



CURS 4
•Ciclul acizilor tricarboxilici
•Lanţul respirator mitocondrial, navete mitocondriale
•Glicozaminoglicani, proteoglicani, glicoproteine

Ciclul acizilor tricarboxilici (citric, Krebs)
– Definiţie, localizare, generalităţi
– Surse de acetil-coA
– Etape, reacţii
– Reglarea ciclului Krebs
– Importanţa ciclului Krebs

Ciclul Krebs – definiţie, localizare
• Definiţie: cale finală de catabolizare a glucidelor, aminoacizilor şi acizilor graşi în condiţii de aerobioză
• = rol foarte important în metabolismul intermediar şi în producerea de energie
• Localizare: reacţiile au loc în mitocondrie (faţă de
glicoliză = citoplasmatică)
– enzimele ciclului = prezente mai ales în matricea
mitocondrială, în proximitatea enzimelor lanţului transportor
de electroni
– celulele fără mitocondrii (hematiile) sunt lipsite de ciclul citric

Ciclul Krebs - generalităţi
• Porneşte de la acetil-CoA:
– metabolit intermediar comun
al catabolismului glucidic, al
acizilor graşi şi AA)
– Substratul bicarbonic (2C) al
ciclului Krebs
• Oxidarea acetil-CoA în
ciclul Krebs la CO2 şi H2O
produce energie, stocată
în molecula de GTP şi a
echivalenţilor reducători
(NADH, H+ şi FADH2) pe
care îi generează
CH3 CO SCoA
Aminoacizi Acizi grasiGlucozã
Colesterol Corpi
cetonici
biosintezã biosintezãAcizi grasi
biosintezã
Ciclul citric
Acizi
biliariHormoni
steroizi
NADH,H+
FADH2
CO2
ATP
lant
respirator

Surse de acetil-CoA
1. Catabolismul glucidic
• Decarboxilarea oxidativă a Py
2. Catabolismul proteic
– Decarboxilarea oxidativă a Py
provenit din AA glucogeni
– Formarea acetil-CoA din AA
cetogeni și mixști
3. Catabolismul lipidic
• Beta-oxidarea acizilor graşi
4. Catabolismul corpilor cetonici
5. Catabolismul etanolului
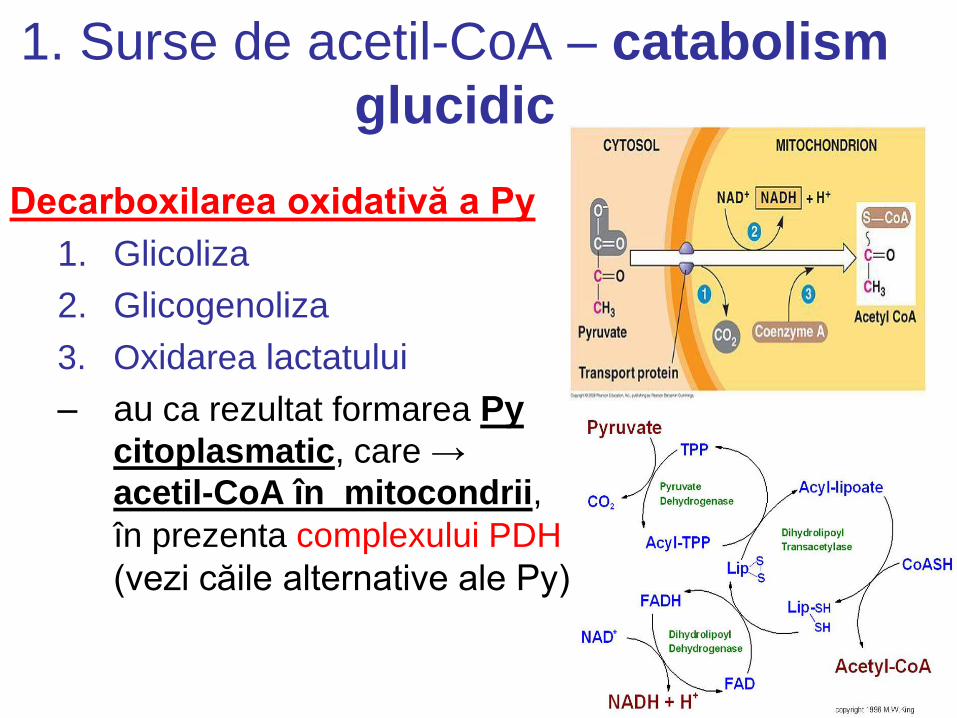
1. Surse de acetil-CoA – catabolism
glucidic
Decarboxilarea oxidativă a Py
1. Glicoliza
2. Glicogenoliza
3. Oxidarea lactatului
– au ca rezultat formarea Py citoplasmatic, care →
acetil-CoA în mitocondrii,
în prezenta complexului PDH
(vezi căile alternative ale Py)

2. Surse de acetil-CoA – catabolism proteic
1. Decarboxilarea oxidativă a Py provenit din AA glucogeni
• AA glucogeni → Py citoplasmatic, care → acetil-CoA în mitocondrii, în
prezenta complexului PDH (vezi căile alternative ale Py)
2. Formarea acetil-CoA din AA cetogeni şi AA mixşti

3. Surse de acetil-CoA – catabolism lipidic
• Prin beta-oxidarea acizilor graşi → acetil-CoA

4. Surse de acetil-CoA – catabolismul corpilor cetonici
• Corpii cetonici sunt produşi în ficat
• Ficatul nu poate utiliza corpii cetonici deoarece nu deţine enzima activatoare a acetoacetatului
• Ţesuturile extrahepatice deţin enzima necesară activării acetoacetatului (sunt capabile să utilizeze corpii cetonici)

Catabolismul corpilor cetonici
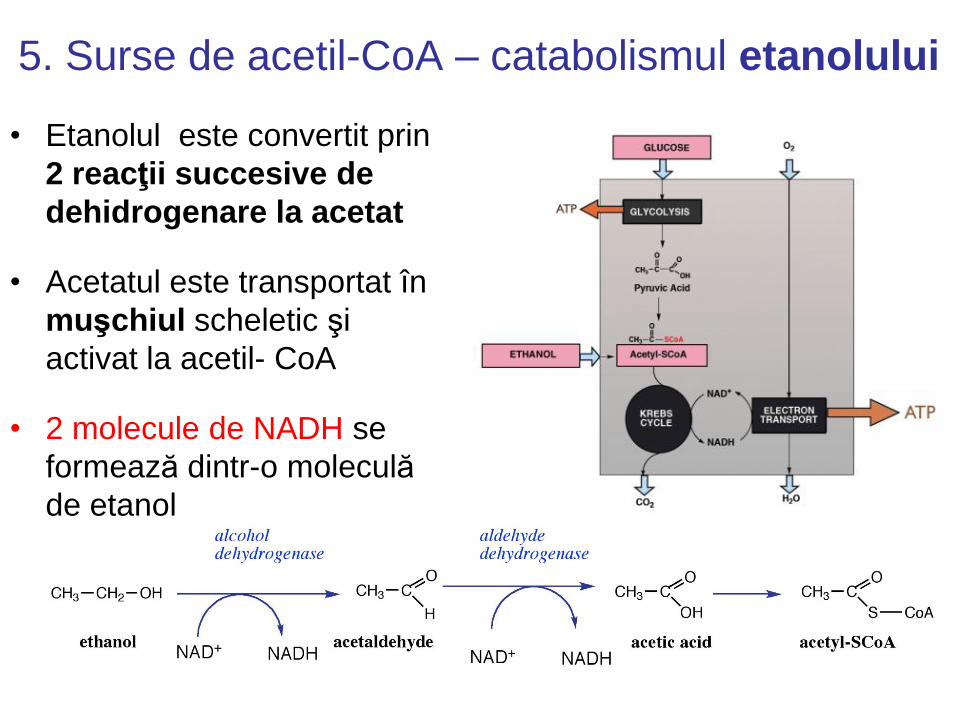
5. Surse de acetil-CoA – catabolismul etanolului
• Etanolul este convertit prin
2 reacţii succesive de
dehidrogenare la acetat
• Acetatul este transportat în
muşchiul scheletic şi
activat la acetil- CoA
• 2 molecule de NADH se
formează dintr-o moleculă
de etanol

