Hurler Syndrome Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources G Souillet 1 , N Guffon 2 , I Maire 3 , M Pujol 1 , P Taylor 2 , F Sevin 2 , N Bleyzac 5 , C Mulier 1 , A Durin 1 , K Kebaili 1 , C Galambrun 1 , Y Bertrand 1 , R Froissart 3 , C Dorche 3 , L Gebuhrer 6 , C Garin 4 , J Berard 4 and P Guibaud 2 1 Department of Paediatric Immuno-Hematology and Bone Marrow Transplantation, Debrousse Hospital, Lyon, France; 2 Paediatric Department, Debrousse Hospital, Lyon, France; 3 Centre of Studies for Metabolic Diseases, Debrousse Hospital, Lyon, France; 4 Orthopaedic Department, Edouard Herriot Hospital, Lyon, France; 5 Pharmacy Department, Debrousse Hospital, Lyon, France; and 6 Histocompatibility Laboratory, Etablissement Franc¸ais du Sang Rho ˆne-Alpes, Lyon, France Summary: Over the last 15 years, we have performed a total of 30 haematopoietic stem cell transplants on 27 children suffering from Hurler’s syndrome. These children were of median age 11 months at the time of diagnosis and 25 months at the time of transplantation. The phenotype was severe in 21 cases (78%). The donor was familial in 13 cases: nine genotypically identical, one phenotypically identical father and three HLA-mismatched donors. Unrelated donors were selected in 17 cases: four phenotypically identical and 13 with 1–4 HLA mis- matches. The conditioning regimen generally consisted of busulphan 600 mg/m 2 plus cyclophosphamide (Endox- an s ) 260 mg/kg and cyclosporin with methotrexate for GvHD prophylaxis. Rabbit anti-thymocyte globulin (Thymoglobuline s ) was given for all unrelated or familial mismatched transplantations. The median nucleated cell dose infused was 6.00 10 8 TNC/kg. No bone marrow (apart from one) was T cell depleted. For first transplants, engraftment was observed in 23/27 patients (pts) (85%). Primary graft failure was observed in 4/27 patients (16%), two were retransplanted from an unrelated donor, one with success. Four patients have died. The primary cause of death was infection in three cases (TRM : 11%) and disease progression in one case, after primary graft failure. Of the 23 living patients, two have disease progression after graft failure and 21 (78%) have functional grafts with a favourable long-term outcome after a median follow-up of 4.7 years, having either full or mixed chimaerism. Among surviving patients with func- tional grafts, 13 (62%) were transplanted from unrelated donors of whom 10 (77 %) had HLA disparities. There was a remarkably low incidence of GvHD. In our experience, haematopoietic stem cell transplantation using an HLA-matched familial donor or an HLA-matched or - mismatched unrelated donor without T cell depletion or irradiation can achieve a favourable outcome in Hurler’s syndrome, with improved cognitive function, but with a limited effect on the corneas and skeleton. Bone Marrow Transplantation (2003) 31, 1105–1117. doi:10.1038/sj.bmt.1704105 Keywords: Hurler’s syndrome; mucopolysaccharidosis; unrelated donor; related donor; haematopoietic stem cell transplantation; HLA; thymoglobuline Hurler’s syndrome (MPS IH) is a mucopolysaccharidosis due to a deficiency of lysosomal enzyme activity of a-l- iduronidase, which is needed to degrade glycosaminogly- cans (GAGS). The consequent accumulation of heparan sulphate and dermatan sulphate substrates leads to the characteristic facial features, hepatosplenomegaly, cardiac and pulmonary disease, progressive mental retardation and skeletal abnormalities, which become evident by 1 year of age in the severe form. The consequence of this progressive autosomal recessive inborn error is premature death by 8 years of age. Since 1980, bone marrow transplantation (BMT) has been used for the treatment of patients with lysosomal storage diseases with the objective of correcting the inborn storage error by replacing the patient’s defective macrophages by marrow-derived donor macrophages. Hobbs et al 1 and Krivit et al 2 showed clearly that BMT could provide metabolic components and cells that can rapidly correct the enzyme deficiency. Since this first report, selected children with Hurler’s syndrome have been transplanted worldwide with a favourable outcome in terms of improvement of neuropsychological functions and long-term survival, 3,4 but with progression of skeletal and corneal abnormalities. The encouraging results obtained with genotypically identical donors 5 prompted some teams to extend to other related donors or to unrelated donors. 6 In a previous study, 7 we reported our experience in 13 patients with Hurler’s syndrome, which included three unrelated bone marrow transplants. Here we report 15 years of cumulative experience for 27 patients suffering from Hurler’s syndrome who received 30 Received 28 May 2002; accepted 13 January 2003 Correspondence: Dr G Souillet, Immuno-Hematology and Bone Marrow Transplantation, Debrousse Hospital, 29, Rue Soeur Bouvier, 69005 Lyon, France Bone Marrow Transplantation (2003) 31, 1105–1117 & 2003 Nature Publishing Group All rights reserved 0268-3369/03 $25.00 www.nature.com/bmt

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Hurler Syndrome
Outcome of 27 patients with Hurler’s syndrome transplanted from either
related or unrelated haematopoietic stem cell sources
G Souillet1, N Guffon2, I Maire3, M Pujol1, P Taylor2, F Sevin2, N Bleyzac5, C Mulier1, A Durin1,K Kebaili1, C Galambrun1, Y Bertrand1, R Froissart3, C Dorche3, L Gebuhrer6, C Garin4, J Berard4
and P Guibaud2
1Department of Paediatric Immuno-Hematology and Bone Marrow Transplantation, Debrousse Hospital, Lyon, France; 2PaediatricDepartment, Debrousse Hospital, Lyon, France; 3Centre of Studies for Metabolic Diseases, Debrousse Hospital, Lyon, France;4Orthopaedic Department, Edouard Herriot Hospital, Lyon, France; 5Pharmacy Department, Debrousse Hospital, Lyon, France; and6Histocompatibility Laboratory, Etablissement Francais du Sang Rhone-Alpes, Lyon, France
Summary:
Over the last 15 years, we have performed a total of 30haematopoietic stem cell transplants on 27 childrensuffering from Hurler’s syndrome. These children wereof median age 11 months at the time of diagnosis and 25months at the time of transplantation. The phenotype wassevere in 21 cases (78%). The donor was familial in 13cases: nine genotypically identical, one phenotypicallyidentical father and three HLA-mismatched donors.Unrelated donors were selected in 17 cases: fourphenotypically identical and 13 with 1–4 HLA mis-matches. The conditioning regimen generally consistedof busulphan 600mg/m2 plus cyclophosphamide (Endox-ans) 260mg/kg and cyclosporin with methotrexate forGvHD prophylaxis. Rabbit anti-thymocyte globulin(Thymoglobulines) was given for all unrelated or familialmismatched transplantations. The median nucleated celldose infused was 6.00� 108 TNC/kg. No bone marrow(apart from one) was T cell depleted. For first transplants,engraftment was observed in 23/27 patients (pts) (85%).Primary graft failure was observed in 4/27 patients(16%), two were retransplanted from an unrelated donor,one with success. Four patients have died. The primarycause of death was infection in three cases (TRM : 11%)and disease progression in one case, after primary graftfailure. Of the 23 living patients, two have diseaseprogression after graft failure and 21 (78%) havefunctional grafts with a favourable long-term outcomeafter a median follow-up of 4.7 years, having either full ormixed chimaerism. Among surviving patients with func-tional grafts, 13 (62%) were transplanted from unrelateddonors of whom 10 (77 %) had HLA disparities. Therewas a remarkably low incidence of GvHD. In ourexperience, haematopoietic stem cell transplantation usingan HLA-matched familial donor or an HLA-matched or -
mismatched unrelated donor without T cell depletion orirradiation can achieve a favourable outcome in Hurler’ssyndrome, with improved cognitive function, but with alimited effect on the corneas and skeleton.Bone Marrow Transplantation (2003) 31, 1105–1117.doi:10.1038/sj.bmt.1704105Keywords: Hurler’s syndrome; mucopolysaccharidosis;unrelated donor; related donor; haematopoietic stem celltransplantation; HLA; thymoglobuline
Hurler’s syndrome (MPS IH) is a mucopolysaccharidosisdue to a deficiency of lysosomal enzyme activity of a-l-iduronidase, which is needed to degrade glycosaminogly-cans (GAGS). The consequent accumulation of heparansulphate and dermatan sulphate substrates leads to thecharacteristic facial features, hepatosplenomegaly, cardiacand pulmonary disease, progressive mental retardation andskeletal abnormalities, which become evident by 1 year ofage in the severe form. The consequence of this progressiveautosomal recessive inborn error is premature death by 8years of age. Since 1980, bone marrow transplantation(BMT) has been used for the treatment of patients withlysosomal storage diseases with the objective of correctingthe inborn storage error by replacing the patient’s defectivemacrophages by marrow-derived donor macrophages.Hobbs et al1 and Krivit et al2 showed clearly that BMTcould provide metabolic components and cells that canrapidly correct the enzyme deficiency. Since this first report,selected children with Hurler’s syndrome have beentransplanted worldwide with a favourable outcome interms of improvement of neuropsychological functions andlong-term survival,3,4 but with progression of skeletal andcorneal abnormalities. The encouraging results obtainedwith genotypically identical donors5 prompted some teamsto extend to other related donors or to unrelated donors.6
In a previous study,7 we reported our experience in 13patients with Hurler’s syndrome, which included threeunrelated bone marrow transplants.Here we report 15 years of cumulative experience for 27
patients suffering from Hurler’s syndrome who received 30Received 28 May 2002; accepted 13 January 2003
Correspondence: Dr G Souillet, Immuno-Hematology and BoneMarrow Transplantation, Debrousse Hospital, 29, Rue Soeur Bouvier,69005 Lyon, France
Bone Marrow Transplantation (2003) 31, 1105–1117& 2003 Nature Publishing Group All rights reserved 0268-3369/03 $25.00
www.nature.com/bmt

haematopoietic cell transplants: 13 from relateddonors (including two cord blood transplants), and 17from unrelated bone marrow donors (UBMDs). Inour centre, the first transplant from an UBMD for Hurler’sdisease was carried out in 1990 from an HLA-mismatc-hed donor with promising results which encouraged usto proceed with such a transplant when an HLA-identicalUBMD was not identified right away. This optionwas proposed in order not to delay BMT. In cases ofUBMDs, Thymoglobulines was added to the standardchemotherapy conditioning regimen, which did not includedonor T-cell depletion or irradiation. The purpose ofthis publication is to update our results for the treatmentof Hurler’s syndrome7 using related and unrelated haema-topoietic cell sources, and to focus on the beneficialeffects and the limitations of the procedure, particularlyin patients with a follow-up longer than 3 years posttransplantation.
