



www.elsevier.com/locate/yabio
Analytical Biochemistry 362 (2007) 108–116
ANALYTICAL
BIOCHEMISTRY
Endotoxin detection in a competitive electrochemical assay: Synthesisof a suitable endotoxin conjugate
Graciela Priano, Diego Pallarola, Fernando Battaglini *
INQUIMAE—Departamento de Quımica Inorganica, Analıtica y Quımica Fısica Facultad de Ciencias Exactas y Naturales,
Universidad de Buenos Aires. Ciudad Universitaria, C1428EHA Buenos Aires, Argentina
Received 9 October 2006Available online 22 December 2006
Abstract
A biotin–lipopolysaccharide (biotin–LPS) conjugate was synthesized from LPS smooth from Salmonella minnesota, yielding a conju-gate with a biotin/LPS ratio equal to 1:1 and endotoxic activity of 0.08 EU ng�1. The conjugate was used in an amperometric competitiveassay to determine endotoxins with endotoxin-neutralizing protein (ENP) as the recognition element. The assay is performed on a mod-ified electrode, involving the covalent binding of carboxymethyl dextran (CMDex) to a cystamine-modified gold electrode and then thecovalent binding of the recognition protein, ENP, to CMDex. The assay is carried out by incubating the modified electrode in an LPSsample to which biotin–LPS was added. Both species compete for the recognition sites on the modified surface. After the incubation stageand a careful rinsing, the electrode is immersed in a solution containing neutravidin–horseradish peroxidase conjugate (N-HRP), whichbinds to the sites containing biotin–LPS on the electrode. The system is rinsed and a current signal is generated by the addition of hydro-gen peroxide and a redox mediator. The assay is able to detect LPS from Salmonella minnesota at concentrations as low as 0.1 ng ml�1,equivalent to 0.07 EU ml�1.� 2006 Elsevier Inc. All rights reserved.
Keywords: Lipopolysaccharide; Competitive assay; Biotin-conjugated lipopolysaccharide
Gram-negative sepsis is a common and serious clinicalproblem. The primary trigger in gram-negative shock syn-drome is due to the lipopolysaccharides (LPSs)1, a majorconstituent of the gram-negative bacteria outer membrane,also termed endotoxins. In events of gram-negative bacte-rial infection, LPS is the well-known activator of thehumoral and cellular components of the host defense sys-tem. Activation of the host defense is essential to fight suchan infection, but uncontrolled stimulation can result in
0003-2697/$ - see front matter � 2006 Elsevier Inc. All rights reserved.
doi:10.1016/j.ab.2006.12.034
* Corresponding author. Fax: +54 11 45763341.E-mail address: [email protected] (F. Battaglini).
1 Abbreviations used: LPS, lipopolysaccharide; ENP, endotoxin-neutral-izing protein; LBP, lipopolysaccharide-binding protein; LPS–HRP, horse-radish peroxidase-labeled lipopolysaccharide conjugate; CMDex,carboxymethyl dextran; N-HRP, neutravidin–horseradish peroxidaseconjugate; LALF, limulus anti-LPS factor; EDTA, ethylenediaminetetra-acetic acid; EDC, l-ethyl-3-[3-(dimethylamino)propyl]carbodiimide; NHS,N-hydroxysuccinimide.
excessive release of inflammatory cytokines, leading to sep-tic shock and death [1,2].
Several institutions have imposed regulations regardingthe maximum allowable presence of LPS. For example,U.S. Pharmacopoeia establishes a limit of 0.25 EU ml�1
for injectable drugs [3], and the Association for theAdvancement of Medical Instrumentation recommends alimit of 2 EU ml�1 for dialysis baths [4].
LPSs are complex lipid-linked carbohydrate negativelycharged molecules. Usually, they are composed of threedistinct regions: a fatty-acylated, highly conserved regioncalled lipid A; a short oligosaccharide, the core region;and an O-antigen portion composed of a polymer ofrepeating oligosaccharide units with a composition thatvaries greatly among gram-negative bacteria. Lipid A isresponsible for many of the pathophysiological effects asso-ciated with gram-negative bacterial infection; therefore, itis the active moiety of LPS [5]. It consists of a hydrophilic,negatively charged bisphosphorylated disaccharide of

Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116 109
glucosamine backbone covalently linked to a hydrophobicdomain of six (Escherichia coli) or seven (Salmonella) acylchains (12–16 carbon atoms) via amide and ester bonds[6–8]. Although LPS itself is chemically inert, the presenceof LPS in blood (endotoxemia) sets off a cascade of exag-gerated host responses affecting the structure and functionof organs and cells, changing metabolic functions, raisingbody temperature, modifying hemodynamics, and causingseptic shock [1].
Endotoxins are detected either by the use of amoebo-cytes from horseshoe crab (the LAL test) or by a live assayon rabbits. Both methods are expensive and troublesome.One of the main challenges currently posed by endotoxindetection is to develop a fast and simple method capableof, for example, performing on-line measurements in puri-fied water lines or in aqueous saline solutions used forhemodialysis or intravenous infusion.
During the past 10 years, great improvements have beenmade in the study of chemical interactions between LPSsand antibacterial agents produced by many organisms.Some of the systems that have been studied include endo-toxin-neutralizing protein (ENP) produced by horseshoecrabs [9], lipopolysaccharide-binding protein (LBP) fromhumans [10], and polymyxin produced by Bacillus poly-
myxia [11]. These findings allow the development of anassay equivalent to assays based on antigen–antibodyinteraction, where the selective recognition is combinedwith a physicochemical change that is used as a transducerelement generating a signal proportional to the concentra-tion of the analyte to be detected.
In a previous work [12], we proposed a competitive elec-trochemical assay using a modified gold electrode with arecombinant ENP from Saccharomyces cerevisiae as a rec-ognition element. Two strategies were used to immobilizethe protein onto the electrode: one based on electrostaticinteractions (electrostatic configuration) and the otherbased on trough covalent binding (covalent configuration).The test was carried out by competition of the LPS in thesample with a horseradish peroxidase-labeled lipopolysac-charide conjugate (LPS–HRP). Sensors constructedaccording to the electrostatic configuration were able todetect the presence of endotoxins in concentrations of0.2 EU ml�1, below the American Pharmacopoeia stan-dard for injectable drugs. Alternatively, the covalent con-figuration showed a broader dynamic range but a higherdetection limit (1.5 EU ml�1).
