Eur. Phys. J. B 71, 185–194 (2009) DOI: 10.1140/epjb/e2009-00272-6 Regular Article T HE EUROPEAN P HYSICAL JOURNAL B Structural, elastic, electronic, optical and thermal properties of c-SiGe 2 N 4 A. Bouhemadou 1, a , Y. Al-Douri 2 , R. Khenata 3 , and K. Haddadi 1 1 Laboratory for Developing New Materials and their Characterization, Department of Physics, Faculty of Science, University of Setif, 19000 Setif, Algeria 2 School of Microelectronic Engineering, University Malaysia Perlis, Block A Kompleks Pusat Pengajian, 02600 Jejawi Arau, Perlis, Malaysia 3 D´ epartement de Technologie, Universit´ e de Mascara, 29000 Mascara, Algeria Received 12 February 2009 / Received in final form 25 June 2009 Published online 18 August 2009 –c EDP Sciences, Societ`a Italiana di Fisica, Springer-Verlag 2009 Abstract. We have investigated the structural, elastic, electronic, optical and thermal properties of c-SiGe2N4 by using the ultrasoft pseudopotential density functional method within the generalized gra- dient approximation. The calculated structural parameters, including the lattice constant, the internal free parameter, the bulk modulus and its pressure derivative are in agreement with the available data. The independent elastic constants and their pressure dependence, calculated using the static finite strain technique, satisfy the requirement of mechanical stability, indicating that c-SiGe2N4 compound could be stable. We derive the shear modulus, Young’s modulus, Poisson’s ratio and Lam´ e’s coefficients for ideal polycrystalline c-SiGe2N4 aggregate in the framework of the Voigt-Reuss-Hill approximation. We estimate the Debye temperature of this compound from the average sound velocity. Band structure, density of states, Mulliken charge populations and pressure coefficients of energy band gaps are investigated. Furthermore, in order to understand the optical properties of c-SiGe2N4, the dielectric function, refractive index, extinction coefficient, optical reflectivity and electron energy loss are calculated for radiation up to 40 eV. Thermal effects on some macroscopic properties of c-SiGe2N4 are predicted using the quasi-harmonic Debye model in which the lattice vibrations are taken into account. We have obtained successfully the variations of the primitive cell volume, volume expansion coefficient, heat capacities and Debye temperature with pressure and temperature in the ranges of 0–40 GPa and 0–2000 K. For the first time, the numerical estimates of the elastic constants and related parameters, and the thermal properties are performed for c-SiGe2N4. PACS. 71.15.Mb Density functional theory, local density approximation, gradient and other corrections – 62.20.de Elastic moduli – 71.20.Nr Semiconductor compounds – 78.20.Ci Optical constants 1 Introduction Solids formed from elements in group-IV and group-V are interesting and important materials with many applica- tions because of their outstanding high temperature and oxidations resistant properties [1]. They crystallize in sev- eral solid phases. The α (P31c, hp28), β (P6 3 /m, hp14) and γ (spinel, Fd-3m, cF56) phases of the well-researched single nitrides C 3 N 4 , Si 3 N 4 , Ge 3 N 4 and Sn 3 N 4 are well- known from experiments [2–6]. The cubic spinel double nitrides c-CSi 2 N 4 , c-CGe 2 N 4 , c-SiGe 2 N 4 , c-GeSi 2 N 4 , etc., are theoretically predicted to have wide direct band gaps (comparable to those of the developed solid state promis- ing optoelectronic materials such as GaN, InN and AlN), a large bulk modulus and a large static dielectric constant. These special properties mean that the cubic spinel double a e-mail: a [email protected] nitrides are candidate materials for potential technological applications [7–9]. Among these double nitrides, the cubic spinel SiGe 2 N 4 has been shown to be of particular interest because it is a stable compound and has a favorable direct band gap [8]. Experimental information on the cubic sipinel SiGe 2 N 4 is scarce. Theoretically, this compound has attracted the attention of some researchers. Ching and co-workers have studied the structural, electronic and optical properties of c-SiGe 2 N 4 by means of the first-principles orthogo- nalized linear combinations of atomic orbitals (OLCAO) method [7–9]. Wang et al. have investigated the struc- tural, electronic and optical properties of SiGe 2 N 4 us- ing an ab initio full potential linearized augmented plane waves (FP-LAPW) code [10]. Neither experimental nor theoretical details regarding the first order elastic con- stants and the thermodynamic properties are available. Moreover, it appears that no earlier calculations about

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Eur. Phys. J. B 71, 185–194 (2009)DOI: 10.1140/epjb/e2009-00272-6
Regular Article
THE EUROPEANPHYSICAL JOURNAL B
Structural, elastic, electronic, optical and thermal propertiesof c-SiGe2N4
A. Bouhemadou1,a, Y. Al-Douri2, R. Khenata3, and K. Haddadi1
1 Laboratory for Developing New Materials and their Characterization, Department of Physics, Faculty of Science, Universityof Setif, 19000 Setif, Algeria
2 School of Microelectronic Engineering, University Malaysia Perlis, Block A Kompleks Pusat Pengajian, 02600 Jejawi Arau,Perlis, Malaysia
3 Departement de Technologie, Universite de Mascara, 29000 Mascara, Algeria
Received 12 February 2009 / Received in final form 25 June 2009Published online 18 August 2009 – c© EDP Sciences, Societa Italiana di Fisica, Springer-Verlag 2009
Abstract. We have investigated the structural, elastic, electronic, optical and thermal properties ofc-SiGe2N4 by using the ultrasoft pseudopotential density functional method within the generalized gra-dient approximation. The calculated structural parameters, including the lattice constant, the internalfree parameter, the bulk modulus and its pressure derivative are in agreement with the available data.The independent elastic constants and their pressure dependence, calculated using the static finite straintechnique, satisfy the requirement of mechanical stability, indicating that c-SiGe2N4 compound could bestable. We derive the shear modulus, Young’s modulus, Poisson’s ratio and Lame’s coefficients for idealpolycrystalline c-SiGe2N4 aggregate in the framework of the Voigt-Reuss-Hill approximation. We estimatethe Debye temperature of this compound from the average sound velocity. Band structure, density of states,Mulliken charge populations and pressure coefficients of energy band gaps are investigated. Furthermore, inorder to understand the optical properties of c-SiGe2N4, the dielectric function, refractive index, extinctioncoefficient, optical reflectivity and electron energy loss are calculated for radiation up to 40 eV. Thermaleffects on some macroscopic properties of c-SiGe2N4 are predicted using the quasi-harmonic Debye modelin which the lattice vibrations are taken into account. We have obtained successfully the variations of theprimitive cell volume, volume expansion coefficient, heat capacities and Debye temperature with pressureand temperature in the ranges of 0–40 GPa and 0–2000 K. For the first time, the numerical estimates ofthe elastic constants and related parameters, and the thermal properties are performed for c-SiGe2N4.
