Selective Protection Against Oxidative Damage in Brain of Mice With a Targeted Disruption of the Neuronal Nitric Oxide Synthase Gene Juan Carlos Martı ´nez-Lazcano, 1 Francisca Pe ´rez-Severiano, 2 Bruno Escalante, 3 Joel Ramı ´rez-Emiliano, 4 Paula Vergara, 4 Rosa O. Gonza ´lez, 5 and Jose ´ Segovia 4 * 1 Escuela Nacional de Ciencias Biolo ´ gicas, Instituto Polite ´cnico Nacional, Me ´xico, D.F. 2 Departamento de Neuroquı ´mica, Instituto Nacional de Neurologı ´a y Neurocirugı ´a MVS, SSA, Me ´xico, D.F. 3 Centro de Investigacio ´ n y de Estudios Avanzados del IPN, Cinvestav-Monterrey, Me ´xico 4 Departamento de Fisiologı ´a, Biofı ´sica y Neurociencias, Centro de Investigacio ´n y de Estudios Avanzados del IPN, Me ´xico, D.F. 5 Departamento de Matema ´ticas, Universidad Auto ´ noma Metropolitana-Iztapalapa, Me ´xico, D.F. Nitric oxide (NO) is an essential messenger molecule in brain, where it is produced in neurons mostly by the ac- tivity of the neuronal isoform of nitric oxide synthase (nNOS). To understand the participation of the different isoforms of NOS in physiological functioning and in path- ological processes, mice with null mutations for each of the NOS isoforms have been generated. In the present paper, we report that there is a selective protection from oxidative damage in the brain of mice with a targeted dis- ruption of the nNOS gene. The cerebellum of these mice shows reduced levels of lipid peroxidation (LP) at the dif- ferent ages tested, compared with wild-type mice, and also a reduction in the formation of reactive oxygen spe- cies (ROS). We observed a decrease of LP in cortex, and no effect on either LP or ROS formation was observed in striatum of knockout mice compared with wild type. We also report increased spontaneous motor activity of knockout mice. The expression and activity of nNOS are crucial to maintain redox status in brain, and we consider that the alteration in oxidative damage may help us to explain the phenotypical characteristics of nNOS knock- out mice and their differential susceptibility to brain insults. V V C 2007 Wiley-Liss, Inc. Key words: nitric oxide; neuronal nitric oxide synthase; lipid peroxidation; reactive oxygen species; oxidative damage Nitric oxide (NO), an unstable diatomic radical, has been recognized as a crucial messenger molecule in the central nervous system (CNS), where it is implicated in neuromodulation, synaptic plasticity, and other funda- mental physiological processes that, in turn, modulate dif- ferent behavioral and neuroendocrine functions. NO is also found in many cell types in peripheric tissues, and it is involved in a variety of biological roles. NO is produced from L-arginine by one of three nitric oxide synthase (NOS) enzymes: neuronal (nNOS), endothelial (eNOS), and inducible (iNOS). Although the three isoforms of NOS have been characterized in brain, neurons produce NO mostly by activation of nNOS, which is constitutively expressed in these cells (Knowles and Moncada, 1994). In brain, nNOS is localized in different discrete populations of cells, including cerebellum, cortex, and striatum (Bredt and Snyder, 1994). Under pathological conditions, NO may promote oxidative damage, through the formation of the highly reactive metabolite peroxynitrite. For this reason, it has been suggested that NO is involved in the pathogene- sis of neurodegenerative processes, such as Huntington’s disease (HD). We have previously shown that oxidative damage parallels the expression of a neurological pheno- type in mice transgenic for the HD mutation and that there are age-dependent changes in NOS activity and expression (Pe ´rez-Severiano et al., 2000, 2002), thus sup- porting the proposed participation of NO as a factor underlying the onset of the disease. Another very impor- tant clinical condition in which oxidative damage is pres- ent is brain ischemia. Immediately after brain ischemia, NO production and release from eNOS are protective, because they inhibit microvascular aggregation and adhe- sion, but, after ischemia develops, NO produced by the overactivation of nNOS contributes to neuronal damage Contract grant sponsor: CONACyT; Contract grant number: 42721-M (to J.S.). *Correspondence to: Dr. Jose ´ Segovia, Departamento de Fisiologı ´a, Bio- fı ´sica y Neurociencias, Centro de Investigacio ´n y de Estudios Avanzados del IPN, Av. Instituto Polite ´cnico Nacional No. 2508, Me ´xico, 07300, D.F. E-mail: jsegovia@fisio.cinvestav.mx Received 28 September 2006; Revised 26 December 2006; Accepted 3 January 2007 Published online 23 March 2007 in Wiley InterScience (www. interscience.wiley.com). DOI: 10.1002/jnr.21261 Journal of Neuroscience Research 85:1391–1402 (2007) ' 2007 Wiley-Liss, Inc.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Selective Protection Against OxidativeDamage in Brain of Mice With a TargetedDisruption of the Neuronal Nitric OxideSynthase Gene
Juan Carlos Martınez-Lazcano,1 Francisca Perez-Severiano,2 Bruno Escalante,3
Joel Ramırez-Emiliano,4 Paula Vergara,4 Rosa O. Gonzalez,5 and Jose Segovia4*1Escuela Nacional de Ciencias Biologicas, Instituto Politecnico Nacional, Mexico, D.F.2Departamento de Neuroquımica, Instituto Nacional de Neurologıa y Neurocirugıa MVS, SSA, Mexico, D.F.3Centro de Investigacion y de Estudios Avanzados del IPN, Cinvestav-Monterrey, Mexico4Departamento de Fisiologıa, Biofısica y Neurociencias, Centro de Investigacion y de Estudios Avanzados delIPN, Mexico, D.F.5Departamento de Matematicas, Universidad Autonoma Metropolitana-Iztapalapa, Mexico, D.F.
Nitric oxide (NO) is an essential messenger molecule inbrain, where it is produced in neurons mostly by the ac-tivity of the neuronal isoform of nitric oxide synthase(nNOS). To understand the participation of the differentisoforms of NOS in physiological functioning and in path-ological processes, mice with null mutations for each ofthe NOS isoforms have been generated. In the presentpaper, we report that there is a selective protection fromoxidative damage in the brain of mice with a targeted dis-ruption of the nNOS gene. The cerebellum of these miceshows reduced levels of lipid peroxidation (LP) at the dif-ferent ages tested, compared with wild-type mice, andalso a reduction in the formation of reactive oxygen spe-cies (ROS). We observed a decrease of LP in cortex, andno effect on either LP or ROS formation was observed instriatum of knockout mice compared with wild type. Wealso report increased spontaneous motor activity ofknockout mice. The expression and activity of nNOS arecrucial to maintain redox status in brain, and we considerthat the alteration in oxidative damage may help us toexplain the phenotypical characteristics of nNOS knock-out mice and their differential susceptibility to braininsults. VVC 2007 Wiley-Liss, Inc.
Key words: nitric oxide; neuronal nitric oxide synthase;lipid peroxidation; reactive oxygen species; oxidativedamage
Nitric oxide (NO), an unstable diatomic radical, hasbeen recognized as a crucial messenger molecule in thecentral nervous system (CNS), where it is implicated inneuromodulation, synaptic plasticity, and other funda-mental physiological processes that, in turn, modulate dif-ferent behavioral and neuroendocrine functions. NO isalso found in many cell types in peripheric tissues, and it isinvolved in a variety of biological roles. NO is producedfrom L-arginine by one of three nitric oxide synthase
(NOS) enzymes: neuronal (nNOS), endothelial (eNOS),and inducible (iNOS).
Although the three isoforms of NOS have beencharacterized in brain, neurons produce NO mostly byactivation of nNOS, which is constitutively expressed inthese cells (Knowles and Moncada, 1994). In brain, nNOSis localized in different discrete populations of cells,including cerebellum, cortex, and striatum (Bredt andSnyder, 1994). Under pathological conditions, NO maypromote oxidative damage, through the formation of thehighly reactive metabolite peroxynitrite. For this reason, ithas been suggested that NO is involved in the pathogene-sis of neurodegenerative processes, such as Huntington’sdisease (HD). We have previously shown that oxidativedamage parallels the expression of a neurological pheno-type in mice transgenic for the HD mutation and thatthere are age-dependent changes in NOS activity andexpression (Perez-Severiano et al., 2000, 2002), thus sup-porting the proposed participation of NO as a factorunderlying the onset of the disease. Another very impor-tant clinical condition in which oxidative damage is pres-ent is brain ischemia. Immediately after brain ischemia,NO production and release from eNOS are protective,because they inhibit microvascular aggregation and adhe-sion, but, after ischemia develops, NO produced by theoveractivation of nNOS contributes to neuronal damage
Contract grant sponsor: CONACyT; Contract grant number: 42721-M (to
J.S.).