Oxaloacetatul şi ciclul Krebs
• Deşi OAA este regenerat în fiecare ciclu, trebuie să fie suplimentat permanent pentru menţinerea generării de produşi intermediari. Surse de OAA:
• Transaminarea acidului aspartic (sub acţiunea ASAT = reversibil) cu formarea acidului glutamic şi OAA.
– Asparagina se dezaminează şi formează acidul aspartic
• Carboxilarea piruvatului, în prezenţa piruvat-carboxilazei (ireversibil):
– Activă între mese, în timpul GNG
– Funcţionează în prezenţa biotinei
– Necesită ATP
– NU FUNCTIONEAZĂ în absenţa acetil-CoA (indică nevoia de acetil-CoA)

Ciclul Krebs – etape, reacții Ciclul Krebs formează un ansamblu de 8 reacţii care se
desfăşoară în matricea mitocondrială, în celulele aerobe
• 2. formare IZOCITRAT (aconitaza-Fe2+)
• 3. formare α-CETOGLUTARAT (izocitrat-DH, NAD+,
Mg2+, Mn2+). Rezultă NADH+H+
• 4. formare SUCCINIL-CoA (α-Cetoglutarat–DH;
necesită coenzime, generează NADH+H+
• 5. formare SUCCINAT ( succinat-tiokinaza); se
produce GTP. GTP este convertit la ATP.
• 6. formare FUMARAT ( succinat-DH); rezultă FADH2
• 7. formare MALAT (fumaraza). Funcţionează ca
navetă pentru OAA în citoplasmă. Poate fi convertit la
Py, OAA şi fumarat.
• 8. formare OAA (malat-DH), se generează NADH+H+
• 1. condensare acetilCoA + OAA → CITRAT (citrat sintaza)

Ciclul Krebs – etape, reacții

1. Formarea citratului • Reacţie ireversibilă de condensare dintre acetil-CoA (2C)
şi OAA (4C) → citrat (6C)
• Enzima implicată: citrat-sintaza, inhibată de: citrat, ATP,
NADH, succinil-CoA
– Citratul inhibă:
• PFK (glicoliză)
– Citratul activează:
• Acetil-CoA-carboxilaza (AG)
CH3 CO SCoA
O C COOH
H2C COOH
OAA
Acetil-CoA
Acid citric
+
H2O CoASH
CS HO C COOH
H2C COOH
H2C COOH

2. Izomerizarea citratului la izocitrat
Acid citric
HO C COOH
H2C COOH
H2C COOH
Aconitazã
(Fe2+)
H2O
Acid cis-aconitic
C COOH
H2C COOH
HC COOH
HC COOH
HO CH COOH
Aconitazã
(Fe2+)
H2O
H2C COOH
Acid izocitric
• Reacţie reversibilă
• Enzima implicată: aconitaza – are un centru Fe-S
– inhibată de fluoro-acetat

3. Decarboxilarea oxidativă a izocitratului cu
formare de α-Cetoglutarat
• Reacţie ireversibilă, etapă limitantă de viteză
• Se formează prima moleculă de CO2 (din cele 2) şi prima de NADH+H+ (din cele 3) din ciclul Krebs
• Enzima implicată: izocitrat-dehidrogenaza:
– activată alosteric de ADP şi Ca2+
– inhibată de ATP şi NADH+H+

4. Decarboxilare oxidativă a α-cetoglutarat
cu formare de succinil-CoA • Reacţie ireversibilă
• Se formează a doua moleculă de CO2 (din cele 2) şi a doua de NADH+H+ (din cele 3) din ciclul Krebs
• Enzima implicată: complexul α-cetoglutarat dehidrogenază
– Asemănator Py→acetil-CoA (complexul PDH), doar că nu este influenţată de reacţii de fosforilare şi defosforilare
– Coenzime: tiamin-pirofosfat, acid lipoic, FAD, NAD+, CoA
– Activată de Ca2+
– Inhibată de ATP, GTP, NADH+H+ şi succinil-CoA
O C COOH
COOH
CH2
Acid -cetoglutaric
(-Kg)
CH2
-Cetoglutarat dehidrogenazãO C SCoA
H2C COOH
CH2
Succinil-CoA
CO2CoASHNAD+
NADH,H+

5. Clivarea succinil-CoA cu formare
de succinat
• Enzima implicată: succinat-tiokinaza
– Scindează legătura macroergică tioester, cuplată cu
fosforilare oxidativă legată de substrat
• Se formează unica moleculă de GTP din ciclul
Krebs care va regenera ATP
O C SCoA
H2C COOH
CH2
Succinil-CoA
H2O CoASH
GDP+Pi GTP
ADPATP
H2C COOH
H2C COOH
Acid succinic
STK

6. Oxidarea succinatului la fumarat
• Reacţie de dehidrogenare reversibilă
• Rezultă unica moleculă de FADH2
• Enzima implicată: succinat dehidrogenaza
– singura enzimă din ciclul Krebs situată în membrana
mitocondrială internă
– aparţine Complexului II a lanţului transportor de e-

7. Hidratarea fumaratului cu formare
de malat
• Reacţie reversibilă
• Enzima implicată: fumaraza
• Fumaratul se mai obţine în:
– ciclul ureei
– cursul sintezei purinelor
– cadrul catabolizării AA
(fenilalanină, tirozină) HO CH COOH
H2O
H2C COOH
COOHC
H HOOC
C
H
Acid fumaric
Fumarazã
Acid L-malic

