Multiprotein Complexes p. 1 BCMP201, 2004 Protein-Protein Interactions References Tsai, C-J., Lin, S. L., Wolfson, H.J., and Nussinov, R. 1997. Studies of protein-protein interfaces: a statistical analysis of the hydrophobic effect. Protein Science 6, 53-64. Janin, J., Miller, S., and Chothia, C. 1988. Surface, Subunit Interfaces, and Interior of Oligomeric Proteins. J. Mol. Biol. 204, 155-164. Some Rationalizations for Multimeric Proteins: • Multienzyme Complexes o Speed reactions o Sequester reactive intermediates o Coordinately regulate activities of subunits (allosteric effects) o Cooperative binding of subunits facilitates rapid assembly/disassembly in response to changed expression levels • Topologically Restricted (Enclosed) Structures o Barrels – proteasome, GroEL chaperone o Rings – sliding clamps, helicases, pores o Shells – virus particles • Filamentous Proteins (actin, tubulin, RecA) o High tensile strength o Dynamic regulation of length and stability in response to NTP hydrolysis Characteristics of Protein-Protein Interaction Surfaces • The principles governing the interactions of proteins are not fully understood. It would be of great value to be able to predict the sites and functional importance of residues that mediate protein-protein interactions, starting with the structures individual proteins and/or their amino acid sequences. • The “hydrophobic effect” is considered to be the main driving force of protein folding and it is probably a major factor in protein-protein interactions (see below). • Protein-protein interaction surfaces are as tightly packed as the interiors of native protein folds (Walls, P.H. & Sternberg, M.J.E. 1992. J. Mol. Biol. 228, 277-297.; Jones, S., and Thornton, J.M. 1996. Proc. Nat’l Acad. Sci. USA 93, 13-20.). The exclusion of water from protein interfaces dictates a close complementarity of the interacting surfaces. • Small molecules tend to bind to cavities, or areas of increased surface roughness on proteins. However, protein interaction surfaces tend to be flat. • Shape and size of the interface correlates with the stability of the interaction – stable/constitutive protein complexes tend to have large interaction surfaces (e.g.,

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Multiprotein Complexes p. 1 BCMP201, 2004
Protein-Protein Interactions References
Tsai, C-J., Lin, S. L., Wolfson, H.J., and Nussinov, R. 1997. Studies of protein-protein interfaces: a statistical analysis of the hydrophobic effect. Protein Science 6, 53-64.
Janin, J., Miller, S., and Chothia, C. 1988. Surface, Subunit Interfaces, and Interior of Oligomeric Proteins. J. Mol. Biol. 204, 155-164.
Some Rationalizations for Multimeric Proteins:
• Multienzyme Complexes
o Speed reactions
o Sequester reactive intermediates
o Coordinately regulate activities of subunits (allosteric effects)
o Cooperative binding of subunits facilitates rapid assembly/disassembly in response to changed expression levels
• Topologically Restricted (Enclosed) Structures
o Barrels – proteasome, GroEL chaperone o Rings – sliding clamps, helicases, pores o Shells – virus particles
• Filamentous Proteins (actin, tubulin, RecA)
o High tensile strength o Dynamic regulation of length and stability in response to NTP hydrolysis
Characteristics of Protein-Protein Interaction Surfaces
• The principles governing the interactions of proteins are not fully understood. It would be of great value to be able to predict the sites and functional importance of residues that mediate protein-protein interactions, starting with the structures individual proteins and/or their amino acid sequences.
• The “hydrophobic effect” is considered to be the main driving force of protein folding and it is probably a major factor in protein-protein interactions (see below).
• Protein-protein interaction surfaces are as tightly packed as the interiors of native protein folds (Walls, P.H. & Sternberg, M.J.E. 1992. J. Mol. Biol. 228, 277-297.; Jones, S., and Thornton, J.M. 1996. Proc. Nat’l Acad. Sci. USA 93, 13-20.). The exclusion of water from protein interfaces dictates a close complementarity of the interacting surfaces.
• Small molecules tend to bind to cavities, or areas of increased surface roughness on proteins. However, protein interaction surfaces tend to be flat.
• Shape and size of the interface correlates with the stability of the interaction – stable/constitutive protein complexes tend to have large interaction surfaces (e.g.,

Multiprotein Complexes p. 2 BCMP201, 2004
>1500-3000 Å2 buried surface area from both subunits vs. roughly half this amount buried by transiently interacting proteins).
Stable interfaces often have a convoluted shape with the subunits more intertwined. The surfaces of proteins that interact transiently are relatively flat (e.g., antigen-antibody combining sites). Is a flat binding interface desireable for the design of drugs that could be used to disrupt protein interactions in vivo?
