




UNIVERSIDADE FEDERAL DE MINAS GERAIS
INSTITUTO DE CIÊNCIAS EXATAS
DEPARTAMENTO DE QUÍMICA
DESENVOLVIMENTO DE CATALISADORES HETEROGÊNEOS PARA
A SÍNTESE DE BIODIESEL
Belo Horizonte
2013
SILVIA DE MORAES PASSOS

SILVIA DE MORAES PASSOS
DESENVOLVIMENTO DE CATALISADORES HETEROGÊNEOS
PARA A SÍNTESE DE BIODIESEL
Monografia de Silvia de Moraes Passos apresentada ao Departamento de Química do Instituto de Ciências Exatas, da Universidade Federal de Minas Gerais, como requisito para a obtenção do título de Bacharel em Química.
Orientadora: Vânya Márcia Duarte Pasa
BELO HORIZONTE
2013

Dedico este trabalho à minha querida mãe.
Além de doar seu amor, ela se sacrifica e se compromete
todos os dias para que meus sonhos se realizem.

AGRADECIMENTOS
A Professora Vânya Pasa, minha orientadora, que me apoiou desde o início na
realização deste trabalho; pela sua paciência, dedicação, compreensão e, acima de tudo, por
tudo aquilo que me transmitiu, tanto a nível acadêmico como humano.
Agradeço também a toda a minha família, em especial aos meus pais Silvio e Evane,
aos meus irmãos Luiz Gustavo e Fernanda Carolina, e ao meu namorado Felipe por todo
amor, compreensão, dedicação e pela paciência.
Aos amigos conquistados ao longo desses anos, sem eles essa jornada seria solitária e
bem mais árdua.
Conny Cerai, Bruno Rodrigues, Ariane Ássimos, Cristina Bernades, Willian Coelho,
Samira Rodabel, Camila Monteiro e claro não poderia faltar a grande companheira Nathália
de Lima. Queridos amigos, sem vocês eu não teria alcançado nada nessa caminha acadêmica.
O apoio, os sorrisos, lanches compartilhados e o mais importante a amizade sincera.
Obrigada pela oportunidade de ter vocês por perto. O meu crescimento humano ao longo
desses dias conquistei junto com vocês.
Sou grata também, à todos os funcionários, professores e amigos do LEC, que sempre
estiveram presente nessa trajetória.
Agradeço o apoio financeiro da Agência Nacional do Petróleo Gás Natural e
Biocombustível – ANP, da Financiadora de Estudos e Projetos (FINEP) e do Ministério da
Ciência e Tecnologia (MCT) por meio do Programa de Formação de Recurso Humanos da
ANP para o setor petróleo e gás - PRH – ANP/MCT.

“O valor das coisas não está no tempo em que elas
duram, mas na intensidade com que acontecem. Por
isso existem momentos inesquecíveis, coisas
inexplicáveis e pessoas incomparáveis” – Fernando
Pessoa

RESUMO
Neste presente trabalho usou-se a rota de redução carbotérmica verde para
redução produção de catalisadores para a síntese de biodiesel via transesterificação do óleo
de soja. Óxidos de cálcio, magnésio e estanho e acetatos de cobre e níquel foram pirolisados
em presença de resina vegetal a uma temperatura máxima de 900°C. Os produtos foram
caracterização por difração de raio X, microscopia eletrônica de varredura e
termogravimetria. Quando foi obtido o metal ao invés do óxido nanoestruturado, percebeu-
se uma etapa complementar de oxidação. Os óxidos CaO, MgO e SnO/SnO2 foram testados
como catalisadores na proporção de 3 %, nas reações de metanólise, sob refluxo e agitação,
usando a razão molar 12:1 de álcool/óleo. O teor de éster foi determinado por H-RMN e
obteve-se uma conversão de 92% para reação de 4 horas com CaO e 100% para reação de 72
horas com MgO.
Palavras-chave: redução carbotérmica,resina vegetal, catalisador e biodiesel

LISTA DE FIGURAS
Figura 1: Transesterificação de triglicerídeos para produção de biodiesel ........................... 16
Figura 2: Esquema simplificado proposto para a nova rota da redução carbotérmica de pós de ZnO. ............................................................................................. 18
Figura 3: Forno tubular horizontal multiprocessado utilizado para reações de redução carbotérmica verde ( Maciel, A. V.; 2010). .......................................................... 24
Figura 4: Representação esquemática para obtenção e caracterização de materiais. ......... 25
Figura 5: Montagem para síntese de biodiesel sob refluxo e agitação mecânica. ................ 28
Figura 6: Curvas (a) TG, (b) DTG e (c) DTA obtidas para CaO, e mistura CaO/resina vegetal (atmosfera de N2 , 950 °C a 10 °C/min). ........................ 31
Figura 7: Difratograma CaO após redução e oxidação. ....................................................... 32
Figura 8: Espectro na região de infravermelho do produto sintetizado a partir da redução e oxidação do CaO. ......................................................................... 33
Figura 9: Imagens do MEV a) e b) CaCO3 sintetizado; c) e d) imagens do CaO sintetizados............... 34
Figura 10: Curvas TG, DTG e DTA obtidas para MgO, e mistura MgO/resina vegetal (atmosfera de N2, 950 °C a 10 °C/min). ............................................................... 35
Figura 11: Difratograma do produto sintetizado via redução carbotérmica verde e oxidação do MgO. ......................................................................................... 37
Figura 12: Espectro na região de infravermelho do produto sintetizado a partir do MgO .............................................................................................................. 37
Figura 13: Imagens do MEV a) e b) produto da redução do MgO comercial; c) e d) imagens do produto após oxidação do MgO. ................................................................... 38
Figura 14: Curvas TG, DTG e DTA obtidas para SnO, e mistura SnO/resina vegetal (atmosfera de N2 , 950 °C a 10 °C/min) ......................................................... 39
Figura 15: Difratograma do produto de redução do SnO2 ................................................... 40
Figura 16: Difratograma do produto de redução seguida de oxidação. ................................ 41
Figura 17:Espectro na região de infravermelho do produto sintetizado SnO ........................ 41
Figura 18:Imagens do MEV a) e b) Sn sintetizado; c) e d) imagens do produto SnO sintetizado. .................................................................................................... 42
Figura 19:Curvas TG, DTG e DTA obtidas para acetato de cobre II, e de sua mistura com resina vegetal (atmosfera de N2 , 950°C a 10°C/min). .............. 44
Figura 20: Difratograma do produto sintetizado após redução carbotérmica verde .............. 45
Figura 21:Espectro na região de infravermelho do produto sintetizado. ............................... 46
Figura 22: Curvas TG, DTG e DTA obtidas para produto da redução carbotérmica ............. 47
Figura 23: Curvas TG, DTG e DTA obtidas para acetato de níquel, e de sua mistura com resina vegetal (atmosfera de N2 , 950°C a 10°C/min). .......................... 48
Figura 24: Difratograma do produto sintetizado via redução carbotérmica verde ................. 49
Figura 25: Curvas TG, DTG e DTA obtidas para o níquel sintetizado. (atmosfera de N2 , 950°C a 10°C/min). ................................................................................ 50

Figura 26: Estudo cinético por CCD da síntese de biodiesel catalisada por CaO sintetizado. .................................................................................................... 51
Figura 27: Separação do Biodiesel da glicerina residual ...................................................... 52
Figura 28: Espectro de RMN Espectros de RMN de 1H do produto de síntese após 4 horas de reação com CaO (200 MHz, CDCl3). ........................................... 53
Figura 29: Espectros de RMN de 1H do produto de síntese após 72 horas de reação com MgO (200 MHz, CDCl3). ............................................................ 54

LISTA DE TABELAS
Tabela 1: Redução das emissões do Biodiesel comparadas às do Diesel de petróleo ......................................................................................................... 15
Tabela 2: Resumos dos eventos determinados nas análises termogravimétrica TG. .......... 30
Tabela 3: Resumos dos eventos determinados nas análises termogravimétrica TG ........... 36
Tabela 4: Resumos dos eventos determinados nas análises termogravimétrica TG ........... 40
Tabela 5: Resumos dos eventos determinados nas análises termogravimétrica TG ............ 45
Tabela 6: Resumos dos eventos determinados nas análises termogravimétrica TG ........... 47
Tabela 7: Resumos dos eventos determinados nas análises termogravimétrica TG ........... 49
Tabela 8: Resumos dos eventos determinados nas análises termogravimétrica TG ........... 50
Tabela 9: - Fórmula utilizada para quantificação do Teor de Éster por RMN de H1 .............. 52

LISTA DE ABREVIATURAS
A – Valor da integração no espectro de RMN
ANP – Agência Nacional do Petróleo
CCD – Cromatografia de Camada Delgada
DRX – Difração de raio X
DTA – Análise Térmica Diferencial
DTG – Termogravimetria Derivada
FTIR – Espectrometria na região do infravermelho
HPA – Hidrocarbonetos Aromáticos Policíclicos
MEV – Microscopia de Varredura Eletrônica
PNPB – Programa Nacional de Produção e Uso de Biodiesel
RMN – Ressonância Magnética Nuclear
TG – Termogravimetria

11
SUMÁRIO
1 INTRODUÇÃO ........................................................................................... 13
1.1 OBJETIVO GERAL .................................................................................... 18
1.2 OBJETIVOS ESPECÍFICOS ...................................................................... 19
1.3 JUSTIFICATIVA ......................................................................................... 19
2 CONCEITOS GERAIS E REVISÃO DA LITERATURA ............................. 20
2.1 CATÁLISE HETEROGÊNEA ..................................................................... 20
2.2 MATERIAIS NANOESTRUTURADOS PARA UTILIZAÇÃO COMO CATALISADORES ..................................................................................... 21
3 PARTE EXPERIMENTAL .......................................................................... 23
3.1 OBTENÇÃO DOS CATALISADORES ....................................................... 23 3.1.1 Materiais..................................................................................................... 23 3.1.2 Metodologia – Preparação dos Materiais de Partida .................................. 23
3.1.3 Síntese dos materiais “nanoestruturados” ................................................. 24 3.1.4 Técnicas de caracterização ........................................................................ 26
3.2 TESTE DOS CATALISADORES – REAÇÃO DE TRANSESTERIFICAÇÃO .......................................................................... 27
3.3 – CARACTERIZAÇÃO DO BIODIESEL – DOSAGEM DO TEOR DE ÉSTERES ............................................................................................ 28
3.3.1 Cromatografia Camada Delgada CCD – Monitoramento da reação de transterificação. ..................................................................................... 29
4 RESULTADOS E DISCUSSÃO ........................................................................... 30
4.1 REDUÇÃO CARBOTÉRMICA VERDE DE ÓXIDOS COM RESINA VEGETAL. .................................................................................................. 30
4.1.1 Resultados de TG/DTG/DTA do óxido de cálcio comercial e sua mistura com resina vegetal ........................................................................ 30
4.1.2 Caracterização do produto sintetizado em forno via redução carbotérmica de CaO comercial com resina vegetal. ................................. 31
4.2 RESULTADOS DE TG/DTG/DTA DO ÓXIDO DE MAGNÉSIO COMERCIAL E SUA MISTURA COM RESINA VEGETAL ........................ 34
4.2.1 Caracterização do produto sintetizado em forno via redução carbotérmica de MgO com resina vegetal. ................................................. 36
4.3 RESULTADOS DE TG/DTG/DTA DO ÓXIDO DE ESTANHO E SUA MISTURA COM RESINA VEGETAL .................................................. 38
4.3.1 Caracterização do produto sintetizado em forno via redução carbotérmica de SnO com resina vegetal. ................................................. 40
4.4 RESULTADOS DE TG/DTG/DTA DO ACETATO DE COBRE II E SUA MISTURA COM RESINA VEGETAL .................................................. 42

