1 FACULTAD DE FARMACIA UNIVERSIDAD COMPLUTENSE TRABAJO FIN DE GRADO TÍTULO: MODIFICACIÓN EN LA LIBERACIÓN RETARDADA DE FÁRMACOS DIRIGIDA A ENFERMEDADES CRÓNICAS DEL COLON Autor: Cristian Daniel Toro Chiluisa Tutor: Dra. Mª Elvira Franco Gil Convocatoria: Julio 2017

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
FACULTAD DE FARMACIA
UNIVERSIDAD COMPLUTENSE
TRABAJO FIN DE GRADO
TÍTULO: MODIFICACIÓN EN LA
LIBERACIÓN RETARDADA DE FÁRMACOS
DIRIGIDA A ENFERMEDADES CRÓNICAS
DEL COLON
Autor: Cristian Daniel Toro Chiluisa
Tutor: Dra. Mª Elvira Franco Gil
Convocatoria: Julio 2017

2
ÍNDICE
RESUMEN.…………………………………………………………………………….3
INTRODUCCIÓN Y ANTECEDENTES…………………………………………….3
OBJETIVOS……………………………………………………………………………6
MATERIAL Y MÉTODOS……………………………………………………………6
RESULTADOS Y DISCUSIÓN……………………………………………………….6
1. SISTEMAS DE LIBERACIÓN PH-DEPENDIENTE…………………………..7
2. SISTEMAS CON LIBERACIÓN DEPENDIENTES DEL TIEMPO DE
TRÁNSITO GASTROINTESTINAL………………………………………….10
3. SISTEMAS DE LIBERACIÓN RETARDADA POR DEGRADACIÓN
ENZIMÁTICA ………………………………………………………………...12
3.1) Profármacos………………………………………………………………12
3.2) Recubrimiento con polímeros biodegradables…………………………...14
3.3) Sistemas matriciales ……………………………………………………..15
3.3.1) Cápsulas E-CDS……………………………………………………15
3.3.2) Comprimidos de lactulosa, sistema CODES……………………….16
3.3.3) Microesferas de quitosano reticulado……………………………...17
3.4) Nuevos sistemas de liberación retardada
3.4.1) Philips’Intelligent pill………………………………………………18
3.4.2) InteliSite® capsule…………………………………………………18
CONCLUSIONES…………………………………………………………………….19
BIBLIOGRAFÍA……………………………………………………………………...20

3
RESUMEN
En este trabajo nos vamos a centrar en los sistemas de liberación retardado a nivel
colónico para las enfermedades tales como, enfermedad de Crohn y colitis ulcerosa. En
la actualidad, las formas farmacéuticas de liberación modificada son de las más
utilizadas en la administración de fármacos, debido a sus múltiples ventajas, como
podemos mencionar: proteger al principio activo de agentes degradantes,
enmascaramiento de características organolépticas desagradables, reducir el efecto
irritante, reducir el número de tomas, etc. De esta manera se mejora la calidad de vida
del paciente, se mejora el cumplimiento del tratamiento y lo optimiza llegando a ser más
eficaz. Además estudiaremos los tres tipos de liberaciones colónicos establecidos
actualmente y sus ejemplos más relevantes.
INTRODUCCIÓN Y ANTECEDENTES
La Real Farmacopea Española describe las formas farmacéuticas de liberación
modificada como aquellas preparaciones en las que la velocidad y el lugar de liberación
de la sustancia o sustancias activas, es diferente del de la forma farmacéutica de
liberación convencional administrada por la misma vía [1]. En este tipo de sistemas
modificados, el agente bioactivo es incorporado a un soporte que generalmente es un
material polimérico o una combinación de varios. La velocidad de liberación de la
sustancia activa desde dicho sistema al medio que la rodea, viene determinada por las
propiedades del propio polímero y, en menor medida, depende de los factores
ambientales, como pueden ser el pH, la temperatura y los fluidos del organismo [3].
Así, las formas farmacéuticas de liberación modificada presentan numerosas ventajas
con respecto a las formas farmacéuticas convencionales, entre las que destacan [5]:
1. Aumentar duración de los efectos de fármacos con una sola dosis, que tenga que
dar un comprimido al día, en vez de tres.
2. Mejorar la relación eficacia-dosis.
3. Aumentar la liberación de fármaco en biofase, reduciendo la concentración de
fármaco en exofase y por tanto reducir el riesgo de efectos secundarios.
4. Aumentar la eficacia de vías de administración
5. Proteger al fármaco del organismo o al organismo del fármaco.

4
El principio activo presente en la forma farmacéutica debe cumplir una serie de
características para ser incluido en la liberación controlada:
1. Estrecho margen terapéutico.
2. Efectos adversos relacionados con la concentración plasmática.
3. Duración de acción corta, requiere tres o más administraciones al día.
4. Riesgo de bajo grado de cumplimiento (tratamientos crónicos).
Por otro lado, los excipientes que se usan para modular esta liberación, van a tener los
siguientes requisitos generales [3]:
1. Biocompatibilidad con la vía de administración.
2. Compatible con el PA
3. Capaz de modular la liberación del PA
4. Estable y preferiblemente biodegradable
5. Características fisicoquímicas específicas: bioadhesión, elasticidad y resistencia.
De esta manera se podrán elaborar sistemas de liberación modificada, siendo el objetivo
principal, conducir a la existencia de una concentración uniforme de fármaco, a la
utilización de dosis más pequeñas y a lograr la ausencia de efectos secundarios , debido
a que con los sistemas convencionales se consigue una liberación inmediata, más
efectos secundarios y de la necesidad de incrementar la dosis o la frecuencia de
administración para asegurar el efecto terapéutico en el paciente crónico; lo cual
produce intoxicaciones o disminuye el cumplimiento [6,7].
La industria farmacéutica ha elaborado diferentes tipos de sistemas modificados para su
administración [1,2, 5]:
1.-Sistemas de liberación sostenida: Liberan inicialmente la cantidad necesaria de
fármaco para tener la respuesta farmacológica deseada de forma rápida y,
posteriormente, en una cantidad adecuada y constante para que la velocidad de
absorción del fármaco sea igual a la velocidad de eliminación durante un tiempo
prolongado, normalmente de 10 a 24 horas. Por lo tanto, estas formas farmacéuticas
presentan una cinética de liberación del principio activo de orden cero, con lo que se
consigue que el nivel plasmático del fármaco se mantenga constante. Un ejemplo de
estos sistemas son los comprimidos osmóticos.
2.-Sistemas de liberación diferida o retardada: diseñadas para salvar el pH gástrico o
para evitar gastrolesividad del fármaco. No prolongan el efecto terapéutico. Ejemplos:
Salazopyrina®(sulfasalazina) , Salofalk®(mesalazina), y Entocord®(budesonida).

