Journal of Genetics, Vol. 97, No. 3, July 2018, pp. 679–701 © Indian Academy of Sciences https://doi.org/10.1007/s12041-018-0955-3 REVIEW ARTICLE Neurodegenerative diseases: model organisms, pathology and autophagy S. N. SURESH 1 , VIJAYA VERMA 2 , SHRUTHI SATEESH 2 , JAMES P. CLEMENT 2 and RAVI MANJITHAYA 1,2 ∗ 1 Molecular Biology and Genetics Unit, and 2 Neuroscience Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bengaluru 560 064, India *For correspondence. E-mail: [email protected]. Received 21 December 2017; revised 9 March 2018; accepted 20 March 2018 Abstract. A proteostasis view of neurodegeneration (ND) identifies protein aggregation as a leading causative reason for damage seen at the cellular and organ levels. While investigative therapies that aim at dissolving aggregates have failed, and the promises of silencing expression of ND associated pathogenic proteins or the deployment of engineered induced pluripotent stem cells (iPSCs) are still in the horizon, emerging literature suggests degrading aggregates through autophagy-related mechanisms hold the current potential for a possible cure. Macroautophagy (hereafter autophagy) is an intracellular degradative pathway where superfluous or unwanted cellular cargoes (such as peroxisomes, mitochondria, ribosomes, intracellular bacteria and misfolded protein aggregates) are wrapped in double membrane vesicles called autophagosomes that eventually fuses with lysosomes for their degradation. The selective branch of autophagy that deals with identification, capture and degradation of protein aggregates is called aggrephagy. Here, we cover the workings of aggrephagy detailing its selectivity towards aggregates. The diverse cellular adaptors that bridge the aggregates with the core autophagy machinery in terms of autophagosome formation are discussed. In ND, essential protein quality control mechanisms fail as the constituent components also find themselves trapped in the aggregates. Thus, although cellular aggrephagy has the potential to be upregulated, its dysfunction further aggravates the pathogenesis. This phenomenon when combined with the fact that neurons can neither dilute out the aggregates by cell division nor the dead neurons can be replaced due to low neurogenesis, makes a compelling case for aggrephagy pathway as a potential therapeutic option. Keywords. neurodegeneration; aggrephagy; small molecule therapeutics. Introduction Cellular homeostasis is achieved through a balance of anabolic and catabolic states. Cells possess several quality control mechanisms to identify, correct or remove dysfunctional or potential toxic cellular compo- nents such as proteins and organelles. For example, inside the cell, at steady state, misfolded proteins are formed con- tinuously and are fixed by chaperones or cleared through the ubiquitin proteasome system, and autophagy related pathways. This maintains the proteostatic equilibrium in cells. Altered cellular states resulting from expression of pathogenic levels or mutant forms of aggregate prone pro- teins overwhelm these quality control systems and their build-up can eventually result in cell death. This is the fate of brain cells in neurodegenerative diseases (NDD). In this review, we unravel this proteostatic central view as a cause for ND, discuss the potential reasons behind the failure of quality control systems with an emphasis on an aggrephagy, autophagy related pathway. Proteostasis Protein quality control machineries ensure the proper folding of newly synthesized proteins for their distinct function. This process is critical as 30% of these new proteins are prone to misfolding (Mymrikov et al. 2017). The cellular quality control measures include unfolding, refolding and/or degradation of the misfolded proteins (Sontag et al. 2017). Not surprisingly, failure of pro- tein quality control poses a threat to the cellular vitality (Balch et al. 2008). This qualitative process of maintain- ing the homeostasis of intracellular pool of functional and ‘healthy’ proteins is called proteostasis. Proteosta- sis as a function for cell survival becomes even more 679

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Journal of Genetics, Vol. 97, No. 3, July 2018, pp. 679–701 © Indian Academy of Scienceshttps://doi.org/10.1007/s12041-018-0955-3
REVIEW ARTICLE
Neurodegenerative diseases: model organisms, pathology and autophagy
S. N. SURESH1, VIJAYA VERMA2, SHRUTHI SATEESH2, JAMES P. CLEMENT2 andRAVI MANJITHAYA1,2∗
1Molecular Biology andGenetics Unit, and 2NeuroscienceUnit, Jawaharlal Nehru Centre for Advanced Scientific Research(JNCASR), Jakkur, Bengaluru 560 064, India*For correspondence. E-mail: [email protected].
Received 21 December 2017; revised 9 March 2018; accepted 20 March 2018
Abstract. A proteostasis view of neurodegeneration (ND) identifies protein aggregation as a leading causative reason for damageseen at the cellular and organ levels. While investigative therapies that aim at dissolving aggregates have failed, and the promises ofsilencing expression of ND associated pathogenic proteins or the deployment of engineered induced pluripotent stem cells (iPSCs)are still in the horizon, emerging literature suggests degrading aggregates through autophagy-related mechanisms hold the currentpotential for a possible cure. Macroautophagy (hereafter autophagy) is an intracellular degradative pathway where superfluous orunwanted cellular cargoes (such as peroxisomes, mitochondria, ribosomes, intracellular bacteria and misfolded protein aggregates)are wrapped in double membrane vesicles called autophagosomes that eventually fuses with lysosomes for their degradation. Theselective branch of autophagy that deals with identification, capture and degradation of protein aggregates is called aggrephagy.Here, we cover the workings of aggrephagy detailing its selectivity towards aggregates. The diverse cellular adaptors that bridgethe aggregates with the core autophagy machinery in terms of autophagosome formation are discussed. In ND, essential proteinquality control mechanisms fail as the constituent components also find themselves trapped in the aggregates. Thus, although cellularaggrephagy has the potential to be upregulated, its dysfunction further aggravates the pathogenesis. This phenomenonwhen combinedwith the fact that neurons can neither dilute out the aggregates by cell division nor the dead neurons can be replaced due to lowneurogenesis, makes a compelling case for aggrephagy pathway as a potential therapeutic option.
Keywords. neurodegeneration; aggrephagy; small molecule therapeutics.
Introduction
Cellular homeostasis is achieved through a balance ofanabolic and catabolic states. Cells possess severalquality control mechanisms to identify, correct orremove dysfunctional or potential toxic cellular compo-nents such as proteins and organelles. For example, insidethe cell, at steady state, misfolded proteins are formed con-tinuously and are fixed by chaperones or cleared throughthe ubiquitin proteasome system, and autophagy relatedpathways. This maintains the proteostatic equilibrium incells. Altered cellular states resulting from expression ofpathogenic levels or mutant forms of aggregate prone pro-teins overwhelm these quality control systems and theirbuild-up can eventually result in cell death. This is thefate of brain cells in neurodegenerative diseases (NDD).In this review, we unravel this proteostatic central view asa cause for ND, discuss the potential reasons behind the
failure of quality control systems with an emphasis on anaggrephagy, autophagy related pathway.
Proteostasis
Protein quality control machineries ensure the properfolding of newly synthesized proteins for their distinctfunction. This process is critical as 30% of these newproteins are prone to misfolding (Mymrikov et al. 2017).The cellular quality control measures include unfolding,refolding and/or degradation of the misfolded proteins(Sontag et al. 2017). Not surprisingly, failure of pro-tein quality control poses a threat to the cellular vitality(Balch et al. 2008). This qualitative process of maintain-ing the homeostasis of intracellular pool of functionaland ‘healthy’ proteins is called proteostasis. Proteosta-sis as a function for cell survival becomes even more
679

680 S. N. Suresh et al.
critical for those cells such as neurons that cannot divideand thus dilute out the toxic protein aggregates (Balchet al. 2008). In addition, with age, the neuronal pro-teostatic machineries become incompetent and pronefor accumulation of damaged organelles and misfoldedproteins (Labbadia and Morimoto 2015; Walther et al.2017).
Protein misfolding is not an uncommon phenomenoninside cells. The presence of misfolded protein activateschaperones to unfold and refold them in an ATP-dependent manner. Misfolded proteins induce heat shockresponse (HSR) and heat shock (HS) transcriptionfactor HSF-1 that include the upregulation of heatshock cognate protein (Hsc70) (Kampinga and Craig2010). The aim of enhancing chaperone expressionand its activity is to prevent protein aggregation.Once the proteins remain misfolded despite theefforts by chaperones, they will form the higher orderstructures such as oligomers and aggregates that willeventually accumulate inside cells. In certain scenarios,when protein aggregates overwhelm the chaperone capac-ity, the available chaperones themselves get trapped inprotein aggregates (Anckar and Sistonen 2011; Ray-chaudhuri et al. 2014). Subsequently, these events alsoaggravate disease pathogenesis. An additional mecha-nism that has been recently described identifies misfoldedproteins as early as they are translating and subse-quently marks them for degradation. Such nascent mis-folded polypeptides are ubiquitinated at K-48 residueas a degradation mark in a process termed as cotrans-lational ubiquitination (CTU) (Wang et al. 2013). Thepolypeptide with this mark will be acted upon by pro-teasome for their effective degradation. CTU happenswithin the active translation complexes (Wang et al.2013).
At the organellar level, accumulation of misfolded pro-teins inside endoplasmic reticulum (ER) lumen inducesER stress, which triggers one of the vital cellular adaptivemechanisms known as unfolded protein response (UPR)(Powers et al. 2009). UPR leads to suppression of activeprotein translation, increase ER chaperone accumulationto unfold and/or degrade these proteins to amelioratethe proteotoxicity (Hetz 2012). Post-mortem brain anal-yses of Alzheimer’s disease (AD), Parkinson’s disease(PD) and Huntington’s disease (HD) have revealed thecorrelation of UPR markers with protein aggregationand onset of disease pathogenesis (Hetz and Mollereau2014).
The fate of misfolded proteins, not refolded bychaperones, are marked for degradation throughubiquitin-proteasome system (UPS) and/or autophagy.UPS degrades ubiquitinated, soluble and short-lived pro-teins. The target protein is tagged by ubiquitin throughthree enzymatic ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin-ligating (E3) reactions(Goldberg 2003). Ubiquitin is attached through its
carboxyl residue to specific lysine residue throughisopeptide linkage. One ubiquitin molecule is a targetfor another ubiquitin molecule and forms a polyubiqui-tin chain at its lysine residues at 48, 63 or at N-terminalmethionine residue (Kirkin et al. 2009). The polyubiquitinsignal at K-48 serves as a proteasome degradation signalwhereas K-63 and N-term methionine signals serve otherfunctions (Kirkin et al. 2009). It is also proposed thatK-63polyubiquitination may target a protein to autophagy, butthe exact mechanism is unclear.UPS has been shown to degrade several neurodegen-
erative disease related proteins such as tau, SOD1 andα-synuclein (Goldberg 2003). Inhibiting UPS accumulatedisease related proteins to aggregate and form inclusionbodies inside cells. Thus, UPS is essential for cells toprevent toxic protein aggregate formation (Lim and Yue2015). It is believed that larger aggregates that cannotbe resolved by UPS are the substrates of the autophagymachinery.
Aggrephagy: definition and modes
In 1960s, the Nobel laureate Christian de Duve observeddouble membrane vesicles entrapping intracellularorganelles and proteins, and coined the term ‘self-eating’or autophagy (De Duve andWattiaux 1966). Nobel laure-ateYoshinoriOhsumi tapped thepowerof yeast genetics toelucidate the molecular mechanism governing autophagyand contributed to the discovery of its conserved naturefrom yeast to mammals (Harnett et al. 2017).Depending on the distinct molecular mechanisms,
autophagy is broadly classified into three types: macroau-tophagy, microautophagy and chaperone-mediated auto-phagy (CMA). Macroautophagy is the most extensivelystudied process that has an indispensable role in maintain-ing cellular andorganismal homeostasis.Duringmacroau-tophagy, a phagophore or isolation membrane forms andfurther expands to form double membrane autophago-somes that engulfs cellular cargoes. These cargoes are partsof cytosol constituting superfluous organelles, pathogenicorganisms, misfolded protein aggregates and/or dam-aged mitochondria (Zaffagnini and Martens 2016). Theseautophagosomes fuse with lysosomes to form autolyso-somes and eventually degrade its intraluminal cargoes.The degraded entities result in building blocks that arerecycled back to the cytosol for fuelling other cellularpathways. Apart from randomly sequestering portions ofcytosol for degradation (general autophagy), macroau-tophagy can be highly selective in the cargoes it captures.The selective autophagy pathway that is involved in clear-ance of misfolded protein aggregates is called aggrephagy(Hyttinen et al. 2014). Here, misfolded proteins that aretagged by ubiquitin are recognized by adaptor proteinssuch as p62, NBR1 and NDP52 which in turn recruitautophagy proteins such as LC3 to facilitate selective cap-ture (figure 1). Thus, aggrephagy is a prominent defense

Neurodegenerative diseases 681
Figure 1. Formation of aggrephagosome. Aggregate proteins are ubiquitinated (Ub) which are recognized by adaptor proteins. p62,NDP-52, NBR1 and optineurin are some of the examples of adaptor proteins that have a ubiquitin-binding domain and additionallyhave LC3 interacting region (LIR) that can recruit LC3 and thereby facilitate cargo recognition and capture.
mechanism against misfolded protein aggregate-mediatedcellular toxicity. Microautophagy is the direct uptakeof cargo by lysosomes through membrane invagination.Chaperone mediated autophagy (CMA) is the selectivedegradative process of proteins that involves Hsc70 butis a vesicle independent process. During CMA, the targetprotein is recognized by Hsc70 through a specific aminoacid motif, KFERQ and delivered to lysosome by inter-acting with LAMP2A in an ATP-dependent manner.
Synaptic dysfunction in NDD
Synaptic plasticity refers to the ability of synapses tomodify their structure and tonus after persistent elec-trical activity and/or signalling. In fact, the number,morphology, position, molecular phenotype and strengthof synapses continue to modify as a function of neurons’requirements. These events take place in the nervous sys-tem during its development, which represents the basis forlearning and memory (Bliss and Collingridge 1993; Citriand Malenka 2008). In case of neurons, in addition to theautophagy process in soma, autophagy occurring at thesynapse, of late, has received much attention as it involvesimmediate turnover of proteins and, thus, affects synap-tic transmission. Mutations in genes involved in severalneurodegenerative diseases such as AD, HD, ALS and
PD affect synaptic proteins levels and functions (Lepetaet al. 2016). Synaptic dysfunction throughout the centraland peripheral nervous systems has shown to be an earlyhallmark of neurodegenerative diseases preceding neu-ronal death and the subsequent onset of clinical symptoms(figure 2; table 1) (Brose et al. 2010). These events have beenvalidated using transgenic mouse models available for dif-ferent neurodegenerative diseases (Trancikova et al. 2011).
Neurodegeneration model systems as tools to studyaggrephagy
Numerous model systems have been utilized to study theaggrephagy and modulate it to clear the protein aggre-gates. It is important to note that the cellular proteostaticmachineries are conserved from simple yeast to highermodel rodents. We will briefly discuss the different modelsystems used to understand the neurodegeneration patho-genesis with a special emphasis on autophagy.
Animal models to study NDD
Historically, mouse has been used as a model to studygenetic mutation-causing diseases in humans, especially