PACS. 71.15.Mb Density functional theory, local density approximation, gradient and other corrections –62.20.de Elastic moduli – 71.20.Nr Semiconductor compounds – 78.20.Ci Optical constants
1 Introduction
Solids formed from elements in group-IV and group-V areinteresting and important materials with many applica-tions because of their outstanding high temperature andoxidations resistant properties [1]. They crystallize in sev-eral solid phases. The α (P31c, hp28), β (P63/m, hp14)and γ (spinel, Fd-3m, cF56) phases of the well-researchedsingle nitrides C3N4, Si3N4, Ge3N4 and Sn3N4 are well-known from experiments [2–6]. The cubic spinel doublenitrides c-CSi2N4, c-CGe2N4, c-SiGe2N4, c-GeSi2N4, etc.,are theoretically predicted to have wide direct band gaps(comparable to those of the developed solid state promis-ing optoelectronic materials such as GaN, InN and AlN),a large bulk modulus and a large static dielectric constant.These special properties mean that the cubic spinel double
a e-mail: a [email protected]
nitrides are candidate materials for potential technologicalapplications [7–9]. Among these double nitrides, the cubicspinel SiGe2N4 has been shown to be of particular interestbecause it is a stable compound and has a favorable directband gap [8].
Experimental information on the cubic sipinel SiGe2N4
is scarce. Theoretically, this compound has attracted theattention of some researchers. Ching and co-workers havestudied the structural, electronic and optical propertiesof c-SiGe2N4 by means of the first-principles orthogo-nalized linear combinations of atomic orbitals (OLCAO)method [7–9]. Wang et al. have investigated the struc-tural, electronic and optical properties of SiGe2N4 us-ing an ab initio full potential linearized augmented planewaves (FP-LAPW) code [10]. Neither experimental northeoretical details regarding the first order elastic con-stants and the thermodynamic properties are available.Moreover, it appears that no earlier calculations about

186 The European Physical Journal B
the strain effect on structural, elastic and electronic prop-erties of this compound have been done.
First-principles calculations offer one of the most pow-erful tools for carrying out theoretical studies of an im-portant number of physical and chemical properties ofthe condensed matter with great accuracy. It is now pos-sible to explain and predict properties of solids whichwere previously inaccessible to experiments. We there-fore think that it is worthwhile to perform first-principlescalculations for the structural, elastic, electronic, opticaland thermal properties of c-SiGe2N4 using the ultra-softpseudo-potential plane-waves (PP-PW) method in orderto provide reference data for the experimentalists and tocomplete existing theoretical works on this compound.
In the following, the paper is divided in three parts.In Section 2, we briefly describe the computational tech-niques used in this study. The most relevant results ob-tained for the structural, elastic, electronic, optical andthermodynamic properties of c-SiGe2N4 compound arepresented and discussed in Section 3. Finally, in Section 4we summarize the main conclusions of our work.
2 Computational method
The first-principles calculations were performed using aplane-wave pseudo-potential total energy method [11]. In-teractions of electrons with ion cores were represented bythe Vanderbilt-type ultrasoft pseudopotential [12] for Si,Ge and N atoms. The exchange-correlation potential wascalculated within the generalized gradient approximation(GGA) of Perdew [13]. The plane-wave basis set cut-offwas set as 280 eV. The special points sampling integra-tion over the Brillouin zone was employed by using theMonkhorst-Pack method with a 5 × 5 × 5 special k-pointmesh [14]. These parameters were sufficient in leadingto well converged total energy, geometrical configurationsand elastic moduli.
The structural parameters of c-SiGe2N4 were de-termined using the Broyden-Fletcher-Goldfarb-Shenno(BFGS) minimization technique [15]; which provides a fastway of finding the lowest energy structure. The tolerancefor geometry optimization were set as the difference of to-tal energy within 5 × 10−6 eV atom−1, maximum ionicHellmann-Feynman force within 0.01 eV A−1 and maxi-mum stress within 0.02 eV A−3.
The elastic coefficients were determined from first-principles calculations by applying a set of given homo-geneous deformations with a finite value and calculatingthe resulting stress with respect to optimizing the inter-nal atomic degrees of freedom [16]. The convergence cri-teria of this optimization were selected as the differenceof the total energy within 1 × 10−6 eV atom−1, the ionicHellmann-Feynman force within 0.002 eV A−1 and themaximum ionic displacement within 1 × 10−4 A. A cu-bic crystal has three different symmetry elements (C11,C12, and C44). One strain pattern, with non-zero first andfourth components, gives stresses related to all three in-dependent elastic coefficients for the cubic system. Threepositive and three negative amplitudes were used for each
strain component with the maximum value of 0.5%, andthen the elastic stiffness coefficients were determined froma linear fit of the calculated stress as a function of thestrain.
