ORIGINAL ARTICLE Re-expression of DNA methylation-silenced CD44 gene in a resistant NB4 cell line: rescue of CD44-dependent cell death by cAMP I Abecassis 1 , J Maes 2 , J-L Carrier 1 , J Hillion 1 , M Goodhardt 2 , K Medjber 1 , L Wany 1 , M Lanotte 1 and A Karniguian 1 1 INSERM U685, IUH, Ho ˆpital Saint-Louis, Paris, France and 2 U662, IUH, Ho ˆpital Saint-Louis, Paris, France In the acute promyelocytic leukemia cell line, NB4, activation of the CD44 receptor triggers apoptosis. This pathway does not operate in the retinoid-maturation-resistant NB4-LR1 subclone. In this work, we show that the CD44 gene is silenced in these cells. The molecular defect involves DNA methylation of cytosine phosphate guanine (CpG) island and underacetylation of histone H3 at CD44 promoter. The methylating inhibitor 5- aza-CdR and cyclic AMP (cAMP) reverse the CD44 gene silencing. Contrary to 5-aza-CdR, cAMP does not induce DNA demethylation or histone modification at the CD44 promoter, whereas an H3pS10/AcK14 dual modification is observed on a global level. cAMP also induces the expression of c-Jun transcription factor and its recruitment at the CD44 promoter. Chromatin immunoprecipitation assays further show the asso- ciation of brahma (Brm), a subunit of SWI/SNF chromatin- remodelling complex involved in the crosstalk between tran- scription and RNA polymerase II (RNA Pol II) processing, as well as the binding of phosphorylated RNA Pol II to the proximal promoter region of CD44. Finally, our study reveals that cAMP re-establishes the CD44-mediated cell death signalling. We propose that one of the actions of cAMP in restoring normal cell phenotype of leukaemia cells may consist in a broad trans-reactivation of silenced genes, despite marked hyper- methylation of their promoters, as illustrated here with CD44 re-expression. Leukemia (2008) 22, 511–520; doi:10.1038/sj.leu.2405071; published online 20 December 2007 Keywords: CD44; gene silencing; cAMP; cell death Introduction CD44 is a multifunctional cell surface molecule differentially expressed, via various alternative spliced isoforms 1 in many cell types. 2,3 These surface molecules are involved in a fascinating number of distinct functions, including cell proliferation, cell survival, differentiation, cell adhesion and migration, angiogen- esis. 4–6 While CD44 is important for the physiological beha- viour of normal cells, it has rapidly been recognized that altered CD44 expression participates in the initiation or progression of tumour cells towards transformed phenotypes and metastasis. Importantly, in the case of myeloid leukaemia, 7,8 targeting CD44 by means of epitope-specific antibodies, cells undergo maturation and/or apoptosis, with anti-tumour features that can be exploited therapeutically. In acute promyelocytic leukaemias (APL), all-trans retinoic acid (ATRA) induces the differentiation of leukemic cells, leading to remission of the disease. This differentiation therapy is however not durable and ATRA resistance often develops. It has been recently reported that in the APL cell model, NB4, 9 anti-CD44 antibodies triggered growth inhibition and cell death. 8,10,11 This feature is of clinical importance for APL cells and CD44 signalling could in particular represent an alternative therapeutic approach for ATRA-resistant APL cells. 12,13 These cells fail to be growth inhibited and do not mature during retinoid treatment. Very likely, they bear secondarily acquired alterations, which have been lastly shown to be overcome by cyclic AMP (cAMP)-dependent signallings. This is typically the case exemplified in vitro by the ATRA-resistant subclone NB4- LR1 cells, which requires a combined cAMP/ATRA treatment to undergo differentiation and cell death. 14–16 The additional defects present in these resistant cells, as well as the molecular mechanism(s) by which cAMP acts to normalize the NB4-LR1 cell phenotype, are still poorly understood, although several proposals supported by independent investigations have been recently forwarded. 17–21 In this study, we demonstrate that there is an altered CD44 gene expression in the ATRA-maturation-resistant NB4-LR1 cells, and characterize the molecular defects behind this lack of CD44 receptor expression as DNA methylation of the CD44 promoter and deacetylation of histones at this locus. Impor- tantly, full reactivation of CD44 gene transcription and synthesis of a functional CD44 receptor protein were rapidly obtained with a DNA methylating inhibitor (5-aza-CdR). Epigenetic events have been reported to be involved in downregulation of genes responsible for drug resistance in cancer cells. 22,23 Interestingly, we show here that cAMP treatment of NB4-LR1 cells reverses CD44 transcription silencing and restores the expression of a functional CD44 receptor, able again to trigger cell death in response to the specific A3D8 anti-CD44 antibody. Given the diverse cellular cAMP effects that, collectively, could contribute to the rescue of the ATRA-maturation program in NB4-resistant cells, we speculate that a broad modification of the chromatin transcription context by PKA signalling could render resistant cells competent for further commitment towards more than a single biological response (for example, cell differentiation, growth arrest, cell death). Materials and methods Reagents ATRA, 8-(4-chlorophenylthio)-cAMP (8-CPT-cAMP), 5-aza-2 0 - deoxycytidine (5-aza-CdR) and trichostatin A (TSA) were purchased from Sigma (St Quentin Fallavier, France). Cell lines NB4 and NB4-LR1 cell lines 16 were routinely cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2 mM L-glutamine, 100 U per milliliter Received 19 July 2006; revised 17 October 2007; accepted 12 November 2007; published online 20 December 2007 Correspondence: Dr A Karniguian, INSERM U-685, 1 Ave Claude Vellefaux, IUH, Ho ˆpital Saint-Louis, Paris 75010, France. E-mail: [email protected] Leukemia (2008) 22, 511–520 & 2008 Nature Publishing Group All rights reserved 0887-6924/08 $30.00 www.nature.com/leu

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL ARTICLE
Re-expression of DNA methylation-silenced CD44 gene in a resistant NB4 cellline: rescue of CD44-dependent cell death by cAMP
I Abecassis1, J Maes2, J-L Carrier1, J Hillion1, M Goodhardt2, K Medjber1, L Wany1, M Lanotte1 and A Karniguian1
1INSERM U685, IUH, Hopital Saint-Louis, Paris, France and 2U662, IUH, Hopital Saint-Louis, Paris, France
In the acute promyelocytic leukemia cell line, NB4, activation ofthe CD44 receptor triggers apoptosis. This pathway does notoperate in the retinoid-maturation-resistant NB4-LR1 subclone.In this work, we show that the CD44 gene is silenced in thesecells. The molecular defect involves DNA methylation ofcytosine phosphate guanine (CpG) island and underacetylationof histone H3 at CD44 promoter. The methylating inhibitor 5-aza-CdR and cyclic AMP (cAMP) reverse the CD44 genesilencing. Contrary to 5-aza-CdR, cAMP does not induce DNAdemethylation or histone modification at the CD44 promoter,whereas an H3pS10/AcK14 dual modification is observed on aglobal level. cAMP also induces the expression of c-Juntranscription factor and its recruitment at the CD44 promoter.Chromatin immunoprecipitation assays further show the asso-ciation of brahma (Brm), a subunit of SWI/SNF chromatin-remodelling complex involved in the crosstalk between tran-scription and RNA polymerase II (RNA Pol II) processing, aswell as the binding of phosphorylated RNA Pol II to the proximalpromoter region of CD44. Finally, our study reveals that cAMPre-establishes the CD44-mediated cell death signalling. Wepropose that one of the actions of cAMP in restoring normalcell phenotype of leukaemia cells may consist in a broadtrans-reactivation of silenced genes, despite marked hyper-methylation of their promoters, as illustrated here with CD44re-expression.Leukemia (2008) 22, 511–520; doi:10.1038/sj.leu.2405071;published online 20 December 2007Keywords: CD44; gene silencing; cAMP; cell death
Introduction
CD44 is a multifunctional cell surface molecule differentiallyexpressed, via various alternative spliced isoforms1 in many celltypes.2,3 These surface molecules are involved in a fascinatingnumber of distinct functions, including cell proliferation, cellsurvival, differentiation, cell adhesion and migration, angiogen-esis.4–6 While CD44 is important for the physiological beha-viour of normal cells, it has rapidly been recognized that alteredCD44 expression participates in the initiation or progression oftumour cells towards transformed phenotypes and metastasis.Importantly, in the case of myeloid leukaemia,7,8 targetingCD44 by means of epitope-specific antibodies, cells undergomaturation and/or apoptosis, with anti-tumour features that canbe exploited therapeutically.
In acute promyelocytic leukaemias (APL), all-trans retinoicacid (ATRA) induces the differentiation of leukemic cells,leading to remission of the disease. This differentiation therapyis however not durable and ATRA resistance often develops. It
has been recently reported that in the APL cell model, NB4,9
anti-CD44 antibodies triggered growth inhibition and celldeath.8,10,11 This feature is of clinical importance for APL cellsand CD44 signalling could in particular represent an alternativetherapeutic approach for ATRA-resistant APL cells.12,13 Thesecells fail to be growth inhibited and do not mature duringretinoid treatment. Very likely, they bear secondarily acquiredalterations, which have been lastly shown to be overcome bycyclic AMP (cAMP)-dependent signallings. This is typically thecase exemplified in vitro by the ATRA-resistant subclone NB4-LR1 cells, which requires a combined cAMP/ATRA treatment toundergo differentiation and cell death.14–16 The additionaldefects present in these resistant cells, as well as the molecularmechanism(s) by which cAMP acts to normalize the NB4-LR1cell phenotype, are still poorly understood, although severalproposals supported by independent investigations have beenrecently forwarded.17–21
In this study, we demonstrate that there is an altered CD44gene expression in the ATRA-maturation-resistant NB4-LR1cells, and characterize the molecular defects behind this lackof CD44 receptor expression as DNA methylation of the CD44promoter and deacetylation of histones at this locus. Impor-tantly, full reactivation of CD44 gene transcription and synthesisof a functional CD44 receptor protein were rapidly obtainedwith a DNA methylating inhibitor (5-aza-CdR). Epigeneticevents have been reported to be involved in downregulationof genes responsible for drug resistance in cancer cells.22,23
Interestingly, we show here that cAMP treatment of NB4-LR1cells reverses CD44 transcription silencing and restores theexpression of a functional CD44 receptor, able again to triggercell death in response to the specific A3D8 anti-CD44 antibody.Given the diverse cellular cAMP effects that, collectively, couldcontribute to the rescue of the ATRA-maturation program inNB4-resistant cells, we speculate that a broad modification ofthe chromatin transcription context by PKA signalling couldrender resistant cells competent for further commitment towardsmore than a single biological response (for example, celldifferentiation, growth arrest, cell death).