12
4.5 RESULTADOS DE TG/DTG/DTA DO ACETATO DE NÍQUEL E SUA MISTURA COM RESINA VEGETAL .................................................. 47
4.6 TESTES DOS CATALISADORES ............................................................. 50 4.6.1 Oxido de Cálcio .......................................................................................... 50 4.6.2 Óxido de Magnésio .................................................................................... 53
5 CONCLUSÕES E SUGESTÕES .......................................................................... 55
REFERÊNCIAS .............................................................................................................. 56
ANEXO A .................................................................................................................. 58

1 INTRODUÇÃO
A década de 70 representou um verdadeiro marco na história energética do Planeta, pois o
homem passou a valorizar as energias, gerando uma nova consciência a respeito de sua
produção e consumo, principalmente energias origiundas de fontes não renováveis. Com o
aumento acelerado e incontido dos preços do petróleo e de uma reflexão sobre o esgotamento
das reservas exploráveis dos combustíveis fósseis, novas fontes alternativas começaram a ser
estudadas para substituir, pelo menos em parte, os derivados de petróleo, tais como diesel e
gasolina. Imergidos nesse cenário, o governo brasileiro implementou o programa PROALCOOL
que objetivava produzir etanol a partir da cana-de-açúcar para uso como combustível em
substituição à gasolina pura, o que reduziria as importações de petróleo. Era conveniente para
este setor a mudança de produção de açúcar para etanol, visto que, o mercado internacional do
açúcar vinha decaindo. Contudo, os preços baixos pagos aos produtores de etanol combustível,
associados à queda no preço do barril de petróleo a partir de 1986, levaram ao insucesso do
programa (Parente, 2004).
A busca incessante por fontes energéticas alternativas ao petróleo e ampliação de
leis mais severas no combate à poluição ambiental têm colocado em evidência a diversificação
energética, como utilização de fontes renováveis para produção de combustíveis menos
poluentes, como o biodiesel.
Em relação ao biodiesel, o Brasil lançou em 2004 o Programa Nacional de Produção
e Uso do Biodiesel ( PNPB), com foco na inclusão social e no desenvolvimento regional, ao gerar
emprego e renda ao produzir biodiesel a partir de diferentes fontes oleaginosas em diferentes
regiões. A comercialização do Biodiesel é realizada por meio de leilões públicos, que oferecem
igualdade de acesso aos fornecedores e não discriminam o porte do produtor de biodiesel,
assegurando a participação da agricultura familiar (Portal Brasil, 2013).
A Lei n° 11.097, publicada em 13 de janeiro de 2005, introduziu o biodiesel na matriz
energética brasileira e ampliou a competência administrativa da ANP, que passou a denominar-
se Agência Nacional do Petróleo, Gás Natural e Biocombustíveis e assumiu atribuições de
estabelecer as normas regulatórias, autorizar e fiscalizar atividades relacionadas à produção,
transporte, transferência, armazenagem, estocagem, importação, exportação, distribuição,
revenda e comercialização, além da avaliação de conformidade e certificação de biocombustíveis
(ANP, 2012).

14
.O biodiesel é definido como uma mistura de mono-alquil ésteres de ácidos graxos
derivados de óleos vegetais ou gordura animal. Em outros termos, é um combustível renovável,
biodegradável e ambientalmente correto, com propriedades similares ao óleo diesel mineral,
obtidos principalmente pela reação de transesterificação de qualquer triglicerídeo com um
álcool de cadeia curta, metanol ou etanol, por exemplo (Parente, 2004).
O biodiesel pode ser utilizado puro ou em mistura com o óleo diesel em qualquer
proporção, visto que é um biocombustível miscível e fisico-quimicamente semelhante ao óleo
diesel mineral, podendo ser usado em motores de ciclo diesel, sem a necessidade de
significativas e onerosas adaptações (Fukuda, 2001). No entanto, esse biocombustível possui
aplicação singular quando em mistura com o óleo diesel de ultrabaixo teor de enxofre, porque
confere a este melhores características de lubricidade. Por ser biodegradável, não-tóxico e
praticamente livre de enxofre e aromáticos, o biodiesel é considerado um combustível ecológico
(Parente, 2004).
No mercado de biocombustíveis convencionou-se adotar a expressão BXX na qual B
significa Biodiesel e XX a proporção do biocombustível misturado ao óleo diesel. Assim, a sigla B2
significa 2% de biodiesel e 98% de óleo diesel e o B5 equivale a 5% de biodiesel e 95% de
petrodiesel. Essas misturas estão aprovadas para uso no território brasileiro e devem ser
produzidas segundo as especificações técnicas definidas pela ANP - Agência Nacional de Petróleo
e Biocombustíveis. Atualmente a legislação brasileira adota o B5 (Holanda, 2004), mas há
interesse que este aumente, uma vez que existe capacidade ociosa no parque industrial
brasileiro.
Por se tratar de uma energia limpa, não poluente, o seu uso motor ciclo diesel
convencional resulta, quando comparado com a queima do diesel de petróleo, em uma redução
substancial de monóxido de carbono e de hidrocarbonetos não queimados, além do fato do
B100 ser completamente livre de enxofre (Knothe, 2007).
A Tabela 1 apresenta a redução da emissão de compostos poluentes no meio
ambiente com a utilização do biodiesel na substituição, ainda que parcial, do diesel de petróleo.

15
Tabela 1: Redução das emissões do Biodiesel comparadas às do Diesel de petróleo
Tipos de Emissão B100 B20
Total de hidrocarbonetos não queimados -67% -20%
Monóxido de carbono -48% -12%
Resíduos Sólidos -47% -12%
Enxofre -100% -20%
Hidrocarbonetos Aromáticos policíclicos (HPA) -80% -13%
HAP Nitrogenados -90% -50%
Óxidos de Nitrogênio +/-10% +/-10%
Gases do Efeito Estufa -78% a 100% -20%
Outro aspecto importante deve-se ao fato desse combustível liberar menos CO2 , um dos
gases responsáveis pelo agravamento do efeito estufa, uma vez que boa parte do dióxido de
carbono resultante do processo de combustão é consumido no plantio das oleaginosas fechando
o ciclo do carbono.
As matérias-primas utilizadas na produção de biodiesel são: óleos vegetais, gordura
animal, óleos e gorduras residuais. Óleos vegetais e gorduras são basicamente compostos de
triglicerídeos, ésteres de glicerol e ácidos graxos. O termo monoglicerídeo ou diglicerídeo refere-
se ao número de ácidos graxos na cadeia formadora do óleo. No óleo de soja, o ácido
predominante é o ácido oleico; no óleo de babaçu, o láurico e no sebo bovino, o ácido esteárico
(Holanda, 2004).
Atualmente existem 64 plantas produtoras de biodiesel autorizadas pela ANP para
operação no país, correspondendo a uma capacidade total autorizada de 20.207,76 m3/dia.
Destas 64 plantas, 61 possuem autorização para comercialização do biodiesel produzido,
correspondendo a 19.159,04 m3/dia de capacidade autorizada para comercialização (ANP, 2013).
Algumas fontes para extração de óleo vegetal, para ser utilizada na produção de
biodiesel, são: sementes de soja, baga de mamona, polpa do dendê, amêndoa do coco de dendê,
amêndoa do coco de babaçu, semente de girassol, polpa e semente de macaúba, caroço de
algodão, grão de amendoim, semente de canola, semente de linhaça, entre outras (Holanda,
2004). Entretanto a produção brasileira está concentrada na soja e gordura animal.
Como relatado anteriormente, o método mais utilizado para obtenção do biodiesel é
de transesterificação, uma reação de um lipídio com um álcool para formação de ésteres e de
glicerol (Fig.1). Como essa reação é reversível, faz-se necessário um excesso de álcool para forçar
o deslocamento do equilíbrio para o lado da formação dos produtos desejados. A estequiometria

16
da reação é de 3:1 (álcool:triglicerídio), contudo na prática, essa relação pode ser de 16:1 para
aumentar a geração do produto (Schuchard, 1998; Marchetti, 2007; Shama, 2008).
Entre os parâmetros que interferem na reação de transesterificação encontra-se o tipo
de catalisador utilizado. O processo catalítico convencional consiste na catálise homogênea, na
qual a massa catalítica permanece dissolvida no meio reacional, devendo ser removida após a
síntese (Schuchard, 1998; Marchetti, 2007; Sharma, 2008).
Os catalisadores mais usados em fase homogênea são os catalisadores alcalinos,
devido a maior velocidade que conferem à reação e maiores rendimentos, não exigindo elevadas
pressões e temperaturas nem razões molares álcool/óleo. Segundo alguns pesquisadores
(Schuchard, 1998; Marchetti, 2007; Sharma, 2008) a catálise homogênea possui algumas
desvantagens, como a necessidade de retirar o resíduo catalítico presente na mistura de ésteres
gerada pela reação. De um modo geral, realiza-se uma lavagem com água dessa fase reacional,
objetivando a remoção dos resíduos de catalisador, álcool e triglicerídeos ainda misturados ao
conjunto de ésteres.
Salienta-se que a remoção do resíduo catalítico possui grande importância, sendo
responsável pela geração de grande quantidade de efluente, estimada em 20% de cada tonelada
de biodiesel sintetizada, o que envolve custos e impacto ambiental (Marchetti, 2007).
Outra limitação da catálise homogênea básica é o uso de óleos com elevada
acidez, que são comuns e baratos. Isto porque estes óleos possuem altos teores de ácidos graxos
livres que em presença de uma base forte, como NaOH, formam sabões, produtos indesejáveis,
que podem danificar os motores dos carros se não forem removidos, além de dificultarem a
separação de fases no processo produtivo. Uma rota alternativa para o uso destes óleos é dividir
o processo em duas etapas, sendo a primeira uma hidrólise ácida, em que os triglicerídeos serão
transformados em ácidos graxos livres e, em uma segunda etapa, a esterificação destes ácidos
graxos (Schuchardt, 2006). Porém, esta divisão que possibilita o uso destes óleos, onera o
C
at.
H2C O COR"
H2C O COR
HC O COR' + 3 CH3CH2OH
H2C OH
H2C OH
HC OH + ROOCH2CH3 + R'OOCH2CH3 + R"OOCH2CH3
Figura 1: Transesterificação de triglicerídeos para produção de biodiesel

17
processo, posto que aumenta a quantidade de operações unitárias envolvidas na produção do
biodiesel.
Além disso, a catálise homogênea impossibilita a reutilização dos catalisadores,
favorece a formação de emulsões no processo de separação do biodiesel e diminui da pureza da
glicerina obtida pela transesterificação com a ocorrência de catalisador residual também nesta
fase. Diante destas dificuldades, a catálise heterogênea tem sido fortemente estudada nos
últimos anos como uma alternativa à transesterificação de óleos e gorduras.
Uma das possibilidades de aumentar a eficiência de um catalisador sólido é
aumentar sua área específica (área/massa), o que pode ser feito com a redução do tamanho das
partículas. Acredita-se que catalisadores com dimensões nanométricas podem ser mais
eficientes. (Endalew, A.K.: 2011)
Os materiais nanoestruturados, ou nanomateriais, por definição são materiais
que apresentam pelo menos, uma de suas dimensões em tamanho nanométrico, ou seja, em
escala 1/1.000.000.000, ou um bilionésimo do metro. Nesta escala de tamanho, os materiais
apresentam novas propriedades físicas, químicas e biológicas, antes não observadas quando em
tamanho micro ou macroscópico. Sendo assim observando o cenário da busca por catalisadores
heterogêneos, e levando em consideração que a nanociência e a nanotecnologia têm atraído
considerável atenção nos últimos anos pela expectativa do impacto que os materiais
nanoestruturados podem causar como catalisadores (Ferreira, 2009).
Os materiais em escala nanométrica apresentam exclusivas e/ou intensificadas
propriedades físicas, eletrônicas, ópticas, catalíticas e magnéticas quando comparadas aos
mesmos materiais em escala micro e macroscópica, e isto é devido à manifestação dos efeitos
quânticos e ao aumento da contribuição relativa dos átomos presentes na superfície destes
materiais (Ferreira, 2009).
O desenvolvimento da tecnologia em escala nanométrica está intimamente
ligado ao domínio de métodos de síntese de materiais nanoestruturados com propriedades
específicas em larga escala e de interesse comercial; ao refinamento das técnicas de
caracterização; à incorporação dos nanomateriais em compósitos e à construção de materiais
nanoeletrônicos e nanodispositivos (Maciel, 2010).
Ao longo dos anos nosso grupo de pesquisa, propôs uma nova rota de redução
carbotérmica de pós de ZnO a Zn nanoestruturado, estudo que foi desenvolvido através das
teses de doutorado das alunas Jacyra D. S. Araújo e da aluna Adriana Veloso Maciel. Nesta rota

