REGULATION OF AMPA RECEPTOR CURRENTS BY MITOCHONDRIAL ATP SENSITIVE K + CHANNELS IN ANOXIC TURTLE NEURONS by George Zivkovic A thesis submitted in conformity with the requirements for the degree of Masters of Science Graduate Department of Cell and Systems Biology University of Toronto © Copyright by George Zivkovic (2010)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

REGULATION OF AMPA RECEPTOR CURRENTS BY MITOCHONDRIAL ATP
SENSITIVE K+ CHANNELS IN ANOXIC TURTLE NEURONS
by
George Zivkovic
A thesis submitted in conformity with the requirements
for the degree of Masters of Science
Graduate Department of Cell and Systems Biology
University of Toronto
© Copyright by George Zivkovic (2010)

ii
Regulation of AMPA receptor currents by mitochondrial ATP
sensitive K+ channels in anoxic turtle neurons
George
Master of Science (2010)
Department of Cell and Systems Biology
University of Toronto
2010
Abstract
Mammalian neurons rapidly undergo excitotoxic cell death during anoxia, while neurons
from the anoxia-tolerant painted turtle can survive without oxygen for hours without apparent
damage. An anoxia-mediated decrease in AMPA receptor currents are an important part of the
turtle‟s natural defence however the mechanism underlying it is unknown. Here I investigate a
mechanism that involves activation of a mitochondrial KATP channel that subsequently signals a
decrease in AMPAR currents. Whole-cell AMPAR currents were stable during normoxia, but
anoxia or pharmacological activation of mKATP channels resulted in a 50% decrease in AMPAR
currents. Conversely, mKATP antagonists blocked the anoxia-mediated decrease. Mitochondrial
KCa channel modulators responded similarly. Blocking the Ca
2+-uniporter also reduced normoxic
AMPAR currents by 40%, and including BAPTA in the recording abolished the anoxia or
agonist-mediated decrease. Therefore, the mKATP channel is involved in the anoxia-mediated
down-regulation of AMPAR activity and is a common mechanism to reduce glutamatergic
excitability.

iii
Acknowledgments
This thesis is the culmination of inspiration, hard work, a passion for science and
continuous support from loved ones throughout my two years as a graduate student. Throughout
my time here, I had the constant support from fellow employers, lab mates, friends and family
members.
Firstly, I would like express my sincere gratitude towards my supervisor, mentor and
friend Les Buck whose knowledge, passion, encouragement, and guidance provided the
inspiration and drive to pursue all of my academic and life ambitions. Not only an amazing
supervisor but also a great person, his continuous support is a large reason for the successful
completion of this work.
I would also like to extend a thank you to my committee members Dr. John Peever and
Dr. Martin Wojtowicz, both of who provided continuous support, feedback and guidance
throughout my time as a graduate student. Furthermore, I would also like to acknowledge Ian
Buglass for the many conversations about sports and life to make this journey more enjoyable.
I am also extremely fortunate to have shared the lab with a group of very intelligent,
supportive, and helpful lab mates that made the two years extremely enjoyable. I would like to
especially thank David “Homeboy” Hogg and Aqsa “A-town” Malik for sharing two wonderful
years of friendship, laughter, stress, and constant debate. I am deeply grateful to you guys and
hope that we remain friends for the many years to come. Petey-boy Hawrish, you‟re presence in
our lab was also greatly appreciated and I hope that you will use your intelligence and drive to
continue our lab tradition of great success and achievement. A final thanks goes out to Young
and Matt, both who helped me a lot when I first entered the lab.
I would also like to thank my family for raising me to pursue my dreams and value
academics at the highest level. Also to my friends Zlatane, Matori, Rui and Tsoki, I couldn‟t
have done it without the constant support and our friendship will remain as strong as Arsenal‟s
unbeaten season. I would like to specially thank Milica Kojic, who being not only my better half
but also my best friend, took care of me and provided the constant support and motivation to
succeed in life. Finally, I would like to leave it by saying one thing that has not only been my
catharsis but also my religion throughout my life: Arsenal F.C is the greatest team the world has
ever seen, its Arsenal „till I die, and there is only one Arsene Wenger!

iv
TABLE OF CONTENTS
ABSTRACT ................................................................................................................................... ii
ACKNOWLEDGMENTS ........................................................................................................... iii
TABLE OF CONTENTS ............................................................................................................ iv
LIST OF TABLES AND FIGURES .......................................................................................... vii
ABBREVIATIONS ........................................................................................................................x
CHAPTER 1: INTRODUCTION .................................................................................................1
1.1 Oxygen in our environment ........................................................................................1
1.2 Varying environments of oxygen availability ............................................................2
1.3 The mammalian brain: Extreme sensitivity to anoxia .............................................3
1.4 Anoxia-tolerance .........................................................................................................9
1.4.1 Criteria for anoxia-tolerance ...........................................................................9
1.4.2 Examples of anoxia-tolerant organisms ........................................................10
1.5 The turtle brain: Extreme tolerance to anoxia........................................................12
1.6 The “Metabolic arrest” hypothesis...........................................................................17
1.7 “Ion channel arrest” ..................................................................................................18
1.7.1 Na+/K
+ ATPase, maintenance of Vm and evidence of channel arrest ...........19

v
1.7.2 Direct evidence of ion channel arrest-NMDAR ...........................................20
1.7.3 AMPAR channel arrest .................................................................................21
1.7.4 “Spike Arrest” ...............................................................................................22
1.8 Putative Mechanisms of channel arrest ...................................................................23
1.8.1 Increased adenosine during anoxia is neuroprotective .................................24
1.8.2 ROS as a signaling molecule in the turtle cortex ..........................................25
1.9 The mitochondrial ATP sensitive potassium (mKATP) channel .............................26
1.9.1 Evidence for the existence of the mKATP channel.........................................28
1.9.2 mKATP channels in NMDAR channel arrest .................................................30
1.9.3 Rationale for study and hypotheses ..............................................................32
CHAPTER 2: Materials and Methods .......................................................................................35
2.1 Animals .......................................................................................................................35
2.2 Dissection and whole-cell patch-clamp protocol .....................................................35
2.3 Evoked AMPA current recordings...........................................................................39
2.4 Pharmacology .............................................................................................................42
2.5 Chemicals ....................................................................................................................41
2.6 Statistical Analysis .....................................................................................................42

vi
CHAPTER 3: RESULTS ............................................................................................................43
3.1 Normoxic and positive control experiments ............................................................43
3.1.1 Normoxic whole-cell evoked AMPAR current measurements ....................44
3.1.2 Positive control experiments using AMPA and NMDAR antagonists .........45
3.2 Pharmacological manipulation of mKATP channels modify AMPAR activity .....47
3.3 Manipulation of mKCa channels also modifies AMPAR activity ...........................50
3.4 Opening of the Ca2+
uniporter abolishes anoxic decreases in AMPAR currents 53
3.5 Ca2+
chelation abolishes the anoxia-mediated decreases in AMPAR currents ....56
CHAPTER 4: DISCUSSION ......................................................................................................59
4.1 Control AMPA and NMDA experiments ................................................................59
4.1.1 Pharmacological AMPA and NMDAR antagonist experiments ..................59
4.2 mKATP channels in ischemic preconditioning and turtle anoxia-tolerance ..........62
4.2.1 IPC and the link to mKATP channels .............................................................62
4.2.2 mKATP channels in the anoxic turtle cortex ..................................................64
4.2.3 mKATP channels regulate AMPA and NMDAR activity during anoxia .......66
4.3 Effects of mitochondrial K+
conductance on whole-cell AMPAR currents ..........68
4.4 Mitochondrial Ca2+
uptake and AMPAR depression .............................................69
4.5 Increased Ca2+
levels and mechanisms of AMPA and NMDAR channel arrest ..70
4.6 AMPAR channel arrest and anoxia tolerance in the western painted turtle .......75

vii
4.7 Conclusions .................................................................................................................76
4.8 Future Directions .......................................................................................................77
CHAPTER 5: APPENDIX ..........................................................................................................79
REFERENCES .............................................................................................................................81

viii
LIST OF TABLES AND FIGURES
CHAPTER 1: INTRODUCTION
Figure 1: Neuronal over-excitation via NMDA and AMPARs and cell death during anoxia .........8
Figure 2: Representative illustration of the relationship between O2 production and availability as
seen in O2 regulators and conformers ..............................................................................................9
Figure 3: Diagrammatic representation of the mechanism by which the painted turtle uses its
shell to sequester H+ and lactate during anoxia .............................................................................15
Figure 4: Schematic illustration of the mechanism by which the mKATP channel regulates [Ca2+
]c
during anoxia and ultimately brings about channel arrest in the AMPA and NMDAR. ...............33
CHAPTER 2: MATERIALS AND METHODS
Figure 5: Turtle cortical sheet model for electrophysiology ..........................................................37
Figure 6: Experimental setup .........................................................................................................38
Figure 7: Whole-cell configuration and evoked AMPAR current recordings ...............................40
CHAPTER 3: RESULTS
Figure 8: Normoxic whole-cell AMPAR current measurements over 90 minutes ........................44
Figure 9: Effect of APV perfusion on whole-cell NMDAR currents ............................................45
Figure 10: Effect of CNQX perfusion on whole-cell AMPAR currents .......................................46
Figure 11: mKATP channel agonists and antagonists regulate AMPAR currents ...........................48
Figure 12: Paired sample trace AMPAR currents for mKATP experiments ...................................49
Figure 13: mKCa channel agonists and antagonists also regulate AMPAR currents .....................51

ix
Figure 14: Paired sample trace AMPAR currents for mKCa experiments .....................................52
Figure 15: Opening of the Ca2+
uniporter abolishes the anoxia-mediated decreases in whole-cell
AMPAR currents ...........................................................................................................................54
Figure 16: Paired sample trace AMPAR currents for Ca2+
uniporter experiments .......................55
Figure 17: Intracellular BAPTA prevents decreases in AMPAR currents during anoxia .............57
Figure 18: Paired sample trace AMPAR currents for BAPTA experiments .................................58
CHAPTER 4: DISCUSSION
Table 1: Anoxia and mKATP channel-mediated decreases in NMDA and AMPAR currents .....67
Figure 19: Diagrammatic representation of Ca2+
-mediated NMDAR channel arrest ....................72
Figure 20: Vehicle (DMSO) application alone did not alter AMPAR currents over 40 minutes of
perfusion ........................................................................................................................................79
Figure 21: mKATP channel openers diazoxide and levcromakalim decrease NMDAR currents
during normoxia in a matter similar to anoxia without pharmacological application ...................80

x
ABBREVIATIONS
5HD 5-hydroxydecanoic acid (mKATP channel antagonist)
8-PT 8-pentyltheophyilline (A1 receptor antagonist)
∆Ψm mitochondrial membrane potential
εPKC epsilon protein kinase C
µM micromolar concentration
μmol-g-h-1
micromol per gram per hour
aCSF artificial cerebrospinal fluid
AMPA α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate
AMPAR α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor
Anox anoxia
ADP adenosine diphosphate
APV (2R)-amino-5-phosphonovaleric acid (NMDA receptor antagonist)
AMP adenosine monophosphate
ATP adenosine triphosphate
[ATP] adenosine triphosphate concentration
BAPTA 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (Ca2+ chelator)

xi
BDM butanedione monoxime (KATP channel antagonist)
CaMK calmodulin-dependent protein kinase
[Ca2+
]c cytosolic Ca2+
concentration
[Ca2+
]m mitochondrial Ca2+ concentration
CBS1 calmodulin binding site
cGKII cGMP-dependent protein kinase II
CNQX 6-cyano-7-nitroquinoxaline-2,3-dione (AMPA receptor antagonist)
DMSO dimethylsulphonic acid
DZX diazoxide (mKATP channel agonist)
ECD excitotoxic cell death
EPSP end-postsynaptic potential
ER endoplasmic reticulum
ETS electron transport chain
GABA γ-aminobutyric acid
[GABA] γ-aminobutyric acid concentration
GABAA γ-aminobutyric acid receptor
GDP guanosine diphosphate

xii
GSH glutathione
GTP guanosine triphosphate
HSP heat shock proteins
IP3 inosine triphosphate
IPC ischemic preconditioning
KATP ATP sensitive K+ channel
Lact lactate
Lev levcromakalim (KATP channel agonist)
MΩ Megaohm
MAGUK membrane-associated guanylate kinase
min minutes
mKATP mitochondrial ATP sensitive K+ channel
mKCa mitochondrial calcium sensitive K+ channel
mmHg millimetre of mercury
mM millimole concentration
mmolg-1
ww-1
millimole per gram per wet weight
mOsm milliosmole

xiii
MPG β mercapto-propyonyl-glycine (ROS scavenger)
mPTP mitochondrial permeability transition pore
mV millivolts
n sample size
nM nanomole
NMDA N-methyl-D-aspartate
NMDAR N-methyl-D-aspartate receptor
NO nitric oxide
NOS nitric oxide synthase
pA picamperes
pO2 partial pressure of oxygen
PP1 protein phosphatase 1
PP2A protein phosphatase 2A
PP2B protein phosphatase 2B
pS picosiemens
pKATP plasmalemal ATP sensitive K+ channel
PKC protein kinase C

xiv
Popen open channel probability
PSD post-synaptic density
O2-
superoxide
PFK phosphofructokinase
PK pyruvate kinase
ROS reactive oxygen species
[ROS] reactive oxygen species concentration
s seconds
S.E.M standard error of mean
SUR sulfonyl urea
SOD superoxide dismutase
t time
TARP transmembrane AMPA regulatory protein
TTX tretrodotoxin (Na+ channel antagonist)
Vm membrane potential
VO2 oxygen utilization

1
CHAPTER 1 INTRODUCTION
1.1 Oxygen in our environment
While diatomic oxygen (O2) only comprises approximately 21% of the air in the Earth’s
atmosphere, it remains an absolute requirement for most of the living organisms that currently
inhabit the planet. About 2.3 billion years ago, cyanobacteria and algae appeared in the world’s
oceans and were responsible for most of the oxygen in our atmosphere, the remaining being
produced by terrestrial plant life some time after (Lane, 2002). These organisms used
photosynthesis to meet their energy requirements, whereby O2 was produced as a by-product of
the reaction and released into the atmosphere (Lane, 2002). Early on, the released O2 was
sequestered by minerals (largely iron), which was essential because O2 exposure was toxic to the
obligate anaerobes inhabiting the Earth. Approximately 2 billion years ago, oxygen began to
accumulate as minerals became saturated and could no longer sequester the released O2,
increasing the concentration of the gas in the atmosphere and leading to a mass extinction of
oxygen intolerant organisms. This increase however also led to the evolution of bacteria and
other organisms that relied on oxygen for their energy production (Lane, 2002).
Most modern day organisms require a continuous supply of oxygen in meet all of their
energy requirements. Vertebrate species developed a variety of respiratory structures to facilitate
the movement of oxygen into their tissues for energy production. For example, the gills of fish
and lungs of mammals provide a pathway through which oxygen can enter organisms and
maximize gas exchange (Buck and Pamenter, 2006). Cells then utilize the dissolved O2 for
cellular respiration in order to produce energy in the form of adenosine triphosphate (ATP)

2
through oxidative phosphorylation. O2 is used as a final electron acceptor in the electron
transport chain where it is reduced to water by cytochrome oxidase and when coupled to
phosphorylation yields 30-36 ATP per mole of glucose.
ATP is the main energy currency of a cell, and cellular respiration produces almost all of
the ATP that the cells need to meet their metabolic demands (Buck and Pamenter, 2006). Most of
the ATP is produced in the mitochondria, but is readily shuttled to different parts of the cell in
order to carry out various functions. These include ion pumping needed for the maintenance of
electrochemical gradients, protein synthesis, and muscle contraction. Since the ATP demands of
a cell are determined by these processes, it is essential that a constant supply of O2 is maintained
in order to meet these needs. Therefore, both aquatic and terrestrial organisms have tightly
coupled ATP supply and demand in order to ensure that enough O2 is constantly delivered to
cells to produce the required ATP.
1.2 Varying environments of oxygen availability
Interestingly, ambient O2 levels can vary drastically across different places on Earth.
Although normal sea level partial pressure of oxygen is typically 156mmHg (normoxia), oxygen-
limiting environments are not uncommon where the partial pressure of O2 (pO2) is reduced
(hypoxia) or complete absent (anoxia) (Buck and Pamenter, 2006). The peak of Mount Everest
has a partial pressure of oxygen roughly one third that at sea level, and the pressure gradient
upon which oxygen moves to facilitate gas exchange is severely compromised. Ice covered lakes
in winter with reduced temperature and light penetration can also be severely hypoxic conditions

3
(Bickler and Buck, 2007). Tide pools also have wide variations in their oxygen availability, as
they undergo a hyperoxic to severely hypoxic transition due to changes in tide from day to night,
respectively (Bickler and Buck, 2007). Finally, flood plains such as the Amazon River basin
dramatically vary in their dissolved O2 concentrations, as the build-up of organic matter in
decreased water levels can establish hypoxic environments during certain periods of the year
(Bickler and Buck, 2007). All of these oxygen-limiting environments provide a stress to
organisms that inhabit them, as their ability to produce the necessary amount of ATP is
compromised. Under hypoxia or anoxia, organisms resort to anaerobic metabolism whereby
glycolysis is the main process of energy production. However, it yields only 2-3 ATP per mole
of glucose or glycogen respectively and thus results in an approximate 10-15 fold decrease in
ATP supply. Although organisms have both short (hyperventilation, tachycardia, vasodilation,
increased anaerobic metabolism) and long term adaptations under hypoxia/anoxia, their survival
under anoxic conditions is in most cases severely compromised.
1.3 The mammalian brain: Extreme sensitivity to anoxia
In general, mammals are extremely anoxia-intolerant, and the human brain is particularly
sensitive to anoxia as it can induce a loss of consciousness within 15-20 seconds (Ramirez et al.,
2007) and suffocation can occur within 3-5 minutes of the onset of anoxia (Lutz et al., 2003).
Every year hundreds of thousands of humans are seriously affected by episodes of low oxygen
availability in instances such as myocardial ischemia, sleep apneas, chronic bronchitis,
emphysema, cerebrovascular accidents, recurrent apneas in premature babies, and sudden infant
death syndrome (Ramirez et al., 2007). The mammalian brain’s extreme sensitivity to low

4
oxygen levels therefore provides a model for understanding the cellular mechanisms associated
with the irreversible damage that is caused by anoxia. The mammalian response to hypoxia
consists of a protective phase where the organism has the ability to survive the first few minutes
of hypoxia without serious damage, and an cytotoxic phase where prolonged oxygen deprivation
leads to irreversible damage in the brain.
With the onset of hypoxia or anoxia, the rate of oxidative phosphorylation begins to slow
down rapidly as the availability of oxygen (being the final electron acceptor in the electron
transport chain) decreases. This decreased ATP production, coupled with a maintained level of
ATP consumption, by cells leads to a more than 90% reduction in cellular ATP levels within 5
minutes of anoxia (Siesjo, 1992a; Siesjo, 1992b). ATP demanding processes become
compromised as the amount of ATP produced is greatly diminished. The Na+/K
+ ATPase is an
ATP driven ion pump that hydrolyses a molecule of ATP to actively move 3 Na+ ions out and 2
K+ ions into the cell. As ATP levels decline, the activity of the Na
+/K
+ ATPase does as well. The
maintenance of ionic electrochemical gradients across the plasma membrane is an important
feature of cells’ physiology and function, and the decreased activity of the pump leads to a loss
of these gradients. This results in a depolarized membrane potential, and the activation of voltage
gated Ca+2
channels in the pre-synaptic terminal that facilitate the influx of Ca+2
into the neuron
(Choi, 1992; Choi, 1994). Consequently, the release of excitatory amino acids (glutamate,
aspartate, serotonin, and dopamine) through exocytosis is facilitated. The process of vesicle
fusion is ATP driven, but the decreased levels of ATP make it unlikely that a sustained level of
amino acid release will continue. Instead, the main mechanism of prolonged over-excitability is
that neurotransmitters remain in the synaptic cleft for abnormally prolonged periods of time
because the main reuptake mechanism through which they are sequestered back into the pre-

5
synaptic neuron, the Na+ dependent co-transporter located on glial cell membranes in close
proximity to the neuron, has now reversed its direction due to the dissipated Na+ gradient. It now
functions to pump neurotransmitters out of cells as shown in Fig. 1 (Choi, 1992; Choi, 1994).
Increased neurotransmitter levels (particularly glutamate) in the synaptic cleft lead to the
subsequent activation of postsynaptic ligand-gated glutamate receptors, such as the N-methyl-D-
aspartate (NMDAR) and the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPAR)
receptors. Both receptors bind glutamate and are involved in the excitatory transmission that
mediates normal information processing and plasticity in neurons (Won et al., 2002). NMDARs
are heterotetramers usually consisting of two NR1 subunits and a mix of either NR2A or B. They
are highly permeable to Ca2+
, as well as Na+ and K
+, and are considered coincident detectors
since their opening depends not only on the binding of glutamate, but also on a depolarized
membrane potential leading to the removal of the Mg2+
block that resides in the receptor pore
(Won et al., 2002). The NR2 subunit binds glutamate, while the NR1 binds the co-agonist
glycine that is necessary for proper channel conductance. AMPARs are also mostly
heterotetramers and contain four subunits (GluR1-4). Each subunit can bind glutamate, and at
least the binding of two glutamate molecules is required to open the channel with subsequent
binding increasing the channel’s conductance. The presence of a GluR2 subunit makes it
energetically unfavourable for Ca2+
to move through the receptor pore, and therefore most
AMPARs in the mammalian cortex are permeable to only Na+ and K
+ (Won et al., 2002).
Normally the concentration of free Ca2+
in the cytoplasm is extremely low, as intra-neuronal
levels are maintained through (1) tight regulation of NMDA and AMPA receptors (2) the uptake
of Ca2+
into the endoplasmic reticulum through the IP3 receptors, (3) activation of plasma

6
membrane Ca2+
ATPases or Na+/Ca
2+ exchanger, (4) the binding of Ca
2+ to target proteins, and
the sequestering of Ca2+
into the mitochondria through uniport mechanisms. (Won et al., 2002).
Over-activation of NMDA and AMPARs during anoxia lead to a massive influx of Ca2+
and Na+
into the post-synaptic neuron, followed by subsequent movement of water and Cl- to counteract
the cation flux. Low oxygen availability also depolarizes the mitochondrial membrane potential
and leads to a decreased driving force for the Ca2+
uniporter to sequester Ca2+
into the
mitochondria, resulting in increased cytosolic Ca2+
concentrations ([Ca2+
]c). Finally, decreased
ATP production leads to increases in [Ca2+
]c through the decreased function of plasmalemal and
ER Ca2+
ATPases. Therefore, the energy failure that occurs during anoxia leads to a massive
Ca2+
overload in the cytoplasm which has detrimental effects to cell integrity and function (Wang
and Qin, 2010). Increased Ca2+
levels promote the production of free radicals and free fatty acids
through the activation of nitric oxide synthase (NOS) and Ca2+
-dependent lipases (Siesjo, 1994).
Ca2+
also activates a calmodulin-dependent protein kinase (CaMK), which can phosphorylate and
alter the activity of enzymes, ion channels, and gene expression. Phospholipase activation by
Ca2+
leads to cleavage of phospholipids in plasmalemal and organellar membranes, resulting in a
loss of cell integrity. Ca2+
activated endonucleases act to destroy cellular DNA. Therefore, a Ca2+
overload followed by subsequent swelling and lysis is consistent with irreversible injury
resulting in necrotic cell death (Fig. 1, Bickler et al., 2002).
Increased levels of intraneuronal free Ca2+
also lead to cell death via the apoptotic pathway
(Choi, 2001). A lack of evidence from microscopic studies raises some doubt as to the actual
extent of the role apoptosis plays in ischemic neurons, since the typical features of apoptosis; cell
shrinkage, apoptotic bodies, nuclear fragmentation and condensation, are not readily observed
(Colbourne et al., 1998). Activation of cytochrome C and caspases through the inhibition of

7
survival factors ( such as Bcl-2) have been suggested (Won et al., 2001). Apoptosis is thought to
be an additional feature of anoxic/ischemic injury, and is more prevalent in the penumbra region
of the infarct (where necrotic cell death pre-dominates) (Choi, 2001).
Overall, the effects of prolonged oxygen deprivation are lethal and anoxic exposure in
mammalian neurons of just several minutes can result in the irreversible damage. However, there
are organisms that can tolerate severe levels of hypoxia and anoxia for extended periods of time
without any apparent excitotoxic insult.

