doi:10.1182/blood-2002-10-3313 Prepublished online January 23, 2003; Robert P Hebbel and Gregory M Vercellotti John D Belcher, Christopher J Bryant, Julia Nguyen, Paul R Bowlin, Miroslaw C Kielbik, John C Bischof, Transgenic sickle mice have vascular inflammation (1174 articles) Red Cells (2497 articles) Hemostasis, Thrombosis, and Vascular Biology (1086 articles) Gene Expression Articles on similar topics can be found in the following Blood collections http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requests Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprints Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtml Information about subscriptions and ASH membership may be found online at: digital object identifier (DOIs) and date of initial publication. the indexed by PubMed from initial publication. Citations to Advance online articles must include final publication). Advance online articles are citable and establish publication priority; they are appeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet Copyright 2011 by The American Society of Hematology; all rights reserved. 20036. the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.org From

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

doi:10.1182/blood-2002-10-3313Prepublished online January 23, 2003;
Robert P Hebbel and Gregory M VercellottiJohn D Belcher, Christopher J Bryant, Julia Nguyen, Paul R Bowlin, Miroslaw C Kielbik, John C Bischof, Transgenic sickle mice have vascular inflammation
(1174 articles)Red Cells � (2497 articles)Hemostasis, Thrombosis, and Vascular Biology �
(1086 articles)Gene Expression �Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. theindexed by PubMed from initial publication. Citations to Advance online articles must include
final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.20036.the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

Title: Transgenic Sickle Mice Have Vascular Inflammation
Running Title: Sickle Mice are Inflamed
Authors: John D. Belcher1, Christopher J. Bryant1, Julia Nguyen1, Paul R. Bowlin1, Miroslaw C. Kielbik1, John C. Bischof2, Robert P. Hebbel1, and Gregory M. Vercellotti1
Institutions: University of Minnesota, Dept. of Medicine, Division of Hematology, Oncology and Transplantation1 and University of Minnesota, Dept. of Mechanical Engineering2, Minneapolis, MN
Support: This work was supported by NHLBI grant HL67367.
Correspondence: John D. Belcher, Ph.D.University of MinnesotaDepartment of MedicineDivision of Hematology, Oncology and Transplantation420 Delaware St SEMMC 480Minneapolis, MN 55455
Telephone: (612) 624-2611Fax: (612) 625-1121Email: [email protected]
Work Counts: Abstract: 238Main Body: 4378
Scientific Heading: Hemostasis, Thrombosis, and Vascular Biology
Acknowledgements: We would like to thank Stephana Choong for breeding and characterizing the transgenic sickle mice used for these studies.
Copyright (c) 2003 American Society of Hematology
Blood First Edition Paper, prepublished online January 23, 2003; DOI 10.1182/blood-2002-10-3313 For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

ABSTRACT
Inflammation may play an essential role in vaso-occlusion in sickle cell disease. Sickle patients have high white counts and elevated levels of serum C-reactive protein (CRP), cytokines and adhesion molecules. In addition, circulating endothelial cells, leukocytes and platelets are activated. We examined four transgenic mouse models expressing human alpha and sickle beta globin genes to determine if they mimic the inflammatory response seen in patients. These mouse models are designated “NY-S”, “Berk-SAntilles”, “NY-S/SAntilles” (NY-S X Berk-SAntilles), and “Berk-S”. The mean white counts were elevated 1.4- to 2.1-fold (p<0.01) in the Berk-SAntilles, NY-S/SAntilles, and Berk-S mice, but not in the NY-S mice compared to normal. Serum amyloid P-component (SAP), an acute phase response protein with 60-70% sequence homology to CRP, was elevated 8.5- to 12.1-fold (p<0.001) in transgenic sickle mice. Similarly, serum interleukin-6 (IL-6) was elevated 1.6- to 1.9-fold (p<0.05). Western blots, confirming immuno-histochemical staining, showed vascular cell adhesion molecule (VCAM), intercellular adhesion molecule (ICAM) and platelet-endothelial cell adhesion molecule (PECAM) were upregulated 3-to 5-fold (p<0.05) in the lungs of sickle mice. Ribonuclease protection assays (RPA) demonstrated VCAM mRNA also was elevated in sickle mice 1.2- to 1.4-fold (p<0.01). Nuclear factor kappa B (NF-κB), a transcription factor critical for the inflammatory response, was elevated 1.9-fold (p<0.006) in NY-S sickle mouse lungs. We conclude that transgenic sickle mice are good models to study vascular inflammation and the potential benefit of anti-inflammatory therapies to prevent vaso-occlusion in sickle cell disease.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

