



www.elsevier.com/locate/heares
Hearing Research 199 (2005) 22–30
Hearing loss associated with enlarged vestibular aqueduct andMondini dysplasia is caused by splice-site mutation in the PDS gene
Jiann-Jou Yang a,b, Chin-Chu Tsai a, Hsiu-Mei Hsu a, Jiun-Yih Shiao c,Ching-Chyuan Su b,d, Shuan-Yow Li a,*
a Genetics Laboratory and Department of Life Sciences, Chung Shan Medical University, No. 110, Sec. 1, Chien-Kuo N. Road,
Taichung 402, Taiwan, ROCb Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan, ROC
c Department of Otorhinolaryngology, Taichung VeteransGeneral Hospital, Taichung, Taiwan, ROCd Tian-Sheng Memorial Hospital, Tong Kang, Pin-Tong, Taiwan, ROC
Received 19 February 2004; accepted 9 August 2004
Available online 6 October 2004
Abstract
Recessive mutations of PDS gene are the common causes of Pendred syndrome and non-syndromic hearing loss associated with
temporal bone abnormalities ranging from isolated enlargement of the vestibular aqueduct (EVA) to Mondini dysplasia. In this
study we evaluate the relationship between EVA and Mondini dysplasia in 10 prelingual deaf patients and PDS gene mutation.
One of three mutations, IVS7 � 2A ! G, IVS16 � 6G ! A or IVS15 + 5G ! A, was identified in the PDS gene in each patient.
In family studies of four probands with the IVS7 � 2A! G mutation, we found that this mutation was inherited from the same
mutant alleles of parental origin. The effect of IVS7 � 2A !G mutation on PDS gene expression was determined by reverse tran-
scription and polymerase chain reaction (RT–PCR). Sequencing of the RT–PCR products revealed that the PDS transcripts from
the allele with IVS7 � 2A !G mutation lose the entire exon 8, resulting in a joining of exons 7 and 9. Deletion of the exon 8 results
in frameshift and premature termination of translation. Haplotype analysis showed a significant haplotype shared among the family
members carrying IVS7 � 2A ! G mutation, suggesting that they may be derived from a common ancestor. Our results provide
evidence that hearing loss with EVA and Mondini dysplasia may be caused by splice-site mutation in the PDS gene.
� 2004 Elsevier B.V. All rights reserved.
Keywords: PDS; EVA; Mondini dysplasia; Prelingual deafness; Pendrin syndrome; Hearing loss
1. Introduction
Hearing loss is a common sensory disorder. The inci-
dence of congenital hearing loss is estimated at 1 in
0378-5955/$ - see front matter � 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.heares.2004.08.007
Abbreviations: EVA, enlargement of the vestibular aqueduct; IVS,
intervening sequence; RT–PCR, reverse transcription and polymerase
chain reaction; DFNB, deafness, autosomal recessive; SSCP, single-
strand conformation polymorphism; PCR, polymerase chain reaction;
PTA, pure tone audiometry* Corresponding author. Tel.: +886 4 2473 0022x1800; fax: +886 4
2475 7412.
E-mail address: [email protected] (S.-Y. Li).
1000, with about an equal number of cases attributed
to environmental and genetic factors (Morton, 1991;
Gorlin, 1995). Environmental causes for hearing loss
are probably in decline as better therapies for bacterial
and viral infections (e.g. vaccines) are created and
acoustic trauma in the workplace is recognized and pre-vented. Ototoxic drugs (e.g. aminoglycosides) are now
also being avoided in genetically susceptible individuals
(Morton, 2002). Of the hearing loss disorders
attributable to genetic causes, nearly 70% are classified
as non-syndromic and the remaining 30% syndromic.
(Resendes, 2001).

J.-J. Yang et al. / Hearing Research 199 (2005) 22–30 23
Mutations of the PDS gene (MIM 605646) on chro-
mosome 7q21-34 can cause sensorineural hearing loss
with thyromegaly (Pendred syndrome, MIM 274600)
or non-syndromic hearing loss without goiter (DFNB4,
MIM 600791) (Everett et al., 1997; Li et al., 1998;
Campbell et al., 2001). PDS gene has 21 exons and con-tains an open reading frame of 2343 base pairs. The
PDS gene product, pendrin, is a highly hydrophobic
protein with 780 amino acid residues (molecular weight
86 kDa) and 11 putative transmembrane domains.
Expression of pendrin has been found in the non-sen-
sory epithelia of inner ears, thyroid folliculocytes, renal
cortical collecting ducts, placental trophoblasts and
uterine endometria (Everett et al., 1999; Royaux et al.,2000; Soleimani et al., 2001; Bidart et al., 2000; Suzuki
et al., 2002).