In spite of the promising results achieved, the systempresents some limitations. One of them is the surface cov-erage, estimated at 0.08 pmol cm�2 LPS–HRP on a cova-lently modified electrode Au/Cys/carboxymethyl dextran(CMDex)/ENP, whereas Limoges and coworkers reporteda surface coverage 50 times higher for a monolayer of neu-travidin–horseradish peroxidase conjugate (N-HRP) [13].Our results suggest a low affinity between the LPS–HRPconjugate and the modified electrode. Several factors havean effect on the amount of LPS–HRP incorporated ontothe electrode. Steric factors are detrimental due to the
molecular size of this conjugate given that it is synthesizedfrom LPS from E. coli 026:B6 [14]. Another factor to takeinto account is the low endotoxic activity of the LPS–HRPdetermined in the homogeneous phase through the LALtest (0.012 EU ng�1), compared with the typical valuesfor LPS (1 EU ng�1), because endotoxic activity correlateswith ENP affinity toward LPS [15].
To improve the characteristics of the conjugate neededfor a competitive assay, it is important that the biologicalproperties and the binding capacity of the LPS portionare not altered by means of the conjugation; on the otherhand, the introduced label needs to be present in a goodratio and to be stable.
Given the complexity of the molecule, the synthesis ofan LPS conjugate is not simple. Endotoxins in solutiondo not constitute a homogeneous sample, and at the con-centrations required for the synthesis aggregates of highmolecular weight are formed [16–18]. To preserve theactive portion of LPS, the covalent binding of low-molecu-lar weight probes to the O-antigenic portion of the LPS hasbeen carried out. The objective is more ambitious than theobjectives presented in previous reports for LPS conjugates[19,20]; in the current case, it is necessary that, on average,all of the LPS molecules are labeled to avoid losing sensi-tivity in the competitive assay.
By means of the controlled oxidation with sodium perio-date, reactive aldehydes could be generated on the saccha-ride portion of the LPS that later was reacted with themarkers through their nucleophile groups [21]. Taking intoaccount the well-developed biotin–avidin technology, aconjugated biotin–LPS was synthesized. The interactionbetween biotin and the protein avidin is one of the stron-gest noncovalent affinity interactions in nature(KD = 10�15 M) [22], and it has been used for a wide rangeof applications, including immunoassays, protein purifica-tion, and diagnostics [23]. The strong interaction isachieved without significant perturbation of the terciaryor quaternary structures of the protein and is stable overa wide range of pH values and temperatures [24]. Some-times, neutravidin is used instead of avidin. One major dif-ference between them is that avidin carries a positivecharge at neutral pH, whereas neutravidin is nearly neutral.
In this work, a biotin–LPS conjugate was synthesizedfrom a purified fraction of LPS from S. minnesota, yieldinga conjugate with improved endotoxic activity and appliedin our recently developed molecular recognition devicebased on ENP [12]. The assay is performed according tothe covalent configuration reported previously, Au/Cys/CMDex/ENP, which involves the covalent binding ofCMDex to a cystamine-modified gold electrode and thenthe covalent binding of the recognition protein (ENP) toCMDex (Scheme 1).
The steps involved in the assay are depicted in Scheme 2.A modified electrode Au/Cys/CMDex/ENP is incubated inan LPS sample to which biotin–LPS was added. Both spe-cies compete for the recognition sites on the modified sur-face. After the incubation stage and a careful rinsing, the

COOH
COOH
COOHHOOC
HOOC
HOOC
HOOC
Au/Cys/CMDex/ENP
+S
NH2
SNH2
COOH
COOH
COOHHOOC
HOOC
OC
Au/Cys/CMDex
SNH2
SNH
EDC
NHS
NH2
NH2
ENP
NH2
NH2NH
ENP
CO
COOH
COHOOC
HOOC
OC
HOOC
SNH2
SNH
NH2
NH2NH
ENPNH2
Scheme 1. Construction of the modified electrode used in this work. CMDex is immobilized by means of an amide bound to the surface of the goldelectrode modified with cystamine, and then ENP is incorporated by means of the same reaction.
N- HRPrinsing
rinsing
H2O2
H2O
ENP LPS Biotin-LPS N-HRP
Scheme 2. Competitive assay between LPS and biotin–LPS. N-HRP is used as a signal-generating element.
110 Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116
electrode is immersed in an N-HRP solution that binds tothe sites containing biotin–LPS on the electrode. The sys-tem is rinsed, and a current signal is generated by the addi-tion of hydrogen peroxide and a redox mediator, HRPsubstrate and cosubstrate. The generated signal will bedirectly proportional to the amount of biotin–LPS incor-porated onto the electrode and will be inversely propor-tional to the analyte present in the sample. Thecompetitive assay was tested for LPS R60 (Ra) fromS. minnesota, and it was able to detect concentrations aslow as 0.1 ng ml�1. This concentration corresponds to0.07 EU ml�1 according to our determination carried outwith the chromogenic LAL test. The results obtained withthis new conjugate represent an important improvement inthe signal generation and dynamic range of the assay.
Materials and methods
Reagents
LPS from S. minnesota was supplied by Sigma, and LPSR60 (Ra) from S. minnesota (MW 4200 Da, endotoxic
activity 0.7 EU ng�1, as determined in our laboratory bythe chromogenic LAL test) was provided by ListLab.(Note: LPS is pyrogen. It may cause fever, and it may beharmful by inhalation, ingestion, or skin absorption. Goodlaboratory techniques should be employed, i.e., wearing labcoat, gloves, and safety glasses. One should work in a well-ventilated area and avoid contact with open wounds.)
The chromogenic LAL test was provided by Biowhitaker.Lysozyme from chicken egg white and biotin–NHS wereprovided by Sigma. Biocytin hydrazide (biotin-LC-hydra-zide) and N-HRP (neutravidin/HRP = 1:1) were providedby Pierce. Residual peroxide test strips were provided bySerim. ENP was supplied by Associates of Cape Code (Sei-kagaku America); this protein is a recombinant version ofthe limulus anti-LPS factor (LALF, pI 8.5, MW 12.2 kDa,105 amino acid residues). Apyrogen water was produced bybidistillation of water distilled previously and passedthrough a MilliQ water system and was collected in apyro-gen glass material (5 h at 180 �C and 2 h at 220 �C); valuesbelow 0.01 EU ml�1 were obtained routinely (determinedby means of the LAL test). Cystamine dihydrochloride,neuraminidase, and galactose oxidase were provided by

Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116 111
Sigma. CMDex (MW 24,200) was obtained from Fluka.All other reagents were of analytical grade. The solubleredox mediator [Os(bpy)2(pyCOOH)Cl]+ (where bpy isbipyridine and pyCOOH is nicotinic acid) was synthesizedin our laboratory as reported previously [25].