Materials and methods
ReagentsATRA, 8-(4-chlorophenylthio)-cAMP (8-CPT-cAMP), 5-aza-20-deoxycytidine (5-aza-CdR) and trichostatin A (TSA) werepurchased from Sigma (St Quentin Fallavier, France).
Cell linesNB4 and NB4-LR1 cell lines16 were routinely culturedin RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA)supplemented with 2 mM L-glutamine, 100 U per milliliter
Received 19 July 2006; revised 17 October 2007; accepted 12November 2007; published online 20 December 2007
Correspondence: Dr A Karniguian, INSERM U-685, 1 Ave ClaudeVellefaux, IUH, Hopital Saint-Louis, Paris 75010, France.E-mail: [email protected]
Leukemia (2008) 22, 511–520& 2008 Nature Publishing Group All rights reserved 0887-6924/08 $30.00
www.nature.com/leu

penicillin (Invitrogen), 0.1 mg ml�1 streptomycin (Invitrogen)and 10% fetal calf serum (Invitrogen, Carlsbad, CA, USA). Cellswere grown at 37 1C in a 5% CO2 atmosphere.
CD44 protein expression in NB4 and NB4-LR1 cells byimmunofluorescence and flow cytometry analysisNB4 and NB4-LR1 cells were incubated with an anti-CD44antibody against an epitope of the standard form (clone J173 orA3D8), or the isotype-matched non-specific mouse immuno-globulin G (IgGs), and Alexa 488-conjugated secondaryantibodies (fluorescein isothiocyanate, FITC; Dako, Trappes,France), in the presence of Texas Red-labelled phalloidin tovisualize the actin filaments. Confocal laser scanning micro-scopy analysis was performed using a Biorad MRC1024confocal imaging system (UK). Immunofluorescent cells werealso analysed by flow cytometry on a FACSCalibur instrument(Becton Dickinson Immunocytometry Systems, de Pont-De-Claix,France) using CellQuest software. Results were expressedas histograms of FITC staining (fluorescence intensity) andquantified as the relative mean fluorescence intensity comparedwith isotype control antibodies. Representative histogramsrepresent an acquisition of 104 events.
RT–PCR analysisTotal RNAs purified from NB4 and NB4-LR1 cells, exposed ornot to 1 mM ATRA or 100 mM 8-CPT-cAMP, for 96 h or 72 h,respectively, were isolated using the RNeasy Kit (Qiagen), andwere reversed transcribed by using 200 U MMLV reversetranscriptase enzyme (Promega). PCR amplification of CD44transcripts was then performed with primers specific for the50- and 30-constant region of CD44, that is, forward primer(in exon 5): 50-TTGTTAACCGTGATGGCACC-30 and reverseprimer (in exon 16): 50-ATTTGGGGTGTCCTTATAG-30. ThePCR reaction was performed for 30 cycles of denaturation at95 1C for 45 s, annealing at 52 1C for 60 s, and extension at 72 1Cfor 75 s, followed by 72 1C for 15 min, using 2.5 U of Ampli taqpolymerase. c-jun and brm transcripts were detected by usingthe following primers: c-jun, forward primer 50-GACTGCAAAGATGGAAACGA-30 and reverse primer 50-GTTGCTGGACTGGATTATCA-30 (253 bp product), brm, forward primer 50-CCAGTAGGCAGGAAACCGAAG-30 and reverse primer 50-GGCTTGCATATGGCGATACA-30 (1 kb product). PCR reactions were runat 94 1C for 30 s, 56 1C for 1 min and 72 1C for 1 min with 29cycles, followed by 10 min at 72 1C. Primers specific forglyceraldehyde-3-phosphate dehydrogenase (GAPDH) genewere used for loading controls: 50-CTCAGACACCATGGGGAAGGTGA-30 (forward) and 50-ATGATCTTGAGGCTGTTGTCATA-30 (reverse).
Restriction enzyme digestion and PCR analysis forCD44 methylationPromoter methylation of CD44 gene was determined bymethylation-sensitive restriction enzyme digestion followed byPCR analysis, as described elsewhere with some modifica-tions.24 Three restriction enzymes exhibiting complementarymethylation sensitivity were used: the methylation-sensitiverestriction enzyme HpaII, its methylation-insensitive isoschizo-mer MspI (control), both acting at CCGG sites, as well asMcrBC, a restriction enzyme that cleaves only methylated DNA(in contrast to HpaII) at G/Am cytosines. Of note, this restrictionenzyme cannot digest the CmCGG HpaII specific sites. PCRs ofthe digested genomic DNA were then performed using
advantage-guanine-cytosine (GC) genomic PCR Kit (Clontech,St Germain-en-Laye, France) in a total volume of 20ml with 1 M
GC melt, 25 pmol of each primer, 0.2 mM desoxyribonucleotidetriphosphates (dNTPs), restriction enzyme-digested DNAs(250 ng) and 2.5 U of advantage Tth polymerase. Amplificationwas carried out on a thermocycler for 94 1C for 45 s, 63 1C for2 min and 72 1C for 2 min for 37 cycles, followed by 72 1C for15 min. Primers were designed according to the publishedsequence of the 50 region of CD44: forward, 50-CTCCCCACCCCTCACTCC-30 and reverse, 50-CCCCGCACCCATCTTGCTG-30.These primers cover the cytosine phosphate guanine (CpG)-richpromoter and exon 1 regions (total of 44 CpG sites), yielding a777 bp product, which corresponds to position �668 to þ 109relative to the translation initiation site. A 20 ml portion of PCRproduct was loaded on 2% agarose gel, stained with ethidiumbromide and scanned using Biomax image analysis software.
5-Aza-CdR treatment of NB4-LR1 cellsExponentially growing NB4-LR1 cells were exposed to themethylating inhibitor agent, 5-aza-CdR, at a concentration of100–250 nM. Cells were mock treated with an identical volumeof dimethyl sulfoxide.
Pyrosequencing assayGenomic DNAs, isolated from NB4 and NB4-LR1 cells,stimulated or not with 8-CPT-cAMP (100mM) or with5-aza-CdR (250 nM), were sodium bisulphite treated with theEZ-96 DNA methylation kit (ZymedResearch, San Francisco,CA, USA). To analyse and quantify the degree of DNAmethylation within CD44 promoter, a 541 bp-modified DNAfragment, covering 41 CpG sites and corresponding to position�541 to þ 1 (relative to the translation initiation site), wasamplified and pyrosequenced by Biotage BA (Uppsala, Swe-den). Percentages of DNA methylation (proportions of methy-lated alleles) at each individual CpG site were determined.
ChIP assays and PCRChromatin immunoprecipitation assays (ChIPs) were performedon growing NB4 and NB4-LR1 cells, treated or not with 8-CPT-cAMP, by using the ChIP assay kit from Upstate Biotechnology(Lake Placid, NY, USA) for histone immunoprecipitations andthe ChIP-IT kit from Active Motif (Rixensart, Belgium) for c-Jun,Brm and RNA polymerase II (RNA Pol II) immunoprecipitations,according to the manufacturer’s instructions. Briefly, cells werecross-linked with 1% formaldehyde, lysed and sonicated toobtain 0.6- to 0.8-kbp genomic DNA. Equal aliquots of theprecleared chromatin lysates were then incubated with pre-immune or no antibody (as negative control), with antibodies tomodified histone H3 and H4 (Upstate Biotechnology) asindicated, or to c-Jun (Active Motif), to Brm (N-19) (Santa Cruz,CA, USA) and to a phosphorylated form of RNA Pol II (serine 5phosphorylation of the C-terminal domain; Active Motif) at 4 1Covernight. To study the DNA/histone complexes, the immuno-precipitated (IP) DNA samples and the input DNA (totalchromatin) were amplified using two pairs of primers that covertwo different 100 bp regions within the CD44 promoter: (1) afirst primer set that corresponds to position �280 to �180 (Aregion): forward, 50-CCGGGAGGGCTGCTACTT-30 and reverse50-GACCTAAGACGGAGGGAGGG-30; (2) a second primer setthat corresponds to the position �547 to �447 (B region):forward, 50-CTCCAGCCGGATTCAGAGAA-30 and reverse,50-CGGCGTCAGGACAGAGGAT-30. Real-time PCR was carried
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
512
Leukemia

out using SYBRGreen PCR Master Mix (Applied Biosystems) on aABI PRISM 7000 thermocycler (Applied Biosystems). The levelof relative enrichment was represented, determined by quantify-ing the PCR product in IP DNA vs input DNA (total chromatin)and normalized against Pax5 gene at 1. Primers specific for Pax5gene are as follows: forward, 50-AGAGAGAGGGCTGGACATGC-30 and reverse, 50-GCCCTGAGACGTTGTGGAGT-30.Means from three independent ChIP experiments were pre-sented with s.e. values. To analyse the CD44 gene that interactswith c-Jun, Brm or RNA Pol II, the IP DNA samples and the inputDNA (total chromatin) were amplified with the appropriateprimer pairs, as follows: c-Jun, forward primer, 50-GGAGGGCTGCTACTTCTTAA-30 and reverse primer, 50-TCGAGGTTGAAAACAGTACC-30 (115 bp product, encompassing the AP-1 siteand corresponding to position �278 to �164, relative to thetranslation initiation site); Brm, forward primer, 50-CCTCTCTCCAGCTCCTCTCCC-30 and reverse primer, 50-TCCGCTGGGCAATGAGGCTG-30 (487 bp product corresponding to position�604 to �118, relative to the translation initiation site); RNAPol II, forward primer, 50-GGAGGGCTGCTACTTCTTAA-30 andreverse primer, 50-GCCACCAAAACTTGTCCATG-30 (297 bpproduct, encompassing the transcription start site and corre-sponding to position �278 to þ 19, relative to the translationinitiation site). Amplifications were carried out at 94 1C for 30 s,56 1C (for c-Jun and RNA Pol II) or 68 1C (for Brm) for 30 s and72 1C for 30 s for 29 cycles, followed by 72 1C for 10 min. As anegative control for c-Jun and Brm ChIP assays, a distalpromoter region of the housekeeping GAPDH gene was PCRamplified, using the following primers: forward primer, 50-TCCTGTTTCATCCAAGCGTG-30 and reverse primer, 50-GACTGTCGAACAGGAGGAG-30 with an annealing temperature at 60 1C,whereas negative primers provided by Active Motif were used asnegative control for RNA Pol II ChIP assays: forward primer,50-ATGGTTGCCACTGGGGATCT-30 and reverse primer,50-TGCCAAAGCCTAGGGGAAGA-30. PCR products wereloaded on 2% agarose gel, stained with ethidium bromide andscanned using Biomax image analysis software.