18
utilizou-se como fonte de carbono uma resina vegetal ao invés das convencionais fontes de
carbono, grafite e nanotubos de carbono, amplamente usados por pesquisadores do mundo
inteiro. Nestas teses foram avaliados diferentes parâmetros de síntese buscando a otimização do
processo e alguns resultados já são de domínio público.
De um modo geral, esta nova rota carbotérmica consiste em promover a
evaporação térmica de uma mistura de pó de ZnO e uma fonte de carbono, no interior de um
tubo de quartzo inserido em um forno elétrico, em temperatura de aproximadamente 900 –
1000°C e em atmosfera inerte. Vapores e um resíduo carbonáceo são gerados, sendo este último
formado a partir do processo de carbonização do precursor de carbono, a resina de Eucalyptus
sp. Em temperaturas elevadas haverá a gaseificação do resíduo carbonáceo, originando mais
gases redutores. Durante o resfriamento do sistema, ocorre condensação lenta do material ao
longo do tubo de reação, permitindo assim, a formação do nanomaterial. A Figura 2 mostra um
esquema simplificado para esta nova rota carbotérmica (Ferreira, 2009).
Figura 2: Esquema simplificado proposto para a nova rota da redução carbotérmica de pós de
ZnO.
Uma vez confirmada a formação de nanoestruturas no trabalho de doutorado da
aluna Jacyra Araújo, e os mecanismos elucidados na tese de aluna Adriana Maciel, este presente
trabalho testa esta rota, já patenteada para desenvolver catalisadores de preferência
nanoestrurados, e testá-los na síntese de transesterificação de biodiesel de soja.
1.1 Objetivo Geral
Este trabalho tem como objetivo geral desenvolver catalisadores heterogêneos
nanoestruturados, para a síntese de biodiesel pela rota metílica, utilizando óleo de soja como
fonte de triglicerídeos. Para tal foram produzidos catalisadores a base de óxidos de cálcio,
estanho, magnésio, cobre e níquel utilizando a rota carbotérmica verde.

19
1.2 Objetivos Específicos
Os objetivos específicos deste trabalho podem ser enumerados da seguinte ordem
de prioridade:
1. Caracterizar todo o material de partida via análise térmica e por espectroscopia na
região do infravermelho.
2. Caracterizar a mistura de resina vegetal e os materiais de partida por análise
termogravimétrica (TG, DTG e DTA).
3. Sintetizar via redução carbotérmica verde, a mistura do item anterior, previamente
caracterizada.
4. Caracterizar o resíduo de síntese utilizando a difração de raio X (DRX), microscopia
eletrônica (MEV), espectroscopia na região do infravermelho e também via análise
térmica (TG, DTG e DTA).
5. Selecionar alguns materiais sintetizados e utilizar na transesterificação de óleo de
soja em metanol na fração molar de 1:3 óleo:álcool, respectivamente.
6. Determinar o teor de éster nos produtos da transesterificação, para avaliar o
potencial de conversão dos catalisadores nanoestruturados descritos anteriormente.
1.3 Justificativa
O desenvolvimento de catalisadores heterogêneos para a síntese de biodiesel é de
grande relevância, pois não permite a formação de sabões e com isto minimiza a formação de
emulsões e reduz o volume de água de lavagem para purificação do biodiesel, levando a um
processo de menor impacto ambiental. Evita a formação de água no processo, o que ocorre com
a catálise homogênea, minimizando a hidrólise do biodiesel formado. Facilita o processo de
purificação do produto, podendo reduzir custos. Pode ainda permitir a esterificação seguida de
transesterificação de óleos de elevada acidez, considerados óleos de menor valor agregado.
As rotas propostas podem levar aos catalisadores com dimensões nanométricas, o
que permitirá uma maior atividade e com isto, uma maior eficiência do processo.

20
2 CONCEITOS GERAIS E REVISÃO DA LITERATURA
2.1 Catálise Heterogênea
Na catálise heterogênea, a massa catalítica fica suportada em sólidos, não se
dissolvendo nos reagentes durante a reação e não formando sabões. Desta forma, os
catalisadores são separados por filtração após a síntese, evitando custos associados às lavagens
exaustivas e consequente geração de águas residuais, podendo ainda ser reciclados e
reutilizados. Alguns estudos mostram também a possibilidade do uso da catálise heterogênea
em óleos ácidos (Jacobson, 2008), pois como o catalisador não está na mesma fase dos
reagentes, não existem metais disponíveis para a formação de sabões.
Na tentativa de obter melhores resultados, Kima e seus colaboradores prepararam
um catalisador sólido quimicamente básico: Na/NaOH/Al2O3. Este catalisador mostrou a mesma
atividade funcional que o catalisador homogêneo NaOH.
Em trabalhos desenvolvidos por Kiss e seus colaboradores são testados reações de
transesterificação de óleos vegetais com metanol, utilizando diferentes sólidos como zeólitas,
resinas de troca iônica e misturas de óxidos metálicos. Foram testados três tipos de zeólitas (H-
ZSM-5, Y e β) e verificou-se que estes produziam uma baixa conversão (1 - 4%) do produto final.
A presença de sílica, alumínio e oxigênio na superfície das zeólitas escondeu os poros, de forma
que o efeito catalítico das zeólitas ocorreu apenas na superfície da estrutura sólida, o que
causou baixas conversões. Quando testaram resinas de troca iônica (formadas a partir de
polímeros), verificaram que estas apresentavam elevada atividade, no entanto ao fim de poucas
horas (2-4h), esta atividade deixou de existir, o que, do ponto de vista industrial, não é
interessante. Analisaram, também, o comportamento da mistura de óxidos metálicos e
escolheram zircônia sulfatada. Este sólido tem a capacidade de ser modificado pela adição de
íons de sulfato, formando-se um catalisador com propriedades ácidas bastante aumentadas.
Este tipo de catalisador é muito utilizado na indústria por ter elevada atividade e seletividade na
esterificação de ácidos graxos livres.
Apesar de inúmeras vantagens da catálise heterogênea em relação à homogênea, o
processo de produção do biodiesel quando se usa catalisadores sólidos tem uma cinética mais
lenta, devido ao menor contato entre reagentes e catalisadores e a etapa de adsorção que
antecede à reação. Estas características do processo induzem ao uso de maiores temperaturas,

21
maiores pressões, maiores razões molares álcool: óleo, maior teor de catalisador e maiores
tempos de reação para que se tenha a mesma conversão alcançada com a catálise homogênea.
2.2 Materiais Nanoestruturados para utilização como catalisadores
Os óxidos metálicos têm sido amplamente estudados como catalisadores
heterogêneos na síntese de biodiesel. Dentre a infinidade de óxidos usados como catalisadores
heterogêneos, alguns são muito promissores, tais como o MgO, CaO, SrO, BaO (Endalew, 2011).
Alguns poucos estudos sobre o óxido de nióbio também foram reportados na literatura
(Gonçalves, 2010).
Os materiais nanoestruturados, ou nanomateriais, por definição são materiais que
apresentam pelo menos, uma de suas dimensões em tamanho nanométrico, ou seja, em escala
1/1.000.000.000, ou um bilionésimo do metro. Nesta escala de tamanho, os materiais
apresentam novas propriedades físicas, químicas e biológicas, antes não observadas quando em
tamanho micro ou macroscópico (Ferreira, 2009).
Os materiais nanoestruturados podem ser feitos de metais, cerâmicas, polímeros,
materiais orgânicos e compósitos destes. Eles incluem, principalmente, nanopartículas,
nanoclusters, nanofios, nanofibras, nanotubos e nanocompósitos. As nanopartículas são
definidas como partículas únicas com diâmetro menor que 100 nm. Os nanofios apresentam
seção transversal menor que 100 nm e o comprimento é da ordem de dezenas de nanômetro e
centenas de micrômetros. Os nanoclusters são aglomerados de partículas e podem apresentar
diâmetro maior que 100 nm. As nanofibras (cilindros sólidos) e nanotubos (ocos) apresentam
duas dimensões, onde o diâmetro é menor que 100 nm e a dimensão axial relativamente maior
(Yang, 2008).
O piche de alcatrão, denominado neste trabalho por resina vegetal, apresenta uma
estrutura macromolecular constituída, principalmente, por unidades de fenóis, guaiacóis e
siringóis, que são resultantes da degradação da madeira, durante a pirólise lenta.
Adicionalmente, também estão presentes hidrocarbonetos alifáticos e aromáticos, éteres
alifáticos, ésteres e cetonas, porém em menores proporções (Prauchner, 2005; Araújo, 2005).
Neste trabalho foi utilizada, resina proveniente de florestas renováveis de
Eucalyptus sp produzido pela Acesita, hoje Aperam.

22
No processo de redução carbotérmica, o material de partida é submetido a um
tratamento térmico, sob temperatura controlada e em atmosfera inerte ou oxidante. Numerosas
reações químicas ocorrem através de diferentes processos de conversões termoquímicas da
fonte de carbono utilizada (Maciel, 2010).

23
3 PARTE EXPERIMENTAL
Serão apresentados os insumos utilizados na preparação dos materiais, as
metodologias adotadas para a fabricação dos catalisadores e serão especificadas as técnicas de
caracterização usadas na caracterização dos produtos sintetizados.
Também será apresentado o teste de atividade de alguns catalisadores na reação de
transesterificação, visando avaliar a eficiência destes.
3.1 Obtenção dos Catalisadores
3.1.1 Materiais
Os reagentes usados no presente trabalho foram: CaO, MgO da marca Vetec com
pureza próxima de 95%, além de SnO2 da Merck com 99% de pureza, e Ni(CH3COO)2.4H2O e
Cu(CH3COO)2.H2O.
Como fonte redutora foi utilizada a resina de Eucalyptus sp produzido a partir da
pirólise lenta de toras de madeira obtidas de florestas plantadas de Eucalyptus sp (Minas Gerais,
Brasil) em fornos de alvenaria da empresa Acesita, hoje Aperam.
O tratamento térmico das amostras ocorreu em um forno elétrico tubular, modelo
AN1080, com câmara de seção cilíndrica com três zonas de aquecimento e um módulo de
controle de temperatura, fabricado pela Analógica Instrumentação e Controle, o forno possui
uma dimensão de ≈120 cm de comprimento.
O gás de arraste utilizado nos processos foi o gás nitrogênio 5.0 Analítico da White
Martins.
3.1.2 Metodologia – Preparação dos Materiais de Partida
Para cada reagente citado, foi preparada uma mistura com a resina vegetal ( 1:1 m/m),
seguido de maceração.
Baseado em trabalhos anteriores do grupo, foram utilizados netas reações em condições
já otimizadas.

