Toxin-Induced Activation of Rho GTP-Binding Protein Increases Bcl-2 Expression and Influences Mitochondrial Homeostasis Carla Fiorentini,* ,1 Paola Matarrese,* Elisabetta Straface,* Loredana Falzano,* Alessia Fabbri,* Gianfranco Donelli,* Andrea Cossarizza,² Patrice Boquet,‡ and Walter Malorni* *Department of Ultrastructures, Istituto Superiore di Sanita ` , Viale Regina Elena 299, 00161 Rome, Italy; ²Department of General Pathology, University of Modena, via Campi 287, Modena, Italy; and ‡Faculte ´ de Me ´decine, INSERM U452, avenue de Valombrose, Nice, France It is now well established that apoptosis plays a piv- otal role in several physiological and pathological sit- uations. Consequently, the mechanisms controlling the cell fate are currently the subject of intense inves- tigation. In this work, we report that an Escherichia coli protein toxin (Cytotoxic Necrotizing Factor 1, CNF1) which activates the Rho GTP-binding protein and prevent apoptosis in epithelial cells was able to: (i) influence the mitochondrial homeostasis and (ii) mod- ulate the expression of proteins belonging to the Bcl-2 family. In particular, the content of the antiapoptotic products Bcl-2 and Bcl-X L resulted to be increased in treated cells, whereas the expression of the proapop- totic protein Bax remained unaltered. CNF1 induces cell spreading via activation of Rho and cell spreading has been reported to promote cell survival. Cytocha- lasin B, which provokes most of the morphological changes typical of CNF1, including cell spreading, but without the involvement of Rho, was unable to coun- teract apoptosis. Altogether our results suggest a link between the Rho GTP-binding protein and the regula- tion of the mitochondrial homeostasis via an effect on the antiapoptotic proteins of the Bcl-2 family. © 1998 Academic Press Key Words: CNF1; Bcl-2; Bcl-X L ; Bax; mitochondria; apoptosis. INTRODUCTION Apoptosis is a physiological form of cell death which plays a major role in tissue development and ho- meostasis. It maintains a correct cell number in the body by balancing cell growth and differentiation with death. The involvement of apoptosis and of its dysregu- lation in the etiology of several pathologies, such as cancer, retroviral infections, autoimmune diseases, neurodegeneration, and aging [1, 2], is currently the subject of intense investigation. The biochemical basis for apoptotic cell death is constitutively present in all mammalian cells, can be activated by a wide variety of extra- and intracellular signals, and involve highly regulated signaling pathways. The induction of apopto- sis is thus characterized by an extreme heterogeneicity of potential triggering signals, which subsequently converge, during the effector phase, into pathways that irrevesibly commit cells to death. The late degradation phase is characterized by typical morphological and molecular markers of apoptosis, such as nuclear chro- matolysis and cytolysis [3], and in vivo end with the removal of apoptotic cells by macrophages which thus prevent inflammation. Several studies indicate that mitochondria play a key role in the early events of the apoptotic cascade [4, 5]. It has been hypothesized that alterations of mito- chondrial permeability transition linked to membrane potential disruption precede nuclear and plasma mem- brane changes associated with apoptosis [6]. In addi- tion, in vitro induction of permeability transition in isolated mitochondria provokes the release of a protein factor [3, 6] capable of inducing nuclear chromatin condensation and fragmentation. This permeability transition appears to be regulated by multiple endog- enous effectors, the most important being members of the Bcl-2 gene family. However, not only permeability transition of mitochondrial membrane, but also varia- tion of mitochondrial membrane potential (Dc m ) can play a key role in the early events of the apoptotic cascade [3– 6]. Thus, compounds influencing mitochon- drial homeostasis and possibly playing a regulatory proapoptotic or antiapoptotic action have recently been analyzed by several research groups in the attempts to exogenously modulate the apoptotic program [for a re- view see 3]. Among factors able to interfere with pathways con- trolling cell survival or death, we have very recently reported that a bacterial toxin from Escherichia coli is capable of protecting epithelial cells against apoptosis [14]. The toxin, named Cytotoxic Necrotizing Factor 1 (CNF1), permanently activates the p21 Rho GTP-bind- 1 To whom correspondence and reprint requests should be ad- dressed. Fax: 139-6-49387140. E-mail: fi[email protected]. 0014-4827/98 $25.00 341 Copyright © 1998 by Academic Press All rights of reproduction in any form reserved. EXPERIMENTAL CELL RESEARCH 242, 341–350 (1998) ARTICLE NO. EX984057

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Toxin-Induced Activation of Rho GTP-Binding Protein Increases Bcl-2Expression and Influences Mitochondrial Homeostasis
Carla Fiorentini,*,1 Paola Matarrese,* Elisabetta Straface,* Loredana Falzano,* Alessia Fabbri,*Gianfranco Donelli,* Andrea Cossarizza,† Patrice Boquet,‡ and Walter Malorni*
*Department of Ultrastructures, Istituto Superiore di Sanita, Viale Regina Elena 299, 00161 Rome, Italy; †Department of GeneralPathology, University of Modena, via Campi 287, Modena, Italy; and ‡Faculte de Medecine,
INSERM U452, avenue de Valombrose, Nice, France
It is now well established that apoptosis plays a piv-otal role in several physiological and pathological sit-uations. Consequently, the mechanisms controllingthe cell fate are currently the subject of intense inves-tigation. In this work, we report that an Escherichiacoli protein toxin (Cytotoxic Necrotizing Factor 1,CNF1) which activates the Rho GTP-binding proteinand prevent apoptosis in epithelial cells was able to: (i)influence the mitochondrial homeostasis and (ii) mod-ulate the expression of proteins belonging to the Bcl-2family. In particular, the content of the antiapoptoticproducts Bcl-2 and Bcl-XL resulted to be increased intreated cells, whereas the expression of the proapop-totic protein Bax remained unaltered. CNF1 inducescell spreading via activation of Rho and cell spreadinghas been reported to promote cell survival. Cytocha-lasin B, which provokes most of the morphologicalchanges typical of CNF1, including cell spreading, butwithout the involvement of Rho, was unable to coun-teract apoptosis. Altogether our results suggest a linkbetween the Rho GTP-binding protein and the regula-tion of the mitochondrial homeostasis via an effect onthe antiapoptotic proteins of the Bcl-2 family. © 1998
Academic Press
Key Words: CNF1; Bcl-2; Bcl-XL; Bax; mitochondria;apoptosis.
