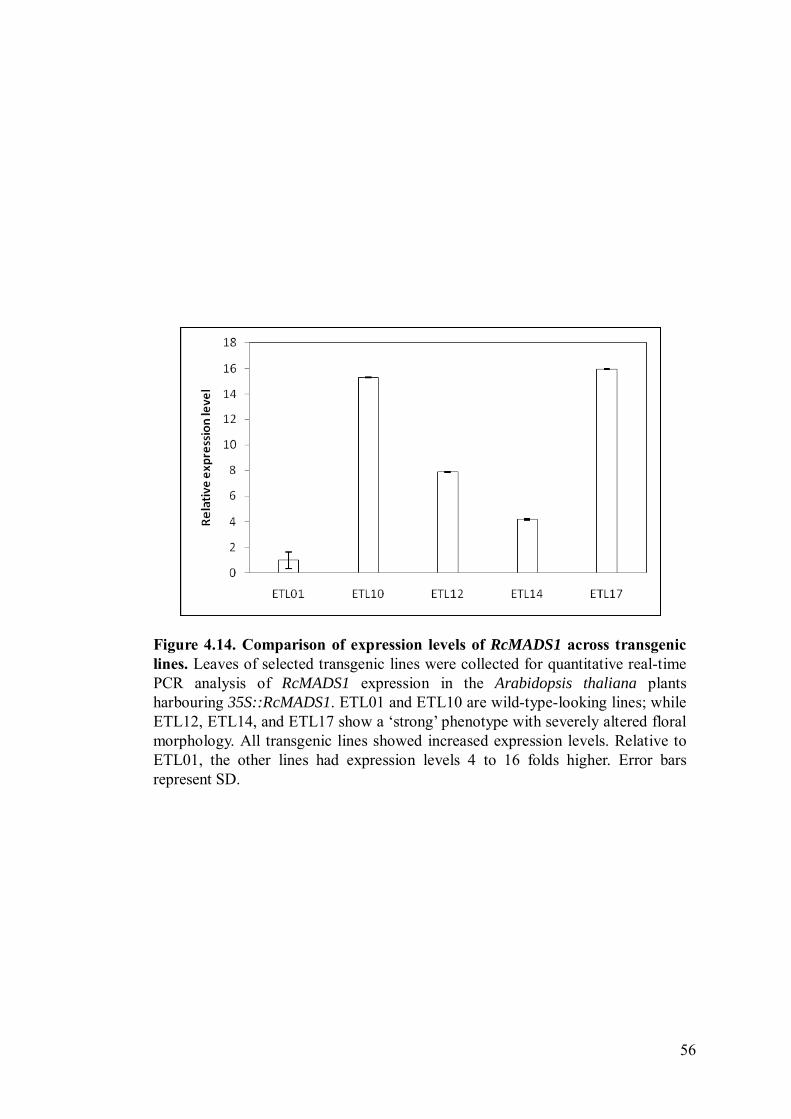
56
Figure 4.14. Comparison of expression levels of RcMADS1 across transgeniclines. Leaves of selected transgenic lines were collected for quantitative real-timePCR analysis of RcMADS1 expression in the Arabidopsis thaliana plantsharbouring 35S::RcMADS1. ETL01 and ETL10 are wild-type-looking lines; whileETL12, ETL14, and ETL17 show a ‘strong’ phenotype with severely altered floralmorphology. All transgenic lines showed increased expression levels. Relative toETL01, the other lines had expression levels 4 to 16 folds higher. Error barsrepresent SD.