The hearing loss associated with PDS mutations
is thought to be related to temporal bone abnormal-
ities, ranging from isolated enlargement of vestibular
aqueduct (EVA) to Mondini dysplasia, a complex
malformation in which the normal cochlear spiral
runs of 2 1/2 turns is replaced by a hypoplastic coilof 1 1/2 turns. Both EVA and Mondini dysplasia
abnormalities can be diagnosed by either computed
tomography or magnetic resonance imaging (Phelps
et al., 1998).
In the Xenopus oocytes and Sf9 cells, pendrin has
been demonstrated to transport chloride and iodine,
and mediate the exchange of chloride and formate (Scott
et al., 1999). PDS mutations have been shown in vitro tocause the disruption of transmembrane anion/base ex-
change activity of pendrin (Scott et al., 2000). To date,
more than 66 different PDS gene mutations have been
described, and they have been found scattered all along
the 21 exons, including the flanking intronic sequences
and coding region (Tsukamoto et al., 2003).
Mutations in PDS gene have also been reported to be
associated with a wide range of phenotypes that are dif-ferent from the typical Pendred syndrome and non-syn-
dromic hearing loss with EVA (Tsukamoto et al., 2003;
Coyle et al., 1998; Van Hauwe et al., 1998; Lopez-Bigas
et al., 1999; Fugazzola et al., 2002). Therefore, identifi-
cation of PDS mutations can help elucidate the relation-
ship between mutant genotypes and phenotypes. In the
present study, we focused on whether the PDS muta-
tions were present in patients with EVA who have hear-ing loss and Mondini dysplasia. To determine the
correlation between phenotypic and genotypic data, we
have examined thePDS gene mutations in 10 prelingual
deaf patients with temporal bone abnormalities of both
EVA and Mondini dysplasia. Our data showed that all
of the patients we examined carried certain mutations
in the introns of PDS gene. Among these mutations,
six were found to be homozygous for a splicing acceptorsite in PDS gene that occurred at position
IVS7 � 2A ! G, one was found to be heterozygous
for IVS7 � 2 A ! G, two were found to be heterozy-
gous for IVS15 + 5G ! A and one heterozygous for
IVS16 � 6G! A. Our results provide evidence that
hearing loss associated with EVA and Mondini dyspla-
sia may be caused by the splice-site mutation in the
PDS gene.
2. Materials and methods
2.1. Subject selection
Ten prelingual deaf patients (from seven unrelated
families) aged between 4 and 13-years-old underwentPDS gene analysis. All 10 were found by computed
tomography of the temporal bone to have bilateral
EVA and bilateral Mondini dysplasia without goiter.
EVA was defined by an enlargement of the vestibular
aqueduct set at >1.5 mm midway between the endo-
lymphatic sac and the vestibule. Mondini dysplasia
was defined by a complex malformation in which the
normal cochlear spiral of 21/2 turns was replaced bya hypoplastic coil of 11/2 turns. Family studies were
performed on nine probands (four from unrelated fam-
ilies) with PDS mutations. Fifty randomly selected nor-
mal hearing individuals were included as control
subjects.
2.2. Detection of PDS mutation
Genomic DNA sample of each subject was ex-
tracted from whole blood using a QIAamp DNA
Blood Kit. (Qiagen) The quality and quantity of puri-
fied genomic DNA were determined by agarose gel
electrophoresis and spectrophotometry. Mutation
screenings were performed by single-strand conforma-
tion polymorphism (SSCP) and direct sequencing on
the PDS coding regions. PDS exons in DNA samplewere first amplified by polymerase chain reaction
(PCR) with intragenic primers as previously described
(Everett et al., 1997). In brief, PCR was conducted in
a volume of 25 ll which contained 100 ng of genomic
DNA, 200 lM dNTP, 0.25 units of proTaq DNA
polymerase (Promega), 200 lM intragenic primers
and 4% DMSO. The PCR products were purified
using a PCR Purification Kit (Qiagen). Reaction prod-ucts were resolved on a 8% non-denatured polyacryla-
mide gel containing 5%w/v glycerol and then
visualized by silver staining. Sequencing was per-
formed on an Applied Biosystems model 310 auto-
mated sequencer (Perkin–Elmer Corporation, Foster
City, CA). Sequence data were compared with the
published sequence of PDS gene using Sequencer 3.1
software program package (Perkin–Elmer).

24 J.-J. Yang et al. / Hearing Research 199 (2005) 22–30
2.3. mRNA extraction and reverse transcription
Total RNA was isolated from peripheral blood
lymphocytes using the Total RNA Extraction Miniprep
System (VIOGEN) according to manufacturer�s instruc-tion. cDNA was synthesized in a reaction of 20 ll whichcontained 2–5 lg RNA, random hexamer primer, and
200 units Improm-IITM Reverse Transcriptase (Promega)
according to the manufacturer�s instructions.