8
Figure 1: Diagramatic representation of neuronal over-excitation through the activation of
NMDA and AMPARs during anoxic-insult leading to excitotoxic cell death (ECD). Decreased
ATP production during anoxia leads to failure of ATP dependent processes such as the Na+/K
+
ATPase. Failure to maintain electrochemical gradients results in membrane depolarization and
subsequent opening of voltage-gated Ca2+
channels. Increased Ca2+
levels facilitate synaptic
release of excitatory neurotransmitters such as glutamate which bind to and activated ligand
gated ionotropic receptors such as the AMPA and NMDAR. Large influx of cations such as Na+
and Ca2+
activate several mediators of necrotic and apoptotic cell death, such as lipases,
proteases and endonucleases leading to ECD.
Na+ Na+ Glu Glu

9
1.4 Anoxia-tolerance
Unlike the sensitivity of the mammalian brain towards low oxygen levels, many
organisms live in environments of variable oxygen availability (explained in sec. 1.2) and have
developed extraordinary capabilities to withstand such stresses for extended time periods.
1.4.1 Criteria for anoxia-tolerance
To survive long-term anoxia, organisms employ several strategies that maintain a stable
energy supply and prevent the excitotoxic insult which is readily observed in the mammalian
brain. These organisms must have, 1) a large glycogen storage capacity, 2) elevated buffering
capacity to compensate for pH changes associated with accumulated acidic anaerobic end-
products, 3) efficient anaerobic metabolic pathways with minimal end-product accumulation, 4)
good antioxidant defence mechanisms and 5) a coordinated reduction in metabolic rate which
includes a matched decrease in cellular ATP supply and demand (Hochachka and Somero,
1984). These organisms are also O2 conformers, as shown below:
(Buck, 2010)
Figure 2: Representative illustration of the relationship between O2 consumption and availability
as seen in O2 regulators (A) and conformers (B). Abreviation: Oxygen utilization (VO2). Both
strategies are explained in text.

10
O2 regulators (Fig. 2A) use physiological adaptations (tachycardia, hyperventilation, and
vasodilation) to maintain their rate of oxygen consumption at a high level even though oxygen
availability decreases. Once oxygen levels drop below a critical level, their O2 consumption
decreases dramatically and falls to zero even before they become completely anoxic. As a result
these organisms cannot tolerate prolonged periods without O2 and will be exposed to anoxic
insult if it is not replenished. O2 conformers (Fig. 2B) conversely adjust their O2 consumption
according to the availability of oxygen, and therefore are more tolerant to hypoxia/anoxia than
O2 regulators. Examples of the above strategies are found in most invertebrates and several
vertebrate species allowing them to tolerate extended periods of anoxia.
1.4.2. Examples of anoxia-tolerant organisms
A wide range of organisms have the remarkable capability of surviving prolonged periods
of hypoxia/anoxia without apparent injury (Bickler et al., 2002). Many invertebrates,
amphibians, reptiles, fish, and even some mammals display various degrees of anoxia tolerance
that makes them a fascinating area of interest for researchers.
The tolerance that lower vertebrates have shown is associated with hypometabolism and
prolonged periods of dormancy with reduced activity levels (Bickler and Buck, 2007).
Invertebrates have experienced a wide variety of environments with varying levels of oxygen
availability even dating back to before the accumulation of oxygen in our atmosphere. For
example, isotopic and chemical analysis of ice cores at the time of the Permian-Triassic
transition showed that ambient oxygen concentrations were only 1/3rd
of what they are today
(Bickler and Buck, 2007). Therefore, the ancestors of modern amphibians, fish, and some
reptiles experienced a selection for hypoxia tolerance that was not present for other organisms

11
(Bickler and Buck, 2007). Among invertebrates, the wild type fruit fly (Drosophila melongaster)
can withstand 4 hours of anoxia with no apparent injury (Krishnan et al., 1997). Among
molluscs, the pond snail (Lymnea stagnalis) is significantly hypoxia-tolerant as hypometabolism
and inactivity is often induced by cold temperatures, and can even survive up to 40 days of
anoxia with a 10% mortality rate after 7 days of recovery (Wijsman et al., 1985). Several
amphibians are also able to tolerate periods of little or no oxygen supply, as the common frog
(Rana temproaria) and the related leopard frog Rana pipiens are able to survive a few days at
low temperatures and up to 3 days of anoxia at room temperature (Christiansen and Penney,
1973; Donohoe et al., 2000). Interestingly, ATP levels in these organisms are reduced by as
much as 50% within 2 hours of anoxia, yet they are able to make a full recovery afterwards (Lutz
and Nilsson, 1997; Nilsson, 2001). However, their recovery is limited as levels of ATP lower
than 50% are usually not tolerated and often lethal.
Fish vary greatly in their levels of hypoxia tolerance with eels, hagfish and carp having
the highest survival capability. The crucian carp (Carrassius carassius) is the most tolerant fish
known to date, being able to endure months of hypoxia under low temperatures (Nilsson and
Renshaw, 2004). Its cousin, the goldfish (Carassius auratus) endures a similar degree of anoxia-
tolerance being able to survive up to 3 months at low temperatures of 10°C and a few days at
room temperatures of 22-25°C (Chen et al., 2005). Interestingly, although they remain anoxic,
the goldfish remains active in the water column unlike the many other anoxia-tolerant species
that display an arrested state of little or no mobility. Both fish are able to convert the end-product
lactate into ethanol and excrete it into the surrounding water, thereby avoiding lactic acidosis
(Bickler and Buck, 2007). Certain species of birds also show some degrees of anoxia tolerance,
such as the Ruppell’s griffin which can fly at altitudes up to 37,000 feet where the partial

12
pressure of oxygen is less than 1/3rd
of the pressure at sea level (Laybourne, 1974). Even in
mammals, the arctic ground squirrel (Spermophillus parryii) can tolerate an approximate 50%
reduction in oxygen-haemoglobin saturation at oxygen partial pressures of 7 mmHg without any
apparent damage (Dave et al., 2009). Finally, the naked mole rat Hetercephalus glaber is thought
to be the most hypoxia tolerant mammal known. It employs a variety of strategies including
living in a chronic hypometabolic state. However, this small mammal can only survive 11 hours
of hypoxia at a oxygen partial pressure of 8 mmHg (Avivi et al., 1999).
The most anoxia-tolerant vertebrate species known to date is a reptile, and it has formed
the basis of much of our understanding of anoxia-tolerance and thus has been studied
extensively.
1.5 The turtle brain: Extreme tolerance to anoxia
The western painted turtle (Chrysemys picta bellii) is the most anoxia-tolerant vertebrate
species known to date. It resides in certain areas of southern Canada west of the great lakes and
in certain parts of the northern United States (Jackson, 2000). Over the winter, freezing
temperatures creating ice-covered ponds force these animals to bury themselves underwater in
typically anoxic mud and remain in a dormant comatose state (Jackson, 2000). In the absence of
oxygen, this species can survive about 1000 times longer than a mammal under warm conditions
and as long as 6 months at decreased temperatures of 1-30C (Ultsch and Jackson, 1982; Herbert
and Jackson, 1985a,b; Lutz, 1992). Being a true facultative anaerobe, this species of turtle can
rely exclusively on anaerobic metabolism to meet all of its energy requirements.

13
As mentioned previously, a good anoxia-tolerant organism must have a large glycogen
storage capacity in order to fuel anaerobic metabolism. The painted turtle, along with the
goldfish, have the largest glycogen storage capacity among all vertebrate species. In the turtle,
glycogen is stored mainly in the liver and accounts for approximately 15% of the turtle’s mass
(Clark and Miller, 1973). Furthermore, anoxia-tolerant organisms must have good buffering
capacity and the ability to sequester or eliminate anaerobic end-products in order to avoid
metabolic acidosis and self-pollution. Although metabolic rate greatly decreases in the anoxic
turtle, lactic acid build-up in the plasma still occurs and blood lactate levels increase to as high as
200mM after several months of anoxic submersion at 2-3°C (Ultsch and Jackson, 1982). Lactate
production is associated with a decrease in pH, since ATP hydrolysis produces a H+ as a by-
product (Hochachka and Mommsen, 1983). However, even though plasma lactate levels increase
in the anoxic turtle, pH remains relatively stable and only decreases from about 8 to slightly
below 7 following months of submergence (Ultsch and Jackson, 1982).
The extraordinary buffering capacity of the turtle allows it to sequester the produced
protons and avoid metabolic acidosis by having elevated plasma bicarbonate (HCO3-) levels of
~40mM (Herbert and Jackson, 1985). The turtle also uses a counter-ion balance strategy in order
to protect against rises in plasma lactate, where increased concentrations of both Ca2+
(50mM)
and Mg2+
(20mM) occur in parallel (Ultsch et al., 1999). Jackson and colleagues (2000)
confirmed that the source of Ca2+
and Mg2+
was the turtle’s skeleton, where an in vitro
preparation of the shell incubated in an acidic solution led to increased release of both cations
which then complexed with lactate to reverse the negative charge of the molecule. Further
experiments were performed to investigate whether turtle bones and shell acted as a site to
sequester lactate during anoxic perfusion (Jackson et al., 1999). Lactate levels in turtles that were

14
dived for four months at temperatures of 3°C increased to 164 and 136 mmolg-1
ww-1
in bone and
shell respectively, indicating that they could be important tissues in preventing the build-up of
lactate during anoxia (Jackson et.al 1999). Jackson and colleagues (2000) further demonstrated
evidence for shell uptake of lactate uptake by showing that a bathing solution containing lactate
turned alkaline once the powder of a turtle shell was administered. Their proposed mechanism
for lactate uptake by the turtle shell is as follows: As lactate and H+
levels increase during
anoxia, they would enter the turtle shell by diffusion. While lactate complexes with Ca2+
, protons
would be buffered by the HCO3-. The latter reaction produces CO2 which then diffuses back into
the plasma and is excreted into the environment (Fig 3). As a result the turtle is capable of
dealing with the increased H+ and lactate production, preventing the end-product accumulation
and metabolic acidosis that is often seen in mammals during anaerobic glycolysis.

15
Bickler and Buck 2007
Figure 3: Diagrammatic representation of the mechanism by which the painted turtle uses its
shell to sequester H+ and lactate during anoxia. Abbrevations: lactate (Lact). The shell acts as a
buffer to avoid metabolic acidosis and end-product accumulation. Lactate is buffered by Ca2+
,
while H+ are sequestered by HCO3
- which is converted to CO2 and then released into the
atmosphere. Similarly, HCO3- and Ca
2+ are also released into the plasma to buffer lactate and H+
accumulation during anoxia.
Another significant challenge when dealing with anoxia is the ability to avoid free-radical
formation and related injury upon re-oxygenation. The painted turtle has an excellent antioxidant
defence mechanism that is used to minimize reperfusion injury (Ramaglia and Buck, 2004). In
the turtle, expression of heat shock proteins (HSP) 72,73, and 90 increase during the anoxic-

16
normoxic transition, but it took more than 18 hours of anoxia to induce a significant increase in
HSP gene expression (Ramaglia et al., 2004; Ramaglia and Buck, 2004). This suggests that
protection from free radical formation may be more significant during re-oxygenation rather than
the anoxic period. Nevertheless, these proteins along with constitutively high levels of
antioxidant enzymes such as glutathione (GSH), catalase, superoxide dismutase (SOD), and alkyl
hydroperoxide reductase allow the turtle to endure anoxic bouts and re-oxygenation without
apparent free-radical damage (Lutz and Milton, 2004; Storey, 1996).
As seen above, the turtle`s ability to employ several strategies in order to cope with
glucose availability, end-product accumulation, acidosis, and free radical formation renders it
tolerant to prolonged oxygen deprivation. However, being able to produce the required energy
for its survival is only one side of the equation. If the ATP demands of the cells do not decrease,
glycolysis will still fall short of meeting the necessary energy requirements. The next two
concepts: metabolic and ion channel arrest, were coined by Peter Hochachka (1986) and have
proven to be the foundation of low-temperature and anoxic survival in anoxia-tolerant species,
including the painted turtle.

17
1.6 The “Metabolic arrest” hypothesis
In 1986, Hochachka outlined his ―metabolic arrest‖ theory on anoxia-tolerance in which
he proposes that decreased energy utilization must accompany decreased energy production to
maintain substrate stores and ionic homeostasis during hypoxia or hypothermia (Hochachka,
1986). The western painted turtle has the remarkable capability to undergo a reversible 85%
decrease in metabolic rate in response to anoxic submergence (Jackson, 1968). Furthermore,
with the combined effects of anoxia and the decreased temperature of 3°C which the turtle
typically encounters over the winter, it can reduce its metabolism up to 99% (Jackson and
Herbert, 1985a). This is due mainly to decreases in protein catabolism (94%), protein synthesis
(92%), urea synthesis (74%) and suppression of Na+/K
+ATPase activity (75%) reduced heart rate
and electrical activity (Buck and Hochachka, 1993; Buck et al., 1993; Land et al., 1993).
Hochachka’s theory predicts that this profound reduction can be achieved by decreasing the rate
of ATP synthesis, a phenomenon known as the reverse Pasteur effect (in contrast to the Pasteur
Effect which indicates an increase in glycolytic rate), in which glycolytic flux is reduced in order
to save glucose and prevent an accumulation of anaerobic end-products (Hochachka, 1986;
Krebs, 1972).
To date there has been substantial support for anoxia-induced metabolic arrest in whole-
animal studies of the painted turtle. As the ATP/ADP ratio changes, these molecules can feed
back to glycolytic enzymes and allosterically modulate their activity. Phosphofructokinase (PFK)
and pyruvate kinase (PK) are two enzymes that catalyze two rate limiting reactions in the
glycolytic pathway. Their decreased activity during anoxia maintains a decreased glycolytic flux
and the reverse Pasteur effect (Bickler and Buck, 2007). Further evidence includes the ability to

18
survive months on anaerobic metabolism alone, a decrease in heart rate from 30 to 0.4 beats per
minute, and severely reduced muscle activity (Jackson and Herbert, 1985a). Whole-animal
calorimetric experiments have also indicated a reversible 90-95% reduction in heat loss (measure
of metabolic activity) during anoxic perfusion (Jackson, 1968). Further studies in brain tissue
and liver hepatocytes based on lactate concentration, oxygen consumption, and glucose
utilization indicated similar reductions in metabolic rate during anoxia (Buck et al., 1993; Doll et
al., 1994). In in situ heart preparations, a 50% decrease in power output during anoxia was also
observed (Farrell et al., 1994).
The remarkable ability of these organisms to suppress their metabolic rate during anoxia
is therefore a result of a reduction in ATP producing pathways which save glucose and minimize
self-pollution by avoiding metabolic acidosis and lactate accumulation. However, if ATP
consuming pathways (ATP demand) do not decrease in parallel in order to maintain ATP
balance, cells would not be able to meet all of their energy requirements and ATP levels would
drastically decline leading to cell death. Along with metabolic arrest, the painted turtle also
utilize a strategy to decrease cellular ATP demand to prevent ATP run-down during anoxia. Two
such strategies are ion channel arrest and spike arrest, outlined below.
1.7 “Ion Channel Arrest”
Peter Hochachka also hypothesized that a requirement for metabolic arrest is an
associated decrease in ion channel density or activity, termed ―ion channel arrest‖ (Hochachka,
1986). He based his hypothesis on two main ideas: 1) hypoxia-tolerant cells possess an

19
inherently low plasma membrane ion channel density and activity, as ectotherms possess
approximately 5 times less ion channels per membrane area than endotherms of similar size; and
2) these organisms possess mechanisms to further depress ion channel density or activity when
exposed to low O2 (Hochachka, 1986). The rationale is that the decreased ion flux leads to
energy conservation by reducing the demand placed on ATP-dependent pumps to maintain
electrochemical gradients across the plasma membrane in all cells, therefore maintain stable ATP
concentrations [ATP], also known as ATP turnover. The primary ion pump responsible for the
establishment and maintenance of cellular electrochemical gradients and consequently
membrane potential is the Na+/K
+ATPase.
1.7.1 Na+/K
+ATPase, maintenance of Vm and indirect evidence of channel arrest
The maintenance of ion gradients is an energetically expensive process with the
Na+/K
+ATPase consuming about 30% of total liver ATP production during normoxia (Buck and
Hochachka, 1993). Given that during anoxia the Na+/K
+ ATPase is the major consumer of
cellular energy, a reduction in ion leak would significantly decrease the ATP cost of maintaining
electrochemical gradients and thereby avoid depolarization and cytotoxic effects. It is therefore
logical that a decrease in ion flux through either reduced channel density or activity would occur
alongside a decrease in Na+/K
+ ATPase activity, resulting in significant energy savings and
thereby promoting anoxic survival at a diminished energy state. Buck and colleagues (1993)
reported a coordinated 75% decrease in Na+/K
+ ATPase activity in painted turtle hepatocytes
during anoxia without significant changes in cell membrane potential. The ATPase activity was
measured by ouabain-inhibitable 86
Rb+ uptake into the cell. Isotopic rubidium is a K
+ substitute
and its radioactivity can be used to measure changes in K+ efflux through the ATPase. The ATP

20
consumption rate of the pump during normoxic conditions was about 19.1μmol-g-h-1
. Isotopic
During anoxia however, the activity of the pump decreased by as much as 75% as the measured
ATP consumption was about 4.8μmol-g-h-1
(Buck et al., 1993). Since the measured membrane
potential did not significantly change during anoxia, the associated decrease in ATPase activity
provided further support for the channel arrest hypothesis.
There has accumulated a considerable amount of evidence in support of an anoxia
induced mechanism of ion channel arrest in various cells of several anoxia-tolerant organisms.
Indirect evidence of channel arrest has contributed to much of our earlier understanding of the
mechanisms by which certain hypoxia-tolerant organisms maintain stable electrochemical
gradients and prevent cytotoxicity. In the cerebellum of the freshwater turtle Pseudemys scripta,
anoxia induces a 42% decrease in voltage-gated Na+ channel density as determined by the
binding of the Na+ channel antagonist [
3H] brevetoxin, and in situ brain studies of the turtle,
Trachemys scripta, anoxia led to a 70% decrease in K+ leakage during anoxia, suggesting
inhibition of K+ channels (Perez-Pinzon et al., 1992; Ghai and Buck, 1999). Furthermore, Chih
and colleagues (1989) were able to show that in the painted turtle that the rate of K+ efflux
decreased by 70% of its normoxic levels when the Na+/K
+ATPase inhibitor ouabain was added
during anoxia. Finally, in frog skeletal muscle hypoxia was also found to mediate a 50%
reduction in Na+/K
+ATPase activity coupled with a decreased Na
+ permeability and maintenance
of membrane potential (Donohoe et al., 2000).
1.7.2 Direct evidence of ion channel arrest-NMDAR
NMDARs are ligand-gated calcium channels that bind glutamate with high affinity (see
1.3). As mentioned, they are highly permeable to Ca2+
, as well as Na+ and K
+, and depend on