Materials and methods
Patient characteristics
From January 1986 to December 2001, we performed 30consecutive transplants on 27 patients with Hurler’ssyndrome: 15 girls and 12 boys aged 11 months (m)(median) at the time of diagnosis (range: 2–87m) and
25months (median) at the time of transplantation (range:14–96 m). In three cases, patients were transplanted twice;from the same identical sibling donor after graft rejection inone case, and from a UBMD after graft failure from thefather as donor in two cases. Patient characteristics areshown in Table 1. Patients no. 10, 16 and patients no. 18, 20are siblings; in the former family, despite the disease in oneof their children, the parents were opposed to prenataldiagnosis and patient no. 16 was diagnosed at birth. In thelatter family, the diagnosis was made in the older child atthe time of birth of the second who also suffered from thedisease.Clinical diagnosis was confirmed by increased excretion
of dermatan and heparan sulphates in the urine8 anddeficiency of a-l-iduronidase in leucocytes.9,10 The threecommon mutations in France (Q70X, P533R, W402X)were sought in all patients and a few patients had completegene sequencing carried out. The phenotypes, evaluated bythe same clinicians in all patients are classified as follows:21 patients (78%) had a severe phenotype characterised byearly diagnosis at a median age of 10 months (range: 2–40m) and/or signs of severe dysostosis and dysmorphia;five patients had an intermediate phenotype with latediagnosis (median 38m), moderate dysmorphia and dysos-tosis and multiple hyperdense areas in the white matter onMRI examination. One patient (no. 4) had a moderatephenotype with a diagnosis made at 7 years 3 monthswith only macrocrania, short stature, very mild dysostosis,
Table 1 Patient’s characteristics
Patients no Ethnic origin Age at diagnosis Age at transplant Date of BMT Phenotype Genotype Current age
1 French 7 m 14 m 31/01/86 Severe W402X/Q70X 17 y 1 m2 French 2 y 3 m 4 y 10 m 27/02/87 Severe Q70X/ ? 19 y 8 m3 French 2 y 3 m 3 y 23/01/90 Severe W402X/134del12 14 y 11 m4 French 7 y 3 m 8 y 05/09/91 Mild P533R/P533R 18 y 2 m5 North African 4 y 8 m 5 y 7 m 18/02/92 Intermediate S633L/S633L5� 6 y 8m (2e) 03/03/93 15 y6 North African 3 y 2 m 6 y 5 m 15/12/92 Intermediate L209R/L209R 15 y 5 m7 French 5 m 20 m 27/01/93 Severe W402X/W402X7� 2 y 10m (2e) 24/03/94 Deceased8 French 7 m 14 m 09/03/93 Severe W402X/W402X 10 y9 North African 4 y 8 m 7 y 3 m 29/01/94 Intermediate P533R/Y581X Deceased10 North African 3 y 4 y 3 m 04/11/94 Intermediate P533R/P533R 11 y 6 m11 French 11 m 4 y 1 m 14/02/95 Severe W402X/ ? 11 y12 French 13 m 23 m 12/01/96 Severe W402X/1124delC 7 y 10 m13 French 7 m 14 m 20/09/96 Severe W402X/Q70X 6 y 5 m14 French 8 m 14 m 17/01/97 Severe W402X/W402X14� 26m (2e) 23/01/98 6 y 1 m15 French 9 m 21 m 07/03/97 Severe W402X/134del 12 6 y 7 m16 North African 2 m 16 m 02/04/97 Intermediate P533R/P533R 6 y 3 m17 French 15 m 2 y 1 m 13/06/97 Severe W402X/W402X 6 y 7 m18 French 3 y 4 m 3 y 10 m 03/04/98 Severe W402X/Q70 X Deceased19 French 6 m 2 y 9 m 03/11/98 Severe W402X/ ? Deceased20 French 2 m 17 m 06/11/98 Severe W402X/Q70X 4 y 6 m21 French 12 m 20 m 20/11/98 Severe W402X/Q70X 4 y 9 m22 French 11 m 26 m 16/04/99 Severe W402X/? 4 y 10 m23 French 26 m 35 m 01/10/99 Severe Q70X/? 5 y 1 m24 French 9 m 25 m 04/11/99 Severe W402X/W402X 4 y 3 m25 North African 15 m 23 m 16/06/00 Severe P533R/? 3 y 6 m26 N. African/French 10 m 22 m 23/02/01 Severe W402X/ ? 2 y 8 m27 French 8 m 19 m 13/04/01 Severe W402X/W402X 2 y 3 m
�=second BMT; ?=allele mutation have not yet been characterised; m=months; y=years.
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1106
Bone Marrow Transplantation

corneal clouding, a normal MRI and an intelligencequotient (IQ) of 103.
Eligibility for BMT
The decision about eligibility of a patient for BMT resultsfrom a detailed multidisciplinary evaluation includingdevelopmental scales, ID/IQ testing, CNS function (includ-ing peripheral nerves and brain), neuroradiology andmetabolic and genetic studies to assess and correlatephenotypic and genotypic severity. The decision to includea patient in a BMT programme is made before it is knownwhether or not a suitable family donor exists. In theabsence of a genotypically identical sibling donor, UBMDsearches are rapidly initiated with the purpose of obtaininga suitable donor before irreversible cerebral damage hasoccurred. Patients were included in a BMT programmeafter local consensus that they had the required intelligencequotient/development quotient (IQ/DQ) and good clinicalstatus. For all patients with the severe phenotype, the IQ/DQ required for graft eligibility is X70.Patients who did not have HLA-identical siblings
were enrolled in an unrelated BMT procedure. Searchesfor an HLA-A, -B, -DR-identical donor were startedsimultaneously on every registry and were often extendedto donors having one or more HLA class Imismatches.
Donor characteristics and HLA data
Patients who received transplants from relatives weregenotyped at the time of the familial investigation for arelated donor. Their HLA typing was determined serolo-gically by standard two-stage complement-dependentmicrolymphocytotoxicity testing.11 For unrelated donors,HLA typing was also initially performed by serology. Atthe end of 1991, patients and donors were typed byserology for class I and molecular biology for class II. In1996, DNA typing for HLA class I alleles was introduced.Currently, we perform DNA typing of alleles at ninepolymorphic loci: alleles at the HLA-A*, -B*, -Cw*,-DRB1*, -B3*, -B4*, -B5*, -DQB1* and -DPB1* identifiedwith the use of the polymerase chain reaction (PCR) withsequence-specific oligonucleotide probes12,13 or PCR-sequence-specific primers. High-resolution typing for classI and/or class II alleles were retrospectively completed forrecipient and donor pairs who had been transplantedbetween 1990 and 1996.The donor was related in 13 cases: nine were genotypi-
cally identical, of whom one was a cord blood transplant,one was a phenotypically identical father and three wereHLA-mismatched donors (father in two cases and cordblood in one).Unrelated donors were selected in 17 cases: four were
phenotypically identical donors and 13 had between oneand four HLA mismatches (DPB1* mismatches not takeninto account). Donor origin, characteristics and HLA dataare detailed in Table 2.
Supportive care
All patients had autologous bone marrow harvestingduring anaesthesia for insertion of indwelling centralvenous catheters. Patients were placed under sterile isolatoror laminar air flow and received sterile care and oral andgut decontamination. All received polyvalent intravenousimmunoglobulins, usually for 6m, and oral prophylaxis ofinfection with penicillin, acyclovir and trimethoprim/sulphamethoxazole until 15m post transplantation. Allblood products were irradiated and selected from CMV-negative donors.