682 S. N. Suresh et al.
Figure 2. Signalling pathways affected in neurodegenerative disorders. Schematic to show impaired synaptic function due to alteredfunction of different signalling pathways in different neurodegenerative disorders. Presence of aggregates in these diseases leads to anincrease in presynaptic release of glutamate orDAupon stimulation that activates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionicacid receptor (AMPAR), DA receptor1 (D1R) and DA receptor2 (D2R), respectively. (a) Activation of AMPARs leads toN-methyl-D-aspartate receptors (NMDAR) activation which is followed by Ca2+ influx. Additionally, voltage gated Ca2+ channel(VGCC) further facilitates the Ca2+ influx leading to an increased intracellular Ca2+ levels at the postsynapse. This increased levelsof Ca2+ results in: (1) excitotoxicity, (2) mitochondrial dysfunction, (3) reactive oxygen species (ROS) activation, (4) CASPASEactivation, (5) apoptosis, and 6) mTOR activation (autophagy inhibition). These events eventually result in neuronal dysfunction,neurodegeneration, and cognitive impairments. (b) Stimulation of metabotropic glutamate receptor5 (mGluR5) by glutamate leadsto activation of inositol 1,4,5-trisphosphate (IP3). In neurodegeneration, the aggregates strongly bind to the IP3 receptor1 (IP3R1),which results in the release of the Ca2+ through IP3R1, in turn, further contributing to already increased intracellular Ca2+ levels,resulting in synaptic dysfunction. (c) DA acting on DA1 receptor activates adenosine 3′,5′-cyclic monophosphate (cAMP), whichfurther activates protein kinase A (PKA). Upon PKA activation, the intracellular Ca2+ levels increase through IP3R1. Addition-ally, PKA activates DA-regulated and cAMP-regulated neuronal phosphoprotein (DARRP-32) and extracellular signal-regulatedkinases (ERK), which alters cAMP response element-binding protein (CREB) activity resulting in altered transcription and cognitiveimpairment. (d) DA acting on DA2 receptor further contributes to the intracellular Ca2+ levels through the activation of IP3R1.This Ca2+ influx simultaneously activates CALCINEURIN followed by glycogen synthase kinase 3 β (GSK-3 β) activation, leadingto the phosphorylation of TAU. This event results in impaired cognition and cell death.
brain related disorders including neurodegenerationdiseases, due to the similarity of mammalian neuronalphysiology and anatomy to human brain. The main ratio-nale to model human disorders in nonhuman organisms isthe identification of fundamental pathogenic mechanismsthat could lead to potential novel therapeutic targets, andthe elevation of efficacy and safety of potential drugs. Aprerequisite for clinical trials of a compound in humansis the successful alleviation of the disease or symptoms inanimal models.The occurrence of neurodegeneration is majorly a spo-
radic event with poorly understood aetiological basis.However, in familial cases of AD, PD, HD and ALS,
the genetic mutations are the underlying causes of diseasephenotypes and these can be recapitulated in animal mod-els. Due to a rapid increase in the availability of the type ofgenetically modifiedmice (Branchi et al. 2003), it is criticalto meticulously characterize the biochemical, pathologi-cal and behavioural features of these mice and comparethem with human phenotypes (Crawley 2008). Generally,laboratories involved in testing the phenotypes of genet-ically modified mice subject these mice to a battery ofbehavioural features to assess cognitive, motor and sen-sory functions. To consider a genetically modified mouseas a disease model, it must fulfil three levels of validityto judge its psychopharmacology (van der Staay et al.

Neurodegenerative diseases 683
Table
1.Su
mmaryof
syna
pticdy
sfun
ctionob
served
indifferentneurod
egenerativediseases.
Properties
ALS
PD
AD
HD
Basal
syna
ptic
tran
smission
(BST
)
Impa
ired
tran
smission
((↓)
presyn
apticrelease)
(Maselli
etal.1
993)
Una
ltered
inmedium
spiny
neuron
s(M
SN’s)
ofLRRK2
over
expressing
mice
(Beccano
-Kelly
etal.2
015)
Altered
BST
was
observed
inApp
695S
wemutan
t,Apoe4
mice(↑),App
Indmice(↓)
(Hsiaet
al.1
999;
Filimon
enko
etal.2
010;
Puzzo
etal.2
017;
Sunet
al.2
017)
InR6/2,
BST
isaltered(↓)
(Cepedaet
al.2
001;
Klapstein
etal.2
001;
Miln
erwoo
det
al.
2006;K
hedrak
ietal.2
017)
Neurotran
smitter
release
Increase
inglutam
ate-indu
ced
excitotoxicity;p
resyna
ptic
tran
smission
deficit(C
oyle
andSchw
arcz
1976;L
ipton
andRosenberg
1994;D
oble
1999;K
iernan
etal.2
011)
Deficiency
ofdo
paminein
the
nigrostriatalsystem,a
ndin
levelsof
somatostatin
(Rinne
etal.1
984)
SVtrafficking
defects(E
spositoet
al.2
012)
Increasedpresyn
apticfunction
(Sun
etal.2
017)
InR6/2,
alteredpresyn
aptic
release(↓),po
sttetanic
potentiation
(↓),an
din
YAC12
8in
12mon
ths(↓)
(Murph
yet
al.2
000;
Cepeda
etal.2
001;
Klapstein
etal.
2001;M
ilnerwoo
det
al.2
006;
And
reet
al.2
011;
Khedrak
iet
al.2
017)
Increasedactivity
ofGABA
and5-HTT(A
nden
1974;
Grabo
wskaan
dMicha
luk
1974;M
ajet
al.1
974;
Hollisteret
al.1
976)
Syna
pticplasticity
Impa
ired
LTPin
Ubqln2P
497H
andSod
1mice
(Geracitan
oet
al.2
003;
Gorrieet
al.2
014)
LTPob
served
inoriginal
LTD-ind
ucingstim
ulus
(Kreitzeran
dMalenka
2005;
Bagetta
etal.2
010)
LTPin
AAV-A
pp/Ps1
(↓),
Apoe4
(↓)(A
udrain
etal.
2016;P
uzzo
etal.2
017;
Sun
etal.2
017)
Impa
ired
LTPan
dLT
Din
R6/2(M
urph
yet
al.2
000)
Cellintrinsic
prop
erties
Altered
excitabilityan
daltered
fastafterhyp
erpo
larization
(fAHP)an
dslow
afterhyp
erpo
larization
(sAHP)in
Sod
1(↑),Tardbp
(↓),Ubqln2P
497H
(↓),
Sod
1A4V
(↑)mice(F
ischer
etal.2
004;
Murrayet
al.2
010;
Zho
uet
al.2
010;
Waing
eret
al.2
014;
Rad
zickietal.
2016;K
imet
al.2
017)
Increasedpo
stsyna
ptic
potentials,intrinsicmem
bran
eprop
erties,p
ostsyn
aptic
respon
sesto
differentagon
ists
ofexcitatory
aminoacid
receptorsno
talteredin
neuron
srecorded
from
dopa
mine-depleted
slices
(Calab
resiet
al.1
993)
Highlydisorgan
ised
syna
pses,
increasedan
ddispersedtonic
glutam
atergiccurrent
amplitud
ein
AAV-A
pp/Ps1
(Aud
rain
etal.2
016)
Altered
inpu
t,resting
mem
bran
epo
tentials,tim
econstantsan
daction
potentialsin
CAG40
KI(↓),
R6/2(↓)an
dYAC128(↓)
(Levineet
al.1
999;
Klapstein
etal.2
001;
Graha
met
al.
2009;C
ummings
etal.2
010)
IncreasedNMDA/A
MPA
ratio
(Hsiaet
al.1
999)
Decreased
inthefrequencyof
mEPSC
andno
chan
gein
amplitud
eon
applicationof
Amyloid-
βin
culture(Y
uet
al.2
012)
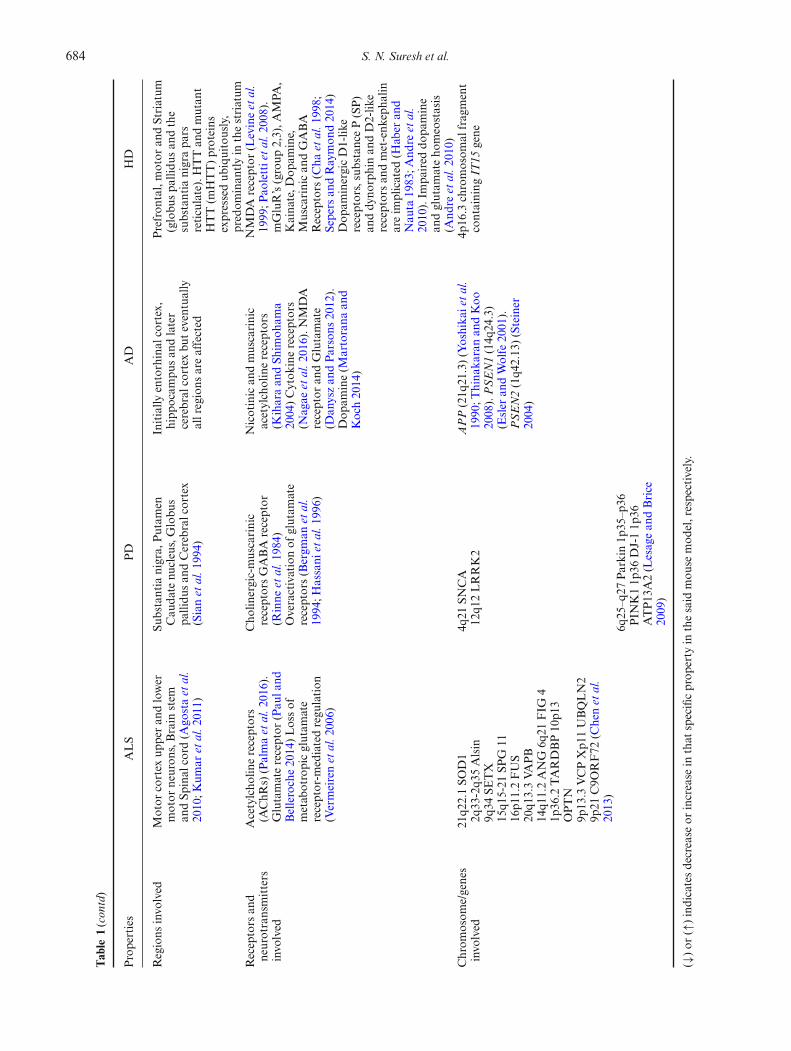
684 S. N. Suresh et al.
Table
1(contd)
Properties
ALS
PD
AD
HD
Regions
invo
lved
Motor
cortex
upperan
dlower
motor
neuron
s,Brain
stem
andSp
inal
cord
(Ago
staet
al.
2010;K
umar
etal.2
011)
Substantia
nigra,
Putam
enCau
date
nucleus,Globu
spa
llidu
san
dCerebralcortex
(Sianet
al.1
994)
Initially
entorhinal
cortex,
hipp
ocam
pusan
dlater
cerebral
cortex
buteventually
allregions
areaffected
Prefron
tal,motor
andStriatum
(globu
spa
llidu
san
dthe
substantia
nigrapa
rsreticulate).HTTan
dmutan
tHTT(m
HTT)proteins
expressedub
iquitously,
predom
inan
tlyin
thestriatum
Receptors
and
neurotransmitters
invo
lved
Acetylcho
linereceptors
(AChR
s)(Palmaet
al.2
016).
Glutamatereceptor
(Pau
land
Belleroche2014)Lossof
metab
otropicglutam
ate
receptor-m
ediatedregu
lation
(Vermeirenet
al.2
006)
Cho
linergic-muscarinic
receptorsGABA
receptor
(Rinne
etal.1
984)
Overactivationof
glutam
ate
receptors(Bergm
anet
al.
1994;H
assani
etal.1
996)
Nicotinican
dmuscarinic
acetylcholinereceptors
(Kiharaan
dSh
imoh
ama
2004)Cytok
inereceptors
(Nagae
etal.2
016).N
MDA
receptor
andGlutamate
(Dan
yszan
dParsons
2012).
Dop
amine(M
artorana
and
Koch2014)
NMDA
receptor
(Levineet
al.
1999;P
aolettietal.2
008).
mGluR’s(group
2,3),A
MPA
,Kaina
te,D
opam
ine,
Muscarinican
dGABA
Receptors
(Cha
etal.1
998;
Sepers
andRaymon
d20
14)
Dop
aminergicD1-lik
ereceptors,substanceP(SP)
anddy
norphinan
dD2-lik
ereceptorsan
dmet-enk
epha
linareim
plicated
(Hab
eran
dNau
ta19
83;A
ndre
etal.
2010).Im
paired
dopa
mine
andglutam
ateho
meostasis
(And
reet
al.2
010)
Chrom
osom
e/genes
invo
lved
21q2
2.1SO
D1
2q33
-2q3
5Alsin
9q34
SETX
15q1
5-21
SPG
1116
p11.2FUS
20q1
3.3VA
PB
14q1
1.2ANG
6q21
FIG
41p
36.2
TARDBP10
p13
OPTN
9p13
.3VCPXp1
1UBQLN2
9p21
C9O
RF72
(Chenet
al.
2013)
4q21
SNCA
12q1
2LRRK2
APP(21q
21.3)(Yoshika
ietal.
1990;T
hina
karanan
dKoo
2008).PSEN1(14q
24.3)
(Esler
andWolfe
2001
).PSEN2(1q4
2.13
)(Steiner
2004)
4p16
.3chromosom
alfragment
containing
IT15
gene
6q25
–q27
Parkin1p
35–p
36PIN
K11p
36DJ-11p
36ATP13
A2(L
esagean
dBrice
2009)
(↓)or
(↑)indicatesdecrease
orincrease
inthat
specificprop
erty
inthesaid
mou
semod
el,respectively.

