Outcome dter Ischemia in the Developing Sheep Brain: An Electroencephalographic and Histologcal Study Chris E. Williams, PhD, Alistair J. Gunn, MD, Carina Mallard, BSc, and Peter D. Gluckman, DSc The role of seizures occurring with perinatal hypoxic-ischemic encephalopathies is unclear. We examined the relation- ships between the time course of parasagittal electroencephalographic (EEG) activity and pathological outcome f‘ollow- ing transient cerebral ischemia, which was induced in 33 chronically instrumented fetal sheep by occluding the carotid arteries after ligation of the vertebral-carotid anastomoses. The EEG was quantified with real-time spectral analysis. Histological outcome was assessed 7i! hours later. After 10 or 20 minutes of ischemia, EEG activity was depressled and then progressively recovered and mild selective neuronal loss was seen. The length of this depression correlated with the duration of ischemia (r = 0.88). After 30 or 40 minutes of ischemia, EEG activity remained depressed for 8 & 2 hours, followed by a rapid transition to low-frequency epileptiform activity that reached maximum intensity at 10 +. 3 hours. By 72 hours, EEG intensity had fallen below control levels. This sequence of prolonged depression, epilepti- form activity, and then loss of intensity was associated with the development of laminar necrosis of the underlying cortex. These electrophysiological sequelae may have prognostic value. The results indicate that after a severe hypoxic- ischemic insult, the parasagittal cortex becomes hyperexcitable before the final loss of activity. Secondary neuronal death may occur in this phase. Williams CE, Gunn AJ, Mallard C, Gluckman PD. Outcome after ischemia in the developing sheep brain: an electroencephalographicand histological study. Ann Neurol 1002;3 1: 14-2 1 Neuronal death is often delayed by hours to days after a transient hypoxic-ischemic insult :in both the mature [l] and the neonatal brain 121. Several authors have speculated that this delayed death is, in part, a conse- quence of postasphyxlal epileptiform activity {3-51. However, despite the common occurrence of seizures with hypoxic-ischemic encephalopathy in newborns T6, 71, in infants with “near-miss” sudden infant death syn- drome (SIDS) {S], and in adults {91, there has been little experimental evidence for a causal association be- tween epileptiform activity and neuronal loss follow- ing a hypoxic-ischemic insult in either adult [lo, 111 or neonad animal preparations. [t remains unclear whether postasphyxial seizures merely reflect the con- sequences of damage or contribute significantly to the loss of neurons. A major problem in neonatology is the rapid and reliable determination of the severity of a hypoxic- ischemic insult so as to allow the appropriate manage- ment of the ensuing encephalopathy {12, 131. Continu- ous electroencephalographic (EEG) analysis was demonstrated to have worthwhile prognostic value {14, 151. However, the relationships between the se- verity of the insult, histological outcome, ancl time course of EEG activity are unclear. Most perinatal asphyxial insults are thought tcl occur as antepartum or intrapartum events { 161, and cerebral damage is thought to be a consequence of asphyxia occurring with a period of impaired cerebral blocid flow or ischemia [17, 181. One of the clinically recognized patterns of damage seen in term infants following as- phyxia is parasagittal cortical damage {6]. These chil- dren are more likely to show physical and mental im- pairment and reduced school performance [7, 101. We recently described an experimental preparation where the chronically instrumented fetal sheep is subjected to transient cerebral ischemia that can induce an encepha- lopathy showing similar characteristics to hypoxic- ischemic brain damage of some asphyxiated term in- fants C20). This preparation offers particular advamtages for continuous electrophysiological monitoring over a long period following hypoxic-ischemic insult. The objective of the present study was to determine the relationships between the severity of a hypoxic- From the Department of Paediatrics, University of Auckland, Auck- land, New :Zealand. Received N ov 8, 1990, and in revised form .4pr 24, 1991. Accepted for publication May 21, 1991. Address correspondence to Dr Williams, Developmental Physiol- ogy, Department of Pardiatrics, University of Auckland, Private Bag, Auckland, New Zealand. 14 Copyright 0 1992 by the American Neurological Association

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Outcome dter Ischemia in the Developing Sheep Brain: An Electroencephalographic
and Histologcal Study Chris E. Williams, PhD, Alistair J. Gunn, MD, Carina Mallard, BSc, and
Peter D. Gluckman, DSc
The role of seizures occurring with perinatal hypoxic-ischemic encephalopathies is unclear. We examined the relation- ships between the time course of parasagittal electroencephalographic (EEG) activity and pathological outcome f‘ollow- ing transient cerebral ischemia, which was induced in 33 chronically instrumented fetal sheep by occluding the carotid arteries after ligation of the vertebral-carotid anastomoses. The EEG was quantified with real-time spectral analysis. Histological outcome was assessed 7i! hours later. After 10 or 20 minutes of ischemia, EEG activity was depressled and then progressively recovered and mild selective neuronal loss was seen. The length of this depression correlated with the duration of ischemia ( r = 0.88). After 30 or 40 minutes of ischemia, EEG activity remained depressed for 8 & 2 hours, followed by a rapid transition to low-frequency epileptiform activity that reached maximum intensity at 10 +. 3 hours. By 72 hours, EEG intensity had fallen below control levels. This sequence of prolonged depression, epilepti- form activity, and then loss of intensity was associated with the development of laminar necrosis of the underlying cortex. These electrophysiological sequelae may have prognostic value. The results indicate that after a severe hypoxic- ischemic insult, the parasagittal cortex becomes hyperexcitable before the final loss of activity. Secondary neuronal death may occur in this phase.