*Correspondence to: Dr. Jose Segovia, Departamento de Fisiologıa, Bio-
fısica y Neurociencias, Centro de Investigacion y de Estudios Avanzados
del IPN, Av. Instituto Politecnico Nacional No. 2508, Mexico, 07300,
D.F. E-mail: [email protected]
Received 28 September 2006; Revised 26 December 2006; Accepted 3
January 2007
Published online 23 March 2007 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/jnr.21261
Journal of Neuroscience Research 85:1391–1402 (2007)
' 2007 Wiley-Liss, Inc.

by the formation of peroxynitrite, a powerful oxidant, thatinduces cell damage by different mechanisms, includinglipid peroxidation (LP; Moro et al., 2004).
It is known that the free radical superoxide (O2�)
reacts with NO, producing the powerful oxidant peroxy-nitrite anion (ONOO�; Beckman and Koppenol, 1996).Peroxinitrite is a nitrating agent capable of attacking andmodifying proteins, lipids, and DNA as well as depletingantioxidant defenses (Torreilles et al., 1999). In addition tothe previously mentioned information, it has been demon-strated that NO triggers apoptosis when it binds to cyto-chrome c oxidase and induces the formation of O2
� in mi-tochondria, generating ONOO�, which inhibits and/ordamages the mitochondrial complexes I, II, IV, and V;aconitase; creatine-kinase; mitochondrial membrane; mi-tochondrial DNA; and mitochondrial SOD, causing Ca2+
release and mitochondrial swelling (Brown, 1999). Addi-tionally (Rubbo et al., 1994), demonstrated a direct rela-tionship between LP and peroxynitrite formation in vitro.All this information supports the concept that increases inNO will produce oxidative stress, which in turn will leadto the generation of reactive oxygen species (ROS), LP,and cell death.
Considering the essential roles that NO plays both innormal brain and in pathological conditions, it becomescritical to understand clearly the role of each of the threeNOS isoforms in brain functioning. There are pharmaco-logical tools that permit the inhibition of the differentNOS isoforms, but their effects are acute, and importantsecondary consequences may appear (Alderton et al.,2001). On the other hand, the use of transgenic animals inbiomedical research has been of great relevance, both forunraveling the participation of different molecules in nor-mal physiological functions and for understanding themechanisms underlying the onset of different diseases, andalso for designing new therapeutic methods to treat them.In this context, animals with null mutations for the differ-ent NOS isoforms have been generated (Huang et al.,1993, 1995; Laubach et al., 1995; Gyurko et al., 2002). Amouse line with a targeted disruption of the nNOS genewas generated that shows very low levels of NOS activityand that presents phenotypic characteristics such as agrossly enlarged stomach (Huang et al., 1993). These micealso exhibit aggressive behavior, related to altered sero-tonin levels (Chiavegatto and Nelson, 2003). Interestingly,these mice are also more resistant to ischemic damage(Huang et al., 1994; Ferriero et al., 1996).
We have previously shown that oxidative damage—determined by LP, formation of free radicals, and NOS ac-tivity and expression—varies in the brains of transgenicmice (Perez-Severiano et al., 2000, 2002, 2004), so weconsidered it relevant to study the degree of oxidativedamage in different areas of the brain of nNOS knockoutmice, as well as behavioral and phenotypical characteris-tics, to understand better the function of nNOS in brain.In the present paper, we report that there is an increase inoxidative damage in different brain areas of older wild-type mice and mice with disruption of the nNOS genebut that the increase is significantly lower in the cortex
and cerebellum of the older knockout mice comparedwith wild-type mice. These results suggest that the lack ofnNOS may protect certain brain areas from oxidativedamage and that this effect may help explain some of thephenotypical characteristics as well as the differential sus-ceptibility to brain insults shown by mice with disruptionof the nNOS gene.
MATERIALS ANDMETHODS
Mouse Genotypes
Male homozygous knockout mice for the nNOS genefrom the B6 129-NOS1tmP1h line (nNOS�/�; Huang et al.,1993), were purchased from the Jackson Laboratory (Bar Har-bor, ME) and back-crossed with wild-type, nNOS+/+,B6CBA female mice to start our colony. All animal procedureswere approved by the Institutional Review Committee and arein accordance with current Mexican legislation, NOM-062-ZOO-1999, and in agreement with the National Institutes ofHealth (NIH, Bethesda, MD) Guide for the care and use of labora-tory animals. Mice were maintained under conditions previouslydescribed (Segovia, 2002). Mouse genotype was determined byPCR analysis. This analysis allowed us to identified mice homo-zygous for the nNOS gene disruption (�/�), heterozygous forthe mutation (+/�), and wild-type (+/+). DNA was obtainedfrom tissue of the left ear, from mice 9–10 weeks old, as previ-ously described (Segovia, 2002). Oligonucleotides that allowedthe identification of the nNOS and neomycin (NEO) geneswere used. The sequences of the primers and the conditionsfor the PCR assays were obtained from the Jackson Labora-tory (JAX Mice Web Site JAX Data Base). Sequences of theprimers that recognize nNOS exon 2 are the following: IMR0406 (sense) TCAGATCTGATCCGAGGAGG, and IMR0407 (antisense) TTCCAGAGCGCTGTCATAGC. For NEO,primers are: IMR 0013 (sense) CTTGGGTGGAGAGGC-TATTC, and IMR 0014 (antisense) AGGTGAGATGACAG-GAGATC.
Behavioral Testing and Phenotype
Male mice homozygous for the disruption of the nNOSgene, (nNOS�/�), heterozygous (nNOS+/�), and wild-typefor the mutation (nNOS+/+) were weighed at 12 and 35 weeksof age. Horizontal and vertical movements were automaticallyrecorded for a single 10-min session for each animal at the agespreviously indicated, as we have previously described (Segovia,2002; Perez-Severiano et al., 2004). To determine aggressivebehavior, we followed the test (resident-intruder paradigm)described by Nelson and collaborators (Nelson et al., 1995;Chiavegatto et al., 2001), with slight modifications. Briefly,homozygous mice from each litter were isolated for 24 hr, andpairs of mice were then placed in the same cage. Animals wereobserved for 15 min, and the period of adaptation was estab-lished. Our criteria to consider that an animal presented aggres-sive behavior were: four combats, longer than 4 sec each one, orthat one of the mice established complete dominance during theperiod of observation. When animals were sacrificed to obtainbrain tissue for biochemical analyses, the stomach was alsodissected out, and its length and width were measured usingcalipers.
1392 Martınez-Lazcano et al.
Journal of Neuroscience Research DOI 10.1002/jnr
NOS Protein Expression
To determine the expression of the three isoforms ofNOS in different brain regions, at the ages indicated, Westernblot assays were performed, as previously described (Perez-Severiano et al., 2002; Segovia and Perez-Severiano, 2004),employing a monoclonal antibody against nNOS and polyclonalantibodies against eNOS and iNOS (Santa Cruz Biotechnology,Santa Cruz, CA) at a final dilution of 1:500. After incubationwith primary antibodies, membranes were incubated with eithera secondary goat anti-mouse peroxidase-labeled antibody or asecondary goat anti-rabbit peroxidase-labeled antibody (Zymed,South San Francisco, CA) diluted 1:6,000 in the blocking solu-tion for 1 hr at room temperature. Blots were washed and pro-tein was developed with the ECL detection system (Perkin-Elmer, Norwalk, CT). Blots were stripped, and, as a control,b-actin levels were determined by using a monoclonal antibody(Garcia-Tovar et al., 2002). Images from films were digitallyacquired with a BioDoc-It System (UVP). and densitometryanalysis was performed in Lab Works 4.0 image acquisition andanalysis software (UVP). Densitometry results of the differentNOS isoforms were normalized with respect to their b-actincontrols and data expressed as normalized optical density (OD)arbitrary units.
NOS Activity
NOS activity was measured based on the stochiometricconversion of L-arginine to NO and L-citrulline (Bredt andSnyder, 1990), as we have previously described, in striata, cor-tex, and cerebella of mice of the three genotypes at two differentages (Segovia, 2002; Perez-Severiano et al., 2002, 2004). Resultswere expressed as nanograms L-citrulline/500 lg protein/min.
LP
At the ages previously indicated, male mice of the threegenotypes were sacrificed by decapitation, and the striata, cor-tex, and cerebella were rapidly dissected out on an ice-cold sur-face and immediately processed. To measure LP, the formationof lipid-soluble fluorescence was monitored based on themethod of Triggs and Willmore (1984), as we have previouslydescribed (Segovia, 2002). Results were expressed as units ofrelative fluorescence (URF) per milligram protein.