INTRODUCTION
Apoptosis is a physiological form of cell death whichplays a major role in tissue development and ho-meostasis. It maintains a correct cell number in thebody by balancing cell growth and differentiation withdeath. The involvement of apoptosis and of its dysregu-lation in the etiology of several pathologies, such ascancer, retroviral infections, autoimmune diseases,neurodegeneration, and aging [1, 2], is currently thesubject of intense investigation. The biochemical basis
for apoptotic cell death is constitutively present in allmammalian cells, can be activated by a wide variety ofextra- and intracellular signals, and involve highlyregulated signaling pathways. The induction of apopto-sis is thus characterized by an extreme heterogeneicityof potential triggering signals, which subsequentlyconverge, during the effector phase, into pathways thatirrevesibly commit cells to death. The late degradationphase is characterized by typical morphological andmolecular markers of apoptosis, such as nuclear chro-matolysis and cytolysis [3], and in vivo end with theremoval of apoptotic cells by macrophages which thusprevent inflammation.
Several studies indicate that mitochondria play akey role in the early events of the apoptotic cascade [4,5]. It has been hypothesized that alterations of mito-chondrial permeability transition linked to membranepotential disruption precede nuclear and plasma mem-brane changes associated with apoptosis [6]. In addi-tion, in vitro induction of permeability transition inisolated mitochondria provokes the release of a proteinfactor [3, 6] capable of inducing nuclear chromatincondensation and fragmentation. This permeabilitytransition appears to be regulated by multiple endog-enous effectors, the most important being members ofthe Bcl-2 gene family. However, not only permeabilitytransition of mitochondrial membrane, but also varia-tion of mitochondrial membrane potential (Dcm) canplay a key role in the early events of the apoptoticcascade [3–6]. Thus, compounds influencing mitochon-drial homeostasis and possibly playing a regulatoryproapoptotic or antiapoptotic action have recently beenanalyzed by several research groups in the attempts toexogenously modulate the apoptotic program [for a re-view see 3].
Among factors able to interfere with pathways con-trolling cell survival or death, we have very recentlyreported that a bacterial toxin from Escherichia coli iscapable of protecting epithelial cells against apoptosis[14]. The toxin, named Cytotoxic Necrotizing Factor 1(CNF1), permanently activates the p21 Rho GTP-bind-
1 To whom correspondence and reprint requests should be ad-dressed. Fax: 139-6-49387140. E-mail: [email protected].
0014-4827/98 $25.00341Copyright © 1998 by Academic Press
All rights of reproduction in any form reserved.
EXPERIMENTAL CELL RESEARCH 242, 341–350 (1998)ARTICLE NO. EX984057

ing protein by deamidation of glutamine 63 [8, 9]. Byactivating Rho, CNF1 promotes actin assembly [10, 11]and cell contractility [12] via stimulation of a numberof kinases in cells [12, 13]. The aim of this study was toinvestigate the cellular events downstream of the acti-vation of Rho which can be involved in the protectionoffered by CNF1 against apoptosis. We herein reportthat CNF1 was able to increase the expression of pro-teins belonging to the Bcl-2 family and to exert a ho-meostatic activity on mitochondria. This may also sug-gest a positive correlation between the activation ofRho and the augmented expression of antiapoptoticproteins.
MATERIALS AND METHODS
Cell Cultures
HEp-2 cells were grown at 37°C in DMEM medium, supplementedwith 5% fetal calf serum (Flow Laboratories, Irvine, UK), 1% nones-sential amino acids, 5 mM L-glutamine, penicillin (100 U/ml), andstreptomycin (100 mg/ml). The subcultures were serially propagatedafter harvesting with 10 mM EDTA and 0.25% trypsin in phosphatebuffer solution (PBS, pH 7.4).
Treatments
CNF1 (10210 M, purified as previously described [10] from the E.coli BM2-1 strain), or 0.5 mg/ml cytochalasin B (CB, Sigma Chemi-cals Co., St. Louis, MO, U.S.A.) were directly added to the culturemedium. After 48 h of treatment, HEp-2 cells were exposed to UVBand then prepared for fluorescence and transmission electron mi-croscopy or for flow cytometry. All the experiments were performedat least three times with triplicate samples for each point.