Williams CE, Gunn AJ, Mallard C, Gluckman PD. Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann Neurol 1002;3 1: 14-2 1
Neuronal death is often delayed by hours to days after a transient hypoxic-ischemic insult :in both the mature [l] and the neonatal brain 121. Several authors have speculated that this delayed death is, in part, a conse- quence of postasphyxlal epileptiform activity {3-51. However, despite the common occurrence of seizures with hypoxic-ischemic encephalopathy in newborns T6, 71, in infants with “near-miss” sudden infant death syn- drome (SIDS) {S], and in adults {91, there has been little experimental evidence for a causal association be- tween epileptiform activity and neuronal loss follow- ing a hypoxic-ischemic insult in either adult [lo, 111 or neonad animal preparations. [t remains unclear whether postasphyxial seizures merely reflect the con- sequences of damage or contribute significantly to the loss of neurons.
A major problem in neonatology is the rapid and reliable determination of the severity of a hypoxic- ischemic insult so as to allow the appropriate manage- ment of the ensuing encephalopathy {12, 131. Continu- ous electroencephalographic (EEG) analysis was demonstrated to have worthwhile prognostic value
{14, 151. However, the relationships between the se- verity of the insult, histological outcome, ancl time course of EEG activity are unclear.
Most perinatal asphyxial insults are thought tcl occur as antepartum or intrapartum events { 161, and cerebral damage is thought to be a consequence of asphyxia occurring with a period of impaired cerebral blocid flow or ischemia [17, 181. One of the clinically recognized patterns of damage seen in term infants following as- phyxia is parasagittal cortical damage {6]. These chil- dren are more likely to show physical and mental im- pairment and reduced school performance [7, 101. We recently described an experimental preparation where the chronically instrumented fetal sheep is subjected to transient cerebral ischemia that can induce an encepha- lopathy showing similar characteristics to hypoxic- ischemic brain damage of some asphyxiated term in- fants C20). This preparation offers particular advamtages for continuous electrophysiological monitoring over a long period following hypoxic-ischemic insult.
The objective of the present study was to determine the relationships between the severity of a hypoxic-
From the Department of Paediatrics, University of Auckland, Auck- land, New :Zealand. Received N ov 8, 1990, and in revised form .4pr 24, 199 1. Accepted for publication May 21, 1991.
Address correspondence to Dr Williams, Developmental Physiol- ogy, Department of Pardiatrics, University of Auckland, Private Bag, Auckland, New Zealand.
14 Copyright 0 1992 by the American Neurological Association

ischemic insult, histological outcome, and evolution of the EEG activity in the developing brain. The severity of a hypoxic-ischemic insult can be described in terms of tissue oxygen deficit C21). Thus by assuming a con- sistent degree of ischemia or reduction in oxygen deliv- ery, different severities of insult can be induced in the fetal sheep by varying the duration of cerebral isch- emia. The parasagittal EEG was continuously analyzed via a real-time spectral analysis technique 122) and si- multaneously recorded on analog chart. Cell death was assessed histologically 3 days after the insult. The data were analyzed according to the extent of neuronal loss, and with respect to the duration of ischemia.
Materials and Methods Surgical Procedures Thirty-three Romney/Suffolk fetal sheep of known gesta- tional age were operated on under halothane anesthesia (2%) using sterile techniques as previously described [20}. Briefly, the head, neck, and forelimbs of the fetus were externalized and catheters were inserted into each of the axillary arteries. Two pairs of shielded stainless-steel electrodes were placed bilaterally on the parietal dura. These were inserted via para- sagittal burr holes that were drilled in the area of the skull overlying the parietal cortex (skull coordinates relative to the bregma: anterior 5 mm and 15 mm, lateral 10 mm). The vertebro-occipital anastomoses between the carotid arteries and vertebral arteries were ligated bilaterally. The vertebral arteries in the sheep d o not supply the brain, but rejoin the common carotid arteries, while the basilar artery is supplied from the circle of Willis. Thus, this procedure restricts verte- bral vascular supply to the carotid arteries. Inflatable occluder cuffs were placed around both carotid arteries. The fetus was returned to the uterus and the uterine and abdominal walls closed. After the operation the ewe was housed in a meta- bolic cage at constant temperature (20°C) and humidity (50%) and given free access to hay and water, supplemented by sheep nuts and alfalfa. Antibiotics (gentamicin, 80 mg; cephalothin, 1 gm) were administered to the ewe daily.
Recording Movement artifacts in the EEG were minimized by using driven shield electrodes. These recording electrodes were constructed from shielded Telfon-insulated wire (AS636 lSSF, Cooner Wire, Chatsworth, CA). The signal was ampli- fied 5,000 times before filtering. Antialiasing filtering was accomplished with eighth-order switched capacitor Butter- worth filters, with the - 3-dB point set to 30 Hz. All experi- ments were continuously recorded on an analog chart re- corder running at 5 mm/min. Intensity (power) spectra were obtained by real-time spectral analysis 122). The EEG was continuously sampled at 1 11 Hz and intensity spectra were analyzed from these data. These spectra were averaged over 5.5-minute intervals, and each average spectrum stored to disk.
Experimental Procedures Experiments were started 72 to 96 hours after surgery. Fe- tuses ranged in gestational age from 119 to 127 days (term
gestation is 147 days). Fetal arterial samples were obtained prior to the start of each experiment and only fetuses exhib- iting arterial blood gases within the normal range for our laboratory (pH > 7.30 and Pao, 1 17 mm Hg) were used. After 4 hours of collecting EEG spectral data (reference pe- riod), the carotid cuffs were inflated with saline solution for either 10, 20, 30, or 40 minutes and then deflated. Spectra were collected continuously for 32 hours following the end of the occlusion. In addition, 4-hour blocks of spectra were collected at 48 and 72 hours after ischemia. Our previous experiments have shown that in about 205% of animals, one of the cuffs immediately fails due to bursting. When this happened in the present study, the second cuff was immedi- ately released and the animals were prospectively used as control animals.
