Virus Research 120 (2006) 36–48 Third genome size category of avian paramyxovirus serotype 1 (Newcastle disease virus) and evolutionary implications Al´ ız Czegl´ edi a,1 , Dorina Ujv´ ari a,1 , Eszter Somogyi a , Enik ˝ o Wehmann a , Ortrud Werner b , B´ ela Lomniczi a,∗ a Veterinary Medical Research Institute of the Hungarian Academy of Sciences, P.O. Box 18, Budapest 1581, Hungary b Institute of Diagnostic Virology, Friedrich-Loeffler-Institutes, Federal Research Centre for Virus Diseases of Animals, D 17493 Greifswald, Boddenblick 5A, Insel Riems, Germany Received 24 June 2005; received in revised form 11 November 2005; accepted 11 November 2005 Available online 12 June 2006 Abstract The goal of the study was to establish if there was a relationship between molecular patterns and virus evolution. Therefore the complete genome sequence of two distinct apathogenic Newcastle disease virus (NDV) strains was determined and a third genome size category, containing 15,198 nucleotides, was recognized. Phylogenetic analysis revealed that two major separations resulting in three genome size categories occurred during the history of NDV. An ancient division in the primordial reservoir (wild waterbird species) led to two basal sister clades, class I and II, with genome sizes 15,198 (due to a 12 nucleotide insert in the phosphoprotein gene) and 15,186 nucleotides, respectively. Ancestors of only class II viruses colonized chicken populations and subsequently converted to virulent forms. These took place more than once and resulted in an early lineage [including genotypes I–IV and H33(W)] with genome size of 15,186 nucleotides. A second division occurred in the 20th century in the secondary (chicken) host. This gave rise to the branching-off of a clade (including recent genotypes V–VIII consisting of only pathogenic viruses) with the concomitant insertion of six nucleotides into the 5 non-coding region of the nucleoprotein gene thereby increasing the genome size to 15,192 nucleotides. © 2006 Elsevier B.V. All rights reserved. Keywords: Newcastle disease virus; Evolution; Genome size; Phylogenetic analysis 1. Introduction Newcastle disease virus (NDV), an avian paramyxovirus, is the causative agent of Newcastle disease (ND), a highly conta- gious disease that may result in 100% morbidity and mortality in chicken flocks (Alexander, 2003). In spite of control mea- sures, including vaccination applied since the 1950s, ND still is a serious problem of the poultry industry. In the past decade the disease was endemic in 57% of the countries rearing poultry while further 23% suffered one or several introduction of the virus (OIE). Sequences have been deposited in the EMBL/GenBank under the following accession numbers: DQ096594–DQ096626, DQ097393–DQ097394. ∗ Corresponding author. Tel.: +36 1 467 4069; fax: +36 1 467 4076. E-mail address: [email protected] (B. Lomniczi). 1 These authors participated equally in the work. Genetic analysis of NDV strains whose isolations spanning a period of 70 years has revealed the existence of at least eight genotypes associated with spatio-temporal and host species delineations (Aldous et al., 2003; Czegl´ edi et al., 2002; Herczeg et al., 1999; Huang et al., 2004; Lee et al., 2004; Liu et al., 2003; Lomniczi et al., 1998; Mase et al., 2002; Tsai et al., 2004; Wehmann et al., 2003b). Although certain genogroups showed region-specific occurrence, temporal distribution was also apparent. For instance, there is evidence that in the decades prior to the 1970s, genotypes II and IV were predominant in North America and Europe, respectively (Czegl´ edi et al., 2002, 2003; Locke et al., 2000). On the other hand, in the 1960s, group VI viruses emerged in epizootics in the Middle East and Asia, while in the early 1970s genogroup V (most likely of South American origin) caused outbreaks in Europe (Herczeg et al., 1999; Wehmann et al., 2003a,b). Most recently several sublin- eages of genotype VII emerged in the Far East and spread to other geographic areas (Aldous et al., 2003; Huang et al., 2004; 0168-1702/$ – see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.virusres.2005.11.009

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Virus Research 120 (2006) 36–48
Third genome size category of avian paramyxovirus serotype 1(Newcastle disease virus) and evolutionary implications�
Alız Czegledi a,1, Dorina Ujvari a,1, Eszter Somogyi a, Eniko Wehmann a,Ortrud Werner b, Bela Lomniczi a,∗
a Veterinary Medical Research Institute of the Hungarian Academy of Sciences, P.O. Box 18, Budapest 1581, Hungaryb Institute of Diagnostic Virology, Friedrich-Loeffler-Institutes, Federal Research Centre for Virus Diseases of Animals,
D 17493 Greifswald, Boddenblick 5A, Insel Riems, Germany
Received 24 June 2005; received in revised form 11 November 2005; accepted 11 November 2005Available online 12 June 2006
Abstract
The goal of the study was to establish if there was a relationship between molecular patterns and virus evolution. Therefore the complete genomesequence of two distinct apathogenic Newcastle disease virus (NDV) strains was determined and a third genome size category, containing 15,198ntgvlsw1©
K
1
tgisitwv
a
0d
ucleotides, was recognized. Phylogenetic analysis revealed that two major separations resulting in three genome size categories occurred duringhe history of NDV. An ancient division in the primordial reservoir (wild waterbird species) led to two basal sister clades, class I and II, withenome sizes 15,198 (due to a 12 nucleotide insert in the phosphoprotein gene) and 15,186 nucleotides, respectively. Ancestors of only class IIiruses colonized chicken populations and subsequently converted to virulent forms. These took place more than once and resulted in an earlyineage [including genotypes I–IV and H33(W)] with genome size of 15,186 nucleotides. A second division occurred in the 20th century in theecondary (chicken) host. This gave rise to the branching-off of a clade (including recent genotypes V–VIII consisting of only pathogenic viruses)ith the concomitant insertion of six nucleotides into the 5′ non-coding region of the nucleoprotein gene thereby increasing the genome size to5,192 nucleotides.
2006 Elsevier B.V. All rights reserved.
eywords: Newcastle disease virus; Evolution; Genome size; Phylogenetic analysis
. Introduction
Newcastle disease virus (NDV), an avian paramyxovirus, ishe causative agent of Newcastle disease (ND), a highly conta-ious disease that may result in 100% morbidity and mortalityn chicken flocks (Alexander, 2003). In spite of control mea-ures, including vaccination applied since the 1950s, ND stills a serious problem of the poultry industry. In the past decadehe disease was endemic in 57% of the countries rearing poultryhile further 23% suffered one or several introduction of theirus (OIE).
� Sequences have been deposited in the EMBL/GenBank under the followingccession numbers: DQ096594–DQ096626, DQ097393–DQ097394.∗ Corresponding author. Tel.: +36 1 467 4069; fax: +36 1 467 4076.
E-mail address: [email protected] (B. Lomniczi).1 These authors participated equally in the work.
Genetic analysis of NDV strains whose isolations spanninga period of 70 years has revealed the existence of at leasteight genotypes associated with spatio-temporal and host speciesdelineations (Aldous et al., 2003; Czegledi et al., 2002; Herczeget al., 1999; Huang et al., 2004; Lee et al., 2004; Liu et al.,2003; Lomniczi et al., 1998; Mase et al., 2002; Tsai et al.,2004; Wehmann et al., 2003b). Although certain genogroupsshowed region-specific occurrence, temporal distribution wasalso apparent. For instance, there is evidence that in the decadesprior to the 1970s, genotypes II and IV were predominant inNorth America and Europe, respectively (Czegledi et al., 2002,2003; Locke et al., 2000). On the other hand, in the 1960s, groupVI viruses emerged in epizootics in the Middle East and Asia,while in the early 1970s genogroup V (most likely of SouthAmerican origin) caused outbreaks in Europe (Herczeg et al.,1999; Wehmann et al., 2003a,b). Most recently several sublin-eages of genotype VII emerged in the Far East and spread toother geographic areas (Aldous et al., 2003; Huang et al., 2004;
168-1702/$ – see front matter © 2006 Elsevier B.V. All rights reserved.oi:10.1016/j.virusres.2005.11.009

A. Czegledi et al. / Virus Research 120 (2006) 36–48 37
Lee et al., 2004; Liu et al., 2003; Mase et al., 2002; Tsai et al.,2004).
NDV is the sole member of serotype 1 of avian paramyx-oviruses (APMV-1). Eight other APMV serogroups are rec-ognized whose natural host includes turkeys, ducks or otherspecies (Alexander, 2003). APMVs are classified in genusAvulavirus, family Paramyxoviridae (Mayo, 2002). Paramyx-oviruses possess single-stranded continuous RNA genomeof negative polarity consisting of over 15,000 nucleotides.The genome of NDV contains six genes in the order of3′-NP-P-M-F-HN-L-5′ that code for six major polypeptides(nucleoprotein, phosphoprotein, matrix, fusion, hemagglutinin-neuraminidase and large, respectively) (Lamb and Kolakofsky,2002).
In addition to virulent NDV strains that are hosted by chick-ens, APMV-1 comprises apathogenic viruses as well that origi-nate from two sources. The first group of avirulent (lentogenic)viruses were discovered in chickens in North America togetherwith intermediately virulent (mesogenic) and virulent (velo-genic) strains that form a single genetic lineage designated geno-type II (Aldous et al., 2003; Alexander, 2003; Ballagi-Pordanyet al., 1996; Sakaguchi et al., 1989).