Conditioning regimen
The preparative regimen was initially based on the Hobbsprotocol14 consisting of chemotherapy alone with thecombination of busulphan (BU) and cyclophosphamide(CY) plus a donor buffy coat infusion (n¼ 10) for relatedtransplants. BU dosage was increased at the time of the firstunrelated transplant : BU from 500 to 600mg/m2 and CYfrom 200 to 260mg/kg was administered without irradia-tion except in one case of second related BMT after graftrejection (no. 5). This patient received CY 200mg/kgcombined with fractionated total body irradiation (TBI)(8Gy). Donor marrow was not T cell depleted beforeinfusion except in one patient (no. 4) who received a BMTdepleted by the use of rabbit complement and anti-CD2,-CD5, -CD7 monoclonal antibodies.Thymoglobulines15 was initially introduced for unre-
lated mismatched BMT because of its antirejection andanti-GvHD effect. It was given prior to transplantation, toobtain a partial in vivo T cell depletion, at a total dose of20mg/kg (5mg/kg at day –7, �5, �3, �1) and this protocolwas subsequently extended to all unrelated transplants,whether mismatched or not.
Pharmacokinetics of BU
In January 1998, we initiated a prospective study tomonitor BU plasma concentrations during the conditioningregimen. BU plasma concentrations were determined byliquid chromatography. For 10 patients transplanted fromUBMD, pharmacokinetic parameters were estimated byusing the USCPACK software program, a Bayesianmethod that allows the use of only two or three bloodsamples. The target area under the curve per dose (AUC)was about 4–6mg � h�1 �ml�1. During the conditioningregimen, four doses were given per day over fourconsecutive days. Daily pharmacokinetic studies wereperformed for each patient with adaptation of the dosageregimen for the remaining doses.16
GvHD prophylaxis
In both related and unrelated donor transplants, patientsreceived a combination of intravenous cyclosporin (CsA),5mg/kg/day beginning on day �2 for 5 days followed byintravenous CsA at 3mg/kg/day and converted to 12.5mg/kg/day orally when oral medication could be tolerated.CsA was continued for 9 months and tapered off over 3months. Intravenous methotrexate (MTX) was adminis-
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1107
Bone Marrow Transplantation
trered at 15mg/m2 on day 1 and at 10mg/m2 on days 3, 6,11. GvHD was graded according to Glucksberg et al17 andconfirmed by appropriate histological studies.
Cell dose
Our policy was to increase the graft cell dose above3.5� 108 TNC/kg recipient body weight for relatedtransplants, and to try to obtain at least 8� 108 TNC/kgfor unrelated transplants. Bone marrow was alwayspreferred to other haematopoietic cell sources. T cellreplete bone marrow was transfused on day 0. All patientsreceived nonfiltered bone marrow. See conditioning regi-men details in Table 3.
Psychological support for patient and family
The psychologist saw parents and children as soon aspossible after the diagnosis as the parents were very muchin need of support. This is when parents were made awareof the disease progression and life expectancy of their child.All patients were psychologically assessed using differenttests according to their developmental level and age. Under
the age of 3 years, we use the Brunet–Lezine locomotor andperformance scale for babies (BLR-E), which gives a DQ.From 3 to 6 years old, we use the Terman–Merril test(T.N.M.L-F), which gives a mental age and a general IQ.After the age of 6, we use the Wechsler Intelligence Scalefor children (WISCR-R), which gives a verbal IQ,performance IQ and an overall general IQ.
Assessment of BMT outcome and patient follow-up
Myeloid engraftment was defined as the first of threeconsecutive days when the absolute neutrophil count(ANC) exceeded 0.5� 109/l. The functionality of the graftwas assessed by measuring leucocyte a-l-iduronidaseenzyme activity, urinary GAG excretion, fingerprintstudies,18 cytogenetic studies when applicable and HLAtyping in mismatched pairs. Complete donor engraftmentwas determined by fingerprint analysis and defined asX95% donor cells at 1 month.Patients were monitored 1 month, 3 months, 6 months
and 1 year after BMT and then yearly. The biologicalfollow-up included fingerprint studies for evaluating thedonor/recipient origin of peripheral leucocytes, enzyme
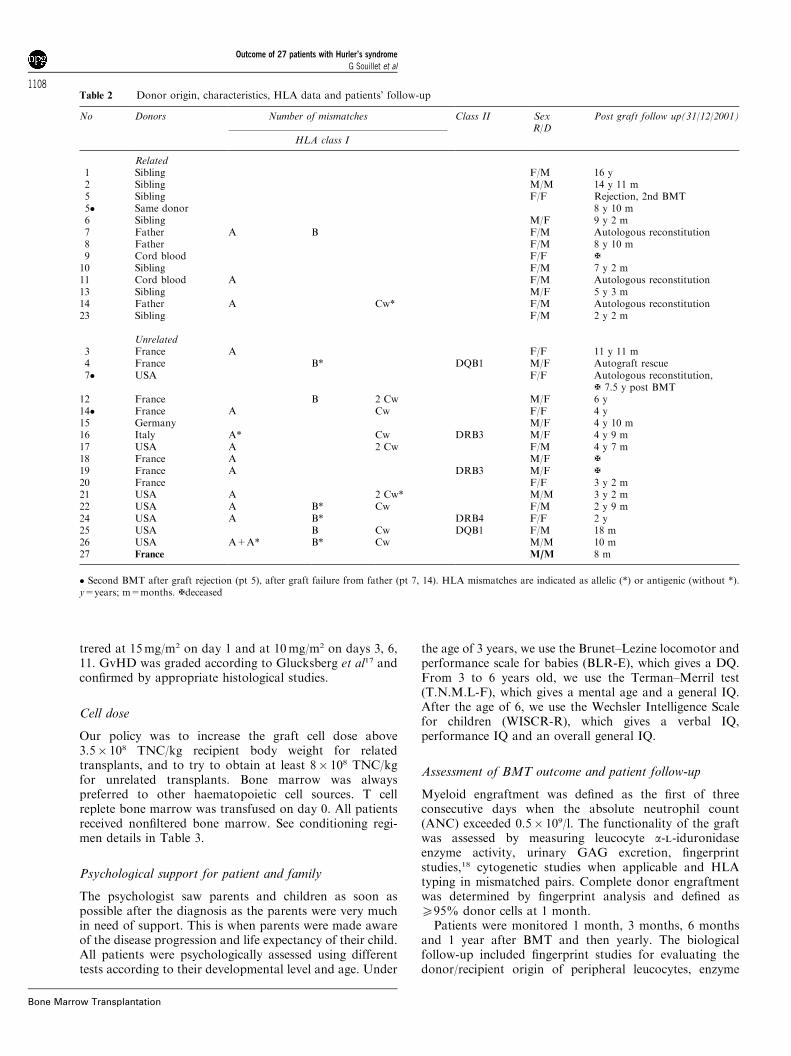
Table 2 Donor origin, characteristics, HLA data and patients’ follow-up
No Donors Number of mismatches Class II SexR/D
Post graft follow up(31/12/2001)
HLA class I
Related1 Sibling F/M 16 y2 Sibling M/M 14 y 11 m5 Sibling F/F Rejection, 2nd BMT5� Same donor 8 y 10 m6 Sibling M/F 9 y 2 m7 Father A B F/M Autologous reconstitution8 Father F/M 8 y 10 m9 Cord blood F/F {10 Sibling F/M 7 y 2 m11 Cord blood A F/M Autologous reconstitution13 Sibling M/F 5 y 3 m14 Father A Cw* F/M Autologous reconstitution23 Sibling F/M 2 y 2 m
Unrelated3 France A F/F 11 y 11 m4 France B* DQB1 M/F Autograft rescue7� USA F/F Autologous reconstitution,
{ 7.5 y post BMT12 France B 2 Cw M/F 6 y14� France A Cw F/F 4 y15 Germany M/F 4 y 10 m16 Italy A* Cw DRB3 M/F 4 y 9 m17 USA A 2 Cw F/M 4 y 7 m18 France A M/F {19 France A DRB3 M/F {20 France F/F 3 y 2 m21 USA A 2 Cw* M/M 3 y 2 m22 USA A B* Cw F/M 2 y 9 m24 USA A B* DRB4 F/F 2 y25 USA B Cw DQB1 F/M 18 m26 USA A+A* B* Cw M/M 10 m27 France M/M 8 m
� Second BMT after graft rejection (pt 5), after graft failure from father (pt 7, 14). HLA mismatches are indicated as allelic (*) or antigenic (without *).y=years; m=months. {deceased
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1108
Bone Marrow Transplantation
activity measurements in leucocytes and serum, andquantitative and qualitative studies of excreted urinaryGAGS.A complete multidisciplinary evaluation was per-
formed yearly including: DQ or IQ determination,cerebral and cervical (for the most recently transplantedpatients) MRI, audiological examinations includingexamination of the external auditory canal and tympanicmembrane, audiogram, orthophonic evaluation andauditory evoked potentials; ophthalmologic examinationswith visual acuity evaluation, slit-lamp examination,intraocular pressure measurement and visual evokedpotentials; nerve conduction velocity of the median nervefor carpal tunnel syndrome if not corrected by surgery,skeletal X-rays and joint mobility evaluation, echocardio-graphy and pulmonary X-ray, orthopaedic and neurologi-cal evaluation.
Informed consent
The risks of the BMT procedure and the expected benefitsand limitations of the transplant were fully explained to thefamily. Parents of all patients provided informed consentfor transplantation.