5
3.-Sistemas de liberación prolongada: fármaco se libera inicialmente en la cantidad
suficiente para producir la acción terapéutica, para después continuar liberándolo de
forma lenta pero a una velocidad que no siempre es igual a la velocidad de eliminación.
Es decir, estas formas farmacéuticas presentan una liberación lenta pero no constante,
observándose un nivel plasmático que varía dentro de la zona terapéutica, describiendo
una curva amplia. Ejemplos: Adalat Oros® y MST Continus® (morfina).
4.-Sistemas flotantes y bioadhesivos: Diseñados para aumentar el período de
residencia gástrico .Ejemplos: Madopar® Retard (levodopa + benserazida).
Con la liberación retardada se conseguirá retrasar el tiempo de liberación, alargando el
tiempo de latencia hasta que el sistema esté en el colon y poder liberar el fármaco en el
órgano diana, por lo tanto, no se obtienen niveles plasmáticos del fármaco hasta que la
forma farmacéutica se encuentre en la zona del tracto digestivo en donde se desea que
se active el sistema. Los sistemas colónicos pueden tener dos objetivos principales, la
absorción colónica para crear un efecto sistémico y la acción local donde la absorción
colónica es escasa. La acción local es uno de los objetivos principales de la enfermedad
de Crohn y la colitis ulcerosa, creando un efecto local antiinflamatorio para reducir los
efectos ocasionados por las enfermedades mencionadas.
La enfermedad de Crohn , es una enfermedad intestinal inflamatoria de origen
autoinmune, genético y ambiental caracterizado por la aparición de brotes inflamatorios
seguidos de periodos de remisión .Generalmente se manifiesta en el íleon y en el
intestino grueso con síntoma como diarrea , dolor abdominal, pérdida de peso y fiebre.
Este tipo de enfermedad puede presentarse en cualquier tramo del tracto gastrointestinal,
desde la boca hasta el recto. El tratamiento sueles ser corticoides, inmunoterapia y
terapia biológica [4].
La colitis ulcerosa es una enfermedad inflamatoria intestinal de origen autoinmune,
ambiental y genético que afecta solamente al intestino grueso y al recto. Se le asocia con
síntomas como diarrea con sangre, dolor abdominal, retortijones y fiebre. Se sabe que la
enfermedad suele comenzar en el recto o en el colon sigmoideo, extendiéndose de forma
parcial o total por el intestino grueso. El tratamiento aplicado suele ser corticoides,
inmunoterapia y terapia biológica [4].
OBJETIVOS
En el trabajo presentado buscaremos dar una idea sobre las formas farmacéuticas más
empleadas en el mercado desde el punto de vista de liberación retardado a nivel