The optical properties may be derived from the knowl-edge of the complex dielectric function ε(ω) = ε1(ω) +iε2(ω). The imaginary part of the dielectric function ε2(ω)is calculated from the momentum matrix elements be-tween the occupied and unoccupied wave functions withinthe selection rules. The real part of dielectric functionε1(ω) can be evaluated from ε2(ω) by the Kramer-Kronigrelation. All the other optical constants, such as the re-fractive index n(ω), the extinction coefficient k(ω), theoptical reflectivity R(ω), the absorption coefficient α(ω)and the energy-loss spectrum L(ω), can be computed fromthe values of ε(ω). For the calculation of the optical prop-erties, which usually requires a dense mesh of uniformlydistributed k-points, the BZ integration was performedusing a 10 × 10 × 10 MP k-mesh.
The study of thermal effects was done within the quasi-harmonic Debye model implemented in the Gibbs pro-gram [17]. For a solid described by an energy-volume(E − V ) relationship in the static approximations, theGibbs program allows us to evaluate the Debye temper-ature, to obtain the Gibbs free energy G(V ; P, T ) and tominimize G for deriving the thermal equation of state(EOS) V (P, T ). Other macroscopic properties related toP and T can be also derived by using standard thermo-dynamic relations. Detailed descriptions of this procedurecan be found in references [17–21].
3 Results and discussion
3.1 Structural properties
Cubic spinels with chemical formula AB2X4 have a closed-packed face-centred-cubic structure, with space groupFd-3m (#227), and their unit cell contains eight AB2X4
unit formulas. The 32 anions (X atoms) occupy the 32esite. The cations occupy either the tetrahedral 8a site(A atoms) or the octahedral 16d site (B atoms). There isonly one internal parameter u, which specifies the devia-tion of the anions in the 〈111〉 direction. The description ofthe atomic positions in spinels is dependent on the choiceof setting for the origin in the Fd-3m space group. Two dif-ferent equipoints with point symmetries −43m and −3mare possible choices for the unit cell origin. In the idealspinel with no anion deviation, uideal = 0.25 or 0.375 fororigins at −3m or −43m symmetry, respectively [22]. TheX atoms are positioned at the (u, u, u) positions, the eightA atoms at (0.125, 0.125, 0.125) and the sixteen atoms at(0.5, 0.5, 0.5). Then its crystal structure is characterizedby two free parameters: the lattice constant a and the in-ternal anion parameter u. In most spinels, u lies between0.24 and 0.275, if the origin of the unit cell is taken at−3m point symmetry. The unit cell of the cubic spinelSiGe2N4 is depicted in Figure 1
The calculated lattice parameter a0 and the internalstructural parameter u, for c-SiGe2N4 as determined from

A. Bouhemadou et al.: Structural, elastic, electronic, optical and thermal properties of c-SiGe2N4 187
Fig. 1. (Color online) The unit cell of the cubic spinel SiGe2N4.
geometry at P = 0 GPa, are given in Table 1 together withthe available results of other calculations for comparison.There is a good agreement between our results and thoseof the previous calculations.
In order to show how the structural parameters be-have under pressure in these compounds, the equilib-rium geometries of c-SiX2O4unit cells were computed atfixed values of applied hydrostatic pressure in the 0 to40 GPa range, with the step of 10 GPa. At each pres-sure, a complete optimization of the structural param-eters is performed. Pressure dependence of the internalparameter, u, is shown in Figure 2a. We clearly ob-serve a quadratic dependence in the considered range ofpressure. The solid curve is a quadratic least-squares fit(u(P ) = u(0) + αP + βP 2). The values of linear andquadratic pressure coefficients of the internal parameteru are found to be equal to −2.36 × 10−5 (GPa)−1 and1.26 × 10−8 (GPa)−2, respectively. c-SiGe2N4 exhibit anegative u vs. P slope, indicating that this spinel underpressure tends to approach more and more the ideal struc-ture characterized by u = 0.25. The obtained pressure-unit cell volume (E − V ) data set was used to obtain theBulk modulus B0 and its pressure derivative B
′by fit-
ting the data to the Birch-Murnaghan equation of state(EOS) [23] (Fig. 2b). The bulk modulus B0 and its pres-sure derivative B
′extracted from the EOS fitting are listed
in Table 1.
3.2 Elastic properties
3.2.1 Elastic constants
In Table 1, we list also our calculated values of the elasticconstants (C11, C12 and C44) for c-SiGe2N4. To date, noexperimental or theoretical data for the elastic constantsof c-SiGe2N4 compound are available to be compared withour theoretical results. Then, our results can provide ref-erence data for future investigations.
Table 1. Calculated lattice constant a0 (in A), bulk modu-lus B0 (in GPa), pressure derivative B′, elastic constants Cij
(in GPa), shear modulus G (in GPa), Young’s modulus E (inGPa), Poisson’s ratios σ, Lame’s coefficients μ and λ (in GPa),pressure derivatives of the elastic moduli, and values of somedirect and indirect band gaps (Γ–Γ, Γ–L, Γ–X, Γ–K, Γ–A, Γ–W) (in eV) for c-SiGe2N4 . These are compared to availabledata.
Present work Other theoretical works
a0 7.9285 8.0871 [8]; 8.1421 [10]
B 283.9a, 279b 277.1 [8]; 241.7 [10]
B′ 3.81a, 4.03b 3.02 [8]; 4.48 [10]
u 0.2541 0.2523 [8]; 0.2535 [10]
C11 494
C12 172
C44 288
E 537
G 228
σ 0.1791
λ 127
μ 228∂C11∂P
5.41
∂C12∂P
3.34
∂C44∂P
1.23
Γ–Γ 2.14 1.58 [8], 1.85 [10]
Γ–L 4.67
Γ–X 5.73
Γ–K 5.70
Γ–W 6.16
a From the EOS fitting; b from the relation: B = 1/3(C11 +2C12).