24
3.1.3 Síntese dos materiais “nanoestruturados”
Baseado em trabalho anteriores do grupo, foram utilizadas nessas reações as condições
já otimizadas.
3.1.3.1 Redução
Inicialmente, o material de partida, previamente preparado e pesado, foi transferido
para um porta-amostra de porcelana e este foi colocado no interior do tubo de quartzo, o qual
estava inserido em um forno tubular horizontal, de modo que o porta-amostra se localizasse no
centro do forno, conforme mostrado na Figura 3.
Em seguida, o tubo de quartzo foi fechado nas duas laterais com tampas de aço
inoxidável e assim mantido até o final do processo. As tampas apresentam orifícios que
possibilitam a entrada e a saída de gases. Um fluxo constante de nitrogênio foi deixado passar
no interior do tubo de reação por vinte minutos que antecederam o acionamento do programa
de aquecimento, com o intuito de eliminar todos os gases ali presentes.
O material de partida foi aquecido até temperatura 900 - 1000 °C, a uma taxa de
aquecimento de 10 °C/min. Depois o forno foi resfriado naturalmente até temperatura
ambiente. O fluxo de nitrogênio foi constante durante todo o processo. O nitrogênio por ser
inerte atua como gás de arraste conduzindo os produtos gasosos gerados in situ a regiões de
temperaturas mais baixas, para que estes possam ser coletados na forma condensada e/ou
apenas eliminados do interior do tubo de reação.
Após resfriamento do tubo, coletou-se o resíduo presente no porta-amostra. O produto
sintetizado foi conduzido para análises de difração de raios X - DRX, microscopia eletrônica de
varredura – MEV e análise termogravimétrica TG/DTG/DTA.
Figura 3: Forno tubular horizontal multiprocessado utilizado para reações de redução
carbotérmica verde ( Maciel, A. V.; 2010).

25
Um estudo complementar a redução carbotérmica verde foi conduzido em micro escala,
usando termobalança, conforme descrito em item 3.1.4.1 visando avaliar as temperaturas em
que ocorrem maiores variações de massa, bem como a extensão destas variações, conforme
Figura 4.
Figura 4: Representação esquemática para obtenção e caracterização de materiais.
3.1.3.2 Oxidação
Os materiais obtidos na etapa de redução carbotérmica verde, a base de magnésio e
estanho foram oxidados em uma temperatura de 600°C e o de cálcio a 900°C. A oxidação se deu
no forno tubular, com taxa de aquecimento 10°C/min e uma isoterma de uma hora. Após o
resfriamento o material foi armazenado em dessecador.

26
3.1.4 Técnicas de caracterização
Os catalisadores foram caracterizados por microscopia eletrônica de varredura- MEV
para análise da morfologia. A composição química foi estudada por difração de raios X, já a
análise termogravimétrica permitiu avaliar a estabilidade térmica dos catalisadores nas
temperaturas de utilização e nos tratamentos térmicos de ativação, antes das reações de
transesterificação.
3.1.4.1 Análise térmica – Termogravimetria (TG) e Análise Térmica Diferencial (DTA)
Amostras dos reagentes comerciais, e de sua mistura com a resina vegetal, foram
submetidas a tratamento térmico em uma termobalança.
Assim, cerca de 3,0 mg de cada amostra foi pesada e transferida para um cadinho de
alumina, o qual foi inserido em uma termobalança (NETZSCH STA 409EP) do Departamento de
Química – UFMG. A temperatura variou entre 25 a 950°C, a uma razão de aquecimento de
10°C/min e sob atmosfera dinâmica de nitrogênio (100 mL/min), sendo obtidas medidas
simultâneas de TG/DTG e DTA.
Os materiais sintetizados em forno tubular também foram submetidos às mesmas
condições e análise térmica.
3.1.4.2 Espectroscopia de absorção na região do infravermelho (IV)
As amostras foram preparadas em forma de pastilhas, misturadas ao KBr em
concentrações apropriadas. As análises de IV foram realizadas em um espectrômetro Spectrum
RX1 da Perkin Elmer, localizado no Departamento de Química da UFMG. Os espectros foram
adquiridos com uma resolução de 4 cm-1 e numa faixa de comprimento de onda de 400-4000 cm-1
.
3.1.4.3 Técnica de Difração de Raios X – DRX
A obtenção da difração de raio X no pó, se deu em um difratômetro, Shimadzu XRD-
6000 usando um cristal de grafite como monocromador, com radiação Cu-Kα com λ= 1,5406 Å.
A faixa de varredura do ângulo de Bragg (2ϴ) para as amostras foi de 10 a 80° com velocidade

27
angular de 0,02°s-1. O equipamento pertence à Universidade Federal de Viçosa - Campus de Rio
Paranaíba. O programa utilizado para análise qualitativa foi o Search-Macht da Crystal Impact V
1.11f e a quantitativa o programa Fullprof, FullProf 2006 and WinPLOTR: New Windows 95/NT
Applications for Diffraction Commission For Powder Diffraction, International Union for
Crystallography, Newsletter N20 (May-August) Summer 1998, Juan Rodriguez-Carvajal, Thierry
Roisnel.
3.1.4.4 Microscopia Eletrônica de Varredura (MEV) As imagens de MEV foram realizadas no Centro de Microscopia da UFMG, utilizou-se o
Microscópio Eletrônico de Varredura - JEOL JSM - 6360LV com detector de elétrons secundários
para alto vácuo, detector de elétrons retroespalhados para alto e baixo vácuo, detector de EDS
(espectrômetro de raio X de energia dispersiva) e detector de EBSD (difração de elétrons
retroespalhados). As amostras foram preparadas com recobrimento de ouro.
3.2 Teste dos Catalisadores – Reação de Transesterificação
Foram realizados testes exploratórios de transterificação para estudos de alguns
catalisadores. As reações foram conduzidas com 3% de catalisador e razão álcool/óleo de 12:1
sob agitação e refluxo, conforme montagem apresentada na Figura 5. Os trabalhos de síntese
foram conduzidos com o óleo de soja, que é a oleaginosa usada em maior extensão no Brasil.

28
Figura 5: Montagem para síntese de biodiesel sob refluxo e agitação mecânica.
3.3 – Caracterização do Biodiesel – Dosagem do Teor de ésteres
No que diz respeito ao monitoramento da quantidade dos ésteres metílicos obtida,
utilizou-se a espectrometria de Ressônancia Magnética Nuclear de Hidrogênio (RMN - 1H) com o
objetivo de monitorar variações no sinal dos hidrogênios (Knothe, 2007). As análises de RMN - 1H
foram realizadas em clorofórmio deuterado (CDCl3) em espectrêmetro Bruker de 200 MHz,
disponível no Departamento de Química da Universidade Federal de Minas Gerais. O número
relativo de hidrogênios foi calculado em cada grupo de integração dos hidrogênios metilênicos
no intervalo de 4,05 a 4,40ppm (Gelbard, 1995). Para tal determinou-se a área dos hidrogênios
do grupo metila e do grupo CH2 em posição alfa à carbonila do éster, de acordo com a Equação
1.
Equação 1.
O processamento envolveu transformada de Fourier utilizando alargamento de linha de
0,3 Hz com 32k pontos, ajuste interativo de fase e ajuste automático de linha de base. As

29
integrais foram corrigidas em termos de inclinação (slope) e horizontalização (bias), sendo
calibradas com o sinal do grupo metilênico ligado à carbonila. A edição do espectro foi realizada
com o programa ACD/NMR Processor Academic Edition: Processor Window.
3.3.1 Cromatografia Camada Delgada CCD – Monitoramento da reação de transterificação.
A reação de transesterificação foi acompanhada por CCD, essa técnica acompanhou o
progresso da síntese, esse monitoramento mostrou-se eficiente, sendo um ótimo indicativo do
estágio da reação e impedindo que a mesma fosse interrompida antes do momento apropriado.
A fase estacionária utilizada foi a sílica, a fase móvel uma solução 9:1 (v/v) de éter
sulfúrico: éter de petróleo. Como revelador foi utilizado o iodo sublimado.

30
4 RESULTADOS E DISCUSSÃO
4.1 Redução Carbotérmica Verde de óxidos com resina vegetal.
4.1.1 Resultados de TG/DTG/DTA do óxido de cálcio comercial e sua mistura com resina vegetal
Para determinar a estabilidade térmica do óxido de cálcio comercial, assim como o de
sua mistura com a resina vegetal foram usadas a análise termogravimétrica (TG)/
termogravimétrica derivada (DTG) e análise térmica diferencial (DTA). Na Figura 6 são
apresentadas as curvas obtidas em atmosfera inerte.
Para amostra de óxido de cálcio comercial tem-se uma perda acentuada na faixa de 370 -
450°C cerca de 23% em massa, que aparece na curva DTA como evento endotérmico e está
associado à liberação de água. Um resíduo bem estável é observado a partir dos 600°C em torno
de 3% de massa. Para mistura CaO com a resina vegetal tem-se três eventos de degração
referentes ao óxido e à resina. O primeiro com uma perda pequena de aproximadamente 3% de
massa, um segundo na faixa de 300 – 500°C com uma perda 10% em massa do material, (evento
exotérmico em 457°C), e uma outra perda de uns 15% de massa na faixa de 700 – 810°C (evento
endotérmico). Este último evento pode estar associado a liberação de CO2 conforme reação que
segue CaCO3 → CaO + CO2.
Deve-se ressaltar que o carbonato de cálcio pode ter sido gerado devido ao grande
volume de CO2 que é liberado na pirólise da resina vegetal, conforme estudos anteriores. (Maciel,
2010).
Na Tabela 2, encontram-se os valores das principais perdas de massa desses materiais.
Tabela 2: Resumos dos eventos determinados nas análises termogravimétrica TG.
Amostra Intervalo de Perda de
temperatura (°C) massa (%)
CaO Comercial 370 – 500
23,03
CaO + resina vegetal 300-500 700 -810
10 15

31
100 200 300 400 500 600 700 800 900
70
75
80
85
90
95
100
-5
-4
-3
-2
-1
0
100 200 300 400 500 600 700 800 900
0,0
0,5
1,0
1,5
2,0
2,5
Temperatura °C
DTA uV/mg TG %DTG %/minCaO Comercialexo
100 200 300 400 500 600 700 800 900
40
50
60
70
80
90
100
110
-3,0
-2,5
-2,0
-1,5
-1,0
-0,5
0,0
100 200 300 400 500 600 700 800 900
-0,2
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
CaO + resina vegetalDTA uV/mg TG %
DTG %/minexo
Figura 6: Curvas (a) TG, (b) DTG e (c) DTA obtidas para CaO, e mistura CaO/resina vegetal
(atmosfera de N2 , 950 °C a 10 °C/min).
4.1.2 Caracterização do produto sintetizado em forno via redução carbotérmica de CaO comercial com resina vegetal.
Após a redução carbotérmica e a oxidação do material esperava-se encontrar óxido de
cálcio. Como pode ser observado no difratograma da Figura 7, o produto encontrado foi de