21
membrane depolarization for their opening in order to remove the Mg2+
block that resides in the
receptor pore. As key mediators of cell death, NMDARs must be regulated in anoxia-tolerant
organisms in order to prevent the excitotoxic effects that are seen in mammals during ischemia.
Indeed, single channel electrophysiological recordings have demonstrated that the NMDAR open
probability (Popen) decreases by 65% in anoxic turtle neurons (Buck and Bickler, 1998). This also
corresponds to a 62% decrease in the NMDAR-mediated changes in [Ca2+
]c (Buck and Bickler,
1995). Furthermore, a 35% decrease in whole-cell NMDA currents in turtle cortical neurons was
observed during anoxia, which was reversible upon re-oxygenation (Shin and Buck, 2003).
Evidence of NMDAR channel arrest was also demonstrated in the anoxia-tolerant goldfish
(Carrasius auratus), where whole-cell currents decreased by 40-50% in response to anoxia
(Wilkie et al., 2008).
1.7.3 AMPAR channel arrest
In addition, recent evidence has indicated that AMPA receptors (AMPAR) also undergo
ion channel arrest in the painted turtle as whole-cell AMPAR currents were shown to decrease
by 60% in response to anoxia (Pamenter et al., 2008a). EPSP frequency and amplitude also
decrease by 28% and 13% respectively (Pamenter et al., 2008a). AMPARs are also ligand-gated
ion channels that bind glutamate and are permeable to Na+, K
+ and Ca
2+, although Ca
2+ flux is
rare due to energetically unfavourable conditions created by the GluR2 subunit. Due to their
ability to mediate fast synaptic transmission through influx of Na+ that consequently induces
downstream Ca2+
influx through NMDARs, they play an important role in excitotoxicity during
ischemic insult. Therefore, AMPAR blockade or decreased activity is thought to be
neuroprotective, especially since it has the ability to decrease excitability earlier than NMDAR

22
down-regulation. Evidence of the importance of AMPARs to ECD comes from experiments
showing that mice over expressing AMPARs are more susceptible than wild-type mice to ECD
(Le et al., 1997). AMPAR blockade has also been shown to be neuroprotective against cell death
caused by seizures and Parkinsonism (Ohmori et al., 1994; Klockgether et al., 1991). Thus,
unlike the potentiation of NMDA and AMPARs that is observed in mammals during anoxia,
turtle NMDA and AMPARs undergo a reversible ion channel arrest during anoxic episodes that
is neuroprotective.
1.7.4 “Spike arrest”
Ion channel arrest is an important strategy employed by anoxia-tolerant organisms such
as the turtle in order to decrease ATP demands and promote survival during anoxia when energy
supply is greatly diminished. Ion channel arrest and the associated decrease in ion flux results in
a suppressed firing frequency or synaptic transmission in neurons. This process is known as
―spike arrest‖ (Sick et al., 1993). In the anoxic turtle cortex, evoked field potentials and EPSPs
are depressed by as much as 80%, and action potential thresholds in the anoxic turtle cerebellar
purkinje fibres increase by 14mV (Lutz and Nilsson, 1997; Perez Pinzon et al., 1992; Buck and
Bickler, 1998). An increased action potential threshold results in increased excitatory inputs
needed to result in an action potential, a strategy that reduces the ATP demands of the cell by
reducing the need to re-establish electrochemical gradients after excitation.
In theory, a decrease in excitability and synaptic transmission can occur either through
channel arrest or through to increased release of inhibitory neurotransmitters. γ-aminobutyric

23
acid (GABA) is a major inhibitory neurotransmitter in mature vertebrate brain, and is therefore a
potential mediator of spike arrest. Previous studies have also indicated that GABA may be
involved in the anoxic response, as Nilsson and colleagues (1990) showed a 60% and 127%
increase in [GABA] in turtle brain after 4 and 13 hours of anoxia respectively. Furthermore,
following 24 hours of anoxic perfusion of turtle cortical neurons, GABAA receptor density was
up-regulated by as much 129% of normoxic values (Lutz and Kabler, 1995). Interestingly, recent
studies have indicated that in the painted turtle brain anoxia causes an increase in whole-cell
conductance (Pamenter et al., unpublished 2009). Although initially thought to be contradictory
to the concept of channel arrest, the increased conductance was found to result from an increase
in brain GABA concentration and activation of postsynaptic GABAA receptors initiating a Cl-
current. The effect of this hyperpolarizing Cl- current is to ―clamp‖ the resting membrane
potential near the Cl-
equilibrium potential, preventing the neuron from depolarizing and
generating an action potential (Pamenter et al., unpublished 2009). Although these findings
conflict somewhat with a general channel arrest theory, they may indicate that in anoxic turtle
brain it is necessary to inhibit excitatory conductance through channel arrest as well as reduce
action potential firing through spike arrest.
1.8 Putative mechanisms of channel arrest
While channel arrest in the anoxic painted turtle is well documented, the endogenous
mechanisms by which a decrease in oxygen availability ultimately brings about a decreased
activity in these ion channels have not yet been characterized. Several pathways have been
proposed to play a key role in the down-regulation of NMDA and AMPA receptors since

24
manipulation of them leads to changes in receptor activity. Adenosine and reactive oxygen
species (ROS) have previously been proposed to be involved in the down-regulation of NMDA
and AMPA receptors since their concentration levels are sensitive to O2 supply.
1.8.1 Increased adenosine during anoxia is neuroprotective
The nucleoside adenosine has been thought to play a role (albeit limited) in the turtle’s
neuroprotective mechanism because of its rapid and reversible (within minutes) increase in
concentration during anoxia. It is referred to as a ―retaliatory metabolite‖ as its increased levels
during anoxic perfusion: 1) produce vasodilation in order to increase substrate availability to
target tissues, 2) stimulate glygenolysis in liver, producing glucose to drive glycolysis and ATP
production, 3) increase the rate of anaerobic glycolysis to meet ATP demands, and 4) decrease
neurotransmitter release and neuronal excitability to reduce cellular energy requirements (Lutz
and Nilsson, 1997; Buck, 2004). Lutz and Kabler (1997) demonstrated that in the freshwater
red-eared slider (Trachemys scripta), brain adenosine levels temporarily increase shortly after the
onset of anoxia, return to baseline after an hour, and increase again approximately 2.5 hours after
the initial peak. These changes can be explained by the early increase in adenosine levels due to
an initial drop in [ATP], followed by a return to baseline.
Evidence of adenosine’s role in neuroprotection also exists in the painted turtle where
extracellular adenosine concentrations increase 10-fold after 2h of anoxia and leads to increased
blood flow to the brain, but antagonizing adenosine A1 receptors leads to a release of K+ from
the cell (as retaining K+ seems to be necessary for tolerance in the turtle) (Nilsson et al., 1990).
Increased adenosine concentrations during normoxia also reduce NMDAR open probability
(Popen) and the NMDA mediated rise in [Ca2+
]c, and this decrease was abolished when perfusing

25
the A1 antagonist 8-pentyltheophyilline (8-PT) (Buck and Bickler, 1998; Perez-Pinzon et al.,
1993). However, antagonising the A1 receptor during anoxia did not abolish the anoxia-mediated
decrease in NMDAR currents suggesting that although neuroprotective, adenosine may not be
the main mediator of ion channel arrest in the painted turtle (Buck and Bickler 1998).
1.8.2 ROS as a signalling molecule in the turtle cortex
ROS have only been recently examined as possible signalling molecules involved
in the coordination of cellular pathways that potentially result in ion channel arrest. ROS are
produced in the mitochondrion by the cytochromes as a result of ―electron leak‖, where only one
of the two electrons is passed along the cytochromes, and the lone electron reacting with oxygen
to form superoxide (O2-). Superoxide is unstable, quickly reduced to hydrogen peroxide by SOD
and moved to the cytosol where it could potentially become a signalling molecule (Vanden Hoek
et al., 1998). In the absence of oxygen, ROS production should decrease. Pamenter and
colleagues (2007) found that [ROS] decreased by as much as 25% within 10 minutes of cyanide
or nitrogen induced anoxia. ROS have the ability to modulate signalling proteins by reversibly
altering specific residues on them, making them good candidate molecules for channel activity
modulation. They also have the ability to modify downstream effectors like protein kinases,
phosphatases, and transcription factors through the oxidation of thiol residues, as well as
modifying cysteine residues on membrane ion channels such as the N-methyl-D-aspartate
receptor (Wang et al., 1997). Ion channel arrest may be initiated through oxidation and reversed
by reducing agents, so the ROS mechanism provides a pathway that is both rapidly activated and
easily reversible (Wang et al., 1997). Mimicking anoxic conditions by scavenging ROS with β
mercapto-propyonyl-glycine (MPG) during normoxia increased action potential (AP) threshold

26
with no change in whole-cell conductance in the turtle cortex (Zivkovic and Buck, unpublished
2008). This indicates that changes in [ROS] may be associated with the neuroprotective
mechanisms in the anoxic turtle cortex. However, recent experiments in the painted turtle
demonstrated that anoxia or NMDAR antagonism decreased nitric oxide (NO, a reactive oxygen
species) production by 27% and 41% respectively, and NMDAR blockade during anoxia reduced
the subsequent NO production by 75 % (Pamenter et al., 2008c). Therefore, the down-regulation
in NMDAR activity and that the turtle’s anoxia tolerance may depend on preventing NO-
mediated damage during the transition in and out of anoxia (Pamenter et al., 2008c). Although
there are conflicting reports regarding the role of ROS during anoxia, it should not be
disregarded as an important candidate cellular signalling molecule that may function as an
oxygen sensor within the cell.
Although both adenosine and ROS may play a modulatory role in the turtle’s
neurprotection against anoxia, a mitochondrial potassium channel has recently been implicated in
channel arrest (Pamenter et al., 2008b). It has been previously linked to ischemic preconditioning
in some animal models and has been the recent focus of the down-regulation of ion channel
activity in the anoxic turtle cortex.
1.9 The mitochondrial ATP sensitive potassium (mKATP) channel
Out of the four different types of potassium channels that have been described in the
mitochondria, the mKATP channel is the most intriguing in that it may be involved in anoxic
neuroprotection in the western painted turtle (Choma et al., 2009; Pamenter et al., 2008b). The

27
mKATP is an inward rectifying K+ channel found in the inner mitochondrial membrane. mKATP
channels have been suggested to play an essential role in mammalian ischemic preconditioning, a
process by which short periods of ischemia provide protection against subsequent ischemic
insults. Ischemic preconditioning may activate several endogenous adaptive mechanisms that
increase the resistance of tissues to ischemic insult (Hanley and Daut, 2005). To date, two
different types of mKATP channels are known: plasmalemal KATP channels (pKATP) which have
been identified and sequenced, and mitochondrial KATP channels which have not (Paucek et al.,
1992).
The structural composition and genetic sequence for the mitochondrial variant remains
unknown but there remains a consensus that it resembles the plasmalemal type. Based on the
similarities with the surface channel and observations regarding the pharmacology and
immunoreactivity of specific antibodies, it is believed that the mKATP channel is composed of
four pore forming subunits (Kir 6.1-6.4) and four sulfonyl urea subunits (SUR) which form
regulatory sites for channel modulation. It is agonized by GTP, GDP, and low levels of AMP,
but inhibited by ATP, ADP, and modulated by a number of intracellular messengers, including
PKC, adenosine and superoxide anions (Bajgar et al., 2001; Busija et al., 2004).
Pharmacological modulators have also been characterized for the channel and used for a wide
range of experiments. One of the key features of these agents is that almost all of the drugs that
agonize the mKATP channel can also open the surface type, with the exception of a few. Common
general KATP agonists are levcromakalim and pinacidil, while the agonists diazoxide and
nicorandil are thought to be mKATP channel specific (Ardehali et al., 2005). The most common
general KATP antagonists are glibenclamide and glipizide, while 5-hydroxydencaoic acid (5HD)
and MCC-134 are considered mKATP specific (Li et al., 2010; Ardehali et al., 2005).

28
1.9.1 Evidence for the existence of the mKATP channel
There is a substantial amount of evidence for the existence of the mKATP channel through
several studies and techniques. Single channel patch clamp studies of isolated rat liver
mitochondria identified a single K+ selective channel with a conductance of ~10pS, which was
inhibited by ATP and ADP (Inoue et al., 1991). Another source of evidence for the existence of
these channels is through protein purification processes that were performed on rat liver
mitochondria, which produced a protein fraction containing a K+ channel with a similar
molecular weight to the pKATP channel (Paucek et al., 1992). Measurements of mitochondrial
matrix volume in isolated mitochondria also provided some evidence to the existence of the
mKATP channel. K+ influx into the mitochondria is usually followed by water, which leads to
subsequent mitochondrial matrix swelling. This is usually accompanied by the movement of K+
out of the mitochondria through the H+/K
+ exchanger, but occasionally the influx transiently
exceeds the counterbalancing activity of the exchanger leading to matrix swelling (Garlid and
Paucek, 2001; Szewcyk et al., 1993). Garlid and colleagues (1997) reported that mitochondrial
matrix swelling increased with diazoxide perfusion, and was reversed with the putative
antagonists 5HD and glibenclamide.
Further evidence for the channel’s existence is derived from flavoprotein fluorescence
experiments on rabbit ventricular myocytes (Hu et al., 1999). The rationale for measuring
flavoprotein fluorescence is that if opening of the mKATP channels causes an increased activity of
the H+/K
+ exchanger and a subsequent dissipation of the proton gradient, the dissipated gradient
accelerates electron transfer along the ETS and leads to a net oxidation of the mitochondrial
cytochromes. Since flavoproteins only fluoresce when they are oxidized, measuring flavoprotein

29
oxidation using several mKATP channel manipulators can provide evidence for the presence of the
channel in the inner mitochondrial membrane. Diazoxide perfusion produced a dose-dependent
increase in flavoprotein fluorescence, and this was blocked with 5HD application (Hu et al.,
1999). Another indicator for the presence of the channel is that there is a slight depolarization of
the mitochondrial membrane potential (∆Ψm) with the application of diazoxide or nicorandil, a
process completely reversed by 5HD or glibenclamide (Debska et al., 2002). Ψm is thought to
depolarize with the opening of the mKATP channel because it leads to increased utilization of the
H+/K
+ exchanger, which partly dissipates the proton gradient. Finally, the most recent and
possibly most convincing evidence for the existence of the mKATP channel comes from Raval and
colleagues (2007) who isolated rat pup liver and hippocampus mitochondria and found the
presence of Kir6.1 and 6.2 subunits using Western blot staining techniques. Although
contamination during the isolation procedure is possible, their experiment indicates the presence
of the pore-forming subunits of the mKATP channel and is the first direct study toward identifying
the channel.
Despite the putatively important role that mKATP channels play in ischemic
preconditioning and neuroprotection, their molecular identity still remains unclear. Until the
gene for the channel is identified and sequenced, the role they play in these processes cannot be
confirmed. Nevertheless, a wide range of studies as shown above coupled with very consistent
pharmacological data indicates the presence of the mKATP channel in the mitochondria whose
protective role in mammalian ischemic preconditioning may also be involved in the mechanisms
of anoxia-tolerance in the painted turtle.

30
1.9.2 mKATP channels in NMDAR channel arrest
The down-regulation of ionotropic glutamate receptors may be due to the opening of the
mKATP channel which has also been linked to ischemic preconditioning in some animal models,
including: cultured rat cortical models following glutamate toxicity (Kis et al., 2003, Kis et al.,
2004), anoxic juvenile mouse brainstem (Muller et al., 2002) and in rat hippocampal and cortical
neurons following anoxia/reperfusion injury (Heurteaux et al., 1995; Semenov et al., 2000).
Pamenter and colleagues (2008b) recently investigated the role of mKATP channels in the down-
regulation of NMDARs in the cortex of the painted turtle during anoxia. Interestingly, they found
that application of the mKATP channel specific agonists diazoxide and levcromakalim decreased
whole-cell NMDAR currents by 40%, similar to anoxic perfusion alone. The anoxia or
diazoxide-mediated decrease in whole-cell NMDAR currents was completely abolished when the
mKATP channel specific antagonists 5HD and glibenclamide were applied, suggesting a strong
link between the mKATP channel and the down-regulation of NMDARs (Pamenter et al., 2008b).
Furthermore, mild elevations in [Ca2+
]c of ~10% were found upon anoxic or diazoxide perfusion
suggesting that the down-regulation of NMDARs during anoxia may be a Ca2+
mediated
mechanism. Pamenter and colleagues (2008b) were further able to determine that the source of
the rise in [Ca2+
]c was mitochondrial, creating a further link between ion channel arrest and the
mKATP channel. Their results also suggest that that mKATP channel activation may be part of a
natural anoxia-tolerance mechanism that ultimately results in decreased whole-cell NMDAR
currents and neuroprotection.
The AMPAR, like the NMDAR, is also an ionotropic glutamate receptor implicated in
mammalian ECD during ischemia. Evidence of the importance of AMPARs to ECD comes from

31
experiments showing that mice over expressing AMPARs are more susceptible than wild-type
mice to ECD (Le et al., 1997). A mechanism to reduce AMPAR currents would be a logical part
of a mechanism to suppress NMDARs (Siesjo et al., 1991). It has been demonstrated that upon
anoxic perfusion AMPARs exhibit this decrease (Pamenter et al., 2008a); however, whether an
mKATP-based mechanism underlies this response remains unknown. Since AMPARs are also
activated by glutamate and play an important role in regulating NMDAR activity, a common
regulatory mechanism would be practical.
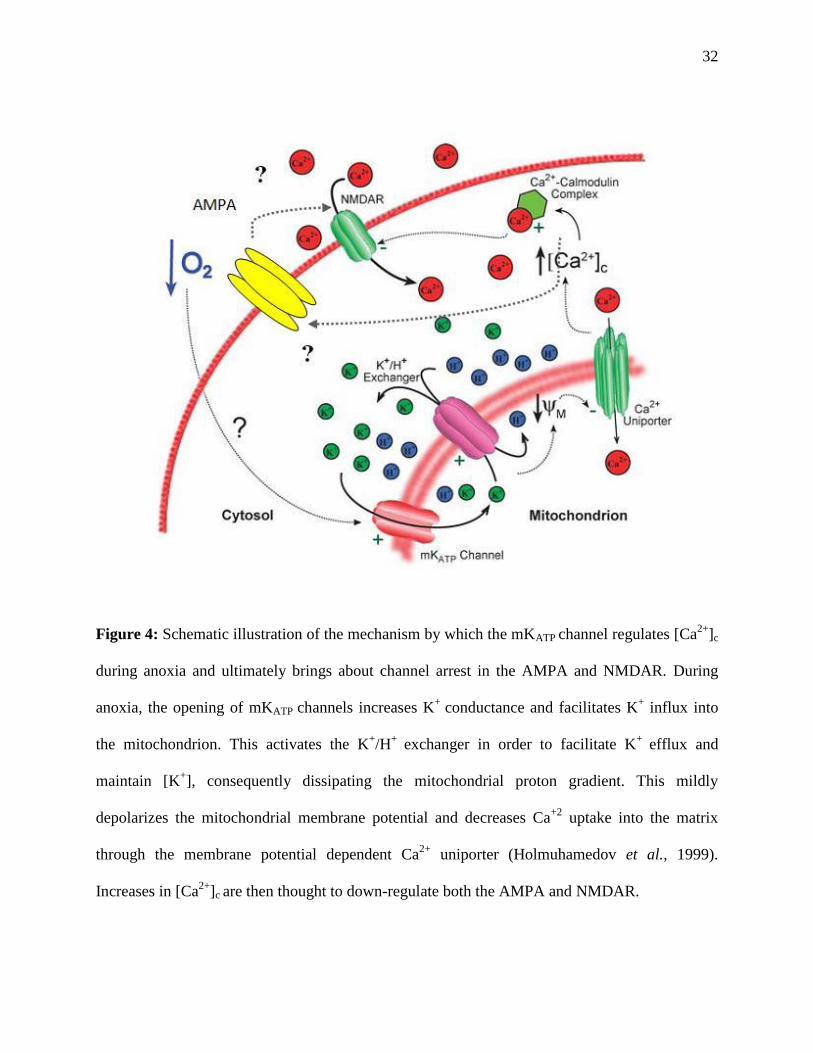
32
Figure 4: Schematic illustration of the mechanism by which the mKATP channel regulates [Ca2+
]c
during anoxia and ultimately brings about channel arrest in the AMPA and NMDAR. During
anoxia, the opening of mKATP channels increases K+
conductance and facilitates K+ influx into
the mitochondrion. This activates the K+/H
+ exchanger in order to facilitate K
+ efflux and
maintain [K+], consequently dissipating the mitochondrial proton gradient. This mildly
depolarizes the mitochondrial membrane potential and decreases Ca+2
uptake into the matrix
through the membrane potential dependent Ca2+
uniporter (Holmuhamedov et al., 1999).
Increases in [Ca2+
]c are then thought to down-regulate both the AMPA and NMDAR.