2.4. cDNA analysis
A fragment containing exons 7, 8 and 9 of the PDS
gene was amplified from cDNA by PCR with primersfrom the coding region (forward 5 0-gtgaggtacttggca-
gatcctt-3 0 and reverse 5 0-cctactgacactgcaatagc-3 0). PCR
reactions were performed in a 25 ll reaction mixture
which contained 1 mM Tris–HCl (pH 9.0), 5 mM
KCl, 150 lM MgCl2, 200 lM dNTP, 0.25 units pro
Taq DNA polymerase (Promega), 100 ng of cDNA,
and 200 lM forward and reverse primers. PCR products
were purified using Gel-MTM Gel Extraction System(VIOGEN). Predenaturation at 95 �C for 5 min, cycled
35 times through the following procedure: denaturation
at 95 �C for 30 s, annealing at 58 �C for 35 s, extension
at 72 �C for 45 s and a final extension step at 72 �C for 7
min. Direct automatic sequencing of PCR products was
performed with the forward and reverse primers using
an Applied Biosystems model 310 automated sequencer.
(Perkin–Elmer) Sequence data were compared as men-tioned above.
2.5. Haplotype analysis
Five polymorphic DNA markers covering the 7q
PDS regions (D7S2549, D7S2420, D7S496, D7S2459,
and D7S2456) were used in the haplotype analysis.
Information about sequence and amplification condi-tions of these markers were obtained from the CEPH-
Genethon set or Genome Data Base (Dib et al., 1996;
http://www.gdb.org). One hundred ng of genomic
DNA samples obtained from siblings and parents of
the study subjects were analyzed with PCR amplification
using fluorescence-tagged primers for each polymorphic
marker. Genotypes were scored semiautomately with an
ABI310 sequencer using GenescanTM/GenotyperTM
(Perkin–Elmer). The haplotype of each study subject
was reconstructed using the GENEHUNTER program
(Kruglyak et al., 1996).
3. Results
Ten prelingual deaf patients from seven unrelatedfamilies were found to have bilateral EVA and bilateral
Mondini dysplasia. These patients were found by com-
puted tomography to have temporal bones abnormali-
ties (Fig. 1) and by pure tone audiometry (PTA) to
have hearing loss to frequencies between 250 and 8000
Hz with a mean threshold of more than 62 dB in both
right and left ears using (Fig. 2).The patients were screened for the mutations in PDS
gene, which cover 21 exons including the flanking intro-
nic sequences and PDS coding region by PCR amplifica-
tion and SSCP. Abnormal migration of SSCP bands
were detected in all of 10 deaf patients (data not shown).
The nature of the abnormal migration was determined
by direct DNA sequence analysis. One of the three
mutations, IVS7 � 2A ! G, IVS16 � 6G ! A andIVS15 + 5G ! A, were identified in the PDS gene in
each patient. The observed mutations are summarized
in Table 1 and Fig. 3. These three mutations were not
detected in the 50 normal hearing control individuals.
Of the 10 patients, PDS gene from 6 patients were
found to be homozygous for a splice acceptor site muta-
tion that occurred at position IVS7 � 2A ! G (Fig.
3(b)), 1 heterozygous mutation for IVS7 � 2A ! G(Fig. 3(c)), 1 heterozygous mutation for IVS16 �6G! A (Fig. 3(e)), and 2 heterozygous for IVS15 +
5G! A (Fig. 3(g)). We also performed family studies
on nine probands with IVS7 � 2A! G mutation. All
were found to be inherited from the same mutant alleles
of their heterozygous parents who had normal hearing
ability (data not shown).
To study whether the PDS mutations (IVS7 �2A! G, or IVS16 � 6G ! A, or IVS15 + 5G! A)
had any effect on the mRNA expression, the levels of
PDS transcripts were determined by a combination of
reverse transcription and PCR (RT–PCR). In
IVS7 � 2A! G mutation, the RT–PCR product gener-
ated by primers located between exons 7 and 9 showed a
212 bp fragment, which is smaller than the 295 bp PCR
fragment obtained from a normal control (data notshown). Sequencing of the 212 bp PCR products re-
vealed that the PDS transcripts from the allele with
IVS7 � 2A! G mutation skipped exon 8 entirely,
resulting in a joining of exons 7 and 9 (Fig. 4(a) and
(b)). The deletion of exon 8 generated a new stop codon
at position 311, which might result in a premature trun-
cated protein of only 310 amino acids (Fig. 4(c)). How-
ever, we did not find any abnormal RT–PCR product bysize of sequence analysis of PDS gene from patients car-
rying mutations of either IVS16 � 6G ! A or IVS15 +
5G! A (data not shown).
