2152 H. S. Rothenfluh, L. Taylor, A. L. M. Bothwell et al. Eur. J. Immunol. 1993. 23: 2152-2159 Harald S. Rothenfluh., Linda TaylorA, Alfred L. M. BothwellO, Gerald W. Both. and Edward J. Steele.. Department of Biological Sciences, University of Wollongong., Wollongong, Division of Biomolecular Engineering, Commonwealth Scientific and Industrial Research Organisation., North Ryde, Section of Immunobiology, Yale Medical Schoolo, New Haven and Division of Cell Biology, John Curtin School of Medical Research., Canberra Somatic hypermutation in 5' flanking regions of heavy chain antibody variable regions" The aim of this study has been to determine the distribution of somatic mutations in the 5' flanking regions of rearranged immunoglobulin heavy chain variable region genes (VDJ).We sequenced the 5' flanking region in 12secondary immune response antibodies produced in C57BL/6j mice against the hapten (Chydroxy- 3-nitropheny1)acetyl (NP) coupled to chicken-y-globulin. In these and previously published sequences, almost 97 % of the mutations occurred in the transcribed region of the gene, and only a minority of genes (5/29) contained mutations upstream of the transcription start (cap) site. No potential germ-line donor was found for a cluster of five base changes previously found in a single heavy chain gene, 3B62. However, the uniqueness of this mutational cluster and its distance from the normally mutated region suggests that the nucleotide changes may not be due to the normal mutator mechanism. Thus, as this was the only instance of somatic mutations that far upstream of the promotedcap site region, the reverse transcriptase model for somatic hypermutation is still a possibility. The data are consistent with a mutational mechanism that requires transcription of the rearranged target V(D)J gene which appears to result in the generation of a positively skewed asymmetrical distribution of somatic mutations. A single mode is centered near theV(D)J and a long tail extends into the 3' non-translatedregion of the J-C intron. Two classes of model could explain this mutation distribution pattern: those where transcription products (RNA, cDNA) are the direct mutational substrates, or those that postulate local unfolding of the chromatin around a V(D)J rearrangement directly exposing the DNA of the transcribed region to specific mutational enzymes. 1 Introduction Somatic hypermutation of mammalianv region genes is an important generator of antibody diversity (reviewed in [ 11). In mice the process is characterized by the introduction of mainly single base substitutions into the genomic DNA of rearranged V region genes and their flanking regions [2-51. It is now established that mutations are introduced during oligoclonal B cell growth in germinal centers [6-81. The transgenic mouse line expressing x light chains developed by Storb and colleagues has illustrated clearly that the mutational substrate is localized to a small region of DNA in and around a V(D)J gene and is accessable to analysis [I 114741 * This research was supported by funds from the University of Wollongong - Commonwealth Scientific and Industrial Research Organization collaborative research grant, a NH&MRC project grant and a University of Wollongong project grant. H. S. Rothenfluh is supported by the Government Employees Medical Research Fund (NSW). A Present address: Bioprocessing Ltd., Medomsley Road, Con- sett, Co. Durham DH8 6TJ, GB Correspondence: Edward J. Steele, Department of Biological Sciences,The University of Wollongong, Northfields Ave. ,Wollon- gong, NSW, 2522, Australia . Abbreviation: CDR Complementarity-determining region Key words: Somatic hypermutation / V regiod5' Flanking region I Murine heavy chain [9-111. It has also been shown that the mutator machinery for both heavy chains and x light chains is focused on the rearranged V(D)J gene irrespective of the J segment utilized [5, 121.The mutator mechanism and the sequences which may trigger it are ill-defined, although the immuno- globulin heavy chain intron enhancer by itself is not sufficient to initiate mutation in a Tcell receptor VDJ transgene [ 131. This implies that other cis-acting elements are required for hypermutation of molecular substrates expressed in B cells. The precise boundaries which define the DNA substrate for mutation are also poorly delineated. Previous work on the substrate for mutation produced different conclusions as to where these boundaries lie [4, 51. Recent results from studies on light chain genes need further investigation. One study suggested that in x light chains sequences 3' of the Cregion may be necessary for the variable region to hypermutate [14]. However the 31 chain transgene used in the negative control experiment [15] may have failed to hypermutate as a result of the use of rat J-C intron and C region sequences. Recently, hl light chain C regions were also shown to undergo somatic hypermutation, suggesting that sequences signalling the termination of mutation are absent in this locus or are contained within the C region because the h chain J-C intron is short compared to the J-C intron of the other loci [16]. An earlier review of available data suggested that the distribution of somatic mutations around rearranged V re- gions may be positively skewed, with a single mode centred on the V(D)J gene and a long tail extending into the J-C intron [17], however it was pointed out that a larger data set was required for a more conclusive analysis. Therefore, to determine accurately the nature of the underlying frequen- 001 4-2980/93/0909-2 152$10 .OO + .25/0 0 VCH Verlagsgesellschaft mbH, D-69451 Weinheim, 1993

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

2152 H. S. Rothenfluh, L. Taylor, A. L. M. Bothwell et al. Eur. J. Immunol. 1993. 23: 2152-2159
Harald S. Rothenfluh., Linda TaylorA, Alfred L. M. BothwellO, Gerald W. Both. and Edward J. Steele..
Department of Biological Sciences, University of Wollongong., Wollongong, Division of Biomolecular Engineering, Commonwealth Scientific and Industrial Research Organisation., North Ryde, Section of Immunobiology, Yale Medical Schoolo, New Haven and Division of Cell Biology, John Curtin School of Medical Research., Canberra
Somatic hypermutation in 5' flanking regions of heavy chain antibody variable regions"
The aim of this study has been to determine the distribution of somatic mutations in the 5' flanking regions of rearranged immunoglobulin heavy chain variable region genes (VDJ). We sequenced the 5' flanking region in 12 secondary immune response antibodies produced in C57BL/6j mice against the hapten (Chydroxy- 3-nitropheny1)acetyl (NP) coupled to chicken-y-globulin. In these and previously published sequences, almost 97 % of the mutations occurred in the transcribed region of the gene, and only a minority of genes (5/29) contained mutations upstream of the transcription start (cap) site. No potential germ-line donor was found for a cluster of five base changes previously found in a single heavy chain gene, 3B62. However, the uniqueness of this mutational cluster and its distance from the normally mutated region suggests that the nucleotide changes may not be due to the normal mutator mechanism. Thus, as this was the only instance of somatic mutations that far upstream of the promotedcap site region, the reverse transcriptase model for somatic hypermutation is still a possibility. The data are consistent with a mutational mechanism that requires transcription of the rearranged target V(D)J gene which appears to result in the generation of a positively skewed asymmetrical distribution of somatic mutations. A single mode is centered near theV(D)J and a long tail extends into the 3' non-translatedregion of the J-C intron. Two classes of model could explain this mutation distribution pattern: those where transcription products (RNA, cDNA) are the direct mutational substrates, or those that postulate local unfolding of the chromatin around a V(D)J rearrangement directly exposing the DNA of the transcribed region to specific mutational enzymes.