INTRODUCTION
Sickle cell disease, one of the most common inherited hematological diseases, is caused
by a single amino acid substitution in the β-globin chain of hemoglobin1. The polymerization of deoxygenated sickle hemoglobin is the primary event in the molecular pathogenesis of sickle cell disease and is responsible for the vaso-occlusive phenomena that are the hallmark of the
disease2. Patients with sickle cell disease suffer widespread end-organ damage due to chronic
vaso-occlusive episodes3. Recent studies using intravital microscopy to visualize blood flow in inflamed cremasteric venules of mice expressing human βS suggest a primary role for leukocytes adherent to endothelium accompanied by sickle red blood cells (RBCs) bound to the adherent
leukocytes in vaso-occlusion4,5. These data imply that activation of endothelium with its associated inflammatory response is necessary for leukocyte adherence and subsequent vaso-occlusion. Patients in sickle cell crisis have multiple indicators of an inflammatory response
including elevated white counts6-9, CRP levels10-13 and cytokines14-17, as well as activated
monocytes13,18, neutrophils19-21, platelets18,22-28 and endothelial cells29,30 in circulation. In vitro, monocytes from sickle patients activate human endothelial cell NF-κB which governs the expression of a wide variety of genes associated with inflammation including adhesion molecules, tissue factor, cytokines, and acute phase proteins13. In vivo, sickle patients have elevated numbers of circulating endothelial cells with markers of activation such as
adhesion molecules and tissue factor on their surface29,30 as well as the abnormal presence of
circulating VCAM in their plasma31-34. The development of transgenic mice that express the human betaS hemoglobin chains
now allows the study of the role of inflammation in vaso-occlusion. We have measured markers
of inflammation in four transgenic sickle mouse models, the “NY-S”35, “Berk-SAntilles”36, “NY-
S/SAntilles”37 and “Berk-S”38 (see Materials and Methods for a description). Some of these
models have been used to study the pathogenesis of vaso-occlusion4,39,40. Sickle mouse
erythrocytes change shape upon de-oxygenation41 and sickle mice display blood flow abnormalities including adhesion of red and white blood cells to post-capillary venules, sludging
in microvessels and decreased blood flow velocity in venules of all diameters4,5,39,40. In response to hypoxia-reoxygenation, sickle mice exhibit an exaggerated inflammatory response
and activate NF-κB in the liver and kidney4,41. Upon pathological analysis of these transgenic sickle mice, there is tissue damage in multiple organs including the kidney, liver, lung and
spleen35-38. We report here that transgenic sickle mice, like human sickle cell patients, have an active
inflammatory response. We hypothesize that anti-inflammatory agents may minimize vaso-occlusion and tissue injury in sickle cell disease. Transgenic sickle mice appear to be good animal models to test this hypothesis.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom
MATERIALS AND METHODS
Reagents were obtained from Sigma Aldrich (St. Louis, MO) unless otherwise indicated.
MiceAll animal experiments were approved by the University of Minnesota’s Institutional
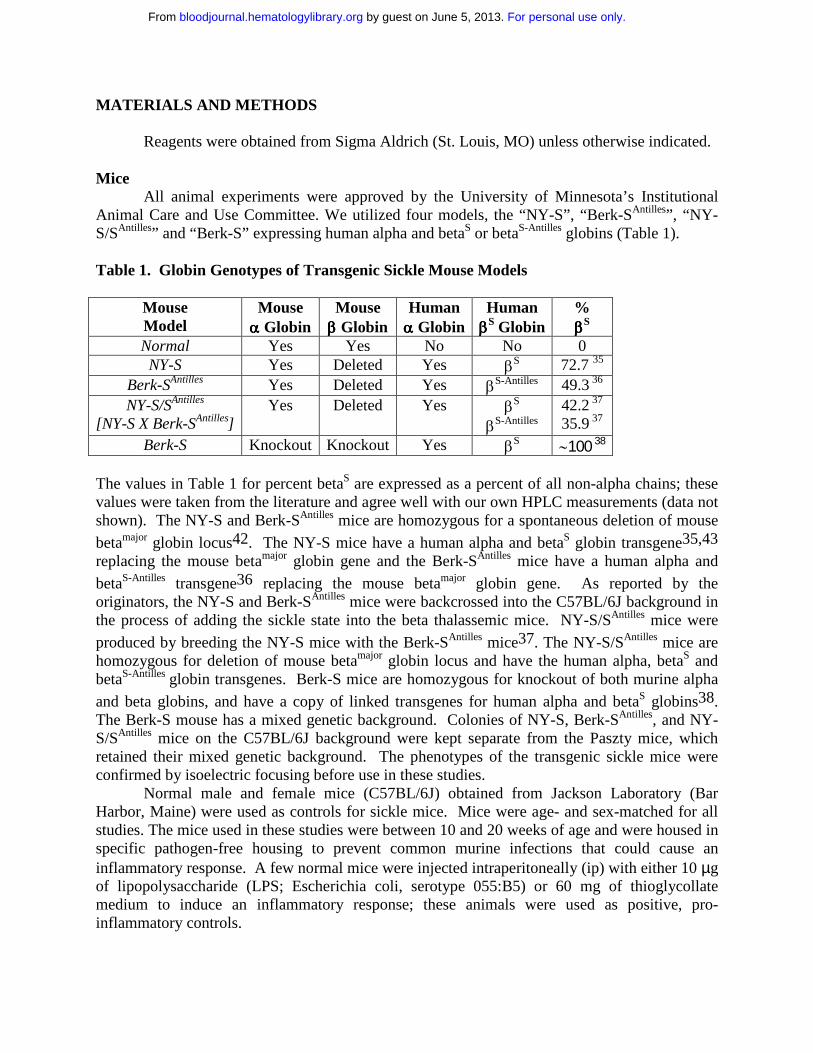
Animal Care and Use Committee. We utilized four models, the “NY-S”, “Berk-SAntilles”, “NY-S/SAntilles” and “Berk-S” expressing human alpha and betaS or betaS-Antilles globins (Table 1).
Table 1. Globin Genotypes of Transgenic Sickle Mouse Models
MouseModel
Mouseαααα Globin
Mouseββββ Globin
Humanαααα Globin
HumanββββS Globin
%ββββS
Normal Yes Yes No No 0NY-S Yes Deleted Yes βS 72.7 35
Berk-SAntilles Yes Deleted Yes βS-Antilles 49.3 36
NY-S/SAntilles
[NY-S X Berk-SAntilles]Yes Deleted Yes βS
βS-Antilles42.2 37
35.9 37
Berk-S Knockout Knockout Yes βS ∼100 38
The values in Table 1 for percent betaS are expressed as a percent of all non-alpha chains; these values were taken from the literature and agree well with our own HPLC measurements (data not shown). The NY-S and Berk-SAntilles mice are homozygous for a spontaneous deletion of mouse
betamajor globin locus42. The NY-S mice have a human alpha and betaS globin transgene35,43
replacing the mouse betamajor globin gene and the Berk-SAntilles mice have a human alpha and
betaS-Antilles transgene36 replacing the mouse betamajor globin gene. As reported by the originators, the NY-S and Berk-SAntilles mice were backcrossed into the C57BL/6J background in the process of adding the sickle state into the beta thalassemic mice. NY-S/SAntilles mice were
produced by breeding the NY-S mice with the Berk-SAntilles mice37. The NY-S/SAntilles mice are homozygous for deletion of mouse betamajor globin locus and have the human alpha, betaS and betaS-Antilles globin transgenes. Berk-S mice are homozygous for knockout of both murine alpha
and beta globins, and have a copy of linked transgenes for human alpha and betaS globins38. The Berk-S mouse has a mixed genetic background. Colonies of NY-S, Berk-SAntilles, and NY-S/SAntilles mice on the C57BL/6J background were kept separate from the Paszty mice, which retained their mixed genetic background. The phenotypes of the transgenic sickle mice were confirmed by isoelectric focusing before use in these studies.
Normal male and female mice (C57BL/6J) obtained from Jackson Laboratory (Bar Harbor, Maine) were used as controls for sickle mice. Mice were age- and sex-matched for all studies. The mice used in these studies were between 10 and 20 weeks of age and were housed in specific pathogen-free housing to prevent common murine infections that could cause an inflammatory response. A few normal mice were injected intraperitoneally (ip) with either 10 µg of lipopolysaccharide (LPS; Escherichia coli, serotype 055:B5) or 60 mg of thioglycollate medium to induce an inflammatory response; these animals were used as positive, pro-inflammatory controls.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

Hematocrits, White Blood Cell (WBC) Counts and WBC DifferentialsBlood was collected from the tail vein of unanesthetized mice after warming the animals
under a heat lamp. Hematocrits were measured after collection of whole blood in heparinized glass capillary tubes. The tubes were sealed and centrifuged at 500 X G for 10 minutes before reading the percentage volume of red cells in the tubes. For white counts, the RBCs were immediately lysed by diluting whole blood 20-fold in 2% acetic acid containing 30 µg/ml EDTA. WBCs were counted on a hemocytometer four times and averaged. A drop of blood was smeared onto a glass slide and stained in Wright-Giemsa solution. The WBC morphologies were examined microscopically and the differentials counted in four separate areas on each slide.
Serum Amyloid P-Component (SAP)Whole blood was collected from the tail vein and allowed to clot for 5-10 minutes. The
blood cells were spun down at 4,000 X G for 4 minutes and the serum was collected and frozen at –800C until use. Serum SAP levels were measured by enzyme immunoassay (EIA) as
previously described44. All samples were measured in replicates of four or six and the resulting values averaged.
Serum IL-6 Serum was collected and stored as described above. IL-6 was measured by EIA according to the manufacturers protocol (Endogen, Woburn, MA).
Immunohistochemistry of Lung TissueMice were asphyxiated with CO2. The lungs were removed and frozen at –80oC in tissue
freezing medium (Baxter Scientific Products, Chicago, IL). Frozen five µm sections were prepared using a cryostat and mounted onto slides. The sections were air-dried for 10 minutes, fixed in acetone for 10 minutes at room temperature and stored at –80oC until immunostaining. Endogenous peroxides were removed by immersing slides in 0.3% H2O2 in PBS, pH 7.4, for 10 minutes. Slides were washed once in TRIS buffered saline (TBS), pH 7.5, containing 0.1% Tween 20. Slides were blocked with 3% BSA in PBS (blocking buffer) for 10 minutes at room temperature. Avidin and biotin binding sites were blocked using specific avidin/biotin blocking reagents according to the manufacturer’s protocol (Vector Laboratories, Burlingame, CA). Biotinylated primary monoclonal antibodies to mouse VCAM-1, ICAM-1, and PECAM-1 (BD Pharmingen, San Diego, CA) were diluted 1:50 in blocking buffer and incubated with thin sections for 1 hour at 37oC in a humidified chamber. Sections were washed 3 times with TBS, 0.1% Tween 20. Bound primary antibodies were visualized using a Vectastain Elite ABC kit containing an avidin/biotin peroxidase complex and a Vector VIP peroxidase substrate kit according to the manufacturer’s protocol (Vector Laboratories). The VIP substrate produces an intense, violet-colored precipitate. Some frozen thin sections from the lungs of NY-S mice were double stained with primary antibodies to VCAM-1 (BD Pharmingen) and von Willebrand factor (Cedarlane Laboratories, Hornby, Ontario). The primary antibodies were visualized with appropriate secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) labeled with fluorescein isothiocyanate (FITC/green) (VCAM) and tetramethyl rhodamine isothiocyanate (TRITC/red) (von Willebrand factor). The nuclei were counterstained with 4’, 6 diamidino-2-phenylindole (DAPI/blue) (Vector Laboratories).
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