Disease-associated haplotypes across the PDS gene
region at chromosome 7q were determined by typing
five microsatellites, including D7S2549, D7S2420,
D7S496, D7S2459, and D7S2456. IVS7 � 2A ! G
linked haplotypes of five microsatellites collected fromsiblings and parents in four unrelated families and ana-
lyzed to distinguish between founder effects and de
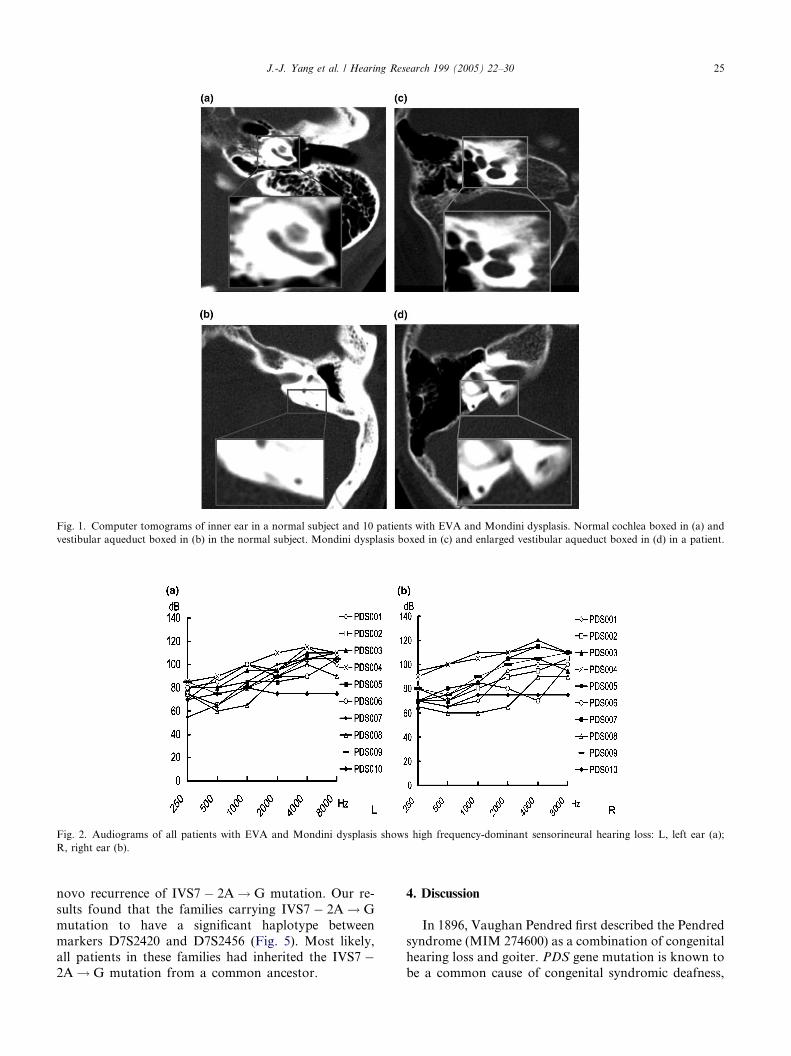
Fig. 1. Computer tomograms of inner ear in a normal subject and 10 patients with EVA and Mondini dysplasis. Normal cochlea boxed in (a) and
vestibular aqueduct boxed in (b) in the normal subject. Mondini dysplasis boxed in (c) and enlarged vestibular aqueduct boxed in (d) in a patient.
Fig. 2. Audiograms of all patients with EVA and Mondini dysplasis shows high frequency-dominant sensorineural hearing loss: L, left ear (a);
R, right ear (b).
J.-J. Yang et al. / Hearing Research 199 (2005) 22–30 25
novo recurrence of IVS7 � 2A! G mutation. Our re-
sults found that the families carrying IVS7 � 2A ! G
mutation to have a significant haplotype between
markers D7S2420 and D7S2456 (Fig. 5). Most likely,all patients in these families had inherited the IVS7 �2A ! G mutation from a common ancestor.
4. Discussion
In 1896, Vaughan Pendred first described the Pendred
syndrome (MIM 274600) as a combination of congenitalhearing loss and goiter. PDS gene mutation is known to
be a common cause of congenital syndromic deafness,

Table 1
Mutation in the 10 prelingual deaf patients with bilateral EVA and bilateral Mondini dysplasis
Number Genotype PTA (dB) Rt. PTA (dB) Lt.
PDS 001 IVS7 � 2A!G/IVS7 � 2A!G 77 95
PDS 002 IVS7 � 2A!G/IVS7 � 2A!G 80 80
PDS 003 IVS7 � 2A!G/IVS7 � 2A!G 86 90
PDS 004 IVS7 � 2A!G/IVS7 � 2A!G 105 100
PDS 005 IVS16 � 6G! A/wt 107 83
PDS 006 IVS7 � 2A!G/IVS7 � 2A!G 80 90
PDS 007 IVS15 + 5G ! A/wt 90 83
PDS 008 IVS15 + 5G ! A/wt 62 72
PDS 009 IVS7 � 2A!G/IVS7 � 2A!G 88 85
PDS 010 IVS7 � 2A!G/wt 72 78
PTA, pure tone audiometry; Rt., right ear ; Lt., left ear.