Once the elastic constants are determined, we wouldlike to compare our results with experiments, or predictwhat an experiment would yield for the elastic constants.A problem arises when single crystal samples cannot beobtained, for then it is not possible to measure the in-dividual elastic constants Cij . Instead, the isotropic bulkmodulus B and shear modulus G are determined. Thesequantities cannot in general be calculated directly fromthe Cij , but we can use our values to place bounds on theisotropic moduli. Reuss found lower bounds for all lattices,while Voigt discovered upper bounds [24]. Hill has shownthat the Voigt and Reuss averages are limits and suggestedthat the actual effective moduli could be approximated bythe arithmetic mean of the two bounds [25]. The isotropicbulk modulus for cubic system is given exactly by:
B = (C11 + 2C12)/3. (1)
The Reuss modulus GR is given by:
GR = 5(C11 − C12)C44
/[4C44 + 3(C11 − C12)] (2)
and the Voigt modulus is given by:
GV = (C11 − C12 + 3C44)/5. (3)

188 The European Physical Journal B
Table 2. The elastic wave velocities (in m/s) for different propagation directions for c-SiGe2N4.
Pressure (GPa) v100L v100
T1 v100T2 v110
L v110T1 v110
T2 v111L v111
T1 v111T2
0 8838 6748 6748 9909 6748 5045 10241 5670 5670
10 9240 6831 6831 10252 6831 5189 10568 5788 5788
20 9545 6875 6875 10515 6875 5272 10819 5855 5855
30 9798 6892 6892 10732 6892 5320 11026 5891 5891
40 10035 6914 6914 10945 6914 5358 11232 5922 5922
Fig. 2. (a) Pressure dependence of the internal structural pa-rameter u. The solid lines are least-square fits of the datapoints to a quadratic polynomial. (b) The calculated pressure-volume relation for c-SiGe2N4. The solid lines are given by theBirch-Murnaghan equation of state with the parameters listedin Table 1.
The shear modulus G is given by:
G = (GR + GV )/2. (4)
We also calculate the Young’s modulus E, Poisson’s rationσ and Lame’s coefficients (μ and λ), which are frequentlymeasured for polycrystalline materials when investigatingtheir hardness. These quantities are related to the bulkmodulus B and the shear modulus G by the followingequations [26]:
E = 9BG/(3B + G) and σ = (3B − E)
/(6B) (5)
λ = σE/((1 + σ)(1 − 2σ) and μ = E
/(2(1 + σ)). (6)
The calculated bulk modulus B, shear modulus G,Young’s modulus E, Poisson’s ratio σ and Lame’s coef-ficients (μ and λ) of the c-SiGe2N4 compound are given
in Table 1. We can see that the calculated value of thebulk modulus B0 from the elastic constants has nearlythe same value as the one obtained from the EOS fitting.These might be an estimate of the reliability and accu-racy of our calculated elastic constants for the c-SiGe2N4
compound.A simple relationship, which empirically links the plas-
tic properties of materials with their elastic moduli, wasproposed by Pugh [27]. The shear modulus G repre-sents the resistance to plastic deformation, while B rep-resents their resistance to fracture [28]. A high B/G ratiois associated with ductility, whereas a low value corre-sponds to the brittle nature. The critical value which sep-arates ductile and brittle materials is around 1.75; i.e.,if B/G > 1.75, the material behaves in a ductile man-ner; otherwise the material behaves in a brittle manner.Frantsevich et al. [29], in a similar fashion, have suggestedB/G ≈ 2.67 as the critical value separating brittle andductile behaviours. We have found that B/G ratios areabout 1.25, classifying this material as brittle.
3.2.2 Elastic wave velocities
From the theoretical elastic constants, we have computedthe elastic wave velocities. The single-crystal elastic wavevelocities, in different directions, are given by the resolu-tion of the Cristoffel equation [30]:
(Cijkl nj nk − ρv2 δil)ul = 0 (7)
Cijkl is the single-crystal elastic constant tensor, n is thewave propagation direction, ρ is the density of material,u is the wave polarization and v is the wave velocity.The solutions of this equation are of two types: a lon-gitudinal wave with polarization parallel to the directionof propagation (vL) and two shear waves (vT1 and vT2)with polarization perpendicular to n. The calculated elas-tic wave velocities along [100], [110] and [111] directionsfor c-SiGe2N4 compound at zero, 10, 20, 30 and 40 GPapressures are shown in Table 2. The longitudinal waves arefastest along [111] and the shear waves are slowest along[110] for c-SiGe2N4.
3.3 Pressure dependence of elastic constants
We next study the pressure dependence of the elastic prop-erties. In Figure 3, we present the variation of the elasticconstants (C11, C12 and C44) and the bulk modulus B

A. Bouhemadou et al.: Structural, elastic, electronic, optical and thermal properties of c-SiGe2N4 189
Fig. 3. (Color online) Pressure dependence of the elasticconstants (C11, C12 and C44) and the bulk modulus B forc-SiGe2N4.
of c-SiGe2N4 as a function of the pressure. We observe alinear dependence in all curves in the considered range ofpressure. In Table 1, we list the results for the pressurederivatives ∂C11/∂P , ∂C12/∂P , ∂C44/∂P and ∂B/∂P . Itis easy to observe that the elastic constants Cij and bulkmodulus B increase when the pressure is enhanced. Fora cubic crystal under pressure P , the generalized elasticstability criteria [24] are:
13(C11 + 2C12 + P ) > 0 ; (C44 − P ) > 0 ;
12(C11 − C12 − 2P ) > 0. (8)
These criteria are satisfied in the studied pressure range,suggesting that c-SiGe2N4 is mechanically stable.
