Send Orders for Reprints to [email protected] Current Physical Chemistry, 2014, 4, 35-44 35 Pharmacophore-based Drug Design of Novel Potential Tau Ligands for Alzheimer's Disease Treatment Susimaire Pedersoli-Mantoani 1, * , Vinicius B. Da Silva 2 , Carlton A. Taft 3 and Carlos H.T.P. Da Silva 1 1 Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Av. do Café, s/n – Monte Alegre, 14040-903, Ribeirão Preto – SP, Brazil; 2 Departamento de Biomedicina e Farmácia, Pontifícia Universidade Católica de Goiás, Rua 232, n° 128, Setor Leste Universitário,74605-140, Goiânia – Brasil; 3 Centro Brasileiro de Pesquisas Físicas, Rua Dr. Xavier Sigaud 150, Urca, 22290-180, Rio de Janeiro, Brazil Abstract: An intracellular hallmark of Alzheimer Disease (AD) is accumulation of hyperphosphorylated tau as tangles of paired helical filaments (PHF). A significant advance in understanding tau’s behaviour isolated came when it was recognized that the protein contains isolated short peptide motifs, embedded in an otherwise hydrophilic environment, which have a high tendency for beta-structure and aggregation, forming the core of the PHF. In a recent work, we used the smallest fragment responsible for aggregation, the hexapeptide 306 VQIVYK 311 , in order to investigate with molecular dynamics simulations possible binding modes of the tau protein fragment with respect to an active flavonoid, which would be responsible for the inhibitory process of aggregation of tau. Considering such results, we have used in this work a selected pharmacophoric model and carried out a pharmacophore-based virtual screening with the purpose of designing novel potential Tau aggregation inhibitors. An initial set of 96 compounds was selected, of which 86 are unpublished regarding Tau anti-aggregation activity and the other 10 compounds are reported as Tau ligands. Prediction of biological activity and pharmaceutical properties indicated four tiophene derivatives as promising Tau aggregation inhibitors for Alzheimer’s disease treatment. Keywords: Aggregation inhibitors, Alzheimer’s disease, ADME/Tox, pharmacophore-based virtual screening, tau protein, tauophathy. INTRODUCTION Alzheimer´s disease is the most common form of dementia in humans, and it is characterized by abnormal accumulation of amyloid beta-peptides (Aβ) in the form of extracellular amyloid plaques as well ashyperphosphorylated tau protein in form of intracellular neurofibrillary tangles (NFT) in the human brain. Therefore, one of the highest priorities in Alzheimer research is to understand the reasons for the pathological aggregation and to find methods to prevent it [1]. Some structural principles governing Aβ aggregation are known in detail, but not concerning the tau protein. Recently, a significant advance in understanding tau’s isolated behaviour came when it was recognized that the protein contains isolated short peptide motifs, beginning of the third repeats (R3) ( 306 VQIVYK 311 ), embedded in an otherwise hydrophilic environment, which have a high tendency for β- structure and aggregation, crucial for formation of the core of the PHF [2]. In the last years, research has focused on developmening inhibitors able to prevent or reverse the process of the NFT formation. Thus, one of the earliest researches with inhibitors *Address correspondence to this author at the Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Av. do Café, s/n – Monte Alegre, 14040-903, Ribeirão Preto – SP, Brazil; Tel: 55 16 3602 4709; Fax: 55 16 3602 4178; E-mail: [email protected] of tau aggregates was described by Claude Wischik, who presented data arguing that the known methylene blue (MB) could reduce the aggregation of tau and thereby slow down the disease [3,4]. Moreover, the Mandelkow research group has reported a large number of inhibitors for Tau aggregation, which belongs to different chemical classes such as anthraquinones, polyphenols, quinoxalines, rhodanines, phenothiazines and phenylthiazolylhydrazides [5-8]. However, with exception of the phenothiazine methylene blue [9], no compound has been tested in vivo for its effect on tau deposition, neurodegeneration, and behavioral impairments. In the last years, virtual screening (VS) approaches have shown an important role in the drug discovery process. The main goal of such methods is to generate chemical diversity through novel active structures, which are typically selected from tens of millions of virtual compounds [10,11]. VS methods are commonly divided into ligand-based VS (LBVS), used in the absence of protein target structure, and structure–based VS (SBVS), used when the receptor target is known. Pharmacophore-based virtual screening (PBVS) is a ligand-based methodology where virtual compounds with a number of conformations are fitted against a pharmacophore model query, which is derived from a set of known bioactive compounds [11]. In this work, from the knowledge of several bioactive compounds capable of preventing the formation of these tau aggregates, it was possible to explore key structural features 1877-9476/14 $58.00+.00 © 2014 Bentham Science Publishers

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Send Orders for Reprints to [email protected]
Current Physical Chemistry, 2014, 4, 35-44 35
Pharmacophore-based Drug Design of Novel Potential Tau Ligands for Alzheimer's Disease Treatment
Susimaire Pedersoli-Mantoani1,*, Vinicius B. Da Silva2, Carlton A. Taft3 and Carlos H.T.P. Da Silva1
1Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Av. do Café, s/n – Monte Alegre, 14040-903, Ribeirão Preto – SP, Brazil; 2Departamento de Biomedicina e Farmácia, Pontifícia Universidade Católica de Goiás, Rua 232, n° 128, Setor Leste Universitário,74605-140, Goiânia – Brasil; 3Centro Brasileiro de Pesquisas Físicas, Rua Dr. Xavier Sigaud 150, Urca, 22290-180, Rio de Janeiro, Brazil
Abstract: An intracellular hallmark of Alzheimer Disease (AD) is accumulation of hyperphosphorylated tau as tangles of paired helical filaments (PHF). A significant advance in understanding tau’s behaviour isolated came when it was recognized that the protein contains isolated short peptide motifs, embedded in an otherwise hydrophilic environment, which have a high tendency for beta-structure and aggregation, forming the core of the PHF. In a recent work, we used the smallest fragment responsible for aggregation, the hexapeptide 306VQIVYK311, in order to investigate with molecular dynamics simulations possible binding modes of the tau protein fragment with respect to an active flavonoid, which would be responsible for the inhibitory process of aggregation of tau. Considering such results, we have used in this work a selected pharmacophoric model and carried out a pharmacophore-based virtual screening with the purpose of designing novel potential Tau aggregation inhibitors. An initial set of 96 compounds was selected, of which 86 are unpublished regarding Tau anti-aggregation activity and the other 10 compounds are reported as Tau ligands. Prediction of biological activity and pharmaceutical properties indicated four tiophene derivatives as promising Tau aggregation inhibitors for Alzheimer’s disease treatment.
