JOURNAL OF VIROLOGY, Feb. 1986, p. 603-613 0022-538X/86/020603-11$02.00/0 Copyright C) 1986, American Society for Microbiology Variant Influenza Virus Hemagglutinin That Induces Fusion at Elevated pH ROBERT W. DOMS,' MARY-JANE GETHING,2t JEAN HENNEBERRY,'2t JUDY WHITE,'t AND ARI HELENIUSl* Department of Cell Biology, Yale University School of Medicine, New Haven, Connecticut 06510,' and Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 117242 Received 16 September 1985/Accepted 31 October 1985 The hemagglutinin (HA) glycoprotein of influenza virus performs two critical roles during infection: it binds virus to cell surface sialic acids, and under mildly acidic conditions it induces fusion of the virion with intracellular membranes, liberating the genome into the cytoplasm. The pH dependence of fusion varies for different influenza virus strains. Here we report the isolation and characterization of a naturally occurring variant of the X31 strain that fuses at a pH 0.2 units higher than the parent strain does and that is less sensitive to the effects of ammonium chloride, a compound known to elevate endosomal pH. The bromelain-solubilized ectodomain of the variant HA displayed a corresponding shift in the pH at which it changed conformation and bound to liposomes. Cloning and sequencing of the variant HA gene revealed amino acid substitutions at three positions in the polypeptide. Two substitutions were in antigenic determinants in the globular region of HAl, and the third occurred in HA2 near the base of the molecule. By using chimeric HA molecules expressed in CV-1 cells from simian virus 40-based vectors, we demonstrated that the change in HA2 was solely responsible for the altered fusion phenotype. This substitution, asparagine for aspartic acid at position 132, disrupted a highly conserved interchain salt bridge between adjacent HA2 subunits. The apparent role of this residue in stabilizing the HA trimer is consistent with the idea that the trimer dissociates at low pH. Furthermore, the results demonstrate that influenza virus populations contain fusion variants, raising the possibility that such variants may play a role in the evolution of the virus. The ability of influenza virus to adapt rapidly to a wide variety of host cells and host defenses is well known. Much of this variability resides in the hemagglutinin (HA), a virally encoded integral membrane protein that has been the object of intense study over the last several years. Through changes in its structure, HA imparts variability to the virus in at least four ways. First, the antigenic epitopes change through antigenic shift and drift (20, 26, 28, 33). Second, a single amino acid substitution in the sialic acid-binding site at the tip of the HA molecule results in alteration or extension of the host range of the virus by enabling it to recognize different sialic acid linkages (19, 21). Third, it has been shown in vitro that the efficiency with which HA undergoes essential host cell-dependent posttranslational proteolytic cleavage can be altered by changes in the structure of HA and that this may also serve to extend the host range of the virus (22). Finally, the pH at which HA catalyzes fusion between viral and endosomal membranes varies among different strains of influenza virus (31). Recently, virus variants which fuse at a higher pH than the parent strains have been selected, and analysis of the HA molecules from these fusion variants has yielded important information on the structural changes in HA that take place at low pH (3, 22). To learn more about the conformational change in HA which leads to membrane fusion, we have recently taken a genetic approach. We have previously described a series of HA mutants that contain site-directed amino acid substitu- tions which alter both the efficiency and pH dependence of * Corresponding author. t Present address: Department of Biochemistry and Howard Hughes Medical Institute, University of Texas Health Science Center, Dallas, TX 75235. t Present address: Department of Pharmacology, University of California at San Francisco, San Francisco, CA 94143. HA-induced membrane fusion (M.-J. Gething, R. W. Doms, D. York, and J. White, J. Cell Biol., in press). Here we report the isolation and characterization of a naturally oc- curring fusion variant which fuses at a pH approximately 0.2 units higher than the wild type. As a result, the variant is more resistant to the effects of agents which elevate the pH in endosomes-the site of virus-cell membrane fusion (18, 35). Sequencing of the variant HA gene revealed three single amino acid substitutions, one of which was solely responsi- ble for the variant phenotype. The location of this substitu- tion in the three-dimensional structure of HA was examined, and the implications for the mechanism of low-pH-induced conformational change as well as for viral pathogenicity are discussed. MATERIALS AND METHODS Preparation of virus. X31, a recombinant influenza virus bearing the HA from the A/Aichi/68 strain (15), was kindly provided by John Skehel, National Institute for Medical Research, Mill Hill, United Kingdom. The virus was propagated by two additional passages in embryonated chicken eggs as described previously (5). Protein concentra- tion was determined by the method of Lowry et al. (16), and the virus was divided into portions and stored at -80°C until use. Plaque purification. Virus was plaque purified in MDCK cells. Confluent monolayers in 50-mm dishes were washed with Dulbecco modified Eagle medium (DMEM) before being infected with serial dilutions of virus in a volume of 250 ,u for 1 h at 37°C. The inoculum was aspirated, and the cells were washed twice with phosphate-buffered saline (PBS). The final wash solution was removed, and 5 ml of freshly prepared 0.9% agarose in DMEM containing 3 ,ug of tolylsulfonyl phenylalanylchloromethylketone (TPCK)- treated trypsin (Sigma Chemical Co., St Louis, Mo.) per ml 603 Vol. 57, No. 2

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF VIROLOGY, Feb. 1986, p. 603-6130022-538X/86/020603-11$02.00/0Copyright C) 1986, American Society for Microbiology
Variant Influenza Virus Hemagglutinin That Induces Fusion atElevated pH
ROBERT W. DOMS,' MARY-JANE GETHING,2t JEAN HENNEBERRY,'2t JUDY WHITE,'t AND ARI HELENIUSl*Department of Cell Biology, Yale University School of Medicine, New Haven, Connecticut 06510,' and Cold Spring
Harbor Laboratory, Cold Spring Harbor, New York 117242
Received 16 September 1985/Accepted 31 October 1985
The hemagglutinin (HA) glycoprotein of influenza virus performs two critical roles during infection: it bindsvirus to cell surface sialic acids, and under mildly acidic conditions it induces fusion of the virion withintracellular membranes, liberating the genome into the cytoplasm. The pH dependence of fusion varies fordifferent influenza virus strains. Here we report the isolation and characterization of a naturally occurringvariant of the X31 strain that fuses at a pH 0.2 units higher than the parent strain does and that is less sensitiveto the effects of ammonium chloride, a compound known to elevate endosomal pH. The bromelain-solubilizedectodomain of the variant HA displayed a corresponding shift in the pH at which it changed conformation andbound to liposomes. Cloning and sequencing of the variant HA gene revealed amino acid substitutions at threepositions in the polypeptide. Two substitutions were in antigenic determinants in the globular region of HAl,and the third occurred in HA2 near the base of the molecule. By using chimeric HA molecules expressed inCV-1 cells from simian virus 40-based vectors, we demonstrated that the change in HA2 was solely responsiblefor the altered fusion phenotype. This substitution, asparagine for aspartic acid at position 132, disrupted a
highly conserved interchain salt bridge between adjacent HA2 subunits. The apparent role of this residue instabilizing the HA trimer is consistent with the idea that the trimer dissociates at low pH. Furthermore, theresults demonstrate that influenza virus populations contain fusion variants, raising the possibility that suchvariants may play a role in the evolution of the virus.
The ability of influenza virus to adapt rapidly to a widevariety of host cells and host defenses is well known. Muchof this variability resides in the hemagglutinin (HA), a virallyencoded integral membrane protein that has been the objectof intense study over the last several years. Through changesin its structure, HA imparts variability to the virus in at leastfour ways. First, the antigenic epitopes change throughantigenic shift and drift (20, 26, 28, 33). Second, a singleamino acid substitution in the sialic acid-binding site at thetip of the HA molecule results in alteration or extension ofthe host range of the virus by enabling it to recognizedifferent sialic acid linkages (19, 21). Third, it has beenshown in vitro that the efficiency with which HA undergoesessential host cell-dependent posttranslational proteolyticcleavage can be altered by changes in the structure of HAand that this may also serve to extend the host range of thevirus (22). Finally, the pH at which HA catalyzes fusionbetween viral and endosomal membranes varies amongdifferent strains of influenza virus (31). Recently, virusvariants which fuse at a higher pH than the parent strainshave been selected, and analysis of the HA molecules fromthese fusion variants has yielded important information onthe structural changes in HA that take place at low pH (3,22). To learn more about the conformational change in HAwhich leads to membrane fusion, we have recently taken agenetic approach. We have previously described a series ofHA mutants that contain site-directed amino acid substitu-tions which alter both the efficiency and pH dependence of
* Corresponding author.t Present address: Department of Biochemistry and Howard
Hughes Medical Institute, University of Texas Health ScienceCenter, Dallas, TX 75235.
t Present address: Department of Pharmacology, University ofCalifornia at San Francisco, San Francisco, CA 94143.