3.3.1 Debye temperature
Once we have calculated the Young’s modulus E, the bulkmodulus B and the shear modulus G, we may obtain theDebye temperature, which is an important fundamentalparameter closely related to many physical properties suchas specific heat and melting temperature. At low temper-atures the vibrational excitations arise solely from acous-tic vibrations. Hence, at low temperature the Debye tem-perature calculated from elastic constants is the same asthat determined from specific heat measurements. One ofthe standard methods to calculate the Debye tempera-ture (θD) is from elastic constants data, since θD may beestimated from the average sound velocity, νm, by the fol-lowing equation [31]:
θD =h
kB
[3
4πVa
] 13
νm (9)
where h is the Plank’s constant, kB the Boltzmann’s con-stant and Va is the atomic volume. The average sound
Table 3. Calculated density (ρ in g/cm3), the longitudinal,transverse and average sound velocity (vl, vt, vm in m/s) andthe Debye temperature (θD in K), calculated from the elasticconstants, for c-SiGe2N4 compound at zero, 10, 20, 30 and40 GPa pressures.
Pressure (GPa) ρ vl vt vm θD
0 6.32 9604 6005 7584 1090a
1079b
10 6.54 9968 6118 7774 1129
20 6.73 10246 6180 7895 1158
30 6.91 10474 6212 7976 1180
40 7.07 10694 6242 8052 1201
a From the elastic constants; b from the quasi-harmonic Debyemodel.
velocity in the polycrystalline material is given by [32]:
νm =[13
(2ν3
t
+1ν3
l
)]− 13
(10)
where νl and νt are the longitudinal and transverse soundvelocity in an isotropic material. These can be obtainedfrom the shear modulus G and the bulk modulus B, bythe use of the Navier’s equation [26]:
νl =(
3B + 4G
3ρ
) 12
and νt =(
G
ρ
) 12
. (11)
The calculated sound velocities and Debye temperature aswell as the density of c-SiGe2N4 at 0, 10, 30 and 40 GPapressures are given in Table 3.
3.4 Electronic properties
3.4.1 Band structure
In Figure 4, we show the electronic band dispersion curvesof c-SiGe2N4 along some high symmetry directions of theBrillouin zone, calculated at equilibrium volume withinthe GGA approximation. The overall band profiles arein fairly good agreement with previous theoretical re-sults [8,10]. The valence band maximum (VBM) and theconduction band minimum (CBM) are located at Γ point,resulting in a direct band gap (Γ–Γ) of 2.14 eV. The cal-culated band gaps are given in Table 1, along with theavailable theoretical results [8,10] for comparison. In viewof Table 1, it is clear that our calculated value for theband gap, using the PP-PW method within the GGA,is larger than that obtained by the FP-LAPW methodwithin GGA [10] and OLCAO method within LDA [8].We expect that this difference in band gap values might bedue to the different theoretical treatments of the exchange-correlation potential and basis function set used in ex-panding the wave functions. It is well known that in theself consistent DFT both LDA and GGA usually under-estimate the energy gap [33,34]. This is mainly due to the
190 The European Physical Journal B
Fig. 4. Electronic band dispersion curves for c-SiGe2N4 alongsome high symmetry directions of the Brillouin zone. Verticallines represent high-symmetry points of the Brillouin zone. Thehorizontal dashed line designates the Fermi level (EF).
fact that they have oversimplified forms that are not suffi-ciently flexible to accurately reproduce both the exchange-correlation energy and its charge derivative. The real bandgap of c-SiGe2N4 must be wider. We notice that no exper-imental data are available for the band gap of this com-pound; all the previous results are obtained via theoreticalmethods.
In order to investigate the effects of the pressure on thesize of the energy gaps of SiGe2N4, the band gap energiesat selected symmetry points are studied as a function ofpressure. Figure 5 shows the plots of the pressure variationof the direct gap (Γ–Γ) and indirect gaps (Γ–L, Γ–X, Γ–K and Γ–W) for c-SiGe2N4. All the calculated band gapsare well fitted to a quadratic function: Eg(P ) = Eg(0) +αP + βP 2, where Eg is in eV, P the pressure in GPa, αand β are the first- and second-order pressure derivatives,respectively. The gaps increase when pressure is enhanced.
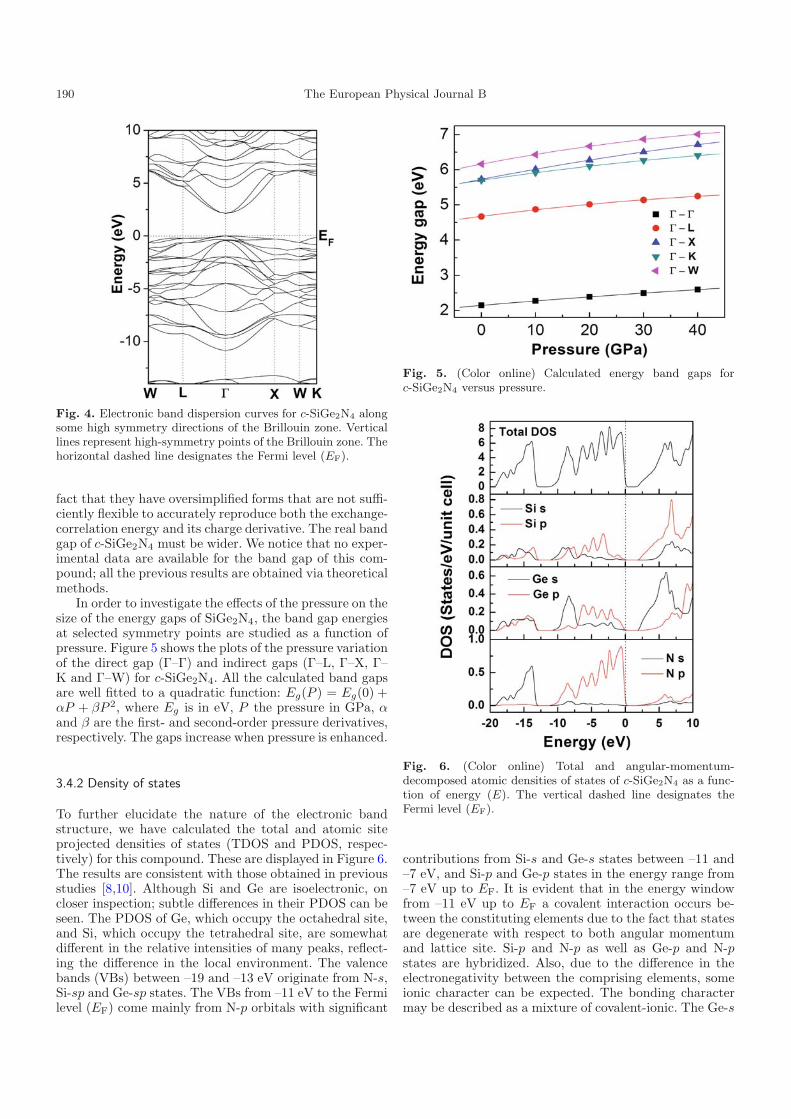
3.4.2 Density of states
To further elucidate the nature of the electronic bandstructure, we have calculated the total and atomic siteprojected densities of states (TDOS and PDOS, respec-tively) for this compound. These are displayed in Figure 6.The results are consistent with those obtained in previousstudies [8,10]. Although Si and Ge are isoelectronic, oncloser inspection; subtle differences in their PDOS can beseen. The PDOS of Ge, which occupy the octahedral site,and Si, which occupy the tetrahedral site, are somewhatdifferent in the relative intensities of many peaks, reflect-ing the difference in the local environment. The valencebands (VBs) between –19 and –13 eV originate from N-s,Si-sp and Ge-sp states. The VBs from –11 eV to the Fermilevel (EF) come mainly from N-p orbitals with significant
Fig. 5. (Color online) Calculated energy band gaps forc-SiGe2N4 versus pressure.