The natural hosts of another group of apathogenic virusesare wild waterbird species living word-wide—their ecologyresembling that of avian influenza virus (Alexander, 1995;Swayne, 2000; Webster et al., 1992). The majority of the lat-ta1BIci1laa
spoKtceivcei
hfNklu
any genetic trait utilisable for delineation above genotypic levelmay be useful in shedding light on the above relationships.One such putative marker, a six nucleotide (nt) insert in the5′ non-coding region (NCR) of the nucleoprotein (NP) gene instrains belonging some recently emerged genotypes has beendescribed (Huang et al., 2004). With this insertion NDV geno-types divided into two genome size categories: 15,192 nt inrecent genogroups (VI–IX) as opposed to 15,186 nt initiallydescribed for genotype II strains (Krishnamurthy and Samal,1998; Romer-Oberdorfer et al., 1999; de Leeuw and Peeters,1999). The evolutionary significance of this trait has remainedunexplored.
The main goal of the current study was to gain insightinto the evolutionary history of APMV-1 strains encompass-ing virus–host relationships from harmless infection to theemergence of disease. For this purpose, the following spe-cific issues were addressed: (i) phylogenetic relationships ofstrains of different virulence and reservoirs; (ii) molecularstructural features utilizable in the grouping above genotypiclevel; and (iii) relationship between genome size classes andthe history of virus–host relationships. To this end, the com-plete nucleotide sequence of the genome of two apathogenicNDV strains belonging to genotype I (vaccine strain PHY-LMV42) and mAb-binding group H or class I (DE-R49/99) weredetermined.
In addition, the 5′NCR of the NP gene of 36 viruses repre-s
2
2
aaeGdcoosftonivfWc
2p
d
er apathogenic isolates were classified into groups G and L bymonoclonal antibody (mAb) binding assay (Alexander et al.,997) and were placed into genotype I (Aldous et al., 2003;allagi-Pordany et al., 1996; Sakaguchi et al., 1989). Genotypestrains, however, were occasionally found in non-immunizedhicken flocks as well. For example, in Australia, where endemicnfection had been maintained for several decades (Westbury,981). Also wild water-birds harbour a highly divergent NDVineage originally designated group H by mAbs (Alexander etl., 1997) or lineage 6 (Aldous et al., 2003) and class I by geneticnalysis (Gould et al., 2003).
The molecular basis for pathogenicity is based on differentubstrate sequences of the F0 precursor protein presented forroteolytic activation: a process indispensable for penetrationf the genome by fusion (Rott and Klenk, 1988; Lamb andolakofsky, 2002). Viruses with monobasic amino acid motif at
he F0 cleavage site are apathogenic because this substrate is sus-eptible only to extracellular trypsin-like proteolytic enzymesxcreted onto the surface of mucous membranes therefore thenfection remains localized and asymptomatic. By contrast,iruses with dibasic motif are pathogenic because this sequencean be activated by ubiquitously present intracellular proteolyticnzymes as well therefore these viruses can elicit generalizednfection in the body.
As to the ecology of APMV-1 strains apparently two majorost systems exist in nature: (i) the wild waterbird species mainlyor apathogenic strains; (ii) chickens, the hosts of pathogenicDV (Alexander, 1995; Awan et al., 1994). However, ournowledge on the evolutionary relationships of APMV-1 virusineages residing in the different reservoirs and those of avir-lent and virulent phenotype is especially limited. Therefore
enting different genotypes was analysed.
. Materials and methods
.1. Viruses
Data of strains whose complete genome was sequenced weres follows. DE-R49/99 was isolated from ducklings in Germanynd identified as group H by mAbs (Dr. O. Werner, National Ref-rence Laboratory for Newcastle Disease Virus, Insel Riems,ermany). PHY-LMV42 is a genotype I apathogenic strain thaterived from a commercial vial used for the production of vac-ine Vitapest (CEVA Sante Animale, Budapest). IT-227/82 isne of the first pigeon PMV-1 strains isolated in Italy at thenset of the epizootic (Ujvari, 2006). Strains, whose partialequences were used in Figs. 1 and 3 (underlined) were obtainedrom the collection of the Veterinary Medical Research Insti-ute, Budapest. From the two novel strains, US-Coot/80s wasbtained from Zagreb (Dr. V. Savic, Poultry Centre, Veteri-ary Institute, Zagreb, Croatia) but was isolated in the 1980sn the United States (originally came from Dr. J. Spalatin, Uni-ersity of Wisconsin, WI); strain DE-WV254K/01 was isolatedrom a shorebird (Calidris alpina) and obtained from Dr. O.
erner. Designations: two-letter country code—local identifi-ation/year.
.2. Preparation of viral RNA, reverse transcription,olymerase chain reaction
These procedures were performed without modification asescribed (Czegledi et al., 2002; Ujvari et al., 2003).

38 A. Czegledi et al. / Virus Research 120 (2006) 36–48
Fig. 1. Phylogenetic tree of representative NDV strains depicting major divisions of classes, sublineages and genotypes. The tree is based on comparison of partialF gene sequence (between nt positions 47 and 420). Viruses whose NP 5′NCR sequences were obtained in the current study are underlined. Complete genomesequences submitted to the EMBL/GenBank (Table 1) but no information is published on the six nt insert are marked by (*). Representative sequences in whichthe presence or absence of the six nt insertion has been reported (Huang et al., 2004) are marked by (+). Avirulent strains are boxed, of which those residing in theprimordial reservoir are set in bold. The putative place of origin of genotypes and examples of resultant epizootics elsewhere (→ in italics) are highlighted on theright.
2.3. Amplification of the 5′ non-coding region of the NPgene by RT-PCR
Primers were selected by the Oligo 5.0 computer pro-gram (National Biosciences Inc., Plymouth, MN, USA). A1631 bp PCR product was amplified with primers NPPf2: 5′TAT CTA TCC AGG CTC AGR TAT GGG TCA CAG 3′ (nt563–592 of the NP gene) and NPPr2: 5′ GAC ARA AGAGAG TTG CTT GCT CCG GTC TTG 3′ (nt 410–439 ofthe P gene). PCR reaction was carried out by Cloned Pfu
DNA Polymerase (Stratagene, CA, USA) according to themanufacturer’s instructions. The annealing temperature was60 ◦C.
2.4. Complete genome amplification of the DE-R49/99 andPHY-LMV42 virus strains by RT-PCR
RT-PCR was conducted with SuperScript II RNase H−Reverse Transcriptase (Invitrogen, Carlsbad, CA), RibonucleaseH (Promega, Madison, WI, USA) and PfuTurbo DNA Poly-

A. Czegledi et al. / Virus Research 120 (2006) 36–48 39
merase (Stratagene, CA, USA), according to the manufacturer’sinstructions. The genomes were amplified in five overlappingportions. Three specific primers were used for the RT andthree primer pairs for the amplification of the inner regionsencompassing 94% of the genome. (All PCR conditions andRT-PCR primers used are available from the authors uponrequest.)
The sequences of the 3′- and 5′ termini of the viral genomewere amplified by 3′- and 5′ RACE. 3′-RACE was carried outby genomic RNA ligation. 250 ng of a DNA oligo was ligatedto the 3′ end of the purified viral RNA (5 �l) in a 40 �l reac-tion mix containing 50 mM Tris–HCl (pH 7.8), 10 mM MgCl2,5 mM DTT, 1mM ATP, 40U RNasin Ribonuclease Inhibitor(Promega, Madison, WI, USA), 20 �l of 40% PEG and 20Uof T4 RNA Ligase (Promega, Madison, WI, USA) at 37 ◦C for30 min. Then the enzyme was denatured at 96 ◦C for 3 min,and the ligated product was precipitated by ethanol and dis-solved in 20 �l double-distilled (dd) DEPC-treated H2O. Fivemicrolitres of the ligated product was used as a template for RT-PCR. The reaction was conducted with SuperScript II RNaseH− Reverse Transcriptase (Invitrogen, Carlsbad, CA), Ribonu-clease H (Promega, Madison, WI, USA) and Cloned Pfu DNAPolymerase (Stratagene, CA, USA), according to the manufac-turer’s instructions. The cDNA and the PCR forward primerwere complementary to the DNA oligo, while the reverse primerwas specific for the virus NP gene. 5′-RACE was carried oututwRoatpPfAC
2.5. Cloning of the amplified products
DNA fragments were extracted from agarose gel. Thenthe PCR products were treated by 2U AmpliTaq DNA Poly-merase (Applied Biosystems Inc., Foster City, CA), for 15 minat 72 ◦C, to introduce 3′ adenine ends. Amplification productswere cloned into the pCR4-TOPO or the pCR-XL-TOPO vec-tor (Invitrogen, Carlsbad, CA) then recombinant plasmids werepurified using the QIAamp Viral RNA Mini Kit (Qiagen, Valen-cia, CA), as per the manufacturer’s protocol.