6
colónico. Por tanto, se tendrán en cuenta los siguientes objetivos:
1. Tipos de sistemas de liberación retardada colónica.
2. Características de las formas farmacéuticas.
3. Los recursos tecnológicos que se emplean para evitar la pérdida de p.a. y que
ejerza su acción local.
4. Nuevos sistemas de liberación retardada dirigida al colon.
MATERIAL Y MÉTODOS
En este trabajo se ha realizado una revisión bibliográfica utilizando una serie de bases
de datos en internet: Bucea, de acceso vía correo institucional de la UCM, artículos de
google y PubMed. La búsqueda de artículos se ha llevado a cabo por lenguaje libre,
analizando además las referencias bibliográficas de artículos ya seleccionados con el fin
de rescatar otros estudios relevantes para su inclusión. Se han aceptado los artículos
considerados más destacados según criterios científicos sin importar el año de
publicación. Finalmente se obtuvieron 22 artículos de referencia descartando los
menos relevantes y descargando en formato PDF los más relevantes. Aparte se
elaboraron tablas y figuras que se incluyeron a lo largo del trabajo.
RESULTADOS y DISCUSIÓN
Todas las formas farmacéuticas se administran por vía oral siendo cápsulas,
comprimidos o micropartículas (pellets) dirigidas al colon para crear un efecto local.
Algunas de las formas farmacéuticas empleadas en los diferentes mecanismos de
acción, suelen ir recubiertas de polímeros gastrorresistentes para [8]:
1. Enmascarar un olor o sabor desagradables (mejora de la cooperación del
paciente en el cumplimiento de la pauta terapéutica).
2. Mejorar la estabilidad del fármaco (protección contra la luz, humedad, etc.).
3. Proteger al organismo de la acción irritante del fármaco (antiinflamatorios que
deben liberarse en el intestino grueso, ya que provocan irritaciones en el
estómago).
4. Permitir regular la liberación del principio activo y, con ello, una liberación
localizada en el colon para ejercer una acción local en la colitis ulcerosa y
enfermedad de Crohn.
Se elaboraron tres mecanismos de liberación retardada dirigida al colon para crear un
efecto local y mejorar los síntomas ocasionados por las enfermedades estudiadas.
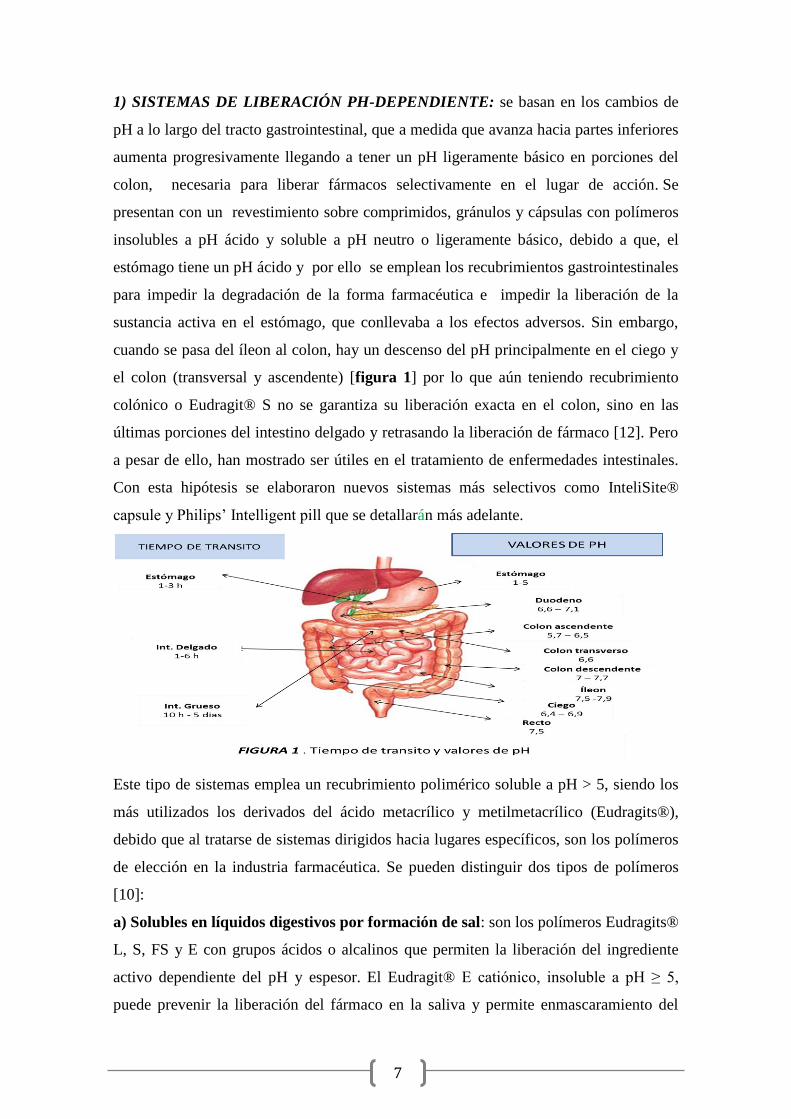
7
1) SISTEMAS DE LIBERACIÓN PH-DEPENDIENTE: se basan en los cambios de
pH a lo largo del tracto gastrointestinal, que a medida que avanza hacia partes inferiores
aumenta progresivamente llegando a tener un pH ligeramente básico en porciones del
colon, necesaria para liberar fármacos selectivamente en el lugar de acción. Se
presentan con un revestimiento sobre comprimidos, gránulos y cápsulas con polímeros
insolubles a pH ácido y soluble a pH neutro o ligeramente básico, debido a que, el
estómago tiene un pH ácido y por ello se emplean los recubrimientos gastrointestinales
para impedir la degradación de la forma farmacéutica e impedir la liberación de la
sustancia activa en el estómago, que conllevaba a los efectos adversos. Sin embargo,
cuando se pasa del íleon al colon, hay un descenso del pH principalmente en el ciego y
el colon (transversal y ascendente) [figura 1] por lo que aún teniendo recubrimiento
colónico o Eudragit® S no se garantiza su liberación exacta en el colon, sino en las
últimas porciones del intestino delgado y retrasando la liberación de fármaco [12]. Pero
a pesar de ello, han mostrado ser útiles en el tratamiento de enfermedades intestinales.
Con esta hipótesis se elaboraron nuevos sistemas más selectivos como InteliSite®
capsule y Philips’ Intelligent pill que se detallarán más adelante.
Este tipo de sistemas emplea un recubrimiento polimérico soluble a pH > 5, siendo los
más utilizados los derivados del ácido metacrílico y metilmetacrílico (Eudragits®),
debido que al tratarse de sistemas dirigidos hacia lugares específicos, son los polímeros
de elección en la industria farmacéutica. Se pueden distinguir dos tipos de polímeros
[10]:
a) Solubles en líquidos digestivos por formación de sal: son los polímeros Eudragits®
L, S, FS y E con grupos ácidos o alcalinos que permiten la liberación del ingrediente
activo dependiente del pH y espesor. El Eudragit® E catiónico, insoluble a pH ≥ 5,
puede prevenir la liberación del fármaco en la saliva y permite enmascaramiento del