Fig. 6. (Color online) Total and angular-momentum-decomposed atomic densities of states of c-SiGe2N4 as a func-tion of energy (E). The vertical dashed line designates theFermi level (EF).
contributions from Si-s and Ge-s states between –11 and–7 eV, and Si-p and Ge-p states in the energy range from–7 eV up to EF. It is evident that in the energy windowfrom –11 eV up to EF a covalent interaction occurs be-tween the constituting elements due to the fact that statesare degenerate with respect to both angular momentumand lattice site. Si-p and N-p as well as Ge-p and N-pstates are hybridized. Also, due to the difference in theelectronegativity between the comprising elements, someionic character can be expected. The bonding charactermay be described as a mixture of covalent-ionic. The Ge-s

A. Bouhemadou et al.: Structural, elastic, electronic, optical and thermal properties of c-SiGe2N4 191
Table 4. Mulliken charges, bond lengths and bond populationsfor the c-SiGe2N4 compound.
Species Charge Bond lengths Bond
(electrons) (A) populations
Si 1.49 N–Si: 1.772 0.79
Ge 1.37 N–Ge: 1.950 0.36
N –1.06
Fig. 7. (Color online) Calculated real part ε1(ω) and imagi-nary part ε2(ω) of the dielectric function ε(ω) for c-SiGe2N4.
and Si-p states dominate the bottom of the conductionbands (CBs) ranging from 2 to about 8 eV.
3.4.3 Charge transfer and bond populations
We have calculated the charge transfer and bond popula-tions in order to understand bonding behaviour. Table 4shows the atomic charges, chemical bond lengths andbond populations calculated by means of the Mullikenanalysis. The charge transfer from Si and Ge atoms tothe neighbouring N atoms is about 1.49 and 1.37 elec-trons, respectively. This suggests a valence state ofSi+1.49(Ge+1.37)2(N−1.06)4. The bond populations indi-cate the overlap degree of the electron cloud of two bond-ing atoms and can be used to access covalent or ionic na-ture of a chemical bond. For the bond populations, thelowest and highest values imply that the chemical bondexhibits strong ionicity and covalency, respectively [35,36].Therefore, we conclude that the bonding behaviour ofc-SiGe2N4 is a combination of covalent and ionic nature.Moreover, the Si-N bond possesses stronger covalent bond-ing than the Ge-N bond which is more ionic.
3.5 Optical properties
The calculated optical properties at the equilibrium lat-tice constant are presented in Figures 7–10, for the energyrange up to 40 eV. Figure 7 shows the real and imagi-nary parts of the dielectric function for c-SiGe2N4. Theoverall real and imaginary parts of the dielectric function
Fig. 8. (Color online) Calculated refractive index n(ω) andextinction coefficient k(ω) for c-SiGe2N4.
Fig. 9. Calculated optical reflectivityR(ω) (a). Calculatedelectron energy loss spectrum L(ω) of c-SiGe2N4 (b).
spectra profiles are in fairly good agreement with previoustheoretical results [8,10]. The calculated imaginary partε2(ω) exhibit two prominent peaks located at 7.65 and9.41 eV, with a magnitude of 12.48 and 9.0, respectively.Comparing to the results reported by Ching et al. [8] andWang e al. [10], our photon energy for the highest peak areslightly bigger than theirs. This result can be explained bythe slightly higher value of our gap. The magnitude of thispeak in the present work is slightly higher than the one ofreference [8] but slightly lower than that of reference [10].There are also some smaller structures superimposed onthe main structures. The most important contribution tothe first prominent peak can probably be attributed to theinterband transition from occupied states, with predomi-nantly N p character, to hybridized unoccupied Si p and

192 The European Physical Journal B
Fig. 10. Pressure dependence of the static optical dielectricconstant ε(0) and refractive index n(0) of c-SiGe2N4.
Ge s states. The second peak is probably related to theinterband transition from occupied states with predomi-nantly N p character to hybridized unoccupied Si p and Gep states. For the real part ε1(ω) spectra, the main peakwith a magnitude of 12.27 is located at about 6.92 eV.We note that since the GGA underestimates the bandgap of semiconductors, the calculated positions of struc-tures in the optical spectra may also be smaller than theexperimental values which are not available at the mo-ment. Our calculated static dielectric constant ε(0) valuefor c-SiGe2N4 equal to 5.84 is to be compared with thecalculated values of 6.20 by Wing et al. [8] and 6.46 byWang and co-workers [10].
