ISSN 00268933, Molecular Biology, 2010, Vol. 44, No. 5, pp. 665–681. © Pleiades Publishing, Inc., 2010. Original Russian Text © I.O. Mazunin, N.V. Volodko, E.B. Starikovskaya, R.I. Sukernik, 2010, published in Molekulyarnaya Biologiya, 2010, Vol. 44, No. 5, pp. 755–772. 665 INTRODUCTION Mitochondria play many roles in the cells, but their most important function is to generate energy via oxi dative phosphorylation (OP). In contrast to other organelles, mitochondria have their own DNA (mtDNA), which codes for several subunits of the complexes involved in OP. Mutations arising in mtDNA may distort energy production and, eventu ally, cause cell death. Such defects of highly differenti ated cells of various human tissues and organs lead to various medical conditions [1–3]. It was known for a long time that defects in energy (ATP) production cause certain neuromuscular syndromes [4, 5], but the cause and effect relations between known disor ders/syndromes and mutations of the mtDNA coding region was established far more recently [6–9]. Mito chondrial disease affects approximately 1 in 10 000 adults in the global population. The review considers the modern views of the structure and organization of the mitochondrial genome and the molecular mechanisms of mitochon drial disorders due to mtDNA mutations. In addition, we compare the molecular methods used to detect mtDNA mutations and experimental strategies aimed at correcting OXPHOS defects. Finally, the means to prevent inheritance of mtDNA mutations are dis cussed, since this is a pressing problem of mitochon drial medicine and, in particular, medical genetic counseling. THE STRUCTURE OF THE MITOCHONDRIAL GENOME Human mtDNA is a 16 568bp circular double stranded molecule and harbors 37 genes, which are involved in energy production taking place in mito chondrial respiratory chain. The gene set includes 13 structural genes, which code for subunits of the OXPHOS complexes, and genes for 22 tRNAs and 2 rRNAs, which are involved in protein synthesis directly in mitochondria. The majority of regulatory regions occur in a noncoding region, which is known as the control region (CR) and is 1122 bp in size. During mtDNA replication, the CR forms a 710bp triple stranded region the socalled displacement loop (Dloop). The coding sequence accounts for a major part of the mitochondrial genome, and only 87 bp correspond to intercistron regions within this sequence. The CR har bors heavystrand (HSP1 and HSP2) and lightstrand (LSP) promoters and a heavystrand replication origin (O H ). A lightstrand replication (O L ) origin is outside the CR. The mtDNA strands are characterized by an asymmetric distribution of G/C pairs. The Grich heavy strand contains both of the rRNA genes, 12 structural genes, and 14 tRNA genes. The remaining eight tRNA genes and one structural gene (ND6) occur in the light strand (Fig. 1a) [12]. Although human mtDNA and prokaryotic DNA share certain structural features (for instance, lack of introns and overlapping genes), the structural organi zation of the mitochondrial genome is far more intri cate [13]. It was found that five to seven mtDNA mol Mitochondrial Genome and Human Mitochondrial Diseases I. O. Mazunin, N. V. Volodko, E. B. Starikovskaya, and R. I. Sukernik Institute of Chemical Biology and Fundamental Medicine, Siberian Branch, Russian Academy of Sciences, Novosibirsk, 630090 Russia; email: [email protected] Received March 25, 2010 Accepted for publication April 8, 2010 Abstract—Today there are described more than 400 point mutations and more than hundred of structural rearrangements of mitochondrial DNA associated with characteristic neuromuscular and other mitochon drial syndromes, from lethal in the neonatal period of life to the disease with late onset. The defects of oxida tive phosphorylation are the main reasons of mitochondrial disease development. Phenotypic diversity and phenomenon of heteroplasmy are the hallmark of mitochondrial human diseases. It is necessary to assess the amount of mutant mtDNA accurately, since the level of heteroplasmy largely determines the phenotypic manifestation. In spite of tremendous progress in mitochondrial biology since the causeandeffect relations between mtDNA mutation and the human diseases was established over 20 years ago, there is still no cure for mitochondrial diseases. DOI: 10.1134/S0026893310050018 Key words: mitochondrial genome, oxidative phosphorylation, mtDNA mutations, heteroplasmy, mitochon drial diseases, mitochondrial respiratory chain defect therapy REVIEWS UDC 576.311.347

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ISSN 0026�8933, Molecular Biology, 2010, Vol. 44, No. 5, pp. 665–681. © Pleiades Publishing, Inc., 2010.Original Russian Text © I.O. Mazunin, N.V. Volodko, E.B. Starikovskaya, R.I. Sukernik, 2010, published in Molekulyarnaya Biologiya, 2010, Vol. 44, No. 5, pp. 755–772.
665
INTRODUCTION
Mitochondria play many roles in the cells, but theirmost important function is to generate energy via oxi�dative phosphorylation (OP). In contrast to otherorganelles, mitochondria have their own DNA(mtDNA), which codes for several subunits of thecomplexes involved in OP. Mutations arising inmtDNA may distort energy production and, eventu�ally, cause cell death. Such defects of highly differenti�ated cells of various human tissues and organs lead tovarious medical conditions [1–3]. It was known for along time that defects in energy (ATP) productioncause certain neuromuscular syndromes [4, 5], but thecause� and� effect relations between known disor�ders/syndromes and mutations of the mtDNA codingregion was established far more recently [6–9]. Mito�chondrial disease affects approximately 1 in 10000adults in the global population.
The review considers the modern views of thestructure and organization of the mitochondrialgenome and the molecular mechanisms of mitochon�drial disorders due to mtDNA mutations. In addition,we compare the molecular methods used to detectmtDNA mutations and experimental strategies aimedat correcting OXPHOS defects. Finally, the means toprevent inheritance of mtDNA mutations are dis�cussed, since this is a pressing problem of mitochon�drial medicine and, in particular, medical geneticcounseling.
THE STRUCTURE OF THE MITOCHONDRIAL GENOME
Human mtDNA is a 16568�bp circular double�stranded molecule and harbors 37 genes, which areinvolved in energy production taking place in mito�chondrial respiratory chain. The gene set includes13 structural genes, which code for subunits of theOXPHOS complexes, and genes for 22 tRNAs and2 rRNAs, which are involved in protein synthesisdirectly in mitochondria. The majority of regulatoryregions occur in a noncoding region, which is known asthe control region (CR) and is 1122 bp in size. DuringmtDNA replication, the CR forms a 710�bp triple�stranded region the so�called displacement loop (D�loop).The coding sequence accounts for a major part of themitochondrial genome, and only 87 bp correspond tointercistron regions within this sequence. The CR har�bors heavy�strand (HSP1 and HSP2) and light�strand(LSP) promoters and a heavy�strand replication origin(OH). A light�strand replication (OL) origin is outsidethe CR. The mtDNA strands are characterized by anasymmetric distribution of G/C pairs. The G�richheavy strand contains both of the rRNA genes, 12structural genes, and 14 tRNA genes. The remainingeight tRNA genes and one structural gene (ND6)occur in the light strand (Fig. 1a) [12].
Although human mtDNA and prokaryotic DNAshare certain structural features (for instance, lack ofintrons and overlapping genes), the structural organi�zation of the mitochondrial genome is far more intri�cate [13]. It was found that five to seven mtDNA mol�
Mitochondrial Genome and Human Mitochondrial DiseasesI. O. Mazunin, N. V. Volodko, E. B. Starikovskaya, and R. I. Sukernik
Institute of Chemical Biology and Fundamental Medicine, Siberian Branch, Russian Academy of Sciences, Novosibirsk, 630090 Russia;
e�mail: [email protected] March 25, 2010
Accepted for publication April 8, 2010
Abstract—Today there are described more than 400 point mutations and more than hundred of structuralrearrangements of mitochondrial DNA associated with characteristic neuromuscular and other mitochon�drial syndromes, from lethal in the neonatal period of life to the disease with late onset. The defects of oxida�tive phosphorylation are the main reasons of mitochondrial disease development. Phenotypic diversity andphenomenon of heteroplasmy are the hallmark of mitochondrial human diseases. It is necessary to assess theamount of mutant mtDNA accurately, since the level of heteroplasmy largely determines the phenotypicmanifestation. In spite of tremendous progress in mitochondrial biology since the cause�and�effect relationsbetween mtDNA mutation and the human diseases was established over 20 years ago, there is still no cure formitochondrial diseases.
DOI: 10.1134/S0026893310050018
Key words: mitochondrial genome, oxidative phosphorylation, mtDNA mutations, heteroplasmy, mitochon�drial diseases, mitochondrial respiratory chain defect therapy
REVIEWS
UDC 576.311.347

666
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
HSP OH
D�loop
T
Cytb
PLSP
E
ND6
ND5
LS
H
ND4
RND3G
COIIIATP6ATP8K
COII
D
COI
W
ND2
MI
ND1
L
Q
OL
ANCY
16S
V12S F
S
ND4L
Matrix
Inner membrane
Intermembrane space
succinate fumarate
Subunits
mtDNA genes:
nDNA genes:
Complex I Complex II Complex III Complex IV Complex V
ND2ND3
ND4LND4
ND5
ND6
H+
e–CoQ
Cytb
Cytc
COICOII
COIII
H2O ADP ATP
7
~39
0
4
1
10
3
10
2
~14
ND1
H+ H+H+
e–e– e–
O2
(a)
(b)
ATP8
ATP6
Fig. 1. (a) Map of the human mitochondrial genome and (b) scheme of oxidative phosphorylation. The genome includes37 genes, of which 13 (ND1�ND6, ND4L, Cytb, COI�COIII, ATP6, and ATP8) code for subunits of the OXPHOS complexes, 2code for rRNAs (12S and 16S), and 22 (designated with Latin capitals) code for tRNAs. The D�loop is a triple�stranded segmentof the mtDNA control region that forms during replication; the control region additionally harbors transcription initiation sitesfor the heavy (HSP) and light (LSP) strands and a heavy�strand translation initiation site (OH). A light�strand translation initia�tion site (OL) is outside the control region. The OXPHOS system includes five complexes. Complex I consists of 46 subunits, ofwhich 7 are encoded by mtDNA and 39 are encoded by nDNA. Complex II consists of four subunits (nDNA). Complex III con�sists of 11 subunits (one encoded by mtDNA and ten encoded by nDNA). Complex IV consists of 13 subunits (three encoded bymtDNA and ten encoded by nDNA). Complex V consists of 16 subunits (2 encoded by mtDNA and 14 encoded by nDNA). Inaddition, there are two electron transporters, CoQ and Cytc. As electrons move along the respiratory chain, protons are pumpedfrom the matrix into the intermembrane space by complexes I, III, and IV and then return to the matrix through complex V. Com�plex V synthesizes ATP from ADP and inorganic phosphate owing to Ψp. Adapted from [2].
ecules of somatic cells form nucleoids, which includehistone�like proteins and proteins involved in tran�scriptional regulation and replication of mtDNA; themajor proteins are mtSSB, POLG, TFAM, and Twin�kle [13–16]. Nucleoids interact with the inner mito�chondrial membrane via the proteins that specificallybind to the mtDNA CR (presumably, with the D�loop) and the inner mitochondrial membrane, thusuniting and stabilizing several mtDNA molecules [17,18]. It is thought that nucleoids have a multilayerstructure with replication and transcription taking
place in the central region and RNA processing andtranslation occurring at the periphery [16]. The nucle�oid organization probably protects mtDNA fromdamage, and the relative arrangement of mtDNAmolecules within a nucleoid facilitates repair via geneconversion. In addition, a nucleoid is considered to bea main unit of mtDNA segregation [15].
There is evidence that mtDNA exchange betweenindividual nucleoids is extremely rare [19], whichindirectly supports a faithful nucleoid hypothesis [20].According to an alternative dynamic nucleoid hypoth�

MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 667
esis, mtDNA moves between nucleoids freely withsubsequent recombination [21].
SPECIFICS OF mtDNA REPLICATION, TRANSCRIPTION, AND TRANSLATION
Two possible models of mtDNA replication are dis�cussed now. According to one model, replication fol�lows a conventional asynchronous mechanism, startsin OH, and proceeds along the heavy strand up to OL;then, the light strand is replicated in the oppositedirection [22, 23]. The alternative model suggests thatreplication similarly starts at OH, but the two strandsare synthesized simultaneously [24, 25]. One oranother replication mechanism is utilized dependingon the cell state. The synchronous mechanism ofmtDNA replication is probably used at the steady�state conditions and is switched to the asynchronousone when the number of mitochondria should beincreased rapidly [24, 25]. Replication is known toinvolve nuclear DNA (nDNA)�encoded proteins:mitochondrial DNA polymerase (POLG), helicase(Twinkle), and single�stranded DNA�binding protein(mtSSB) [26, 27].
Transcription of mtDNA starts from two promoterson the heavy strand (HSP1 and HSP2) and from onepromoter on the light strand (LSP). Transcriptionfrom LSP yields a polycistronic RNA, which consistsof eight tRNAs and one mRNA for an ND6 subunit.The transcripts synthesized from HSP1 and HSP2include the 14 other tRNAs, 2 rRNAs, and 12mRNAs. The amount of the transcript including tworRNAs and two tRNAs (short transcript synthesizedfrom HSP1) is one order of magnitude higher thanthat of the other transcript. Maturation of individualmRNAs has its specifics: the mRNAs are excised fromthe polycistronic transcript via recognition of second�ary structures in the tRNAs whose genes are betweenthe structural genes [28]. Polyadenylation is a key stepin the expression of mitochondrial mRNAs, sincepolyadenylation provides certain mRNAs with a stopcodon (UAA), which is absent from pre�mRNA [29].The main proteins of the transcription machineryinclude mitochondrial RNA polymerase (POLRMT);mitochondrial transcriptional activators A (TFAM)B1 (TFB1M), and B2 (TFB2M); and a transcriptiontermination factor (mTERF) [30–32].
The mtDNA�encoded proteins are synthesized inthe matrix on mitochondrial ribosomes (mitoribo�somes), which have fewer rRNAs and more ribosomalproteins as compared with bacterial or eukaryoticribosomes. The translation machinery of human mito�chondria includes two initiation factors (IF2 and IF3),three elongation factors (EFG, EFTs, and EFTu), andat least one termination factor (mtRF1). Some of thespecifics of mitochondrial translation are that a uniquegenetic code is used, there are 22 tRNAs, and mRNAslack caps, which are necessary for mRNA to recognizethe binding site on the ribosome [33–35].
OXIDATIVE PHOSPHORYLATION
OXPHOS is one of the fundamental metabolicreactions, and it takes place on the inner mitochon�drial membrane. The reaction consists in a coupling ofelectron transport with ATP production. TheOXPHOS system includes five protein complexes,each consisting of several subunits (Fig. 1b). Ineukaryotes, electrons are transported via the respira�tory chain from NADH through complex I (NADH�coenzyme Q reductase) or from succinate throughcomplex II (succinate�CoQ reductase) and then, con�secutively, through the integral membrane electrontransporter CoQ, complex III (ubiquinol�cytochromec reductase), the electron transporter cytochrome c(Cytc), and complex IV (cytochrome c oxidase) tomolecular oxygen. [39–42]. This creates an electro�chemical potential difference (Ψp, a proton gradient)across the inner membrane. The energy stored in theform of Ψp is utilized by complex V (ATP synthase).As protons transported back into the matrix throughthe proton channel (the Fо–subunit of ATP synthase),ADP is phosphorylated with inorganic phosphate toyield ATP [42]. Substrate oxidation and oxygen reduc�tion are, thus, coupled with ATP production.
The OXPHOS complexes were found to driftwithin the inner membrane as a single molecularsupercomplex termed the respirasome, rather than asindividual structures [43–45]. The proportion of thecomplexes in the respirasome is probably species spe�cific [46–49]. It is clear that a true respirasome, whichis capable of transferring electrons from NADH tomolecular oxygen, is a supercomplex that includescomplexes I, II, III, and IV and the specific transport�ers CoQ and Cytc [50]. An ATP synthasome washypothesized to include complex V, the inorganicphosphate transporter, and adenine nucleotide trans�locase (ANT) at a 1 : 1 : 1 ratio [51, 52]. However, thereis evidence that these components function indepen�dently [53].
Although the Mitchell theory is commonlyaccepted [54], the mechanism of proton transfer fromthe matrix into the intermembrane space is still poorlyunderstood, and the structures of the complexes thatare involved in this process were not identified. How�ever, a comparison of the OXPHOS complexes for dif�ferent species showed that proton and electron trans�ports involve the mtDNA�encoded subunits.
In addition to producing ATP, OXPHOS acts as anendogenous source of reactive oxygen species (ROS),including (superoxide), Н2О2 (hydrogen perox�
ide), and ОН– (hydroxyl radical) [55–57]. Superoxideforms mostly in complexes I and III [39, 58, 59].Mitochondrial Mn�dependent superoxide dismutaseor Cu–Zn�dependent superoxide dismutase convert
to H2O2, which is converted to H2O by glutathioneperoxidase. In addition, H2O2 may be converted toOH– in the presence of Fe2+ and Сu2+. is capable
O2–
O2–
O2–

668
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
of reacting with NO (nitric oxide), which is producedin mitochondria endogenously by mitochondrial NOsynthase [60, 61]. The reaction yields ONOO– (perox�ynitrite). Complex IV was shown to play a role in theproduction of reactive nitrogen species [56]. ChronicROS exposure of the cell leads to oxidative damage toproteins, lipids, and nucleic acids, while acute ROSexposure inactivates the Fe–S centers of the enzy�matic OXPHOS complexes and aconitase, which isinvolved in the tricarboxylic acid (TCA) cycle [62],thus, decreasing the ATP production [63, 64]. Highlyreactive ONOO– nitrifies tyrosine residues in sur�rounding proteins, damaging complex I and mito�chondrial superoxide dismutase [65, 66]. In addition,complex I may be nitrosylated at the sulfhydryl groups,which inhibits its activity [67]. The effect of ROS onmtDNA leads to accumulation of multiple mutations,decreases the OXPHOS rate, and further stimulatesROS accumulation. Taken together, these alterationseventually distort the cell function and cause pro�grammed cell death, or apoptosis [68–70].
PATHOGENIC mtDNA MUTATIONS AND MITOCHONDRIAL DISEASES
The mutation rate of mtDNA is approximately 17�fold higher than in nDNA [71]. This is determined bya set of factors, including the specific structural orga�nization of the mitochondrial genome, the functionalstate of ribonucleotide reductase, replication errors,and mutations of the nuclear genes, whose proteinproducts function in mitochondria. The greatest con�tribution is made by ROS [72]. The pathway from amutation arising in mtDNA to the clinical manifesta�tion of disease is poorly understood. It is thought thatmtDNA mutations lead to ROS accumulation,change calcium metabolism, activate mitochondrialpermeability transition pores (mtPTPs), and, eventu�ally, trigger apoptosis. This scenario is probably char�acteristic of neurodegenerative processes due tomtDNA mutations [73, 74].
To date, mitochondrial diseases have been wellcharacterized both clinically and biochemically [75–80]. However, a molecular analysis of mtDNA is nec�essary for diagnosing the disease and evaluating theprognosis for affected patients and the risk for healthycarriers. Diseases are commonly described using aclassification based on the mtDNA region affected bya mutation. Accordingly, pathogenic mtDNA muta�tions are divided into (1) mutations of the structuralgenes, (2) mutations of the rRNA and tRNA genes,and (3) structural rearrangements involving largemtDNA segments.
Pathogenic Mutations of the Structural Genes
Pathogenic mutations that change the nucleotidesequences of the mtDNA structural genes are divided
into four groups depending on the affected OXPHOScomplex.
Mutations of the mitochondrial genes for complex I.The greatest number of pathogenic mutations wereidentified in the structural genes of complex I [81, 82].As of February 9, 2010, the MITOMAP database [10]contained data on a total of 110 pathogenic mutations,including 33 mutations of ND1, 12 of ND2, 6 of ND3,5 of ND4L, 14 of ND4, 22 of ND5, and 18 of ND6.
Leber’s hereditary optic neuropathy (LHON)(atrophy) is the most common mitochondrial diseaseand is due to mutations of mtDNA structural genes;the ND genes are affected in most cases (Table 1).Clinically, LHON is characterized by degeneration ofthe retinal ganglion cell layer and atrophy of the opticnerve. Approximately 95% of LHON cases in the Cau�casian population are due to three primary risk muta�tions: A3460G (ND1), G11778A (ND4), andT14484C (ND6). Many rare LHON�associated muta�tions were identified in the mtDNA genes, and theirnumber is continuously increasing [83, 84]. LHON isone of a few mitochondrial diseases where the expres�sion of a pathogenic mutation was correlated with acertain phyletic lineage (mtDNA haplogroup). Thus,the G11778A and T14484C mutations are often asso�ciated with haplogroup J, while the G3460A mutationis associated with haplogroup K [85, 86]. In particular,we found that the G3460A mutation identified inSiberia is associated with haplogroups deriving frommacrohaplogroup M, which occurs at the highest fre�quency in indigenous Siberian populations (Altaians,Tuvinians, and Buryats), while the G11778A mutationis expressed when occurring in haplogroups of clusterTJ according to published data [85, 87].
Leigh syndrome (LS) is another common diseaseassociated with a mutation of the ND genes. LS is aprogressive neurodegenerative condition that affectsthe brainstem and basal ganglia and is accompanied bythe formation of characteristic symmetrical necroticchanges. Similar symptoms are caused by mutations ofCOIII, ATP6, and certain tRNA genes [88].
Mutations of the mitochondrial genes for complexIII. In total, 29 pathogenic mutations were identifiedin the Cytb gene; these mutations usually cause myop�athy [10, 81]. In addition, Cytb mutations are associ�ated with encephalomyopathy, cardiomyopathy, tubu�lopathy, and LHON [89–91].
Mutations of mitochondrial genes for complex IV.At present, there are 33 known pathogenic mutationsof COI, 14 mutations of COII, and 13 mutations ofCOIII [10]. Most patients with mutations of thesegenes develop neuromuscular syndromes, while cer�tain mutations are associated with LHON and sen�sorineural hearing loss (SNHL) [91, 92], and cer�tain COI mutations are associated with prostatecancer [93].
Mutations of mitochondrial genes for complex V. Atotal of 19 pathogenic mutations were identified in
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 669
ATP6, which codes for an ATP synthase subunit [10].Only one mutation, A8381G, was identified in theATP8 gene, and this mutation leads to maternallyinherited diabetes mellitus and sensorineural deafness(MIDD) [94].
The most common disorder associated with theT8993G mutation of ATP6 is a complex of symptomsincluding neuropathy, ataxia, and retinitis pigmentosa(NARP). It is of interest that the T8993G mutation isexpressed as NARP when mutant mtDNA accountsfor 70–90% of total cell mtDNA and as maternallyinherited Leigh syndrome (MILS) when the propor�tion is 90–95%. Similar syndromes are associated withthe mutations T8993C, T9176G, and T9176C [95].The T8993G and T8993C pathogenic mutations,which change highly conserved Leu156 to Pro or Arg,respectively, decrease the proton flow through ATPsynthase by 30% [96]. The pathogeny of the diseasemay be affected by a certain mitochondrial haplo�group [97].
Pathogenic Mutations of the rRNA and tRNA Genes
Mutations of the genes for rRNAs and tRNAsinvolved in protein biosynthesis in mitochondria maycause several mitochondrial diseases [10].
Pathogenic mutations of the rRNA genes. In total,16 pathogenic mutations were found to alter the struc�ture of the 12S rRNA; no mutation leading to diseasewas identified in the 16S rRNA gene.
The G1555A transition is the most common inrRNA and is phenotypically expressed as SNHL. Themutation affects the highly conserved region of the12S rRNA, which is a component of the small riboso�mal subunit. The mutation changes the aminoglyco�side�binding site of the 12S rRNA, and the patientsare, consequently, sensitive to ototoxic aminoglyco�sides [98]. All other pathogenic mutations of the 12SrRNA gene also lead to SNHL [99].
Pathogenic mutations of the tRNA genes. Approxi�mately two�thirds (166) of the pathogenic point muta�tions of mtDNA occur in the tRNA genes. Mutationsaffecting various tRNAs are expressed as various clin�ical syndromes. The most common mutations affectthe tRNALeu and tRNALys genes.
The A3243G mutation is diagnosed in approxi�mately 80% of cases of mitochondrial encephalopathywith stroke�like episodes and lactic acidosis(MELAS). This transition affects the tertiary structuretRNALys, as well as methylation, acetylation, and tau�rine modification of the anticodon, thus distortingtranslation [100]. It is of interest that the A3243Gmutation is usually heteroplasmic. The proportion ofmutant and wild�type mtDNAs greatly varies amongtissues; the portion of mutant mtDNA is the highest inmuscle tissue and brain cells and the lowest in bloodleukocytes [100, 101]. The mutant mtDNA portionmay increase with age in all tissues except blood cells,which is apparently due to specific selection [102].Apart from MELAS, the disorders associated with the
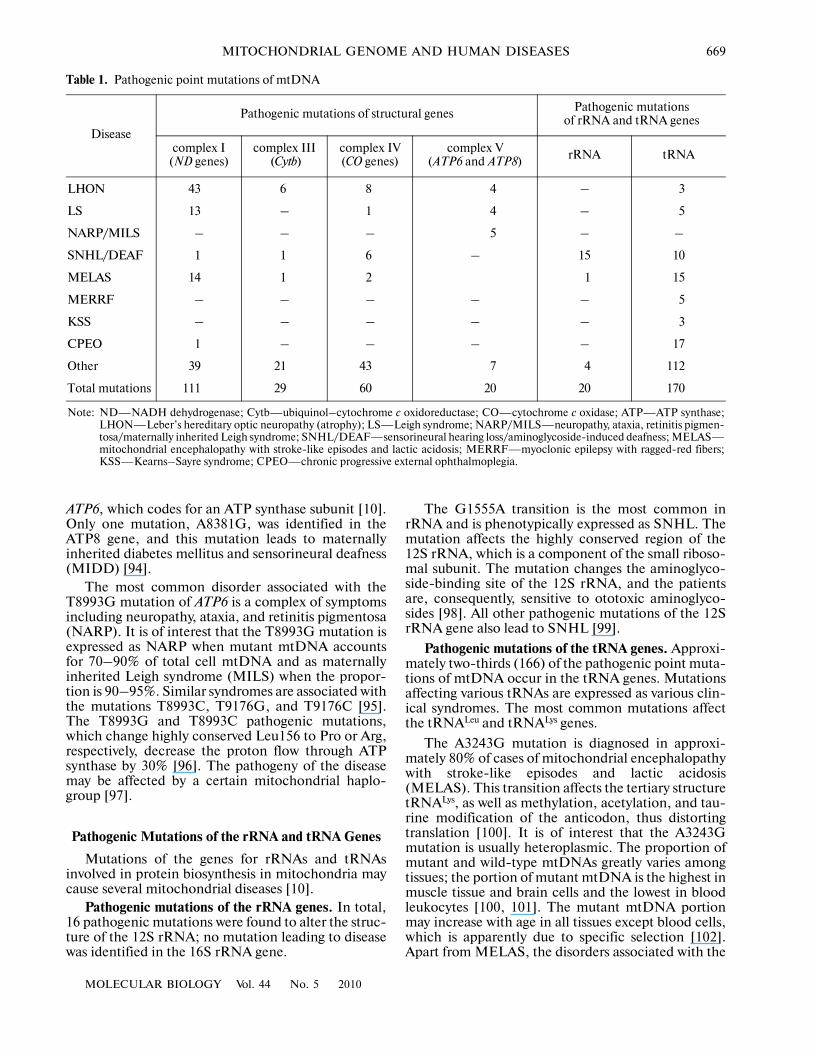
Table 1. Pathogenic point mutations of mtDNA
Disease
Pathogenic mutations of structural genes Pathogenic mutations of rRNA and tRNA genes
complex I (ND genes)
complex III (Cytb)
complex IV (CO genes)
complex V (ATP6 and ATP8) rRNA tRNA
LHON 43 6 8 4 – 3
LS 13 – 1 4 – 5
NARP/MILS – – – 5 – –
SNHL/DEAF 1 1 6 – 15 10
MELAS 14 1 2 1 15
MERRF – – – – – 5
KSS – – – – – 3
CPEO 1 – – – – 17
Other 39 21 43 7 4 112
Total mutations 111 29 60 20 20 170
Note: ND—NADH dehydrogenase; Cytb—ubiquinol–cytochrome c oxidoreductase; CO—cytochrome c oxidase; ATP—ATP synthase;LHON—Leber’s hereditary optic neuropathy (atrophy); LS—Leigh syndrome; NARP/MILS—neuropathy, ataxia, retinitis pigmen�tosa/maternally inherited Leigh syndrome; SNHL/DEAF—sensorineural hearing loss/aminoglycoside�induced deafness; MELAS—mitochondrial encephalopathy with stroke�like episodes and lactic acidosis; MERRF—myoclonic epilepsy with ragged�red fibers;KSS—Kearns–Sayre syndrome; CPEO—chronic progressive external ophthalmoplegia.