Keywords: Aggregation inhibitors, Alzheimer’s disease, ADME/Tox, pharmacophore-based virtual screening, tau protein, tauophathy.
INTRODUCTION
Alzheimer´s disease is the most common form of dementia in humans, and it is characterized by abnormal accumulation of amyloid beta-peptides (Aβ) in the form of extracellular amyloid plaques as well ashyperphosphorylated tau protein in form of intracellular neurofibrillary tangles (NFT) in the human brain. Therefore, one of the highest priorities in Alzheimer research is to understand the reasons for the pathological aggregation and to find methods to prevent it [1]. Some structural principles governing Aβ aggregation are known in detail, but not concerning the tau protein. Recently, a significant advance in understanding tau’s isolated behaviour came when it was recognized that the protein contains isolated short peptide motifs, beginning of the third repeats (R3) (306VQIVYK311), embedded in an otherwise hydrophilic environment, which have a high tendency for β-structure and aggregation, crucial for formation of the core of the PHF [2]. In the last years, research has focused on developmening inhibitors able to prevent or reverse the process of the NFT formation. Thus, one of the earliest researches with inhibitors
*Address correspondence to this author at the Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Av. do Café, s/n – Monte Alegre, 14040-903, Ribeirão Preto – SP, Brazil; Tel: 55 16 3602 4709; Fax: 55 16 3602 4178; E-mail: [email protected]
of tau aggregates was described by Claude Wischik, who presented data arguing that the known methylene blue (MB) could reduce the aggregation of tau and thereby slow down the disease [3,4]. Moreover, the Mandelkow research group has reported a large number of inhibitors for Tau aggregation, which belongs to different chemical classes such as anthraquinones, polyphenols, quinoxalines, rhodanines, phenothiazines and phenylthiazolylhydrazides [5-8]. However, with exception of the phenothiazine methylene blue [9], no compound has been tested in vivo for its effect on tau deposition, neurodegeneration, and behavioral impairments. In the last years, virtual screening (VS) approaches have shown an important role in the drug discovery process. The main goal of such methods is to generate chemical diversity through novel active structures, which are typically selected from tens of millions of virtual compounds [10,11]. VS methods are commonly divided into ligand-based VS (LBVS), used in the absence of protein target structure, and structure–based VS (SBVS), used when the receptor target is known. Pharmacophore-based virtual screening (PBVS) is a ligand-based methodology where virtual compounds with a number of conformations are fitted against a pharmacophore model query, which is derived from a set of known bioactive compounds [11]. In this work, from the knowledge of several bioactive compounds capable of preventing the formation of these tau aggregates, it was possible to explore key structural features
1877-9476/14 $58.00+.00 © 2014 Bentham Science Publishers

36 Current Physical Chemistry, 2014, Vol. 4, No. 1 Pedersoli-Mantoani et al.
of each class of compounds and to perform several PBVS experiments. In order to identify prototype inhibitors of tau assembly that exhibit favourable combinations of potency, selectivity, drug-like and ADME/Toxproperties, compounds were selected from different databases as novel and potential candidate inhibitors of tau aggregates.
METHODOLOGY
Data Preparation
The 50 Tau aggregation inhibitors here investigated were selected from five chemical classes:rhodanines, phenylthiazolylhydrazide, flavonoids, phenothiazines and polyphenols [6-8, 12], which have IC50 lower than 50µM. (supplementary material).The two-dimensional chemical structures of these inhibitors were sketched using the ChemSketch 11.0 [13], and subsequently converted in 3D and optimized using the Discovery Studio 3.1 software [14].
Pharmacophore Perception
Pharmacophoric models were generated from two independent softwares, the PharmaGist webserver [15] and the pharmacophore modeling module from Discovery Studio. PharmaGist webserver generates 3D pharmacophores from a set of molecules that are known to bind to a common target receptor. The method efficiently searches for possible pharmacophores and reports the highest-scoring ones [16]. The candidate pharmacophores are detected by multiple flexible alignment of the input ligands, where the flexibility of the ligands is treated explicitly and in a deterministic manner in the alignment process. Another key advantage of the method is the ability to detect pharmacophores common to subsets of input ligands, a characteristic that makes PharmaGist tolerant to outliers and to several binding modes [15]. The pharmacophore modeling module from Discovery Studio was also used, applying the HipHop algorithm to generate common feature-based pharmacophore models for Tau ligands. A minimum of 3 to a maximum of 6 features including hydrogen-bond acceptor (HBA), hydrogen bond donor (HBD), hydrophobic (HBic), and ring aromatic (RingArom) features were selected for generating the quantitative pharmacophore model [17]. Ten pharmacophore models with significant statistical parameters were thus generated. The best model was selected on the basis of the highest scored models in consensus obtained with PharmaGist and Discovery Studio. Moreover, structure-activity relationships of bioactive compounds with respect to the binding models here proposed were investigated with Molecular Dynamics simulations. Individual phamacophore models regarding each class of Tau aggregation inhibitors were obtained.
Compounds Databases
For the discovery of novel potential Tau ligands, the following chemical databases were screened from the following Pharmacophoric models: CNS-ChemBrigde, DIVERSet-ChemBrigde, PremiumSet-ChemBrigde, Molecular Weightset-ChemBrigde [18], MayBridgeScreening Collection [19], and CNS-ZINC [20].
Molecular Interaction Fields(MIF) and Molecular Dynamics Simulations
MIFs were obtained using the GRID22b [21] software and aromatic carbon, dry, carbonyl oxygen, phenolic hydroxyl oxygen, thioestersulfur and sp2 nitrogenas chemical probes. In sequence, bioactive flavonoids were oriented with respect to the hexapeptide after analysis of MIFs. In sequence, molecular dynamics simulations were performed and the trajectories obtained were analysed using the InsightII package [22].
