Cellular Microbiology (2003) 5 (11), 797–807 doi:10.1046/j.1462-5822.2003.00320.x © 2003 Blackwell Publishing Ltd Blackwell Science, LtdOxford, UKCMICellular Microbiology 1462-5822Blackwell Publishing Ltd, 20035 11797807 Original Article M. A. Ingersoll et al.ShiA attenuates inflammation Received 9 May, 2003; revised 4 July, 2003; accepted 4 July, 2003. *For correspondence at the Max Planck Institute for Infection Biology; E-mail [email protected]; Tel. ( + 49) 30 28460 300; Fax ( + 49) 30 28460 301. †These two authors contributed equally to this work. The ShiA protein encoded by the Shigella flexneri SHI-2 pathogenicity island attenuates inflammation Molly A. Ingersoll, 1,2† Jeremy E. Moss, 2† Yvette Weinrauch, 2 Peter E. Fisher, 3 Eduardo A. Groisman 4 and Arturo Zychlinsky l,2 * 1 Max Planck Institute for Infection Biology, 21/22 Schumannstraße, 10117 Berlin, Germany. 2 Skirball Institute and Department of Microbiology, New York University Medical Center, 540 First Avenue, New York, NY 10016, USA. 3 Department of Pathology, Columbia University, 630 West 168th Street, New York, NY 10032, USA. 4 Howard Hughes Medical Institute, Department of Molecular Microbiology, Washington University School of Medicine, 660 S. Euclid Ave, St. Louis, MO 63110, USA. Summary Shigella spp. are the aetiologic agents of dysentery, a severe diarrhoeal syndrome characterized by acute inflammation in the colon. The inflammatory response, which includes recruitment of polymorpho- nuclear leukocytes (PMN), damages the colonic mucosa and exacerbates the infection. Shigella encodes a pathogenicity island (PAI), SHI-2, which is localized in a region of the chromosome linked to the induction of inflammation. Surprisingly, SHI-2 deletion mutants induce a stronger inflammatory response than wild-type Shigella as measured by increased villus blunting, increased PMN infiltration and induction of apoptosis in a rabbit ileal loop model of shigellosis. Mutational analysis mapped the hyper- inflammatory phenotype to a single gene, shiA . Sim- ilar to SHI-2 deletion mutants, infection with a shiA mutant strain induces dramatically elevated levels of inflammation when compared to the wild-type strain. Furthermore, infection with a wild-type strain contain- ing multiple copies of shiA results in fewer infiltrating PMN and apoptotic cells, as well as preservation of a normal villus architecture at the site of infection, thus acting in a dominant fashion over the pro-inflamma- tory mechanisms of Shigella . The molecular mecha- nism of action of ShiA is independent of any in vitro phenotype associated with Shigella virulence. Our data suggest that ShiA allows Shigella to attenuate the host inflammatory response in a novel manner. Introduction Shigella spp. are the causative agents of bacillary dysen- tery. Disease symptoms range from mild diarrhoea to an acute inflammatory disease of the colon characterized by bloody, mucopurulent stools (Sansonetti, 1992). Shigella infection is predominantly found in developing areas of the world where it is responsible for the deaths of more than one million infants per year (Kotloff et al ., 1999). Shigella infection is highly inflammatory and remains localized to the intestine of the host. After ingestion, Shi- gella traverse the intestinal epithelial barrier through spe- cialized antigen sampling cells, called M-cells, at the level of the colon (Perdomo et al ., 1994a; Sansonetti and Pha- lipon, 1999; Wassef et al ., 1989). Following transcytosis, Shigella is phagocytosed by resident tissue macrophages (Jarry et al ., 1989; Soesatyo et al ., 1990). Rapidly there- after, the bacteria escape into the macrophage cytoplasm by destroying the phagosomal membrane (Finlay and Falkow, 1988; Maurelli and Sansonetti, 1988). The pro-inflammatory reaction induced by Shigella infection includes the delivery of the bacterial effector protein IpaB through the type three secretion system (TTSS) to the cytoplasm of macrophages. There, IpaB activates caspase-1, leading to apoptosis and the release of large amounts of the active pro-inflammatory cytokines interleukin (IL)-1 b and IL-18 (Hilbi et al ., 1997; 1998). Shigella released from the apoptotic macrophages can invade the epithelial cell layer from the basolateral side (Perdomo et al ., 1994a). Infection of the epithelial cells results in the secretion of IL-8 (Sansonetti et al ., 1999). IL-1 b , IL-18 and IL-8 appear to be the primary inducers of acute colonic inflammation in shigellosis (Sansonetti et al ., 1995; 1999; 2000). The major mediators of this characteristic inflammatory reaction are host polymorpho- nuclear cells (PMN). After recruitment to the site of infec- tion, PMN disrupt the epithelial cell tight junctions, providing Shigella with broader access to the basolateral surface of the epithelial cells. Thus, the PMN initially exac- erbate the infection (Perdomo et al ., 1994b). Eventually,

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Cellular Microbiology (2003)
5
(11), 797–807 doi:10.1046/j.1462-5822.2003.00320.x
© 2003 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKCMICellular Microbiology 1462-5822Blackwell Publishing Ltd, 20035
11797807
Original Article
M. A. Ingersoll et al.ShiA attenuates inflammation
Received 9 May, 2003; revised 4 July, 2003; accepted 4 July, 2003.*For correspondence at the Max Planck Institute for Infection Biology;E-mail [email protected]; Tel. (
+
49) 30 28460 300;Fax (
+
49) 30 28460 301. †These two authors contributed equally tothis work.
The ShiA protein encoded by the
Shigella flexneri
SHI-2 pathogenicity island attenuates inflammation
Molly A. Ingersoll,
1,2†
Jeremy E. Moss,
2†
Yvette Weinrauch,
2
Peter E. Fisher,
3
Eduardo A. Groisman
4
and Arturo Zychlinsky
l,2
*
1
Max Planck Institute for Infection Biology, 21/22 Schumannstraße, 10117 Berlin, Germany.
2
Skirball Institute and Department of Microbiology, New York University Medical Center, 540 First Avenue, New York, NY 10016, USA.
3
Department of Pathology, Columbia University, 630 West 168th Street, New York, NY 10032, USA.
4
Howard Hughes Medical Institute, Department of Molecular Microbiology, Washington University School of Medicine, 660 S. Euclid Ave, St. Louis, MO 63110, USA.
Summary
Shigella
spp. are the aetiologic agents of dysentery,a severe diarrhoeal syndrome characterized byacute inflammation in the colon. The inflammatoryresponse, which includes recruitment of polymorpho-nuclear leukocytes (PMN), damages the colonicmucosa and exacerbates the infection.
Shigella
encodes a pathogenicity island (PAI), SHI-2, whichis localized in a region of the chromosome linked tothe induction of inflammation. Surprisingly, SHI-2deletion mutants induce a stronger inflammatoryresponse than wild-type
Shigella
as measured byincreased villus blunting, increased PMN infiltrationand induction of apoptosis in a rabbit ileal loop modelof shigellosis. Mutational analysis mapped the hyper-inflammatory phenotype to a single gene,
shiA
. Sim-ilar to SHI-2 deletion mutants, infection with a
shiA
mutant strain induces dramatically elevated levels ofinflammation when compared to the wild-type strain.Furthermore, infection with a wild-type strain contain-ing multiple copies of
shiA
results in fewer infiltratingPMN and apoptotic cells, as well as preservation of anormal villus architecture at the site of infection, thusacting in a dominant fashion over the pro-inflamma-tory mechanisms of
Shigella
. The molecular mecha-
nism of action of ShiA is independent of any
in vitro
phenotype associated with
Shigella
virulence. Ourdata suggest that ShiA allows
Shigella
to attenuatethe host inflammatory response in a novel manner.