2.6. Sequence analysis
DNA sequences were determined at Genotype GmbH,Hirschhorn, Medigenomix GmbH, Martinsried/Munchen, Ger-many and at the Agricultural Biotechnology Centre, Godollo,Hungary. In case of the complete genome sequencing, primer-walking technology was used. Data and accession numbersof complete genome sequences of NDV strains that were uti-lized in the present study are presented in Table 1. Addi-tional complete L gene sequences used in Fig. 2 were takenfrom the EMBL/GenBank (Wise et al., 2004). Thirteen novelF gene (between nt positions 47–420) and twenty NP 5′NCR(between nt position 593 of the NP and nt 409 of the Pgene) sequences determined in this study and used (under-ltd2sCSBabTmf
TD
D Ac
C DQAYAYNCAFY1AYAYAYAYAYAJAFNC
C DQ
sing the 5′-RACE kit (Invitrogen, Carlsbad, CA) accordingo the manufacturer’s instruction. In brief, first strand cDNAas synthesized by SuperScript II Reverse Transcriptase andNase mix using a primer, which is specific to the L genef the virus strain. The cDNA was purified by GlassMAX,nd a polyC tail was added to the 3′ end of the cDNA usingerminal deoxynucleotidyl transferase (TdT) and dCTP. TheolyC-tailed cDNA was then used as a template for PCR. TheCR was carried out with a forward primer, which was specificor the virus L gene and an abridged anchor reverse primer bympliTaq DNA Polymerase (Applied Biosystems Inc., Fosterity, CA).
able 1ata of the complete genome of NDV strains
ivision Genotype Virus strain
lass II I PHY-LMV42/66AU-1334/02Ulster 2C/67
II B-1/46LaSota/46Clone 30
IV Herts/33 (deLeeuw)V US(FL)/Largo/71
US(CA)/211472/02US(FL)/44083/93
VI US(CA)/1083Fontana/72IT-227/82
VII CN/ZJ-1/00CN/SF-1/02
lass I DE-R49/99
ined) in Figs. 1 and 3, respectively, have been deposited inhe EMBL/GenBank. Source of other NDV sequences wasescribed previously (Huang et al., 2004; Czegledi et al., 2002,003; Ujvari et al., 2003 and references therein). Publishedequences of APMV-6 (GenBank accession no. AY029299,hang et al., 2001), APMV-2 (GenBank accession no. D13977,ugita et al., 2000, unpublished results) and APMV-3b (Gen-ank accession no. AY137207, Shihmanter et al., 2005) werelso used for comparison. The sequence data were alignedy the MegAlign program in the Lasergene package (DNAS-AR Inc., Madison, WI 53715, USA), using the CLUSTAL Wultiple alignment algorithm. Phylogenetic analysis was per-
ormed using the TREECON for Windows 1.3b software (Van
cession number References
097394 Current study935490 Gould and Kattenbelt (2005), unpublished results562991 Wise et al. (2004)002617 Sellers and Seal (2005), unpublished results077761 de Leeuw and Peeters (1999)8898 Romer-Oberdorfer et al. (1999)741404 deLeeuw et al. (2005)562990 ⎫⎬
⎭ Wise et al. (2004)562987562986562988880277 Ujvari (2006)431744 Huang et al. (2004)005036 Zou et al. (2005)
097393 Current study
40 A. Czegledi et al. / Virus Research 120 (2006) 36–48
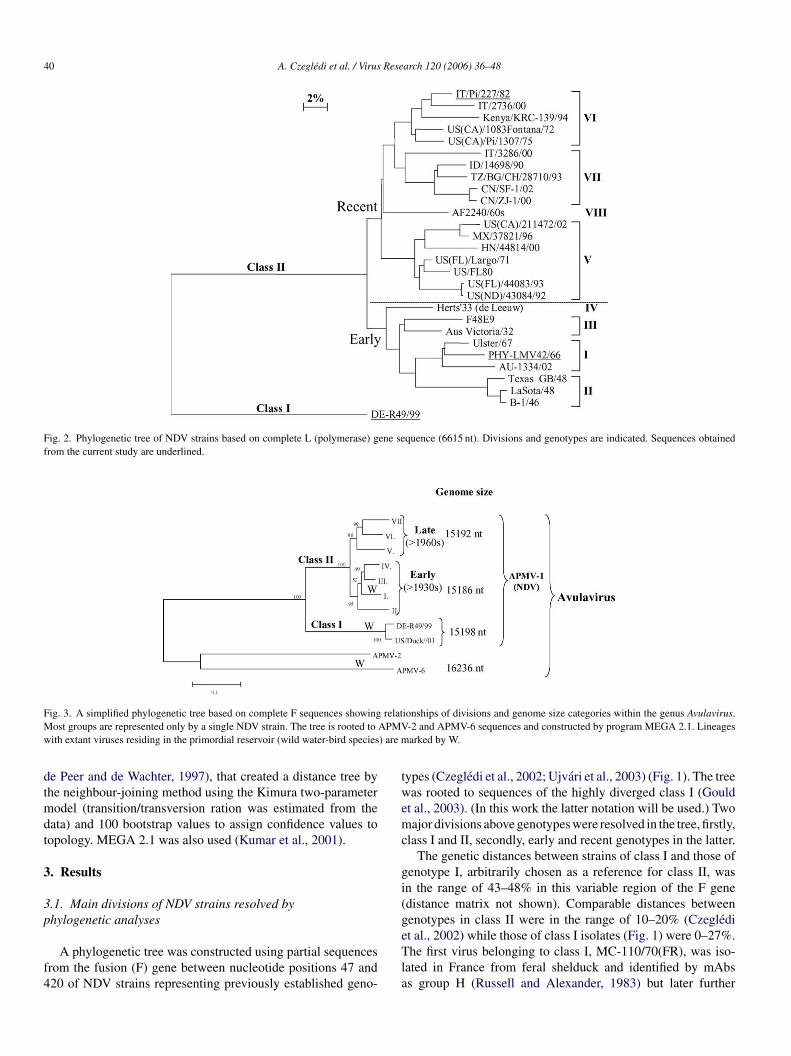
Fig. 2. Phylogenetic tree of NDV strains based on complete L (polymerase) gene sequence (6615 nt). Divisions and genotypes are indicated. Sequences obtainedfrom the current study are underlined.
Fig. 3. A simplified phylogenetic tree based on complete F sequences showing relationships of divisions and genome size categories within the genus Avulavirus.Most groups are represented only by a single NDV strain. The tree is rooted to APMV-2 and APMV-6 sequences and constructed by program MEGA 2.1. Lineageswith extant viruses residing in the primordial reservoir (wild water-bird species) are marked by W.
de Peer and de Wachter, 1997), that created a distance tree bythe neighbour-joining method using the Kimura two-parametermodel (transition/transversion ration was estimated from thedata) and 100 bootstrap values to assign confidence values totopology. MEGA 2.1 was also used (Kumar et al., 2001).
3. Results
3.1. Main divisions of NDV strains resolved byphylogenetic analyses
A phylogenetic tree was constructed using partial sequencesfrom the fusion (F) gene between nucleotide positions 47 and420 of NDV strains representing previously established geno-
types (Czegledi et al., 2002; Ujvari et al., 2003) (Fig. 1). The treewas rooted to sequences of the highly diverged class I (Gouldet al., 2003). (In this work the latter notation will be used.) Twomajor divisions above genotypes were resolved in the tree, firstly,class I and II, secondly, early and recent genotypes in the latter.
The genetic distances between strains of class I and those ofgenotype I, arbitrarily chosen as a reference for class II, wasin the range of 43–48% in this variable region of the F gene(distance matrix not shown). Comparable distances betweengenotypes in class II were in the range of 10–20% (Czeglediet al., 2002) while those of class I isolates (Fig. 1) were 0–27%.The first virus belonging to class I, MC-110/70(FR), was iso-lated in France from feral shelduck and identified by mAbsas group H (Russell and Alexander, 1983) but later further

A. Czegledi et al. / Virus Research 120 (2006) 36–48 41
isolations were made from wild water-birds in the region ofthe North Sea, the Far East, Australia, New Zealand (Aldouset al., 2003; Collins et al., 1998; Gould et al., 2003) and theUnited States as indicated by isolate US-Coot/80s, analysedhere. Two strains that were isolated in live bird markets (LBM)in the United States (Seal et al., 2005), the sequences of whichare also used here for comparison, as well as two strains thatwere obtained from Germany: DE-R49/99 from ducklings andDE-WV254K/01 from a shorebird.
Class II contains eight established genotypes each linkedto well-defined epizootics described previously (Aldous et al.,2003; Czegledi et al., 2002; Herczeg et al., 1999; Huang et al.,2004; Lee et al., 2004; Liu et al., 2003; Lomniczi et al., 1998;Mase et al., 2002; Tsai et al., 2004; Ujvari et al., 2003; Wehmannet al., 2003a,b). However, a major separation divided class II intotwo sublineages, which was supported by 100% bootstrap value.One of these, termed early, comprised genetic groups I–IV andH33(W) [named after its single member, strain Herts’33(W)].These groups included all the available virulent strains in classII known to be responsible for early ND outbreaks with datesof isolation going back as early as the 1930s. Examples includeCAL-11914/43 (isolated in California in 1943) in genotype II(Czegledi et al., 2003), JP-Sato/30 (Japan, 1930; Mase et al.,2002), AUS-Victoria/32 in genotype III and Italien(Milano)/45in genotype IV. Extant avirulent APMV-1 strains (boxed inFig. 1) are contained only in genotypes I and II. Genotype Ico(si1o(vw(lisslhshawtbGgb
cni1
VI, VIIa and VIII, respectively (Aldous et al., 2003; Czegledi etal., 2002).