8. Oxidarea malatului la OAA
• Reacţie de dehidrogenare reversibilă
• Enzima implicată: malat dehidrogenază
• A treia reacţie de formare a NADH din ciclul Krebs
HO CH COOH
H2C COOH
Acid L-malic
NAD+ NADH,H+
MDH O C COOH
H2C COOH
OAA
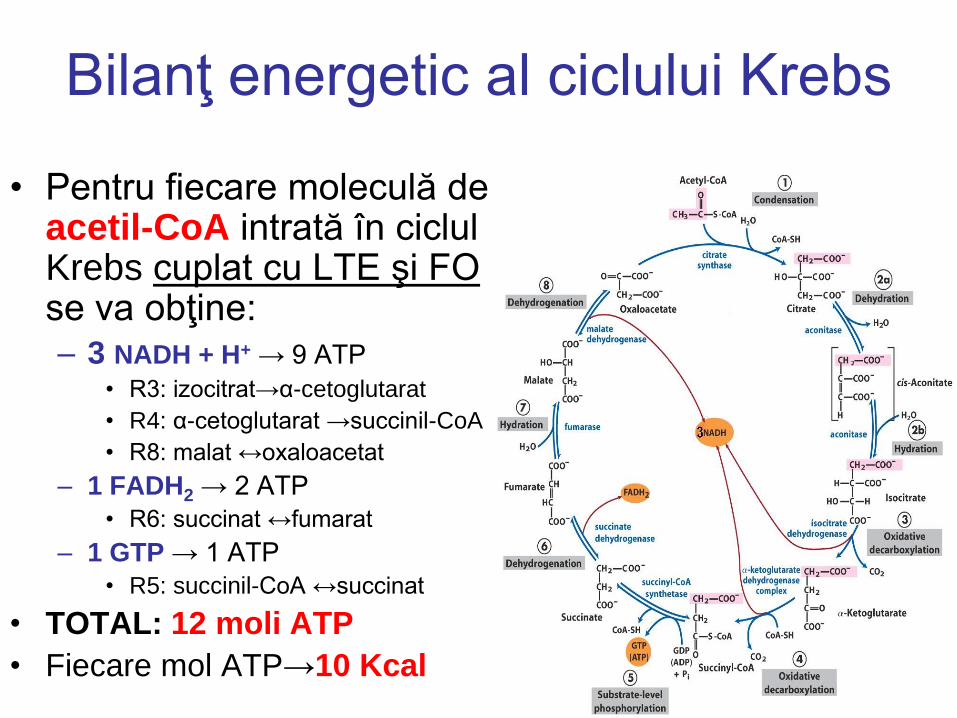
Bilanţ energetic al ciclului Krebs
• Pentru fiecare moleculă de acetil-CoA intrată în ciclul Krebs cuplat cu LTE şi FO se va obţine: – 3 NADH + H+ → 9 ATP
• R3: izocitrat→α-cetoglutarat
• R4: α-cetoglutarat →succinil-CoA
• R8: malat ↔oxaloacetat
– 1 FADH2 → 2 ATP
• R6: succinat ↔fumarat
– 1 GTP → 1 ATP
• R5: succinil-CoA ↔succinat
• TOTAL: 12 moli ATP
• Fiecare mol ATP→10 Kcal

Bilanţ energetic al arderii complete
a unei molecule de glucoză
• glicoliza aerobă→ 2 Py: – 2 ATP
– 2 NADH+H+ → 4 sau 6 ATP
– subtotal: 6-8 ATP
• Decarboxilare oxidativă (2 Py → 2 acetil-CoA): – 2 NADH+H+→ 6 ATP
– subtotal: 6 ATP
• Ciclul Krebs: – din 2 acetil-CoA→ 2 x 12 ATP
– subtotal: 24 ATP
TOTAL: 36-38 ATP:
• 36 ATP prin naveta glicerol-3-P
• 38 ATP prin naveta malat-aspartat

Reglarea ciclului Krebs
1. Reglare generală (în funcţie de necesităţi) via lanţul
respirator mitocondrial responsabil de generarea de ATP prin
oxidarea coenzimelor reduse (NADH+H+, FADH2)
• Nivelul ↑ ATP sau NADH+H+, va inhiba ciclul la nivelul
enzimelor:
• citrat sintetaza
• izocitrat dehidrogenaza
• α-cetoglutarat dehidrogenaza
• Invers, ↓ ATP (sau ↑ ADP) = stimul important al ciclului Krebs
Ciclul Krebs este inhibat în următoarele condiţii:
– Înfometare (lipsa glucidelor)
– Diabetul zaharat (lipsa insulinei)
– Condiţii anaerobe (lipsa mitocondriilor sau oxigenului)

Reglarea ciclului Krebs
2. Reglarea la nivelul enzimelor: CS, IC-
DH, αCG-DH. Aceste enzime sunt
inhibate de ATP şi NADH+H+.
– citrat-sintaza, inhibată de: citrat,
ATP, NADH+H+, succinil-CoA
– izocitrat-dehidrogenaza:
• Activată alosteric de ADP şi Ca2+
• Inhibată de ATP, NADH+H+
– α-cetoglutarat dehidrogenaza
• Activată de Ca2+
• Inhibată de ATP, GTP, NADH+H+
şi succinil-CoA

Importanţa ciclului Krebs
1. = cale catabolică comună a oxidării tuturor principiilor
alimentare (Glucide, AA, acizi graşi)
2. = sursă majoră de energie celulară (cuplat cu lanţul respirator
generează o mare cantitate de energie sub formă de ATP -
12 ATP), exceptând celulele fără mitocondrii
3. asigură conexiunea dintre metabolismele glucidic – lipidic –
proteic

Importanţa ciclului Krebs • Este o cale amfibolică: caracter dual, atât
catabolic cât şi anabolic:
– Catabolic –prin producere de energie
– Anabolic – oferă substraturi proceselor anabolice:
• sinteza Glu pe calea GNG [în înfometare, OAA (via malat) → PEP]
• Procese de transaminare (prin intermediul α-cetoglutaratului)
• Sinteza de aminoacizi (Asp din OAA; Glutamat din α-cetoglutarat; Ala din Py)
• Sinteza de neurotransmiţători [în creier:α-KG→Glutamat→ GABA (acid γ-aminobutiric)]
• Sinteza de acizi graşi (postprandial via citrat. Pentru a obţine acetil-CoA citosolic, celulele transportă citrat în citosol, unde este clivat la acetil-CoA şi OAA de către citrat-liază)
• Ciclul Krebs este sursa majoră de succinil-CoA folosită pentru:
– Sinteza hemoglobinei
– Activarea corpilor cetonici
– Detoxifiere prin conjugare (sulfonamide)
OAA Glutamat
NADH,H+
NAD+
CITOSOL
MITOCONDRIE
MDH(citosol)
Malat
Malat
MDH(mitocondrie)
OAA
NAD+
NADH,H+
-Kg
-Kg Aspartic
Glutamat
Aspartic
Transaminare
GOT
Transaminare
GOT

Transaminarea şi relaţia cu ciclul Krebs
• Transaminarea este reacţia dintre un AA şi un α- cetoacid în
care NH2 este transferat cu producerea unui nou α- cetoacid şi
a unui nou AA.
• Enzimele se numesc transaminaze şi fac legătura dintre
metabolismul glucidic şi proteic prin intermediul ciclului Krebs

Transaminazele de interes clinic
A. Alanin transaminaza (ALT, ALAT, GPT)
– Catalzează transferul NH2 al alaninei la acidul α-cetoglutaric, formând acid glutamic si acid piruvic.
B. Aspartat transaminaza (AST, ASAT, GOT)
– Catalizeaza transferul NH2 al acidului aspartic la acidul α-cetoglutaric, formând acid glutamic şi acid oxaloacetic.
• Coenzimele pentru transaminaze: piridoxal fosfat (forma activă a vit B6)
• Ţesuturile bogate în transaminaze: muşchiul cardiac, ficatul, muşchiul scheletal.