UVB Exposure
HEp-2 cells were exposed to UVB irradiation in phosphate-buff-ered saline (PBS) using a Philips TL 20 W/12 lamp as previouslydescribed [15]. The plastic petri dishes containing the cells wereplaced without covers at the vertical distance of 10 cm from thecenter of the tube to UVB. In order to eliminate UVC radiation, aKodak filter (Kodacell TL 401) having an optical density of less than0.4 for wavelengths below 285 nm was employed and was placed onthe petri dishes during exposure. In these conditions, the UVB radi-ant flux density to the cells was 2.2 Wm22, as verified by an OsramCentra UV meter. The filter used in our experiments has an opticaldensity of 2.5 at 285 nm, 3 at 280 nm, and above 4 for wavelenthsbelow 270 nm. Therefore we can estimate that the contamination byUVC to cells does not exceed 0.003% of the total UVB. All the resultsreported herein by evaluating cell integrity, mitochondrial mem-brane potential, and the expression of apoptosis-related moleculeswere referred to cells still adhering to the substrate analyzed 24 hafter UV exposure.
Fluorescence Microscopy
HEp-2 cells were grown on 13-mm-diameter glass coverslips inseparate wells (5 3 104 cells/well) in a 37°C incubator containing anatmosphere of 95% air and 5% CO2. Following toxin and UVB treat-ments, both control and treated cells were fixed with 3.7% formal-dehyde in PBS with 2% bovine serum albumin (BSA) for 10 min atroom temperature. After washing in the same buffer, cells werepermeated with 0.5% Triton X-100 (Sigma) in PBS for 10 min atroom temperature. To analyze the nuclei, cells were stained with
Hoechst 33258 (Sigma, working dilution 1:1000) at 37°C for 30 minas previously described [16]. Finally, after washings, coverslips weremounted with glycerol-PBS (2:1) and analyzed with a Nikon Micro-phot fluorescence microscope.
Flow Cytometry
Analysis of mitochondrial mass and membrane potential (Dcm).For the analysis of mitochondrial mass, HEp-2 cells were incubatedat 37°C for 30 min with 5 mM nonylacridine orange (NAO, MolecularProbes Inc., U.S.A.) [17]. After washing with cold PBS, samples wereimmediately analyzed on a FACScan flow cytometer (Becton–Dickinson, San Jose, U.S.A.) equipped with a 488-nm argon laser.Data were recorded and analyzed by a Hewlett–Packard computerusing the Lysys II software (Becton–Dickinson). This procedure wasalso followed for all the cytofluorimetrical analyses described below.Dcm was studied in control and CNF1-treated cells by using 10 mg/mlof 5,59,6,69-tetrachloro-1,19,3,39-tetraethylbenzimidazol-carbocya-nine iodide (JC-1, Molecular Probes) as previously described [18].JC-1 is a molecule able to selectively enter mitochondria, and existsin monomeric form emitting at 527 nm after excitation at 488 nm.However, depending on the membrane potential, JC-1 can formJ-aggregates that are associated with a large shift in the emission(590 nm), and can be easily detected in the orange channel (FL2)present in the most common flow cytometers [18]. JC-1 is bothqualitative (considering the shift from green to orange JC-1 fluores-cence emission) and quantitative (considering the pure fluorescenceintensity, which can be detected in both FL1 and FL2 channels) [19].
Detection of intracellular antigens. Control and CNF1-treatedHEp-2 cells were pelleted, fixed in 70% ice-cold methanol, andwashed twice with cold PBS. For Bcl-X labeling a polyclonal antibody(Santa Cruz Biotechnology, Inc., U.S.A.) recognizing Bcl-XL [20] wasused. For analyzing Bcl-2 and Bax expression in the same cell pop-ulation, a double-staining procedure was performed incubating cellswith a mixture of a monoclonal anti-Bcl-2 (Chemicon International,Inc., Temecule, U.S.A.) and a polyclonal anti-Bax (pAb) (Santa Cruz).All antibodies were used at a final concentration of 0.1 mg/ml for 1 hat 4°C. After several washings cells were incubated with a mixture ofFITC-labeled anti-rabbit pAb and PE-labeled anti-mouse mAb (Sig-ma) for 30 min at 37°C and then washed and analyzed.
Immunoblotting
HEp-2 cells were seeded into 100-mm petri dishes. After 24 h,subconfluent cell layers were incubated with 10210 M CNF1 for 0, 4,24, and 48 h. At each time point, cells were washed once withD-MEM serum free and twice with cold PBS. All further steps wereperformed at 4°C. Cells were scraped in lysis buffer (150 mM Hepes,50 mM NaCl, 10 mM EDTA, 1% Triton X-100) supplemented with 1tablet of complete protease inhibitor (Boehringer-Mannheim, Ger-many). One hundred micrograms of total proteins were separated foreach sample by SDS–PAGE on a 12% gel and electrophoreticallytransferred to nitrocellulose membrane (Amersham, Buckingham-shire, UK). Immunoblots were probed with monoclonal anti Bcl-2(Chemicon International, working dilution 1:1000) and polyclonalantibodies against Bax and Bcl-xL (Santa Cruz, working dilution1:1000). Bound antibodies were detected with horseradish peroxi-dase-conjugated secondary antibodies, including anti-rabbit IgG andanti-mouse IgG (Sigma), followed by enhanced chemoluminescencesubstrate (ECL) and autoradiography.