8
gusto. Las formas aniónicas de Eudragits® (FS, L y S) se emplean como materiales
gastrorresistentes y favorecer la liberación de la sustancia activa en porciones inferiores,
por ello son polímeros de elección para proteger al fármaco del pH acido del estómago.
Estos polímeros son insolubles a valores de pH inferiores a 6 (Eudragits® L) y 7
(Eudragits® FS, S), pero se disuelven rápidamente tras la desprotonización de los
grupos carboxílicos a valores superiores de pH, que se alcanzaría a medida que el
sistema avance por el tubo digestivo, hacia el lugar específico de liberación del fármaco.
b) Insolubles pero permeable en fluidos digestivos: Los polímeros Eudragits® RL y
RS con polímeros catiónicos, permiten una liberación tiempo dependiente del
ingrediente activo por hinchamiento y son independientes del pH. [11].
PRODUCTO DISPONIBILIDAD pH
EUDRAGIT ® L 100-55 Polvo Disolución pH > 5,5
EUDRAGIT ® L 30 D-55 Dispersión acuosa Disolución pH > 5,5
EUDRAGIT ® L 100 Polvo Disolución pH > 6
EUDRAGIT ® L 12,5 Solución orgánica Disolución pH > 6
EUDRAGIT ® S 100 Polvo Disolución pH > 7
EUDRAGIT ® S 12,5 Solución orgánica Disolución pH > 7
EUDRAGIT® FS 100 Polvo Disolución pH > 7
EUDRAGIT ® FS 30 D Dispersión acuosa Disolución pH > 7
EUDAGRIT ® E Polvo Disolución pH 1 -5
TABLA 1. Polímeros utilizados en los sistemas de liberación colónica dependientes de pH.
Entre los sistemas de liberación pH-dependiente más destacados se encuentra el Oros-ct
®, que consta de una capa gelatinosa convencional en cuyo interior hay varias unidades
de Oros push-pull ®. Cada una de las unidades lleva un recubrimiento colónico que
ayuda a conservar su integridad hasta el colon, bloqueando el paso de agua hacia el
interior del sistema. Al ingerir el sistema, la cápsula se disuelve, liberando las unidades
de Oros push-pull ® que pasaran al duodeno sin perder su integridad. Continua por el
tracto digestivo llegando al colon donde empezará a disolverse el recubrimiento
colónico al encontrarse con un pH > 6, dejando los sistemas Oros push-pull ® listos
para ejercer su acción a nivel colónico. De manera que entra el agua en el
compartimento osmótico, se disuelve el excipiente osmótico produciendo un aumento
de presión osmótica y al tener una membrana flexible entre los dos compartimentos
(osmótico y reservorio), empuja a la sustancia activa liberándolo por el orificio del
sistema [9,15]. Las unidades Oros-ct ® pueden mantener una liberación constante
durante 24 horas en el colon o pueden liberar el fármaco en un periodo corto de
aproximadamente cuatro horas.

9
FIGURA 2. Sistema oros-ct , liberación colónica
Este tipo de sistemas presenta un inconveniente debido a la presencia de ácidos grasos y
sales biliares , en porciones inferiores del tubo digestivo , que pueden acidificar el
medio en el que se encuentre el sistema Oros-ct® impidiendo la liberación del fármaco
al lugar de acción , lo que conlleva a tener tiempos de latencia más prolongados y una
mala especificidad del sitio de acción. Pero a pesar de esto, los sistemas dependientes
de pH utilizados actualmente, han mostrado beneficios en el tratamiento de la
enfermedad inflamatoria del intestino, a pesar de su baja especificidad.
Otros estudios llevados a cabo por Leopold Eikeler (2006), descubrió que el pH
luminal durante los ataques agudos de la enfermedad inflamatoria intestinal cae a
valores de orden de 2,3 a 4,7 lo que constituyó el punto de partida para la preparación
de comprimidos de dexametasona recubiertos con un polímero soluble en ácido. El
sistema consta de tres capas: una capa externa entérico con HPMC, una capa intermedia
con HPC y una capa interior de Eudragit ® E soluble a pH ácido (Figura 3 ). La
presencia de una capa intermedia inhibe los fenómenos de interacción entre las otras dos
capas, debido al hecho de que HPC es un polímero inerte. Al ingerir el sistema,
mantiene su integridad hasta el duodeno donde empieza a disolverse. Tras la disolución
del recubrimiento entérico, la capa interna de Eudragit ® E promueve la liberación de
dexametasona en sitios de inflamación donde el pH es ácido, y en la que estos
polímeros se disuelven [15].
FIGURA 3. Sistemas de liberación pH dependiente por Leopold Eikeler

10
2) SISTEMAS CON LIBERACIÓN DEPENDIENTE DE TIEMPO TRÁNSITO
GASTROINTESTINAL: se basa en el tiempo que tarda el sistema en llegar al colon
impulsado por el peristaltismo intestinal. En este caso se recurre a un polímero entérico
que se disuelve una vez que la formulación alcanza el intestino delgado, lugar en el que
comienza un periodo de latencia que se hace coincidir con el tiempo de permanencia o
tránsito en el intestino delgado (3 ± 1 h) [9,15]. En el tiempo de tránsito, el polímero
gelificable incluido en la forma farmacéutica captará agua pasando de un estado sólido
a semisólido. Este es el caso de Pentasa ®, que consta de gránulos comprimidos
resultantes de la compresión del ácido 5-aminosalicílico recubierto con etilcelulosa. Una
vez en contacto con los fluidos del tracto gastrointestinal, el comprimido se desintegra y
libera los microgránulos recubiertos. El polímero gelificable captará agua pasando de un
estado sólido a semisólido, por tanto, la liberación de ácido 5-aminosalicílico se hace
por un proceso de erosión y difusión a través de la capa de gel. Sin embargo, esta
estrategia tiene poca especificidad ya que la liberación de ácido 5-aminosalicílico se
inicia en el estómago o el intestino delgado que es normalmente detectable 30 a 60
minutos después de la ingestión de Pentasa ®, no liberando el principio activo en el
colon y perdiendo especificidad [15]. Posteriormente se propuso otro sistema
dependiente del tiempo, que es uno de los más empleados actualmente y es el sistema
Pulsincap®. Se basa en el mismo principio de la formulación descrita anteriormente y
se compone de: 1) un cuerpo insoluble a todo el pH, que se elimina con las heces
totalmente inalterado 2) una tapa recubierta de un polímero gastrorresistente, soluble a
pH >5 3) tapón de hidrogel (polímero gelificable) localizado en la boca del cuerpo
(Figura 4). Al ingerir el sistema llega al estómago y al contener la tapa un polímero
gastrorresistente mantiene su integridad en el estómago. Continua por el tubo digestivo,
llegando al duodeno que se encuentra con un pH >6, empezando a disolverse el
recubrimiento de la tapa. El tapón de hidrogel, empieza a captar agua del medio
fisiológico pasando de estado sólido a semisólido. Este tiempo de captación de agua,
coincide con el tiempo de tránsito del sistema por el intestino delgado, generando un
tiempo de latencia para poder liberar el fármaco en el colon. Tras pasar el tapón a estado
semisólido, sale del cuerpo fácilmente y se produce la liberación del fármaco en el
colon. Esta liberación está controlada por el grado de hinchamiento del hidrogel que a
su vez es independiente del pH, y debe proceder durante 3 a 4 horas, es decir, el tiempo
requerido para que este sistema alcance el colon [2,7, 15].