57
insertion and expression in these transgenic lines. Using ETL01 as the control, the
other transgenic lines showed expression levels 4 to 16 folds higher, after
normalisation. This shows that increased levels of RcMADS1 may have an effect on
floral development.
In Arabidopsis thaliana, AGL24 and SVP regulate and are regulated by a host
of genes in the control of floral meristem identity. These genes include AP1, CO, FLC,
FT, LFY, SOC1, and SEP3. To examine the effect of ectopic expression of RcMADS1
in Arabidopsis thaliana and elucidate the putative function of this gene, we performed
quantitative real-time PCR to compare the expression of all these genes that
RcMADS1 may interact with: AGL24, AP1, CO, FLC, FT, LFY, SEP3, SOC1, and
SVP. This preliminary survey showed that FLC was upregulated (Figure 4.15A) while
FT was downregulated (Figure 4.15B) in transgenic Arabidopsis lines displaying the
‘strong’ phenotype (i.e. ETL12, ETL14, and ETL17), whereas gene expression of
FLC and FT in wild-type-looking transgenic lines (ETL01 and ETL10) were not
significantly different from that in Columbia wild-type plants. All other genes studied
did not show any changes in expression levels.
A decrease in FT expression levels would most probably be due to an increase
in FLC expression levels, since FLC acts as a repressor of FT (Michaels et al., 2003).
In Arabidopsis thaliana, FLC acts in tandem with SVP to repress FT expression,
which would otherwise activate SOC1 and AP1 expression (Liu et al., 2009a), and
FLC itself also directly represses SOC1 expression. SOC1 and AGL24 directly
upregulate the expression of one another, and together form a protein complex which
activates LFY. AGL24 therefore directly and indirectly upregulates SOC1 and LFY
respectively, but the data did not show increases in SOC1 and LFY expression levels,
indicating that RcMADS1 does not quite behave like AGL24 in regulating native

58
Figure 4.15. Effect of RcMADS1 ectopic expression on FLC and FT. Leaves ofselected transgenic lines were collected for quantitative real-time PCR analysis ofgenes that may be affected by RcMADS1 ectopic expression. (A) FLC expressionhad increased 8 to 12 folds in transgenic lines displaying severe alterations infloral morphology (ETL12, ETL14, and ETL17), while remaining relativelyunchanged in transgenic lines that are wild-type-looking (ETL01 and ETL10). (B)FT expression was contrastingly decreased, about 2 to 5 folds compared toColumbia wild type, in ETL12, ETL14, and ETL17; FT expression levels wererelatively unchanged in ETL01 and ETL10. Error bars represent SD.
A
B

59
Arabidopsis thaliana genes downstream of AGL24, as might be expected of a putative
homologue of AGL24. While an increased level of SVP (or its homologue) may
explain the increase of FT through the action of the SVP–FLC complex, but it does
not explain the increased expression of FLC itself.
The respective changes in gene expression of FLC and FT were both
correlated to increased expression of RcMADS1 in the ‘strong’ transgenic lines,
indicating that RcMADS1 acts in a dosage-dependent manner, much like AGL24.
However, since the material used were leaves harvested after bolting had occurred,
not much conclusive data can be gained. Almost all of the pertinent interactions occur
in the shoot apical meristems during floral transition (Liu et al., 2009a).
AGL24 does not appear to upregulate FLC natively in Arabidopsis thaliana
(Michaels et al., 2003), whereas RcMADS1 does, as shown in the quantitative real-
time PCR data (Figure 4.15). While RcMADS1 and AGL24 are similar in amino acid
sequence, and in phenotypic effect during ectopic expression or overexpression in
Arabidopsis thaliana, it seems possible that RcMADS1 may interact with protein
partners in a manner rather different from AGL24. This could also indicate a different
native function in Rafflesia, despite the similarities in phenotypes between
35S::AGL24 and 35S::RcMADS1 when ectopically expressed in Arabidopsis thaliana.

60
CHAPTER 5
GENERAL DISCUSSION AND FUTURE WORK
5.1. RcMADS1 may be involved in regulation of flowering time
In Arabidopsis thaliana, AGL24 is an integrator of flowering signals leading
to a precise regulation of floral meristem specification (Liu et al., 2009a). RcMADS1,
a putative homologue of AGL24 from Rafflesia cantleyi may have such a function,
similar to genes from other species in the StMADS11 clade, such as INCO from
Antirrhinum majus (Masiero et al., 2004), BM1 from Hordeum vulgare (Trevaskis et
al., 2007), and OsMADS22 and OsMADS47 from Oryza sativa (Fornara et al., 2008).
Ectopic expression of RcMADS1 resulted in early flowering and floral
reversion, with altered floral morphology that are largely similar to results obtained in
similar studies overexpressing AGL24 in Arabidopsis thaliana (Yu et al., 2002; Yu et
al., 2004). Moreover, the effects of ectopic expression of RcMADS1 is dosage-
dependent, as is the case for AGL24 (Yu et al., 2002). This suggests RcMADS1 is a
functional homologue of AGL24 in Rafflesia cantleyi.
However, the preliminary quantitative real-time PCR data suggests that there
are some differences between RcMADS1 and AGL24 when interacting with
intermediates in the regulatory network of AGL24 in Arabidopsis thaliana. AGL24
has not been known to upregulate FLC. Other than that, there is no enough data for
quantitative real-time PCR at this stage to draw more definite conclusions.
StMADS11-like genes are quite diverse in function. Besides regulating
flowering time and specification and maintenance of floral meristems in Arabidopsis
thaliana (Liu et al., 2009a), there are StMADS11-like genes with other functions in
development. For example, PkMADS1 is involved in shoot induction and phyllotaxy