Western blot analysisExponentially growing NB4 or NB4-LR1 cells were exposed toATRA (1 mM), 8-CPT-cAMP (100 mM) or to adequate vehicle, asindicated. Cells were lysed in Laemmli buffer and samples wereresolved by SDS–PAGE on 10–20% polyacrylamide gels foranalysis of histones, or on 10% polyacrylamide gels for analysesof CD44, c-Jun or Brm, and electrotransferred onto 0.2 mmnitrocellulose membrane sheets (BA85, Whatman Schleicher &Schuell, Dassel, Germany). Membranes were then probed withthe appropriate specific antibodies to modified histones (UpstateBiotechnology), to c-Jun (sc-45, Santa Cruz), to p-c-Jun(Ser63/73) (sc-16312, Santa Cruz) or to Brm (N-19, Santa Cruz), asindicated. Anti-mouse, anti-rabbit or anti-goat IgGs, conjugatedto horse peroxidase (Dako) were used as secondary antibodies,and immunoreactive bands were revealed by a chemilumines-cence detection kit (ECL, Amersham, Buckinghamshire, UK).Immunoblots were further scanned to quantify band intensitiesusing Biomax image analysis software.
Morphology and DAPI analysis of NB4-LR1 cells inresponse to A3D8 anti-CD44 antibodyExponentially growing NB4-LR1 cells were exposed to 8-CPT-cAMP(100mM) or to adequate vehicle, for 48 h. 8-CPT-cAMP-primedcells were then further treated either with 8-CPT-cAMP alone orin co-treatment with A3D8 anti-CD44 antibody (Sigma) at a
concentration of 2 mg ml�1 for 96 h. As control, mocked NB4-LR1 were also treated with the A3D8 anti-CD44 antibody(2 mg ml�1) alone for the same time. Cells were then centrifugedby cytospin techniques onto glass sides, stained in May-Grunwald-Giemsa solution and analysed for cell morphologyby means of differential interference contrast microscopy, usinga � 63 objective, or fixed with 4% paraformaldehyde (PFA) andstained with 4,6-diamidino-2-phenylindole (DAPI, Sigma) tovisualize the nuclei. Fluorescent images were acquired using aLeica fluorescence microscope with a � 63 objective (excitationwavelength 350 nm).
Annexin-V FITC analysisAnnexin-V analysis was performed on NB4-LR1 cells treated, asdescribed above, with A3D8 or 8-CPT-cAMP alone, and in co-treatment with 8-CPT-cAMP and A3D8, using the Annexin-VFITC kit from AbCys (France) according to the manufacturer’sinstructions. Cells were then analysed by flow cytometry on aFACSCalibur instrument (Becton Dickinson ImmunocytometrySystems) using CellQuest software. Results were expressed ashistograms of FITC staining (fluorescence intensity) and quanti-fied as the relative mean fluorescence intensity. Representativehistograms represent an acquisition of 104 events.
Results and discussion
Defects in CD44 expression in NB4-LR1 cellsIt has been previously shown that A3D8, an anti-CD44 antibodydirected against epitope 1, induces typical morphologicalchanges in NB4 cells followed by cell death.8,25 By contrast,our data indicated that the maturation-resistant NB4-LR1subclone failed to respond to A3D8 anti-CD44 signalling. Wewanted to elucidate the origin of this defect. One explanationfor this unresponsiveness might be that the CD44 gene issilenced in the resistant cells. We therefore analysed compara-tively CD44 expression in the parental NB4 cells and in NB4-LR1 cells, at CD44 transcript and protein level.
CD44 transcripts, as evaluated by reverse transcription(RT)–PCR, were strongly expressed in NB4 cells, while, underthe same conditions, only a faint signal was detected inNB4-LR1 cells (Figure 1a). Comparative fluorescence-activatedcell sorting (FACS) analyses of NB4 and NB4-LR1 cells(Figure 1b) showed that, in concordance with PCR data forCD44 transcripts, NB4-LR1 cells had hardly detectable CD44protein expression at the outer plasmatic membrane, while, asexpected, the CD44-specific fluorescence profile for NB4 cellsshowed a marked shift to the right (42 log). Confocal analyseswere also performed and confirmed the defective pattern ofCD44 expression in NB4-LR1: the lack of surface CD44expression was due to the absence of CD44 protein synthesisby these cells and did not result from a modified subcellulardistribution (Figure 1c). Altogether, these data clearly demon-strated that the NB4-LR1 unresponsiveness to CD44 wascorrelated to silencing of the CD44 gene.
DNA hypermethylation and histone deacetylation atpromoter of CD44 geneSeveral mechanisms could explain CD44 gene silencing inNB4-LR1 cells. Analysis of the 1.1 kb CD44 promoter sequencerevealed a GC-rich domain (64%)26,27 containing four CCGGmotifs for HpaII/MspI enzymatic digestion (featured in Figure 2b,bottom). Therefore, genomic DNAs from NB4 and NB4-LR1
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
513
Leukemia
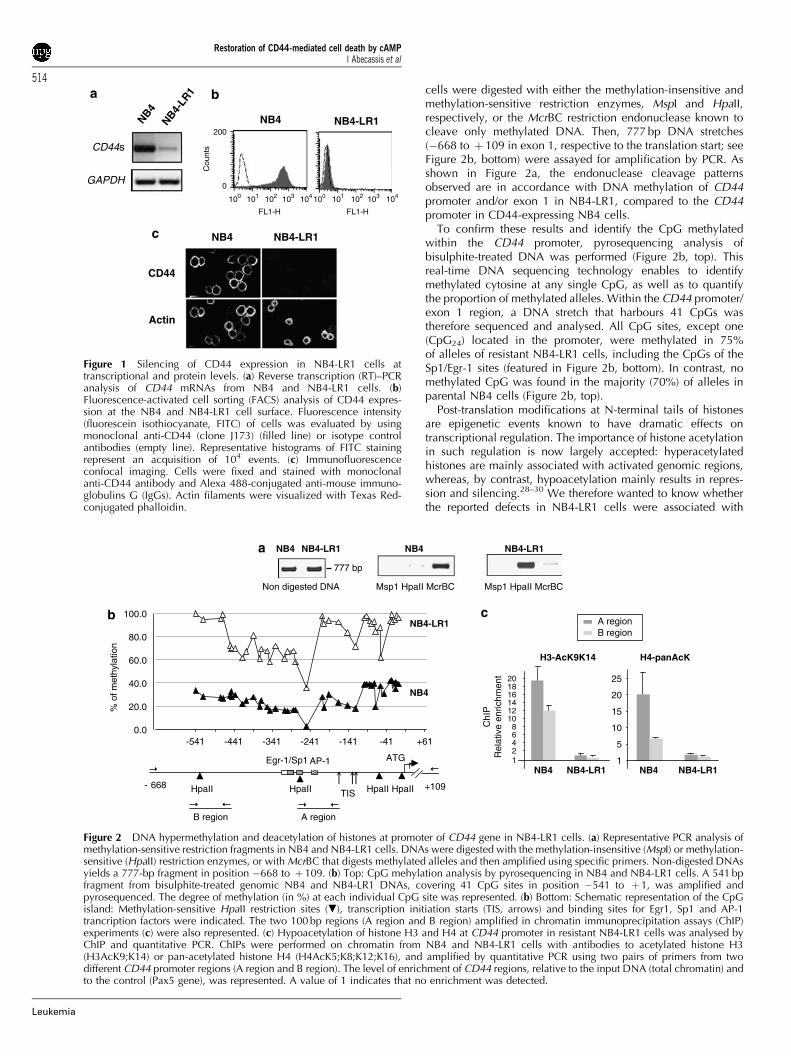
cells were digested with either the methylation-insensitive andmethylation-sensitive restriction enzymes, MspI and HpaII,respectively, or the McrBC restriction endonuclease known tocleave only methylated DNA. Then, 777 bp DNA stretches(�668 to þ 109 in exon 1, respective to the translation start; seeFigure 2b, bottom) were assayed for amplification by PCR. Asshown in Figure 2a, the endonuclease cleavage patternsobserved are in accordance with DNA methylation of CD44promoter and/or exon 1 in NB4-LR1, compared to the CD44promoter in CD44-expressing NB4 cells.