Aspecte esenţiale despre ciclul Krebs
1. ciclul Krebs are loc numai în celulele cu mitocondrii şi are nevoie de
enzimele lantului respirator mitocondrial pentru a produce energie (ATP)
2. Glucidele sunt sursa de Py, principalul furnizor de OAA, esenţial în iniţierea
ciclului Krebs.
3. Insulina (stimulează glicoliza, furnizând Py) stimulează enzimele ciclului
Krebs
4. Oxigenul este esenţial pentru un lanţ respirator intact şi activ
5. Ciclul Krebs este inactivat în inaniţie (lipsa glucidelor), diabet zaharat
(deficit de insulină), anaerobioză (lipsa mitocondriilor sau oxigenului).
6. Efect Pasteur = fenomen prin care ciclul Krebs este inhibat de prezenţa în
exces a oxigenului. (Mai puţine molecule de Glu sunt oxidate în aerobioză
decât in anaerobioză. Astfel, în prezenţa oxigenului nu este nevoie de
oxidarea mai multor molecule de Glu, iar rata glicolizei este mai mică decât
în ţesuturile anaerobiotice)

Lanţul respirator mitocondrial
-Lanţul transportor de electroni
-Cuplarea cu fosforilarea oxidativă
-Surse de NADH + H+ şi FADH2
-Navete mitocondriale
-Inhibitori şi decuplanţi ai lanţului respirator mitocondrial
-Boli mitocondriale

Lanţul respirator mitocondrial
– Elementele care constituie Lanţul
transportor de electroni (e-) (LTE)
– Unitatea de fosforilare oxidativă (FO)
• Intermediarii reacţiilor de oxidare din
căile metabolice (ex. glicoliza)
cedează e- către coenzime specifice:
– NAD+ (nicotinamid-adenil-dinucleotidul)
– FAD (flavin-adenin-dinucleotidul)
• Rezultând formele reduse (bogate în
energie):
– NADH + H+
– FADH2
Lanţul respirator mitocondrial = ansamblu de proteine
localizate în membrana internă mitocondrială, alcătuit din :

• e- parcurg LTE şi îşi pierd o mare
parte a energiei libere
• O parte a energiei este captată şi
depozitată prin sinteza de ATP
(←ADP + Pi) = procesul de
fosforilare oxidativă (FO)
• Restul energiei este utilizat în reacţii
auxiliare:
– transportul Ca2+ în mitocondrii
– termogeneză
• Aceste forme reduse (NADH + H+ şi FADH2 ) cedează 2e- unui grup
specializat de transportori de e- = lanţ transportori de e- (LTE)

Lanţul transportor de electroni
• LTE = cale comună finală prin care e- sunt
transferaţi pe molecula de O2
• Localizat în membrana internă a mitocondriei
• Compus din:
– 5 complexe proteice (enzimatice) fixe:
• complexele I→IV aparţin LTE
• Complexul V este unitatea de FO
– 2 elemente mobile:
• Coenzima Q (ubiquinona) de natură lipidică
• Citocromul c, de natură proteică

Lanţul transportor de electroni
• Complexul I format din: – NADH-dehidrogenaza (coenzimă FMN)
– Proteine cu Fe şi S = centre Fe-S (Fe4S4 sau
Fe2S2), cu rol de a delocaliza e-, modificând
orbitalii acestora
• Complexul III (ubiquinol-citocrom c-reductaza) format din: – Citocromii b şi c1
– Proteine cu Fe şi S
• Complexul II format din: – Succinat-dehidrogenaza (coenzimă FAD)
– Proteine cu Fe şi S

Lanţul transportor de electroni
• Complexul IV (citocrom c-oxidaza) format din: – Citocromii a şi a3
– loni de Cu şi Zn
– Singurul transportor de e- în care Fe hemului conţine un situs de
conjugare capabil să reacţioneze direct cu O2.
• La acest nivel: e- transportaţi, O2 şi protonii liberi se unesc şi formează H2O
Cele 4 complexe au rol de a separa şi transporta e- şi protonii proveniţi din
substrate cu scopul de a crea un gradient de protoni între matricea
mitocondrială şi spaţiul intermembranar
Citocromii: – conţin o grupare hem (nucleu porfirinic şi 1 atom Fe)
– Fe este transformat reversibil Fe2+(feros) ↔ Fe3+ (feric) (! ≠ hemoglobină)
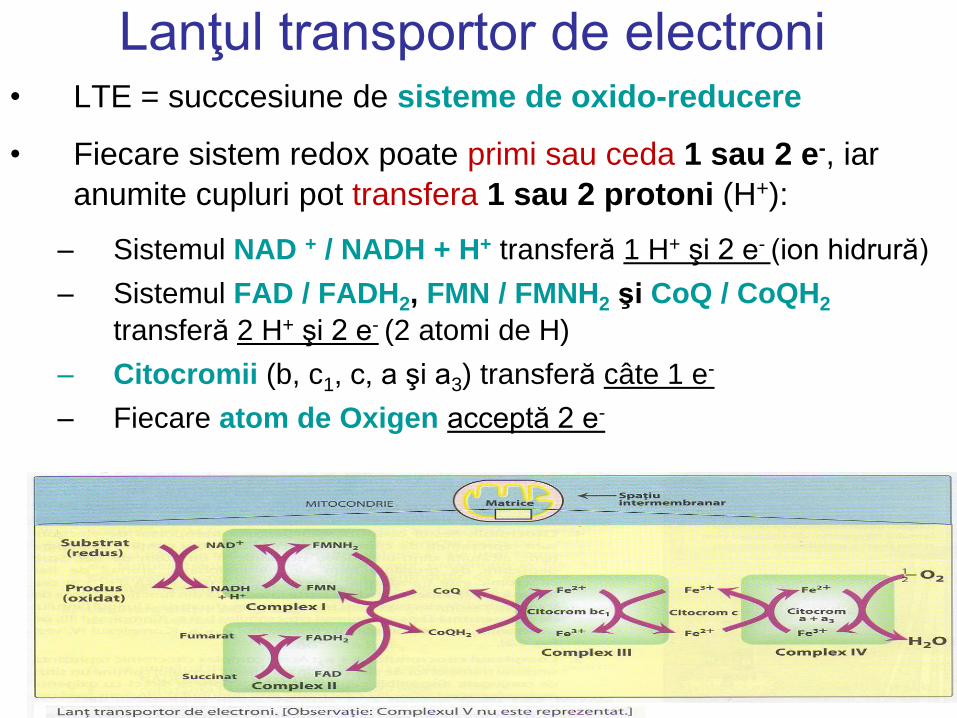
Lanţul transportor de electroni • LTE = succcesiune de sisteme de oxido-reducere
• Fiecare sistem redox poate primi sau ceda 1 sau 2 e-, iar
anumite cupluri pot transfera 1 sau 2 protoni (H+):
– Sistemul NAD + / NADH + H+ transferă 1 H+ şi 2 e- (ion hidrură)
– Sistemul FAD / FADH2, FMN / FMNH2 şi CoQ / CoQH2
transferă 2 H+ şi 2 e- (2 atomi de H)
– Citocromii (b, c1, c, a şi a3) transferă câte 1 e-
– Fiecare atom de Oxigen acceptă 2 e-

Lanţul transportor de electroni
• Există:
1.Un transfer al e- şi H+ (numiţi echivalenţi
reducători) până la CoQ
2.Transfer exclusiv de e- (via citocromi) de la
CoQH2→O2

