Send Orders for Reprints to [email protected] MicroRNA, 2013, 2, 91-101 91 2211-5366/13 $58.00+.00 © 2013 Bentham Science Publishers Non Coding RNA in Muscle Differentiation and Disease Mariangela Morlando 1 , Alessandro Rosa 1 , Elisa Caffarelli 2,3 , Alessandro Fatica 1 and Irene Bozzoni* ,1,2,3 1 Dept. of Biology and Biotechnology "Charles Darwin"; 2 Institute of Molecular Biology and Pathology of C.N.R.; 3 Center for Life Nano Science @Sapienza, Istituto Italiano di Tecnologia, Sapienza University of Rome, P.le A. Moro 5, 00185 Rome, Italy Abstract: Non coding RNAs have provided in the last decades a very exciting research field with the discovery that a largely unexplored fraction of our genome encodes for RNA without protein coding activity. Here we revise the current knowledge of how non coding RNAs impact on muscle differentiation and homeostasis in normal and disease conditions and how they can provide powerful tools for therapeutic interventions and disease diagnosis. Moreover, we discuss new insights into additional mechanisms of post-transcriptional regulation involving a new class of long non coding RNAs shown to impact on the distribution of microRNA molecules on their mRNA targets. Keywords: Duchenne muscular dystrophy, dystrophin, lncRNA, microRNA, muscle differentiation, ncRNA. INTRODUCTION The central dogma referring to genes as templates for protein synthesis belongs to a past time of molecular biol- ogy. In the post-genomic era, a careful examination of which DNA sequences are transcribed into RNA in different eu- karyotic species has opened an intriguing debate about the exact definition of a gene and the relationship between the number of genes and the complexity of higher organisms’ biology. Moreover, the discovery of abundant transcription of RNAs that do not code for proteins provided an important new perspective on the role of RNA in gene regulation. Deep sequencing analysis of the transcriptome has indeed shown that the fraction of the genome that is used as a tem- plate for RNA synthesis is much higher than the 1-2% which is known to contain codogenic capacity, even though most of this unannotated transcription remains to be characterized. Moreover, the abundance of overlapping transcripts, either on the same strand or on the opposite one, suggests a new view of genomic organization, where gene borders are less defined and the usage of the same sequence for different purposes seems common. Therefore, it is now becoming largely accepted that the non-coding portion of the genome rather than its coding counterpart is likely to account for the greater complexity of higher eukaryotes and that it hides many yet unknown and unexplored functions [1]. Non coding RNAs belong to several groups and are in- volved in many cellular processes: while the knowledge on the biogenesis and function of small non coding RNAs (sncRNAs) has incredibly expanded in the last decades, from the initial identification of tRNAs, snRNAs, snoRNAs and *Address correspondence to this author at the Dept. of Biology and Bio- technology "Charles Darwin”, Sapienza University of Rome, P.le A. Moro 5, 00185 Rome – Italy; Tel: +39-0649912202; Fax: +39-0649912500; E-mail: [email protected] more recently microRNAs (miRNAs, see [2]), very little is still known about the long non coding counterpart of the transcriptome. Long non coding RNAs (lncRNAs) bio- chemically resemble mRNAs, however, instead of templat- ing protein synthesis they function in the control of genetic regulatory circuitries. Among the sncRNA class, miRNAs have recently at- tracted most of the scientists’ interest. Initially regarded as a curious peculiarity of nematodes, miRNAs are now consid- ered as major regulators of development, metabolism, differ- entiation and homeostasis in all multicellular organisms [3- 6]. The recognition that miRNAs are involved in several human diseases, including cancer [7], contributed to foster the interest in this class of small non coding RNAs. In this review, we discuss recent findings showing how different features of ncRNAs have provided very useful tools for improving our knowledge on the molecular mechanisms controlling muscle differentiation and related disorders. In particular, we will describe the use of ncRNAs as: i) thera- peutic molecules enabling the modification of gene expres- sion in a sequence specific way and the rescue of defective functions; ii) diagnostic molecules identifying specific pathogenetic alterations; iii) important gene expression modulators able to control muscle differentiation and ho- meostasis. MUSCLE DIFFERENTIATION During embryonic development, skeletal muscles form from a subpopulation of mesoderm cells, in which the mus- cle fate is dictated by the ordered activation of several spe- cific transcription factors. A crucial role is played by the paired box genes, Pax3 and Pax7, and the MADS box tran- scriptional regulators SRF and MEF2 that act in combinato- rial cooperation with myogenic bHLH factors, such as MyoD, Myf5, Mrf4 and myogenin. These myogenic factors, in turn, regulate the expression of downstream structural genes.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Send Orders for Reprints to [email protected]
MicroRNA, 2013, 2, 91-101 91
2211-5366/13 $58.00+.00 © 2013 Bentham Science Publishers
Non Coding RNA in Muscle Differentiation and Disease
Mariangela Morlando1, Alessandro Rosa
1, Elisa Caffarelli
2,3, Alessandro Fatica
1 and
Irene Bozzoni*,1,2,3
1Dept. of Biology and Biotechnology "Charles Darwin";
2Institute of Molecular Biology and Pathology of C.N.R.;
3Center for Life Nano Science @Sapienza, Istituto Italiano di Tecnologia, Sapienza University of Rome, P.le A. Moro 5,
00185 Rome, Italy
Abstract: Non coding RNAs have provided in the last decades a very exciting research field with the discovery that a
largely unexplored fraction of our genome encodes for RNA without protein coding activity. Here we revise the current
knowledge of how non coding RNAs impact on muscle differentiation and homeostasis in normal and disease conditions
and how they can provide powerful tools for therapeutic interventions and disease diagnosis. Moreover, we discuss new
insights into additional mechanisms of post-transcriptional regulation involving a new class of long non coding RNAs
shown to impact on the distribution of microRNA molecules on their mRNA targets.
Keywords: Duchenne muscular dystrophy, dystrophin, lncRNA, microRNA, muscle differentiation, ncRNA.
INTRODUCTION
The central dogma referring to genes as templates for protein synthesis belongs to a past time of molecular biol-ogy. In the post-genomic era, a careful examination of which DNA sequences are transcribed into RNA in different eu-karyotic species has opened an intriguing debate about the exact definition of a gene and the relationship between the number of genes and the complexity of higher organisms’ biology. Moreover, the discovery of abundant transcription of RNAs that do not code for proteins provided an important new perspective on the role of RNA in gene regulation.
Deep sequencing analysis of the transcriptome has indeed shown that the fraction of the genome that is used as a tem-plate for RNA synthesis is much higher than the 1-2% which is known to contain codogenic capacity, even though most of this unannotated transcription remains to be characterized. Moreover, the abundance of overlapping transcripts, either on the same strand or on the opposite one, suggests a new view of genomic organization, where gene borders are less defined and the usage of the same sequence for different purposes seems common.
Therefore, it is now becoming largely accepted that the non-coding portion of the genome rather than its coding counterpart is likely to account for the greater complexity of higher eukaryotes and that it hides many yet unknown and unexplored functions [1].
Non coding RNAs belong to several groups and are in-volved in many cellular processes: while the knowledge on the biogenesis and function of small non coding RNAs (sncRNAs) has incredibly expanded in the last decades, from the initial identification of tRNAs, snRNAs, snoRNAs and
*Address correspondence to this author at the Dept. of Biology and Bio-
technology "Charles Darwin”, Sapienza University of Rome, P.le A. Moro
5, 00185 Rome – Italy; Tel: +39-0649912202; Fax: +39-0649912500;
E-mail: [email protected]
more recently microRNAs (miRNAs, see [2]), very little is still known about the long non coding counterpart of the transcriptome. Long non coding RNAs (lncRNAs) bio-chemically resemble mRNAs, however, instead of templat-ing protein synthesis they function in the control of genetic regulatory circuitries.
Among the sncRNA class, miRNAs have recently at-tracted most of the scientists’ interest. Initially regarded as a curious peculiarity of nematodes, miRNAs are now consid-ered as major regulators of development, metabolism, differ-entiation and homeostasis in all multicellular organisms [3-6]. The recognition that miRNAs are involved in several human diseases, including cancer [7], contributed to foster the interest in this class of small non coding RNAs.
In this review, we discuss recent findings showing how different features of ncRNAs have provided very useful tools for improving our knowledge on the molecular mechanisms controlling muscle differentiation and related disorders. In particular, we will describe the use of ncRNAs as: i) thera-peutic molecules enabling the modification of gene expres-sion in a sequence specific way and the rescue of defective functions; ii) diagnostic molecules identifying specific pathogenetic alterations; iii) important gene expression modulators able to control muscle differentiation and ho-meostasis.