Transmission Electron Microscopy (TEM)
For ultrastructural analyses cells were fixed with 2.5% glutaral-dehyde in 0.1 M cacodylate buffer, postfixed with 1% osmium tetrox-ide in the same buffer for 1 h, pH 7.4, dehydrated through gradedethanols and embedded in Agar 100 resin. Ultrathin sections ob-tained by using an LKB Ultratome Nova were stained with uranyl
342 FIORENTINI ET AL.

acetate and lead citrate and examined with a Zeiss 10C electronmicroscope.
Statistical Analyses
Values are given as the mean of four separete experiments 6standard deviations. Student’s t test for correlated samples wasused. A P value of less than 0.01 was considered significant.Correlation has been evaluated by using ‘‘Statistics’’ program forMacintosh by a specific paired correlation test. Concerning flowcytometry, the statistical significance was calculated by using theKolmogorov–Smirnov (K/S) test included in Lysys II software(Becton–Dickinson).
RESULTS
CNF1 Protects HEp-2 Cells from UVB-InducedApoptosis
We have recently reported that UVB radiation in-duces cytoskeleton-dependent surface blebbing andcell death by apoptosis in epithelial cells [15] andthat such an effect can be counteracted by CNF1[14]. Cells stained with Hoechst, a fluorescent probewhich specifically binds DNA and evidences chroma-tin structure and clumping typical of apoptosis [16]are shown in Fig. 1. HEp-2 control cells show a singlerounded nucleus per cell (Fig. 1a). CNF1 treatment,although causing multinucleation [21], did not inter-fere with chromatin organization and all nuclei areregularly shaped (Fig. 1b). After exposure to UVBradiation, adhering cells, thereafter maintained for24 h in fresh medium, displayed signs of injury withtypical nuclear clumping and condensed or frag-mented chromatin (Fig. 1c). Pretreatment withCNF1 for 48 h led to a decrease in UVB-inducedeffects, most cells displaying normally shaped nuclei(Fig. 1d). Quantitative and statistical analyses ofthese phenomena (Fig. 1e) performed on four differ-ent experiments showed that CNF1 offered a signif-icant protection against the UVB-induced apoptosis(P , 0.01).
CNF1 Protects HEp-2 Cells against the Dropof Mitochondrial Membrane PotentialProvoked by Radiation
It has recently been reported that mitochondriaplay a key role in apoptosis [3] and that drugs capa-ble of improving actin assembly can protect cellsfrom apoptosis also by regulating mitochondrial in-tegrity and function [19]. We thus performed a flowcytometry study to verify the possible influence ofCNF1 on mitochondrial homeostasis by analyzingthe Dcm. The data obtained showed that, in cellsexposed to CNF1 (Fig. 2b), the Dcm was unchangedwith respect to untreated cells (Fig. 2a). By contrast,after UVB treatment (Fig. 2c), the percentage of cellswith depolarized mitochondria, i.e., those present in
the lower right part of the panel, was remarkablyincreased. Treatment with CNF1 before UVB irradi-ation reduced the mitochondrial membrane depolar-ization induced by UVB as shown by the contour plot(Fig. 2d). A quantitative evaluation of the percentageof cells with depolarized mitochondria was also per-formed. CNF1 toxin was capable of significantly re-ducing this percentage when administered to UVB-treated cells (Fig. 2e).
CNF1 Influences the Expression of Bcl-2-RelatedGene Products
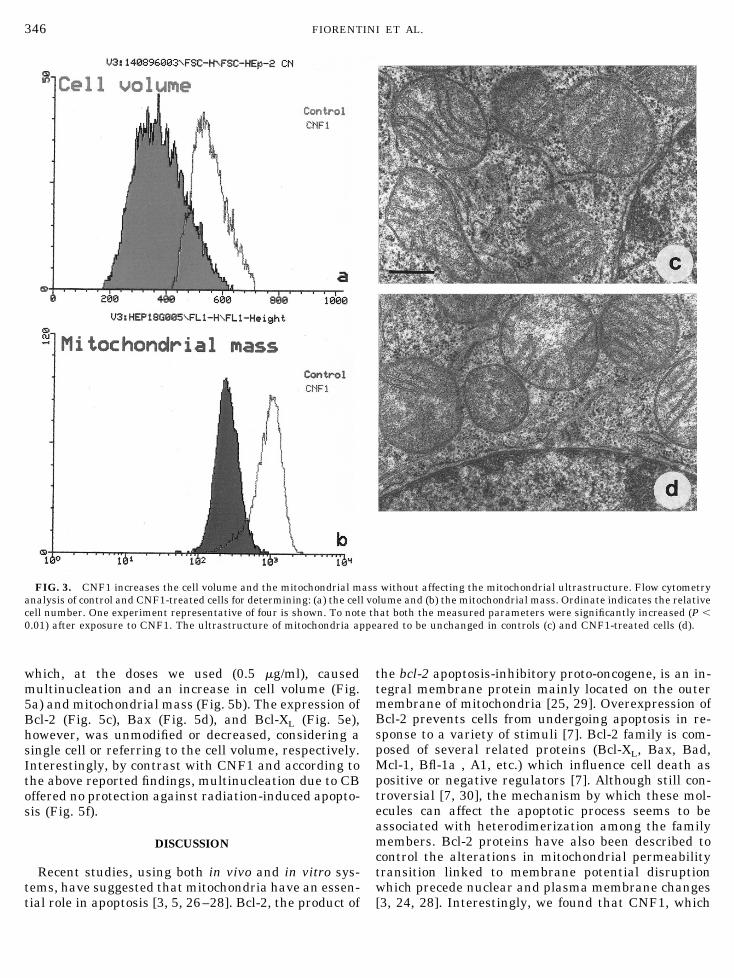
How can CNF1 influence the mitochondrial mem-brane potential? The toxin is known to causemultinucleation by impairing the formation of theactin contractile ring, without strictly interferingwith the mitotic process per se [21]. Cells exposed toCNF1 for 48 h, became significantly larger (Fig. 3a)and consequently showed an increase in the totalmitochondrial mass (Fig. 3b), as measured by NAO,a probe which selectively binds mitochondria [17,22]. The mitochondrial ultrastructure, however, wasnot modified by CNF1, large mitochondria with wellorganized cristae and an electron-dense matrix