Statistical analysis
All 27 patients who received BMT for the treatment ofHurler’s disease were included in this study. The reportedoutcomes are as of 31 December 2001. The overall survival(including following the second graft) was evaluated by theKaplan–Meier method, and the log-rank test was used tocompare the probability of survival and graft functionalityfor the whole group, as well as for related or unrelatedrecipient–donor pairs, and by degree of HLA matching.All statistical analyses were performed using SPSS forWindows (version 9.0, SPSS, Chicago, IL, USA).
Results
BU dose adjustment at time of conditioning regimen
For seven out of the 10 patients included in this clinicaltrial, BU dosage regimens were decreased from 7 to 24%with regards to our previous dosage (600mg/m2) (Table 3).Dosage was increased for two patients who needed a totaldose of 688 and 612mg/m2. For the final patient, the dosewas unchanged. We observed an intraindividual variabilityof BU clearance for two-thirds of the children during the
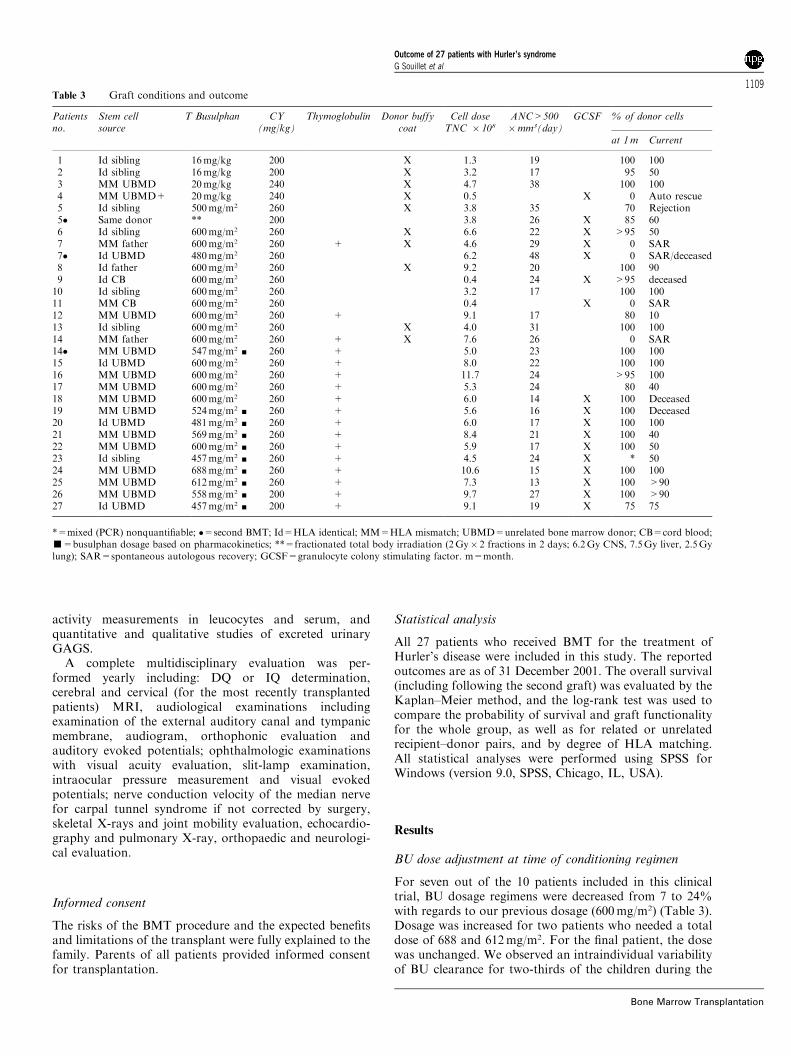
Table 3 Graft conditions and outcome
Patientsno.
Stem cellsource
T Busulphan CY(mg/kg)
Thymoglobulin Donor buffycoat
Cell doseTNC � 108
ANC>500�mm3(day)
GCSF % of donor cells
at 1m Current
1 Id sibling 16mg/kg 200 X 1.3 19 100 1002 Id sibling 16mg/kg 200 X 3.2 17 95 503 MM UBMD 20mg/kg 240 X 4.7 38 100 1004 MM UBMD+ 20mg/kg 240 X 0.5 X 0 Auto rescue5 Id sibling 500mg/m2 260 X 3.8 35 70 Rejection5� Same donor ** 200 3.8 26 X 85 606 Id sibling 600mg/m2 260 X 6.6 22 X >95 507 MM father 600mg/m2 260 + X 4.6 29 X 0 SAR7� Id UBMD 480mg/m2 260 6.2 48 X 0 SAR/deceased8 Id father 600mg/m2 260 X 9.2 20 100 909 Id CB 600mg/m2 260 0.4 24 X >95 deceased10 Id sibling 600mg/m2 260 3.2 17 100 10011 MM CB 600mg/m2 260 0.4 X 0 SAR12 MM UBMD 600mg/m2 260 + 9.1 17 80 1013 Id sibling 600mg/m2 260 X 4.0 31 100 10014 MM father 600mg/m2 260 + X 7.6 26 0 SAR14� MM UBMD 547mg/m2 ’ 260 + 5.0 23 100 10015 Id UBMD 600mg/m2 260 + 8.0 22 100 10016 MM UBMD 600mg/m2 260 + 11.7 24 >95 10017 MM UBMD 600mg/m2 260 + 5.3 24 80 4018 MM UBMD 600mg/m2 260 + 6.0 14 X 100 Deceased19 MM UBMD 524mg/m2 ’ 260 + 5.6 16 X 100 Deceased20 Id UBMD 481mg/m2 ’ 260 + 6.0 17 X 100 10021 MM UBMD 569mg/m2 ’ 260 + 8.4 21 X 100 4022 MM UBMD 600mg/m2 ’ 260 + 5.9 17 X 100 5023 Id sibling 457mg/m2 ’ 260 + 4.5 24 X * 5024 MM UBMD 688mg/m2 ’ 260 + 10.6 15 X 100 10025 MM UBMD 612mg/m2 ’ 260 + 7.3 13 X 100 >9026 MM UBMD 558mg/m2 ’ 200 + 9.7 27 X 100 >9027 Id UBMD 457mg/m2 ’ 200 + 9.1 19 X 75 75
*=mixed (PCR) nonquantifiable; �=second BMT; Id=HLA identical; MM=HLA mismatch; UBMD=unrelated bone marrow donor; CB=cord blood;’=busulphan dosage based on pharmacokinetics; **=fractionated total body irradiation (2Gy� 2 fractions in 2 days; 6.2Gy CNS, 7.5Gy liver, 2.5Gylung); SAR=spontaneous autologous recovery; GCSF=granulocyte colony stimulating factor. m=month.
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1109
Bone Marrow Transplantation

4 days of therapy. All these patients had successfulengraftment and no cases of veno-occlusive disease wereobserved during the post transplant period. In ourinstitution, BU clearance disposition for these 10 patientswith Hurler’s disease is 0.32770.164 l/h�1 kg�1, which ishigher than that observed in other patients (n¼ 35)suffering from other diseases: 0.23270.066 l/h�1 kg�1
(P¼ 0.025) (Bleyzac, personal communication).
Immediate tolerability of Thymoglobulines
As expected, all patients experienced fever at least onceduring the first infusion of Thymoglobulines and themajority developed a rash and hypotension. Some patientsdeveloped hypertension.
Engraftment
The median total nucleated cell dose infused per kg was6.00� 108 (range: 1.3–13.3); this calculation excludes thepatients who received T-cell depleted marrow (n¼ 1) orcord blood cells (n¼ 2).Engraftment was evaluable for all patients and initially
observed in 23 out of 27 patients (85%). The median timeto neutrophil engraftment was 21 days (range 16–48 days).Four patients had primary graft failure (16%): the patient(no. 4) who received T cell depleted marrow from an HLA-mismatched UBMD, two patients transplanted from twoclass I HLA antigen-mismatched fathers (no. 7, 14) and onepatient who received an HLA-A-mismatched related cordblood transplant (no. 11). The first (no. 4) receivedautologous transplant rescue. The others demonstratedautologous reconstitution. Among them, two (no. 7, 14)were retransplanted from UBMD, successfully in onepatient (no. 14).Patient no. 5 had secondary graft rejection 6 months
after full engraftment from an HLA geno-identical sibling.A second BMT from the same donor was performedsuccessfully, before the introduction of Thymoglobulines
into our strategy, with intensification of the conditioningregimen (irradiation).
Complications and GvHD
One patient (no. 26) had a severe episode of arterialhypertension. Another patient (no. 4) had intracranialhypertension, which resolved after shunting. Three patients(no. 16, 21, 22) suffered severe haemolytic anaemia thatresolved with corticosteroid treatment after tapering CsA,and, for one patient, after the use of the anti-CD20antibody rituximab (Mabtheras).Acute GvHD occurred in five patients (22%) following
their first transplant, of grade I (n¼ 3) and grade II (n¼ 2).The three patients who underwent a second BMT did notdevelop acute GvHD. None had chronic GvHD.
Transplant-related mortality
Four patients have died (15%). Three died of infection afterfull engraftment; invasive aspergillosis in one patient (no. 9)at day 26, one case of interstitial pneumonia (RSV) at day
28 (no. 18) and one case of septicaemia (Escherichia cali) atday 89 (no. 19). The transplant-related mortality (TRM)rate is equal to 11%. One patient (no. 7) died of diseaseprogression 7.5 years after failure of two transplants (froma mismatched father and from an identical UBMD withoutThymoglobulines for the second BMT) at the age of 9years and 2 months.