To confirm these results and identify the CpG methylatedwithin the CD44 promoter, pyrosequencing analysis ofbisulphite-treated DNA was performed (Figure 2b, top). Thisreal-time DNA sequencing technology enables to identifymethylated cytosine at any single CpG, as well as to quantifythe proportion of methylated alleles. Within the CD44 promoter/exon 1 region, a DNA stretch that harbours 41 CpGs wastherefore sequenced and analysed. All CpG sites, except one(CpG24) located in the promoter, were methylated in 75%of alleles of resistant NB4-LR1 cells, including the CpGs of theSp1/Egr-1 sites (featured in Figure 2b, bottom). In contrast, nomethylated CpG was found in the majority (70%) of alleles inparental NB4 cells (Figure 2b, top).
Post-translation modifications at N-terminal tails of histonesare epigenetic events known to have dramatic effects ontranscriptional regulation. The importance of histone acetylationin such regulation is now largely accepted: hyperacetylatedhistones are mainly associated with activated genomic regions,whereas, by contrast, hypoacetylation mainly results in repres-sion and silencing.28–30 We therefore wanted to know whetherthe reported defects in NB4-LR1 cells were associated with
NB4-LR1NB4
GAPDH
CD44s
NB4-LR
1
NB4
CD44
Actin
NB4 NB4-LR1
200
0
Cou
nts
100 101 104103102100 101 104103102
FL1-HFL1-H
Figure 1 Silencing of CD44 expression in NB4-LR1 cells attranscriptional and protein levels. (a) Reverse transcription (RT)–PCRanalysis of CD44 mRNAs from NB4 and NB4-LR1 cells. (b)Fluorescence-activated cell sorting (FACS) analysis of CD44 expres-sion at the NB4 and NB4-LR1 cell surface. Fluorescence intensity(fluorescein isothiocyanate, FITC) of cells was evaluated by usingmonoclonal anti-CD44 (clone J173) (filled line) or isotype controlantibodies (empty line). Representative histograms of FITC stainingrepresent an acquisition of 104 events. (c) Immunofluorescenceconfocal imaging. Cells were fixed and stained with monoclonalanti-CD44 antibody and Alexa 488-conjugated anti-mouse immuno-globulins G (IgGs). Actin filaments were visualized with Texas Red-conjugated phalloidin.
A regionB region
H4-panAcK
NB4 NB4-LR11
5
10
15
20
25
NB4-LR1NB4
H3-AcK9K14
ChI
P
Rel
ativ
e en
richm
ent
12
8
46
101214161820
ATGEgr-1/Sp1 AP-1
HpaIITIS+109- 668 HpaIIHpaIIHpaII
B region A region
% o
f met
hyla
tion
0.0
20.0
40.0
60.0
80.0
100.0
-541 -441 -341 -241 -141 -41 +61
NB4
NB4-LR1
NB4
Msp1 HpaII McrBCNon digested DNA
777 bp
NB4 NB4-LR1
Msp1 HpaII McrBC
NB4-LR1
Figure 2 DNA hypermethylation and deacetylation of histones at promoter of CD44 gene in NB4-LR1 cells. (a) Representative PCR analysis ofmethylation-sensitive restriction fragments in NB4 and NB4-LR1 cells. DNAs were digested with the methylation-insensitive (MspI) or methylation-sensitive (HpaII) restriction enzymes, or with McrBC that digests methylated alleles and then amplified using specific primers. Non-digested DNAsyields a 777-bp fragment in position �668 to þ109. (b) Top: CpG mehylation analysis by pyrosequencing in NB4 and NB4-LR1 cells. A 541 bpfragment from bisulphite-treated genomic NB4 and NB4-LR1 DNAs, covering 41 CpG sites in position �541 to þ 1, was amplified andpyrosequenced. The degree of methylation (in %) at each individual CpG site was represented. (b) Bottom: Schematic representation of the CpGisland: Methylation-sensitive HpaII restriction sites (.), transcription initiation starts (TIS, arrows) and binding sites for Egr1, Sp1 and AP-1trancription factors were indicated. The two 100 bp regions (A region and B region) amplified in chromatin immunoprecipitation assays (ChIP)experiments (c) were also represented. (c) Hypoacetylation of histone H3 and H4 at CD44 promoter in resistant NB4-LR1 cells was analysed byChIP and quantitative PCR. ChIPs were performed on chromatin from NB4 and NB4-LR1 cells with antibodies to acetylated histone H3(H3AcK9;K14) or pan-acetylated histone H4 (H4AcK5;K8;K12;K16), and amplified by quantitative PCR using two pairs of primers from twodifferent CD44 promoter regions (A region and B region). The level of enrichment of CD44 regions, relative to the input DNA (total chromatin) andto the control (Pax5 gene), was represented. A value of 1 indicates that no enrichment was detected.
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
514
Leukemia
altered acetylation of histones at the CD44 locus. Histoneacetylation at the CD44 promoter region was analysed in NB4and NB4-LR1 cells comparatively, by ChIP experiments usingantibodies against acetylated histone H3 and H4. As shown inFigure 2c, CD44 promoter regions are strongly immunopreci-pitated with anti-acetylated H3 and H4 antibodies in NB4 cells.In contrast, a very low level of acetylation is found in NB4-LR1cells for the CD44 promoter. These results suggest that atranscriptionally repressive chromatin characterized by thepresence of strongly underacetylated histones is indeed asso-ciated with methylated DNA at the CD44 gene promoter, inresistant NB4-LR1 cells.
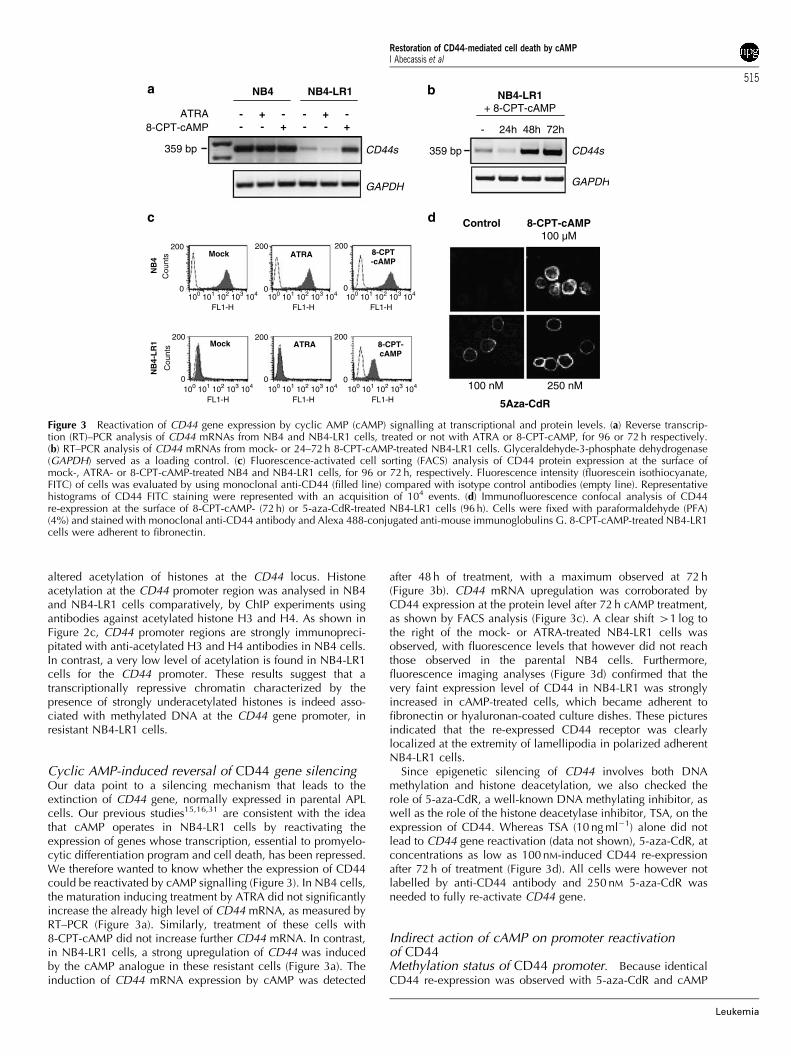
Cyclic AMP-induced reversal of CD44 gene silencingOur data point to a silencing mechanism that leads to theextinction of CD44 gene, normally expressed in parental APLcells. Our previous studies15,16,31 are consistent with the ideathat cAMP operates in NB4-LR1 cells by reactivating theexpression of genes whose transcription, essential to promyelo-cytic differentiation program and cell death, has been repressed.We therefore wanted to know whether the expression of CD44could be reactivated by cAMP signalling (Figure 3). In NB4 cells,the maturation inducing treatment by ATRA did not significantlyincrease the already high level of CD44 mRNA, as measured byRT–PCR (Figure 3a). Similarly, treatment of these cells with8-CPT-cAMP did not increase further CD44 mRNA. In contrast,in NB4-LR1 cells, a strong upregulation of CD44 was inducedby the cAMP analogue in these resistant cells (Figure 3a). Theinduction of CD44 mRNA expression by cAMP was detected
after 48 h of treatment, with a maximum observed at 72 h(Figure 3b). CD44 mRNA upregulation was corroborated byCD44 expression at the protein level after 72 h cAMP treatment,as shown by FACS analysis (Figure 3c). A clear shift 41 log tothe right of the mock- or ATRA-treated NB4-LR1 cells wasobserved, with fluorescence levels that however did not reachthose observed in the parental NB4 cells. Furthermore,fluorescence imaging analyses (Figure 3d) confirmed that thevery faint expression level of CD44 in NB4-LR1 was stronglyincreased in cAMP-treated cells, which became adherent tofibronectin or hyaluronan-coated culture dishes. These picturesindicated that the re-expressed CD44 receptor was clearlylocalized at the extremity of lamellipodia in polarized adherentNB4-LR1 cells.