Western Blots of Lung VCAM, ICAM, and PECAM Protein ExpressionMice were asphyxiated with CO2 and the left lobes of the lungs were removed and frozen
in liquid N2. Lung tissue homogenate was prepared as previously described45. The lung tissue, frozen in liquid N2, was broken into small pieces with a hammer between layers of aluminum foil, then transferred to a mortar and reduced to a fine powder in liquid N2. The thawed powder was homogenized on ice in 5 ml of 0.6% Nonidet P-40 (Calbiochem, La Jolla, CA), 150 mM NaCl, 10 mM HEPES pH 7.9, 1 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride in a 15 ml Dounce tissue homogenizer (Wheaton, Millville, NJ) with 10 strokes of the tight fitting pestle B. Cell debris was removed by centrifuging the crude homogenate at 500 X G for 30 seconds at 4oC. The lung supernatant was frozen at –80oC. The lung homogenate protein concentration was determined by the BCA protein assay (Pierce, Rockford, IL) after protein precipitation with
trichloroacetic acid46. Lung homogenate DNA concentrations were determined by a fluorometric DNA dye-binding assay (Bio Rad, Hercules, CA). The bis-benzimide dye (Hoechst 33258) binds specifically to dsDNA. RNA does not interfere significantly with the assay. For Western blotting, lung homogenates, containing one microgram of lung DNA per well, were subjected to SDS-PAGE (7.5%). After SDS-PAGE, the homogenates were transferred electrophoretically to PVDF membranes, and immunoblotting was performed with goat anti-VCAM- 1, ICAM-1 and PECAM-1 IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Sites of primary antibody binding were visualized with alkaline phosphatase-conjugated donkey anti-goat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). The final detection of immunoreactive bands was performed using a chemifluorescent detection substrate (Amersham Biosciences, Piscataway, NJ).
RNA Extraction and Ribonuclease Protection Assay (RPA) of VCAM mRNATotal RNA was extracted from the right lobe of the lungs of sickle and normal mice using
an RNAqueousTM kit (Ambion, Austin, TX). RNA protection assays (RPA) were performed with 10 µg of extracted mouse lung RNA using an RPAIIITM kit (Ambion) and VCAM-1 and GAPDH antisense RNA probe templates (BD Biosciences Pharmingen, San Diego, CA) labeled with [32P]UTP using a T7 MaxiscriptTM kit (Ambion). The 32P-labeled antisense VCAM and GAPDH RNA probes were hybridized to lung mRNA overnight at 56oC and then digested with RNAase at 30oC for 45 minutes. Protected RNA fragments were separated by electrophoresis on a 5% acrylamide/8 M urea/ TBE gel.
Electrophoretic Mobility Shift Assay (EMSA) for NF-κκκκB Expression in LungsWhole lung cell extracts were prepared from lung homogenates (described above). Lung
homogenates were diluted 1:5 v/v in lysis buffer (Active Motif, Carlsbad, CA) containing 20 mM HEPES, pH 7.5, 350 mM NaCl, 20% glycerol, 1% Igepal-CA630, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 50 mM DTT and a protease inhibitor cocktail. Samples were incubated in lysis buffer 10 minutes on ice and then centrifuged at 16,000 X G for 20 minutes. The supernatants were collected and stored at –80oC until use. Lung extracts were incubated with end-labeled 32P-dsDNA containing a consensus murine NF-κB DNA binding sequence (underlined base pairs): 5’-AGTTGAGGGGACTTTCCCAGGC-3’ (Santa Cruz Biotechnology). DNA-protein binding reactions contained lung extracts from 260 ng of lung homogenate DNA and 70 fmol of radiolabelled NF-κB consensus dsDNA. Reactions were carried out in 20 mM HEPES, pH 7.9, 5 mM KCl, 0.5 mM EDTA, 5% glycerol, 1 mM dithiothreitol, 0.5 mM PMSF, 1 mg/mL BSA, 0.1% NP-40, and 250 ng of poly dI/dC. Binding reactions were incubated 30
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

minutes at room temperature. Reaction mixtures were separated on a 6% non-denaturing polyacrylamide gel using 0.5X TBE running buffer. To confirm the identity of NF-κB bands, some reactions were run with an excess of unlabeled NF-κB dsDNA for competition experiments or with antibodies to the p50 or p65 subunit of NF-κB for supershift experiments (Santa Cruz Biotechnology). The mouse NF-κB EMSA bands contained both the p50 and p65 subunits (data not shown).
Quantitation of Western Blots, RPA and NF-κκκκB GelsFluorescent western blots and radioactive RPA and EMSA gels exposed to a phosphor
screen were scanned on a Storm 840 gel and blot imaging system (Molecular Dynamics, Sunnyvale, CA). The Storm system provides a linear response to fluorescence and phosphorescence signal intensity. Fluorescent or radioactive bands on each image were quantified with ImageQuant 5.0 software (Molecular Dynamics) using volume quantitation and background subtraction. Volume quantitation calculates the volume under the surface created by a 3-D plot of the pixel locations and pixel intensities of each band.
StatisticsResults from control and transgenic sickle mice were compared using a Students t-test on
SigmaStat 2.0 for Windows (SPSS Inc, Chicago, IL).
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

RESULTS
White blood cell counts (Table 2) were elevated in all of the transgenic sickle mouse models compared to normal except the NY-S mice. White counts were elevated 144, 164 and206% of normal in the Berk-SAntilles (p < 0.01), NY-S/SAntilles (p < 0.001) and Berk-S (p < 0.001) mice, respectively. Elevated white counts reflected significant monocytosis and neutrophilia in the sickle mice (Table 3). There was a 2-fold increase in monocytes in NY-S mice compared to normal mice despite similar total white counts (Tables 2 and 3). The Berk-S mice had a significantly lower hematocrit than normal (p < 0.001); hematocrits of the other transgenic sickle mice (Table 2) were normal.
Table 2. White Counts and Hematocrits in Transgenic Sickle Mice
MouseModel n
Mean + SDWBC/µµµµL (X 10-3) n
Mean + SDHematocrit (%)
Normal 14 8.7 + 2.3 7 45.6 + 2.0NY-S 10 8.6 + 1.4 4 45.5 + 1.9
Berk-SAntilles 10 12.5 + 4.1** 5 48.4 + 2.7NY-S/SAntilles 12 14.3 + 3.8*** 3 46.3 + 5.1
Berk-S 8 17.9 + 7.6*** 4 30.9 + 6.7***
**p < 0.01***p < 0.001
Table 3. WBC Differentials in Transgenic Sickle Mice (WBC/µµµµL X 10-3)
MouseModel n Neutrophils Lymphocytes Monocytes Eosinophils BasophilsNormal 3 1.31 + 0.56 6.08 + 0.59 0.69 + 0.09 0.17 + 0.02 0.02 + 0.04NY-S 3 0.75 + 0.06 5.69 + 0.88 1.46 + 0.11*** 0.43 + 0.32 0.42 + 0.18*
Berk-SAntilles 3 3.11 + 0.18** 5.89 + 0.36** 3.33 + 1.09* 0.74 + 0.44 0.27 + 0.02***
Berk-S 3 5.11 + 2.03* 10.76 + 3.72* 2.33 + 1.05* 0.66 + 0.59 0.29 + 0.40
*p < 0.05**p < 0.01***p < 0.001
SAP is a well-documented acute-phase reactant in mice with extensive (60-70%)
sequence homology with human CRP47,48. Serum SAP was elevated 8- to 12-fold in transgenic sickle mice compared to normal control mice (p < 0.001). SAP also was elevated 5- to 6-fold in normal mice 24 hours after an ip injection of either 10 µg LPS (p < 0.05) or 60 mg thioglycollate medium (p < 0.001), respectively (Table 4). Similarly, serum IL-6 (Table 5), recognized as the principal regulator of most acute-phase response genes, was elevated in untreated NY-S, Berk-SAntilles and NY-S/SAntilles mice and in LPS-treated (10 µg ip) normal mice compared to untreated normal control mice (p < 0.05).
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