• Tsai and Nussinov (1997) studied protein interfaces in 376 structurally non-redundant complexes of proteins (Janin et al. 1988. reached similar conclusions from a smaller dataset of oligomeric proteins):
o Charged residues highly preferred on the surface vs. the buried core of proteins (no surprise here). Polar residues somewhat more abundant on surface than core – about 2:1 preference for burying hydrophobic vs. hydrophilic residues.
o The amino acid composition of protein interaction surfaces mirrors the overall amino acid composition of these proteins.
o Hydrophobic residues predominate in both the surfaces buried in protein interactions and in the buried core of protein monomers. However, interaction surfaces are considerably more polar than the protein core. Charged and polar residues are far more common at subunit interfaces, esp. Arg, Lys, Glu, and Gln. Proline is overrepresented at subunit interfaces versus the interior.
o A comparison of exposed surface residues with those buried in protein interaction surfaces is more revealing. Interaction surfaces are enriched for Leu, Val, Tyr, and Phe. The distribution of charged and polar residues on the surfaces of proteins is biased - these residues are more prevalent in the exposed surfaces and less frequently found in interacting surfaces. From this, we can infer that although the hydrophobic effect is an important contributor to the strength of protein-protein interactions, it plays a larger role in stabilizing the native fold of each subunit than in the interactions between protein chains. (What type of protein interactions [stable vs. transient] are likely to be more dominated by the hydrophobic effect? Why?
• Specificity of protein-protein interactions: Based on what you know about these interfaces, what factors could contribute to the specificity of macromolecular interactions?
• Rate of protein interactions: If two proteins can interact only in a specific orientation, how could their association rate be increased beyond that achieved by random collisions of subunits?

Multiprotein Complexes p. 3 BCMP201, 2004
Hot Spots for Protein Interactions Bogan, A.A., and Thorn, K.S. 1998. Anatomy of Hot Spots in Protein Interfaces. J. Mol. Biol. 280, 1-9.
• The residues located at protein interfaces do not make equal energetic contributions to protein-protein interactions. The results of extensive mutagenesis of some protein interfaces suggests that the energy landscape is very uneven. Single residues can contribute a large portion of the binding energy. These dominant residues are “hot spots” of protein-protein interactions.
• Although the extent of buried surface in various protein interfaces correlates with the molecular weights of the interacting proteins, the binding strength does not correlate with the size of the buried interface. Would you expect this behavior if the hydrophobic effect is the primarily determinant of the strength of protein-protein interactions?
• Bogan and Thorn compiled a database of alanine-scanning mutations at protein interfaces, and defined hot spots as those residues when substituted with alanine that have a large effect (∆∆ G ≥ 2 kcal mol-1) on binding energy.
• Hot spots are generally located near the center of protein-protein interfaces, away from solvent. However, many other residues that are occluded from solvent are not hot spots.
• Since most protein (transient) interaction surfaces are fairly flat and lacking invaginations or pockets, the exclusion of a hot spot residue from water requires a “seal” formed by the surrounding residues of the protein interface.
• The residues most frequently forming a hotspot are tryptophan (21%), arginine (13 %), and tyrosine (12 %). What properties of these residues might explain their overrepresentation at the critical positions of protein interfaces?
Other Factors That Contribute to the Strength and Specificity of Protein-Protein Interactions
• Bound water molecules are part of many (most?) protein-protein interfaces. In crystal structures, they participate in hydrogen bonding networks that bridge between subunits. In NMR studies, interfacial waters have been shown to have short residence times, with interfacial waters exchanging rapidly with bulk solvent (see Janin, J. 1999. Structure 7, R277-R279).
• Protein interaction surfaces may be partly or wholly disordered in the absence of a partner. The energetic consequences of coupled folding and binding can contribute to specificity.
Evolution of Multimeric Proteins – A Domain Swapping Hypothesis
Bennett, M.J., Sclunegger, M.P., and Eisenberg, D. 1995. 3D Domain Swapping: A Mechanism for Oligomer Assembly. Protein Science 4, 2455-2468.
Newcomer, M.E. 2002. Protein Folding and Three-Dimensional Domain Swapping: A Strained Relationship? Current Opinion in Structural Biology 12, 48-53.

Multiprotein Complexes p. 4 BCMP201, 2004
How did multimeric proteins evolve from simple precursors? • The “sticky billiard ball” model seems unlikely. Domain swapping creates
proximity that allows specific interactions to gradually evolve through multiple amino acid substitutions.
• Essential elements of domain swapping: o 1) formation of open hinge conformation o 2) docking of two proteins o 3) evolution of the protein interface
Example – Swapped domains of a RnaseA dimer:
Liu, Y., Hart, J., Schlunegger, M.P., and Eisenberg, D. 1998. The crystal structure of a 3D domain-swapped dimer of RnaseA at 2.1 Å resolution. Proc. Natl. Acad. Sci. USA 95, 3437-3442.