11
FIGURA 4. Sistema Pulsincap® dirigido hacia el colon
Pero estos sistemas, al igual que los sistemas dependientes de pH, presentan una serie
de limitaciones, debido a las variaciones del tiempo de vaciado gástrico en seres
humanos, de manera que el tiempo de llegada al colon de las formas de dosificación no
se puede predecir con exactitud, resultando una baja disponibilidad a nivel colónico. Las
desventajas de este sistema son [15]:
El tiempo de vaciado gástrico varía entre los sujetos o de una manera
dependiente del tipo y la cantidad de ingesta de alimentos.
El movimiento gastrointestinal, especialmente la ausencia de peristaltismo o la
contracción en el estómago daría lugar a cambio en el tránsito gastrointestinal
del medicamento, aumentando el tiempo de latencia.
Se ha observado tránsito acelerado a través de diferentes regiones del colon en
pacientes con EII, síndrome carcinoide, diarrea, y colitis ulcerosa.
Pero a pesar de las limitaciones y su baja especificidad, los sistemas dependientes de
tiempo y pH utilizados han demostrado beneficio en las enfermedades estudiadas.
3) SISTEMAS DE LIBERACIÓN RETARDADA POR DEGRADACIÓN
ENZIMÁTICA: Estos sistemas usan el metabolismo bacteriano para promover la
liberación del fármaco específico en el colon mediante el uso de profármacos,
formulaciones recubiertas de polímeros biodegradables o sistemas matriciales
(hidrogeles). Es una estrategia muy específica debido a que estas bacterias están
presentes solamente en el colon, en los que intervienen fundamentalmente
polisacaridasas, azorreductasas y glucosidadas.
3.1) Profármacos: El uso de profármacos dentro de los sistemas basados en la
microflora del colon, implica la formación de moléculas farmacológicamente inactivas
con diferentes características fisicoquímicas de la molécula activa. Estos sistemas
deberán cumplir los siguientes requisitos: A) no se absorban en el intestino delgado y,
una vez que alcanzan el colon, se convierten en el compuesto farmacológicamente

12
activo a través de la acción metabólica de la microflora bacteriana colónica , y poder
ejercer su efecto farmacológico en el colon para las enfermedades estudiadas. B) Deben
ser hidrófilos o de alto peso molecular, para limitar su absorción en las partes
superiores del tracto gastrointestinal y ejercer su acción a nivel colónico.
Pero como todo tipo de sistemas estudiados presentan una serie de inconvenientes: 1)
su aplicación se limita a ciertos fármacos, por tanto, no son sistemas universales. 2) no
ofrecen protección contra el medio ácido de las regiones superiores del tracto
gastrointestinal, no siendo tan eficaces como otros sistemas 3) no permite la
administración de grandes cantidades de fármaco [15].
PRINCIPIO ACTIVO LIGANDO DEL PROFÁRMACO
Acido 5-aminosalicílico Sulfapiridina
Acido 5-aminosalicílico Polímeros bioadhesivos (HPMA)
Dexametasona Glucósidos
Budesonida Glucurónido
Naproxeno Dextrano
Dexametasona Dextrano
Metilprednisolona Dextrano
Acido 5-aminosalicílico Ciclodextrina
Acido 5-aminosalicílico Polímeros acrílicos
TABLA 2. Composición de prófarmacos utilizados en la liberación colónica
Estos sistemas, para liberar el principio activo emplean dos tipos de reacciones:
a) Azo-reducción: reacción catalizada por azorreductasas presentes en la microflora
bacteriana del colon, para liberar la forma activa. El ejemplo clásico de un profármaco
que sufre azo-reducción es la sulfasalazina. Se forma mediante la combinación de
sulfapiridina, salicilato y ácido 5-aminosalicílico unido a través de un enlace azoico. El
ácido 5-aminosalicílico es muy eficaz en el tratamiento de la enfermedad inflamatoria
intestinal, que si se administra solo por vía oral , se somete a una absorción intensa en
las partes superiores del tracto gastrointestinal, no ejerciendo acción local en el colon .
Por el contrario, cuando se administra en forma de sulfasalazina, al menos el 85% de
una dosis oral pasa intacto a través del estómago y el intestino delgado, llegando dosis
al colon. Pero tiene un inconveniente ya que tras el metabolismo llevado a cabo por las
azorreductasas, libera la sulfapirida que produce efectos secundarios por una absorción
sistémica, tales como, diarrea y pancreatitis. Por ello, surgió la necesidad de desarrollar
nuevos prófarmacos menos tóxicos tales como balsalazina , olsalazina e ipsalazina. Otro