1 Introduction
Somatic hypermutation of mammalianv region genes is an important generator of antibody diversity (reviewed in [ 11). In mice the process is characterized by the introduction of mainly single base substitutions into the genomic DNA of rearranged V region genes and their flanking regions [2-51. It is now established that mutations are introduced during oligoclonal B cell growth in germinal centers [6-81. The transgenic mouse line expressing x light chains developed by Storb and colleagues has illustrated clearly that the mutational substrate is localized to a small region of DNA in and around a V(D)J gene and is accessable to analysis
[I 114741
* This research was supported by funds from the University of Wollongong - Commonwealth Scientific and Industrial Research Organization collaborative research grant, a NH&MRC project grant and a University of Wollongong project grant. H. S. Rothenfluh is supported by the Government Employees Medical Research Fund (NSW).
A Present address: Bioprocessing Ltd., Medomsley Road, Con- sett, Co. Durham DH8 6TJ, GB
Correspondence: Edward J. Steele, Department of Biological Sciences,The University of Wollongong, Northfields Ave. ,Wollon- gong, NSW, 2522, Australia .
Abbreviation: CDR Complementarity-determining region
Key words: Somatic hypermutation / V regiod5' Flanking region I Murine heavy chain
[9-111. It has also been shown that the mutator machinery for both heavy chains and x light chains is focused on the rearranged V(D)J gene irrespective of the J segment utilized [5, 121.The mutator mechanism and the sequences which may trigger it are ill-defined, although the immuno- globulin heavy chain intron enhancer by itself is not sufficient to initiate mutation in a Tcell receptor VDJ transgene [ 131. This implies that other cis-acting elements are required for hypermutation of molecular substrates expressed in B cells.
The precise boundaries which define the DNA substrate for mutation are also poorly delineated. Previous work on the substrate for mutation produced different conclusions as to where these boundaries lie [4, 51. Recent results from studies on light chain genes need further investigation. One study suggested that in x light chains sequences 3' of the Cregion may be necessary for the variable region to hypermutate [14]. However the 31 chain transgene used in the negative control experiment [15] may have failed to hypermutate as a result of the use of rat J-C intron and C region sequences. Recently, h l light chain C regions were also shown to undergo somatic hypermutation, suggesting that sequences signalling the termination of mutation are absent in this locus or are contained within the C region because the h chain J-C intron is short compared to the J-C intron of the other loci [16].
An earlier review of available data suggested that the distribution of somatic mutations around rearranged V re- gions may be positively skewed, with a single mode centred on the V(D)J gene and a long tail extending into the J-C intron [17], however it was pointed out that a larger data set was required for a more conclusive analysis. Therefore, to determine accurately the nature of the underlying frequen-
001 4-2980/93/0909-2 152$10 .OO + .25/0 0 VCH Verlagsgesellschaft mbH, D-69451 Weinheim, 1993

Eur. J. Immunol. 1993. 23: 2152-2159 Distribution of somatic mutations in 5' flanking regions 2153
cy distribution and 5' boundary for somatic hypermutation we have now sequenced an additional 12 somatically mutated, rearranged VH regions and their 5' flanking regions to a point approximately 520 bp upstream of the transcription start (cap) site. All 12 immunoglobulins were produced in C57BL/6j mice during a secondary immune response against the hapten NP, and all have been previous- ly characterized and some are clonally related [18, 191. We show here that almost all mutations fall within the trans- cribed region of the V genes, with very few mutational events occurring in the region 5' of the cap site.
We previously reported on a cluster of five nucleotide changes 2 375 bp upstream of the cap site in the heavy chain of the hybridoma 3B62 [4], which also contains a codon deletion within theVgene [19].The proposition that the somatic hypermutator is linked to or dependent on transcription [4, 5,201 does not predict the occurrence of nucleotide changes so far upstream of the cap site. In addition it was suggested that 3B62 may have been derived by gene conversion [19]. It was therefore imperative to determine the origin of the upstream changes. We will discuss the relevance of these nucleotide changes to the mechanism of somatic hypermutation. In addition, we analyze the asymmetrical nature of the mutation frequency distribution as well as the nature and frequency of "selec- tion-free" mutations [21], i.e. changes found in non- translated regions of V(D)J genes that are not affinity- selected by antigen. Such changes may reflect the mutation profile of the enzymatic machinery responsible for somatic hypermutation [21,22].
2 Materials and methods
2.1 PCR amplification
DNA isolated from twelve hybridomas from C57BL/6j mice immunized with the hapten NP [18, 191 was used for amplification with the polymerase chain reaction (PCR, [23]). The sequences and positions of the primers used in PCR amplifications are shown in Fig. 1. The 5' ends of the upstream primers contained a SalI restriction site while the 5' ends of the downstream primers contained an EcoRI restriction site to facilitate cloning. The sequences were determined for the entire rearranged heavy chain variable region to a point approximately 520 bp upstream of the cap site. The coding V region sequences of hybridomas H1-7,
H1-72 [18] and those of hybridomas 3A112, 3C52, 3D61 [19] were previously determined from the mRNA.
PCR amplifications were performed in a 25-p1 reaction mixture containing 7-100 ng hybridoma DNA, 2 pmol each primer, 0.2 mM of each deoxynucleoside triphosphate in 10 mM Tris-HC1 (pH 8.3), 50 mM KC1,2-3 mM MgCl2 and 0.001 YO (w/v) gelatin when 2.5 U of Taq DNA polymerase (Cetus Corp.) was used, or in 20 mM Tris-HC1 (pH 8.2), 10 mM KC1, 6 mM (N&)2S04, 2 mM MgC12, 0.1 YO Triton X-100 and 10 ng/p1 BSA when 2.5 U of Pfu DNA polymer- ase (Stratagene) was used. PCR amplification of 3A112, 3C52,3D61 and H1-72 hybridoma DNA was performed as previously described [4]. The other hybridoma DNA were amplified in a HBTRl thermal cycler (Hybaid Ltd., Middlesex, GB). All reagents except for the primers and
H1-8, H1-27, H1-29, H1-30, H1-39, H1-45, H1-51,
the DNA polymerase were added to a 0.5-ml microcentri- fuge tube and then overlaid with mineral oil. The DNA was denatured at 95 "C for 5 min. After the initial denaturation, a mixture containing the primers and the DNA polymerase was added. The DNA was amplified by 40 cycles of denaturation at 95 "C for 30 s, annealing at 60°C for 30 s, and extension at 72 "C for 1 min 20 s if Taq DNA polyme- rase was used, or at 75°C for 1 min 20 s if Pfu DNA polymerase was used.To complete any unfinished products, a 5-min extension step was employed after the last cycle. The PCR products were analyzed by electrophoresis on 0.6% agarose gels, and the appropriate bands were excised.
