ORIGINAL RESEARCH PAPER TNF-a suppression by glutathione preconditioning attenuates hepatic ischemia reperfusion injury in young and aged rats Arumugam Suyavaran • Chitteti Ramamurthy • Ramachandran Mareeswaran • Ariraman Subastri • Polaki Lokeswara Rao • Chinnasamy Thirunavukkarasu Received: 5 September 2014 / Accepted: 7 November 2014 Ó Springer Basel 2014 Abstract Background and aim Hepatic ischemia reperfusion (I/R) stimulates Kupffer cells and initiates injury through tumor necrosis factor-a (TNF-a) upregulation. Aim of this study was to compare the variable effects of reduced glutathione (GSH) pre-treatment on I/R liver injury in young and aged rats. Methods Wistar male rats were sorted into young (groups I–III) and aged (groups IV–VI). All groups except sham (groups I and IV) were subjected to 90-min ischemia and 2-h reperfusion. The treatment groups received 200 mg/kg bwt (groups III and VI) of GSH, 30 min prior to I/R. Variable effects of GSH were studied by transaminase activities, thiobarbituric acid-reactive substances (TBARS), GSH level, GSH/oxidized GSH (GSSG) ratio, TNF-a level, apoptotic markers and confirmed by histopathological observations. Results Our findings revealed that I/R inflicted more liver damage in aged rats than young rats. The GSH treatment prior to surgery significantly lowered the serum transami- nase activities, hepatic TBARS level and effectively restored the GSH/GSSG ratio in both young and aged rats more remarkably in the mitochondria. Western analysis depicted that the GSH treatment effectively suppressed TNF-a expression and apoptotic markers in both young and aged rats. These findings were further confirmed by ter- minal deoxynucleotide transferase dUTP nick end labeling assay and histopathological observations of liver sections of young and aged rats. Conclusion Restoration of GSH/GSSG ratio through GSH pre-conditioning inhibits TNF-a and apoptosis in hepatic I/R injury. Hence, GSH pre-conditioning may be utilized in both young and aged individuals during liver transplantation/surgery for better post-operative outcomes. Keywords Ischemia/reperfusion Á TNF-a Á Glutathione Á Ageing Á GSH/GSSG ratio Introduction The hepatic inflow from portal triad is occluded during transection procedures to prevent severe blood loss and reduce the morbidity [1]. Pringle maneuver is the procedure by which the portal triad is clamped either partially or fully until surgical intervention and then released to re-establish the blood flow [2, 3]. The occlusion of blood flow during this procedure leads to ischemia of the hepatic parenchyma which leads to accumulation of toxic metabolites further worsening the hypoxic condition. The ischemia reperfusion (I/R) injury occurs due to oxidative stress, during restoration of blood supply to hypoxic organ. Such sudden release of blood supply, also results in reperfusion of toxic metabolites and inflammatory substances into the parenchyma [4]. Ear- lier studies have shown that I/R-induced oxidative damage is associated with increased rate of acute liver graft failure and chronic liver dysfunction after liver transplantation [4]. The I/R is unavoidable in hepatic surgical procedures including liver transplantation. Hence, therapeutic strategies to curtail I/R injury are being considered for the better clinical care of hepatic disorders. The reperfusion injury has been largely attributed to activation of Kupffer cells (KC) and generation of reactive oxygen species (ROS) [5, 6]. It has been shown earlier that, A. Suyavaran Á C. Ramamurthy Á R. Mareeswaran Á A. Subastri Á P. Lokeswara Rao Á C. Thirunavukkarasu (&) Department of Biochemistry and Molecular Biology, Pondicherry University, Puducherry 605014, India e-mail: [email protected] Inflamm. Res. DOI 10.1007/s00011-014-0785-6 Inflammation Research 123

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL RESEARCH PAPER
TNF-a suppression by glutathione preconditioning attenuateshepatic ischemia reperfusion injury in young and aged rats
Arumugam Suyavaran • Chitteti Ramamurthy •
Ramachandran Mareeswaran • Ariraman Subastri •
Polaki Lokeswara Rao • Chinnasamy Thirunavukkarasu
Received: 5 September 2014 / Accepted: 7 November 2014
� Springer Basel 2014
Abstract
Background and aim Hepatic ischemia reperfusion (I/R)
stimulates Kupffer cells and initiates injury through tumor
necrosis factor-a (TNF-a) upregulation. Aim of this study
was to compare the variable effects of reduced glutathione
(GSH) pre-treatment on I/R liver injury in young and aged
rats.
Methods Wistar male rats were sorted into young (groups
I–III) and aged (groups IV–VI). All groups except sham
(groups I and IV) were subjected to 90-min ischemia and
2-h reperfusion. The treatment groups received 200 mg/kg
bwt (groups III and VI) of GSH, 30 min prior to I/R.
Variable effects of GSH were studied by transaminase
activities, thiobarbituric acid-reactive substances (TBARS),
GSH level, GSH/oxidized GSH (GSSG) ratio, TNF-a level,
apoptotic markers and confirmed by histopathological
observations.
Results Our findings revealed that I/R inflicted more liver
damage in aged rats than young rats. The GSH treatment
prior to surgery significantly lowered the serum transami-
nase activities, hepatic TBARS level and effectively
restored the GSH/GSSG ratio in both young and aged rats
more remarkably in the mitochondria. Western analysis
depicted that the GSH treatment effectively suppressed
TNF-a expression and apoptotic markers in both young and
aged rats. These findings were further confirmed by ter-
minal deoxynucleotide transferase dUTP nick end labeling
assay and histopathological observations of liver sections
of young and aged rats.
Conclusion Restoration of GSH/GSSG ratio through
GSH pre-conditioning inhibits TNF-a and apoptosis in
hepatic I/R injury. Hence, GSH pre-conditioning may be
utilized in both young and aged individuals during liver
transplantation/surgery for better post-operative outcomes.
Keywords Ischemia/reperfusion � TNF-a � Glutathione �Ageing � GSH/GSSG ratio
Introduction
The hepatic inflow from portal triad is occluded during
transection procedures to prevent severe blood loss and
reduce the morbidity [1]. Pringle maneuver is the procedure
by which the portal triad is clamped either partially or fully
until surgical intervention and then released to re-establish
the blood flow [2, 3]. The occlusion of blood flow during this
procedure leads to ischemia of the hepatic parenchyma
which leads to accumulation of toxic metabolites further
worsening the hypoxic condition. The ischemia reperfusion
(I/R) injury occurs due to oxidative stress, during restoration
of blood supply to hypoxic organ. Such sudden release of
blood supply, also results in reperfusion of toxic metabolites
and inflammatory substances into the parenchyma [4]. Ear-
lier studies have shown that I/R-induced oxidative damage is
associated with increased rate of acute liver graft failure and
chronic liver dysfunction after liver transplantation [4]. The
I/R is unavoidable in hepatic surgical procedures including
liver transplantation. Hence, therapeutic strategies to curtail
I/R injury are being considered for the better clinical care of
hepatic disorders.
The reperfusion injury has been largely attributed to
activation of Kupffer cells (KC) and generation of reactive
oxygen species (ROS) [5, 6]. It has been shown earlier that,
A. Suyavaran � C. Ramamurthy � R. Mareeswaran � A. Subastri �P. Lokeswara Rao � C. Thirunavukkarasu (&)
Department of Biochemistry and Molecular Biology,
Pondicherry University, Puducherry 605014, India
e-mail: [email protected]
Inflamm. Res.
DOI 10.1007/s00011-014-0785-6 Inflammation Research
123

upon reperfusion-induced activation KC releases inflam-
matory mediators such as tumor necrosis factor-a (TNF-a),
interleukins and chemokines into sinusoidal space, which
further leads to ROS generation mediating proinflamma-
tory gene activation resulting in further damage to hepatic
parenchyma [7–10].
Aged liver does not show significant morphological
changes when compared to young liver. However, reduced
blood flow and bile flow have been shown to be associated
with liver ageing [11, 12]. The extent to which aged liver
can withstand oxidative insult is a mystery, since the
enzymatic and non-enzymatic antioxidant machinery in
aged is not as efficient as in young [13, 14]. Reports have
shown that ROS accumulation in aged liver mitochondria
affects its DNA and membrane integrity leading to apop-
tosis and organ dysfunction [15].