beingwell visible in both control and treated cells (Figs. 3cand 3d). Related to mitochondria are several Bcl-2-family-related gene products that regulate apopto-sis, either negatively or positively [7, 23, 24]. By flowcytometry, we investigated whether the intracellularquantitative expression of some Bcl-2-related pro-teins could be modulated by CNF1. In particular, wemeasured the expression of the death antagonistmolecules Bcl-2 and Bcl-XL and of the agonist proteinBax. Considering the situation in the single cell, theexpression of Bcl-2 and Bcl-XL resulted to be signif-icantly increased (P , 0.01), whereas no significantchange in Bax expression was observed after 48 h oftreatment with CNF1 (Figs. 4a– 4c). However, takinginto account the increase in cell volume and mito-chondrial mass in cells exposed to CNF1, Bcl-2, andBcl-XL resulted to be unchanged while Bax was de-creased. Whatever we consider the problem, it re-mains that the ratio between death antagonists(Bcl-2 and Bcl-XL) versus the death agonist Bax is infavor of the former proteins in CNF1-treated sam-ples. These findings were supported by the doublestaining contour plots of Bax and Bcl-2 antigensobtained by flow cytometry analysis and indicating adecreased ratio between the two molecules (Figs. 4dand 4e). This is relevant considering that Bcl-2-re-lated proteins undergo selective dimerization pro-cesses and that the ratio of death antagonists toagonists determines whether a cell will respond to anapoptotic signal [7]. These results were further con-firmed by Western blot analysis (Fig. 4f), showing
343CNF1 INCREASES Bcl-2 AND Bcl-XL EXPRESSION

that a clear increase in the amount of both Bcl-2 andBcl-xL proteins occurred already after 4 h of CNF1exposure, this increase becoming much more evidentafter 24 and 48 h of CNF1 treatment (Fig. 4f, lines 2,3, and 4, respectively). By contrast, Bax protein didnot show the same clearly evident increase of the twoantiapoptotic proteins, the amount of Bax remainingsubstantially unchanged (Fig. 4f).
Cytochalasin B, Although Increasing the Cell Volumeand the Mitochondrial Mass, Does Not ProtectHEp-2 Cells against Radiation-Induced Apoptosis
We then studied whether the formation of giantmultinucleated cells containing a higher mitochondrialmass could offer, by itself, protection against apoptosis.For this purpose, we used CB, an actin-disrupting drug
FIG. 1. CNF1 treatment prevents UVB-induced apoptosis in HEp-2 cells. Fluorescence microscopy of cells stained with Hoechst 33258:(a) control cells; (b) cells treated for 48 h with CNF1; (c) UVB-treated cells; (d) cells treated for 48 h with CNF1 and then with UVB. (e)Percentages of apoptotic cells after different treatments. It is noteworthy that CNF1 is capable of significantly decreasing the percentage ofapoptosis (P , 0.01). The values are the means 6 SD from four separate experiments.
344 FIORENTINI ET AL.

FIG. 2. CNF1 prevents mitochondrial membrane depolarization induced by UVB. Cytofluorimetric analysis of HEp-2 cells stained withJC-1: (a) control cells; (b) cells treated with CNF1; (c) UVB irradiated cells; (d) cells treated first with CNF1 and then irradiated. In abscissaFL1 (green fluorescence, log. scale), in ordinate FL2 (orange fluorescence, log. scale). One experiment representative of four is shown. (e)Percentage of cells with depolarized mitochondria. Statistical analysis indicates a significant protection (P , 0.01) offered by CNF1.
which, at the doses we used (0.5 mg/ml), causedmultinucleation and an increase in cell volume (Fig.5a) and mitochondrial mass (Fig. 5b). The expression ofBcl-2 (Fig. 5c), Bax (Fig. 5d), and Bcl-XL (Fig. 5e),however, was unmodified or decreased, considering asingle cell or referring to the cell volume, respectively.Interestingly, by contrast with CNF1 and according tothe above reported findings, multinucleation due to CBoffered no protection against radiation-induced apopto-sis (Fig. 5f).