Since epigenetic silencing of CD44 involves both DNAmethylation and histone deacetylation, we also checked therole of 5-aza-CdR, a well-known DNA methylating inhibitor, aswell as the role of the histone deacetylase inhibitor, TSA, on theexpression of CD44. Whereas TSA (10 ng ml�1) alone did notlead to CD44 gene reactivation (data not shown), 5-aza-CdR, atconcentrations as low as 100 nM-induced CD44 re-expressionafter 72 h of treatment (Figure 3d). All cells were however notlabelled by anti-CD44 antibody and 250 nM 5-aza-CdR wasneeded to fully re-activate CD44 gene.
Indirect action of cAMP on promoter reactivationof CD44Methylation status of CD44 promoter. Because identicalCD44 re-expression was observed with 5-aza-CdR and cAMP
GAPDH
ATRA8-CPT-cAMP
359 bp CD44s
NB4 NB4-LR1
- +- +
++-
--
- -
8-CPT-cAMP 100 µM
Control
250 nM100 nM
5Aza-CdR
CD44s
GAPDH
359 bp
- 24h 48h 72h
NB4-LR1 + 8-CPT-cAMP
NB
4
Mock ATRA 8-CPT -cAMP
NB
4-L
R1 8-CPT-
cAMPATRAMock
-
200
0100 101 102 103 104
200
0100 101 102 103 104
200
0100 101 102 103 104
FL1-H FL1-H FL1-H
200
0100 101 102 103 104
200
0100 101 102 103 104
200
0100 101 102 103 104
FL1-H FL1-H FL1-H
Cou
nts
Cou
nts
Figure 3 Reactivation of CD44 gene expression by cyclic AMP (cAMP) signalling at transcriptional and protein levels. (a) Reverse transcrip-tion (RT)–PCR analysis of CD44 mRNAs from NB4 and NB4-LR1 cells, treated or not with ATRA or 8-CPT-cAMP, for 96 or 72 h respectively.(b) RT–PCR analysis of CD44 mRNAs from mock- or 24–72 h 8-CPT-cAMP-treated NB4-LR1 cells. Glyceraldehyde-3-phosphate dehydrogenase(GAPDH) served as a loading control. (c) Fluorescence-activated cell sorting (FACS) analysis of CD44 protein expression at the surface ofmock-, ATRA- or 8-CPT-cAMP-treated NB4 and NB4-LR1 cells, for 96 or 72 h, respectively. Fluorescence intensity (fluorescein isothiocyanate,FITC) of cells was evaluated by using monoclonal anti-CD44 (filled line) compared with isotype control antibodies (empty line). Representativehistograms of CD44 FITC staining were represented with an acquisition of 104 events. (d) Immunofluorescence confocal analysis of CD44re-expression at the surface of 8-CPT-cAMP- (72 h) or 5-aza-CdR-treated NB4-LR1 cells (96 h). Cells were fixed with paraformaldehyde (PFA)(4%) and stained with monoclonal anti-CD44 antibody and Alexa 488-conjugated anti-mouse immunoglobulins G. 8-CPT-cAMP-treated NB4-LR1cells were adherent to fibronectin.
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
515
Leukemia
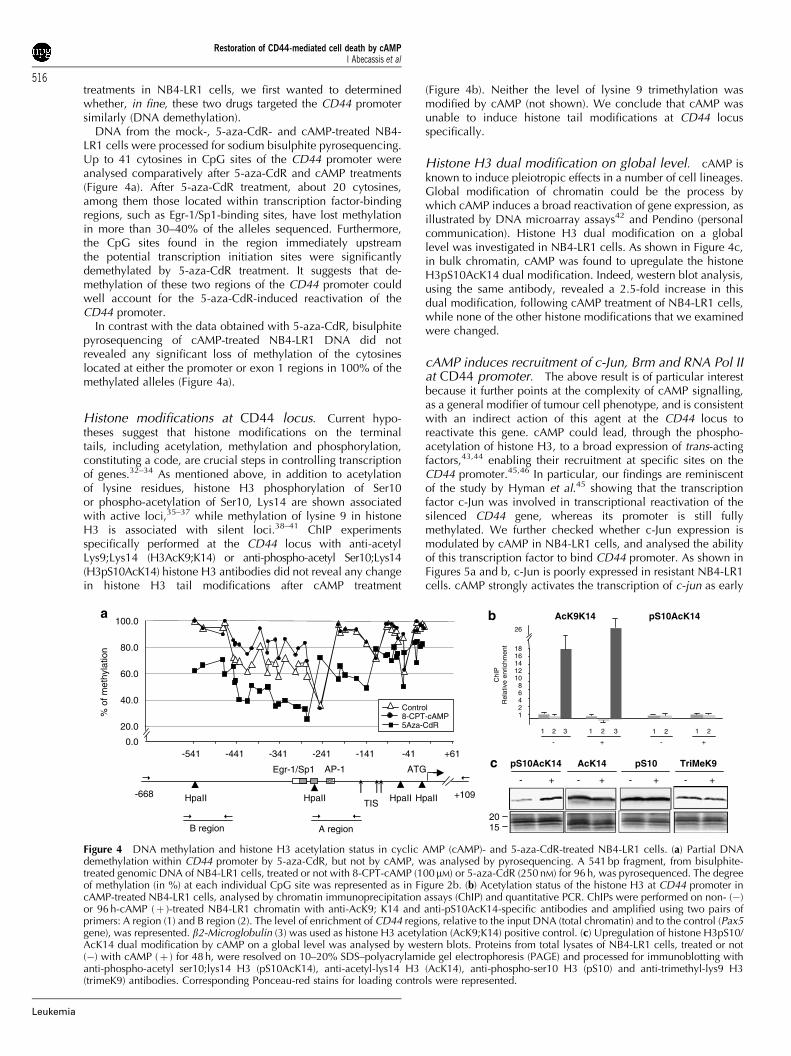
treatments in NB4-LR1 cells, we first wanted to determinedwhether, in fine, these two drugs targeted the CD44 promotersimilarly (DNA demethylation).
DNA from the mock-, 5-aza-CdR- and cAMP-treated NB4-LR1 cells were processed for sodium bisulphite pyrosequencing.Up to 41 cytosines in CpG sites of the CD44 promoter wereanalysed comparatively after 5-aza-CdR and cAMP treatments(Figure 4a). After 5-aza-CdR treatment, about 20 cytosines,among them those located within transcription factor-bindingregions, such as Egr-1/Sp1-binding sites, have lost methylationin more than 30–40% of the alleles sequenced. Furthermore,the CpG sites found in the region immediately upstreamthe potential transcription initiation sites were significantlydemethylated by 5-aza-CdR treatment. It suggests that de-methylation of these two regions of the CD44 promoter couldwell account for the 5-aza-CdR-induced reactivation of theCD44 promoter.
In contrast with the data obtained with 5-aza-CdR, bisulphitepyrosequencing of cAMP-treated NB4-LR1 DNA did notrevealed any significant loss of methylation of the cytosineslocated at either the promoter or exon 1 regions in 100% of themethylated alleles (Figure 4a).
Histone modifications at CD44 locus. Current hypo-theses suggest that histone modifications on the terminaltails, including acetylation, methylation and phosphorylation,constituting a code, are crucial steps in controlling transcriptionof genes.32–34 As mentioned above, in addition to acetylationof lysine residues, histone H3 phosphorylation of Ser10or phospho-acetylation of Ser10, Lys14 are shown associatedwith active loci,35–37 while methylation of lysine 9 in histoneH3 is associated with silent loci.38–41 ChIP experimentsspecifically performed at the CD44 locus with anti-acetylLys9;Lys14 (H3AcK9;K14) or anti-phospho-acetyl Ser10;Lys14(H3pS10AcK14) histone H3 antibodies did not reveal any changein histone H3 tail modifications after cAMP treatment
(Figure 4b). Neither the level of lysine 9 trimethylation wasmodified by cAMP (not shown). We conclude that cAMP wasunable to induce histone tail modifications at CD44 locusspecifically.
Histone H3 dual modification on global level. cAMP isknown to induce pleiotropic effects in a number of cell lineages.Global modification of chromatin could be the process bywhich cAMP induces a broad reactivation of gene expression, asillustrated by DNA microarray assays42 and Pendino (personalcommunication). Histone H3 dual modification on a globallevel was investigated in NB4-LR1 cells. As shown in Figure 4c,in bulk chromatin, cAMP was found to upregulate the histoneH3pS10AcK14 dual modification. Indeed, western blot analysis,using the same antibody, revealed a 2.5-fold increase in thisdual modification, following cAMP treatment of NB4-LR1 cells,while none of the other histone modifications that we examinedwere changed.