32
carbonato de cálcio, CaCO3. O carbonato foi formado porque a degradação da resina vegetal
gera uma quantidade significativa de CO2.
Figura 7: Difratograma CaO após redução e oxidação.
Após o conhecimento da composição química predominante no composto, pela técnica
DRX, se fez a confirmação nos espectros de absorção na região do IV. Através da espectrometria
na região do infravermelho, se pode confirmar ou excluir a existência de grupos funcionais no
produto sintetizado.
Observa-se na Figura 8 uma banda muito intensa em 1444 cm-1 e uma banda forte em
874 cm-1, ambas características do grupo carbonato. As bandas de menor intensidade são
referentes ao estiramento O-H da água (3424 cm-1), dióxido de carbono (2360 – 2342 cm-1),
dentre outras.
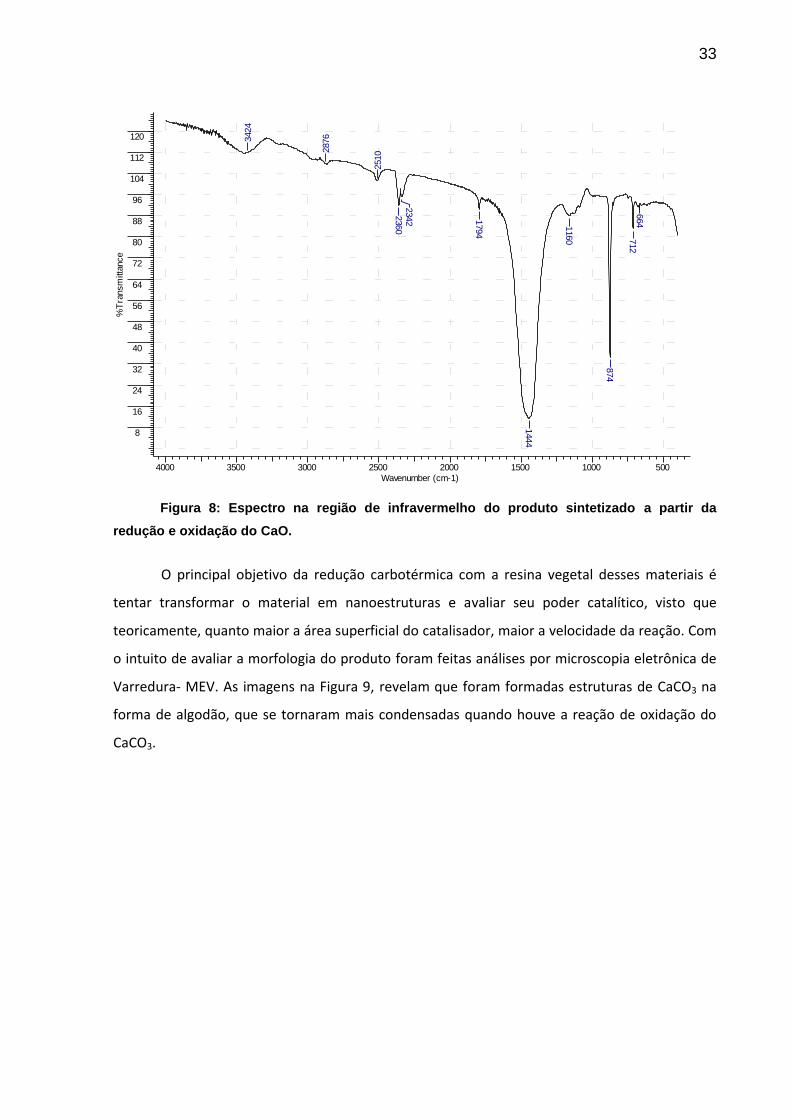
33
4000 3500 3000 2500 2000 1500 1000 500
Wavenumber (cm-1)
8
16
24
32
40
48
56
64
72
80
88
96
104
112
120
%T
ransm
itta
nce
3424
2876
2510
2360
2342 1794
1444
1160
874
712
664
Figura 8: Espectro na região de infravermelho do produto sintetizado a partir da
redução e oxidação do CaO.
O principal objetivo da redução carbotérmica com a resina vegetal desses materiais é
tentar transformar o material em nanoestruturas e avaliar seu poder catalítico, visto que
teoricamente, quanto maior a área superficial do catalisador, maior a velocidade da reação. Com
o intuito de avaliar a morfologia do produto foram feitas análises por microscopia eletrônica de
Varredura- MEV. As imagens na Figura 9, revelam que foram formadas estruturas de CaCO3 na
forma de algodão, que se tornaram mais condensadas quando houve a reação de oxidação do
CaCO3.

34
Figura 9: Imagens do MEV a) e b) CaCO3 sintetizado; c) e d) imagens do CaO sintetizados.
4.2 Resultados de TG/DTG/DTA do óxido de magnésio comercial e sua mistura com
resina vegetal
Nesta etapa do trabalho, pode se avaliar a estabilidade térmica do óxido de magnésio
comercial, assim como o de sua mistura com resina vegetal a partir da análise termogravimétrica
(TG)/ termogravimétrica derivada (DTG) e a análise térmica diferencial (DTA).
Como ilustrado nas curvas na Figura 10, foram observados vários eventos de degradação
do óxido de magnésio, provavelmente pela presença de água adsorvida, além da fase hidróxido
de magnésio que se decompõe a 350°C, conforme reação: Mg(OH)2 → MgO + H2O

35
Para a mistura óxido de magnésio e resina vegetal os eventos se sobrepuseram e houve
uma geração de resíduo de cerca de 38% em massa. A perda de massa de 60% refere-se à
degradação da resina e do MgO e suas impurezas.
100 200 300 400 500 600 700 800 900
78
80
82
84
86
88
90
92
94
96
98
100 200 300 400 500 600 700 800 900
-1,8
-1,6
-1,4
-1,2
-1,0
-0,8
-0,6
-0,4
-0,2
0,0
0,2
0,4
100 200 300 400 500 600 700 800 900
0,2
0,4
0,6
0,8
1,0
1,2
1,4
DTA uV/mg DTG %/min TG %
Temperatura °C
exo
Oxido Magnésio
100 200 300 400 500 600 700 800 900
30
40
50
60
70
80
90
100
100 200 300 400 500 600 700 800 900
0,0
0,5
1,0
1,5
2,0
2,5
3,0
100 200 300 400 500 600 700 800 900
-2,0
-1,5
-1,0
-0,5
0,0
Temperatura °C
exo
TG %DTG %/minDTA uV/mg
Oxido de Magnésio + Resina Vegetal
Figura 10: Curvas TG, DTG e DTA obtidas para MgO, e mistura MgO/resina vegetal
(atmosfera de N2, 950 °C a 10 °C/min).

36
Os valores das principais perdas de massa desses materiais são mostrados na Tabela 3.
Deve se observar o comportamento térmico do óxido comercial em mistura com o resina
vegetal, tem um evento exotérmico acentuado, devido à degradação da cadeia carbônica da
resina, enquanto que o comportamento apenas do óxido no intervalo bem próximo tem-se um
fenômeno endotérmico de desidratação.
Tabela 3: Resumos dos eventos determinados nas análises termogravimétrica TG
Amostra Intervalo de Perda de
temperatura (°C) massa (%)
MgO Comercial 269 - 408 15
MgO + resina vegetal 256 - 667 47
Para o óxido de magnésio comercial, percebe-se um primeiro evento pouco expressivo,
com uma perda pequena de massa na faixa de temperatura de 165-254°C, em sequência tem-se
uma perda acentuada de massa cerca de 15% entre 255 – 408°C, referente ao Mg(OH)2. Perdas
sucessivas ocorrem menos expressivas referentes a resina vegetal. No final há apenas 21% de
resíduo, que é o óxido de magnésio.
O hidróxido de magnésio sugerido é devido adsorção de água do óxido de magnésio
comercial.
Após o estudo do comportamento térmico da mistura, iniciou-se a síntese no forno
conforme descrito na metodologia, após a redução e oxidação o produto da pirólise foi
caracterizado.
4.2.1 Caracterização do produto sintetizado em forno via redução carbotérmica de MgO com resina vegetal.
Após estudo quantitativo da amostra por DRX, verificou-se que o produto esperado foi
obtido. Foram determinados mais de 91% de MgO cristalino na amostra. Como ilustrado no
difratograma da Figura 11.

37
Figura 11: Difratograma do produto sintetizado via redução carbotérmica verde e
oxidação do MgO.
O espectro de absorção de infravermelho do produto esperado serviu para identificar
grupos funcionais presentes. Pode ser visualizada na Figura 12, as bandas 2340 e 2360 cm-1,
referentes ao dióxido de carbono adsorvido. São apresentadas bandas em 1458 e 3700 cm-1
referentes ao Mg – O.
4000 3500 3000 2500 2000 1500 1000 500
Wavenumber (cm-1)
8
16
24
32
40
48
56
64
72
80
88
96
104
112
120
%T
ransm
itta
nce
3700
2360
2340
1722
1458
1094
860
666
416
Figura 12: Espectro na região de infravermelho do produto sintetizado a partir do MgO

38
As imagens de MgO após redução e após oxidação são representadas na Figura 13.
Observa-se que a morfologia lembra algodão, havendo uma maior abertura das estruturas após
oxidação. Isto sugere uma maior área superficial, que precisa ser quantificada.
Figura 13: Imagens do MEV a) e b) produto da redução do MgO comercial; c) e d) imagens do
produto após oxidação do MgO.
4.3 Resultados de TG/DTG/DTA do óxido de estanho e sua mistura com resina vegetal
Como observado na Figura 14, o dióxido de estanho comercial, é muito estável, apresenta
pequenas perdas de massa associadas às moléculas de águas ou algumas impurezas. O
SnO em mistura com a resina vegetal apresenta perdas mais consideráveis de massa ao

39
longo do aquecimento, sendo um perda pequena de 3% em 100 – 200°C, e outra
significativa cerca de 27% na faixa de 200 – 560°C, além de uma perda de 14% em massa
entre 570 – 710°C. Essas duas perdas na faixa de 200 – 750°C oriunda de evento
endotérmico, esperado nas reduções carbotérmicas.
93,0
93,5
94,0
94,5
95,0
95,5
96,0
100 200 300 400 500 600 700 800 900
0,2
0,3
0,4
0,5
0,6
0,7
-0,6
-0,5
-0,4
-0,3
-0,2
-0,1
0,0
0,1
0,2
0,3
TG %
SnO comercial
Temperatura °C
DTA uV/mg
exo
DTG %/min
100 200 300 400 500 600 700 800 900
50
60
70
80
90
100
110
100 200 300 400 500 600 700 800 900
-1,6
-1,4
-1,2
-1,0
-0,8
-0,6
-0,4
-0,2
0,0
0,2
0,4
100 200 300 400 500 600 700 800 900
0,0
0,5
1,0
1,5
2,0
2,5
TG %DTG %/minDTA uV/mgexo
Temperatura °C
SnO2 + resina vegetal
Figura 14: Curvas TG, DTG e DTA obtidas para SnO, e mistura SnO/resina vegetal
(atmosfera de N2 , 950 °C a 10 °C/min)
Para a mistura do óxido comercial e a resina vegetal, o evento endotérmico até 400°C
refere-se á degradação das cadeias carbônicas da resina. Para temperaturas entre 400 e 800°C
40
ocorre a gaseificação do carbono residual segundo a equação C+ CO2 →2 CO e redução do
estanho SnO2 + 2CO2 →Sn2 + CO2.
Tabela 4: Resumos dos eventos determinados nas análises termogravimétrica TG
Amostra Intervalo de Perda de
temperatura (°C) massa (%)
SnO + resina vegetal 200 – 560
570 – 710 27 14
Posteriormente houve a oxidação do estanho Sn, ou seja: 2 Sn + 1,5 O2 → SnO + SnO2
4.3.1 Caracterização do produto sintetizado em forno via redução carbotérmica de SnO com resina vegetal.
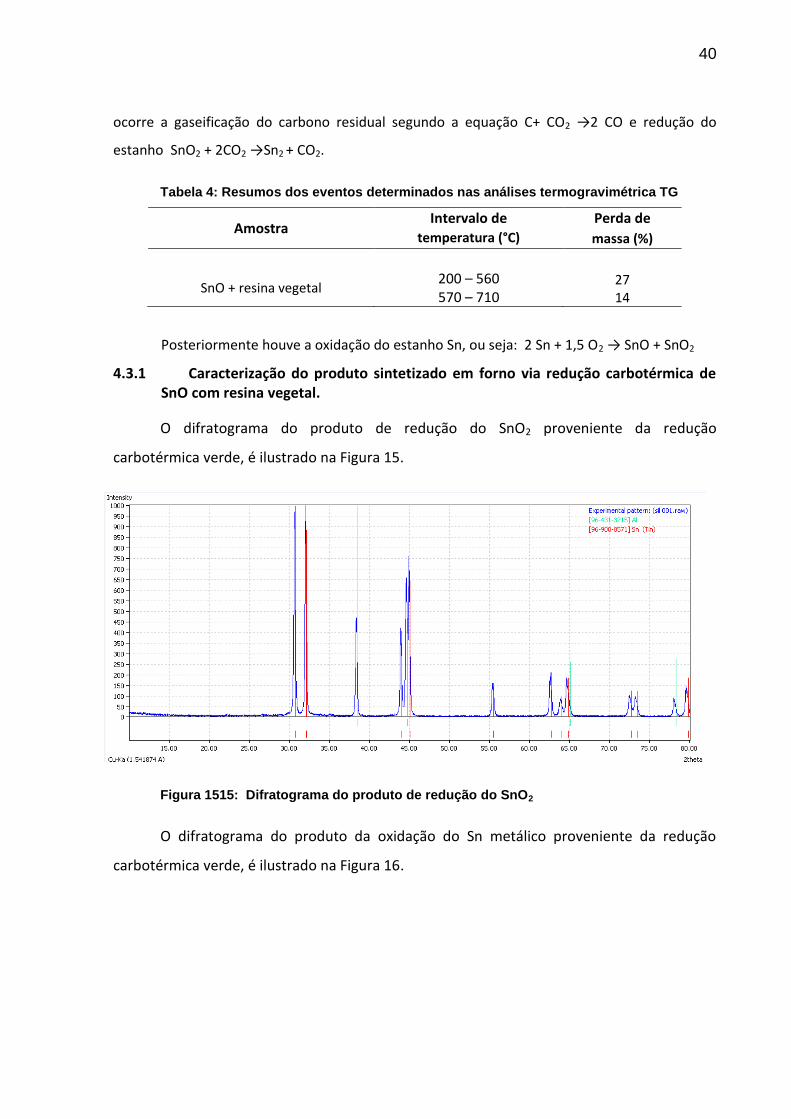
O difratograma do produto de redução do SnO2 proveniente da redução
carbotérmica verde, é ilustrado na Figura 15.
Figura 1515: Difratograma do produto de redução do SnO2
O difratograma do produto da oxidação do Sn metálico proveniente da redução
carbotérmica verde, é ilustrado na Figura 16.