Reduced glutathione (GSH) an endogenous antioxidant
which protects the liver from various intrinsic oxidants
formed during cellular metabolism and also external oxi-
dant sources by neutralizing them and maintaining the
cellular homeostasis [16, 17]. The GSH from GSH trans-
porters of hepatocytes has been shown to act against KC-
generated ROS, thereby protecting the hepatic infrastruc-
ture [18, 19]. The endogenous GSH is usually depleted
during pre-operative starvation which makes the hepatic
parenchyma vulnerable to I/R injury and the risk is higher
in aged liver [20]. Mitochondrial GSH pool acts as the
primary defense against peroxide-induced stress and pro-
tects against oxidative damage [21]. We suspect that
depletion of liver mitochondrial GSH pool with ageing
may lead to defective detoxification of mitochondrial ROS
paving way to cumulative oxidative damage and increased
susceptibility to hepatic I/R damage.
The earlier studies on GSH pre-conditioning show its
effectiveness in reduction of hepatic I/R injury and improved
survival rates [22, 23]. Though these studies elucidate the
potential protective effect of GSH administration against
hepatic I/R injury, there is a greater lacuna in the under-
standing of variable age-dependent response of GSH pre-
conditioning in I/R injury. Hence, we devised the present
study emphasizing the protective effect of GSH pre-treat-
ment against I/R injury in aged rat liver compared with that of
young rat. GSH was preferred to other antioxidants, since it is
an intrinsic antioxidant and its mode of action and metabo-
lism have been extensively studied [19, 21–23].
Materials and methods
Animals
Male Wistar (approved by the Institutional Animal Ethics
Committee, Pondicherry University, Puducherry, India)
young rats of age 6 weeks weighing 140 ± 20 g and aged
rats of age 24 months weighing 300 ± 30 g at the time of
surgery were used for the study. Animals were maintained
in central animal facilities of Pondicherry University. They
were allowed free access to food and water ad libitum until
8 h before surgery. Animals were treated and experimented
as per the guidelines of the Committee for the Purpose of
Control and Supervision of Experiments on Animals,
Government of India.
Experimental design
The animals were divided into six groups (n = 6):
Group I: Young sham (without surgical procedure).
Group II: Young I/R—subjected to saline pre-treatment
via intraperitoneal (i.p.), 30 min before I/R surgery (90-
min ischemia and 2-h reperfusion).
Group III: Young rats pre-treated with GSH (200 mg/kg
bwt, i.p.) 30 min before I/R surgery.
Group IV: Aged sham (without surgical procedure).
Group V: Aged I/R—subjected to saline pre-treatment
(i.p.) 30 min before I/R surgery.
Group VI: Aged rats pre-treated with GSH (200 mg/kg
bwt, i.p.) 30 min before I/R surgery.
The rats were anesthetized by ketamine/xylazine
(100 mg/kg bwt/10 mg/kg bwt, i.p) and then subjected to
midline laparotomy [24]. Hepatic I/R was achieved by
subjecting the rats to hepatic ischemia by partial hepatic
occlusion (covers 70 % of hepatic parenchyma) for 90 min
and released for 2 h for reperfusion. The hepatic pedicle
was identified and the branch left of porta-hepatis which
supplies median and left lobe were clamped with micro-
vascular clamp for 90 min (ischemia) and released exactly
for 2 h (reperfusion). This method prevented congestion of
mesenteric venous drain by permitting decompression of
portal supply through right and caudate lobes [25]. About
2 ml of blood was collected for serum analysis from vena
cava caudalis before killing the rats. Liver tissues were
washed with 0.9 % saline and divided into three portions.
First portion was wrapped in aluminum foil and stored at
-80 �C, for biochemical assays. Second portion was fixed
in neutral buffered formalin for histopathological studies
and the third portion was saved for western blot analysis.
The 2-h reperfusion model was chosen for the analysis
of reperfusion injury in young and aged rats because KC
activation and the release of proinflammatory cytokines are
initiated at this early stage, which later progresses to induce
hepatic fibrosis [26]. The molecular changes during the
early phase of reperfusion are critical since they determine
the ultimate fate of the cells. We assume that the effective
suppression of these changes in ischemia reperfusion may
yield better post-operative outcome.
A. Suyavaran et al.
123

Serum analysis
Hemolysis-free serum from rats was subjected to ALT and
AST analysis using commercially available kit (Cayman
chemical company, MI, USA). The GSH and oxidized
GSH (GSSG) in serum were estimated by HT glutathione
assay kit from Trevigen (Trevigen, Inc, MD, USA).
Hepatic GSH, GSSG and TBARS levels
Tissues were homogenized with 0.25 M sucrose–phosphate
buffer solution (50 mM, pH 7.4) at 4 �C to obtain 10 %
homogenate (1 g tissue in 10 ml of ice cold buffer) in a
motorized homogenizer. Mitochondrial fraction was sepa-
rated by differential centrifugation method [27]. Lipid
peroxidation level in mitochondrial and cytosol fractions
was estimated in terms of the thiobarbituric acid-reactive
substances (TBARS) using malondialdehyde as standard
[28] and GSH and GSSG were estimated by using com-
mercially available kit (Trevigen, Inc, MD, USA).
Western blot analysis
The expression levels of active caspase-3, N-terminal cleavage
fragment of poly (ADP-ribose) polymerases-1 (PARP-1) and
TNF-a were analyzed by Western blot. The samples (30 lg of
protein) were subjected to 12 % SDS-PAGE and then trans-
ferred onto a polyvinylidene fluoride membrane. The
membranes were blocked with 1 % BSA in TBST (Tris-buf-
fered saline, 0.01 % Tween 20) for 2 h at RT and then incubated
with appropriate primary antibodies for 2 h at RT, washed
(3 9 15 min each) with TBST and incubated with appropriate
secondary antibody. The membranes were subjected to
enhanced chemiluminescence reaction and densitometric anal-
yses of the blots were done by Image J—image analysis
software (NIH, Bethesda, USA).b-Actin expression was used as
an internal control to confirm equal protein loading. The anti-
bodies for caspase 3, PARP-1, TNF-a and b–Actin were
purchased from Santacruz biotechnology Inc, USA.
Terminal deoxynucleotide transferase dUTP nick end
labeling (TUNEL) assay
The extent of DNA fragmentation in hepatocytes was ana-
lyzed by TUNEL staining using TACS 2 TdT-Fluor in situ
apoptosis detection kit (Trevigen Inc, MD, USA). Minimum
of ten different fields were observed for each slide.
Assessment of serum and liver TNF-a level
The TNF-a level was quantified by using InvitrogenTM
ELISA kit (KRC3011). Values are expressed as pg/ml in
serum and pg/mg protein in liver tissue.
Histopathological assessment
Liver tissues fixed in neutral buffered formalin were
dehydrated in ascending series of alcohol, cleared in xylene
and embedded in paraffin. The sections (4 lm thickness)
were cut using Leica RM2125 rotatory microtome (LeicaTM,
Germany) and fixed onto gelatin–formaldehyde coated
slides. The slides were deparaffinized in xylene and stained
using hematoxylin and eosin (H&E). They were pictured in
109 objective field using an Olympus CX40 microscope
with ProgresTM image capture setup.
Statistical analysis
Statistical analyses was performed using one-way analysis
of variance (ANOVA) followed by Tukey’s multiple test.
Differences were considered to be significant at P B 0.05
against control. Data were presented as mean ± SD
(standard deviation).