Fig. 3. Sequence analysis of PDS genomic DNA mutation in 10 prelingual deaf patients associated with bilateral EVA and bilateral Mondini
dysplasis. Genomic sequence of the PDS gene from normal individuals (a), (d), and (f). A splice acceptor site mutation occurrs at position
IVS7 � 2A! G, including homozygous of IVS7 � 2A!G (b) and heterozygous of IVS7 � 2A! G. (c). Two of PDSmutations, IVS16 � 6G! A (e)
and IVS15 + 5G ! A (g), respectively are found to be heterozygous. Vertical arrows indicate changes of the nucleotide changed. wt: wild type.
26 J.-J. Yang et al. / Hearing Research 199 (2005) 22–30

Fig. 4. Sequence analysis of normal and IVS7 � 2A!G mutated PDS gene from RT–PCR. (a) The cDNA sequence of the region corresponds to
exons 7–9. Black bar indicates sequence of exon 8. (b) The IVS7 � 2A! G mutation in the cDNA result in deletion of exon 8. (c) Predicted protein
sequences coded from PDS gene of either normal or IVS7 � 2A !G mutation.
J.-J. Yang et al. / Hearing Research 199 (2005) 22–30 27
which accounts for 4–10% of congenital deafness in chil-
dren (Pendred, 1986; Gorlin, 1995). Park et al. (2003)
have shown that PDS gene mutations make up approx-
imately 5% of all the cases of prelingual deafness in east
Asia and 5% of recessive hearing loss in south Asia.
PDS gene product, Pendrin, is expressed in the endo-lymphatic duct and sac in the inner ear and regulates
endolymph secretion and resorption (Everett et al.,
1999). Therefore, Pendrin defects result in neuroepithel-
ial damage which in turn cause inner ear malformations
where the upper coils of the cochlea form a common
cavity (Mondini malformation) and the vestibular aque-
ducts are dilated (Johnsen et al., 1986; Phelps et al.,
1998). These inner ear anomalies lead to a generally pro-found sensorineural hearing loss, beginning prelingually
and having a mean threshold of more than 60 dB in
PTA as a mean threshold (Kabakkaya et al., 1993;
Fugazzola et al., 2002).
One previous genetic analysis has confirmed that
mutations in PDS cause a broader phenotypic spec-
trum, ranging from typical Pendred syndrome to
non-syndromic hearing loss associated with EVA(Usami et al., 1999). Campbell et al. (2001) have indi-
cated that mutations in PDS cause both Pendred syn-
drome and DFNB4. Pendred syndrome and non-
syndromic hearing loss can be differentiated clinically
by the association of goiter with the former, while
morphological abnormalities of inner ear, such as
EVA and Mondini dysplasia, are common to bothdiseases. Classically, hearing loss is congenital, and
thyromegaly is developed in the second decade (Phelps
et al., 1998; Campbell et al., 2001). Each of the 10 pa-
tients we studied exhibited severe prelingual hearing
loss with bilateral EVA and bilateral Mondini dyspla-
sia, but none of them had a goiter. Being between
4- and 13-years-old, they may have been at such an
earlier stage of disease that a had not yet developed.Another possibility is that the full expression of Pen-
dred syndrome requires gene modifications, environ-
mental factors, or a combination of these two
mechanisms. We will continue to follow these patients
into the second decade and monitor their phenotypical
changes. Whether these mutations, IVS7 � 2A ! G,
IVS16 � 6G! A and IVS15 + 5G ! A, predispose
people to the development of thyroid nodules remainsto be established.

Fig. 5. Haplotypes analysis of prelingual deaf patients with
IVS7 � 2AG mutation of PDS gene in four unrelated families. The
haplotypes linked to splice site mutation, IVS7 � 2A! G, are boxed.
x indicates crossover.
28 J.-J. Yang et al. / Hearing Research 199 (2005) 22–30
More than 66 different PDS gene mutations have
been described (Coyle et al., 1996; Everett et al., 1997;
Coucke et al., 1999; Yong et al., 2001; Tsukamoto
et al., 2003). Three frequent mutations, L236P, T416P,
and IVS8 + 1G ! A, are the most commonly reported
mutations in patients in the West, and they accountfor more than 50% of cases of Pendred syndrome (Van
Hauwe et al., 1998). Eleven mutations have been de-
tected in families with non-syndromic hearing loss asso-
ciated with EVA (Tsukamoto et al., 2003). Another
study from Japan has reported another mutation,
T410M, in non-syndromic hearing loss with EVA
(Kitamura et al., 2000). Park et al. (2003) found
H273R mutation to be a prevalent allele that could befound in a majority of PDS mutations in Korean and
Japanese populations. Nevertheless, we did not observe
any of the above mutations except for IVS7 � 2A! G.