13
mecanismo propuesto, fue el desarrollo de nuevos prófarmacos resultantes de la unión
mediante enlaces azoicos, de ácido 5-aminosalicílico con polímeros bioadhesivos
(HPMA). Estos polímeros interaccionan con receptores de las células epiteliales,
facilitando la intervención de azorreductasas y prolongar la liberación del fármaco en el
colon impidiendo su eliminación con las heces. Pero se requieren dosis muy altas de
fármaco para alcanzar dosis terapéuticas en el colon.
b) Hidrólisis: este tipo de reacción transforma diferentes profármacos gracias a
enzimas específicas del colon. Podemos tener [15]: 1) Conjugación con glucósidos:
esta estrategia se ha utilizado para la liberación de los corticosteroides en el colon y para
el tratamiento local de la enfermedad inflamatoria intestinal. El profármaco es
dexametasona -D-glucósido que resiste al paso precolónico y por su gran peso
molecular no puede ser absorbido. Una vez llega al colon, la β-glucuronidasa
microbiana hidroliza el compuesto liberando budesonida. 2) Conjugación con
glucurónidos: se han observado que estos conjugados son más hidrófilos que los
glucósidos con lo que su absorción en el intestino delgado es menor y las
glucuronidasas tiene una baja actividad enzimática en intestino delgado, por lo que
favorece su liberación en el colon. El profármaco es D-glucurónido budesonida.
3) Conjugado con dextranos: la especie Bacteroides secretan algunos dextranasas y
están presentes en la flora fecal de los pacientes que sufren de colitis ulcerosa y
enfermedad de Crohn. Esto permitió el desarrollo de profármaco, que se obtuvo
mediante la unión de naproxeno, metilprednisolona o dexametasona con dextrano,
observándose algunas ventajas, tales como, no degradación enzimática en el intestino
delgado, no absorción sistemática y activación rápida mediante la hidrólisis por las
bacterias del colon. 4) Conjugación con ciclodextrinas: las ciclodextrinas son
oligosacáridos cíclicos susceptibles a la fermentación por polisacaridasas presentes en el
colon. Estos compuestos se hidrolizan fácilmente en el colon y se absorben sólo
ligeramente durante su paso a través del estómago y el intestino delgado. Estas dos
características han llevado al desarrollo de profármacos de ácido 5-aminosalicílico
enlazadas a ciclodextrinas. 5) Conjugación con polímeros acrílicos: otra estrategia
utilizada para la administración de fármacos específica en el colon se basa en la unión
de polímeros acrílicos con ácido 5-aminosalicílico que dan lugar a los profármacos que
tienen enlaces amida o éster biodegradables. En ambientes con bajo pH, estos
polímeros tienen una baja capacidad de captación de agua, reduciendo la posibilidad de
hidrólisis. Sin embargo, cuando el pH aumenta (colon), el grado de hinchamiento se

14
hace más significativa, haciéndolos más susceptibles a la hidrólisis, liberando el
fármaco en el lugar de acción.
3.2) Recubrimiento con polímeros biodegradables: Otra estrategia para liberar
fármacos en el colon es recurrir al empleo de polímeros tanto naturales como
semisintéticos. Por ello se emplea recubrimientos de las formas farmacéuticas mediante
polímeros que serán degradados por enzimas liberadas de la microflora bacteriana del
colon y así liberar la forma activa que ejercerá su acción en el colon. Estos polímeros
empleados en el recubrimiento deberán cumplir una serie de características: 1) proteger
el contenido de fármaco del pH ácido y garantizar la liberación rápida de la forma activa
en el colon. 2) ser susceptible a la degradación por bacterias colónicas 3) ser soluble o
dispersable en disolventes apropiados para formulaciones de revestimiento 4) mostrar
capacidad de formar película 5) estar exento de toxicidad.
Los polímeros naturales son los más empleados por la industria farmacéutica ya que
cuentan con la ventaja de haber sido autorizados como excipientes natural y aparte son
abundantes, baratos y se pueden modificar químicamente, no son tóxicos y son
biodegradables. Pero algunos de ellos tienen propiedades plásticas deficientes y tienden
al hinchamiento favoreciendo su disolución. Para evitarlo se añaden polímeros
semisintéticos hidrófobos en una proporción adecuada que se determina
experimentalmente (TABLA 3).
POLÍMERO TIPO ORIGEN CARACTERÍSTICAS
amilosa natural vegetal Soluble en medio acuoso y degradable por
amilasas
pectina natural vegetal Alta solubilidad acuosa y degradable por
enzimas colónicas
quitosano natural animal Soluble en medio ácido, hinchable, degradable por quitinasas
goma xantana natural microbiano Hinchable y degradable
Condroitín-sulfato
natural animal Soluble en medio acuoso, hinchable, degradable por enzimas microbianas
HPMC Semisintético Vegetal Hinchable y soluble, retarda la liberación
etilcelusosa Semisintético vegetal Hidrófoba y retarda la liberación
TABLA 3. Polímeros naturales y semisintéticos empleados en recubrimientos de formas
farmacéuticas y en sistemas matriciales.
3.3) Sistemas matriciales: Los polisacáridos que se utilizan en la matriz de la
formulación son la pectina, goma guar, el quitosano y sulfato de condroitina. Estos
polisacáridos tienen una alta hidrofilicidad, lo que supone una limitación importante,

15
por lo que su uso está restringido a fármacos poco solubles. Para ello se recurren a la
reticulación de los polímeros para reducir su solubilidad y poder emplearlos en sistemas
dirigidos al colon. Al administrar este sistema por vía oral, el polímero empiezan a
captar agua que entra entre las cadenas, favoreciendo el hinchamiento del polímero .De
esta manera, se produce una relajación de las cadenas, entrando en contacto con
azorreductasas presentes en la flora colónica. Actúan sobre los grupos azoicos de la
cadena, rompiéndolos y favoreciendo la liberación del principio activo. Pero como todo
de sistemas presenta una serie de inconvenientes y son: 1) La administración conjunta
de antibióticos puede destruir las azorreductasas e impedir la liberación del fármaco en
el colon 2) El tipo de comida ingerida puede reducir las azorreductasas. Actualmente,
los sistemas más empleados son:
3.3.1) Cápsulas E-CDS: el sistema consta de: a) cápsula con capa de polímero entérico
b) micropartículas de quitosano alrededor de la cápsula. El quitosano se disuelve
fácilmente a pH ácido (Tabla 3), por ello, es protegido con el polímero entérico del pH
acido del estómago para impedir su degradación c) núcleo de fármaco más excipientes.
Al ingerir la cápsula, llega al estómago y mantiene su integridad por la cubierta entérica.
Cuando llega al intestino delgado, al haber un pH>5, el polímero entérico se ioniza y
empieza a disolverse. A medida que se disuelve, el quitosano puede entrar en contacto
con el agua y se va hinchando, gelificando y adquiriendo adherencia. Con estas
características, las micropartículas de quitosano se pegan unas con otras y forman una
capa de quitosano sobre la cápsula gelatinosa. Este recubrimiento se va formando a lo
largo del intestino delgado, lo que requiere un tiempo de tránsito. Pero cuando llegan al
colon, el recubrimiento de quitosano entra en contacto con la flora bacteriana donde las
azoredectasas rompen los enlaces reticulantes de quitosano y liberan el principio activo
en el colon. Este sistema es muy eficaz ya que aparte de emplear la degradación
enzimática, lo combina con la liberación pH-dependiente y el tiempo de tránsito, lo que
le hace un método más específico a nivel del colon [14].