Survival
Overall, 23 (85%) patients survived. In all, 21 had afunctional graft (78%) 8 months to 15 years post BMT;in two cases this was following a second transplant (no. 5,no. 14). Two patients are alive with disease progression.The overall actuarial survival at 3 years is 85% with nodifference according to donor origin (familial 90%;unrelated 82.35, log rank 0.39) or HLA identity (HLAmatch 85.71%, HLA mismatch 84.62%, log rank 0.77)(Figure 1a–c).
Post transplant outcome
Biological outcome
For all patients with durable engraftment (n¼ 21), full(n¼ 11) or mixed (n¼ 10) chimaerism was obtained with afunctional graft. Enzyme activity measurements in recipientleucocytes were in agreement with the fingerprint resultsand homozygous or heterozygous donor status. Table 4reports the biological data in patients successfully engraftedfor more than 3 years. GAG urinary excretion (Table 4)was elevated before grafting and consisted mainly ofheparan and dermatan sulphate. After successful engraft-ment, excretion decreased to the upper limit of age-matchedcontrol values, but a low amount of dermatan sulphate anda hardly detectable fraction of heparan sulphate wereobserved on electrophoresis. No difference could beobserved on GAG excretion between patients engraftedwith a homozygous or heterozygous donor and betweenfully engrafted or chimeric patients.
Clinical outcome
For all engrafted patients, we observed the resolution ofhepatosplenomegaly during the first 3 months. Within 3–6months, persistent rhinorrhoea, obstruction of upper air-ways and hearing dramatically improved. Coarse facialfeatures resolved progressively. Cardiac murmurs or valvedysplasia remained stable and no myocardiopathy oc-curred. No pulmonary insufficiency was observed. Theeffect of BMT on skeletal and neuropsychological devel-opment is reported on the 15 patients (11 with severephenotype) who had stable engraftment with more than 3years of follow-up, with full (n¼ 8) or mixed (n¼ 7)chimaerism. Seven of these patients were transplantedfrom genotypically identical siblings and eight fromunrelated donors; all after the age of 14 months.
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1110
Bone Marrow Transplantation

Joint mobility
In the physiotherapist measurements, the 15 patients wereassessed before BMT and yearly. In every patient, there wasan improvement in joint mobility. Their improved shouldermovements and knee extension are the most impressivemeasurements. For shoulder antepulsion, 85% of patients
had a regular and analogous progression whatever the timefrom BMT (Figure 2). For one child, progression isstationary despite a 7-year follow-up and one other patientlost 101 of antepulsion with a 10-year follow-up. Thechanges are similar for knee extension. All patients, but oneimproved (Figure 3) including one patient who had a 01 ofknee extension at the time of BMT, and only one patientworsened, losing 51 of extension.
Orthopaedic management is shown in Table 5
Linear growth was maintained for some years and then fellto �1 s.d. for most patients (n¼ 10) and lower than �2 s.d.for six patients. Growth remained normal for patient no. 15and above the normal for patients no. 13, 20, 21. Whilegrowth was generally maintained or increased at the time ofBMT, it became evident that growth worsened with time,independent of the presence (nos. 3, 8) or absence ofkyphosis (nos. 2, 5). Growth velocity curves for boys andgirls are shown in Figures 4 and 5.Dorsolumbar spine kyphosis was present in 10 cases,
severe in eight and associated with scoliosis in three cases(no. 1, 12, 21). All except the two patients with slightkyphosis wore an adapted jacket for a prolonged period.Kyphoscoliosis worsened in patients no. 1 and no. 12 andrequired surgery. Patient no. 1 had posterior spinal fusionand anterior spinal fusion, respectively, 10 and 11 yearspost BMT; patient no. 12 had posterior spinal fusion 18months post BMT with improvement in both these cases.Spinal arthrodesis is planned for patients no. 13 and 21.Patient no. 2 (height - 3 s.d.) required a C1-C2 arthrodesisbecause of a cervical spine dislocation without othervertebral abnormality. In the case of the other patients,thoraco-lumbar kyphosis became stable with brace treat-ment. Despite the absence of kyphosis, patients no. 6 andno. 10 had poor growth (respectively �1.2 and �1.5 s.d.).Carpal tunnel syndrome developed in 10 out of these 15
patients (67%), all of whom who underwent decompressionsurgery before grafting in four cases, post BMT in six cases,with a favourable outcome in all cases.Genu valgum deformity was noted in 11/15 of our
patients (73%) with progressive increasing valgus defor-mity. Epiphyseal stapling was performed in two cases, 5and 8 years post BMT, respectively.Hip dysplasia was present in 10 cases. Nine patients
underwent surgical orthopaedic treatment, femoral varusosteotomy in seven cases, femoral varus osteotomy andiliac Salter osteotomy in one case, hip abutment alone inone case. Currently, no patient reports hip pain or has a hipdislocation despite acetabular and femoral capital epiphy-seal dysplasia. Patients are stabilised and no hip subluxa-tion has occurred.
Neuropsychological outcome
Psychological outcome is reported for the same 15 patients(Table 6). Long-term follow- up MRIs of the brain didnot show progressive cerebral atrophy or hydrocephalus.Before grafting, five children had a normal IQ/DQ (¼ 100).For the others (n¼ 10), IQ/DQ varied from 66 to 99. Thefollow-up shows that all children made progress initially,
Figure 1 Survival curves: (a) overall survival for the whole group; (b)overall survival function of donor origin (related/unrelated); (c) overallsurvival function of HLA identity (match/mismatch).
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1111
Bone Marrow Transplantation
they then developed difficulties and showed a decline inequivalent age score, and then appeared to stabilize(Figure 6). For five children: IQ/DQX100, for 10 children,IQ/DQ was between 77 and 99. No patient had severemental retardation. All the patients are going to school.
They all adapted well to nursery school, but learningdifficulties began to be obvious on entering elementaryschool. Owing to their broad hands and stubby digits, somehave problems in drawing and writing, but they learn toadapt well with time. The majority of them have languagetroubles and require speech therapy. Among five patients
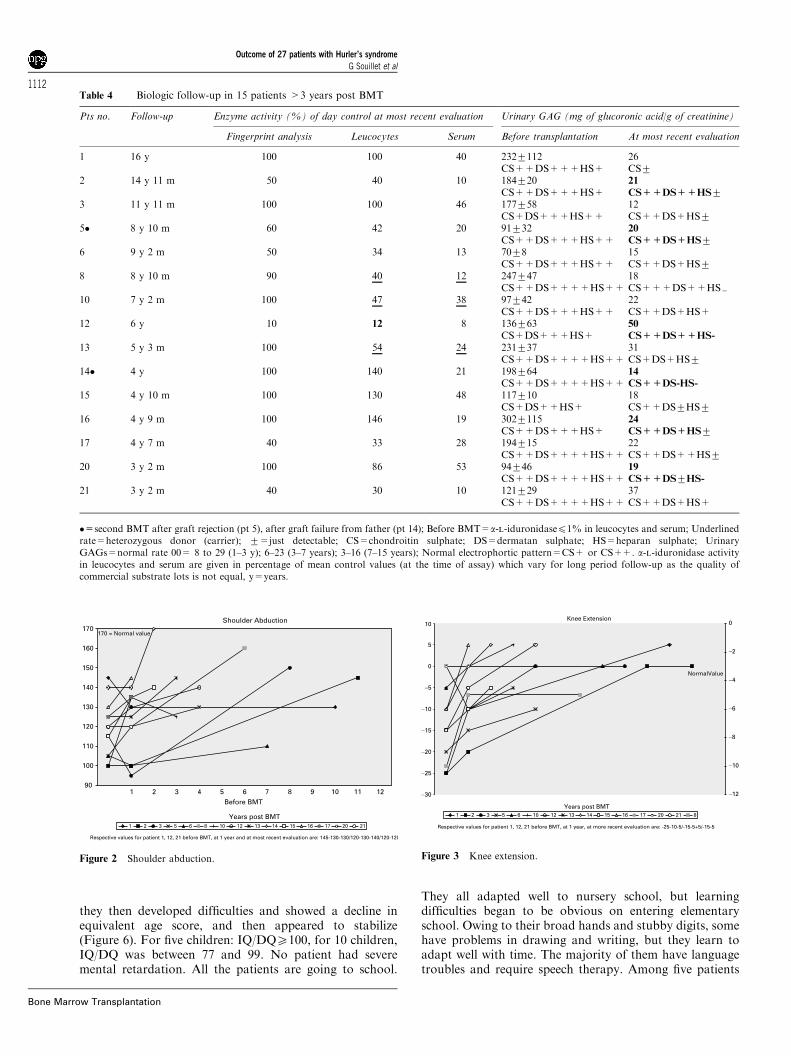
Table 4 Biologic follow-up in 15 patients >3 years post BMT
Pts no. Follow-up Enzyme activity (%) of day control at most recent evaluation Urinary GAG (mg of glucoronic acid/g of creatinine)
Fingerprint analysis Leucocytes Serum Before transplantation At most recent evaluation
1 16 y 100 100 40 2327112 26CS++DS+++HS+ CS7
2 14 y 11 m 50 40 10 184720 21
CS++DS+++HS+ CS++DS++HS73 11 y 11 m 100 100 46 177758 12
CS+DS+++HS++ CS++DS+HS75� 8 y 10 m 60 42 20 91732 20
CS++DS+++HS++ CS++DS+HS76 9 y 2 m 50 34 13 7078 15
CS++DS+++HS++ CS++DS+HS78 8 y 10 m 90 40 12 247747 18
CS++DS++++HS++ CS+++DS++HS=10 7 y 2 m 100 47 38 97742 22
CS++DS+++HS++ CS++DS+HS+12 6 y 10 12 8 136763 50
CS+DS+++HS+ CS++DS++HS-
13 5 y 3 m 100 54 24 231737 31CS++DS++++HS++ CS+DS+HS7
14� 4 y 100 140 21 198764 14
CS++DS++++HS++ CS++DS-HS-
15 4 y 10 m 100 130 48 117710 18CS+DS++HS+ CS++DS7HS7
16 4 y 9 m 100 146 19 3027115 24
CS++DS+++HS+ CS++DS+HS717 4 y 7 m 40 33 28 194715 22
CS++DS++++HS++ CS++DS++HS720 3 y 2 m 100 86 53 94746 19
CS++DS++++HS++ CS++DS7HS-
21 3 y 2 m 40 30 10 121729 37CS++DS++++HS++ CS++DS+HS+
�=second BMT after graft rejection (pt 5), after graft failure from father (pt 14); Before BMT=a-l-iduronidasep1% in leucocytes and serum; Underlinedrate=heterozygous donor (carrier); 7=just detectable; CS=chondroitin sulphate; DS=dermatan sulphate; HS=heparan sulphate; UrinaryGAGs=normal rate 00= 8 to 29 (1–3 y); 6–23 (3–7 years); 3–16 (7–15 years); Normal electrophortic pattern=CS+ or CS++. a-l-iduronidase activityin leucocytes and serum are given in percentage of mean control values (at the time of assay) which vary for long period follow-up as the quality ofcommercial substrate lots is not equal, y=years.