ROS
ROS formation was evaluated in striata, cortex, and cere-bella of homozygous, heterozygous, and wild-type male mice,at the ages previously specified. A fluorometric assay, based onthe oxidation of 2070-dichlodihydrofluorescein diacetate(DCFH-DA; Molecular Probes, Eugene, OR) to 2070-dichloro-fluorescein (DCF; Ali et al., 1992), modified for brain tissue(Perez-Severiano et al., 2004), was employed. Results wereexpressed as nanomoles DCF formed per milligram protein/min.
Statistical Analysis
A cross-tabulation v2 test was performed to analyzeaggressive behavior patterns. For all other data, two-wayANOVAs followed by contrast analyses to test for differences
among genotypes were employed. P< 0.05, was considered sig-nificant. The SPSS 13 package was employed for all analyses.
RESULTS
Phenotypical Characteristics
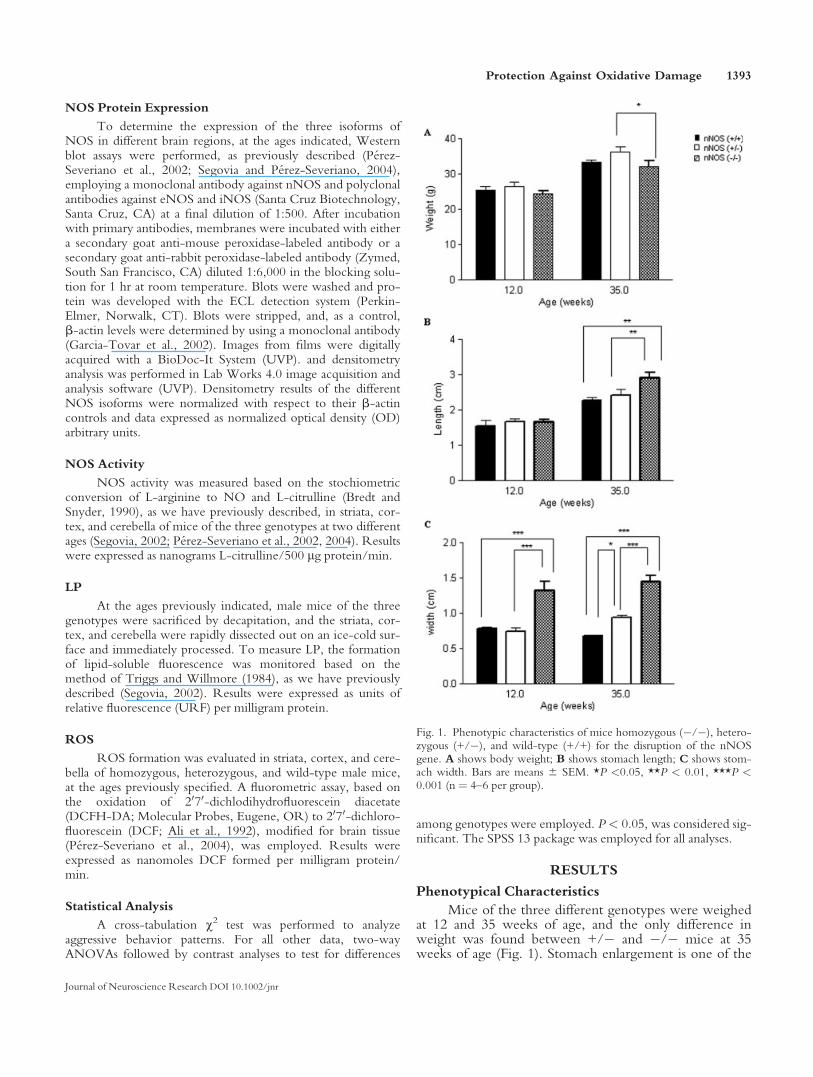
Mice of the three different genotypes were weighedat 12 and 35 weeks of age, and the only difference inweight was found between +/� and �/� mice at 35weeks of age (Fig. 1). Stomach enlargement is one of the
Fig. 1. Phenotypic characteristics of mice homozygous (�/�), hetero-zygous (+/�), and wild-type (+/+) for the disruption of the nNOSgene. A shows body weight; B shows stomach length; C shows stom-ach width. Bars are means 6 SEM. *P <0.05, **P < 0.01, ***P <0.001 (n ¼ 4–6 per group).
Protection Against Oxidative Damage 1393
Journal of Neuroscience Research DOI 10.1002/jnr
more obvious characteristics of the nNOS knockout mice(Huang et al., 1993). We determined this parameter inmice from our colony and observed that knockout mice,at the two ages used in this work, showed significantlyenlarged stomachs, measured as width of the stomach,compared with both wild-type and heterozygous mice,differences being as large as 114% in comparing (�/�)mice with wild-type mice at 35 weeks of age. When meas-uring the length of the stomach, knockout mice were alsosignificantly longer than both wild-type and heterozygousat 35 weeks of age, but differences were not as marked asfor stomach width, with 26% difference between wild-type and knockout mice of 35 weeks of age (Fig. 1).
We also determined aggressive behavior andobserved, as had been previously described (Nelson et al.,1995; Chiavegatto et al., 2001), that all male knockoutmice presented this type of behavior (6 of 6 as assessed bythe resident-intruder test, and none of the wild-type micepresented this behavior, 0 of 12). In this case, heterozygousmice also presented an intermediate behavior; 30% (4 of
13) of the male mice showed aggressive behavior. Thedata presented in Table I, illustrate that there is a signifi-cant difference in the proportion of mice showing aggres-sive behavior depending on the genotype.
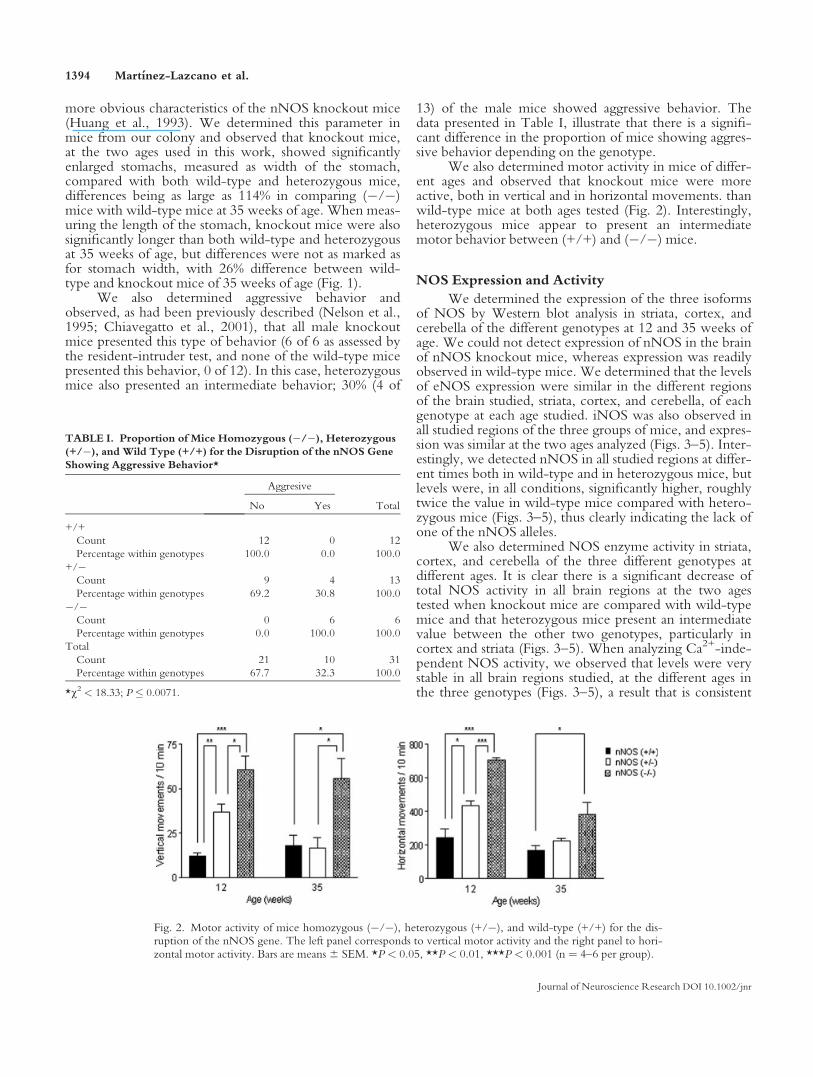
We also determined motor activity in mice of differ-ent ages and observed that knockout mice were moreactive, both in vertical and in horizontal movements. thanwild-type mice at both ages tested (Fig. 2). Interestingly,heterozygous mice appear to present an intermediatemotor behavior between (+/+) and (�/�) mice.