Introduction
Shigella
spp. are the causative agents of bacillary dysen-tery. Disease symptoms range from mild diarrhoea to anacute inflammatory disease of the colon characterized bybloody, mucopurulent stools (Sansonetti, 1992).
Shigella
infection is predominantly found in developing areas of theworld where it is responsible for the deaths of more thanone million infants per year (Kotloff
et al
., 1999).
Shigella
infection is highly inflammatory and remainslocalized to the intestine of the host. After ingestion,
Shi-gella
traverse the intestinal epithelial barrier through spe-cialized antigen sampling cells, called M-cells, at the levelof the colon (Perdomo
et al
., 1994a; Sansonetti and Pha-lipon, 1999; Wassef
et al
., 1989). Following transcytosis,
Shigella
is phagocytosed by resident tissue macrophages(Jarry
et al
., 1989; Soesatyo
et al
., 1990). Rapidly there-after, the bacteria escape into the macrophage cytoplasmby destroying the phagosomal membrane (Finlay andFalkow, 1988; Maurelli and Sansonetti, 1988).
The pro-inflammatory reaction induced by
Shigella
infection includes the delivery of the bacterial effectorprotein IpaB through the type three secretion system(TTSS) to the cytoplasm of macrophages. There, IpaBactivates caspase-1, leading to apoptosis and the releaseof large amounts of the active pro-inflammatory cytokinesinterleukin (IL)-1
b
and IL-18 (Hilbi
et al
., 1997; 1998).
Shigella
released from the apoptotic macrophages caninvade the epithelial cell layer from the basolateral side(Perdomo
et al
., 1994a). Infection of the epithelial cellsresults in the secretion of IL-8 (Sansonetti
et al
., 1999).IL-1
b
, IL-18 and IL-8 appear to be the primary inducersof acute colonic inflammation in shigellosis (Sansonetti
et al
., 1995; 1999; 2000). The major mediators of thischaracteristic inflammatory reaction are host polymorpho-nuclear cells (PMN). After recruitment to the site of infec-tion, PMN disrupt the epithelial cell tight junctions,providing
Shigella
with broader access to the basolateralsurface of the epithelial cells. Thus, the PMN initially exac-erbate the infection (Perdomo
et al
., 1994b). Eventually,

798
M. A. Ingersoll
et al.
© 2003 Blackwell Publishing Ltd,
Cellular Microbiology
,
5
, 797–807
however, PMN efficiently kill
Shigella
(Mandic-Mulec
et al
.,1997) and are therefore crucial for resolving the infection.
The
Shigella
invasion and cytotoxicity effector proteinsare encoded on a large virulence plasmid, which is essen-tial for pathogenicity (Sansonetti
et al
., 1982). In additionto the genes encoded on the large virulence plasmid,three chromosomal loci,
his
,
purE
, and
arg-mtl
arerequired for the full inflammatory reaction characteristic of
Shigella
infection (Falkow
et al
., 1963; Sansonetti
et al
.,1983).
The SHI-2 pathogenicity island (PAI) (Moss
et al
., 1999;Vokes
et al
., 1999; Ingersoll
et al
., 2002) is located withinthe
arg-mtl
region, at the
selC
locus. All strains of
S.flexneri
contain SHI-2, whereas most other groups of
Shi-gella
possess, at least, fragments of the island. SHI-2contains the aerobactin operon (
iucA-D, iutA
), whoseproducts mediate bacterial acquisition of iron, as well asseven novel ORFs,
shiA-G
, lacking substantial similarityto published sequences (Moss
et al
., 1999; Vokes
et al
.,1999).
Shigella
strains unable to synthesize aerobactinare slightly attenuated in the rabbit ileal loop infectionmodel (Nassif
et al
., 1987), but the presence of the aero-bactin operon alone is not sufficient to account for therequirement of the
arg-mtl
locus for induction of inflamma-tion. Thus, we investigated whether the seven novel genesencoded in SHI-2 are required for inflammation.
In this study, we investigated the role of the SHI-2 PAIgenes in
Shigella
pathogenicity in the rabbit ligated ilealloop model (Voino-Tasenetsky and Khavkin, 1964; Gots
et al
., 1974). We established that the ShiA protein atten-uates the inflammation induced by
Shigella
, suggesting arole for the protein in reducing the host inflammatoryresponse to
Shigella
infection.
Results
SHI-2 deletion mutants cause increased inflammation in infected rabbit ileal loops
Inflammation in response to
Shigella
is characterized byoedema, PMN accumulation, erythema, haemorrhage,and sometimes ulceration of the tissue (Anand
et al
.,1986). Inflammatory processes in intestinal tissues resultin villus blunting where the extent of shortening correlateswith the severity of the inflammatory response (Sansonetti
et al
., 1999). To test the role of SHI-2 in
Shigella
inducedinflammation
in vivo
, rabbit ligated ileal loops wereinfected with wild-type
Shigella flexneri
5a strain M90Tand
D
17.8 (Table 1, Fig. 1). Six hours post infection, tissuesamples were collected and analysed. Villi from loopsinfected with
D
17.8 displayed a severe inflammatoryresponse and were more blunted than loops infected withthe wild-type strain (data not shown). In the
D
17.8 strain,the
shiA
through
G
genes, as well as the aerobactin genes
iucA
,
iucB
, and part of
iucC
, were deleted (Fig. 1). This
strain was unable to grow overnight in the presence of theiron chelator ethylenediamine-di(
o
-hydroxyphenol aceticacid), confirming the strain’s inability to synthesize aero-bactin (Lawlor
et al
., 1987). The inflammatory phenotypedid not correlate with the previously observed attenuationof the aerobactin mutant (Nassif
et al
., 1987). Thus, todistinguish whether the absent
shi
genes or the deficientaerobactin system was responsible for the observedhyper-inflammatory phenotype, we tested strain
D
10.5,lacking only
shiA-C
(Table 1, Fig. 1), and determined that,similar to
D
17.8, this mutant also caused increased villusblunting, as compared to a wild-type strain (Fig. 2.
P
<
0.001). These results suggest that genes
shiA-C
, butnot the aerobactin operon, contribute to the hyper-inflam-matory phenotype observed with
D
17.8 mutant.
ShiA,
in trans
, complements the hyper-inflammatory phenotype of
D
10.5
To define the gene or genes required for the mutant’shyper-inflammatory phenotype, we complemented
D
10.5with a plasmid encoding
shiA-C
. Infections with
D
10.5pshiA-C (Fig. 2D) resulted in villus blunting similar to thatcaused by M90T carrying an empty vector (Fig. 2B), indi-cating the involvement of at least one of these genes inthe inflammation phenotype. We confirmed that pshiA-Cwas maintained
in vivo
even in the absence of ampicillin,the resistance marker in the plasmid, in 25 independentisolates from the infected loops.
We cloned
shiA
,
shiB
and
shiC
independently intopUC19 and transformed them into
D
10.5 to test their indi-vidual role in modulating inflammation. Villi from loopsinfected with
D
10.5/pshiA (Fig. 2E) were strikingly similarin morphology and length to villi from loops infected withnon-virulent CRN (Fig. 2A) and, hence, less blunted thanthose infected with wild-type M90T (
P
<
0.001). These
Table 1.