670
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
A3243G mutation include myoclonic epilepsy withragged�red fibers (MERRF), chronic progressiveexternal ophthalmoplegia (CPEO), Kearns–Sayresyndrome (KSS), SNHL, and LS [100].
Another common mutation is the A8344G transi�tion in the tRNALys gene; the mutation is associatedwith MERRF in 80% of cases [103]. The mutationaffects a highly conserved nucleotide in the pseudouri�dine loop of tRNA, thus blocking mitochondrial proteinsynthesis. The disease is not expressed phenotypicallyuntil the heteroplasmy level reaches 85–90% [104].
Structural Rearrangements Involving Large mtDNA Segments
Deletions of mtDNA underlie certain mitochon�drial diseases and, probably, play a key role in the agingof postmitotic tissues. Two models of the origin ofmtDNA deletions are considered now [105]. Onemodel suggests that deletions arise during mtDNAreplication via the asynchronous mechanism. Theother model postulates that deletions form duringdouble�strand DNA repair in mtDNA. Deletions usu�ally arise sporadically and are not transmitted to thenext generation [106].
An extended 4977�bp mtDNA deletion (region8488–13 460) is considered to be the most commoncause of KSS, which is characterized by progressiveexternal ophthalmoplegia, retinitis pigmentosa, andearly onset (until 20 years of age) [107].
The main cause of CPEO is either one extendeddeletion or multiple short deletions. CPEO is charac�terized by progressive paralysis of the oculomotormuscles, leading to impaired eye mobility and ptosis[108].
Pearson’s syndrome (PS) is a rare disease thatoccurs in early childhood and is characterized by sid�eroblastic anemia, pancytopenia, and exocrine insuf�ficiency of the pancreas. The disease is extremelysevere and early leads to a fatal outcome; survivingpatients develop the clinical symptoms of KSS. All tis�sues and organs usually contain a large portion ofmtDNA with deletions in these syndromes [109, 110].
Each of the three above syndromes is associatedwith mtDNA deletions of certain sizes and localiza�tions, which should be considered in diagnosis [111].
PATHOGENIC MUTATIONS OF nDNA AND MITOCHONDRIAL DISEASES
Since approximately 2000 genes of the nucleargenome are involved in the biogenesis of mitochondria[112, 113], it is clear that nDNA lesions may alsocause mitochondrial diseases. Defects in nDNA arefar more diverse than mtDNA defects and includemutations of the genes involved in OP, the mitochon�drial protein synthesis machinery, mitochondrialimport and export, movements of mitochondria, theirfusion and division, mtDNA transcription and repli�
cation, various enzymatic cycles (TCA cycle and fattyacid β�oxidation), and other metabolic pathwaysrelated to the function of mitochondria [82, 114, 115].These nDNA defects and the associated mitochon�drial diseases, which clinically differ from the classicalmitochondrial diseases, are beyond the scope of thisreview.
FACTORS AFFECTING THE MANIFESTATION OF MITOCHONDRIAL DISEASES
The observed diversity of clinical symptoms inmitochondrial diseases is due to several factors, suchas heteroplasmy, a threshold effect, and a bottleneckeffect.
The multiplicity of mtDNA copies in the cell oftenleads to heteroplasmy, where several mtDNA variantsoccur in one mitochondrion, one cell, or one organ incontrast to homoplasmy, where all mtDNA copies areidentical [116]. When the cell is divided, mitochondriaare distributed between the daughter cells at randomvia mitotic segregation, and the daughter cells mayconsequently differ in the level of heteroplasmy [117].The rate of a shift towards mutant or wild�typemtDNA in daughter (somatic) cells presumablydepends on the nucleoid composition of the parentalcell. Both mutant and wild�type mtDNAs may appearin one nucleoid (a heteroplasmic nucleoid) or in dif�ferent nucleodis (homoplasmic nucleoids). When amaternal cell contains heteroplasmic nucleoids theheteroplasmy level changes insignificantly in thedaughter cells. When maternal nucleoids arehomoplasmic, the heteroplasmy level may dramati�cally differ between the daughter cells and depends onselection and genetic drift [118, 119].
The heteroplasmy level of a pathogenic mtDNAmutation usually determines the severity of the mito�chondrial disease [120]. The disease does not manifestitself until the mutant mtDNA portion increases overa certain level; the phenomenon is known as thethreshold effect. For instance, the threshold portion ofmtDNA with the A8344G mutation is 85–90% in thecase of MERRF. The T8993G mutation leads to onedisease (NARP) when its content (heteroplasmy level)reaches 70–90% and another disease (MILS) at ahigher (90–95%) heteroplasmy level [121].
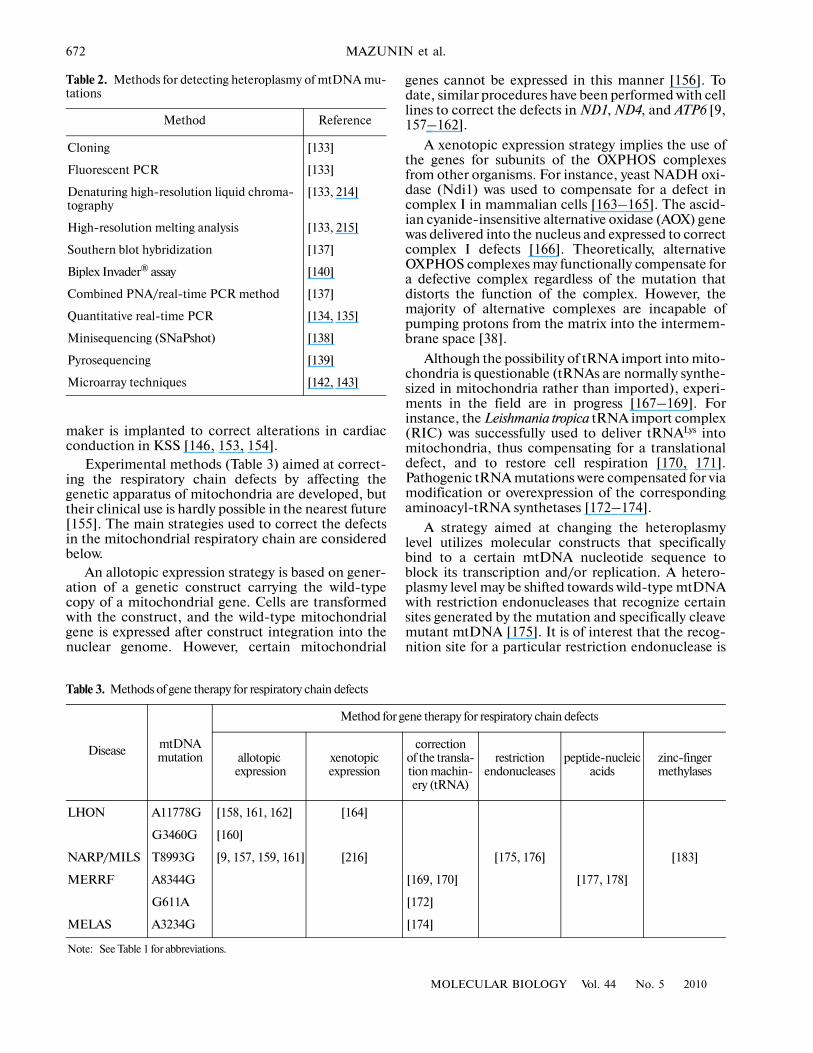
With a few exceptions, mammalian mtDNA isinherited maternally. Mature eggs contain at least100000 mtDNA copies, approximately one or twocopies per mitochondrion [122–124]. In spite of themultiplicity of mtDNA copies in the egg, new mtDNAvariants may appear as early as the next generation.Rapid segregation of new mtDNA variants (D�loopmutations) in cattle occurred within a few generations[125]. Based on these findings, a bottleneck wasassumed for one of the egg development stages(Fig. 2). Further ultrastructural studies showed thatfertilization is followed by a number of zygote divi�sions without a division of mitochondria (and, accord�

MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 671
ingly, without mtDNA replication); i.e., the mito�chondrial pool is halved at each cell division. Forinstance, a mouse primary germline cell containsapproximately 10 mitochondria [126, 127]. Thus, whenprecursor germline cells form, their mitochondriaaccount for only a minor portion (0.01%) of the initialmitochondrial pool of the zygote [128–130]. It isthought that the mitochondrial pool characteristic of amature egg is restored owing to only some of the mito�chondrial subpopulations of primordial follicles [131].
Since the number of mitochondria characteristic ofa mature egg originates from a highly limited mito�chondrial pool of primary germline cells; newlyformed mitochondria are homogeneous (or almosthomogeneous) in composition. In other words, a fun�damental evolutionary effect of the genetic bottleneckis to maintain mtDNA homoplasmy and to minimizeheteroplasmy [132].
DETECTION OF mtDNA MUTATIONS AND ESTIMATION OF THE HETEROPLASMY
LEVEL
Since the level of heteroplasmy is known to deter�mine the phenotypic expression of a mutation to asubstantial extent, it is necessary to estimate the pro�portion of mutant mtDNA when performing a molec�ular analysis. It should be noted that estimation of theheteroplasmy level already includes the identificationof a mutation, while the methods used to detect amutation fail to estimate its heteroplasmy in somecases.
The main methods used to estimate the hetero�plasmy level for mtDNA mutations are summarized inTable 2. Cloning, which produces reliable quantitativeresults, is the most laborious and time consuming.Fluorescent PCR yields more accurate results and isless laborious, but small deletions or insertions remainundetectable in this case. Denaturing high�resolutionliquid chromatography yields reproducible results forall types of heteroplasmic mutations (deletions, inser�tions, and point mutations). This method estimatesthe heteroplasmy level more accurately than cloningand fluorescent PCR [133]. Real�time PCR was alsoemployed in detecting and quantifying mtDNA muta�tions, and excellent results were obtained with bothhydrolysable probes (TaqMan) and the intercalatingdye SYBR [134]. Molecular beacons were proposedfor detecting mtDNA mutations and estimating thelevel of their heteroplasmy [135]. Modification of theTaqMan system to utilize specific primers was used toestimate the heteroplasmy level for the A3243G muta�tion [136]. A comparison of three methods estimatingthe heteroplasmy level—DNA sequencing, Southernblot hybridization, and a combination of the peptidenucleic acid (PNA) technique with real�time PCR—showed that PNA/real�time PCR more accurately(quantitatively) distinguishes mutant and wild�typemtDNAs as compared with sequencing, while South�
ern blotting fails to reveal the actual level of hetero�plasmy [137]. The most accurate estimates are pro�duced by three methods: SNaPshot [138], pyrose�quencing [139], and Biplex Invader [140]. While theyare comparable in accuracy, Biplex Invader is the sim�plest to use and SNaPshot is the most expensive [141].As detection of mtDNA mutations becomes a routinetest, preference is given to microarray techniques,which make it possible to simultaneously test manymtDNA samples for the most common pathogenicmutations and to establish the level of heteroplasmyfor each individual mutation [142, 143].
THERAPY OF RESPIRATORY CHAIN DEFECTS
Mitochondrial diseases are still incurable. Symp�tomatic therapies used in medicine include the use ofmedications, special diets [144–149], and physicalexercise [150–153]. Certain pathologies due tomtDNA mutations are corrected surgically. Forinstance, cochlear implants are used for sensorineuralhearing loss in MELAS, SNHL, and KSS, and a pace�
Number of mitochondria per cell
100000
1000
10
200
5000
Fertilizedovicell
Blastocyst
Primarygermcells
Oogonia
Primordialfollicle
Matureovicell
Bottleneck
100000
50
20000
4000
400
Fig. 2. Changes in the number of mitochondria duringfemale germline cell development in mice. The number ofmitochondria is indicated for each developmental stage. Abottleneck is observed at the stage of primary germline celldevelopment. An approximate number of germline cells inmice is indicated on the right. Adapted from [127].
672
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
maker is implanted to correct alterations in cardiacconduction in KSS [146, 153, 154].
Experimental methods (Table 3) aimed at correct�ing the respiratory chain defects by affecting thegenetic apparatus of mitochondria are developed, buttheir clinical use is hardly possible in the nearest future[155]. The main strategies used to correct the defectsin the mitochondrial respiratory chain are consideredbelow.
An allotopic expression strategy is based on gener�ation of a genetic construct carrying the wild�typecopy of a mitochondrial gene. Cells are transformedwith the construct, and the wild�type mitochondrialgene is expressed after construct integration into thenuclear genome. However, certain mitochondrial
genes cannot be expressed in this manner [156]. Todate, similar procedures have been performed with celllines to correct the defects in ND1, ND4, and ATP6 [9,157–162].
A xenotopic expression strategy implies the use ofthe genes for subunits of the OXPHOS complexesfrom other organisms. For instance, yeast NADH oxi�dase (Ndi1) was used to compensate for a defect incomplex I in mammalian cells [163–165]. The ascid�ian cyanide�insensitive alternative oxidase (AOX) genewas delivered into the nucleus and expressed to correctcomplex I defects [166]. Theoretically, alternativeOXPHOS complexes may functionally compensate fora defective complex regardless of the mutation thatdistorts the function of the complex. However, themajority of alternative complexes are incapable ofpumping protons from the matrix into the intermem�brane space [38].
Although the possibility of tRNA import into mito�chondria is questionable (tRNAs are normally synthe�sized in mitochondria rather than imported), experi�ments in the field are in progress [167–169]. Forinstance, the Leishmania tropica tRNA import complex(RIC) was successfully used to deliver tRNALys intomitochondria, thus compensating for a translationaldefect, and to restore cell respiration [170, 171].Pathogenic tRNA mutations were compensated for viamodification or overexpression of the correspondingaminoacyl�tRNA synthetases [172–174].
A strategy aimed at changing the heteroplasmylevel utilizes molecular constructs that specificallybind to a certain mtDNA nucleotide sequence toblock its transcription and/or replication. A hetero�plasmy level may be shifted towards wild�type mtDNAwith restriction endonucleases that recognize certainsites generated by the mutation and specifically cleavemutant mtDNA [175]. It is of interest that the recog�nition site for a particular restriction endonuclease is
Table 2. Methods for detecting heteroplasmy of mtDNA mu�tations
Method Reference
Cloning [133]
Fluorescent PCR [133]
Denaturing high�resolution liquid chroma�tography
[133, 214]
High�resolution melting analysis [133, 215]
Southern blot hybridization [137]
Biplex Invader® assay [140]
Combined PNA/real�time PCR method [137]
Quantitative real�time PCR [134, 135]
Minisequencing (SNaPshot) [138]
Pyrosequencing [139]
Microarray techniques [142, 143]
Table 3. Methods of gene therapy for respiratory chain defects
Disease mtDNA mutation
Method for gene therapy for respiratory chain defects
allotopic expression
xenotopic expression
correction of the transla�tion machin�ery (tRNA)
restriction endonucleases
peptide�nucleic acids
zinc�finger methylases
LHON A11778G [158, 161, 162] [164]
G3460G [160]
NARP/MILS T8993G [9, 157, 159, 161] [216] [175, 176] [183]
MERRF A8344G [169, 170] [177, 178]
G611A [172]
MELAS A3234G [174]
Note: See Table 1 for abbreviations.

MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 673
not necessarily unique, since mutant and wild�typemtDNAs may differ in the number of restriction sites[176]. A promising approach to changing the propor�tion of mutant and wild�type mtDNAs is to use PNAs,which are linear polymers of N�(2�aminoethyl)�gly�cine substituted with nucleic base derivatives at nitro�gen of the aminoethyl group and are capable of nonco�valent interactions with DNA and RNA bases. PNAsspecifically bind to mutant mtDNA, blocking its rep�lication [177–181]. Modified PNAs known as cellmembrane crossing oligomers (CMCO) have a higherpolarity than PNAs and better penetrate into mito�chondria [182]. In addition, zinc�finger proteinsproved capable of binding to a certain nucleotidesequence in mtDNA [183].
A transfer of normal mitochondria from stem andsomatic cells into cells with defective mitochondria torestore cell respiration is a promising strategy for treat�ing mitochondrial diseases [184–186].
An interesting approach to treatment of mitochon�drial diseases is based on delivering DNA or a proteindirectly into defective mitochondria. For this purpose,DNA or proteins are packaged into liposomes, whichserve as lipid�soluble capsules. Such capsules haveaffinity for the mitochondrial membrane, specificallybind to and fuse with mitochondria, and release theircontents into the mitochondrial matrix [187, 188].
As with other hereditary disorders, the main diffi�culty in therapy of mitochondrial diseases is that tar�geted delivery of a necessary compound to all mito�chondria of all (or certain) human cells is impossibleas of yet. Thus, the only alternative available now is toprevent transmission of pathogenic mtDNA muta�tions from a mother to her children.
STRATEGIES AIMED AT PREVENTING TRANSMISSION OF PATHOGENIC mtDNA
MUTATIONS
To prevent mutant mtDNA transmission to theprogeny is the most pressing problem of mitochondrialmedicine at the current stage of its development [189].Oocyte donation may help to prevent the transmissionof mutant mtDNA to the next generation. An embryois obtained via in vitro fertilization (IVF) andimplanted into the uterus, and the child does not havethe mother’s mitochondrial disease as a result [189,190]. It should be noted that the mother’s female rela�tives are not recommended as ovicell donors, sincethey may carry the pathogenic mtDNA mutation.
Prenatal diagnosis (PND) via sampling fetal mate�rial for laboratory testing [191] has substantial limita�tions because mtDNA mutations are nonuniformlyspread through different tissues and organs. Criteriafor PND of mitochondrial diseases were established[192]. According to the criteria, reliable interpretationof the PND results is possible only for the mutationsthat display a significant correlation between the het�eroplasmy level and disease severity and are uniformly
spread through all tissues, and whose heteroplasmylevel remains unchanged throughout life. As wasfound, only the T8993G/C mutations meet theserequirements.
Preimplantation genetic diagnosis (PGD) is thediagnosis of genetic abnormalities in embryos prior totheir implantation into the uterus. PGD is performedwith individual cells of embryos obtained via IVF[193]. Both the polar body and one or two blastomersof an early embryo (up to the eight�cell stage) are suit�able for PGD, since all of these cells have the samelevel of heteroplasmy. The heteroplasmy level is farmore efficient to estimate with blastomers than withthe polar body [194]. The embryo is implanted into theuterus only when pathogenic mutations are com�pletely absent or the heteroplasmy level is low, sincethe PND criteria [192] are applicable for PGD as well.It should be noted that the procedure was performedonly in two cases [195, 196].
An apparent advantage of PGD over PND is thatthe pregnancy may be preserved [197–199].
Cytoplasmic transport is a transfer of normallyfunctioning mitochondria from another ovicell orzygote to an ovicell with defective mitochondria inorder to reduce the portion of defective mitochondriaand to compensate for the defect in energy production[200]. However, disappointing results were obtainedwhen a donor cytoplasm was experimentally trans�ferred into a defective ovicell: the incidence of chro�mosome aberrations in newborns was significantlyhigher than average [201, 202]. It was found thatmRNAs, proteins, and other factors are transferredwith the cytoplasm in addition to mitochondria, con�tributing to a new surrounding of the nuclear genome[203, 204].
Nuclear transport is theoretically feasible at variousstages of ovicell or zygote development when thematerial to be transferred is (i) a germinal vesicle,(ii) the chromosomes of a mature ovicell, (iii) pronu�clei, or (iv) the nucleus of one of the blastomers [205–209]. It should be noted in connection with the ethicalproblems of cloning that the first three variants are notassociated with cloning, since nDNA is not doubled atthe corresponding stages. However, the use of a blas�tomer nucleus is cloning by definition, which is pro�hibited in humans (United Nations Declaration onHuman Cloning 59/280 dated March 8, 2005, and theBill on Continuing the Ban on Human Cloning, Rus�sia, January 22, 2010).
In a recent study, nuclear material (sprindle�chromo�some complex of a mature primate (Macaca mulatta))ovicell in metaphase II was successfully transferredinto an enucleated ovicell [210]. As a mtDNA analysisshowed, mitochondria were not transferred togetherwith the chromosomes. A unique feature of the proce�dure is selection of a proper stage (metaphase II) whenthe ovicell karyoplast is free from mitochondria.Another method to prevent the transfer of mitochon�dria along with nDNA is to destroy them [209].