Each sheep was killed 3 days after the insult by an infusion of pentobarbital; the fetus was immediately removed and the brain perfused through the common carotid arteries with 500 ml of normal saline solution followed by 500 ml of 10% formalin and then 500 ml of 10% formalin/lO%i, sucrose. The brains were embedded in paraffin, coronally subserially sectioned to 8 k m thick, and then stained with thionin-acid fuchsin 1231. Every 40th section was mounted and examined by light microscopy. Each selected region was assessed under a magnification of 100 x before detailed assessment at a mag- nification of 400 x . Neurons with ischemic cell change were identified according to the criteria of Brown and Brierley C24). Each region was scored in multiple preselected areas for the proportion of dead neurons by two independent as- sessors, one of whom was blinded to the experiment. The correlation between scores obtained by the two observers was Y = 0.88 (p < 0.00001, n = 654). The cortical regions were selected from sections of the temporoparietal cortex corresponding with the positions of the EEG electrodes. The “parasagittal” cortex was defined to include all of the gyrus bordering the sagittal sulcus, and the medial half of the later- ally adjacent gyrus. The ‘‘lateral’’ cortex was defined as includ- ing all of the most lateral gyrus on the section.
The average values from two sections and from both the right and the left hemispheres were used in all cases except for the thalami where values for all the nuclei in three sec- tions were averaged, and the hippocampus where values for the dorsal and ventral gyri were averaged. A 6-point damage scale was used. Each region was scored for damage as follows: 096, 1 to 1096, 10 to 5092, 50 to 9096, 90 to 9996, and 100% neurons dead. To convert this to a ratio scale and thus allow parametric statistics to be performed on this 6-point scale 1251, the nominal damage score was taken as the mid- point of each of these ranges: 0, 5 , 30, 70, 95, and 100, respectively.
Analysis Two animals were rejected from the duration analysis due to only partial suppression of the EEG during the occlusion. None were rejected from the evaluation of histological out- come. Intensity spectra were obtained as previously de- scribed {221. The descriptive measurements and peak inten- sity, peak time, peak duration, and depression time were made on the smoothed time series. For smoothing a digital Blackman low-pass filter (Asystant + , Asystant Software
Williams et al: EEG and Outcome after Ischemia 15

Technologies, Inc, Rochester, NY) with a cutoff of 0.1 cy- clesipoint was applied to the log-transformed dat-a to mini- mize short-term fluctuations of less than 25 minutes. Peak measurements were made by a maxima-detecting algorithm applied from 0 to 30 hours after the insult. The duration of the depressed period was measured as the time after insult until EEG intensity increased above - 5 dB of the preocclu- sion intensity.
For statistical analysis all calculations were made on loga- rithms of the intensity (dB). This transformation gives a bet- ter approximation to the normal distribution {26:t. The log- transformed intensity was normalized with respect to the mean valm of the first 4 hours of recording, that is, prior to the occlusion. This normalization removes variability in amplitude between animals that is a consequence of electrode positioning.
After descriptive measurements were made, animals show- ing a parasagittal damage score of less than 50 were defined as mildly damaged arid those that had a score greater than 50 as severely damaged. EEG measurements following different durations of ischemia were compared with analysis of vari- ance and the post hoc comparisons made with Fisher’s least- significance-difference test. Comparisons of EEG parameters between mild and severely damaged groups were made with unpaired t tests. Significance levels were adjusted using Bonferroni’s correctnon for multiple comparisons. Data are expressed as mean 2. standard deviation (SD).
Results The metabolic status and gestational age given in the Table were similar for all durations of ischemia.
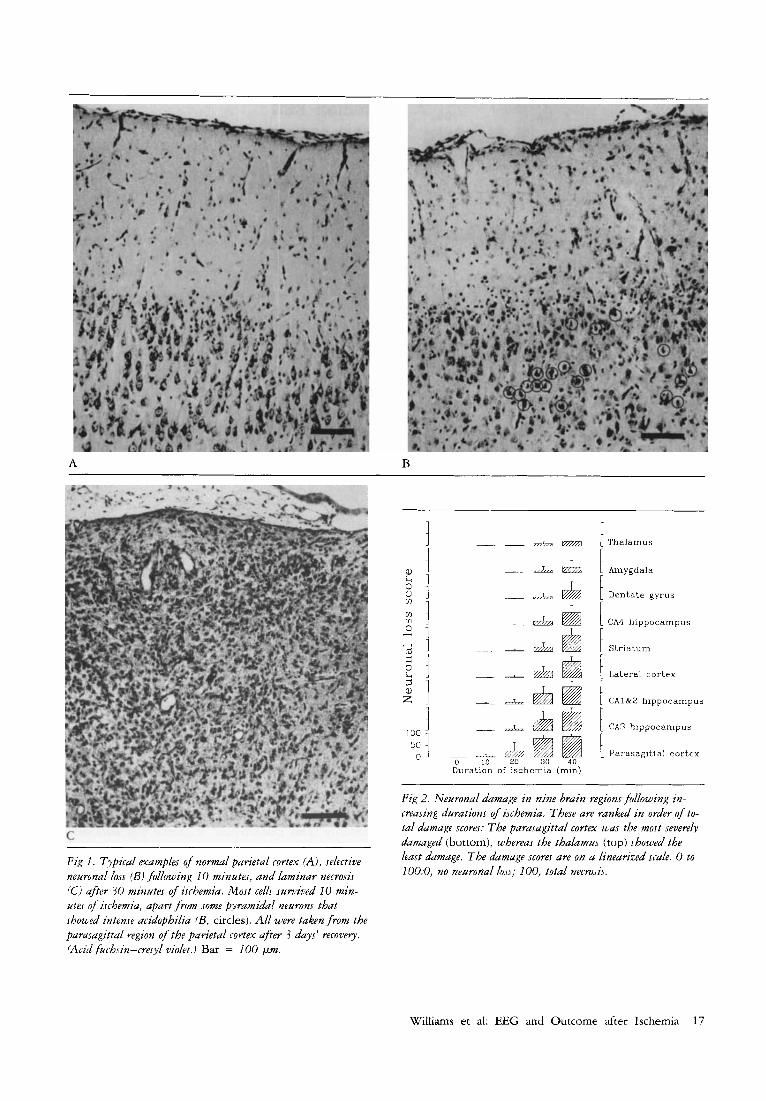
Density of Neuronarl Damage versus Durution of Ischemta All regions showed increased neuronal loss as the dura- tion of the insult lengthened (Figs 1 and 2). The tdistri- bution of damage was similar to that described pre- viously [20], with predominant parasagittal damage. In control animals, acidophilic neurons were absent in all areas examined. Mild selective neuronal loss was seen after 10 or 20 minutes of ischemia (see Fig 1). In con- trast, occlusions of 30 minutes produced severs cell loss (laminar necrosis) within the parasagittal cortex (see Fig 1) and moderate damage in the hippocampal regions cornu amrnonis 1 (CA1) and CA3. In addition, lateral cortex, striatum, and the hippocampal CA4 re- gion showed a mild loss of neurons. Cell loss was not
Physdogii-ul Purumeters Measured before Ischemia
seen in the substantia nigra pars reticulata. Forty min- utes of ischemia resulted in severe neuronal death in the parasagittal and lateral cortex, striatum, and most of the hippocampus (see Fig 2). The granule cells of the dentate gyms tended to have only mild damage. The amygdala and thalamic nuclei were comparatively spared and only mild neuronal damage could be de- tected.