DISCUSSION
Recent studies, using both in vivo and in vitro sys-tems, have suggested that mitochondria have an essen-tial role in apoptosis [3, 5, 26–28]. Bcl-2, the product of
the bcl-2 apoptosis-inhibitory proto-oncogene, is an in-tegral membrane protein mainly located on the outermembrane of mitochondria [25, 29]. Overexpression ofBcl-2 prevents cells from undergoing apoptosis in re-sponse to a variety of stimuli [7]. Bcl-2 family is com-posed of several related proteins (Bcl-XL, Bax, Bad,Mcl-1, Bfl-1a , A1, etc.) which influence cell death aspositive or negative regulators [7]. Although still con-troversial [7, 30], the mechanism by which these mol-ecules can affect the apoptotic process seems to beassociated with heterodimerization among the familymembers. Bcl-2 proteins have also been described tocontrol the alterations in mitochondrial permeabilitytransition linked to membrane potential disruptionwhich precede nuclear and plasma membrane changes[3, 24, 28]. Interestingly, we found that CNF1, which
FIG. 3. CNF1 increases the cell volume and the mitochondrial mass without affecting the mitochondrial ultrastructure. Flow cytometryanalysis of control and CNF1-treated cells for determining: (a) the cell volume and (b) the mitochondrial mass. Ordinate indicates the relativecell number. One experiment representative of four is shown. To note that both the measured parameters were significantly increased (P ,0.01) after exposure to CNF1. The ultrastructure of mitochondria appeared to be unchanged in controls (c) and CNF1-treated cells (d).
346 FIORENTINI ET AL.

promotes cell survival [14], was able to shift the ratiobetween some of the antiapoptotic (Bcl-2 and Bcl-XL)and proapoptotic (Bax) products in favor of the deathantagonists. This suggests that the toxin, by modulat-ing the expression of proteins of Bcl-2 family, mayoperate on one of the main regulatory systems which
drive a cell toward death or survival: the mitochondrialfunction [7, 25]. Multinucleation and increase in cellvolume and mitochondrial mass have been found afterCNF1 treatment. However, these effects do not appearto be involved in the inhibition of apoptosis. This wasdemonstrated by the experiments carried out with CB
FIG. 4. CNF1 modulates the expression of Bcl-2-related proteins. Flow cytometric analysis of (a) Bax, (b) Bcl-2, and (c) Bcl-xL. Note theincreased expression of Bcl-2 (b) and Bcl-XL (c) in CNF1-treated cells. Analyses by double staining procedure of Bax and Bcl-2 molecules in(d) control and (e) CNF1-treated cells indicate an increased Bcl-2 expression. (f) Analysis of Bcl-2, Bcl-xL, and Bax proteins by Western blotafter 0, 4, 24, and 48 h of CNF1 exposure (lines 1, 2, 3, and 4, respectively). Augmenting the time of exposure to CNF1, a clear increase inthe amount of Bcl-2 and Bcl-xL proteins was evident, while the amount of Bax protein did not undergo the same dramatic increase.
347CNF1 INCREASES Bcl-2 AND Bcl-XL EXPRESSION

toxin, which provokes the same morphological changesof CNF1 without modifying the expression of Bcl-2,Bax, and Bcl-XL and failing in offering protectionagainst apoptotic cell death. The relative increase inBcl-2 and Bcl-XL expression induced by CNF1 was alsoconsistent with the protection against the drop in mi-
tochondrial membrane potential due to CNF1 activity.It is known, in fact, that hyperexpression of Bcl-2 pre-vents both Dcm fall and apoptosis induced by differentstimuli [3].
How can CNF1 control the observed changes in pro-tein expression and the mitochondrial homeostasis?
FIG. 5. CB increases cell volume and mitochondrial mass without influencing apoptosis. Flow cytometric analyses of (a) cell volume; (b)mitochondrial mass; expressions of (c) Bcl-2, (d) Bax, (e) Bcl-XL in control and CB-treated cells. To note that although cell volume andmitochondrial mass do increase following toxin treatment, no significant change in the expression of Bcl-2-related proteins was detected. Allthis is in accordance with the lack of protection against radiation-induced apoptosis offered by CB.
348 FIORENTINI ET AL.

We have recently reported that the antioxidant N-ace-tylcysteine, which shares with CNF1 the property ofimproving actin assembly, also protects cells from ap-optosis [16, 31] and influences mitochondrial integrityand function [20]. Upon entry into the cell cytosol,CNF1 activates the Rho GTP-binding protein which inturn activates a number of kinases [12, 13], thus lead-ing to the promotion of actin assembly and cell spread-ing in epithelial cells [11, 12]. Although cell spreadinghas been reported to promote cell survival [32], ourdata show that the spreading induced by CB, in con-trast to that provoked by CNF1, was uneffective incounteracting apoptosis. We have, however, to under-line that CNF1 is able to induce cell spreading via Rhoactivation differing in this respect from CB whose cel-lular effects do not involve Rho-dependent pathways.This indicates Rho GTPase as a key molecule whichcontrols cell survival and death. Small G-proteins be-longing to the Rho family are involved in a number ofcell activities, such as the regulation of actin assembly[33] or the control of gene transcription [34], and maylead to apoptosis once inactivated [35, 36]. It has re-cently been reported that Rho plays a selective role inearly thymic development as a critical determinant forproliferation and cell survival signals [37]. Moreover,proteins exerting a regulatory function on Rho, such asGDI, are substrates for ICE-like proteases [38]. Modu-lation of Bcl-2 expression in cells by activating or in-activating Rho might thus represent a key event intriggering cell death or survival. It is intriguing, infact, the possibility that a direct link might exist be-tween the Rho-dependent cell spreading which inhibitsapoptosis [32] and the increase in Bcl-2 and Bcl-XLcontent. Accordingly, we have very recently reportedthe positive correlation between CNF1-induced cell ac-tivities, such as spreading and phagocytic behavior andthe cell survival in terms of resistance to apoptoticinduction [14].