2.2 Cloning and screening of PCR products
The 3A112, 3C52, 3D61 and H1-72 PCR fragments were electroeluted and purified as previously described [4]. The other PCR fragments were electroeluted directly into Centricon-100 microconcentrators (Amicon) which were then used to concentrate and purify the DNA fragments.
The 3A112,3C52 and 3D61 fragments were sub-cloned and screened as described [4]. The H1-72 fragment was phos- phorylated, concatamerized by ligation and then restricted with EcoRI and SalI (Boehringer-Mannheim) prior to ligation into vector M13mp19 [24]. The other PCR frag- ments were ligated into vector pUC19. After ligation, DNA was transformed into Escherichia coli JM109 follow- ing heat inactivation of the T4 DNA ligase (Boehringer- Mannheim).The M13mp19 transformants were plated onto 5-bromo-4-chloro-3-indolyl-@-~-galactopyranoside (X-Gal; Boehringer-Mannheim)/isopropyl-@-D-thiogalac- topyranoside (IPTG; Boehringer-Mannheim) minimal plates in order to allow selection of clear recombinant plaques, whereas the pUC19 transformants were plated onto IPTGK-GaYampicillin (100 pg/mL, Boehringer- Mannheim) minimal plates to allow selection of white colonies. Hybridization selection of positive recombinant clones was carried out as described [25].
2.3 PCR amplification with radiolabel incorporation
A modification of a PCR technique allowing the incorpo- ration of a radionucleotide [26] was used to produce radioactive DNA fragments. The PCR reaction mixes were prepared as described above using Taq DNA polymerase (Cetus Corp.). Primers 1 and 2 in Fig. 1 were used to PCR amplify the probe from cloned V ~ 3 B 4 4 DNA [27,28]. The probe was amplified by 30 cycles of denaturation (95 "C), annealing (55 "C) and extension (72"C), following which another 2.5 U of Taq DNA polymerase were added. After another five cycles, 20 pCi of [a-32P] dATP (Bresatec Ltd., Thebarton 5031, South Australia) was added and PCR amplification was then allowed to continue for a further 5 cycles. A sample was electrophoresed on a 1.5 YO agarose gel and the remainder was extracted once with chloroform then precipitated with isopropanol.
2.4 DNA preparation and sequencing
Double-stranded DNA from hybridization positive recom- binants was prepared with a modification of the alkaline

2154 H. S. Rothenfluh, L. Bylor, A. L. M. Bothwell et al. Eur. J. Immunol. 1993. 23: 2152-2159
lysis method [29]. Single-stranded DNA was extracted from the bacteriophage particles with phenol, phenol-chloro- form (1: l ) , chloroform and ether, then precipitated with isopropanol. Single-stranded DNA was sequenced with the dideoxy method [30] using T7 DNA polymerase (Pharma- cia). Double-stranded templates were sequenced with the Taq DNA polymerase cycle sequencing system (Promega) for 40 cycles of denaturation (95 "C), annealing (60°C) and extension (72 "C).
2.5 Southern blot hybridization
The DNA was cut with the appropriate restriction enzymes and then separated on a 2 % agarose gel. The DNA fragments were transferred onto a Hybond-N+ nylon membrane (Amersham) using the alkaline transfer tech- nique previously described [25].The probe was PCR ampli- fied as described above. The membrane was prehybridized in 1.25 mM EDTA, 0.525 m~ Na2HPO4, 7 % SDS and 50 pg/ml sonicated salmon sperm DNA at 55 "C for 1 h.The heat-denatured probe was then added and the membrane was allowed to hybridize overnight. The next day the membrane was washed twice with 1 mM EDTA, 50 mM Na2HPO4, 5 YO SDS at room temperature. Following this the membrane was washed three times with 1 mM EDTA, 100 mM NazHPO4, 1 YO SDS at 55 "C. The membrane was then exposed to Hyperfilm-NP (Amersham) at -70 "C with intensifying screens.
3 Results
3.1 Fidelity of Taq and Pfu DNA polymerases
In this study we have been concerned with the accurate determination of somatic mutation frequencies in the 5' upstream region of rearranged heavy chain V genes. To this end we have employed strategies that allow the identifica- tion of errors introduced into the DNA during PCR amplification. Between three and five different subclones of each rearranged gene were sequenced in order to identify any non-reproducible mutations which may have been induced by Thq or Pfu DNA polymerase. A total of 28 254 bases of DNA amplified with Taq DNA Polymerase was
Table 1. Comparison of the error rates of Taq DNA polymerase and Pfu DNA polymerase
- 96 error rate 0.081 % ltansi tions
TC AG GA CT
CG CA AT
Transversions
sequenced revealing 23 base changes which were probably induced by the enzyme. Twenty of these were transitions (Table 1). The preference for transitions over transversions and the predominance of T.A to C-G transitions (Table 1) is in agreement with previous reports [23,31,32].
Pfu DNA polymerase was used to PCR amplify 9 175 bases that were sequenced. We did not find any unique Pfu DNA polymerase-induced errors (Table 1) indicating that this enzyme is better suited for the accurate amplification of DNA than Taq DNA polymerase.
3.2 Search for the 3B62 RsaI site in C57BL/6j germ-line DNA
To determine whether the mutation that introduced a RsaI restriction site at position 141 in 3B62 (Fig. 2 in [4]) was present in a germ-line gene, we amplified the region flanked by primers 1 and 4 (Fig. 1) from C57BL/6j genomic liver DNA using PCR with the incorporation of a radionucleo- tide. We also amplified the same fragment from cloned V~3B62 DNA and from cloned V~3B44 which was used as
3B44 C57BL 3B62
- 1107
- 560
- 237 - 154 - 109
Figure 2. Autoradiograph showing the radiolabeled, RsaI-res- tricted PCR products amplified using the primers 1 and 4 from the positive (3B62) and negative (3B44) controls and from C57BL16j mouse liver DNA (C57BL).The diagnostic 139-bp fragment in the 3B62 lane is marked by an arrow-head.
L Vu DJu
Figure 1 . Positions of the primers used for PCR amplifications. The primers are represented by arrows. A indicates the codon deletion in 3B62. Primer 5 was used to amplify J H ~ rearrangements, primer 6 for the amplification of J H - ~ rearrangements and primer 7 was used to amplify from 3C52,3A112 and 3D61 hybridoma DNA. Sequence (and positions) of the primers used (numbering system as in [4]): 1: 5'-GCGGTCGACGTGATGCAATATTCIGTTGA- C-3' (11-32); 2: S'-TGGAAA'TTCATCCCAT-3' (370-387); 3: 5'-GCGAA'TTCCACIGTACITAATATCACTATA-3' (898-922); 4: 5'-GCGAA'TTCAGAGTCCTCAGATGTCAGGC- T-3' (991-1001); 5 : 5'-GCGAA'TTCGAGGTTGTAAGGACI'C- ACCI-3' (1411-1430); 6: 5'-GCGAA'TTCAAGACCTGGAGA- GGCCA'TTCT-3' (2370-2390); 7: 5'-GCGAATT'CGTCCCTAT- GCATCGGATACI'GTATA-3' (2734-2758).