16
FIGURA 5.Cápsulas E-CDS, degradación enzimática en el colon
3.3.2) Comprimidos de lactulosa, sistemas CODES: sistema formado por: a) capa de
polímero entérico o gastrorresistente b) capa de polímero permeable que permite la
entrada y salida del disolvente y se disuelve a pH acido (Eudragit® E) c) núcleo de
fármaco no hidrosoluble más lactulosa (se trata de un azúcar sintético diseñado para no
ser digerido por las enzimas digestivas pero sí para ser degradado por la flora bacteriana
del colon). Al ingerir el comprimido, se mantiene su integridad en el estómago por el
empleo del polímero entérico. A continuación, llega al intestino delgado donde se ioniza
y se disuelve el recubrimiento gastrorresistente. Al disolverse, entra agua del medio
fisiológico por la membrana permeable hasta que hay suficiente disolvente para disolver
lo que hay dentro de la forma farmacéutica. El fármaco al ser no hidrosoluble, no se
disuelve con el agua del medio fisiológico que ha entrado, pero la lactulosa sí. La
lactulosa requiere un tiempo de tránsito a lo largo del intestino delgado para disolverse
y llegar al colon .Cuando esta disuelta puede salir por la capa del polímero permeable y
entra en contacto con la microflora bacteriana del colon, degradándolo a productos
ácidos. De esta manera, se crea un ambiente ácido en torno al sistema de liberación,
favoreciendo la disolución de polímero permeable y quedar libre el fármaco que
ejercerá un efecto local [9]. Al igual que el anterior sistema mencionado, combina la
degradación enzimática, tiempo de tránsito y pH. Sin embargo, es probable que ocurra
una reacción de condensación de tipo Maillard entre azúcares reductores, tales como
lactulosa, con fármacos que contienen grupos amino primarios o secundario dando lugar
a productos cancerígenos. Para evitar esto, la lactulosa necesita ser sustituida por un
compuesto alternativo que no debe ser absorbido en el intestino delgado para prevenir
retrasos en el tiempo de tránsito, debe ser altamente soluble en agua para asegurar una
fácil penetración a través de la membrana de polímero Eudragit® E y debe degradarse
rápidamente en ácidos orgánicos en el colon por microflora de colon. La alternativa es
el isomalt, un disacárido formado por la unión de glucomanitol y glucosorbitol que no
se absorbe ni se metaboliza en el estómago y el intestino delgado, sino que se degrada
por la microflora del colon a glucosa y manitol / sorbitol, los cuales se degradan
posteriormente en ácidos orgánicos, evitando la reacción tipo Maillard [13].

17
F FIGURA 6. Sistema CodesTM
3.3.3) Microesferas de quitosano reticulado: las microesferas se incorporan a una
cápsula de liberación convencional, cada microesfera consta de: a) recubrimiento
colonico (soluble a pH>7) b) mezcla de quitosano y etilcelulosa c) núcleo con el
fármaco más excipientes. Al ingerir la cápsula, en el estómago se disuelve liberando de
manera convencional las microesferas de quitosano. Al tener un recubrimiento colónico,
mantiene su integridad por todo el intestino delgado ya que hay un pH menor de 7.
Cuando llega al colon se disuelve el recubrimiento y el quitosano entra en contacto con
el medio colónico, actuando las azoreductasa sobre los enlaces reticulados, liberando el
principio activo [7,18].
3.4) Nuevos sistemas de liberación retardada: Debido a los inconvenientes de los
sistemas anteriormente dichos, se han desarrollado nuevos sistemas para liberar el
fármaco de manera más específica en el colon.
3.4.1) Philips’Intelligent pill: es una pequeña cápsula, similar a una píldora de cámara.
Fue diseñado para ser administrado vía oral y luego pasar a través del tracto
digestivo. Antes de la ingesta, se programa para administrar el fármaco de una manera
controlada de acuerdo con un perfil de liberación que se va a crear por paciente. El
dispositivo puede determinar su ubicación precisa en el tracto intestinal midiendo la
acidez de su entorno y, cuando alcanza un área definida para la liberación de fármaco,
comienza a liberar con precisión usando una bomba controlada por microprocesador[17]
FIGURA 7. Philips’ Intelligent pill, nuevo sistema de liberación retardada

18
3.4.2) InteliSite® capsule: es un dispositivo de liberación de fármacos activado por
radio control, capaz de incluir formulaciones tanto sólidas como líquidas y que no se
desintegra pasando sin daño por todo el tubo digestivo. Un radiomarcador incluido en el
depósito de fármaco, permite determinar la ubicación exacta de la cápsula en una región
específica del tracto gastrointestinal y permite la activación por el control
remoto cuando la cápsula alcanza la posición deseada en el tracto gastrointestinal, los
contenidos del depósito de fármaco de la cápsula se expulsan a través de un mecanismo
de liberación activo cargado con resorte [16].
FIGURA 8. InteliSite® capsule
En España, se comercializa sistemas de liberación retardada ya que son baratos y fáciles
de emplear. Se presentan en forma de comprimidos, cápsulas y microgránulos
autorizados por la AEMPS para su empleo en el tratamiento de colitis ulcerosa y
enfermedad de Crohn. A continuación se detallarán las formas comerciales más
empleadas en el mercado hoy en día (TABLA 4) [12].
PRINCIPIO ACTIVO NOMBRE COMERCIAL TRANSPORTADOR
Mesalazina
Claversal® Eudragit® L
Lixacol® / Asacol® Eudragit® S
Pentasa® Microgránulos etilcelulosa
Salofalk® Eudragit® L con matriz granular
Sulfasalazina Salazopyrina® Eudragit® S con sistema multimatricial
Budesonida Entocord® Eudragit®
Intestifalk® Eudragit® RL – RS
Eudragit® L100 – S100 TABLA 4. Formas farmacéuticas retardadas comercializadas en España.