Table 4. Serum Amyloid P (SAP) in Transgenic Sickle Mice
MouseModel n
Mean + SDSAP (µµµµg/mL)
Normal 19 20 + 11Normal + LPS 2 100 + 40*
Normal + thioglycollate 3 124 + 12***
NY-S 10 241 + 91***
Berk-SAntilles 10 189 + 100***
NY-S/SAntilles 11 169 + 108***
Berk-S 8 242 + 146***
*p < 0.05***p < 0.001
Table 5. Serum IL-6 in Transgenic Sickle Mice
MouseModel n
Mean + SDIL-6 (pg/mL)
Normal 9 45 + 21Normal + LPS 2 >400*
NY-S 7 73 + 31*
Berk-SAntilles 8 88 + 41*
NY-S/SAntilles 4 78 + 2*
*p < 0.05
Frozen thin sections of lungs were prepared from three mice in each model and immunostained with specific IgG against mouse VCAM, ICAM, PECAM, von Willebrand factor and non-specific control IgG. VCAM, ICAM and PECAM staining was visually increased in normal mouse lungs 18 hours after an ip injection of 10 µg LPS and in all transgenic sickle mouse lungs relative to untreated normal control mice (Fig. 1A). VCAM staining (green) in NY-S sickle mouse lungs was often co-localized with an endothelial cell marker, von Willebrand factor (red) (Fig 1B). The co-localized green and red fluorescence appears yellow (Fig 1B) indicating VCAM was expressed on the vascular endothelial cells in the lungs. ICAM staining in the lungs was diffuse throughout the parenchyma and was not concentrated as heavily around the blood vessels like VCAM and PECAM (Fig 1A). Different ICAM antibodies gave a similar diffuse staining pattern (data not shown). Interestingly, red and white blood cells are visible in the blood vessels of NY-S/SAntilles mice (Fig 1A, see VCAM and PECAM stains). Some of the visible leukocytes appear to be marginated.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom
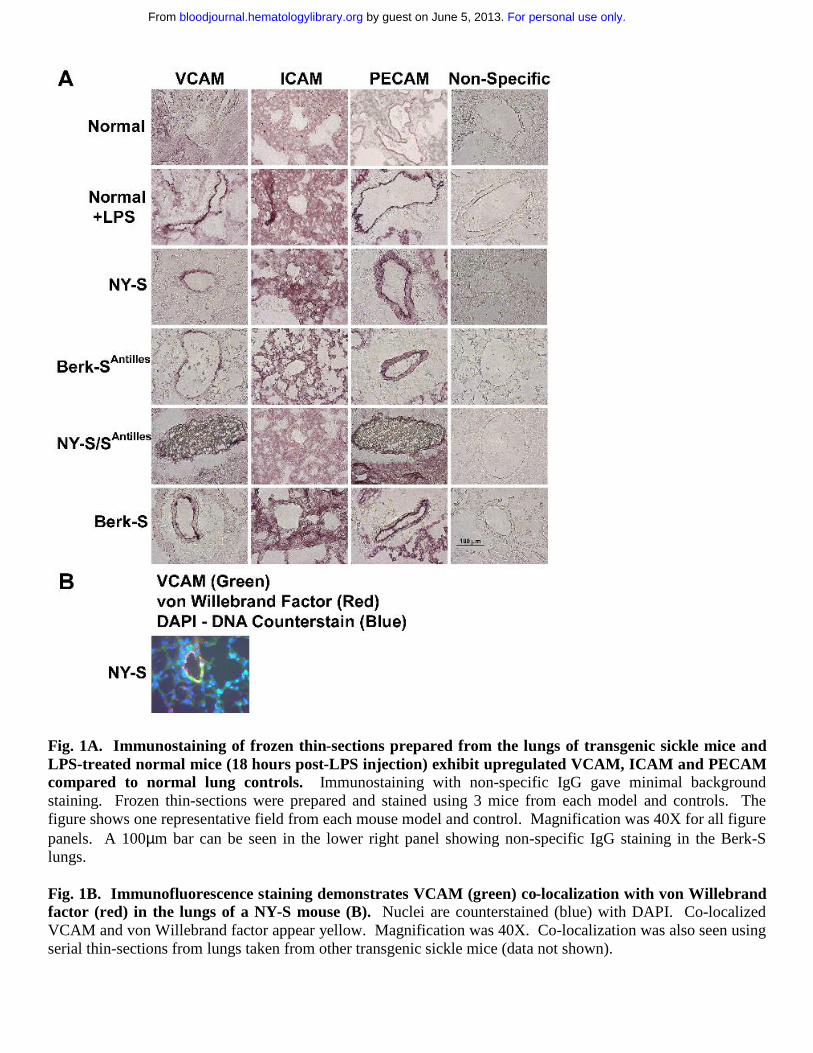
Fig. 1A. Immunostaining of frozen thin-sections prepared from the lungs of transgenic sickle mice and LPS-treated normal mice (18 hours post-LPS injection) exhibit upregulated VCAM, ICAM and PECAM compared to normal lung controls. Immunostaining with non-specific IgG gave minimal background staining. Frozen thin-sections were prepared and stained using 3 mice from each model and controls. The figure shows one representative field from each mouse model and control. Magnification was 40X for all figure panels. A 100µm bar can be seen in the lower right panel showing non-specific IgG staining in the Berk-S lungs.
Fig. 1B. Immunofluorescence staining demonstrates VCAM (green) co-localization with von Willebrand factor (red) in the lungs of a NY-S mouse (B). Nuclei are counterstained (blue) with DAPI. Co-localized VCAM and von Willebrand factor appear yellow. Magnification was 40X. Co-localization was also seen using serial thin-sections from lungs taken from other transgenic sickle mice (data not shown).
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