33
1.9.3 Rationale for study
The involvement of AMPARs in neuroprotection and their link to the mKATP channel has
not yet been investigated. The primary aim of this study was to investigate whether
pharmacological manipulation of mKATP channel activity regulates whole-cell evoked AMPAR
currents during normoxia and anoxia in turtle cortex. Since the mKATP channel has already been
shown to down-regulate the NMDAR during anoxia, it remains important to determine whether
the AMPAR is regulated in a similar way. Furthermore, it will also be important to determine
whether the response is Ca2+
dependent similar to the NMDAR.
The neuroprotective role that the mKATP channel is thought to confer during anoxia is due
to its ability to regulate Ca2+
uptake into the mitochondrial matrix and prevent mitochondrial
Ca2+
overload. The opening of mKATP channels increases K+
conductance and facilitates K+
influx into the mitochondrion. This activates the K+/H
+ exchanger in order to facilitate K
+ efflux
and maintain the [K+], consequently dissipating the mitochondrial proton gradient. This mildly
depolarizes the mitochondrial membrane potential and decreases Ca+2
uptake into the matrix
through the membrane potential dependent Ca2+
uniporter (Holmuhamedov et al., 1999). Mild
increases in [Ca2+
]c are then thought to down-regulate the AMPAR (as well as the NMDAR)
through a mechanism not yet known. See fig. 4 for a schematic illustration of this mechanism.
Based upon the previous evidence showing mKATP regulation of the NMDAR, I propose the
following:

34
Hypotheses:
1. Agonists of mKATP channels will reduce normoxic AMPAR currents and antagonists
will prevent the anoxia-mediated decrease in AMPAR currents. Using whole-cell
electrophysiological recordings, measure evoked AMPA EPSCs following anoxic exposure
and/or pharmacological manipulation of the mKATP channel.
2. Opening of the Ca2+
sensitive mitochondrial K+ (mKCa) will lead to decreases in
normoxic AMPAR currents similar to mKATP experiments. A positive control experiment
to confirm the role of mitochondrial K+ uptake on whole-cell AMPAR activity using the well
known and sequenced mKCa channel.
3. Increasing mitochondrial Ca+2
uptake through the uniporter will abolish the anoxia-
mediated decrease in AMPAR currents. Using whole-cell recordings, the effects of
decreased mitochondrial Ca+2
uptake through the Ca+2
uniporter on whole-cell AMPAR
currents will be measured.
4. Sequestering Ca+2
with BAPTA will prevent the anoxia-mediated decreases in whole-
cell AMPAR currents. Measure whole-cell AMPAR currents during normoxia and anoxia
with the inclusion of a calcium chelator in the recording pipette to investigate whether the
down-regulation of AMPAR during anoxia is Ca+2
dependent.

35
CHAPTER 2 MATERIALS AND METHODS
2.1 Animals
This study was approved by the University of Toronto Animal Care committee and
conforms to the relevant guidelines for the care and handling of experimental animals as outlined
in the Guide to the care and Use of Experimental animals as determined by the Canadian
Council on Animal Care.
Adult western painted turtles were purchased from Niles Biological Inc. (Sacramento,
CA, USA). Animals were housed in an indoor aquarium equipped with a flow-through de-
chlorinated freshwater system, a heating lamp, and a basking platform. The water temperature
was maintained at approximately 17°C, and the air temperature between 20-22°C. The turtles
were given continuous access to food, and were kept on a 12h:12h light-dark photoperiod.
2.2 Dissection and whole-cell patch-clamp recording protocol
All experiments were performed at a room temperature of 22°C. The basic protocols for
cortical sheet dissection and whole-cell patch clamp recordings under normoxic and anoxic
conditions are described elsewhere (Shin and Buck, 2003). Briefly, turtles were decapitated and
whole brains were rapidly excised from the cranium within 30 seconds of decapitation (Fig. 5).
Six cortical sheets were isolated from whole brains and bathed in artificial cerebrospinal fluid
(aCSF; mM: 107 NaCl, 2.6 KCl, 1.2 CaCl2, 1 MgCl2, 2 NaH2PO4 (2H2O), 26.5 NaHCO3, 10
glucose, 5 imidazole, pH 7.4; osmolarity 285-290 mOsm).

36
Cortical sheets were placed in an RC-26 chamber with a P1 platform (Warner
Instruments, CT, USA) (Fig. 6). The chamber was gravity perfused with aCSF at a rate of 2-3
mL min-1
(Fig. 6). Normoxic aCSF was gassed with 95% O2-5%CO2, while a gas mixture of 95%
N2-5%CO2 was used to achieve anoxic conditions. In order to maintain anoxic conditions in the
bath, perfusion tubes from the IV bottle were double jacketed and the outer jacket gassed with
95% N2-5%CO2. The anoxic aCSF reservoir was bubbled for 30 minutes prior to the start of an
experiment. To maintain anoxic conditions in the bath a plastic cover with a hole for the
recording electrode was placed over the recording chamber, and the space between the cover and
the bath was gently gassed with 95% N2-5%CO2. The aCSF reservoir and recording chamber
were constantly gassed with 95% N2-5%CO2 throughout the entire anoxic experiment. The
partial pressure of oxygen (PO2) was measured using a Clark-type electrode and decreased from
approximately 610 mmHg PO2 (hyperoxia) to 0.5 mmHg PO2 (anoxia) within 5 minutes of gassing
which is the limit of detection for the PO2 electrode and not significantly different from the anoxic
aCSF reservoir (data not shown).
Whole-cell patch clamp recordings were obtained using 3-6MΩ borosilicate glass
electrodes. Electrodes contained a solution containing (in mM): 8 NaCl, 0.0001 CaCl2, 10 Na
HEPES, 110 Kgluconate, 1 MgCl2, 0.3 NaGTP, 2 NaATP, adjusted to a pH of 7.4 using
methanesulfonic acid; osmolarity 280-290 mOsm. Cell-attached 5-20GΩ seals were obtained
using the blind-patch technique described elsewhere (Blanton et al., 1989). To break into the
cell, the recording electrode potential was clamped to -70mV and a sharp pulse of suction was
applied. When the configuration became whole-cell, typical access resistance ranged from 10-30
MΩ and any patches were discarded if access resistance varied by more than 20%. From the
voltage-clamp configuration, an Axopatch 1D amplifier was switched to the zero-current
37
position and the membrane potential was read from the main meter. The cell-attached patch was
allowed to stabilize for 2 min before the resting membrane potential was determined, which
ranged from -55 to -85mV. All data were collected at 2 kHz using an Axopatch-1D amplifier, a
CV-4 headstage and a Digidata 1200 interface and analyzed using the Clampex 7 software (Axon
Instruments, Union City, CA) (Fig. 6).
Figure 5: Turtle cortical sheet model for electrophysiology. After decapitation and removal of
the turtle cortex, rostral, caudal and midline cuts were made to the telencephalon, which opened
up the thin cortical sheet. The interior portion of the tissue (as shown by the staining) has densely
packed perikaryal layer of pyramidal and stellate neurons that can be used for
electrophysiological experiments.

38
Figure 6: Experimental setup (see explanation in 2.2). Tissues were gravity perfused with
saline throughout the entire experiment. Normoxic aCSF was gassed with 95% O2-5%CO2, while
a gas mixture of 95% N2-5%CO2 was used to achieve anoxic conditions. In order to maintain
anoxic conditions in the bath, perfusion tubes from the IV bottle were double jacketed and the
outer jacket gassed with 95% N2-5%CO2. Recordings were digitized and analyzed using the
pClamp software. All data were collected at 2 kHz using an Axopatch-1D amplifier, a CV-4
headstage and a Digidata 1200 interface and analyzed using the Clampex 7 software.

39
2.3 Evoked AMPA current recordings
Normoxic experiments consisted of an O2/CO2 aCSF perfusion as explained above. A
fast-step perfusion system (VC-6 perfusion valve controller and SF-77B fast-step perfusion
system, Warner Instruments) was used to deliver 1µM tetrodotoxin (TTX) and 50µM α-amino-3-
hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) (Fig. 7). The concentration of AMPA used
was selected based on previous experiments done in the turtle cortex (Pamenter et. al 2008a).
Higher concentrations (AMPA 100-200 µM) resulted in large currents that became deleterious to
the cell, as assessed by membrane potential run-down and cell death. An AMPA concentration of
50µM produced repeatable currents that did not affect the integrity of the neurons throughout the
course of the 90 minute experiment. In bulk perfusate, 25 µM (2R)-amino-5-phosphonovaleric
acid (APV) was added to prevent NMDAR mediated currents that may result from AMPAR
activation. Prior to each recording, cortical sheets were perfused with TTX for 5 minutes to
prevent spontaneous and evoked action potentials. AMPAR current-voltage relationships in the
cortex of the painted turtle were previously determined, with the reversal potential being ~ 4mV
(Pamenter et al., 2008a). For all experiments, the attached cells were clamped at a holding
potential of -80mV, and AMPA was applied onto cortical sheets using the fast-perfusion system
until a current was measured (2-10 seconds, depending on the proximity of the drug delivery
system to the neuron being recorded from) (Fig. 7). The same AMPA application time was then
used for every recording performed on the same neuron within a single experiment.
Control evoked AMPAR currents were recorded at the start of the experiment (t=0min)
and at t=10 minutes. The initial current recording at t=0 min was set to 100% and all subsequent
recordings from that experiment were normalized to that control value, while the second control

40
value at 10 minutes was used for statistical analysis. Following the two recordings, the cells were
perfused with anoxic aCSF or aCSF containing specific pharmacologic compounds for 40
minutes and evoked AMPAR currents recorded at 20 minute intervals. Tissue was then re-
perfused with control normoxic aCSF for 40 minutes and currents recorded at 20 minute
intervals following the change in aCSF perfusion.
Figure 7: Whole-cell configuration and evoked AMPAR current recordings (see 2.3 for
explanation). Whole-cell patch clamp recordings were obtained using 3-6 MΩ borosilicate glass
electrodes. Cell-attached 5-20 GΩ seals were obtained using the blind-patch technique described
elsewhere (Blanton et al., 1989). To break into the cell, the recording electrode potential was
clamped to -70 mV and a sharp pulse of suction was applied For all experiments, the attached
cells were clamped at a holding potential of -80 mV, and AMPA was applied onto cortical sheets
using the fast-perfusion system until a current was measured (2-10 seconds, depending on the
proximity of the drug delivery system to the neuron being recorded from)

41
2.4 Pharmacology
All pharmacological compounds used in the whole-cell AMPAR experiments were
dissolved in bulk perfusate and applied onto the cortical neurons as specified in the Results.
AMPA receptors were stimulated with AMPA (50 µM) and blocked with CNQX (30 µM).
Mitochondrial KATP channels were activated using levcromakalim (100µM) or the channel-
specific agonist diazoxide (100µM), and blocked with glibenclamide (80µM) or the channel-
specific antagonist 5HD (100 µM). We have previously measured that at the concentrations used,
diazoxide does not affect state-3 respiration in painted turtle mitochondria (Pamenter et al.,
2008b). Diazoxide is nearly 2000 times more selective for the mitochondrial KATP channel than
for the plasmalemal KATP channel and is not a significant activator of the pKATP channel at the
concentrations used (Garlid et al., 1996; Garlid et al., 1997). Similarly, 5HD does not have a
significant effect on pKATP channels but is an effective blocker of mKATP channels (McCullough
et al., 1991, Garlid et al., 1997). The mitochondrial Ca2+
uniporter was activated by spermine
(500 µM) and blocked by ruthedium red (40nM) (Allshire et. al 1985). For experiments
involving calcium chelation, 1,2-bis(o-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid)
(BAPTA, 5mM) was added to the recording electrode solution (Shin et. al 2005). Finally,
mitochondrial Ca2+
sensitive K+ channels (mKCa) were activated by NS1619 (50 µM) and
blocked by paxilline (1 µM) (Sato et al., 2005).
2.5 Chemicals
All chemicals were obtained from Sigma Chemical (Oakville, ON, Canada). Diazoxide,
levcromakalim, NS1619, glibenclamide and paxilline were initially dissolved in

42
dimethylsulphonic acid (DMSO) before being dissolved in the bulk aCSF, not exceeding 1% v/v.
DMSO application alone did not affect AMPA-R mediated currents (see Fig. 20 in appendix).
2.6 Statistical Analysis
AMPAR mediated whole cell current data were collected using a repeat measures design
and analyzed following root arcsine transformation using a two-way ANOVA with a Student-
Newman-Keuls (all pair-wise) post hoc test to compare within and against treatment and control
values. Significance was determined at P<0.05, and all data are expressed as mean ± standard
error of mean (S.E.M). All statistical analyses were performed using SigmaStat ver.11.0. All
recordings at t=0 min were set to 100%, and all subsequent recordings from that experiment were
normalized to that control value, while the second control value at 10 minutes was used to test
for significance between the values obtained throughout the experiment.

43
CHAPTER 3 RESULTS
3.1 Normoxic and Positive Control Experiments
The experimental 90 minute time frame was chosen to maintain consistency with all of
the other AMPA experiments conducted for the purpose of the study. Whole-cell evoked
AMPAR currents did not change throughout 90 minutes of recording from the turtle cortex (Fig
8, n=8 p<0.05). The purpose of this experiment was to also determine whether the chosen
[AMPA] would produce stable and repeatable evoked currents. An AMPA concentration of
50µM produced repeatable currents that did not affect the integrity of the neurons throughout the
course of the 90 minute experiment.
To confirm that both NMDA and AMPARs in turtle neurons respond similarly to their
artificial mammalian glutamate analogs NMDA and AMPA, selective antagonists for each were
perfused to determine if the recorded currents could be abolished. Whole-cell NMDAR currents
were completely abolished following 30 minutes perfusion of the selective NMDAR antagonist
2-amino-5-phosphonovaleric acid (APV; 25 µM, p<0.05) (Fig. 9). Similarly, application of the
specific AMPAR antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 30 µM, p<0.05)
abolished whole-cell AMPAR currents after 60 minutes (Fig. 10).
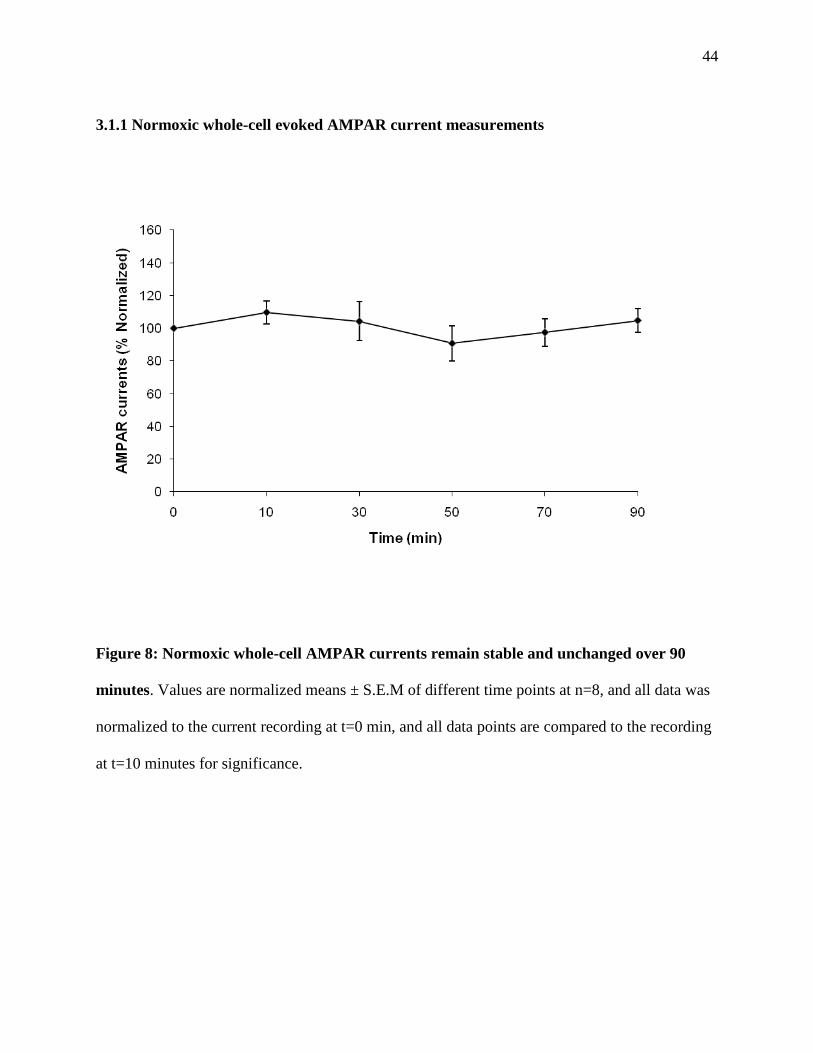
44
3.1.1 Normoxic whole-cell evoked AMPAR current measurements
Figure 8: Normoxic whole-cell AMPAR currents remain stable and unchanged over 90
minutes. Values are normalized means ± S.E.M of different time points at n=8, and all data was
normalized to the current recording at t=0 min, and all data points are compared to the recording
at t=10 minutes for significance.

45
3.1.2 Positive Control Experiments using AMPA and NMDAR antagonists
Figure 9: The selective NMDAR antagonist 2-amino-5-phosphonovaleric acid (APV) abolishes
whole-cell NMDA currents after 30 minutes of perfusion. Lines are representative NMDAR
current traces at control (t=0 min), and following perfusion of APV for 15 min and 30 min.

46
Figure 10: The selective AMPAR antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX)
abolishes whole-cell AMPA currents after 60 minutes of perfusion. Lines are representative
AMPAR current traces at control (t=0 min), and following perfusion of CNQX after 10 min and
20 min.

47
3.2 Pharmacological manipulation of the mKATP channel modifies AMPAR activity
Under normoxia, evoked AMPAR currents did not change significantly over 90 minutes
of recording, but decreased by 46 ± 6.4% and 52 ± 4.9% after 20 minutes and 40 minutes of
anoxic perfusion, respectively (n=7, p<0.05, Fig 11, Fig.12 B). Activation of the mKATP channel
during normoxia using the specific agonist diazoxide decreased the AMPAR current by 41 ±
7.2% and 46 ± 4.2% after 20 and 40 minutes of perfusion, respectively (n=8, p<0.05, Fig 11, Fig
12 C). Similarly, activation of the KATP channel using the general agonist levcromakalim resulted
in a 47 ± 6.7 % and 41 ± 4.8 % decrease in AMPAR current amplitude following 20 and 40
minutes of normoxic perfusion respectively (n=7, p<0.05, Fig 11, Fig 12D). Diazoxide and
levcromakalim mediated decreases in AMPAR activity were not significantly different from the
anoxia mediated decrease (p <0.001). The reduction in AMPAR current amplitude observed after
either diazoxide and levcromakalim perfusion was abolished by the perfusion of the general
mKATP channel antagonist glibenclamide or specific antagonist 5HD (n=7, p<0.05, Fig 11, Fig
12F, G and I). The mKATP antagonised currents were not significantly different from normoxic
control recordings. During anoxia, perfusion with either 5HD or glibenclamide blocked the
anoxia-mediated decrease in AMPAR currents and these were not significantly different from
normoxic control, or normoxic currents with either 5HD or glibenclamide (n=7, p<0.05, Fig 11,
Fig 12E and H).

48
Norm
oxic
Anoxic
Dia
zoxi
de
Levcr
omak
alim
Anox + 5
HD
DZX +
5HD
Lev +
5HD
Anox + G
b
DZX +
Gb
AM
PA
R c
urr
en
ts (
% N
orm
alize
d)
0
20
40
60
80
100
120
140
160
*
*
* * * *
††
† †
‡‡
‡
‡
‡‡
Figure 11: mKATP channel agonists and antagonists regulate whole-cell AMPAR currents.
Normalized whole-cell AMPAR currents at t=30 min (black bars) and t=50 min (grey bars) of
treatment. Continuous line represents normoxic controls, while dashed line represents anoxic
controls. Symbols indicate values significantly different from normoxic control (*), anoxic
control (†), or drug treatment control (‡) (p<0.05). All data are expressed as means ± S.E.M
(n=7). Abbreviations: Anoxia (anox), 5-Hydroxydecanoic Acid (5HD), diazoxide (DZX),
levcromakalim (Lev), and glibenclamide.

49
Figure 12: Paired sample trace AMPAR currents at t=10 min (control) and at t=50 min
following 40 minutes of anoxic or pharmacological drug perfusion. Abbreviations: Anoxia
(anox), 5-Hydroxydecanoic Acid (5HD), diazoxide (DZX), levcromakalim (Lev), and
glibenclamide.
A B C
D E F
G H I

50
3.3 Pharmacological manipulation of the mKCa channel also modifies AMPAR activity
Opening of the mKATP is proposed to increase mitochondrial K+ uptake; however, the
channel has not be cloned and positively identified in the mitochondrial membrane. Therefore,
we used pharmacological modulators of the mitochondrial calcium sensitive K+ (mKCa) channel.
Although the mKCa channel is not thought to be involved in the anoxic response, this channel has
been cloned and sequenced and has a known pharmacology that can serve as a control to confirm
the role of mitochondrial K+ influx (Sato et al., 2005). Administration of the mKCa channel
agonist NS1619 decreased whole-cell AMPAR currents by 44.2 ± 9.1 % and 56 ± 3.4% after 20
and 40 minutes of perfusion, respectively (n=8, p<0.05, Fig 13, Fig 14A). This decrease was
abolished when NS1619 was applied with the mKCa channel antagonist paxilline, as AMPAR
currents did not change throughout the 40 minute perfusion (n=8, p<0.05, Fig 13, Fig 14B). The
mKATP channel antagonist 5HD did not abolish the decrease in AMPAR currents when applied
with NS1619, as AMPAR currents decreased by 37 ± 10.9% and 58.5 ± 8.1% (n=8, p<0.05, Fig
13, Fig 14C). Similarly, the mKATP-mediated decrease in AMPAR currents seen following
diazoxide perfusion was not blocked after administration of the mKCa channel antagonist
paxilline, where AMPAR currents decreased by 46.8 ± 9.9% and 39.1 ± 7.2% after 20 and 40
minutes of perfusion, respectively (n=8, p<0.05, Fig 13, Fig 14D).