The above separations between NDV geno-groups were con-firmed by phylogenetic analysis performed with the other fivegenes of the virus (not shown). Only the analysis based onthe most conservative region, the polymerase gene L that com-prises 43% of the genome (coding region 6615 nt) is shown herefor comparison (Fig. 2). Though only a limited number of fullsequences of the L gene are available, the tree has indicated verysimilar relationships of genetic groups to that of the relativelyvariable portion of the F gene.
3.2. A shared derived character delineating early andrecent NDV genotypes of class II
Initially, based on entire genome sequence, 15,186 ntwas reported for several strains belonging to genotype II(Krishnamurthy and Samal, 1998; Romer-Oberdorfer et al.,1999; de Leeuw and Peeters, 1999). The second size category,15,192 nt, was also based on the entire genome sequence of agenotype VII virus isolated from geese in China (Huang et al.,2004). The increase was due to six nt insertion after nt position1647 in the 5′NCR of the NP gene, the presence of which wasascertained in 16 strains (belonging to groups VI–VIII and IX,the latter comprising strains only from China) but it was absentin six viruses belonging to genotypes I, II, IV and V.
teStrtiicrhnI1a(stgie
ttTetPt[
omprise viruses that reside either in world-wide populationsf feral waterbirds (bold setting) or occasionally in chickensAldous et al., 2003; Gould et al., 2003; Westbury, 1981). Oneuch avirulent virus, which was similar to those that had endem-cally infected chicken flocks in Australia acquired virulence in998 (Gould et al., 2001, 2003; Westbury, 2001). The sequencef a derived velogenic strain, AU-1334/01, is shown in the tree.It was chosen as a representative of 10 very closely relatedirulent Australian isolates whose complete genomic sequencesere deposited in the EMBL/GenBank by Gould and Kattenbelt
2005), unpublished results; Table 1.) Interestingly, genotype IIentogenic strains were found only in chickens in North American the 1940s and have been used as live vaccines (LaSota, B-1)ince (Alexander, 2003). As one of the earliest known isolates,train Herts’33, is a historically important NDV strain: it was iso-ated in 1933 from an outbreak in Hertfordshire (England) andas been maintained in the Addlestone (Weybridge) laboratoryince then (Russell and Alexander, 1983). The isolate designatedere Herts’33(W) is the sole representative of an extinct groupnd its identity with that of the original (Weybridge) isolateas documented previously (Czegledi et al., 2003). We believe
hat the authenticity of other strains bearing similar notation butelonging to genotype IV [e.g. Herts/33 (de Leeuw et al., 2005,enBank accession no. AY741404) or HER/33 of unknown ori-in (Toyoda et al., 1989, GenBank accession no. M24702)] cane questioned.
The other major sublineage of class II, referred to as recent,omprised genotypes V–VIII with isolation dates of membersot earlier than the mid-1960s. For example, the earliest isolatesnclude: Essex’70 (UK, 1970), Warwick’66 (UK, 1966), ID-/88 (Indonesia) and ZA-5/68 (South Africa) in genotypes V,
In order to gain complementary information independent ofree analysis on the relationships of the early and recent sublin-ages, the insert was examined in a number of additional strains.equences between nt position 593 of the NP and nt 409 of
he P gene of 23 novel viruses (9 in the early and 14 in theecent groups) were determined in the current study while fur-her sequences previously not evaluated for the presence of thensert were taken from the EMBL/GenBank (sources are shownn Table 1). In general, the absence or presence of the characterorresponded with the division to early and recent genotypes,espectively. Additional new data were as follows. On the oneand, representatives of two early genotypes III and H33(W),ot examined previously, as well as further viruses of groups–II and IV equally lacked the 6 nt insert thereby predicting a5,186 nt long genome size for these groups. Consistent with thebove, a rare present-day survivor of one of the early genotypesIV), MA-13/02, isolated in 2002 in Morocco, equally lacked theix nt insert (Fig. 1). In addition, the entire genome sequence ofhe vaccine strain PHY-LMV42/66 confirmed the placement ofenotype I viruses in the 15,186 nt size category. It was of specialnterest that the virulent descendants of the Australian avirulentndemic strains equally had this size (Table 1).
On the other hand, it was an unexpected finding that unlikeo that reported by Huang et al. (2004), genotype V strains,oo, possessed the six extra nt in the 5′NCR of the NP gene.he finding was based on 5 additional sequences of genotype Vxamined here. Thereby the combined size of the 5′-NCR plushe downstream intergenic region (IGR) between the NP and
genes has been increased from 212 to 218 nt, consequentlyhe predicted genome size from 15,186 to 15,192 nt (Table 2).It has to be noted that in some instances the poly-A of the
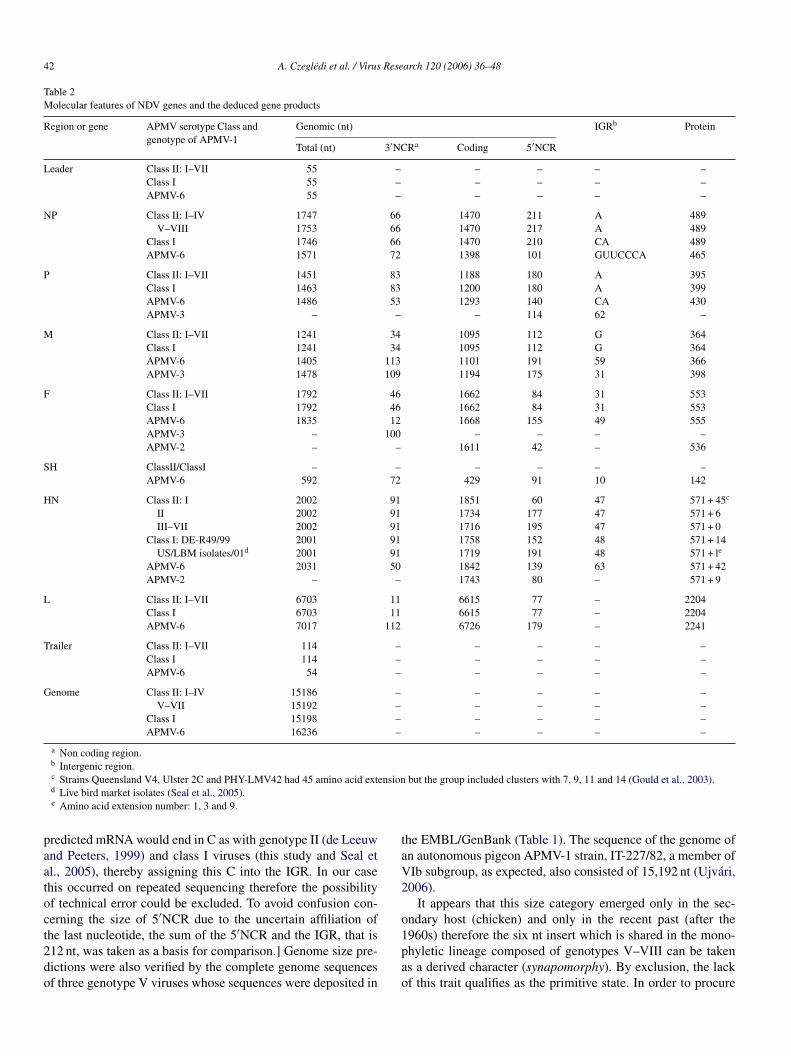
42 A. Czegledi et al. / Virus Research 120 (2006) 36–48
Table 2Molecular features of NDV genes and the deduced gene products
Region or gene APMV serotype Class andgenotype of APMV-1
Genomic (nt) IGRb Protein
Total (nt) 3′NCRa Coding 5′NCR
Leader Class II: I–VII 55 – – – – –Class I 55 – – – – –APMV-6 55 – – – – –
NP Class II: I–IV 1747 66 1470 211 A 489V–VIII 1753 66 1470 217 A 489
Class I 1746 66 1470 210 CA 489APMV-6 1571 72 1398 101 GUUCCCA 465
P Class II: I–VII 1451 83 1188 180 A 395Class I 1463 83 1200 180 A 399APMV-6 1486 53 1293 140 CA 430APMV-3 – – – 114 62 –
M Class II: I–VII 1241 34 1095 112 G 364Class I 1241 34 1095 112 G 364APMV-6 1405 113 1101 191 59 366APMV-3 1478 109 1194 175 31 398
F Class II: I–VII 1792 46 1662 84 31 553Class I 1792 46 1662 84 31 553APMV-6 1835 12 1668 155 49 555APMV-3 – 100 – – – –APMV-2 – – 1611 42 – 536
SH ClassII/ClassI – – – – – –APMV-6 592 72 429 91 10 142
HN Class II: I 2002 91 1851 60 47 571 + 45c
II 2002 91 1734 177 47 571 + 6III–VII 2002 91 1716 195 47 571 + 0
Class I: DE-R49/99 2001 91 1758 152 48 571 + 14US/LBM isolates/01d 2001 91 1719 191 48 571 + le
APMV-6 2031 50 1842 139 63 571 + 42APMV-2 – – 1743 80 – 571 + 9
L Class II: I–VII 6703 11 6615 77 – 2204Class I 6703 11 6615 77 – 2204APMV-6 7017 112 6726 179 – 2241
Trailer Class II: I–VII 114 – – – – –Class I 114 – – – – –APMV-6 54 – – – – –
Genome Class II: I–IV 15186 – – – – –V–VII 15192 – – – – –
Class I 15198 – – – – –APMV-6 16236 – – – – –
a Non coding region.b Intergenic region.c Strains Queensland V4, Ulster 2C and PHY-LMV42 had 45 amino acid extension but the group included clusters with 7, 9, 11 and 14 (Gould et al., 2003).d Live bird market isolates (Seal et al., 2005).e Amino acid extension number: 1, 3 and 9.