Biotin–lysozyme was synthesized in our laboratory frombiotin–NHS and lysozyme [21]. The modified protein pre-sents a biotin/lysozyme ratio equal to 1, as determined bymeans of the test for quantification of biotin (EZ BiotinQuantitation Kit [Pierce]).
Synthesis of LPS conjugate
LPS purification
LPS smooth from S. minnesota was chromatographical-ly purified at room temperature with a column of SephacrylHiPrep 16/60 (S-200 HR, Amersham) in an AKTA Explor-er fast phase liquid chromatography system. As elutionbuffer, 10 mM Tris buffer (pH 8.0) containing 0.2 M NaCl,1 mM ethylenediaminetetraacetic acid (EDTA), 0.02%sodium azide, and 0.25% sodium deoxycholate was used.LPS (10–30 mg) was dissolved in 1 ml of buffer. The samplewas injected and eluted isocratically at 0.5 ml/min. Thenfractions containing LPS were dialyzed extensively against10 mM Tris buffer (pH 8.0), with the purpose of removingthe salts and the detergent, and then against distilledwater. The dialyzed fractions were lyophilized and storedat �20 �C.
Enzymatic treatment
The purified LPS was treated with neuraminidase andgalactose oxidase as reported previously [26]. Reactionprogress was determined by following the peroxide genera-tion with hydrogen peroxide test strips.
Periodate oxidation
To find the best conditions for the LPS conjugation,dansyl hydrazine (Invitrogen) was reacted with purifiedLPS by changing the NaIO4 concentration, the time reac-tion, and the temperature. The reaction yield was deter-mined by a fluorescence assay, exciting at 319 nm andfollowing emission at 499 nm. To compare the reactivityof the saccharide moieties present in LPS with other poly-saccharides, dextran (MW 69 kDa) was used as a reference.
Finally, the conjugation reaction was carried outbetween biotin-LC-hydrazide and purified LPS (endotoxicactivity 0.4 EU ng�1). LPS (2 mg) was dissolved in 4 ml of100 mM buffer acetic/acetate (pH 5) and was sonicated for20 min. Later, NaIO4 was added up to a final concentra-tion of 150 mM. The oxidation was carried out for 3 h atroom temperature and in the dark. The reaction wasstopped by the addition of ethylene glycol. After a dialysisprocess (MWCO 3500), 4 mg of biotin-LC-hydrazide(Pierce) was added to the oxidized LPS and the reactionmixture was incubated for 2 h at room temperature withagitation in the dark. Later, 250 ll of 450 mM NaCNBH3
was added and the reduction reaction was continued for
40 min. Finally, the conjugated biotin–LPS was dialyzedand lyophilized. The biotin/LPS ratio was determined bymeans of the test for quantitation of biotin, the EZ BiotinQuantitation Kit, and the LAL test was used to character-ize the conjugate endotoxic activity.
Electrochemical measurements
A standard three-electrode system was used togetherwith a purpose-built operational amplifier potentiostat(TEQ-02). The system consisted of a working electrode, aplatinum mesh counter electrode, and an Ag/AgCl refer-ence electrode. Working electrodes were freshly preparedfor every analysis.
Amperometric assays
The time-based measurements were performed at fixedelectrode potential, 50 mV versus Ag/AgCl. For all of theassays described below, the electrodes were carefully rinsedand introduced into the electrochemical cell containing0.2 M KNO3 and 50 mM Tris buffer solution (pH 7.5).[Os(bpy)2Cl(pyCOOH)]Cl was added to a final concentra-tion of 40 lM. The modified electrode was connected tothe potentiostat and left to equilibrate with the solution,and hydrogen peroxide was added to give a final concentra-tion of 1 mM. An immediate change in the current wasobserved and reached a constant value due to the cathodiccatalytic current produced, which was proportional to thesurface concentration of N-HRP. The current informedfor each concentration is the average of two independentexperiments unless otherwise stated.
Construction of modified electrodes
Gold flags (1 cm2) were used as electrodes. The gold sur-face was cleaned by immersion in 30% H2O2/98% H2SO4
solution (1:3, piranha solution) for 3 h (Note: This solutionis highly corrosive and reacts violently with organic mate-rials. Precautions must be taken at all times when handlingthis solution.) Clean electrodes were rinsed with MilliQwater, and the cleanliness of the surface was checked bycyclic voltammetry in 1.8 M H2SO4 between 0 and+1.6 V versus Ag/AgCl at 0.05 V s�1. When the cleaningstep was verified satisfactorily, the electrodes were readyto be modified.
Cystamine adsorption (Au/Cys)
Clean gold electrodes were immersed in 20 mM cysta-mine dihydrochloride in absolute ethanol/water (9:1) for2 h under smooth stirring. Fresh solutions were used ineach preparation.
Carboxymethyldextran modification (Au/Cys/CMDex)
CMDex was bound to cystamine through the formationof an amide bond. CMDex (10 mg ml�1) was dissolved in150 mM l-ethyl-3-[3-(dimethylamino)propyl]carbodiimide

112 Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116
(EDC) and 27 mM N-hydroxysuccinimide (NHS) solutionin 10 mM Pipes buffer (pH 6.5). Then Au/Cys electrodeswere immersed and left overnight at room temperatureunder stirring. When no subsequent modification proceed-ed, CMDex-modified electrodes were immersed in a 1 Methanolamine solution to quench the activated carboxylategroups. Afterward, the CMDex-modified electrodes wererinsed with distilled water, immersed in 1 M NaCl solution,and rinsed with apyrogen water.
ENP modification (Au/Cys/CMDex/ENP)
To incorporate the specific recognition molecule, theAu/Cys/CMDex electrodes were immersed in a solutioncontaining 100 mM EDC and 50 mM NHS in Pipes buffer(pH 6.5) for 30 min to activate the carboxylate groups.Then they were rinsed with apyrogen water and immersedin 0.8 lM ENP solution in 10 mM Hepes buffer (pH 8.0).Finally, the electrodes were immersed in a 1 M ethanol-amine solution to quench the activated carboxylate groupsand were rinsed with apyrogen water.