HA-induced membrane fusion (M.-J. Gething, R. W. Doms,D. York, and J. White, J. Cell Biol., in press). Here wereport the isolation and characterization of a naturally oc-curring fusion variant which fuses at a pH approximately 0.2units higher than the wild type. As a result, the variant ismore resistant to the effects of agents which elevate the pHin endosomes-the site of virus-cell membrane fusion (18,35). Sequencing of the variant HA gene revealed three singleamino acid substitutions, one of which was solely responsi-ble for the variant phenotype. The location of this substitu-tion in the three-dimensional structure ofHA was examined,and the implications for the mechanism of low-pH-inducedconformational change as well as for viral pathogenicity arediscussed.
MATERIALS AND METHODS
Preparation of virus. X31, a recombinant influenza virusbearing the HA from the A/Aichi/68 strain (15), was kindlyprovided by John Skehel, National Institute for MedicalResearch, Mill Hill, United Kingdom. The virus waspropagated by two additional passages in embryonatedchicken eggs as described previously (5). Protein concentra-tion was determined by the method of Lowry et al. (16), andthe virus was divided into portions and stored at -80°C untiluse.
Plaque purification. Virus was plaque purified in MDCKcells. Confluent monolayers in 50-mm dishes were washedwith Dulbecco modified Eagle medium (DMEM) beforebeing infected with serial dilutions of virus in a volume of 250,u for 1 h at 37°C. The inoculum was aspirated, and the cellswere washed twice with phosphate-buffered saline (PBS).The final wash solution was removed, and 5 ml of freshlyprepared 0.9% agarose in DMEM containing 3 ,ug oftolylsulfonyl phenylalanylchloromethylketone (TPCK)-treated trypsin (Sigma Chemical Co., St Louis, Mo.) per ml
603
Vol. 57, No. 2

604 DOMS ET AL.
was added at between 41 and 45°C. The agarose was allowedto solidify for several minutes, and the cells were incubatedat 37°C for 2 days. To stain the resulting plaques, 3 ml of0.9% agarose in DMEM containing 0.01% neutral red was
added. After being stained, the plaques were picked with a
micropipette, suspended in DMEM, and stored at -80'Cuntil further use.
Preparation and radiolabeling of BHA. Bromelain-releasedHA (BHA) was obtained by a minor modification (5) of thetechnique of Brand and Skehel (2). The resulting protein wasjudged to be >95% pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) andCoomassie blue staining. BHA was labeled with lodogen(Pierce Chemical Co., Rockford, Ill.) by the technique ofFraker and Speck (8). After being labeled, the 125I-BHA was
chromatographed on a 2-ml column which contained 1 ml ofDowex-1-chloride (Sigma Chemical Co., St. Louis, Mo.) ontop of 1 ml of ricin-conjugated Sepharose (Pharmacia FineChemicals, Piscataway, N.J.) prepared as previously de-scribed (5). The column was washed with PBS until theeluate contained a minimal amount of radioactivity. 125i-BHA was eluted from the column with 0.2 M galactose inPBS, collected in a volume of 1.5 ml, and used withoutadditional purification.
Assays for conversion and liposome binding. Conversion ofBHA to the low-pH conformation was measured by a
protease sensitivity assay as previously described (5). Tomeasure the kinetics of conversion to the low-pH form, 1 mlof 125I-BHA was acidified to pH 4.8. At various times afteracidification, 100-,u samples were removed, neutralizedimmediately, and digested with proteinase K. The amount ofprotein in the low-pH form was determined by trichloroace-tic acid (TCA) precipitation.To measure the efficiency and pH dependence of BHA
binding to liposomes, trace amounts of 125I-BHA in a volumeof 5 ,ul were added to 95 ,ul of liposomes (6 mM lipid)prepared as previously described (29) with a lipid composi-tion of phosphatidylcholine, phosphatidylethanolamine,sphingomyelin, cholesterol, and phosphatidic acid in a molarratio of 1:1:1:1.5:0.3. All lipids were from Avanti (Birming-ham, Ala.), except cholesterol, which was from Sigma. Thesolution was incubated at 370C, and the pH was adjusted bythe addition of acid. After 15 min, the solution was neutral-ized, diluted to 30% (wt/vol) with sucrose, and overlaid with300 ,u1 of 25% sucrose and 200 ptl of 10% sucrose (both wt/volin PBS) in a Beckman 700-pd tube. After centrifugation at200,000 x g for 3 h, seven 100-pI fractions were taken fromthe top by careful pipetting from the interface. The amountof 125I-BHA protein in each fraction was determined bycounting with a Beckman Gamma 5500 counter. Counts inthe top half of the tube were defined as liposome-boundBHA, and those in the lower half were defined as unbound.See Doms et al. (5) for further characterization of thissystem.
Hemolysis assays. Human or guinea pig erythrocytes(RBC) were collected and washed three times in 15 volumesof MES (morpholineethanesulfonic acid)-saline. The RBCwere diluted to a final concentration of 1.0% (vol/vol) anddivided into 900-pI samples. Virus solution (100 ,ul; eitherallantoic fluid or purified virus) was added to each RBCsample, and the samples were mixed and warmed to 37°C.Amounts of 0.5 N acetic acid were added to the samples toadjust the pH to the desired value. The solution was neu-
tralized by adding an equal volume of 0.5 N NaOH precisely15 min after acidification. The RBC were pelleted, and thesupernatant was assayed for the presence of hemoglobin by
measuring adsorption at 540 nm. Total (100%) hemolysis wasdefined by the amount of hemoglobin released in the pres-ence of 0.1% Triton X-100. All virus solutions were dilutedso that only 50% of the RBC were lysed at the optimum pHfor hemolysis. We found that the ratio of virus to RBC mustbe controlled to determine the pH dependence of hemolysiswith accuracy. In addition, RBC should be used within 24 hof collection.
Cell fusion assays. To determine the pH dependence ofvirus-induced fusion, several assays were used. Fusion fromwithout was done essentially as described by White et al.(32). Confluent monolayers of CV-1 cells were washed withice-cold PBS, after which 1 to 5 ,ug of virus in DMEMwithout serum was added at 4°C. The virus was allowed tobind to the cell surface for 1 h with gentle agitation. The cellswere washed with cold medium to remove unbound virusand then placed in prewarmed (37°C) fusion medium (PBSwith 10 mM MES and 10 mM HEPES [N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid]) of theappropriate pH for 3 min. The fusion medium was aspiratedand replaced with normal growth medium. After 3 h, thecells were fixed in methanol and stained with Giemsa stain.The extent of fusion was determined by calculating thenumber of nuclei present in polykaryons, which we definedas cells with three or more nuclei. Under optimal conditionsnearly 100% of the cells fused, forming a giant syncytium.The kinetics of fusion were also determined, and we foundthat longer incubation in fusion medium did not result in anincrease in polykaryon formation. Cell-cell fusion fromwithin was done 12 h after infection with virus. Slightlysubconfluent monolayers of CV-1 cells in 30-mm dishes wereinfected with 1 ,ug of virus in a volume of 0.6 ml. After 1 h,2 ml of normal growth medium was added; 12 h later the cellswere washed with PBS and then incubated with 1 ml oftrypsin (5 ,ug/ml) for 10 min at 37°C. The trypsin wasaspirated, and a fivefold excess of soybean trypsin inhibitor(Sigma) was added. After 3 min, the cells were placed infusion medium for 3 min, incubated in complete growthmedium for 3 h, and processed for light microscopy asdescribed above.
Fusion activity was also assayed by binding and fusingRBC to the surface of CV-1 cells infected with eitherinfluenza virus (see above) or simian virus 40 (SV40)-basedvectors containing the HA gene (see below). Either 12 h afterinfection with influenza virus or 65 h after infection withSV40-HA recombinant viruses, the cells were washed andthe HA was activated by mild trypsinization. Guinea pigRBC were bound to the surface of the infected cells for 15min at 37°C. Prewarmed fusion medium of defined pH wasadded for 3 min, after which the cells were placed in growthmedium for 30 min. The cells were fixed in methanol,washed in PBS, and stained for the presence of cytoplasmichemoglobin with 1% benzidine in 12% acetic acid. Hydrogenperoxide was added to a final concentration of 2% immedi-ately before staining. The cells were stained for 30 s and thenwashed extensively with PBS. The extent of RBC-cell fusionwas determined by scoring for the presence of the charac-teristic blue staining of the cytoplasm, which indicatedfusion between the RBC and the CV-1 cell, with subsequentdiffusion of hemoglobin into the CV-1 cytoplasm.Recombinant DNA techniques. Buffers and reaction condi-
tions for restriction enzymes, T4 DNA ligase, and theKlenow fragment of DNA polymerase were those listed bythe commercial source (New England BioLabs, Beverly,Mass.). Reverse transcriptase was obtained from Beard, andRNasin was obtained from Promega Biotech. The oligonu-
J. VIROL.

FUSION VARIANT OF INFLUENZA VIRUS HA 605
cleotide containing the ClaI restriction site was obtainedfrom Collaborative Research, Inc., Lexington, Mass. Prep-aration of plasmid DNAs and DNA fragments, PAGE, andother standard recombinant DNA techniques were carriedout as described by Maniatis et al. (17) and in other refer-ences cited there. Transformation of Escherichia coli DH-1cells was done by the method of Hanahan (11).