Relevant bacterial strains and plasmids.
Strain Description
M90T Streptomycin resistant derivative of wild-type
Shi-gella flexneri. All Shigella strains used in this study are derivatives of this strain (Allaoui et al., 1992).
CRN Spontaneous Congo Red negative derivative of M90T (avirulent, non-invasive, non-cytotoxic)
D17.8 Deletion in nt 764–17829 of SHI-2D10.5 Deletion in nt 764–10538 of SHI-2DshiA Chloramphenicol cassette inserted into shiA
Plasmid Description
pshiA-C Nt 764–10246 of SHI-2 cloned into the Sac I-Kpn IpLCshiA pWSK29 (low copy number) containing shiApHCshiA pUC19 (high copy number) containing shiA
Unless otherwise stated, all strains contain an empty pUC19 orpWSK29 vector.
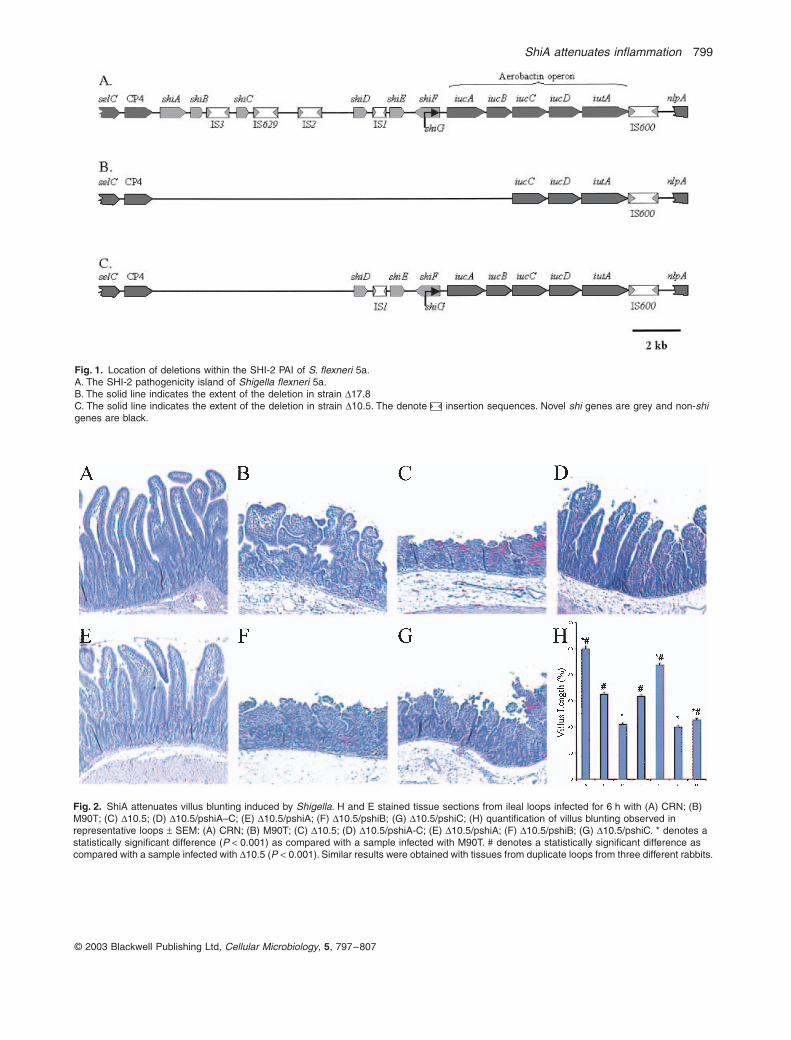
ShiA attenuates inflammation 799
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
Fig. 1. Location of deletions within the SHI-2 PAI of S. flexneri 5a.A. The SHI-2 pathogenicity island of Shigella flexneri 5a.B. The solid line indicates the extent of the deletion in strain D17.8C. The solid line indicates the extent of the deletion in strain D10.5. The denote insertion sequences. Novel shi genes are grey and non-shi genes are black.
Fig. 2. ShiA attenuates villus blunting induced by Shigella. H and E stained tissue sections from ileal loops infected for 6 h with (A) CRN; (B) M90T; (C) D10.5; (D) D10.5/pshiA–C; (E) D10.5/pshiA; (F) D10.5/pshiB; (G) D10.5/pshiC; (H) quantification of villus blunting observed in representative loops ± SEM: (A) CRN; (B) M90T; (C) D10.5; (D) D10.5/pshiA-C; (E) D10.5/pshiA; (F) D10.5/pshiB; (G) D10.5/pshiC. * denotes a statistically significant difference (P < 0.001) as compared with a sample infected with M90T. # denotes a statistically significant difference as compared with a sample infected with D10.5 (P < 0.001). Similar results were obtained with tissues from duplicate loops from three different rabbits.

800 M. A. Ingersoll et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
data clearly indicate that shiA alone is sufficient to com-plement D10.5, restoring wild-type levels of inflammationto the hyper-inflammatory strain. Villus blunting in loopsinfected with D10.5 (Fig. 2C), D10.5/pshiB (Fig. 2F), andD10.5/pshiC (Fig. 2G) was significantly greater than thatobserved in loops infected with wild-type M90T carryingan empty vector (Fig. 2. P < 0.001 for all strains). Theseresults indicate that neither shiB nor shiC play a role ininflammation and suggest that shiA plays a critical role insuppressing the host inflammatory responses to Shigellainfection in vivo.
ShiA is sufficient to attenuate villus blunting
To ascertain if the ShiA protein was sufficient to accountfor the enhanced villus blunting observed with strainsD17.8 and D10.5, we constructed a shiA disruption mutant(Table 1, Fig. 1). The mutant strain was complementedwith both a low copy number plasmid expressing shiA(pLCshiA) and a high copy number plasmid expressingshiA (pHCshiA). Infections with DshiA (Fig. 3C) resultedin significantly more villus blunting (P < 0.001) than withM90T (Fig. 3B). Villus epithelium, after infection withDshiA/pLCshiA (Fig. 3D), displayed similar to wild-typelevels of inflammation. Villi infected with DshiA/pHCshiA(Fig. 3F) or M90T/pHCshiA (Fig. 3G) demonstrated asubstantial decrease in blunting relative to those infectedwith DshiA (Fig. 3C) (P < 0.001) and M90T (Fig. 3B)
(P < 0.001) and were similar to villi infected with CRN(Fig. 3A). Whereas the low copy number shiA plasmid wassufficient to complement the hyper-inflammatory pheno-type, by lessening the degree of inflammation, the highcopy shiA plasmid served to almost entirely suppress thephenotype, resulting in virtually no inflammation. Thus, theshiA gene in trans abrogates the hyper-inflammatory phe-notype of the shiA disruption mutant strain, in a dose-dependent manner, confirming its role in repressing theinflammation induced by wild-type bacteria.