Lysozyme and biotin–lysozyme modification (Au/Cys/
CMDex/lys and Au/Cys/CMDex/biot–lys)The incorporation of lysozyme and biotin–lysozyme onto
the electrodes was carried out with a procedure similar tothat used for the construction of Au/Cys/CMDex/ENP elec-trodes. The Au/Cys/CMDex electrodes were immersed in asolution 100 mM EDC and 50 mM NHS in 10 mM Pipesbuffer (pH 6.5) for 30 min with the purpose of activatingthe carboxylate groups. Then they were rinsed with apyrogenwater and immersed in solutions of 0.08, 0.8, 8, and 80 lMlysozyme or biotin–lysozyme in 10 mM Hepes buffer(pH 8.0). Finally, the electrodes were immersed in a solutionof 1 M ethanolamine to block the activated carboxylategroups and were rinsed with apyrogen water.
Biotin modification (Au/Cys/CMDex/biot)
The Au/Cys/CMDex electrodes were immersed in a solu-tion 100 mM EDC and 50 mM NHS in 10 mM Pipes buffer(pH 6.5) for 30 min. Then they were rinsed with apyrogenwater and immersed in solutions of 0.08, 0.8, 8, and 80 lMbiotin-LC-hydrazide in 10 mM Pipes buffer (pH 7.0). Final-ly, the electrodes were immersed in a solution 1 M ethanol-amine and were rinsed with apyrogen water.
N-HRP adsorption
The adsorption of the conjugate containing the enzymeHRP on the different electrodes was carried out by immer-sion of 25 lg ml�1 N-HRP in 150 mM NaCl and 20 mMTris buffer (pH 7.4) for 120 min. After the incubation,the electrodes were rinsed with abundant MilliQ water.
Competitive assay
The competitive test was carried out by immersion ofthe Au/Cys/CMDex/ENP electrodes (0.8 lM) in solutions
with different concentrations of LPS Ra from S. minnesota
(previously sonicated) and with a fixed biotin–LPS concen-tration of 10 lg ml�1 for 60 min. The solutions were pre-pared in 150 mM NaCl and 20 mM Tris buffer (pH 7.4).After the incubation, the electrodes were exhaustivelyrinsed with the same buffer and then with apyrogen waterand were incubated separately in 25 lg ml�1 N-HRP in150 mM NaCl and 20 mM Tris buffer (pH 7.4) for120 min. Each modified electrode was used solely for oneLPS concentration, and the current informed for each con-centration is the average of three independent experiments.
Results and discussion
In the assay presented here, ENP was chosen as the rec-ognition element. This election adheres to some importantcharacteristics, it binds to and neutralizes LPS fromnumerous different gram-negative strains in both in vitroand in vivo assays [27–29], it exists in a recombinant ver-sion from S. cerevisiae, and it is highly stable. The lyophi-lized powder can be stored at �20 �C indefinitely, and asolution in water can be stored at 2–8 �C for 1 month orfrozen at �20 �C for several months.
Another two key components of the assay are the biotin-labeled LPS and the signaling element N-HRP. To design abalanced competitive assay, biotin–LPS should haveappropriate endotoxic activity and a good biotin/LPSratio. Therefore, the synthesis of biotin–LPS that fulfillsthose characteristics must be achieved. On the other hand,certain nonspecific N-HRP adsorption onto the electrodesurface (free or modified with a protein, e.g., the recogni-tion element) has been observed. Thus, the experimentalconditions (e.g., the incorporation of the recognition pro-tein onto the electrode) were optimized to obtain a goodsignal/background ratio. All of this work was carried outbefore the implementation of this new version of the com-petitive assay for endotoxins.
Synthesis of LPS conjugate
Because the lipid A region constitutes the active pyro-genic portion of LPS and is involved in the specific recog-nition toward ENP [9,28,29], a suitable method forconjugation needs to preserve that portion of the moleculestructurally and conformationally. Therefore, it would beconvenient if the label were bound to a distant region suchas the core or, even better, the O-antigen region. Being con-stituted by carbohydrates, a typical method is to attach thelabel to the hydroxyl groups previously oxidized to alde-hydes. Two techniques were proposed: an enzymatic oxida-tion with neuraminidase and galactose oxidase [26] and theoxidation with periodate [19].
The enzymatic method is based on the fact that galac-tose oxidase can form C-6 aldehydes on terminal D-galac-tose or N-acetyl-D-galactose residues. When galactoseresidues are penultimate to sialic acid residues, anotherenzyme (neuraminidase) must be used to remove the sialic

Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116 113
acid sugars and expose galactose as the terminal residue.Therefore, the combined action of both enzymes generatesC-6 aldehyde derivative from the galactose that can be con-jugated later with amino or hydrazide groups [21]. The oli-gosaccharide core of LPS possesses galactose and sialicacid residues [30]. The galactose oxidase catalyzes the pri-mary alcohol reaction to aldehydes, according to the fol-lowing reaction (where GO is galactose oxidase):
RCH2OHþO2!GO
RCHOþH2O2
This method was tested with LPS from S. minnesota.The reaction can be easily followed by measuring the pro-duction of H2O2 [31] with stripes for residual peroxidedetection (limit of detection 1 ppm). For samples contain-ing LPS smooth and LPS R60, both of S. minnesota, thehydrogen peroxide production was below the detectionlimit, whereas for samples containing galactose in equalproportion, a concentration greater than 10 ppm of perox-ide could be determined; therefore, it seems that the galac-tose oxidase is not able to access the LPS galactose residuesfor the oxidation to proceed. Although the enzyme workedwith very good yield in monosaccharides, the yield of thereaction was practically nil in a polymer such as the LPS.
The other method is based on the periodate cleavage ofC–C bonds that possess adjacent hydroxyls to form highlyreactive aldehydes. Acting on the conditions during theoxidation, the amount of modified carbohydrate residuescan be controlled [21]. Thus, different NaIO4 concentra-tions, times, and temperatures of the oxidation reactionwere tested to tailor the aldehyde generation in the LPS.Dansyl hydrazine was used to find the best conditions forthe conjugation because this probe allows following thereaction yield directly by fluorescence, being quite suitablefor handling small samples.