NOS Expression and Activity
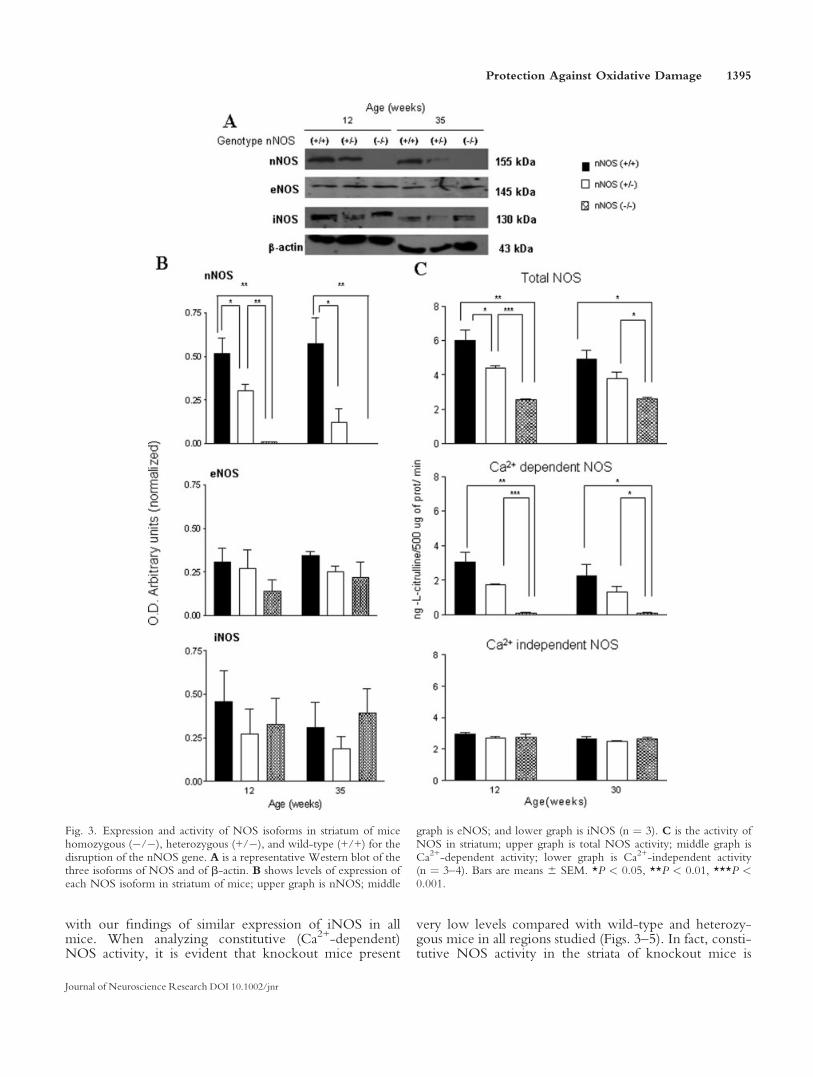
We determined the expression of the three isoformsof NOS by Western blot analysis in striata, cortex, andcerebella of the different genotypes at 12 and 35 weeks ofage. We could not detect expression of nNOS in the brainof nNOS knockout mice, whereas expression was readilyobserved in wild-type mice. We determined that the levelsof eNOS expression were similar in the different regionsof the brain studied, striata, cortex, and cerebella, of eachgenotype at each age studied. iNOS was also observed inall studied regions of the three groups of mice, and expres-sion was similar at the two ages analyzed (Figs. 3–5). Inter-estingly, we detected nNOS in all studied regions at differ-ent times both in wild-type and in heterozygous mice, butlevels were, in all conditions, significantly higher, roughlytwice the value in wild-type mice compared with hetero-zygous mice (Figs. 3–5), thus clearly indicating the lack ofone of the nNOS alleles.
We also determined NOS enzyme activity in striata,cortex, and cerebella of the three different genotypes atdifferent ages. It is clear there is a significant decrease oftotal NOS activity in all brain regions at the two agestested when knockout mice are compared with wild-typemice and that heterozygous mice present an intermediatevalue between the other two genotypes, particularly incortex and striata (Figs. 3–5). When analyzing Ca2+-inde-pendent NOS activity, we observed that levels were verystable in all brain regions studied, at the different ages inthe three genotypes (Figs. 3–5), a result that is consistent
Fig. 2. Motor activity of mice homozygous (�/�), heterozygous (+/�), and wild-type (+/+) for the dis-ruption of the nNOS gene. The left panel corresponds to vertical motor activity and the right panel to hori-zontal motor activity. Bars are means6 SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (n ¼ 4–6 per group).
TABLE I. Proportion of Mice Homozygous (�/�), Heterozygous
(+/�), and Wild Type (+/+) for the Disruption of the nNOS Gene
Showing Aggressive Behavior*
Aggresive
TotalNo Yes
+/+
Count 12 0 12
Percentage within genotypes 100.0 0.0 100.0
+/�Count 9 4 13
Percentage within genotypes 69.2 30.8 100.0
�/�Count 0 6 6
Percentage within genotypes 0.0 100.0 100.0
Total
Count 21 10 31
Percentage within genotypes 67.7 32.3 100.0
*v2 < 18.33; P � 0.0071.
1394 Martınez-Lazcano et al.
Journal of Neuroscience Research DOI 10.1002/jnr
with our findings of similar expression of iNOS in allmice. When analyzing constitutive (Ca2+-dependent)NOS activity, it is evident that knockout mice present
very low levels compared with wild-type and heterozy-gous mice in all regions studied (Figs. 3–5). In fact, consti-tutive NOS activity in the striata of knockout mice is
Fig. 3. Expression and activity of NOS isoforms in striatum of micehomozygous (�/�), heterozygous (+/�), and wild-type (+/+) for thedisruption of the nNOS gene. A is a representative Western blot of thethree isoforms of NOS and of b-actin. B shows levels of expression ofeach NOS isoform in striatum of mice; upper graph is nNOS; middle
graph is eNOS; and lower graph is iNOS (n ¼ 3). C is the activity ofNOS in striatum; upper graph is total NOS activity; middle graph isCa2+-dependent activity; lower graph is Ca2+-independent activity(n ¼ 3–4). Bars are means 6 SEM. *P < 0.05, **P < 0.01, ***P <0.001.
Protection Against Oxidative Damage 1395
Journal of Neuroscience Research DOI 10.1002/jnr
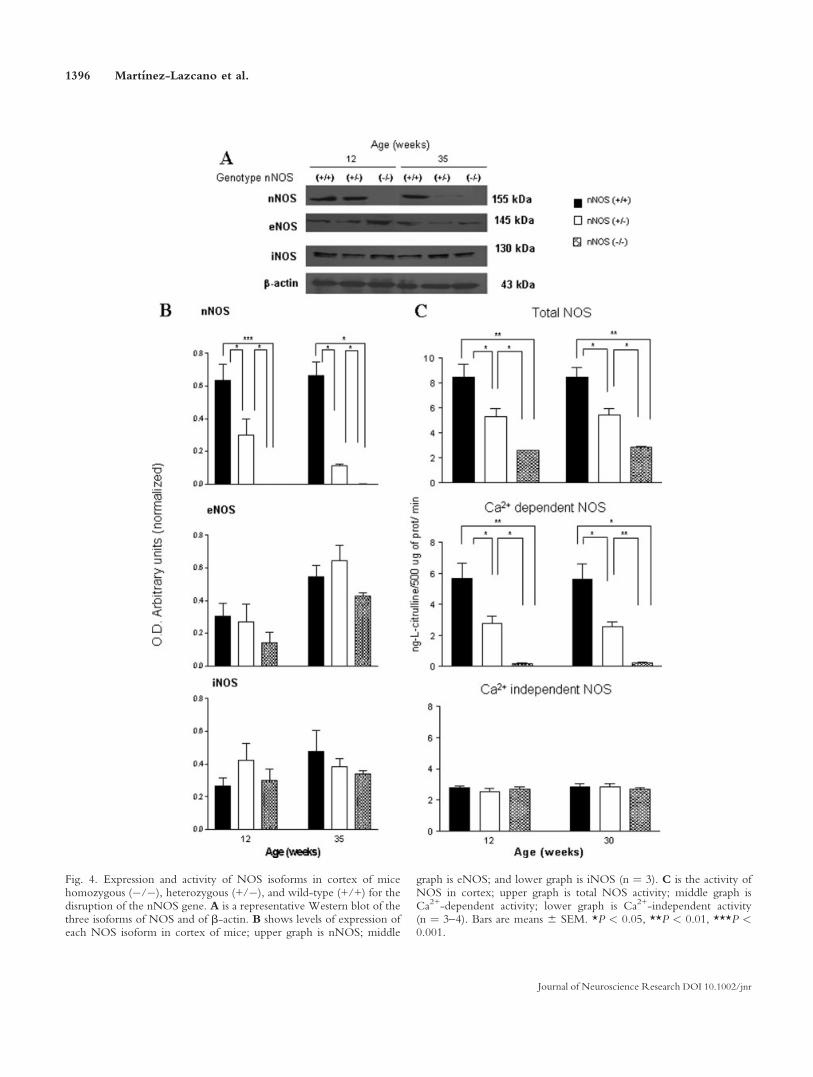
Fig. 4. Expression and activity of NOS isoforms in cortex of micehomozygous (�/�), heterozygous (+/�), and wild-type (+/+) for thedisruption of the nNOS gene. A is a representative Western blot of thethree isoforms of NOS and of b-actin. B shows levels of expression ofeach NOS isoform in cortex of mice; upper graph is nNOS; middle
graph is eNOS; and lower graph is iNOS (n ¼ 3). C is the activity ofNOS in cortex; upper graph is total NOS activity; middle graph isCa2+-dependent activity; lower graph is Ca2+-independent activity(n ¼ 3–4). Bars are means 6 SEM. *P < 0.05, **P < 0.01, ***P <0.001.