Neurodegenerative diseases 685
2009).Ananimalmodel should score highon the followingvalidities: face validity, i.e., resemblances of behaviouralphenotypes of mouse model to that of human disorder;construct validity, i.e., closely reconstructs and mimics theunderlying cause of the disease or disorder; and predic-tive validity, i.e., treatments alleviate symptoms in mouseand human. A successful mouse model should fulfil faceand construct validity before being tested for therapeutics(predictive validity). Due to these varied features and theirbehavioural readouts, the mouse models of the respectivehuman diseases are highly useful in NDD research and inpreclinical studies.
Parkinson disease
PD is characterized by progressive degeneration of nigros-triatal dopaminergic neurons, leading to loss of motorfunction, rigidity, postural instability, tremor, andbradyki-nesia as described in the 1800’s by JamesParkinson (Mhyreet al. 2012; Rouse et al. 2000). Both familial (5–10% ofall cases) and acquired (90–95% of all cases) forms ofParkinsonism are usually caused by defects in dopamine(DA) metabolism. Decrease in DA inputs from basal gan-glia (BG) result in impaired control of motor circuitsthat ultimately leads to clinical symptoms (Spillantiniet al. 1997). For the PD mouse model, the transgen-ics express pathogenic mutant version of PD associatedgenes such as α-Synuclein, Parkin, Pink1 and Park7. α-Synuclein transgenic mice exhibit Lewy bodies that arethe histopathological hallmark of PD. These animals exertage-related progressive movement deficits, which is associ-ated with dopaminergic neuronal loss. However, not alltransgenic mutants exert significant dopaminergic neu-ronal loss.Activity-dependent modifications in synaptic efficacy,
such as long-term depression (LTD) and long-term poten-tiation (LTP) represent the key cellular substrates foradaptive motor control and procedural memory (Blissand Collingridge 1993). DA acting on D1-like and D2-like receptors play a critical role in driving the above-mentioned forms of synaptic plasticity in striatum. D1and D2 receptor signalling pathways converge in oppositemanners on a common target, DARPP-32. The involve-ment of DA in these phenomena has been thoroughlyestablished by the study of synaptic plasticity in striatalneurons recorded from rodent models of PD (Calabresiet al. 1992a,b). The absence of synaptic plasticity andabnormal synapse structure implied the cellular basisunderlying the abnormalities in striatal output within theBG, and consequently resulting in PD symptoms.Abnormalities in the subcellular distribution of N-
methyl-D-aspartate receptor (NMDAR)subunitGRIN2Brepresent one of the major changes that take placeat corticostriatal glutamatergic synapses. In fact, stud-ies using dyskinetic mouse models, increased levels of
GRIN2A and lower levels of GRIN2B were observed inextrasynaptic sites which altered binding ofNMDAR sub-units with their cargo proteins such as Synapse-associatedprotein 97 (SAP97) and SAP102 (Gardoni et al. 2006;Sheng and Sala 2001).Moreover, activation ofDARPP-32(DA-regulated and cAMP-regulated phosphoprotein-32)results in increased opening of the L-type Ca2+ chan-nels promoting the transition of medium spiny neurons(MSN) to a higher level of excitability, which in turnsphosphorylates AMPARs (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDARs providing amechanism for the direct control of glutamatergic trans-mission byDA signalling, that is altered in PD (Greengardet al. 1998; Vergara et al. 2003). This is further evidentfrom the studies that show the DA-denervation augmentsneuronal excitability in the striatum leading to excitabilityof striatal neurons, which is caused due to the increasedglutamatergic cortical inputs to the striatum (Calabresiet al. 1993; Centonze et al. 2004). The increased glutamatelevel in the synaptic cleft (Herrera-Marschitz et al. 1994) isconsequently responsible for the overactivity ofNMDARsand AMPARs on MSNs, which correlate with the motorbehaviour abnormalities observed in a rat model of PD.Thus, functional changes of the striatal neurons may alterthe output signals from the striatum to the other struc-tures of the BG that leads to pathophysiological changesobserved in PD.Evidences also suggest that the genes linked toPDplay a
critical role in regulating proper presynaptic and synapticvesicular transport, modification of DA flow and alteredpresynaptic plasticity (Dihanich andManzoni 2011).OnceParkinsonism is well established, i.e. parkinsonian state,most BG mechanisms are insufficient and cortical mech-anisms become important (Dihanich and Manzoni 2011;Blesa et al. 2017). At the postsynaptic level, decreased acti-vation of D2 receptors leads to a disinhibition of voltage-gated ion channels and increased influx of Ca2+ that leadsto degeneration of the synapses observed in PD-affectedanimals and in PD patients (Nitsch and Riesenberg 1995;Arbuthnott et al. 2000; Calautti et al. 2007). In DA-denervated striatum, NMDAR subunit GRIN1, and itsinteracting protein at synapse, PSD-95, levels are selec-tively reduced in the postsynapse (Lundblad et al. 2004).
Amyotrophic lateral sclerosis
ALS is a devastating neurodegenerative disease charac-terized by progressive loss of upper motor neurons in themotor cortex (cortical layer of pyramidal cells of corticallayer V), and of lower motor neurons in the brainstem andspinal cord as first described by Jean-Martin Charcot in1869 (Kumar et al. 2011). ALS typically affects adults inmid-life, with an incidence of 1–2 per 100,000 per year(Rosen 1993). Most ALS patients have no affected fam-ily members and are considered to have sporadic ALS.

686 S. N. Suresh et al.
Familial ALS occurs in 5–10% of cases and hasbreak anautosomal dominant inheritance.Mutations in genes suchas TARDP (TDP-43), FUS and SOD1 drive the famil-ial forms of ALS. In 20% of familial cases, mutations inthe superoxide dismutase-1 (SOD1) gene on chromosome21q were identified (ALS1) (Rosen 1993). The recessive,juvenile ALS2, is caused by mutations in ALSIN, whichcodes for a protein containing guanine exchange factordomains (Hadano et al. 2001; Yang et al. 2001). Micetransgenics overexpressing wild type or mutant TDP-43proteins cause TDP-43 inclusion bodies and loss of motorneurons with behavioural impairments. In addition, miceoverexpressing mutant SOD-1 develops inclusion bodies,neuronal loss, gliosis, tremor, hindlimb paralysis with sig-nificant decrease in lifespan.The pathological hallmark of ALS is the degeneration
of lower motor neurons in the brainstem and spinal cord,upper motor neurons in the motor cortex, and of the cor-ticospinal tracts, accompanied by reactive gliosis (Peharet al. 2005). The exact pathogenic mechanism underly-ing the selective motor neuron death in ALS is yet to beelucidated, although many possible mechanisms in spo-radic ALS, and SOD1-linked ALS have been proposed(Brown and Robberecht 2001; Cleveland and Rothstein2001; Julien 2001; Heath and Shaw 2002). One of the com-mon reasons for neuronal degeneration is because of theoverstimulation of glutamate receptors induced excitotox-icity (Lipton and Rosenberg 1994).
Evidences from the previous studies have strengtheneda link between glutamate-mediated toxicity and sporadicALS. Motor neurons express Ca2+-permeable AMPARs,GRIA2 subunit, and are, therefore, particularly vulnera-ble toAMPAR-mediated excitotoxicity (vonLewinski andKeller 2005). Transgenic mice expressing GRIA2 subunitswith an asparagine at the Q/R site (conferred a two-foldincrease in the Ca2+ permeability of AMPARs) (Feld-meyer et al. 1999), and showed late-onset of degenerationof spinal neurons and a decline in motor function (Kuneret al. 2005). Crossing these mice with those carrying amutation linked to familial ALS accelerated disease pro-gression and motor decline (Kuner et al. 2005), and thephenotype is exaggerated when GRIA2 is deleted (VanDamme et al. 2005).
The NMDAR-mediated neurotoxicity and subsequentoverload of mitochondrial Ca2+ and ROS productionhas been shown to take place in cultured motor neu-rons (Carriedo et al. 2000). Further, the NMDAR/Ca2+mediated excitotoxicity has been demonstrated in theneuronal loss observed in spinal neurons obtained fromhuman low molecular weight Neuro filament (NF) pro-tein (hNfl+/+) mice, an ALS mouse model (Nicholls et al.2007; Sanelli et al. 2007; Kambe et al. 2011). Compellingevidence supports the fact that excessive Ca2+ influxthrough NMDARs targets mitochondria which resultsin excitotoxicity in ALS-related MN death (Peng et al.1998). It is intriguing to hypothesize that regain control
of the NMDAR functionality will in turn affect AMPARfunction, due to the close interaction between these recep-tors, thereby further hampering the ALS-related excito-toxic drive.Apart from the extensive loss of motor neurons, there is
degeneration of midbrain dopaminergic cells and reducedtyrosine hydroxylase positive neurons which has beendescribed in both familial and sporadic forms of ALS(Andreassen et al. 2000; Kostic et al. 1997), and functionalalteration of the voltage-dependent Na2+ channel (Zonaet al. 1998). The dysfunction in the DA neurons is shownby investigating the corticostriatal synaptic plasticity inmice overexpressing the human SOD1 and mutating the(Gly933Ala) form (G93A) of the same enzyme (Lovingeret al. 1993; Calabresi et al. 2000).
The pivotal role of DA in corticostriatal LTD is wellestablished. For example, injection of 6-hydroxydopaminein rodent model leads to DA denervation that modifies thecorticostriatal plasticity (Calabresi et al. 1992a,b). More-over, a decreased striatal D2 receptor binding in sporadicALShas been described and is likely to occur because of anexcessive glutamatergic corticostriatal neurotransmission(Vogels et al. 1999, 2000). This pattern of degenerationis consistent with the observation that elevated levels ofSOD1 are expressed in BG cells, including the midbraindopaminergic neurons (Pardo et al. 1995). In conclu-sion, perturbations in these systems may cause the cell tobecome more susceptible to excitotoxic damage.There is spectrum of synaptic changes that occur in
ALS during the process of anterior horn neuron degen-eration. One such probable cause is due to the decreasesin cell body area, number of synapses, and synaptic con-tact length which is evident even during the early stage ofthe disease. It is noteworthy that despite decreases in cellbody area, synaptic numbers and synaptic contact length,the length of the active zone was not reduced in the nor-mally appearing neurons of the ALS patients (Sasaki andMaruyama 1994). The continuous loss of synapses thatis observed from myriad studies implies a decrease in theglobal connectivity of the motor system and a decreasedpotential for motoneuronal interaction.Theneuromuscular junction (NMJ) is the synapsewhere
the axon terminal of a motor neuron (MN) meets themotor endplate. Recent studies suggest that distal degen-eration in the skeletal muscle plays a key role in theprogression of ALS. Several studies using SOD1G93Amice have shown that NMJ degeneration occurs in theinitial stages of disease progression, long before MN loss(Brooks et al. 2004; Fischer andGlass 2010;Kanning et al.2010).
Huntington’s disease
HD is a progressive neurological disorder characterizedby chorea (uncontrolled dance like movement), cognitive

Neurodegenerative diseases 687
disturbances, depression and other psychiatric symptoms(Harper 1996). It is inherited in an autosomal dominantfashion, and severity of the disease depends on numberof CAG repeats (Jones et al. 1997; Walker 2007). BothHTT and mutant HTT (mHTT) proteins are ubiquitouslyexpressed, predominantly in the striatum, although it isthe least affected in HD, and in moderate levels in otherparts of the brain (Bhide et al. 1996). HTT is shown tobe expressed in the brain from midgestational period, andthe expression of mHTT was observed as early as 10-weekold infant’s brain suggesting an implication for proper neu-ronal development and function, especially when the brainis vulnerable to excitotoxic injury (DiFiglia 1997).
To further understand the function of HTT and mHTTin normal neuronal function and in diseased states respec-tively, several mouse models of HD has been created(Menalled and Chesselet 2002; Levine et al. 2004; Cepedaet al. 2010; Menalled and Brunner 2014). There are twotypes of mouse models of HD: ‘transgenics’, in which themutant gene or part of it, is inserted randomly into themouse genome, leading to the expression of mutant pro-tein along with the endogenous normal HUNTINGTIN;and ‘knock-ins’, in which the mutant gene is inserted intothe mouseHdh (mouseHuntington) gene, leading to eitherhomozygotes or heterozygotes for the mutation (for fur-ther informationplease referMenalledandChesselet 2002;Menalled and Brunner 2014). Myriad of transgenic micewith expanded polyQ repeats like R6/1, R6/2 and N171-Q82 have been generated that recapitulate the symptomsof HD. All these models develop aggregate inclusions,neuronal loss and motor-coordination deficits. The firstand most widely used transgenic mouse model of HDwas generated by overexpression of exon 1 of the humangene encodingHUNTINGTIN(IT15)with141-157CAG-repeat expansions, which was termed as R6 (Mangiariniet al. 1996). Unlike Htt homozygous knock-out (KO)(embryonically lethal but notHtt conditional Knock-out)(Nasir et al. 1995; Wang et al. 2016), R6 mice survive for13 weeks. Apart from the motor deficits, these differentmouse models exhibit altered synaptic function and cog-nitive deficits.Researchovermany years suggests that cognitive defects
appear long before the onset of overt motor dysfunctionin HD (Paulsen et al. 2008). These studies have shown thatsynaptic dysfunction precedes cell death by many yearsin humans (Murer et al. 2002). It is observed that loss ofmedium-shaped spiny (MSN) projection neurons in stria-tum, and pyramidal neurons from cortex is considered asprominent pathological characteristics of HD (Von sattelet al. 1985; Milnerwood et al. 2010; Milnerwood and Ray-mond 2010).
Impaired DA homeostasis is one of the major conse-quences ofHTT (includingHtt) mutation that contributesto the impaired information processing from the corti-cal inputs to striatum (Andre et al. 2010). Dopaminergicneurons from substantia nigra and ventral tegmental
area (VTA) project to the dorsal striatum to regulateglutamatergic neurons (direct pathway; rich in D1 recep-tors; facilitates movement), whereas, indirect pathwayprojects to external globus pallidus (rich in D2 recep-tors; suppresses movement) (Andre et al. 2010; Sepersand Raymond 2014), which are found to be degeneratedin HD. On the contrary, Andre et al. (2010) have shownthat hyperactivation of the nigrostriatal pathwaymay elicitthe characteristics of chorea observed in HD. Therefore,agents affecting dopamine transmission are used to mod-ulate HD (Murer et al. 2002).
Proper glutamatergic function of these neurons is drivenmajorly by glutamate (excitatory neurotransmitter), and,thus have an increased sensitivity towards NMDARactivation leading to neuronal death, especially in HD(McGeer et al. 1977; Fonnum et al. 1981; Schwarcz et al.1983). By the timemotor symptomswere observed,major-ity of striatal glutamatergic neurons and spine densitieswere lost (Li 1999;Cepeda et al. 2007, 2010). Thus, reducedglutamatergic signalling in the striatum could lead to lossof spines and, eventually, contribute to motor deficits.To study the functional insights of HD pathology, elec-
trophysiological studies have dissected out intrinsic andsynaptic neuronal properties (Sepers and Raymond 2014).Studies have shown that, in R6/2 mice, basal synaptictransmission and presynaptic release were progressivelyaltered (Cepeda et al. 2001; Klapstein et al. 2001; Milner-wood et al. 2006; Khedraki et al. 2017). PresymptomaticR6/2 mice showed increased neuronal input resistanceand lower stimulus intensity to evoke action potentials(rheobase), whereas, symptomatic R6/2 mice exhibitedincreased resting membrane potentials, input resistanceand decreased membrane time constants and synapticplasticity. Taken together, these findings indicate that pas-sive and active membrane and synaptic properties ofmedium-sized spiny neurons are altered in the R6/2 trans-genic.Many studies have observed an early augmentation of
NMDAR activity in MSNs of HD mouse (Milnerwoodand Raymond 2007; Cepeda et al. 2010; Cummings et al.2010). Further, overexpression of GluN2B subunit in HDhas been shown to exacerbate the phenotype and pathol-ogy. Studies have shown that brief stimulation of synapticNMDAR (containing GluN1/GluN2A) leads to survivalsignalling via brain derived neurotrophic factor (BDNF)activation (Martire et al. 2013) but activation of extrasy-naptic NMDAR (containing GluN1/GluN2B) is neuro-toxic and leads to cell death (Hardingham et al. 2002).These studies suggest that hyperactivation of extrasynap-tic NMDAR leads to neuronal loss in HD.In another mouse model of HD, yeast artificial chro-
mosome (YAC)-Htt (YAC128), containing entire humanHTT to have 128 CAG-repeat expansions (Slow et al.2003), it was found that the probability of release at D1-presynapse was increased in 1.5 months, but decreased at12 months. However, D2-presynapse release probability