Fig 2. Western Blots confirm upregulated adhesion molecule expression in the lungs of transgenic sickle mice and LPS-treated normal mice (18 hours post-LPS injection) compared to normal lung controls.Lung homogenates were prepared from 3 mice in each group. Homogenate proteins, representing 1 µg of lung DNA per lane, were separated by SDS PAGE, transferred electrophoretically to PVDF membranes, and immunoblotted with either goat anti-VCAM, ICAM and PECAM IgG. Sites of primary antibody binding were visualized with alkaline phosphatase-conjugated donkey anti-goat IgG. The final detection of immunoreactive bands was performed using a chemifluorescent detection substrate. Protein bands corresponding to each adhesion molecule were quantified by fluorescence densitometry. The figure shows the adhesion molecule bands from one representative lung from each model and a summary bar graph. The bar graph plots the mean +SD adhesion molecule expression for each mouse model as a percentage of normal control mice (n=3).
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom
The increased adhesion molecule staining suggested the lungs of transgenic sickle mice were inflamed. Western blots were run on lung homogenates to confirm and quantify increased adhesion molecule expression in transgenic sickle mice. The lungs of sickle mice had a significantly higher hemoglobin and protein content than normal mice as judged by their red color and the protein to DNA ratios of the lung homogenates (data not shown, p < 0.05). Therefore, equal amounts (1µg) of lung homogenate DNA were loaded onto gel wells to normalize each lung sample for DNA, a better surrogate of cell number than protein. On Western blots, VCAM, ICAM and PECAM were upregulated 3- to 5-fold (p < 0.05) in normal mice 18 hours after LPS treatment (10 µg ip) and in transgenic NY-S, Berk-SAntilles, NY-S/SAntilles and Berk-S mice (Fig. 2). Some of the ICAM and PECAM Westerns appeared to show a doublet closely related in molecular weight. Both bands were included in the densitometry quantification. Different antibodies to ICAM and PECAM gave the same banding pattern suggesting the doublets were related in sequence (data not shown). These data confirm and quantify the upregulation of adhesion molecules in transgenic sickle mouse lungs seen by immunohistochemistry.
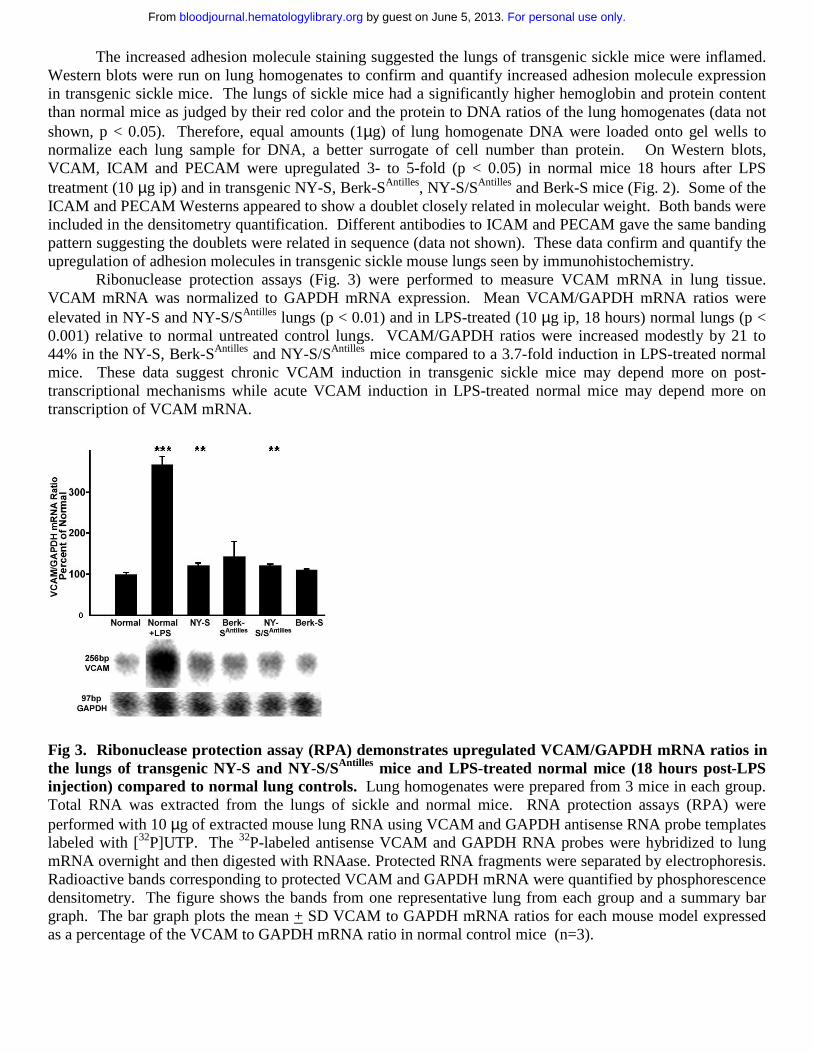
Ribonuclease protection assays (Fig. 3) were performed to measure VCAM mRNA in lung tissue. VCAM mRNA was normalized to GAPDH mRNA expression. Mean VCAM/GAPDH mRNA ratios were elevated in NY-S and NY-S/SAntilles lungs (p < 0.01) and in LPS-treated (10 µg ip, 18 hours) normal lungs (p < 0.001) relative to normal untreated control lungs. VCAM/GAPDH ratios were increased modestly by 21 to 44% in the NY-S, Berk-SAntilles and NY-S/SAntilles mice compared to a 3.7-fold induction in LPS-treated normal mice. These data suggest chronic VCAM induction in transgenic sickle mice may depend more on post-transcriptional mechanisms while acute VCAM induction in LPS-treated normal mice may depend more on transcription of VCAM mRNA.
Fig 3. Ribonuclease protection assay (RPA) demonstrates upregulated VCAM/GAPDH mRNA ratios in the lungs of transgenic NY-S and NY-S/SAntilles mice and LPS-treated normal mice (18 hours post-LPS injection) compared to normal lung controls. Lung homogenates were prepared from 3 mice in each group. Total RNA was extracted from the lungs of sickle and normal mice. RNA protection assays (RPA) were performed with 10 µg of extracted mouse lung RNA using VCAM and GAPDH antisense RNA probe templates labeled with [32P]UTP. The 32P-labeled antisense VCAM and GAPDH RNA probes were hybridized to lung mRNA overnight and then digested with RNAase. Protected RNA fragments were separated by electrophoresis. Radioactive bands corresponding to protected VCAM and GAPDH mRNA were quantified by phosphorescence densitometry. The figure shows the bands from one representative lung from each group and a summary bar graph. The bar graph plots the mean + SD VCAM to GAPDH mRNA ratios for each mouse model expressed as a percentage of the VCAM to GAPDH mRNA ratio in normal control mice (n=3).
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom
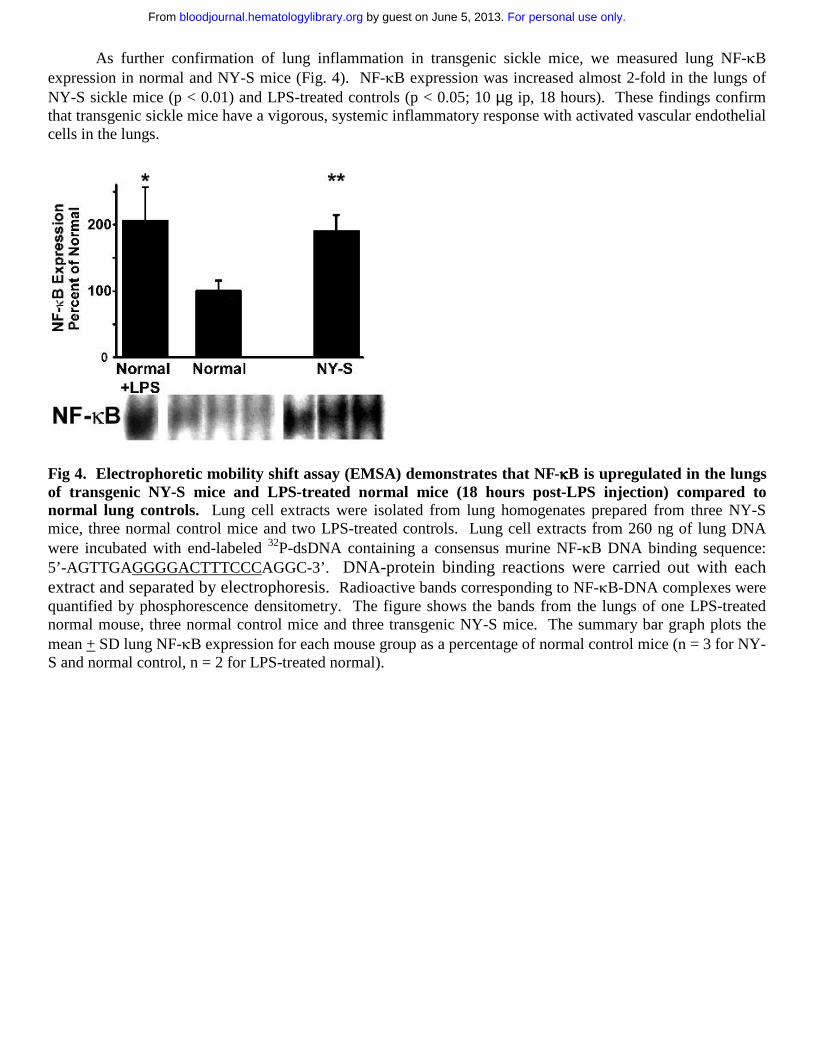
As further confirmation of lung inflammation in transgenic sickle mice, we measured lung NF-κB expression in normal and NY-S mice (Fig. 4). NF-κB expression was increased almost 2-fold in the lungs of NY-S sickle mice (p < 0.01) and LPS-treated controls (p < 0.05; 10 µg ip, 18 hours). These findings confirm that transgenic sickle mice have a vigorous, systemic inflammatory response with activated vascular endothelial cells in the lungs.
Fig 4. Electrophoretic mobility shift assay (EMSA) demonstrates that NF-κκκκB is upregulated in the lungs of transgenic NY-S mice and LPS-treated normal mice (18 hours post-LPS injection) compared to normal lung controls. Lung cell extracts were isolated from lung homogenates prepared from three NY-S mice, three normal control mice and two LPS-treated controls. Lung cell extracts from 260 ng of lung DNA were incubated with end-labeled 32P-dsDNA containing a consensus murine NF-κB DNA binding sequence: 5’-AGTTGAGGGGACTTTCCCAGGC-3’. DNA-protein binding reactions were carried out with each extract and separated by electrophoresis. Radioactive bands corresponding to NF-κB-DNA complexes were quantified by phosphorescence densitometry. The figure shows the bands from the lungs of one LPS-treated normal mouse, three normal control mice and three transgenic NY-S mice. The summary bar graph plots the mean + SD lung NF-κB expression for each mouse group as a percentage of normal control mice (n = 3 for NY-S and normal control, n = 2 for LPS-treated normal).
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