MUSCLE DIFFERENTIATION
During embryonic development, skeletal muscles form from a subpopulation of mesoderm cells, in which the mus-cle fate is dictated by the ordered activation of several spe-cific transcription factors. A crucial role is played by the paired box genes, Pax3 and Pax7, and the MADS box tran-scriptional regulators SRF and MEF2 that act in combinato-rial cooperation with myogenic bHLH factors, such as MyoD, Myf5, Mrf4 and myogenin. These myogenic factors, in turn, regulate the expression of downstream structural genes.

92 MicroRNA, 2013, Vol. 2, No. 2 Morlando et al.
At the onset of myogenesis in the embryo, signals from adjacent tissues induce, in the developing dorsal somite, the transcription of the muscle fate determination genes Myf5/Mrf4, which in turn activate MyoD [8]. This first wave of myogenesis determines the formation of the early myo-tome and is followed by a second wave in which Pax3 and Pax7 regulate the entry of cells into the myogenic pro-gramme by inducing Myf5 and MyoD, which in turn deter-mine the commitment to the myogenic lineage [9]. Down-stream of Myf5 and MyoD, myogenin and Mrf4 are involved in the establishment of the terminal muscle phenotype. As myoblasts terminally differentiate, they irreversibly exit the cell cycle, fuse to form multinucleated myotubes and start expressing structural genes such as M-cadherin, myosin heavy and light chains, muscle creatine kinase and dystro-phin.
This transcriptional network has been known for a long time [10]; however, an unexpected second layer of regulation emerged in the last years as muscle-associated miRNAs came to the scene [11-18]. Striated muscle cells express spe-cific miRNAs, whose expression is restricted to this tissue. They belong to two groups: the miR-1 and miR-133 clusters and the miR-208/499 family. The first group includes three clusters, each containing a miR-1 and a miR-133 component: miR-1-1/miR-133a-2 on mouse chromosome 2, miR-1-2/miR-133a-1 on mouse chromosome 18 and miR-206/miR-133b on mouse chromosome 1 [12]. The second group is represented by miRNAs hosted in introns of Myosin heavy chains (MHC) genes (Myomirs): miR-208 ( MHC), miR-208b ( MHC) and miR-499 (MYH7b) [16].
The transcription regulation of the miR-1/133 loci is un-der the control of the myogenic factors MyoD, myogenin, Mef2 and SRF [11, 13, 19, 20]. In particular, it has been shown that in the somites the expression of miR-1-1 and miR-133a-2 is driven by Mef2, whereas MyoD regulates the expression of miR-1-2 and miR-133a-1. In the heart, SRF acts together with its co-activator myocardin to strongly ac-tivate both clusters [11]. The discovery of long transcripts spanning across these loci suggested that each miR-1 and miR-133 couple could originate by processing from a com-mon bicistronic precursor [12]. However, two distinct cis-regulatory elements have been described upstream to each miRNA coding region [20]. Moreover, recent findings on the miR-206/133b cluster support the hypothesis that a more complex biosynthetic pathway underlies the expression of the two members of the cluster [19] (see below).
Despite belonging to the same clusters, the two members of the miR-1/miR-133 pairs have been shown to play distinct roles in modulating skeletal muscle proliferation and differ-entiation. Whereas miR-1/206 promotes differentiation of cultured myoblasts, miR-133 controls proliferation [12]. Mechanistically, this is achieved by targeting multiple genes involved in myogenesis. Among miR-1/206 relevant targets, histone deacetylase 4 (HDAC4) [12], the p180 subunit of DNA polymerase (Pola1) [21], connexin43 (Cx43) [22], utrophin (Utrn) [23] and Notch3 [24] have established roles during terminal differentiation, being involved in the main-tenance of a proliferative state. Whereas miR-1 is expressed in both skeletal and cardiac muscle, miR-206 is specifically restricted to skeletal muscle where it plays a crucial role in
the differentiation of activated satellite cells by targeting Pax7 mRNA [25] (see below). Conversely, the control of proliferation mediated by miR-133 is exerted by the repres-sion of SRF [12] and the Mef2c co-activator, Maml1 [19], among other target genes.
While the miR-1/133 clusters play a role in both skeletal muscle and heart, the expression of miR-208/499 is restricted to cardiomyocites [26]. It has been shown that these Myo-mirs are involved in multiple aspects of the regulation of cardiac stress response, such as the - to - myosin heavy chain (MHC) switch occurring upon heart injury and in car-diomyocyte hypertrophy and fibrosis (reviewed in [16]). Mechanistically, downregulation of the target THRAP1 (thy-roid hormone receptor coregulator) could contribute to the miR-208-mediated activation of -MHC in response to stress [26].
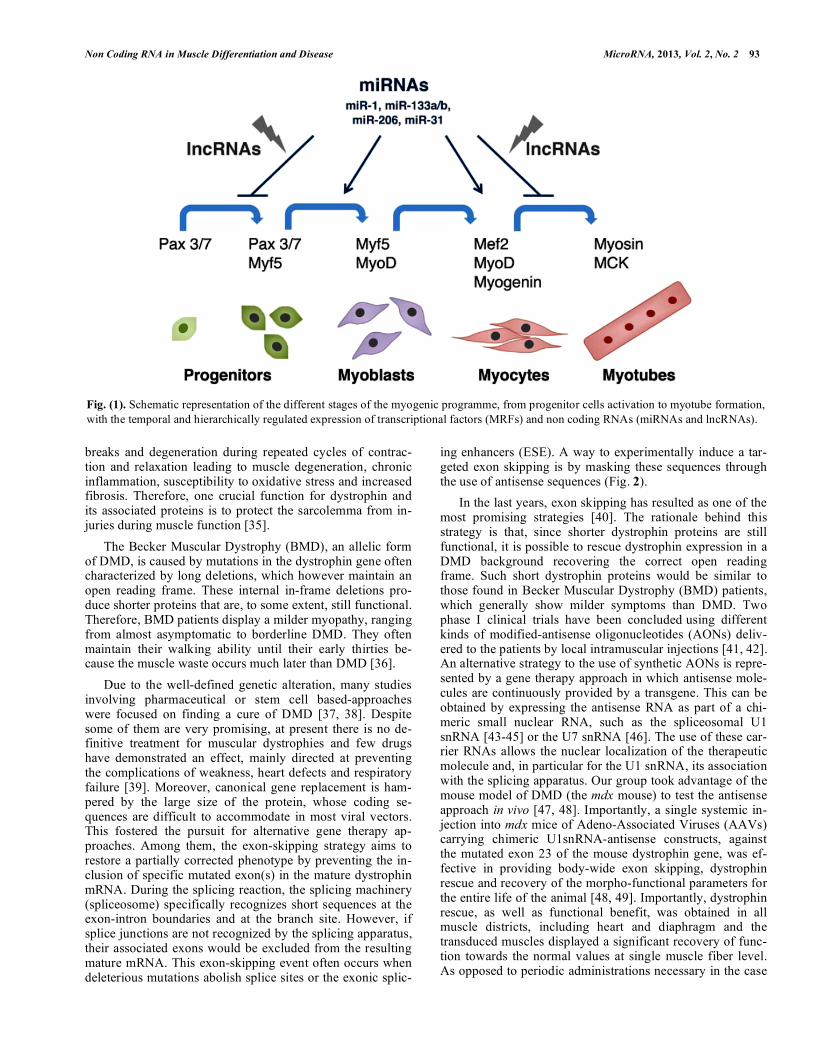
Besides the master regulators of myogenesis known so far (transcriptional factors and miRNAs - [11, 27]), recent discoveries added new players, belonging to a previously disregarded class of transcripts, named long non-coding RNAs (lncRNAs), which may play a relevant role in the con-trol of gene expression. The interplay between transcrip-tional factors and non-coding RNAs, including miRNAs and lncRNAs, establishes a hierarchically organized regulatory network underlying muscle differentiation (Fig. 1). Impor-tantly, the alteration of each component of this network may play major roles in muscle pathology, such as muscular dys-trophies. Therefore, both coding and non-coding genes might represent relevant therapeutic targets for gene therapy ap-proaches. To this regard, while several miRNAs were found to be deregulated in human muscular disorders [28], suggest-ing an important role of miRNAs in the onset of the pathol-ogy, so far only a few example of lncRNAs have been re-ported [29] (see below).