Pharmacophore-Based Virtual Screening
The virtual screening was performed with the Screen Library module from Discovery Studio [15], and the best pharmacophore models obtained were used as constraint to screen the chemical database. Initially, for each molecule in thedatabase, the fast conformer generation method produced 255 conformers with a maximum energy tolerance of 20 kcal/mol above the minimum global. Two database searching options such as Fast/Flexible and Best/Flexible are employed in Discovery Studio. In sequence, compounds with best fit to the pharmacophore models were selected. Then, further screening virtual experiments were carried out using the Best/Flexible option. Setting the “Maximum Omitted Features” option to zero, the best pharmacophore model obtained was used to screen the databases for those compounds that fit all features of the pharmacophore model. The calculations of the values were based on how well the chemical substructures match the location constraints of the pharmacophoric features and their distance deviation from the feature centers. High fit values indicate good matches. Following this criterium, the best ranked molecules regarding the pharmacophoric constraints thus employed were filtered for each one of the six pharmacophore models investigated.
Tanimoto Similarity
2DTanimoto similarity searching procedure was performed using the BindingDB Webserver [23]. The most active molecules representing each class of tau ligands were set as reference, and the screened molecules presenting Tanimoto index of 0.6 or higher, in comparison with at least one of the reference molecules, were selected. For molecules presenting Tanimoto index lower than 0.6, only the top-scored regarding the pharmacophoric constraints were selected.
Biological Activity Prediction
The determination of the possible biological activity spectrum related to the 86 selected compounds was performed using the PASS (Prediction of Activity Spectra of Substances) server [24]. PASS is a tool to find out possible biological effects of a compound based entirely on the structural formula using MNA descriptors, suggesting that the biological activity is a function of the chemical structure. The PASS algorithm method estimate biological activity by comparing the structure of a new compound entity with training set of approximately 46.000 well known bioactive compounds. Only the activities predicted with Pa

Pharmacophore-based Drug Design of Novel Potential Tau Ligands Current Physical Chemistry, 2014, Vol. 4, No. 1 37
(probability to be active) > Pi (probability to be inactive) for the selected compound were considered [24].
Prediction of Pharmaceutical Properties
QikProp was used to predict pharmaceutical properties of 16 selected compounds, candidate to tau aggregation inhibitors. QikProp calculates physically significant descriptors and pharmaceutically relevant properties of organic molecules, either individually or in batches. In addition to such predictions, QikProp provides ranges for comparing particular properties of a molecule with those of 95% of known drugs [25].
Toxicity Predictions
Toxicity predictions were performed with the knowledge-based expert system of the DEREK software [26, 27] for specific toxicity endpoints, including carcinogenicity, chromosome damage, genotoxicity, mutagenicity, neuro- toxicity, hepatotoxicity, teratogenicity, irritancy, reproductive toxicity, respiratory sensitization, thyroid toxicity and skin sensitisation.
RESULTS AND DISCUSSION
At least five molecular classes of chemical compounds presenting Tau aggregation inhibitory activity are reported, including rhodanines, phenylthiazolyl-hydrazides, flavonoids, phenothiazines and polyphenols (Fig. 1). All of them provide compounds with benzene rings in one or both molecular extremities and a variable number of polar substituents able to act as hydrogen bond donor and/or acceptor groups. Experimental evidences suggest that the inhibition mechanism of Tau aggregation involves π –stacking inter- actions of a benzene ring of the bioactive molecules with the tyrosine benzene ring (Y310) of the hexapeptide sequence [28], evidencing the importance of a benzene ring as active moiety of such molecules.
Pharmacophore Modelling
Pharmacophore modelling procedures were used to identify the main characteristics of the tau aggregation
inhibitors in order to design noveldrug candidates in Alzheimer’s disease. First, bioactive molecules of the five molecular classes considered (Fig. 1) were found in literature, and the one presenting IC50 lower than 50 µM were then selected for pharmacophore perception calculations, comprising a total of 50 compounds (supplementary material). In order to obtain reliable pharmacophoric models for the five classes of Tau aggregation inhibitors, two independent softwares were used, the PharmaGist webserver [16] and the pharmacophore modeling module from Discovery Studio package [15]. For PharmaGist pharmacophore perception, the 50 molecules selected were used as input. The consensual model obtained includes rhodanines, phenylthiazolylhydrazide, flavonoids, phenothiazines and polyphenols, and it is composed of features including one aromatic ring and three hydrogen bond acceptor groups (Fig. 2A). The pharmacophoric model obtained for anti-aggregation Tau inhibitors using Discovery Studio software showed a slightly similar configuration when compared with the one obtained using the PharmaGist webserver. Such a model is composed by an aromatic group and two hydrogen bond acceptor groups (Fig. 2B). The characteristics of the pharma- cophore models obtained using both methods privilege the flavonoids, where the position of the acceptors groups with respect to the aromatic ring is optimal for flavonoids regarding the pharmacophoric models. There are few studies reporting the possible binding modes of Tau ligands as well as relevant structure-activity relationships for some of these compounds. For phenylthiazolyl-hydrazides, the aromatic group responsible for π –stacking interactions with the hexapeptide tyrosine is in the alpha-carbonyl position [6, 8], whereas for rhodanines, which have an unique aromatic region, the presence of a furan ring is important for obtaining lower IC50 values [7, 8]. For flavonoids, Han et al., [28] developed a broader and detailed study which identified that the benzopyronemoiety is the aromatic region responsible for π–stacking interactions, and the carbonyl group that favours the conjugated system formed between rings would be the basic factor for the inhibitory process. Circular Dichroism (CD) experiments clearly demonstrated that the binding of flavonoids to R3
Fig. (1). Chemical classes of tau aggregation inhibitors reported in literature [3-8].