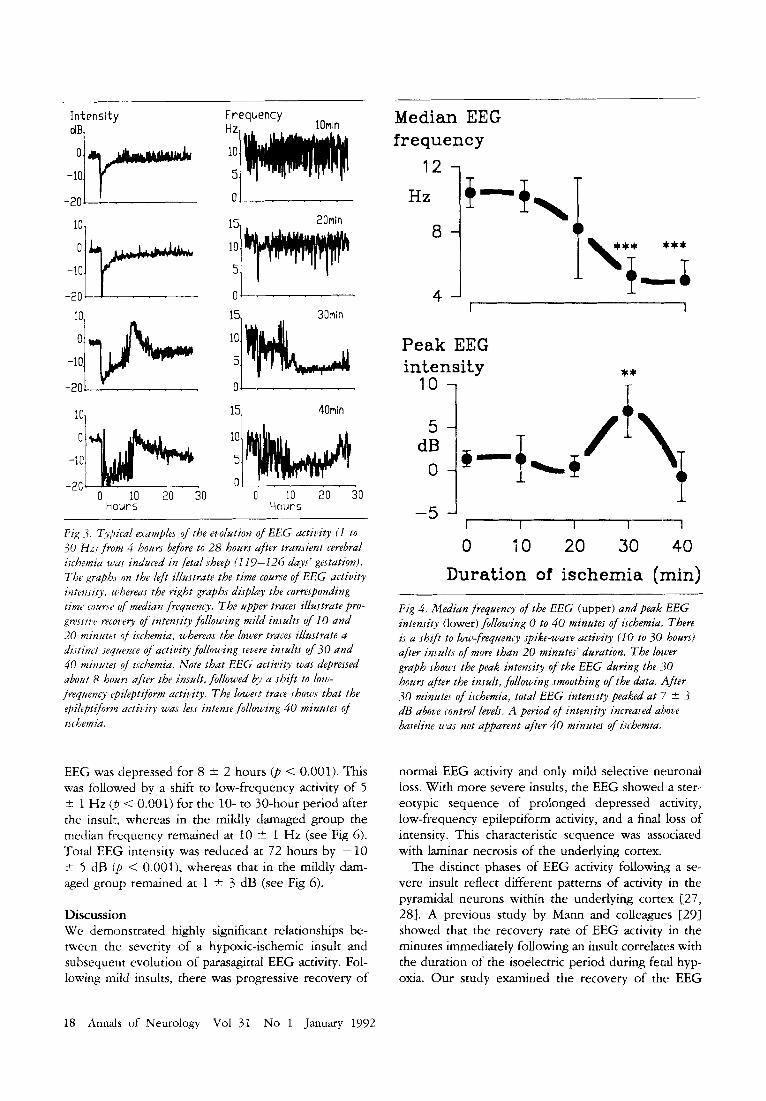
Relations hips between Electrophys io logy and Duration of the Ischemia All fetuses showed preinsult EEG amplitudes between 80 and 150 pV. During the insult, the EEG rapidly (< 10 seconds) became isoelectric. Immediately following release of the cuffs, EEG activity was severely de- pressed (Fig 3). The length of time the EEG was de- pressed was linearly related to the duration of ischemia ( r = 0.88, p < 0.00001, n = 31). After the shorter insults of 10 and 20 minutes’ duration, EEG activity progressively recovered (Figs 3, 4 , and 5 ) . However, after longer insults of 30 to 40 minutes’ duration, there appeared to be a biphasic time course with a transition to low-frequency epileptiform activity at 8 ? 2 hours after the insult (see Fig 3). This low-frequency activity peaked in intensity at 10 2 3 hours and then gradually resolved, losing intensity to less than that of the control group by 72 hours (see Fig 5) . After 30 minutes of ischemia, EEG activity peaked in intensity at 7 2 3 ciB above preinsult levels (p < 0.005) (see Fig 4). This increased activity remained above preinsult levels for 6 +- 5 hours. The peak intensity after 40 minutes of ischemia was smaller ( - 1 t 3 dB, p < 0.001) than the post-30-minute peak (see Fig 4). The EEG at 72 hours showed a significantly reduced median frequency in the 40-minute group (p < 0.05) (see Fig 5) .