The ShiA knockout increases the number of apoptotic cells in infected villi
Given that villi from loops infected with DshiA were signif-icantly shorter than those infected with wild-type Shigella,we assessed whether ShiA modulated the number ofintestinal apoptotic cells. Ileal loop tissue sections werestained using TUNEL to detect cells with fragmentedDNA, a biochemical hallmark of apoptosis. Villi from loopsinfected with M90T showed a greater number of TUNELpositive cells as compared to the uninfected (data notshown) or CRN infected tissues (Fig. 4A). Apoptotic cellswere localized to the epithelium and the lamina propria.Interestingly, villi infected with DshiA (Fig. 4C) containedsignificantly (P < 0.001) more apoptotic cells than villi fromloops infected with M90T (Fig. 4B). In DshiA infections,dying cells were scattered throughout the lamina propria
Fig. 3. Shigella shiA mutants cause enhanced villus blunting. H and E stained tissue sections from ileal loops infected for 6 h with (A) CRN; (B) M90T; (C) DshiA; (D) DshiA/pLCshiA; (E) M90T/pLCshiA; (F) DshiA/pHCshiA; (G) M90T/pHCshiA. (H) Evaluation of villus blunting observed in representative loops ± SEM: (A) CRN; (B) M90T; (C) DshiA; (D) DshiA/pLCshiA; (E) M90T/pLCshiA; (F) DshiA/pHCshiA; (G) M90T/pHCshiA. *denotes a statistically significant difference (P < 0.001) as compared with a sample infected with M90T. # denotes a statistically significant difference as compared with a sample infected with DshiA (P < 0.001). Similar results were obtained with tissues from duplicate loops from three different rabbits.
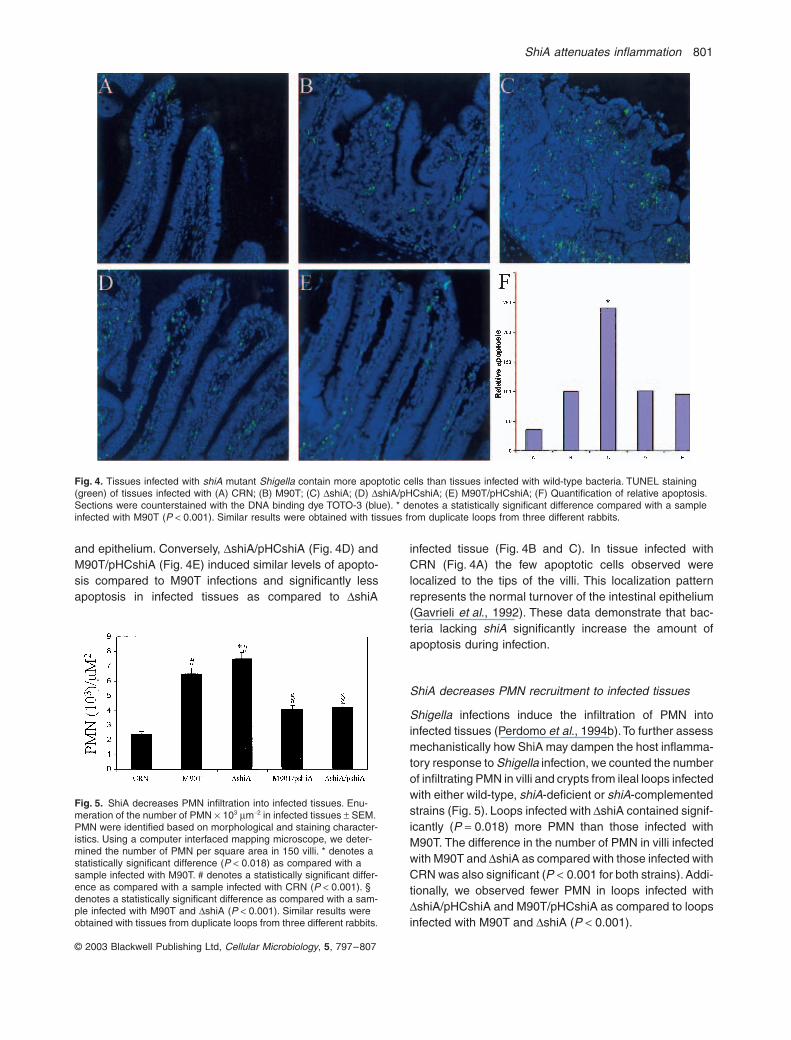
ShiA attenuates inflammation 801
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
and epithelium. Conversely, DshiA/pHCshiA (Fig. 4D) andM90T/pHCshiA (Fig. 4E) induced similar levels of apopto-sis compared to M90T infections and significantly lessapoptosis in infected tissues as compared to DshiA
infected tissue (Fig. 4B and C). In tissue infected withCRN (Fig. 4A) the few apoptotic cells observed werelocalized to the tips of the villi. This localization patternrepresents the normal turnover of the intestinal epithelium(Gavrieli et al., 1992). These data demonstrate that bac-teria lacking shiA significantly increase the amount ofapoptosis during infection.
ShiA decreases PMN recruitment to infected tissues
Shigella infections induce the infiltration of PMN intoinfected tissues (Perdomo et al., 1994b). To further assessmechanistically how ShiA may dampen the host inflamma-tory response to Shigella infection, we counted the numberof infiltrating PMN in villi and crypts from ileal loops infectedwith either wild-type, shiA-deficient or shiA-complementedstrains (Fig. 5). Loops infected with DshiA contained signif-icantly (P = 0.018) more PMN than those infected withM90T. The difference in the number of PMN in villi infectedwith M90T and DshiA as compared with those infected withCRN was also significant (P < 0.001 for both strains). Addi-tionally, we observed fewer PMN in loops infected withDshiA/pHCshiA and M90T/pHCshiA as compared to loopsinfected with M90T and DshiA (P < 0.001).
Fig. 4. Tissues infected with shiA mutant Shigella contain more apoptotic cells than tissues infected with wild-type bacteria. TUNEL staining (green) of tissues infected with (A) CRN; (B) M90T; (C) DshiA; (D) DshiA/pHCshiA; (E) M90T/pHCshiA; (F) Quantification of relative apoptosis. Sections were counterstained with the DNA binding dye TOTO-3 (blue). * denotes a statistically significant difference compared with a sample infected with M90T (P < 0.001). Similar results were obtained with tissues from duplicate loops from three different rabbits.
Fig. 5. ShiA decreases PMN infiltration into infected tissues. Enu-meration of the number of PMN ¥ 103 mm-2 in infected tissues ± SEM. PMN were identified based on morphological and staining character-istics. Using a computer interfaced mapping microscope, we deter-mined the number of PMN per square area in 150 villi. * denotes a statistically significant difference (P < 0.018) as compared with a sample infected with M90T. # denotes a statistically significant differ-ence as compared with a sample infected with CRN (P < 0.001). § denotes a statistically significant difference as compared with a sam-ple infected with M90T and DshiA (P < 0.001). Similar results were obtained with tissues from duplicate loops from three different rabbits.

802 M. A. Ingersoll et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
The localization of the PMN in the tissues was depen-dent upon the infecting strain. In villi infected with non-invasive CRN, PMN were almost entirely confined to thecrypts, overlying the muscularis mucosae. In infectionswith invasive Shigella strains, PMN were found frequentlyin clusters, in the crypts, the lamina propria, and theepithelium. Importantly, these data suggest that ShiAaffected the number, but not the localization of infiltratingPMN.
ShiA does not affect invasion or localization of the bacteria in tissues
We determined whether ShiA affected the number orlocalization of bacteria in infected tissues. We quantifiedcolony forming units (cfu) using a gentamicin protectionassay (Menard, 1994) of infected loops. The number ofcfu in tissues infected with each of the invasive strains ofShigella were similar within each experiment, while tissueinfected with CRN contained approximately two orders ofmagnitude fewer bacteria (Fig. 6F).