674
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
CONCLUSIONS
In spite of the substantial advances made since thecause�and�effect relations had been establishedbetween mtDNA mutations and human diseases [6–9], mitochondrial diseases are still incurable now.First, this is determined by gaps in the understandingof the biogenesis of mitochondria. The available dataon the structure and function of mitochondria arecontinuously corrected and augmented with thedevelopment of new physico�chemical, moleculargenetic, and bioinformatics methods. In addition,there is an abyss between molecular and pathophysio�logical studies because humans remain the only sub�ject apart from mouse models (mitomice) and celllines [211–213], and this imposes many limitationsbecause of the possible risk of health injury or life�threatening consequences. However, there are meansto avoid the inheritance of a pathogenic mitochondrialmutation or to defer the onset of a disease due mito�chondrial dysfunction.
ACKNOWLEDGMENTS
We are grateful to Professor G.M. Dymshits (Insti�tute of Cytology and Genetics, Siberian Branch, Rus�sian Academy of Sciences) and Dr. K.Yu. Popadin(Institute of Information Transmission Problems,Russian Academy of Sciences) for reading the manu�script and helpful comments.
This work was supported by the Russian Founda�tion for Basic Research (project no. 09�04�00183a).
REFERENCES
1. Sukernik R.I., Derbeneva O.A., Starikovskaia E.B.,Volodko N.V., Mikhailovskaia I.E., Bychkov I.Yu,Lott M., Brown M., and Wallace D. 2002. The mito�chondrial genome and human mitochondrial diseases.Russ. J. Genet. 38, 161–170.
2. DiMauro S., Schon E.A. 2008. Mitochondrial disor�ders in the nervous system. Annu. Rev. Neurosci. 31,91–123.
3. Di Donato S. 2009. Multisystem manifestations ofmitochondrial disorders. J. Neurol. 256, 693–710.
4. Ernster L., Ikkos D., Luft R. 1959. Enzymic activitiesof human skeletal muscle mitochondria: A tool in clin�ical metabolic research. Nature. 184, 1851–1854.
5. Luft R., Ikkos D., Palmieri G., Ernster L., Afzelius B.1962. A case of severe hypermetabolism of monthyroidorigin with a defect in the maintenance of mitochon�drial respiratory control: A correlated clinical, bio�chemical, and morphological study. J. Clin. Ivest. 41,1776–1804.
6. Holt I.J., Harding A.E., Morgan Hughes J.A. 1988.Deletions of muscle mitochondrial DNA in patientswith mitochondrial myopathies. Nature. 331, 717–719.
7. Wallace D.C., Singh G., Lott M.T., Hodge J.A.,Shurr T.G., Lezza A.M., Elsas L.J. 2nd, Nikoskelainen E.K.1988. Mitochondrial DNA mutation associated with
Leber’s hereditary optic neuropathy. Science. 242,1427–1430.
8. van den Ouweland J.M., Lemkes H.H., Ruitenbeek W.,Sandkuijl L.A., de Vijlder M.F., Struyvenberg P.A., vande Kamp J.J., Maassen J.A. 1992. Mutation in mito�chondrial tRNA(Leu)(UUR) gene in a large pedigreewith maternally transmitted type II diabetes mellitusand deafness. Nature Genet. 1, 368–371.
9. Tatuch Y., Christodoulou J., Feigenbaum A., Clarke J.T.,Wherret J., Smith C., Rudd N., Petrova�Benedict R.,Robinson B.H. 1992. Heteroplasmic mtDNA muta�tion (T—G) at 8993 can cause Leigh disease when thepercentage of abnormal mtDNA is high. Am. J. Hum.Genet. 50, 852–858.
10. MITOMAP: A Human Mitochondrial Genome Data�base. http://www.mitomap.org, 2009.
11. Schaefer A.M., McFarland R., Blakely E.L., He L.,Whittaker R.G., Taylor R.W., Chinnery P.F., Turnbull D.M.2008. Prevalence of mitochondrial DNA disease inadults. Ann. Neurol. 63, 35–39.
12. Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H.,Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P.,Roe B.A., Sanger F., Schreier P.H., Smith A.J., Staden R.,Young I.G. 1981. Sequence and organization of thehuman mitochondrial genome. Nature. 290, 457–465.
13. Spelbrink J.N. 2010. Functional organization of mam�malian mitochondrial DNA in nucleoids: History,recent developments, and future challenges. IUBMBLife. 62, 19–32.
14. Iborra F.J., Kimura H., Cook P.R. 2004. The func�tional organization of mitochondrial genomes inhuman cells. BMC Biol. 24, 2–9.
15. Holt I.J., He J., Mao C.C., Boyd�Kirkup J.D., Mar�tinsson P., Sembongi H., Reyes A., Spelbrink J.N.2007. Mammalian mitochondrial nucleoids: Organiz�ing an independently minded genome. Mitochondrion.7, 311–321.
16. Bogenhagen D.F., Rousseau D., Burke S. 2008. Thelayered structure of human mitochondrial DNAnucleoids. J. Biol. Chem. 8, 3665–3675.
17. He J., Mao C.C., Reyes A., Sembongi H., Di Re M.,Granycome C., Clippingdale A.B., Fearnley I.M.,Harbour M., Robinson A.J., Reichelt S., Spelbrink J.N.,Walker J.E., Holt I.J. 2007. The AAA+ proteinATAD3 has displacement loop binding properties andis involved in mitochondrial nucleoid organization.J. Cell. Biol. 15, 141–146.
18. Di Re M., Sembongi H., He J., Reyes A., Yasukawa T.,Martinsson P., Bailey L.J., Goffart S., Boyd�Kirkup J.D.,Wong T.S., Fersht A.R., Spelbrink J.N., Holt I.J. 2009.Nucleic Acids Res. 37, 5701–5713.
19. Gilkerson R.W., Schon E.A., Hernandez E., David�son M.M. 2008. Mitochondrial nucleoids maintaingenetic autonomy but allow for functional comple�mentation. J. Cell. Biol. 30, 1117–1128.
20. Jacobs H.T., Lehtinen S.N., Spelbrink J.N. 2000. Nosex please, we’re mitochondria: A hypothesis on thesomatic unit of inheritance of mammalian mtDNA.BioEssays. 22, 564–572.
21. D’Aurelio M., Gajewski C.D., Lin M.T., Mauck W.M.,Shao L.Z., Lenaz G., Moraes C.T., Manfredi G. 2004.

MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 675
Heterologous mitochondrial DNA recombination inhuman cells. Hum. Mol. Genet. 15, 3171–3179.
22. Clayton D.A. 1982. Replication of animal mitochon�drial DNA. Cell. 28, 693–705.
23. Clayton D.A. 2003. Mitochondrial DNA replication:what we know. IUBMB Life. 55, 213–217.
24. Holt I.J., Lorimer H.E., Jacobs H. T. 2000. Coupledleading� and lagging�strand synthesis of mammalianmitochondrial DNA. Cell. 100, 515–524.
25. Fish J., Raule N., Attardi G. 2004. Discovery of amajor D�loop replication origin reveals two modes ofhuman mtDNA synthesis. Science. 306, 2098–2101.
26. Korhonen J.A., Pham X.H., Pellegrini M., Falkenberg M.2004. Reconstitution of a minimal mtDNA replisomein vitro. in vitro. EMBO J. 23, 2423–2429.
27. Holt I. 2009. Mitochondrial DNA replication andrepair: All a flap. Trends Biochem. Sci. 34, 358–365.
28. Ojala D., Montoya J., Attardi G. 1981. tRNA punctu�ation model of RNA processing in human mitochon�dria. Nature. 290, 470–474.
29. Nagaike T., Suzuki T., Ueda T. 2008. Polyadenylationin mammalian mitochondria: Insights from recentstudies. Biochim. Biophys. Acta. 1779, 266–269.
30. Asin�Cayuela J., Gustafsson C.M. 2007. Mitochon�drial transcription and its regulation in mammaliancells. Trends Biochem. Sci. 32, 111–117.
31. Scarpulla R. C. 2008. Transcriptional paradigms inmammalian mitochondrial biogenesis and function.Physiol. Rev. 88, 611–638.
32. Sologub M.Yu., Kochetkov S.N., Temiakov D.E.2009. Transcription and its regulation in mammalianand human mitochondria. Mol. Biol. 43, 198–210.
33. Spremulli L.L., Coursey A., Navratil T., Hunter S.E.2004. Initiation and elongation factors in mammalianmitochondrial protein biosynthesis. Prog. NucleicAcids Res. Mol. Biol. 77, 211–261.
34. Coenen M.J., Antonicka H., Ugalde C., Sasarman F.,Rossi R., Heister J.G., Newbold R.F., Trijbels F.J., vanden Heuvel L.P., Shoubridge E.A., Smeitink J.A. 2004.Mutant mitochondrial elongation factor G1 and com�bined oxidative phosphorylation deficiency. N. Engl. J.Med. 351, 2080–2086.
35. Rorbach J., Soleimanpour�Lichaei R., LightowlersR.N., Chrzanowska�Lightowlers Z.M. 2007. How domammalian mitochondria synthesize proteins? Bio�chem. Soc. Trans. 35, 1290–1291.
36. Hatefi Y. 1985. The mitochondrial electron transportand oxidative phosphorylation system. Annu. Rev. Bio�chem. 54, 1015–1069.
37. Moser C.C., Farid T.A., Chobot S.E., Dutton P.L.2006. Electron tunneling chains of mitochondria. Bio�chim. Biophys. Acta. 1757, 1096–1109.
38. Lenaz G., Genova M.L. 2010. Structure and organiza�tion of mitochondrial respiratory complexes: A newunderstanding of an old subject. Antioxid. Redox Sig�nal. 12, 961–1008.
39. Zickermann V., Dröse S., Tocilescu M.A., Zwicker K.,Kerscher S., Brandt U. 2008. Challenges in elucidat�ing structure and mechanism of proton pumpingNADH:ubiquinone oxidoreductase (complex I).J. Bioenerg. Biomembr. 40, 475–483.
40. Hunte C., Palsdottir H., Trumpower B.L. 2003. Pro�tonmotive pathways and mechanisms in the cyto�chrome bc1 complex. FEBS Lett. 12, 39–46.
41. Belevich I., Verkhovsky M.I. 2008. Molecular mecha�nism of proton translocation by cytochrome c oxidase.Antioxid. Redox Signal. 10, 1–29.
42. von Ballmoos C., Wiedenmann A., Dimroth P. 2009.Essentials for ATP synthesis by F1F0 ATP synthases.Annu. Rev. Biochem. 78, 649–672.
43. Schaegger H. 2001. Respiratory Chain Supercom�plexes. IUBMB Life. 52, 119–128.
44. Wittig I., Carrozzo R., Santorelli F.M., Schägger H.2006. Supercomplexes and subcomplexes of mito�chondrial oxidative phosphorylation. Biochim. Bio�phys. Acta. 1757, 1066–1072.
45. Schäfer E., Dencher N.A., Vonck J., Parcej D.N.2007. Three�dimensional structure of the respiratorychain supercomplex I1III2IV1 from bovine heartmitochondria. Biochemistry. 46, 12579–12585.
46. Vonck J., Schaefer E. 2009. Supramolecular organiza�tion of protein complexes in the mitochondrial innermembrane. Biochim. Biophys. Acta. 1793, 117–124.
47. Wittig I., Schaegger H. 2009. Supramolecular organi�zation of ATP synthase and respiratory chain in mito�chondrial membranes. Biochim. Biophys. Acta. 1787,672–680.
48. Lenaz G., Genova M.L. 2009. Structural and func�tional organization of the mitochondrial respiratorychain: A dynamic super�assembly. Int. J. Biochem.Cell. Biol. 41, 1750–1772.
49. Dudkina N.V., Kou il R., Peters K., Braun H.P.,Boekema E.J. 2010. Structure and function of mito�chondrial supercomplexes. Biochim. Biophys. Acta. Inpress.
50. Acín�Pérez R., Fernández�Silva P., Peleato M.L.,Pérez�Martos A., Enriquez J.A. 2008. Respiratoryactive mitochondrial supercomplexes. Mol. Cell. 32,529–539.
51. Ko Y.H., Delannoy M., Hullihen J., Chiu W., Peder�sen P.L. 2003. Mitochondrial ATP synthasome: Cris�tae�enriched membranes and a multiwell detergentscreening assay yield dispersed single complexes con�taining the ATP synthase and carriers for Pi andADP/ATP. J. Biol. Chem. 278, 12305–12309.
52. Chen C., Ko Y., Delannoy M., Ludtke S.J., Chiu W.,Pedersen P.L. 2004. Mitochondrial ATP synthasome:Three�dimensional structure by electron microscopyof the ATP synthase in complex formation with carri�ers for Pi and ADP/ATP. J. Biol. Chem. 279, 31761–31768.
53. Chinopoulos C., Adam�Vizi V. 2010. Mitochondria asATP consumers in cellular pathology. Biochim. Bio�phys. Acta. 1802, 221–227.
54. Mitchell P. 1961. Coupling of phosphorylation to elec�tron and hydrogen by a chemi�osmotic type of mech�anism. Nature. 191, 144–148.
55. Murphy M. P. 2009. How mitochondria produce reac�tive oxygen species. Biochem. J. 417, 1–13.
56. Poyton R.O., Ball K.A., Castello P.R. 2009. Mito�chondrial generation of free radicals and hypoxic sig�naling. Trends Endocrinol. Metab. 20, 332–340.
r
ˆ