38 Current Physical Chemistry, 2014, Vol. 4, No. 1 Pedersoli-Mantoani et al.
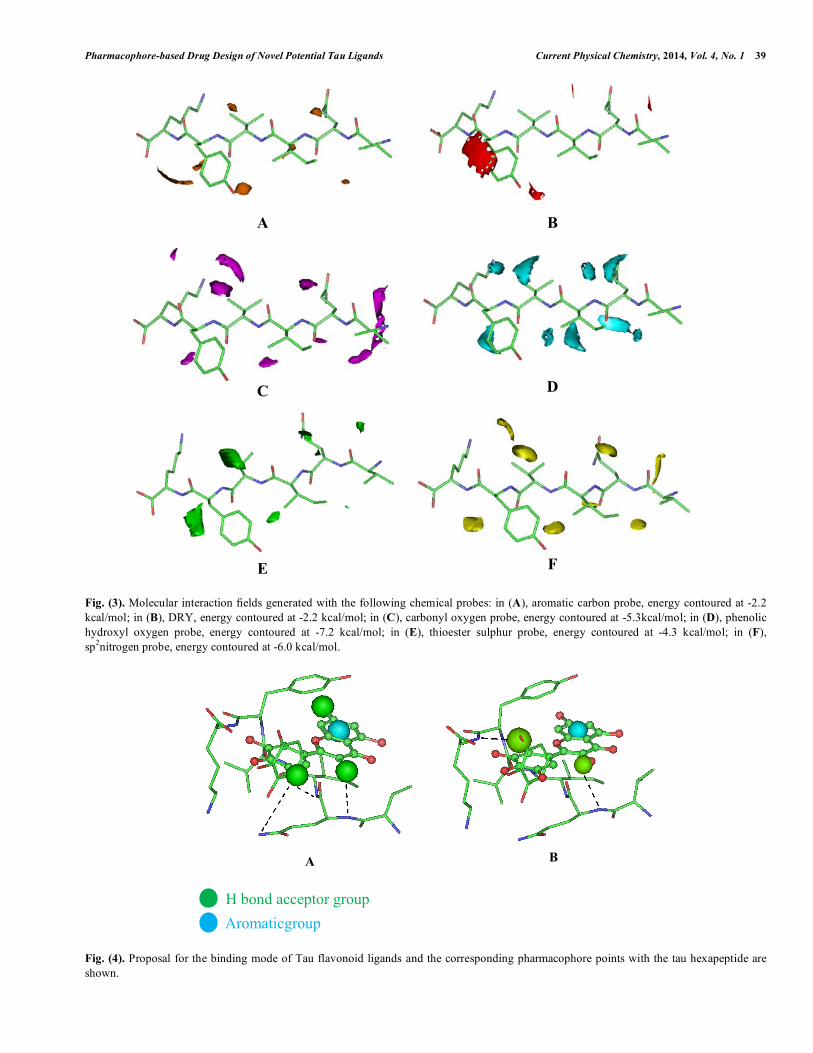
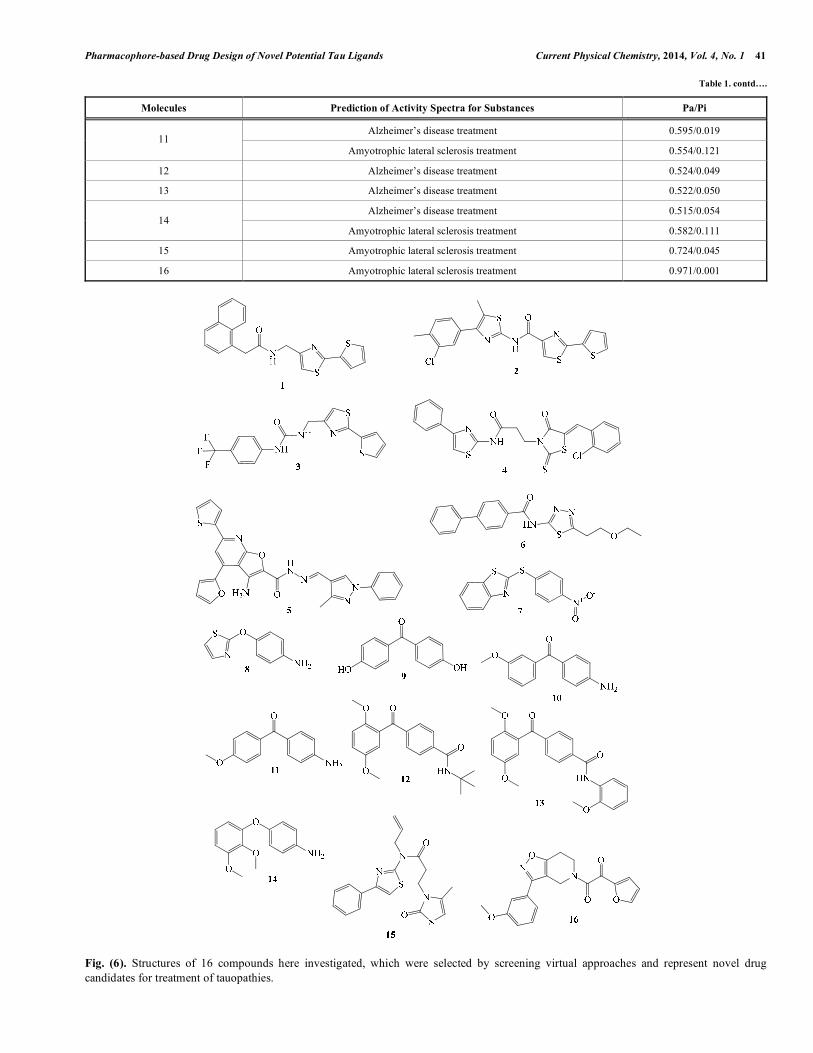
causes a partial elimination of the random coil structure of the protein, suggesting that the freedom in rotation or folding of peptide chain gradually degenerates when the organic molecules bind to the peptide. In addition, a greater number of hydroxyl groups favour a greater number of flavonoid-peptide interactions [28]. After the pharmacophore perception, molecular interaction fields (MIFs) were generated for the hexapeptide structure in order to guide the spatial orientation of the bioactive compounds (Fig. 3). MIFs were obtained using four relevant chemical probes, such as described in the methodology section, representing aromatic, hydrophobic and hydrogen bond (acceptor and donor groups) interactions. Then, the initial orientation of each bioactive compound was obtained by visual inspection, by selecting the best fittings of the functional groups with the MIFs. Molecular dynamics trajectory [29], showed an induced conformational change of the tyrosine hexapeptide residue towards the aromatic ring system of flavonoid inhibitors (Fig. 4), in agreement with CD results reported by Han et al., [28]. The main interactions described also agree with both the two pharmacophoric models (Fig. 4) obtained for Tau ligands, indicating a likely binding mode for flavonoids and other tau ligands in general. The pharmacophoric models here proposed independently show that the aromatic feature and one of the hydrogen bond acceptor groups (HBA1) of both models are placed in the same position regarding the hexapeptide (Fig. 4). The aromatic group could be able to perform a π-stacking interaction with the tyrosine residue, whereas theHBA1 group could be able to perform a hydrogen bond with the main chain amine group from the glutamine hexapeptide residue. HBA2 and HBA4 could also interact with main chain and side chain amine groups from the hexapeptide by hydrogen bond. Individual models regarding each class of Tau aggregation inhibitors were also obtained. For this task all compounds representing each class were submitted separately for pharmacophore perception procedures. The highest scored consensual models obtained using PharmaGist and Discovery Studio were selected (Fig. 5).