We localized the different Shigella strains by indirectimmunofluorescence of the infected loops using an anti-S. flexneri serotype 5a primary antibody to identify thebacteria and Cy-3 conjugated goat antimouse to visualize
the primary antibody. As shown in Fig. 6, M90T (b), DshiA(c), DshiA/pHCshiA (d), and M90T/pHCshiA (e) had asimilar distribution in the tissues. As expected, no bacteriawere observed in sections of tissues from loops infectedwith non-invasive bacteria (Fig. 6A).
ShiA is dispensable for several key Shigellavirulence phenotypes
Shigella lacks a ‘true’ animal model and in vitro assaysserve to further dissect virulence mechanisms. To under-stand the basis of our in vivo findings on a cellular andmolecular level, we investigated Shigella virulence asso-ciated phenotypes of the wild-type and shiA-mutantstrains in vitro.
Using Western blot analysis, no significant differencewas observed among M90T, D17.8, D10.5, DshiA, DshiA/pHCshiA and M90T/pHCshiA in expression and secretionof the effector proteins, necessary for invasion and cyto-toxicity, through the TTSS (data not shown). Invasion intoepithelial cells and intracellular growth (data not shown)were both similar in all strains tested. M90T, D17.8, D10.5,DshiA, DshiA/pHCshiA, M90T/pHCshiA, DshiA/pLCshiAand M90T/pLCshiA all killed J774 cells, thioglycollate-elicited peritoneal macrophages, human peripheral blood
Fig. 6. Shigella localization within infected intestinal tissue is independent of ShiA. Immunostaining for Shigella LPS (red) in tissues infected with (A) CRN; (B) M90T; (C) DshiA; (D) DshiA/pHCshiA; (E) M90T/pHCshiA. The tissue is counterstained with the DNA binding dye TOTO-3 and nuclei appear blue. Similar results were obtained with tissues from duplicate loops from three different rabbits.

ShiA attenuates inflammation 803
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
leukocytes and human PMN with similar efficiency andkinetics (data not shown). The supernatants of thesestrains also induced death at a similar rate in THP-1 andJ774 cells, indicating that ShiA did not significantly alterthe secreted profiles of the strains (Aliprantis et al., 2001).Furthermore, no difference in the toxicity of these strainstowards the epithelial cells HeLa and HEK293 andNIH3T3 fibroblasts was detected (data not shown). Asexpected, CRN did not secrete effectors, could not invadeand cause cytotoxicity, and its supernatant was not cyto-toxic towards any of the cell lines tested (data not shown).Together, these findings indicate that the mechanism ofShiA action does not have an obvious effect on the TTSSand its effectors.
ShiA does not affect the induction ofpro-inflammatory mediators
The severity of dysentery is determined in part by theresponse of the host immune system, including activationof transcriptional activators and production of pro-inflam-matory cytokines. Therefore, we assayed for activation ofthe transcriptional activator NF-kB in epithelial cells usinga luciferase reporter system. NF-kB activation levelsin epithelial cells infected with M90T, DshiA and M90T/pHCshiA were comparable (data not shown). Epithelialcells secreted similar levels of IL-8, induced by NF-kB,as shown by ELISA when infected with the same threestrains (data not shown). We quantified the pro-inflammatory cytokines IL-lb, TNF-a, IFNg, IL-6, and anti-inflammatory IL-10 in supernatants of primary mousemacrophages infected with M90T, CRN, Dl7.8, DshiA andDshiA/pHCshiA. All but CRN triggered the secretion ofmature IL-lb and TNF-a to an equal extent and with com-parable kinetics (data not shown). Consistent with previ-ous observations (Zychlinsky et al., 1994a, b, c), no IL-lb,but a significantly higher level of TNF-a was found in thesupernatants of cells infected with CRN. IFNg, IL-6, andIL-10 were not detected in any of the supernatants.
Comparison of M90T, D17.8 and D10.5 in the Sérenytest (Murayama et al., 1986) illustrated that ShiA does noteffect the ability of Shigella to induce keratoconjuctivitisas each infection induced equivalent levels of inflamma-tion and resolved with equal kinetics (data not shown).
Although PMN exacerbate the infection at first, they areultimately responsible for clearing the Shigella infection.We found the oxidative burst of PMN from healthy humandonors to be similar when PMN were exposed to M90T,M90T/pHCshiA and DshiA (data not shown). A PMNextract, enriched in granule proteins from rabbit or humanPMN, killed M90T, M90T/pHCshiA and DshiA equally wellover a range of extract concentrations (data not shown).No significant differences in sensitivity to killing by PMNwere observed among M90T, M90T/pHCshiA and DshiA
(data not shown). These data indicate that ShiA doesnot exert an observable effect on TTSS-independentinduction of host cytokine production, keratoconjuctivitisinduction, and sensitivity to killing by PMN and theircomponents.
ShiA is localized to the membrane fraction ofthe bacterium
We localized the protein to the membrane of the bacteria.Spheroplast production from the bacteria was followed bysonication and then ultracentrifugation to separate thebacterial lysates into three fractions. As shown in Fig. 7,the protein appears exclusively in the membrane fraction(M) of the wild-type bacteria. The protein was neverdetected in the extracellular medium under any condition,indicating that the protein is not secreted (data notshown). These data raise the possibility that ShiA may notact alone, but in concert with other bacterial and hostproteins in order to downregulate the immune response.
Discussion
ShiA attenuates inflammation induced by Shigella in vivo
Shigella, the causative agent of dysentery, poses a seri-ous public health problem in developing nations. SHI-2is a recently described pathogenicity island, harbouringseven novel genes (Moss et al., 1999; Vokes et al.,1999; Ingersoll et al., 2002). We postulated these genesof SHI-2 might play a role in promoting an inflammatoryresponse given that this island is located in a region ofthe Shigella chromosome required for the induction ofinflammation (Falkow et al., 1963; Sansonetti et al.,1983). Surprisingly, we found that SHI-2 deletionmutants cause increased inflammation in infected rabbitileal loops. ShiA, in trans, complemented the hyper-inflammatory phenotype of the SHI-2 deletion mutantsrestoring wild-type levels of inflammation to tissues
Fig. 7. ShiA is localized to the membrane fraction of Shigella. Wild-type M90T and DshiA strains were fractionated into three parts. Frac-tions and whole bacterial lysates (W) were subjected to SDS-PAGE and immunoblotted with a-RecA, a marker for cytoplasm (C), a-MBP, a marker for periplasm (P), a-IcsA, a marker for membranes (M) and a-ShiA. ShiA appears in the membrane fraction (M) of wild type M90T only and is absent in the membrane fraction (M) of the DshiA strain. (W) denotes wild type M90T whole bacterial lysates.

804 M. A. Ingersoll et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
infected with these strains. shiA on a plasmid was suffi-cient to attenuate villus blunting, decrease the numberof apoptotic cells and decrease PMN recruitment toinfected tissues. We found that a few copies of shiA,encoded on a low copy plasmid, complemented the shiAdisruption. Over-expressing ShiA, from a high copy plas-mid, blocked the inflammation induced by wild-type Shi-gella. Thus, it seems that the amount of ShiA producedby Shigella modulates the potency of the inflammatoryresponse. This observation is illustrated in Fig. 3 whereas the amount of ShiA expressed increased, the amountof tissue damage observed decreased. The effect ofShiA is dominant over the inflammatory phenotypes ofthe plasmid and chromosomal virulence determinants.These data demonstrate that ShiA modulates theinflammatory response to Shigella and may be criticalfor suppressing host inflammatory responses to thepathogen long enough for the bacteria to replicate anddisseminate.
How does ShiA work?