Synthesis and cloning of cDNA copies of wild-type andmutant X31 HA virion RNAs. Purified X31 virus (5 mg in 300RI of PBS) was disrupted on ice in 2 ml of lysis buffer (10 mMTris chloride, pH 8.6, 140 mM NaCl, 1.5 mM MgCl2, 0.5%Nonidet P-40, and 0.015% Macaloid). Virion proteins werethen digested for 45 min at 37°C with proteinase K after theaddition of 2 ml of a solution containing 200 mM Trischloride, pH 7.5, 300 mM NaCl, 25 mM EDTA, 2% SDS,and 1 mg of enzyme. The reaction mixture was then ex-tracted twice with phenol and once with chloroform andprecipitated twice with ethanol. The yield of purified viralRNA was approximately 50 ,ug.
First-strand cDNA synthesis from 7 ,ug of viral RNAtemplate was performed essentially as described by Maniatiset al. (17), except that a specific oligonucleotide primercomplementary to the 3' terminus of influenza virus RNAs[d(AGCAAAAGCAGG), kindly supplied by G. Winter,Medical Research Council Laboratory of Molecular Biology,Cambridge, England] replaced the oligo(dT) primer, andtreatment with methylmercuric hydroxide was omitted. Sec-ond-strand cDNA synthesis was done with reverse tran-scriptase as described by Maniatis et al. (17). The double-stranded cDNA was then ligated with phosphorylated ClaIlinkers as previously described (17) to attach the linkers atthe end of the DNA corresponding to the 3' terminus of theoriginal virus RNA. The reaction mixture was then dilutedso that the composition of the buffer was 10 mM Trischloride (pH 7.5)-60 mM NaCI-6 mM MgCI2 and heated to70°C for 10 min to inactivate the ligase enzyme. The ligatedDNA was digested with ClaI (60 U for 5 h at 37°C) togenerate a single ClaI sticky end on each cDNA. It haspreviously been shown (27) that the X31 virus HA genenucleotide sequence contains a single BamHI restriction siteat nucleotide 1618, a position that corresponds in the aminoacid sequence to the junction between the externalectodomain and the transmembrane region. Digestion of thecDNA with 20 U of BamHI should therefore generate aClaI-BamHI fragment of HA cDNA of approximately 1,600base pairs. The digested cDNA mixture was chromato-graphed on a 1-ml column of Sepharose CL-4B in 10 mMTris chloride (pH 7.5)-300 mM NaCl-1 mM EDTA, and150-1.I fractions were collected. Portions of these fractionswere separated by electrophoresis on a 6% polyacrylamidegel, and the fractions containing cDNAs longer than 1,000base pairs were identified by autoradiography. These frac-tions were pooled and concentrated by ethanol precipitationbefore ligation to plasmid pXf3 (17) digested with ClaI andBamHI. E. coli DH-1 cells were then transformed with theligation mixtures. Bacterial colonies containing X31 virusHA sequences were identified by hybridization with a frag-ment of wild-type X31 HA cDNA (9) labeled with 32P by nicktranslation as the probe (17). Plasmid DNAs were preparedfrom individual colonies, and clones containing the appro-priately sized ClaI-BamHI fragment were identified by re-striction digestion. The identity of plasmids containing thevariant X31 virus HA gene nucleotide sequences wereconfirmed by more extensive endonuclease digestion withreference to the known restriction map (27), and the HAgene sequences were purified and cloned into M13 bac-
teriophage vectors (23) for dideoxy sequencing analysis bythe chain termination technique (24) with either the M13universal primer or oligonucleotides complementary to se-quences spaced along the X31 virus cDNA.
Cloning of the remaining nucleotides, corresponding to thetransmembrane domain and cytoplasmic sequences of thewild-type X31 virus HA gene, is described in detail else-where (C. Doyle, J. Sambrook, and M.-J. Gething, manu-script in preparation). The XhoI-BamHI fragment (nucleo-tides 1302 to 1618) of the HA gene was purified for use as aprimer to synthesize cDNA from the viral RNA template.After second-strand synthesis, the hairpin loop was cleavedwith S1 nuclease and Sall linkers were ligated to the cDNA,which was then digested with Sall and BamHI. The cDNApool containing fragments approximately 400 to 500 basepairs long was ligated to M13mpl8 (replicative form) cutwith Sall and BamHI and then transfected into E. coli TG-1cells. Plaques containing the X31 virus HA gene sequenceswere identified by DNA sequence analysis. The BamHI-SalIfragment containing the sequences encoding thetransmembrane and cytoplasmic tail domains of X31 HAwas excised from double-stranded M13 DNA and insertedinto an SV40 vector after the sequences encoding theectodomain of the protein (Doyle et al., manuscript inpreparation).SV40 vectors designed to express wild-type and mutant X31
HAs. A vector (see Fig. 7) was constructed to express thefull-length gene encoding the wild-type X31 HA (Doyle etal., manuscript in preparation). The vector closely resemblesSVEHA3, which has been used to express the cloned HAfrom the A/Japan/305/57 strain of influenza virus (10).pSVEXHA contained the ClaI-SalI X31 virus fragmentinserted between the HpaII (nucleotide 346) and BamHI(nucleotide 2533) sites of SV40 DNA, so that the HA genesequences replaced the late region of the SV40 genome,which encodes the capsid proteins. For amplification andmanipulation of the DNA sequences, the genome was in-serted through the unique KpnI site in the SV40 sequenceinto plasmid pK iB (7). These plasmid sequences wereremoved by dige ion with KpnI, and the SV40 DNA carry-ing the X31 HA g. .e was purified by gel electrophoresis andrecircularized b) dilute ligation before transfection intoCV-1 cells to generate recombinant virus stocks (7, 10).To generate recombinant SV40-HA genomes containing
the mutant HA gene sequences, the plasmid containing thecloned cDNA from the variant X31 virus was cleaved withClaI and BamHI to generate a fragment encoding the HAectodomain with three amino acid changes, with Sacl andXhoI to generate a fragment encoding sequences containingthe two amino acid changes in HAl, and with XhoI andBamHI to generate a fragment encoding sequences contain-ing the sole amino acid change in HA2 (see Fig. 6). Thesefragments were purified and inserted into the pSVEXHAvector, replacing the equivalent wild-type sequences. Virusstocks that expressed the various combinations of mutantHAs were generated as described above.
Immunoprecipitation of wild-type and mutant HA proteins.At 36 h postinfection with the various SV40-HA virusstocks, CV-1 cells were labeled with [35S]methionine (50 ,uCiper 5-cm dish) for 10 min at 37°C. The monolayers were thenwashed once with Tris-saline solution, and cell extracts wereprepared in 50 mM Tris hydrochloride (pH 8.0) containing1% Nonidet P-40. Samples of the cell extracts were im-munoprecipitated with a polyclonal anti-X31 HA rabbitantiserum or with monoclonal antibodies (Mabs) directedagainst various epitopes on the X31 HA molecule. The Mabs
VOL. 57, 1986
606 DOMS ET AL.
were kindly provided by R. Webster, St. Jude Children'sResearch Hospital, Memphis, Tenn. The precipitated HAproducts were separated by SDS-PAGE and visualized byautoradiography.