Table 1 shows the estimated dansyl/LPS ratios obtainedfrom different conditions. As a reference, an experimentcarried out with dextran (MW 69 kDa) is presented. Theoxidation of dextran with periodate proceeds with goodyield even at smooth conditions, with a dansyl/dextranratio of 10 being obtained (row a). When the LPS was usedfor the conjugation, under similar conditions (row b) andin somewhat more aggressive conditions (row c), ratiossmaller than 1 were obtained. These results can be
Table 1Oxidation reaction conditions of carbohydrates present in LPS or dextranand labeling ratios
Conjugate Oxidation conditions Ratioa
NaIO4
(mM)Temperature(�C)
Time(h)
a Dansyl–dextran
1 0 10 10
b Dansyl–LPS 7 0 14 0c Dansyl–LPS 7 20 24 0.8d Dansyl–LPS 150 45 16 3e Biotin–LPS 150 20 3 1
a Moles of probe/moles of polymer (LPS average MW 20 kDa).
explained based on the different behavior of each speciesin aqueous solution. LPSs form aggregates of high molec-ular weight at the concentrations used in these reactions.A better ratio can be obtained in more severe conditions;however, it leads to a complete loss of the endotoxic activ-ity (row d).
Finally, a biotin–LPS conjugate was synthesized at theconditions detailed in Table 1 (row e), allowing the intro-duction of a number of labels retaining the endotoxic activ-ity. The characterization shows a biotin/LPS ratio equal to1:1 considering a 20-kDa average molecular mass for theLPS smooth [17,32].
N-HRP adsorption
Nonspecific adsorption can be an important interferencein recognition assays. Before evaluating biotin–LPS in acompetitive assay, the N-HRP nonspecific adsorption wasstudied on different surfaces, all of them related to the stag-es of construction of the sensor: gold (Au), cystamine-mod-ified gold (Au/Cys), the last modified with CMDex (Au/Cys/CMDex), and electrodes modified with lysozyme(Au/Cys/CMDex/lys). Also, electrodes modified covalentlywith the specific counterpart biotin (Au/Cys/CMDex/biot)and with biotin–lysozyme (Au/Cys/CMDex/biot–lys) weretested to determine the specificity of the proposed assay.Lysozyme was used due to its similarity with the specificrecognition protein, ENP, related to its isoelectric pointand size [33–35]. Therefore, a similar behavior can beexpected for its incorporation onto the matrix of CMDex.Thus, for convenience, lysozyme was used in those experi-ments where the specificity of the biotin–neutravidin bind-ing was determined.
Some conclusions can be extracted from the results pre-sented in Fig. 1. A comparison among gold (Au), cysta-mine-modified gold (Au/Cys), and the last containingdextran (Au/Cys/CMDex) evidenced the ability of CMDexto decrease the nonspecific protein adsorption in additionto providing an adapted surface to immobilize covalentlya recognition agent [36,37]. Polysaccharides, such asCMDex, can provide heavily hydrated, densely packed,neutral, and conformationally mobile chains, renderingmodified surfaces whose properties made them effective atprotein and cell rejection. Numerous works have shownthat surface-bound polysaccharides can prevent nonspecificprotein adsorption on surfaces [38]. As shown in Fig. 1, thecurrents obtained for dextran-modified electrodes areapproximately four times smaller than those for the othertested electrodes.
The further attachment of biotin to these electrodesincreases the values obtained for the catalytic current dueto the incorporation of a specific component in the elec-trode, as can be concluded from the comparison of Au/Cys/CMDex and Au/Cys/CMDex/biot electrodes.
The incorporation of the protein onto the surfaceincreases the hydrophobicity of the modified electrode;consequently, the N-HRP finds a friendly surface to
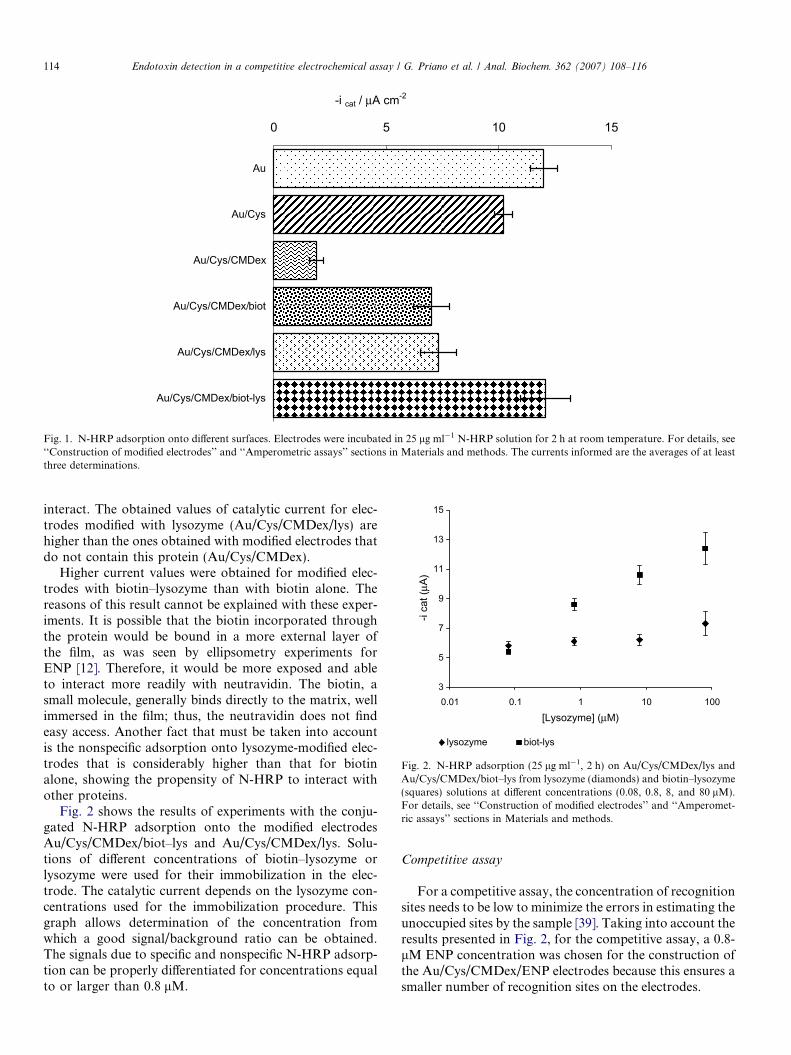
-i cat / μA cm-2
0 5 10 15
Au
Au/Cys
Au/Cys/CMDex
Au/Cys/CMDex/biot
Au/Cys/CMDex/lys
Au/Cys/CMDex/biot-lys
Fig. 1. N-HRP adsorption onto different surfaces. Electrodes were incubated in 25 lg ml�1 N-HRP solution for 2 h at room temperature. For details, see‘‘Construction of modified electrodes’’ and ‘‘Amperometric assays’’ sections in Materials and methods. The currents informed are the averages of at leastthree determinations.