In conclusion, the findings reported in this worksuggest the existence of regulatory pathways linkingeither the Rho-dependent actin assembly or the Rho-dependent gene transcription control to the expressionof Bcl-2 and related proteins in cells. Subsequently, theactivation of Rho by CNF1 can be somehow associatedto the observed maintenance of mitochondrial ho-meostasis.
This work was partially supported by the Italian National Re-search Council (CNR), Grants 97.04906.ST74 (to C.F.) and97.04814.ST74 (to W.M.).
REFERENCES
1. Martins, L. M., and Earnshow W. C. (1997). Apoptosis: Aliveand kicking in 1997. Trends Cell Biol. 7, 111–114.
2. Webb, S. J., Harrison, D. J., and Wyllie, A. H. (1997). Apoptosis:An overview of the process and its relevance in disease. Adv.Pharmacol. 41, 1–34.
3. Kroemer, G., Zamzami, N., and Susis, S. A. (1997). Mitochon-drial control of apoptosis. Imm. Today 18, 44–51.
4. Kroemer, G., Petit, P. X., Zamzami, N., Vayssiere, J. L., andMignotte, B. (1995). The biochemistry of programmed celldeath. FASEB J. 9,1277–1287.
5. Richter, C., Schweizer, M., Cossarizza, A., and Franceschi, C.(1996). Control of apoptosis by the cellular ATP level. FEBSLett. 378, 107–110.
6. Petit, P. X., Susin, S. A., Zanzami, N., Mignotte, B., and Kroe-mer G. (1996). Mitochondria and programmed cell death: Backto the future. FEBS Lett. 396, 7–13.
7. Reed, J. C. (1997). Double identity for proteins of the Bcl-2family. Nature 387, 773–776
8. Flatau, G., Lemichez, E., Gauthier, M., Chardin, P., Paris, S.,Fiorentini, C., and Boquet, P. (1997). Toxin-induced activationof the G protein p21 Rho by deamidation of glutamine. Nature387, 729–733.
9. Schmidt, G., Sehr, P., Wilm, M., Selzer, J., Mann, M., andAktories, K. (1997). Glu 63 of Rho is deamidated by Escherichiacoli cytotoxic necrotizing factor -1. Nature 387, 725–729.
10. Falzano, L., Fiorentini, C., Donelli, G., Michel, E., Kocks, C.,Cossart, P., Cabanie, L., Oswald, E., and Boquet, P. (1993).Induction of phagocytic behaviour in human epithelial cells byE. coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 9,1247–1254:
11. Fiorentini, C., Donelli, G., Matarrese, P., Fabbri, A., Paradisi,S., and Boquet, P. (1995). Escherichia coli cytotoxic necrotizingfactor 1: Evidence for induction of actin assembly by constitu-tive activation of the p21 Rho GTPase. Infect. Immun. 63,3936–3944.
12. Fiorentini, C., Fabbri, A., Flatau, G., Donelli, G., Matarrese, P.,Lemichez, E., Falzano, L., and Boquet, P. (1997). Escherichiacoli cytotoxic necrotizing factor 1 (CNF1): A toxin which acti-vates the Rho GTPase. J. Biol. Chem., 272, 19532–19537.
13. Lacerda, H. M., Pullinger, G. D., Lax, A. J., and Rozengurt, E.(1997). Cytotoxic necrotizing factor 1 from Escherichia coli anddermonecrotic toxin from Bortetella bronchiseptica induce p21(rho)-dependent tyrosine phosphorylation of focal adhesion ki-nase and paxillin in Swiss 3T3 cells. J. Biol. Chem. 272, 9587–9596.
14. Fiorentini C., Fabbri, A., Matarrese, P., Falzano, L., Boquet P.,and Malorni, W. (1997). Hinderance of apoptosis and phagocyticbehaviour induced by E. coli cytotoxic necrotizing factor 1(CNF1): Two related activities in epithelial cells. Biochem. Bio-phys. Res. Comm. 241, 341–346.
15. Malorni, W., Donelli, G., Straface, E., Santini, M. T., Paradisi,S., and Giacomoni, P. U. (1994). Both UVA and UVB inducecytoskeleton-dependent surface blebbing in epidermoid cells. J.Photochem. Photobiol. B: Biology 26, 265–270.
16. Malorni, W., Rivabene, R., Santini, M. T., and Donelli, G.(1993). N-acetylcysteine inhibits apoptosis and decreases viralparticles in HIV-chronically infected U937 cells. FEBS Lett.327, 75–78.
17. Maftah, A., Petit, J. M., Ratinaud, M. H., and Julien, R. (1989).10-N nonyl acridine orange: A fluorescent probe which stainsmitochondria independently of their energetic state. Biochem.Biophys. Res. Commun. 164, 185–190.