676
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
57. Poyton R.O., Castello P.R., Ball K.A., Woo D.K.,Pan N. 2009. Mitochondria and hypoxic signaling: Anew view. Ann. NY. Acad. Sci. 1177, 48–56.
58. St�Pierre J., Buckingham J.A., Roebuck S.J.,Brand M.D. 2002. Topology of superoxide productionfrom different sites in the mitochondrial electrontransport chain. J. Biol. Chem. 277, 44784–44790.
59. Sun J., Trumpower B.L. 2003. Superoxide anion gen�eration by the cytochrome bc1 complex. Arch. Bio�chem. Biophys. 419, 198–206.
60. Giulivi C. 2003. Characterization and function ofmitochondrial nitric�oxide synthase. Free Radic. Biol.Med. 34, 397–408.
61. Giulivi C. 2007. Mitochondria as generators and tar�gets of nitric oxide. Novartis Fdn Symp. 287, 92–100.
62. Melov S., Coskun P., Patel M., Tuinstra R., Cottrell B.,Jun A.S., Zastawny T.H., Dizdaroglu M., Goodman S.I.,Huang T.T., Miziorko H., Epstein C.J., Wallace D.C.1999. Mitochondrial disease in superoxide dismutase 2mutant mice. Proc. Natl. Acad. Sci. USA. 96, 846�851.
63. Andreyev A.Y., Kushnareva Y.E., Starkov A.A. 2005.Mitochondrial metabolism of reactive oxygen species.Biokhimiya. 70, 200–214.
64. Fariss M.W., Chan C.B., Patel M., van Houten B.,Orrenius S. 2005. Role of mitochondria in toxic oxida�tive stress. Mol. Interv. 5, 94–111.
65. Yamakura F., Taka H., Fujimura T., Murayama K.1998. Inactivation of human manganese�superoxidedismutase by peroxynitrite is caused by exclusive nitra�tion of tyrosine 34 to 3�nitrotyrosine. J. Biol. Chem.273, 14085–14089.
66. Riobó N.A., Clementi E., Melani M., Boveris A.,Cadenas E., Moncada S., Poderoso J.J. 2001. Nitricoxide inhibits mitochondrial NADH:ubiquinonereductase activity through peroxynitrite formation.Biochem. J. 359, 139–145.
67. Clementi E., Brown G.C., Feelisch M., Moncada S.1998. Persistent inhibition of cell respiration by nitricoxide: Crucial role of S�nitrosylation of mitochondrialcomplex I and protective action of glutathione. Proc.Natl. Acad. Sci. USA. 95, 7631–7636.
68. Kroemer G., Petit P., Zamzami N., Vayssière J.L.,Mignotte B. 1995. The biochemistry of programmedcell death. FASEB J. 9, 1277–1287.
69. Perkins G., Bossy�Wetzel E., Ellisman M.H. 2009.New insights into mitochondrial structure during celldeath. Exp. Neurol. 218, 183–192.
70. Scorrano L. 2009. Opening the doors to cytochrome c:Changes in mitochondrial shape and apoptosis. Int. J.Biochem. Cell. Biol. 41, 1875–1883.
71. Wallace D.C., Ye J.H., Neckelmann S.N., Singh G.,Webster K.A., Greenberg B.D. 1987. Sequence analy�sis of cDNAs for the human and bovine ATP synthasebeta subunit: mitochondrial DNA genes sustain seven�teen times more mutations. Curr. Genet. 12, 81–90.
72. Todorov I.N., Todorov G.I. 2009. The multifactornature of high mutation frequency in mtDNA ofmammalian somatic cells. Biokhimiya. 74, 1184–1194.
73. Turnbull H.E., Lax N.Z., Diodato D., Ansorge O.,Turnbull D.M. 2010. The mitochondrial brain: From
mitochondrial genome to neurodegeneration. Bio�chim. Biophys. Acta. 1802, 111–121.
74. Abramov A.Y., Smulders�Srinivasan T.K., Kibry D.M.,Acin�Perez R., Enriquez J.A., Lightowlers R.N.,Duchen M.R., Turnbull D.M. 2010. Mechanism ofneurodegeneration of neurons with mitochondrialDNA mutations. Brain. 133, 797–807.
75. Munnich A., Rustin P. 2001. Clinical spectrum anddiagnosis of mitochondrial disorders. Am. J. Med.Genet. 106, 4–17.
76. Taylor R.W., Schaefer A.M., Barron M.J., McFarland R.,Turnbull D.M. 2004. The diagnosis of mitochondrialmuscle disease. Neuromuscul. Disord. 14, 237–245.
77. Haas R.H., Parikh S., Falk M.J., Saneto R.P.,Wolf N.I., Darin N., Cohen B.H. 2007. Mitochondrialdisease: A practical approach for primary care physi�cians. Pediatrics. 120, 1326–1333.
78. Haas R.H., Parikh S., Falk M.J., Saneto R.P., Wolf N.I.,Darin N., Wong L.J., Cohen B.H., Naviaux R.K.2008. The in�depth evaluation of suspected mitochon�drial disease. Mol. Genet. Metab. 94, 16–37.
79. Rahman S., Hanna M.G. 2009. Diagnosis and therapyin neuromuscular disorders: Diagnosis and new treat�ments in mitochondrial diseases. J. Neurol. Neurosurg.Psychiatry. 80, 943–953.
80. McFarland R., Turnbull D.M. 2009. Batteries notincluded: Diagnosis and management of mitochon�drial disease. J. Intern. Med. 265, 210–228.
81. Wong L.J. 2007. Pathogenic mitochondrial DNAmutations in protein�coding genes. Muscle Nerve. 36,279–293.
82. Rotig A. 2010. Genetic bases of mitochondrial respira�tory chain disorders. Diabetes Metab. 36, 97–107.
83. Carelli V., La Morgia C., Valentino M.L., Barboni P.,Ross�Cisneros F.N., Sadun A.A. 2009. Retinal gan�glion cell neurodegeneration in mitochondrial inher�ited disorders. Biochim. Biophys. Acta. 1787, 518–528.
84. Yu�Wai�Man P., Griffiths P.G., Hudson G., Chinnery P.F.2009. Inherited mitochondrial optic neuropathies.J. Med. Genet. 46, 145–158.
85. Brown M.D., Starikovskaya E., Derbeneva O.,Hosseini S., Allen J.C., Mikhailovskaya I.E., Sukernik R.I.,Wallace D.C. 2002. The role of mtDNA background indisease expression: A new primary LHON mutationassociated with Western Eurasian haplogroup J. Hum.Genet. 110, 130–138.
86. Hudson G., Carelli V., Spruijt L., Gerards M., Mow�bray C., Achilli A., Pyle A., Elson J., Howell N., LaMorgia C., Valentino M.L., Huoponen K., Savontaus M.L.,Nikoskelainen E., Sadun A.A., Salomao S.R.,Belfort R.Jr., Griffiths P., Man P.Y., de Coo R.F., Hor�vath R., Zeviani M., Smeets H.J., Torroni A., Chin�nery P.F. 2007. Clinical expression of Leber hereditaryoptic neuropathy is affected by the mitochondrialDNA�haplogroup background. Am. J. Hum. Genet. 81,228–233.
87. Volodko N.V, L’vova M.A, Starikovskaya E.B., Der�beneva O.A., Bychkov I.Yu., Mikhailovskaya I.E.,Pogozheva I.V., Fedotov F.F., Soyan G.V., Procaccio V.,Wallace D.C., Sukernik R.I. 2006. Spectrum of patho�genic mtDNA mutations in Leber’s hereditary opticneuropathy families from Siberia. Genetika. 42, 78–87.

MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 677
88. Finsterer J. 2008. Leigh and Leigh�like syndrome inchildren and adults. Pediatr. Neurol. 39, 223–235.
89. Johns D.R., Neufeld M.J. 1991. Cytochrome b muta�tions in Leber hereditary optic neuropathy. Biochem.Biophys. Res. Commun. 181, 1358–1364.
90. Valnot I., Kassis J., Chretien D., de Lonlay P., Parfait B.,Munnich A., Kachaner J., Rustin P., Rötig A. 1999. Amitochondrial cytochrome b mutation but no muta�tions of nuclearly encoded subunits in ubiquinol cyto�chrome c reductase (complex III) deficiency. Hum.Genet. 104, 460–466.
91. Abu�Amero K.K., Bosley T.M. 2006. Mitochondrialabnormalities in patients with LHON�like optic neur�opathies. Invest. Ophthalmol. Vis. Sci. 47, 4211–4220.
92. Lévêque M., Marlin S., Jonard L., Procaccio V., Rey�nier P., Amati�Bonneau P., Baulande S., Pierron D.,Lacombe D., Duriez F., Francannet C., Mom T., Jour�nel H., Catros H., Drouin�Garraud V., Obstoy M.F.,Dollfus H., Eliot M.M., Faivre L., Duvillard C.,Couderc R., Garabedian E.N., Petit C., Feldmann D.,Denoyelle F. 2007. Whole mitochondrial genomescreening in maternally inherited non�syndromichearing impairment using a microarray resequencingmitochondrial DNA chip. Eur. J. Hum. Genet. 15,1145–1155.
93. Petros J.A., Baumann A.K., Ruiz�Pesini E., Amin M.B.,Sun C.Q., Hall J., Lim S., Issa M.M., Flanders W.D.,Hosseini S.H., Marshall F.F., Wallace D. C. 2005.mtDNA mutations increase tumorigenicity in prostatecancer. Proc. Natl. Acad. Sci. USA. 102, 719–724.
94. Perucca�Lostanlen D., Narbonne H., Hernandez J.B.,Staccini P., Saunieres A., Paquis�Flucklinger V., Vial�ettes B., Desnuelle C. 2000. Mitochondrial DNA vari�ations in patients with maternally inherited diabetesand deafness syndrome. Biochem. Biophys. Res. Com�mun. 3, 771–775.
95. Schon E.A., Santra S., Pallotti F., Girvin M.E. 2001.Pathogenesis of primary defects in mitochondrial ATPsynthesis. Semin. Cell. Dev. Biol. 12, 441–448.
96. Solaini G., Harris D.A., Lenaz G., Sgarbi G., Baracca A.2008. The study of the pathogenic mechanism of mito�chondrial diseases provides information on basicbioenergetics. Biochim. Biophys. Acta. 1777, 941–945.
97. D’Aurelio M., Vives�Bauza C., Davidson M.M.,Manfredi G. 2010. Mitochondrial DNA backgroundmodifies the bioenergetics of NARP/MILS ATP6mutant cells. Hum. Mol. Genet. 19, 374–386.
98. Cortopassi G., Hutchin T. 1994. A molecular and cel�lular hypothesis for aminoglycoside�induced deafness.Hear Res. 78, 27–30.
99. Kokotas H., Petersen M.B., Willems P.J. 2007. Mito�chondrial deafness. Clin. Genet. 71, 379�391.
100. Finsterer J. 2007. Genetic, pathogenetic, and pheno�typic implications of the mitochondrial A3243GtRNALeu(UUR) mutation. Acta Neurol. Scand. 116,1–14.
101. Ma Y., Fang F., Yang Y., Zou L., Zhang Y., Wang S., Xu Y.,Pei P., Qi Y. 2009. The study of mitochondrial A3243Gmutation in different samples. Mitochondrion. 9, 139–143.
102. Sue C.M., Quigley A., Katsabanis S., Kapsa R., Crim�mins D.S., Byrne E., Morris J.G. 1998. Detection of
MELAS A3243G point mutation in muscle, blood andhair follicles. J. Neurol. Sci. 161, 36–39.
103. Silvestri G., Ciafaloni E., Santorelli F.M., Shanske S.,Servidei S., Graf W.D., Sumi M., DiMauro S. 1993.Clinical features associated with the A G transitionat nucleotide 8344 of mtDNA (“MERRF mutation”).Neurology. 43, 1200–1206.
104. Shoffner J.M., Lott M.T., Lezza A.M., Seibel P., Ball�inger S.W., Wallace D. C. 1990. Myoclonic epilepsyand ragged�red fiber disease (MERRF) is associatedwith a mitochondrial DNA tRNA(Lys) mutation. Cell.6, 931–937.
105. Krishnan K.J., Reeve A.K., Samuels D.C., Chinnery P.F.,Blackwood J.K., Taylor R.W., Wanrooij S., Spelbrink J.N.,Lightowlers R.N., Turnbull D.M. 2008. What causesmitochondrial DNA deletions in human cells? NatureGenet. 40, 275–279.
106. Chinnery P.F., DiMauro S., Shanske S., Schon E.A.,Zeviani M., Mariotti C., Carrara F., Lombes A.,Laforet P., Ogier H., Jaksch M., Lochmüller H., Hor�vath R., Deschauer M., Thorburn D.R., Bindoff L.A.,Poulton J., Taylor R.W., Matthews J.N., Turnbull D.M.2004. Risk of developing a mitochondrial DNA dele�tion disorder. Lancet. 364, 592–596.
107. Maceluch J.A., Niedziela M. 2006. The clinical diag�nosis and molecular genetics of Kearns�Sayre syn�drome: A complex mitochondrial encephalomyopa�thy. Pediatr. Endocrinol. Rev. 4, 117–137.
108. van Goethem G., Martin J.J., Van Broeckhoven C.2003. Progressive external ophthalmoplegia character�ized by multiple deletions of mitochondrial DNA:Unraveling the pathogenesis of human mitochondrialDNA instability and the initiation of a genetic classifi�cation. Neuromolecular Med. 3, 129–146.
109. Rötig A., Cormier V., Blanche S., Bonnefont J.P.,Ledeist F., Romero N., Schmitz J., Rustin P., Fischer A.,Saudubray J.M. 1990. Pearson’s marrow�pancreassyndrome. A multisystem mitochondrial disorder ininfancy. J. Clin. Invest. 86, 1601–1608.
110. Lee H.F., Lee H.J., Chi C.S., Tsai C.R., Chang T.K.,Wang C.J. 2007. The neurological evolution of Pearsonsyndrome: Case report and literature review. Eur. J.Paediatr. Neurol. 11, 208–214.
111. López�Gallardo E., López�Pérez M.J., Montoya J.,Ruiz�Pesini E. 2009. CPEO and KSS differ in the per�centage and location of the mtDNA deletion. Mito�chondrion. 9, 314–317.
112. Dimmer K.S., Rapaport D. 2008. Proteomic view ofmitochondrial function. Genome Biol. 9, 209.
113. Ruiz�Romeo C., Blanco F.J. 2009. Mitochondrialproteomics and its application in biomedical research.Mol. BioSyst. 5, 1130–1142.
114. Jacobs H.T, Turnbull D.M. 2005. Nuclear genes andmitochondrial translation: A new class of genetic dis�ease. Trends Genet. 21, 312–314.
115. Zhu X., Peng X., Guan M�X., Yan Q. 2009. Patho�genic mutations of nuclear genes associated with mito�chondrial disorders. Acta Biochim. Biophys. Sinica. 41,179–187.
116. Kmiec B., Woloszynska M., Janska H. 2006. Hetero�plasmy as a common state of mitochondrial genetic