19
CONCLUSIONES
La liberación retardada a nivel colónico es uno de los mecanismos más estudiados en la
actualidad ya que ayuda a reducir los efectos secundarios a nivel del tracto digestivo
superior, emplear menos dosis de principio activo y dirigirlo hacia el colon en las
enfermedades inflamatorias intestinales. Por tanto se puede decir que:
1.-Los tipos de sistemas de liberación estudiadas en este trabajo son: a) sistemas
dependientes de pH b) sistemas de liberación dependiente del tiempo de tránsito
intestinal c) sistemas de liberación por degradación enzimática
2.-Las formas farmacéuticas se administra por vía oral y son comprimidos, cápsulas o
microgránulos destinados a aumentar la liberación de fármaco en biofase, aumentar la
eficacia de vías de administración, proteger al fármaco del organismo, reducir la dosis
de fármaco y favorecer su liberación específica en el colon para crear un efecto local .
3.-Los recursos tecnológicos que se emplean para evitar la pérdida de p.a. y que ejerza
su acción local son: a) sistemas dependientes de pH, emplean recubrimientos con
polímeros insolubles a pH ácido y solubles a pH básico, como por ejemplo, Eudragit
que son los más empleados hoy en día. b) sistemas dependientes de tiempo-tránsito,
emplean polímeros entéricos para evitar la pérdida de principio activo en estómago y
además emplean un polímero gelificable capaz de captar agua a lo largo del intestino
delgado (tiempo tránsito) favoreciendo la liberación de fármaco en el colon c) sistemas
de degradación enzimática, emplean polímeros biodegradables en sistemas matriciales o
sistemas de recubrimiento y ligandos en profármacos, donde las enzimas específicas de
la microflora del colon favorecerá la liberación de fármaco en el lugar de acción.
4.-Nuevos sistemas de liberación específica son InteliSite® capsule y Philips’
Intelligent pill que favorece una liberación más específica en el colon, basada por un
control externo mediante radiocontrol o una programación previa a la administración,
respectivamente.
Por tanto se puede concluir que los sistemas basados en la actividad enzimática, y
especialmente los que consisten en polisacáridos, son más específicos y tienen baja
toxicidad. Los sistemas dependientes del tiempo y del pH comercializados han
demostrado ser beneficiosos en el tratamiento de la enfermedad inflamatoria del
intestino, a pesar de su baja especificidad. Además se están creando nuevos sistemas de
liberación modificada para evitar una pérdida de principio activo a lo largo de tracto
digestivo, una liberación de fármaco más segura, eficaz y económica.

20
BIBLIOGRAFÍA
1.- Domínguez JM. Nuevas formas farmacéuticas de liberación modificada. Sescam
Castilla la Mancha. 2007; 14(1):5-7
2.-Lastres García JL. Nuevos sistemas orales de liberación modificada. Schironia. 2002;
15(1): 63-71
3.-Saez Virginia, Estibaliz Hernáez, Lucio Sanz Angulo. Sistema de liberación
controlada de medicamentos. Revista iberoamericana. 2002; 3(3): 1-16
4.- S Tobón, S Vinaccia, JM Quiceno. Aspectos psicopatológicos en la enfermedad de
Crohn y la colitis ulcerosa. Revistas urosario. 2007; 25(2): 83-97
5.-Crispin PL. Estudio comparativo de pectina e hidroxipropolmetilcelulosa en la
formulación de comprimidos matriciales. 2012; 12(2): 3-155
6.- Asghar L, Chure B. C., Chandran, S. Colon specific delivery of indomethacin: effect
of incorporating pH sensitive polymers in xanthan gum matrix bases. AAPS
PharmSciTech. 2009; 10(2): 418 – 429.
7.- Vila Jato, J. Tecnología Farmacéutica II, vol II: formas farmacéuticas. Síntesis (Madrid), 1997: 410 – 416. 8.- Suñé Negre JM. Nuevas aportaciones galénicas a las formas de administración.
Sanired. 2005; 3(2): 30-65
9.- Anil K. Philip, Betty Philip. Colon targeted drug delivery systems: a review on
primary and novel approaches. Oman Medical Journal. 2010, 25.
10.- Eudragit® Polímeros Acrílicos para Formas Farmacéuticas Sólidas Orales [en línea].
Alemania: Evonik Industries; 2011. [acceso 01/05/2017].
URL://eudragit.evonik.com/sites/lists/HN/Documents/evonik-brochure-eudragit-ES.pdf
11.-Livia CL. Utilidad de algunos copolimeros acrilicos para el control de liberación de
fármacos en pellets matriciales. Santiago de Compostela; 2009
12- Philip A, Philip B. Colon targeted drug delivery systems. A review on primary and
novel approaches. OMJ 2010; 25(2): 70 -78
13.- Dehghan M, Mohan V, Mohammed S, Darwis Y, Rizwan M, Mundada V.
Assessment of isomalt for colon-specific drug delivery system and its comparison with
lactulose. AAPS PharmSciTech. 2013; 14(1): 54 – 59
14.- Ali Ashgar L, Chandran S. Multiparticulate formulation approach to colon specific
drug delivery: current perspectives. J. Pharm. Pharmaceut. Sci. 2006; 9(3): 327 – 338
15.- Ana Cristina Freire; Fridrun Podczec , João Sousa. Liberação específica de
fármacos no cólon por via oral. Scielo. 2006; 42(3): 15-23
16.- Takada K., Murakami M. US20056890547. 2005.
17.- Philips' intelligent pill targets drug development and treatment for digestive tract
diseases[en linea]. Inglaterra : Medical express; 2008. [ acceso 05/05/17]
URL://medicalxpress.com/news/2008-11-philips-intelligent-pill-drug-treatment.html 18.- Omwancha W, Mallipeddi R, Valle B, Neau S. Chitosan as a pore former in coated
beads for colon specific drug delivery of 5-ASA. Int. J. Pharm. 2013; 441(1-2): 343 –
351.
Related Documents