The refractive index and the extinction coefficient aredisplayed in Figure 8. The static refractive index n(0) isfound to have the value 2.42. It increases with energy inthe transparency region, reaching a peak in the ultravioletat about 7.10 eV. It then decreases to a minimum level at23.34 eV. The origin of the structures in the imaginarypart of the dielectric function also explains the structuresin the refractive index.
The electron energy loss function L(ω) is an impor-tant factor describing the energy loss of a fast electrontraversing in a material. The prominent peaks in L(ω)spectra represent the characteristic associated with theplasma resonance (a collective oscillation of the valenceelectrons) and the corresponding frequency is the so-calledplasma frequency ωP . The peaks of L(ω) correspond tothe trailing edges in the reflection spectra; R(ω); for in-stance, the prominent peak of L(ω) located at about25.13 eV (Fig. 9b) is at an energy corresponding to theabrupt reduction of R(ω) (Fig. 9a). Our calculated plasma
Fig. 11. (Color online) Variation of the normalized volumewith pressure (a) and with temperature (b) for c-SiGe2N4. V0
is the equilibrium volume at T = 0 K.
frequency ωp is in reasonable agreement with those previ-ously reported [8,10].
Figure 10 shows the pressure dependence of the staticdielectric constant ε(0) and static refractive index n(0).As can be seen, the increase of the static dielectric con-stant and static refractive index with pressure is practi-cally quadratic.
3.6 Thermodynamic properties
To investigate the thermodynamic properties of thec-SiGe2N4 compound under high temperature and highpressure, we apply the quasi-harmonic Debye approxima-tion. As a first step, a set of total energy calculation versusprimitive cell volume (E−V ) was carried out, in the staticapproximation. The results are then fitted with a numeri-cal EOS in order to determine its structural parameters atP = 0 and T = 0, and to derive the macroscopic propertiesas a function of P and T from standard thermodynamicrelations.
In Figure 11, we present the normalized volume-temperature diagram of c-SiGe2N4 at several pressures.The primitive cell volume increases with increasing tem-perature but the rate is more important for temperaturerange above 400 K. On the other side, as the pressure Pincreases, the relative volume V/V0 decreases at a giventemperature. The effect of increasing temperature on thelattice parameter is just the same as decreasing pressure.
In Figure 12, we present the variations of the volumeexpansion coefficient α as function of temperature, α−T ,and pressure. It is shown that, at a given pressure, α in-creases sharply with the increase of temperature up to700 K. When T > 700 K, α gradually approaches a linearincrease with enhanced temperature and the propensityof increment becomes moderate, which means that thetemperature dependence of α is very small at high tem-perature. For a given temperature, α decreases drastically

A. Bouhemadou et al.: Structural, elastic, electronic, optical and thermal properties of c-SiGe2N4 193
Fig. 12. (Color online) Variation of the bulk modulus withtemperature at some pressures for c-SiGe2N4.
Fig. 13. (Color online) Variation of the heat capacities CV
with temperature at some pressures for c-SiGe2N4.
with the increase of pressure. At 300 K and zero pressure,α = 1.01886× 105 K−1.
Figure 13 shows the bulk modulus variation versustemperature at a given pressure. One can notice thatthe bulk modulus is nearly constant from 0 to 200 Kand decreases linearly with increasing temperature forT > 400 K. The compressibility increases with increas-ing temperature at a given pressure and decreases withpressure at a given temperature. These results are due tothe fact that the effect of increasing pressure on the ma-terial is the same as that of the decreasing temperature.At 300 K and zero pressure, B = 272 GPa.
The variation of the heat capacities CV versus temper-ature at 0, 20 and 40 GPa pressures is shown in Figure 14.It is found that when T < 1500 K, the heat capac-ity CV is depending on both temperature and pressure.At higher temperature (T > 1500 K) CV tends to thePetit and Dulong limit, which is common to all solids athigh temperature. At high temperature CV approaches173 J mol−1 K−1. At zero pressure and 300 K, CV is99 Jmol−1 K−1.
Fig. 14. (Color online) Variations of the volume expansioncoefficient α as function of temperature at some pressures forc-SiGe2N4.
Figure 15 displays the dependence of the Debye tem-perature θD on temperature and pressure. It can be seenthat θD is nearly constant from 0 to 200 K and decreaseslinearly with increasing temperature for T > 200 K. Itis also shown that when the temperature is constant, theDebye temperature increases almost linearly with appliedpressure. Our calculated θD at zero pressure and zero tem-perature is 1079 K, which is in good agreement with thevalue computed accurately in terms of the elastic con-stants (see Tab. 3). This might be an indication that thequasi-harmonic Debye model is a very reasonable alterna-tive to account for the thermal effects with no expensivetask in terms of computational time.
4 Conclusions
This study reports a detailed investigation of the struc-tural, electronic, elastic, optical and thermal propertiesof the c-SiGe2N4 compound using the PP-PW methodwithin the GGA. The most relevant results are summa-rized as follows:
1. The calculated ground state properties such as latticeparameters, bulk modulus and its pressure derivativeagree quite well with the available theoretical results.
2. The calculated elastic constants showed thatc-SiGe2N4 is elastically stable.
3. Some elastic moduli and related properties for idealpolycrystalline c-SiGe2N4 aggregate are predicted.
4. The calculated band structures show that c-SiGe2N4
is a direct band gap material. Our calculated funda-mental gap is 2.14 eV and increases with increasingpressure. We note that since the GGA underestimatesthe band gap of semiconductors, the calculated bandgap value may be smaller than the experimental valuewhich is not available at the moment. This type of un-derestimate is a well-known feature of DFT, mainlywhen applied to semiconductors and insulators; it oc-curs because one is incorrectly interpreting the true

194 The European Physical Journal B
Fig. 15. (Color online) Variations of the Debye tempera-ture θD as function of pressure (a) and temperature (b) forc-SiGe2N4.
unoccupied states of the system with the correspond-ing Kohn-Sham states of DFT.