Relationships between Electrophysiology and Pathological Outcome To resolve the relationships between the time course of the parasagittal EEG and final neuronal loss, the animals were separated into two groups: those that had severe parasagittal damage (score average of 82 2 13) and those that showed mild selective o r no neuronal loss (score average of 4 * 7) at 72 hours after the insult. Those that showed severe parasagittal damage had a distinct sequence of EEG activity (Figs 3, 6). The
~
Duration Pco, Po2 Lactate Gestation ( m i d n p H (mm Hg) (mm Hg) (mmol!L) !days)
0 5 7.36 2 0.04 53 * 8 21 * 5 2.4 2 1.0 123 5 2 10 6 7.33 * 0.03 57 t 4 22 2 2 2.6 IT 0.6 123 * 3
123 IT 2 20 4 7.35 t 0.01 54 * 2 21 2 4 2.4 t 0.7 123 rt 1 30 9 7.35 2 0.02 51 * 4 20 2 5 2.5 * 0.6 124 ? 2
-
40 9 7.35 2 0.03 55 2 5 23 2 5 2.8 r 0.6
16 Annals of Neurology Vol 31 No 1 January 1992
A
c
Fig 1. Typical examples of n o m l parietal cortex (A), selective neuronal loss (Bi fallowing 10 minutes, and laminar necrosis lCi after 30 minutes of ischemia. Most cellj survived 10 min- utes of i.rchemia, apart from some pyramidal neurons that showed intense acidophilia (B, circles). All were taken from the parasagittal region of the parietal cortex afrer 3 days' recovely. (Acid fuchsin-cresyl violet.) Bar = 100 pm.
B
- ~ A & t Thalamus 1 r ' 1 0
0 W l
~ &L & 1 Amygdala
- & & 1 Dentate gyrus T r
& & CA4 hippocampus
+ @ \ Lateral cortex
I - & & & 1 CA1&2 hippocampus
100 & & [ C A 3 hippocampus
5:1 0 A 10 A 20 @ 30 @I 40 Duration of ischemia (min)
1 Parasagittal cortex
Fig 2. Neuronal damage in nine brain regions follwing m- creasing durations of ischemia. These are ranked in order of to- tal damage scores: The parasagittal cortex was the most severely damaged (bottom), whereas the thalamus (top) showed the least damage. The damage scores are on a linearized scale. 0 to 100:0, no neuronal lo.is; 100. total necrosis.
Williams et al: EEG and Outcome after Ischemia 17
~~
I n tens i ty
d7 -10 O:'Y"""
101 0-
-10.
In
- 2 O u ' ' ' ' '
I n
Frequency C I 7 lOmin
i? 20min
15. 30min
-10.
-2n. 1- "
0 10 20 30 hours
0 1 . . .
40min
0 10 20 30 Hours
Fig -3, Typii-al examples of the evolution of EEG activity (I t o 30 H z ) fmm 4 hours before to 28 hours ajiter transient cerebral isihemia zr'as induced in fetal sheep (1 19-226 days' gestation). The grapbs on the ldt illustrate the time coume of E E G activity intensity. whereas the right graphs displhy the corresponding time courrt' of median frequency. The upper traces illustrate pro- gressiw recovery of intensity following mild i n d t s of I0 and 20 minutes of ischemia, whereas the lower traces illustrate a distinct sequence of actitv'ty following severe in.tults of 30 and 40 minutes of ischemia. Note that EEG activity was depressed about 8 hours after the insalt, followed by a shift t o low- frequency rpileptifomz aitivity. The lowesi traiz shows that the rpilepti/orm activity was less intense following 40 minutes of ischemia.
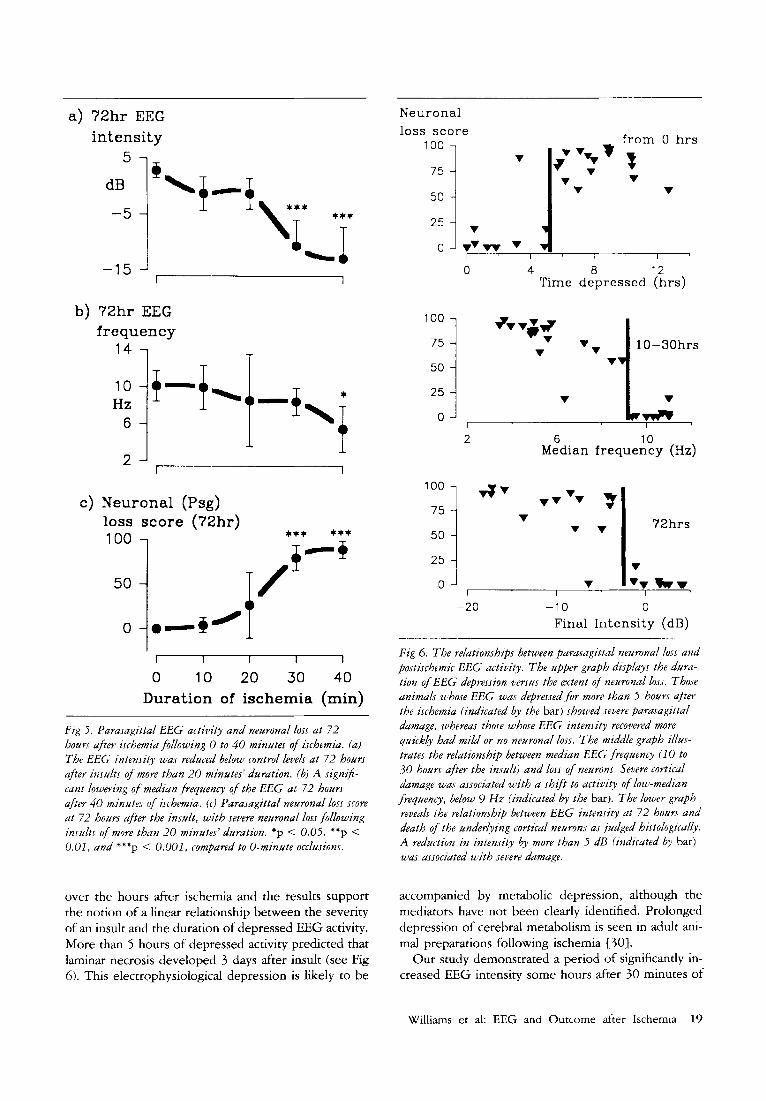
EEG was depressed for 8 k 2 hours (p < 0.001). This was followed by a shift to low-frequency activity of 5 % 1 Hz @ < 0.001) for the 10- to 30-hour period after the insult, whereas in the mildly damaged group the median frequency remained at 10 re 1 Hz (see Fig 6). Total EEG intensity was reduced air 72 hours by - 10 % 5 dB ( p < 0.001), whereas that in the mildly dam- aged group remained at 1 * 3 dB (see Fig 6).