3
5
7
9
11
13
15
0.01 0.1 1 10 100
[Lysozyme] (μM)
-i ca
t (μA
)
lysozyme biot-lys
Fig. 2. N-HRP adsorption (25 lg ml�1, 2 h) on Au/Cys/CMDex/lys andAu/Cys/CMDex/biot–lys from lysozyme (diamonds) and biotin–lysozyme(squares) solutions at different concentrations (0.08, 0.8, 8, and 80 lM).For details, see ‘‘Construction of modified electrodes’’ and ‘‘Amperomet-ric assays’’ sections in Materials and methods.
114 Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116
interact. The obtained values of catalytic current for elec-trodes modified with lysozyme (Au/Cys/CMDex/lys) arehigher than the ones obtained with modified electrodes thatdo not contain this protein (Au/Cys/CMDex).
Higher current values were obtained for modified elec-trodes with biotin–lysozyme than with biotin alone. Thereasons of this result cannot be explained with these exper-iments. It is possible that the biotin incorporated throughthe protein would be bound in a more external layer ofthe film, as was seen by ellipsometry experiments forENP [12]. Therefore, it would be more exposed and ableto interact more readily with neutravidin. The biotin, asmall molecule, generally binds directly to the matrix, wellimmersed in the film; thus, the neutravidin does not findeasy access. Another fact that must be taken into accountis the nonspecific adsorption onto lysozyme-modified elec-trodes that is considerably higher than that for biotinalone, showing the propensity of N-HRP to interact withother proteins.
Fig. 2 shows the results of experiments with the conju-gated N-HRP adsorption onto the modified electrodesAu/Cys/CMDex/biot–lys and Au/Cys/CMDex/lys. Solu-tions of different concentrations of biotin–lysozyme orlysozyme were used for their immobilization in the elec-trode. The catalytic current depends on the lysozyme con-centrations used for the immobilization procedure. Thisgraph allows determination of the concentration fromwhich a good signal/background ratio can be obtained.The signals due to specific and nonspecific N-HRP adsorp-tion can be properly differentiated for concentrations equalto or larger than 0.8 lM.
Competitive assay
For a competitive assay, the concentration of recognitionsites needs to be low to minimize the errors in estimating theunoccupied sites by the sample [39]. Taking into account theresults presented in Fig. 2, for the competitive assay, a 0.8-lM ENP concentration was chosen for the construction ofthe Au/Cys/CMDex/ENP electrodes because this ensures asmaller number of recognition sites on the electrodes.
Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116 115
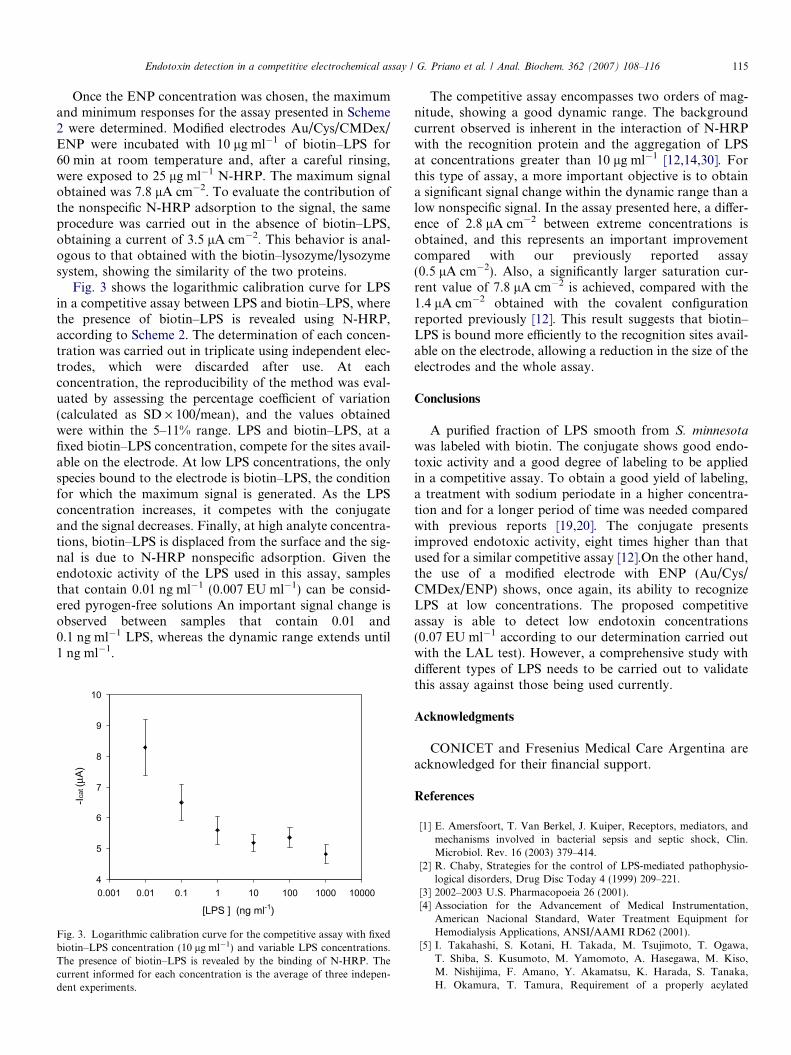
Once the ENP concentration was chosen, the maximumand minimum responses for the assay presented in Scheme2 were determined. Modified electrodes Au/Cys/CMDex/ENP were incubated with 10 lg ml�1 of biotin–LPS for60 min at room temperature and, after a careful rinsing,were exposed to 25 lg ml�1 N-HRP. The maximum signalobtained was 7.8 lA cm�2. To evaluate the contribution ofthe nonspecific N-HRP adsorption to the signal, the sameprocedure was carried out in the absence of biotin–LPS,obtaining a current of 3.5 lA cm�2. This behavior is anal-ogous to that obtained with the biotin–lysozyme/lysozymesystem, showing the similarity of the two proteins.