RESULTS
Identification and isolation of variant influenza viruses. Theacid-induced conformational change in influenza virus HAand its water-soluble ectodomain, BHA, can be monitoredby changes in protease sensitivity (5, 25). The neutral-pHform is highly resistant to proteinase K, whereas acid-treated molecules are cleaved to TCA-soluble fragments (5).When 125I-BHA derived from the parent X31 virus wassubjected to buffers of decreasing pH, half-maximal conver-sion to the proteinase K-sensitive form occurred at pH 5.3,with nearly complete conversion occurring at pH 5.0 (Fig.1). However, we consistently observed that some of theprotein (10 to 15%) was converted between pH 5.6 and 5.8,suggesting heterogeneity in the response to pH. Prolongedincubation (up to 1 h at 37°C) of the protein at either pH 5.6or 5.8 did not result in further conversion, suggesting that theheterogeneity was not kinetic but rather stemmed fromdifferences in the pH sensitivity of BHA molecules.That the BHA preparation contained populations of mol-
ecules with different pH thresholds for conversion to theproteinase K-sensitive form was confirmed by using twoconsecutive acid treatments. A solution of 125I-BHA fromthe uncloned virus stock was first acidified to pH 5.3 for 15min and then neutralized. When a sample was removed, itwas found that 52% of the BHA remained resistant to
A
B C loonm
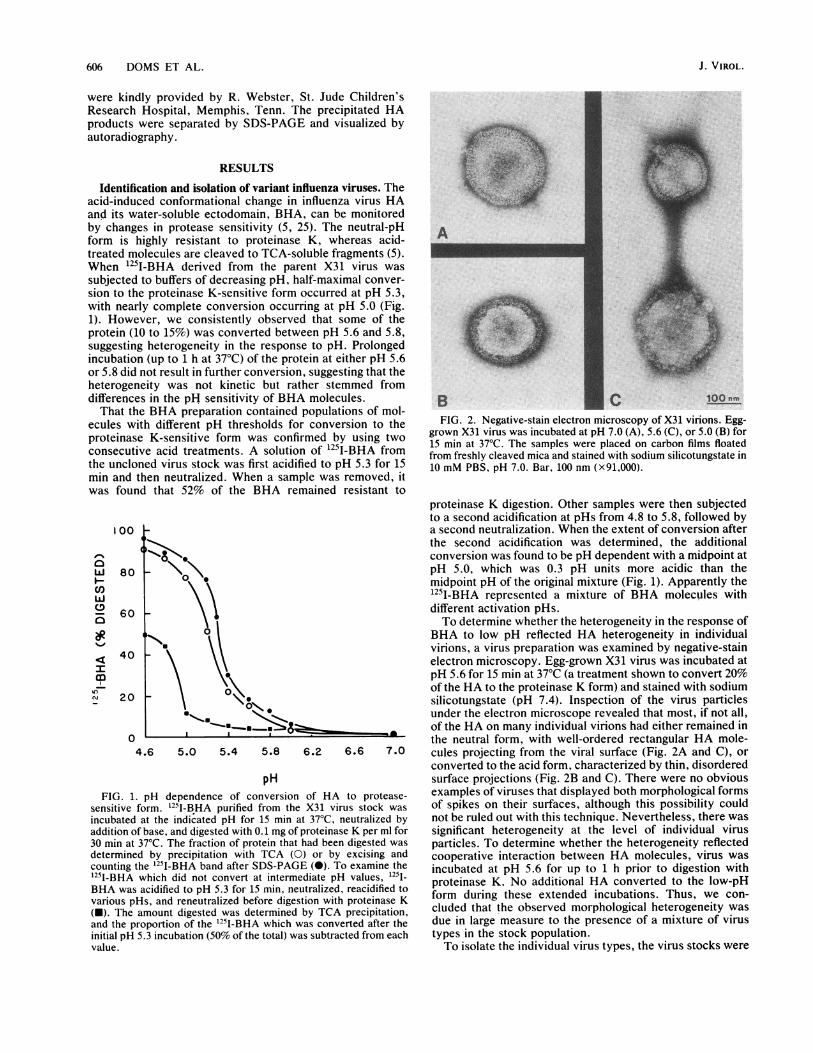
FIG. 2. Negative-stain electron microscopy of X31 virions. Egg-grown X31 virus was incubated at pH 7.0 (A), 5.6 (C), or 5.0 (B) for15 min at 37°C. The samples were placed on carbon films floatedfrom freshly cleaved mica and stained with sodium silicotungstate in10 mM PBS, pH 7.0. Bar, 100 nm (x91,000).
100
80
60
40
20
0
Zk
10.N C
4.6 5.0 5.4 5.8 6.2 6.6
pHFIG. 1. pH dependence of conversion of HA to protease-
sensitive form. 1251-BHA purified from the X31 virus stock was
incubated at the indicated pH for 15 min at 37°C, neutralized byaddition of base, and digested with 0.1 mg of proteinase K per ml for30 min at 37°C. The fraction of protein that had been digested was
determined by precipitation with TCA (0) or by excising andcounting the '25I-BHA band after SDS-PAGE (0). To examine the1251I-BHA which did not convert at intermediate pH values, 1251_BHA was acidified to pH 5.3 for 15 min, neutralized, reacidified tovarious pHs, and reneutralized before digestion with proteinase K(U). The amount digested was determined by TCA precipitation,and the proportion of the 125I-BHA which was converted after theinitial pH 5.3 incubation (50% of the total) was subtracted from eachvalue.
proteinase K digestion. Other samples were then subjectedto a second acidification at pHs from 4.8 to 5.8, followed bya second neutralization. When the extent of conversion afterthe second acidification was determined, the additionalconversion was found to be pH dependent with a midpoint atpH 5.0, which was 0.3 pH units more acidic than themidpoint pH of the original mixture (Fig. 1). Apparently the125I-BHA represented a mixture of BHA molecules withdifferent activation pHs.To determine whether the heterogeneity in the response of
BHA to low pH reflected HA heterogeneity in individualvirions, a virus preparation was examined by negative-stainelectron microscopy. Egg-grown X31 virus was incubated atpH 5.6 for 15 min at 37°C (a treatment shown to convert 20%of the HA to the proteinase K form) and stained with sodiumsilicotungstate (pH 7.4). Inspection of the virus particlesunder the electron microscope revealed that most, if not all,of the HA on many individual virions had either remained inthe neutral form, with well-ordered rectangular HA mole-cules projecting from the viral surface (Fig. 2A and C), orconverted to the acid form, characterized by thin, disorderedsurface projections (Fig. 2B and C). There were no obviousexamples of viruses that displayed both morphological formsof spikes on their surfaces, although this possibility couldnot be ruled out with this technique. Nevertheless, there wassignificant heterogeneity at the level of individual virusparticles. To determine whether the heterogeneity reflectedcooperative interaction between HA molecules, virus wasincubated at pH 5.6 for up to 1 h prior to digestion withproteinase K. No additional HA converted to the low-pHform during these extended incubations. Thus, we con-cluded that the observed morphological heterogeneity wasdue in large measure to the presence of a mixture of virustypes in the stock population.To isolate the individual virus types, the virus stocks were
01aCICO
(Da
0
I
mC
J. VIROL.
FUSION VARIANT OF INFLUENZA VIRUS HA 607
those for fusion from without (Fig. 4) and for the7,r o\ \ conformational change (see below). The reason for theo/ \ \ \ difference in the pH threshold for fusion from without and
\ \ \ fusion from within is not known at present, although it may\\* reflect differences in posttranslational processing between
HA produced in CV-1 cells and HA produced in allantoicSTOCK membranes. Alternatively, it may reflect differences in the
o \ \ local environment close to the plasma membrane of the cell.C22 Nevertheless, the results are consistent in that A31 virus
A3 1 induced fusion at a pH 0.2 units higher than that observed forC22 and the stock virus whether assayed by fusion from
50&\n within, fusion from without, or hemolysis.Effect of ammonium chloride on infectivity of variant and
wild-type viruses. To determine whether the differencesobserved between the wild-type and variant viruses by invitro fusion assays were manifested during the entry of the
5.0 5.2 5.4 5.6 5.8 6.0 7.0 virions into cells, CV-1 monolayers were incubated witheach virus stock in the presence of increasing concentrations
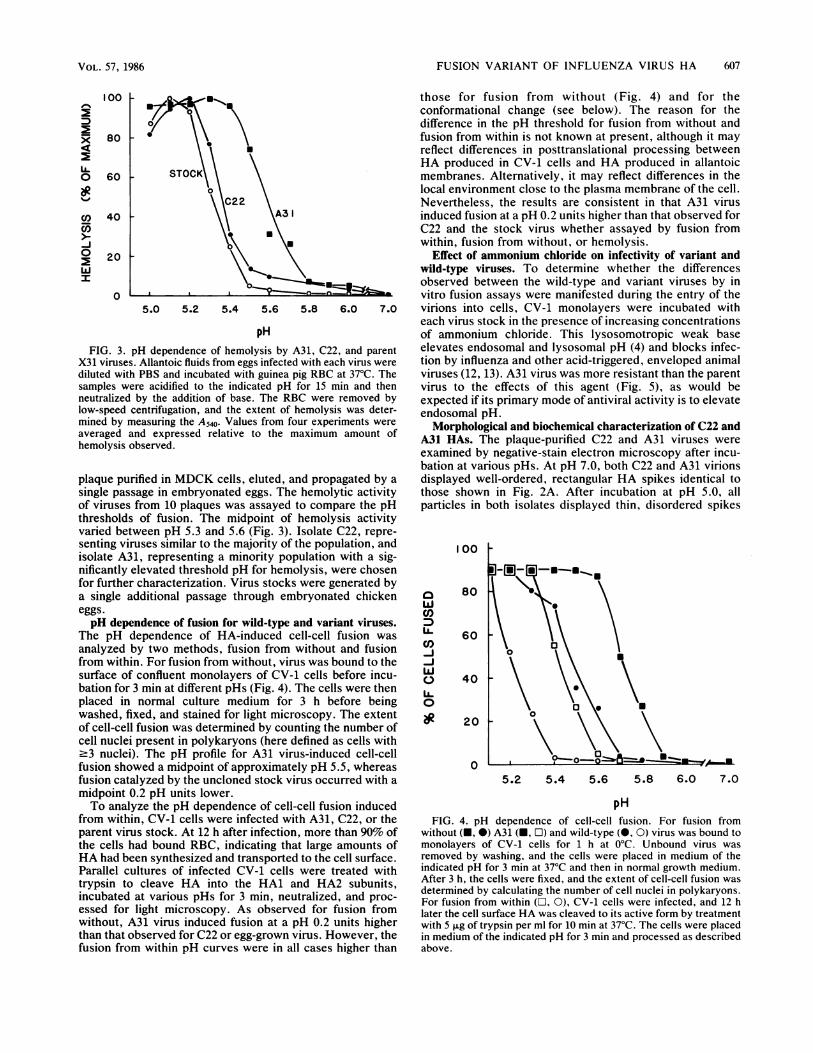
pH of ammonium chloride. This lysosomotropic weak baseH dependence of hemolysis by A31, C22, and parent elevates endosomal and lysosomal pH (4) and blocks infec-Allantoic fluids from eggs infected with each virus were tion by influenza and other acid-triggered, enveloped animalPBS and incubated with guinea pig RBC at 37°C. The viruses (12, 13). A31 virus was more resistant than the parente acidified to the indicated pH for 15 min and then virus to the effects of this agent (Fig. 5), as would be)y the addition of base. The RBC were removed by expected if its primary mode of antiviral activity is to elevatevntrifugation, and the extent of hemolysis was deter- endosomal pH.-asuring the A540. Values from four experiments weredesltmMorphological and biochemical characterization of C22 and
served. A31 HAs. The plaque-purified C22 and A31 viruses wereexamined by negative-stain electron microscopy after incu-bation at various pHs. At pH 7.0, both C22 and A31 virions
fied in MDCK cells, eluted, and propagated by a displayed well-ordered, rectangular HA spikes identical totge in embryonated eggs. The hemolytic activity those shown in Fig. 2A. After incubation at pH 5.0, allrom 10 plaques was assayed to compare the pH particles in both isolates displayed thin, disordered spikes
thresholds of fusion. The midpoint of hemolysis activityvaried between pH 5.3 and 5.6 (Fig. 3). Isolate C22, repre-senting viruses similar to the majority of the population, andisolate A31, representing a minority population with a sig-nificantly elevated threshold pH for hemolysis, were chosenfor further characterization. Virus stocks were generated bya single additional passage through embryonated chickeneggs.pH dependence of fusion for wild-type and variant viruses.