41
Figura 16: Difratograma do produto de redução seguida de oxidação.
Os resultados de DRX mostram que o produto obtido foi de 97% de SnO e cerca de 3 %
de SnO2
No espectro de absorção na região do IV, Figura 17, vizualiza-se a banda intensa em
torno de 666cm-1 referente à ligação Sn – O.
4000 3500 3000 2500 2000 1500 1000 500
Wavenumber (cm-1)
0
8
16
24
32
40
48
56
64
72
80
%T
ransm
itta
nce
2360
2342
1570
1360
1096
668 620
Figura 17:Espectro na região de infravermelho do produto sintetizado SnO
As amostras analisadas por MEV, ilustradas na Figura 18, mostram uma estrutura mais
densa para o Sn se comparada com o SnO. Não se observou a formação de esferas de estanho
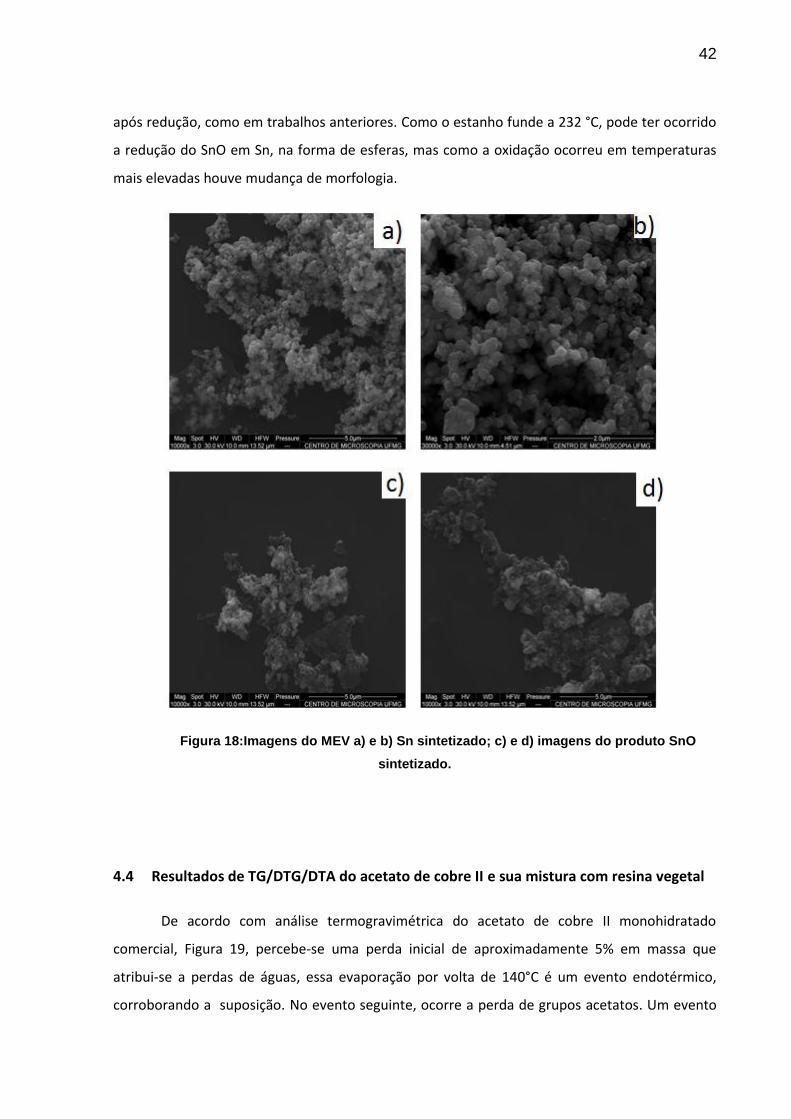
42
após redução, como em trabalhos anteriores. Como o estanho funde a 232 °C, pode ter ocorrido
a redução do SnO em Sn, na forma de esferas, mas como a oxidação ocorreu em temperaturas
mais elevadas houve mudança de morfologia.
Figura 18:Imagens do MEV a) e b) Sn sintetizado; c) e d) imagens do produto SnO
sintetizado.
4.4 Resultados de TG/DTG/DTA do acetato de cobre II e sua mistura com resina vegetal
De acordo com análise termogravimétrica do acetato de cobre II monohidratado
comercial, Figura 19, percebe-se uma perda inicial de aproximadamente 5% em massa que
atribui-se a perdas de águas, essa evaporação por volta de 140°C é um evento endotérmico,
corroborando a suposição. No evento seguinte, ocorre a perda de grupos acetatos. Um evento

43
exotérmico como revela a DTA em torno de 301,6°C. O resíduo formado no final do processo
cerca de 950°C corresponde apenas 2% em massa do composto analisado. Já a mistura com a
resina vegetal, tem-se um comportamento semelhante, porém as perdas de massa são menos
expressivas, sendo elas mais graduais por volta de 250- 300°C uma perda de 44%, referente á
degradação do acetato, e na faixa de 301- 700°C uma perda de 33% são percebidos pela
degradação da resina vegetal. Além disso, um evento endotérmico é percebido em 512,4°C
atribuindo à gaseificado do carbono, seguida de redução de cobre, conforme equações
C + CO2 → 2 CO2
CO + CuO → Cu + CO2

44
200 400 600 800 1000
-1
0
1
2
3
4
5
6
200 400 600 800 1000
40
50
60
70
80
90
100
110
200 400 600 800 1000
-14
-12
-10
-8
-6
-4
-2
0
2
Temperatura (°C)
Temperatura (°C)
Temperatura (°C)
TG %DTG %/minDTA uV/mgAcetado de Cobre II
exo
200 400 600 800
-1
0
1
2
3
4
5
6
200 400 600 800
0
20
40
60
80
100
200 400 600 800
-8
-7
-6
-5
-4
-3
-2
-1
0
1
Temperatura (°C)
Acetato de Cobre II + piche vegetal
DTA uV/mg DTG %/min TG %
exo
Temperatura (°C)
Figura 19:Curvas TG, DTG e DTA obtidas para acetato de cobre II, e de sua mistura com
resina vegetal (atmosfera de N2 , 950°C a 10°C/min).
45
Tabela 5: Resumos dos eventos determinados nas análises termogravimétrica TG
Amostra Intervalo de Perda de
temperatura (°C) massa (%)
Cu(CH3COO)2.H2O Comercial 205 – 303 50,96
Cu(CH3COO)2.H2O + resina vegetal 250 - 300 301 - 700
44,24 32,98
Este material em especial, não foi oxidado após a redução carbotérmica, este resíduo da
reação permaneceu depositado no porta amostra e foi analisado via DRX para estudos de sua
composição.
Por ainda ter algum material oriundo da resina vegetal, visto que, não foi submetido
assim como os demais materiais, a outro processo térmico na presença de oxigênio, o resultado
do raio X mostram a predominância do cobre metálico (Figura 20). Há também a formação das
espécies Cu2O, CuO.
Figura 20: Difratograma do produto sintetizado após redução carbotérmica verde
Os dados de FTIR não mostraram absorções significativas de compostos orgânicos
remanecentes da resina vegetal., como pode ser visto na Figura 21.