ncRNAS AS THERAPEUTIC MOLECULES: AN-TISENSE RNA AND EXON SKIPPING
Duchenne Muscular Dystrophy (DMD) is a X-linked se-vere progressive myopathy that affects 1 in 3500 live males [30]. This fatal disease is caused by deletions and point mu-tations disrupting the translational reading frame of the dys-trophin gene. This huge gene (2.5-Mb) encodes for several proteins, produced by alternative splicing and usage of dif-ferent promoters. The full-length structural 427-kDa isoform plays a key role in maintaining muscle fiber integrity. It comprises four domains: an N-terminal actin binding do-main, a central “rod” domain (26 spectrin-like repeats), a cystein-rich domain and a C-terminal domain [31]. In mus-cles, dystrophin is localized near the cytoplasmic face of the sarcolemma [32]. In this position, the C-terminus of dystro-phin, containing a dystroglycan-binding domain (DgBD), assembles the dystrophin-Associated Protein Complex (DAPC), containing - and - dystroglycans, - - - - and - sarcoglycans, sarcospan, - and - syntrophins, neuronal
nitric oxide syntase (nNOS) and dystrobrevins [33]. In dys-trophic muscle fibers of DMD patients and in animal models, the absence of dystrophin leads to a dramatic destabilization of the DAPC and reduced levels of DAPs [34]. In absence of such bridge over the sarcolemma, muscle fibers become more sensitive to mechanical damage and susceptible to
Non Coding RNA in Muscle Differentiation and Disease MicroRNA, 2013, Vol. 2, No. 2 93
breaks and degeneration during repeated cycles of contrac-tion and relaxation leading to muscle degeneration, chronic inflammation, susceptibility to oxidative stress and increased fibrosis. Therefore, one crucial function for dystrophin and its associated proteins is to protect the sarcolemma from in-juries during muscle function [35].
The Becker Muscular Dystrophy (BMD), an allelic form of DMD, is caused by mutations in the dystrophin gene often characterized by long deletions, which however maintain an open reading frame. These internal in-frame deletions pro-duce shorter proteins that are, to some extent, still functional. Therefore, BMD patients display a milder myopathy, ranging from almost asymptomatic to borderline DMD. They often maintain their walking ability until their early thirties be-cause the muscle waste occurs much later than DMD [36].
Due to the well-defined genetic alteration, many studies involving pharmaceutical or stem cell based-approaches were focused on finding a cure of DMD [37, 38]. Despite some of them are very promising, at present there is no de-finitive treatment for muscular dystrophies and few drugs have demonstrated an effect, mainly directed at preventing the complications of weakness, heart defects and respiratory failure [39]. Moreover, canonical gene replacement is ham-pered by the large size of the protein, whose coding se-quences are difficult to accommodate in most viral vectors. This fostered the pursuit for alternative gene therapy ap-proaches. Among them, the exon-skipping strategy aims to restore a partially corrected phenotype by preventing the in-clusion of specific mutated exon(s) in the mature dystrophin mRNA. During the splicing reaction, the splicing machinery (spliceosome) specifically recognizes short sequences at the exon-intron boundaries and at the branch site. However, if splice junctions are not recognized by the splicing apparatus, their associated exons would be excluded from the resulting mature mRNA. This exon-skipping event often occurs when deleterious mutations abolish splice sites or the exonic splic-
ing enhancers (ESE). A way to experimentally induce a tar-geted exon skipping is by masking these sequences through the use of antisense sequences (Fig. 2).
In the last years, exon skipping has resulted as one of the most promising strategies [40]. The rationale behind this strategy is that, since shorter dystrophin proteins are still functional, it is possible to rescue dystrophin expression in a DMD background recovering the correct open reading frame. Such short dystrophin proteins would be similar to those found in Becker Muscular Dystrophy (BMD) patients, which generally show milder symptoms than DMD. Two phase I clinical trials have been concluded
using different
kinds of modified-antisense oligonucleotides (AONs) deliv-ered to the patients by local intramuscular injections [41, 42]. An alternative strategy to the use of synthetic AONs is repre-sented by a gene therapy approach in which antisense mole-cules are continuously provided by a transgene. This can be obtained by expressing the antisense RNA as part of a chi-meric small nuclear RNA, such as the spliceosomal U1 snRNA [43-45] or the U7 snRNA [46]. The use of these car-rier RNAs allows the nuclear localization of the therapeutic molecule and, in particular for the U1 snRNA, its association with the splicing apparatus. Our group took advantage of the mouse model of DMD (the mdx mouse) to test the antisense approach in vivo [47, 48]. Importantly, a single systemic in-jection into mdx mice of Adeno-Associated Viruses (AAVs) carrying chimeric U1snRNA-antisense constructs, against the mutated exon 23 of the mouse dystrophin gene, was ef-fective in providing body-wide exon skipping, dystrophin rescue and recovery of the morpho-functional parameters for the entire life of the animal [48, 49]. Importantly, dystrophin rescue, as well as functional benefit, was obtained in all muscle districts, including heart and diaphragm and the transduced muscles displayed a significant recovery of func-tion towards the normal values at single muscle fiber level. As opposed to periodic administrations necessary in the case
Fig. (1). Schematic representation of the different stages of the myogenic programme, from progenitor cells activation to myotube formation,
with the temporal and hierarchically regulated expression of transcriptional factors (MRFs) and non coding RNAs (miRNAs and lncRNAs).

47� 51� 52�
CUA AGC UGA AUG CAA�
47� 51� 52�
CUA AG� CUG AAU� G CAA�
out of frame mRNA�
���
47� 51� 52�UGA�UG
pre-mRNA�
CUA AGG CAA�47� 52�
in frame mRNA�
47� 52�47474747
DAPC�nNOS�
5’�
7 5557 527 5277 525477 577 557 5247777 524777 57 5
DYS�
CUA AG� CUG AAU� G CAA�
47� 51� 52�
pre-mRNA�
DMD background� exon skipping strategy�
94 MicroRNA, 2013, Vol. 2, No. 2 Morlando et al.
of AONs, this suggested that one single AAV treatment could be effective in conferring a long-term benefit, an im-portant feature to lay the basis for antisense-mediated gene therapy in DMD patients.
miRNAs AND DUCHENNE MUSCULAR DYSTRO-PHY
miRNAs and lncRNAs are integrated in the molecular networks controlling normal muscle differentiation and their alteration is linked to muscle pathology. The work from our lab and others in cellular and animal model systems of Duchenne Muscular Dystrophy contributed to elucidate the role of single network components during muscle differentia-tion and regeneration [50]. To this aim, rescue of the Dystro-phin protein in deficient cells by exon-skipping represents a useful tool to dissect molecular networks underlying the pa-thology.
Several miRNAs were previously described to be deregu-lated in human muscular disorders [28], suggesting an im-portant role of miRNAs in the onset of these pathologies. Particular emphasis was given to Duchenne Muscular Dys-trophy, which is one of the most diffuse and severe muscle degenerative processes.
Many pieces of evidence suggest that besides their well-defined structural role, dystrophin and its associated proteins could have regulatory functions. The subcellular localization of DAPC, near the sarcolemma, places the proteins of the complex in a proper position to function as sensors for ex-tracellular stimuli. Several signalling molecules interact with components of the DAPC [51-53]. Therefore, this complex could function as a scaffold for the localization of signalling mediators. In a fiber lacking dystrophin, and an appropriate DAPC, the malfunction of these signalling events would
contribute to Duchenne pathogenesis. To this regard, it is in-teresting to note that the pathological features of DMD mus-cles were ameliorated in mdx mice also when dystrophin res-cue occurred at very low levels [49, 54], suggesting that par-tial protein re-localization to the membrane could be only part of the story. Regarding non-structural regulatory func-tions, a role of the DAPC in the intracellular Nitric Oxide (NO) pathway has been established. The association between the sarcolemmal neuronal Nitric Oxide Synthase (nNOS) and DAPC seems to be particularly important, since its disrup-tion leads to impaired NO production in dystrophic muscles [51]. Nitric Oxide directly controls Histone Deacetylases (HDACs), impinging on the regulation of chromatin state. In neurons, a NO-dependent S-nitrosylation of HDAC2 induced the activation of genes for neuronal development [55]. Moreover, HDAC2 activity resulted to be specifically in-creased in dystrophin-deficient mdx mice, which are defec-tive for the NO-pathway [56]. In line with this, the dys-trophic phenotype of mdx mice was relieved by the rescue of nNOS expression or by treatment with deacetylase inhibitors [57, 58].