1. Transferul de e- şi H+ (echivalenţi reducători)
până la CoQ
• NADH + H+ este preluat de Complexul I şi oxidat la NAD sub acţiunea NADH-dehidrogenazei
• concomitent coenzima FMN se reduce → FMNH2 care cedează echivalenţi reducători către CoQ rezultând CoQH2 (ubiquinol)
• Succinatul este preluat de complexul II şi oxidat la fumarat sub acţiunea succinat-dehidrogenazei
• concomitent FAD se reduce → FADH2 care cedează echivalenţi reducători către CoQ rezultând CoQH2
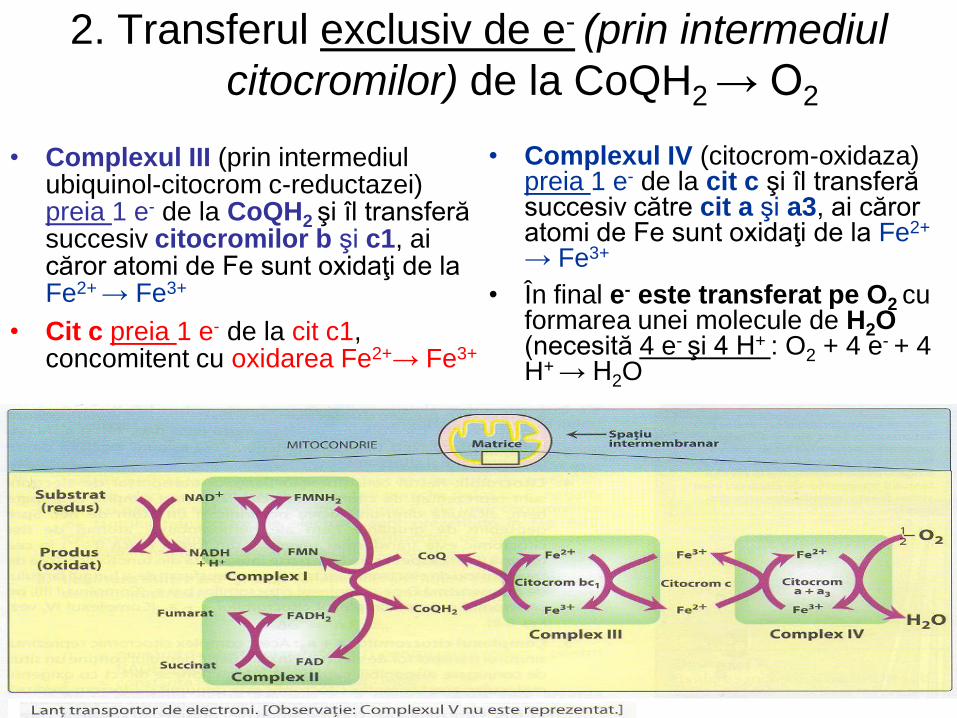
2. Transferul exclusiv de e- (prin intermediul
citocromilor) de la CoQH2 → O2
• Complexul III (prin intermediul ubiquinol-citocrom c-reductazei) preia 1 e- de la CoQH2 şi îl transferă succesiv citocromilor b şi c1, ai căror atomi de Fe sunt oxidaţi de la Fe2+ → Fe3+
• Cit c preia 1 e- de la cit c1, concomitent cu oxidarea Fe2+→ Fe3+
• Complexul IV (citocrom-oxidaza) preia 1 e- de la cit c şi îl transferă succesiv către cit a şi a3, ai căror atomi de Fe sunt oxidaţi de la Fe2+
→ Fe3+
• În final e- este transferat pe O2 cu formarea unei molecule de H2O (necesită 4 e- şi 4 H+ : O2 + 4 e- + 4 H+ → H2O

Cuplarea LTE cu FO
• Complexul V = ATP-sintetaza – este unitatea de FO
– aici are loc disiparea energiei gradientului de H+ generat de
LTE şi utilizarea sa pentru fosforilarea ADP → ATP
• Moleculele de ATP părăsesc matricea mitocondrială prin
intermediul unui transportor specializat: ATP/ADP-translocaza.
– Această translocază asigură transportul în sens invers al ADP, în
vederea refosforilării

Cuplarea LTE cu FO - teoria chemiosmotică
• Teoria chemiosmotică (Mitchel) explică cuplarea LTE cu FO:
– Energia generată de transportul echivalenţilor reducători de-a lungul LTE este recuperată şi utilizată pentru pomparea protonilor la nivelul complexelor I, III şi IV
– Aceasta generează un gradient electrochimic între cele 2 feţe ale membranei interne mitocondriale încărcate diferit (matricea mitocondrială - , spaţiul intermembranar +)
– Protonii concentraţi în spaţiul intermembranar au tendinţa de a reveni în interiorul matricei mitocondriale, aceasta fiind imposibilă datorită impermeabilităţii membranei interne

Cuplarea LTE cu FO - teoria chemiosmotică
– Fluxul de protoni se va concentra la nivelul complexului V
(ATP-sintetazei), al cărui canal (domeniul F0) străbate
membrana internă
– Acest flux determină rotaţia domeniului F0 şi o serie de
modificări conformaţionale ale domeniului extramembranar
F1 (ATP-sintetaza propriu-zisă) care va:
• stimula activitatea catalitică şi sinteza ATP (din ADP şi Pi)
• disiparea gradientelor electrochimice şi de pH

Cuplarea LTE cu FO - aplicaţie
• Oxidarea 1 mol NADH + H+ generează un
flux de protoni, realizat prin funcţionarea a
3 pompe de protoni de la nivelul
complexelor I, III şi IV, permiţând sinteza a
3 moli de ATP
• Oxidarea a 1 mol FADH2 generează un
flux mai redus de protoni (datorită
pompării acestora la nivelul complexelor III
şi IV), permiţând sinteza a 2 moli de ATP

Surse de NADH + H+ pentru LTE
• Surse mitocondriale de NADH + H+ : – Ciclul Krebs sursă majoră (fiecare acetil-CoA→ 3 molecule NADH + H+)
– Oxidare Py → acetil-CoA, sub acţiunea complexului PDH
– Catabolism lipide (oxidarea OH-acil-CoA rezultat din β-oxidarea AG)
– Catabolism corpi cetonici (oxidarea 3-OH-butirat → acetoacetat)
– Catabolism proteine (dezaminare oxidativă glutamat sau glicină)
– Catabolism alcool (acetaldehidă →acetat sub acţiunea acetaldehid-DH)
• Surse citoplasmatice de NADH + H+ : – Oxidarea GA-3P↔1,3-DPG în cursul glicolizei
– Conversia lactat ↔piruvat sub acţiunea LDH
– Oxidarea alcoolului (etanol→acetaldehidă sub acţiunea Alcool-DH)
!! NADH + H+ citoplasmatic NU poate traversa membrana mitocondrială (nu există transportor specializat)
Pentru oxidarea acestuia intervin navetele mitocondriale care preiau e- şi H+ şi îi transportă în interiorul mitocondriilor:
1. Naveta glicerol-3-fosfat
2. Naveta malat-aspartat