51
NS16
19
NS16
19+Pax
illin
e
DZX+Pax
illin
e
NS16
19+5H
D
AM
PA
R c
urr
en
ts (
% n
orm
alize
d)
0
20
40
60
80
100
120
140
160
180
*
*
* *
††
Figure 13: mKCa channel agonists and antagonists regulate whole-cell AMPAR currents.
Normalized whole-cell AMPAR currents at t=30 min (black bars) and t=50 min (grey bars) of
treatment. Continuous line represents normoxic controls, while dashed line represents anoxic
controls. Symbols indicate values significantly different from normoxic control (*), or drug
treatment control (†) (p<0.05). All data are expressed as means ± S.E.M (n=8). Abbreviations:
diazoxide (DZX), 5-hydroxydecanoic acid (5HD).
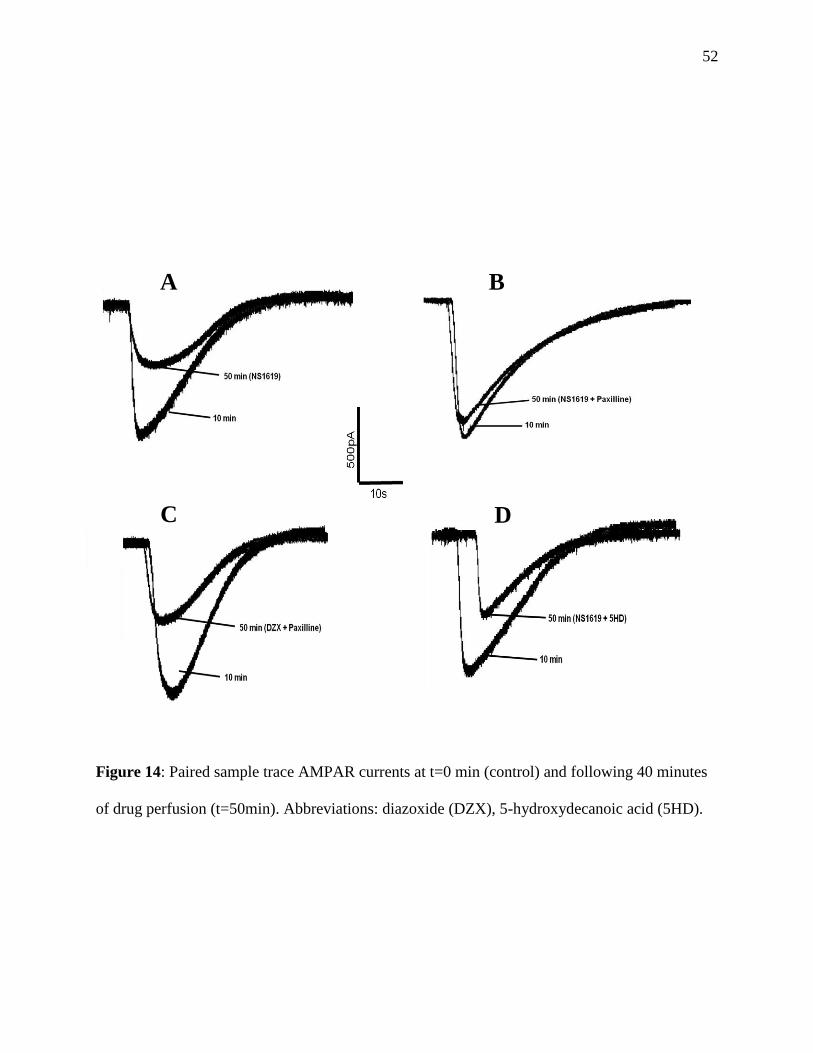
52
Figure 14: Paired sample trace AMPAR currents at t=0 min (control) and following 40 minutes
of drug perfusion (t=50min). Abbreviations: diazoxide (DZX), 5-hydroxydecanoic acid (5HD).
A B
C D

53
3.4 Opening of the Ca2+
uniporter abolishes the anoxia-mediated decrease in whole-cell
AMPAR currents
Mild uncoupling of the mitochondria through the mKATP channel is believed to dissipate
the proton gradient and reduce Ca2+
uptake through the uniporter, resulting in an increase in
[Ca2+
]c. To confirm that a moderate increase in [Ca+2
]c mediates a decrease in AMPAR currents
during anoxia, we measured whole-cell AMPAR currents in the presence of pharmacological
manipulators of the mitochondrial Ca2+
uniporter. Following perfusion of the Ca2+
uniporter
antagonist ruthenium red, whole-cell AMPAR currents decreased by 41.4 ± 7.9% and 37.9 ±
4.8% after 20 and 40 minutes, respectively (n=7, p<0.05, Fig 15, Fig 16A). The decrease in
AMPAR currents during anoxia was abolished using the Ca2+
uniporter agonist spermine, where
after 40 minutes of perfusion, whole-cell AMPAR currents did not change significantly (n=7,
p<0.05, Fig 15, Fig 16 C). Furthermore, application of spermine for 40 minutes during normoxia
did not alter AMPAR currents (n=7, p<0.05, Fig 15, Fig 16 B). The diazoxide mediated decrease
(Fig 11, Fig. 12 C) in whole-cell AMPAR currents was abolished when the drug was applied
along with the uniporter agonist spermine (n=7, p<0.05, Fig 15, Fig 16D). Lastly, spermine
application along with the mKATP channel specific antagonist 5HD had no effect on normoxic
whole-cell AMPAR currents throughout the entire perfusion period (n=7, p<0.05, Fig 15, Fig
16E).

54
Ruth
eniu
m R
ed
Sperm
ine
Anox+Sper
min
e
DZX+Sper
min
e
Sperm
ine+
5HD
AM
PA
R c
urr
en
ts (
% n
orm
ali
zed
)
0
20
40
60
80
100
120
140
160
* *
†
†‡
‡
Figure 15: Opening of the Ca+2
uniporter abolishes the anoxia-mediated decreases in
whole-cell AMPAR currents. Normalized whole-cell AMPAR currents at t=30 min (black bars)
and t=50 min (grey bars) of treatment. Continuous line represents normoxic controls, while
dashed line represents anoxic controls. Symbols indicate values significantly different from
normoxic control (*), anoxic control (†), or drug treatment control (p<0.05, refer to Fig. 2) (‡).
All data are expressed as means ± S.E.M (n=7). Abbreviations: anoxia (Anox), 5-
hydroxydecanoic acid (5HD), diazoxide (DZX).

55
Figure 16: Paired sample AMPAR current traces at t=10 min (control) and at t=50 min
following 40 minutes of anoxic or drug perfusion. Abbreviations: anoxia (Anox), 5-
hydroxydecanoic acid (5HD), diazoxide (DZX).
A B
C
E
D

56
3.5 Calcium chelation abolishes the anoxia-mediated decrease in whole-cell AMPAR
currents
Increases in [Ca2+
]c during anoxia are associated with the decrease in NMDAR activity in
the turtle, therefore we investigated whether it has a similar effect on AMPAR currents during
anoxia. Normoxic AMPAR currents were not affected by the addition of the calcium chelator
BAPTA to the recording electrode (5mM; n=7, p<0.05, Fig 17, Fig 18 A); however, inclusion of
BAPTA blocked the anoxia-mediated decrease in AMPAR currents (n=7, p<0.05, Fig 17, Fig 18
B). Furthermore, BAPTA blocked the diazoxide-mediated decrease in AMPAR currents and
these data were not significantly different those observed with BAPTA alone (n=7, p<0.05, Fig
17, Fig 18 C).
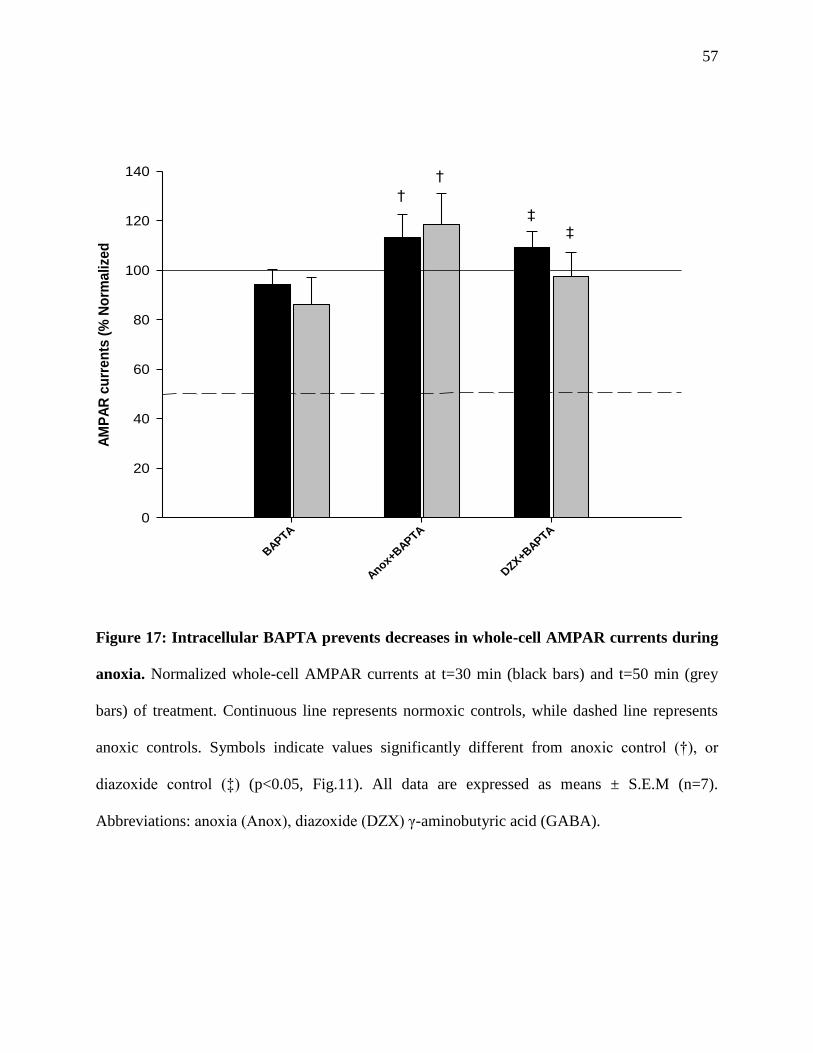
57
BAPTA
Anox+BAPTA
DZX+B
APTA
AM
PA
R c
urr
en
ts (
% N
orm
alize
d
0
20
40
60
80
100
120
140
†
†
‡
‡
Figure 17: Intracellular BAPTA prevents decreases in whole-cell AMPAR currents during
anoxia. Normalized whole-cell AMPAR currents at t=30 min (black bars) and t=50 min (grey
bars) of treatment. Continuous line represents normoxic controls, while dashed line represents
anoxic controls. Symbols indicate values significantly different from anoxic control (†), or
diazoxide control (‡) (p<0.05, Fig.11). All data are expressed as means ± S.E.M (n=7).
Abbreviations: anoxia (Anox), diazoxide (DZX) γ-aminobutyric acid (GABA).

58
Figure 18: Paired sample trace AMPAR currents at t=0 min (control) and following 40 minutes
of anoxic or drug perfusion (t=50min). Abbreviations: anoxia (Anox), diazoxide (DZX) γ-
aminobutyric acid (GABA).
A B
C

59
CHAPTER 4 DISCUSSION
4.1 Control AMPA and NMDA experiments
In order to study the effects of anoxia and/or pharmacological manipulation of the mKATP
channel on whole-cell AMPAR activity, it was first important to demonstrate that stable
recordings and measurements could be obtained and maintained for the 90 minute experimental
time frame. An AMPA concentration ([AMPA]) of 50µM produced repeatable currents that did
not affect the integrity of the neurons throughout the course of the 90 minute experiment (Fig 8),
as it was previously shown that higher concentrations of 100-200 µM often lead to membrane
potential run down and cell death (Pamenter et. al 2008b). Pamenter and colleagues (2008a)
further demonstrated that AMPA reversal potential in turtle neurons was ~3mV, which is similar
to that of mammalian AMPARs (Maruo et al., 2006). Over the 90 minute experiment, AMPAR
currents were measured at 20 minute intervals following measurements at t=0 and t=10 minutes
in order to mimic the experimental protocol for the anoxic and mKATP channel experiments (Fig
7). When access resistance changed by > 20 % or the membrane potential depolarized by more
than 20mV under normoxic conditions, the patches were discarded and not used. Measured
currents did not change significantly over the entire experiment, and the patch used for control
remained stable with a change in access resistance of < 20% and no membrane potential run-
down.
4.1.1 Pharmacological AMPA and NMDAR antagonist experiments
To confirm the specificity of AMPA to reptilian AMPARs, specific AMPAR antagonists
were used to show that the measured currents could be abolished. When the AMPAR

60
specific antagonist CNQX (30 µM) was applied, the measured AMPAR currents were abolished
after 60 minutes of perfusion (Fig 9). Although most of the current amplitude was abolished after
30 minutes, the time of diffusion, specificity of the drug, placement of the fast-step perfusion
pipette in relation to the patch, and the drug concentration used may have contributed to the
length of time it took to completely abolish the AMPAR current. Since the experimental setup
uses a fast flow perfusion system, the drug may have taken longer to reach the neuronal receptors
since it is being cleared at a rapid rate by the suction pump. Furthermore, dissected whole-tissue
slices were used for all of the designed experiments, and when deeper layers of neurons were
being recorded from it may have taken longer to reach the cells and saturate the receptors by way
of diffusion. The proximity of the perfusion pipette to the recording electrode may also have
contributed to the time it took to completely abolish the AMPAR current. One would assume that
the closer the recording neuron was to the perfusion pipette, the less time it would have taken for
the drug to diffuse and abolish the current. Finally, the drugs used are designed for mammalian
AMPAR specificity which may have decreased the affinity of the agents for the receptors.
Although the turtle and mammalian AMPARs likely share similarity in both structure and
function, sequence homology may have differences which have an effect on the binding affinity
of the antagonists for the receptors (Kiefer and Carr, 2000; Fowler et al., 1999; Pamenter et al.,
2008a). Nevertheless, by decreasing and eventually blocking AMPAR currents using CNQX it
confirmed that the measured currents were AMPAR specific and that the mammalian analog
AMPA and the blocker CNQX were good pharmacological agents for electrophysiological
experiments in the western painted turtle.
The designed AMPA experiments also take into account that upon AMPA activation, the
subsequent depolarization can result in the removal of the Mg2+
block that resides in the

61
NMDAR pore. This can lead to NMDAR opening and increased conductance if the endogenous
glutamate present in the tissue binds to the NR2 subunit of the receptor. Consequently, the
measured currents following AMPA application may not solely represent AMPAR conductance
due to the downstream activation of NMDARs. It was therefore important to abolish NMDAR
currents throughout the duration of the AMPA experiments in order to make sure that only
AMPAR currents were measured. Similar to CNQX perfusion, the NMDAR antagonist APV
abolished the measured NMDAR currents after 30 minutes of perfusion (Fig 8). Interestingly, it
only took half of the time for APV to abolish whole-cell NMDAR currents than it took for
CNQX to abolish AMPAR currents at similar concentrations. This may indicate that: APV is
more specific to NMDARs than CNQX is to AMPARs in the turtle, that it took less time for the
drug to diffuse into the tissue because of its lower molecular mass and less bulky 3-dimensional
structure, or that the density of NMDARs in the cortex of the turtle may be lower than that of
AMPARs. In mammalian neurophysiology literature the concentrations of APV and CNQX
often do not exceed 1µM, so the high concentrations of the drugs for these experiments were
used to account for the above possibilities and confirm that the measured currents were solely
reflective of the drugs applied. Regardless of the time taken, application of APV resulted in the
complete reduction of NMDAR currents. As a result the tissues were pre-incubated in APV for
30 minutes prior to each experiment, and all bulk perfusates contained APV throughout the
entire experiment time course ensuring that only AMPAR currents were being measured.
Several of the pharmacological agents used in the experimennts had to be dissolved in
DMSO. Sham experiments using vehicle only (DMSO) were performed to make sure that the
measured AMPAR currents were not affected by including DMSO in the bulk perfusate.
AMPAR currents measured during DMSO inclusion were not significantly different from control

62
(Fig. 20 in Appendix 5.1), Therefore, DMSO was a good solute and vehicle for all drug
experiments.
4.2 mKATP channels in ischemic preconditioning and turtle anoxia-tolerance
Although the mammalian brain is extremely sensitive to oxygen availability and suffers
ECD within minutes of anoxia or ischemia, it can survive short bouts of ischemia without
apparent neuronal structural damage or functional impairment. Short periods of ischemic insults
do not have a cumulative deleterious effect but can rather be protective against subsequent longer
bouts of ischemia, a mechanism called ischemic preconditioning (IPC) (Janoff, 1964; Murray et
al., 1986).
4.2.1 IPC and the link to mKATP channels
IPC reduces the severity of subsequent prolonged ischemic episodes both through a rapid
adaptive response that comes into effect within minutes, and an induced long-term tolerance
lasting approximately 3 days which likely involves changes in gene expression and protein
synthesis (Gidday, 2006; Yellon and Downey, 2003). It is assumed that IPC activates several
endogenous intracellular mechanisms that may increase tolerance to injury, and that activation of
these mechanisms does not prevent, but rather delays cell death (Hanley and Daut, 2005).
Despite their lack of molecular identity, through biochemical and electrophysiological studies
researchers have provided evidence for the presence of mKATP channels in the inner
mitochondrial membrane of the various cell types. The mKATP channel has to date been described
in brain (Bajgar et al., 2001; Debska et al., 2001; Kulawiak and Bednarczyk, 2005), liver (Inoue
et al., 1991), heart (Paucek et al., 1992), skeletal muscle (Pan et al., 1995; Pang et al., 1997;

63
Badhwar et al., 2004; Debska et al., 2002), renal (Cancherini et al., 2003), human T-
lymphocytes (Dahlem et al., 2004) and amoeba mitochondria (Kicinska et al., 2007).
While several physiological and chemical cues have been identified to be involved in IPC
induction, the mechanisms by which it is brought about are not well understood. The mKATP
channel has recently been linked to IPC due to its ability to prevent mitochondrial Ca2+
overload
and subsequent activation of the mitochondrial permeability transition pore (mPTP). In mammals
ischemia results in a large depolarization of the Ψm which leads to the formation of mPTPs that
release apoptotic activators and initiate cell death (Baines, 2007). The mPTP is a non-selective
high conductance pore that forms at the junction between the inner and outer mitochondrial
membranes. Opening of the pore during ischemia is thought facilitate cell death through the
release of apoptotic factors and the spill-over of mitochondrial contents. In mammals, the mKATP
channel has been proposed to reduce Ca2+
overload into the mitochondria and prevent the
activation of mPTPs through a εPKC–mediated pathway, thereby conferring some protection
(Hansson et al., 2010; Liu et al., 1998; Hausenloy et al., 2004). Further evidence for the role of
mKATP channels in mammalian IPC is that when the mKATP channel agonists diazoxide or
pinacidil are applied under normoxic conditions the protective effects of IPC can be mimicked,
and this effect can be blocked using the mKATP channel antagonist 5HD (O’Rourke, 2004). The
fact that mKATP channel-mediated protection against anoxia has been demonstrated in rat cortical
neurons exposed to glutamate toxicity (Kis et al., 2003; Kis et al., 2004), in rat hippocampal
neurons following anoxia/reperfusion injury (Heurteaux et al., 1995; Semenov et al., 2000), and
in rodent cerebral artery occlusion experiments (Shimizu et al., 2002), suggests that an adaptive
neuroprotective mechanism may be shared among organisms, but enhanced in anoxia-tolerant
species.