Shoulder Abduction
90
100
110
120
130
140
150
160
170
Before BMT1 3 5 8 10 11 12
Years post BMT
170 = Normal value
2 4 76 9
Figure 2 Shoulder abduction.
Knee Extension
−30
−25
−20
−15
−10
−5
0
5
10
Years post BMT
−12
−10
−8
−6
−4
−2
0
NormalValue
Figure 3 Knee extension.
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1112
Bone Marrow Transplantation

currently over 12 years old, only one remains at normalschool level (no. 1). The others go to a special school. Theyhave memory and concentration deficits. Three out of fiveare self-conscious about their physical appearance, theothers enjoy their life and they all hope to study for a job inthe future. Social life has improved in every patient becauseof a better physical appearance and the improvement ofjoint mobility.
Vision
Only three patients had normal vision. Others had impairedvision and glasses were required for the majority ofpatients. Before BMT, all patients had visible cornealclouding; following BMT improvement was seen in fivepatients, but one patient needed corneal transplantation.Glaucoma was observed in two patients. No cataracts wereobserved.
Table 5 Orthopaedic management in 15 patients >3 years post BMT
Ptsno.
Follow-up Growth(s.d.)
Kyphosis Surgeryyear
Carpal tunnelsyndrome
Decompressionsurgery
Genu valgumintermalleolardistance
Hipdisplasia
Surgeryyear
Femoralosteotomy
1 16 y �2.5 Severe Arthrodesis + 9 y 5 cm No(96 ; 97)
2 14 y 11 m �3 No C1-C2instability
Arthrodesis + 6 y 5 cm No
(96)3 11 y 11 m �4.5 Severe + 2 y 9 cm + + +
Surgery 98 * 1998, 20015� 8 y 10 m �2.3 No + Before BMT 14 cm No
surgery 97, 996 9 y 2 m �1.2 No + Before BMT No + + &
20008 8 y 10 m �3.5 Slight + 2 y 5 cm + + +
200010 7 y 2 m �1.5 No + 1 y No +12 6 y �3.3 Severe Arthrodesis + Before BMT 7 cm + + +
(97) (left) 2 y (right) 200013 5 y 3 m +0.3 Severe Planed No 10 cm + + ’
2002 200014� 4 y �1.8 Severe + 1 y No + + +
Jacket 200115 4 y 10 m Mean Severe No 7 cm + +
Jacket 200116 4 y 9 m �1 No No No No17 4 y 7 m �0.5 Moderate + 5 cm + + +
Jacket 200020 3 y 2 m +0.5 Slight No 3 cm + + +
200121 3 y 2 m +1 Severe Planed + Before BMT 5 cm + + +
Jacket 2002 2000
�=second BMT after graft rejection (pt 5), after graft failure from father (pt 14); decompression surgery for carpal tunnel syndrome=number of years post-BMT; patients 1, 12, 21 had severe associated scoliosis; *=physeal stapling; ’=femoral osteotomy+innominate osteotomy; &=hip abutment only.
Growth Velocity (girls) n = 8
60
70
80
90
100
110
120
130
140
150
160
170
180
1 10 11 12 13 14 15 16 17 18Age (year)
Hei
gh
t (c
m)
-2SD
+2SD
98765432
Figure 4 Growth velocity (girls).
Growth velocity (boys) n = 7
60
70
80
90
100
110
120
130
140
150
160
170
180
190
1 3 5 7 9 10 11 12 13 14 15 16 17 18Age (year)
Hei
gh
t (c
m)
−2SD
+2SD
2 4 6 8
Figure 5 Growth velocity (boys).
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1113
Bone Marrow Transplantation

Hearing
Hearing impairment is still present in eight out of the 15patients (53 %), has normalised in four and improved andstablised in four patients (62.5 %) after BMT.
Discussion
Allogeneic BMT represents the only method of durablyreplacing genetically abnormal stem cells with normaldonor stem cells. It is thus the treatment of choice forpatients with severe Hurler’s syndrome for whom death isusually expected in the first decade of life. As most of thechildren did not have a genotypically identical siblingdonor, alternative donors were identified for transplant.In our protocol, UBMDs have been used with increasingfrequency. This study reports a 4.7 years median follow-upin 27 patients who underwent transplantation frommatched or mismatched donors, either related or unrelated,
in a single paediatric centre. There were 13 related and 14unrelated transplants but a high number of HLA mis-matches were present in the unrelated transplant group.Since the beginning of our experience,7 unrelated transplantoutcome has been encouraging19 despite the absence of themost stringent HLA matching. That is why, currently, if amatched sibling donor is not available, our policy is to lookfor an UBMD, this being preferred to alternatives such ascord blood transplantation or haplo-identical transplanta-tion. Out of our group of patients, 16 transplants wereperformed with HLA disparities between recipient anddonor; 76% of unrelated transplants and 23% of relatedtransplants being mismatched.Patient selection was conducted carefully after a full
evaluation and consensus regarding disease severity. All butone patient included had an IQ/DQ score X70, and 67 %of patients with a severe phenotype were under the age of 2years at the time of BMT. Severe phenotype correlates wellwith severe genotype and high levels of urinary GAGS.Homozygotes for the nonsense W402X mutation (patientsno. 7, 8, 14, 17, 24, 27 – Table 1) or compoundheterozygotes for the two nonsense mutations W402Xand Q70X (patients no. 1, 13, 18, 20, 21 – Table 1) were alldiagnosed very early in life, at 2 and 15 months of age,except patient no. 18, diagnosed at 3 years 4 months, whodied. This finding confirms previous reports of a goodcorrelation between genotyping and phenotyping in thesecases.20–22 Genotype–phenotype correlations are moredifficult for missense mutations. This difficulty is illustratedby P533R homozygous patients whose phenotype wasassociated with either mild (patient no. 4) intermediate(sibling no. 10 and no. 16) or severe Hurler’s phenotype.23
Patients with urinary GAGS levels greater than 100mg ofglycuronic acid/g of creatinine had a severe phenotype andgenotype. Consequently, these easily and accurately mea-surable physiological parameters should be assessed aftertransplants have successfully engrafted (Table 4).While neuropsychological improvement stabilised after
the progress generally observed during the first 2 yearsfollowing BMT, none of the children deteriorated. These
Table 6 Neuropsychological outcome in 15 patients >3 years post BMT
Ptsno.
Follow-up IQ atBMT
IQ current Scholastic Socialadjustment
MRI Cornealclouding
Visualacuity
Hearingimpairment
1 16 y 75 103 Normal Very good Normal Improved 6/24 Normalized2 14 y 11 m 95 88 Working after apprenticeship Very good Normal + * 6/30 Normalized3 11 y 11 m 66 90 Normal Very good Normal + 6/30 Normalized5� 8 y 10 m 100 85 Especially school Good Unchanged Improved 6/6 No6 9 y 2 m 79 94 Apprenticeship Acceptable Unchanged + 6/24 No8 8 y 10 m 100 95 1 y late Good Slight atrophy + 6/24 Improved10 7 y 2 m 100 100 Normal Very good Unchanged + 6/10 No12 6 y 80 105 Elementary school Good Unchanged Improved 6/24 Normalized13 5 y 3 m 100 99 Elementary school Good Unchanged + 6/24 No14� 4 y 100 100 Nursery school Very good Normal Improved 6/10 Improved15 4 y 10 m 90 86 Nursery school Good Unchanged + 6/6 Improved16 4 y 9 m 99 77 Nursery school Satisfying Stroke + 6/6 No17 4 y 7 m 70 91 Nursery school Very good Normal + 6/6 No20 3 y 2 m 93 100 Nursery school Very good Normal Improved Subnormal Improved21 3 y 2 m 83 83 Nursery school Good Normal Stable 6/6 No
�=second BMT after graft rejection (pt 5), after graft failure from father (pt 14); *=corneal graft; y=years; m=months.