Multiprotein Complexes p. 5 BCMP201, 2004
Predicting Protein-Protein Interactions: Detecting protein function and protein-protein interactions from genome sequences. Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. 1999. Science 285, 751-753.
Guerois, R., Nielsen, J.E., and Serrano, L. 2002. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J. Mol. Biol. 320, 369-387.
Kortemme, T., and Baker, D. 2002. A Simple Physical Model for Binding Energy Hot Spots in Protein-Protein Complexes. Proc. Nat’l. Acad. Sci. USA 99, 14116-14121.
• Rosetta Stone Hypothesis(Marcotte&Eisenberg):

Multiprotein Complexes p. 6 BCMP201, 2004
Multiprotein Complexes: The β-Clamp Loader – A Molecular Monkey Wrench Reviews
Ellison, V., and Stillman, B. 2001. Opening of the Clamp: An Intimate View of an ATP-Driven Biological Machine. Cell 106, 655-660.
Bloom, L.B., and Goodman, M.F. 2001. A Sliding Clamp Monkey Wrench. Nature Struct. Biol. 8, 829-831.
Neuwald, A.F., Araind, L., Spouge, J.L., and Koonin, E.V. 1999. AAA+: A Class of Chaperone-like ATPases Associated with the Assembly, Operation, and Disassembly of Protein Complexes. 1999. Genome Res. 9, 27-43.
Replicative DNA polymerases are highly processive:
• limited window of time for replication. • different mechanisms of processivity for different polymerases.
o Phage T7 DNA polymerase – E. coli thioredoxin
o Herpes Simplex Virus DNA polymerase - UL42 protein
o E. coli DNA pol III, phage T4 DNA pol, mammalian replicative pols – sliding clamps
PCNA
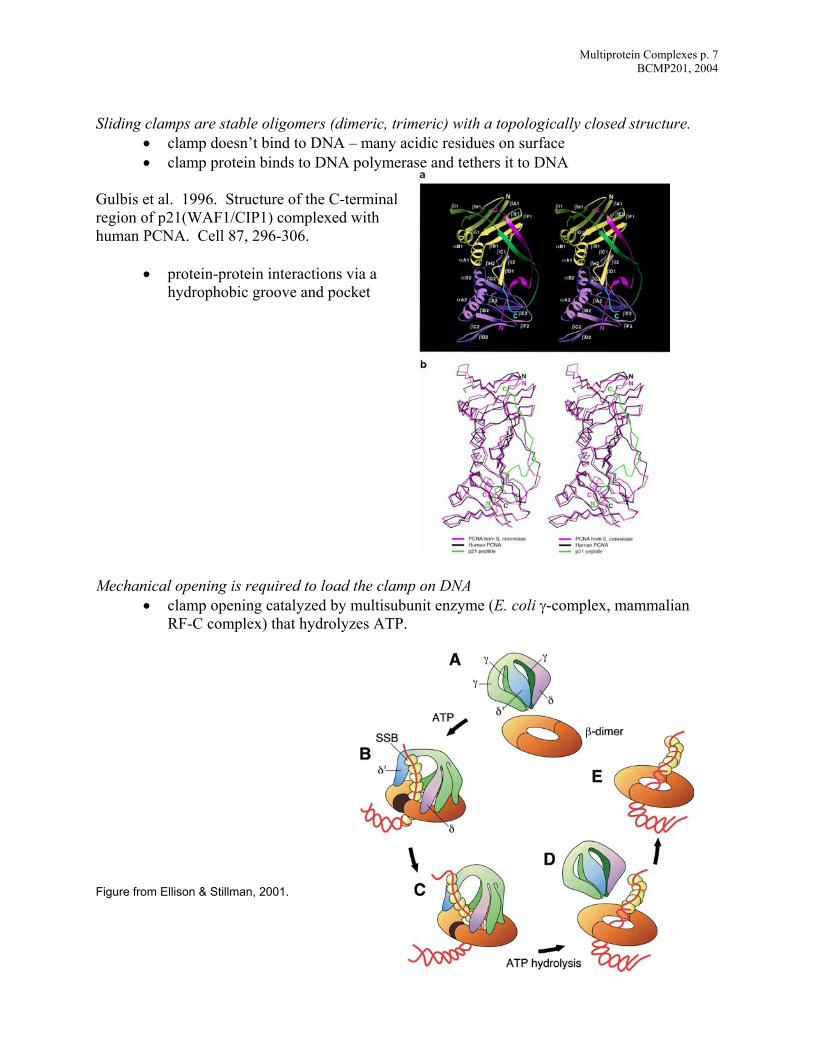
Multiprotein Complexes p. 7 BCMP201, 2004
Sliding clamps are stable oligomers (dimeric, trimeric) with a topologically closed structure.