Eur. J. Immunol. 1993.23: 2152-2159 Distribution of somatic mutations in 5' flanking regions 2155
required. The fusion partner used in producing the hybri- doma 3B62 was derived from a BALBlc mouse. To address the possibility that the five base substitutions may have been derived by a post-fusion recombination event with a homologous BALBlc gene, we sequenced ten genes ampli- fied from C57BL/6j mouse liver DNA as well as ten genes amplified from BALB/c mouse liver DNA. The sequences spanning the region in which the five base changes occurred in 3B62 are shown for these genes in Fig. 4. The data indicate that not a single one of the five changes was found in the amplified DNA fragments.
the negative control as it lacks an RsaI site at position 141. RsaI restriction of these radioactive PCR products yielded the pattern shown in Fig. 2. The region amplified from V~3B62 contains two additional RsaI restriction sites producing restriction fragments of 522 bp, 255 bp and 103 bp in addition to the RsaI site introduced by the upstream mutation, which generates the 139-bp fragment. The presence of a RsaI site in the V~3B44 PCR product produced a 909-bp and a 110-bp fragment. The 1018-bp fragment in all three lanes indicates the presence of some undigested PCR products. However, the diagnostic 139-bp fragment was absent from the PCR products amplified from both the V~3B44 and genomic DNA, indicating that none of the amplified germ-line genes contained the sequence coding for this upstream RsaI site in hybridoma 3B62.
3.3 Search for the gene carrying the 3B62 codon deletion in C57BL/6j germ-lie DNA
We designed a primer that spanned the region of the codon deletion and several other unique mutations in theVH gene of 3B62 (primer 3, Fig. 1) and used this with primer 1 to amplify C57BL/6j genomic liver DNA, as well as the positive control (cloned V~3B62) and the negative control (V~3B44) DNA. DNA of the expected size (929 bp) was only amplified from 3B62 DNA (Fig. 3a). Bands of the correct sue were not detected for V~3B44 and genomic DNA even following a 48-h over-exposure of the dried agarose gel. To confirm that this was not due to the insensitivity of this PCR assay, we seeded C57BL/6j genomic liver DNA with between 1 and loo00 copies of cloned 3B62 DNA and repeated the PCR amplification. The results shown in Fig. 3b indicate that using these conditions we detected a product from as few as ten template molecules after one round of amplification. Thus, the specific codon deletion characteristic of V~3B62 is absent from C57BL/6j germ-line DNA.
3.4 Sequence analysis of germ-lie V, genes related to the V~186.2 gene
The above strategies were only useful for determining the presence of the base change that created the RsaI site in the DNA. To detect whether any or all of the five changes occurred in germ-line VH genes, sequence information was
-Itm
6 7 - 1
c57-3 c57-4 5 7 - 5
5 7 - 6 5 7 - 7 5 7 - 8 5 1 - 9 -7-10
3 1 ~ 1
cs7-a
0-Un 386a 5 7 - 1
0 7 - 3 5 7 - 4 5 7 - 5
5 7 4 5 7 - 7 5 7 4
0 7 - 9 E57-10
-1
-3 a w 4 -5 a w 6 a w 7 -0 a w e a w l 0
6 7 - 1
a w a
.......... .......... .......... .......... .......... .......... .......... .......... .......... .......... ..........
r n m T c m h ~ A m w 2 & w c .c ............................ .............................. .............................. .............................. ...... T... .................... ........................... 0.. .............................. .............................. .............................. ...... T... .................... a .......................... c..
m u w ~ a a ~ ~ a . n r r r r o r .............................. ....... A.. .................... ....... A.. .................... ....... 0.. .......... ..A. ...... ....... A.. .......... C ......... .. A....o.. .................... ....... A,. .................... ....... A.. .................... ...... A., .................... ....... A.. .......... C . . . . . . ... ...,--. A A ........ A. ... C...A..
......... T ................................ A....o.. .......... ............................................... A.. .......... ......... T ..................................... A.. .......... ............................................... A.. ..........
............................................... A.. .......... ............................................................
........................................ ..A.A..o.. .......... ............................................... A.. .......... ............................................... A.. .......... .......................................... A....O.. .......... 50 60 70 80 90 100
-5T
... E . . . . . .
..........
.......... .......... .......... .......... . ..c ...... ... C. . . . . . ... E......
......... A
..........
.......... .......... .......... .......... .......... .......... .......... .......... .......... .......... 119
?oEAuMm ..... T.... .......... .......... .......... .......... .......... .......... .......... .......... .......... .......... ..A ....... ...... a... .......... .......... .......... ...... a... ...... a,.. .......... .......... ..........
130
1 '-mmmaTm- .......... c ................... ... T...... ..A ................. ... T...... ..A ............ A.... ... T...... .............. R.... ... T...... ..A ............ A.... .......... ..A ................. ... T...... ..Ab....... .......... ... T...... ..A ................. ... T...... ..A....... .......... ... T...... ..A ............ A,.... ... T...A.. .. A....C.. .......... ... T.....C .............. R.... ... T...... ..A ........... R.... ... T...... ..A....... .......... ... T...... .................... .. .T. ...... A.. ............... ... T...... .................... .............................. ... T...... .................... ... T...... ..A ....... C ......... ... T...... .......... ....cI.... , 140 150 160
,lurmmlm .......... .......... .......... ......... A .......... .......... .......... .......... .......... .......... .......... .......... .......... .......... .......... ........ T. .......... .......... .......... .......... ..........
I 170 180
a b lo4 lo3 lo2 10
Figure4 Partial DNA sequences for the upstream region of VH186.2 related germ-line genes. The sequences were obtained from DNA amplified from C57BL/6j mouse liver DNA ((257 clones 1-10) and from BALB/c mouse liver DNA (BALB clones 1-10) and are compared to the V~I86.2 germ-line sequence. The V"3B62 sequence for this region is also shown. C57 clones 4 and 9, C57 clones 6-8 and BALBIc clones 6 and 8 have identical sequences over the region shown, however they are from different germ-line V genes (data not shown).The vertical arrow indicates the position of the diagnostic RsaI site found in the distal S'flanking region of 3B62. Symbols: ".": identical to the germ-line sequence; "-": deletion of a base.
1
Figure 3. (a) Autoradiograph of the radio- labeled DNA amplified with the 3B62 codon deletion-specific PCR from the positive (3B62) and negative (3B44) controls and from C57BL/6j mouse liver DNA (C57BL). (b) Autoradiograph showing the DNA amplified from C57BL/6j mouse liver DNA seeded with 104, 103, 102, 10 and 1 copy of cloned 3B62 DNA. The first two lanes are from a 20-min exposure, the last three lanes were exposed for 20 h.