Results
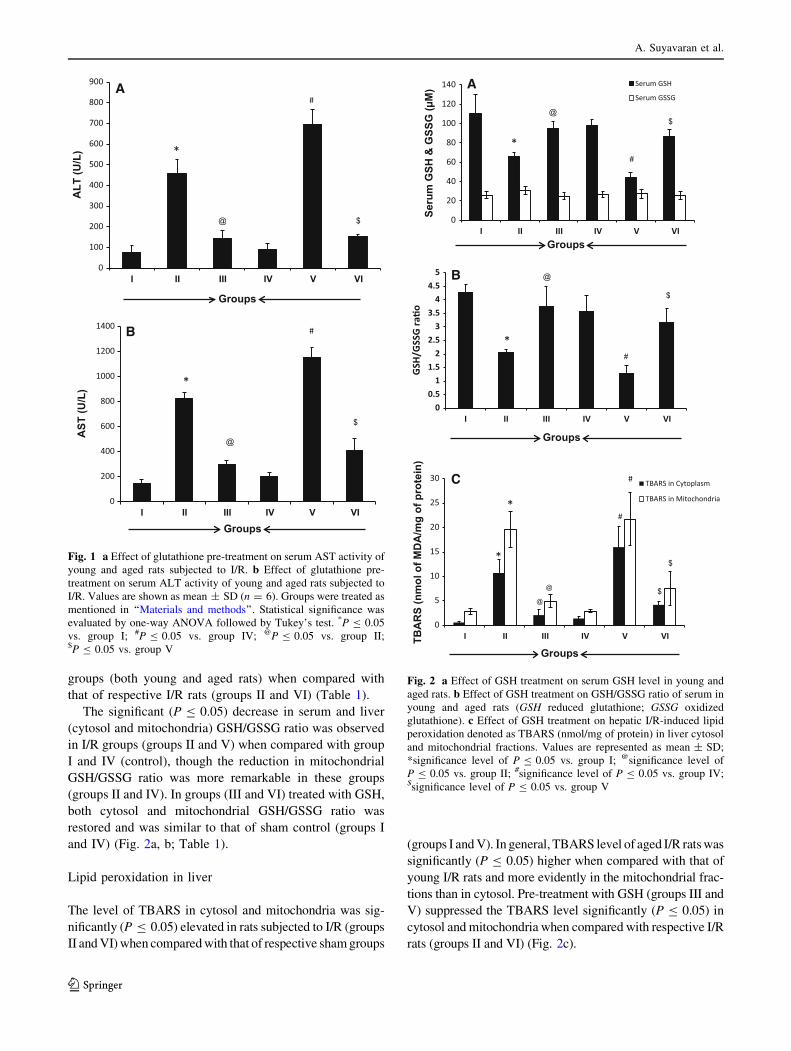
Alteration in liver marker enzyme activities
The ALT and AST activities were significantly (P B 0.05)
elevated in I/R (group II) rats when compared with sham
(group I) rats. The transaminase activities in GSH-treated
group (group III) were significantly lowered when com-
pared with I/R group (group II) (P B 0.05). The similar
trend was observed in aged rats subjected to I/R and pre-
treated with GSH. However, the ALT and AST activities
were elevated in aged I/R (group V) (P B 0.05) when
compared with young I/R (group II) rats (Fig. 1a, b).
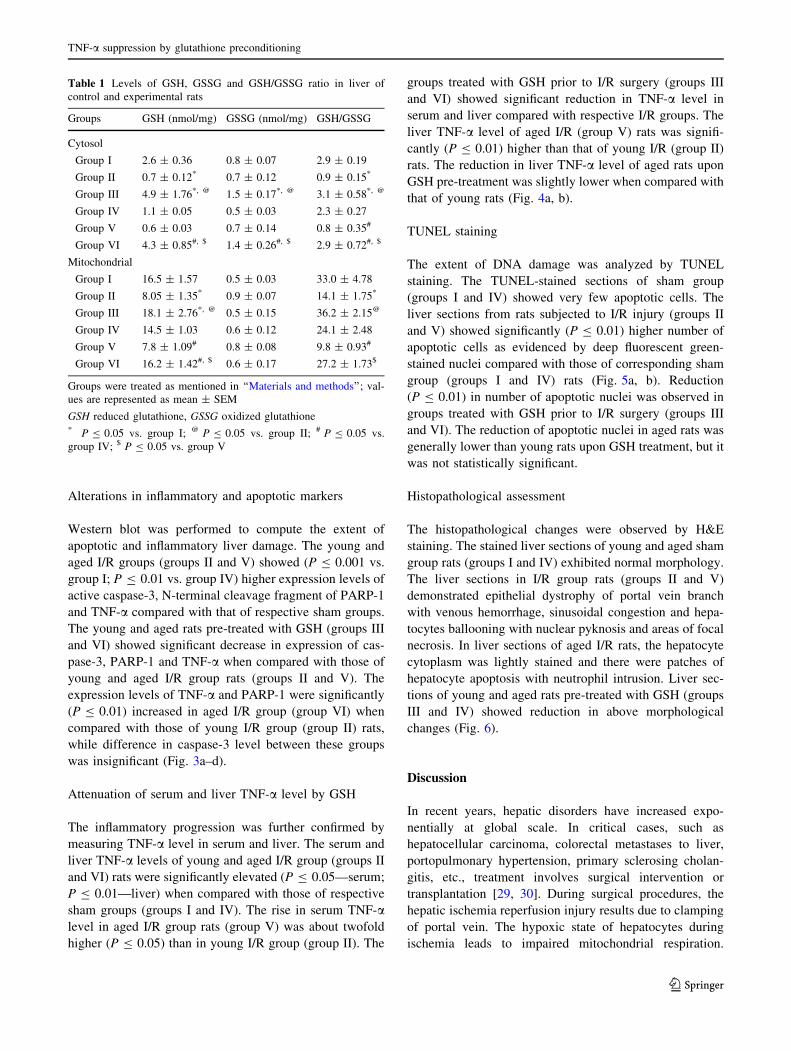
GSH, GSSG and its ratio in serum and liver tissue
The serum GSH level was significantly (P B 0.05) lower in
I/R groups (groups II and V) compared with sham groups
(group I and IV). However, GSH level was significantly
(P B 0.05) decreased in aged I/R rats (group V) when
compared with young I/R (group II) rats. Recovery
(P B 0.05) in serum GSH level was observed in GSH-
treated groups both in young and aged rats (groups III and
VI). No significant changes were observed in GSSG level
in all groups (Fig. 2a, b).
In general, the mitochondrial GSH level in liver tissue
was about tenfold higher than that of cytosol fraction. The
liver tissue cytosol and mitochondrial GSH level of I/R
groups (groups II and V) were significantly (P B 0.05)
lower than respective sham groups (groups I and IV).
Significantly (P B 0.05) elevated level of GSH was
observed in cytosol and mitochondria of all GSH-treated
TNF-a suppression by glutathione preconditioning
123
groups (both young and aged rats) when compared with
that of respective I/R rats (groups II and VI) (Table 1).
The significant (P B 0.05) decrease in serum and liver
(cytosol and mitochondria) GSH/GSSG ratio was observed
in I/R groups (groups II and V) when compared with group
I and IV (control), though the reduction in mitochondrial
GSH/GSSG ratio was more remarkable in these groups
(groups II and IV). In groups (III and VI) treated with GSH,
both cytosol and mitochondrial GSH/GSSG ratio was
restored and was similar to that of sham control (groups I
and IV) (Fig. 2a, b; Table 1).
Lipid peroxidation in liver
The level of TBARS in cytosol and mitochondria was sig-
nificantly (P B 0.05) elevated in rats subjected to I/R (groups
II and VI) when compared with that of respective sham groups
(groups I and V). In general, TBARS level of aged I/R rats was
significantly (P B 0.05) higher when compared with that of
young I/R rats and more evidently in the mitochondrial frac-
tions than in cytosol. Pre-treatment with GSH (groups III and
V) suppressed the TBARS level significantly (P B 0.05) in
cytosol and mitochondria when compared with respective I/R
rats (groups II and VI) (Fig. 2c).