DISCUSSION
Like human sickle cell patients, transgenic sickle mice that express the human alpha and betaS and/or betaS-Antilles globins exhibit numerous markers of an inflammatory response. Leukocytosis was present in three of the four transgenic sickle mouse models, manifested primarily as monocytosis and neutrophilia. SAP, an acute phase response protein and a mouse homologue to human CRP, was dramatically elevated in all four transgenic sickle mouse models compared to normal. Correspondingly, serum IL-6, a pro-inflammatory cytokine, was significantly elevated in all three of the sickle mouse models that were measured. These systemic markers of inflammation were, in part, a reflection of endothelial cell activation that could be seen in both large and small blood vessels in the lungs. VCAM, ICAM and PECAM were upregulated in the lungs of the four transgenic sickle mouse models compared to normal as seen by Western blots and immunohistochemistry. Ribonuclease protection assays (RPA) revealed modest increases in VCAM mRNA in the lungs of NY-S and NY-S/SAntilles mice. Furthermore,
NF-κB, a transcription factor critical for the inflammatory response49, was significantly increased in the lungs of NY-S sickle mice compared to normal controls. These data on the inflammatory response in transgenic sickle mice mirror the clinical data from patients and strengthen the concept that the sickle beta globin gene mutation promotes an inflammatory response.
There were few statistically significant differences in inflammation markers between the four sickle mouse models despite differences in human beta-S and beta-SAntilles globin expression
and differences in the severity of their organ pathology35-38. The NY-S mice had significantly lower white counts relative to the other three sickle models, but had higher serum SAP levels than the Berk-SAntilles and NY-S/SAntilles models. These findings can be reconciled by the fact that the NY-S mice, despite normal white counts, had absolute monocytosis – more than twice that of normal control mice (Table 3). Hepatic SAP production is stimulated by cytokines secreted by
activated monocytes/macrophages50, which may help to explain the elevated SAP levels in the transgenic sickle mice. Serum IL-6 and lung adhesion molecule expression were similar in the four models. In general, each of the four transgenic sickle mouse models had similar inflammatory responses and all had an inflammatory response greater than normal control mice.
Over the past two decades the role of the endothelium and its interactions with sickle red and white blood cells has led to a revised paradigm for the understanding of vaso-occlusive
phenomena in sickle cell disease3-5,51-55. Interactions of sickle red and white blood cells with
vascular endothelium depend on a variety of factors56 including agents that promote the expression of adhesion molecules on the vessel wall. These agents such as inflammatory cells, cytokines, oxidants, hypoxic stress and infection result in an adhesive, inflammatory phenotype that augments sickle red and white blood cell adherence to the endothelium. Clinical conditions, including infections, surgery, and pregnancy, (all states that are “stressors” and in some aspects
pro-inflammatory) are associated with more vaso-occlusive crises8. The acute chest syndrome frequently is associated with an atypical pneumonia, fever, chest pain and a rise in the white
count and soluble VCAM8,9,34. In these situations, pro-inflammatory factors such as cytokines could further activate endothelium and promote changes in vascular tone and permeability, anticoagulant-procoagulant balance, changes in leukocyte trafficking, induction of acute phase reactants and promotion of red and white cell adhesion to an activated endothelium. High dose intravenous dexamethasone ameliorates acute chest syndrome in children and adolescents with
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