678
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
information in plants and animals. Curr. Genet. 50,149–159.
117. Wonnapinij P., Chinnery P.F., Samuels D. C. 2008.The distribution of mitochondrial DNA heteroplasmydue to random genetic drift. Am. J. Hum. Genet. 83,582–593.
118. Gilkerson R.W., Schon E.A. 2008. Nucleoid auton�omy: An underlying mechanism of mitochondrialgenetics with therapeutic potential. Commun. Integr.Biol. 1, 34–36.
119. Gilkerson R.W. 2009. Mitochondrial DNA nucleoidsdetermine mitochondrial genetics and dysfunction.Int. J. Biochem. Cell Biol. 41, 1899–1906.
120. Lightowlers R.N., Chinnery P.F., Turnbull D.M.,Howell N. 1997. Mammalian mitochondrial genetics:Heredity, heteroplasmy and disease. Trends Genet. 13,450–455.
121. DiMauro S., Schon E.A. 2001. Mitochondrial DNAmutations in human disease. Am. J. Med. Genet. 106,18–26.
122. van Blerkom J. 2008. Mitochondria as regulatoryforces in oocytes, preimplantation embryos and stemcells. Reprod. Biomed. Online. 16, 553–569.
123. van Blerkom J. 2009. Mitochondria in early mamma�lian development. Semin. Cell Dev. Biol. 20, 354–364.
124. Ramalho�Santos J., Varum S., Amaral S., Mota P.C.,Sousa A.P., Amaral A. 2009. Mitochondrial function�ality in reproduction: from gonads and gametes toembryos and embryonic stem cells. Hum. Reprod.Update. 15, 553–572.
125. Hauswirth W.W., Laipis P.J. 1982. MitochondrialDNA polymorphism in a maternal lineage of Holsteincows. Proc. Natl. Acad. Sci. USA. 79, 4686–4690.
126. Jansen R.P., de Boer K. 1998. The bottleneck: Mito�chondrial imperatives in oogenesis and ovarian follic�ular fate. Mol. Cell. Endocrinol. 145, 81–88.
127. Shoubridge E.A., Wai T. 2007. Mitochondrial DNAand the mammalian oocyte. Curr. Top. Dev. Biol. 77,87–111.
128. Cao L., Shitara H., Horii T., Nagao Y., Imai H., Abe K.,Hara T., Hayashi J., Yonekawa H. 2007. The mito�chondrial bottleneck occurs without reduction ofmtDNA content in female mouse germ cells. NatureGenet. 39, 386–390.
129. Cree L.M., Samuels D.C., de Sousa Lopes S.C.,Rajasimha H.K., Wonnapinij P., Mann J.R., Dahl H.H.,Chinnery P.F. 2008. A reduction of mitochondrialDNA molecules during embryogenesis explains therapid segregation of genotypes. Nature Genet. 40, 249–254.
130. Stewart J.B., Freyer C., Elson J.L., Larsson N.G.2008. Purifying selection of mtDNA and its implica�tions for understanding evolution and mitochondrialdisease. Nature Rev. Genet. 9, 657–662.
131. Wai T., Teoli D., Shoubridge E.A. 2008. The mito�chondrial DNA genetic bottleneck results from repli�cation of a subpopulation of genomes. Nature Genet.40, 1484–1488.
132. Cummins J.M. 2001. Mitochondria: Potential roles inembryogenesis and nucleocytoplasmic transfer. Hum.Reprod. Update. 7, 217–228.
133. Kurelac I., Lang M., Zuntini R., Bartoletti S.A., San�tamaria M., Attimonelli M., Gasparre G., Romeo G.2009. Experimental and critical assessment of sixmethodological approaches to quantify heteroplasmyof mitochondrial mutations. Abstr. Eur. Hum. Genet.Conf., Vienna, Austria, May 23–26, 2009.
134. Wong L.J., Bai R.K. 2006. Real�time quantitativepolymerase chain reaction analysis of mitochondrialDNA point mutation. Methods Mol. Biol. 335, 187–200.
135. Poe B.G., Navratil M., Arriaga E.A. 2007. Absolutequantitation of a heteroplasmic mitochondrial DNAdeletion using a multiplex three�primer real�time PCRassay. Anal. Biochem. 362, 193–200.
136. Bai R.K., Wong L.J. 2006. Detection and quantifica�tion of heteroplasmic mutant mitochondrial DNA byreal�time amplification refractory mutation systemquantitative PCR analysis: A single�step approach.Clin. Chem. 50, 996–1001.
137. Thèves C., Keyser�Tracqui C., Crubézy E., Salles J.P.,Ludes B., Telmon N. 2006. Detection and quantifica�tion of the age�related point mutation A189G in thehuman mitochondrial DNA. J. Forensic Sci. 51, 865–873.
138. Alvarez�Iglesias V., Barros F., Carracedo A., Salas A.2008. Minisequencing mitochondrial DNA patho�genic mutations. BMC Med. Genet. 10, 9–26.
139. White H.E., Durston V.J., Seller A., Fratter C.,Harvey J.F., Cross N. C. 2005. Accurate detection andquantitation of heteroplasmic mitochondrial pointmutations by pyrosequencing. Genet. Test. 9, 190–199.
140. Mashima Y., Nagano M., Funayama T., Zhang Q.,Egashira T., Kudho J., Shimizu N., Oguchi Y. 2003.Rapid quantification of the heteroplasmy of mutantmitochondrial DNAs in Leber’s hereditary optic neu�ropathy using the Invader technology. Clin. Biochem.37, 268–276.
141. Pati N., Schowinsky V., Kokanovic O., Magnuson V.,Ghosh S. 2004. A comparison between SNaPshot,pyrosequencing, and biplex invader SNP genotypingmethods: Accuracy, cost, and throughput. J. Biochem.Biophys. Methods. 60, 1–12.
142. Du W., Li W., Chen G., Cao H., Tang H., Tang X.,Jin Q., Sun Z., Zhao H., Zhou W., He S., Lv Y.,Zhao J., Zhang X. 2009. Detection of known base sub�stitution mutations in human mitochondrial DNA ofMERRF and MELAS by biochip technology. Biosens.Bioelectron. 24, 2371–2376.
143. Nishigaki Y., Ueno H., Coku J., Koga Y., Fujii T.,Sahashi K., Nakano K., Yoneda M., Nonaka M.,Tang L., Liou C.W., Paquis�Flucklinger V., Harigaya Y.,Ibi T., Goto Y.I., Hosoya H., Dimauro S., Hirano M.,Tanaka M. 2010. Extensive screening system using sus�pension array technology to detect mitochondrialDNA point mutations. Mitochondrion. 10, 300–308.
144. Chinnery P., Majamaa K., Turnbull D., Thorburn D.2006. Treatment for mitochondrial disorders.Cochrane. Database. Syst. Rev. 1, CD004426.
145. Dimauro S., Rustin P. 2009. A critical approach to thetherapy of mitochondrial respiratory chain and oxida�tive phosphorylation diseases. Biochim. Biophys. Acta.1792, 1159–1167.

MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 679
146. Parikh S., Saneto R., Falk M.J., Anselm I., Cohen B.H.,Haas R., Medicine Society TM. 2009. A modernapproach to the treatment of mitochondrial disease.Curr. Treat. Options Neurol. 11, 414–430.
147. Finsterer J. 2010. Treatment of mitochondrial disor�ders. Eur. J. Paediatr. Neurol. 14, 29–44.
148. Finsterer J., Segall L. 2010. Drugs interfering withmitochondrial disorders. Drug. Chem. Toxicol. 33, 138–151.
149. Kerr D. S. 2010. Treatment of mitochondrial electrontransport chain disorders: A review of clinical trialsover the past decade. Mol. Genet. Metab. 99, 246–255.
150. Taivassalo T., Haller R.G. 2004. Implications of exer�cise training in mtDNA defects: Use it or lose it? Bio�chim. Biophys. Acta. 1659, 221–231.
151. Taivassalo T., Haller R.G. 2005. Exercise and trainingin mitochondrial myopathies. Med. Sci. Sports Exerc.37, 2094–2101.
152. Gardner J.L., Craven L., Turnbull D.M., Taylor R.W.2007. Experimental strategies towards treating mito�chondrial DNA disorders. Biosci. Rep. 27, 139–150.
153. Edmonds J.L. Jr. 2004. Surgical and anesthetic man�agement of patients with mitochondrial dysfunction.Mitochondrion. 4, 543–548.
154. Footitt E.J., Sinha M.D., Raiman J.A., Dhawan A.,Moganasundram S., Champion M. P. 2008. Mito�chondrial disorders and general anaesthesia: A caseseries and review. Br. J. Anaesth. 100, 436–441.
155. Chinnery P.F., Bindoff L.A., Europen neuromuscularcenter. 2003. 116th ENMC international workshop:The treatment of mitochondrial disorders, 14th�16thMarch 2003, Naarden, The Netherlands. Neuromus�cul. Disord. 13, 757–764.
156. Oca�Cossio J., Kenyon L., Hao H., Moraes C. T.2003. Limitations of allotopic expression of mitochon�drial genes in mammalian cells. Genetics. 165, 707–720.
157. Manfredi G., Fu J., Ojaimi J., Sadlock J.E., Kwong J.Q.,Guy J., Schon E.A. 2002. Rescue of a deficiency inATP synthesis by transfer of MTATP6, a mitochondrialDNA�encoded gene, to the nucleus. Nature Genet. 30,394–399.
158. Guy J., Qi X., Pallotti F., Schon E.A., Manfredi G.,Carelli V., Martinuzzi A., Hauswirth W.W., Lewin A.S.2002. Rescue of a mitochondrial deficiency causingLeber hereditary optic neuropathy. Ann. Neurol. 52,534–542.
159. Zullo S.J., Parks W.T., Chloupkova M., Wei B., Weiner H.,Fenton W.A., Eisenstadt J.M., Merril C.R. 2005. Sta�ble transformation of CHO Cells and human NARPcybrids confers oligomycin resistance (oli(r)) followingtransfer of a mitochondrial DNA�encoded oli(r)ATPase6 gene to the nuclear genome: A model systemfor mtDNA gene therapy. Rejuvenation Res. 8, 18–28.
160. Bonnet C., Augustin S., Ellouze S., Bénit P., Bouaita A.,Rustin P., Sahel J.A., Corral�Debrinski M. 2008. Theoptimized allotopic expression of ND1 or ND4 genesrestores respiratory chain complex I activity in fibro�blasts harboring mutations in these genes. Biochim.Biophys. Acta. 1783, 1707–1717.
161. Bonnet C., Kaltimbacher V., Ellouze S., Augustin S.,Bénit P., Forster V., Rustin P., Sahel J.A., Corral�
Debrinski M. 2007. Allotopic mRNA localization tothe mitochondrial surface rescues respiratory chaindefects in fibroblasts harboring mitochondrial DNAmutations affecting complex I or V subunits. Rejuve�nation Res. 10, 127–144.
162. Ellouze S., Augustin S., Bouaita A., Bonnet C., Simo�nutti M., Forster V., Picaud S., Sahel J.A., Corral�Debrinski M. 2008. Optimized allotopic expression ofthe human mitochondrial ND4 prevents blindness in arat model of mitochondrial dysfunction. Am. J. Hum.Genet. 83, 373–387.
163. Seo B.B., Wang J., Flotte T.R., Yagi T., Matsuno�Yagi A.2000. Use of the NADH�quinone oxidoreductase(NDI1) gene of Saccharomyces cerevisiae as a possiblecure for complex I defects in human cells. J. Biol.Chem. 275, 37774–37778.
164. Park J.S., Li Y.F., Bai Y. 2007. Yeast NDI1 improvesoxidative phosphorylation capacity and increases pro�tection against oxidative stress and cell death in cellscarrying a Leber’s hereditary optic neuropathy muta�tion. Biochim. Biophys. Acta. 1772, 533–542.
165. Marella M., Seo B.B., Yagi T., Matsuno�Yagi A. 2009.Parkinson’s disease and mitochondrial complex I: Aperspective on the Ndi1 therapy. J. Bioenerg.Biomembr. 41, 493–497.
166. Hakkaart G.A., Dassa E.P., Jacobs H.T., Rustin P.2006. Allotopic expression of a mitochondrial alterna�tive oxidase confers cyanide resistance to human cellrespiration. EMBO Rep. 7, 341–345.
167. Kolesnikova O.A., Entelis N.S., Mireau H., Fox T.D.,Martin R.P., Tarassov I.A. 2000. Suppression of muta�tions in mitochondrial DNA by tRNAs imported fromthe cytoplasm. Science. 289, 1931–1933.
168. Entelis N.S., Kolesnikova O.A., Martin R.P., Tarassov I.A.2001. RNA delivery into mitochondria. Adv. Drug.Deliv. Rev. 49, 199–215.
169. Kolesnikova O.A., Entelis N.S., Jacquin�Becker C.,Goltzene F., Chrzanowska�Lightowlers Z.M., Light�owlers R.N., Martin R.P., Tarassov I. 2004. NuclearDNA�encoded tRNAs targeted into mitochondria canrescue a mitochondrial DNA mutation associated withthe MERRF syndrome in cultured human cells. Hum.Mol. Genet. 13, 2519–2534.
170. Mahata B., Mukherjee S., Mishra S., Bandyopadhyay A.,Adhya S. 2006. Functional delivery of a cytosolictRNA into mutant mitochondria of human cells. Sci�ence. 314, 471–474.
171. Mukherjee S., Mahata B., Mahato B., Adhya S. 2008.Targeted mRNA degradation by complex�mediateddelivery of antisense RNAs to intracellular humanmitochondria. Hum. Mol. Genet. 17, 1292–1298.
172. Ling J., Roy H., Qin D., Rubio M.A., Alfonzo J.D.,Fredrick K., Ibba M. 2007. Pathogenic mechanism ofa human mitochondrial tRNAPhe mutation associ�ated with myoclonic epilepsy with ragged red fiberssyndrome. Proc. Natl. Acad. Sci. USA. 104, 15299–15304.
173. Rorbach J., Soleimanpour�Lichaei R., Lightowlers R.N.,Chrzanowska�Lightowlers Z.M. 2007. How do mam�malian mitochondria synthesize proteins? Biochem.Soc. Trans. 35, 1290–1291.