A
B
0
100
200
300
400
500
600
700
800
900
I II III IV V VI
ALT
(U/L
)
#
*
@ $
0
200
400
600
800
1000
1200
1400
I II III IV V VI
AST
(U/L
) *
@
#
$
Groups
Groups
Fig. 1 a Effect of glutathione pre-treatment on serum AST activity of
young and aged rats subjected to I/R. b Effect of glutathione pre-
treatment on serum ALT activity of young and aged rats subjected to
I/R. Values are shown as mean ± SD (n = 6). Groups were treated as
mentioned in ‘‘Materials and methods’’. Statistical significance was
evaluated by one-way ANOVA followed by Tukey’s test. *P B 0.05
vs. group I; #P B 0.05 vs. group IV; @P B 0.05 vs. group II;$P B 0.05 vs. group V
A
B
00.5
11.5
22.5
33.5
44.5
5
I II III IV V VIGS
H/GS
SG ra
�o
0
20
40
60
80
100
120
140
I II III IV V VI
Seru
m G
SH &
GSS
G (μ
M) Serum GSH
Serum GSSG
@
*
$
#
*
@
#
$
Groups
Groups
0
5
10
15
20
25
30
I II III IV V VITBA
RS
(nm
ol o
f MD
A/m
g of
pro
tein
)
TBARS in Cytoplasm
TBARS in Mitochondria
*
*
@
@
#
#
$
$
C
Groups
Fig. 2 a Effect of GSH treatment on serum GSH level in young and
aged rats. b Effect of GSH treatment on GSH/GSSG ratio of serum in
young and aged rats (GSH reduced glutathione; GSSG oxidized
glutathione). c Effect of GSH treatment on hepatic I/R-induced lipid
peroxidation denoted as TBARS (nmol/mg of protein) in liver cytosol
and mitochondrial fractions. Values are represented as mean ± SD;
*significance level of P B 0.05 vs. group I; @significance level of
P B 0.05 vs. group II; #significance level of P B 0.05 vs. group IV;$significance level of P B 0.05 vs. group V
A. Suyavaran et al.
123
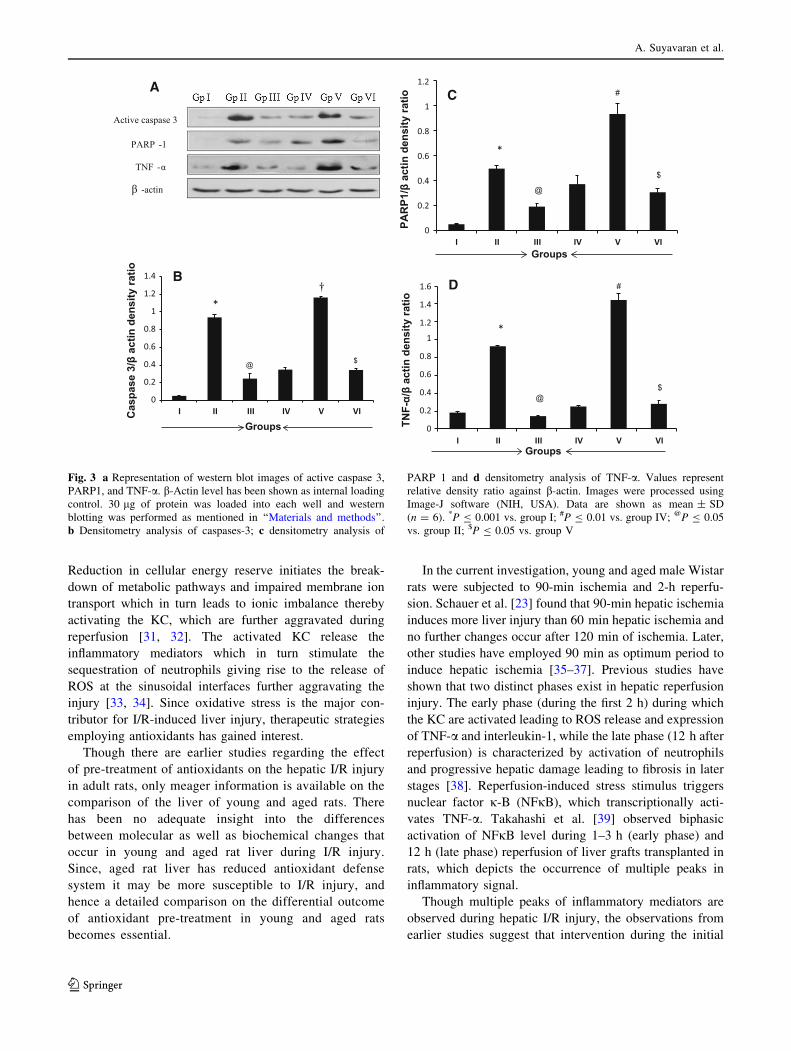
Alterations in inflammatory and apoptotic markers
Western blot was performed to compute the extent of
apoptotic and inflammatory liver damage. The young and
aged I/R groups (groups II and V) showed (P B 0.001 vs.
group I; P B 0.01 vs. group IV) higher expression levels of
active caspase-3, N-terminal cleavage fragment of PARP-1
and TNF-a compared with that of respective sham groups.
The young and aged rats pre-treated with GSH (groups III
and VI) showed significant decrease in expression of cas-
pase-3, PARP-1 and TNF-a when compared with those of
young and aged I/R group rats (groups II and V). The
expression levels of TNF-a and PARP-1 were significantly
(P B 0.01) increased in aged I/R group (group VI) when
compared with those of young I/R group (group II) rats,
while difference in caspase-3 level between these groups
was insignificant (Fig. 3a–d).
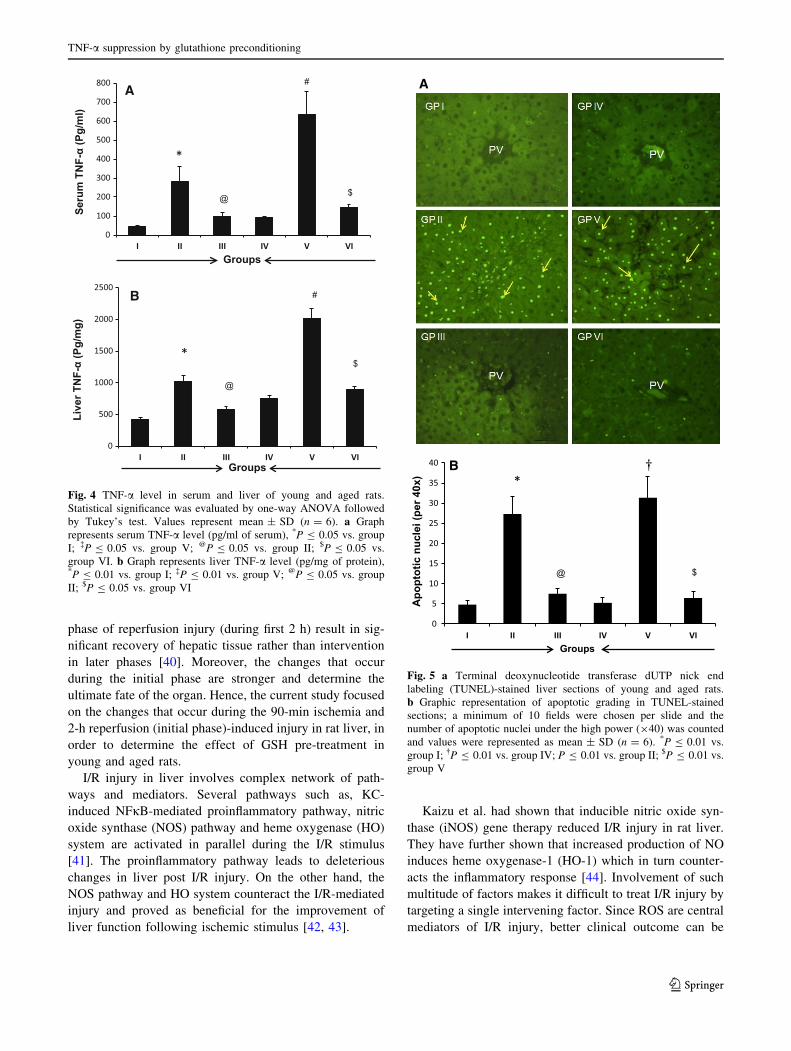
Attenuation of serum and liver TNF-a level by GSH
The inflammatory progression was further confirmed by
measuring TNF-a level in serum and liver. The serum and
liver TNF-a levels of young and aged I/R group (groups II
and VI) rats were significantly elevated (P B 0.05—serum;
P B 0.01—liver) when compared with those of respective
sham groups (groups I and IV). The rise in serum TNF-alevel in aged I/R group rats (group V) was about twofold
higher (P B 0.05) than in young I/R group (group II). The
groups treated with GSH prior to I/R surgery (groups III
and VI) showed significant reduction in TNF-a level in
serum and liver compared with respective I/R groups. The
liver TNF-a level of aged I/R (group V) rats was signifi-
cantly (P B 0.01) higher than that of young I/R (group II)
rats. The reduction in liver TNF-a level of aged rats upon
GSH pre-treatment was slightly lower when compared with
that of young rats (Fig. 4a, b).