• clamp doesn’t bind to DNA – many acidic residues on surface • clamp protein binds to DNA polymerase and tethers it to DNA
Gulbis et al. 1996. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 87, 296-306.
• protein-protein interactions via a hydrophobic groove and pocket
Mechanical opening is required to load the clamp on DNA
• clamp opening catalyzed by multisubunit enzyme (E. coli γ-complex, mammalian RF-C complex) that hydrolyzes ATP.
Figure from Ellison & Stillman, 2001.
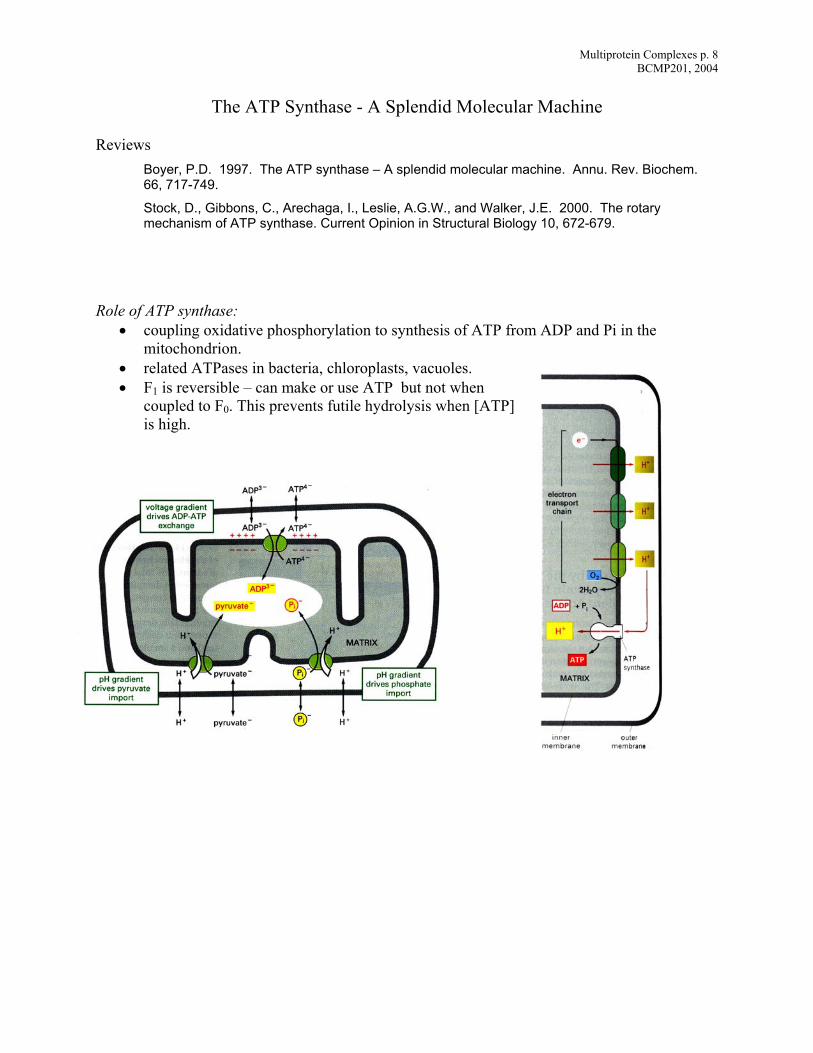
Multiprotein Complexes p. 8 BCMP201, 2004
The ATP Synthase - A Splendid Molecular Machine Reviews
Boyer, P.D. 1997. The ATP synthase – A splendid molecular machine. Annu. Rev. Biochem. 66, 717-749.
Stock, D., Gibbons, C., Arechaga, I., Leslie, A.G.W., and Walker, J.E. 2000. The rotary mechanism of ATP synthase. Current Opinion in Structural Biology 10, 672-679.
Role of ATP synthase:
• coupling oxidative phosphorylation to synthesis of ATP from ADP and Pi in the mitochondrion.
• related ATPases in bacteria, chloroplasts, vacuoles. • F1 is reversible – can make or use ATP but not when
coupled to F0. This prevents futile hydrolysis when [is high.
ATP]

Multiprotein Complexes p. 9 BCMP201, 2004
Bilobed architecture of F0F1-ATP synthase. (figure from Elston, T., Wang, H., and Oster, G. 1998. Energy Transduction in ATP synthase. Nature 391, 510-513.)