Besides its therapeutic applications, the antisense RNA-mediated exon skipping strategy to rescue dystrophin func-tion represents a valuable tool for functional studies. Indeed, lack of dystrophin in DMD myoblasts can be rescued at dif-ferent levels by exon skipping, providing a useful system to dissect the functional role of variable amounts of the protein in muscle cell physiology. To this regard, our group demon-strated that while in DMD the absence of dystrophin triggers the delocalization and inactivation of nNOS, exon-skipping mediated rescue of dystrophin led to nNOS re-localizion on the membrane and its activation. In the absence of dystro-phin and nNOS, HDAC2 remained bound to the chromatin of specific target genes, while restoration of nNOS on the
Fig. (2). Antisense-mediated exon skipping. In most of the cases, DMD mutations disrupt the translational open reading frame of the dystro-
phin gene. In the indicated example, the deletion of exons 48-50 produces an out of frame mRNA with the generation of a premature stop
codon (UGA, red triplet), which induces mRNA degradation through the nonsense mediated decay (NMD) machinery. The absence of Dys-
trophin leads to the destabilization of the DAPC complex and nNOS at the sarcolemma. The exon skipping strategy (right panel), employing
an antisense molecule against the splice junctions of exon 51, induces its exclusion from the mature mRNA. The effective skipping of the
exon 51 restores the open reading frame generating a shorter but still functional Dystrophin able to re-localize DAPC and nNOS at the sar-colemma.

miR-1/miR-29……�
promoter�
WT muscle fibers�
nucl
eus�
cyto
plas
m�
DAPC�
nNOS�DYS�
HDAC2�
S S NO NO
Duchenne muscle fibers�
nucl
eus�
cyto
plas
m�
oxidative stress / fibrosis�
HDAC2�
S S
Non Coding RNA in Muscle Differentiation and Disease MicroRNA, 2013, Vol. 2, No. 2 95
membrane correlated with HDAC2 nitrosylation and its re-lease from the chromatin [25].
Furthermore, we correlated this regulatory pathway with the expression of a specific set of miRNAs controlling mo-lecular circuitries with a crucial role in DMD physiopathol-ogy. With respect to control myoblasts, in dystrophic cells the absence of HDAC2 nitrosylation led to its persistence on specific miRNA promoters and subsequent transcriptional repression; notably, upon recue of dystrophin, HDAC2 was nitrosylated and the same group of miRNAs, previously re-pressed, were up-regulated (Fig. 3).
A similar control was also shown in human DMD myoblasts rescued for dystrophin synthesis through exon skipping. HDAC2 persisted onto miRNA promoters in the absence of dystrophin, resulting in a decrease of acetylation of the lysine 9 of histone H3 (H3K9-Ac) and low levels of miRNA expression. On the contrary, upon rescue of dystro-phin synthesis, HDAC2 was released from miRNA promot-ers, H3K9-Ac increased and transcription resumed. There-fore, a regulatory pathway comprising dystrophin, nNOS and HDAC2 modulates the expression of specific miRNAs in muscle cells and the disruption of this circuitry in DMD could impair the proper expression of these miRNAs (Fig. 3).
miRNAs deregulated by loss of dystrophin/nNOS signal-ling could be grouped in two classes: muscle specific (miR-1 and miR-133) and more ubiquitous (miR-29 and miR-30) ones. Interestingly, these miRNAs are poorly expressed in mdx mice, but recovered at levels close to wild-type mice when dystrophin was rescued at less than 10% of its normal value. Both groups, by different routes, correlate with several
pathogenetic traits of DMD. Dystrophic muscles are report-edly susceptible to oxidative stress. In DMD muscles, miR-1 down-regulation caused the up-regulation of a direct target, Glucose-6-Phosphate Dehydrogenase (G6PD). G6PD is a key enzyme in the control of the redox state of the cell; how-ever, in dystrophic fibers, where higher expression levels of NADPH oxidase are detected, G6PD has an aberrant action in radical production; in fact in mdx muscles, most of the NADPH is utilized to convert O2 into O
-2. Therefore, the low
levels of miR-1 in dystrophic muscles worsen the oxidative stress conditions of the fibers. Moreover, detoxification of the superoxide radical could further reduce NO levels al-ready low in mdx. Local over-expression of miR-1 in mdx muscles determined down-regulation of G6PD and in turn increase of HDAC2 nitrosylation, proving the existence of a feed forward circuitry between G6PD and miR-1 through NO levels and nitrosylation of HDAC2. These data indicated that the regulatory network dependent on the Dys/nNOS pathway has physiological relevance in controlling the oxido-reductase state of the injured muscle.
The relevant role of the identified dystrophin-dependent miRNAs in the DMD pathogenesis was also tested for mir-29 that was causally linked to the fibrotic state of the dys-trophic fiber. Down-regulation of miR-29 in mdx muscles favoured fibrosis that was instead reduced when its levels were restored. Indeed, miR-29 is known to repress collagen and elastin mRNAs responsible for fibrotic tissue deposition [25].
In the overall, these data suggested that nNOS activity plays an important role in muscle homeostasis via its ability to directly regulate the expression of specific HDAC2 target
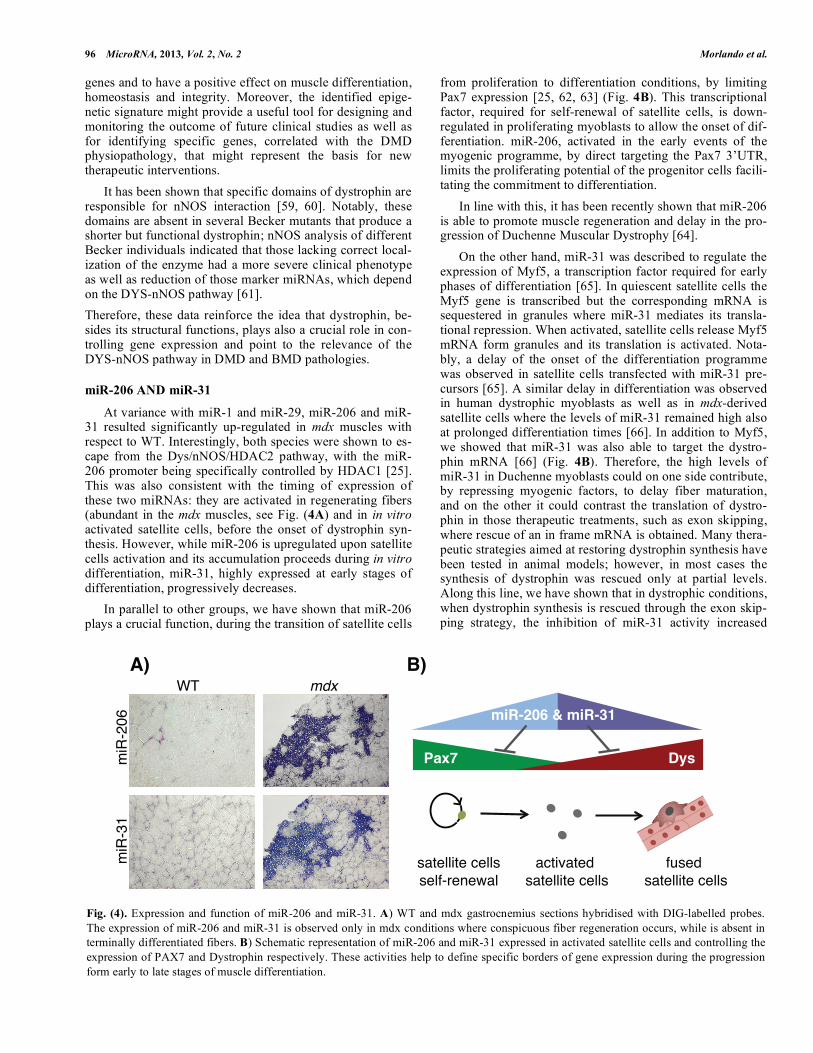
Fig. (3). Regulatory pathway involving Dystrophin, nNOS and HDAC2. In WT muscle fibers (left panel) the presence of Dystrophin allows
the correct localization and function of the DAPC complex and the stabilization and activation of the nNOS enzyme. Increased NO levels,
produce he nitrosylation of HDAC2 cysteine residues 262 and 274. These modifications exclude HDAC2 from promoter regions and allow
the expression of several genes; among them miR-1 and miR-29 were identified. The lack of Dystrophin in Duchenne conditions (right
panel) results in the delocalization of DAPC and nNOS with subsequent reduction of intracellular NO levels. Therefore unmodified HDAC2
persists on the promoters leading to transcriptional repression.
96 MicroRNA, 2013, Vol. 2, No. 2 Morlando et al.
genes and to have a positive effect on muscle differentiation, homeostasis and integrity. Moreover, the identified epige-netic signature might provide a useful tool for designing and monitoring the outcome of future clinical studies as well as for identifying specific genes, correlated with the DMD physiopathology, that might represent the basis for new therapeutic interventions.