Pharmacophore-based Virtual Screening
Virtual screening simulations were performed using the pharmacophore models obtained (Figs. 2 and 5) as constraints. The 1,000th best ranked molecules regarding the pharmacophoric constraints were selected/filtered for each one of the six pharmacophore models obtained (Figs. 2 and 5). In order to reduce the final virtual screening list, all the molecules were submitted to a 2DTanimoto similarity searching procedure, where the most active molecules representing each class of Tau ligands were used as reference, and the screened molecules with Tanimoto index of 0.6 or higher in comparison with at least one of the reference molecules were selected. For molecules with Tanimoto index lower than 0.6, only the top-scored regarding the pharmacophoric constraints were selected. After this procedure, 96 compounds were then obtained, where 86 are unpublished regarding Tau anti-aggregation activity. The other 10 compounds are reported in literature as Tau ligands [8, 30].
Biological Activity and ADME/Tox Prediction
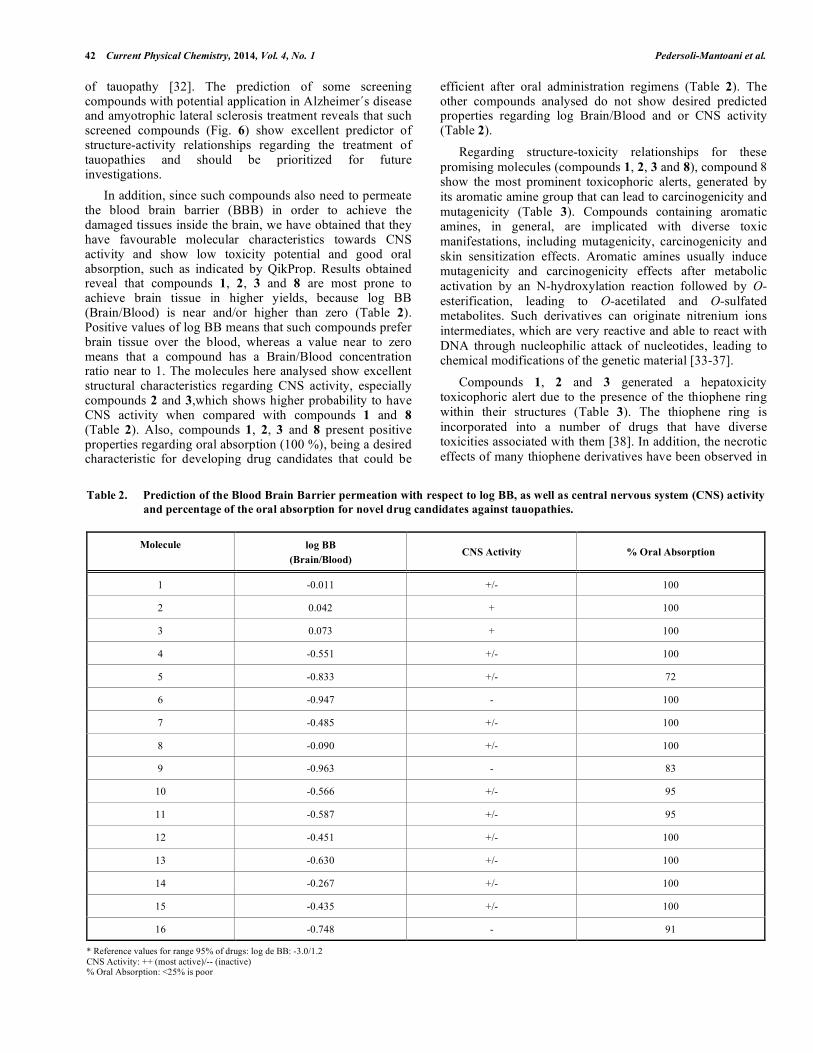
In the attempt to select the most promising Tau anti-aggregation candidates among the resulting list of 86 screened ones, MNA(Multilevel Neighborhoods of Atoms) descriptors were computed through the PASS webserver [24] for predicting which compounds could be anti-Alzheimer’s disease drug candidates. Considering the known bioactive molecules as reference (Table 1), during the validation step of the PASS method, 75% of the Tau ligands were predict as anti-Alzheimer’s disease and/or for amyotrophic lateral sclerosis treatment, considering Pa > Pi. For screened molecules, at least 16 show significant Pa > Pi for Alzheimer’s disease and or amyotrophic lateral sclerosis treatment, and hence were selected for further analysis (Table 1; Fig. 6). Amyotrophic lateral sclerosis is a neurodegenerative disease characterized by motor neurons degeneration [31]. Scientific evidences suggest a relation of amyotrophic lateral sclerosis with Parkinson disease and dementia by mutations on the gene that codify Tau protein. This aspect indicates the classification of the amyotrophic lateral sclerosis as a form
Fig. (2). Pharmacophore models for Tau aggregation inhibitors, obtained using PharmaGist (A) and Discovery Studio (B) softwares. Pharmacophore features are color-coded in (A) light-blue for hydrophobic feature and green for hydrogen-bond acceptor and in (B) orange for aromatic group and green for hydrogen-bond acceptor.
A B
Pharmacophore-based Drug Design of Novel Potential Tau Ligands Current Physical Chemistry, 2014, Vol. 4, No. 1 39
Fig. (3). Molecular interaction fields generated with the following chemical probes: in (A), aromatic carbon probe, energy contoured at -2.2 kcal/mol; in (B), DRY, energy contoured at -2.2 kcal/mol; in (C), carbonyl oxygen probe, energy contoured at -5.3kcal/mol; in (D), phenolic hydroxyl oxygen probe, energy contoured at -7.2 kcal/mol; in (E), thioester sulphur probe, energy contoured at -4.3 kcal/mol; in (F), sp2nitrogen probe, energy contoured at -6.0 kcal/mol.