In an effort to unravel ShiA¢s mechanism of action, weevaluated its effects on the known virulence mechanismsof Shigella. Shigella virulence depends upon effectorsencoded by the large virulence plasmid and the chromo-some, as well as on the host response to the infection.Interestingly, ShiA affects a novel pathogenic mechanism,since mutations in shiA do not affect known Shigella plas-mid virulence factors or their secretion. This is confirmedby the ability of the shiA mutant to invade and cause celldeath in multiple cells types in in vitro assays. Similar toour in vitro observation, we detected similar levels of inva-sion in vivo. In contrast, we observed an increased levelof apoptosis in DshiA infected tissue, suggesting that ShiAhas a more indirect effect on apoptosis. ShiA does notregulate the induction of several pro-inflammatory media-tors including NF-kB or the production of IL-1b, TNF-a, IL-6 or IFN-g by macrophages or IL-8 in epithelial cells invitro. Production of IL-10, an anti-inflammatory molecule,is equally unaffected. Finally, shiA deletion mutants are assusceptible as wild-type strains to killing by PMN and theircomponents. We evaluated, in vivo, the host inflammatoryresponse, including apoptosis and PMN migration toinfected tissue. ShiA decreased the amount of apoptosisin infected tissues and the number of PMN recruited tothe site of infection. Localization and invasion of the bac-teria were similar in all strains tested, thus these actionscannot serve as explanations for these effects of ShiA.The mechanism of the modulation is almost certainly indi-rect, as we have localized ShiA to the inner membrane ofthe bacterium and found that it is not secreted into thecellular milieu. Altogether, ShiA has no direct effect on anyknown virulence mechanism of Shigella.
Why does ShiA downregulate inflammation?
Wild-type Shigella induces a dramatic inflammation in theintestine. Inflammation allows for the colonization of intes-tinal tissues (Perdomo et al., 1994b) and causes a diar-rhoeal syndrome that promotes spread of the organism toother hosts. Although inflammation is clearly important inthe pathogenesis of shigellosis, we propose that there isan optimal level of inflammation for Shigella. Shigella mayneed to encode an anti-inflammatory gene as an overlyexuberant inflammatory response, such as that caused bya shiA deletion, may ultimately be detrimental to the bac-teria. A rapid, massive influx of PMN to the site of infectionmay serve to completely destroy the bacteria before theyhave the opportunity to establish an infection, therebydecreasing their overall survival fitness. Alternatively, anexcessive inflammation may kill the host before the bac-teria have the opportunity to disseminate. The integratedfunctioning of ShiA and other virulence proteins may allowShigella to achieve an optimal level of inflammation, wherethey can stave off the immune response long enough tomultiply, but still cause significant diarrhea to spread intothe environment. Thus, although not necessary for infec-tion, ShiA allows Shigella to elegantly fine-tune the inflam-matory reaction to confer upon itself a selective advantage.
Overall, our results illustrate our incomplete understand-ing of Shigella virulence. This suggests that a novel mech-anism of virulence manipulation may exist, one observablein the animal, yet unseen by in vitro systems, that allowsShigella to optimize its success rate in infection.
Experimental procedures
Strains
Strains and plasmids used are listed in Table 1. Shigella’s abilityto bind Congo Red correlates with the presence of the virulenceplasmid (Qadri et al., 1988). A spontaneous Congo Red Negativederivative of a wild-type streptomycin resistant Shigella strain(CRN) was cloned and used as an avirulent control. CRN doesnot invade epithelial cells or kill macrophages, two functions thatrequire the virulence plasmid of Shigella (data not shown).
Bacterial growth conditions
Escherichia coli and Shigella strains were grown at 37∞C in LuriaBroth (LB) and Tryptic Soy Broth (TSB) respectively. Ampicillin(100 mg ml-1), streptomycin (100 mg ml-1), or chloramphenicol(34 mg ml-1 for bacteria containing high copy number plasmids,8 mg ml-1 for SHI-2 deletion mutant strains) were added to themedia where indicated.
Cell lines and growth conditions
The murine macrophage-like cell line mouse J774, thioglycollate-elicited peritoneal macrophages, NIH 3T3 mouse fibroblasts, the

ShiA attenuates inflammation 805
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
human cervical carcinoma cell line HeLa, the human kidneyepithelial cell line HEK293 and the human monocyte cell lineTHP-1 were grown in a humidified incubator at 37∞C with 5% CO2
in RPMI 1640 medium (Gibco BRL) supplemented with 5–10%(v/v) decomplemented fetal calf serum (Gibco BRL), 2 mMglutamine, and 50 mg ml-1 penicillin and streptomycin.
Deletion of SHI-2 genes from the Shigella chromosome
The suicide vector (pCVD442) system described by Donnen-berg and Kaper (1991) was used to generate SHI-2 deletions.SM10Îpir E. coli (Simon et al., 1983) was used for transforma-tions with pCVD442 and its derivatives. Briefly, the 3.6 kb EcoRV fragment from pJM4 (Moss et al., 1999) corresponding toSHI-2 nucleotide (nt) 17829–21468 was cloned into the Sma Isite of pCVD442. The Sph I-Sac I fragment of pJM1 (Mosset al., 1999), ranging from 883 bp upstream of SHI-2 to nt 764of SHI-2, was subsequently cloned into the Sph I and Sac Isites. A chloramphenicol resistance cassette (cml) frompKRP10 (Reece and Phillips, 1995) was cloned into the Sac Isite, resulting in pJMD17.8. SM10lpir/pJMD17.8 was conjugallymated with M90T (streptomycin resistant derivative) (Allaouiet al., 1992). Ampicillin sensitive, streptomycin, chlorampheni-col, and sucrose resistant clones were selected. These cloneswere screened by PCR and analysed by Southern blot forexchange of the intact region of SHI-2 on the Shigella chromo-some with the disrupted copy on the suicide plasmid, identify-ing deletion mutant D17.8.
To delete the 5¢ 10.5 kb of SHI-2 from Shigella, the Eco47 IIIfragment of pJM4 corresponding to nt 10538–11841 was clonedinto the Sma I site on pCVD442. The Sph I-Sac I fragment ofpJM1 was ligated into the Sph I and Sac I sites. The cml cassettefrom pKRP10 was cloned into the Sac I site. Selection for deletionmutants (D10.5) was performed as described above.
To delete the shiA gene, the Sma I (end of Mini Mu cloningvector) to Sal I (nt 6170 in SHI-2) fragment from pJM4 was ligatedinto pCVD442. The cml cassette from pKRP10 was cloned intothe Sph I site in shiA (nucleotide 2409 of SHI-2). Mutants withan insert in shiA (DshiA) were selected as described above.
Complementation analysis was performed using shiA-, shiB-,or shiC-containing pUC19, 100–200 copies per cell (Yanisch-Perron et al., 1985) or shiA-containing pWSK29, 6–8 copies percell (Wang and Kushner, 1991) transformed into the appropriatemutant strains.
Rabbit ileal loop assay
All experimental protocols were approved by the institutionalanimal care and use committee (IACUC).