46
4000 3500 3000 2500 2000 1500 1000 500
Wavenumber (cm-1)
5
10
15
20
25
30
35
40
45
%T
ransm
itta
nce
3212
2360
2342
1558
1450
1212
1002
630
Figura 21:Espectro na região de infravermelho do produto sintetizado.
O produto sintetizado também foi analisado por termogravimenteria para estudar sua
estabilidade térmica, conforme mostra a Figura 22. Como era de se esperar seu comportamento
foi muito semelhante ao da mistura com a resina, pois ainda era presente muitos compostos do
agente redutor.
Devido as presentes espécies Cu2O e CuO, atribui-se ao evento endotérmico na faixa de
800 – 900 °C a redução do cobre tendo perda de massa a saída de CO.
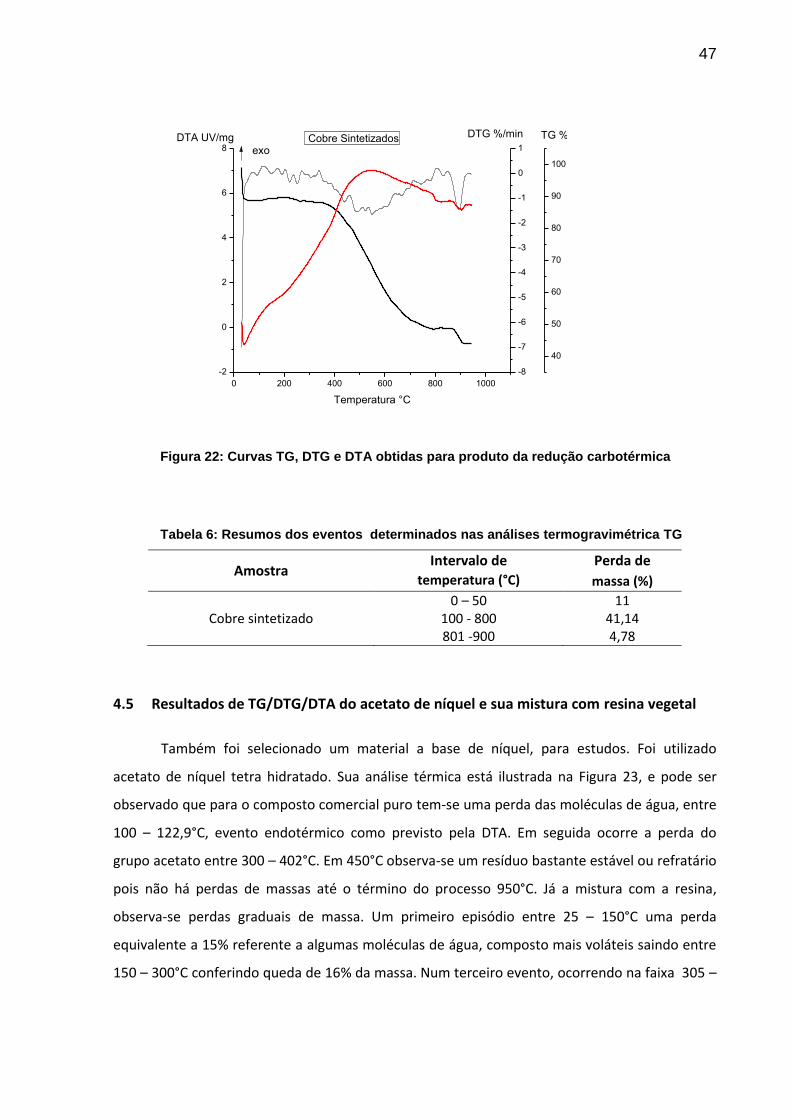
47
40
50
60
70
80
90
100
-8
-7
-6
-5
-4
-3
-2
-1
0
1
0 200 400 600 800 1000
-2
0
2
4
6
8
TG %
DTG %/min
Temperatura °C
Cobre SintetizadosDTA UV/mg
exo
Figura 22: Curvas TG, DTG e DTA obtidas para produto da redução carbotérmica
Tabela 6: Resumos dos eventos determinados nas análises termogravimétrica TG
Amostra Intervalo de Perda de
temperatura (°C) massa (%)
Cobre sintetizado 0 – 50
100 - 800 801 -900
11 41,14 4,78
4.5 Resultados de TG/DTG/DTA do acetato de níquel e sua mistura com resina vegetal
Também foi selecionado um material a base de níquel, para estudos. Foi utilizado
acetato de níquel tetra hidratado. Sua análise térmica está ilustrada na Figura 23, e pode ser
observado que para o composto comercial puro tem-se uma perda das moléculas de água, entre
100 – 122,9°C, evento endotérmico como previsto pela DTA. Em seguida ocorre a perda do
grupo acetato entre 300 – 402°C. Em 450°C observa-se um resíduo bastante estável ou refratário
pois não há perdas de massas até o término do processo 950°C. Já a mistura com a resina,
observa-se perdas graduais de massa. Um primeiro episódio entre 25 – 150°C uma perda
equivalente a 15% referente a algumas moléculas de água, composto mais voláteis saindo entre
150 – 300°C conferindo queda de 16% da massa. Num terceiro evento, ocorrendo na faixa 305 –

48
500°C uma perda de 33% em sequencia uma perda de 22% entre 501 – 650°C. E no final tem-se
um resíduo correspondente a apenas 1% de massa.
0 200 400 600 800 1000
40
50
60
70
80
90
100
110
-14
-12
-10
-8
-6
-4
-2
0
2
0 200 400 600 800 1000
-1
0
1
2
3
4
5
6
TG %Acetato de Niquel comercial
DTG %/min
Temperatura °C
DTA uV/mg
exo
0
20
40
60
80
100
-8
-7
-6
-5
-4
-3
-2
-1
0
1
0 200 400 600 800 1000
-1
0
1
2
3
4
5
6
TG %Acetato de niquel + piche vegetal
DTG %/min
Temperatura °C
DTA uV/mgexo
Figura 23: Curvas TG, DTG e DTA obtidas para acetato de níquel, e de sua mistura com
resina vegetal (atmosfera de N2 , 950°C a 10°C/min).

49
Tabela 7: Resumos dos eventos determinados nas análises termogravimétrica TG
Amostra Intervalo de Perda de
temperatura (°C) massa (%)
Ni(CH3COO)2.4H2O Comercial 100 – 122,9
300 - 402 35,13 37,58
Ni(CH3COO)2.4H2O + resina vegetal
25 - 150
305 - 500 505 – 650
14,82 33,07 22,20
Assim como o material de cobre, o produto da redução com resina vegetal a base de
níquel foi estudado logo após sua reação. Ou seja, não tem-se a etapa de oxidação após o
procedimento. Com tudo, foi realizado estudo do comportamento térmico desse produto, assim
como sua morfologia por DRX. Apresentando em sua composição níquel metálico como pode
ser observado no difratograma da Figura 24, bem como a presença de carbono grafite.
Figura 24: Difratograma do produto sintetizado via redução carbotérmica verde
Na Figura 25, percebe-se que pela curva TG, e confirmados pela DTG, tem se uma leve
perda de massa em 4% até 150°C, houve outra pequena porção cerca de 3% de 150 – 450°C.
Observou-se também uma perda perda de 39% em massa na faixa de 550 – 800°C, e um evento
endotérmico em 610°C, totalizando um perda total de 93 % de massa.
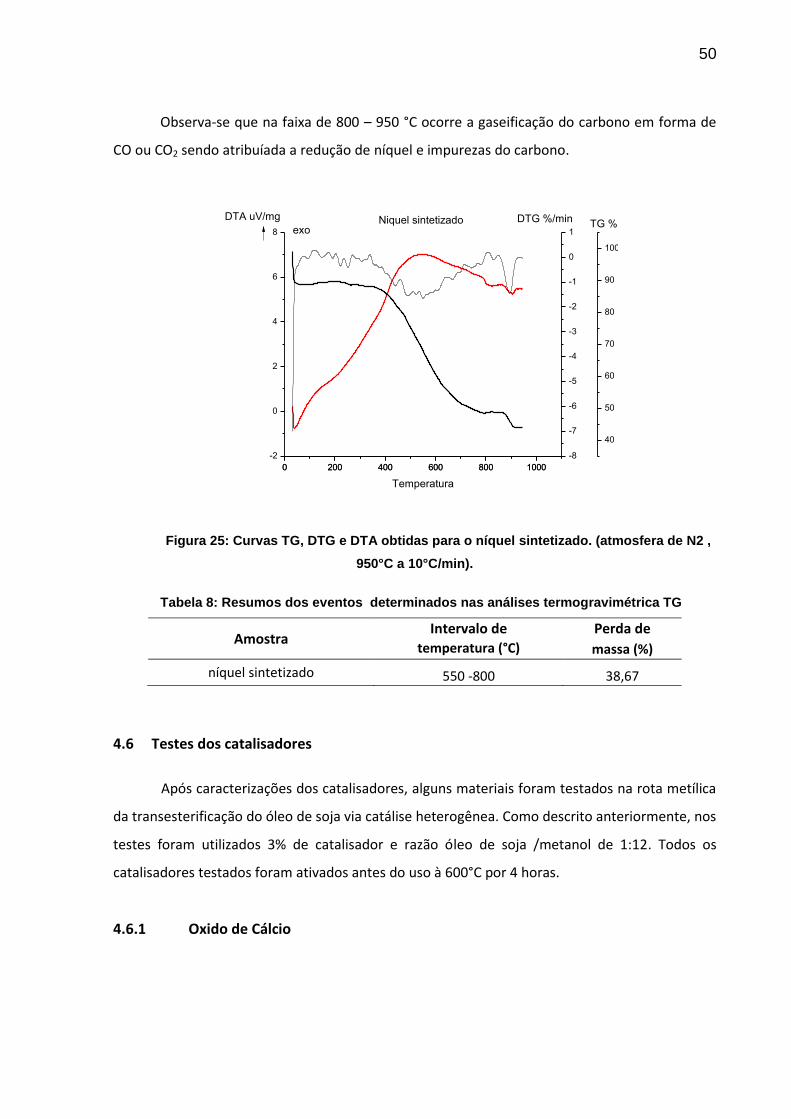
50
Observa-se que na faixa de 800 – 950 °C ocorre a gaseificação do carbono em forma de
CO ou CO2 sendo atribuíada a redução de níquel e impurezas do carbono.
0 200 400 600 800 1000
-2
0
2
4
6
8
0 200 400 600 800 1000
40
50
60
70
80
90
100
-8
-7
-6
-5
-4
-3
-2
-1
0
1
Temperatura
DTA uV/mg
exoNiquel sintetizado TG %
DTG %/min
Figura 25: Curvas TG, DTG e DTA obtidas para o níquel sintetizado. (atmosfera de N2 ,
950°C a 10°C/min).
Tabela 8: Resumos dos eventos determinados nas análises termogravimétrica TG
Amostra Intervalo de Perda de
temperatura (°C) massa (%)
níquel sintetizado 550 -800 38,67
4.6 Testes dos catalisadores
Após caracterizações dos catalisadores, alguns materiais foram testados na rota metílica
da transesterificação do óleo de soja via catálise heterogênea. Como descrito anteriormente, nos
testes foram utilizados 3% de catalisador e razão óleo de soja /metanol de 1:12. Todos os
catalisadores testados foram ativados antes do uso à 600°C por 4 horas.
4.6.1 Oxido de Cálcio
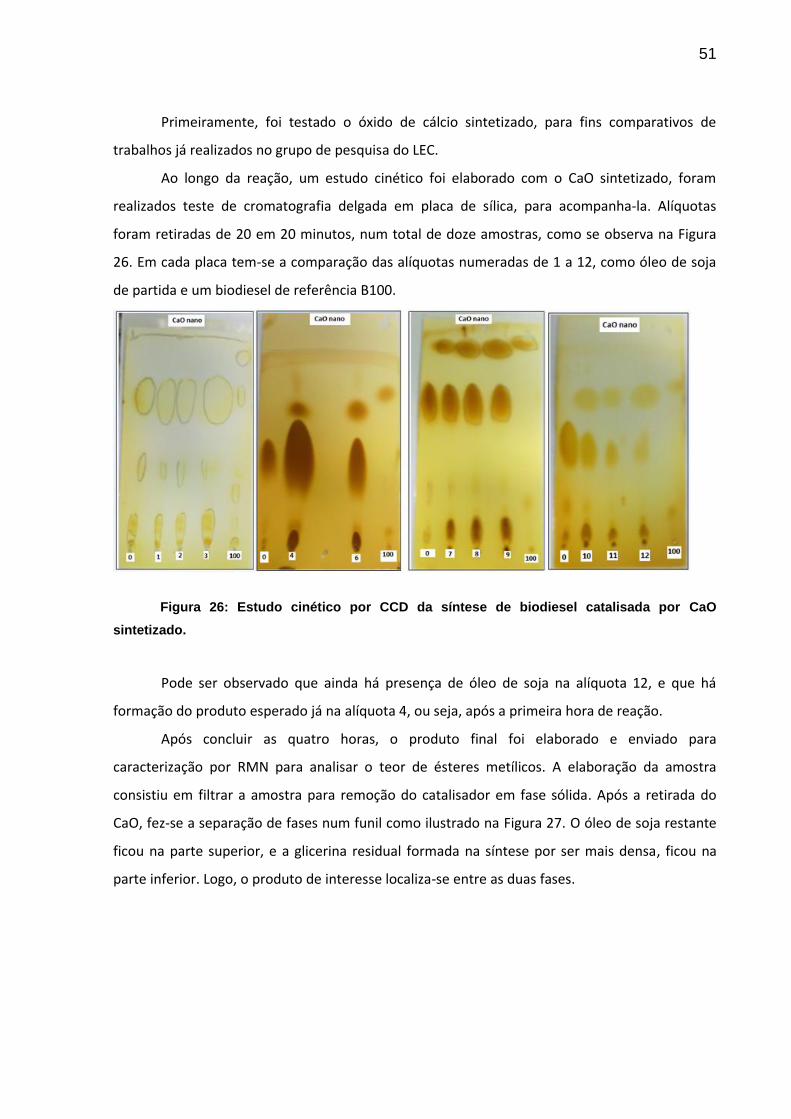
51
Primeiramente, foi testado o óxido de cálcio sintetizado, para fins comparativos de
trabalhos já realizados no grupo de pesquisa do LEC.
Ao longo da reação, um estudo cinético foi elaborado com o CaO sintetizado, foram
realizados teste de cromatografia delgada em placa de sílica, para acompanha-la. Alíquotas
foram retiradas de 20 em 20 minutos, num total de doze amostras, como se observa na Figura
26. Em cada placa tem-se a comparação das alíquotas numeradas de 1 a 12, como óleo de soja
de partida e um biodiesel de referência B100.
Figura 26: Estudo cinético por CCD da síntese de biodiesel catalisada por CaO
sintetizado.
Pode ser observado que ainda há presença de óleo de soja na alíquota 12, e que há
formação do produto esperado já na alíquota 4, ou seja, após a primeira hora de reação.
Após concluir as quatro horas, o produto final foi elaborado e enviado para
caracterização por RMN para analisar o teor de ésteres metílicos. A elaboração da amostra
consistiu em filtrar a amostra para remoção do catalisador em fase sólida. Após a retirada do
CaO, fez-se a separação de fases num funil como ilustrado na Figura 27. O óleo de soja restante
ficou na parte superior, e a glicerina residual formada na síntese por ser mais densa, ficou na
parte inferior. Logo, o produto de interesse localiza-se entre as duas fases.