cAMP induces recruitment of c-Jun, Brm and RNA Pol IIat CD44 promoter. The above result is of particular interestbecause it further points at the complexity of cAMP signalling,as a general modifier of tumour cell phenotype, and is consistentwith an indirect action of this agent at the CD44 locus toreactivate this gene. cAMP could lead, through the phospho-acetylation of histone H3, to a broad expression of trans-actingfactors,43,44 enabling their recruitment at specific sites on theCD44 promoter.45,46 In particular, our findings are reminiscentof the study by Hyman et al.45 showing that the transcriptionfactor c-Jun was involved in transcriptional reactivation of thesilenced CD44 gene, whereas its promoter is still fullymethylated. We further checked whether c-Jun expression ismodulated by cAMP in NB4-LR1 cells, and analysed the abilityof this transcription factor to bind CD44 promoter. As shown inFigures 5a and b, c-Jun is poorly expressed in resistant NB4-LR1cells. cAMP strongly activates the transcription of c-jun as early
pS10AcK14 AcK14
1520
TriMeK9pS10
1 21 21 2 3 1 2 3
ChI
P
Rel
ativ
e en
richm
ent
8
1246
1012141618
26
AcK9K14 pS10AcK14
- + - +0.0
20.0
40.0
60.0
80.0
100.0
8-CPT-cAMPControl
5Aza-CdR
-541 -441 -341 -241 -141 -41 +61
% o
f met
hyla
tion
+109-668
ATGEgr-1/Sp1 AP-1
HpaIITIS HpaIIHpaIIHpaII
B region A region
-- -- ++++
Figure 4 DNA methylation and histone H3 acetylation status in cyclic AMP (cAMP)- and 5-aza-CdR-treated NB4-LR1 cells. (a) Partial DNAdemethylation within CD44 promoter by 5-aza-CdR, but not by cAMP, was analysed by pyrosequencing. A 541 bp fragment, from bisulphite-treated genomic DNA of NB4-LR1 cells, treated or not with 8-CPT-cAMP (100mM) or 5-aza-CdR (250 nM) for 96 h, was pyrosequenced. The degreeof methylation (in %) at each individual CpG site was represented as in Figure 2b. (b) Acetylation status of the histone H3 at CD44 promoter incAMP-treated NB4-LR1 cells, analysed by chromatin immunoprecipitation assays (ChIP) and quantitative PCR. ChIPs were performed on non- (�)or 96 h-cAMP (þ )-treated NB4-LR1 chromatin with anti-AcK9; K14 and anti-pS10AcK14-specific antibodies and amplified using two pairs ofprimers: A region (1) and B region (2). The level of enrichment of CD44 regions, relative to the input DNA (total chromatin) and to the control (Pax5gene), was represented. b2-Microglobulin (3) was used as histone H3 acetylation (AcK9;K14) positive control. (c) Upregulation of histone H3pS10/AcK14 dual modification by cAMP on a global level was analysed by western blots. Proteins from total lysates of NB4-LR1 cells, treated or not(�) with cAMP (þ ) for 48 h, were resolved on 10–20% SDS–polyacrylamide gel electrophoresis (PAGE) and processed for immunoblotting withanti-phospho-acetyl ser10;lys14 H3 (pS10AcK14), anti-acetyl-lys14 H3 (AcK14), anti-phospho-ser10 H3 (pS10) and anti-trimethyl-lys9 H3(trimeK9) antibodies. Corresponding Ponceau-red stains for loading controls were represented.
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
516
Leukemia

as 24 h (Figure 5a), and induces the accumulation of the proteinat 48 h (Figure 5b, top). Furthermore, cAMP causes thephosphorylation of c-Jun at both Ser63 and Ser73, a dualphosphorylation, which, located within the transactivationdomain of the protein, is known to potentiate the AP-1 activity
(Figure 5b). To determine whether the newly synthesizedc-Jun interacts with the CD44 promoter, we performed ChIPexperiments (Figure 5c). Sheared chromatins from mocked- and48 h-cAMP-treated NB4-LR1 cells were immunoprecipitatedwith anti-c-Jun antibodies and recovered DNA was subjected to
3h 24h 48h 72h
-
GAPDH
c-Jun
3h 24h 48h 72h
GAPDH
p-c-Jun (ser63/73)
8-CPT-cAMP
39kDa
39kDa
contr
ol
c-Jun
Ab
Input
contr
ol
c-Jun
Ab
Input
NB4-LR1 NB4-LR1 + 8-CPT-cAMP
CD44
GAPDH
contr
ol
c-Jun
Ab
Input
contr
ol
c-Jun
Ab
Input
NB4-LR1 NB4-LR1 + 8-CPT-cAMP
GAPDH
-
c-jun
8-CPT-cAMP
253 bp
Figure 5 Cyclic AMP (cAMP) induces the expression of c-Jun and its recruitment at the CD44 promoter in NB4-LR1 cells. (a) Reverse transcription(RT)–PCR analysis of c-jun mRNAs from mock- or 3- to 72 h-cAMP-treated NB4-LR1 cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)served as a loading control. (b) Western blot analyses of c-Jun expression and phosphorylation. Proteins from total lysates of NB4-LR1 cells, treatedor not (�) with cAMP for 3–72 h, were resolved on 10% SDS–polyacrylamide gel electrophoresis (PAGE) and processed for immunoblotting withanti-c-Jun antibody (top) or anti-Ser63/Ser73 phosphorylated c-Jun (bottom). GAPDH expression served as a loading control. (c) Chromatinimmunoprecipitation assays (ChIP) assay for interaction of c-Jun with the CD44 promoter. Equal amounts of chromatin from mock- or 48 h-cAMP-treated NB4-LR1 cells were immunoprecipitated (ChIP) with no antibody (control), or specific antibody to c-Jun (c-Jun Ab). PCR reactions werecarried out using primers encompassing the AP-1 site within the CD44 promoter (115 bp product). PCRs with input chromatin were shown aspositive control. A region of the GAPDH promoter was amplified as background control (c, bottom).
Input
contr
ol
Brm A
b
Input
contr
ol
Brm A
b
NB4-LR1 NB4-LR1 + 8-CPT-cAMP
Input
contr
ol
Brm A
b
Input
contr
ol
Brm A
b
NB4-LR1 NB4-LR1 + 8-CPT-cAMP
CD44 GAPDH
RNA Pol
II Ab
contr
ol
contr
ol
RNA Pol
II Ab
Input
Input
NB4-LR1 NB4-LR1 + 8-CPT-cAMP
CD44 Negative control
contr
ol
RNA Pol
II Ab
Input
NB4-LR1 + 8-CPT-cAMP
24h 48h 72h
8-CPT-cAMP
-
210 kDa
GAPDH
Brm
24h 48h 72h
8-CPT-cAMP
-
Brm
GAPDH
1 kb
Figure 6 Cyclic AMP (cAMP) induces the recruitment of Brm and RNA Pol II at the CD44 promoter in NB4-LR1 cells. (a) Reverse transcription(RT)–PCR analysis of Brm mRNAs from mocked- or 3 to 72 h-cAMP-treated NB4-LR1 cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)served as a loading control. (b) Western blot analyses of Brm expression. Proteins from total lysates of NB4-LR1 cells, treated or not (�) with cAMPfor 3–72 h, were resolved on 10% SDS–polyacrylamide gel electrophoresis (PAGE) and processed for immunoblotting with anti-Brm antibody.GAPDH expression served as a loading control. (c) Chromatin immunoprecipitation (ChIP) assay for interaction of Brm with the CD44 promoter.Equal amounts of chromatin from mock- or 48 h-cAMP-treated NB4-LR1 cells were immunoprecipitated (ChIP) with no antibody (control), orspecific antibody to Brm (Brm Ab). PCR reactions were carried out using primers within the CD44 promoter (487 bp product). PCRs with inputchromatin were shown as positive control. A region of the GAPDH promoter was amplified as background control (GAPDH). (d) ChIP assay forinteraction of RNA PoI II within the CD44 proximal promoter region. Equal amounts of chromatin from mock- or 48 h-cAMP-treated NB4-LR1 cellswere immunoprecipitated (ChIP) with no antibody (control), or anti-RNA Pol II (RNA Pol II Ab). PCR reactions were carried out using primersencompassing the transcription start region (297 bp product). PCRs with input chromatin were shown as positive control. Negative primers servedas background control (negative control).
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
517
Leukemia
PCR, using primers specific for a region of CD44 promoterencompassing the AP-1 site (�278/�164). As shown inFigure 5c, c-Jun interacts with the CD44 promoter in48 h-cAMP-treated NB4-LR1 cells, but not in the absence oftreatment, or in 24 h-treated cells (data not shown). It isinteresting to note that the cytosine at position �250, locatedjust upstream from the AP-1 site, is found unmethylated bypyrosequencing in 70% of the alleles in NB4-LR1 cells, treatedor not by cAMP (see in Figures 2b and 4a).
The human SWI/SNF subunit brahma (Brm) is known to be achromatin-remodelling factor. It has been recently described tocontribute to the crosstalk between transcription and RNA Pol IIprocessing and to be directly involved in the re-activation ofCD44 in a carcinoma cell line where CD44 transcription wassilenced by hypermethylation of CpG island within its promo-ter.46,47 In NB4-LR1 cells, Brm is expressed at transcriptional andprotein levels (Figures 6a and b), without modulation of itsexpression by cAMP. ChIP assays reveal that, in 48 h-cAMP-treated cells, Brm protein interacts with CD44 promoter, whereasno interaction is detectable in untreated cells (Figure 6c). Bindingof phosphorylated RNA Pol II to the proximal promoter region ofCD44, encompassing the transcription start site, is also observedin the same chromatin samples, whereas RNA Pol II is poorlypaused at the promoter region of the inactive CD44 gene inabsence of 8-CPT-cAMP treatment (Figure 6d).
Altogether, our results suggest that, in NB4-LR1 cells, cAMPleads to the recruitment of trans-acting factors at the CD44promoter, to the re-expression of CD44 gene and CD44
receptor, then, ultimately, to a functional CD44 signallingpathway. To check this hypothesis, we wish to determinewhether cAMP could restore a CD44-dependent cell deathresponse in these cells.