predicted mRNA would end in C as with genotype II (de Leeuwand Peeters, 1999) and class I viruses (this study and Seal etal., 2005), thereby assigning this C into the IGR. In our casethis occurred on repeated sequencing therefore the possibilityof technical error could be excluded. To avoid confusion con-cerning the size of 5′NCR due to the uncertain affiliation ofthe last nucleotide, the sum of the 5′NCR and the IGR, that is212 nt, was taken as a basis for comparison.] Genome size pre-dictions were also verified by the complete genome sequencesof three genotype V viruses whose sequences were deposited in
the EMBL/GenBank (Table 1). The sequence of the genome ofan autonomous pigeon APMV-1 strain, IT-227/82, a member ofVIb subgroup, as expected, also consisted of 15,192 nt (Ujvari,2006).
It appears that this size category emerged only in the sec-ondary host (chicken) and only in the recent past (after the1960s) therefore the six nt insert which is shared in the mono-phyletic lineage composed of genotypes V–VIII can be takenas a derived character (synapomorphy). By exclusion, the lackof this trait qualifies as the primitive state. In order to procure

A. Czegledi et al. / Virus Research 120 (2006) 36–48 43
further information on the possible ancestry of this trait, thehighly diverged class I was also examined. Due to the exces-sive divergence between class I and II viruses, especially in theuntranslated regions, the sequences encompassing the positionof the insert of strain DE-R49/99 and the three US LBM-isolatesof class I were aligned manually using end nucleotides and thegap as reference points (not shown). The combined length of NP5′NCR and IGR consisted of only 212 nt similarly to viruses inthe early genotypes of class II.
3.3. A third genome size class of APMV-1 and phylogeneticrelationships of the genome size categories
While the manuscript was being prepared it was reported thatsome viruses belonging to class I clade, isolated in LBMs in theUnited States, possessed a 12 nt insert in the coding region ofthe P gene after positions 2381 but a large part of the L genesequence and the 3′ leader region were not determined (Seal etal., 2005).
Based on complete sequence, the genomic RNA of strainDE-R49/99 consisted of 15,198 nt (Tables 1 and 2), which alsocomplied with the ‘rule of six’ (Kolakofsky et al., 1998). Withthis, class I APMV-1 viruses have been recognized as a thirdgenome size group, in addition to 15,186 nt of the early [I–IVand H33(W)] and 15,192 nt of the recent (V–VIII) genotypes ofclass II. The presence of a 12-nt insert, TGG GAG ACG GGG,ictWa
ugtrscaPr1l
3
uooT62rrst
Apart from the two insertions no other supra-group charac-ters of contiguous sequence were found. There were, however,molecular motifs that could be utilized as genetic markers totrack phylogeny at different levels and/or on varying timescales.Of these we have focussed on those that qualified as sharedderived characters of recent genotypes. For example, upon ear-lier characterization of NDV strains with restriction enzymes(RE) based on a 75% portion (1349 nt) of the F gene, RE cleav-age sites HinfI 1064, BstOI 1260 and 1478 as well as the loss ofRsaI 872 shared only among the recent genotypes V–VIII wererevealed (Ballagi-Pordany et al., 1996; Herczeg et al., 1999;Czegledi et al., 2002; Wehmann et al., 2003b). It is noteworthythat none of these sites occurred in class I viruses (not shown).Likewise, looking at the F polypeptide, six positions, partlyconservative (S139A, Q195R, T341S) partly non-conservative(E104G, T288N, E482A), displayed substitutions in which thederived amino acids were common in the recent geno-groupswhereas the primitive ones in the early genotypes (I–IV). Andagain, the latter were also shared with those of class I strains(not shown).
The size of HN of NDV strains is highly variable due to theposition of the stop codon, which gave rise to different sizesof the predicted protein product, the shortest one consisting of571 amino acids (Sakaguchi et al., 1989; Gould et al., 2003).Analysing the HN sequences of several dozen of strains, thisbasic value was found in geno-groups III–VIII consisting exclu-s6lLladoF(sr1s
o(aWatisutappKms
n the coding region of the P gene of strain DE-R49/99 has beenonfirmed (not shown). This sequence was identical with that ofhe US LBM-isolates and the predicted amino acid sequence was
ETG (Seal et al., 2005). The insertion occurred after aminocid position 165 in the P gene.
When an outline tree of the genus Avulavirus was constructedsing only a single full F gene sequence representing eachenotype and APMV-2 and APMV-6 for rooting the tree, thehree genome size delineations of APMV-1 were again clearlyesolved (Fig. 3). The branching pattern of the tree reflectedimilar asymmetry with respect to genetic distances of the sizeategories as that of other trees: The most basal division broughtbout class I with genome size of 15,198 nt (15,186 + 12 in the
gene) and class II. In the latter a contemporary separationesulted in two further size categories: 15,186 nt of early and5,192 nt (15,186 + 6 in the 5′NCR of NP gene) of recent sub-ineages.
.4. Other molecular features associated with grouping
Features such as length of genes, predicted gene products andntranslated regions have been utilized in grouping of paramyx-viruses (Chang et al., 2001; Lamb and Kolakofsky, 2002). Somef these data are listed for the three genome size lineages inable 2. Data of a fully sequenced avian paramyxovirus (APMV-) and individual genes of APMV-2 and APMV-3 (Chang et al.,001; Shihmanter et al., 2005; Sugita et al., 2000, unpublishedesults) are also presented for comparison. There was a cor-espondence between the antigenic property of HN, on whicherotype distinction of APMVs is based (Alexander, 2003), andhe gross structural features shown in Table 2.
ively of viscerotropic velogenic strains. Genotype II strains hadamino acids extension regardless of whether the strain was
entogenic, mesogenic or velogenic (Sakaguchi et al., 1989).ikewise, both the putative avirulent progenitor and its viru-
ent derivatives of genotype I from Australia shared nine aminocid extension (Gould et al., 2003). On the other hand, viruseserived from the primordial reservoir, whether belong to class Ir class II, displayed a great diversity of sizes of the extension.or example, in genotype I: 7, 9, 11, 14 and 45 amino acidsGould et al., 2003). PHY-LMV42/66 belonged to the 45 exten-ion category. In class I viruses: whereas values −1 and 45 wereeported for isolates derived in Australia (Gould et al., 2003),, 3 and 9 for three recent US LBM-isolates (Seal et al., 2005),train DE-R49/99 displayed a sixth version, 14.
A substantial variation of the proteolytic cleavage site motifsf the F0 protein exists for both avirulent and virulent strainsCollins et al., 1993; Gould et al., 2003). Typical monobasicnd dibasic sequences, respectively, are presented in Table 3.
hereas two groups (class I and genotype I of class II) ofpathogenic viruses with monobasic cleavage site motifs (primi-ive state) are residing in the primordial reservoir or occasionallyn chickens, those of genotype II appear to be established exclu-ively in chickens. All class I strains of low virulence examinedp to now (Aldous et al., 2003; Collins et al., 1993) includinghe three recent US LBM-isolates (Seal et al., 2005) as wells DE-WV254K/01 and US-Coot/80s in the current work dis-layed the motif 112ERQER↓LVG119, only strain DE-R49/99ossessed 112GRQGR↓LVG119 instead. A unique substitution,113R, distinguished genotype I from II and rendered theirotifs group-specific. By contrast, strains with dibasic cleavage
ite motifs (derived state) occur in all geno-groups of which,

44 A. Czegledi et al. / Virus Research 120 (2006) 36–48
Table 3F gene proteolytic cleavage site sequences of NDV strains in the different reservoirs
aCollins et al. (1993, 1998); bGould et al. (2003). ( ) Colonization of chickens, conformed; ( ) colonization of chickens, inferred; ( ) virulence conversion,observed; ( ) virulence conversion, assumed.
however, those of class I, genotype I and II seem to representacquisitions independent from each other and the remainder. Thedibasic sequence of genotypes III and IV differed from that ofV–VIII by an R115K substitution. Individual intragroup varia-tions such as an I118V change in genotype V or substitutionsR112G and Q114K in some subgroups of pigeon adapted (VIb)viruses (not shown) made the picture varied (Werner et al., 1999;Ujvari et al., 2003).