61
in Paulownia kawakamii (Prakash and Kumar, 2002); MPF2-like genes are involved
in development of the inflated calyx syndrome in Solanaceae (Hu and Saedler, 2007);
OsMADS22, OsMADS47, and OsMADS55 are involved in regulation of
brassinosteroid responses in Oryza sativa (Fornara et al., 2008). The study of the rice
StMADS11-like genes suggested that heterologous overexpression of StMADS11-like
genes in Arabidopsis thaliana causes the same phenotypes as that of overexpression
of AGL24 and SVP genes. This could be due to inappropriate protein–protein
interactions that disturb normal flower development (Fornara et al., 2008), and is no
indication of the real function of such genes in the native non-Arabidopsis thaliana
plants. This finding was underscored by the fact that the StMADS11-like genes from
rice did not complement agl24 and svp mutants (Fornara et al., 2008). While the
phenotypes observed from transgenic plants harbouring 35S::RcMADS1 are shown to
be similar to those overexpressing AGL24, and shows that RcMADS1 is a StMADS11-
like gene with possibly similar function to AGL24, it is not conclusive evidence for
the actual native function in Rafflesia cantleyi since all StMADS11-like genes cause
similar floral reversions when overexpressed in Arabidopsis thaliana.
5.2. Temporal and spatial expression of RcMADS1 in Rafflesia cantleyi
Ideally, there has to be more information on when, where, and how RcMADS1
is expressed natively. This would allow us to have a better understanding of the
possible mechanisms of regulation RcMADS1 may be controlling in Rafflesia cantleyi.
Due to technical difficulties in isolating good quality RNA and DNA in appreciable
quantities, it was not possible to obtain data on temporal and spatial expression
patterns of RcMADS1. Additionally, it would be difficult to study the stage before
floral transition, given that Rafflesia is holoparasitic — the vegetative tissues are

62
threadlike and embedded within the host plant tissues, and therefore are difficult to
isolate.
5.3. Future work
The quantitative real-time PCR data for the effects of ectopically expressing
RcMADS1 in Arabidopsis thaliana is quite preliminary in nature, using leaf tissue
after bolting had occurred. For a clearer picture of how RcMADS1 interacts with other
flowering time genes and floral development genes, changes in gene expression levels
of target genes should be studied during the floral transition stage. This would allow
more appropriate examination of the respective changes of various members of the
regulatory network involved in integrating flowering signals, forming the
inflorescence meristems, and in acquiring and maintaining floral meristem identity.
Protein–protein interaction studies can also be carried out to elucidate the differences
(in interaction with protein partners) between AGL24 and RcMADS1.
More importantly, temporal and spatial expression patterns of RcMADS1 (and
possibly other MADS-box genes identified from Rafflesia) should be studied, in order
to better understand the role and function in Rafflesia itself.
5.4. Conclusions
Here, we report the cloning and characterisation of a MADS-box gene from
Rafflesia cantleyi. This cDNA has been named RcMADS1. The full-length cDNA was
cloned using a degenerate PCR approach. RcMADS1 shows high sequence similarity
to several MADS-box genes, in particular SVP and AGL24, all of which belong to the
StMADS11 clade. Ectopic expression of this gene (35S::RcMADS1) in a heterologous
system, namely Arabidopsis thaliana, resulted in a floral phenotype similar to that of

63
35S::AGL24. Quantitative real-time PCR analysis of selected target genes in the
regulatory network of AGL24 and SVP were performed in transgenic Arabidopsis
thaliana harbouring 35S::RcMADS1. To the best of our knowledge, this is the first
report of cloning and partial functional characterisation of a floral pathway gene from
Rafflesia.