2156
3.5 Southern blot analysis of C57BL/6j genomic DNA
To detect any possible germ-line donor genes that could serve as donor sequences for 3B62, 5 pg of C57BL/6j liver DNA and the two control DNA were digested with HincII and RsaI. The RsaI site in 3B62 is flanked by two HincII sites which are also present in the V~186.2 germ-line gene. If the RsaI site is also present in a related germ-line gene, a 112-bp fragment and a 129-bp fragment should be prod- uced,whereas if the RsaI site is absent, the two HincII sites will produce a 241-bp fragment. Clearly, the genomic and V~3B44 DNA both yielded the 241-bp fragment (Fig. 5).
H. S. Rothenfluh, L. Taylor, A. L. M. Bothwell et al. Eur. J. Immunol. 1993. 23: 2152-2159
3B62 C578L 3844
2000 - 1429'
766 - 560 - 470 - 237 - 154 - 109 -
Figure 5. Southern blot hybridization of a radioactive probe PCR amplified from cloned 3B44 DNA with homologous DNA frag- ments produced by HincII and RsaI restriction of positive (3B62) and negative (3B44) control DNA and from C57BL16j mouse liver DNA (C57BL).
Only V~3B62 DNA yielded the 112-1129-b~ products. Since the probe extended 108 bp beyond the 3' HincII restriction site, the larger molecular weight band detected in all three lanes (Fig. 5) reflects hybridization of the probe to the downstream HincII or RsaI fragment. Collectively, the above data make it unlikely that a direct precursor of the 3B62 gene exists in the C57BL/6j germ-line DNA.
3.6 Somatic mutation upstream of the cap site is a rare event
The coding sequences for the rearranged heavy chain variable regions from the 12 hybridomas were previously determined [18, 191 and we have extended and corrected these data to provide more information on the frequency of upstream mutation (Fig. 6 and 7). Our data disagree with the previously published sequences at a number of sites (see the legend to Fig. 6). Hybridomas H1-27 and H1-30 thus have identical VDJ coding and 5' non-coding regions. Since the 3' flanking regions of these two hybridomas may differ, we tentatively named the consensus sequence H1-27/30 (Figs. 6 and 7), and scored the mutations found in this sequence only once in all subsequent analyses (Table 2, Fig. 8). These discrepancies do not alter the conclusions drawn in the papers in which the mRNA sequences of these hybridomas were published [18, 191.
To determine the frequency of mutation in the 5' nontrans- lated regions, we extended these sequences to approxi- mately 520 bp 5' to the cap site by sequencing each DNA three to five times. Of the eleven different hybridomas sequenced in that region, onlyone, H1-7,was mutated 5' to the cap site (Fig. 6).Three mutations occurred upstream of, but within 200 bp of the cap site: a G+T transversion at
........ me,. 380 540 550 650 750 770 800 820 830 840
~ 8 6 . 2 n r m c u x ATACWAAA mcyx.prui - nwmrac rrn;ocma crnum cm-n
........................................ ............... T...... ....... 0.. ........ ................................... G.. . ........ - 3c5a ......................................................... c.. ............... c ..........
............................................. ............................................. ............................................. _ _ _ _ _ _ _ --------------=DR1---------------
850 860 890 910 910 940 950 970 990 - - -T Y.'LOOTOOT)L CTupTICuI UIDUrua: cmcAmcw ccm- Q2AQZFaGc vH186.2
3c5a .. G ....................................................................................... HI-72 . ............................ .. 3N12 . ........... T ................ 3D61 . .................................. c.
Figure 6. The condensed sequence of the 5' flanking regions and V, gene elements of various hybridomas (Hl-7, HI-8, H1-27, H1-30, H1-29, H1-39, H1-45, H1-51, H1-72,3A112,3C52 and 3D61) compared with the V~186.2 germ-line sequence [4]. The sequences are divided into blocks of ten nucleotides and only the blocks containing mutations in at least one gene are shown.The hybridomas in families 1 and 3 were grouped together to highlight clonal relationships [ 181. In codons 3,74 and 106 of H1-7,3 and 29 of H1-8,lOO of HI-27.16, 84,87 and lOOof H1-30,61 and 108 of H1-39,108 of H1-45 and 66 and 85 of H1-72 [HI, and in codons 24 of 3C52 and 38 of 3D61[ 191 the sequences determined by us differ from those determined by others ([18, 191, also see update in the EMBL data base).The positions of CDRl and CDR2 are indicated by dashes above the nucleotide sequence. Other annotations are described in the legend to Fig. 4.The numbering is as shown in Fig. 2 of [4].
Eur. J. Immunol. 1993. 23: 2152-2159 Distribution of somatic mutations in 5' flanking regions 2157
mis6.a TAllwmRAQn
81-1 .......... cc 81.8 .......... cc ni-ai130 .......... cc
cTLl6.1 - -C "&---, .......... ..cIc-- - ............................................ .......... ..cIc--- - ............................................ ..... T.... ..cIc----- - ............................................ 81-51 .......... CC .......... ..cIc---- . ............................................
....... OR -. .......... m- . -. ......... ... T. .. OR -. .......... m- . ............ ....... .......... ...- . ......... ___ 81-19 81-39 81-45
3.251 .......... R ........ 1. oLx0o.a- . ................................................
TMXCC .......... .......T-. . ------..T. ..-.. . .. C ..... T. .ID..--- .
Figure 7. Sequences of the hybridomaVH-D-JH junctions (Hl-7, H1-8, H1-27, H1-30, H1-29, H1-39, H1-45, H1-51, H1-72,3A112, 3C52 and 3D61) compared with the germ-line sequences of the V~186.2, D n 1 6 . ~ and JH.genes. The sequence H1-27/30 represents the VH-DJH junction sequence of hybridomas H1-27 and H1-30, which are identical.The additional sequences at some of theVH-D junctions are N regions. The symbols used are as described in the legend to Fig. 4.