• can separate globular F1 head (α3β3γδε) from F0 membrane component (ab2c12 for E. coli ATPase).
Cloning and sequencing of the α- and β-subunits of bovine heart ATP synthase:
Conserved sequences of α- and β-subunits suggests structural similarity.
Two conserved sequence motifs present in a vast number of NTP binding proteins (Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J. 1982. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases, and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945-951):
• Walker Type I (“A box”) sequence – GX4GK[T,S] • Walker Type II (“B box”) sequence – Xφ4DEX
The case for rotary catalysis by F1-ATPase.
• positive cooperativity of the catalytic activities (ATP hydrolysis) of 3 active sites. • negative cooperativity of substrate binding to these sites. • γ- subunit unlikely to contact α3β3 symmetrically • crosslinking of γ-subunit to β-subunit blocks activity
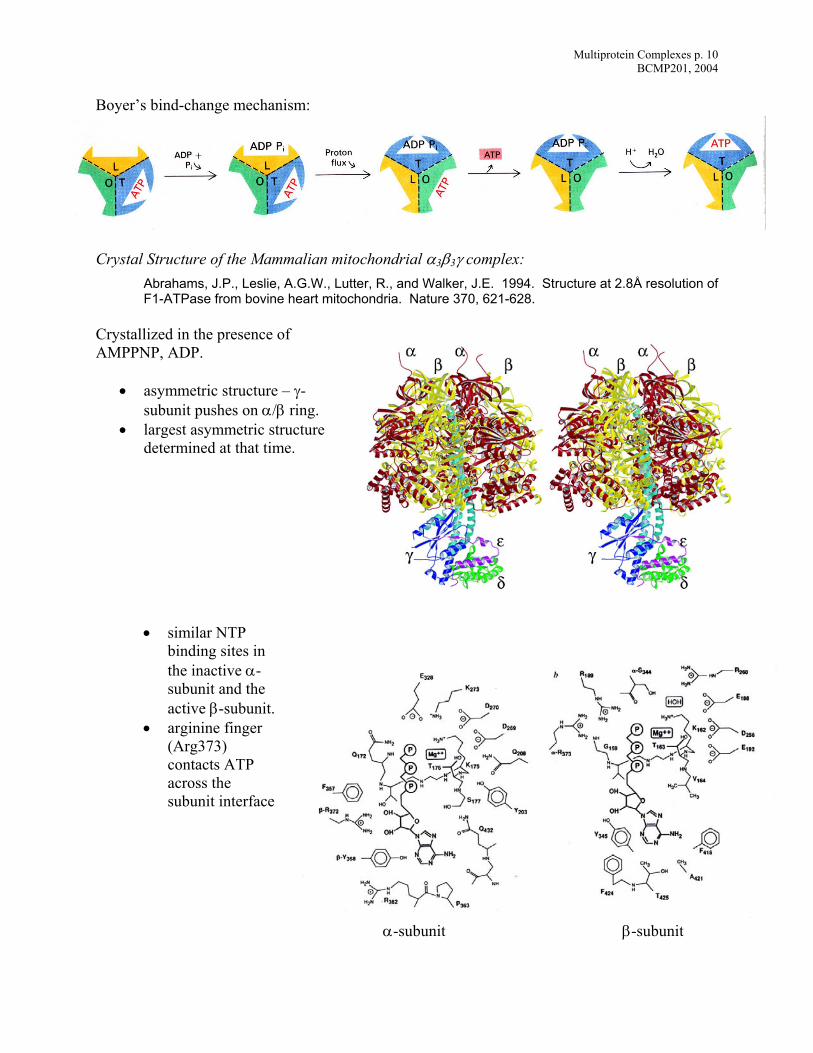
Multiprotein Complexes p. 10 BCMP201, 2004
Boyer’s bind-change mechanism:
Crystal Structure of the Mammalian mitochondrial α3β3γ complex:
Abrahams, J.P., Leslie, A.G.W., Lutter, R., and Walker, J.E. 1994. Structure at 2.8Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621-628.
Crystallized in the presence of AMPPNP, ADP.
• asymmetric structure – γ-subunit pushes on α/β ring.
• largest asymmetric structure determined at that time.