64
References Alvarez-Buylla, E. R., S. Pelaz, S. J. Liljegren, S. E. Gold, G. Burgeff, G. S. Ditta,
L. Ribas de Pouplana, L. Martinez-Castilla, and M. F. Yanofsky. 2000. An
ancestral MADS-box gene duplication occurred before the divergence of
plants and animals. Proceedings of the National Academy of Sciences of the
United States of America 97: 5328–5333.
Angenent, G. C., J. Franken, M. Busscher, A. van Dijken, J. L. van Went, H. J. M.
Dons, A. J. van Tunen. 1995. A novel class of MADS box genes is involved in
ovule development in petunia.. The Plant Cell 7: 1569–1582.
Barcelona, J. F., P. B. Pelser, D. S. Balate, and L. L. Co. 2009. Taxonomy, ecology,
and conservation status of Philippine Rafflesia (Rafflesiaceae). Blumea 54:
77–93.
Barkman, T. J., S.-H. Lim, K. Mat Salleh, and J. Nais. 2004. Mitochondrial DNA
sequences reveal photosynthetic relatives of Rafflesia, the world’s largest
flower. Proceedings of the National Academy of Sciences of the United States
of America 101: 787–792.
Beaman, R. S., P. J. Decker, and J. H. Beaman. 1988. Pollination of Rafflesia
(Rafflesiaceae). American Journal of Botany 75: 1148–1162.
Becker, A., and G. Theissen. 2003. The major clades of MADS-box genes and their
role in the developmen and evolution of flowering plants. Molecular
Phylogenetics and Evolution 29: 464–489.
Brill, E. M., and J. M. Watson. 2004. Ectopic expression of a Eucalyptus grandis SVP
orthologue alters the flowering time of Arabidopsis thaliana. Functional Plant
Biology 31: 217–224.
Cho, S., S. Jang, S. Chae, K. M. Chung, Y. Moon, G. An, and S. K. Jang. 1999.
Analysis of the C-terminal region of Arabidopsis thaliana APETALA1 as a
transcription activation domain . Plant Molecular Biology 40: 419–429.
Clough, S. J., and A. F. Bent. 1998. Floral dip: a simplified method for
Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant
Journal 16: 735–743.
Coen, E. S., and E. M. Meyerowitz. 1991. War of the whorls: genetic interactions
controlling flower development. Nature: 353: 31–37.

65
Colombo, L., J. Franken, E. Koetje, J. van Went, H. J. M. Dons, G. C. Angenent, and
A. J. van Tunen. 1995. The petunia MADS box gene FBPI7 determines ovule
identity. The Plant Cell 7: 1859–1868.
Davis, C. C., M. Latvis, D. L. Nickrent, K. J. Wurdack, and D. A. Baum. 2007. Floral
gigantism in Rafflesiaceae. Science 315: 1812.
Di Stilio, V. S., E. M. Kramer, and D. A. Baum. 2005. Floral MADS box genes and
homeotic gender dimorphism in Thalictrum dioicum (Ranunculaceae) — a
new model for the study of dioecy. The Plant Journal 41: 755–766.
Ditta, G., A. Pinyopich, P. Robles, S. Pelaz, and M. F. Yanofsky. 2004. The SEP4
gene of Arabidopsis thaliana functions in floral organs and meristem identity.
Current Biology 14: 1935–1940.
Doyle, J. J., and J. L. Doyle. 1987. A rapid DNA isolation from small amount of fresh
leaf tissue. Phytochemical Bulletin 19: 11–15.
Egea-Cortines, M., H. Saedler, and H. Sommer. 1999. Ternary complex formation
between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA
is involved in the control of floral architecture in Antirrhinum majus. The
EMBO Journal 18: 5370–5379.
Fan, H. Y., Y. Hu, M. Tudor, and H. Ma. 1997. Specific interactions between the K
domains of AG and AGLs, members of the MADS domain family of DNA
binding proteins. The Plant Journal 12: 999–1010.
Favaro, R., A. Pinyopich, R. Battaglia, M. Kooiker, L. Borghi, G. Ditta, M. F.
Yanofsky, M. M. Kater, and L. Colombo. 2003. MADS-box protein
complexes control carpel and ovule development in Arabidopsis. The Plant
Cell 15: 2603–2611.
Fornara, F., V. Gregis, N. Pelucchi, L. Colombo, and M. Kater. 2008. The rice
StMADS11-like genes OsMADS22 and OsMADS47 cause floral reversions in
Arabidopsis without complementing the svp and agl24 mutants. Journal of
Experimental Botany 59: 2181–2190.
Goloboff, P., S. Farris, and K. Nixon. 2000. T.N.T. Tree Analysis Using New
Technology (BETA). ver. 1.0. Published by the authors, Tucumán, Argentina.
Haughn, G. W., and C. R. Somerville. 1988. Genetic control of morphogenesis in
Arabidopsis. Developmental Genetics 9: 73–89.
Honma, T., and K. Goto. 2001. Complexes of MADS-box proteins are sufficient to
convert leaves into floral organs. Nature 409: 525–529.