F i 4 Y 5 4 I
- 1000 1000 2600 3000 4-00 Nucleotides from cap site
- c l t t c w P L VJHl JH2 JH3 JH4 E
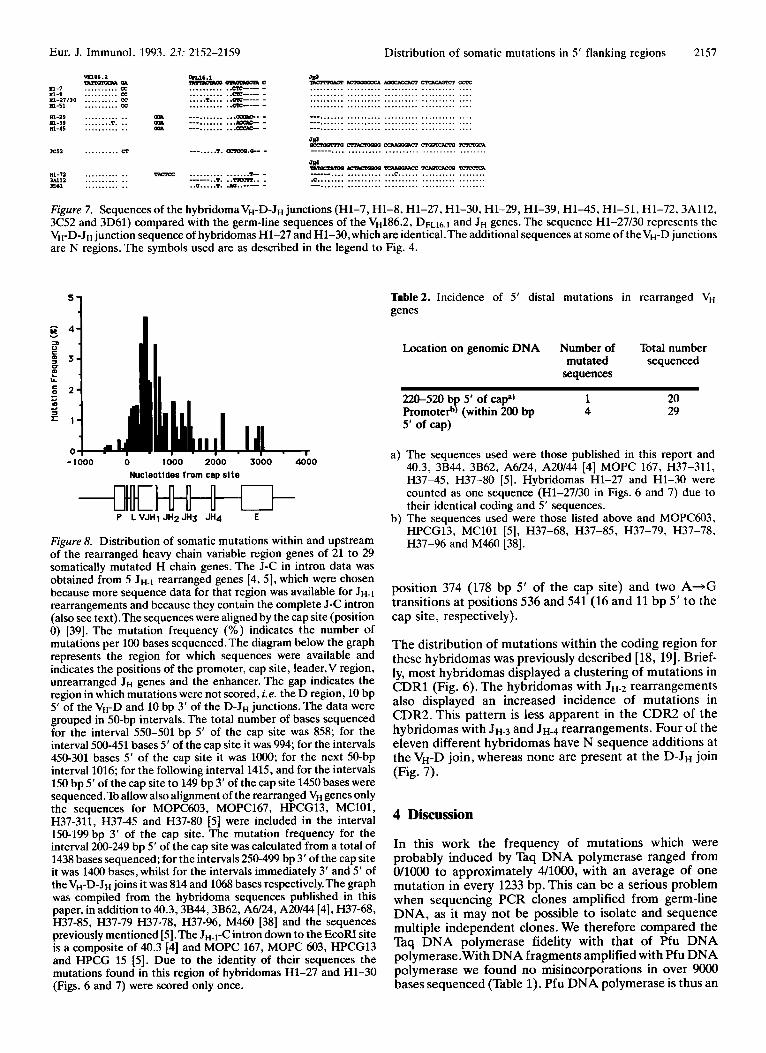
Figure 8. Distribution of somatic mutations within and upstream of the rearranged heavy chain variable region genes of 21 to 29 somatically mutated H chain genes. The J-C in intron data was obtained from 5 JH-, rearranged genes [4, 51, which were chosen because more sequence data for that region was available for J H - ~ rearrangements and because they contain the complete J-C intron (also see text).The sequences were aligned by the cap site (position 0) [39]. The mutation frequency (%) indicates the number of mutations per 100 bases sequenced.The diagram below the graph represents the region for which sequences were available and indicates the positions of the promoter, cap site, leader,V region, unrearranged JH genes and the enhancer. The gap indicates the region in which mutations were not scored, i .e. the D region, 10 bp 5' of the VH-D and 10 bp 3' of the D-JH junctions. The data were grouped in 50-bp intervals. The total number of bases sequenced for the interval 550-501 bp 5' of the cap site was 858; for the interval 500-451 bases 5' of the cap site it was 994; for the intervals 450-301 bases 5' of the cap site it was 1OOO; for the next 50-bp interval 1016; for the following interval 1415, and for the intervals 150 bp 5' of the cap site t o 149 bp 3' of the cap site 1450 bases were sequenced.To allow also alignment of the rearranged VH genes only the sequences for MOPC603, MOPC167, HPCG13, MClOl, H37-311, H37-45 and H37-80 [5] were included in the interval 150-199 bp 3' of the cap site. The mutation frequency for the interval 200-249 bp 5' of the cap site was calculated from a total of 1438 bases sequenced; for the intervals 250-499 bp 3' of the cap site it was 1400 bases, whilst for the intervals immediately 3' and 5' of theVH-D-JH joins it was 814 and 1068 bases respectively.The graph was compiled from the hybridoma sequences published in this paper, in addition to 40.3,3B44,3B62, A6/24, A20144 [4], H37-68, H37-85, H37-79 H37-78, H37-96, M460 [38] and the sequences previously mentioned [5].The JH-l-C intron down to the EcoFU site is a composite of 40.3 [4] and MOPC 167, MOPC 603, HPCG13 and HPCG 15 [5]. Due to the identity of their sequences the mutations found in this region of hybridomas H1-27 and H1-30 (Figs. 6 and 7) were scored only once.
Table2. Incidence of 5' distal mutations in rearranged V, genes
Location on genomic DNA Number of Total number mutated sequenced
sequences
220-520 b 5' of =pa' 1 20 Promote$ P (within 200 bp 4 29 5' of cap)
a) The sequences used were those published in this report and
H37-45, H37-80 [5]. Hybridomas H1-27 and H1-30 were counted as one sequence (Hl-27/30 in Figs. 6 and 7) due to their identical coding and 5' sequences.
b) The sequences used were those listed above and MOPC603,
40.3, 3B44, 3B62, A6/24, A20/44 [4] MOPC 167, H37-311,
HPCG13, MClOl [5], H37-68, H37-85, H37-79, H37-78, H37-96 and M460 [38].
position 374 (178 bp 5' of the cap site) and two A+G transitions at positions 536 and 541 (16 and 11 bp 5' to the cap site, respectively).
The distribution of mutations within the coding region for these hybridomas was previously described [18, 191. Brief- ly, most hybridomas displayed a clustering of mutations in CDRl (Fig. 6). The hybridomas with J H - ~ rearrangements also displayed an increased incidence of mutations in CDR2. This pattern is less apparent in the CDR2 of the hybridomas with J H - ~ and J H - ~ rearrangements. Four of the eleven different hybridomas have N sequence additions at the VH-D join, whereas none are present at the D-JH join (Fig. 7).
4 Discussion
In this work the frequency of mutations which were probably induced by Taq DNA polymerase ranged from O/lOOO to approximately 4/1000, with an average of one mutation in every 1233 bp. This can be a serious problem when sequencing PCR clones amplified from germ-line DNA, as it may not be possible to isolate and sequence multiple independent clones. We therefore compared the Taq DNA polymerase fidelity with that of Pfu DNA polymerase.With DNA fragments amplified with Pfu DNA polymerase we found no misincorporations in over 9OOO bases sequenced (Table 1). Pfu DNA polymerase is thus an

2158 H. S. Rothenfluh, L. Taylor, A. L. M. Bothwell et al. Eur. J. Immunol. 1993. 23: 2152-2159
enzyme ideally suited for the types of mutation analysis carried out in this work, especially when amplifying and sequencing independent clones from genomic DNA.