688 S. N. Suresh et al.
was not altered in these mice (Cummings et al. 2010).Further studies have shown that the synaptic transmis-sion and function are altered in YAC128 mouse model ofHD (Miller et al. 2008; Joshi et al. 2009). Similar obser-vations were found in other mouse models of HD (Levineet al. 1999, 2004; Klapstein et al. 2001; Graham et al. 2009;Pouladi et al. 2009; Cummings et al. 2010; Akopian et al.2016; Kolodziejczyk and Raymond 2016).
Alzheimer’s disease
AD is the most common cause of senile dementia anda devastating neurodegenerative disease. Formation ofsenile plaques, neurofibrillary tangles, massive loss ofsynapses, and eventual neuronal cell death are the patho-logical hallmarks of AD. The pathogenesis is associatedwith increased β-amyloid (Aβ) levels in the brain generatedby proteolysis of amyloid precursor protein (APP) withthe help of Presenilin1 (PSEN1), and part of γ- secretasecomplex (Strooper et al. 1998; Selkoe 2002). Mutations inAPP and Presenilin1 or both genes result in an autosomaldominant form of AD. To understand the mechanism ofAD, temporal evolution, and to translate it for therapeuticpurposes, several transgenicmousemodels ofADhas beenused till date. There are currently 127 animalmodels ofADin use having wide variety of mutations (single, double andtriple mutations) that are implicated in AD (Higgins andJacobsen 2003; LaFerla and Green 2012; www.alzforum.org). Recently, very robust mouse model, 5xFAD, hav-ing five familial AD mutations, causes relatively early andaggressive presentation of AD, has been widely used ascompared to other models. It recapitulates the followingdisease phenotypes: β-amyloidosis, plaques, neurite dys-trophy, dendritic spine loss, neuroinflammation, neuronalloss and age-dependent cognition decline. Following arethe limitations of this models: (i) evidences from positron-emission tomography (PET) suggest that mouse plaquesare significantly differ from human biochemically, and (ii)another caveat is the neuronal loss is not profound as thatof humans (Sasaguri et al. 2017). A detailed review onvarious animal models of AD can reviewed here (LaFerlaand Green 2012; Pozueta et al. 2013; Sasaguri et al. 2017).These transgenic models show synaptic dysfunction, and,thus, provide an opportunity to understand the mech-anisms of neurophysiological deficits observed in AD,mainly the synaptic and cognitive decline (Rowan et al.2003; Spires-Jones and Knafo 2012). Abnormalities insynaptic function in AD was observed more than fourdecades ago by Gonatas et al. (1967). Since then, thereare many studies that focussed on reinforcing the idea thatloss of synaptic function is the main characteristics of AD.Indeed, this was further confirmed by quantitative ultra-structural and immunohistochemical post-mortem studiesfrom human AD patients with symptoms ranging frommild-cognitive impairments to early-mild AD (Masliah
et al. 1989; Terry et al. 1991; Scheff and Price 1993). Mostof the electrophysiological studies of amyloid depositingmouse models have investigated alterations in synapticstrengthbetweenhippocampalpyramidal neuronsbymea-suring basal synaptic strength, synaptic transmission, andneuronal intrinsic properties (Nistico et al. 2012). A studyfrom a mouse model of AD expressing single or doubletransgenic mutations showed normal basal synaptic trans-mission but impaired LTP in early stage of AD (Liu 2008),while in another study using App695Swe mutation, nor-mal LTP with deficits in basal synaptic transmission wasobserved for both 12-month as well as 18-month old mice(Fitzjohn et al. 2001). These initial studies have shown thatdisruption in synaptic function is themajor cause of cogni-tive decline in AD. Studies have shown that AD pathologyincreases in an age dependant manner (Hsia et al. 1999;Sun et al. 2017). By using Apoe4mouse (four months andolder), alteration in short as well as long-term plasticityhas been observed (Sun et al. 2017). A similar age depen-dent decline in basal synaptic function was observed inother models (Hsia et al. 1999; Puzzo et al. 2017). Thesestudies clearly show that the decline in basal synaptic func-tion is an early indicator cognitive decline observed in laterstages of AD. To further understand whether amyloido-genic processing of APP is responsible for amyloid-β andtau toxicity, toxic proteins are administered in acute brainslices prepared from wild type (WT) as well as BACE1KO mice. No affect in basal synaptic transmission wasobserved inWT or BACE1 (β secretase) KOmice but LTPwas impaired in both genotypes, suggesting that the toxi-city did not depend on APP processing alone (Puzzo et al.2017). Apart from the alterations in synaptic function,abnormal dendritic spine structures were also observed inAD.Dendritic spines are specialized anatomical structureson neuronal cells that serve as the postsynaptic componentfor the excitatory dendritic spines. Several studies usingdifferent mouse models of AD have identified severe den-dritic spine loss and plaque-associated structural plasticitydeficits in AD. This is extensively reviewed here (Rowanet al. 2003; Spires-Jones and Knafo 2012; Pozueta et al.2013). Based on these studies, basal synaptic function canbe used as a biomarker to detect AD in an early stage ofdevelopment.
Drosophila melanogaster
It is a multicellular eukaryote that exhibits both histo-logical and behavioural phenotype associated with neu-rodegeneration. Flies expressing the proteins such asα-synuclein, β-amyloid, polyglutamine repeat, tau reca-pitulate many of the histological and behavioural diseasephenotypes associated with neurodegeneration. In thismodel, the toxic proteins are overexpressed in retinaleading to rough-eye phenotype that forms a platform,which is used extensively for genetic and chemical screens

Neurodegenerative diseases 689
(Luheshi et al. 2007). The advantages working with fliesare short doubling time, multicellularity, availability ofgenetic resources and ability to express proteins in a tissue-specific manner. Also, the fly model can unravel the basicpathophysiological mechanisms underlying neurodegen-eration. It is often used synergisticallywith another diseasemodel to validate the basic mechanisms. For example, asmall molecule screen performed on fly model identifiedmGLUR5 (GPCR) as a druggable target that is abun-dantly expressed in brain (Chang et al. 2008). Recently,regulation of protein QPCT (glutaminyl cyclase) has beenshown to curb neurodegeneration symptoms in fly modeland mammalian cells (Jimenez-Sanchez et al. 2015).
Caenorhabditis elegans
It is one of the widely used models to study neurodegen-eration for more than a decade. Pros are the availability ofgenetic resources of this tiny transparent animal where all302 neurons are mapped for their interactions and theirshort generation time (Nussbaum-Krammer and Mori-moto 2014). Genetic strains were constructed that expresstoxic protein aggregates such as Aβ, α-synuclein, TDP43and polyglutamine repeats in either whole body or tis-sue specific manner. They not only form visible aggregatesbut also show the behaviour phenotype such as paralysis.Number of genetic and chemical screens was conductedto identify modulators that rescue the behavioural phe-notype. This led to identification of small molecules thatcleared toxic protein aggregates andwere further validatedin higher model systems (Narayan et al. 2014). Success-ful genetic screens too have identified many genes such asLRRK2, the reduction of its kinase activity rescued thedopaminergic deficit motor phenotype (Yao et al. 2013).Various regulators of HSR have been tested in the wormmodel to treat proteotoxicity. Its transparent tissue enablesfor high content imaging. More importantly, they alsoshow a typical ageing symptoms making them an idealtool to investigate the interplay of metabolism, ageing andneurodegeneration comprehensively (Chung et al. 2008).
Cellular models
Yeast
The budding yeast (Saccharomyces cerevisiae) is a simpleyet powerful tool to study the cellular pathophysiolog-ical mechanisms underlying the protein toxicity (Smithand Snyder 2006). It recapitulates the toxicity of proteinaggregates as that of neurons. This is because the funda-mental cellular features and vital cellular pathways areconserved (Khurana and Lindquist 2010). For instance,the membrane-bound organelles such as mitochondria,Golgi, lysosome, endoplasmic reticulum and so on existin yeast. More importantly, the unfolded protein response
is conserved as that of the mammalian cells. The majoradvantages of yeast are short doubling time, existence ofgenetic sources, amenable to genetic manipulation andscalable to high throughput genetic and chemical screens.Also, around 3500 genes are found to be homologues ofthose of human cells (Botstein et al. 1997; Kachroo et al.2015). Pioneering work by Susan Lindquist and her col-leagues have shown that yeast cells expressing aggregateprone genes and their variants that have been identifiedin ND have been used to understand their pathobiology(Outeiro and Lindquist 2003). The ER-Golgi traffick-ing defect due to α-synuclein overexpression leads tocellular toxicity was first noted in yeast and further val-idated in mammalian cells (Cooper et al. 2006; Winslowet al. 2010). Several studies including ours have identi-fied novel peptidomimetics and small molecule autophagymodulators that ameliorate ND related aggregate pro-teotoxicity (Sarkar et al. 2007b; Khurana and Lindquist2010; Rajasekhar et al. 2015). Some of the obvious limi-tation of the yeast model is that it cannot recapitulate thecomplexities of neuronal network andmulticellularity thatis present at the organ level.
iPSC
iPSC technology enables researchers to generate humanspecific, disease relevant cell type such as neurons directlyfrom patient fibroblasts. The advancement of gene editingtechniques such as CRISPR-Cas9 has simplified geneticknock out/in studies in iPSC-derived neurons to under-stand the pathogenesis of neurodegeneration. The poten-tial of this model system to shed light on basic diseasepathology mechanisms is massive (Narayan et al. 2014).iPSC lines for AD, PD, Niemann-Pick disease type Cand ALS disease have been generated for their biologicalinvestigations (Chung et al. 2013; Ryan et al. 2013; Kiski-nis et al. 2014). Consequently, genetic or small moleculescreens can be performed using iPSC. The current limi-tations are (i) hurdle in scaling up for high throughputscreening due to their relative slow propagation rates,(ii) extensive and time-consuming neuronal differentiationprotocols. However, they can be used to validate the datagenerated from nonhuman disease models. For example,yeast screen yielded theNAB2 compoundwhere it rescuedthe disease phenotype and validated its disease modifyingmechanisms in iPSC (Chung et al. 2013).
Current understanding on the mechanistic insights ofaggrephagy
These and the other model systems have contributedimmensely to unravel molecular aspects of the process ofaggrephagy. And how aggrephagy is perturbed by variousprotein aggregates is detailed below.

690 S. N. Suresh et al.
Substrate recognition
Protein aggregate (substrate) recognition by receptors/adaptors such as Alfy, p62/SQSTM1, Optineurin andCue5/Tollip, is one of the first steps of autophagy for itsclearance (Stolz et al. 2014). The selectivity of autophagiccargo depends on specific receptors/adaptors that recog-nize it by degradation marks. Autophagic receptors bindto the ubiquitinated substrates through their respectivedomains to capture the aggregates. They also tether themto autophagic machinery like LC3 to enable autophago-some formation around the aggregates. The brief descrip-tion pertaining to autophagic receptors are as follows:
Alfy : This multidomain scaffolding protein recognizesubiquitinated protein aggregates for their clearance. Itcontains followingdomains: (i) Pleckstrinhomology (PH)-BEACH domain that binds to p62, (ii) FYVE domainfor PI3P binding, and (iii) WD40 repeats that interactswithAtg5. It colocalizes with ubiquitin positive p62 aggre-gates (Isakson et al. 2013). It stabilizes the LC3 and p62interaction and also recruits Atg5 for the formation ofautophagosomes that facilitated huntingtin polyQ aggre-gate clearance (Filimonenko et al. 2010).
p62/SQSTM1 : p62 is the adaptor for aggrephagy (Pankivet al. 2007), mitophagy (Geisler et al. 2010), pexophagy(Kim et al. 2008b) and xenophagy (Bah and Vergne 2017).It possesses several domains namely: (i) UBA domainthat binds ubiquitin, (ii) PB1 domain for aggregation ofcargoes, and (iii) LC3 interacting region (LIR) motif forLC3 binding (Lim and Yue 2015). It also has nuclearlocalization signal (NLS) and nuclear export signal (NES)for nucleocytoplasmic shuttling (Pankiv et al. 2007). p62mutations have been linked to sporadic and familial ALSthrough impaired clearance of TDP-43 and SOD1 (Fectoet al. 2011). Paget’s disease of bone patientswithmutationsin p62 are predisposed to ALS, a disease that is character-ized by p62 positive inclusions in neurons. Mutant p62contributes to pathogenesis through multitude of mech-anisms and one of them is via over activation of nuclearfactor-κB (NF-κB) signalling (Chamoux et al. 2013).
Optineurin : It binds to ubiquitinated proteins through itsUBA and NF-kB essential modulator (NEMO) domains(Kachaner et al. 2012). On its other end, it binds toLC3 through LIR motif. Genetic mutations of optineurinare also associated with ALS. Optineurin mutation isassociatedwith rareALSmutations leading toTARDNA-binding protein 43 (TDP-43) inclusions (Ito et al. 2011).
Cue5 : Cue5 is the recently identified yeast autophagyreceptor that binds to ubiquitin through its CUE domainand Atg8 through its AIM1 and AIM2 motifs (Lu et al.2014). Its mammalian homologue is Tollip which is essen-tial for the huntingtin polyQ clearance.
Valosin-containing protein (VCP) : It is a member ofAAA+ family of ATPase and is involved in the sortingof ubiquitinated cargoes through endolysosomal path-way. VCP mutation is associated with Paget’s disease ofbone and fronto temporal dementia (FTD). Overexpres-sion or knockdown of VCP in cells leads to appearance ofautophagosomes that are immature with accumulation ofubiquitin positive cargoes (Ritz et al. 2011).
Autophagosome formation
mTOR complex 1 (TORC1) negatively regulates auto-phagy. Various stimuli in neurons such as ATP, ERstress, and specific amino acids repress TORC1 to acti-vate ULK complex (ULK1, FIP200, ULK2, Atg101 andAtg13) (Mizushima 2010). Phosphorylation of ULK1 atits active site leads to subsequent phosphorylation ofother components of complex for initiating autophagy(Chan et al. 2009). Although ULK1 phosphorylationis primarily regulated by TORC1, it can be also inde-pendently modulated by AMP-activated protein kinase(AMPK) (Khan and Kumar 2012). Also, ULK1 phos-phorylation leads to the translocation of class III PI3kinase complex (beclin1, Atg14, Vps15 and Vps34) to iso-lation membranes (also known as omegasome formationat the ER) by phosphorylating one of its components,AMBRA (Di Bartolomeo et al. 2010). Vps34 activity gen-erates phosphatidylinositol-3-phosphate (PI3P) to bind toits effectors such as WD repeat domain phosphoinositideinteracting 1 (WIPI1) and WD repeat domain phospho-inositide interacting 2 (WIPI2) (Jaber and Zong 2013).This is to catalyse the ubiquitination like reactions at theisolation membrane that aid autophagosome formation.Atg5–Atg12–Atg16L complex is formed on the isolationmembrane in presence of Atg7 and Atg10. This Atg5–Atg12–Atg16L catalyses the covalent attachment of phos-phatidylethanolamine to LC3 that facilitates the closure ofautophagosome membranes (Ichimura et al. 2000). Atg9aims to supply membranes for the growing autophago-somes by shuttling from various sources such as endo-plasmic reticulum-mitochondria contact sites (Hamasakiet al. 2013), Golgi (Geng and Klionsky 2010) and plasmamembrane (Ravikumar et al. 2010) to autophagosomeformation site. ER has been suggested as a membranesource for autophagosomes formed at synapse but themain source is not yet clear (Maday and Holzbaur 2014).In neurons, how autophagy induced is still an enigma.
Conventionally, the key trigger for autophagy is starva-tion that induces autophagy in numerous cell types (Kimet al. 2008a). However, even 48 h of starvation did notinduce GFP-LC3 puncta in brain but profoundly inducedautophagy in the liver (Mizushima et al. 2004). On theother hand, while caloric restriction induced autophagyin cortical, Purkinje and motor neurons, nutrient starva-tion failed to induce autophagy in cultured hippocampal