sickle cell disease57. This anti-inflammatory agent can act on inflammation in a variety of ways including decreasing the production of cytokines, decreasing the activation of leukocytes and inhibiting NF-κB activation. Unfortunately, many of the symptoms rebounded after the drug was discontinued57. These data suggest that anti-inflammatory agents may be useful in the treatment or prevention of sickle cell crises.
Even in the steady state, absolute monocytosis is seen in nearly all sickle patients58.
Moreover, sickle leukocytes have abnormal adhesion and activation13,19,20,59-61. White counts are significantly elevated and are highly correlated with stroke in children with sickle cell
anemia62. High white count may even cause sickle crisis63. Several case reports have linked granulocyte colony-stimulating factor (G-CSF) with induction of severe or fatal vaso-occlusive
crisis63-65. In the large multicenter trial of hydroxyurea to ameliorate sickle cell crisis, it was noted that a rise in hemoglobin F levels inversely correlated with the frequency of crises, acute
chest syndrome, leg ulcers and early mortality62,66-69. However, before hemoglobin F increases, there is a marked drop in the white blood count and the reduction of total white blood
count is a predictor of clinical response to hydroxyurea68. Thus, the white cell appears to be playing an important role in vaso-occlusive crisis. Recently, intravital microscopy of blood flow in the cremasteric venules of mice expressing human sickle beta globin demonstrated a primary
role for adherent leukocytes in producing vaso-occlusion in inflamed venules5. These studies showed sickle RBCs binding to adherent leukocytes to produce vaso-occlusion in inflamed venules. Moreover, sickle mice deficient in P- and E-selectins, which display defective recruitment of leukocytes to the vessel wall, were protected from vaso-occlusion. Thus, interactions between leukocytes and endothelial cells in the vessel wall may be of primary importance in understanding and preventing vaso-occlusion.
It is unclear from these studies whether the vascular inflammation is a primary response to the polymerization of sickle hemoglobin or a secondary response to tissue injury or infection. Infection was not a likely contributor to inflammation in these studies as the mice were housed in specific pathogen-free housing with careful monitoring and precautions taken against common murine pathogens. The inflammatory response could be a response to both tissue injury and sickle hemoglobin polymerization. The cellular and molecular events that translate the beta globin gene mutation into an inflammatory response remain speculative. Spontaneous oxygen
radical formation in the sickle RBC70 is one potential mechanism that could lead to an inflammatory response. Oxygen free-radicals, especially the hydroxyl radical, can activate NF-
κB71 leading to inflammatory gene expression. Interactions between sickle RBCs, hemoglobin
and leukocytes could amplify inflammatory cytokine production19,72 leading to activation of the
endothelium13 and subsequent leukocyte and RBC adhesion leading to vaso-occlusion.Another pro-inflammatory family of molecules is the endothelins. Endothelin-1 is a potent
vaso-constrictor and pro-inflammatory agonist that is significantly elevated in sickle cell
disease73-75. Endothelial cells increase their production of endothelin-1 in response to hypoxia76 and after incubation with sickled, but not unsickled, RBCs77. Ischemia-reperfusion injury is another potential mechanism that could promote vascular inflammation and tissue injury. Sickle cell disease involves repeated, lifelong, and perhaps near constant development of
transient ischemic episodes3. Reperfusion of tissues after interruption of their blood supply can
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

cause free-radical generation and lead to vigorous inflammatory responses and subsequent tissue
damage78. Transgenic sickle mice exhibit biochemical footprints consistent with excessive free-radical generation even at ambient air and following transient induction of enhanced
sickling4,41. An ongoing reperfusion injury would be consistent with the chronic inflammatory response seen in sickle cell patients and transgenic sickle mice. Novel anti-inflammatory and anti-oxidant treatments may provide fruitful therapies for sickle cell disease. Transgenic sickle mice appear to be good models to study the potential benefit of anti-inflammatory therapies to prevent vaso-occlusion in sickle cell disease.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

REFERENCES
1. Pauling L, Itano HA, Singer SJ. Sickle cell anemia, a molecular disease. Science. 1949;100:543-548.2. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762-769.3. Embury SH, Hebbel RP, Steinberg MH, Mohandas N. Pathogenesis of Vasoocclusion. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, eds. Sickle Cell Disease: Basic Principles and Clincal Practice. Vol 21. New York: Raven Press, Ltd.; 1994:311-326.4. Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411-420.5. Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99:3047-3051.6. Buchanan GR, Bowman WP, Smith SJ. Recurrent cerebral ischemia during hypertransfusion therapy in sickle cell anemia. J Pediatr. 1983;103:921-923.7. Awogu AU. Leucocyte counts in children with sickle cell anaemia usefulness of stable state values during infections. West Afr J Med. 2000;19:55-58.8. Castro O, Brambilla DJ, Thorington B, Reindorf CA, Scott RB, Gillette P, Vera JC, Levy PS. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84:643-649.9. Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89:1787-1792.10. Hedo CC, Aken'ova YA, Okpala IE, Durojaiye AO, Salimonu LS. Acute phase reactants and severity of homozygous sickle cell disease. J Intern Med. 1993;233:467-470.11. Singhal A, Doherty JF, Raynes JG, McAdam KP, Thomas PW, Serjeant BE, Serjeant GR. Is there an acute-phase response in steady-state sickle cell disease? Lancet. 1993;341:651-653.12. Stuart J, Stone PC, Akinola NO, Gallimore JR, Pepys MB. Monitoring the acute phase response to vaso-occlusive crisis in sickle cell disease. J Clin Pathol. 1994;47:166-169.13. Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451-2459.14. Francis RB, Jr., Haywood LJ. Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. J Natl Med Assoc. 1992;84:611-615.15. Malave I, Perdomo Y, Escalona E, Rodriguez E, Anchustegui M, Malave H, Arends T. Levels of tumor necrosis factor alpha/cachectin (TNF alpha) in sera from patients with sickle cell disease. Acta Haematol. 1993;90:172-176.16. Croizat H. Circulating cytokines in sickle cell patients during steady state. Br J Haematol. 1994;87:592-597.17. Kuvibidila S, Gardner R, Ode D, Yu L, Lane G, Warrier RP. Tumor necrosis factor alpha in children with sickle cell disease in stable condition. J Natl Med Assoc. 1997;89:609-615.18. Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol. 2002;24:81-88.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

19. Hofstra TC, Kalra VK, Meiselman HJ, Coates TD. Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood. 1996;87:4440-4447.20. Fadlon E, Vordermeier S, Pearson TC, Mire-Sluis AR, Dumonde DC, Phillips J, Fishlock K, Brown KA. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998;91:266-274.21. Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol. 1999;66:411-415.22. Kenny MW, George AJ, Stuart J. Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism. J Clin Pathol. 1980;33:622-625.23. Westwick J, Watson-Williams EJ, Krishnamurthi S, Marks G, Ellis V, Scully MF, White JM, Kakkar VV. Platelet activation during steady state sickle cell disease. J Med. 1983;14:17-36.24. Beurling-Harbury C, Schade SG. Platelet activation during pain crisis in sickle cell anemia patients. Am J Hematol. 1989;31:237-241.25. Papadimitriou CA, Travlou A, Kalos A, Douratsos D, Lali P. Study of platelet function in patients with sickle cell anemia during steady state and vaso-occlusive crisis. Acta Haematol. 1993;89:180-183.26. Wun T, Paglieroni T, Tablin F, Welborn J, Nelson K, Cheung A. Platelet activation and platelet-erythrocyte aggregates in patients with sickle cell anemia. J Lab Clin Med. 1997;129:507-516.27. Wun T, Paglieroni T, Field CL, Welborn J, Cheung A, Walker NJ, Tablin F. Platelet-erythrocyte adhesion in sickle cell disease. J Investig Med. 1999;47:121-127.28. Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, Klein NJ. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. 2000;111:474-481.29. Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. 1997;337:1584-1590.30. Solovey A, Gui L, Key NS, Hebbel RP. Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest. 1998;101:1899-1904.31. Duits AJ, Pieters RC, Saleh AW, van Rosmalen E, Katerberg H, Berend K, Rojer RA. Enhanced levels of soluble VCAM-1 in sickle cell patients and their specific increment during vasoocclusive crisis. Clin Immunol Immunopathol. 1996;81:96-98.32. Saleh AW, Duits AJ, Gerbers A, de Vries C, Hillen HF. Cytokines and soluble adhesion molecules in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 1998;100:26-31.33. Saleh AW, Hillen HF, Duits AJ. Levels of endothelial, neutrophil and platelet-specificfactors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 1999;102:31-37.34. Stuart MJ, Setty BN. Sickle cell acute chest syndrome: pathogenesis and rationale for treatment. Blood. 1999;94:1555-1560.35. Fabry ME, Costantini F, Pachnis A, Suzuka SM, Bank N, Aynedjian HS, Factor SM, Nagel RL. High expression of human beta S- and alpha-globins in transgenic mice: erythrocyte abnormalities, organ damage, and the effect of hypoxia. Proc Natl Acad Sci U S A. 1992;89:12155-12159.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