The pH dependence of HA-induced cell-cell fusion wasanalyzed by two methods, fusion from without and fusionfrom within. For fusion from without, virus was bound to thesurface of confluent monolayers of CV-1 cells before incu-bation for 3 min at different pHs (Fig. 4). The cells were thenplaced in normal culture medium for 3 h before beingwashed, fixed, and stained for light microscopy. The extentof cell-cell fusion was determined by counting the number ofcell nuclei present in polykaryons (here defined as cells with.3 nuclei). The pH profile for A31 virus-induced cell-cellfusion showed a midpoint of approximately pH 5.5, whereasfusion catalyzed by the uncloned stock virus occurred with amidpoint 0.2 pH units lower.To analyze the pH dependence of cell-cell fusion induced
from within, CV-1 cells were infected with A31, C22, or theparent virus stock. At 12 h after infection, more than 90% ofthe cells had bound RBC, indicating that large amounts ofHA had been synthesized and transported to the cell surface.Parallel cultures of infected CV-1 cells were treated withtrypsin to cleave HA into the HAl and HA2 subunits,incubated at various pHs for 3 min, neutralized, and proc-essed for light microscopy. As observed for fusion fromwithout, A31 virus induced fusion at a pH 0.2 units higherthan that observed for C22 or egg-grown virus. However, thefusion from within pH curves were in all cases higher than
100
0
w
CO
C)
-iw
C.)
LL.
0
80
60
40
20
0
I[E, mN\o o0.2 54 56 58
5.2 5.4 5.6 5.8 6.0 7.0
pHFIG. 4. pH dependence of cell-cell fusion. For fusion from
without (-, 0) A31 (-, O) and wild-type (0, 0) virus was bound tomonolayers of CV-1 cells for 1 h at 0°C. Unbound virus wasremoved by washing, and the cells were placed in medium of theindicated pH for 3 min at 37°C and then in normal growth medium.After 3 h, the cells were fixed, and the extent of cell-cell fusion was
determined by calculating the number of cell nuclei in polykaryons.For fusion from within (L1, 0), CV-1 cells were infected, and 12 hlater the cell surface HA was cleaved to its active form by treatmentwith 5 ,ug of trypsin per ml for 10 min at 37°C. The cells were placedin medium of the indicated pH for 3 min and processed as describedabove.
100
80
60
40
20
x
2cm0
CO
0wI
0
FIG. 3. p1X31 viruses.diluted withsamples werneutralized blow-speed cemined by meaveraged anhemolysis ob
plaque purilsingle passaof viruses fi
VOL. 57, 1986
608 DOMS ET AL.
100
0z
m
m0
zo
80
60
40
20
1 2 3 4 5 6
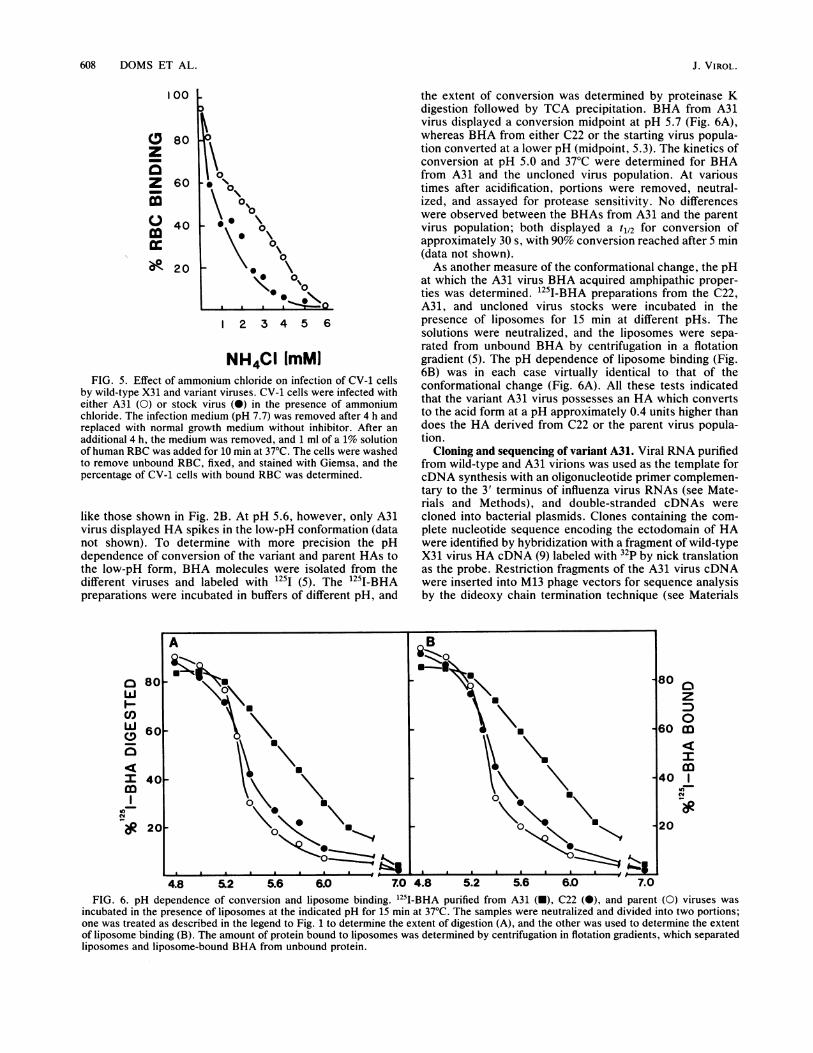
NH4CI ImMIFIG. 5. Effect of ammonium chloride on infection of CV-1 cells
by wild-type X31 and variant viruses. CV-1 cells were infected witheither A31 (0) or stock virus (0) in the presence of ammoniumchloride. The infection medium (pH 7.7) was removed after 4 h andreplaced with normal growth medium without inhibitor. After anadditional 4 h, the medium was removed, and 1 ml of a 1% solutionof human RBC was added for 10 min at 37°C. The cells were washedto remove unbound RBC, fixed, and stained with Giemsa, and thepercentage of CV-1 cells with bound RBC was determined.