It has been shown that specific domains of dystrophin are responsible for nNOS interaction [59, 60]. Notably, these domains are absent in several Becker mutants that produce a shorter but functional dystrophin; nNOS analysis of different Becker individuals indicated that those lacking correct local-ization of the enzyme had a more severe clinical phenotype as well as reduction of those marker miRNAs, which depend on the DYS-nNOS pathway [61].
Therefore, these data reinforce the idea that dystrophin, be-sides its structural functions, plays also a crucial role in con-trolling gene expression and point to the relevance of the DYS-nNOS pathway in DMD and BMD pathologies.
miR-206 AND miR-31
At variance with miR-1 and miR-29, miR-206 and miR-31 resulted significantly up-regulated in mdx muscles with respect to WT. Interestingly, both species were shown to es-cape from the Dys/nNOS/HDAC2 pathway, with the miR-206 promoter being specifically controlled by HDAC1 [25]. This was also consistent with the timing of expression of these two miRNAs: they are activated in regenerating fibers (abundant in the mdx muscles, see Fig. (4A) and in in vitro activated satellite cells, before the onset of dystrophin syn-thesis. However, while miR-206 is upregulated upon satellite cells activation and its accumulation proceeds during in vitro differentiation, miR-31, highly expressed at early stages of differentiation, progressively decreases.
In parallel to other groups, we have shown that miR-206 plays a crucial function, during the transition of satellite cells
from proliferation to differentiation conditions, by limiting Pax7 expression [25, 62, 63] (Fig. 4B). This transcriptional factor, required for self-renewal of satellite cells, is down-regulated in proliferating myoblasts to allow the onset of dif-ferentiation. miR-206, activated in the early events of the myogenic programme, by direct targeting the Pax7 3’UTR, limits the proliferating potential of the progenitor cells facili-tating the commitment to differentiation.
In line with this, it has been recently shown that miR-206 is able to promote muscle regeneration and delay in the pro-gression of Duchenne Muscular Dystrophy [64].
On the other hand, miR-31 was described to regulate the expression of Myf5, a transcription factor required for early phases of differentiation [65]. In quiescent satellite cells the Myf5 gene is transcribed but the corresponding mRNA is sequestered in granules where miR-31 mediates its transla-tional repression. When activated, satellite cells release Myf5 mRNA form granules and its translation is activated. Nota-bly, a delay of the onset of the differentiation programme was observed in satellite cells transfected with miR-31 pre-cursors [65]. A similar delay in differentiation was observed in human dystrophic myoblasts as well as in mdx-derived satellite cells where the levels of miR-31 remained high also at prolonged differentiation times [66]. In addition to Myf5, we showed that miR-31 was also able to target the dystro-phin mRNA [66] (Fig. 4B). Therefore, the high levels of miR-31 in Duchenne myoblasts could on one side contribute, by repressing myogenic factors, to delay fiber maturation, and on the other it could contrast the translation of dystro-phin in those therapeutic treatments, such as exon skipping, where rescue of an in frame mRNA is obtained. Many thera-peutic strategies aimed at restoring dystrophin synthesis have been tested in animal models; however, in most cases the synthesis of dystrophin was rescued only at partial levels. Along this line, we have shown that in dystrophic conditions, when dystrophin synthesis is rescued through the exon skip-ping strategy, the inhibition of miR-31 activity increased
Fig. (4). Expression and function of miR-206 and miR-31. A) WT and mdx gastrocnemius sections hybridised with DIG-labelled probes.
The expression of miR-206 and miR-31 is observed only in mdx conditions where conspicuous fiber regeneration occurs, while is absent in
terminally differentiated fibers. B) Schematic representation of miR-206 and miR-31 expressed in activated satellite cells and controlling the
expression of PAX7 and Dystrophin respectively. These activities help to define specific borders of gene expression during the progression
form early to late stages of muscle differentiation.
miR
-31�
miR
-206
�
WT� mdx�
satellite cells�self-renewal �
activated� satellite cells�
fused� satellite cells�
Pax7� Dys�
miR-206 & miR-31�
A)� B)�

Non Coding RNA in Muscle Differentiation and Disease MicroRNA, 2013, Vol. 2, No. 2 97
dystrophin production. Since in a compromised muscle the contribution to dystrophin production by regenerating fibers is quite relevant, miR-31 repression in this compartment can represent an improvement to current therapeutic treatments. Rescue of consistent levels of dystrophin will also have addi-tional benefits such as the completion of the muscle fiber maturation process.
miRNAs AS SERUM BIOMARKERS FOR DUCHENNE MUSCULAR DYSTROPHY
One of the major problems in Duchenne Muscular Dys-trophy is the lack of reliable biomarkers able to evaluate the progression of the disease and, in the future, to compare the benefit of different therapeutic treatments. The measurement of serum Creatine Kinase (CK) levels, even if it is the first evaluation made when DMD is suspected, is not a reliable assessment to estimate the progression of the disease and the outcomes of a therapeutic approach. Several independent factors can influence the serum CK levels such as the age of the patients [67], pharmacological treatments and physical activity [68]. Moreover, clinical evaluation of the treatment outcomes by Magnetic resonance imaging was shown not to correlate with the levels of the CK in the serum [69].
It has been recently reported that miRNAs expressed in specific body compartments can be released in the blood as a consequence of different types of injuries and that they can represent sensitive biomarkers for several diseases, including cancer [70]. The surprising high stability of miRNAs in the serum was justified by the observation that miRNAs could be part of exosomal particles [71].
In line with this, we demonstrated that, as a consequence of intense muscle degeneration occurring in DMD patients, specific miRNAs expressed in muscle cells (miR-1, miR-206 and miR-133) are released into the blood [72].
Analyses performed on young ambulant patients (<6 years), for which the quantification of disease severity was assessed by North Star Ambulatory Assessment (NSAA) [73], have demonstrated that miR-1, miR-206 and miR-133 are specifically enriched in the serum of these patients in comparison to healthy controls. Moreover, independently from age, a very clear inverse correlation between the level of these miRNAs and the NSAA scores was observed: high levels of miRNAs corresponded to low ambulant activity, correlating with the severity of muscle damage, while low levels were found in less severe DMD forms [72]. Therefore, miRNA quantification may be considered a diagnostic marker more reliable than CK for the study of DMD pro-gression.
Notably, 20/40-folds enrichment of the serum levels of miR-1 and miR-206 were detected also in mdx mice with re-spect to WT. However, when exon skipping was applied to mdx animals the circulating muscle miRNAs were strongly reduced to almost wild type levels. These results indicated that the recovery of muscle integrity, as a consequence of exon skipping, is accompanied by a drastic decrease in miRNA release in the bloodstream. The conspicuous release of miR-1 and miR-206, observed in mdx conditions, was confirmed also in WT animals treated with cardiotoxin, a drug normally used to induce an acute muscle damage [72].
These results suggest that this method can be applied as an objective parameter for measuring the outcomes and effec-tiveness of different therapeutic interventions. This seems particularly useful since, even though several therapeutic ap-proaches are now entering clinical trial, a unifying method for assessing the benefit of the different treatments is still lacking.
lncRNAs IN MUSCLE DIFFERENTIATION AND DIS-EASE
High throughput transcriptome analysis over the last few years has disclosed that the mammalian genome is perva-sively transcribed into many different complex families of RNAs; even among the polyadenylated species, half resulted to be non-protein-coding RNAs. So far, a large range of functions has been attributed to these long non coding RNAs (lncRNAs) [1, 74-76]. It is clear that several lncRNA mole-cules associate with chromatin structures to regulate tran-scription [77-82], others exert their control at the post-transcriptional level [83-86]. In the cytoplasm, lncRNAs were described to transactivate STAU1-mediated mRNA de-cay by duplexing with 3' UTRs via Alu elements [87] or, in the case of pseudogenes, to compete for miRNA binding, thereby modulating the derepression of miRNA targets [88, 89]. Moreover, lncRNAs have also been shown to play a role in muscle disease: DBE-T non-coding RNA was found to de-repress a class of genes involved in Facioscapulohumeral muscular dystrophy (FSHD) by recruiting Trithorax group proteins to their genomic locus [29].