Fig. 3 shows the logarithmic calibration curve for LPSin a competitive assay between LPS and biotin–LPS, wherethe presence of biotin–LPS is revealed using N-HRP,according to Scheme 2. The determination of each concen-tration was carried out in triplicate using independent elec-trodes, which were discarded after use. At eachconcentration, the reproducibility of the method was eval-uated by assessing the percentage coefficient of variation(calculated as SD · 100/mean), and the values obtainedwere within the 5–11% range. LPS and biotin–LPS, at afixed biotin–LPS concentration, compete for the sites avail-able on the electrode. At low LPS concentrations, the onlyspecies bound to the electrode is biotin–LPS, the conditionfor which the maximum signal is generated. As the LPSconcentration increases, it competes with the conjugateand the signal decreases. Finally, at high analyte concentra-tions, biotin–LPS is displaced from the surface and the sig-nal is due to N-HRP nonspecific adsorption. Given theendotoxic activity of the LPS used in this assay, samplesthat contain 0.01 ng ml�1 (0.007 EU ml�1) can be consid-ered pyrogen-free solutions An important signal change isobserved between samples that contain 0.01 and0.1 ng ml�1 LPS, whereas the dynamic range extends until1 ng ml�1.
4
5
6
7
8
9
10
0.001 0.01 0.1 1 10 100 1000 10000
[LPS ] (ng ml-1)
-Icat
(μA)
Fig. 3. Logarithmic calibration curve for the competitive assay with fixedbiotin–LPS concentration (10 lg ml�1) and variable LPS concentrations.The presence of biotin–LPS is revealed by the binding of N-HRP. Thecurrent informed for each concentration is the average of three indepen-dent experiments.
The competitive assay encompasses two orders of mag-nitude, showing a good dynamic range. The backgroundcurrent observed is inherent in the interaction of N-HRPwith the recognition protein and the aggregation of LPSat concentrations greater than 10 lg ml�1 [12,14,30]. Forthis type of assay, a more important objective is to obtaina significant signal change within the dynamic range than alow nonspecific signal. In the assay presented here, a differ-ence of 2.8 lA cm�2 between extreme concentrations isobtained, and this represents an important improvementcompared with our previously reported assay(0.5 lA cm�2). Also, a significantly larger saturation cur-rent value of 7.8 lA cm�2 is achieved, compared with the1.4 lA cm�2 obtained with the covalent configurationreported previously [12]. This result suggests that biotin–LPS is bound more efficiently to the recognition sites avail-able on the electrode, allowing a reduction in the size of theelectrodes and the whole assay.
Conclusions
A purified fraction of LPS smooth from S. minnesotawas labeled with biotin. The conjugate shows good endo-toxic activity and a good degree of labeling to be appliedin a competitive assay. To obtain a good yield of labeling,a treatment with sodium periodate in a higher concentra-tion and for a longer period of time was needed comparedwith previous reports [19,20]. The conjugate presentsimproved endotoxic activity, eight times higher than thatused for a similar competitive assay [12].On the other hand,the use of a modified electrode with ENP (Au/Cys/CMDex/ENP) shows, once again, its ability to recognizeLPS at low concentrations. The proposed competitiveassay is able to detect low endotoxin concentrations(0.07 EU ml�1 according to our determination carried outwith the LAL test). However, a comprehensive study withdifferent types of LPS needs to be carried out to validatethis assay against those being used currently.
Acknowledgments
CONICET and Fresenius Medical Care Argentina areacknowledged for their financial support.
References
[1] E. Amersfoort, T. Van Berkel, J. Kuiper, Receptors, mediators, andmechanisms involved in bacterial sepsis and septic shock, Clin.Microbiol. Rev. 16 (2003) 379–414.
[2] R. Chaby, Strategies for the control of LPS-mediated pathophysio-logical disorders, Drug Disc Today 4 (1999) 209–221.
[3] 2002–2003 U.S. Pharmacopoeia 26 (2001).[4] Association for the Advancement of Medical Instrumentation,
American Nacional Standard, Water Treatment Equipment forHemodialysis Applications, ANSI/AAMI RD62 (2001).
[5] I. Takahashi, S. Kotani, H. Takada, M. Tsujimoto, T. Ogawa,T. Shiba, S. Kusumoto, M. Yamomoto, A. Hasegawa, M. Kiso,M. Nishijima, F. Amano, Y. Akamatsu, K. Harada, S. Tanaka,H. Okamura, T. Tamura, Requirement of a properly acylated

116 Endotoxin detection in a competitive electrochemical assay / G. Priano et al. / Anal. Biochem. 362 (2007) 108–116
b(1 fi 6)-D-glucosamine disaccharide biphosphate structure for effi-cient manifestation of full endotoxic and associated bioactivities oflipid A, Infect. Immunol. 65 (1987) 57–68.
[6] E.T. Rietschel, T. Kirikae, U.F. Schade, A.J. Ulmer, O. Holst,H. Brade, G. Schmidt, U. Mamat, H-D. Gimmecke, S. Kusumoto,U. Zahringer, The chemical structure of bacterial endotoxin inrelation to bioactivity, Immunobiology 187 (1993) 169–190.
[7] U. Zahringer, B. Lindner, E.T. Rietschel, Molecular structure of lipidA, the endotoxic center of bacterial lipopolysaccharides, Adv.Carbohydr. Chem. Biochem. 50 (1994) 211–276.
[8] A.H. Taylor, G. Heavner, M. Nedelman, D. Sherris, E. Brunt,D. Knight, J. Ghrayeb, Lipopolysaccharide (LPS) neutralizingpeptides reveal a lipid A binding site of LPS binding protein, J. Biol.Chem. 270 (1995) 17934–17938.
[9] A. Hoess, S. Watson, G.R. Siber, R. Liddington, Crystal structure ofan endotoxin-neutralizing protein from the horseshoe crab, limulusanti-LPS factor, at 1.5 A resolution, EMBO J. 12 (1993) 3351–3356.
[10] J. Weiss, P. Eslbach, I. Olsson, H. Oderberg, Purification andcharacterization of a potent bactericidal and membrane active proteinfrom the granules of human polymorphonuclear leukocytes, J. Biol.Chem. 253 (1978) 2664–2672.