52
Figura 27: Separação do Biodiesel da glicerina residual
4.6.1.1 Determinação do Teor de Ésteres
O teor de ésteres do biodiesel foi determinado por RMN de 1H utilizando CDCl3 como
solvente e Espectrômetro Bruker AVANCE DPX 200 nas mesmas condições descritas no subitem
3.3. Para cálculo dos teores foram determinadas algumas áreas específicas (A), sendo uma delas
referente ao hidrogênio alfa carbonílico, comum a mono, di e triglicerídeos e biodiesel, e a outra
referente a hidrogênio específico dos ésteres de metanol. A fórmula utilizadas, bem como o
deslocamento químico do sinal, são mostrados na Tabela 9.
Tabela 9: - Fórmula utilizada para quantificação do Teor de Éster por RMN de H1
Tipo de Éster Alquílico Cálculo do Teor de Éster Posição do Sinal (ppm)
H-Met Hα
Metílico % E = (2AH-Met x 100)/3AHα 3,70 - 3,64 2,30 - 2,20 Disponível em Gelbard, 1995.
Para auxiliar no cálculo do teor de éster, a identificação do hidrogênio que deu origem ao
sinal químico do H-Met do álcool utilizado na síntese apresenta desdobramento spin-spin 1, que
representa um singleto, pois não existe hidrogênios ao carbono vizinho, o valor de integração é
3, a origem química desde sinal é RO-CH3.

53
Na Figura 28 tem-se o espectro de RMN H1 do produto da reação de transesterificação
utilizando CaO sintetizado como catalisador.
Figura 28: Espectro de RMN Espectros de RMN de 1H do produto de síntese após 4
horas de reação com CaO (200 MHz, CDCl3).
Com base nos cálculos, percebeu-se um teor de ésteres de 92%, nesta síntese. Em
comparação com CaO comercial, seria mais apropriado a síntese durar mais algum tempo ou
melhorar as condições, por exemplo, aumentando a temperatura para proporcionar maior
energia cinética entre catalisador, moléculas de álcool e óleo.
4.6.2 Óxido de Magnésio
O óxido de magnésio é único em sua basicidade, com um pKa próximo de 16,
sendo a ligação entre o magnésio e o oxigênio coordenada octaédricamente. Explorando essa
natureza básica do MgO, planejou-se a síntese de biodiesel nas mesmas condições em que foi
realizado o outro teste anteriormente descrito.
Ao decorrer 4 horas de reação verificou-se através de CCD, que nas condições
descritas ainda não se tinha o produto esperado, o perfil do produto analisado está nitidamente
igual ao óleo de soja indicado por 0 na figura. Com isso, foram sendo acompanhadas hora a hora,
a reação e depois após 48 horas ainda era presente muito óleo de soja sem reagir. Por motivos

54
práticos, ao completar 72 horas, foi desligada a reação e o produto analisado via RMN para
avaliar o catalisador em teste.
O teor de éster do biodiesel catalisado por MgO (sintetizado) foi analisado por RMN H1,
de forma similar a análise anteriormente descrita no item 4.6. De acordo com a tabela 9 foi
realizado o cálculo de teor de éster.
O espectro de RMN do produto sintetizado após 72 horas de reação com MgO é
apresentado na Figura 29.
5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Inte
nsity
0.480.08 0.080.080.08 0.060.050.04
5.2
6
3.5
7
2.7
0
Figura 29: Espectros de RMN de 1H do produto de síntese após 72 horas de reação
com MgO (200 MHz, CDCl3).
O teor de ésteres calculado foi de 100%, isso não quer dizer que o rendimento da reação
foi ótimo e sim que a conversão do catalisador é excelente. Para melhor comparação seria
necessário analisar via cromatografia gasosa para saber o real rendimento da síntese. Mas para
fins exploratórios, percebe-se que ainda se deve estudar rotas de síntese para esse catalisador.
A ANP, em sua resolução n° 7 de 19 de março de 2008 define a cromatografia gasosa
como o ensaio adequado para definir a pureza do biodiesel, pois nesse método leva-se em
consideração a massa de biodiesel em relação à massa total de amostra. Já a caracterização via
RMN, dá uma idéia da conversão da transesterificação, ou seja, qual o percentual de
triglicerídeos que se transformou em éster.

55
5 CONCLUSÕES E SUGESTÕES
O trabalho realizado teve um caráter exploratório de aplicação da Rota Carbotérmica
Verde visando a obtenção de materiais contudo cálcio, magnésio, estanho, cobre e níquel que
são potenciais catalisadores da reação de transesterificação para produção de biodiesel.
Não foi possível quantificar as dimensões das estruturas sintetizadas por MEV, mas os
resultados mostram que foram obtidos materiais porosos, como morfologia tipo algodão.
Foram obtidos óxidos de cálcio (CaO), magnésio (MgO), estanho (SnO), sendo testados os
CaO e MgO como catalisadores. Os resultados mostram que o CaO tem maior eficiência por
permitir elevada conversão em 4 horas de reação ( teor de 92%). O MgO levou a um produto de
alta pureza (teor de éster 100%) com um tempo elevado de 72 horas. Este último resultado
poderá ser melhorado usando MgO sem tratamento térmico a 600°C e em temperaturas
superiores da de refluxo do metanol.
O trabalho mostrou o potencial desta rota deverá ter continuidade.

56
REFERÊNCIAS
ANP, Agência Nacional do Petróleo, Gás Natural e Biocombustívei. Especificação do Biodiesel – resolução ANP nº 7 de 19 de março de 2008. Disponível em <http://nxt.anp.gov.br/nxt/resolucoes_anp/2008/> Acesso em 11 de Novembro de 2012.
Araújo, R.C.S.; Pasa, V.M.D.; Melo, B.N.; Effects of biopitch on the properties of flexible polyurethane foams, European Polymer Journal, 2005, 41, 1420-1428.
Endalew, A.K.; Kiros, Y.;Zanzi, R.; Inorganic Heterogeneous Catalysts for Biodiesel Prodion from Vegetable Olis, Biomass and Bioenergy, 2011, 35, 3787- 3809.
Ferreira, H.S.; Rangel, M.C.; Nanotecnologia: Aspectos gerais e potencial de aplicações em catálise, Química Nova, 2009, 32, 1860-1870.
Fukuda, H.; Akihko, A.; Noda, H.; Biodiesel Fuel Production by Transesterification of Oils; Journal of Bioscience and Bioengineering, 2001, 92, 405-416.
Gelbard, G.; Brès, O.; Vargas, R.M.; Vielfaure, F.; Scuchardt; U. 1H nuclear magnetic resonance determination of the yield of the transesterification of rapessed oil with methanol, Journal of American Oil Chemical Society, 1995, 72, 1239-1241.
Gonçalves, J.A.; Ramos, A.L.D.; Rocha, L.L.L.; Domingos, A.K.; Monteiro, R.S.; Peres, J.S.; Furtado, N.C.; Taft, C.A.; Aranda, D.A.; Niobium Oxide Solid Catalyst: Esterification of Fatty Acids and Modeling for Biodiesl Prodution, Journal of Physical Organic Chemistry, 2010, 54-64.
Holanda, A.; Biodiesel e Inclusão Social; Brasília, Câmara dos Deputados, Coordenação de publicações; 2004.
Jacobson, K.; Gopinath, R.; Meher, L.C.; Dalai, A.K.; Solid Acid Catalyzed Biodiesel Production from Waste Cooking oil. Applied Catalysis B: Environmental, 2008, 85, 86-91.
Kima, H, J,; Kanga, B.S.; Kima, M.J.; Park, Y.M.; Kimb, D.K.; Lee, J.S.; Lee, K.Y.; Transesterification
of Vegetable Oil to Biodiesel Using Heterogeneous Base Catalyst. Catalysis Today, 2004, 93-95,
315-320.
Kiss, A. A.; Dimian, A. C.; Rothenberg, G.; Solid Acid Catalysts for Biodiesel Production. Towards Sustainable Energy. 2005, 348, 75-81.
Knothe, G.; Dunn, R.O.; Bagby, M.O.; The use of vegetable oils and their derivatives as alternative diesel fuels, 2007. Disponível em:< http://www.biodiesel.org/resources > Acesso em 30 de fevereiro de 2013.
Maciel, A.V.; Mussel, W.N.; Pasa, V.M.D.; A Novel Synthesis of Nanostructured ZnO via Thermal Oxidation of Zn Nanowires Obtained by a Green Route. Materials Sciences and Applications, 2010, 1, 279-284.

57
Marchetti, J.M.; Miguel, V.U.; Errazu; A.F.; Possible Methods for Biodiesel Production, Renewable & Sustentable Energy Reviews, 2007, 11, 1300-1311.
Parente, J.E.S.; Biodiesel: Uma Aventura Tecnológica num País Engraçado. Fortaleza. Unigráfica, 2004.
Prauchner, M.J.; Pasa, V.M.D.; Otani, S.; Otani, C.; Biopitch-based general purpose carbon fibers: processing and properties, Carbon, 2005, 43, 591-597.
Portal Brasil. Produção de Biodiesel gerou mais de R$ 2 bilhões para agricultura familiar, disponível em <http://www.brasil.gov.br> Acesso em 28 de março de 2013.
Schuchardt, U.; Sercheli, R.; Vargas, R,M.; Transesterification of Vegetable Oils: A Review, Journal of Brazilian Chemical Society, 1998, 9, 199-210. Schuchardt, U.; Garcia, C.M.; Teixeira, S.; Marciniuk, L.L.; Matérias-primas Alternativas para Produção de Biodiesel por Catálise Ácida, I Congresso da Rede Brasileira de Tecnologia de Biodiesel, Book Abrstract, 2006, 2, 300-303.
Sharma, Y.C.; Singh, B.; Upadhyay, S.N.; Advancements in Development and Characterization of Biodiesel: A review, Fuel, 2008, 87, 2355-2373.
Yang, W.; Peters, J. I.; Inhaled Nanoparticles - A Current Review, International Journal of Pharmaceutics, 2008, 356, 239-247.

58
ANEXO A
O espectro de RMN 1H do biodiesel podem ser interpretados de acordo com a figura
abaixo, atribuições dos hidrogênios.





