4. Discussion
The main goal of the present study was to address the hypoth-esis that Newcastle disease as we see it today is the result of ahistorical process. Therefore, in addition to phylogenetic anal-yses of NDV strains some molecular features of the virus werealso examined using cladistic approach. Furthermore, in orderto infer the order of events that have led to the present situation,issues independent from phylogeny, namely, association withhost and formation of reservoirs were also considered.
The salient findings of the current work involved the recogni-tion of a third genome size category and two principal divisionsin the history of APMV-1, which corresponded with these cate-gories.
The full-length sequence of the genomic RNA of a classI strain (DE-R49/99) consisted of 15,198 nt. Initially 15,186wI1sgTtfiaVg
t
minated some of the phases which both the virus and itsinteraction with the hosts presumably passed through duringevolution.
A contemporary case of virulence transition has providedcompelling evidence for the temporal relationship of two evo-lutionary coupled states of the virus. The emergence of virulentNDV strains from local apathogenic viruses in the late 1990s inAustralia (Gould et al., 2001, 2003) can serve as a model for theemergence of ND in the field. The incidence was preceded bya long (but unknown) period of infection of immunologicallynaıve chicken populations by genotype I apathogenic strainswhose close relatives are prevalent in wild aquatic birds in theregion. The period of endemic infection, only discovered inthe mid-sixties (Westbury, 1981) partly overlapped the sharpincrease of poultry production (from 20 to 80 million chickensbetween 1960 and 1998) in Australia (FAO). It can be concludedthat the observed direction of virulence change under field con-ditions is consistent with the notion that the dibasic cleavagesite sequence motifs of the F gene is a derived state therefore,by exclusion, the monobasic is the primitive one.
Knowledge as regards the timing of the emergence of NDderives from information on the association of virus with thehost and on the formation of host system capable of maintain-ing chains of infection. Although infection with APMV-1 couldbe indicated in more than 200 bird species (Kaleta and Baldauf,1988), whether or not these can serve as reservoirs has remaineduabr(cisca
fe
as reported for several NDV strains belonging to genotypeI (Krishnamurthy and Samal, 1998; Romer-Oberdorfer et al.,999; de Leeuw and Peeters, 1999) but recently a genotype IVtrain designated Herts/33 (de Leeuw et al., 2005) and furtherenotype I viruses have been added to this category (Table 1).he second size category, 15,192 nt, was originally based on
he entire genome sequence of a genotype VII virus isolatedrom geese in China. The increase in length was due to a six ntnsert in the 5′NCR of the NP gene (Huang et al., 2004). Lately,nother goose isolate (Zou et al., 2005) and a pigeon type groupIb strain (Ujvari, 2006) have been added to the list of entireenome sequences.
The correspondence displayed among molecular patterns,ree branching and virus–host (reservoir) associations have illu-
nclear. Reservoir is defined as “an ecological system in whichn infectious agent survives indefinitely” (Ashford, 2003). It cane assumed that only two such host systems exist; namely, natu-al communities of wild water-bird species constitutes an ancientprimordial) reservoir for apathogenic strains whereas the oneonsisting of chickens, created “artificially” during civilization,s the scene for virulence conversion and maintenance of virulenttrains. It is to be noted that a number of parallel contemporaryases have been reported with different avian influenza A virusess well (Capua and Alexander, 2004).
The virus inhabitants of these reservoirs adopted radically dif-erent life styles. In the primary host primitive forms of virusesxist in symptomless infection. Their long-term survival has

A. Czegledi et al. / Virus Research 120 (2006) 36–48 45
been achieved through a minimal program of infectivity, whichis based on restricted cleavability of the F0 polypeptide dueto the presence of monobasic substrate for extracellular prote-olytic enzymes. The cleavage is limited only to a single cycle andfusion takes place only between the virus and the cell membrane(Rott and Klenk, 1988; Lamb and Kolakofsky, 2002).
Historically apathogenic inhabitants of the natural reservoircould be responsible for seeding the man-made (or secondary)host system. But even the earliest estimate of the emergenceof pathogenic NDV strains reflects a relatively short period ofvirus–host association because domestication of chickens canbe put to approximately 5000 b.c. (Fumihito et al., 1996). Evenso, one can conclude that many early colonization events couldfail to end in disease because for stable chain of infection to bemaintained by a virus with such a highly adverse effect on itshost, chicken populations had to reach a critical mass in bothflock size and area density. In the chicken host, viruses thatacquired dibasic substrate sequence susceptible for ubiquitouslypresent intracellular proteolytic enzymes have as well widenedtheir tropism through cell-to-cell fusion that resulted in systemicinfection, the basis of pathogenicity (Rott and Klenk, 1988).By now, the secondary reservoir, the largest single species hostsystem amounting to some 16 billion chickens in the world,in spite of the high pathogenicity of the inhabitants, maintainsseveral genetic lineages (Aldous et al., 2003; Czegledi et al.,2002; Herczeg et al., 1999; Huang et al., 2004; Lee et al., 2004;Le
trnc
rpttacg
Fpg
Iomc
admttm
in class I viruses can be treated as an autapomorphy, a strongmarker of this particular lineage.
The other major division could have taken place as recentlyas the 20th century in the secondary host giving rise to thebranching-off of a recent clade (genotypes V–VIII) from theearly [I–IV and H33(W)] lineage with a concomitant insertionof six nt into the 5′NCR of the NP gene.
Other studies have arrived at different conclusions as to theseparations above genotypic level. Phylogenetic analyses of theM, P and L gene sequences classified NDV strains into twomajor clades, one of which included viruses isolated in the USprior to 1970, while the other comprised virulent strains circu-lating worldwide (Locke et al., 2000; Seal et al., 2000; Wise etal., 2004). Interestingly, strain Australia Victoria/32 was singledout as the possible progenitor for this second clade. In our stud-ies the first clade corresponded to genotype II, while AustraliaVictoria/32 was placed in geno-group III (Lomniczi et al., 1998).By applying sequences of the attachment proteins (F and HN),however, four potential clusters were created by either distance,maximum likelihood or parsimony analysis (Seal, 2004). (Forconvenience, genotype designations corresponding to those inFig. 1 are also given.) These are as follows: (i) Old (prior to1970) lentogenic US isolates (genotype II in our grouping); (ii)old mesogenic and velogenic US isolates (genotype II); (iii)apathogenic isolates outside the US (genotype I); and (iv) allhighly virulent strains worldwide (genotypes III–VIII). It is tobe(reIa2
sbciuietfds
togtdstbss
iu et al., 2003; Lomniczi et al., 1998; Mase et al., 2002; Sealt al., 2000; Tsai et al., 2004; Wehmann et al., 2003b).
Phylogenetic analyses of extant NDV strains have revealedwo major separation events during the history of APMV-1esulting in three genome size classes (Fig. 3) and the distinct-ess of these supragroup delineations is supported by derivedharacters as well.
An ancient division occurred in the primordial reservoiresulting in two sister clades, class I and II, and, most likely,redated the genesis of pathogenic viruses. It is assumed thathe life strategy of the extant apathogenic viruses reflects that ofhe ancient members. In a way it is surprising, however, that thencient reservoir harbours only two major apathogenic APMV-1lades. Several arguments can be presented to support the sug-estion that this is an ancient separation.
First; the exceptionally high genetic distance (∼35% for fullgene) between these lineages is consistent with a much longer
eriod of association with the primary host than that of any otherenotypes (I–VIII) with the secondary (chicken) host.
Second; both of the highly diverged avirulent lineages (classand genotype I of class II) lack the six nt insert in the 5′NCRf NP gene, a shared derived character (synapomorphy) of aonophyletic group consisting of recent epizootic viruses in
hickens.Third; besides being distinguished by a plethora of single
mino acid substitution (not shown), they most conspicuouslyiffer in a four amino acid in-del in the P gene. In the lack ofore deeply branched-off NDV lineages, there is no way of
elling whether the presence or absence of the P gene insert ishe primitive state. If the former were the case, it could be sur-
ised that genotype I has lost this trait. Nevertheless its presence
e noted that the discrepancy is most likely due to the differ-nce in analysis. In trees, basic separation of class II genotypesI–VIII) often occurs between genotype II and the remainderegardless of the mode of analysis (Czegledi et al., 2002; Ujvarit al., 2003, and references therein), whereas rooting to classor more distant outgroups reveals the division between earlynd recent genotypes (see also Gould et al., 2003; Wise et al.,004).
The results of phylogenetic and character analysis are con-istent with the notions that, on the one hand, primitive mem-ers of only class II viruses were responsible for colonizinghicken populations and on the other hand there is direct andndirect evidence that both colonization and conversion to vir-lence in chickens occurred more than once in the past. Thiss consistent with the observation that dibasic sequence can beither acquired (e.g. in genotype I and II) or inherited (geno-ype V–VIII, Table 3). The plausibility that events in the pastollowed this scenario is supported by observations reflectingifferent stages of virus–host association in the formation of theecondary reservoir.