• similar NTP binding sites in the inactive α-subunit and the active β-sarginine f
ubunit. • inger
TP
rface
α-subunit β-subunit
(Arg373) contacts Aacross the subunit inte
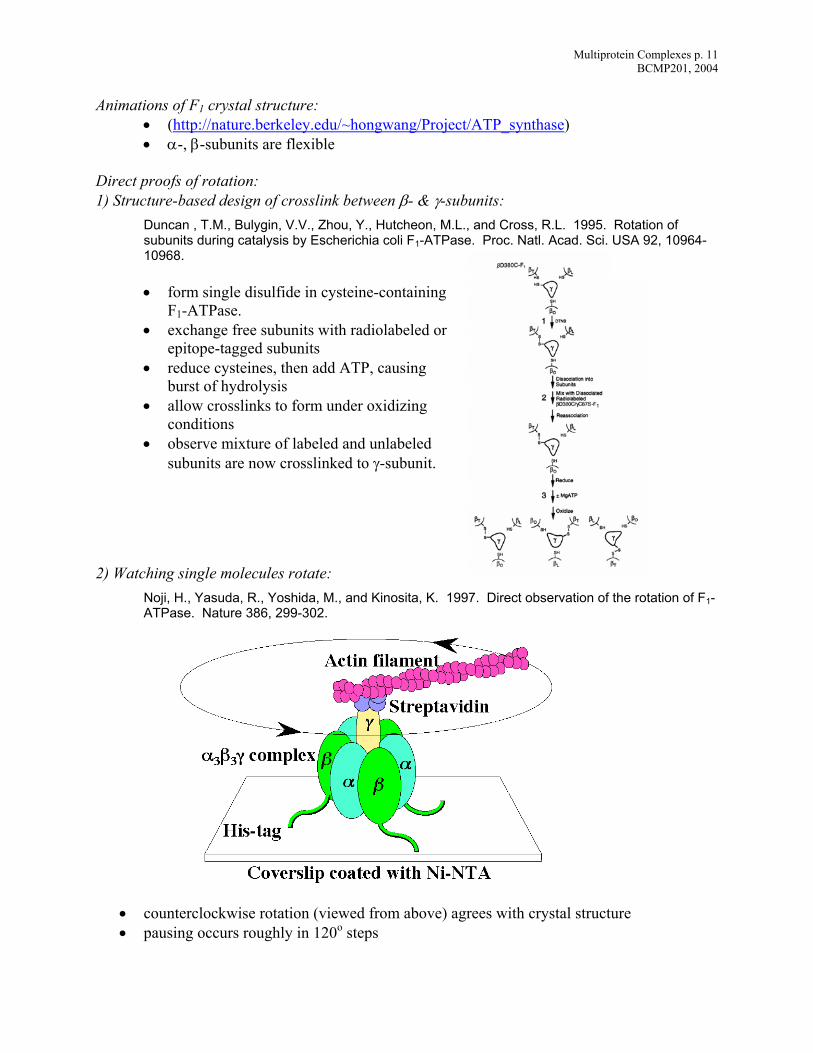
Multiprotein Complexes p. 11 BCMP201, 2004
Animations of F1 crystal structure: • (http://nature.berkeley.edu/~hongwang/Project/ATP_synthase) • α-, β-subunits are flexible
Direct proofs of rotation: 1) Structure-based design of crosslink between β- & γ-subunits:
Duncan , T.M., Bulygin, V.V., Zhou, Y., Hutcheon, M.L., and Cross, R.L. 1995. Rotation of subunits during catalysis by Escherichia coli F1-ATPase. Proc. Natl. Acad. Sci. USA 92, 10964-10968.
• form single disulfide in cysteine-containing
F1-ATPase. • exchange free subunits with radiolabeled or
epitope-tagged subunits • reduce cysteines, then add ATP, causing
burst of hydrolysis • allow crosslinks to form under oxidizing
conditions • observe mixture of labeled and unlabeled
subunits are now crosslinked to γ-subunit. 2)
1997. Direct observation of the rotation of F1-
• counterclockwise rotation (viewed from above) agrees with crystal structure • pausing occurs roughly in 120o steps
Watching single molecules rotate: Noji, H., Yasuda, R., Yoshida, M., and Kinosita, K.ATPase. Nature 386, 299-302.