680
MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MAZUNIN et al.
174. Park H., Davidson E., King M. P. 2008. Overexpressedmitochondrial leucyl�tRNA synthetase suppresses theA3243G mutation in the mitochondrialtRNA(Leu(UUR)) gene. RNA. 14, 2407–2416.
175. Tanaka M., Borgeld H.J., Zhang J., Muramatsu S.,Gong J.S., Yoneda M., Maruyama W., Naoi M., Ibi T.,Sahashi K., Shamoto M., Fuku N., Kurata M.,Yamada Y., Nishizawa K., Akao Y., Ohishi N., Miya�bayashi S., Umemoto H., Muramatsu T., Furukawa K.,Kikuchi A., Nakano I., Ozawa K., Yagi K. 2002. Genetherapy for mitochondrial disease by delivering restric�tion endonuclease SmaI into mitochondria. J. Biomed.Sci. 9, 534–541.
176. Bacman S.R., Williams S.L., Hernandez D., Moraes C.T.2007. Modulating mtDNA heteroplasmy by mito�chondria�targeted restriction endonucleases in a 'dif�ferential multiple cleavage�site' model. Gene Ther. 14,1309–1318.
177. Taylor R.W., Chinnery P.F., Turnbull D.M., Lightowl�ers R.N. 1997. Selective inhibition of mutant humanmitochondrial DNA replication in vitro by peptidenucleic acids. Nature Genet. 15, 212–215.
178. Chinnery P.F., Taylor R.W., Diekert K., Lill R., Turn�bull D.M., Lightowlers R.N. 1999. Peptide nucleicacid delivery to human mitochondria. Gene Ther. 6,1919–1928.
179. Muratovska A., Lightowlers R.N., Taylor R.W., Turn�bull D.M., Smith R.A., Wilce J.A., Martin S.W., Mur�phy M. P. 2001. Targeting peptide nucleic acid (PNA)oligomers to mitochondria within cells by conjugationto lipophilic cations: Implications for mitochondrialDNA replication, expression and disease. NucleicAcids Res. 29, 1852–1863.
180. Taylor R.W., Wardell T.M., Smith P.M., Muratovska A.,Murphy M.P., Turnbull D.M., Lightowlers R.N. 2001.An antigenomic strategy for treating heteroplasmicmtDNA disorders. Adv. Drug. Deliv. Rev. 49, 121–125.
181. Flierl A., Jackson C., Cottrell B., Murdock D., Seibel P.,Wallace D. C. 2003. Targeted delivery of DNA to themitochondrial compartment via import sequence�conjugated peptide nucleic acid. Mol. Ther. 7, 550–557.
182. Kyriakouli D.S., Boesch P., Taylor R.W., Lightowlers R.N.2008. Progress and prospects: gene therapy for mito�chondrial DNA disease. Gene Ther. 15, 1017–1023.
183. Minczuk M., Papworth M.A., Kolasinska P., Murphy M.P.,Klug A. 2006. Sequence�specific modification ofmitochondrial DNA using a chimeric zinc fingermethylase. Proc. Natl. Acad. Sci. USA. 103, 19689–19694.
184. Spees J.L., Olson S.D., Whitney M.J., Prockop D.J.2005. Mitochondrial transfer between cells can rescueaerobic respiration. Proc. Natl. Acad. Sci. USA. 103,1283–1238.
185. Csordás A. 2006. Mitochondrial transfer betweeneukaryotic animal cells and its physiologic role. Reju�venation Res. 9, 450–454.
186. Dani M.A., Dani S.U. 2010. Improving upon nature’ssomatic mitochondrial DNA therapies. Med. Hypothe�ses. 74, 1021–1025.
187. Yamada Y., Harashima H. 2008. Mitochondrial drugdelivery systems for macromolecule and their thera�
peutic application to mitochondrial diseases. Adv.Drug. Deliv. Rev. 60, 1439–1462.
188. Boddapati S.V., D’Souza G.G., Weissig V. 2010. Lipo�somes for drug delivery to mitochondria. Methods Mol.Biol. 605, 295–303.
189. Brown D.T., Herbert M., Lamb V.K., Chinnery P.F.,Taylor R.W., Lightowlers R.N., Craven L., Cree L.,Gardner J.L., Turnbull D.M. 2006. Transmission ofmitochondrial DNA disorders: Possibilities for thefuture. Lancet. 368, 87–89.
190. Poulton J., Kennedy S., Oakeshott P., Wells D. 2009.Preventing transmission of maternally inherited mito�chondrial DNA diseases. Br. Med. J. 338, b94.
191. South S.T., Chen Z., Brothman A.R. 2008. Genomicmedicine in prenatal diagnosis. Clin. Obstet. Gynecol.51, 62–73.
192. Poulton J., Turnbull D.M. 2000. 74th ENMC interna�tional workshop: Mitochondrial diseases. 19⎯20November 1999, Naarden, the Netherlands. Neuro�muscul. Disord. 10, 460–462.
193. Geraedts J.P., De Wert G.M. 2009. Preimplantationgenetic diagnosis. Clin. Genet. 76, 315–325.
194. Dean N.L., Battersby B.J., Ao A., Gosden R.G.,Tan S.L., Shoubridge E.A., Molnar M.J. 2003. Pros�pect of preimplantation genetic diagnosis for heritablemitochondrial DNA diseases. Mol. Hum. Reprod. 9,631–638.
195. Bickerstaff H., Flinter F., Yeong C.T., Braude P. 2001.Clinical application of preimplantation genetic diag�nosis. Hum. Fertil. (Cambridge). 4, 24–30.
196. Steffann J., Frydman N., Gigarel N., Burlet P.,Ray P.F., Fanchin R., Feyereisen E., Kerbrat V., Tach�djian G., Bonnefont J.P., Frydman R., Munnich A.2006. Analysis of mtDNA variant segregation duringearly human embryonic development: A tool for suc�cessful NARP preimplantation diagnosis. J. Med.Genet. 43, 244–247.
197. Bredenoord A.L., Dondorp W., Pennings G., De Die�Smulders C.E., De Wert G. 2008. PGD to reducereproductive risk: The case of mitochondrial DNA dis�orders. Hum. Reprod. 23, 2392–2401.
198. Bredenoord A.L., Pennings G., Smeets H.J., de Wert G.2008. Dealing with uncertainties: Ethics of prenataldiagnosis and preimplantation genetic diagnosis toprevent mitochondrial disorders. Hum. Reprod.Update. 14, 83–94.
199. Bredenoord A., Dondorp W., Pennings G., de Die�Smulders C., Smeets B., de Wert G. 2009. Preimplan�tation genetic diagnosis for mitochondrial DNA disor�ders: ethical guidance for clinical practice. Eur. J.Hum. Genet. 17, 1550–1559.
200. Brenner C.A., Barritt J.A., Willadsen S., Cohen J.2000. Mitochondrial DNA heteroplasmy after humanooplasmic transplantation. Fertil. Steril. 74, 573–578.
201. Cohen J., Scott R., Alikani M., Schimmel T., Munné S.,Levron J., Wu L., Brenner C., Warner C., Willadsen S.1998. Ooplasmic transfer in mature human oocytes.Mol. Hum. Reprod. 4, 269–280.
202. Barritt J.A., Brenner C.A., Malter H.E., Cohen J.2001. Mitochondria in human offspring derived fromooplasmic transplantation. Hum. Reprod. 16, 513–516.

MOLECULAR BIOLOGY Vol. 44 No. 5 2010
MITOCHONDRIAL GENOME AND HUMAN DISEASES 681
203. Hawes S.M., Sapienza C., Latham K.E. 2002. Ooplas�mic donation in humans: The potential for epigenicmodifications. Hum. Reprod. 17, 850–852.
204. Harvey A.J., Gibson T.C., Quebedeaux T.M., Bren�ner C.A. 2007. Impact of assisted reproductive tech�nologies: A mitochondrial perspective of cytoplasmictransplantation. Curr. Top. Dev. Biol. 77, 229–249.
205. Taylor R.W., Turnbull D.M. 2005. MitochondrialDNA mutations in human disease. Nature Rev. Genet.5, 389–402.
206. Fulka H., Fulka J. Jr. 2007. The use of micromanipu�lation methods as a tool to prevention of transmissionof mutated mitochondrial DNA. Curr. Top. Dev. Biol.77, 187–211.
207. Fulka J. Jr., Fulka H., John J. C. 2007. Transmission ofmitochondrial DNA disorders: Possibilities for theelimination of mutated mitochondria. Cloning StemCells. 9, 47–50.
208. Bredenoord A.L., Pennings G., de Wert G. 2008. Oop�lasmic and nuclear transfer to prevent mitochondrialDNA disorders: Conceptual and normative issues.Hum. Reprod. Update. 14, 669–678.
209. Fulka H., Langerova A., Barnetova I., Novakova Z.,Mosko T., Fulka J. Jr. 2009. How to repair the oocyteand zygote? J. Reprod. Dev. 55, 583–587.
210. Tachibana M., Sparman M., Sritanaudomchai H.,Ma H., Clepper L., Woodward J., Li Y., Ramsey C.,
Kolotushkina O., Mitalipov S. 2009. Mitochondrialgene replacement in primate offspring and embryonicstem cells. Nature. 461, 367–372.
211. Khan S.M., Smigrodzki R.M., Swerdlow R.H. 2007.Cell and animal models of mtDNA biology: progressand prospects. Am. J. Physiol. Cell. Physiol. 292, 658–669.
212. Vempati U.D., Torraco A., Moraes C.T. 2008. Mousemodels of oxidative phosphorylation dysfunction anddisease. Methods. 46, 241–247.
213. Wallace D.C., Fan W. 2009. The pathophysiology ofmitochondrial disease as modeled in the mouse. GenesDev. 23, 1714–1736.
214. Lim K.S., Naviaux R.K., Wong S., Haas R.H. 2008.Pitfalls in the denaturing high�performance liquidchromatography analysis of mitochondrial DNAmutation. J. Mol. Diagn. 10, 102–108.
215. Dobrowolski S.F., Hendrickx A.T., van den Bosch B.J.,Smeets H.J., Gray J., Miller T., Sears M. 2009. Identi�fying sequence variants in the human mitochondrialgenome using high�resolution melt (HRM) profiling.Hum. Mutat. 30, 891–898.
216. Ojaimi J., Pan J., Santra S., Snell W.J., Schon E.A. Analgal nucleus�encoded subunit of mitochondrial ATPsynthase rescues a defect in the analogous humanmitochondrial�encoded subunit. Mol. Biol. Cell. 13,3836–3844.
Related Documents