Male New Zealand White rabbits (>2.7 kg) were fasted 24 hbefore surgery. Rabbits were anesthetized with 50 mg kg-1 ket-amine and 10 mg kg-1 xylazine. Anesthesia was maintained bymask with 2.5% isoflourane and 4 l min-1 of 100% oxygen. Mid-line incisions were made in the rabbit and approximately 2 inchlong small intestinal loops were created by ligation while preserv-ing intestinal blood supply, as described previously (Voino-Tasenetsky and Khavkin, 1964; Gots et al., 1974). Loops werethen injected with 500 ml of a saline solution containing 4 ¥ 109
bacteria ml-1. After 6 h, rabbits were euthanized and the intes-tines removed for examination. Strips from individual loops were
cut for histopathological examination or to count the number ofcolony forming units (cfu) within the tissues. Bacterial enumera-tion was performed as described previously (Sansonetti et al.,1999). Histological samples were fixed for 2 days in 10% neutralbuffered formalin, embedded in paraffin, and processed usingstandard techniques for haematoxylin and eosin (H and E) orimmunofluorescence staining. Each strain was tested in at leastthree different animals. Samples were confirmed by analysis ofduplicate loops within a single rabbit.
Histopathological analysis and quantification
To quantify villus length, digital images of at least 50 villi persample from different sections of tissue in infected loops wereanalysed using Adobe Photoshop software.
To enumerate the number of PMN in the tissues, cells werecounted in H and E stained sections from 25 villi in each of sixinfected loops. PMN were identified based on their multilobedmorphology and staining characteristics (Sansonetti et al., 1999).The area of villi was calculated using a computer interfacedmapping microscope (Alvarez-Buylla and Vicario, 1988).
Terminal dUTP Nick End Labeling (TUNEL) reactionswere performed according to the manufacturer’s instructions(Promega, Madison, WI) on thin sections of tissue after deparaf-finization and rehydration. The sections were counterstained withthe DNA binding dye TOTO-3 (5 mM, Molecular Probes, Eugene,OR).
Immunohistochemical staining for bacterial LPS was per-formed on thin cut sections of tissue after deparaffinization andrehydration. After blocking in 5% rabbit serum, tissues were incu-bated with 5 mg ml-1 anti-S. flexneri serotype 5a primary antibody(kind gift of Dr Armelle Phalipon, Institut Pasteur, Paris, France)at 4∞C overnight in PBS containing 1% Saponin and 3% BovineSerum Albumin. Cy-3 conjugated goat anti-mouse (JacksonImmunoResearch, West Grove, PA) was used as a secondaryantibody. Images were obtained using a Leica ConfocalMicroscope.
All statistical analyses were performed using a one-tailedt-test.
In vitro Shigella virulence assays
Epithelial cell invasion and intracellular growth of bacteria weredetermined as previously described (Menard, 1994). Cytotoxicityin macrophages was measured using the CytoTox96 kit(Promega, Madison, WI) as described previously (Chen, 1996).
Cytokine assays
Thioglycollate elicited macrophages from Balb/C mice wereinfected at an MOI of 100:1. 30, 90, 135, and 180 min postinfection, supernatants were harvested for detection of extracel-lular cytokines by enzyme-linked immunosorbent assay (ELISA)as described previously (Zychlinsky et al., 1994a). HeLa cellswere infected at an MOI of 100:1 and supernatants harvested at30 min, 2 h and 4 h were assayed for IL-8 activity by ELISA.Cytokine assays for mouse IL-10 (M1000), IFNg (MIF00), IL-6(M6000), TNF-a (MTA00), IL-1b (MLB00) and human IL-8(DY208) were performed according to the manufacturer’s speci-fications (R and D Systems, Minneapolis, MN).

806 M. A. Ingersoll et al.
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
Bacterial fractionation
Overnight cultures of M90T and DshiA were subcultured 1:100in TSB, shaking at 37∞ for 2 h. Approximately 1 ¥ 108 bacteriawere washed once with 50 mM Tris pH 7.4. The bacteria werethen resuspended in spheroplast buffer (100 mM Tris pH 8.0,500 mM sucrose, 0.5 mM EDTA pH 8.0, 5 mg lysozyme) andincubated at room temperature for 15 min (min). Spheroplastformation was confirmed by bacterial lysis in a 5 mM EDTAsolution and no lysis in a 50 mM Tris pH 7.4, 20 mM MgSO4
solution. MgSO4 was added to a final concentration of 20 mMand the bacteria were spun at 4500 r.p.m. for 10 min. The super-natant contained the periplasmic protein fraction. The bacteriawere then resuspended in PBS, sonicated three times, 15 s each,and spun again at 4500 r.p.m., 10 min. The supernatant con-tained the cytoplasmic protein fraction. The pellet was resus-pended in PBS and spun at 80 000, 30 min, 4∞C. The pellet wasthe membrane fraction. The periplasmic and cytoplasmic frac-tions were subjected to methanol/chloroform protein concentra-tion. The three fractions were subjected to SDS-PAGE andimmunoblotted with a-RecA, a marker for cytoplasm, a-MBP,a marker for periplasm, a-IcsA, a marker for membranes anda-ShiA.
Acknowledgements
We thank David Weiss and Björn Albrecht for thoughtful discus-sions and critical reading of this manuscript.
References
Aliprantis, A.O., Weiss, D.S., Radolf, J.D., and Zychlinsky, A.(2001) Release of Toll-like receptor-2-activating bacteriallipoproteins in Shigella flexneri culture supernatants. InfectImmun 69: 6248–6255.
Allaoui, A., Sansonetti, P.J., and Parsot, C. (1992) MxiJ, alipoprotein involved in secretion of Shigella Ipa invasins, ishomologous to YscJ, a secretion factor of the Yersinia Yopproteins. J Bacteriol 174: 7661–7669.
Alvarez-Buylla, A., and Vicario, D.S. (1988) Simple micro-computer system for mapping tissue sections with the lightmicroscope. J Neurosci 25: 165–173.
Anand, B.S., Malhorta, V., Bhattacharya, S.K., Datta, P.,Sen, D., Bhattacharya, M.K., et al. (1986) Rectal histologyin acute bacillary dysentery. Gastroenterology 90: 654–660.
Chen, Y., Smith, M.R., Thirumalai, K., and Zychlinsky, A.(1996) A bacterial invasin induces macrophage apoptosisby directly binding ICE. EMBO J 15: 3853–3860.
Donnenberg, M.S., and Kaper, J.B. (1991) Construction of aeae deletion mutant of enteropathogenic Escherichia coliby using a positive-selection suicide vector. Infect Immun59: 4310–4317.
Falkow, S., Schneider, H., Baron, L.S., and Formal, S.B.(1963) Virulence of Escherichia-Shigella genetic hybridsfor the guinea pig. J Bacteriol 86: 1251–1258.
Finlay, B.B., and Falkow, S. (1988) Comparison of the inva-sion strategies used by Salmonella cholerae-suis, Shigellaflexneri and Yersinia enterocolitica to enter cultured animalcells: endosome acidification is not required for bacterialinvasion or intracellular replication. Biochimie 70: 1089–1099.
Gavrieli, Y., Sherman, Y., and Ben-Sasson, S.A. (1992) Iden-tification of programmed cell death in situ via specific label-ing of nuclear DNA fragmentation. J Cell Biol 119: 493–501.
Gots, R.E., Formal, S.B., and Giannella, R.A. (1974)Indomethacin inhibition of Salmonella typhimurium, Shi-gella flexneri, and cholera-mediated rabbit ileal secretion.J Infect Dis 130: 280–284.
Hilbi, H., Chen, Y., Thirumalai, K., and Zychlinsky, A. (1997)The interleukin 1beta-converting enzyme, caspase 1, isactivated during Shigella flexneri-induced apoptosis inhuman monocyte-derived macrophages. Infect Immun 65:5165–5170.
Hilbi, H., Moss, J.E., Hersh, D., Chen, Y., Arondel, J., Ban-erjee, S., et al. (1998) Shigella-induced apoptosis is depen-dent on caspase-1 which binds to IpaB. J Biol Chem 273:32895–32900.