Navete mitocondriale
• Membrana externă mitocondrială este permeabilă pentru
majoritatea intermediarilor metabolici
• Membrana internă mitocondrială este permeabilă doar pentru
O2, CO2, NH3, corpi cetonici şi acizi graşi < 12 atomi de C
• Transferul intermediarilor metabolici de-o parte şi de alta a
membranei interne se face prin activitatea unor transportori
specifici, dependenţi de:
– aportul energetic
– diferenţele de pH sau de potenţial electric
• Moleculele care nu dispun de transportori specifici pot fi
transferate prin conversia prealabilă în intermediari care pot
fi transportaţi în interiorul mitocondriilor

Navetele glicerol-3-fosfat şi malat-aspartat
• Membranele mitocondriale sunt impermeabile
pentru NADH citoplasmatic şi nu dispun de un
transportor pentru acesta
• Aceste navete:
– transferă e- proveniţi de la NADH + H+, sub forma ionului
hidrură (H:-), spre un acceptor care beneficiază de un
transportor mitocondrial
– cuprind reacţii de oxidoreducere care au ca scop
regenerarea NAD+ citoplasmatic, esenţial pentru
desfăşurarea neîntreruptă a glicolizei

a. Naveta glicerol-3-fosfat
• Utilizează cele 2
enzime sinergice
(citoplamatică şi
mitocondrială) a
glicerol-3-PDH
• În final, se pierde 1
ATP prin această
navetă:
C O
CH2 OH
CH2 O P
H C OH
CH2 O P
DHAP Glicerol- P
CH2 OH
NADH,H+NAD+
Glicerol- PDH
din citosol
CITOSOL
MITOCONDRIE
CH2 OH
H C OH
CH2 O P
Glicerol- P
Glicerol- PDH
mitocondrialã
C O
CH2 O P
DHAP
CH2 OHFADFADH2
• 1 NADH + H+ (citoplasmatic) → 1 FADH2 mitocondrial preluat de LTE la nivelul CoQ → 2ATP
Faţă de
• 1 NADH + H+ (mitocondrial) este preluat de LTE la nivelul Complexului I → 3ATP

b. Naveta malat-aspartat
• Utilizează reacţii de:
– dehidrogenare sub
acţiunea MDH
(malat-DH)
– transaminare sub
acţiunea GOT
(glutamat-
oxaloaacetat-
transaminazei)
OAA Glutamat
NADH,H+
NAD+
CITOSOL
MITOCONDRIE
MDH(citosol)
Malat
Malat
MDH(mitocondrie)
OAA
NAD+
NADH,H+
-Kg
-Kg Aspartic
Glutamat
Aspartic
Transaminare
GOT
Transaminare
GOT
• NADH + H+ regenerat mitocondrial este preluat ca substrat al
Complexului I al LTE și cuplat cu FO → 3ATP

Naveta citrat-malat-piruvat
• Citratul intramitocondrial poate fi exportat în citoplasmă prin intermediul unui transportor specific
• Acest mecanism permite ieşirea grupărilor acetil din mitocondrii (acetil-CoA nu are un transportor specializat)
• Regenerarea acetil-CoA citoplasmatic este punctul de plecare pentru biosinteza acidului palmitic, în perioadele alimentare

Naveta malat-oxaloacetat si ciclul
fumarat-aspartat
Naveta malat-oxaloacetat utilizează izoenzimele citoplasmatică şi mitocondrială ale MDH (malat-DH) şi transportorul malat-cetoglutarat
• Scop = exportul citoplasmatic al OAA şi NADH mitocondrial
• rol esenţial în iniţierea GNC
Ciclul fumarat-aspartat regenerează aspartatul necesar ureogenezei
• stabileşte o punte între ciclul Krebs şi ciclul ureogenezei hepatice, prin
intermediul malat şi OAA mitocondrial

Surse de FADH2 pentru LTE
• Intră în LTE la nivelul CoQ pe care o reduce →
CoQH2
• Se formează în interiorul mitocondriilor prin:
– Oxidarea Glicerol-3-P → DHAP (naveta Gli-3-P)
– Oxidarea succinat → fumarat (ciclul Krebs)
– Oxidarea acil-CoA (beta-oxidarea AG)

Inhibitori ai lanţului respirator mitocondrial
• Complexul I: barbiturice, rotenona.
• Complexul II: malonat (similar succinatului, existând o inhibiţie competitivă)
• Complexul III: antimicina A (la niv. cit b), myxothiazol (la niv. centrelor Fe-S)
• Complexul IV: CO, H2S, HCN → deces
• Complexul V: oligomicină, arsenic
• Coenzima Q: doxorubicina (chimioterapic)
• Translocaza: atractilat

Decuplanţi ai lanţului respirator mitocondrial
• Decuplează LTE de FO: – 2,4-dinitrofenol
– Fenil-hidrazone
– Anumite antibiotice
– Dicumarol (anticoagulant)
– Termogenina
• Rezultat: energia din lanțul respirator mitocondrial → energie pentru termoreglare
• Ex.: termogenina:
– proteină eliberată la nivelul terminaţiilor nervoase simpatice şi ţesutului brun (dezvoltat la nou-născuţi şi animalele care hibernează)
– Sub acţiunea frigului sau după o masă, eliberarea termogeninei duce la consumarea energiei lanţului respirator pentru producerea de căldură

Bolile mitocondriale
• cauzate de mutaţiile genelor care codifică
proteinele sau ARN mitocondrial
• Proteinele localizate în mitocondrii pot fi
codificate de gene:
– mitocondriale
– nucleare (proteinele = transferate apoi în mitocondrii
după sinteza citoplasmatică)

Bolile mitocondriale -particularităţi
• Transmiterea mendeliană
– dacă mutaţia este la nivelul unei gene nucleare
• Transmiterea matrilineară
– dacă mutaţia afectează genomul mitocondrial (ADN mitocondrial este transmis exclusiv pe linie maternă)
• Segregarea replicativă
= repartiţia aleatorie a mitocondriilor afectate şi a celor normale în urma
diviziunii celulare → mare varietate de simptome clinice
• Acumularea progresivă a mutaţiilor ADN mitocondrial (urmare a acumulării radicalilor liberi, formaţi permanent la nivelul lanţului respirator)
– manifestările clinice apar după atingerea unui prag, în organe cu necesităţi energetice ridicate (miocard, muşchii scheletici şi respiratori, sistemul nervos)

Bolile mitocondriale – manifestări:
Bolile cauzate de anomalii ale lanţului respirator pot debuta în :
• perioada neonatală, în cazul deficienţelor grave
• copilărie, dacă activitatea enzimatică reziduală permite sintetiza unui număr minim de molecule de ATP
Pacienţii prezintă:
• encefalopatie metabolică
• hipotonie severă
• cardiomiopatie
• steatoză hepatică şi insuficienţă renală
• acidoza lactică datorită:
– inhibării retroactive a ciclului Krebs şi conversiei integrale PY → lactat
• accentuarea cetogenezei → hipercetonemia şi raportul beta-hidroxibutirat / acetoacetat

Bolile mitocondriale – exemple:
• Sindromul OXPHOS
– boli cauzate de disfuncţia unuia dintre complexele lanţului respirator
• Sindromul MERRF (myoclonic epileptic ragged red fiber disease)
– afecţiune neurodegenerativă
– produsă de mutaţii ale genei mitocondriale pentru ARNt-Liz →perturbarea biosintezei mitocondriale a proteinelor
• Sindromul MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes)
– afecţiune neurodegenerativă letală
– produsă de mutaţii ale mai multor gene mitocondriale, inclusiv cele pentru ARN de transfer