TUNEL staining
The extent of DNA damage was analyzed by TUNEL
staining. The TUNEL-stained sections of sham group
(groups I and IV) showed very few apoptotic cells. The
liver sections from rats subjected to I/R injury (groups II
and V) showed significantly (P B 0.01) higher number of
apoptotic cells as evidenced by deep fluorescent green-
stained nuclei compared with those of corresponding sham
group (groups I and IV) rats (Fig. 5a, b). Reduction
(P B 0.01) in number of apoptotic nuclei was observed in
groups treated with GSH prior to I/R surgery (groups III
and VI). The reduction of apoptotic nuclei in aged rats was
generally lower than young rats upon GSH treatment, but it
was not statistically significant.
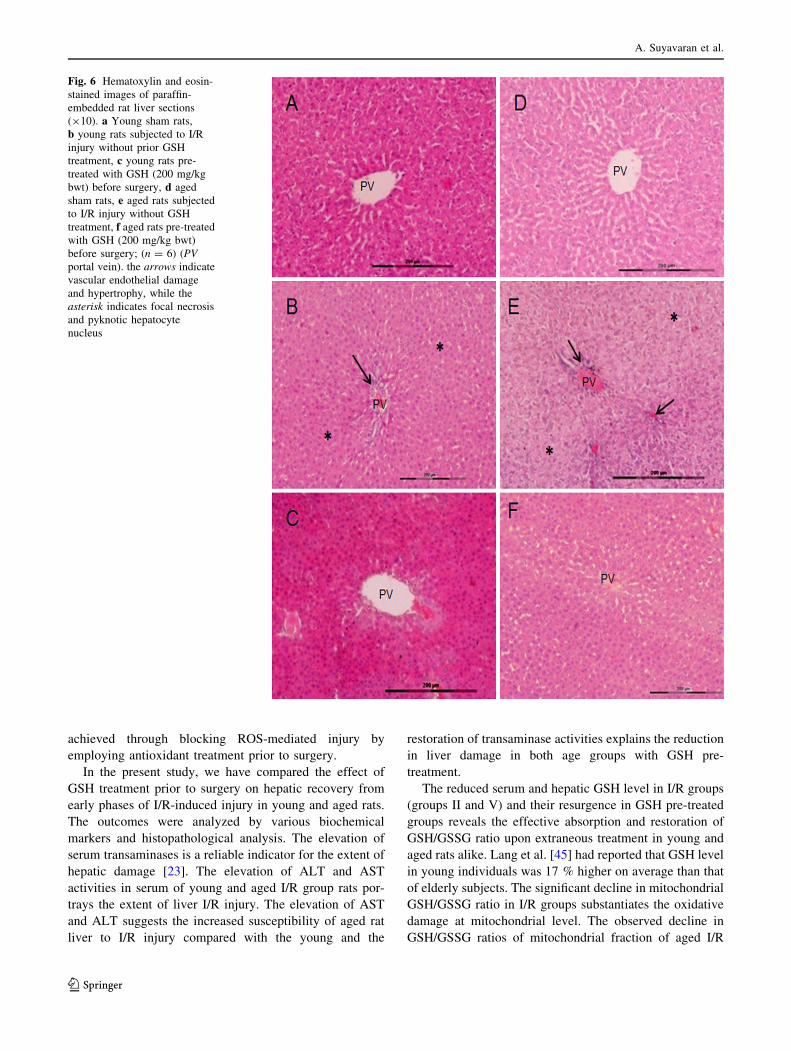
Histopathological assessment
The histopathological changes were observed by H&E
staining. The stained liver sections of young and aged sham
group rats (groups I and IV) exhibited normal morphology.
The liver sections in I/R group rats (groups II and V)
demonstrated epithelial dystrophy of portal vein branch
with venous hemorrhage, sinusoidal congestion and hepa-
tocytes ballooning with nuclear pyknosis and areas of focal
necrosis. In liver sections of aged I/R rats, the hepatocyte
cytoplasm was lightly stained and there were patches of
hepatocyte apoptosis with neutrophil intrusion. Liver sec-
tions of young and aged rats pre-treated with GSH (groups
III and IV) showed reduction in above morphological
changes (Fig. 6).
Discussion
In recent years, hepatic disorders have increased expo-
nentially at global scale. In critical cases, such as
hepatocellular carcinoma, colorectal metastases to liver,
portopulmonary hypertension, primary sclerosing cholan-
gitis, etc., treatment involves surgical intervention or
transplantation [29, 30]. During surgical procedures, the
hepatic ischemia reperfusion injury results due to clamping
of portal vein. The hypoxic state of hepatocytes during
ischemia leads to impaired mitochondrial respiration.
Table 1 Levels of GSH, GSSG and GSH/GSSG ratio in liver of
control and experimental rats
Groups GSH (nmol/mg) GSSG (nmol/mg) GSH/GSSG
Cytosol
Group I 2.6 ± 0.36 0.8 ± 0.07 2.9 ± 0.19
Group II 0.7 ± 0.12* 0.7 ± 0.12 0.9 ± 0.15*
Group III 4.9 ± 1.76*, @ 1.5 ± 0.17*, @ 3.1 ± 0.58*, @
Group IV 1.1 ± 0.05 0.5 ± 0.03 2.3 ± 0.27
Group V 0.6 ± 0.03 0.7 ± 0.14 0.8 ± 0.35#
Group VI 4.3 ± 0.85#, $ 1.4 ± 0.26#, $ 2.9 ± 0.72#, $
Mitochondrial
Group I 16.5 ± 1.57 0.5 ± 0.03 33.0 ± 4.78
Group II 8.05 ± 1.35* 0.9 ± 0.07 14.1 ± 1.75*
Group III 18.1 ± 2.76*, @ 0.5 ± 0.15 36.2 ± 2.15@
Group IV 14.5 ± 1.03 0.6 ± 0.12 24.1 ± 2.48
Group V 7.8 ± 1.09# 0.8 ± 0.08 9.8 ± 0.93#
Group VI 16.2 ± 1.42#, $ 0.6 ± 0.17 27.2 ± 1.73$
Groups were treated as mentioned in ‘‘Materials and methods’’; val-
ues are represented as mean ± SEM
GSH reduced glutathione, GSSG oxidized glutathione* P B 0.05 vs. group I; @ P B 0.05 vs. group II; # P B 0.05 vs.
group IV; $ P B 0.05 vs. group V
TNF-a suppression by glutathione preconditioning
123
Reduction in cellular energy reserve initiates the break-
down of metabolic pathways and impaired membrane ion
transport which in turn leads to ionic imbalance thereby
activating the KC, which are further aggravated during
reperfusion [31, 32]. The activated KC release the
inflammatory mediators which in turn stimulate the
sequestration of neutrophils giving rise to the release of
ROS at the sinusoidal interfaces further aggravating the
injury [33, 34]. Since oxidative stress is the major con-
tributor for I/R-induced liver injury, therapeutic strategies
employing antioxidants has gained interest.
Though there are earlier studies regarding the effect
of pre-treatment of antioxidants on the hepatic I/R injury
in adult rats, only meager information is available on the
comparison of the liver of young and aged rats. There
has been no adequate insight into the differences
between molecular as well as biochemical changes that
occur in young and aged rat liver during I/R injury.
Since, aged rat liver has reduced antioxidant defense
system it may be more susceptible to I/R injury, and
hence a detailed comparison on the differential outcome
of antioxidant pre-treatment in young and aged rats
becomes essential.