IVS7 � 2A! G homozygous mutation was found in
6 out of the 10 patients examined in our study. This
mutation is also commonly found among different Asian
populations, such as Korean, Japanese and Chinese, but
it has not been observed in the Western populations
(Park et al., 2003). Our family studies found all ofIVS7 � 2A! G homozygous mutations to be inherited
from the same mutant allele of their IVS7 � 2A ! G
heterozygous parents, who had normal hearing. In our
family studies, we did not analyze all of the probands
because we had not collected DNA samples from
our patients. Surprisingly, we also detected the
IVS7 � 2A! G heterozygous mutation in one patient
(PDS 010) with hearing loss. Other mutations,IVS15 + 5G ! A and IVS16 � 6G ! A, represented
heterozygous mutations in which a second mutation
could not be identified in PDS gene. The significance
of IVS15 + 5GA and IVS16 � 6G ! A heterozygous
mutations is not clear but they do not seem to be com-
mon polymorphisms. Therefore, patients carrying the
heterozygous mutation are more likely to develop hear-
ing loss in the presence of additional genes or environ-mental factors.
The IVS7 � 2A ! G mutation, which changes the
conserved nucleotide of the acceptor splice site, most
probably affects the splicing of PDS gene (Krawczak
et al., 1992). Using RT–PCR, we proved that this
mutation caused the deletion of exon 8 entirely and
generated a truncated protein consisting of only 310
amino acids, a result similar to that reported forIVS8 � 4GA mutation of PDS gene, the skipping of
exon 9 of PDS gene (Massa et al., 2003). The patho-
genic potential of mutations on both IVS16 � 6G ! A
and IVS15 + 5G ! A is unknown since their effect on
splicing has not been determined. However, other
genes whose mutations occur at similar positions in
intronic regions have been found to cause various lev-
els of normal and aberrant transcripts, with the pres-ence of transcripts differing from one tissue to another
(Krawczak et al., 1992; Larriba et al., 1998). There-
fore, we presume these mutations may affect the
mRNA stability of PDS or normal protein function
of pendrin. Moreover, the possibility that the interac-
tion of PDS gene mutations with other genes resulted
in hearing loss cannot be ruled out. It is also possible
that these two nucleotide changes represent a rare oruncommon polymorphism not associated with the dis-
ease phenotype in these patients.
In haplotype analysis, we found that the patients car-
rying IVS7 � 2A ! G mutation shared the same haplo-
type. We suggest that IVS7 � 2A! G mutation may
have arisen from ancestral founder chromosomes. How-
ever, such haplotype has not been found to have any
link to IVS7 � 2A! G in different Asian populations(Korean, Chinese and Japanese) (Park et al., 2003).
We reason that IVS7 � 2A! G may be an older

J.-J. Yang et al. / Hearing Research 199 (2005) 22–30 29
founder mutation which has undergone ancestral recom-
bination events with the flanking marker.
Our evaluation of PDS mutations provide an impor-
tant base for improving the clinical diagnosis of deaf
patients with both EVA and Mondini dysplasia. More-
over, the fact that PDS gene is also expressed in thesyncytiotrophoblast cells of the placenta (Bidart et al.,
2000) shows that it may used in prenatal genetic analysis
in the future.
Acknowledgements
We thank all the subjects who participated in the pre-sent project. We also thank Chieh-Tien Shih for his
technical assistance. This work is supported by National
Science Council, ROC (NSC 91-2745-P-040-002, and
NSC 91-2320-B-040-015).
References
Bidart, J.M., Lacroix, L., Evain-Brion, D., Caillou, B., Lazar, V.,
Frydman, R., Bellet, D., Filetti, S., Schlumberger, M., 2000.
Expression of Na+/I� symporter and Pendred syndrome genes in
trophoblast cells. J. Clin. Endocrinol. Metab. 85, 4367–4372.
Campbell, C., Cucci, R.A., Prasad, A., Green, G.E., Edeal, J.B., Galer,
C.E., Karniski, L.P., Sheffield, V.C., Smith, R.J.H., 2001. Pendred
syndrome, DFNB4, and PDS/SLC26A4 identification of eight
novel mutation and possible genotype–phenotype correlations.
Hum. Mutat. 17, 403–411.
Coucke, P.J., Van Hauwe, P., Everett, L.A., 1999. Identification of two
different mutations in the PDS gene in an inbred family with
Pendred syndrome. J. Med. Genet. 36, 475–477.
Coyle, B., Coffey, R., Armour, J.A., Gausden, E., Hochberg, Z.,
Grossman, A., Britton, K., Pembrey, M., Reardon, W., Trembath,
R., 1996. Pendred syndrome (goiter and sensorineural hearing loss)
maps to chromosome 7 in the region containing the nonsyndromic
deafness gene DFNB4. Nat. Genet. 12, 423–426.