IQ/DQ FOLLOW UP
60
70
80
90
100
110
120
Before BMT1 6 10 11 12
Post transplant follow up
5432 987
Figure 6 IQ/DQ follow-up.
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1114
Bone Marrow Transplantation

favourable long-term outcomes support the efficacy ofBMT itself, whatever the donor origin, and whatever thedisease severity. However, Peters has shown that childrentransplanted from unrelated donors after 2 years of agedemonstrate a different and less favourable trajectory ofdevelopment than those transplanted before the age of 2.6
The significant association between age at BMT and theIQ/DQ score was found in his study reporting transplanta-tion results from related donors.5 Thus, in children with agood initial IQ/DQ who are aged less than or equal to 2.4years at the time of BMT, the risk for cognitivedeterioration appears relatively low, in comparison withhistorical controls who were not transplanted.24 Theefficacy of related or unrelated BMT to prevent theinevitable dementia associated with the disease is con-firmed. These reports suggested that the optimal neurop-sychological outcome following BMT would occur whenchildren were less than 2 years of age and had an IQ/DQgreater than 70 which underlines the importance ofan early diagnosis and clinical decision-making. In ourstudy, the transplant procedure was not delayed bythe necessity of finding UBMDs. The median intervalbetween search initiation and unrelated donor recruitmentwas 1.7 months.19 Currently, finding an UBMD is not achallenge, but finding a perfectly HLA-matched donor forcertain patients, particularly for non-caucasian children,can be. Given the urgency to proceed to the transplant,we therefore accepted partially compatible donors.19
Identification of a suitable donor of haematopoietic stemcells includes not only HLA typing and matching whenpossible but also donor enzyme level estimations. Neuro-psychological function is significantly lower when thedonor is a carrier, or recipient engraftment is less thancomplete from a homozygous enzymatically normal do-nor.5 Using haematopoietic stem cells from an obligatecarrier in haplo-identical grafting5 from partially mis-matched related donors25 or from genotypically matchedsibling carriers gives less favourable outcomes than using ahomozygous enzymatically normal sibling or normalunrelated donor. The latter also have the advantage ofgiving a larger graft cell dose. In our experience, as forother investigators, two or three loci haplo-identical relatedBMTs have less favourable outcomes than unrelatedBMTs.The major obstacle, using both related or unrelated
transplants, is to find the optimal method of obtainingdurable engraftment. The transplant procedure must besufficiently immunosuppressive and myeloablative to givethe highest likelihood of donor cell engraftment and thelowest chance of rejection and GvHD while minimizingtoxicity to fragile organ systems secondary to pathologicalaccumulation of dermatan sulphate, particularly in liver,lung, heart and brain. A consensus regarding the optimalpreparation for Hurler’s syndrome is lacking. We opted forthe combination BU-CY without irradiation or T celldepletion and with the addition of Thymoglobulines inunrelated transplants.Several studies have shown a wide inter- and intrapatient
variability of BU disposition in children26 depending onage, disease, drug interactions, etc. They all concluded thatimprovement in BMT results, in terms of optimal efficacy
and controlled toxicity, requires a reduction in interpatientvariability in systemic exposure.27 In order to regulate BUpharmocokinetics, specific monitoring was instituted in-cluding a first dose and a daily Bayesian forecasting of BUplasma levels.16 In this study, the rate of full engraftmentand veno-occlusive disease (VOD)-free survival was higherthan in other studies, while doses were decreased in sevenpatients, increased in two and unchanged in only one out of10 patients with Hurler’s syndrome (a subset of the studydescribed here). Clearance of BU was increased incomparison to non-Hurler’s diseases.28
To prevent GvHD in unrelated transplants, we opted forthe use of Thymoglobulines. The use of Thymoglobulines
induces in vivo T cell depletion involving both donor Tlymphocytes and residual host T lymphocytes resistant tothe conditioning regimen. Thymoglobulines has a potentcontrol effect on immunoactivation of T cells involved bothin graft rejection and acute GvHD. In Peters’ study6 of theoutcome of unrelated donor BMT in 40 children withHurler’s syndrome, neither T-lymphocyte depletion of thebone marrow nor irradiation appeared to influence thelikelihood of engraftment. In all, 25 of the patients initiallyengrafted, with an estimated 49% of patients being alive at2 years, 63% alloengrafted and 37% autoengrafted. Intotal, 10 of 16 patients are alive at 1 year who received abone marrow cell doseX3.5� 108 cells/kg engrafted, whileonly three of 11 patients receiving a lower bone marrow celldose engrafted. Peters speculates that in this patientpopulation, insufficient myeloablative and/or immunosup-pressive therapy was the main cause of graft rejection(16%). In our experience, engraftment (85%) and rejectionare not major problems, nor indeed is GvHD (22%). Weobserved a low incidence of aGvHD (three grade I, twograde II) and an absence of chronic GvHD despite a highnumber of HLA mismatches (Table 2). In the 40transplanted patients, 25 initially engrafted, the probabilityof grade II to IV aGvHD was 30% and the probability ofextensive chronic GvHD was 18%.Durable engraftment in our patients led to a dramatic
reduction of GAGS in urine to the upper limit of age-matched control values with a low amount of dermatansulphate and a hardly detectable fraction of heparansulphate. No difference could be observed between patientsengrafting from related or unrelated donors, homozygousenzymatically normal donors or carriers, or between fullyengrafted vs mixed donor/recipient chimaerism. In fullyengrafted patients, leucocyte a-l-iduronidase enzyme levelsremained within the expected range of their respectivedonors.Survival of engrafted patients is radically different from
that of patients who do not undergo transplantation orwho experienced BMT failure. As hypothesised by Hobbs,1
donor leucocyte precursors provide a natural source of themissing a-l-iduronidase enzyme able to correct lysosomalengorgement of the cells. BMT dramatically improves themetabolism and clearance of GAGS from highly perfusedorgans such as adenoids, tonsils, spleen, liver, lung andheart, Virchow-Robin space, but with a limited effect onthe corneas and no effect on skeletal tissue.As in other reports,3–7,25,29 in all our engrafted patients
substantial clinical improvement of somatic disease was
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1115
Bone Marrow Transplantation

evident with resolution of hepatosplenomegaly, improve-ment in facial appearance, hearing, maintenance of normalheart and pulmonary function and prevention of hydro-cephalus with improvements in Virchow–Robin spaceabnormalities.In contrast, dysostosis multiplex progressed with abnor-
mal growth of the skeleton. In the musculoskeletal tissues,growth and cellular maturation is deficient with systemicdisturbance of bone modelling and focal failures ofossification particularly at the upper two lumbar vertebralbodies, at the superolateral roof of the acetabulum and thelateral margin of the proximal tibial metaphasis.30,31 Lineargrowth by the age of 10 years was retarded with apparentretarded trunk growth. Increasing genu valgum was notprevented. Orthopaedic problems persisted in engraftedpatients and orthopaedic surgery was required for genuvalgum, kyphoscoliosis, acetabular dysplasia, trigger digitsand median nerve release for carpal tunnel syndrome if notperformed before grafting. Eight patients had a femoralosteotomy of whom one also had a Salter osteotomy, andone patient required hip abutment only, with an excellentresult. Our patients seem to be currently stabilised withoutpain or hip subluxation. No gradual musculoskeletaldeterioration has been observed under orthopaedic man-agement with the help of intensive prolonged physio-therapy. The orthopaedic management of these patientsremains a research problem. In Field’s experience,30 10 of11 children showed gradual musculoskeletal deterioration.The hip dysplasia was not prevented by proximal femoralosteotomies. In Masterson’s experience,31 bilateral hip-containment surgery was performed in five cases at a meanfollow-up of 17 months. Innominate osteotomy wouldappear to be an essential part of the surgical procedure withimproved cover of the femoral head. In the Minneapolisexperience, Ogilvie and Peters (Orthopaedic aspects ofMPS syndrome, personal communication, ASH meeting2001) and Pemberton reported an improvement in thepatients’ orthopaedic outcomes by the use of varus femoralosteotomy. In our experience, we have systematicallyperformed preoperative tridimensional scanner reconstruc-tion of the pelvis and upper femur, which has shown severeacetabular dysplasia with anterior and posterior defects.For that reason, we do not advise innominate pelvisosteotomy combined with varus femoral osteotomy toobtain good hip containment. On the other hand, aPemberton osteotomy seems to be more logical to obtainan anatomical acetabular hip containment. After a firstexperience of varus femoral osteotomy alone (combinedwith hip abutment at the end of skeletal growth), we nowplan to start a new surgical procedure combining Pember-ton osteotomy and varus femoral osteotomy followingOlgivie and Peters recommendations.The reported neurological outcome is variable and
related to the heterogeneous natural course of the disease,IQ/DQ and age before the graft. Quality of life is globallyimproved despite skeletal problems; joint mobility isimproved and clawing of the digits is limited compared tothe degree seen in untreated cases; however, the digitsremain stubby, and the hands remain abnormally broad inthose with the severe phenotype. Family and patients enjoya better physical appearance, better socialisation and the
possibility of a normal scholastic life even with the severephenotype (no. 1). Nevertheless, we will have to wait untilthe children in this cohort have reached adulthood beforetheir quality of life and social adjustment can be properlyassessed. Undoubtedly, improved quality of life after BMTwill occur as a result of earlier diagnosis and earlytransplantation during the first year of life facilitated bydose-adaptation of BU.Today, stem cell transplantation remains the only
effective intervention in severe Hurler’s syndrome, even ifpreliminary results from human a-l-iduronidase enzymereplacement are encouraging and demonstrate comparablequality of life outcomes in those with the moderatephenotype.32 Enzyme replacement cannot be applied insevere forms because the substitute enzyme cannot cross theblood–brain barrier.Ongoing analysis of patients with Hurler’s disease who
are untreated indicates a median survival of slightly lessthan 5 years of age and the progressive deterioration leadsto a minuscule survival rate after 10 years. Unrelated donorgrafts and mismatched procedures are more difficulttechnically than identical related transplantations but arefeasible, and successful with a low transplant-relatedmortality rate. We would recommend that such transplantsbe offered to the patients and their families but be carriedout only in specialised centres with appropriate paediatricmetabolic expertise.