Neurodegenerative diseases 691
neurons (Kaushik et al. 2011). These contradicting resultsmay mirror the neuronal specificity in inducing and reg-ulating autophagy. Several studies have shed light about‘other’ ways of modulating autophagy in the brain.Recently, the neuronal activity such as acute stimuli
can trigger autophagy. Upon high frequency stimulation,the Atg8 puncta is increased at NMJ (presynapse) inD. melanogaster (Vanhauwaert et al. 2017). Although themolecular players and its mechanism(s) are not yet elu-cidated, calcium signalling can be a key player. Duringexocytosis driven by action potential, there is remarkableincrease in calcium levels at presynapse (Rizzoli 2014).Calcium can either enhance or inhibit autophagy depend-ing on the context and calcium responsive proteins areabundant at presynapse. It is noteworthy to mention thatinvestigating calcium-mediated autophagy at presynapsecan shed light on the novel mechanistic aspect of neuronalautophagy. Interestingly, the presynaptic resident proteinssuch as Endophilin A, Basoon, Synaptojanin 1 regulateautophagy by interacting with autophagy-related proteins(Vijayan and Verstreken 2017).Regulation of protein synthesis by TORC1 is impli-
cated in synaptic plasticity (Kovacs et al. 2007). However,TORC1-mediated autophagy induction at presynapse hasnot been studied extensively. Autophagy mediated degra-dation of GABA receptors and AMPA receptors (that areinvolved in synaptic plasticity) at postsynapse induce long-term depression (Shehata et al. 2012).
In PD model, α-synuclein perturbs Rab1a-mediatedAtg9 translocation to isolation membrane leading toimpairment of omegasome formation (Cooper et al. 2006).Atg9 trafficking is also abnormal in vacuolar proteinsorting-associated protein 35 (VPS35) D620N mutationthat results in autosomal dominant case of PD (Zhou et al.2017). VPS35 (component of retromer complex) recruitsactin nucleation-promoting WASP and Scar homologue(WASH) complex to endosomes. Mutation in VPS35perturbs this recruitment to cause the abnormal Atg9 traf-ficking to impair autophagy (Zavodszky et al. 2014). TheE122D Atg5 mutation has been identified in childhoodataxia (Kim et al. 2016). This disease is characterizedby loss of motor coordination and cerebellar hypoplasia.This mutation decreases the affinity of Atg5 to Atg12 andreduces the autophagosome biogenesis and its flux (Kimet al. 2016).
β-Propeller protein-associated neurodegeneration(BPAN), the static encephalopathy of childhood with neu-rodegeneration in adulthood (SENDA) is caused by denovo mutations in WDR45 (Haack et al. 2013). WDR45gene encodes WIPI4, the key protein that bridges PI3Pproduction and LC3 lipidation for autophagosome mat-uration (Zhao et al. 2015). Neuronal specific KO ofWDR45 recapitulates someBPANdisease phenotype suchas impaired motor coordination, cognitive deficit, axonalswelling with accumulation of ubiquitin positive aggre-gates (Saitsu et al. 2013).
Hexanucleotide repeats in C9ORF72 gene is one of themost common causes of ALS. Different mechanisms havebeen proposed for this disease pathogenesis and pertur-bation of autophagy is one of them. C9ORF72 interactswith ULK1 and affects the phagophore initiation com-plex (Webster et al. 2016). In addition, C9ORF72 is a partof WD repeat domain 41 (WDR41) and Smith–Magenissyndrome chromosome region, candidate 8 (SMCR8)complex that is involved in both vesicular traffickingand autophagy. This complex is a guanosine diphos-phate (GDP)/guanosine triphosphate (GTP) exchangefactor (GEF) that activates the small GTPases Rab39 andRab8 for autophagosome formation (Amick andFerguson2017).
Wild-type huntingtin protein acts as a scaffold forautophagosome formation whereas its mutant fails torecognize the cargo leading to ‘empty’ autophagosomes(Martinez-Vicente et al. 2010). It also impairs autophago-some transport and further accumulation of autophagicsubstrates (Wong andHolzbaur 2014).Mutant huntingtininteracts with Rhes that are selectively expressed in stria-tum to inactivate it. Rhes interacts with beclin 1 to induceautophagy by reducing the bcl2-beclin 1 interaction (Sub-ramaniam et al. 2009). Further,mTOR, is also sequesteredin HD and spino-cerebellar ataxia 7 (SCA7) aggregates(Jimenez-Sanchez et al. 2012).
Autophagosome–lysosome fusion
Autophagosomes fuse with lysosomes to generateautolysosomes that digest the toxic protein aggregates. Inneurons, autophagosomes generated in axons are trans-ported to the lysosomes that are abundant near perin-uclear microtubule-organizing centre (MTOC). Micro-tubules and motor proteins such as dynein–dynactincomplex are involved in retrograde transport ofautophagosomes towards neuronal cell body. Mutationsin dynein (axonal Charcot–Marie–Tooth hereditary neu-ropathy type 2) and dynactin (motor neuron disease)motor protein complexes are implicated in neurodegenera-tion pathogenesis indicating the importance of retrogradetransport of autophagosomes to fuse with lysosomes(Ferrucci et al. 2011). Interestingly, decreased dyneinactivity resulted in the accumulation of autophagosomes,autolysosomes and p62 positive inclusions in motor neu-rons before the onset of symptoms where they degenerateupon clinical manifestations (Sasaki 2011).Amphisome formation (a fusion product of endosomes
with autophagosome) is perturbed in familial ALS andFTD and is associated with loss-of-function mutationsin charged multivesicular body protein 2B (CHMP2B).Mutant CHMP2B neurons accumulate ubiquitinpositive inclusion by disrupting ESCRT machinery activ-ity (Skibinski et al. 2005). Autophagy and endolyso-somal pathways have significant crosstalk pertaining to

692 S. N. Suresh et al.
usage of cellular protein machineries. Mutations in anendolysosomal pathway affects autophagy and vice versa(Otomo et al. 2011). Endosomal maturation proteinRab7 is essential for autophagic flux and its mutationis associated with bone related osteopetrosis (lysosomalperturbation) and neurodegenerative CMT2 (autophagyflux perturbation) diseases (Tabata et al. 2010). Further,Rab7 significantly influences lysosome positioning. ALS2protein mutation, an activator of Rab5 perturbs ALS2mediated Rab5 activation that hampers amphisome for-mation as observed in familial cases of ALS (Otomo et al.2011). The coordinated crosstalk of endolysosomal andautophagy pathways to clear toxic protein aggregates inhealthy cells goes awry in neurodegeneration.In neuronal specific interferon-β KO of transgenic
mouse model, the autophagosome–lysosome fusion isaffected with concomitant accumulation of ubiquitin pos-itive inclusion in neurons that leads tomotor coordinationand behavioural impairments (Ejlerskov et al. 2015).
Regulation of lysosomal activity
Autophagosomes fuse with lysosomes for the clearance ofits contents. For this process to be efficient, the uncompro-mised lysosomal activity is essential. The lysosomal pH isoptimally (∼4.5–5) maintained by proton pump vacuolarATPase for the activity of hydrolytic enzymes (proteases,peptidases, lipases and nucleases). Ionic balances of lyso-somes are regulated by presence of numerous ion channels(calcium, chloride and potassium). Lysosomal associatedmembrane protein-1 (LAMP1) and LAMP2 prevents theself-digestion of lysosomes (Nixon 2013). The functionalintegrity of lysosomes is compromised in most neurode-generative disorders (Kroemer and Jaattela 2005).
Lysosomes serve as key players in the initial stage ofautophagy induction. Upon perceiving the autophagyinducing stimuli, the TORC1 localized on the lysosomalmembrane is repressed and that further allows activa-tion of transcription factor EB (TFEB) to translocate tothe nucleus. TFEB activates genes involved in lysosomalbiogenesis and autophagy process. TORC1 regulation ofTFEB is achieved through Rag-GTPase pathway (Kimet al. 2008a). TORC1 and TFEB activities are tightlycontrolled by the presence of intracellular and lysosomalamino acid contents. If amino acids are limiting inside thecells, for instance as in starvation, the TORC1 activityis repressed and is followed by translocation of dephos-phorylated TFEB to nucleus resulting in upregulation ofautophagy. On the other hand, the surplus amino acidslead to reactivation of TORC1 to terminate the autophagyinduction (Avruch et al. 2009). The androgen receptorpolyQ repeat mutant (implicated in SBMA) interacts withTFEB and suppresses autophagy at transcription level(Cortes et al. 2014). SCA3 transgenic animals show lowlevels of sirtuin 1 (deacetylates numerous autophagy and
its related proteins), parkin and beclin 1 (Cunha-Santoset al. 2016).Zinc-finger protein with KRAB and SCAN domains 3
(ZKSCAN3) is repressor of autophagy and has oppositefunction that of TFEB. Inhibition of mTOR enhances itsaccumulation in the cytosol. Downregulating ZKSCAN3induces autophagosome and lysosomal biogenesis(Chauhan et al. 2013). Apart from this, around 20 dif-ferent transcription factors are reported to regulate theautophagy at gene transcription level depending on thecontext of stimuli. For instance, p53, forkhead box O3(FOXO3) and microphthalmia-associated transcriptionfactor (MITF), all profoundly affect autophagy geneexpression (Li et al. 2017).
Lysosomal storage disorders (LSDs) illustrates thenexus between lysosomal dysfunction and neurodegener-ation. In LSDs, the unavailability of functional lysosomalhydrolytic enzymes leads to defective autophagy thatman-ifests disease pathology in brain. In Niemann Pick type C(NPC) disease, themutations ofNPC1 and/orNPC2 geneslead to defective cholesterol trafficking where profoundautophagic vacuole are observed in neurons (Elrick et al.2012). In neuronal ceroid lipofuscinosis, the mutation inCLN3 gene reduces autolysosome formation, whereas inCathepsin D mutation that causes the same disease wheredeclined lysosomal proteolysis is observed (Nixon 2004).
Therapeutic strategies for ND
Current therapeutic interventions for neurodegenerationinclude three different strategies. First, preventing thetoxic protein aggregate build-up. For instance, the anti-senseoligonucleotides ameliorated the amyotrophic lateralsclerosis disease phenotype under preclinical settings andshowed significant safety in phase I clinical trial (Niz-zardo et al. 2016). Second, dissolving the already culmi-nated toxic protein aggregates inside the neurons. Variousgroups identified small molecules and peptides that canbring about disintegration of the aggregate. This has beena widely used strategy for many years as a promisingantineurodegeneration therapeutic. For AD, few smallmolecule candidates that dissolve the amyloid plaques arecurrently in the clinical trials inUSAandEurope.The thirdand recent approach is to promote cellular pathways tocapture and degrade the protein aggregates by either ubiq-uitin proteasome (UPS) or autophagy systems. Numerousgroups have demonstrated that neurodegeneration couldbe ameliorated by upregulating the activities of cellu-lar clearance systems such as proteasome or autophagythrough smallmolecules or peptides.Many smallmoleculescreens have been reported that have identified the drug-gable candidate(s) (target proteins or pathways). Forexample, Rilmenidine, an autophagy inducer is currentlyin clinical trial as a HD therapeutic in Europe.