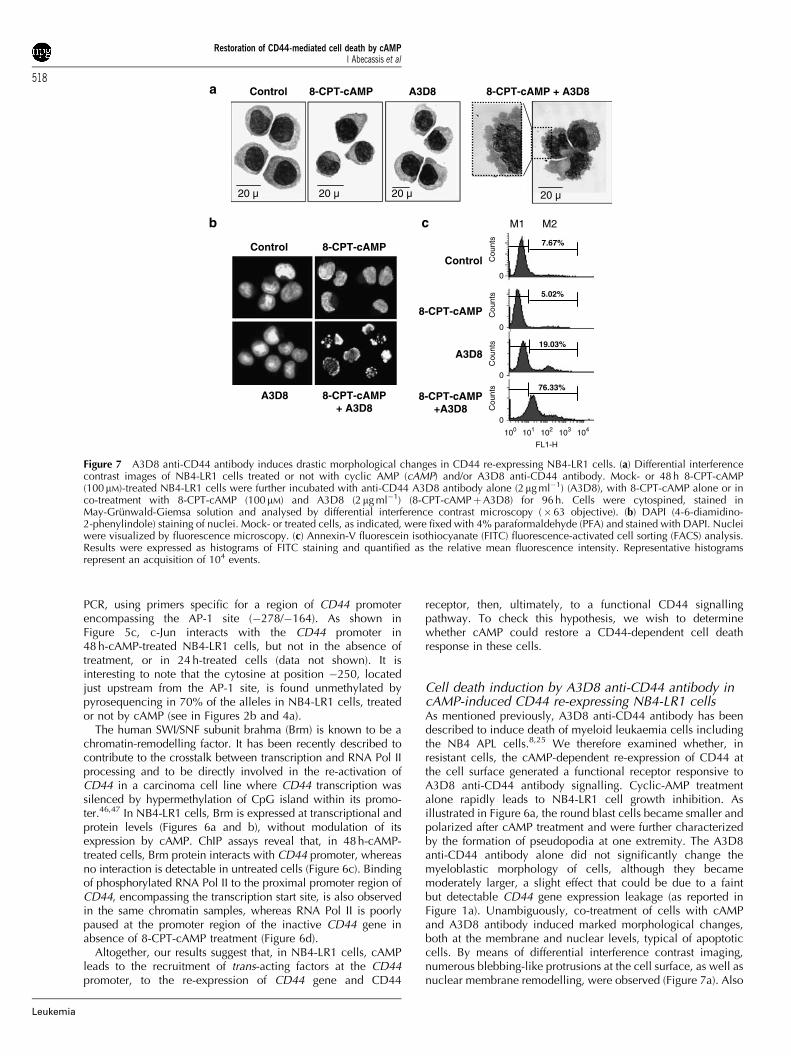
Cell death induction by A3D8 anti-CD44 antibody incAMP-induced CD44 re-expressing NB4-LR1 cellsAs mentioned previously, A3D8 anti-CD44 antibody has beendescribed to induce death of myeloid leukaemia cells includingthe NB4 APL cells.8,25 We therefore examined whether, inresistant cells, the cAMP-dependent re-expression of CD44 atthe cell surface generated a functional receptor responsive toA3D8 anti-CD44 antibody signalling. Cyclic-AMP treatmentalone rapidly leads to NB4-LR1 cell growth inhibition. Asillustrated in Figure 6a, the round blast cells became smaller andpolarized after cAMP treatment and were further characterizedby the formation of pseudopodia at one extremity. The A3D8anti-CD44 antibody alone did not significantly change themyeloblastic morphology of cells, although they becamemoderately larger, a slight effect that could be due to a faintbut detectable CD44 gene expression leakage (as reported inFigure 1a). Unambiguously, co-treatment of cells with cAMPand A3D8 antibody induced marked morphological changes,both at the membrane and nuclear levels, typical of apoptoticcells. By means of differential interference contrast imaging,numerous blebbing-like protrusions at the cell surface, as well asnuclear membrane remodelling, were observed (Figure 7a). Also
Control 8-CPT-cAMP A3D8
20 µ 20 µ 20 µ
8-CPT-cAMP + A3D8
20 µ
Control 8-CPT-cAMP
A3D8 8-CPT-cAMP + A3D8
Control
8-CPT-cAMP
A3D8
8-CPT-cAMP +A3D8
7.67%
5.02%
19.03%
76.33%
M1 M2
100 101 102 103 104
FL1-H
0
0
0
0
Cou
nts
Cou
nts
Cou
nts
Cou
nts
Figure 7 A3D8 anti-CD44 antibody induces drastic morphological changes in CD44 re-expressing NB4-LR1 cells. (a) Differential interferencecontrast images of NB4-LR1 cells treated or not with cyclic AMP (cAMP) and/or A3D8 anti-CD44 antibody. Mock- or 48 h 8-CPT-cAMP(100mM)-treated NB4-LR1 cells were further incubated with anti-CD44 A3D8 antibody alone (2mg ml�1) (A3D8), with 8-CPT-cAMP alone or inco-treatment with 8-CPT-cAMP (100mM) and A3D8 (2mg ml�1) (8-CPT-cAMPþA3D8) for 96 h. Cells were cytospined, stained inMay-Grunwald-Giemsa solution and analysed by differential interference contrast microscopy (�63 objective). (b) DAPI (4-6-diamidino-2-phenylindole) staining of nuclei. Mock- or treated cells, as indicated, were fixed with 4% paraformaldehyde (PFA) and stained with DAPI. Nucleiwere visualized by fluorescence microscopy. (c) Annexin-V fluorescein isothiocyanate (FITC) fluorescence-activated cell sorting (FACS) analysis.Results were expressed as histograms of FITC staining and quantified as the relative mean fluorescence intensity. Representative histogramsrepresent an acquisition of 104 events.
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
518
Leukemia

nuclei completely lost their round homogenous appearance andpresented profound modifications before cell structures werefinally dismantled. DAPI staining of the nuclei further showed achromatin gathering at the periphery of the nucleus or afragmented morphology of nuclear bodies (Figure 7b). Inconcordance with these data, FACS analysis of Annexin-Vexpression, a marker of apoptosis, showed a significant positivestaining of NB4-LR1 cells co-treated with 8-CPT-cAMP andA3D8 (Figure 7c), with a fluorescence profile comparable to thatobserved in A3D8-treated parental NB4 cells (data not shown).The slight effect observed with A3D8 alone could be due, asmentioned above, to the faint basal expression of CD44 in thesecells.
These results demonstrate that cAMP induces the expressionof a functional CD44 receptor at the surface of resistant cells,which is able to trigger cell death signalling induced by theA3D8 antibody, as in NB4 APL cells.
Conclusion
Here, we report for the first time that the ATRA-maturation-resistant NB4-LR1 cells are additionally resistant to CD44-dependent cell death. The molecular bases of the CD44-deathunresponsiveness and its rescue have been deciphered:
(1) These cells are deficient in the expression of CD44(Figure 1), an adhesion and signalling receptor known tobe a key actor in the maturation of myeloid progenitors.48
(2) The defect involves alterations in DNA and chromatinstructure, that is, hypermethylation of the 50-regionCpG island and de-acetylation of histones associated withthe promoter site of the CD44 gene, two epigeneticmechanisms, which could be responsible for CD44 generepression. This result is supported by the finding that themethylating inhibitor 5-aza-CdR restores CD44 expressionand this re-activation is associated with partial demethyla-tion of the promoter (Figure 4a).
(3) cAMP, like the demethylating agent 5-aza-CdR, restorestranscriptional competence of the highly methylatedCD44 gene, but importantly it occurred despite that theCD44 promoter remained highly methylated.
(4) cAMP signalling could favour broad chromatin changes (H3dual modification H3pS10AcK14) and transcription patterns(c-Jun, CD44), benefiting anti-tumour signalling (cell death).
This work has revealed a new and unique phenotypic featureof NB4-LR1 cells: a double (ATRA-maturation and CD44-dependent death) resistant cell line, whose defects in thetriggering of two distinct biological responses are cured by thesame signal (cAMP). As we reported earlier, NB4-LR1 cells showdefects in this crosstalk and pharmacological cAMP signalling isnecessary for retinoid induction of maturation.14–16 At present,little is known about the role of cAMP and its crosstalk withretinoids to induce the maturation and the apoptosis of APL. Themechanisms by which cAMP acts seem to be complex and mayinvolve different signalling pathways and targets, includingtargeting transcription effectors, such as CREB, CBP300, PML-RARaor RXR receptors by direct or indirect phosphorylation,18–20,49 orchanges in nuclear bodies17,50 or late and indirect re-expressionof a large list of genes,43,51,52 including c-Jun and CD44.
Given the reported features shared by cAMP/retinoid (cellmaturation) and cAMP/CD44 (cell death) cooperation, namely,the biological and biochemical effects are late and need asustained signalling to develop, it is more clear that themechanisms involved do not depend on a discrete set of
cAMP/PKA targets, but rather result from a broad transcriptionalreprogrammation of a multi-resistant tumour cell.
More specifically, the finding of a new cooperation pathway,that is, cAMP/anti-CD44 co-signalling, which is able to inducedeath of an otherwise maturation-resistant tumour cell, could beof prime importance for the development of new combinationtherapies in resistant APL.
Acknowledgements
We are grateful to Monica Pettersson (Biotage BA, Sweden) forpyrosequencing assays. We thank Niclas Setterblad and FredericBrau for performing confocal microscopy analysis (Serviced’Imagerie Cellulaire, IUH, Hopital Saint-Louis). This work wassupported by grants from ‘Ligue Nationale contre le Cancer’,‘Association pour la Recherche contre le Cancer’, ‘Fondation pourla Recherche Medicale’ and ‘Fondation de France’. I Abecassisreceived fellowships from MRT, ‘Association pour la Recherchecontre le Cancer’ (ARC) and ‘Societe Francaise d’Hematologie’.
References
1 Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI.Genomic structure of DNA encoding the lymphocyte homingreceptor CD44 reveals at least 12 alternatively spliced exons. ProcNatl Acad Sci USA 1992; 89: 12160–12164.
2 Ghaffari S, Smadja-Joffe F, Oostendorp R, Levesque JP, DoughertyG, Eaves A et al. CD44 isoforms in normal and leukemichematopoiesis. Exp Hematol 1999; 27: 978–993.
3 Lesley J, Hyman R, Kincade PW. CD44 and its interaction withextracellular matrix. Adv Immunol 1993; 54: 271–335.
4 Herrlich P, Morrison H, Sleeman J, Orian-Rousseau V, Konig H,Weg-Remers S et al. CD44 acts both as a growth- and invasiveness-promoting molecule and as a tumor-suppressing cofactor. Ann NYAcad Sci 2000; 910: 106–118; discussion 118–120.
5 Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. CD44 incancer. Crit Rev Clin Lab Sci 2002; 39: 527–579.
6 Ponta H, Sherman L, Herrlich PA. CD44: from adhesion moleculesto signalling regulators. Nat Rev Mol Cell Biol 2003; 4: 33–45.