like those shown in Fig. 2B. At pH 5.6, however, only A31virus displayed HA spikes in the low-pH conformation (datanot shown). To determine with more precision the pHdependence of conversion of the variant and parent HAs tothe low-pH form, BHA molecules were isolated from thedifferent viruses and labeled with 1251 (5). The 125I-BHApreparations were incubated in buffers of different pH, and
the extent of conversion was determined by proteinase Kdigestion followed by TCA precipitation. BHA from A31virus displayed a conversion midpoint at pH 5.7 (Fig. 6A),whereas BHA from either C22 or the starting virus popula-tion converted at a lower pH (midpoint, 5.3). The kinetics ofconversion at pH 5.0 and 37°C were determined for BHAfrom A31 and the uncloned virus population. At varioustimes after acidification, portions were removed, neutral-ized, and assayed for protease sensitivity. No differenceswere observed between the BHAs from A31 and the parentvirus population; both displayed a t1/2 for conversion ofapproximately 30 s, with 90% conversion reached after 5 min(data not shown).As another measure of the conformational change, the pH
at which the A31 virus BHA acquired amphipathic proper-ties was determined. 1251-BHA preparations from the C22,A31, and uncloned virus stocks were incubated in thepresence of liposomes for 15 min at different pHs. Thesolutions were neutralized, and the liposomes were sepa-rated from unbound BHA by centrifugation in a flotationgradient (5). The pH dependence of liposome binding (Fig.6B) was in each case virtually identical to that of theconformational change (Fig. 6A). All these tests indicatedthat the variant A31 virus possesses an HA which convertsto the acid form at a pH approximately 0.4 units higher thandoes the HA derived from C22 or the parent virus popula-tion.
Cloning and sequencing of variant A31. Viral RNA purifiedfrom wild-type and A31 virions was used as the template forcDNA synthesis with an oligonucleotide primer complemen-tary to the 3' terminus of influenza virus RNAs (see Mate-rials and Methods), and double-stranded cDNAs werecloned into bacterial plasmids. Clones containing the com-plete nucleotide sequence encoding the ectodomain of HAwere identified by hybridization with a fragment of wild-typeX31 virus HA cDNA (9) labeled with 32P by nick translationas the probe. Restriction fragments of the A31 virus cDNAwere inserted into M13 phage vectors for sequence analysisby the dideoxy chain termination technique (see Materials
0w
I-
COw
4D'a
A
801
601
401
201
I
80 C)zD0
60 M
ICD
40 1
20
4.8 52 5.6 6.0 7.0 4.8 5.2 5.6 6.0 7.0FIG. 6. pH dependence of conversion and liposome binding. '25I-BHA purified from A31 (-), C22 (0), and parent (0) viruses was
incubated in the presence of liposomes at the indicated pH for 15 min at 37°C. The samples were neutralized and divided into two portions;one was treated as described in the legend to Fig. 1 to determine the extent of digestion (A), and the other was used to determine the extentof liposome binding (B). The amount of protein bound to liposomes was determined by centrifugation in flotation gradients, which separatedliposomes and liposome-bound BHA from unbound protein.
J. VIROL.

FUSION VARIANT OF INFLUENZA VIRUS HA 609
TABLE 1. Location of amino acid substitutions in the HA fromfusion variant A31
HA subunit Amino acid (codon)(residue Location on HA
no.) Wild type A31
HAl (144) Gly (GGT) Asp (GAT) Loop region (epitope A)HAl (215) Pro (CCG) Leu (CTG) Interface region (epitope D)HA2 (132) Asp (GAC) Asn (AAC) Base of HA2 (forms
interchain salt bridgewith Arg-124 onadjacent, subunit)
and Methods). Deduction of the amino acid sequence of A31HA from the nucleotide sequence of the gene revealed thatthree residues were altered from those in the wild-typeprotein (Table 1). Two of the substitutions were in the HAlsubunit, and the third amino acid change was in HA2. Bothsubstitutions in HAl occurred in previously defined anti-
HAI
genic epitopes; Gly-144 to Asp-144 in antigenic site A (33) inthe loop region of HAl, and Pro-215 to Leu-215 in antigenicregion D, which is located close to the trimer interface regionof HAl (34). The first substitution has been observed in avariety of influenza virus isolates of the H3 subtype (1).However, to the best of our knowledge, the mutation atresidue 215 has not previously been described in H3 HAmolecules. The third mutation changed Asp-132 to Asn-132in the HA2 subunit.
Expression of wild-type and variant HAs in CV-1 cells withSV40-based vectors. To determine which of the three muta-tions was responsible for the observed shift in fusion phe-notype, it was necessary to express and assay HA proteinsthat contained various combinations of the altered aminoacids. Hybrid HA genes were constructed that contained allthree mutations (muHA12), only the two substitutions inHAl (muHA1), or only the single substitution in HA2(muHA2). The cDNA from the variant A31 virus gene wasdigested with ClaI plus BamHI, SacI plus XhoI, or XhoI plus
HA2Cleavage
sitesignalHydrophobicNH2 terminus
X31HA
CytoplasmicAnchor tail
H Hcs ~o o I
- s _X_ E
I/I'
I,71N +
/
pKSB
SV40T Antigen
N A / XhoIBamHI
SaIIFIG. 7. Schematic diagram of X31 HA and the SV40-based vector used to express HA in CV-1 cells. The linear map (top) relates the
various functional regions of the HA molecule to the restriction sites in the gene and shows the positions of the altered residues found in thevariant A31 HA. Cross-hatched boxes, Hydrophobic sequences; open boxes, antigenic regions; solid boxes, glycosylation sites; *, alteredresidues. The restriction fragments excised from the A31 HA cDNA and inserted into the SVEXHA vector (described in Materials andMethods) replacing the equivalent wild-type HA sequences are shown by double-headed arrows beneath the linear map.
VOL. 57, 1986
I1,
I'-
610 DOMS ET AL.
'4?<
-- 3 23 23'~F E F
_.,- C\M
Fr1 1-23 :2 3 23F~F F
-D .7E C
92 K
68 K -- -
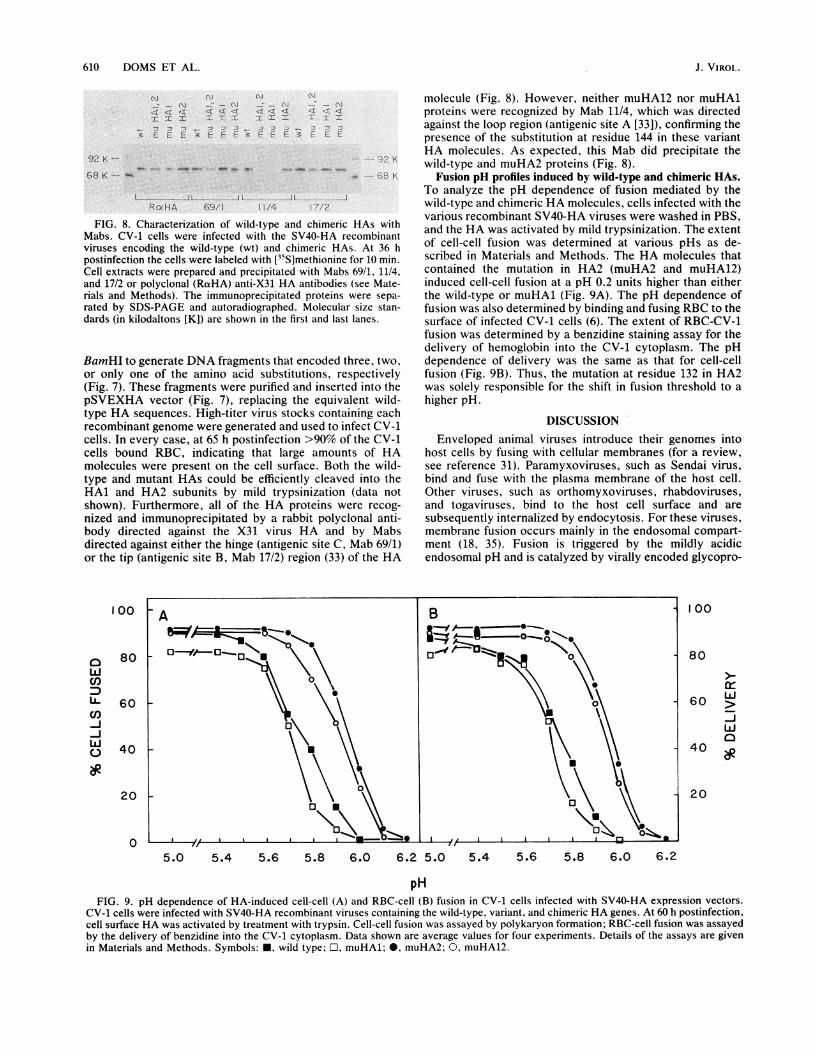
l i ..J LRoeHA 69/1 11/4
FIG. 8. Characterization of wild-typeMabs. CV-1 cells were infected with thviruses encoding the wild-type (wt) andpostinfection the cells were labeled with [3Cell extracts were prepared and precipitaand 17/2 or polyclonal (RaHA) anti-X31 Irials and Methods). The immunoprecipitrated by SDS-PAGE and autoradiographdards (in kilodaltons [K]) are shown in th
BamHI to generate DNA fragments tlor only one of the amino acid subs(Fig. 7). These fragments were purificpSVEXHA vector (Fig. 7), replacin
]_,\, molecule (Fig. 8). However, neither muHA12 nor muHAl<tnX I <rl proteins were recognized by Mab 11/4, which was directed
against the loop region (antigenic site A [33]), confirming thepresence of the substitution at residue 144 in these variantHA molecules. As expected, this Mab did precipitate the
..2. K wild-type and muHA2 proteins (Fig. 8).Fusion pH profiles induced by wild-type and chimeric HAs.