Fig. (4). Proposal for the binding mode of Tau flavonoid ligands and the corresponding pharmacophore points with the tau hexapeptide are shown.
A
B
C
D
E F
A B
H bond acceptor group Aromaticgroup

40 Current Physical Chemistry, 2014, Vol. 4, No. 1 Pedersoli-Mantoani et al.
Fig. (5). Pharmacophore models obtained for known Tau aggregation inhibitors, discriminated by chemical classes. Pharmacophore features are color-coded with light-blue for hydrophobic feature, magenta for hydrogen-bond donor, green for hydrogen-bond acceptor and orange for aromatic group.
Table 1. Molecules selected with virtual screening approaches as potential prototypes for Alzheimer’s disease treatment and
amyotrophic lateral sclerosis, which have Pa>Pi.
Molecules Prediction of Activity Spectra for Substances Pa/Pi
1 Amyotrophic lateral sclerosis treatment 0.696/0.061
2 Amyotrophic lateral sclerosis treatment 0.662/0.079
3 Amyotrophic lateral sclerosis treatment 0.716/0.050
4 Amyotrophic lateral sclerosis treatment 0.613/0.099
5 Amyotrophic lateral sclerosis treatment 0.764/0.064
6 Amyotrophic lateral sclerosis treatment 0.689/0.064
7 Amyotrophic lateral sclerosis treatment 0.804/0.100
8 Amyotrophic lateral sclerosis treatment 0.609/0.100
9 Alzheimer’s disease treatment 0.608/0.015
Alzheimer’s disease treatment 0.604/0.016 10
Amyotrophic lateral sclerosis treatment 0.479/0.156
rhodanines phenylthiazolylhydrazide
Flavonoids
phenothiazines polyphenols
Pharmacophore-based Drug Design of Novel Potential Tau Ligands Current Physical Chemistry, 2014, Vol. 4, No. 1 41
Table 1. contd….
Molecules Prediction of Activity Spectra for Substances Pa/Pi
Alzheimer’s disease treatment 0.595/0.019 11
Amyotrophic lateral sclerosis treatment 0.554/0.121
12 Alzheimer’s disease treatment 0.524/0.049
13 Alzheimer’s disease treatment 0.522/0.050
Alzheimer’s disease treatment 0.515/0.054 14
Amyotrophic lateral sclerosis treatment 0.582/0.111
15 Amyotrophic lateral sclerosis treatment 0.724/0.045
16 Amyotrophic lateral sclerosis treatment 0.971/0.001
Fig. (6). Structures of 16 compounds here investigated, which were selected by screening virtual approaches and represent novel drug candidates for treatment of tauopathies.
42 Current Physical Chemistry, 2014, Vol. 4, No. 1 Pedersoli-Mantoani et al.
of tauopathy [32]. The prediction of some screening compounds with potential application in Alzheimer´s disease and amyotrophic lateral sclerosis treatment reveals that such screened compounds (Fig. 6) show excellent predictor of structure-activity relationships regarding the treatment of tauopathies and should be prioritized for future investigations. In addition, since such compounds also need to permeate the blood brain barrier (BBB) in order to achieve the damaged tissues inside the brain, we have obtained that they have favourable molecular characteristics towards CNS activity and show low toxicity potential and good oral absorption, such as indicated by QikProp. Results obtained reveal that compounds 1, 2, 3 and 8 are most prone to achieve brain tissue in higher yields, because log BB (Brain/Blood) is near and/or higher than zero (Table 2). Positive values of log BB means that such compounds prefer brain tissue over the blood, whereas a value near to zero means that a compound has a Brain/Blood concentration ratio near to 1. The molecules here analysed show excellent structural characteristics regarding CNS activity, especially compounds 2 and 3,which shows higher probability to have CNS activity when compared with compounds 1 and 8 (Table 2). Also, compounds 1, 2, 3 and 8 present positive properties regarding oral absorption (100 %), being a desired characteristic for developing drug candidates that could be
efficient after oral administration regimens (Table 2). The other compounds analysed do not show desired predicted properties regarding log Brain/Blood and or CNS activity (Table 2).
Regarding structure-toxicity relationships for these promising molecules (compounds 1, 2, 3 and 8), compound 8 show the most prominent toxicophoric alerts, generated by its aromatic amine group that can lead to carcinogenicity and mutagenicity (Table 3). Compounds containing aromatic amines, in general, are implicated with diverse toxic manifestations, including mutagenicity, carcinogenicity and skin sensitization effects. Aromatic amines usually induce mutagenicity and carcinogenicity effects after metabolic activation by an N-hydroxylation reaction followed by O-esterification, leading to O-acetilated and O-sulfated metabolites. Such derivatives can originate nitrenium ions intermediates, which are very reactive and able to react with DNA through nucleophilic attack of nucleotides, leading to chemical modifications of the genetic material [33-37].
Compounds 1, 2 and 3 generated a hepatoxicity toxicophoric alert due to the presence of the thiophene ring within their structures (Table 3). The thiophene ring is incorporated into a number of drugs that have diverse toxicities associated with them [38]. In addition, the necrotic effects of many thiophene derivatives have been observed in
Table 2. Prediction of the Blood Brain Barrier permeation with respect to log BB, as well as central nervous system (CNS) activity and percentage of the oral absorption for novel drug candidates against tauopathies.