18. Cossarizza, A., Baccarini-Contri, M., Kalashnikova, G., andFranceschi, C. (1993). A new method for the cytofluorimetricanalysis of mitochondrial membrane potential using the J-ag-gregate forming lipophilic cation 5,59,6,69-tetrachloro-1,19,3,39-
349CNF1 INCREASES Bcl-2 AND Bcl-XL EXPRESSION

tetraethybenzamidazolcarbocyanine iodide (JC-1). Biochem.Biophys. Res. Commun. 197, 40–45.
19. Cossarizza, A., Franceschi, C., Monti, D., Salvioli, S., Bellesia,E, Rivabene, R., Biondo, L., Rainaldi, G., Tinari, A., and Mal-orni, W. (1995). Protective effect of N-acetylcysteine in TumorNecrosis Factor-a-induced apoptosis in U937 cells: The role ofmitochondria. Exp. Cell Res. 220, 232–240.
20. Akbar, A., Borthwick, N., Wickremasinghe, R., Panayiotidis,P., Pilling, D., Bofill, M., and Krajewski, S. (1996). Interleu-kin-2 receptor common g-chain signalling cytokines regulateactivated T cell apoptosis in response to growth factor with-drawal: Selective induction of anti-apoptotic (bcl-2, bcl-xL)but not pro-apoptotic (bax, bcl-xs). Eur. J. Immunol. 26,294 –299.
21. Fiorentini, C., Arancia, G., Caprioli, A., Falbo,V., Ruggeri,F. M., and Donelli, G. (1988). Cytoskeletal changes induced inHep-2 cells by the cytotoxic necrotizing factor of Escherichiacoli. Toxicon 26, 1047–1056.
22. Ratinaud, M. H., Leprat, P., and Julien, R. (1988). In situ flowcytometric analysis of nonyl acridine orange-stained mitochon-dria from splenocytes. Cytometry 9, 206–212.
23. Nunez, G., and Clarke, M. F. (1994). The Bcl-2 family of pro-teins: Regulators of cell death and survival. Trends Cell Biol. 4,399–403.
24. Vander Heiden, M. G., Chandel, N. S., Williamson, E. K., Schu-macker, P. T., and Thompson, C. B. (1997). Bcl-xL regulates themembrane potential and volume homeostasis of mitochondria.Cell 91, 627–637.
25. Kroemer, G. (1997). The proto-oncogene Bcl-2 and its role inregulating apoptosis. Nature Med. 3, 614–620.
26. Liu, X., Kuin, C. N., Yang, J., Jemmerson, R., and Wang, X.(1996). Induction of apoptotic program in cell-free extracts:Requirement for dATP and cytocrome c. Cell 86, 147–157.
27. Kluck, R. M., Bossy-Wetzel, E., Green, D. R., and Newmeyer,D. D. (1997). The release of cytocrome c from mitochondria: Aprimary site for Bcl-2 regulation of apoptosis. Science 275,1132–1136.
28. Yang, J., Liu, X., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J.,Peng, T.-I, Jones, D. P., and Wang, X. (1997). Prevention ofapoptosis by Bcl-2: Release of cytocrome c from mitochondriablocked. Science 275, 1129–1132.
29. Hockenbery, D. M., Oltvai, Z. N., Yin, X. M., Milliman, C. L.,and Korsmeyer, S. J. (1993). Bcl-2 functions in an antioxidantpathway to prevent apoptosis. Cell 75, 241–251.
30. Hsu, Y. T., and Youle R. J. (1997). Nonionic detergents inducedimerization among members of Bcl-2 family. J. Biol. Chem.272, 13829–13834
31. Malorni, W., Rivabene, R., and Matarrese, P. (1995). The anti-oxidant N-acetyl-cysteine protects cultured cells from menadi-one-induced cytopathology. Chem. Biol. Interact. 96, 113–123.
32. Ruoslahti, E. (1997). Stretching is good for a cell. Science 276,1345–1346.
33. Machesky, L. M., and Hall, A. (1996). Rho: A connection be-tween membrane receptor signalling and the cytoskeleton.Trends Cell Biol. 6, 304–310.
34. Hill, C., Wynne, J., and Treisman, R. (1995). The Rho familyGTPases RhoA, Rac1 and Cdc42Hs regulate transcriptionalactivation by SRF. Cell 81, 1159–1170.
35. Moorman, J. P., Bobak, D. A., and Hahn, C. S. (1996). Inacti-vation of the small GTP binding protein Rho induces multinu-cleated cell formation and apoptosis in murine T lymphomaEL4. J. Immunol. 156, 4146–4153.
36. Fiorentini, C., Donelli, G., Nicotera, P., and Thelestam, M.(1993). Closridium difficile toxin A elicits Ca1-independent cy-totoxic effects in cultured normal rat intestinal crypt cells.Infect. Immun. 61, 3988–3993.
37. Henning, S. W., Galandrini, R., Hall, A., and Cantrell, D. A.(1997). The GTPase Rho has a critical regulatory role in thymusdevelopment. EMBO J. 16, 2397–2407.
38. Kolesnitchenko, V., King, L., Riva, A., Tani, Y., Korsmeyer,S. J., and Cohen, D. I. (1997). A major human immunodefi-ciency virus type 1-initiated killing pathway distinct from ap-optosis. J. Virol. 71, 9753–9763.
Received July 16, 1997Revised version received December 23, 1997
350 FIORENTINI ET AL.
Related Documents