Neurodegenerative diseases 693
Chemical modulation of autophagy
As mentioned earlier that upregulation of autophagygene expression enhances the longevity of several modelorganisms (Pyo et al. 2013). Convincing evidences demon-strate that dysfunctional autophagy contribute to thepathogenesis of neurodegeneration (Nixon 2013). Impor-tantly, autophagy can clear the toxic misfolded proteinaggregates. Mechanisms that govern degradation of toxicmisfolded protein aggregates is thus a therapeuticallyattractive target (Sarkar et al. 2007b). Numerous stud-ies demonstrated that pharmacological upregulation ofautophagy to clear protein aggregates is beneficial in ame-liorating neurodegeneration.
mTOR dependent autophagy modulators
mTORdependentmodulators induce autophagy by repre-ssing mTOR and it can be of ATP competitive (e.g.,Torin1) or nonATP competitive (e.g., rapamycin). mTORcan be found in two complexes such as TORC1 (negativeregulator of autophagy) and TORC2 (positive regulator ofautophagy). Rapamycin and their analogues are relativelysafer owing to its nonATP competitivemode of inhibition.The allosteric TORC1 inhibitor, rapamycin was the firstidentified small molecule autophagy inducer. Rapamycininteracts with FK506-binding protein 12 (FKBP12) toinhibit the TORC1 activity (Lorenz and Heitman 1995).Rapamycin has been demonstrated to be neuroprotectivein various diseases such as PD, HD, AD and FTD inan autophagy dependent manner (Jahrling and Laberge2015). Limited absorption of rapamycin led to the devel-opment of its analogues (rapalogues) such as everolimus,temsirolimus, and ridaforolimus. Rapalogues are underrigorous investigation for their therapeutic potential inneurodegeneration. For instance, food and drug adminis-tration (FDA) approved everolimus as a tuberous sclerosistherapeutic (Bissler et al. 2017). Torin1 inhibits bothTORC1 and TORC2 in concentration dependent man-ner (Ma et al. 2013). There are classes of modulators thatindirectly inhibit mTOR activity. For instance, metformin(type II diabetes therapeutic) inhibits mTOR throughregulating AMPK pathway. Another AMPK activator,Nilotinib also exerts neuroprotection by inhibitingmTORactivity and is currently under phase II clinical trial for PDtherapy (Karuppagounder et al. 2014). PI103 belong todual mTOR/class III PI3Kinase modulators that inhibitboth mTOR and AKT pathway to induce autophagy.However, due to its rapid in vivometabolism, PI103 is cur-rently unsuitable for neurodegeneration therapy (Hassanet al. 2013).
mTOR independent autophagy modulators
mTOR independent autophagy modulators are thosethat induce autophagy without repressing the mTOR
activity. mTOR being the epicentre for cellular growthsignalling whose chronic inhibition exerts significant tox-icity. These inhibitors are preferred therapeutically assignificant side effects such as impaired wound healingand immunosuppression that are classically associatedwith mTOR inhibitors can be avoided. Thus, these arealso more suited for long-term drug administration. Smallmolecule screening for FDA approved drugs revealedthat several modulators ameliorated neurodegenerationpathogenesis in various model systems in mTOR inde-pendent manner. This study revealed the involvement oftwo cyclical pathways namely Gs α /calpain/Ca2+ (Car-denas et al. 2010) and EPAC/cAMP/PLC ε /Ins(1,4,5)P3(Williams et al. 2008) that are important for the mTORindependent clearance of aggregates.Rilmenidine, the imidazoline receptor inhibitor, enhan-
ces autophagy by reducing cAMP levels. It has been shownto ameliorate neurodegeneration in variousmodel systemssuch as primary neurons and transgenic mouse models ofHD. It is important to note that rilmenidine is currentlyundergoing safety trials for HD in Europe (Williams et al.2008).
Other drugs identified to regulate autophagy throughthe cyclical pathway are lithium, valproic acid and car-bamazepine. Carbamazepine and valproic acid repressinositol synthesis, while lithium inhibits inositolmonophosphatase to reduce Ins(1,4,5)P3 levels and thusinduce autophagy (Williams et al. 2008).
Modulators ofGs α /calpain/Ca2+pathway suchas vera-pamil and amiodarone along with calpain inhibitors suchas calpeptin and calpastatin have shown to exert neuro-protective potential by degrading the protein aggregatesthrough an autophagy-mediated mechanism (Williamset al. 2008).Trehalose clearsα-synuclein andmutant huntingtin pro-
tein aggregates in autophagy dependentmanner in variousmodel systems (Sarkar et al. 2007a). It has been shown tobe neuroprotective in tauopathy models as well (Schaefferet al. 2012). It extends life span, curbs motor and cognitivedeficits in transgenic HD andALSmousemodels (Tanakaet al. 2004).
Perspectives
The aetiological components, as well as the molecularand cellular points of convergence in ND, remain elu-sive and hence optimizing a defined therapeutic approachhas been challenging. Cellular mechanisms that directlyimpact synaptic function, including autophagy, endoso-mal trafficking, and mitochondrial homeostasis, are likelyto constitute effective interventional targets in therapy infuture. It is becoming increasingly evident from recent lit-erature that clearing protein aggregates through proteinquality control systems such as aggrephagy hold thera-peutic promise for curbing neurodegenerative diseases.

694 S. N. Suresh et al.
Acknowledgements
We thank members of the autophagy lab (JNCASR),Mridhula Giridharan and Aparna Hebbar for critical reading ofthe manuscript. We apologize to researchers whose work couldnot be included due to constraint in space. We acknowledgeWellcome Trust/DBT India Alliance Intermediate Fellowship(500159-Z-09-Z), DST-SERB Grant (EMR/2015/001946) andLSRB-DRDO Grant (LSRB-31012017-2018) to RM, DST-SERB Grant (EMR/2015/001946) to JC, and JNCASR intra-mural funds.
References
Agosta F., Chio A., Cosottini M., De Stefano N., Falini A.,Mascalchi M. et al. 2010 The present and the future of neu-roimaging in amyotrophic lateral sclerosis.Am. J. Neuroradiol.31, 1769–1777.
Akopian G. J., Barry C., Cepeda and Levine M. S. 2016 Alteredmembrane properties and firing patterns of external globuspallidus neurons in the R6/2 mouse model of Huntington’sdisease. J. Neurosci. Res. 94, 1400–1410.
Amick J. and Ferguson S. M. 2017 C9orf72: at the intersectionof lysosome cell biology and neurodegenerative disease. Traf-fic 18, 267–276.
Anckar J. and Sistonen L. 2011 Regulation of HSF1 functionin the heat stress response: implications in aging and disease.Annu. Rev. Biochem. 80, 1089–1115.
Anden N. E. 1974 Inhibition of the turnover of the braindopamine after treatment with the gammaaminobutyrate:2-oxyglutarate transaminase inhibitor aminooxyacetic acid.Naunyn Schmiedebergs Arch. Pharmacol. 283, 419–424.
Andre V. M., Cepeda C. and Levine M. S. 2010 Dopamine andglutamate in Huntington’s disease: a balancing act.CNSNeu-rosci. Ther. 16, 163–178.
Andre V.M., Cepeda C., Fisher Y. E., HuynhM., BardakjianN.,Singh S. et al 2011 Differential electrophysiological changes instriatal output neurons inHuntington’s disease. J.Neurosci. 31,1170–1182.
Andreassen O. A., Dedeoglu A., Klivenyi P., Beal M. F. andBush A. I. 2000 N-acetyl-L-cysteine improves survival andpreserves motor performance in an animal model of familialamyotrophic lateral sclerosis. Neuroreport 11, 2491–2493.
Arbuthnott G. W., Ingham C. A. and Wickens J. R. 2000Dopamine and synaptic plasticity in the neostriatum. J.Anat. 196, 587–596.
Audrain M., Fol R., Dutar P., Potier B., Billard J. M., FlamentJ. et al. 2016 Alzheimer’s disease-like APP processing in wild-type mice identifies synaptic defects as initial steps of diseaseprogression.Mol. Neurodegener. 11, 5.
Avruch J., Long X., Ortiz-Vega S., Rapley J., Papageorgiou A.and Dai N. 2009 Amino acid regulation of TOR complex 1.Am. J. Physiol. Endocrinol. Metab. 296, E592–E602.
Bah A. and Vergne I. 2017Macrophage autophagy and bacterialinfections. Front Immunol. 8, 1483.
Balch W. E., Morimoto R. I., Dillin A. and Kelly J. W. 2008Adapting proteostasis for disease intervention. Science 319,916–919.
Bagetta V., Ghiglieri V., Sgobio C., Calabresi P. and Picconi B.2010 Synaptic dysfunction in Parkinson’s disease. Biochem.Soc. Trans. 38, 493–497.
Beccano-Kelly D. A., Volta M., Munsie L. N., Paschall S. A.,Tatarnikov I. and Co K. 2015 LRRK2 overexpression altersglutamatergic presynaptic plasticity, striatal dopamine tone,
postsynaptic signal transduction, motor activity and memory.Hum. Mol. Genet. 24, 1336–1349.
Bergman H., Wichmann T., Karmon B. and DeLong M. R.1994 The primate subthalamic nucleus. II. Neuronal activityin theMPTPmodel of Parkinsonism. J. Neurophysiol. 72, 507–520.
Bhide P. G., Day M., Sapp E., Schwarz C., Sheth A., Kim J.et al. 1996 Expression of normal and mutant huntingtin in thedeveloping brain. J. Neurosci. 16, 5523–5535.
Bissler J. J., Kingswood J. C., RadzikowskaE., ZonnenbergB.A.,BelousovaE., FrostM.D. et al. 2017Everolimus long-termusein patients with tuberous sclerosis complex: four-year updateof the EXIST-2 study. PLoS One 12, e0180939.
Blesa J., Trigo-Damas I., Dileone M., Del Rey N. L., Hernan-dez L. F. and Obeso J. A. 2017 Compensatory mechanismsin Parkinson’s disease: circuits adaptations and role in diseasemodification. Exp. Neurol. 298, 148–161.
Bliss T. V. andCollingridgeG. L. 1993A synapticmodel ofmem-ory: long-term potentiation in the hippocampus. Nature 361,31–39.
Botstein D., Chervitz S. A. and Cherry J. M. 1997 Yeast as amodel organism. Science 277, 1259–1260.
Branchi I., Bichler Z., Berger-Sweeney J. and Ricceri L. 2003Animal models of mental retardation: from gene to cognitivefunction. Neurosci. Biobehav. Rev. 27, 141–153.
BrooksK. J.,HillM.D.,HockingsP.D. andReidD.G. 2004MRIdetects early hindlimbmuscle atrophy in Gly93Ala superoxidedismutase-1 (G93A SOD1) transgenic mice, an animal modelof familial amyotrophic lateral sclerosis.NMRBiomed. 17, 28–32.
Brose N., O’Connor V. and Skehel P. 2010 Synaptopathy: dys-function of synaptic function? Biochem. Soc. Trans. 38, 443–444.
Brown Jr. R. H., and Robberecht W. 2001 Amyotrophic lateralsclerosis: pathogenesis. Semin. Neurol. 21, 131–139.
Calabresi P., Maj R., Pisani A., Mercuri N. B. and Bernardi G.1992a Long-term synaptic depression in the striatum: physio-logical and pharmacological characterization. J. Neurosci. 12,4224–4233.
Calabresi P., Pisani A., Mercuri N. B. and Bernardi G. 1992bLong-termPotentiation in the striatum is unmasked by remov-ing the voltage-dependent magnesium block of NMDA recep-tor channels. Eur. J. Neurosci. 4, 929–935.
Calabresi P., Mercuri N. B., Sancesario G. and Bernardi G. 1993Electrophysiology of dopamine-denervated striatal neurons.Implications for Parkinson’s disease. Brain 116, 433–452.
Calabresi P., Centonze D., Gubellini P., Marfia G. A., Pisani A.,SancesarioG. et al. 2000Synaptic transmission in the striatum:fromplasticity toneurodegeneration.Prog.Neurobiol.61, 231–265.
Calautti C., Naccarato M., Jones P. S., Sharma N., Day D. D.,Carpenter A. T. et al. 2007 The relationship between motordeficit and hemisphere activation balance after stroke: A 3TfMRI study. Neuroimage 34, 322–331.
Cardenas C., Miller R. A., Smith I., Bui T., Molgo J., Muller M.et al. 2010 Essential regulation of cell bioenergetics by consti-tutive InsP3 receptorCa2+ transfer tomitochondria.Cell 142,270–283.
Carriedo S.G., Sensi S. L., YinH. Z. andWeiss J. H. 2000AMPAexposures induce mitochondrial Ca(2+) overload and ROSgeneration in spinal motor neurons in vitro. J. Neurosci. 20,240–250.
Centonze D., Battista N., Rossi S., Mercuri N. B., Finazzi-AgroA., Bernardi G. et al. 2004 A critical interaction betweendopamine D2 receptors and endocannabinoids mediates theeffects of cocaine on striatal gabaergic Transmission. Neu-ropsychopharmacology 29, 1488–1497.