• Glicozaminoglicani, proteoglicani, glicoproteine
– Definiție, structură
– Clasificare
– Caracteristici, rol
– Importanță medicală

Glicozaminoglicani, proteoglicani,
glicoproteine
Definiții:
• Glicozaminoglicanii (sau mucopolizaharidele) =
heteropolizaharide liniare, încărcate negativ, componenţi
ai matricei extracelulare
• Asociate cu o cantitate de proteine, formează proteoglicanii
– conţin carbohidraţi în cantitate mare
• Spre deosebire, glicoproteinele conţin în structura lor
predominant proteine, carbohidraţii fiind în cantitate
redusă

Glicozaminoglicani
Structură:
• sunt compuşi din unităţi
dizaharidice repetitive de tipul
(glucid acid – glucid aminat)n
– Glucidul acid este:
• acidul D-glucuronic, sau
• acidul L-iduronic
– Glucidul aminat este:
• D-glucozamina, sau
• D-galactozamina
– Glucidul aminat este frecvent
acetilat sau poate fi sulfatat.

Proteoglicani Structură:
• lanţul polizaharidic (unitatea
dizaharidică repetitivă) se leagă de
structura proteică cel mai frecvent prin
intermediul unei regiuni de legătură
reprezentată de o unitate trizaharidică:
xiloză-galactoză-galactoză:
– aceasta face legătura între unitatea
dizaharidică repetitivă a glicozaminoglicanilor
şi gruparea -OH a unui rest de serină din
structura proteinei
• monomerii de proteoglicani astfel formaţi
se ataşează unei molecule de acid
hialuronic pentru a forma ulterior
agregate de proteoglicani

Glicoproteinele Structură:
• sunt proteine care au ataşate covalent
catene carbohidrate (de tip oligozaharidic):
– în general scurte (2-10 unităţi glucidice,
dar pot fi şi mai lungi)
– ramificate (nu liniare)
– fără unităţi dizaharidice repetitive în
structură
– ataşate de proteine prin legături de tip N-
sau O-glicozidic:
• În cazul legăturii N-glicozidice, catena
glucidică se leagă la gruparea –NH2 a
unei asparagine din structura proteinei
• În cazul legăturii O-glicozidice, catena
carbohidrată se leagă la gruparea –OH a
unei serine sau treonine din structura
proteinei

Glicoproteinele
Roluri:
• Sunt componente ale matricei extracelulare
• Rol de lubrefianţi (fiind componenţi ai mucusurilor)
• Rol în comunicarea intercelulară
• Rol de apărare (imunoglobulinele, etc)
• Rol în antigenicitatea de suprafaţă (antigenele de grup sanguin)
• Rol de hormoni (gonadotropinele, etc)
• Rol în coagularea sângelui (fibrinogenul, etc)
• Rol de transport (ceruloplasmina, feritina, haptoglobina etc)