64
4.2.2 mKATP channels in the anoxic turtle cortex
Although the mechanisms are yet to be revealed, many aspects of the pathways by which
the mKATP channels are involved in mammalian IPC are similar to the proposed mechanisms of
channel arrest in the western painted turtle. As explained above, in mammalian IPC the mKATP
channel is thought to prevent Ca2+
overload by reducing the drive for the Ca2+
uniporter
(Holmuhamedov et al., 1998; Rousou et al., 2004). Rat hearts exposed to ischemia showed 4-
fold increases in [Ca2+
]c and 10-fold increases in mitochondrial Ca2+ concentration ([Ca2+
]m)
which led to ECD (Wang et al., 2001). However, treatment with diazoxide led to further
cytosolic elevations in Ca2+
but an 80% reduction in [Ca2+
]m that seemed to be protective.
Therefore, elevations in mitochondrial Ca2+
concentrations ([Ca2+
]m) during ischemia are toxic
and one of the main causes of cell death.
While evidence of mKATP channels exists in rats across various cell types, their presence
in the turtle cortex and their role during anoxia has not been extensively studied. Earlier studies
indicated the presence of KATP channels distributed throughout the turtle brain using a radioactive
glibenclamide density assay (Jian et al., 1992). Pamenter and colleagues (2008b) through
fluorescent labelling also recently confirmed that ATP-sensitive K+ channels are present in the
turtle cortex. However, their role in the turtle’s remarkable tolerance to anoxia has been poorly
investigated and contains conflicting reports. Where some have reported that antagonism of KATP
channels with glibenclamide reduced K+ efflux during early anoxia, others have reported little or
no change (Pek-Scott and Lutz, 1998; Jiang et al., 1992). However, these authors used
glibenclamide only for the first hour of anoxia, then reverted to 2,3-buanedione monoxime
(BDM) for the following 3 hours. The use of BDM however may have led to these conflicting

65
results as it directly reduced K+ flux by inhibiting Kv2.1 channels in neuroendocrine cells
(Lopatin and Nichols, 1993). Furthermore, it also inhibits glycine-gated Cl-
channels, L-type
Ca2+
channels, and activates GABA receptors increasing their conductance (Brightman et al.,
1995; Allen et al., 1998; Ye and McArdle, 1996).
A few studies however have indirectly indicated the potential protective role of mKATP
channels in relation to NMDARs during ischemia. KATP activation was shown to decrease
glutamate release in the anoxic turtle cortex (Milton and Lutz, 1998; Milton et al., 2002;
Thompson et al., 2007). ECD was prevented in rat hippocampal cultures during ischemia when
neurons were treated with an mKATP channel agonist, and this process was reversed by the
general KATP channel antagonist glyburide (Abele and Miller, 1990). In the western painted
turtle, a 10% increase in [Ca2+
]c was measured during anoxia which is likely associated with
decreased Ca2+
uptake into the mitochondria (Pamenter et al., 2008b). [Ca2+
]c is tightly regulated
in cells through binding to various substances and transport across extracellular and intracellular
membranes (Saris and Carafoli, 2005). Furthermore, the mitochondria is a major sink for Ca2+
in
the cell and is an active [Ca2+
]c regulator.
Mitochondrial Ca2+
uptake primarily occurs through the Ca2+
uniporter, which is driven
by the electrochemical gradient across the inner mitochondrial membrane (Saris and Carafoli,
2005). Although large increases in [Ca2+
]c are toxic to the cell and lead to ECD during ischemia,
the moderate increase in [Ca2+
]c seen in the anoxic turtle cortex is thought to be neuroprotective
and central to channel arrest of the NMDA and AMPARs (Pamenter et al., 2008b). This
increase is thought to occur as a result of opening of mKATP channels, which initiate a cascade of
events that ultimately decreases the uptake of Ca2+
into the mitochondria via the Ca2+
uniporter.

66
In fact, application of mKATP channel agonists diazoxide and levcromakalim led to similar
increases in [Ca2+
]c compared to anoxia alone (Pamenter et al., 2008b). This increase was
abolished when the specific antagonists 5HD or glibenclamide were applied.
4.2.3 mKATP channels regulate NMDA and AMPAR activity during anoxia
NMDAR and AMPARs have both been previously shown to undergo channel arrest in
the anoxic turtle cortex (Shin and Buck, 2003; Pamenter et al., 2008a). Thus far, the mKATP
channel has been linked to NMDARs as a key mediator of channel arrest. Pamenter and
colleagues (2008b) demonstrated that upon perfusion of the mKATP channel agonists diazoxide or
levcromakalim, whole-cell NMDAR currents decreased by 42 and 55% respectively. To confirm
these results, diazoxide and levcromakalim were again used to decrease normoxic NMDAR
activity. Both agents decreased whole-cell NMDAR currents by amounts similar to anoxic
perfusion alone, and these decreases were completely reversible upon re-oxygenation (Appendix
5.2; Table 1). In this study, AMPARs are also confirmed to undergo channel arrest during
anoxia in a way that is similar to NMDARs and that their down-regulation is also modulated by
the opening of the mKATP channels (Fig 11). Similar to anoxic perfusion alone, application of
mKATP channel specific agonists diazoxide or levcromakalim resulted in significantly reduced
AMPAR current amplitudes (Fig 11). These reductions were also similar to what the decreases in
whole-cell NMDAR currents during pharmacological mKATP channel manipulation. When either
of these agents was used together with the mKATP channel antagonists 5HD or glibenclamide, the
reduction was blocked (Fig 11). Therefore, it is likely during anoxia that opening of mKATP
channels increases K+ conductance, uncouples and depolarizes mitochondria leading to an
increase in [Ca2+
]c that subsequently leads to a reduction in both AMPAR and NMDAR currents.

67
Table 1: Anoxia and mKATP channel-mediated decreases in NMDA and AMPARs after 40
minutes of treatment. An asterisk denotes values significantly different from normoxic control.
Treatment NMDAR currents (% Normalized) AMPAR currents (% Normalized)
Normoxia (40 min) 109 ± 7 104 ± 12
Anoxia (40 min) 41 ± 8.1 * 48 ± 4.9 *
Diazoxide (40 min) 55 ± 3.9 * 54 ± 4.2 *
Levcromakalim (40 min) 50 ± 12 * 59 ± 4.8 *
Comparing the decreases in whole-cell currents between NMDA and AMPARs in
response to anoxia or opening of the mKATP channel, it is evident that both receptors respond
similarly to anoxic or pharmacological application. Furthermore, the decreases in currents for
both receptors were reversed after either re-oxygenation or perfusion of the mKATP channel
antagonists 5HD or glibenclamide (Fig 10; Pamenter et al., 2008b). Considering the evidence for
the presence of mKATP channels in the turtle cortex and the consistency of our pharmacological
mKATP experiments, mKATP channels seem to be key mediators in the down-regulation of
AMPAR activity in the anoxic turtle cortex. This suggests that anoxia brings about changes in
the cell which activate secondary messenger pathways that lead to the opening of the mKATP
channel, facilitating K+ uptake into the mitochondria and initiating a cascade of events ultimately
leading to a decrease in AMPAR conductance.

68
4.3 Effects of mitochondrial K+ conductance on whole-cell AMPAR currents
The opening of the mKATP is proposed to increase mitochondrial K+ uptake but the
channel has not yet been cloned and positively identified in the mitochondrial membrane.
Nevertheless, the proposed influx of K+
into the mitochondria during anoxia in the painted turtle
is important in initiating the channel arrest response. Caesium (a K+ channel antagonist)
experiments performed by Pamenter and colleagues (2008b) further confirmed that K+ channels
were specifically involved in the anoxia-mediated depression of NMDARs, as caesium perfusion
during anoxia completely abolished the decrease. To test the effects of increased mitochondrial
K+
conductance on whole cell-AMPAR currents, pharmacological modulators of the
mitochondrial calcium sensitive K+ (mKCa) channel were used. Although the mKCa channel is
not thought to be involved in the turtle’s anoxic response, this channel has been cloned and
sequenced and has a known pharmacology that can serve as a control to confirm the role of
mitochondrial K+ influx (Sato et al., 2005).
Activation of the mKCa channel using the specific agonist NS1619 reduced AMPAR
currents in a similar fashion to anoxia and the mKATP channel antagonists diazoxide and
levcromakalim. This decrease was abolished when NS1619 was applied with the selective mKCa
channel antagonist paxilline (Fig 13). Blocking the channel while simultaneously activating the
mKATP channel did not abolish the decrease in AMPAR current, indicating that the mKATP
channel can play a specific role in the anoxic regulation of AMPAR. This strengthens the
hypothesis that increased K+ conductance into the mitochondria through the mKATP channel is
responsible for mediating downstream mitochondrial Ca2+
uptake and subsequent depression of
AMPA and NMDARs during anoxia.

69
4.4 Mitochondrial Ca2+
uptake and AMPAR depression
In the 1950’s, it was first discovered that Ca2+
and several other bivalent cations were
taken up by mitochondria (Carafoli, 2003). The mitochondria is now known to be a major Ca2+
sink in the cell, and acts to tightly regulate free [Ca2+
]c. As mentioned earlier, the main
mechanism for Ca2+
uptake is via the Ca+2
uniporter. Relating to the mKATP channel, it was
previously demonstrated that increased K+
uptake through the mKATP channel uncouples
mitochondrial respiration by 10-20%, a range that is similar to the measurements performed in
mammalian mitochondria (Pamenter et al., 2008b; Murata et al., 2001). The link between
mitochondrial uncoupling via mKATP channels and AMPAR channel arrest is the decreased
activity of the Ca2+
uniporter (Pamenter et al., 2008b). Uncoupling leads to increased activity of
the H+/K
+ exchanger in order to maintain electrochemical gradients, and reduces the driving
force for mitochondrial Ca2+
uptake via the Ca2+
uniporter, leading to mildly elevated [Ca2+
]c
levels. Since the mitochondria are a major Ca2+
sink, decreased uptake would lead to increased
release and a rise in [Ca2+
]c.
It is now thought that the Ca2+
uniporter is involved in this response (Fig 15). Ruthenium
red is a potent uniporter antagonist that prevents Ca2+
uptake into the mitochondria and therefore
increases [Ca2+
]c. In rat cortical slices, blocking the uniporter with ruthenium red decreased
NMDAR activity and reduced glutamate-mediated Ca2+
influx (Kannurpatti et al., 2000). As
predicted, the neurons of the painted turtle also showed a similar response as normoxic
application of ruthenium red resulted in a decrease in AMPAR current similar to the anoxia-
mediated decrease. (Fig 15). Furthermore, activation of the uniporter with spermine prevented
the anoxia or diazoxide-mediated decrease in AMPAR currents. Thus, during anoxia a dissipated

70
proton gradient across the mitochondria leads to decreased Ca2+
uptake through the uniporter
which ultimately leads to a 10% increase in [Ca2+
]c (Pamenter et al., 2008b). Unlike the
damaging accumulation that occurs during ECD in mammalian neurons (Choi, 1992), mildly
elevated [Ca2+
]c levels in the painted turtle neurons is neuroprotective (Bickler and Buck, 2007).
4.5 Increased Ca2+
levels and downstream mechanisms of AMPAR and NMDAR channel
arrest
The moderate increases in [Ca2+
]c that occur during anoxia seem to be critical in the
down-regulation of both AMPA and NMDARs in the western painted turtle, since sequestering
Ca2+
with BAPTA abolished the anoxia and diazoxide-mediated decreases in whole-cell
NMDAR and AMPAR currents (Pamenter et al., 2008b; Fig 17). Although the idea of increased
Ca2+
levels during anoxia being neuroprotective seems somewhat paradoxical, it is not surprising
that Ca2+
is involved in the mechanism of down-regulation due to its multitude of functions
within cells including gene expression, enzymatic function, protein phosphorylation, muscle
contraction, cellular respiration, neurotransmitter release, and many more. In particular, the
formation of Ca2+
-calmodulin complex is known to regulate the activity of a large number of ion
channels and receptors through the activation of protein kinases and phosphatases.
Mammalian NMDARs are known to be regulated by Ca2+
-calmodulin (Hisatune et al.,
1997), histamine (Bekkers, 1993), arachidonic acid (Miller et al., 1992), adenosine (de
Mendonca et al., 1995), pH (Tang et al., 1990), ethanol (Wirkner et al., 1999), and cytoskeletal
elements (Rosenmund and Westbrook, 1993). At the synapse, NMDARs are anchored to the

71
cytoskeleton through large protein complexes via the C-termini of the NR1 and NR2 subunits
(Collins et al., 2006). Furthermore, these NMDAR-protein complexes facilitate their localization
to specific areas of neurons, such as the post-synaptic density (PSD) which connect it to a variety
of downstream signalling molecules that modify receptor activity (Waxman and Lynch, 2005). In
fact, the C-termini of both subunits are usually associated with a group of proteins called
membrane-associated guanylate kinases (MAGUKs) which include PSD-95, PSD 93, and
SAP102, all of which contain several PDZ-protein interaction domains that bind to other protein
complexes (Waxman and Lynch, 2005). This dense and highly organized structure facilitates the
recruitment of cytoplasmic transducing enzymes (kinases and phosphatases) and Ca2+
dependent
signalling molecules that have the ability to rapidly and actively regulate the activity of
NMDARs (Shin and Buck, 2005). NMDAR activity has previously been shown to be attenuated
by protein phosphatase 1 (PP1) and 2A (Wang et al., 1994), as well as the Ca2+
-calmodulin
dependent protein phosphatase 2B (PP2B), otherwise known as calcineurin (Mulkey et al., 1994;
Wang and Kelly, 1996; Wang and Kelly, 1997). Furthermore, Shin and Buck (2005)
demonstrated that the presence of okadaic acid and calyculin A, inhibitors of PP1 and PP2A, as
well as the Ca2+
-chelator BAPTA potentiated NMDAR currents during normoxia and abolished
the anoxia-mediated decrease in NMDAR currents in the turtle. Therefore, NMDAR activity can
be regulated by specific protein phosphatases that are dependent on Ca2+
-calmodulin, creating a
potential link between [Ca2+
]c elevation and NMDAR depression in the anoxic turtle.
Although the mechanism is not yet fully understood, increases in [Ca2+
]c during anoxia
are thought to activate such phosphatases and decrease NMDAR activity in the turtle brain. The
proposed mechanism by Shin and colleagues (2005) is outlined in Fig 19.
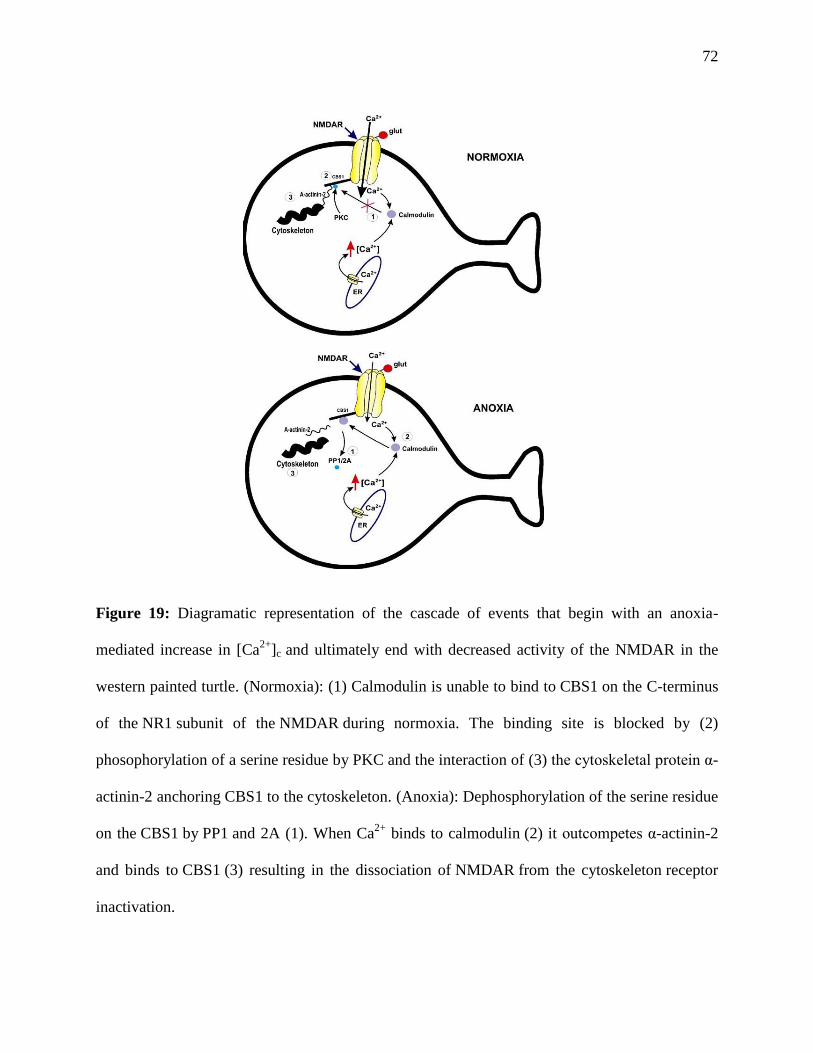
72
Figure 19: Diagramatic representation of the cascade of events that begin with an anoxia-
mediated increase in [Ca2+
]c and ultimately end with decreased activity of the NMDAR in the
western painted turtle. (Normoxia): (1) Calmodulin is unable to bind to CBS1 on the C-terminus
of the NR1 subunit of the NMDAR during normoxia. The binding site is blocked by (2)
phosophorylation of a serine residue by PKC and the interaction of (3) the cytoskeletal protein α-
actinin-2 anchoring CBS1 to the cytoskeleton. (Anoxia): Dephosphorylation of the serine residue
on the CBS1 by PP1 and 2A (1). When Ca2+
binds to calmodulin (2) it outcompetes α-actinin-2
and binds to CBS1 (3) resulting in the dissociation of NMDAR from the cytoskeleton receptor
inactivation.

73
Figure 19 demonstrates NMDAR activity in two neurons, one during normoxia, and the
other during a transition to anoxia. It was previously shown that during normoxia the serine
residue on the calmodulin binding site which resides on the NR1 subunit of the NMDAR is
phosphorylated by protein kinase C (PKC) (1) (Ehlers et al., 1996). This inhibits the Ca2+
dependent binding of calmodulin to the calmodulin binding site (CBS1) (2), which results in
calmodulin being outcompeted by α-actinin (3). Thus, NMDAR activity remains functional
since it stays tethered to the cytoskeleton. During anoxia, the proposed opening of the mKATP
channel leads to an increase in [Ca2+
]c. Subsequent activation of PP1 and PP2A dephosphorylate
a serine residue on the CBS1 of the NR1 C-terminus (1), permitting Ca2+
activated calmodulin to
bind to CBS1 (2) (Shin et al., 2005). This facilitates NMDAR dissociation from the cytoskeleton
and a decrease in NMDAR currents (3). Although it is currently not certain that phosphatases
and Ca2+
-calmodulin effectively work in tandem to modulate NMDAR activity, the mechanism
is supported by good pharmacological data (Shin and Buck 2005) and provides a regulatory link
between the Ca+2
increases and NMDAR activity decreases which occur in the anoxic turtle
cortex.
Mammalian AMPAR subunits are translated and assembled in the rough endoplasmic
reticulum, followed by recruitment to the plasma membrane. Their assembly in the ER and
subsequent trafficking to the membrane is influenced by RNA editing of the GluR2 subunit at
the arginine site (Greger and Esteban, 2007). At the site of synthesis and post-translational
modification, they co-assemble with transmembrane AMPA regulatory proteins (TARPs), which
act as auxiliary subunits required for AMPAR maturation, trafficking and channel activity
modulation (Korber et al., 2007; Tomita, 2010). Stargazin, a member of the TARP family, was
first identified in mutant gene Stargazer mice that exhibited symptoms of epilepsy and ataxia as a

74
result of a lack of functional AMPARs in the brain (Osten and Stern-Bach, 2006; Chen et al.,
2000; Schnell et al., 2002). This protein contains 5 isoforms, all which share homology with
muscle voltage-gated Ca2+
channels (Burgess et al., 1999; Klugbauer et al., 2000). Their C-
termini interact with the PDZ domains (such as SAP 97) of PSD-95 and other MAGUKs, similar
to NMDARs (Chen et al., 2000; Schnell et al., 2002). Through this interaction, TARPs help
promote AMPAR recruitment and clustering at the plasma membrane in excitatory synapses
(Chen et al., 2000; Chetkovich et al., 2002; Schnell et al., 2002; Tomita et al., 2003; Elias et al.,
2006).
AMPAR subunits are subject to phosphorylation by specific kinases that modulate their
stability in the synapse and channel conductance. The GluR1 subunit can be phosphorylated at
three serine residues, all of which are located in the intracellular C-terminus. Serine 831 can be
regulated by PKC and calmodulin-activated protein kinase (CAMKII) (Roche et al., 1996;
Mammen et al., 1997), serine 818 is regulated by PKC (Boehm et al., 2006), and serine 845 is
an active substrate for protein kinase A (PKA) and cGMP-dependent protein kinase II (cGKII)
(Roche et al., 1996; Serulle et al., 2007). More importantly however, these sites are also subject
to de-phosphorylation by PP1 and PP2A, potentially leading to internalization of the receptor and
decreased conductance. Phosphorylation of serine residues 831 and 845 by protein kinase A has
been shown to increase channel open probability and peak current amplitude (Wang et al., 2005)
Conversely, dephosphorylation of these residues by PP1 puts the receptor into a ―low acitivity‖
state, decreasing its conductance and open probability (Wang et al., 2005).
As a result AMPARs, similar to the NMDAR, can be regulated through differential
phosphorylation and a common regulatory mechanism of down-regulation may exist in the turtle.
Another possibility is that a change in the interaction between stargazing and the AMPAR can

75
also modulate AMPAR currents, as stargazin can modulate AMPAR channel gating by slowing
glutamate-induced AMPAR deactivation and desensitization, thereby increasing AMPAR
conductance (Priel et al., 2005; Tomita et al., 2005; Nicoll et al., 2006). Since the anoxia-
mediated decrease in AMPARs occurs very rapidly, differential phosophorylation/de-
phosphorylation is a good strategy for AMPAR depression. Long-term anoxia may well include
internalization of the receptor as a further strategy of AMPAR depression in the turtle, as it was
already demonstrated with the NMDAR (Bickler et al., 2000; Thompson et al., 2007). It is also
possible that there is crosstalk between the receptors during anoxia in the turtle, since the activity
of the NMDAR depends significantly on Na+ influx through the AMPAR. Studies in rat
hippocampal slices have demonstrated that modulation of AMPAR results in an opposing change
in NMDAR currents through a Ca2+
independent mechanism, further stating that since glutamate
is an activator for both receptors it may be beneficial to have a regulatory mechanism between
the receptors (Bai et al., 2002). Since both NMDA and AMPARs are tethered to post-synaptic
densities or the synaptic region by association with PSD-95, their close proximity to each other
suggests a common regulatory mechanism that is rapidly and reversibly activated (Colledge et
al., 2003; Dosemeci et al., 2007).
4.6 AMPAR channel arrest and anoxia-tolerance in the western painted turtle
The anoxia-mediated depression of AMPAR that occurs in the turtle cortex not only
contributes to a reduced excitatory input, but also to decreased NMDAR activity, ATP savings,
reduced Na+/K
+ ATPase activity, and overall reduced metabolic demand (Bickler and Buck,
2007). Along with AMPA and NMDA receptors, voltage-gated Na+ channels are known to
undergo ion channel arrest in the anoxic turtle cortex (Pamenter et al., 2008b; Perez-Pinzon et