Molecule log BB (Brain/Blood)
CNS Activity % Oral Absorption
1 -0.011 +/- 100
2 0.042 + 100
3 0.073 + 100
4 -0.551 +/- 100
5 -0.833 +/- 72
6 -0.947 - 100
7 -0.485 +/- 100
8 -0.090 +/- 100
9 -0.963 - 83
10 -0.566 +/- 95
11 -0.587 +/- 95
12 -0.451 +/- 100
13 -0.630 +/- 100
14 -0.267 +/- 100
15 -0.435 +/- 100
16 -0.748 - 91
* Reference values for range 95% of drugs: log de BB: -3.0/1.2 CNS Activity: ++ (most active)/-- (inactive) % Oral Absorption: <25% is poor
Pharmacophore-based Drug Design of Novel Potential Tau Ligands Current Physical Chemistry, 2014, Vol. 4, No. 1 43
rodents [39]. The thiophene ring is known to undergo metabolic activation mediated by cytochrome P450 enzymes resulting in the formation an S-oxide metabolite. This intermediately possesses a sufficiently electrophilic carbon, which may be subjected to a Michael type addition with cellular nucleophiles [40]. Some drugs, containing thiophene ring have also been reported to act as suicide substrates of cytochrome P450 enzymes following S-oxidation [41, 42]. In this way, compounds 1, 2, 3 and 8 are interesting molecules selected in silico as novel Tau ligand candidates because they show desired pharmacophoric properties for biological activity and good pharmacotherapeutic profile including potential oral absorption, CNS activity and permeation of the blood brain barrier, providing novel structural basis for designing new bioactive molecules with potential application in Alzheimer’s disease treatment. The presence of the toxicophoric groups within these compounds
is not a guarantee that the toxic effects described will be demonstrated, but they constitute alerts that should be investigated experimentally in order to determine the importance of the aromatic amine and the thiophene groups for the development of toxic mechanisms, giving insights for future chemical modifications able to optimize the pharmaceutical profile of such molecules [27].
CONCLUSIONS
Compounds 1, 2, 3 and 8 are promising thiophene derived drug candidates for tauopathies regarding their potential inhibitory activity against Tau aggregation, thus demonstrated by pharmacophoric approaches, pharmacotherapeutic profiles and favourable structure-activity relationships with respect to tau protein. The major in silico drawback identified for such molecules is the presence of toxicophoric groups that may cause carcinogenicity, mutagenicity and hepatoxicity. Nevertheless, the positive chemical characteristics of such molecules generate a novel structural concept that can be furtheroptimized for development of novel Tau ligands with adequate pharmaceutical properties, particularly in view of the fact that any Tau ligand has still been approved for tauopathies treatment.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We acknowledge financial support from CNPq, CAPES, FAPERJ and FAPESP.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
REFERENCES [1] Mukrasch, M.D.; Biernat, J.; von Bergen, M.; Griesinger, C.;
Mandelkow, E.; Zweckstetter, M. Sites of tau important for aggregation populate beta-structure and bind to microtubules and polyanions. J. Biol. Chem., 2005, 280(26), 24978-24986.
[2] Jeganathan, S.; Von Bergen, M.; Mandelkow, E.; Mandelkow E. The natively unfolded character of tau and its aggregation to alzheimer-like paired helical filaments. Biochemistry, 2008, 47, 10526-10539.
[3] Hattori, M.; Sugino, E.; Minoura, K.; In, Y.; Sumida, M.; Taniguchi, T.; Tomoo, K.; Ishida, T. Different inhibitory response of cyaniding and methylene blue for filament formation of tau microtubule-binding domain. Biochem. Bioph. Res. Commun., 2008, 374, 158-163.
[4] Wischik, C.M.; Edwards, P.C.; Lai, R.Y.K.; Roth, M.; Harrington, C. R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. U.S.A., 1996, 93, 11213-11218.
[5] Pickhardt, M.; Gazova, Z.; von Bergen, M.; Khlistunova, I.; Wang, Y.P.; Hascher, A.; Mandelkow, E.M.; Biernat, J.; and Mandelkow, E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J. Biol. Chem., 2005, 280, 3628-3635.
[6] Pickhardt, M.; Larbig, G.; Khlistunova, I.; Coksezen, A.; Meyer, B., Mandelkow, E.M.; Schmidt, B.; Mandelkow, E. Phenylthiazolyl- hydrazide and its derivatives are potent inhibitors
Table 3. Toxicity prediction by identification of toxicophoric groups in the structures of the novel drug candi- datesagainst tauopathies.
Molecule Toxicity Prediction Toxicophoric Group
1 Hepatotoxicity Thiophene
2 Hepatotoxicity Thiophene
3 Hepatotoxicity Thiophene
4 -- --
5
Carcinogenicity
Hepatotoxicity
Skin sensitization
Furan
Furan/Thiophene
Hydrazine
6 --- ---
7
Carcinogenicity
Chromosome damage
Hepatotoxicity
Mutagenicity
Aromatic nitro
8 Carcinogenicity
Mutagenicity Aromatic amine
9 Phototoxicity
Skin sensitization
Diaryl Ketone
Phenol
10 Phototoxicity Diaryl Ketone
11 Phototoxicity Diaryl Ketone
12 Phototoxicity Diaryl Ketone
13 Phototoxicity Diaryl Ketone
14 Carcinogenicity
Mutagenicity Aromatic amine
15 --- ---
16 Hepatotoxicity
Skin sensitization
Furan
Phenol

44 Current Physical Chemistry, 2014, Vol. 4, No. 1 Pedersoli-Mantoani et al.
of tau aggregation and toxicity in vitro and in cells. Biochemistry, 2007, 46, 10016-10023.
[7] Bulic, B.; Pickhardt, M.; Khlistunova, I.; Biernat, J.; Mandelkow, E. M.; Mandelkow, E.; Waldmann, H. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew. Chem. Int. Ed., 2007, 46, 9215-9219.
[8] Bulic, B.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E. Tau protein and tau aggregation inhibitors. Neuropharmacology, 2010, 59, 276-289.
[9] Harrington, C.R.; Ricard, J.E.; Horsley, D.; Harrington, K.A.; Hindley, K.P.; Riedel, G.; Theuring, F.; Seng, K.M.; Wischik, C. M. Methylthioninium chloride (MTC) acts as a tau aggregation inhibitor in a cellular model and reverses tau pathology in transgenic mice models of Alzheimer’s disease. Int. Conf. Alz. Dis. Abs., 2008.
[10] Muegge, I.; Oloff, S. Advances in virtual screening. Drug Discov. Today, 2006, 3(4), 405-11.
[11] Sun, H. Pharmacophore-based virtual screening. Curr. Med. Chem., 2008, 15(10), 1018-1024.
[12] Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem., 2005, 280, 7614-7623.