Neurodegenerative diseases 695
Cepeda C., Ariano M. A., Calvert C. R., Flores-Hernandez J.,Chandler S. H., Leavitt B. R. et al. 2001 NMDA receptorfunction in mouse models of Huntington disease. J. Neurosci.Res. 66, 525–539.
Cepeda C., Wu N., Andre V. M., Cummings D. M. and LevineM.S. 2007The corticostriatal pathway inHuntington’s disease.Prog. Neurobiol. 81, 253–271.
Cepeda C., Cummings D. M., Andre V. M., Holley S. M. andLevineM. S. 2010 Genetic mouse models of Huntington’s dis-ease: focus on electrophysiological mechanisms.ASNNeuro 2,e00033.
Cha J. H., Kosinski C. M., Kerner J. A., Alsdorf S. A., Mangia-rini L., Davies S. W. et al. 1998 Altered brain neurotransmitterreceptors in transgenic mice expressing a portion of an abnor-mal human huntington disease gene. Proc. Natl. Acad. Sci.USA 95, 6480–6485.
Chamoux E., McManus S., Laberge G., Bisson M. and Roux S.2013 Involvement of kinase PKC-zeta in the p62/p62(P392L)-driven activation of NF-kappaB in human osteoclasts.Biochim. Biophys. Acta 1832, 475–484.
Chan E. Y., Longatti A., McKnight N. C. and Tooze S. A. 2009Kinase-inactivated ULK proteins inhibit autophagy via theirconserved C-terminal domains using an Atg13-independentmechanism.Mol. Cell. Biol. 29, 157–171.
Chang S., Bray S. M., Li Z., Zarnescu D. C., He C., Jin P.et al. 2008 Identification of small molecules rescuing fragileX syndrome phenotypes in Drosophila. Nat. Chem. Biol. 4,256–263.
Chauhan S., Goodwin J. G., Chauhan S., Manyam G., Wang J.,KamatA.M. et al. 2013ZKSCAN3 is amaster transcriptionalrepressor of autophagy. Mol. Cell 50, 16–28.
Chen S., Sayana P., Zhang X. and Le W. 2013 Genetics of amy-otrophic lateral sclerosis: an update. Mol. Neurodegener. 8,28.
Chung K., Crane M. M. and Lu H. 2008 Automated on-chiprapid microscopy, phenotyping and sorting ofC. elegans.Nat.Methods 5, 637–643.
Chung C. Y., Khurana V., Auluck P. K., Tardiff D. F., Maz-zulli J. R., Soldner F. et al. 2013 Identification and rescue ofalpha-synuclein toxicity in Parkinson patient-derived neurons.Science 342, 983–987.
Citri A. and Malenka R. C. 2008 Synaptic plasticity: multipleforms, functions, and mechanisms. Neuropsychopharmacol-ogy 33, 18–41.
Cleveland D. W. and Rothstein J. D. 2001 From Charcot to LouGehrig: deciphering selectivemotor neuron death inALS.Nat.Rev. Neurosci. 2, 806–819.
Cooper A. A., Gitler A. D., Cashikar A., Haynes C. M., HillK. J., Bhullar B. et al. 2006 Alpha-synuclein blocks ER-Golgitraffic and Rab1 rescues neuron loss in Parkinson’s models.Science 313, 324–328.
Cortes C. J., Ling S. C., Guo L. T., Hung G., Tsunemi T.,Ly. L. et al. 2014 Muscle expression of mutant androgenreceptor accounts for systemic and motor neuron disease phe-notypes in spinal and bulbar muscular atrophy. Neuron 82,295–307.
Coyle J. T. and Schwarcz R. 1976 Lesion of striatal neuroneswith kainic acid provides a model for Huntington’s chorea.Nature 263, 244–246.
Crawley J. N. 2008 Behavioral phenotyping strategies for mutantmice. Neuron 57, 809–818.
Cummings D. M., Cepeda C. and Levine M. S. 2010 Alterationsin striatal synaptic transmission are consistent across geneticmouse models of Huntington’s disease.ASNNeuro. 2, e00036.
Cunha-Santos J., Duarte-Neves J., Carmona V., Guarente L.,Pereira de Almeida L. and Cavadas C. 2016 Caloric restrictionblocks neuropathology andmotor deficits inMachado-Joseph
disease mouse models through SIRT1 pathway. Nat. Com-mun. 7, 11445.
Danysz W. and Parsons C. G. 2012 Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine—searching for the connections. Br. J. Pharmacol. 167, 324–352.
DeDuveC. andWattiauxR. 1966Functions of lysosomes.Annu.Rev. Physiol. 28, 435–492.
De Strooper B., Saftig P., Craessaerts K., Vanderstichele H.,Guhde G., Annaert W. et al. 1998 Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein.Nature 391, 387–390.
Di Bartolomeo S., Corazzari M., Nazio F., Oliverio S., LisiG., Antonioli M. et al. 2010 The dynamic interaction ofAMBRA1 with the dynein motor complex regulates mam-malian autophagy. J. Cell Biol. 191, 155–168.
DiFigliaM. 1997Clinical genetics, II.Huntington’s disease: fromthe gene to pathophysiology. Am. J. Psychiatry 154, 1046.
Dihanich S. and C. Manzoni 2011 LRRK2: a problem lurkingin vesicle trafficking? J. Neurosci. 31, 9787–9788.
Doble A. 1999 The role of excitotoxicity in neurodegenerativedisease: implications for therapy. Pharmacol. Ther. 81, 163–221.
Ejlerskov P., Hultberg J. G., Wang J., Carlsson R., Ambjorn M.,KussM. et al. 2015Lackof neuronal IFN-beta-IFNARcauseslewy body- and Parkinson’s disease-like dementia. Cell 163,324–339.
ElrickM. J., YuT., ChungC. andLiebermanA. P. 2012 Impairedproteolysis underlies autophagic dysfunction inNiemann-picktype C disease. Hum. Mol. Genet. 21, 4876–4887.
Esler W. P. and Wolfe M. S. 2001 A portrait of Alzheimersecretases–new features and familiar faces. Science 293, 1449–1454.
EspositoG.,AnaClaraF. andVerstrekenP. 2012 Synaptic vesicletrafficking and Parkinson’s disease. Dev. Neurobiol. 72, 134–144.
Fecto F., Yan J., Vemula S. P., Liu E., YangY., ChenW. et al. 2011SQSTM1mutations in familial and sporadic amyotrophic lat-eral sclerosis. Arch Neurol. 68, 1440–1446.
Feldmeyer D., Kask K., Brusa R., Kornau H. C., Kolhekar R.,RozovA. et al.1999Neurological dysfunctions inmice express-ing different levels of the Q/R site-unedited AMPAR subunitGluR-B. Nat. Neurosci. 2, 57–64.
FerrucciM., Fulceri F., Toti L., Soldani P., SicilianoG., PaparelliA. et al. 2011 Protein clearing pathways in ALS. Arch. Ital.Biol. 149, 121–149.
Filimonenko M., Isakson P., Finley K. D., Anderson M., JeongH., Melia T. J. et al. 2010 The selective macroautophagicdegradation of aggregated proteins requires the PI3P-bindingprotein Alfy. Mol. Cell 38, 265–279.
Fischer L. R. and Glass J. D. 2010 Oxidative stress induced byloss of Cu, Zn-superoxide dismutase (SOD1) or superoxide-generating herbicides causes axonal degeneration in mouseDRG cultures. Acta Neuropathol. 119, 249–259.
Fischer L. R., Culver D. G., Tennant P., Davis A. A., Wang M.,Castellano-Sanchez A. 2004 Amyotrophic lateral sclerosis is adistal axonopathy: evidence inmice andman.Exp.Neurol.185,232–240.
FitzjohnS.M.,MortonR.A.,KuenziF.,RosahlT.W., ShearmanM., Lewis H. et al. 2001 Age-related impairment of synaptictransmission but normal long-term potentiation in trans-genic mice that overexpress the human APP695SWE mutantform of amyloid precursor protein. J. Neurosci. 21, 4691–4698.
Fonnum F., Storm-Mathisen J. and Divac I. 1981 Biochemicalevidence for glutamate as neurotransmitter in corticostriataland corticothalamic fibres in rat brain. Neuroscience 6, 863–873.

696 S. N. Suresh et al.
Gardoni F., Picconi B., Ghiglieri V., Polli F., Bagetta V., BernardiG. et al. 2006 A critical interaction between NR2B andMAGUK in L-DOPA induced dyskinesia. J. Neurosci. 26,2914–2922.
Geisler S., HolmstromK.M., SkujatD., Fiesel F. C., Rothfuss O.C., Kahle P. J. et al. 2010 PINK1/Parkin-mediated mitophagyis dependent on VDAC1 and p62/SQSTM1.Nat. Cell Biol. 12,119–131.
Geng J. and Klionsky D. J. 2010 The Golgi as a potential mem-brane source for autophagy. Autophagy 6, 950–951.
Geracitano R., Paolucci E., Prisco S., Guatteo E., Zona C., Lon-gone P. et al. 2003 Altered long-term corticostriatal synapticplasticity in transgenic mice overexpressing human CU/ZNsuperoxide dismutase (GLY(93)–> ALA) mutation. Neuro-science 118, 399–408.
Goldberg A. L. 2003 Protein degradation and protection againstmisfolded or damaged proteins. Nature 426, 895–899.
Gonatas N. K., Anderson W. and Evangelista I. 1967 The con-tribution of altered synapses in the senile plaque: an electronmicroscopic study in Alzheimer’s dementia. J. Neuropathol.Exp. Neurol. 26, 25–39.
Gorrie G. H., Fecto F., Radzicki D., Weiss C., Shi Y., Dong H.et al. 2014 Dendritic spinopathy in transgenic mice express-ingALS/dementia-linkedmutantUBQLN2.Proc. Natl. Acad.Sci. USA 111, 14524–14529.
Grabowska, M. and Michaluk J. 1974 On the role of serotoninin apomorphine-induced locomotor stimulation in rats. Phar-macol. Biochem. Behav. 2, 263–266.
Graham R. K., Pouladi M. A., Joshi P., Lu G., Deng Y., Wu N.P. et al. 2009 Differential susceptibility to excitotoxic stress inYAC128 mouse models of Huntington disease between initia-tion and progression of disease. J. Neurosci. 29, 2193–2204.
Greengard P., NairnA. C., Girault J. A., Ouimet C. C., SnyderG.L., FisoneG. et al. 1998 TheDARPP-32/protein phosphatase-1 cascade: a model for signal integration.Brain Res. Brain Res.Rev. 26, 274–284.
Haack T. B., Hogarth P., Gregory A., Prokisch H. and HayflickS. J. 2013 BPAN: the only X-linked dominant NBIA disorder.Int. Rev. Neurobiol. 110, 85–90.
Haber S. N. and Nauta W. J. 1983 Ramifications of the globuspallidus in the rat as indicated by patterns of immunohisto-chemistry. Neuroscience 9, 245–260.
Hadano S., Hand C. K., Osuga H., Yanagisawa Y., Otomo A.,Devon R. S. et al. 2001 A gene encoding a putative GTPaseregulator is mutated in familial amyotrophic lateral sclerosis2. Nat. Genet. 29, 166–173.
Hamasaki M., Furuta N., Matsuda A., Nezu A., YamamotoA., Fujita N. et al. 2013 Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393.
Hardingham G. E., Fukunaga Y. and Bading H. 2002 Extrasy-naptic NMDARs oppose synaptic NMDARs by triggeringCREB shut-off and cell death pathways.Nat. Neurosci. 5, 405–414.
Harnett M. M., Pineda M. A., Latre de Late P., Eason R. J.,Besteiro S., Harnett W. et al. 2017 From Christian de Duveto Yoshinori Ohsumi: more to autophagy than just dining athome. Biomed. J. 40, 9–22.
Harper P. S. 1996New genes for old diseases: the molecular basisof myotonic dystrophy and Huntington’s disease. The Lum-leian lecture 1995. J. R. Coll. Phys. Lond. 30, 221–231.
Hassani O. K., Mouroux M. and Feger J. 1996 Increased sub-thalamic neuronal activity after nigral dopaminergic lesionindependent of disinhibition via the globus pallidus. Neuro-science 72, 105–115.
Hassan B., Akcakanat A., Holder A. M. andMeric-Bernstam F.2013 Targeting the PI3-kinase/Akt/mTOR signaling pathway.Surg. Oncol. Clin. N. Am. 22, 641–664.
Heath P. R. and Shaw P. J. 2002 Update on the glutamater-gic neurotransmitter system and the role of excitotoxic-ity in amyotrophic lateral sclerosis. Muscle Nerve 26, 438–458.
Herrera-Marschitz M., Loidl C. F., You Z. B., Andersson K.,Silveira R., O’Connor W. T. et al. 1994 Neurocircuitry of thebasal ganglia studied by monitoring neurotransmitter release.Effects of intracerebral and perinatal asphyctic lesions. Mol.Neurobiol. 9, 171–82.
Hetz C. 2012 The unfolded protein response: controlling cell fatedecisions under ER stress and beyond. Nat. Rev. Mol. CellBiol. 13, 89–102.
Hetz C. and Mollereau B. 2014 Disturbance of endoplasmicreticulumproteostasis in neurodegenerative diseases.Nat. Rev.Neurosci. 15, 233–249.
Higgins G. A. and H. Jacobsen 2003 Transgenic mouse mod-els of Alzheimer’s disease: phenotype and application. Behav.Pharmacol. 14, 419–438.
Hollister A. S., Breese G. R., Kuhn C. M., Cooper B. R. andSchanberg S. M. 1976 An inhibitory role for brain serotonin-containing systems in the locomotor effects of d-amphetamine.J. Pharmacol. Exp. Ther. 198, 12–22.
Hsia A. Y., Masliah E., McConlogue L., Yu G. Q., Tatsuno G.,Hu K. et al. 1999 Plaque-independent disruption of neuralcircuits inAlzheimer’s diseasemousemodels.Proc.Natl. Acad.Sci. USA 96, 3228–3233.
Hyttinen J. M., Amadio M., Viiri J., Pascale A., Salminen A.and K. Kaarniranta 2014 Clearance of misfolded and aggre-gated proteins by aggrephagy and implications for aggregationdiseases. Ageing Res. Rev. 18, 16–28.
Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y.,IshiharaN. et al. 2000Aubiquitin-like systemmediates proteinlipidation. Nature 408, 488–492.
Isakson P., Holland P. and Simonsen A. 2013 The role of ALFYin selective autophagy. Cell Death Differ. 20, 12–20.
ItoH.,NakamuraM.,KomureO.,AyakiT.,WateR.,MaruyamaH. et al. 2011 Clinicopathologic study on an ALS familywith a heterozygous E478G optineurin mutation. Acta Neu-ropathol. 122, 223–229.
Jaber N. and Zong W. X. 2013 Class III PI3K Vps34: essentialroles in autophagy, endocytosis, and heart and liver function.Ann. N. Y. Acad. Sci. 1280, 48–51.
Jahrling J. B. and Laberge R.M. 2015Age-related neurodegener-ation prevention through mTOR inhibition: potential mech-anisms and remaining questions. Curr. Top. Med. Chem. 15,2139–2151.
Jimenez-Sanchez M., Thomson F., Zavodszky E. and Rubin-szteinD.C. 2012Autophagy andpolyglutamine diseases.Prog.Neurobiol. 97, 67–82.
Jimenez-SanchezM.,LamW.,HannusM., SonnichsenB., Imari-sioS.,FlemingA. et al.2015 siRNAscreen identifiesQPCTasadruggable target forHuntington’s disease.Nat. Chem. Biol. 11,347–354.
Jones A. L., Wood J. D. and Harper P. S. 1997 Huntington dis-ease: advances in molecular and cell biology. J. Inherit. Metab.Dis. 20, 125–138.
Joshi P. R., Wu N. P., Andre V. M., Cummings D. M., CepedaC., Joyce J. A. et al. 2009 Age-dependent alterations of corti-costriatal activity in the YAC128 mouse model of Huntingtondisease. J. Neurosci. 29, 2414–2427.
Julien J. P. 2001 Amyotrophic lateral sclerosis. Unfolding the tox-icity of the misfolded. Cell 104, 581–591.
Kachaner D., Genin P., Laplantine E. and Weil R. 2012 Towardan integrative view of Optineurin functions. Cell Cycle 11,2808–2818.
Kachroo A. H., Laurent J. M., Yellman C. M., Meyer A. G.,Wilke C. O. and Marcotte E. M. 2015 Evolution systematic