In the current investigation, young and aged male Wistar
rats were subjected to 90-min ischemia and 2-h reperfu-
sion. Schauer et al. [23] found that 90-min hepatic ischemia
induces more liver injury than 60 min hepatic ischemia and
no further changes occur after 120 min of ischemia. Later,
other studies have employed 90 min as optimum period to
induce hepatic ischemia [35–37]. Previous studies have
shown that two distinct phases exist in hepatic reperfusion
injury. The early phase (during the first 2 h) during which
the KC are activated leading to ROS release and expression
of TNF-a and interleukin-1, while the late phase (12 h after
reperfusion) is characterized by activation of neutrophils
and progressive hepatic damage leading to fibrosis in later
stages [38]. Reperfusion-induced stress stimulus triggers
nuclear factor j-B (NFjB), which transcriptionally acti-
vates TNF-a. Takahashi et al. [39] observed biphasic
activation of NFjB level during 1–3 h (early phase) and
12 h (late phase) reperfusion of liver grafts transplanted in
rats, which depicts the occurrence of multiple peaks in
inflammatory signal.
Though multiple peaks of inflammatory mediators are
observed during hepatic I/R injury, the observations from
earlier studies suggest that intervention during the initial
0
0.2
0.4
0.6
0.8
1
1.2
1.4
I II III IV V VICas
pase
3/β
act
in d
ensi
ty ra
tio
*
@ $
A
B
Active caspase 3
PARP -1
β -actin
TNF -α
Groups
D
C
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
I II III IV V VI
TNF-
α/β
actin
den
sity
ratio
*
@
#
$
0
0.2
0.4
0.6
0.8
1
1.2
I II III IV V VI
PAR
P1/β
act
in d
ensi
ty ra
tio
*
@
#
$
Groups
Groups
Fig. 3 a Representation of western blot images of active caspase 3,
PARP1, and TNF-a. b-Actin level has been shown as internal loading
control. 30 lg of protein was loaded into each well and western
blotting was performed as mentioned in ‘‘Materials and methods’’.
b Densitometry analysis of caspases-3; c densitometry analysis of
PARP 1 and d densitometry analysis of TNF-a. Values represent
relative density ratio against b-actin. Images were processed using
Image-J software (NIH, USA). Data are shown as mean ± SD
(n = 6). *P B 0.001 vs. group I; #P B 0.01 vs. group IV; @P B 0.05
vs. group II; $P B 0.05 vs. group V
A. Suyavaran et al.
123
phase of reperfusion injury (during first 2 h) result in sig-
nificant recovery of hepatic tissue rather than intervention
in later phases [40]. Moreover, the changes that occur
during the initial phase are stronger and determine the
ultimate fate of the organ. Hence, the current study focused
on the changes that occur during the 90-min ischemia and
2-h reperfusion (initial phase)-induced injury in rat liver, in
order to determine the effect of GSH pre-treatment in
young and aged rats.
I/R injury in liver involves complex network of path-
ways and mediators. Several pathways such as, KC-
induced NFjB-mediated proinflammatory pathway, nitric
oxide synthase (NOS) pathway and heme oxygenase (HO)
system are activated in parallel during the I/R stimulus
[41]. The proinflammatory pathway leads to deleterious
changes in liver post I/R injury. On the other hand, the
NOS pathway and HO system counteract the I/R-mediated
injury and proved as beneficial for the improvement of
liver function following ischemic stimulus [42, 43].
Kaizu et al. had shown that inducible nitric oxide syn-
thase (iNOS) gene therapy reduced I/R injury in rat liver.
They have further shown that increased production of NO
induces heme oxygenase-1 (HO-1) which in turn counter-
acts the inflammatory response [44]. Involvement of such
multitude of factors makes it difficult to treat I/R injury by
targeting a single intervening factor. Since ROS are central
mediators of I/R injury, better clinical outcome can be
A
B
0
100
200
300
400
500
600
700
800
I II III IV V VI
Seru
m T
NF-
α (P
g/m
l)
$
#
@
*
0
500
1000
1500
2000
2500
I II III IV V VI
Live
r TN
F-α
(Pg/
mg)
*
@
#
$
Groups
Groups
Fig. 4 TNF-a level in serum and liver of young and aged rats.
Statistical significance was evaluated by one-way ANOVA followed
by Tukey’s test. Values represent mean ± SD (n = 6). a Graph
represents serum TNF-a level (pg/ml of serum), *P B 0.05 vs. group
I; �P B 0.05 vs. group V; @P B 0.05 vs. group II; $P B 0.05 vs.
group VI. b Graph represents liver TNF-a level (pg/mg of protein),*P B 0.01 vs. group I; �P B 0.01 vs. group V; @P B 0.05 vs. group
II; $P B 0.05 vs. group VI
B
0
5
10
15
20
25
30
35
40
I II III IV V VI
Apo
ptot
ic n
ucle
i (pe
r 40x
) *
@ $
A
Groups
Fig. 5 a Terminal deoxynucleotide transferase dUTP nick end
labeling (TUNEL)-stained liver sections of young and aged rats.
b Graphic representation of apoptotic grading in TUNEL-stained
sections; a minimum of 10 fields were chosen per slide and the
number of apoptotic nuclei under the high power (940) was counted
and values were represented as mean ± SD (n = 6). *P B 0.01 vs.
group I; �P B 0.01 vs. group IV; P B 0.01 vs. group II; $P B 0.01 vs.
group V
TNF-a suppression by glutathione preconditioning
123
achieved through blocking ROS-mediated injury by
employing antioxidant treatment prior to surgery.
In the present study, we have compared the effect of
GSH treatment prior to surgery on hepatic recovery from
early phases of I/R-induced injury in young and aged rats.
The outcomes were analyzed by various biochemical
markers and histopathological analysis. The elevation of
serum transaminases is a reliable indicator for the extent of
hepatic damage [23]. The elevation of ALT and AST
activities in serum of young and aged I/R group rats por-
trays the extent of liver I/R injury. The elevation of AST
and ALT suggests the increased susceptibility of aged rat
liver to I/R injury compared with the young and the
restoration of transaminase activities explains the reduction
in liver damage in both age groups with GSH pre-
treatment.
The reduced serum and hepatic GSH level in I/R groups
(groups II and V) and their resurgence in GSH pre-treated
groups reveals the effective absorption and restoration of
GSH/GSSG ratio upon extraneous treatment in young and
aged rats alike. Lang et al. [45] had reported that GSH level
in young individuals was 17 % higher on average than that
of elderly subjects. The significant decline in mitochondrial
GSH/GSSG ratio in I/R groups substantiates the oxidative
damage at mitochondrial level. The observed decline in
GSH/GSSG ratios of mitochondrial fraction of aged I/R
Fig. 6 Hematoxylin and eosin-
stained images of paraffin-
embedded rat liver sections
(910). a Young sham rats,
b young rats subjected to I/R
injury without prior GSH
treatment, c young rats pre-
treated with GSH (200 mg/kg
bwt) before surgery, d aged
sham rats, e aged rats subjected
to I/R injury without GSH
treatment, f aged rats pre-treated
with GSH (200 mg/kg bwt)
before surgery; (n = 6) (PV
portal vein). the arrows indicate
vascular endothelial damage
and hypertrophy, while the
asterisk indicates focal necrosis
and pyknotic hepatocyte
nucleus
A. Suyavaran et al.
123

groups (group VI) and its recovery with GSH treatment in
aged groups are comparable to the similar outcome in
young rats, thereby suggesting the mitochondrial GSH
replenishing efficiency of extraneous GSH administration
prior to hepatic surgery across age barrier.
Lipid peroxidation is the key mechanism by which ROS
execute the oxidative damage in liver [46–48]. Moreover,
mitochondrial lipid peroxidation due to oxidative injury
has been described as the major culprit in the hepatic I/R
injury [49]. Ageing has been shown to be associated with
increased hepatic lipid peroxidation in aged subjects [50].
In our study, TBARS level was elevated in the aged rats
subjected to hepatic I/R, but were subdued in the rats
treated with GSH prior to surgery.