Acknowledgements
We thank the assistant physicians, nurses and medical staff ofthe Immuno-haematology & Paediatric Bone Marrow Trans-plant Unit, Debrousse Hospital, for their support and commit-ment (Prof Philippe, Dr K Kebaili, Dr R Mardini) and thephysicians and technicians of Histocompatibility Laboratory inLyon, for their efforts and efficient collaboration (Dr AEljaafari, Dr D Rigal, Dr JP Tremisi, JP Bourgeot, Ph Debost,S Rey). All of them have contributed to help the patients. Wealso address special thanks to Prof C Peters and Dr N Whitakerfor their constructive and useful suggestions in reviewing themanuscript.
References
1 Hobbs JR, Hugh-Jones K, Barrett AJ et al. Reversal of clinicalfeatures of Hurler’s disease and biochemical improvement aftertreatment by bone marrow transplantation. Lancet 1981; 2:709–712.
2 Krivit W, Sung JH, Lockman LA, Shapiro EG. Centralnervous system reconstitution after bone marrow transplanta-tion for lysosomal and peroxisomal storage diseases. In: RichRR, Fleisher TA, Schwartz BD, Shearer WT, Strober W (eds).Principles of Clinical Immunology, Vol 2. Mosby: St Louis,MO, 1995, p. 1852.
3 Whitley CB, Belani KG, Chang PN et al. Long-term outcomeof Hurler syndrome following bone marrow transplantation.Am J Med Genet 1993; 46: 209–218.
4 Hoogerbrugge PM, Brouwer OF, Bordigoni P et al. A for theEuropean group for Bone Marrow Transplantation.Allogeneic bone marrow transplantation for lysosomal storagediseases. Lancet 1995; 345: 1398–1402.
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1116
Bone Marrow Transplantation

5 Peters C, Shapiro EG, Anderson J et al. Hurler Syndrome. II.Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation infifty-four children. Blood 1998; 91: 2601–2608.
6 Peters C, Balthazor M, Shapiro EG et al. Outcome ofunrelated donor bone marrow transplantations in 40 childrenwith Hurler syndrome. Blood 1996; 87: 4894–4902.
7 Guffon N, Souillet G, Maire I et al. Follow-up of nine patientswith Hurler syndrome after bone marrow transplantation.J Pediatr 1998; 133: 119–125.
8 Piraud M, Boyer S, Mathieu M, Maire I. A diagnosis ofmucopolysaccharidoses in a clinically selected population byurinary glycosaminoglycan analysis: a study of 2000 urinesamples. Clin Chim Acta 1993; 221: 171–781.
9 Hall CW, Neufeld EF. a-l-iduronidase activity in cultured skinfibroblasts and amniotic fluid cells. Arch Biochem Biophys1973; 158: 817–821.
10 Young EP. Prenatal diagnosis of Hurler disease by analysis ofa-l-iduronidase in chorionic villi. J Inher Metab Dis 1992; 15:224–230.
11 Gebuhrer L, Betuel H, Lambert J et al. A division of HLA-DQw1 associated with DR1, DR1x-DQw1, DR2, short,DRw10, and DRw14. I. Definition by alloantiserum LY1327.Human Immunol 1987; 18: 235–245.
12 Tiercy JM, Gorski J, Betuel H et al. DNA typing of DRw6subtypes : correlation with DRB1 and DRB3 allelic sequencesby hybridization with oligonucleotide probes. Human Immunol1989; 24: 1–14.
13 Tiercy JM, Morel C, Freidel AC et al. Selection of unrelateddonors for bone marrow transplantation is improved by HLAclass II genotyping with oligonucleotide hybridization. ProcNatl Acad Sci USA 1991; 1988: 7121–7125.
14 Hobbs JR. The evolution of displacement bone marrowtransplantation for inborn errors. In: Hobbs JR (ed.).Correction of Certain Genetic Diseases by Transplantation.The COGENT Fund: London, 1989, pp. 1–22.
15 Souillet G. Thymoglobulin: conditioning in children prior tobone marrow transplantation, a single center retrospectivestudy. In: Thymoglobulin – anti-thymocyte globulin (rabbit).The role of Thymoglobulin in Alternative Donor HaematopoieticStem Cell Transplantation. Clinical monograph, SangStat2000, pp. 35–41.
16 Bleyzac N, Souillet G, Magron P et al. Improved clinicaloutcome of paediatric bone marrow recipients using a test doseand Bayesian pharmacokinetic individualization of busulfandosage regimen. Bone Marrow Transplant 2001; 28: 743–751.
17 Glucksberg H, Storb R, Fefer A. Clinical manifestations ofGVHD in human recipients of marrow from HLA matchedsibling donors. Transplantation 1994; 18: 295–301.
18 Wong Z, Wilson V, Jeffreys AJ, Thein SL. Characterization ofa panel of highly variable minisatellites cloned from humanDNA. Ann Hum Genet 1996; 51: 269–288.
19 Souillet G, Rey S, Bertrand Y et al. Outcome of unrelatedbone marrow donor search in 174 children resulting in 45patients transplanted in HLA match and mismatch situation.Bone Marrow Transplant 2000; 26: 31–43.
20 Bunge S, Kleijer WJ, Steglich C et al. Mucopolysaccharidosistype I: identification of 8 novel mutations and determination ofthe frequency of the two common alpha-l-iduronidasemutations (W402X and Q70X) among European patients.Hum Mol Genet 1994; 3: 861–866.
21 Scott HS, Bunge S, Gal A et al. Molecular genetics ofmucopolysaccharidosis type I: diagnostic, clinical, and bio-logical implications. Hum Mutat 1995; 6: 288–302.
22 Beesley CE, Meaney CA, Greenland G et al. Mutationalanalysis of 85 mucopolysaccharidosis type I families: frequencyof known mutations, identification of 17 novel mutations andin vitro expression of missense mutations. Hum Gen 2001; 109:503–511.
23 Scott SH, Litjens T, Nelson PV et al. a-l-iduronidasemutations (Q70X and P533R) associate with a severe Hurlerphenotype. Hum Mutat 1992; 1: 333–339.
24 Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuro-psychological outcomes of several storage diseases with andwithout bone marrow transplantation. J Inherit Metab Dis1995; 18: 41.
25 Fleming DR, Henslee-Downey PJ et al. The use of partiallyHLA mismatched donors for allogeneic transplantation inpatients with mucopolysaccharidosis-I. Pediatr Transplant1998; 2: 250–253.
26 Bolinger AM, Zangwill AB, Slattery JT et al. An evaluationof engraftment, toxicity and busulfan concentration inchildren receiving bone marrow transplantation forleukaemia or genetic disease. Bone Marrow Transplant 2000;25: 925–930.
27 Vassal G, Deroussent A, Challine D et al. Is 600mg/m2
appropriate dosage of busulfan in children undergoing bonemarrow transplantation? Blood 1992; 79: 2475–2479.
28 Vassal G, Fisher A, Challine D et al. Busulfan dispositionbelow the age of three: alteration in children with lysosomalstorage disease. Blood 1993; 82: 1030–1034.
29 Vellodi A, Young EP, Cooper A. Bone marrow transplanta-tion for mucopolysaccharidosis type I: experience of twoBritish centres. Arch Dis Child 1997; 76: 92–99.
30 Field RE, Buchana JAF, Copplemans MGJ, Aichroth PM.Bone marrow transplantation in Hurler syndrome: effect onskeletal development. J Bone Joint Surg Br 1994; 76B:975–981.
31 Masterson EL, Murphy PG, O’Meara A et al. Hip dysplasia inHurler’s syndrome : orthopaedic management after bonemarrow transplantation. J Pediatr Orthop 1996; 16: 731–733.
32 Kakkis ED, Muenzer J, Tiller GE et al. Enzyme-replacementtherapy in mucopolysaccharidosis I. N Engl J Med 2001; 344:182–188.
Outcome of 27 patients with Hurler’s syndromeG Souillet et al
1117
Bone Marrow Transplantation
Related Documents