1396 Martınez-Lazcano et al.
Journal of Neuroscience Research DOI 10.1002/jnr
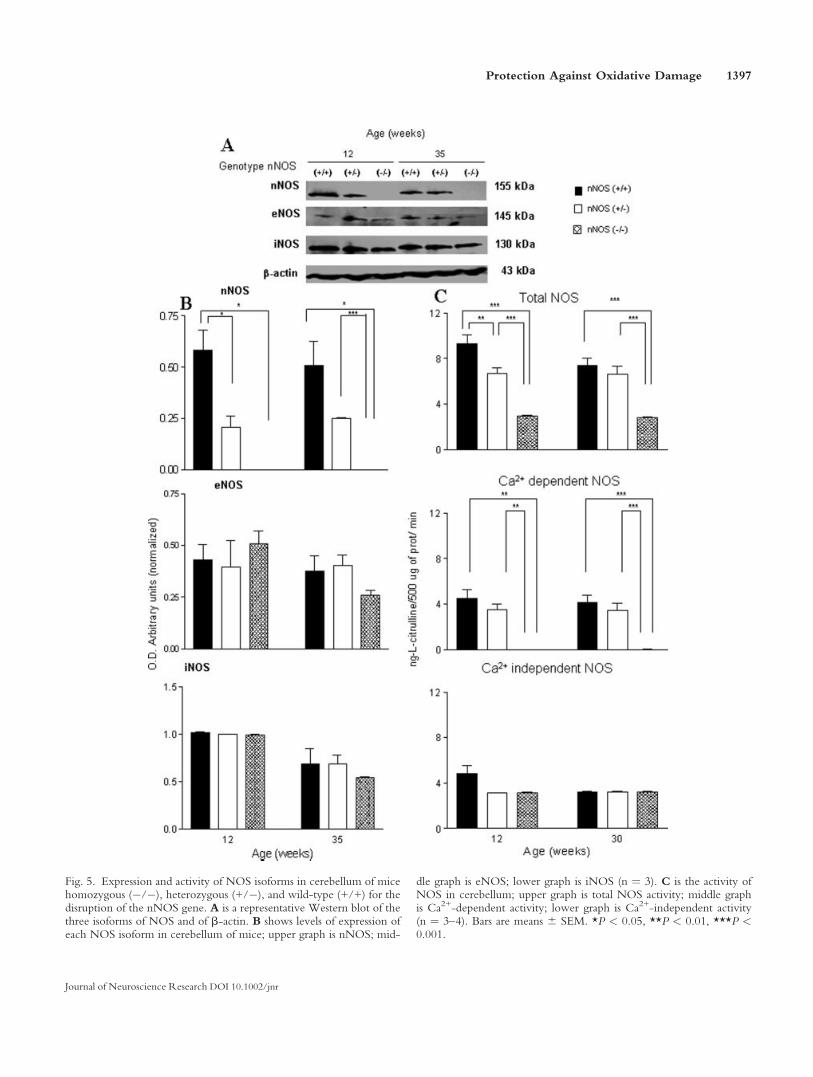
Fig. 5. Expression and activity of NOS isoforms in cerebellum of micehomozygous (�/�), heterozygous (+/�), and wild-type (+/+) for thedisruption of the nNOS gene. A is a representative Western blot of thethree isoforms of NOS and of b-actin. B shows levels of expression ofeach NOS isoform in cerebellum of mice; upper graph is nNOS; mid-
dle graph is eNOS; lower graph is iNOS (n ¼ 3). C is the activity ofNOS in cerebellum; upper graph is total NOS activity; middle graphis Ca2+-dependent activity; lower graph is Ca2+-independent activity(n ¼ 3–4). Bars are means 6 SEM. *P < 0.05, **P < 0.01, ***P <0.001.
Protection Against Oxidative Damage 1397
Journal of Neuroscience Research DOI 10.1002/jnr
approximately 5% of that of wild-type controls, 3% of thatin cortex, and almost undetectable (>1%) compared withcerebella from +/+ mice. Again we observed an interme-diate level of activity of heterozygous mice, particularly incortex and striata (Figs. 3–5). We believe that all or mostNOS constitutive or Ca2+-dependent activity is due tonNOS, with little or no participation of eNOS, becausethe three genotypes showed similar levels of eNOSexpression and significant differences in the expression of
nNOS, which is consistent with the differences in enzymeactivity.
Oxidative Damage
To determine oxidative damage in the brain of micewith nNOS gene knockout, we measured LP and forma-tion of ROS in the striata, cortex, and cerebella of malemice of the three genotypes at the two ages previouslydescribed. Our results show that there is a very highincrease of LP in striata, cortex, and cerebella of mice of
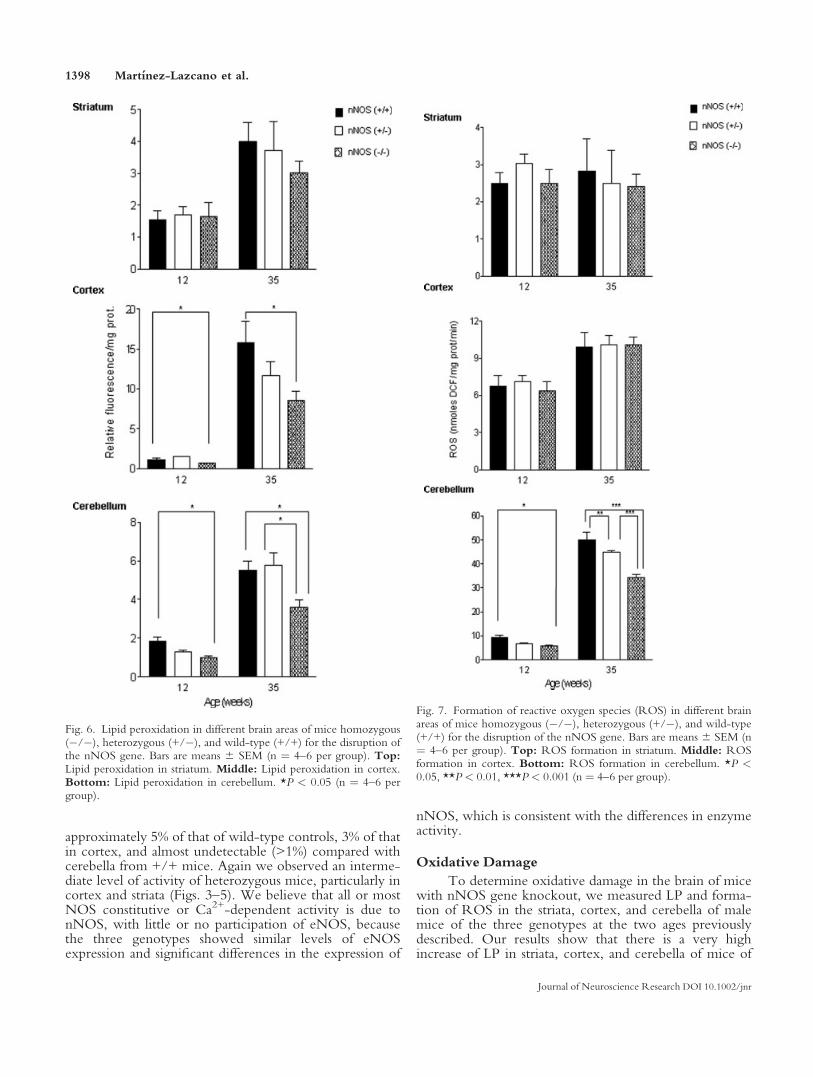
Fig. 6. Lipid peroxidation in different brain areas of mice homozygous(�/�), heterozygous (+/�), and wild-type (+/+) for the disruption ofthe nNOS gene. Bars are means 6 SEM (n ¼ 4–6 per group). Top:Lipid peroxidation in striatum. Middle: Lipid peroxidation in cortex.Bottom: Lipid peroxidation in cerebellum. *P < 0.05 (n ¼ 4–6 pergroup).