7 Charrad RS, Li Y, Delpech B, Balitrand N, Clay D, Jasmin C et al.Ligation of the CD44 adhesion molecule reverses blockage ofdifferentiation in human acute myeloid leukemia. Nat Med 1999;5: 669–676.
8 Charrad RS, Gadhoum Z, Qi J, Glachant A, Allouche M, Jasmin Cet al. Effects of anti-CD44 monoclonal antibodies on differentiationand apoptosis of human myeloid leukemia cell lines. Blood 2002;99: 290–299.
9 Lanotte M, Martin-Thouvenin V, Najman S, Balerini P, Valensi F,Berger R. NB4, a maturation inducible cell line with t(15;17)marker isolated from a human acute promyelocytic leukemia (M3).Blood 1991; 77: 1080–1086.
10 Allouche M, Charrad RS, Bettaieb A, Greenland C, Grignon C,Smadja-Joffe F. Ligation of the CD44 adhesion molecule inhibitsdrug-induced apoptosis in human myeloid leukemia cells. Blood2000; 96: 1187–1190.
11 Gadhoum Z, Leibovitch MP, Qi J, Dumenil D, Durand L, Leibovitch Set al. CD44: a new means to inhibit acute myeloid leukemia cellproliferation via p27Kip1. Blood 2004; 103: 1059–1068.
12 Gallagher RE. Retinoic acid resistance in acute promyelocyticleukemia. Leukemia 2002; 16: 1940–1958.
13 Melnick A, Licht JD. Deconstructing a disease: RARalpha, itsfusion partners, and their roles in the pathogenesis of acutepromyelocytic leukemia. Blood 1999; 93: 3167–3215.
14 Benoit G, Roussel M, Pendino F, Segal-Bendirdjian E, Lanotte M.Orchestration of multiple arrays of signal cross-talk and combina-torial interactions for maturation and cell death: another vision oft(15;17) preleukemic blast and APL-cell maturation. Oncogene2001; 20: 7161–7177.
15 Ruchaud S, Duprez E, Gendron MC, Houge G, Genieser HG,Jastorff B et al. Two distinctly regulated events, priming andtriggering, during retinoid-induced maturation and resistance of
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
519
Leukemia

NB4 promyelocytic leukemia cell line. Proc Natl Acad Sci USA1994; 91: 8428–8432.
16 Roussel MJ, Lanotte M. Maturation sensitive and resistant t(15;17)NB4 cell lines as tools for APL physiopathology: nomenclature ofcells and repertory of their known genetic alterations andphenotypes. Oncogene 2001; 20: 7287–7291.
17 Duprez E, Lillehaug JR, Naoe T, Lanotte M. cAMP signalling isdecisive for recovery of nuclear bodies (PODs) during maturationof RA-resistant t(15;17) promyelocytic leukemia NB4 cells expres-sing PML-RAR alpha. Oncogene 1996; 12: 2451–2459.
18 Zhu Q, Zhang JW, Zhu HQ, Shen YL, Flexor M, Jia PM et al.Synergic effects of arsenic trioxide and cAMP during acutepromyelocytic leukemia cell maturation subtends a novel signalingcross-talk. Blood 2002; 99: 1014–1022.
19 Boisvert FM, Kruhlak MJ, Box AK, Hendzel MJ, Bazett-Jones DP.The transcription coactivator CBP is a dynamic component of thepromyelocytic leukemia nuclear body. J Cell Biol 2001; 152:1099–1106.
20 Parrella E, Gianni M, Cecconi V, Nigro E, Barzago MM, RambaldiA et al. Phosphodiesterase IV inhibition by piclamilast potentiatesthe cytodifferentiating action of retinoids in myeloid leukemiacells. Cross-talk between the cAMP and the retinoic acid signalingpathways. J Biol Chem 2004; 279: 42026–42040.
21 Kamashev D, Vitoux D, De The H. PML-RARA-RXRoligomers mediate retinoid and rexinoid/cAMP cross-talk in acutepromyelocytic leukemia cell differentiation. J Exp Med 2004; 199:1163–1174.
22 Razin A. CpG methylation, chromatin structure and genesilencing-a three-way connection. EMBO J 1998; 17: 4905–4908.
23 Baker EK, El-Osta A. The rise of DNA methylation and theimportance of chromatin on multidrug resistance in cancer. ExpCell Res 2003; 290: 177–194.
24 Yamada Y, Watanabe H, Miura F, Soejima H, Uchiyama M,Iwasaka T et al. A comprehensive analysis of allelic methylationstatus of CpG islands on human chromosome 21q. Genome Res2004; 14: 247–266.
25 Maquarre E, Artus C, Gadhoum Z, Jasmin C, Smadja-Joffe F,Robert-Lezenes J. CD44 ligation induces apoptosis via caspase-and serine protease-dependent pathways in acute promyelocyticleukemia cells. Leukemia 2005; 19: 2296–2303.
26 Shtivelman E, Bishop JM. Expression of CD44 is repressed inneuroblastoma cells. Mol Cell Biol 1991; 11: 5446–5453.
27 Gardiner-Garden M, Frommer M. CpG islands in vertebrategenomes. J Mol Biol 1987; 196: 261–282.
28 Berger SL. Histone modifications in transcriptional regulation. CurrOpin Genet Dev 2002; 12: 142–148.
29 Eberharter A, Becker PB. Histone acetylation: a switch betweenrepressive and permissive chromatin. Second in review series onchromatin dynamics. EMBO Rep 2002; 3: 224–229.
30 Grunstein M. Histone acetylation in chromatin structure andtranscription. Nature 1997; 389: 349–352.
31 Benoit G, Altucci L, Flexor M, Ruchaud S, Lillehaug J,Raffelsberger W et al. RAR-independent RXR signalinginduces t(15;17) leukemia cell maturation. EMBO J 1999; 18:7011–7018.
32 Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk.Curr Opin Cell Biol 2003; 15: 172–183.
33 Peterson CL, Laniel MA. Histones and histone modifications. CurrBiol 2004; 14: R546–R551.
34 Strahl BD, Allis CD. The language of covalent histone modifica-tions. Nature 2000; 403: 41–45.
35 Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM,Allis CD. Synergistic coupling of histone H3 phosphorylation and
acetylation in response to epidermal growth factor stimulation.Mol Cell 2000; 5: 905–915.
36 Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD et al.Phosphorylation of serine 10 in histone H3 is functionally linked invitro and in vivo to Gcn5-mediated acetylation at lysine 14. MolCell 2000; 5: 917–926.
37 Nowak SJ, Corces VG. Phosphorylation of histone H3 correlateswith transcriptionally active loci. Genes Dev 2000; 14:3003–3013.
38 Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T.Methylation of histone H3 lysine 9 creates a binding site for HP1proteins. Nature 2001; 410: 116–120.
39 Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI.Role of histone H3 lysine 9 methylation in epigeneticcontrol of heterochromatin assembly. Science 2001; 292:110–113.
40 Zhang Y, Reinberg D. Transcription regulation by histonemethylation: interplay between different covalent modificationsof the core histone tails. Genes Dev 2001; 15: 2343–2360.
41 Kouzarides T. Histone methylation in transcriptional control. CurrOpin Genet Dev 2002; 12: 198–209.
42 Zambon AC, Zhang L, Minovitsky S, Kanter JR, Prabhakar S,Salomonis N et al. Gene expression patterns define key transcrip-tional events in cell-cycle regulation by cAMP and protein kinaseA. Proc Natl Acad Sci USA 2005; 102: 8561–8566.
43 Li J, Guo Y, Schroeder FA, Youngs RM, Schmidt TW, Ferris C et al.Dopamine D2-like antagonists induce chromatin remodeling instriatal neurons through cyclic AMP-protein kinase A and NMDAreceptor signaling. J Neurochem 2004; 90: 1117–1131.
44 Salvador LM, Park Y, Cottom J, Maizels ET, Jones JC, Schillace RVet al. Follicle-stimulating hormone stimulates protein kinase A-mediated histone H3 phosphorylation and acetylation leading toselect gene activation in ovarian granulosa cells. J Biol Chem2001; 276: 40146–40155.
45 Hyman R. Lack of a consistent relationship between demethylationof the CD44 promoter and CD44 expression. Immunogenetics2002; 53: 914–924.
46 Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M,Knudsen ES et al. SWI/SNF chromatin-remodelling factors inducechanges in DNA methylation to promote transcriptional activation.Cancer Res 2005; 65: 3542–3547.
47 Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunitBrm is a regulator of alternative splicing. Nat Struct Mol Biol 2006;13: 22–29.
48 Schmits R, Filmus J, Gerwin N, Senaldi G, Kiefer F, Kundig T et al.CD44 regulates hematopoietic progenitor distribution, granulomaformation, and tumorigenicity. Blood 1997; 90: 2217–2233.
49 Rochette-Egly C. Dynamic combinatorial networks in nuclearreceptor-mediated transcription. J Biol Chem 2005; 280:32565–32568.
50 Gianni M, Terao M, Norio P, Barbui T, Rambaldi A,Garattini E. All-trans retinoic acid and cyclic adenosine mono-phosphate cooperate in the expression of leukocyte alkalinephosphatase in acute promyelocytic leukemia cells. Blood 1995;85: 3619–3635.
51 Kim J, Jia L, Stallcup MR, Coetzee GA. The role of protein kinase Apathway and cAMP responsive element-binding protein inandrogen receptor-mediated transcription at the prostate-specificantigen locus. J Mol Endocrinol 2005; 34: 107–118.
52 Doucas V, Tini M, Egan DA, Evans RM. Modulation of CREBbinding protein function by the promyelocytic (PML) oncoproteinsuggests a role for nuclear bodies in hormone signaling. Proc NatlAcad Sci USA 1999; 96: 2627–2632.
Restoration of CD44-mediated cell death by cAMPI Abecassis et al
520
Leukemia
Related Documents