Multiprotein Complexes p. 12 BCMP201, 2004
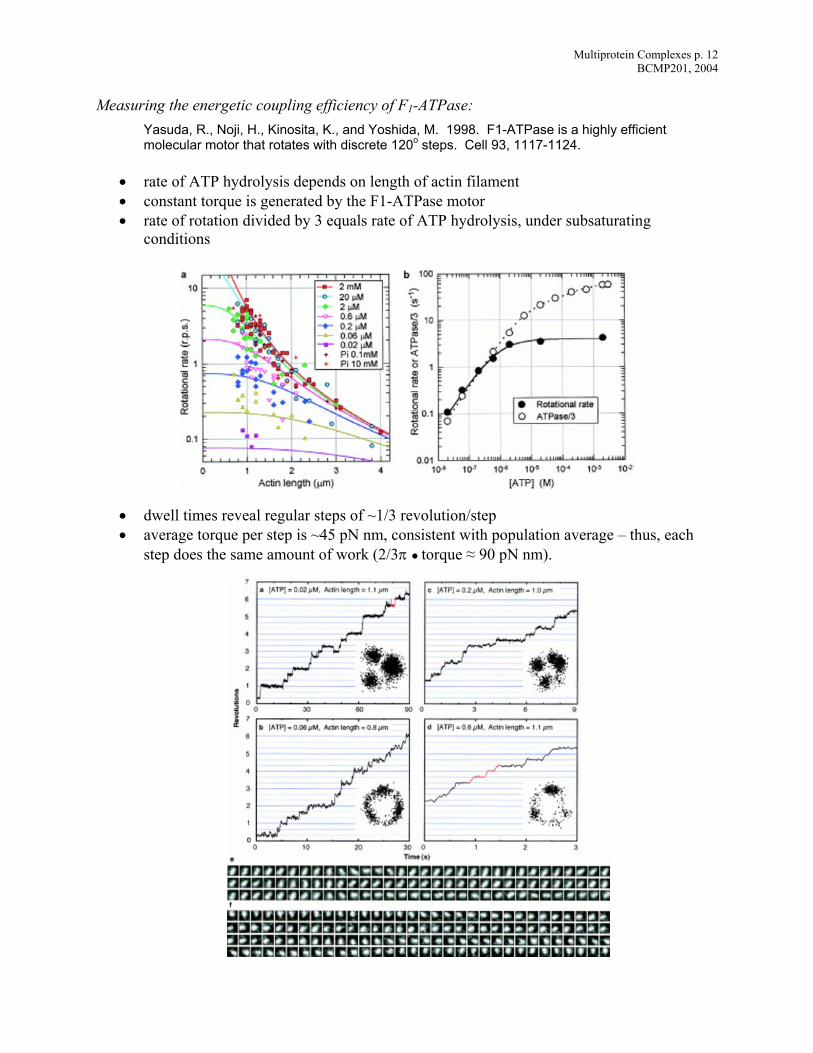
Measuring the energetic coupling efficiency of F1-ATPase: s highly efficient l discrete 120o steps. Cell 93, 1117-1124.
ysis depends on length of actin filament
•
• dwell times reveal regular steps of ~1/3 revolution/step • average torque per step is ~45 pN nm, consistent with population average – thus, each
step does the same amount of work (2/3π ● torque ≈ 90 pN nm).
Ya uda, R., Noji, H., Kinosita, K., and Yoshida, M. 1998. F1-ATPase is amo ecular motor that rotates with
• rate of ATP hydrol• constant torque is generated by the F1-ATPase motor
rate of rotation divided by 3 equals rate of ATP hydrolysis, under subsaturating conditions
Multiprotein Complexes p. 13 BCMP201, 2004
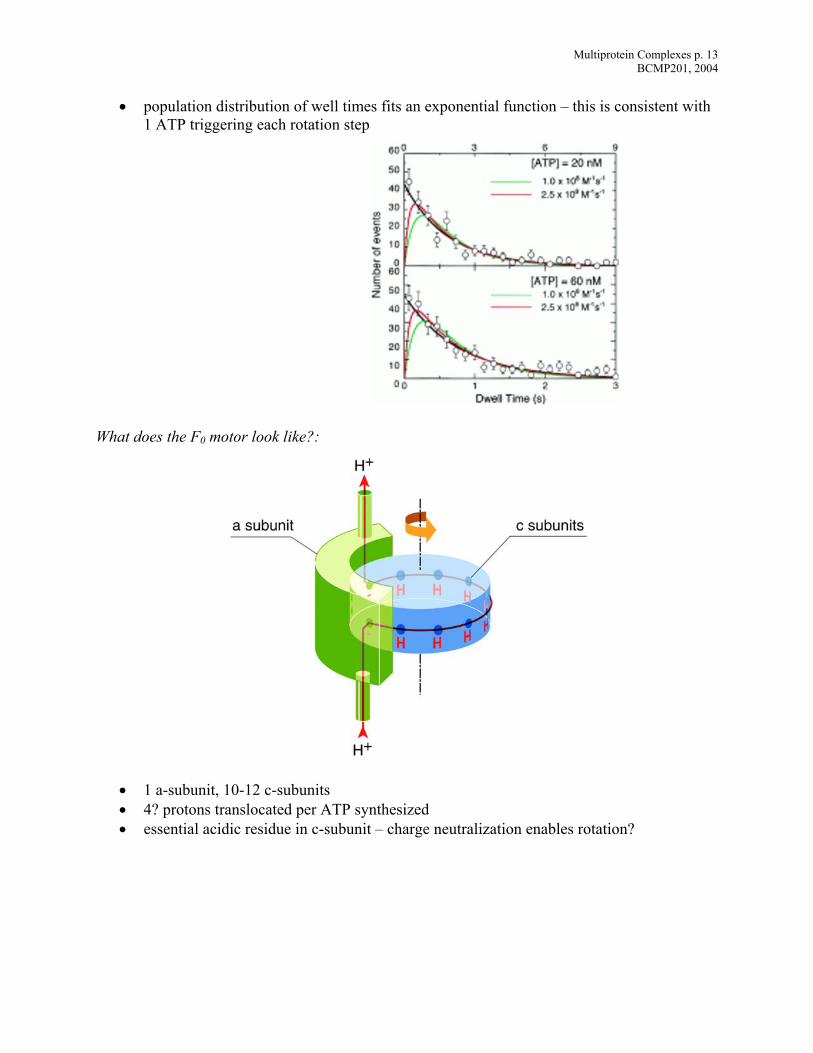
• population distribution of well times fits an exponential function – this is consistent with
hat does the F0 motor look like?:
• 1 a-subunit, 10-12 c-subunits • 4? protons translocated per ATP synthesized • essential acidic residue in c-subunit – charge neutralization enables rotation?