[13] Chem. Sketch, version 11.0, http://chemskecth.softonic.com.br [14] Discovery Studio, version 3.1, Accelrys Software Inc., San Diego,
CA, 2011. [15] Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.;
Wolfson, H.J. Pharma Gist: a webserver for ligand-based pharma- cophore detection. Nucleic Acids Res., 2008, 36, W223-W228.
[16] Inbar, Y.; Schneidman-Duhovny, D.; Dror, O.; Nussinov, R. Wolfson, H.J. Deterministic pharmacophore detection via multiple exible alignment of drug-like molecules. In Speed, T.P.; Huang, H., (eds), Research in Computational Molecular Biology (RECOMB), 11th Ann. Internat. Conf. Springer, Oakland, CA, USA, 2007, 4453, 412-429.
[17] Lu, S.H.; Wu, J. W.; Liu, H.L.; Zhao, J.H.; Liu, K.T.; Chuang, C.K.; Lin, H.Y.; Tsai, W.B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophoremodeling, virtual screening, and molecular docking studies. J. Biom. Sci., 2011, 18(8), 1-13.
[18] Chembridge Corporation, San Diego, CA, USA, 2011. [19] Waltham, M.A. Maybridge Screening Collection; Thermo Fisher
Scientific, 2004. [20] Irwin, J.J.; Shoichet, B.K. ZINC - a free database of commercially
available compounds for virtual screening. J. Chem. Inf. Comp. Sci., 2005, 45, 177-82.
[21] Goodford, P.J. A computational procedure for determining energetically favourable binding sites on biologically important macromolecules. J. Med. Chem., 1985, 28, 849-857.
[22] InsightII, version 2005, Accelrys: CA, USA, 2005. [23] Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. Binding DB:
a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res., 2007, 35, D198-D201.
[24] Poroikov, V.V.; Filimonov, D.A.; Ihlenfeldt, W.D.; Gloriozova, T. A.; Lagunin, A.A.; Borodina, Y.V.; Stepanchikova, A.V.; Nicklaus, M.C. PASS Biological Activity Spectrum Predictions in the Enhanced Open NCI Database Browser. J. Chem. Inf. Comput. Sci., 2003, 43, 228-236.
[25] QikProp, version 3.4, Schrödinger, L.L.C., New York, NY, 2011 [26] Derek for Windows, version 10.0.2, Lhasa ltd., UK, Leeds, 2007. [27] Ridings, J.E.; Barratt, M.D.; Cary, R.; Earnshaw, C.G.; Eggington,
C.E.; Ellis, M.K.; Judson, P.N.; Langowski, J.J.; Marchant, C.A.; Payne, M.P.; Watson, W.P.; Yih, T.D. Computer prediction of possible toxic action from chemical structure: an update on the DEREK system. Toxicology, 1996, 106, 267-279.
[28] Han, L.; Shi, S.; Zheng, L.; Yang, D.; Yao, T.; Ji, L. Flavonoids inhibit heparin-induced aggregation of the third repeat (r3) of microtubule-binding domain of alzheimer’s tau protein. Bull. Chem. Soc. Jpn., 2010, 83, 911-922.
[29] Pedersoli-Mantoani, S.; da Silva, C.H.T.P.; Silva, V.B. In silico binding mode proposed of flavonoid ligands of tau protein with interest in alzheimer's disease. Curr. Bioact. Compd., 2013, 9, 21-26.
[30] Bulic, B.; Pickhardt, M.; Schmidt, B.; Mandelkow, E.; Waldmann, H.; Mandelkow, E. Development of tau aggregation inhibitors for alzheimer’s disease. Angew. Chem. Int. Ed., 2009, 48, 1740-1752.
[31] Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med., 2001, 344, 1688-1700.
[32] Shaw, P.J. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry, 2005, 76 (8), 1046-1057.
[33] Verna, L.; Whysner, J.; Willliams, G.M. 2-Acetylaminofluorene mechanistic data and risk assessment: DNA reactivity, enhanced cell proliferation and tumor initiation. Pharmacol. Therapeut., 1996, 71, 83-105.
[34] Miller, E.C. Some current perspectives on chemical carcinogenesis in humans and experimental animals: presidential address. Cancer Res., 1978, 38, 1479-1496.
[35] Andersen, R.A.; Enomoto, M.; Miller, E.C.; Miller, J.A. Carcinogenesis and inhibition of the Walker 256 tumor in the rat by trans-4-acetylaminostilbene, its N-hydroxy metabolite, and related compounds. Cancer Res., 1964, 24, 128-143.
[36] Miller, J.A. Carcinogenesis by chemicals: an overview - G.H.A. Clowes memorial lectura. Cancer Res., 1970, 30, 559-576.
[37] Turesky, R.J.; Lang, N.P.; Butler, M.A.; Teitel, C.H.; Kadlubar, F.F. Metabolic activation of carcinogenic heterocyclic aromatic amines by human liver and colon. Carcinogenesis, 1991, 12, 1839-1845.
[38] Greene, N.; Fisk, L.; Naven, R.T.; Note, R.R.; Patel, M.L.; Pelletier, D.J. Developing structure-activity relationships for the prediction of hepatotoxicity. Chem. Res. Toxicol., 2010, 23, 1215-1222.
[39] McMurtry, R.J.; Mitchell, J.R. Renal and hepatic necrosis after metabolic activation of 2-substituted furans and thiophenes, including furosemide and cephaloridine. Toxicol. Appl. Pharmacol., 1977, 42, 285-300.
[40] Dalvie, D.K.; Kalgutkar, A.S.; Khojasteh-Bakht, S.C.; Obach, R.S.; O’Donnell, J.P. Biotransformation reactions of five membered aromatic heterocyclic rings. Chem. Res. Toxicol., 2002, 15, 269-299.
[41] Beaune, P.H.; Lecoeur, S. Immunotoxicology of the liver: adverse reactions to drugs. J. Hepatol., 1997, 26, 37-42.
[42] O’Donnell, J.P.; Dalvie, D.K.; Kalgutkar, A.S.; Obach, R.S. Mechanism-based inactivation of human recombinant P4502C9 by the nonsteroidal anti-inflammatory drug suprofen. Drug Metab. Dispos., 2003, 31, 1369-1377.
Received: December 31, 2011 Revised: May 31, 2012 Accepted: July 31, 2012
Related Documents