[11] C.J. Thomas, B.P. Gangadhar, N. Surolia, A. Surolia, Kinetics andmechanism of the recognition of endotoxin by polymyxin B, J. Am.Chem. Soc. 120 (1998) 12428–12434.
[12] G. Priano, F. Battaglini, Use of an antimicrobial protein forendotoxin detection in a competitive electrochemical assay, Anal.Chem. 77 (2005) 4976–4984.
[13] B. Limoges, J-M. Saveant, D. Yazidi, Quantitative analysis of catalysisand inhibition at horseradish peroxidase monolayers immobilized onan electrode surface, J. Am. Chem. Soc. 125 (2003) 9192–9203.
[14] N. Santos, A. Silva, M. Castanho, J. Martins-Silva, C. Saldanha,Evaluation of lipopolysaccharide aggregation by light scatteringspectroscopy, Chem. Bio. Chem. 4 (2003) 96–100.
[15] T.J. Novitsky, R.J. Ridge, J.L. Sloyer, U.S. patent 6,171,807 (2001).[16] D. Petsch, F.J. Anspach, Endotoxin removal from protein solutions,
J. Biotechnol. 76 (2000) 97–119.[17] J. Shands, P. Chun, The dispersion of gram-negative lipopolysaccha-
ride by deoxycholate: Subunit molecular weight, J. Biol. Chem. 255(1980) 1221–1226.
[18] S. Snyder, D. Kim, T. McIntosh, Lipopolysaccharide bilayer struc-ture: effect of chemotype, core mutations, divalent cations, andtemperature, Biochemistry 38 (1999) 10758–10767.
[19] J.M. Luk, A. Kumar, R. Tsang, D. Staunton, Biotinylated lipopoly-saccharide binds to endotoxin receptor in endothelial and monocyticcells, Anal. Biochem. 232 (1995) 217–224.
[20] A. Visintin, E. Latz, B.G. Monks, T. Espevik, D.T. Golenbock,Lysines 128 and 132 enable lipopolysaccharide binding to MD-2,leading to Toll-like receptor-4 aggregation and signal transduction,J. Biol. Chem. 278 (2003) 48313–48320.
[21] G. Hermanson, Bioconjugate Techniques, Academic Press, SanDiego, CA, 1996.
[22] N.M. Green, Avidin: I. The use of [14C]biotin for kinetic studies andfor assay, Biochem. J. 89 (1963) 585–591.
[23] M. Wilchek, E.A. Bayer, Foreword and introduction to the book(strept)avidin–biotin system, Biomol. Eng. 16 (1999) 1–4.
[24] C. Rosano, P. Arioso, M. Bolognesi, The X-ray three-dimensionalstructure of avidin, Biomol. Eng. 16 (1999) 5–12.
[25] E. Corton, C. Danilowicz, F. Battaglini, Osmium complexes bearingfunctional groups: Building blocks for integrated chemical systems,J. Electroanal. Chem. 445 (1998) 89–94.
[26] K. Triantafilou, M. Triantafilou, N. Fernandez, Lipopolysaccharide(LPS) labeled with Alexa 488 hydrazide as a novel probe for LPSbinding studies, Cytometry 41 (2000) 316–320.
[27] H.S. Warren, M.L. Glennon, N. Wainwright, S.F. Amato,K.M. Black, S.J. Kirsch, G.R. Riveau, R.I. Whyte, W.M. Zapol,T.J. Novitsky, Binding and neutralization of endotoxin by limulusantilipopolysaccharide factor, Infect. Immunol. 60 (1992) 2506–2513.
[28] C. Ried, C. Wahl, T. Miethke, G. Wellnhofer, C. Landgraf,J. Schneider-Mergener, A. Hoess, High affinity endotoxin-bindingand neutralizing peptides based on the crystal structure of recombi-nant limulus anti-lipopolysaccharide factor, J. Biol. Chem. 271 (1996)28120–28127.
[29] D. Bannerman, M. Fitzpatrick, D. Anderson, A. Bhattarcharjee,T. Novitsky, J. Hasday, A. Cross, S. Goldblum, Endotoxin-neutral-izing protein protects against endotoxin-induced endothelial barrierdysfunction, Infect. Immunol 66 (1998) 1400–1407.
[30] K. Brandenburg, J. Andra, M. Muller, M.H.J. Koch, P. Garidel,Physicochemical properties of bacterial glycopolymers in relation tobioactivity, Carbohydr. Res. 338 (2003) 2477–2489.
[31] M.M. Whittaker, D.P. Ballou, J.W. Whittaker, Kinetic isotope effectsas probes of the mechanism of galactose oxidase, Biochemistry 37(1998) 8426–8436.
[32] C.A. Aurell, A. Wistrom, Critical aggregation concentrations ofgram-negative bacterial lipopolysaccharides (LPS), Biochem. Bio-phys. Res. Commun. 253 (1998) 119–123.
[33] L.R. Wetter, H.F. Deutsch, Immunological studies on egg whiteproteins: IV. Immunochemical and physical studies of lysozyme,J. Biol. Chem. 192 (1951) 237–242.
[34] R.E. Canfield, The amino acid sequence of egg white lysozyme,J. Biol. Chem. 238 (1963) 2698–2707.
[35] A.J. Sophianopoulos, C.K. Rhodes, D.N. Holcomb, K.E. Van Holde,Physical studies of lysozyme: I. Characterization, J. Biol. Chem. 237(1962) 1107–1112.
[36] U. Jonson, M. Malmqvist, Real time biospecific interaction analysis,Adv. Biosens. 2 (1992) 291–336.
[37] R. Polzius, T. Schneider, F. Bier, U. Bilitewski, W. Koschinski,Optimization of biosensing using grating couplers: Immobilizationon tantalum oxide waveguides, Biosens. Bioelectron. 11 (1996) 503–514.
[38] E. Osterberg, K. Bergstrom, K. Holmberg, T.P. Schuman,J.A. Riggs, N.L. Burns, J.M. Van Alstine, J.M. Harris, Protein-rejecting ability of surface-bound dextran in end-on and side-onconfigurations: comparison to PEG, J. Biomed. Mater. Res. 29(1995) 741–747.
[39] C. Davies, in: D. Wild (Ed.), The Immunoassay Handbook, 2nd ed.,Nature Publishing Group, New York, 2001, chap. 1.






