Ingersoll, M., Groisman, E.A., and Zychlinsky, A. (2002)Pathogenicity islands of Shigella. Curr Top Microbiol Immu-nol 264: 49–65.
Jarry, A., Robaszkiewicz, M., Brousse, N., and Potet, F.(1989) Immune cells associated with M cells in the follicle-associated epitehlium of Peyer’s patches in the rat. CellTissue Res 255: 293–298.
Kotloff, K.L., Winickoff, J.P., Ivanoff, B., Clemens, J.D., Swer-dlow, D.L., Sansonetti, P.J., et al. (1999) Global burden ofShigella infections: implications for vaccine developmentand implementation of control strategies. Bull World HealthOrgan 77: 651–666.
Lawlor, K., Daskaleros, P., Robinson, R., and Payne, S.(1987) Virulence of iron transport mutants of Shigella flex-neri and utilization of host iron compounds. Infect Immun55: 594–599.
Mandic-Mulec, I., Weiss, J., and Zychlinsky, A. (1997) Shi-gella flexneri is trapped in polymorphnuclear leukocyte vac-uoles and efficiently killed. Infect Immun 65: 110–115.
Maurelli, A.T., and Sansonetti, P.J. (1988) Genetic determi-nants of Shigella pathogenicity. Ann Rev Microbiol 42:127–150.
Menard, R., Sansonetti, P., and Parsot, C. (1994) The secre-tion of the Shigella flexneri Ipa invasins is activated byepithelial cells and controlled by IpaB and IpaD. EMBO J13: 5293–5302.
Moss, J.E., Cardozo, T.J., Zychlinsky, A., and Groisman, E.A.(1999) The selC-associated SHI-2 pathogenicity island ofShigella flexneri. Mol Microbiol 33: 74–83.
Murayama, S.Y., Sakai, T., Makino, S., Kurata, T., Sasakawa,C., and Yoshikawa, M. (1986) The use of mice in theSereny test as a virulence assay of shigellae and entero-invasive Escherichia coli. Infect Immun 51: 696–698.
Nassif, X., Mazert, M.C., Mounier, J., and Sansonetti, P.J.(1987) Evaluation with an iuc:: Tn10 mutant of the role ofaerobactin production in the virulence of Shigella flexneri.Infect Immun 55: 1963–1969.
Perdomo, O.J., Cavaillon, J.M., Huerre, M., Ohayon, H.,Gounon, P., and Sansonetti, P.J. (1994a) Acute inflamma-tion causes epithelial invasion and mucosal destruction inexperimental shigellosis. J Exp Med 180: 1307–1319.
Perdomo, J.J., Gounon, P., and Sansonetti, P.J. (1994b)Polymorphonuclear leukocyte transmigration promotesinvasion of colonic epithelial monolayer by Shigella flexneri.J Clin Invest 93: 633–643.
Qadri, F., Hossain, S.A., Ciznar, I., Haider, K., Ljungh, A.,Wadstrom, T., and Sack, D.A. (1988) Congo red binding

ShiA attenuates inflammation 807
© 2003 Blackwell Publishing Ltd, Cellular Microbiology, 5, 797–807
and salt aggregation as indicators of virulence in Shigellaspecies. J Clin Microbiol 26: 1343–1348.
Reece, K.S., and Phillips, G.J. (1995) New plasmids carryingantibiotic-resistance cassettes. Gene 165: 141–142.
Sansonetti, P.J. (1992) Molecular and cellular biology ofShigella flexneri invasiveness: from cell assay sys-tems to shigellosis. Curr Top Microbiol Immunol 180:1–19.
Sansonetti, P.J., and Phalipon, A. (1999) M cells as ports ofentry for enteroinvasive pathogens: mechanisms of inter-action, consequences for the disease process. SeminImmunol 11: 193–203.
Sansonetti, P.J., Kopecko, D.J., and Formal, S.B. (1982)Involvement of a plasmid in the invasive ability of Shigellaflexneri. Infect Immun 35: 852–860.
Sansonetti, P.J., Hale, T.L., Dammin, G.J., Kapfer, C., Col-lins, H.H., and Formal, S.B. (1983) Alterations in the patho-genicity of Escherichia coli K-12 after transfer of plasmidand chromosomal genes from Shigella flexneri. InfectImmun 39: 1392–1402.
Sansonetti, P.J., Arondel, J., Cavaillon, J.M., and Huerre, M.(1995) Role of interleukin-1 in the pathogenesis of experi-mental shigellosis. J Clin Invest 96: 884–892.
Sansonetti, P.J., Arondel, J., Huerre, M., Harada, A., andMatsushima, K. (1999) Interleukin-8 controls bacterial tran-sepithelial translocation at the cost of epithelial destructionin experimental shigellosis. Infect Immun 67: 1471–1480.
Sansonetti, P., Phalipon, A., Arondel, J., Thirumalai, K., Ban-erjee, S., Akira, S., et al. (2000) Caspase-1 activation of IL-1b and IL-18 are essential for Shigella flexneri inducedinflammation. Immunity 12: 581–590.
Simon, R., Priefer, U., and Puhler, A. (1983) A broad hostrange mobilization system for in vivo genetic engineering:transposon mutagenesis in Gram-negative bacteria. Bio/Technology 1: 784–791.
Soesatyo, M., Biewenga, J., Kraal, G., and Sminia, T. (1990)
The localization of macrophage subsets and dendritic cellsin the gastrointestinal tract of the mouse with special ref-erence to the presence of high endothelial venules. Animmuno- and enzyme-histochemical study. Cell Tissue Res259: 587–593.
Voino-Tasenetsky, M.V., and Khavkin, T.N. (1964) A study ofintraepithelial localization of dysentery causative agentswith the aid of flourescent antibodies. J Microbiol 12: 98–100.
Vokes, S.A., Reeves, S.A., Torres, A.G., and Payne, S.M.(1999) The aerobactin iron transport system genes in Shi-gella flexneri are present within a pathogenicity island. MolMicrobiol 33: 63–73.
Wang, R.F., and Kushner, S.R. (1991) Construction of ver-satile low-copy-number vectors for cloning, sequencingand gene expression in Escherichia coli. Gene 100: 195–199.
Wassef, J.S., Keren, D.F., and Mailloux, J.L. (1989) Role ofM cells in initial antigen uptake and in ulcer formation inrabbit intestinal loop model of shigellosis. Infect Immun 57:858–863.
Yanisch-Perron, C., Vieira, J., and Messing, J. (1985)Improved M13 phage cloning vectors and host strains:nucleotide sequences of the M13mp18 and pUC19 vec-tors. Gene 33: 103–119.
Zychlinsky, A., Fitting, C., Cavaillon, J.M., and Sansonetti,P.J. (1994a) Interleukin-1 is released by murine macroph-ages during apoptosis induced by Shigella flexneri. J ClinInvest 94: 1328–1332.
Zychlinsky, A., Kenny, B.M.,nard, R., Pr,vost, M.C., Holland,I.B., and Sansonetti, P.J. (1994b) IpaB mediates macroph-age apoptosis induced by Shigella flexneri. Mol Microbiol11: 619–627.
Zychlinsky, A., Perdomo, J.J., and Sansonetti, P.J. (1994c)Molecular and cellular mechanisms of tissue invasion byShigella flexneri. A N Y Acad Sci 739: 197–208.
Related Documents