We have recently contributed to this field by identifying a lncRNA, linc-MD1, playing a relevant role in controlling the correct timing of in vitro muscle differentiation [19]. Linc-MD1 is a conserved (between mouse and human) mus-cle-specific long non-coding RNA localised in the cytoplasm of differentiating myoblasts. It derives from the genomic lo-cus containing the miR-206 and miR-133b coding regions, which spans 13kb in chromosome 1 of the mouse genome. 5’ RACE experiments showed the existence in this genomic locus of two promoters with different activation timing: in proliferating myoblasts only the PROX promoter is active that drives the expression of miR-206. No miR-133b is pro-duced under these conditions, likely due to efficient miR-206 Drosha processing and subsequent downstream transcrip-tional termination. In fact, several reports have shown that Drosha cleavage occurs co-transcriptionally [90] and that, as a consequence of processing of an upstream miRNA, Xrn2 exonuclease activity induces transcriptional termination thus reducing transcription on downstream sequences [91]. In line with this, Chromatin IP experiments indeed indicated that in proliferation conditions no RNA Polymerase II is detected downstream of miR-206, on the miR-133b coding region. Upon differentiation, a drastic reorganization in the architec-ture of mir-206/mir-133b locus occurs: 3-C analysis indi-cated that the DIST promoter interacts with the PROX one and that upon interaction it becomes activated [19]. Gene loops have been already described as transcription-dependent DNA structures representing specific domains of active chromatin [92, 93]. In this case, looping correlated with an enhancement of miR-206 expression and transcription from the DIST promoter of a long non coding transcript (linc-MD1) spanning the entire region. From this RNA both miR-
MEF2C�
MAML1�
pA�miR-133�
miR-133�linc-MD1�
muscle genes�muscle genes�
MAML1�
MEF2C�
98 MicroRNA, 2013, Vol. 2, No. 2 Morlando et al.
NAs can be produced, miR-206 from the second intron and miR-133b from the third exon.
Since linc-MD1 contains the miR-133b stem loop struc-ture potentially cleaved by Drosha it has to be assumed that the polyadenylated species accumulating in the cytoplasm represents a proportion of the transcripts escaping nuclear Drosha processing. So far nothing is known about the mechanism underlying the regulation of the Drosha activity on linc-MD1 RNA able to generate the proper ratio between miR-133b processing and the export of the unprocessed pre-cursor to the cytoplasm. This is a relevant question to be ad-dressed in the future in order to clarify whether it represents a more general phenomenon occurring for many of the lncRNAs previously described as pri-miRNA species.
In the cytoplasm, the unprocessed linc-MD1 was shown to act as a “sponge” molecule for two specific miRNAs, miR-135 and miR-133 [19]. By this mode of action linc-MD1 can impact the distribution of these two miRNAs on their natural mRNAs targets imposing an additional level to the post-transcriptional gene regulation occurring during muscle differentiation. A similar mechanism has been al-ready described for the so called “competing endogenous RNAs” (ceRNAs, see [88]), which cross-regulate specific mRNAs by competing for miRNA binding.
The Mastermind-like 1 gene, MAML1 and Myocyte-specific enhancer factor 2C, MEF2C, two relevant factors activating the expression of specific genes required for mus-cle development and function [94-97], have been identified as natural targets of the "sponged" miR-133 and miR-135, respectively. According with linc-MD1 being a sponge for these two miRNAs, modulation of linc-MD1 expression, by either RNAi mediated down-regulation or by ectopic over-expression, affected MAML1 and MEF2C levels thus im-pinging on muscle differentiation [19] (Fig. 5). Notably down-regulation or overexpression of linc-MD1 was shown
to delay or facilitate the differentiation programme of mouse myoblasts, respectively.
When human primary myoblasts from Duchenne patients (DMD) were analyzed, a strikingly down-regulation of linc-MD1 was found [19]. In comparison to control cells, DMD myoblasts showed a reduced ability to undergo terminal dif-ferentiation, accompanied by a reduced and retarded accu-mulation of muscle-specific markers such as myogenin (MyoG) and myosin heavy chain (MHC). The low levels of linc-MD1 explained the absence of sponge activity; in fact, the unaffected accumulation of miR-135, determined low levels of MEF2C; viceversa, the strong down-regulation of miR-133, occurring in Duchenne condition, correlated with the up-regulation of MAML1. In Duchenne cells the rescue of linc-MD1 through lentiviral-mediated expression pro-duced the expected sponge activity with the recovery of both MAML1 and MEF2C expression and the partial rescue of the correct timing of the differentiation programme. A simi-lar behaviour of these markers was also identified during dif-ferentiation of satellite cells derived from WT and mdx ani-mals [19]. In overall, these data reinforced the hypothesis of a consistent contribution of linc-MD1 to the muscle differen-tiation program and pointed to the relevance of its down-regulation in the pathogenesis of Duchenne Muscular Dys-trophy.
From the above data it can be concluded that a specific type of communication exists between coding and non cod-ing RNAs for their ability to bind miRNAs; lncRNAs, by competing for specific miRNA binding, can protect from re-pression mRNA targets of the same miRNAs. This cross-talk, recently described for mRNA (ceRNA hypothesis) [88], strongly depends on the relative concentration and affinity of ceRNAs and the miRNAs that are sequestered. Moreover, the sponge activity of lncRNAs would also be affected by the number and affinity of miRNA binding sites they contain, as
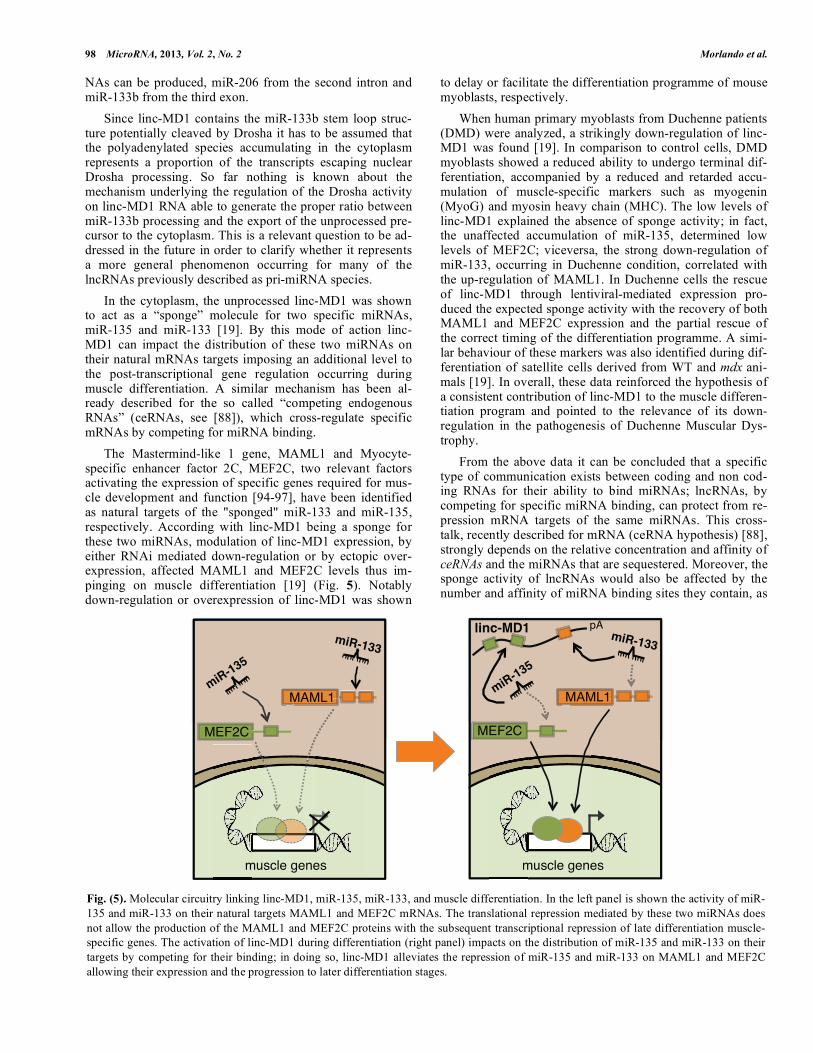
Fig. (5). Molecular circuitry linking linc-MD1, miR-135, miR-133, and muscle differentiation. In the left panel is shown the activity of miR-
135 and miR-133 on their natural targets MAML1 and MEF2C mRNAs. The translational repression mediated by these two miRNAs does
not allow the production of the MAML1 and MEF2C proteins with the subsequent transcriptional repression of late differentiation muscle-
specific genes. The activation of linc-MD1 during differentiation (right panel) impacts on the distribution of miR-135 and miR-133 on their
targets by competing for their binding; in doing so, linc-MD1 alleviates the repression of miR-135 and miR-133 on MAML1 and MEF2C
allowing their expression and the progression to later differentiation stages.

Non Coding RNA in Muscle Differentiation and Disease MicroRNA, 2013, Vol. 2, No. 2 99
well as by their accessibility to miRNA molecules, which may be also controlled by specific RNA binding proteins. In the case studied, binding site affinity prediction and copy number quantification (unpublished data) revealed that the balance of this circuitry is in favour of linc-MD1 since it is more abundant and has better miRNA binding sites than MAML1 and MEF2C mRNAs. Moreover, the cellular levels of linc-MD1 are similar to those of miR-135 indicating that this species is not in excess over its targets.