66
Hu, J.-Y., and H. Saedler. 2007. Evolution of the inflated calyx syndrome in
Solanaceae. Molecular Biology and Evolution 24: 2443–2453.
Huang, H., M. Tudor, T. Su, Y. Zhang, and H. Ma. 1996. DNA binding properties of
two Arabidopsis MADS doma proteins: binding consensus and dimer
formation. Plant Cell 8: 81–94.
Huang, K., J. M. Louis, L. Donaldson, F. L. Lim, A. D. Sharrocks, and G. M. Clore.
2000. Solution structure of the MEF2A-DNA complex: structural basis for the
modulation of DNA bending and specificity by MADS-box transcription
factors. EMBO Journal 19: 2615–2628.
Jack, T. 2001. Relearning our ABCs: new twists on an old model. Trends in Plant
Science 6: 310–316.
Jager, M., A. Hassanin, M. Manuel, H. Le Guyader, and J. Deutsch. 2003. MADS-
box genes in Ginkgo biloba and the evolution of the AGAMOUS family.
Molecular Biology and Evolution 20: 842–854.
Kaufmann, K., R. Melzer, and G. Theissen. 2005. MIKC-type MADS-domain
proteins: structural modularity, protein interactions and network evolution in
land plants. Gene 347: 183–198.
Kim, S., M.-J. Yoo, V. A. Albert, J. S. Farris, P. S. Soltis, and D. E. Soltis. 2004.
Phylogeny and diversification of B-function MADS-box genes in angiosperms:
evolution and functional implications of a 260 million-year-old duplication.
American Journal of Botany 91: 2102–2118.
Kramer, E. M., R. L. Dorit, and V. F. Irish. 1998. Molecular evolution of genes
controlling petal and stamen development: duplication and divergence within
the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics 149:
765–783.
Krizek, B. A., and J. C. Fletcher. 2005. Molecular mechanisms of flower development:
an armchair guide. Nature Reviews Genetics 6: 688–698.
Kuijt, J. 1969. The biology of parasitic flowering plants. University of California
Press: Berkeley.
Li, D., C. Liu, L. Shen, Y. Wu, H. Chen, M. Robertson, C. A. Helliwell, T. Ito, E.
Meyerowitz, and H. Yu. 2008. A repressor complex governs the integration of
flowering signals in Arabidopsis. Developmental Cell 15: 110–120.