Previous attempts to define the boundaries of somatic hypermutation [4, 51 identified the need for more sequence data in the 5‘ and 3‘ flanking regions in order to allow more accurate definition of these boundaries. In this work we have sequenced an additional 12 heavy chain 5’ flanking regions (Fig. 6 and 7). These additional sequences confirm and extend the previous finding that somatic mutations are rarely found beyond the promoterkap site regions (Fig. 8, [4, 51). In fact, out of a total of twenty 5’ flanking region sequences distal to the cap site, only a single VDJ gene, 3B62, has mutations beyond the promoter (Table 2).
In the heavily mutated 3B62 gene a cluster of five nucleotide changes was found 2 375 bp upstream of the cap site. Since the location of these changes falls well outside the transcribed region they are incompatible with models where the nucleic acid transcripts are the major substrate [20]. We therefore investigated the potential origin of this mutation cluster. For two mutations immediately down- stream of the codon deletion in the V region of V~3B62, a possible germ-line donor, H17, has been reported [19]. In H17 this region of homology is adjacent to a palindromic sequence [ 191,which may have promoted a gene conversion event [33].
However, PCR and restriction analyses of germ-line DNA failed to detect a possible donor for the cluster of five upstream base changes found in 3B62 (Figs. 2, 3 and 5) . Sequence analysis of ten C57BL/6j and ten BALBk V~186.2 related genes also failed to detect any of the five base changes (Fig. 4). These results suggest that the five upstream nucleotide changes may not have a germ-line donor and that they arose by a chance somatic mutational event or as a post-fusion technical artifact. Indeed, if this upstream region was a target for somatic mutation one would expect it to be mutated in a significant proportion of antibody genes. The distal 5’ flanking region of four other heavily mutated antibody heavy chains, 3B44 [4], MOPC167, H37-311 and H37-80 [5] are free of any nucleotide changes. Therefore the uniqueness and isolation of these nucleotide substitutions suggests that they may not be due to the normal somatic hypermutator mechanism and that they cannot be taken as evidence against RNAkDNA- based mutator models [34]. Some of the rearranged V region sequences presented in this work display low levels of somatic mutation, however any conclusions drawn were not based solely on our data set but from a collection of all known sequences (see Fig. 8 and Table 2), the statistical reliability of which is now strengthened signifi- cantly by the additional 12 sequences presented in this work. In order to determine whether mutations may be present further upstream of the regions sequenced to date, we are currently extending the upstream flanking sequences of several genes (including 3B62).
A composite graph of a limited data set showing the distribution in mutation frequencies across a VDJH.~ gene suggested that the distribution of mutations around VDJ genes may be asymmetrical and that the mutation frequen- cy 5’ of the cap site was at least an order of magnitude lower than that 3’ of the cap site (reviewed in [17]). An important
outcome of this data review was that more sequences of 5’ flanking regions was obviously required. Adding the sequences published in this work to that earlier data set clearly illustrates the asymmetrical distribution of mutation frequency around aVDJH-, gene (Fig. 8).We have done this because it shows the total L-VDJ-C target area and because for the distal 3‘ flanking region most sequence data comes from J H . ~ rearranged genes [4,5]. The mode of the muta- tion distribution curve is centred on the rearranged VDJ which is in agreement with previous work where it was found that mutations are focused on the VDJ unit regard- less of which J element was rearranged [ 12, 51. If a mutation distribution graph for four J H - ~ ) rearranged VDJ sequences [5] down to the enhancer region is plotted, the general pattern is the same as the one seen for the J H - ~ rearranged genes (data not shown).The larger data set also emphasizes the clear difference in mutation rates between regions immediately 5’ and 3‘ to the cap site.We have discussed the implications of these observations in relation to possible molecular models for somatic hypermutation elsewhere ~ 7 1 .
The region containing the promoter and the cap site has now been sequenced for 29 heavy chain genes, and only 4 of these contain any mutations in that region (Table 2). Out of a total of 407 mutations scored in the sequences used in Fig. 8, only 14 were found upstream of the cap site. Therefore in heavy chain genes, almost 97% of somatic mutations fall within the transcribed region, and the bulk of these genes are free of mutations upstream of the cap site. This strongly supports the proposition that, at least in heavy chain genes, the mutator mechanism is linked to or dependent on transcription [4, 5, 171. Whatever molecular mechanism is invoked to explain somatic hypermutation, it must be able to account for both the positively skewed, asymmetrical distribution of mutations around rearranged V regions and the sharp decline in the mutation frequency around the cap site.
It has been proposed that analysis of the nature of base changes found in murine immunoglobulin non-coding regions may reflect the real mutation profile of the mutator mechanism, as these changes are assumed to be “selection free” [21]. For Xenopus heavy chains a base substitution analysis of “selection-free’’ mutations revealed that there is a significant bias towards mutating G.C base pairs [35]. A similar analysis of 160 nucleotide substitutions found in non-translated regions of murine heavy chains [4, 51 and 154 nucleotide substitutions found in x light chains [5 , 12, 341, taking into account the base composition of the sequences, did not detect a statistically significant bias for the alteration of any particular base(s) (data not shown). However, the data does indicate a bias towards transitions over transversions in both the heavy chain and the x light chain, and among the transversions a preference for A+T, T+A, A+C and G+T in both chains. The heavy chain data is in agreement with a previous analysis of 75 “non-selected” base substitutions found in 5 heavy chains [41.
In summary, our data on the distribution of somatic mutations in 5’ non-translated regions demonstrates that most (- 97 %) of somatic mutations are introduced into the transcribed region of the genomic DNA of heavy chain VDJ regions.This is consistent with our proposition that in

Eur. J. lmmunol. 1993. 23: 2152-2159 Distribution of somatic mutations in 5’ flanking regions 2159
the great bulk of genes the cap site may be the 5’ boundary for somatic hypermutation [ 171. One reasonable hypothesis would be to assign the “statistical” 5’ boundary for somatic hypermutation to the cap site, i .e. the transcription unit would then be considered the primary substrate for the mutator, extending 3‘ in the H chain locus to the enhancer region [5]. This view would be consistent with mutator models which are dependent on the transcriptional state of rearranged variable region genes [4, 5, 17, 201. The model proposed by Lebecque and Gearhart [5] predicts that the promoter region is the usual delimiting boundary on the 5’ side of the gene. This model differs from that proposed by our laboratory [4, 17, 201,which predicts that RNA/cDNA is the main nucleic acid substrate where nucleotide changes are first introduced [20], whereas Lebecque and Gearhart predict that mutations are first introduced into genomic DNA [5].The observation that the 5‘ boundary of mutation generally does not exceed the transcription unit may be suggestive of a model involving transcription but is by itself insufficient evidence for a conclusion that mutations are introduced on the transcript or its associated cDNA.
There are therefore two major classes of model to explain the mutation distribution pattern, the first where nucleic acid transcription products are the direct mutational sub- strate; the other postulating local unfolding of chromatin around a V(D)J allowing access of the mutational machi- nery to the DNA of the transcribed region. Despite the increased rigor of the sequence data now available it is still not possible to distinguish between these two classes of model.