Discussion We demonstrated highly significant relationships be- tween the severity of a hypoxic-ischemic insult and subsequent evolution of parasagittal EEG activity. Fol- lowing mild insults, there was progressive recovery of
Median EEG frequency
12 1 Hz
8
1 4 '
Peak EEG intensity
l o 1 ** T
I I I 1 -5 ' 0 10 20 30 40
Duration of ischemia (min)
Fig 4. Median frequency of the EEG (upper) and peak EEG intensity (lower) following 0 t o 40 minutes of ischemia. There ic a shift t o low,frequency .spike-wave activity (I0 t o 30 hours) after insults of more than 20 minutes' duration. The lmuer graph shows the peak intensity of the EEG during the 30 hours after the insult, following smoothing of the data. '4fter .30 minutes of ischemia, total EEG intensity peaked at 7 * .S dB above control levels. A period of intensity increased ahove baseline was not apparent after 40 minutes of ischemia.
normal EEG activity and only mild selective neuronal loss. With more severe insults, the EEG showed a ster- eotypic sequence of prolonged depressed activity, low-frequency epileptiform activity, and a final loss of intensity. This characteristic sequence was associated with laminar necrosis of the underlying cortex.
The distinct phases of EEG activity following a se- vere insult reflect different patterns of activity in the pyramidal neurons within the underlying cortex (27 , 28). A previous study by Mann and colleagues C29) showed that the recovery rate of EEG activity in the minutes immediately following an insult correlates with the duration of the isoelectric period during fetal hyp- oxia. Our study examined the recovery of the EEG
18 Annals of Neurology Vol 31 No 1 January 1992
a) 72hr EEG intensity
5 1-
Neuronal loss score
100 1
-5 1 - -b I I
b) 72hr EEG frequency
-r T
I
100
59
0
c) Neuronal (Psg) loss score (72hr) *** ***
I I I I 1 0 10 20 30 40
Duration of ischemia (min)
Fig 5 . Parasagittal EEG activity and neuronal loss at 72 hours after ischemia following (3 t o 40 minutes of ischemia. (a) The EEG intensity was reduced below control levels at 72 hours after insults o f more than 20 minutes' duration. (b) A sign$- cant lowering of median frequency of the EEG at 72 hours after 40 minutes of ischemia. (r) Parasagittal neuronal loss score at 72 hours after the insult, with severe neuronal loss following insults of more than 20 minutes' duration. *p < 0.05. **p < 0.01, and ***p < 0.001, compared to 0-minute occlusions.
over the hours after ischemia and the results support the notion of a linear relationship between the severity of an insult and the duration of depressed EEG activity. More than 5 hours of depressed activity predicted that laminar necrosis developed 3 days after insult (see Fig 6). This electrophysiological depression is likely to be
50 7 5 1 2: i ;w
0
100
75
50
25
0
100
75
50
25
0
from 0 hrs
3 v
v
-7
4 8 12 Time depressed (hrs)
I
v
10-30hrs
v v vt *
2 6 10 Median frequency (Hz)
v I I
-20 -10 0 Final Intensity (dB)
Fig 6. The relationships between parasagittal neuronal loss and postischemic E E G activity. The upper graph dzsplays the dura- tion of EEG depression versus the extent of neuronal loss. Those animals whose E E G was depressed for mare than 5 hours aftrr the ischemia (indicated by the bar) showed severe parasagittal dumage, whereas those whose EEG intensity recovered more quickly had mild or no neuronal loss. The middle graph illus- trates the relationship between median E E G frequency (10 to 30 hours after the insult) and loss of neurons. Severe cortical damage was associated with a shift t o activity of low-median frequency, below 9 H z (indicated by the bar). The lower graph reveuls the relationship between EEG intensity at 72 hours and death o f the underlying cortical neurons as judged histologically. A reduction in intensity by more than 5 dB (indicated by bar) was associated with severe dumage.
accompanied by metabolic depression, although the mediators have not been clearly identified. Prolonged depression of cerebral metabolism is seen in adult ani- mal preparations following ischemia C30).
Our study demonstrated a period of significantly in- creased EEG intensity some hours after 30 minutes of
Williams et al: EEG and Outcome after Ischemia 19

ischemia. This intense low-frequency activity, apparent as epileptiform or spike-wave activity [20}, is 1- .nown to be coupled with increased burst firing of the pyramidal neurons [31]. The peak intensity of this activity was biphasically related to the duration of the ischemia. (see Fig 4). The longest insults showed a lesser secortdary increase in intensity, perhaps because more parasagittal neurons were damaged during the longer primary in- sult 1321.
The mechanisms that trigger the critical Iransition from EEG depression to epileptiform activity arc: not known, but potential causes are worth considering. It has been suggested that epileptiform activity occurs with activation of polysynaptic feedback pat.hways as a consequence of diffuse hyperexcitability of cortical neurons [31, 333. There are several possible causes for the apparent increase in excitability of this population of neurons: First, it has been suggested that it is a consequence of a loss of inhibitory neurons (gamma- aminobutyric acid [IGABAIergic) 134, 353. Second, in- creased field (ephaptic) coupling between celils due to loss of extracellular space has been shown to increase excitability {36]; however, this is unlikely in this prepa- ration since the secondary loss of extracellular space occurs after the onset of epileptiform activity 1321. Al- ternatively, increased excitatory input may be a conse- quence of greater activity at the excitatory amino acid receptors. Excitotoxic activity is known to be induced after an insult in the developing brain f37). T,his could result from either an accumulation of excitatory amino acids in the extraccllular space or an upregulation of excitatory amino acid receptors 1381.