Fig. 7. Formation of reactive oxygen species (ROS) in different brainareas of mice homozygous (�/�), heterozygous (+/�), and wild-type(+/+) for the disruption of the nNOS gene. Bars are means 6 SEM (n¼ 4–6 per group). Top: ROS formation in striatum. Middle: ROSformation in cortex. Bottom: ROS formation in cerebellum. *P <0.05, **P < 0.01, ***P < 0.001 (n ¼ 4–6 per group).
1398 Martınez-Lazcano et al.
Journal of Neuroscience Research DOI 10.1002/jnr

the three phenotypes at 35 weeks of age compared with12-week-old animals, as shown in Figure 6 (P < 0.001,two-way ANOVA). However, there are significant differ-ences in the levels of LP among the brain regions studied.There is no significant difference in LP between striata ofknockout and control mice at either age tested independ-ently, indicating that the lack of nNOS does not provideprotection against oxidative damage in the striatum (Fig.6). In cortex, levels of LP in younger mice are very lowand similar among the different genotypes, although �/�mice show a higher level of LP at 12 weeks comparedwith wild-type animals. A dramatic increase, over tenfold,in LP occurs in the cortex of the older mice (35 weeksold), and we observed that there is significant 46%decrease in LP in knockout mice compared with wild-type mice, whereas heterozygous mice present an interme-diate value of LP (Fig. 6). For cerebellum we observed aconsistent pattern of LP formation; levels of knockoutmice were significantly lower at both 12 and 35 weeks ofage compared with wild-type control, suggesting thatthere is a continuous and persistent protection from oxida-tive damage in this brain region induced by the lack of thenNOS isoform (Fig. 6).
As another measure of oxidative damage in brain, wealso determined the formation of ROS in the differentbrain regions, at the ages previously indicated. We foundno significant changes in the formation of ROS in thestriatum; there is an age-dependent increase in ROS in thecortex of older mice (P < 0.001, two-way ANOVA), butno difference was observed between the genotypes ateither age tested (Fig. 7). For cerebellum we determinedan age-dependent increase in ROS formation (P < 0.001,two-way ANOVA) but that ROS formation was signifi-cantly lower in �/� mice compared with +/+ mice atboth ages studied and that +/� mice presented intermedi-ate values (Fig. 7).
DISCUSSION
NO is an essential messenger molecule both in theCNS and in peripheric tissues that is involved in many dif-ferent physiological functions from synaptic plasticity tocontrol of vascular relaxation, but it can also participate inpathological processes, such as neurodegenerative diseasesand brain ischemia (Christopherson and Bredt, 1997;Ashwal et al., 1998). NO is produced by three isoforms ofNOS, and it is of great relevance to a full understanding ofthe functions of NO in brain, as well as its role in disease,to define clearly the biological task of each of the NOSisoforms. A very powerful approach to study the functionof individual molecules in an organism is the generation ofanimals with null mutations. Knockout mice for thenNOS gene have been previously described (Huang et al.,1993), which show very low levels of NOS activity andpresent phenotypical characteristics such as an enlargedstomach and aggressive behavior (Chiavegatto and Nelson,2003; Huang et al., 1993; Nelson et al., 1995).
NO is implicated in oxidative damage, mainlythrough the formation of the highly reactive metabolite
peroxynitrite (Stamler, 1994), so we considered it relevantto determine the consequences of the lack of nNOS in theredox status in different areas of the brain of knockoutmice at different ages. We were also interested in studyingphenotypical characteristics of the different mouse geno-types, to determine whether those traits could be relatedto the alterations of the redox status of the brain.
We established a colony of knockout mice, and thePCR genotyping analysis allowed us to identify wild-typeand heterozygous and homozygous mice for the nNOSmutation, that were born with a mendelian frequency.Knockout and heterozygous male mice were viable andfertile. We confirmed in our colony the expression of phe-notypical traits previously described for nNOS�/� mice,such as stomach enlargement and the presence of aggres-sive behavior (Huang et al., 1993; Nelson et al., 1995; Fig.1, Table I). We established that the enlargement of thestomach of �/� mice was already present at 12 weeks ofage and persisted in 35-week-old mice. Aggressive behav-ior was evident in all knockout mice tested, starting at 12weeks of age. Interestingly, about half of the heterozygousmice also showed this behavior, suggesting a partial com-pensation when only one gene is active (Table I). Eliassonet al. (1997) reported that the lack of nNOS mRNA invirtually all nuclei of the hypothalamus, and the more than95% reduction in the number of the cells in the dorsome-dial nucleus of the amygdala containing nNOS mRNA,regions known to mediate male-specific aggression andsexual behavior (Nelson et al., 1995), may account for thedramatic behavior changes observed in nNOS knockoutmice.
It has been reported that there is no motor impair-ment in nNOS knockout mice (Itzhak et al., 1998); how-ever, we observed that knockout mice are much moreactive than both wild-type controls and heterozygousmice at the two ages tested, although knockout and heter-ozygous mice decreased their horizontal movements asthey became older (Fig. 2). Our tests were performed dur-ing the day, and there is a report indicating that there aredecreases in the motor activity of knockout mice at nightcompared with controls (Kriegsfeld et al., 1999). Theacute administration of NOS inhibitors decreases locomo-tion and induces catalepsy in rats, an effect that is additiveto that induced by haloperidol, whereas subchronic dosesof the NOS inhibitors induce tolerance to the catalepticeffect, thus hinting that there are plastic effects involvedand that NO may modulate motor behavior by interferingwith dopaminergic, serotoninergic, and cholinergic neu-rotransmission in the striatum (Del Bel et al., 2005), aregion that we report is not protected from oxidative dam-age in nNOS knockout mice. A recent report shows thatthe inhibition of NOS activity at a specific period in earlypostnatal life induces increased spontaneous locomotoractivity in adult rats, suggesting a role of NO in the deve-lopment of regions that control motor behavior (Mejoradaet al., 2006). It will be important to analyze further thestructure and function of the regions of knockout micestudied in this work and include others such as hippocam-pus that may be involved in this conduct, to determine
Protection Against Oxidative Damage 1399
Journal of Neuroscience Research DOI 10.1002/jnr

fully the mechanism responsible for the altered motorbehavior, insofar as it is known that the aggressive behav-ior of nNOS knockout mice is related to the serotoniner-gic system (Chiavegatto and Nelson, 2003).
In agreement with previous reports (Huang et al.,1993), we could not detect the presence of nNOS byWestern blot assays in any of the brain regions studied inknockout mice (Figs. 3–5). On the other hand, we wereable to detect the expression of eNOS in the three geno-types studied here, a fact that is consistent with theoriginal report indicating that the expression of eNOS issimilar in knockout and wild-type mice (Huang et al.,1993). We also report the presence of iNOS in the dif-ferent brain regions of knockout, heterozygous, andwild-type mice. Although iNOS expression is tradition-ally associated with inflammatory responses in brain,there are several reports showing its expression in normalrodent and human brain tissue (Park et al., 1996, 2000;Uttenthal et al., 1998; Ferrini et al., 2001; Siles et al.,2002). We consider that the results of NOS expressionare consistent with our findings regarding NOS activity.Knockout mice show significantly decreased levels oftotal NOS activity compared with normal controls in allbrain regions analyzed and at the different ages but simi-lar levels of Ca2+-independent activity, that is, the activ-ity produced by iNOS. The disparity in NOS activity isdue to differences in constitutive or Ca2+-dependent ac-tivity. In this case, we can observe that enzyme activityin knockout mice is very small compared with that inwild-type controls. We believe that constitutive NOSactivity in brain is due completely or almost completelyto the presence and enzymatic activity of nNOS, becausein all genotypes there is a readily detectable level ofexpression of eNOS; however, the changes in enzymeactivity correspond with the changes in the expression ofnNOS. We also believe that iNOS expression and Ca2+-independent NOS activity do not participate in main-taining redox status in the brain under basal conditions,because their levels of expression and activity are similarin all regions studied in the three genotypes, yet there isa selective protection from oxidative damage in cortexand cerebella of nNOS knockout mice (Figs. 3–5). Het-erozygous mice present intermediate results betweenknockout and wild-type subjects, indicating the effect ofthe lack of one of the nNOS alleles.