1 ATP triggering each rotation step
W
Multiprotein Complexes p. 14 BCMP201, 2004
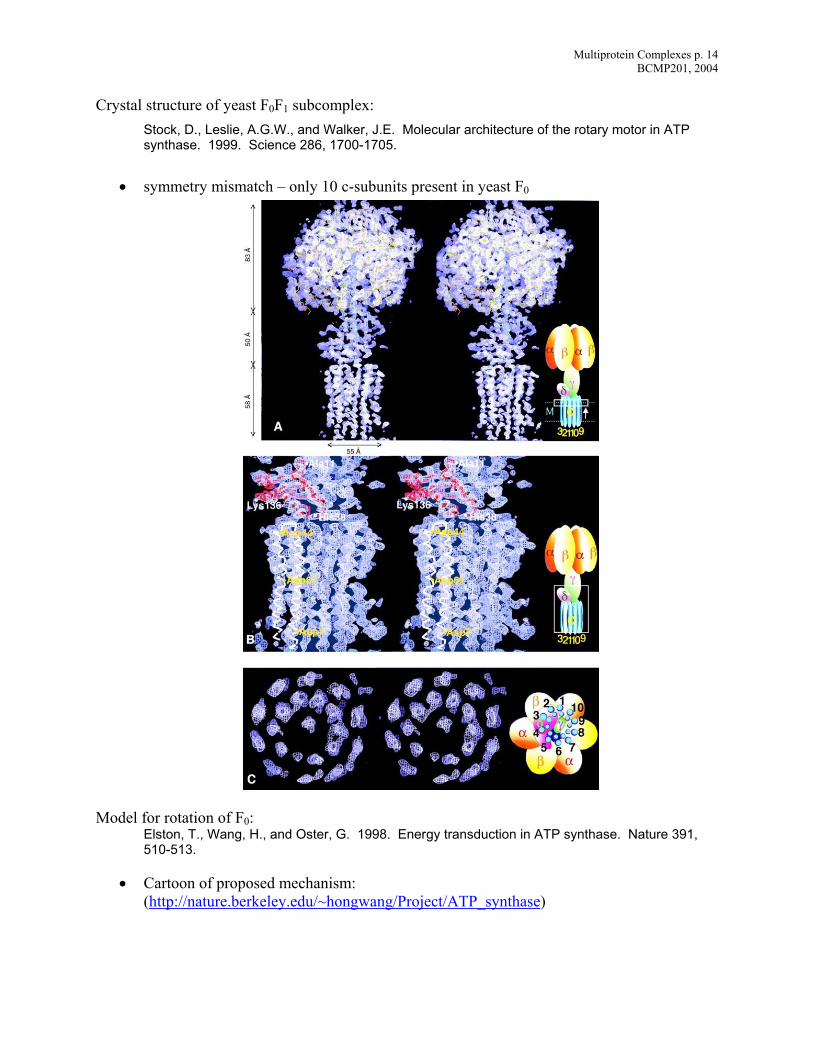
Cry l.E. Molecular architecture of the rotary motor in ATP
synthase. 1999. Science 286, 1700-1705.
• symmetry mismatch – only 10 c-subunits present in yeast F0
Mo l
ture 391, 510-513.
• Cartoon of proposed mechanism: (http://nature.berkeley.edu/~hongwang/Project/ATP_synthase
sta structure of yeast F0F1 subcomplex: Stock, D., Leslie, A.G.W., and Walker, J
de for rotation of F : 0Elston, T., Wang, H., and Oster, G. 1998. Energy transduction in ATP synthase. Na
)
Related Documents