First. The history of a biologically heterogeneous (owing tohe simultaneous occurrence of lentogenic, mesogenic and vel-genic strains representing the whole spectrum of virulence) butenetically more homogeneous group of NDV strains belongingo genotype II (Sakaguchi et al., 1989; Lomniczi et al., 1998)iscovered in North-America in the 1940s, is indicative of acenario similar to that seen in Australia. The only difference ishat colonization with genotype II must have taken place longefore the above date, which allowed enough time for virulenttrains to evolve, while descendants of the apathogenic seedingtock were still persistent in chickens. It is suggested that the

46 A. Czegledi et al. / Virus Research 120 (2006) 36–48
simultaneous occurrence of avirulent and virulent strains of thesame genotype in chickens serves as an evolutionary signaturefor an independent seeding with that particular genotype andreflects an early stage of colonization of the secondary host.Assuming that group II strains are the descendants of virusesthat once constituted a separate lineage in the primordial reser-voir, the absence of these isolates in wild waterbirds is surprisingand may reflect either under-representation or even extinction.
Second. Epidemiological data independent of phylogenyhave unequivocally qualified the six nt insert as a shared derivedcharacter of the monophyletic group consisting of genotypesV–VIII, consequently, its absence can be taken, by exclusion,as an evidence for the primitive state. This is best illustrated bythe finding that the virulent descendants of genotype I virusesin Australia, as expected, also lacked the six-nt insert in the5′NCR of NP gene, which greatly strengthened the value of thisphylogenetic trait (Table 1).
Third. While it can be inferred that some of the velogenicviruses of the early genotypes could emerge directly and inde-pendently from distinct apathogenic lines that had stably estab-lished in naıve chicken populations (genotype II, earlier on andgenotype I, very recently), a different hypothesis can be put for-ward for the origin of viruses (in groups V–VIII) that emergedin epizootics of the vaccination era that is after the 1950s. Apartform supragroup features (the six nt insert, phylogeneticallyrelated cleavage site motifs of the F polypeptide and commonrwpgstp
avtuNdtmsgbti2ftslvsIht
means, the introduction of vaccination in the 1950s, albeit thehost species remained the same, brought about such a change interms of susceptibility that was tantamount to a shift at reservoirlevel. It is noteworthy that recent genotypes (V–VIII) emergedin epizootics approximately one to two decades following vac-cination against ND had become widespread. It is not clear,however, where and when their progenitor had emerged beforethey entered epizootics. Further investigations are needed to elu-cidate if immunological selection played a part in the generationand spread of these viruses. We have preliminary results thatrecent NDV strains are more efficient in spreading among immu-nised chickens than viruses from early epizootics (manuscriptin preparation).
Acknowledgements
Parts of this work were supported by grants from the NationalScience Fund (OTKA nos. T 038 254 and T 045 200) and CEVA-Phylaxia, CEVA Sante Animale, Budapest, Hungary.
References
Aldous, E.W., Mynn, J.K., Banks, J., Alexander, D.J., 2003. A molecularepidemiological study of avian paramyxovirus type 1 (Newcastle diseasevirus) isolates by phylogenetic analysis of a partial nucleotide sequenceof the fusion protein gene. Avian Pathol. 32, 239–257.
A
A
A
A
A
B
C
C
C
C
C
C
estriction enzyme cleavage sites), there is indirect evidence asell for the notion that their most recent common ancestor wasathogenic. On the one hand, no avirulent members of recentenotypes were ever found in a period when modern diagnosticervice was in place. On the other hand, it appears that there is lit-le chance for a primitive apathogenic virus to colonize chickenopulations with immunity in large regions.
There are a number of problems, however, concerning thepparently rapid diversification and origin of recent epizooticiruses. (i) We calculated the rate of change at the variable por-ion of the F gene and an approximate 1% per decade for strainsnder epizootic situations was found (Czegledi et al., 2002).evertheless, this rate is unlikely to be realistic beyond epi-emiological timescale (that is a couple of decades) because dueo the rapid saturation of mutable sites in RNA viruses assess-
ent from distances results in underestimation of time. (ii) NDVtrains belonging to recent genotypes displayed wide genetic andeographical separation as early as the 1960s. Where they hadeen before they emerged in epizootics? A similarly contradic-ory observation was made on the emergence of autonomous NDn the pigeon (a tertiary host) at the early 1980s (Ujvari et al.,003). (iii) Thus the origin of recent genotypes is not clear. Theact that they are clearly not the direct descendants of any ofhe known early (II–IV) groups and no avirulent relatives existuggests that their most recent common ancestor could be a viru-ent virus. The progenitor in all probability derived from an earlyirus pool in chickens, which harboured virulent members. Theame pool could serve as a source for the ancestors of genotypesII, IV and H33(W), as well as for a number of other groups thatave gone extinct since. Isolates that would be indispensableo elucidate the sequence of earlier events are missing. By all
lexander, D.J., 1995. The epidemiology and control of avian influenza andNewcastle disease. J. Comp. Pathol. 112, 105–126.
lexander, D.J., 2003. Newcastle disease, other avian paramyxoviruses, andpneumovirus infections. In: Saif, J.M., Barnes, H.J., Glisson, J.R., Fadly,A.M., McDougald, L.R., Swayne, D.E. (Eds.), Diseases of Poultry, 11thed. Iowa State University Press, Ames, Iowa, pp. 63–99.
lexander, D.J., Manvell, R.J., Lowings, J.P., Frost, K.M., Collins, M.S., Rus-sell, P.H., Smith, J.E., 1997. Antigenic diversity and similarities detectedin avian paramyxovirus type 1 (Newcastle disease virus) isolates usingmonoclonal antibodies. Avian Pathol. 26, 399–418.
shford, R.W., 2003. When is a reservoir not a reservoir? Emerg. Infect.Dis. 9, 1495–1496.
wan, M.A., Otte, M.J., James, A.D., 1994. The epidemiology of Newcastledisease in rural poultry: a review. Avian Pathol. 23, 405–423.
allagi-Pordany, A., Wehmann, E., Herczeg, J., Belak, S., Lomniczi, B.,1996. Identification and grouping of Newcastle disease virus strains byrestriction site analysis of a region from the F gene. Arch. Virol. 141,243–261.
hang, P.-C., Hsieh, M.-L., Shien, J.-H., Graham, D.A., Lee, M.-S., Shieh,H.K., 2001. Complete nucleotide sequence of avian paramyxovirus type6 isolated from ducks. J. Gen. Virol. 82, 2157–2168.
apua, I., Alexander, D.J., 2004. Avian influenza: recent developments. AvianPathol. 33, 393–404.
ollins, M.S., Bashiruddin, J.B., Alexander, D.J., 1993. Deduced amino acidsequences at the fusion protein cleavage site of Newcastle disease virusshowing variation in antigenicity and pathogenicity. Arch. Virol. 128,363–370.
ollins, M.S., Franklin, S., Strong, I., Meulemans, G., Alexander, D.J., 1998.Antigenic and phylogenetic studies on a variant Newcastle disease virususing anti-fusion protein monoclonal antibodies and partial sequencing ofthe fusion protein gene. Avian Pathol. 27, 90–96.
zegledi, A., Herczeg, J., Hadjiev, G., Doumanova, L., Wehmann, E., Lom-niczi, B., 2002. The occurrence of five major Newcastle disease virusgenotypes (II, IV, V, VI and VIIb) in Bulgaria between 1959 and 1996.Epidemiol. Infect. 129, 679–688.
zegledi, A., Wehmann, E., Lomniczi, B., 2003. On the origins and rela-tionships of Newcastle disease virus vaccine strains Hertfordshire andMukteswar, and virulent strain Herts’33. Avian Pathol. 32, 271–276.

A. Czegledi et al. / Virus Research 120 (2006) 36–48 47
de Leeuw, O., Peeters, B., 1999. Complete nucleotide sequence of Newcas-tle disease virus: evidence for the existence of a new genus within thesubfamily Paramyxovirinae. J. Gen. Virol. 80, 131–136.
de Leeuw, O.S., Koch, G., Hartog, L., Ravenshorst, N., Peeters, B.P.H., 2005.Virulence of Newcastle disease virus is determined by the cleavage siteof the fusion protein and by both the stem region and globular headof the hemagglutinin-neuraminidase protein. J. Gen. Virol. 86, 1759–1769.
Fumihito, A., Miyake, T., Takada, M., Shingu, R., Endo, T., Gojobori, T.,Kondo, N., Ohno, S., 1996. Monophyletic origin and unique dispersalpatterns of domestic fowls. Proc. Natl. Acad. Sci. U.S.A. 93, 6792–6795.
Gould, A.R., Hannson, E., Selleck, K., Kattenbelt, J.A., Mackenzie,M., Della-Porta, A.J., 2003. Newcastle disease virus fusion andhaemagglutinin-neuraminidase gene motifs as markers for viral lineage.Avian Pathol. 32, 361–373.
Gould, A.R., Kattenbelt, J.A., Selleck, P., Hansson, E., Della-Porta, A., West-bury, H.A., 2001. Virulent Newcastle disease in Australia: molecularepidemiological analysis of viruses isolated prior to and during the out-breaks of 1998–2000. Virus Res. 77, 51–60.
Herczeg, J., Wehmann, E., Bragg, R.R., Travassos Dias, P.M., Hadjiev, G.,Werner, O., Lomniczi, B., 1999. Two novel genetic groups (VIIb andVIII) responsible for recent Newcastle disease outbreaks in SouthernAfrica, one (VIIb) of which reached Southern Europe. Arch. Virol. 144,2087–2099, http://www.fao.org, http://www.oie.int.