76
al., 1992). The western painted turtle’s remarkable anoxia tolerance is therefore attributed to a
number of strategies, and each of these mechanisms seems to be important in the turtle’s survival
during anoxia. The rationale for the down-regulation of these receptors is an increased excitatory
input would lead to ATP run-down and potentially deleterious neuronal effects. The fact that
anoxia-mediated decreases in both AMPA and NMDA receptor currents is mediated by the
mKATP channel suggests that mitochondria are an important component of the cascade of events
leading up to channel arrest.
4.7 Conclusions
Taken together, the findings from this study suggest that AMPAR down-regulation
during anoxia is regulated by the mKATP channel. Similar to the down-regulation of the
NMDAR, opening of the mKATP channel during normoxia leads to a coordinated decrease in
AMPAR currents (Fig 11). Furthermore, blocking the channel with 5HD or glibenclamide during
anoxia or diazoxide-treated normoxia abolishes the decrease completely. The mechanism is
dependent on the influx of K+ into the mitochondria as confirmed by experiments with the mKCa
channel, which facilitates mild uncoupling and reduced Ca2+
uptake (Fig 13). Blocking
mitochondrial Ca2+
uptake by the uniporter with the antagonist ruthenium red also decreased
whole-cell AMPAR currents, while the anoxia-mediated decrease was blocked by opening the
uniporter with spermine (Fig 15). This indicates that the mitochondria is the main source of
increased [Ca2+
]c and that the reduced Ca2+
uptake is important in the anoxic response. Finally,
Ca2+
is critical in the mechanism for downstream AMPAR and NMDAR depression, since
sequestering it with BAPTA abolished the anoxic response (Fig 17). This mechanism is
fundamental to the turtle’s remarkable anoxia tolerance, as the down-regulation of AMPAR and

77
NMDAR through the mKATP channel may indicate a common mechanism of decreasing
excitatory input in turtle neurons that prevents ECD and allows for extended survival. While
mKATP channels are considered key mediators in mammalian IPC-mediated protection, they are
also found in the turtle brain and are important mediators in the endogenous mechanism of
channel arrest during anoxia, suggesting that a common adaptive mechanism exists between
ischemic preconditioning and the prevention of ECD. Therefore, even though clinical trials using
NMDAR and AMPAR antagonists have failed due to their lack of efficacy and detrimental side
effects (Ikonomidou and Turski, 2002; Muir, 2006), understanding key aspects of the turtle’s
endogenous mechanism will help us determine more effective therapeutic and preventative
strategies against ischemia and greatly reduce the remarkably high number of deaths and serious
injuries caused by it.
4.8 Future Directions
Elucidating the mechanism by which [Ca2+
]c ultimately brings about channel arrest in
both receptors will provide further insights into the turtle’s remarkable anoxia tolerance and will
further give clinicians putative clinical therapeutic strategies against ischemic insult. By
determining the definite role that intracellular Ca2+
levels play in AMPA and NMDAR
depression, a better understanding of channel arrest in the turtle will be achieved and will help us
appreciate how the turtle brain is endogenously able to protect itself at times of limited oxygen
availability. Furthermore, recent studies have indicated the potential role of protein kinase C
epsilon (εPKC) in cardioprotection. This kinase is thought to translocate to the mitochondria and
directly modulate the mKATP channel, and this interaction may preserve neurons after ischemia
(Ohnuma et al., 2002; Jaburek et al., 2006; Raval et al., 2007). Thus, investigating the possible

78
involvement of εPKC in the turtle’s mechanism of neuroprotection may help build a more
comprehensive picture of the strategies employed to tolerate periods of anoxia.
It was also previously mentioned that the goldfish (sec. 1.4.2), among many other
organisms, can also endure a similar degree of anoxia-tolerance being able to survive up to 3
months at low temperatures of 10°C and a few days at room temperatures of 22-25°C (Chen et
al., 2005). More importantly, evidence of NMDAR channel arrest was also demonstrated in the
goldfish, where whole-cell currents decreased by 40-50% in response to anoxia (Wilkie et al.,
2008). Whether this down-regulation is associated with the opening of the mKATP channel is
unknown. Therefore, investigating the potential involvement of mKATP channels in another
anoxia-tolerant species may provide further insight into the adaptive mechanisms of anoxia-
tolerance that occur across a variety of species.
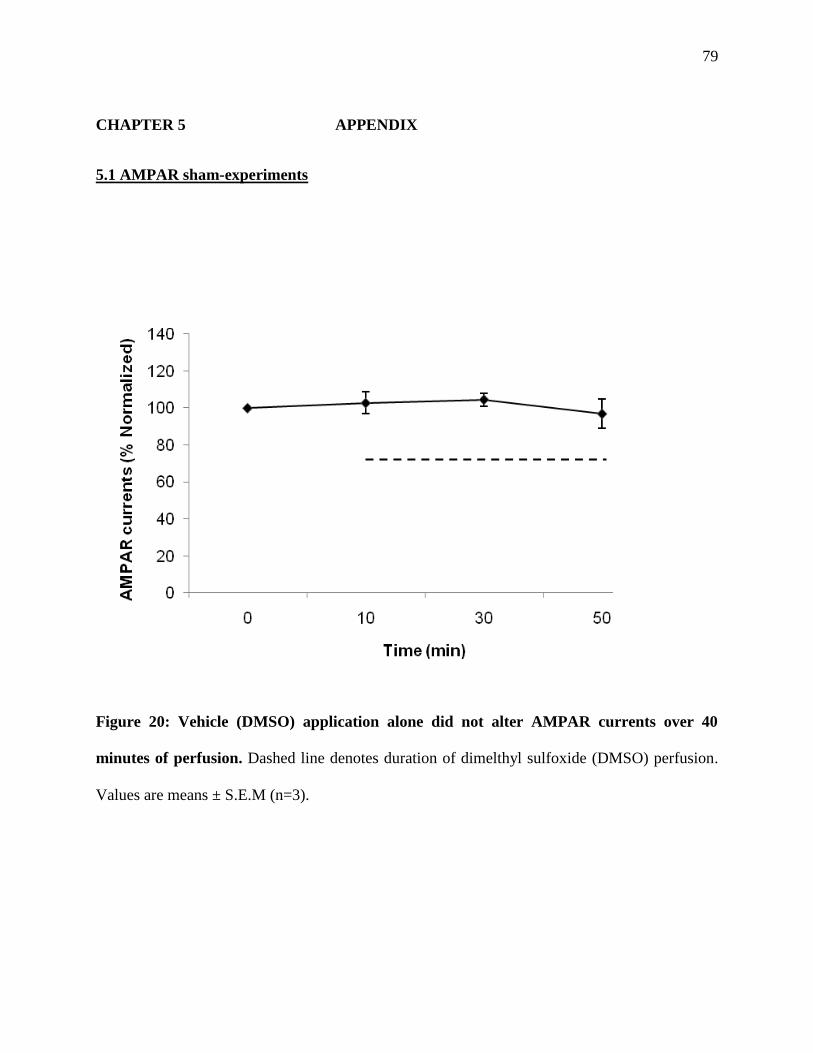
79
CHAPTER 5 APPENDIX
5.1 AMPAR sham-experiments
Figure 20: Vehicle (DMSO) application alone did not alter AMPAR currents over 40
minutes of perfusion. Dashed line denotes duration of dimelthyl sulfoxide (DMSO) perfusion.
Values are means ± S.E.M (n=3).

80
5.2 Whole-cell NMDAR experiments using mKATP channel agonists
Figure 20: mKATP channel openers diazoxide (100µM, n=7) and levcromakalim (100µM,
n=6) decrease NMDA EPSC’s during normoxia, in a manner similar to anoxia (n=7)
without pharmacological application. Values represent normalized whole-cell NMDAR
currents from turtle cortical neurons during control, anoxic, or pharmacological drug perfusion.
All data are means ± S.E.M of different time points in the range of n=6-7. An asterisk (*)
denotes values significantly different from the control at t=10 minutes.

81
REFERENCES
Abele AE, Miller RJ (1990). Potassium channel activators abolish excitotoxicity in cultured
hippocampal pyramidal neurons. Neurosci Lett. 115(2-3): 195-200.
Allen TJ, Mikala G, Wu X, Dolphin AC (1998). Effects of 2,3-butanedione monoxime (BDM)
on calcium channels expressed in Xenopus oocytes. J Physiol. 508 ( Pt 1): 1-14.
Allshire A, Bernardi P, Saris NE (1985). Manganese stimulates calcium flux through the
mitochondrial uniporter. Biochem Biophys Acta 807: 202-209.
Ardehali H, O’Rourke B (2005). Mitochondrial KATP channels in cell survival and death. J
Mol Cell Cardiol 39: 7-16.
Avivi A, Resnick MB, Nevo E, Joel A, Levy AP (1999). Adaptive hypoxic tolerance in the
subterranean mole rat Spalax ehrenbergi: the role of vascular endothelial growth factor. FEBS
Lett. 11;452(3): 133-40.
Badhwar A, Forbes TL, Lovell MB, Dungey AA, McCarter SD, Scott JR, DeRose G, Harris
KA, Potter RF (2004). Chronic lower extremity ischemia: a human model of ischemic
tolerance. Can J Surg. 47(5): 352-8.
Bai D, Muller RU, Roder JC (2002). Non-ionotropic cross-talk between AMPA and NMDA
receptors in rodent hippocampal neurones. J Physiol. 543(Pt 1):23-33.
Baines CP (2007). The mitochondrial permeability transition pore as a target of cardioprotective
signaling. Am J Physiol Heart Circ Physiol. 293(2): H903-4.

82
Bajgar R, Seetharaman S, Kowaltowski AJ, Garlid KD, Paucek P (2001). Identification and
properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J
Biol Chem 276(36): 33369-74.
Bekkers JM (1993). Enhancement by histamine of NMDA-mediated synaptic transmission in
the hippocampus. Science. 261(5117): 104-6.
Bickler PE, Buck LT (2007). Hypoxia tolerance in reptiles, amphibians, and fishes: life with
variable oxygen availability. Annu Rev Physiol 69:145-170.
Bickler PE, Donohoe PH, Buck LT (2002). Molecular adaptations for survival during anoxia:
lessons from lower vertebrates. Neuroscientist 8(3): 234-242.
Blanton MG, Lo Turco JJ, Kriegstein AR (1989). Whole recordings from neurons in slices of
reptilian and mammalian cerebral cortex. J Neurosci Methods 30: 203-210.
Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R (2006). Synaptic
incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on
GluR1. Neuron. 51(2): 213-25.
Brightman T, Ye JH, Ortiz-Jimenez E, Flynn EJ, Wu WH, McArdle JJ (1995). 2,3-
Butanedione monoxime protects mice against the convulsant effect of picrotoxin by facilitating
GABA-activated currents. Brain Res. 24;678(1-2): 110-6.
Buck LT (2004). Adenosine as a signal for ion channel arrest in anoxia-tolerant organisms.
Comp Biochem Physiol B Biochem Mol Biol 139(3): 401-14

83
Buck LT, Bickler PE (1995). Role of adenosine in NMDA receptor modulation in the cerebral
cortex of an anoxia-tolerant turtle (Chrysemys picta belli). J Exp Biol 198(Pt 7): 1621-8.
Buck LT, Bickler PE (1998). Adenosine and anoxia reduce N-methyl-D-aspartate receptor open
probability in turtle cerebrocortex. J Exp Biol 201(2): 289-97.
Buck LT, Hochachka PW (1993). Anoxic suppression of Na(+)-K(+)-ATPase and constant
membrane potential in hepatocytes: support for channel arrest. Am J Physiol 265(5): R1020-5.
Buck LT, Hochachka PW, Schön A, Gnaiger E (1993). Microcalorimetric measurement of
reversible metabolic suppression induced by anoxia in isolated hepatocytes. Am J Physiol 265(5
2): R1014-9.
Buck LT, Pamenter ME (2006). Adaptive responses of vertebrate neurons to anoxia--matching
supply to demand. Respir Physiol Neurobiol 54(1-2):226-40.
Burgess DL, Biddlecome GH, McDonough SI, Diaz ME, Zilinski CA, Bean BP, Campbell
KP, Noebels JL (1999). beta subunit reshuffling modifies N- and P/Q-type Ca2+ channel
subunit compositions in lethargic mouse brain. Mol Cell Neurosci. 13(4): 293-311.
Busija D, Lacza Z, Rajapakse N, Shimizu K, Kis B (2004) Targeting mitochondrial ATP-
sensitive potassium channels—a novel approach to neuroprotection. Brain Res Rev 46: 282-294.
Cancherini DV, Trabuco LG, Rebouças NA, Kowaltowski AJ (2003). ATP-sensitive K+
channels in renal mitochondria. Am J Physiol Renal Physiol. 285(6):F1291-6
Carafoli E (2003). Historical review: mitochondria and calcium: ups and downs of an unusual
relationship. Trends Biochem Sci. 28(4):175-81.

84
Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt
DS, Nicoll RA (2000). Stargazin regulates synaptic targeting of AMPA receptors by two distinct
mechanisms. Nature. 408(6815): 936-43.
Chen J, Zhu JX, Wilson I, Cameron JS (2005). Cardioprotective effects of K ATP channel
activation during hypoxia in goldfish Carassius auratus. J. Exp. Biol. 208: 2765-2772.
Chetkovich DM, Chen L, Stocker TJ, Nicoll RA, Bredt DS (2002). Phosphorylation of the
postsynaptic density-95 (PSD-95)/discs large/zona occludens-1 binding site of stargazin
regulates binding to PSD-95 and synaptic targeting of AMPA receptors. J Neurosci. 22(14):
5791-6.
Chih CP, Feng ZC, Rosenthal M, Lutz PL, Sick TJ (1989). Energy metabolism, ion
homeostasis, and evoked potentials in anoxic turtle brain. Am J Physiol 257(4): R854-60.
Chih CP, Rosenthal M, Sick TJ (1989). Ion leakage is reduced during anoxia in turtle brain: a
potential survival strategy. Am J Physiol 257(6):R1562-4.
Choi DW (1992). Excitotoxic cell death. J Neurobiol 23: 1261-1276.
Choi DW (1994). Calcium and excitotoxic neuronal injury. Ann N Y Acad Sci 747: 162-71.
Choi DW (2001). Excitotoxicity, apoptosis and ischemic stroke. J. Biochem. Mol. Biol 34(1): 8-
13.
Choma K, Bednarczyk P, Koszela-Piotrowska I, Kulawiak B, Kudin A, Kunz WS, Dołowy
K, Szewczyk A (2009). Single channel studies of the ATP-regulated potassium channel in brain
mitochondria. J Bioenerg Biomembr 41(4) 323-34.

85
Christiansen J, Penney D (1973). Anaerobic glycolysis and lactic acid accumulation in cold-
submerged Rana pipiens. J. Comp. Physiol 87: 237-245.
Clark VM, Miller AT Jr. (1973). Studies on anaerobic metabolism in the fresh-water turtle
(Pseudemys scripta elegans). Comp Biochem Physiol A 44(1): 55-62.
Colbourne F, Sutherland GR, Auer RN (1998). Electron microscopic evidence against
apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 19(11): 4200-
4211.
Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF,
Scott JD (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface
expression. Neuron 40 (3): 595-607.
Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, Choudhary JS,
Grant SG (2006). Molecular characterization and comparison of the components and
multiprotein complexes in the postsynaptic proteome. J Neurochem. 97 Suppl 1:16-23.
Dahlem YA, Horn TF, Buntinas L, Gonoi T, Wolf G, Siemen D (2004). The human
mitochondrial KATP channel is modulated by calcium and nitric oxide: a patch-clamp approach.
Biochim Biophys Acta. 1656(1): 46-56.
Dave KR, Anthony Defazio R, Raval AP, Dashkin O, Saul I, Iceman KE, Perez-Pinzon
MA, Drew KL (2009). Protein kinase C epsilon activation delays neuronal depolarization during
cardiac arrest in the euthermic arctic ground squirrel. J Neurochem. 110(4): 1170-9.

86
Debska G, Kicinska A, Skalska J, Szewczyk A, May R, Elger CE, Kunz WS (2002).
Opening of potassium channels modulates mitochondrial function in rat skeletal muscle. Biochim
Biophys Acta 1556(2-3): 97-105.
de Mendonça A, Sebastião AM, Ribeiro JA. Inhibition of NMDA receptor-mediated currents
in isolated rat hippocampal neurones by adenosine A1 receptor activation. Neuroreport. 6(8):
1097-100.
Doll C, Hochachka P, Hand S (1994). A microcalorimetric study of turtle cortical slices:
insights into brain metabolic depression. J Exp Biol 191(1): 141-53.
Donohoe PH, West TG, Boutilier RG (2000). Factors affecting membrane permeability and
ionic homeostasis in the cold-submerged frog. J. Exp. Biol 203: 405-414.
Dosemeci A, Makusky AJ, Jankowska-Stephens E, Yang X, Slotta DJ, Markey SP (2007).
Composition of the synaptic PSD-95 complex. Mol Cell Proteomics 6 (10): 1749-1760.
Ehlers MD, Zhang S, Bernhadt JP, Huganir RL (1996). Inactivation of NMDA receptors by
direct interaction of calmodulin with the NR1 subunit. Cell. 84(5): 745-55.
Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA (2007). Synapse-specific and
developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding
proteins. Neuron. 52(2): 307-20.
Farrell A, Franklin C, Arthur P, Thorarensen H, Cousins K (1994). Mechanical performance
of an in situ perfused heart from the turtle Chrysemys scripta during normoxia and anoxia at 5 C
and 15 C. J Exp Biol: 191(1):207-29.

87
Fowler M, Medina L, Reiner A (1999). Immunohistochemical localization of NMDA- and
AMPA-type glutamate receptor subunits in the basal ganglia of red-eared turtles. Brain Behav
Evol. 54(5):276-89.
Garlid KD, Paucek P (2001). The mitochondrial potassium cycle. IUBMB Life 52(3-5): 153-8.
Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D'Alonzo AJ, Lodge
NJ, Smith MA, Grover GJ (1997). Cardioprotective effect of diazoxide and its interaction with
mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res
81(6): 1072-82.
Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA (1996). The mitochondrial
KATP channel as a receptor for potassium channel openers. J Biol Chem 271: 8796-8799.
Ghai HS, Buck LT (1999). Acute reduction in whole cell conductance in anoxic turtle brain.
Am J Physiol 277(3): R887-93.
Gidday JM (2006). Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 7(6):
437-48.
Greger IH, Esteban JA (2007). AMPA receptor biogenesis and trafficking. Curr Opin
Neurobiol. 17(3): 289-97.
Hanley PJ, Daut J (2005). K(ATP) channels and preconditioning: a re-examination of the role
of mitochondrial K(ATP) channels and an overview of alternative mechanisms. J Mol Cell
Cardiol 39(1): 17-50.

88
Hansson MJ, Morota S, Teilum M, Mattiasson G, Uchino H, Elmér E (2010). Increased
potassium conductance of brain mitochondria induces resistance to permeability transition by
enhancing matrix volume. J Biol Chem. 285(1): 741-50.
Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR (2004). Preconditioning protects by
inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 287(2):
H841-9.
Herbert CV, Jackson DC (1985a). Termperature effects on the responses to prolonged
submergence in the turtle Chrysemys picta bellii I. Blood acid-base and ionic changes during and
following anoxic submergence. Physiol Zool 58(6): 655-669.
Herbert CV, Jackson DC (1985b). Termperature effects on the responses to prolonged
submergence in the turtle Chrysemys picta bellii II. Metabolic rate, blood acid-base and ionic
changes, and cardiovascular functionin aerated and anoxic water. Physiol Zool 58(6): 670-681.
Heurteaux C, Lauritzen I, Widmann C, Lazdunski M (1995). Essential role of adenosine,
adenosine A1 receptors, and ATP sensitive K+ channels in cerebral ischemic preconditioning.
Proc Natl Acad Sci U S A 92: 4666-4670.
Hisatsune C, Umemori H, Inoue T, Michikawa T, Kohda K, Mikoshiba K, Yamamoto T
(1997). Phosphorylation-dependent regulation of N-methyl-D-aspartate receptors by calmodulin.
J Biol Chem. 272(33): 20805-10.
Hochachka PW, Somero GN (2002). Biochemical adaptation. Mechanism and process in
physiological evolution. Oxford University Press. New York. Pp 101-102.