Generation of NO participates in oxidative damagethrough the formation of peroxynitrite, but the effect ofNO in protection or as a participant of oxidative damageremains controversial (Lipton et al., 1993; Rubbo et al.,1994; Espey et al., 2000), and the final protective or harm-ful effect may depend on the nature of the insult to theorganism (Stamler, 1994). In this paper, we examinedthe degree of oxidative damage in three brain regions atthe two different ages studied. We observed, in all regionsanalyzed, an increase in LP in older mice (Fig. 6), in ac-cordance with previous reports showing an age-dependentincrease in LP and oxidative damage of proteins (carbonylformation) in brain (Head et al., 2002; Navarro and Bove-ris, 2004; McCann et al., 2005).
However, we noticed a selective protection fromoxidative damage in cerebellum and cortex of nNOSknockout mice, whereas no effect was seen in striatum.Levels of LP were lower in the cerebellum of knockoutmice compared with wild-type mice at both ages tested,implyng a continuous and permanent decrease of oxidativedamage in this brain region, a conclusion further sup-ported by the decrease of ROS formation in the sameregion of �/� mice (Figs. 6, 7). A decrease of LP levelswas also observed in cortex, but no protection from ROSformation was detected in this region (Figs. 6, 7). Finally,no significant differences in either LP or ROS formationwere detected in the striata of knockout mice comparedwith either homozygous or wild-type mice (Figs. 6, 7).Different levels of NOS activity in diverse brain regions ofknockout mice have been observed, with the lowest levelsin the cerebellum (0.75% compared with wild-type con-trol) and the highest remaining NOS activity in striatum(7% of control and 5% of control in cortex; Huang et al.,1993). Our experiments yield similar results, with remain-ing constitutive NOS activity of 5% in striata, 3% in cor-tex, and almost undetectable (>1%) in cerebella of knock-out mice compared with the respective wild-type controls(Figs. 3–5). This remaining NOS activity may be the basisfor the dissimilar levels of oxidative damage observedamong the diverse brain regions examined in this report.Moreover, the cerebellum has a higher metabolic activitythan the striatum, and this activity produces more free rad-icals (Gredilla et al., 2001; Lopez-Torres et al., 2002), sothe effects of the lack of the enzyme, as well as the effectsinduced by aging, may be more evident in this region.
We have previously observed (Perez-Severianoet al., 2000, 2004) that, in brain regions where transgenicmice showed very high levels (174% over wild-type con-trol) of LP, increases in ROS formation were considerablylower (18% over control). These data are in accordancewith the more discrete changes in ROS we observed inthe present experiments compared with changes in LP.
Considering the essential physiological roles playedby NO in the organism, it is likely that compensatorymechanisms enter into action when one of the NOS iso-forms is not present. This may explain the complex phe-notype shown by knockout and heterozygous mice, withthe latter presenting intermediate phenotypes betweenknockout and wild-type mice in various behavioral tests,as well as in brain redox status. This compensation may beobtained by differentially regulating the expression and/oractivities of the other NOS isoforms, i.e., eNOS and/oriNOS or even by the involvement of other messengermolecules, such as CO (Snyder et al., 1998). Our resultsindicate that a possible compensatory effect is not meditedby differentially regulating the expression of eNOS oriNOS, because the levels of the proteins are similar in thebrain of mice of the three different genotypes (Figs. 3–5).There are also different nNOS splice variants (Eliassonet al., 1997) that can be active, thus providing low buteffective levels of NO produced by nNOS. It has beenreported that there is an important up-regulation of thenNOSb form in the striatum of knockout mice and also a
1400 Martınez-Lazcano et al.
Journal of Neuroscience Research DOI 10.1002/jnr

significant, but smaller, up-regulation of this isoform incortex, whereas it is not expressed in the cerebellum ofknockout mice (Eliasson et al., 1997), a fact that mayaccount for the dissimilar levels (although all very low) ofenzyme activity we observed in these brain regions. Thismay explain the differences we observed in oxidative dam-age in different brain areas, which directly relate nNOSactivity with basal redox status, in that we detected noprotection from oxidative damage in striatum, with thehighest levels of expression of the b isoform, a permanentdecrease of oxidative damage in cerebellum, where thereis a complete elimination of nNOS, and a partial protec-tion in cortex, where there is an intermediate level ofexpression of the nNOS splice variant, results that are con-sistent with the levels of NOS activity that we find in thiswork. We have observed that there is no detectableexpression of nNOS in knockout mice, as determined byWestern blot analysis, but we cannot exclude the presenceof other nNOS isoforms not revealed by the antibody weare currently using or that the tissue amounts of nNOS arebelow the detection level of our assay. Our results alsosuggest that the other isoforms of NOS, eNOS and iNOS,are not participating in the maintenance of the redox statusin the brains of nNOS knockout mice.
NO produced from nNOS is implicated in the oxida-tive damage induced by brain ischemia (Moro et al., 2004),and nNOS knockout mice are more resistant to this insult(Huang et al., 1994). Perhaps this protective effect may berelated to lower levels of oxidative damage in brain. Theeffects of the lack of NO are complex and some of them canbe expressed only when more than one of the NOS isoen-zymes are knocked out, as occurs in hippocampus, whereboth nNOS and eNOS activities must be eliminated toabolish long-term potentiation completely (Son et al.,1996). Furthermore, it appears that a certain level of stress,or external stimulus, is necessary to uncover fully the conse-quences of the null mutation, as is the case for the presenta-tion of aggressive behavior, which is exacerbated by the iso-lation of the mice (Chiavegatto and Nelson, 2003). Thesedata suggest that stress or other external influences may affectthe balance induced by compensatory mechanisms that can-not adequately respond to these challenges. Nevertheless,further studies are necessary to determine the consequencesof the nNOS null mutation in other metabolic pathways.
Here we report that there is a selective protectionfrom oxidative damage in different brain areas of micewith a targeted disruption of the nNOS gene, under basalconditions. This selective protection from oxidative dam-age may be part of the molecular alterations underlyingthe expression of the nNOS knockout mouse phenotype,and it might also play a role in determining their suscepti-bility to different brain insults. Moreover, our results indi-cate than the nNOS isoform is crucial in determining andmaintaining basal redox status in brain.
ACKNOWLEDGMENTS
We thank Dr. Alberto Castillo (UAM) for adviceregarding statistical analysis, Morayma Guerrero for keep-
ing and maintaining transgenic mice lines, and RubenSanchez for laboratory support.
REFERENCES
Alderton WA, Cooper CE, Knowles RG. 2001. Nitric oxide synthases:
structure, function and inhibition. Biochem J 357:593–615.
Ali SF, LeBel CP, Bondy SC. 1992. Reactive oxygen species formation as a
biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxi-
cology 13:637–648.
Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ, Faraci FM. 1998. Core
and penumbral nitric oxide synthase activity during cerebral ischemia and
reperfusion—editorial comment. Stroke 29:1037–1047.
Beckman JS, Koppenol WH. 1996. Nitric oxide, superoxide, and peroxy-
nitrite: the good, the bad, and ugly. Am J Physiol Cell Physiol 271:
C1424–C1437.
Bredt DS, Snyder SH. 1990. Isolation of nitric oxide synthetase, a calmodu-
lin-requiring enzyme. Proc Natl Acad Sci U S A 87:682–685.
Bredt DS, Snyder SH. 1994. Nitric oxide: a physiologic messenger mole-
cule. Annu Rev Biochem 63:175–795.
Brown GC. 1999. Nitric oxide and mitochondrial respiration. Biochim
Biophys Acta 1411:351–369.
Chiavegatto S, Nelson RJ. 2003. Interaction of nitric oxide and serotonin
in aggressive behavior. Horm Behav 44:233–241.
Chiavegatto S, Dawson VL, Mamounas LA, Koliatsos VE, Dawson TM,
Nelson RJ. 2001. Brain serotonin dysfunction accounts for aggression in
male mice lacking neuronal nitric oxide synthase. Proc Natl Acad Sci
U S A 98:1277–1281.
Christopherson KS, Bredt DS. 1997. Nitric oxide in excitable tissues: physi-
ological roles and disease. J Clin Invest 100:2424–2429.
Del Bel EA, Guimaraes FS, Bermudez E, Gomes MZ, Schiaveto DS, Pado-
van-Neto FE, Tumas V, Barion-Cavalcanti AP, Lazzarini M, Nucci-
da-Silva LP, de Paula-Souza D. 2005. Role of nitric oxide on motor
behavior. Cell Mol Neurobiol 25:371–392.