67
Liu, C., H. Chen, H. L. Er, H. M. Soo, P. P. Kumar, J.-H. Han, Y. C. Liou, and H. Yu.
2008. Direct interaction of AGL24 and SOC1 integrates flowering signals in
Arabidopsis. Development 135: 1481–1491.
Liu, C., Z. Thong, and H. Yu. 2009a. Coming into bloom: the specification of floral
meristems. Development 136: 3379–3391.
Liu, C., W. Xi, L. Shen, C. Tan, and H. Yu. 2009b. Regulation of floral patterning by
flowering time genes. Developmental Cell 16: 711–722.
Liu, L., M. J. White, and T. H. MacRae. 1999. Transcription factors and their genes in
higher plants: functional domains, evolution and regulation. European Journal
of Biochemistry 262: 247–257.
Lodhi, M. A., G.-N. Ye, N. F. Weeden, and B. I. Reisch. 1994. A simple and efficient
method for DNA extraction from grapevine cultivars and Vitis species.
Martin, C., and J. Paz-Ares. 1997. MYB transcription factors in plants. Trends in
Genetics 13: 67–73.
Masiero, S., M. A. Li, I. Will, U. Hartmann, H. Saedler, P. Huijser, Z. Schwartz-
Sommer, and H. Sommer. 2004. INCOMPOSITA: a MADS-box gene
controlling prophyll development and floral meristems identity in Antirrhinum.
Development 131: 5981–5990.
Meijer, W. 1997. Rafflesiaceae, in Flora Malesiana I, 13: 1–42.
Messenguy, F., and E. Dubois. 1993. Genetic evidence for a role for MCM1 in the
regulation of arginine metabolism in Saccharomyces cerevisiae. Molecular
and Cellular Biology 13: 2586–2592.
Messenguy, F., and E. Dubois. 2003. Role of MADS box proteins and their cofactors
in combinatorial control of gene expression and cell development. Gene
316: 1–21.
Michaels, S. D., G. Ditta, C. Gustafson-Brown, S. Pelaz, M. Yanofsky, and R. M.
Amasino. 2003. AGL24 acts as a promoter of flowering in Arabidopsis and is
postively regulated by vernalization. The Plant Journal 33: 867–874.
Nam, J., C. W. dePamphilis, H. Ma, and M. Nei. 2003. Antiquity and evolution of the
MADS-box gene family controlling flower development in plants. Molecular
Biology and Evolution 20: 1435–1447.
Ng, M., and M. F. Yanofsky. 2001. Function and evolution of the plant MADS-box
gene family. Nature Reviews Genetics 2: 186–195.

68
Nickrent, D. L., and E. M. C. Starr. 1994. High rates of nucleotide substitution in
nuclear small subunit (18S) rDNA from holoparasitic flowering plants.
Journal of Molecular Evolution 39: 62–70.
Nickrent, D. L., R. J. Duff, and D. A. Konings. 1997. Structural analyses of plastid-
derived 16S rRNAs in holoparasitic angiosperms. Plant Molecular Biology 34:
731–743.
Norman, C., M. Runswick, R. Pollock, and R. Treisman. 1988. Isolation and
properties of cDNA clones encoding SRF, a transcription factor that binds to
the c-fos serum response element. Cell 55: 989–1003.
Pabo, C. O., and R. T. Sauer. 1992. Transcription factors: structural families and
principles of DNA recognition. Annual Review of Biochemistry 61: 1053–1095.
Passmore, S., R. Elble, and B. K. Tye. 1989. A protein involved in minichromosome
maintenance in yeast binds a transcriptional enhancer conserved in eukaryotes.
Genes and Development 3: 921–935.
Pelaz, S., G. S. Ditta, E. Baumann, E. Wisman. and M. F. Yanofsky. 2000. B and C
floral organ identity functions require SEPATALLA MADS-box genes. Nature
405: 200–203.
Pelaz, S., C. Gustafson-Brown, S. E. Kohalmi, W. L. Crosby, and M. F. Yanofsky.
2001. APETALA1 and SEPALLATA3 interact to promote flower development.
The Plant Journal 26: 385–394.
Pellegrini, L., S. Tan, and T. J. Richmond. 1995. Structure of serum response factor
core bound to DNA. Nature 376: 490–498.
Prakash, A. P., and P. P. Kumar. 2002. PkMADS1 is a novel MADS box gene
regulating adventitious shoot induction and vegetative shoot development in
Paulownia kawakamii. The Plant Journal 29: 141–151.
Riechmann, J. L., B. A. Krizek, and E. M. Meyerowitz. 1996a. Dimerization
specificity of Arabidopsis MADS domain homoeotic proteins APETALA1,
APETALA3, PISTILLATA, and AGAMOUS. Proceedings of the National
Academy of Sciences of the United States of America 93: 4793–4798.
Riechmann, J. L., M. Wang, and E. M. Meyerowitz. 1996b. DNA-binding properties
of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3,
PISTILLATA and AGAMOUS. Nucleic Acids Research 24: 3134–3141.