89
Hochachka PW, Mommsen TP (1983). Protons and anaerobiosis. Science 219(4591): 1391-
1397.
Hochachka PW (1986). Defense strategies against hypoxia and hypothermia. Science
231(4735): 234-231.
Holmuhamedov EL, Jovanovic S, Dzeja PP, Jovanovic A, Terzic A (1999). ATP sensitive K+
channel openers prevent Ca+2 overload in rat cardiac mitochondria. J Physiol 519: 347-360.
Hu H, Sato T, Seharaseyon J, Liu Y, Johns DC, O'Rourke B, Marbán E (1999).
Pharmacological and histochemical distinctions between molecularly defined sarcolemmal
KATP channels and native cardiac mitochondrial KATP channels. Mol Pharmacol 55(6):1000-5.
Inoue I, Nagase H, Kishi K, Higuti T (1991). ATP-sensitive K+ channel in the mitochondrial
inner membrane. Nature 352(6332): 244-7.
Ikonomidou C, Turski L (2002). Why did NMDA receptor antagonists fail clinical trials for
stroke and traumatic brain injury? Lancet Neurol 1: 383-386.
Jabůrek M, Costa AD, Burton JR, Costa CL, Garlid KD (2006). Mitochondrial PKC epsilon
and mitochondrial ATP-sensitive K+ channel copurify and coreconstitute to form a functioning
signaling module in proteoliposomes. Circ Res. 99(8):878-83.
Jackson DC (1968). Metabolic depression and oxygen depletion in the diving turtle. J Appl
Physiol 24(4): 503-9.

90
Jackson (2000). Living without oxygen: lessons from the freshwater turtle. Comp Biochem
Physiol 24(4): 503-509.
Jackson DC, Crocker CE, Ultsch GR (2000). Bone and shell contribution to lactic acid
buffering of submerged turtles Chrysemys pica bellii at 3 degrees C. Am J Physiol Regul Integr
Comp Physiol 278(6): R1564-1571.
Jackson DC, Goldberger Z, Visuri S, Armstrong RN (1999). Ionic exchanges of turtle shell in
vitro and their relevance to shell function in the anoxic turtle. J Exp Biol. 202: 513-520.
Janoff A (1964). Alterations in lysosomes(intracellular enzymes) during shock; effects of
preconditioning (tolerance) and protective drugs. Int Anesthesiol Clin.2: 251-69.
Jiang C, Xia Y, Haddad GG (1992). Role of ATP-sensitive K+ channels during anoxia: major
differences between rat (newborn and adult) and turtle neurons. J Physiol. 448:599-612.
Kannurpatti SS, Joshi PG, Joshi NB (2000). Calcium sequestering ability of mitochondria
modulates influx of calcium through glutamate receptor channel. Neurochem Res. 25(12):1527-
36.
Keifer J, Carr MT (2000). Immunocytochemical localization of glutamate receptor subunits in
the brain stem and cerebellum of the turtle Chrysemys picta. J Comp Neurol 427(3):455-68.
Kicinska A, Swida A, Bednarczyk P, Koszela-Piotrowska I, Choma K, Dolowy K, Szewczyk
A, Jarmuszkiewicz W (2007). ATP-sensitive potassium channel in mitochondria of the
eukaryotic microorganism Acanthamoeba castellanii. J Biol Chem. 282(24):17433-41.

91
Kis B, Nagy K, Snipes JA, Rajapakse NC, Horiguchi T, Grover GJ, Busija DW (2003).
Diazoxide induces delayed preconditioning in curltured rat cortical neurons. J Neurochem 87:
969-980.
Kis B, Nagy K, Snipes JA, Rajapakse NC, Horiguchi T, Grover GJ, Busija DW (2004). The
mitochondrial KATP channel opener BMS-191095 induces neuronal preconditioning.
Neuroreport 15: 345-349.
Klockgether T, Turski L, Honoré T, Zhang ZM, Gash DM, Kurlan R, Greenamyre JT
(1991). The AMPA receptor antagonist NBQX has antiparkinsonian effects in monoamine-
depleted rats and MPTP-treated monkeys. Ann Neurol 30(5): 717-23.
Klugbauer N, Dai S, Specht V, Lacinová L, Marais E, Bohn G, Hofmann F (2000). A family
of gamma-like calcium channel subunits. FEBS Lett. 470(2): 189-97.
Körber C, Werner M, Kott S, Ma ZL, Hollmann M (2007). The transmembrane AMPA
receptor regulatory protein gamma 4 is a more effective modulator of AMPA receptor function
than stargazin (gamma 2). J Neurosci. 27(31): 8442-7
Krishnan SN, Sun YA, Mohsenin A, Wyman RJ, Haddad G (1997). Behavioural and
electrophysiologic responses of Drosophila melongaster to prolonged periods of anoxia. J Insect
Physiol 43: 203-210.
Krebs HA (1972). The Pasteur effect and the relations between respiration and fermentation.
Essays Biochem: 8:1-34.
Kulawiak B, Bednarczyk P (2005). Reconstitution of brain mitochondria inner membrane into
planar lipid bilayer. Acta Neurobiol Exp (Wars). 65(3): 271-6.

92
Land SC, Buck LT, Hochachka PW (1993).Response of protein synthesis to anoxia and
recovery in anoxia-tolerant hepatocytes. Am J Physiol 265(1): R41-8.
Lane, N (2002). Oxygen. The molecule that made the world. New York: Oxford University Press
Inc.
Layborne R (1974). Collision between a vulture and an aircraft at an altitude of 37, 000 feet.
Wilson Bull. 86: 461-462.
Le D, Das S, Wang YF, Yoshizawa T, Sasaki YF, Takasu M, Nemes A, Mendelsohn M,
Dikkes P, Lipton SA, Nakanishi N (1997). Enhanced neuronal death from focal ischemia in
AMPA-receptor transgenic mice. Brain Res Mol Brain Res 52: 235-241.
Li X, Rapedius M, Baukrowitz T, Liu GX, Srivastava DK, Daut J, Hanley PJ (2010). 5-
Hydroxydecanoate and coenzyme A are inhibitors of native sarcolemmal KATP channels in
inside-out patches. Biochim Biophys Acta 1800(3): 385-91.
Liu Y, Sato T, O'Rourke B, Marban E (1998). Mitochondrial ATP-dependent potassium
channels: novel effectors of cardioprotection? Circulation. 97(24): 2463-9.
Lopatin AN, Nichols CG (1993) 2,3-Butanedione monoxime (BDM) inhibition of delayed
rectifier DRK1 (Kv2.1) potassium channels expressed in Xenopus oocytes. J Pharmacol Exp
Ther. 265(2): 1011-6.
Lutz PL (1992). Mechanisms for anoxic survival in the vertebrate brain. Annual Rev Physiol 54:
601-618.
Lutz PL, Kabler S (1995). Up-regulation of GABAA receptor during anoxia in the turtle brain.
Am. J. Physiol. 37: R1332-133.

93
Lutz PL, Kabler S (1997). Release of adenosine and ATP in the brain of the freshwater turtle
(Trachemys scripta) during long-term anoxia. Brain Res 769(2): 281-286.
Lutz PL, Milton SL (2004). Negotiating brain anoxia survival in the turtle. J Exp Biol 207
(18):3141-7.
Lutz PL, Nilsson GE (1997). The brain without oxygen. Landes Bioscience. Austin, TX.
Lutz PL, Prentice HM, Milton SL (2003). Is turtle longevity linked to enhanced mechanisms
for surviving brain anoxia and reoxygenation? Exp Gerontol 38(7): 797-800.
Mammen AL, Kameyama K, Roche KW, Huganir RL (1997). Phosphorylation of the alpha-
amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by
calcium/calmodulin-dependent kinase II. J Biol Chem. 272(51): 32528-33.
Maruo K, Yamamoto S, Kanno T, Yaguchi T, Maruo S, Yashiya S, Nishizaki T (2006).
Tunicamycin decreases the probability of single-channel openings for N-methyl-D-aspartate and
alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. Neuroreport 17: 313-
317.
McCullogh JR, Normandin DE, Conder ML, Sleph PG, Dzwonczyk S, Grover GJ (1991).
Specific block of the anti-ischemic actions of cromakalim by sodium 5-hydroxydecanoate. Circ
Res 69: 949-958.
Miller B, Sarantis M, Traynelis SF, Attwell D (1992). Potentiation of NMDA receptor
currents by arachidonic acid. Nature. 355(6362): 722-5.

94
Milton SL, Lutz PL (1998). Low extracellular dopamine levels are maintained in the anoxic
turtle (Trachemys scripta) striatum. J Cereb Blood Flow Metab. 18(7):803-7.
Milton SL, Thompson JW, Lutz PL (2002). Mechanisms for maintaining extracellular
glutamate levels in the anoxic turtle striatum. Am J Physiol Regul Integr Comp Physiol. 282(5):
R1317-23.
Muir KW (2006). Glutamate-based therapeutic approaches: clinical trials with NMDA
antagonists. Curr Opin Pharmacol. 6(1):53-60. Epub 2005 Dec 15.
Mulkey RM, Endo S, Shenolikar S, Malenka RC (1994). Involvement of a
calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature.
369(6480):486-8.
Muller M, Brockhuase J, Ballanyi K (2002). ATP-independent anoxic activation of ATP-
sensitive K+
channels in dorsal vagal neurons of juvenile mice in situ. Neuroscience 109: 313-
328.
Murata M, Akao M, O'Rourke B, Marbán E (2001). Mitochondrial ATP-sensitive potassium
channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible
mechanism of cardioprotection. Circ Res. 89(10):891-8.
Murray CE, Jennings RB, Reimer KA (1986). Preconditioning with ischemia: a delay of lethal
cell injury in ischemic myocardium. Circulation 74: 1124-1136.
Nicoll RA, Tomita S, Bredt DS (2006). Auxiliary subunits assist AMPA-type glutamate
receptors. Science. 311(5765): 1253-6.
Nilsson GE (2001). Surviving anoxia with the brain turned on. News Physiol. Sci. 16: 217-221.

95
Nilsson GE, Alfaro AA, Lutz PL (1990). Changes in turtle brain neurotransmitters and related
substances during anoxia. Am J Physiol 259(2): R376-84.
Nilsson GE, Renshaw GM (2004). Hypoxic survival strategies in two fishes: extreme anoxia
tolerance in the North European crucian carp and natural hypoxic preconditioning in a coral-reef
shark. J. Exp. Biol. 69-199-206.
Ohmori J, Sakamoto S, Kubota H, Shimizu-Sasamata M, Okada M, Kawasaki S, Hidaka
K, Togami J, Furuya T, Murase K (1994). 6-(1H-imidazol-1-yl)-7-nitro-2,3(1H,4H)-
quinoxalinedione hydrochloride (YM90K) and related compounds: structure-activity
relationships for the AMPA-type non-NMDA receptor. J Med Chem 37(4): 467-75.
Ohnuma Y, Miura T, Miki T, Tanno M, Kuno A, Tsuchida A, Shimamoto K (2002).
Opening of mitochondrial K(ATP) channel occurs downstream of PKC-epsilon activation in the
mechanism of preconditioning. Am J Physiol Heart Circ Physiol. 283(1):H440-7.
O'Rourke B (2004). Evidence for mitochondrial K+ channels and their role in cardioprotection.
Circ Res. 94(4): 420-32.
Osten P, Stern-Bach Y (2006). Learning from stargazin: the mouse, the phenotype and the
unexpected. Curr Opin Neurobiol. 16(3): 275-80.
Pamenter ME, Hogg DW, Buck LT (2008c). Endogenous reductions in N-methyl-d-aspartate
receptor activity inhibit nitric oxide production in the anoxic freshwater turtle cortex. FEBS Lett
582(12):1738-42.
Pamenter ME, Richards MD, Buck LT (2007). Anoxia-induced changes in reactive oxygen
species and cyclic nucleotides in the painted turtle. J Comp Physiol B 177(4):473-81.

96
Pamenter ME, Shin DS, Buck LT (2008a). AMPA recepters undergo channel arrest in the
anoxic turtle brain. Am J Physiol Regul Integr Comp Physiol 294: 606-613.
Pamenter ME, Shin DS, Cooray M, Buck LT (2008b). Mitochondrial ATP-sensitive K+
channels regulate NMDAR activity in the cortex of the anoxic western painted turtle. J. Physiol
586.4: 1043-1058.
Pang CY, Yang RZ, Zhong A, Xu N, Boyd B, Forrest CR (1995). Acute ischaemic
preconditioning protects against skeletal muscle infarction in the pig. Cardiovasc Res. 29(6):
782-8.
Pang CY, Neligan P, Zhong A, He W, Xu H, Forrest CR (1997). Effector mechanism of
adenosine in acute ischemic preconditioning of skeletal muscle against infarction. Am J Physiol.
273(3 Pt 2): R887-95.
Paucek P, Mironova G, Mahdi F, Beavis AD, Woldegiorgis G, Garlid KD (1992).
Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+
channel from rat liver and beef heart mitochondria. J Biol Chem 267(36): 26062-9.
Pék-Scott M, Lutz PL (1998). ATP-sensitive K+ channel activation provides transient
protection to the anoxic turtle brain. Am J Physiol. 275(6 Pt 2):R2023-7.
Pérez-Pinzón MA, Lutz PL, Sick TJ, Rosenthal M (1993). Adenosine, a "retaliatory"
metabolite, promotes anoxia tolerance in turtle brain. J Cereb Blood Flow Metab 13(4):728-32.
Pérez-Pinzón MA, Rosenthal M, Sick TJ, Lutz PL, Pablo J, Mash D (1992). Downregulation
of sodium channels during anoxia: a putative survival strategy of turtle brain. Am J Physiol
262(4): R712-5.

97
Priel A, Kolleker A, Ayalon G, Gillor M, Osten P, Stern-Bach Y (2005). Stargazin reduces
desensitization and slows deactivation of the AMPA-type glutamate receptors. J Neurosci.
25(10): 2682-6.
Ramaglia V, Buck LT (2004). Time-dependent expression of heat shock proteins 70 and 90 in
tissues of the anoxic western painted turtle. J Exp Biol 207(21): 3775-84.
Ramaglia V, Harapa GM, White N, Buck LT (2004). Bacterial infection and tissue-specific
Hsp72, -73 and -90 expression in western painted turtles. Comp Biochem Physiol C Toxicol
Pharmacol. 138(2): 139-48.
Ramirez J-M, Folkow LP, Blix AS (2007). Hypoxia tolerance in mammals and birds: From the
wilderness to the clinic. Annu Rev of Physiol 69:113-143.
Raval AP, Dave KR, DeFazio RA, Perez-Pinzon MA (2007). epsilonPKC phosphorylates the
mitochondrial K(+) (ATP) channel during induction of ischemic preconditioning in the rat
hippocampus. Brain Res 1184: 345-53.
Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL (1996). Characterization of
multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 16(6): 1179-88.
Rosenmund C, Westbrook GL (1993). Calcium-induced actin depolymerization reduces
NMDA channel activity. Neuron. 10(5): 805-14.
Rousou AJ, Ericsson M, Federman M, Levitsky S, McCully JD (2004). Opening of
mitochondrial KATP channels enhances cardioprotection through the modulation of
mitochondrial matrix volume, calcium accumulation, and respiration. Am J Physiol Heart Circ
Physiol. 287(5): H1967-76.

98
Saris NE, Carafoli E (2005). A historical review of cellular calcium handling, with emphasis on
mitochondria. Biochemistry (Mosc). 70(2):187-94.
Sato T, Saito T, Saegusa N, Nakaya H (2005). Mitochondrial Ca2+
activated K+
channels in
cardiac myocytes: a mechanism of the cardioprotective effect and modulation by protein kinase
A. Circulation 111: 198-203.
Semenov DG, Samoilov Zielonka MO, Lazarewiscz P (2000). Responses to reversible anoxia
of intracellular free and bound Ca+2
in rat cortical slices. Resuscitation 44: 207-214.
Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB (2007).
A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 56(4): 670-88.
Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA (2002). Direct
interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl
Acad Sci U S A99(21): 13902-7.
Shin DS, Buck LT (2003). Effect of anoxia and pharmacological anoxia on whole-cell NMDA
receptor currents in cortical neurons from the western painted turtle. Physiol Biochem Zool 76:
41-51.
Shin DS, Wilkie MP, Pamenter ME, Buck LT (2005). Calcium and protein phosphatase 1/2A
attenuate N-methyl-d-aspartate receptor activity in the anoxic turtle cortex. Comp Biochem
Physiol A Mol Integr Physiol 142: 50-57.
Sick T, Perez-Pinzon M, Lutz PL (1993). Maintaining coule metabolism and membrane
function in anoxic brain: A comparison between the turtle and the rat. Boca Raton: CRC Press
pp. 351-364.

99
Siesjo BK (1992a). Pathophysiology and treatment of focal cerebral ischemia. Part I:
Pathophysiology. J Neurosurg 77(2): 169-184.
Siesjo BK (1992b). Pathophysiology and treatment of focal cerebral ischemia. Part II:
Mechanisms of damage and treatment. J Neurosurg 77(3): 337-354.
Siesjo BK, Memezawa H, ML (1991). Neurocytotoxicity: pharmacological implications.
Fundam Clin Pharmacol 5: 755-767.
Storey KB (1996). Oxidative stress: animal adaptations in nature. Braz J Med Biol Res 29(12):
1715-33.
Szewczyk A, Mikołajek B, Pikuła S, Nałecz MJ (1993). Potassium channel openers induce
mitochondrial matrix volume changes via activation of ATP-sensitive K+ channel. Pol J
Pharmacol 45(4): 437-43.
Tang CM, Dichter M, Morad M (1990). Modulation of the N-methyl-D-aspartate channel by
extracellular H+. Proc Natl Acad Sci U S A. 87(16): 6445-9.
Thompson JW, Prentice HM, Lutz PL (2007). Regulation of extracellular glutamate levels in
the long-term anoxic turtle striatum: coordinated activity of glutamate transporters, adenosine, K
(ATP) (+) channels and GABA. J Biomed Sci. 14(6): 809-17.
Tomita S (2010). Regulation of ionotropic glutamate receptors by their auxiliary subunits.
Physiology (Bethesda). 25(1): 41-9.

100
Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS
(2005). Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature.
435(7045): 1052-8.
Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS (2003).
Functional studies and distribution define a family of transmembrane AMPA receptor regulatory
proteins. J Cell Biol. 161(4): 805-16.
Ultsch GR, Jackson DC (1982). Long-term submergence at 3 degrees C of the turtle Chrysemys
picta bellii in normoxic and severely hypoxic water. III. Effects of changes in ambient PO2 and
subsequent air breathing. J Exp Biol. 97: 87-99.
Ultsch GR, Carwile ME, Crocker CE, Jackson DC (1999). The physiology of hibernation
among painted turtles: the eastern painted turtle Chrysemys picta picta. Physiol Biochem Zool
72(4): 493-501.
Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT (1998). Reactive oxygen
species released from mitochondria during brief hypoxia induce preconditioning in
cardiomyocytes. J Biol Chem 273: 18092–18098.
Wang JQ, Arora A, Yang L, Parelkar NK, Zhang G, Liu X, Choe ES, Mao L (2005).
Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 32(3):
237-49.
Wang L, Cherednichenko G, Hernandez L, Halow J, Camacho SA, Figueredo V, Schaefer
S (2001). Preconditioning limits mitochondrial Ca(2+) during ischemia in rat hearts: role of
K(ATP) channels. Am J Physiol Heart Circ Physiol. 280(5): H2321-8.

101
Wang JH, Kelly PT (1996). The balance between postsynaptic Ca2+
dependent protein kinase
and phosphatase activities controlling synaptic strength. Learn. Mem. 3: 170-181.
Wang JH, Kelly PT (1997). Postsynaptic calcineurin activity down-regulates synaptic
transmission by weakening intracellular Ca2+
signalling mechanisms in hippocampal CA1
neurons. J. Neurosci. 17: 4600-4611.
Wang ZW, Nara M, Wang YX, Kotlikoff MI (1997). Redox regulation of large conductance
Ca(2+)-activated K+ channels in smooth muscle cells. J Gen Physiol 110(1):35-44.
Wang LY, Orser BA, Brautigan DL, MacDonald JF (1994). Regulation of NMDA receptors
in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 369(6477):230-2.
Wang Y, Qin ZH (2010). Molecular and cellular mechanisms of excitotoxic neuronal death.
Apoptosis: Epub ahead of print.
Waxman EA, Lynch DR (2005). N-methyl-D-aspartate receptor subtypes: multiple roles in
excitotoxicity and neurological disease. Neuroscientist. 11(1):37-49.
Wilkie MP, Pamenter ME, Alkabie S, Carapic D, Shin DS, Buck LT (2008). Evidence of
anoxia-induced channel arrest in the brain of the goldfish (Carassius auratus). Comp Biochem
Physiol C Toxicol Pharmacol 148(4): 355-62
Wijsman, TCM, van der Lugt HC, Hoogland, HP (1985). Anaerobic metabolism in the
freshwater snail Lymnaea stagnalis: haemolymph as a reservoir of D-lactate and succinate.
Comp. Biochem. Physiol. 81: 889-895.
Wirkner K, Poelchen W, Köles L, Mühlberg K, Scheibler P, Allgaier C, Illes P (1999).
Ethanol-induced inhibition of NMDA receptor channels. Neurochem Int. 35(2): 153-62.

102
Won SJ, Kim DY, Gwag BJ (2002). Cellular and molecular pathways of ischemic neuronal
death. J Biochem Mol Bio. 35(1): 67-86.
Ye JH, McArdle JJ (1996). 2,3-Butanedione monoxime modifies the glycine-gated chloride
current of acutely isolated murine hypothalamic neurons. Brain Res. 30;735(1):20-9.
Yellon DM, Downey JM (2003). Preconditioning the myocardium: from cellular physiology to
clinical cardiology. Physiol Rev. 83(4): 1113-51.
Related Documents