In the current study, we have observed significantly high
expression of TNF-a, active caspase-3 and PARP-1 (N-
terminal fragment) in rats subjected to hepatic I/R injury
without GSH treatment. TNF-a has been observed to play a
major role in the onset of I/R-mediated oxidative hepatic
injury. Previous studies have elucidated the mechanism of
TNF-a-induced activation of apoptosis and its role in
hepatic injury [51–53]. Circulating neutrophils have been
shown to elicit TNF-a-induced caspase-3 activation [54].
PARP-1 cleavage fragments are a signature of apoptotic
progression [55]. Our observation of correlated increase in
TNF-a, caspase-3 and PARP-1 confirms a positive loop of
apoptotic signaling involved in hepatic I/R injury. The rise
in serum and liver tissue TNF-a level upon hepatic I/R
injury and its reduction in GSH pre-treated rats, as shown
by ELISA further supports the inflammatory process aug-
mented by I/R liver injury.
The increase in TUNEL-positive nuclei in young and
aged rats of I/R group elucidates the DNA damage inflicted
by I/R-mediated oxidative damage. Earlier studies have
reported similar increase in TUNEL-positive cells upon
reperfusion followed by ischemia [56, 60]. The effective-
ness of GSH pre-treatment prior to surgery in reducing
oxidative DNA damage is demonstrated by reduction in
number of TUNEL-positive cells in GSH-treated young
and aged groups.
Liver sections of aged I/R groups showed vascular
endothelial degradation and patches of hepatocyte necrosis
with neutrophil intrusion, while hepatocytes ballooning and
pyknotic nucleus was observed in young rats subjected to
hepatic I/R injury. The liver damage in I/R injury has been
proven earlier to be extensively due to the triggering of
inflammatory mediators released by KC stimulation and
the resulting release of inflammatory mediators like TNF-
a, IL1-a and IL6 [57, 58]. The elevated expression of TNF-
a has been shown to be the culprit for P-selectin upregu-
lation and neutrophil recruitment [59]. Suppression of
TNF-a-mediated apoptotic signaling by GSH pre-treatment
can be explained by attenuation of KC simulation by
lowering the oxidative stress stimulus.
Conclusions
We have observed that the administration of GSH prior to
hepatic I/R surgery protects both the young and aged rats
from I/R-induced oxidative damage. The pre-treatment
with GSH significantly reduced the apoptosis and TNF-aby restoration of GSH/GSSG ratio at mitochondrial level,
in young and aged rats. These findings suggest that GSH
supplementation prior to surgery would be an efficient
therapeutic strategy and can be used synergistically with
other treatments to yield better post-operative outcomes,
thus irrespective of age factor.
Acknowledgments The authors duly acknowledge the funding
support from Indian Council of Medical Research (ICMR Ref: 52/13/
2007) and Department of Science and Technology (NO.SR/FT/LS-63/
2011 & DST-FIST), New Delhi, India. Author A. Suyavaran
acknowledges UGC, New Delhi, India, for financial support in the
form of Junior Research Fellowship [CSIR-UGC-JRF; S. No. F.17-
115/98 (SA-I)].
References
1. Carden DL, Granger DN. Pathophysiology of ischemia–reperfu-
sion injury. J Pathol. 2000;190:255–66.
2. Smyrniotis VE, Kostopanagiotou GG, Contis JC, Farantos CI,
Voros DC, Kannas DC, et al. Selective hepatic vascular exclusion
vs. Pringle maneuver in major liver resections: prospective study.
World J Surg. 2003;27:765–9.
3. Man K, Fan ST, Ng IO, Lo CM, Liu CL, Wong J. Prospective
evaluation of Pringle maneuver in hepatectomy for liver tumors
by a randomized study. Ann Surg. 1997;226:704–11.
4. Kupiec-Weglinski JW, Busuttil RW. Ischemia and reperfusion in
liver transplantation. Transpl Proc. 2005;37:1653–6.
5. Rymsa B, Wang JF, de Groot H. O2.- release by activated Kupffer
cells upon hypoxia–reoxygenation. Am J Physiol.
1991;261:G602–7.
6. Tomiyama K, Ikeda A, Ueki S, Nakao A, Stolz DB, Koike Y,
et al. Inhibition of Kupffer cell-mediated early proinflammatory
response with carbon monoxide in transplant-induced hepatic
ischemia/reperfusion injury in rats. Hepatology.
2008;48:1608–20.
7. Essani NA, McGuire GM, Manning AM, Jaeschke H, et al.
Endotoxin-induced activation of the nuclear transcription factor
kappa-B and expression of E-selectin messenger RNA in hepa-
tocytes, Kupffer cells and endothelial cells in vivo. J Immunol.
1996;156:2956–63.
8. Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2
formation by rat heart mitochondria on substrate availability and
donor age. J Bioenerg Biomembr. 1997;29:89–95.
9. Miquel J, Economos CA, Fleming J, Johnson JE Jr. Mitochon-
drial role in cell aging. Exp Gerontol. 1980;15:575–91.
10. Kim HG, Hong SM, Kim SJ, Paark HJ, Lee YY, Moon JS, et al.
Age related changes in the activity of antioxidant and redox
enzymes in rats. Mol Cells. 2003;16:278–84.
TNF-a suppression by glutathione preconditioning
123

11. Le Couteur DG, McLean AJ. The aging liver. Drug clearance and
an oxygen diffusion barrier hypothesis. Clin Pharmacokinet.
1998;34:359–73.
12. Strolin BM, Whomsley R, Baltes EL. Differences in absorption,
distribution, metabolism and excretion of xenobiotics between
the paediatric and adult populations. Expert Opin Drug Metab
Toxicol. 2005;1:447–71.
13. Mallikarjuna K, Shanmugham KR, Nishanth K, Mc Wu, Hou
CW, Kuo CH, et al. Alcohol-induced deterioration in primary
antioxidant and glutathione family enzymes reversed by exercise
training in the liver of old rats. Alcohol. 2010;44:523–9.
14. Castro MR, Suarez E, Kraiselburd E, Isidro A, Paz J, Ferder L,
et al. Aging increases mitochondrial DNA damage and oxidative
stress in liver of rhesus monkeys. Exp Gerontol. 2012;47:29–37.
15. Sastre J, Pallardo FV, Pla R, Pellin A, Juan G, O’Connor JE, et al.
Aging of the liver: age-associated mitochondrial damage in intact
hepatocytes. Hepatology. 1996;24:1199–205.
16. Kosower NS, Kosower EM. The glutathione status of cells. Int
Rev Cytol. 1978;54:109–60.
17. Han D, Hanawa N, Saberi B, Kaplowitz N. Mechanisms of liver
injury. III. Role of glutathione redox status in liver injury. Am J
Physiol Gastrointest Liver Physiol. 2006;291:G1–7.
18. Brass CA, Roberts TG. Hepatic free radical production after cold
storage: Kupffer cell-dependent and independent mechanism in
rats. Gasteroenterology. 1995;108:1167–75.
19. Fernandez-Checa JC, Yi JR, Ruiz CG, Ookhtens M, Kaplowitz N.
Plasma membrane and mitochondrial transport of hepatic reduced
glutathione. Semin Liver Dis. 1996;16:147–58.
20. Selzner M, Selzner N, Jochum W, Graf R, Clavien PA. Increased
ischemic injury in old mouse liver: an ATP-dependent mecha-
nism. Liv Transpl. 2007;13:382–90.
21. Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glu-
tathione: transport and role in disease and toxicity. Toxicol Appl
Pharmacol. 2005;204:263–73.
22. Xue F, Wang G, Pang Z, Liu C, Liang T. Protective effect of
glutathione against liver warm ischemia–reperfusion injury in rats
is associated with regulation of P-selectin and neutrophil infil-
tration. Anat Rec. 2008;291:1016–22.
23. Schauer RJ, Gerbes AL, Vonier D, Meissner H, Michl P, Leiderer
R, et al. Glutathione protects the rat liver against reperfusion
injury after prolonged warm ischemia. Ann Surg.
2004;239:220–31.
24. Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD,
Linden J. Protection from ischemic liver injury by activation of
A–A adenosine receptors during reperfusion: inhibition of che-
mokine induction. Am J Physiol Gastrointest Liver Physiol.
2004;286:G285–93.