Based on these findings, it is possible to conclude that also long non-coding RNAs play a relevant role in the com-plex network of regulatory interactions governing muscle terminal differentiation and disease. Moreover, the discovery of linc-MD1 as a sponge for miRNAs introduces a further level of complexity to the network of interactions that con-trol gene expression at the post-transcriptional level and it might facilitate the prediction and identification of new regu-latory networks acting through miRNA competition.
CONFLICT OF INTEREST
The authors confirm that this article content has no con-
flicts of interest.
ACKNOWLEDGEMENTS
This work was partially supported by grants from: Tele-thon (GGP11149), Parent Project Italia, EU project SI-ROCCO (LSHG-CT-2006-037900), AIRC, IIT “SEED”, FIRB and PRIN.
REFERENCES
[1] Mattick JS. The central role of RNA in human development and cognition. FEBS Lett 2011; 585(11): 1600-16.
[2] Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to
lin-14. Cell 1993; 75(5): 843-54. [3] Bartel DP. MicroRNAs: target recognition and regulatory func-
tions. Cell 2009; 136: 215-33. [4] Bushati N, Cohen SM. MicroRNA Functions. Annu Rev Cell Dev
Biol 2007; 23(1): 175-205. [5] Rosa A, Brivanlou AH. MicroRNAs in early vertebrate develop-
ment. Cell Cycle 2009; 8(21). [Epub Ahead of Print]. [6] Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic
disorders. Nat Rev Mol Cell Biol 2012; 13(4): 239-50. [7] Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnos-
tics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med 2012; 4(3): 143-59.
[8] Buckingham M, Montarras D. Skeletal muscle stem cells. Curr Opin Genet Dev 2008; 18(4): 330-36.
[9] Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 2005; 16(4-5): 585-95.
[10] Molkentin JD, Olson EN. Defining the regulatory networks for muscle development. Curr Opin Genet Dev 1996; 6(4): 445-53.
[11] Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogene-
sis. Nature 2005; 436(7048): 214-20. [12] Chen J-F, Mandel EM, Thomson JM, et al. The role of microRNA-
1 and microRNA-133 in skeletal muscle proliferation and differen-tiation. Nat Genet 2006; 38(2): 228-33.
[13] Rao PK, Kumar RM, Farkhondeh M, Myogenic factors that regu-late expression of muscle-specific microRNAs. Proc Natl Acad Sci
USA 2006; 103(23): 8721-6. [14] Zhao Y, Ransom JF, Li A, et al. Dysregulation of cardiogenesis,
cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007; 129(2): 303-17.
[15] Ivey KN, Muth A, Arnold J, et al. MicroRNA regulation of cell
lineages in mouse and human embryonic stem cells. Cell Stem Cell 2008; 2(3): 219-29.
[16] van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles. Trends Genet 2008; 24(4): 159-66.
[17] Williams AH, Liu N, van Rooij E, Olson EH. MicroRNA control of muscle development and disease. Curr Opin Cell Biol 2009; 21:
461-69. [18] Ge Y, Chen J. MicroRNAs in skeletal myogenesis. Cell Cycle
2011; 10(3): 441-8. [19] Cesana M, Cacchiarelli D, Legnini I, et al. A long noncoding RNA
controls muscle differentiation by functioning as a competing en-dogenous RNA. Cell 2011; 147(2): 358-69.
[20] Liu S, Liu S, Zhu X, et al. Nonnatural protein-protein interaction-pair design by key residues grafting. Proc Nat Acad Sci 2007; 104:
5330-5. [21] Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-
specific microRNA miR-206 promotes muscle differentiation. J Cell Biol 2006; 174(5): 677-87.
[22] Anderson C, Catoe H, Werner R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res
2006; 34(20): 5863-71. [23] Rosenberg MI, Georges SA, Asawachaicharn A, Analau E, Tap-
scott SJ. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J Cell Biol 2006; 175(1): 77-85.
[24] Gagan J, Dey BK, Layer R, Yan Z, Dutta A. Notch3 and Mef2c proteins are mutually antagonistic via Mkp1 protein and miR-1/206
microRNAs in differentiating myoblasts. J Biol Chem 2012; 287(48): 40360-70.
[25] Cacchiarelli D, Martone J, Girardi E. MicroRNAs Involved in Mo-lecular Circuitries Relevant for the Duchenne Muscular Dystrophy
Pathogenesis Are Controlled by the Dystrophin/nNOS Pathway. Cell Metab 2010; 12(4): 341-51.
[26] Van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expres-
sion by a microRNA. Science 2007; 316(5824): 575-9. [27] Buckingham M, Vincent SD. Distinct and dynamic myogenic
populations in the vertebrate embryo. Curr Opin Genet Dev 2009; 19(5): 444-53.
[28] Eisenberg I, Eran A, Nishino I, et al. Distinctive patterns of mi-croRNA expression in primary muscular disorders. Proc Natl Acad
Sci USA 2007; 104(43): 17016-21. [29] Cabianca DS, Casa V, Bodega B, et al. A long ncRNA links copy
number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012; 149(4): 819-31.
[30] Emery AE. Population frequencies of inherited neuromuscular dis-eases--a world survey. Neuromuscul Disord 1991; 1(1): 19-29.
[31] Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988;
53(2): 219-28. [32] Zubrzycka-Gaarn EE, Bulman DE, Karpati G, et al. The Duchenne
muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 1988; 333(6172):466-9.
[33] Ervasti JM, Campbell KP. Membrane organization of the dystro-phin-glycoprotein complex. Cell 1991; 66(6): 1121-31.
[34] Matsumura K, Campbell KP. Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle
Nerve 1994; 17(1): 2-15. [35] Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dys-
trophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 1993; 90(8): 3710-4.
[36] Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients
bearing partial deletions of the DMD locus. Genomics 1988; 2(1): 90-5.
[37] Cossu G, Sampaolesi M. New therapies for Duchenne muscular dystrophy: challenges, prospects and clinical trials. Trends Mol
Med 2007; 13(12): 520-6. [38] Aartsma-Rus A. Antisense-mediated modulation of splicing: thera-
peutic implications for Duchenne muscular dystrophy. RNA Biol 2010; 7(4): 453-61.
[39] Hilton T, Orr RD, Perkin RM, Ashwal S. End of life care in Duchenne muscular dystrophy. Pediatr Neurol 1993; 9(3): 165-77.
[40] van Deutekom JC, Bremmer-Bout M, Janson AA, et al. Antisense-induced exon skipping restores dystrophin expression in DMD pa-
tient derived muscle cells. Hum Mol Genet 2001; 10(15): 1547-54.

100 MicroRNA, 2013, Vol. 2, No. 2 Morlando et al.
[41] van Deutekom JC, Janson AA, Ginjaar IB. Local dystrophin resto-
ration with antisense oligonucleotide PRO051. N Engl J Med 2007; 357(26): 2677-86.
[42] Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658
in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol
2009; 8(10): 918-28. [43] De Angelis FG, Sthandier O, Berarducci B, et al. Chimeric snRNA
molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and
restoration of a dystrophin synthesis in Delta 48-50 DMD cells. Proc Natl Acad Sci USA 2002; 99(14): 9456-61.
[44] Brun C, Suter D, Pauli C, et al. U7 snRNAs induce correction of mutated dystrophin pre-mRNA by exon skipping. Cell Mol Life Sci
2003; 60(3): 557-66. [45] Incitti T, De Angelis FG, Cazzella V, et al. Exon skipping and
duchenne muscular dystrophy therapy: selection of the most active U1 snRNA antisense able to induce dystrophin Exon 51 Skipping.
Mol Ther 2010; 18(9): 1675-82. [46] Goyenvalle A, Vulin A, Fougerousse F, et al. Rescue of dystrophic
muscle through U7 snRNA-mediated exon skipping. Science 2004; 306(5702): 1796-9.
[47] Denti MA, Rosa A, D'Antona G, et al. Chimeric adeno-associated virus/antisense U1 small nuclear RNA effectively rescues dystro-
phin synthesis and muscle function by local treatment of mdx mice. Hum Gene Ther 2006; 17(5): 565-74.
[48] Denti MA, Rosa A, D'Antona G, et al. Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc Natl
Acad Sci USA 2006; 103(10): 3758-63. [49] Denti MA, Incitti T, Sthandier O, et al. Long-Term Benefit of
Adeno-Associated Virus/Antisense-Mediated Exon Skipping in Dystrophic Mice. Hum Gene Ther 2008; 19(6): 601-8.