36. Rubin EM, Witkowska HE, Spangler E, Curtin P, Lubin BH, Mohandas N, Clift SM. Hypoxia-induced in vivo sickling of transgenic mouse red cells. J Clin Invest. 1991;87:639-647.37. Fabry ME, Sengupta A, Suzuka SM, Costantini F, Rubin EM, Hofrichter J, Christoph G, Manci E, Culberson D, Factor SM, et al. A second generation transgenic mouse model expressing both hemoglobin S (HbS) and HbS-Antilles results in increased phenotypic severity. Blood. 1995;86:2419-2428.38. Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876-878.39. Kaul DK, Fabry ME, Costantini F, Rubin EM, Nagel RL. In vivo demonstration of red cell-endothelial interaction, sickling and altered microvascular response to oxygen in the sickle transgenic mouse. J Clin Invest. 1995;96:2845-2853.40. Embury SH, Mohandas N, Paszty C, Cooper P, Cheung AT. In vivo blood flow abnormalities in the transgenic knockout sickle cell mouse. J Clin Invest. 1999;103:915-920.41. Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS, Hebbel RP. Reperfusion injury pathophysiology in sickle transgenic mice. Blood. 2000;96:314-320.42. Skow LC, Burkhart BA, Johnson FM, Popp RA, Popp DM, Goldberg SZ, Anderson WF, Barnett LB, Lewis SE. A mouse model for beta-thalassemia. Cell. 1983;34:1043-1052.43. Fabry ME, Nagel RL, Pachnis A, Suzuka SM, Costantini F. High expression of human beta S- and alpha-globins in transgenic mice: hemoglobin composition and hematological consequences. Proc Natl Acad Sci U S A. 1992;89:12150-12154.44. Burlingame RW, Volzer MA, Harris J, Du Clos TW. The effect of acute phase proteins on clearance of chromatin from the circulation of normal mice. J Immunol. 1996;156:4783-4788.45. Deryckere F, Gannon F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. BioTechniques. 1994;16:405.46. Brown RE, Jarvis KL, Hyland KJ. Protein measurement using bicinchoninic acid: elimination of interfering substances. Anal Biochem. 1989;180:136-139.47. Pepys MB, Dash AC. Isolation of amyloid P component (protein AP) from normal serum as a calcium-dependent binding protein. Lancet. 1977;1:1029-1031.48. Osmand AP, Friedenson B, Gewurz H, Painter RH, Hofmann T, Shelton E. Characterization of C-reactive protein and the complement subcomponent C1t as homologous proteins displaying cyclic pentameric symmetry (pentraxins). Proc Natl Acad Sci U S A. 1977;74:739-743.49. Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405-455.50. Le PT, Mortensen RF. In vitro induction of hepatocyte synthesis of the acute phase reactant mouse serum amyloid P-component by macrophages and IL 1. J Leukoc Biol. 1984;35:587-603.51. Hebbel RP, Moldow CF, Steinberg MH. Modulation of erythrocyte-endothelial interactions and the vasocclusive severity of sickling disorders. Blood. 1981;58:947-952.52. Hebbel RP, Schwartz RS, Mohandas N. The adhesive sickle erythrocyte: cause and consequence of abnormal interactions with endothelium, monocytes/macrophages and model membranes. Clin Haematol. 1985;14:141-161.53. Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc Natl Acad Sci U S A. 1989;86:3356-3360.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

54. Francis RB, Jr., Johnson CS. Vascular occlusion in sickle cell disease: current concepts and unanswered questions. Blood. 1991;77:1405-1414.55. Hebbel RP, Mohandas N. Sickle cell adherence. In: Embury S, Hebbel RP, Mohandas N, Steinberg MH, eds. Sickle cell disease: basic principles and clinical practice. New York: Raven Press; 1994:217-230.56. Hebbel RP, Vercellotti GM. The endothelial biology of sickle cell disease. J Lab Clin Med. 1997;129:288-293.57. Bernini JC, Rogers ZR, Sandler ES, Reisch JS, Quinn CT, Buchanan GR. Beneficial effect of intravenous dexamethasone in children with mild to moderately severe acute chest syndrome complicating sickle cell disease. Blood. 1998;92:3082-3089.58. Mahoney DH, Jr., Fernbach DJ. Monocyte functions in sickle cell disorders. Am J Pediatr Hematol Oncol. 1983;5:409-411.59. Boghossian SH, Nash G, Dormandy J, Bevan DH. Abnormal neutrophil adhesion in sickle cell anaemia and crisis: relationship to blood rheology. Br J Haematol. 1991;78:437-441.60. Mohamed AO, Hashim MS, Nilsson UR, Venge P. Increased in vivo activation of neutrophils and complement in sickle cell disease. Am J Trop Med Hyg. 1993;49:799-803.61. Mollapour E, Porter JB, Kaczmarski R, Linch DC, Roberts PJ. Raised neutrophil phospholipase A2 activity and defective priming of NADPH oxidase and phospholipase A2 in sickle cell disease. Blood. 1998;91:3423-3429.62. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639-1644.63. Abboud M, Laver J, Blau CA. Granulocytosis causing sickle-cell crisis. Lancet. 1998;351:959.64. Adler BK, Salzman DE, Carabasi MH, Vaughan WP, Reddy VV, Prchal JT. Fatal sickle cell crisis after granulocyte colony-stimulating factor administration. Blood. 2001;97:3313-3314.65. Grigg AP. Granulocyte colony-stimulating factor-induced sickle cell crisis and multiorgan dysfunction in a patient with compound heterozygous sickle cell/beta+ thalassemia. Blood. 2001;97:3998-3999.66. Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, Milner PF, Orringer EP, Phillips G, Jr., Platt OS, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. 1992;79:2555-2565.67. Platt OS. Sickle cell paths converge on hydroxyurea. Nat Med. 1995;1:307-308.68. Charache S, Barton FB, Moore RD, Terrin ML, Steinberg MH, Dover GJ, Ballas SK, McMahon RP, Castro O, Orringer EP. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive "switching" agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore). 1996;75:300-326.69. Howard LW, Kennedy LD. Hydroxyurea in the treatment of sickle-cell anemia. Ann Pharmacother. 1997;31:1393-1396.70. Hebbel RP, Eaton JW, Balasingam M, Steinberg MH. Spontaneous oxygen radical generation by sickle erythrocytes. J Clin Invest. 1982;70:1253-1259.71. Shi X, Dong Z, Huang C, Ma W, Liu K, Ye J, Chen F, Leonard SS, Ding M, Castranova V, Vallyathan V. The role of hydroxyl radical as a messenger in the activation of nuclear transcription factor NF-kappaB. Mol Cell Biochem. 1999;194:63-70.72. McFaul SJ, Bowman PD, Villa VM. Hemoglobin stimulates the release of proinflammatory cytokines from leukocytes in whole blood. J Lab Clin Med. 2000;135:263-269.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom

73. Hammerman SI, Kourembanas S, Conca TJ, Tucci M, Brauer M, Farber HW. Endothelin-1 production during the acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med. 1997;156:280-285.74. Werdehoff SG, Moore RB, Hoff CJ, Fillingim E, Hackman AM. Elevated plasma endothelin-1 levels in sickle cell anemia: relationships to oxygen saturation and left ventricular hypertrophy. Am J Hematol. 1998;58:195-199.75. Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M, McMillen MA. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood. 1998;92:2551-2555.76. Faller DV. Endothelial cell responses to hypoxic stress. Clin Exp Pharmacol Physiol. 1999;26:74-84.77. Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. 1995;96:1145-1151.78. Zimmerman BJ, Granger DN. Mechanisms of reperfusion injury. Am J Med Sci. 1994;307:284-292.
For personal use only. by guest on June 5, 2013. bloodjournal.hematologylibrary.orgFrom
Related Documents