Clasificarea glicozaminoglicanilor
Sunt 6 tipuri majore de glicozaminoglicani:
1. Condroitin sulfați
2. Keratan sulfați
3. Acid hialuronic
4. Dermatan sulfați
5. Heparină
6. Heparan sulfați
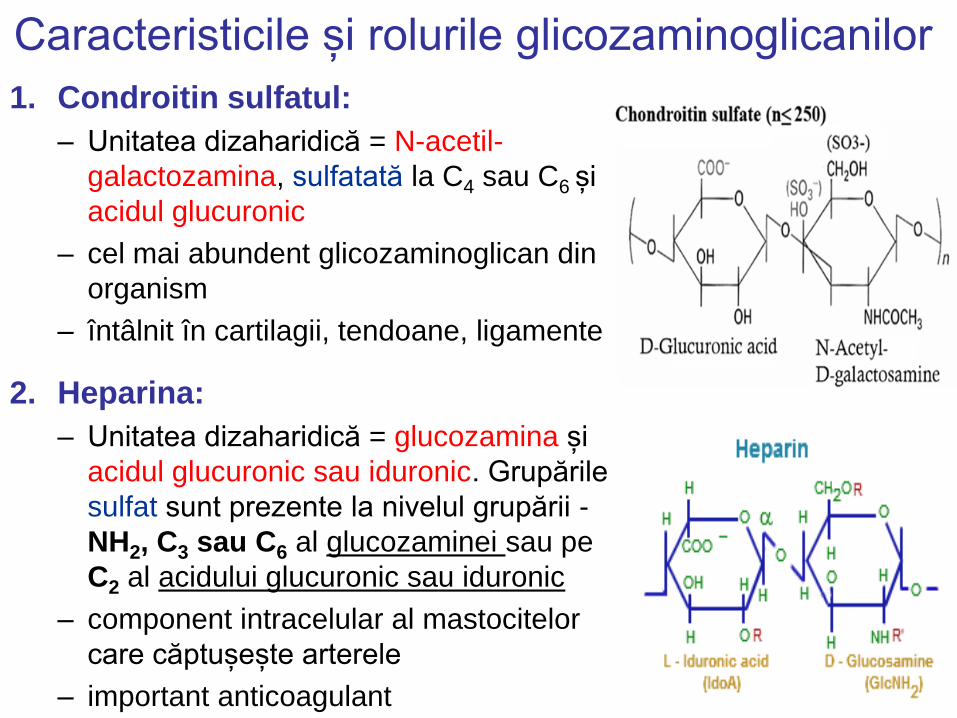
Caracteristicile și rolurile glicozaminoglicanilor
1. Condroitin sulfatul:
– Unitatea dizaharidică = N-acetil-
galactozamina, sulfatată la C4 sau C6 și
acidul glucuronic
– cel mai abundent glicozaminoglican din
organism
– întâlnit în cartilagii, tendoane, ligamente
2. Heparina:
– Unitatea dizaharidică = glucozamina și
acidul glucuronic sau iduronic. Grupările
sulfat sunt prezente la nivelul grupării -
NH2, C3 sau C6 al glucozaminei sau pe
C2 al acidului glucuronic sau iduronic
– component intracelular al mastocitelor
care căptușește arterele
– important anticoagulant
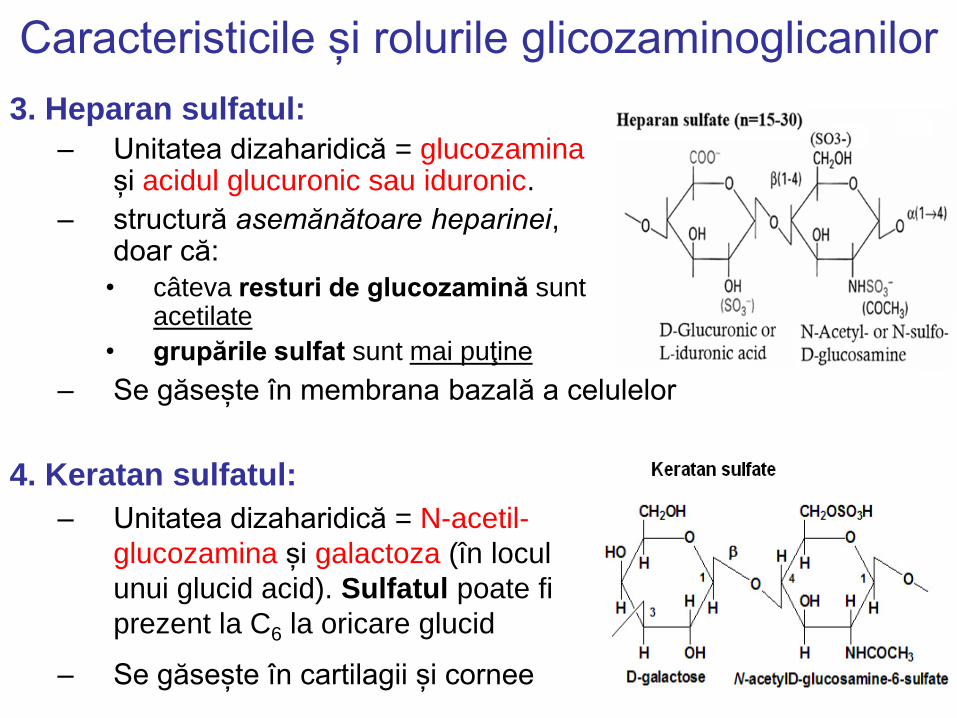
Caracteristicile și rolurile glicozaminoglicanilor
3. Heparan sulfatul:
– Unitatea dizaharidică = glucozamina și acidul glucuronic sau iduronic.
– structură asemănătoare heparinei, doar că:
• câteva resturi de glucozamină sunt acetilate
• grupările sulfat sunt mai puţine
– Se găsește în membrana bazală a celulelor
4. Keratan sulfatul:
– Unitatea dizaharidică = N-acetil-
glucozamina și galactoza (în locul
unui glucid acid). Sulfatul poate fi
prezent la C6 la oricare glucid
– Se găsește în cartilagii și cornee
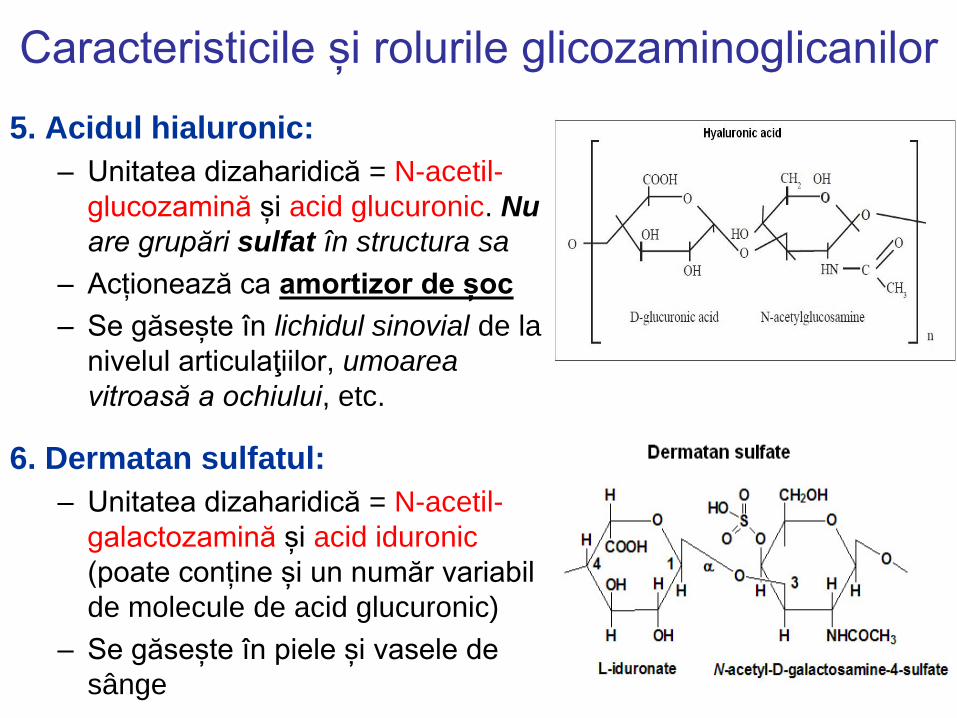
Caracteristicile și rolurile glicozaminoglicanilor
5. Acidul hialuronic:
– Unitatea dizaharidică = N-acetil-
glucozamină și acid glucuronic. Nu
are grupări sulfat în structura sa
– Acționează ca amortizor de șoc
– Se găsește în lichidul sinovial de la
nivelul articulaţiilor, umoarea
vitroasă a ochiului, etc.
6. Dermatan sulfatul:
– Unitatea dizaharidică = N-acetil-
galactozamină și acid iduronic
(poate conține și un număr variabil
de molecule de acid glucuronic)
– Se găsește în piele și vasele de
sânge

Importanţa medicală
Mucopolizaharidozele
• boli ereditare, caracterizate printr-o acumulare intralizozomală de
glicozaminoglicani (GAG) la nivelul diferitelor ţesuturi
• Cauza = deficienţa unor enzime lizozomale (hidrolaze) implicate în
degradarea GAG (sinteza lor nu este afectată), → acumularea GAG
• Se transmit AR (excepţie tipul II -sindromul Hunter, transmis X-linkat
• Manifestările clinice importante:
– Deformări scheletale
– Trăsături faciale grosiere
– Retardare mentală
– Opacifiere corneeană
– Modificări de creştere

Mucopolizaharidozele
• Tipul I – Sindromul Hurler:
– Enzima deficitară este α-L-iduronidaza
– Dermatan sulfatul şi heparan sulfatul apar
în urină
– formă foarte gravă de boală, cu retardare
mentală foarte severă, asociată cu deformări
scheletale si opacifiere corneeană importante
• Tipul II – Sindromul Hunter:
– Enzima deficitară este iduronat sulfataza
– Dermatan sulfatul şi heparan sulfatul apar
în urină
– grad variabil de severitate, iar opacifierea
corneeană este absentă

Mucopolizaharidozele
• Tipul III – Sindromul Sanfilippo:
– Enzimele deficitare: heparan sulfataza, N-acetil
glucozaminidaza
– Heparan sulfatul apare în urină
– Sunt cunoscute 4 forme de boală (fiecare cu enzima
sa deficitară specifică)
• Tipul IV – Sindromul Morquio:
– Enzimele deficitare: galactozamin sulfataza, β-D-
galactozidaza
– Sunt descrise 2 forme de boală
– Keratan sulfatul şi condroitin sulfatul apar în urină

Mucopolizaharidozele
• Tipul V – Sindromul Scheie:
– considerat a fi o formă mai puţin severă a tipului I (sindromul Hurler)
– Enzima deficitară: α-L-iduronidaza
– Clinic: deformări scheletale moderate, retardare mentală absentă
– Dermatan sulfatul şi heparan sulfatul apar în urină
• Tipul VI – Sindromul Maroteaux-Lamy:
– Enzima deficitară: N-acetil-β-D-Galactozamino-4-sulfataza
– Fară retardare mentală dar cu severe deformări scheletale
– Dermatan sulfatul apare în urină
• Tipul VII – Sindromul Sly:
– Enzima deficitară: β-glucuronidaza
– Dermatan sulfatul şi heparan sulfatul apar în urină






