gL To analyze the pH dependence of fusion mediated by the7 2 wild-type and chimeric HA molecules, cells infected with the
and chimeric HAs withvarious recombinant SV40-HA viruses were washed in PBS,aeSV4-HA recombinant and the HA was activated by mild trypsinization. The extent
Ichimeric HAs. At 36 h of cell-cell fusion was determined at various pHs as de-35S]methionine for 10 min. scribed in Materials and Methods. The HA molecules thatited with Mabs 69/1, 11/4, contained the mutation in HA2 (muHA2 and muHA12)HA antibodies (see Mate- induced cell-cell fusion at a pH 0.2 units higher than either:ated proteins were sepa- the wild-type or muHAl (Fig. 9A). The pH dependence ofied. Molecular size stan- fusion was also determined by binding and fusing RBC to thee first and last lanes. surface of infected CV-1 cells (6). The extent of RBC-CV-1
fusion was determined by a benzidine staining assay for thedelivery of hemoglobin into the CV-1 cytoplasm. The pH
iat encoded three, two, dependence of delivery was the same as that for cell-cellstitutions, respectively fusion (Fig. 9B). Thus, the mutation at residue 132 in HA2-d and inserted into the was solely responsible for the shift in fusion threshold to ag the equivalent wild- higher pH.
type HA sequences. High-titer virus stocks containing eachrecombinant genome were generated and used to infect CV-1cells. In every case, at 65 h postinfection >90% of the CV-1cells bound RBC, indicating that large amounts of HAmolecules were present on the cell surface. Both the wild-type and mutant HAs could be efficiently cleaved into theHAl and HA2 subunits by mild trypsinization (data notshown). Furthermore, all of the HA proteins were recog-nized and immunoprecipitated by a rabbit polyclonal anti-body directed against the X31 virus HA and by Mabsdirected against either the hinge (antigenic site C, Mab 69/1)or the tip (antigenic site B, Mab 17/2) region (33) of the HA
100 KA
a
-JP-wDI,
80
60
40
20
0
DISCUSSIONEnveloped animal viruses introduce their genomes into
host cells by fusing with cellular membranes (for a review,see reference 31). Paramyxoviruses, such as Sendai virus,bind and fuse with the plasma membrane of the host cell.Other viruses, such as orthomyxoviruses, rhabdoviruses,and togaviruses, bind to the host cell surface and aresubsequently internalized by endocytosis. For these viruses,membrane fusion occurs mainly in the endosomal compart-ment (18, 35). Fusion is triggered by the mildly acidicendosomal pH and is catalyzed by virally encoded glycopro-
B I 00
0e~~~8
o ~~~~60>iLii
40
20
5.0 5.4 5.6 5.8 6.0 6.2 5.0 5.4 5.6 5.8 6.0 6.2
pHFIG. 9. pH dependence of HA-induced cell-cell (A) and RBC-cell (B) fusion in CV-1 cells infected with SV40-HA expression vectors.
CV-1 cells were infected with SV40-HA recombinant viruses containing the wild-type, variant, and chimeric HA genes. At 60 h postinfection,cell surface HA was activated by treatment with trypsin. Cell-cell fusion was assayed by polykaryon formation; RBC-cell fusion was assayedby the delivery of benzidine into the CV-1 cytoplasm. Data shown are average values for four experiments. Details of the assays are givenin Materials and Methods. Symbols: *, wild type; El, muHA1; 0, muHA2; 0, muHA12.
J. VIROL.

FUSION VARIANT OF INFLUENZA VIRUS HA 611
HAI
NmHA2
FIG. 10. Location of amino acid substitutions in variant A31. Adrawing of the ectodomain of an HA monomer, depicting itsthree-dimensional structure as determined by Wilson et al. (34).Arrows, Beta structures; barrels, alpha helices. In the wild-type X31HA, the negatively charged amino acid at position 132 (glutamic acidin the protein analyzed by Wilson et al. [34] and Verhoeyen et al.[27], but aspartic acid in our wild-type protein [P. J. G. Gallagherand M.-J. Gething, unpublished data]) forms an intersubunit saltbridge with the arginine residue at position 124 in a second HA2subunit. In variant virus A31, the aspartic acid at position 132 isreplaced with an asparagine, disrupting the salt bridge. The othersubstitutions in the A31 HA (at residues 144 and 215 in HA1) are
also shown. In another fusion variant described by Daniels et al. (3),a substitution at position 9 in HA2 also appears to destabilize the saltbridge between residues 132 and 124.
teins. The pH dependence of fusion varies among virus typesand, in the case of influenza virus, among strains, with theoptimal pH for fusion generally falling within the range frompH 5.0 to 6.2 for endocytosed viruses (31).To gain insight into the molecular mechanisms of the
low-pH-induced fusion reactions, virus mutants have been
isolated which fuse with pH optima different from those oftheir respective parents. The first such fusion mutant was avariant of Semliki Forest virus isolated after mutagenesis(14). This virus, fus-1, fuses at a pH optimum 0.7 pH unitslower than that of the wild type. Subsequently, Rott andco-workers (22) have shown that variants of the X31 strain ofinfluenza virus selected for their ability to undergo activationcleavage and grow in MDCK cells have an elevated fusionpH threshold (approximately 0.7 pH units higher than thewild type). Similar influenza virus variants have been se-lected by growth in the presence of amantadine, a compoundthat raises endosomal pH (3). Variant viruses were obtainedthat fused at pHs 0.1 to 0.7 units higher than the parentstrain. Sequence analysis of the HA from each variantidentified amino acid residues that play a role in the pHdependence of fusion.Here we report the isolation and characterization of a
naturally occurring fusion variant of the X31 strain ofinfluenza virus which was plaque purified directly from awild-type stock population. The virus was neither mutagen-ized nor subjected to any selective pressure; rather, it wasfound by morphological and biochemical examination of thestock virus preparation. At this time we do not know thefrequency with which this or other fusion variants occur inthe X31 stock population. The finding does suggest, how-ever, that fusion variants of a virus strain may be circulatingin the natural population and that this variability may repre-sent yet another means by which influenza virus can adapt toa variety of hosts.Once the variant and its wild-type counterpart had been
examined in detail, we found a strong correlation betweenthe pH dependence of the fusion reaction, the changedmorphology of the HA spike, the biochemical manifestationof the conformational change, and the ability of the HA tobind to a target membrane. In all cases, the pHs at whichthese low-pH-induced changes occurred were higher for thevariant than for the wild-type HA, even though the absolutemagnitude of the increase in pH was somewhat variable. Forexample, although variant A31 induced 50% cell-cell fusionat a pH 0.2 units higher than the fusion midpoint of thewild-type virus, there was a 0.4-unit difference between thepHs at which 50% of the variant and wild-type BHAsunderwent conformational change. These results are, how-ever, in agreement with our previous work, which suggeststhat 50% fusion occurs when approximately 75% conversionof the HA to the low-pH conformation has been reached (5).If the point at which 75% conversion of the variant andwild-type BHAs is compared, a 0.2-unit pH difference isseen.
That the variant fusion phenotype of the A31 virus wasdue to a difference in the HA and not some other viralprotein was proven by cloning both the variant and wild-typeHAs, expressing them in simian cells, and analyzing theirfusion phenotypes. Here again, a 0.2-unit difference in thepH optima for fusion was observed. Since the amino acidsequence of the A31 HA differed from that of the wild typeHA at three positions, chimeric genes were constructed andused to express HA molecules that contained subsets of theamino acid changes. Analysis of the fusion activity of thechimeric proteins clearly established that the substitution ofan asparagine for the aspartic acid residue at position 132 inthe HA2 polypeptide was solely responsible for the alter-ation in fusion phenotype. Examination of the three-dimen-sional structure of HA (34) revealed that residue 132 waslocated near the base of the molecule, approximately 2 nmfrom where the polypeptide enters the lipid bilayer of the
VOL. 57, 1986

612 DOMS ET AL.
viral envelope or the plasma membrane (Fig. 10). In thewild-type HA, Asp-132 forms a salt bridge with Arg-124 in anadjacent HA2 subunit in the trimer (3). The residues in-volved in this salt link are highly conserved in the HAs frommany different strains of influenza virus, suggesting that thisis an important structural interaction. Interestingly, anamino acid substitution in a fusion variant described previ-ously, in which Phe-9 of HA2 was replaced with Leu-9, mayexert its effects at least in part by destabilizing this interac-tion (3). Both the substitutions at residues 9 and 132, as wellas others described by Daniels et al. (3), affect residueswhich are involved in stabilizing the HA trimer at neutralpH. Substitutions which disrupt these interactions maydestabilize the trimer, allowing it to undergo the fusion-inducing conformational change at a higher pH. Thesefindings are consistent with the hypothesis that the HAtrimer undergoes at least partial dissociation at acid pH (3, 5,25), although direct evidence for this has yet to be obtained.