Huang, Y., Wan, H.Q., Liu, H.Q., Wu, Y.T., Liu, X.F., 2004. Genomicsequence of an isolate of Newcastle disease virus isolated from an out-break in geese: a novel six nucleotide insertion in the non-coding regionof the nucleoprotein gene. Arch. Virol. 149, 1445–1457.
Kaleta, E.F., Baldauf, C., 1988. Newcastle disease in free-living and petbirds. In: Alexander, D.J. (Ed.), Newcastle disease. Kluwer Academic
K
K
K
L
L
L
L
L
M
M
Romer-Oberdorfer, A., Mundt, E., Mebatsion, T., Buchholz, U.J., Metten-leiter, T.C., 1999. Generation of recombinant lentogenic Newcastle diseasevirus from cDNA. J. Gen. Virol. 80, 2987–2995.
Rott, R., Klenk, H.-D., 1988. Molecular basis of infectivity and pathogenicityof Newcastle disease virus. In: Alexander, D.J. (Ed.), Newcastle Disease.Kluwer Academic Publishers, Boston, pp. 98–112.
Russell, P.H., Alexander, D.J., 1983. Antigenic variation of Newcastle diseasevirus strains detected by monoclonal antibodies. Arch. Virol. 75, 243–253.
Sakaguchi, T., Toyoda, T., Gotoh, B., Inocencio, N.M., Kuma, K., Miyata, T.,Nagai, Y., 1989. Newcastle disease virus evolution. I. Multiple lineagesdefined by sequence variability of the hemagglutinin-neuraminidase gene.Virology 169, 260–272.
Seal, B.S., 2004. Nucleotide and predicted amino acid sequence analysisof the fusion protein and hemagglutinin-neuraminidase protein genesamong Newcastle disease virus isolates. Phylogenetic relationships amongParamyxovirinae based on attachment glycoprotein sequences. Funct.Integr. Genomics 4, 246–257.
Seal, B.S., King, D.J., Meinersman, R.J., 2000. Molecular evolution of theNewcastle disease virus matrix protein gene and phylogenetic relation-ships among the paramyxoviridae. Virus Res. 66, 1–11.
Seal, B.S., Wise, M.G., Pedersen, J.C., Senne, D.A., Alvarez, R., Scott,M.S., King, D.J., Yu, Q., Kapczynski, D.R., 2005. Genomic sequences oflow-virulence avian paramyxovirus-1 (Newcastle disease virus) isolatesobtained from live-bird markets in North America not related to com-monly utilized commercial vaccine strains. Vet. Microbiol. 106, 7–16.
Shihmanter, E., Panashin, A., Lipkind, M., 2005. Nucleotide sequence of thematrix protein gene of avian paramyxovirus, serotype 3b: evidence onanother member of the suggested new genus of the subfamily Paramyx-ovirinae. Comp. Immunol. Microbiol. Infect. Dis. 28, 37–51.
Swayne, D.E., 2000. Understanding the ecology and epidemiology of avian
T
T
U
U
V
W
W
W
W
Publishers, Boston, pp. 197–246.olakofsky, D., Pelet, T., Garcin, D., Hausmann, S., Curran, J., Roux, L.,
1998. Paramyxovirus RNA synthesis and the requirement for hexamergenome length: the rule of six revisited. J. Virol. 72, 891–899.
rishnamurthy, S., Samal, S.K., 1998. Nucleotide sequence of the trailer,nucleocapsid protein gene and intergenic regions of Newcastle diseasevirus strain Beaudette C and completion of the entire genome sequence.J. Gen. Virol. 79, 2419–2424.
umar, S., Tamura, K., Jakobsen, I.B., Nei, M., 2001. MEGA2: molec-ular evolutionary genetics analysis software. Bioinformatics 17, 1244–1245.
amb, R.A., Kolakofsky, D., 2002. Paramyxoviridae: the viruses andtheir replication. In: Fields, B.B., Kniepe, D.M., Howley, P.M. (Eds.),Fundamental Virology. Lippincott-Raven Publishers, Philadelphia, pp.1305–1340.
ee, Y.J., Sung, H.W., Choi, J.G., Kim, J.H., Song, C.S., 2004. Molecularepidemiology of Newcastle disease viruses isolated in South Korea usingsequencing of the fusion protein cleavage site region and phylogeneticrelationships. Avian Pathol. 33, 482–491.
iu, X.F., Wan, H.Q., Ni, X.X., Wu, Y.T., Liu, W.B., 2003. Pathotypical andgenotypical characterization of strains of Newcastle disaese virus isolatedfrom outbreaks in chicken and goose flocks in some regions of Chinaduring 1985–2001. Arch. Virol. 148, 1387–1403.
omniczi, B., Wehmann, E., Herczeg, J., Ballagi-Pordany, A., Kaleta, E.F.,Werner, O., Meulemans, G., Jorgensen, P.H., Mante, A.P., Gielkens,A.L.J., Capua, I., Damoser, J., 1998. Newcastle disease outbreaks inrecent years in Western Europe were caused by an old (VI) and a novelgenotype (VII). Arch. Virol. 143, 49–64.
ocke, D.P., Sellers, H.S., Crawford, J.M., Schultz-Cherry, S., King, D.J.,Meinersmann, R.J., Seal, B.S., 2000. Newcastle disease virus phospho-protein gene analysis and transcriptional editing in avian cells. Virus Res.69, 55–68.
ase, M., Imai, K., Sanada, Y., Sanada, N., Yuasa, N., Imada, T., Tsukamoto,K., Yamaguchi, S., 2002. Phylogenetic analysis of Newcastle disease virusgenotypes isolated in Japan. J. Clin. Microbiol. 40, 3826–3830.
ayo, M.A., 2002. Virus Taxonomy—Houston 2002. Arch. Virol. 147,1071–1076.
influenza viruses: implications for zoonotic potential. In: Brown, C.,Bolin, C. (Eds.), Emerging Diseases of Animals. ASM Press, Washington,DC, pp. 101–130.
oyoda, T., Sakaguchi, T., Hirota, H., Gotoh, B., Kuma, K., Miyata, T.,Nagai, Y., 1989. Newcastle disease virus evolution. II. Lack of generecombination in generating virulent and avirulent strains. Virology 169,273–282.
sai, H.J., Chang, K.H., Tseng, C.H., Frost, K.M., Manvell, R.J., Alexan-der, D.J., 2004. Antigenic and genotypical characterization of Newcastledisease virus isolated in Taiwan between 1969 and 1996. Vet. Microbiol.104, 19–30.
jvari, D., 2006. Complete nucleotide sequence of IT-227/82, an avianparamyxovirus type-1 strain of pigeons (Columba livia). Virus Genes32, 49–57.
jvari, D., Wehmann, E., Kaleta, E.F., Werner, O., Savic, V., Nagy, E., Czifra,G., Lomniczi, B., 2003. Phylogenetic analysis reveals extensive evolutionof avian paramyxovirus type 1 strains of pigeons (Columba livia) andsuggests multiple species transmission. Virus Res. 96, 63–73.
an de Peer, Y., de Wachter, R., 1997. Construction of evolutionary distancetrees with TREECON for Windows: accounting for variation in nucleotidesubstitution rate among sites. Comput. Appl. Biosci. 13, 227–230.
ebster, R.G., Bean, W.J., Gorman, O.T., Chambers, T.M., Kawaoka, Y.,1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56,152–179.
ehmann, E., Czegledi, A., Werner, O., Kaleta, E.F., Lomniczi, B., 2003a.Occurrence of genotypes IV, V, VI and VIIa in Newcastle disease out-breaks in Germany between 1939 and 1995. Avian Pathol. 32, 157–163.
ehmann, E., Ujvari, D., Mazija, H., Velhner, M., Ciglar-Grozdanic, I., Savic,V., Jermolenko, G., Cac, Z., Prukner-Radovcic, E., Lomniczi, B., 2003b.Genetic analysis of Newcastle disease virus strains isolated in Bosnia-Herzegovina, Croatia, Slovenia and Yugoslavia, reveals the presence ofonly a single genotype, V, between 1979 and 2002. Vet. Microbiol. 94,269–281.
erner, O., Romer-Oberdorfer, A., Kollner, B., Manvell, R.J., Alexander,D.J., 1999. Characterization of avian paramyxovirus type 1 strains isolatedin Germany during 1992–1996. Avian Pathol. 28, 79–88.

48 A. Czegledi et al. / Virus Research 120 (2006) 36–48
Westbury, H., 2001. Newcastle disease virus: an evolving pathogen? AvianPathol. 30, 5–11.
Westbury, H.A., 1981. Newcastle disease virus in Australia. Aust. Vet. J. 57,292–298.
Wise, M.G., Sellers, H.S., Alvarez, R., Seal, B.S., 2004. RNA-dependentRNA polymerase gene analysis of worldwide Newcastle disease virus
isolates representing different virulence types and their phylogenetic rela-tionship with other members of the paramyxoviridae. Virus Res. 104,71–80.
Zou, J., Shan, S., Yao, N., Gong, Z., 2005. Complete genome sequence andbiological characterizations of a novel goose paramyxovirus-SF02 isolatedin China. Virus Genes 30, 13–21.
Related Documents