25. Camargo CA Jr, Madden JF, Gao W, Selvan RS, Clavien PA.
Interleukin-6 protects liver against warm ischemia/reperfusion
injury and promotes hepatocyte proliferation in the rodent.
Hepatology. 1997;26:1513–20.
26. Ozkan E, Yardimci S, DUlundu E, et al. Protective potential of
montelukast against hepatic ischemia/reperfusion injury in rats.
J Surg Res. 2010;159:588–94.
27. Sahoo A, Chainy GB. Alterations in the activities of cerebral
antioxidant enzymes of rat are related to aging. Int J Dev Neu-
rosci. 1997;15:939–48.
28. Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal
tissues by thiobarbituric acid reaction. Anal Biochem.
1979;95:351–8.
29. Anaya DA, Becker NS, Abraham NS. Global graying, colorectal
cancer and liver metastasis: new implications for surgical man-
agement. Crit Rev Oncol Hematol. 2011;77:100–8.
30. Goldberg DS, Fallon MB. Model for end-stage liver disease-
based organ allocation: managing the exceptions to the rules. Clin
Gastroenterol Hepatol. 2013;11:452–3.
31. Clavien PA, Harvey PR, Strasberg SM. Preservation and reper-
fusion injuries in liver allografts. An overview and synthesis of
current studies. Transplantation. 1992;53:957–78.
32. Rosser BG, Gores GJ. Liver cell necrosis: cellular mechanisms
and clinical implications. Gastroenterology. 1995;108:252–75.
33. Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide
generation by Kupffer cells and priming of neutrophils during
reperfusion after hepatic ischemia. Free Radic Res Commun.
1991;15:277–84.
34. Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced
oxidant stress and ischemia–reperfusion injury in rat liver. Am J
Physiol. 1991;260:G355–62.
35. Xu SQ, Li YH, Hu SH, Chen K, Dong LY. Effect of Wy14643 on
hepatic ischemia reperfusion injury in rats. World J Gastroen-
terol. 2008;14:6936–42.
36. Wu C, Wang P, Rao J, Wang Z, Zhang C, Lu L, Zhang F.
Triptolide alleviates hepatic ischemia/reperfusion injury by
attenuating oxidative stress and inhibiting NF-jB activity in
mice. J Surg Res. 2011;166:e205–13.
37. Lin CM, Lee JF, Chiang LL, Chen CF, Wang D, Su CL. The
protective effect of curcumin on ischemia–reperfusion induced
liver injury. Transplant Proc. 2012;44:974–7.
38. Glantzounis GK, Salacinski HJ, Yang W, Davidson BR, Seifalian
AM. The contemporary role of antioxidant therapy in attenuating
the ischemia–reperfusion injury: a review. Liver Transpl.
2005;11:1031–47.
39. Takahashi Y, Ganster RW, Gambotto A, Shao L, Kaizu T, Wu T,
et al. Role of NF-kappaB on liver cold ischemia–reperfusion
injury. Am J Physiol Gastrointest Liv Physiol.
2002;283:G1175–84.
40. Crockett ET, Galligan JJ, Uhal BD, Harkema J, Roth R, Pandya
K. Protection of early phase hepatic ischemia–reperfusion injury
by cholinergic agonists. BMC Clin Pathol. 2006;6:3.
41. Vardanian AJ, Busuttil RW, Kupiec-Weglinski JW. Molecular
mediators of liver ischemia and reperfusion injury: a brief review.
Mol Med. 2008;14:337–45.
42. Hines IN, Harada H, Flores S, Gao B, McCord JM, Grisham MB.
Endothelial nitric oxide synthase protects the post-ischemic liver:
potential interactions with superoxide. Biomed Pharmacother.
2005;59:183–9.
43. Tsuchihashi S, Zhai Y, Bo Q, Busuttil RW, Kupiec Weglinski
JW. Heme-oxygenase-1 mediated cytoprotection against liver
ischemia and reperfusion injury: inhibition of type-1 interferon
signaling. Transplantation. 2007;83:1628–34.
44. Kaizu T, Ikeda A, Nakao A, Takahashi Y, Tsung A, Kohmoto J,
et al. Donor graft adenoviral iNOS gene transfer ameliorates rat
liver transplant preservation injury and improves survival.
Hepatology. 2006;43:464–73.
45. Lang CA, Naryshkin S, Schneider DL, Mills BJ, Lindeman RD.
Low blood glutathione in healthy aging adults. J Lab Clin Med.
1992;120:720–5.
46. Glantzounis GK, Salacinski HJ, Yang W, Davidson BR, Seifalian
AM. The contemporary role of antioxidant therapy in attenuating
liver ischemia–reperfusion injury: a review. Liver Transpl.
2005;11:1031–47.
47. Zhang W, Wang M, Xie HY, Zhou L, Meng XQ, Shi J, et al. Role
of reactive oxygen species in mediating hepatic ischemia–reper-
fusion injury and its therapeutic application in liver
transplantation. Transplant Proc. 2007;39:1332–7.
48. Knight TR, Fariss MW, Farhood A, Jaeschke H. Role of lipid
peroxidation as a mechanism of liver injury after acetaminophen
overdose in mice. Toxicol Sci. 2003;76:229–36.
49. Mukhopadhyay P, Horvath B, Zsengeller Z, Batkai S, Cao Z,
Kechrid M, et al. Mitochondrial reactive oxygen species gener-
ation triggers inflammatory response and tissue injury associated
with hepatic ischemia–reperfusion: therapeutic potential of
A. Suyavaran et al.
123

mitochondrially targeted antioxidants. Free Radic Biol Med.
2012;53:1123–38.
50. Farahmand SK, Samini F, Samini M, Samarghandian S. Safarnal
ameliorates antioxidant enzymes and suppresses lipid peroxida-
tion and nitric oxide formation in aged male rat liver.
Biogerontology. 2013;14:63–71.
51. Okajima K, Harada N, Kushimoto S, Uchiba M. Role of micro-
thrombus formation in the development of ischemia/reperfusion-
induced liver injury in rats. Thromb Haemost. 2002;88:473–80.
52. Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al.
The mitochondrial-targeted compound SS-31 re-energizes
ischemic mitochondria by interacting with cardiolipin. J Am Soc
Nephrol. 2013;24:1250–61.
53. Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa
JC. Mitochondrial glutathione, a key survival antioxidant. Anti-
oxid Redox Signal. 2009;11:2685–700.
54. Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A
novel TNFR1-triggered apoptosis pathway mediated by class IA
PI2Ks in neutrophils. Blood. 2011;117:5953–62.
55. Chaitanya GV, Alexander JS, Babu PP. PARP-1 cleavage frag-
ments: signatures of cell-death proteases in neurodegeneration.
Cell Commun Signal. 2010;8:31. doi:10.1186/1478-811X-8-31.
56. Schmitt-Graff A, Kruger S, Bochard F, Gabbiani G, Denk H.
Modulation of alpha smooth muscle actin and desmin expression
in perisinusoidal cells of normal and diseased human livers. Am J
Pathol. 1991;138:1233–42.
57. Kim do Y, Chung SI, Ro SW, Paik YH, Lee JI, Jung MK, et al.
Combined effects of an antioxidant and Caspase inhibitor on the
reversal of hepatic fibrosis in rats. Apoptosis. 2013;18:1481–91.
58. Menger MD, Richter S, Yamauchi J. Vollmar. Role of micro-
circulation in hepatic ischemia/reperfusion injury. Hepato
Gastroenterol. 1999;46:1452–7.
59. Peralta C, Fernandez L, Panes J, Prats N, Sans M, Pique JM, et al.
Preconditioning protects against systemic disorders associated
with hepatic ischemia–reperfusion through blockade of tumor
necrosis facto-induced P-selectin up-regulation in the rat. Hepa-
tology. 2001;33:100–13.
60. Serbetci K, Uysal O, Erkasap N, Koken T, Baydemir C, Erkasap
S. Anti-apoptotic and antioxidant effect of leptin on CCl4 induced
acute liver injury in rats. Mol Biol Rep. 2012;39:1173–80.
TNF-a suppression by glutathione preconditioning
123
Related Documents