We found that the shift to epileptiform activity was inevitably associated with laminar necrosis, as opposed to mild selective neiironal loss of the underlying cortex in the fetal sheep. Several authors have speculated that postischemic epileptiform activity destroys forebrain neurons via excessive excitation after the primary insult [3-51. However, there is conflicting evidence. A close temporal association between loss of parietal cortical neurons and the onset of convulsions has been dernon- strated in the adult hyperglycemic rat following ce- rebral ischemia [ 3 5 , 391. In contrast, other studies indicated that there is an absence of postischemic epi- leptiform activity during the development of neuronal loss in adult animd preparations f 10, 11).
More than 2 hours of intense epileptiforni activity can cause edema and rapid neuronal death in adult animals 140, 41). Seizures lasting more than 30 min- utes following asphyxia in human neonates are associ- ated with poor neurological outcome 142, 431. During epileptic seizures, the cortical metabolic rate increases by 1.5 to 2.5 times r441. It has been suggeljted that epileptic damage occurs as a consequence of local met- abolic demands exceeding the limited availability of energy substrate in vulnerable neurons, and .not local
hypoxia-ischemia f41 , 451. Thus, postischemic epilepti- form activity is likely to increase local metabolic de- mands within an already compromised brain: This in- creased load may contribute to the cell dysfunction and death. Indeed, secondary intracellular edema appears to be a consequence of postischemic epileptiform activ- ity in this preparation [32]. In contrast, it has been suggested that postischemic seizures may stimulate functional recovery by reversing the depression 1461. A recent study revealed that these seizures can stimu- late expression of c-fos, a proto-oncogene; this is thought to mediate aspects of cell growth and differen- tiation 1471. Hence, further studies are needed to de- termine the precise consequences of the epileptiform activity.
Severe asphyxia can lead to hypoxic-ischemic en- cephalopathies; however, many asphyxiated infants re- cover without neurological abnormalities. Thus the neurological consequences of a perinatal asphyxic in- sult are difficult to assess quickly: Commonly used indi- ces of asphyxia at birth such as low Apgar scores, acide- mia, and the presence of amniotic meconium are poor predictors of neurological outcome 12, 131. Improved early diagnosis of those at risk of developing brain damage may facilitate therapeutic intervention to re- duce the severity of neurological deficits. Some of the electrophysiological criteria described here, such as the duration of EEG depression, may have prognostic value. The developments of online systems for data reduction of the EEG [223 offers a practical approach to such an assessment.
Continuous EEG analysis revealed that a distinct se- quence of parietal EEG activity was associated with hypoxic-ischemic encephalopathy in the fetal sheep. This information may prove useful for the rapid deter- mination of the severity and progression of hypoxic- ischemic damage. The development of intense epilepti- form activity indicates that the parasagittal cortex passes through a phase of postinsult hyperexcitability before the final loss of activity. Further studies are needed to determine the consequences of these sei- zures.
This research was funded by grants from the Medical Research Council of New Zealand, the Neurological Foundation, and the Na- tional Children’s Health Research Foundation.
We thank Dr Sue Davis for reviewing the manuscript
References 1. Pulsinelli W, Brierley J, Plum F. Temporal profile of neuronal
damage in a model of transient forebran ischemia. Ann Neurol 1982;11:491-498
2. Massarweh W, Vinters H, Schwam P, e t al. Delayed neuronal necrosis in neonatal hypoxia-ischemia. SOC Neurosci Abstr 1987; 13: 10
20 Annals of Neurology Vol 31 No 1 January 1992

3. Jorgensen M, Johnsen F, Diemer N. Removal of the entorhinal cortex protects hippocampal CA-1 neurons from ischemic dam- age. Acta Neuropathol (Bed) 1987;73:189-194
4. Brown A, Levy D, Kublik M, et al. Selective chromatolysis of neurons in the gerbil brain: a possible consequence of “epileptic” activity produced by common carotid artery occlusion. Ann Neurol 1979;5: 12 7 - 1 38
5 . Meyer F. Calcium, neuronal hyperexcitability and ischemic in- jury. Brain Res 1989;14:227-243
6. Hill A, Volpe J. Seizures, hypoxic-ischemic brain injury, and inrraventricular hemorrhage in the newborn. Ann Neurol
7. Mulligan J, Painter M, ODonoghue P. Neonatal asphyxia. 11: neonatal mortality and long term sequelea. J Pediatr 1980;
8. Aubourg P, Dulac 0, Plouin P, Diebler C. Infantile status epi- lepticus as a complication of “near-miss” sudden infant death. Dev Med Child Neurol 1985;27:40-48
9. Snyder B, Houser W, Loewenson R, et al. Neurologic prognosis after cardiopulmonary arrest. 111: seizure activity. Neurology 1980;30:1292-1297
10. Armstrong D, Neil1 K, Crain B, Nadler J. Absence of electro- graphic seizures after transient forebrain ischemia in the Mongo- lian gerbil. Brain Res 1989;476:174-178
11. Suzuki R, Yamaguchi T, Choh-Luh L, Klatzo I. The effects of 5-minute ischemia in Mongolian gerbils. 11. Changes of sponta- neous neuronal activity in cerebral cortex and CA1 sector of hippocampus. Acta Neuroparhol (Berl) 1983;60:2 17-222
12. Dixjhoorn M, Visser G, Fidler V, et al. Apgar score, meconium and acidaemia at birth in relation to neonatal neurological mor- bidity. Br J Obstet Gynaecol 1986;93:217-222
13. Dennis J. Johnson A, Mutch L, et al. Acid-base status at birth and neurodevelopmental outcome at four and one-half years. Am J Obstet Gynecol 1989;171:213-220
14. Connell J, Oozer R, De Bries L, et al. Continuous EEG monitor- ing of neonatal seizures: diagnostic and prognostic considera- tions. Arch Dis Child 1989;64:452-458
15. Bjerre I, Hellstrom-Westas L, Rosen I, Svenningsen N. Moni- toring of cerebral function after severe asphyxia in infancy. Arch Dis C hild 1983; 58:997- 1002
16. Low J, Robertson D, Simpson L. Temporal relationships of neu- ropathologic conditions caused by perinatal asphyxia. Obstet Gynecol 1989;160:608-614
17. Volpe J, Herscovitch P, Perlman J. Positron emission tomogra- phy in the asphyxiated term newborn: parasagittal impairment of cerebral blood flow. Ann Neurol 1985;17:287-296 Silverstein F, Buchanann K, Johnston M. Pathogenesis of hyp- oxic ischemic injury in a perinatal rodent model. Neurosci Lett
19. Robertson C, Finer N, Grace M. School performance of survi- vors of neonatal encephalopathy associated with birth asphyxia at term. J Pediatr 1989;114:753-760
20. Williams C, Gunn A. Gluckman P, Synek B. Delayed seizures occurring with hypoxic-ischemic encephalography in the fetal sheep. Pediarr Res 1990;27:561-565
21. Emoto S, Kitner D, Feyzi J, Gilboe D. Relating cerebral isch- emia and hypoxia to insult intensity. J Neurochem 1988;50: 1952-1958
22. Williams C, Gluckman P. Real-time spectral intensity analysis of the EEG on a microcomputer. J Neurosci Methods
23. Smith M, Auer R, Siesjo B. The density and distribution of ischemic brain injury in the rat following 2-10 min of forebrain ischemia. Ann Neuropathol 1984;64:3 19-332
24. Brown A, Brierley J. Anoxic-ischaemic cell change in rat brain light microscopic and fine structural observations. J Neurol Sci
1981;lO: 109-121
96:903-907
18.