In addition to providing information on the molecularmechanism of membrane fusion, variants of influenza virussuch as the one described in this paper should in the futureprovide useful probes for elucidating the endocytic pathway.By infecting cells with viruses which fuse at slightly differentpHs, it will be possible to measure the kinetics and extent ofintracellular acidification by following the conformationalchange in the variant HAs with established biochemical andimmunological criteria.
ACKNOWLEDGMENTS
We wish to thank Margaret Kielian for advice on plaquepurification, Don Wiley for helpful discussions on the positions andfunctions of the amino acid substitutions in the structure of HA, andRobert Webster for his gift of antibodies. We also thank Anne Biedlerfor expert technical assistance and Margie Moench and Mike Okklerfor their artwork.The work was supported by Public Health Service grants A118582
(to A. H. for work at Yale) and Al 19630 (to M.-J.G. for work at ColdSpring Harbor) from the National Institutes of Health. J.W. wassupported by a Swebelius Cancer Research Award.
LITERATURE CITED1. Both, G. W., M. J. Sleigh, N. J. Cox, and A. P. Kendal. 1983.
Antigenic drift in influenza virus H3 hemagglutinin from 1968 to1980: multiple evolutionary pathways and sequential amino acidchanges at key antigenic sites. J. Virol. 48:52-60.
2. Brand, C. M., and J. J. Skehel. 1972. Crystalline antigen fromthe influenza virus envelope. Nature (London) New Biol.238:145-147.
3. Daniels, R. S., J. C. Downie, A. J. Hay, M. Knossow, J. J.Skehel, M. L. Wang, and D. C. Wiley. 1985. Fusion mutants ofthe influenza virus hemagglutinin glycoprotein. Cell 40:431-439.
4. DeDuve, C., T. DeBarsy, B. Poole, A. Trouet, P. Tulkens, and F.Van Hoof. 1974. Lysosomotropic agents. Biochem. Pharmacol.23:2495-2531.
5. Doms, R. W., A. H. Helenius, and J. White. 1985. Membranefusion, activity of the influenza virus hemagglutinin. J. Biol.Chem. 260:2973-2981.
6. Doxsey, S. J., J. Sambrook, A. Helenius, and J. White. 1985. Anefficient method for introducing macromolecules into livingcells. J. Cell Biol. 101:19-27.
7. Doyle, C., M. G. Roth, J. Sambrook, and M.-J. Gething. 1985.Mutations in the cytoplasmic domain of influenza virus hemag-glutinin affect different stages of intracellular transport. J. CellBiol. 100:704-714.
8. Fraker, P. J., and J. C. Speck, Jr. 1978. Protein and cellmembrane iodinations with a sparingly soluble chloroamide,1,3,4,6-tetrachloro-3a,6a-diphenylglycoluracil. Biochem. Bio-
phys. Res. Commun. 80:849-857.9. Gething, M.-J., J. Bye, J. J. Skehel, and M. D. Waterfield. 1980.
Cloning and DNA sequence of double-stranded copies ofhaemagglutinin genes from H2 and H3 strains elucidates anti-genic shift and drift in human influenza virus. Nature (London)287:301-306.
10. Gething, M.-J., and J. Sambrook. 1981. Cell surface expressionof influenza hemagglutinin from a cloned DNA copy of the RNAgene. Nature (London) 293:620-625.
11. Hanahan, D. 1983. Studies on transformation of Escherichia coliwith plasmids. J. Mol. Biol. 166:557-580.
12. Jensen, E. M., E. E. Force, and J. B. Ungar. 1961. Inhibitoryeffect of ammonium ions on influenza virus in tissue culturecells. Proc. Soc. Exp. Biol. Med. 107:447-451.
13. Jensen, E. M., and 0. C. Liu. 1963. Inhibitory effect of simplealiphatic amines on influenza virus in tissue culture. Proc. Soc.Exp. Biol. Med. 112:456-459.
14. Kielian, M. C., S. Keranen, L. Kaariainen, and A. Helenius.1984. Membrane fusion mutants of Semliki Forest virus. J. CellBiol. 98:139-145.
15. Kilbourne, E. D. 1969. Future influenza vaccines and the use ofgenetic recombinants. Bull. W.H.O. 41:643-645.
16. Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall.1951. Protein measurement with the Folin phenol reagent. J.Biol. Chem. 193:265-275.
17. Maniatis, T., E. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
18. Marsh, M. 1984. The entry of enveloped viruses into cells byendocytosis. Biochem. J. 218:1-10.
19. Naeve, C. W., V. S. Hinshaw, and R. G. Webster. 1984.Mutations in the hemagglutinin receptor-binding site can changethe biological properties of an influenza virus. J. Virol.51:567-569.
20. Palese, P., and J. F. Young. 1982. Variation of influenza A, B,and C viruses. Science 215:1468-1473.
21. Rogers, G. N., J. C. Paulson, R. S. Daniels, J. J. Skehel, andD. C. Wiley. 1983. Single amino acid substitutions in influenzahaemagglutinin change receptor binding specificity. Nature(London) 304:76-78.
22. Rott, R., M. Orlich, H.-D. Klenk, M. L. Wang, J. J. Skehel, andD. C. Wiley. 1984. Studies on the adaptation of influenza virusesto MDCK cells. EMBO J. 3:3329-3332.
23. Sanger, F., A. R. Coulson, B. G. Barrell, A. J. H. Smith, andB. A. Roe. 1980. Cloning in single-stranded bacteriophage as anaid to rapid DNA sequencing. J. Mol. Biol. 143:161-178.
24. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain terminating inhibitors. Proc. Nat]. Acad. Sci.USA 74:5436-5467.
25. Skehel, J. J., P. M. Bayley, E. B. Brown, S. R. Martin, M. D.Waterfield, J. M. White, I. A. Wilson, and D. C. Wiley. 1982.Changes in the conformation of influenza hemagglutinin at thepH optimum of virus mediated membrane fusion. Proc. Natl.Acad. Sci. USA 79:968-972.
26. VanRompuy, L., W. M. Jou, M. Verhoeyen, D. Huylebroeck,and W. Fiers. 1983. Molecular variation of influenza surfaceantigens. Trends Biochem. Sci. 8:414-417.
27. Verhoeyen, M., R. Fang, W. M. Jou, R. Devos, D. Huylebroeck,E. Saman, and W. Fiers. 1980. Antigenic drift between thehemagglutinin of the Hong Kong influenza virus strainsA/Aichi/2/68 and A/Victoria/3/75. Nature (London) 286:771-776.
28. Webster, R. G., W. G. Laver, G. M. Air, and G. C. Schild. 1982.Molecular mechanisms of variation of influenza viruses. Nature(London) 296:115-121.
29. White, J., and A. Helenius. 1980. pH-dependent fusion betweenthe Semliki Forest virus membrane and liposomes. Proc. Natl.Acad. Sci. USA 77:3273-3277.
30. White, J., J. Kartenbeck, and A. Helenius. 1982. Membranefusion activity of influenza virus. EMBO J. 1:217-222.
31. White, J., M. Kielian, and A. Helenius. 1983. Membrane fusionproteins of enveloped animal viruses. Q. Rev. Biophys.16:151-195.
J. VIROL.

FUSION VARIANT OF INFLUENZA VIRUS HA
32. White, J., K. Matlin, and A. Helenius. 1981. Cell fusion bySemliki Forest, infhuenza, and vesicular stomatitis viruses. J.Cell Biol. 89:674-679.
33. Wiley, D. C., I. A. Wilson, and J. J. Skehel. 1981. Structuralidentification of the antibody-binding sites of Hong Kong influ-enza haemagglutinin and their involvement in antigenic varia-
tion. Nature (London) 289:373-378.34. Wilson, I. A., J. J. Skehel, and D. C. Wiley. 1981. Structure of
the haemagglutinin membrane glycoprotein of influenza virus at3A resolution. Nature (London) 289:366-373.
35. Yoshimura, A., and S.-I. Ohnishi. 1984. Uncoating of influenzavirus in endosomes. J. Virol. 51:497-504.
VOL. 57, 1986 613
Related Documents