Eliasson MJ, Blackshaw S, Schell MJ, Snyder SH. 1997. Neuronal nitric
oxide synthase alternatively spliced forms: Prominent functional localiza-
tions in the brain. Proc Natl Acad Sci U S A 94:3396–3401.
Espey MG, Miranda KM, Feelisch M, Fukuto J, Grisham MB, Vitek MP,
Wink DA. 2000. Mechanisms of cell death governed by the balance
between nitrosative and oxidative stress. Ann N Y Acad Sci 899:209–221.
Ferriero DM, Holtzman DM, Black SM, Sheldon RA. 1996. Neonatal
mice lacking neuronal nitric oxide synthase are less vulnerable to
hypoxic-ischemic injury. Neurobiol Dis 3:64–71.
Ferrini M, Wang C, Swerdloff RS, Sinha Hikim AP, Rajfer J, Gonzalez-
Cadavid NF. 2001. Aging-related increased expression of inducible nitric
oxide synthase and cytotoxicity markers in rat hypothalamic regions asso-
ciated with male reproductive function. Neuroendocrinology 74:1–11.
Garcia-Tovar CG, Luna J, Mena R, Soto-Zarate CI, Cortes R, Perez A,
Leon-Avila G, Mornet D, Rendon A, Hernandez JM. 2002. Dystrophin
isoform Dp7l is present in lamellipodia and focal complexes in human
astrocytoma cells U-373 MG. Acta Histochem 104:245–254.
Gredilla R, Barja G, Lopez-Torres M. 2001. Thyroid hormone-induced
oxidative damage on lipids, glutathione and DNA in the mouse heart.
Free Radic Res 35:417–425.
Gyurko R, Leupen S, Huang PL. 2002. Deletion of exon 6 of the neuronal
nitric oxide synthase gene in mice results in hypogonadism and infertility.
Endocrinology 143:2767–2774.
Head E, Liu J, Hagen TM, Muggenburg BA, Milgram NW, Ames BN,
Cotman CW. 2002. Oxidative damage increases with age in a canine
model of human brain aging. J Neurochem 82:375–381.
Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. 1993. Tar-
geted disruption of the neuronal nitric oxide synthase gene. Cell 75:
1273–1286.
Protection Against Oxidative Damage 1401
Journal of Neuroscience Research DOI 10.1002/jnr

Huang PL, Huang Z, Mashimo H, Bloch K, Moskowitz MA, Bevan J,
Fishman MC. 1995. Hypertension in mice lacking the gene for endothe-
lial nitric oxide synthase. Nature 377:239–242.
Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz
MA. 1994. Effects of cerebral ischemia in mice deficient in neuronal nitric
oxide synthase. Science 265:1883–1885.
Itzhak Y, Ali SF, Martin JL, Black MD, Huang PL. 1998. Resistence of
neuronal nitric oxide synthase-deficient mice to cocaine-induced loco-
motor sensitization. Psychopharmacology 140:378–386.
Knowles RG, Moncada S. 1994. Nitric oxide synthases in mammals. Bio-
chem J 298:249–258.
Kriegsfeld LJ, Eliasson MJL, Demas GE, Blackshaw S, Dawson TM, Nelson
RJ, Snyder SH. 1999. Nocturnal motor coordination deficits in neuronal
nitric oxide synthase knock-out mice. Neuroscience 89:311–315.
Laubach VE, Shesely EG, Smithies O, Sherman PA. 1995. Mice lacking
inducible nitric oxide synthase are not resistant to lipopolysaccharide-
induced death. Proc Natl Acad Sci U S A 92:10688–10692.
Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J,
Singel DJ, Stamler JS. 1993. A redox-based mechanism for the neuropro-
tective and neurodestructive effects of nitric oxide and related nitroso-
compounds. Nature 12:626–632.
Lopez-Torres M, Gredilla R, Sanz A, Barja G. 2002. Influence of aging and
long-term caloric restriction on oxygen radical generation and oxidative
DNA damage in rat liver mitochondria. Free Radic Biol Med 32:882–
889.
McCann SM, Mastronardi C, De Laurentiis SA, Rettori V. 2005. The ni-
tric oxide theory of aging revisited. Ann N Y Acad Sci 1057:64–84.
Mejorada A, Aguilar-Alonso P, Leon-Chavez BA, Flores G. 2006.
Enhanced locomotor activity in adult rats with neonatal administration of
N-omega-nitro-L-arginine. Synapse 60:264–270.
Moro MA, Cardenas A, Hurtado O, Leza JC, Lizasoain I. 2004. Role of
nitric oxide after brain ischaemia. Cell Calcium 36:265–275.
Navarro A, Boveris A. 2004. Rat brain and liver mitochondria develop oxi-
dative stress and lose enzymatic activities on aging. Am J Physiol Regul
Integr Comp Physiol 287:R1244–R1249.
Nelson RJ, Demas GE, Huang PL, Fishman MC, Dawson VL, Dawson
TM, Snyder SH. 1995. Behavioural abnormalities in male mice lacking
neuronal nitric oxide synthase. Nature 378:383–386.
Park CS, Park R, Krishna G. 1996. Constitutive expression and structural
diversity of inducible isoform of nitric oxide synthase in human tissues.
Life Sci 59:219–225.
Park CS, Krishna G, Ahn MS, Kang JH, Chung WG, Kim DJ, Hwang HK,
Lee JN, Paik SG, Cha YN. 2000. Differential and constitutive expression
of neuronal, inducible, and endothelial nitric oxide synthase mRNAs and
proteins in pathologically normal human tissues. Nitric Oxide 4:459–471.
Perez-Severiano F, Rios C, Segovia J. 2000. Striatal oxidative domage par-
allels the expression of a neurological phenotype in mice transgenic for
the mutation on Huntington’s disease. Brain Res 862:234–237.
Perez-Severiano F, Escalante B, Vergara P, Rıos C, Segovia J. 2002. Age-
dependent changes in nitric oxide synthase activity and protein expression
in striata of mice transgenic for the Huntington’s disease mutation. Brain
Res 951:34–42.
Perez-Severiano F, Santamaria A, Pedraza-Chaverri J, Medina-Campos
ON, Rıos C, Segovia J. 2004. Increased formation of reactive oxygen
species, but no changes in glutathione peroxidase activity, in striata of
mice transgenic for the Huntington’s disease mutation. Neurochem Res
29:729–733.
Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk
M, Freeman BA. 1994. Nitric oxide regulation of superoxide and peroxy-
nitrite-dependent lipid peroxidation. Formation of novel nitrogen-con-
taining oxidized lipid derivatives. J Biol Chem 269:26066–26075.
Segovia J. 2002. Transgenic model for the study of oxidative damage in
Huntington’s disease. Methods Enzymol 353:365–373.
Segovia J, Perez-Severiano F. 2004. Oxidative damage in Huntington’s dis-
ease. Methods Mol Biol 277:321–334.
Siles E, Martinez-Lara E, Canuelo A, Sanchez M, Hernandez R, Lopez-
Ramos JC, Del Moral ML, Esteban FJ, Blanco S, Pedrosa JA, Rodrigo J,
Peinado MA. 2002. Age-related changes of the nitric oxide system in the
rat brain. Brain Res 956:385–392.
Snyder SH, Jaffrey SR, Zakhary R. 1998. Nitric oxide and carbon monox-
ide: parallel roles as neural messengers. Brain Res Brain Res Rev 26:167–
175.
Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kan-
del ER. 1996. Long-term potentiation is reduced in mice that are doubly
mutant in endothelial and neuronal nitric oxide synthase. Cell 87:1015–
1023.
Stamler JS. 1994. Redox signaling: nitrosylation and related target interac-
tions of nitric oxide. Cell 78:931–936.
Torreilles F, Salman-Tabcheh S, Guerin M, Torreilles J. 1999. Neurodege-
nerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev
30:153–163.
Triggs WJ, Willmore LJ. 1984. In vivo lipid peroxidation in rat brain fol-
lowing intracortical Fe2+ injection. In vivo lipid peroxidation in rat brain
following intracortical Fe2+ injection. J Neurochem 976–980.
Uttenthal LO, Alonso D, Fernandez AP, Campbell RO, Moro MA, Leza
JC, Lizasoain I, Esteban FJ, Barroso JB, Valderrama R, Pedrosa JA, Pei-
nado MA, Serrano J, Richart A, Bentura ML, Santacana M, Martinez-
Murillo R, Rodrigo J. 1998. Neuronal and inducible nitric oxide synthase
and nitrotyrosine immunoreactivities in the cerebral cortex of the aging
rat. Microsc Res Techniq 43:75–88.
1402 Martınez-Lazcano et al.
Journal of Neuroscience Research DOI 10.1002/jnr
Related Documents