Coyle, B., Reardon, W., Herbrick, J.A., Tsui, L.C., Gausden, E., Lee,
J., Coffey, R., Grueters, A., Grossman, A., Phelps, P.D., Luxon,
L., Kendall-Taylor, P., Scherer, S.W., Trembath, R.C., 1998.
Molecular analysis of the PDS gene in Pendred Syndrome
(sensorineural hearing loss and goiter). Hum. Mol. Genet. 7,
1105–1112.
Dib, C., Faure, S., Fizames, C., Samnson, D., Drouot, N., Vignal, A.,
Millasseau, P., Marc, S., Hazan, J., Seboun, E., Lathrop, M.,
Gyapay, G.., Morissette, J., Weissenbach, J., 1996. A comprehen-
sive genetic map of the human genome based on 5264 microsat-
ellites. Nature 14, 152–154.
Everett, L.A., Glaser, B., Beck, J.C., Idol, J.R., Buchs, A., Heyman,
M., Adawi, F., Hazani, E., Nassir, E., Baxevanis, A., Sheffield,
V.C., Green, E.D., 1997. Pendred syndrome is cause by mutation in
a putative sulphate transporter gene (PDS). Nat. Genet. 17, 411.
Everett, L.A., Morsli, H., Wu, D.K., Green, E.D., 1999. Expression
pattern of the mouse ortholog of the Pendred�s syndrome gene
(Pds) suggests a key role for pendrin in the inner ear. Proc. Natl.
Acad. Sci. USA 969, 727–732.
Fugazzola, L., Cerutti, N., Mannavola, D., Crino, H., Cassio, H.,
Gasparoni, P., Vannucch, G., Beck-Peccoz, P., 2002. Differential
diagnosis between pendred and pseudo-pendred syndromes:
clinical, radiological and molecular studies. Pediatr. Res. 51, 479–
484.
Gorlin, R.J., 1995. Genetic hearing loss associated with endocrine and
metabolic disorders. In: Gorlin, R.J. (Ed.), Hereditary Hearing
Loss and Its Syndromes. Oxford University Press, New York, pp.
337–339.
Johnsen, T., Jorgensen, M.B., Johnsen, S., 1986. Mondini cochlea in
Pendred�s syndrome. A historical study. Acta Oto-Laryngol. 102,
239–247.
Kabakkaya, Y., Bakan, E., Yigitoglu, M.R., Gokce, G., Dogan, M.,
1993. Pendred�s syndrome. Ann. Oto-Laryngol. 102, 285–288.
Kitamura, K., Takahashi, K., Noguchi, Y., Kuroishikawa, Y.,
Tamagawa, Y., Ishikawa, K., Ichimura, K., Hagiwara, H., 2000.
Mutation of the Pendred syndrome gene (PDS) in patients with
large vestibular aqueduct. Acta Oto-Laryngol. 120, 137–141.
Krawczak, M., Reiss, J., Cooper, D.N., 1992. The mutational
spectrum of single base-pair substitutions in mRNA splice
junctions of human genes: causes and consequences. Hum. Genet.
90, 41–45.
Kruglyak, L., Daly, M.J., Reeve-Daly, M.P., Lander, E.S., 1996.
Parametric and nonparametric linkage analysis: a unified multi-
point approach. Am. J. Hum. Genet. 58, 1347–1363.
Larriba, S., Bassas, L., Gimenez, J., Ramos, M.D., Segura, A., Nunes,
V., Estivill, X., Casals, T., 1998. Testicular CFTR splice variants in
patients with congenital absence of the vas deferens. Hum. Mol.
Genet. 7, 1739–1744.
Li, X.C., Everett, L.A., Lalwani, A.K., Desmukh, D., Friedman, T.B.,
Green, E.D., Wilcox, E.R., 1998. A mutation in PDS causes non-
syndromic recessive deafness. Nat. Genet. 18, 215–217.
Lopez-Bigas, N., Rabionet, R., de Cid, R., Govea, N., Gasparini, P.,
Zelante, L., Arbones, M.L., Estivill, X., 1999. Splice-site mutation
in the PDS gene may result in intrafamilial variability for deafness
in Pendred syndrome. Hum. Mutat. 14, 520–526.
Massa, G., Jaenen, N., de Varebeke, S.J., Peeters, N., Wuyts, W.,
2003. Solitary thyroid nodule as presenting symptom of Pendred
syndrome caused by a novel splice-site mutation in intron 8 of the
SLC26A4 gene. Eur. J. Pediatr. 162, 674–677.
Morton, N.E., 1991. Genetic epidemiogy of hearing impairment. Ann.
N.Y. Acad. Sci. 630, 16–31.
Morton, C.C., 2002. Genetics, genomics and gene discovery in the
auditory system. Hum. Mol. Genet. 11, 1229–1240.