There are other additional criticisms of the direct RNA- based model. In one well defined multicopy VJ, transgene system only certain transcribed transgenes and not others appear to mutate [34]. If this turns out to be a general result, it is difficult to explain why a particular copy should be targeted if it were a simple matter of mutated retrotran- scripts acting as diffusible intermediates [U. Storb, person- al communication]. Finally, the direct RNA-based model [20] also requires additional assumptions to account for the small number of changes just upstream of the cap site, e.g. initiation of transcription from minor upstream start sites [36,37] or homologous recombination-induced changes, to explain the low frequency of changes between the cap site and the promoter.
We thank U. Weiss for critical reading of the manuscript and K . Rajewsky and his group for supplying us with clones of various V, genes, hybridomas and C57BLI6j DNA. We also wish to thank L. Du Pasquier and L. Claflin for critical discussions.
The nucleotide sequence data reported in this paper have been submitted to the EMBL. GenBank and DDBJ Nucleotide Sequence databases and assigned accession number L09566- L09597.
Received January 29. 1993: in final revised form May 14, 1993.
5 References
1 Steele, E. J. (Ed.) Somatic hypermulalion in V-regions, CRC
2 Gearhart, P. J. and Bogenhagen, D. F., Proc. Natl. Acad. Sci. Press, Boca Raton 1991.
USA 1983. 80: 3439.
3 Chien, N. C., Pollock, R. R. , Desaymard, C. and Scharff,
4 Both, G. W.,Taylor, L., Pollard, J. W. and Steele, E. J., Mol.
5 Lebecque, S. G. and Gearhart, P. J., J. Exp. Med. 1990. 172:
6 MacLennan, I., Nature 1991. 354: 352. 7 Jacob, J., Kelsoe, G. , Rajewsky, K. and Weiss, U., Nature 1991.
8 Berek, C., Berger, A. and Apel, M., Cell 1991. 67: 1121. 9 Storb, U., Pinkert, C.,Arp, B., Engler,P., Gollahon, K., Manz,
J., Brady,W. and Brinster, R. L., J. Exp. Med. 1986.164: 627. 10 Storb, U., Ritchie, K. A., O’Brien, R. L., Arp, B. and
Brinster, R., Immunol. Rev. 1986. 89: 85. 11 O’Brien, R. L., Brinster, R. L. and Storb, U., Nature 1987.
326: 405. 12 Weber, J. S., Berry, J., Litwin, S. and Claflin. L., J. Immunol.
1991. 146: 3218. 13 Hackett, J. Jr., Stebbins, C., Rogerson, B., Davis, M. M. and
Storb, U., J. Exp. Med. 1992. 176: 225. 14 Sharpe, M. J., Milstein, C., Jarvis, J. M. and Neuberger. M. S.,
EMBO J. 1991. 10: 2139. 15 Sharpe, M. J., Neuberger, M. S., Pannell, R., Surani, M. A.
and Milstein, C., Eur. J. Immunol. 1990. 20: 1379. 16 Motoyama, N., Okada, H. and Azuma,T., Proc. Natl. Acad.
Sci. USA 1991. 88: 7933. 17 Steele, E. J., Rothenfluh, H. S. and Both, G. W., Immunol.
Cell Biol. 1992. 70: 129. 18 Blier, I? R. and Bothwell, A. L. M., J. Immunol. 1987. 139:
3996. 19 Cumano, A. and Rajewsky, K., EMBO J. 1986. 10: 2459. 20 Steele, E. J. and Pollard, J. W., Mol. Immunol. 1987.24: 667. 21 Kaartinen, M., Kulp, S. and Makela. O., in Steele, E. J. (Ed.),
Somatic hypermutation in V-regions, CRC Press, Boca Raton 1991, p. 105.
22 Kunkel,T. A. 1991, in Steele, E. J. (Ed.), Somatic hypermuta- tion in V-regions, CRC Press, Boca Raton 1991, p. 159.
23 Saiki, R. K., Gelfand, D. H., Stoffel, S.. Scharf, S. J., Higuchi, R., Horn, G. T., Mullis, K. B. and Erlich, H. A., Science 1988. 239: 487.
24 Jung,V, Pestka, S. B. and Pestka, S., Nucleic Acids Res. 1990. 18: 6156.
25 Sambrook, J., Fritsch, E. F. and Maniatis, T., Molecular cloning. A laboratory manual, 2nd Edit., Cold Spring Harbor Laboratory Press, Cold Spring Harbor 1989.
26 Schowalter, D. B. and Sommer, S. S., Anal. Biochem. 1989. 177: 90.
27 Roes, J., Huppi, K., Rajewsky, K. and Sablitzky, F., J. Immunol. 1989. 142: 1022.
28 Siekevitz, M., Kocks, C., Rajewsky, K. and Dildrop, R., Cell 1987. 48: 757.
29 Maniatis, T., Fritsch, E. F. and Sambrook, J., Molecular cloning. A laboratory manual, 1st Edit., Cold Spring Harbor Laboratory Press, Cold Spring Harbor 1982.
30 Sanger, F., Nicklen, S. and Coulson, A. R., Proc. Natl. Acad. USA 1977. 74: 5463.
31 Tindall, K. R. and Kunke1,T. A., Biochemistry 1988. 27: 6008. 32 Keohavong, I? and Thilly, W. G., Proc. Natl. Acad. Sci. USA
33 Krawinkel, U., Zoebelein, G. and Bothwell, A. L. M., Nucleic
34 Rogerson, B., Hackett Jr., J., Peters, A., Haasch, D. and Storb,
35 Wilson, M., Hsu, E., Marcuz, A., Courtet, M., Du Pasquier, L.
36 Dougherty, J. F! and Temin, H. M., Proc. Natl. Acad. Sci. USA
37 Dornburg, R. and Temin, H. M., Mol. Cell. Biol. 1988.8: 2328. 38 Clarke, S., Rickert, R., Kopke Wloch, M., Staudt, L.,
39 Ballard, D. W. and Bothwell, A., Proc. Natl. Acad. Sci. USA
M. D., J. Exp. Med. 1988. 167: 954.
Cell. Biol. 1990. 10: 5187.
1717.
354: 389.
1989. 86: 9253.
Acids Res. 1986. 9: 3871.
U., EMBO J. 1991. 10: 4331.
and Steinberg, C., EMBO J. 1992. 11: 4337.
1987. 84: 1197.
Gerhard, W. and Weigert, M., J. Immunol. 1990. 145: 2286.
1986. 83: 9626.
Related Documents


![local piRNA biogenesis · presumably through phased piRNA biogenesis [37–39] , since transcripts derived from unique genomic regions flanking the TE will not be the direct target](https://static.cupdf.com/doc/110x72/5f51f802441d0649ae60009e/local-pirna-biogenesis-presumably-through-phased-pirna-biogenesis-37a39-since.jpg)