69
Schwarz-Sommer, Z,. P. Huijser, W. Nacken, H. Saedler, and H. Sommer. 1990.
Genetic control of flower development by homeotic genes in Antirrhinum
majus. Science 250: 931–936.
Shore, P., and A. D. Sharrocks, 1995. The MADS-box family of transcription factors.
European Journal of Biochemistry 229: 1–13.
Sommer, H., J.-P. Beltrán, P. Huijser, H. Pape, W.-E. Lönnig, H. Saedler, and Z.
Schwarz-Sommer. 1990. Deficiens, a homeotic gene involved in the control of
flower morphogenesis in Antirrhinum majus: the protein shows homology to
transcription factors. EMBO Journal 9: 605–613.
Soltis, D. E., P. S. Soltis, V. A. Albert, D. G. Oppenheimer, C. W. dePamphilis, H.
Ma, M. W. Frohlich, and G. Theissen. 2002. Missing links: the genetic
architecture of flower and floral diversification. Trends in Plant Science 7: 22–
31.
Soltis, D. E., H. Ma, M. W. Frohlich, P. S. Soltis, V. A. Albert, D. G. Oppenheimer,
N. S. Altman, C. dePamphilis, and J. Leebens-Mack. 2007. The floral genome:
an evolutionary history of gene duplication and shifting patterns of gene
expression. Trends in Plant Science 12: 358–367.
Theissen, G., J. T. Kim, and H. Saedler. 1996. Classification and phylogeny of the
MADS-box multigene family suggest defined roles of MADS-box gene
subfamilies in the morphological evolution of eukaryotes. Journal of
Molecular Evolution 43: 484–516.
Theissen, G., A. Becker, A. Di Rosa, A. Kanno, J. T. Kim, T. Münster, K.-U. Winter,
and H. Saedler. 2000. A short history of MADS-box genes in plants. Plant
Molecular Biology 42: 115–149.
Theissen, G. 2001. Development of floral organ identity: stories from the MADS
house. Current Opinion in Plant Biology 4: 75–85.
Theissen, G., and Saedler H. (2001) Floral quartets. Nature, 409: 469-471.
Trevaskis, B., M. Tadege, M. N. Hemming, W. J. Peacock, E. S. Dennis, and C.
Sheldon. 2007. Short vegetative phase-like MADS-box genes inhibit floral
meristems identity in barley. Plant Physiology 143: 225–235.
Weigel, D., and E. M. Meyerowitz. 1994. The ABCs of floral homeotic genes. Cell
78: 203–209.

70
Yanofsky, M. F., H. Ma, J. L. Bowman, G. N. Drews, K. A. Feldmann, and E. M.
Meyerowitz. 1990. The protein encoded by Arabidopsis homeotic gene
agamous resembles transcription factors. Nature 346: 35–39.
Yu, H., Y. Xu, E. L. Tan, and P. P. Kumar. 2002. AGAMOUS-LIKE 24, a dosage-
dependent mediator of the flowering signals. Proceedings of the National
Academy of Sciences of the United States of America 99: 16336–16341.
Yu, H., T. Ito, F. Wellmer, and E. M. Meyerowitz. 2004. Repression of AGAMOUS-
LIKE 24 is a crucial step in promoting flower development. Nature Genetics
36: 157–161.
Yu, Y.-T., R. E. Breitbart, L. B. Smoot, Y. Lee, V. Mahdavi, and B. Nadal-Ginard.
1992. Human myocyte-specific enhancer factor 2 comprises a group of tissue-
restricted MADS box transcription factors. Genes and Development 6: 1783–
1798.
Zhang, P., H. T. Tan, K. H. Pwee, and P. P. Kumar. 2004. Conservation of class C
function of floral organ development during 300 million years of evolution
from gymnosperms to angiosperms. The Plant Journal 37: 566–577.





