198449~27 1-277
1990;32:9-13
197 1;16:59-84
25. Siegal S. Nonparametric statistics for the behavioural sciences. New York McGraw-Hill, 1956
26. Gasser T, Bacher P, Mocks J. Transformations towards the nor- mal distribution of broad band spectral parameters of the EEG. Electroencephalogr Clin Neurophysiol 1982;53:119-124
27. Mitzdorf U. Current source-density method and application in cat cerebral coi-rex: investigation of evoked potentials and EEG phenomena. Physiol Rev 1985;65:37-100
28. Barth D, Di S, Baumgartner C. Laminar cortical interactions during epileptic spikes studied with principal component analy- sis and physiological modelling. Brain Res 1989;484: 13-35
29. Mann L, Prichard J, Symmes D. EEG, ECG, and acid-base ob- servations during acute fetal hypoxia. Am J Obstet Gynecol
30. Kozuka M, Smith M, Siesjo B. Preischemic hyperglycemia en- hances postischemic depression of cerebral metabolic rate. J Cereb Blood Flow Metab 1989;9:478-490
31. Gloor P, Fariello R. Generalized epilepsy: some of its cellular mechanisms differ from those of focal epilepsy. Trends Neu- rosci 1988;11:63-68
32. Williams C, Gunn A, Gluckman P. The time course of inrracel- lular edema and epileptiform activity following perinatal hypoxia-ischemia in the sheep. Stroke 1991;22:516-521
33. Taylor C. How do seizures begin? Clues from hippocampal slices. Trends Neurosci 1988;11:357-378
34. Sloper J, Johnson P, Powell T. Selective degeneration of in- terneurons in the motor cortex of infant monkeys following controlled hypoxia: a possible cause of epilepsy. Brain Res 1980; 198:204-209
35. Smith M, Warner D, Siesjo B. Morphological lesions in the brain preceding the development of postischemic seizures. Acta Neuropathol (Berl) 1988;76:253-264
36. Andrew R, Fagan M, Ballyk B, Rosen A. Seizure susceptibility and the osmotic state. Brain Res 1989;498:175-180
37. Hattori H, Morin A, Schwartz P, et al. Posthypoxic treatment with MK-801 reduces hypoxic-ischemic damage in the neonatal rat. Neurology 1989;39:7 13-7 18
38. Baudry N, Smith E, Lynch G. Micromolar calcium stimulates proteolysis and glutamate binding in rat brain synaptic mem- branes. Science 1982;212:937-938
39. Warner D, Smith M, Siesjo B. Ischemia in normo- and hypergly- cemic rats: effects on brain water and electrolytes. Stroke
40. Meldrum B, Brierley J. Prolonged epileptic seizures in primates. Ischemic cell change and its relation to ictal physiological events. Arch Neurol 1973;28:10-17
41. Nevander G, Ingvar M, Auer R, Siesjo B. Starus epilepticus in well-oxygenated rats causes neuronal necrosis. Ann Neurol
42. Mellits E, Holden K, Freeman J. Neonatal seizures 11. A mulri- variate analysis of factors associated with outcome. Pediatrics
43. Tudehope D, Harris A, Hawes D, Hayes M. Clinical spectrum and outcome of neonatal convulsions. Aust Paediatr J 1988; 24:249-2 53
44. Meldrum B, Nilsso B. Cerebral blood flow and metabolic rate early and late in prolonged epileptic seizures induced by bicucul- line. Brain 1976;99:523-542
45. Watson G, Rader R, Lanthorn T. Epileptiform activity in vitro can produce long-term synaptic failure and persistent neuronal depolarization. Brain Res 1989;498:81-88
46. Schallert T, Hernandez T, Barth T. Recovery of function after brain damage: severe and chronic disruption by diazepam. Brain Res 1986;379:104-111
47. Gunn A, Dragunow M, Faull R, Gluckman P. Effects of hypoxia-ischemia and seizures on neuronal and glial-like c-fos protein levels in the infant rat. Brain Res 1990;S31:105-116
1970;106:39-5 1
1987;18:464-47 1
1985;18:281-290
1982;70:177-185
Williams e t al: EEG and Outcome after Ischemia 21
Related Documents