5. The density of states and Mulliken charge populationsanalysis show that the bonding in c-SiGe2N4 is of acovalent-ionic nature.
6. The optical constants such as the dielectric function,refractive index, extinction coefficient, reflectivity andloss energy function have been studied.
7. The quasi-harmonic Debye model is successfully ap-plied to determine the thermal properties of c-SiGe2N4
in the 0–2000 K temperature range.8. To the best of our knowledge, there are no previous
reports on the elastic constants, thermodynamic prop-erties and the effect of pressure on the electronic struc-ture. It is our ambition that these results will sparkexperimental interest to this compound.
One of us (Y.A.) would like to mention that his contribution tothis work is supported from University Malaysia Perlis, grantNo. 9001-00073.
References
1. S.-D. Mo, L. Ouyang, W.Y. Ching, I. Tanaka, Y. Koyama,R. Riedel, Phys. Rev. Lett. 83, 5046 (1999)
2. I. Kohastu, J.W. McCauley, Mater. Res. Bull. 9, 917(1974)
3. H.F. Priest, F.C. Burns, G.L. Priest, E.C. Skaar, J. Am.Ceram. Soc. 56, 395 (1973)
4. O. Borgen, H.M. Seip, Acta Chem. Scand. 15, 1789 (1961)5. S. Wild, P. Grieveson, K.H. Jack, Spec. Ceram. 5, 385
(1972)6. A. Zerr, G. Miehe, G. Serghiou, M. Schwartz, E. Kroke,
R. Riedel, H. Fuess, P. Kroll, R. Boehler, Nature 400, 340(1999)
7. W.Y. Ching, S.-D. Mo, L. Ouyang, P. Rulis, J. Am. Ceram.Soc. 85, 75 (2002)
8. W.Y. Ching, S.-D. Mo, L. Ouyang, Phys. Rev. B 63,245110 (2001)
9. W.Y. Ching, S.-D. Mo, I. Tanaka, M. Yoshiya, Phys. Rev.B 63, 064102 (2001)
10. H. Wang, Y. Chen, Y. Kaneta, S. Iwata, Eur. Phys. B 59,155 (2007)
11. M.D. Segall, P.J.D. Lindan, M.J. Probert, C.J. Pickard,P.J. Hasnip, S.J. Clark, M.C. Payne, J. Phys.: Condens.Matter 14, 2717 (2002)
12. D. Vanderbilt, Phys. Rev. B 41, 7892 (1990)13. J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77,
3865 (1996)14. H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13, 5188 (1976)15. T.H. Fischer, J. Almlof, J. Phys. Chem. 96, 9768 (1992)16. V. Milman, M.C. Warren, J. Phys.: Condens. Matter 13,
241 (2001)17. M.A. Blonco, E. Francisco, V. Luana, Comput. Phys.
Commun. 158, 57 (2004)18. M.A. Blonco, A.M. Pendas, E. Francisco, J.M. Recio, R.
Franco, J. Molec. Struct. Theochem. 368, 245 (1996)19. M. Florez, J.M. Recio, E. Francisco, M.A. Blonco, A.M.
Pendas, Phys. Rev. B 66, 144112 (2002)20. E. Francisco, J.M. Recio, M.A. Blonco, A.M. Pendas, J.
Phys. Chem. 102, 1595 (1998)21. E. Francisco, M.A. Blonco, G. Sanjurjo, Phys. Rev. B 63,
049107 (2001)22. S.M. Hosseini, Phys. Stat. Sol. (b) 245, 2800 (2008)23. F. Birch, J. Geophys. Res. B 83, 1257 (1978)24. M.J. Mehl, B.M. Klein, D.A. Papaconstantopoulos,
Intermetallic Compounds: Principle and Practice, Vol. I:Principles, edited by J.H. Westbrook, R.L. Fleischeir (JohnWiley and Sons, London, 1995), Chap. 9, pp. 195–210
25. R. Hill, Proc. Phys. Soc. London A 65, 349 (1952)26. E. Schreiber, O.L. Anderson, N. Soga, Elastic Constants
and Their Measurement (McGraw-Hill, New York, 1973)27. S.F. Pugh, Philos. Mag. 45, 823 (1954)28. G. Vaitheeswaran, V. Kanchana, R.S. Kumar, A.L.
Cornelius, M.F. Nicol, A. Savane, A. Delin, B. Johansson,Phys. Rev. B 76, 014107 (2007)
29. I.N. Frantsevich, F.F. Voronov, S.A. Bokuta, ElasticConstants and Elastic Moduli of Metals and InsulatorsHandbook, edited by I.N. Frantsevich (Naukova Dumka,Kiev, 1983), pp. 60–180
30. B.B. Karki, L. Stixrude, S.J. Clark, M.C. Warren, G.J.Ackland, J. Crain, Am. Mineral. 82, 51 (1997)
31. P. Wachter, M. Filzmoser, J. Rebizant, Physica B 293, 199(2001)
32. O.L. Anderson, J. Phys. Chem. Solids 24, 909 (1963)33. G. Onida, L. Reining, A. Rubio, Rev. Modern Phys. 74,
601 (2002)34. S. Fahy, K.J. Chang, S.G. Louis, M.L. Cohen, Phys. Rev.
B 35, 7840 (1989)35. M.D. Segall, C.J. Pickard, R. Shah, M.C. Payne, Phys.
Rev. B 54, 16317 (1996)36. J. Sun, X.F. Zhou, Y.X. Fan, J. Chen, H.T. Wang, Phys.
Rev. B 73, 045108 (2006)
Related Documents