Park, H.J., Shaukat, S., Liu, X.Z., Hahn, S.H., Naz, S., Ghosh, M.,
Kim, H.N., Moon, S.K., Abe, S., Tukamoto, K., Riazuddin, S.,
Kabra, M., Erdenetungalag, R., Radnaabazar, J., Khan, S.,
Pandya, A., Usami, S.I.., Nance, W.E., Wilcox, E.R., Riazuddin,
S., Griffith, A.J., 2003. Origins and frequencies of SLC26A4 (PDS)
mutations in east and south Asians: global implications for the
epidemiology of deafness. J. Med. Genet. 40, 242–248.
Pendred, V., 1986. Deaf mutism and goiter. Lancet 2, 532.
Phelps, P.D., Coffey, R.A., Trembath, R.C., Luxon, L.M., Grossman,
A.B., Britton, K.E., Kendall-Taylor, P., Graham, J.M., Cadge,
B.C., Stephens, S.G.D., Pembrey, M.E., Reardon, W., 1998.
Radiological malformations of the ear in Pendred syndrome. Clin.
Radiol. 53, 268–273.
Resendes, B.L., Williamson, R.E., Morton, C.C., 2001. At the speed of
sound: gene discovery in the auditory system. Am. J. Hum. Genet.
69, 923–935.
Royaux, I.E., Suzuki, K., Mori, A., Katoh, R., Everett, L.A., Kohn,
L.D., Green, E.D., 2000. Pendrin, the protein encoded by the
Pendred syndrome gene (PDS), is an apical porter of iodide in the
thyroid and is regulated by thyroglobulin in FRTL-5 cells.
Endocrinology 141, 839–845.
Scott, D.A., Wang, R., Kreman, T.M., Sheffield, V.C., Karnishki,
L.P., 1999. The Pendred syndrome gene encodes a chloride-iodide
transport protein. Nat. Genet. 2, 440–443.
Scott, D.A., Wang, R., Kreman, T.M., Andrew, M., McDonald, J.M.,
Bishop, J.R., Smith, R.J.H., Karniski, L.P., Sheffield, V.C., 2000.
Functional differences of the PDS gene product are associated with
phenotypic variation in patients with Pendred syndrome and

30 J.-J. Yang et al. / Hearing Research 199 (2005) 22–30
non-syndromic hearing loss (DFB4). Hum. Mol. Genet. 9, 1709–
1715.
Soleimani, M., Greeley, T., Petrovic, S., Wang, Z., Amlal, H., Kopp,
P., Burnham, C.E., 2001. Pendrin: an apical Cl�/OH�/HCO3�
exchanger in the kidney cortex. Am. J. Physiol. Renal. Physiol. 280,
F356–F364.
Suzuki, K., Royaux, I.E., Everett, L.A., Mori-Aoki, A., Suzuki, S.,
Nakamura, K., Sakai, T., Katoh, R., Toda, S., Green, E.D., Kohn,
L.D., 2002. Expression of PDS/Pds, the Pendred syndrome gene, in
endometrium. J. Clin. Endocrinol. Metab. 987, 938.
Tsukamoto, K., Suzuki, H., Harada, D., Namba, A., Abe, S., Usami,
S., 2003. Distribution and frequencies of PDS (SLC26A4)
mutations in Pendred syndrome and nonsyndromic hearing
loss associated with enlarged vestibular aqueduct: a unique
spectrum of mutations in Japanese. Eur. J. Hum. Genet. 11, 916–
922.
Usami, S., Abe, S., Weston, M.D., Shinkawa, H., Camp, G.V.,
Kimberling, W.J., 1999. Non-syndromic hearing loss associated
with enlarged vestibular aqueduct is caused by PDS mutations.
Hum. Genet. 104, 188–192.
Van Hauwe, P., Everett, L.A., Coucke, P., Scott, D.A., Kraft, M.L.,
Ris-Stalpers, C., Bolder, C., Otten, B., de Vijider, J.J., Dietrich,
N.L., 1998. Two frequent missense mutations in Pendred syn-
drome. Hum. Mol. Genet. 7, 1099–1104.
Yong, A.M.L., Goh, S.S., Zhao, Y., Eng, P.H.K., Koh, L.K.H.,
Khoo, D.H.G., 2001. Two Chinese families with Pendred�ssyndrome- Radiological imaging of the ear and molecular analysis
of the Pendrin gene. J. Clin. Endocrinol. Metab. 86, 3907–3911.













![Aqueduct el[1]](https://static.cupdf.com/doc/110x72/557ea115d8b42ac5658b47e0/aqueduct-el1.jpg)





![Aqueduct en[1]](https://static.cupdf.com/doc/110x72/557e9f8ed8b42a1d048b535e/aqueduct-en1.jpg)