[50] Marrone AK, Shcherbata HR. Dystrophin orchestrates the epigenetic profile of muscle cells Via miRNAs. Front Genet 2011; 2: 64.
[51] Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal mus-
cle sarcolemma in Duchenne muscular dystrophy. Cell 1995; 82(5): 743-52.
[52] Yang B, Jung D, Rafael JA, Chamberlain JS, Campbell KP. Identi-fication of alpha-syntrophin binding to syntrophin triplet, dystro-
phin, and utrophin. J Biol Chem 1995; 270(10): 4975-8. [53] Abramovici H, Hogan AB, Obagi C, Topham MK, Gee SH. Dia-
cylglycerol kinase-zeta localization in skeletal muscle is regulated by phosphorylation and interaction with syntrophins. Mol Biol Cell
2003; 14(11): 4499-511. [54] Ghahramani SMM, Graham IR, Athanasopoulos T, et al. RNAi-
mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum Mol Genet
2008; 17(17): 2622-32. [55] Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-
Nitrosylation of histone deacetylase 2 induces chromatin remodel-ling in neurons. Nature 2008; 455(7211): 411-5.
[56] Colussi C, Mozzetta C, Gurtner A, et al. HDAC2 blockade by ni-tric oxide and histone deacetylase inhibitors reveals a common tar-
get in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci USA 2008; 105(49): 19183-7.
[57] Brunelli S, Sciorati C, D'Antona G, et al. Nitric oxide release com-bined with nonsteroidal antiinflammatory activity prevents muscu-
lar dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci USA 2007; 104(1): 264-9.
[58] Minetti GC, Colussi C, Adami R, et al. Functional and morpho-logical recovery of dystrophic muscles in mice treated with deace-
tylase inhibitors. Nat Med 2006; 12(10): 1147-50. [59] Wells KE, Torelli S, Lu Q, et al. Relocalization of neuronal nitric
oxide synthase (nNOS) as a marker for complete restoration of the dystrophin associated protein complex in skeletal muscle. Neuro-
muscul Disord 2003; 13(1): 21-31. [60] Lai Y, Thomas GD, Yue Y, et al. Dystrophins carrying spectrin-
like repeats 16 and 17 anchor nNOS to the sarcolemma and en-hance exercise performance in a mouse model of muscular dystro-
phy. J Clin Invest 2009; 119(3): 624-35. [61] Cazzella V, Martone J, Pinnarò C, et al. Exon 45 skipping through
U1-snRNA antisense molecules recovers the Dys-nNOS pathway and muscle differentiation in human DMD myoblasts. Mol Ther
2012; 20(11): 2134-42.
[62] Dey BK, Gagan J, Dutta A. MiR-206 and -486 induce myoblast
differentiation by downregulating Pax7. Mol Cell Biol 2011; 31(1): 203-14.
[63] Chen J-F, Tao Y, Li J, et al. MicroRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentia-
tion by repressing Pax7. J Cell Biol 2010; 190(5): 867-79. [64] Liu N, Williams AH, Maxeiner JM, et al. MicroRNA-206 pro-
motes skeletal muscle regeneration anddelays progression of Duchenne muscular dystrophy in mice. J Clin Invest 2012; 122(6):
2054-65. [65] Crist CG, Montarras D, Buckingham M. Muscle satellite cells are
primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell
Stem Cell 2012; 11(1): 118-26. [66] Cacchiarelli D, Incitti T, Martone J, et al. MiR-31 modulates dys-
trophin expression: new implications for Duchenne muscular dys-trophy therapy. EMBO Rep 2011; 12(2): 136-41.
[67] Zatz M, Rapaport D, Vainzof M, et al. Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as com-
pared with Becker (BMD) muscular dystrophy. J Neurol Sci 1991; 102(2):190-6.
[68] Malm C, Nyberg P, Engstrom M, et al. Immunological changes in human skeletal muscle and blood after eccentric exercise and mul-
tiple biopsies. J Physiol (Lond) 2000; 529: 243-562. [69] Kim HK, Laor T, Horn PS. T2 mapping in Duchenne muscular
dystrophy: distribution of disease activity and correlation with clinical assessments. Radiology 2010; 255(3): 899-908.
[70] Schöler N, Langer C, Döhner H, Buske C, Kuchenbauer F. Serum microRNAs as a novel class of biomarkers: a comprehensive re-
view of the literature. Exp Hematol 2010; 38(12): 1126-30. [71] Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs
as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 2008; 105(30): 10513-8.
[72] Cacchiarelli D, Legnini I, Martone J, et al. MiRNAs as serum bio-markers for Duchenne muscular dystrophy. EMBO Mol Med 2011;
3(5): 258-65. [73] Mazzone E, Martinelli D, Berardinelli A, et al. North Star Ambula-
tory Assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord
2010; 20(11): 712-6. [74] Nagano T, Fraser P. No-nonsense functions for long noncoding
RNAs. Cell 2011; 145(2): 178-81. [75] Wang KC, Chang HY. Molecular mechanisms of long noncoding
RNAs. Mol Cell 2011; 43(6): 904-14. [76] Rinn JL, Chang HY. Genome Regulation by Long Noncoding
RNAs. Annu Rev Biochem 2012; 81: 145-66. [77] Chaumeil J, Le Baccon P, Wutz A, Heard E. A novel role for Xist
RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 2006; 20(16):
2223-37. [78] Rinn JL, Kertesz M, Wang JK, et al. Functional demarcation of
active and silent chromatin domains in human HOX loci by non-coding RNAs. Cell 2007; 129(7): 1311-23.
[79] Nagano T, Mitchell JA, Sanz LA, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin.
Science 2008; 322(5908): 1717-20. [80] Mohammad F, Mondal T, Guseva N, Pandey GK, Kanduri C.
Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 2010; 137(15): 2493-9.
[81] Bertani S, Sauer S, Bolotin E, Sauer F. The noncoding RNA Mis-tral activates Hoxa6 and Hoxa7 expression and stem cell differen-
tiation by recruiting MLL1 to chromatin. Mol Cell 2011; 43(6): 1040-6.
[82] Guil S, Soler M, Portela A, et al. Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat Struct Mol Biol 2012; 19(7):
664-70. [83] Beltran M, Puig I, Peña C, et al. A natural antisense transcript regu-
lates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev 2008; 22(6): 756-69.
[84] Tripathi V, Ellis JD, Shen Z, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR
splicing factor phosphorylation. Mol Cell 2010; 39(6): 925-38. [85] Yoon J-H, Abdelmohsen K, Gorospe M. Post-transcriptional gene
regulation by long noncoding RNA. J Mol Biol 2012. [Epub Ahead of Print].

Non Coding RNA in Muscle Differentiation and Disease MicroRNA, 2013, Vol. 2, No. 2 101
[86] Carrieri C, Cimatti L, Biagioli M, et al. Long non-coding antisense
RNA controls Uchl1 translation through an embedded SINEB2 re-peat. Nature 2012; 491(7424): 454-7.
[87] Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature
2011; 470(7333): 284-8. [88] Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA Hy-
pothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011; 146(3): 353-8.
[89] Poliseno L, Salmena L, Zhang J. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature
2010; 465(7301): 1033-8. [90] Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proud-
foot NJ. Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 2008; 15(9): 902-9.
[91] Ballarino M, Pagano F, Girardi E, et al. Coupled RNA processing and transcription of intergenic primary microRNAs. Mol Cell Biol
2009; 29(20): 5632-8. [92] Tan-Wong SM, French JD, Proudfoot NJ, Brown MA. Dynamic
interactions between the promoter and terminator regions of the
mammalian BRCA1 gene. Proc Natl Acad Sci USA 2008; 105(13):
5160-5. [93] West AG, Fraser P. Remote control of gene transcription. Hum
Mol Genet 2005; 14: R101-11. [94] Shen H, McElhinny AS, Cao Y, et al. The Notch coactivator,
MAML1, functions as a novel coac- tivator for MEF2C-mediated transcription and is required for normal myogen- esis. Genes Dev
2006; 20(6): 675-88. [95] Wilson-Rawls J, Molkentin JD, Black BL, Olson EN. Activated
notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol Cell Biol 1999; 19(4):
2853-62. [96] Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac
morphogenesis and myogenesis by transcription factor MEF2C. Science 1997; 276(5317): 1404-7.
[97] Potthoff MJ, Arnold MA, McAnally J, Richardson JA, Bassel-Duby R, Olson EN. Regulation of skeletal muscle sarcomere integ-
rity and postnatal muscle function by Mef2C. Mol Cell Biol 2007; 27(23): 8143-51.
Received: December 19, 2012 Revised: May 13, 2013 Accepted: May 31, 2013
Related Documents