Neurodegenerative diseases 697
humanization of yeast genes reveals conserved functions andgenetic modularity. Science 348, 921–925.
Kambe Y., Nakamichi N., Takarada T., Fukumori R., NakazatoR., Hinoi E. et al. 2011 A possible pivotal role of mitochon-drial free calcium in neurotoxicity mediated by N-methyl-d-aspartate receptors in cultured rat hippocampal neurons.Neurochem. Int. 59, 10–20.
Kampinga H. H. and Craig E. A. 2010 The HSP70 chaperonemachinery: J proteins as drivers of functional specificity. Nat.Rev. Mol. Cell Biol. 11, 579–592.
Kanning K. C., Kaplan A. and Henderson C. E. 2010 Motorneuron diversity in development and disease. Annu. Rev. Neu-rosci. 33, 409–440.
Karuppagounder S. S., Brahmachari S., Lee Y., Dawson V. L.,DawsonT.M. andKoH. S. 2014The c-Abl inhibitor, nilotinib,protects dopaminergic neurons in a preclinical animal modelof Parkinson’s disease. Sci. Rep. 4, 4874.
Kaushik S., Rodriguez-Navarro J. A., Arias E., Kiffin R., SahuS., Schwartz G. J. et al. 2011 Autophagy in hypothalamicAgRP neurons regulates food intake and energy balance. CellMetab. 14, 173–183.
KhanS.H. andKumarR. 2012Roleof an intrinsicallydisorderedconformation in AMPK-mediated phosphorylation of ULK1and regulation of autophagy.Mol. Biosyst. 8, 91–96.
Khedraki A., Reed E. J., Romer S. H., Wang Q., Romine W.,Rich M. M. et al. 2017 Depressed synaptic transmission andreduced vesicle release sites inHuntington’s disease neuromus-cular junctions. J. Neurosci. 37, 8077–8091.
Khurana V. and Lindquist S. 2010Modelling neurodegenerationin Saccharomyces cerevisiae: why cookwith baker’s yeast?Nat.Rev. Neurosci. 11, 436–449.
Kiernan M. C., Vucic S., Cheah B. C., Turner M. R., EisenA., Hardiman O. et al. 2011 Amyotrophic lateral sclerosis.Lancet 377, 942–955.
Kihara T. and Shimohama S. 2004 Alzheimer’s disease andacetylcholine receptors. Acta Neurobiol. Exp. 64, 99–105.
Kim E., Goraksha-Hicks P., Li L., Neufeld T. P., and Guan K.L. 2008a Regulation of TORC1 by Rag GTPases in nutrientresponse. Nat. Cell Biol. 10, 935–945.
Kim M., Sandford E., Gatica D., Qiu Y., Liu X., Zheng Y. et al.2016Mutation inATG5 reduces autophagy and leads to ataxiawith developmental delay. Elife 5, pii: e12245.
KimP.K.,HaileyD.W.,MullenR.T. andLippincott-Schwartz J.2008b Ubiquitin signals autophagic degradation of cytosolicproteins and peroxisomes. Proc. Natl. Acad. Sci. USA 105,20567–20574.
Kim J., Hughes E. G., Shetty A. S., Arlotta P., Goff L. A.,Bergles D. E. and Brown S. P. 2017 Changes in the excitabil-ity of neocortical neurons in a mouse model of amyotrophiclateral sclerosis are not specific to corticospinal neurons andare modulated by advancing disease. J. Neurosci. 37, 9037–9053.
Kirkin V., McEwan D. G., Novak I. and Dikic I. 2009 A role forubiquitin in selective autophagy.Mol. Cell 34, 259–269.
Kiskinis E., Sandoe J., Williams L. A., Boulting G. L., MocciaR.,Wainger B. J. et al. 2014 Pathways disrupted in humanALSmotor neurons identified through genetic correction ofmutantSOD1. Cell Stem Cell 14, 781–795.
Kreitzer A. C. and Malenka R. C. 2005 Dopamine modulationof state-dependent endocannabinoid release and long-termdepression in the striatum. J. Neurosci. 25, 10537–10545.
Klapstein G. J., Fisher R. S., Zanjani H., Cepeda C., Jokel E. S.,Chesselet M. F. et al. 2001 Electrophysiological and morpho-logical changes in striatal spiny neurons in R6/2 Huntington’sdisease transgenic mice. J. Neurophysiol. 86, 2667–2677.
Kolodziejczyk K. and Raymond L. A. 2016 Differential changesin thalamic and cortical excitatory synapses onto striatal spiny
projectionneurons in aHuntingtondiseasemousemodel.Neu-robiol. Dis. 86, 62–74.
Kostic V., Jackson-Lewis V., de Bilbao F., Dubois-Dauphin M.and Przedborski S. 1997 Bcl-2: prolonging life in a transgenicmouse model of familial amyotrophic lateral sclerosis. Sci-ence 277, 559–562.
Kovacs K. A., Steullet P., Steinmann M., Do K. Q., MagistrettiP. J., Halfon O. et al. 2007 TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity.Proc. Natl. Acad. Sci. USA 104, 4700–4705.
Kroemer G. and Jaattela M. 2005 Lysosomes and autophagy incell death control. Nat. Rev. Cancer 5, 886–897.
Kumar D. R., Aslinia F., Yale S. H. and Mazza J. J. 2011 Jean-Martin charcot: the father of neurology. Clin. Med. Res. 9,46–49.
Kuner R., Groom A. J., Bresink I., Kornau H. C., Stefovska V.,MullerG. et al. 2005Late-onsetmotoneuron disease caused bya functionally modified AMPA receptor subunit. Proc. Natl.Acad. Sci. USA 102, 5826–5831.
Labbadia J. and Morimoto R. I. 2015 Repression of the heatshock response is a programmed event at the onset of repro-duction.Mol. Cell 59, 639–650.
LaFerlaF.M.andGreenK.N. 2012Animalmodels ofAlzheimerdisease. Cold Spring Harb. Perspect Med. 2, pii: a006320
Lepeta K., LourencoM. V., Schweitzer B. C., Martino Adami P.V., Banerjee P., Catuara-Solarz S. et al. 2016 Synaptopathies:synaptic dysfunction in neurological disorders—a review fromstudents to students. J. Neurochem. 138, 785–805.
Lesage S. andBriceA. 2009 Parkinson’s disease: frommonogenicforms to genetic susceptibility factors. Hum. Mol. Genet. 18,R48–59.
LevineM. S., CepedaC., HickeyM.A., Fleming S.M. andChes-selet M. F. 2004 Genetic mouse models of Huntington’s andParkinson’s diseases: illuminating but imperfect. Trends Neu-rosci. 27, 691–697.
Levine M. S., Klapstein G. J., Koppel A., Gruen E., Cepeda C.,Vargas M. E. et al. 1999 Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockinmousemodels of Huntington’s disease. J. Neurosci. Res. 58, 515–532.
Li L., Zviti R., Ha C., Wang Z. V., Hill J. A. and Lin F. 2017ForkheadboxO3 (FoxO3) regulates kidney tubular autophagyfollowing urinary tract obstruction. J. Biol. Chem. 292, 13774–13783.
LiX. J. 1999The early cellular pathologyofHuntington’s disease.Mol. Neurobiol. 20, 111–124.
Lim J. and Yue Z. 2015 Neuronal aggregates: formation, clear-ance, and spreading. Dev. Cell 32, 491–501.
Lipton S. A. and Rosenberg P. A. 1994 Excitatory amino acidsas a final common pathway for neurologic disorders. N. Engl.J. Med. 330, 613–622.
Lorenz M. C. and Heitman J. 1995 TOR mutations conferrapamycin resistance by preventing interaction with FKBP12-rapamycin. J. Biol. Chem. 270, 27531–27537.
Lovinger D. M., Tyler E. C. and Merritt A. 1993 Short- andlong-term synaptic depression in rat neostriatum. J. Neuro-physiol. 70, 1937–1949.
Lu K., Psakhye I. and Jentsch S. 2014 Autophagic clearance ofpolyQ proteins mediated by ubiquitin-Atg8 adaptors of theconserved CUET protein family. Cell 158, 549–563.
Luheshi L. M., Tartaglia G. G., Brorsson A. C., Pawar A. P.,Watson I. E., Chiti F. et al. 2007 Systematic in vivo analysisof the intrinsic determinants of amyloid Beta pathogenicity.PLoS Biol. 5, e290.
Lundblad M., Picconi B., Lindgren H. and Cenci M. A. 2004 Amodel of L-DOPA-induced dyskinesia in 6-hydroxydopamine

698 S. N. Suresh et al.
lesioned mice: relation to motor and cellular parameters ofnigrostriatal function. Neurobiol. Dis. 16, 110–123.
Ma N., Liu Q., Zhang L., Henske E. P. and Ma Y. 2013 TORC1signaling is governedby twonegative regulators infissionyeast.Genetics 195, 457–468.
Maday S. and Holzbaur E. L. 2014 Autophagosome biogenesisin primary neurons follows an ordered and spatially regulatedpathway. Dev. Cell 30, 71–85.
Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A.,Hetherington C. et al. 1996 Exon 1 of the HD gene with anexpanded CAG repeat is sufficient to cause a progressive neu-rological phenotype in transgenic mice. Cell 87, 493–506.
Maj J., Pawlowski L. and Sarnek J. 1974 The role of brain 5-hydroxytryptamine in the central action of L-DOPA. Adv.Biochem. Psychopharmacol. 10, 253–256.
Martorana A. and Koch G. 2014 Is dopamine involved inAlzheimer’s disease? Front. Aging Neurosci. 6, 252.
Martinez-Vicente M., Talloczy Z., Wong E., Tang G., Koga H.,Kaushik S. et al. 2010 Cargo recognition failure is responsiblefor inefficient autophagy in Huntington’s disease. Nat. Neu-rosci. 13, 567–576.
Martire A., Pepponi R., Domenici M. R., Ferrante A., Chiodi V.and Popoli P. 2013 BDNF prevents NMDA-induced toxicityin models of Huntington’s disease: the effects are genotypespecific and adenosine A2A receptor is involved. J. Neu-rochem. 125, 225–235.
Maselli R. A., Wollman R. L., Leung C., Distad B., PalombiS. and Richman D. P. 1993 Neuromuscular transmissionin amyotrophic lateral sclerosis. Muscle Nerve 16, 1193–1203.
Masliah E., Terry R. D., DeTeresa R. M. and Hansen L. A.1989 Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci.Lett. 103, 234–239.
McGeer P. L., McGeer E. G., Scherer U. and Singh K. 1977 Aglutamatergic corticostriatal path? Brain Res. 128, 369–373.
Menalled L. and Brunner D. 2014 Animal models of Hunting-ton’s disease for translation to the clinic: best practices. Mov.Disord. 29, 1375–1390.
Menalled L. B. andChesseletM. F. 2002Mousemodels ofHunt-ington’s disease. Trends Pharmacol. Sci. 23, 32–39.
Mhyre T. R., Boyd J. T., Hamill R. W. and Maguire-Zeiss K. A.2012 Parkinson’s disease. Subcell Biochem. 65, 389–455.
Miller B. R., Walker A. G., Shah A. S., Barton S. J. and RebecG. V. 2008 Dysregulated information processing by mediumspiny neurons in striatum of freely behaving mouse models ofHuntington’s disease. J. Neurophysiol. 100, 2205–2216.
MilnerwoodA. J., CummingsD.M.,DalleracG.M., Brown J.Y.,Vatsavayai S. C., Hirst M. C. et al. 2006 Early development ofaberrant synaptic plasticity in a mouse model of Huntington’sdisease. Hum. Mol. Genet. 15, 1690–1703.
Milnerwood A. J., Gladding C. M., Pouladi M. A., KaufmanA. M., Hines R. M., Boyd J. D. et al. 2010 Early increasein extrasynaptic NMDA receptor signaling and expressioncontributes to phenotype onset in Huntington’s disease mice.Neuron 65, 178–190.
Milnerwood A. J. and Raymond L. A. 2007 Corticostriatalsynaptic function in mouse models of Huntington’s disease:early effects of huntingtin repeat length and protein load. J.Physiol. 585, 817–831.
MilnerwoodA. J. andRaymondL.A. 2010Early synaptic patho-physiology in neurodegeneration: insights from Huntington’sdisease. Trends Neurosci. 33, 513–523.
Mizushima N. 2010 The role of the Atg1/ULK1 complex inautophagy regulation. Curr. Opin. Cell Biol. 22, 132–139.
Mizushima N., Yamamoto A., Matsui M., Yoshimori T. andOhsumi Y. 2004 In vivo analysis of autophagy in response to
nutrient starvation using transgenic mice expressing a fluores-cent autophagosome marker.Mol. Biol. Cell 15, 1101–1111.
Murphy K. P., Carter R. J., Lione L. A., Mangiarini L., MahalA., Bates G. P. et al. 2000 Abnormal synaptic plasticity andimpaired spatial cognition in mice transgenic for exon 1 of thehuman Huntington’s disease mutation. J. Neurosci. 20, 5115–5123.
Murray L. M., Talbot K. and Gillingwater T. H. 2010 Review:neuromuscular synaptic vulnerability in motor neurone dis-ease: amyotrophic lateral sclerosis and spinal muscular atro-phy. Neuropathol. Appl. Neurobiol. 36, 133–156.
Murer M. G., Tseng K. Y., Kasanetz F., Belluscio M. andRiquelme L. A. 2002 Brain oscillations, medium spiny neu-rons, and dopamine. Cell Mol. Neurobiol. 22, 611–632.
Mymrikov E. V., Daake M., Richter B., Haslbeck M. and Buch-ner J. 2017 The chaperone activity and substrate spectrum ofhuman small heat shock proteins. J. Biol. Chem. 292, 672–684.
Nagae T., Araki K., Shimoda Y., Sue L. I., Beach T. G. andKonishi Y. 2016 Cytokines and cytokine receptors Involved inthe pathogenesis of Alzheimer’s disease. J. Clin. Cell. Immunol.. 7, 441.
NarayanP., Ehsani S. andLindquist S. 2014Combating neurode-generative disease with chemical probes and model systems.Nat. Chem. Biol. 10, 911–920.
Nasir J., Floresco S. B., O’Kusky J. R., Diewert V. M., RichmanJ.M., Zeisler J. et al. 1995 Targeted disruption of the Hunting-ton’s disease gene results in embryonic lethality and behavioralandmorphological changes in heterozygotes.Cell 81, 811–823.
Nicholls D. G., Johnson-Cadwell L., Vesce S., Jekabsons M. andYadavaN. 2007Bioenergetics ofmitochondria in culturedneu-rons and their role in glutamate excitotoxicity. J. Neurosci.Res. 85, 3206–3212.
Nistico R., Pignatelli M., Piccinin S., Mercuri N. B. andCollingridge G. 2012 Targeting synaptic dysfunction inAlzheimer’s disease therapy.Mol. Neurobiol. 46, 572–587.
Nitsch C. and R. Riesenberg 1995 Synaptic reorganisationin the rat striatum after dopaminergic deafferentation: anultrastructural study using glutamate decarboxylase immuno-cytochemistry. Synapse 19, 247–263.
Nixon R. A. 2004 Niemann-Pick type C disease and Alzheimer’sdisease: the APP-endosome connection fattens up. Am. J.Pathol. 164, 757–761.
Nixon R. A. 2013 The role of autophagy in neurodegenerativedisease. Nat. Med. 19, 983–997.
Nizzardo M., Simone C., Rizzo F., Ulzi G., Ramirez A., RizzutiM. et al. 2016Morpholino-mediated SOD1 reduction amelio-rates an amyotrophic lateral sclerosis disease phenotype. Sci.Rep. 6, 21301.
Nussbaum-KrammerC. I. andMorimotoR. I. 2014Caenorhab-ditis elegans as a model system for studying non-cell-autonomous mechanisms in protein-misfolding diseases. Dis.Model Mech. 7, 31–39.
Otomo A., Kunita R., Suzuki-Utsunomiya K., Ikeda J. E. andHadano S. 2011 Defective relocalization of ALS2/alsin mis-sense mutants to Rac1-induced macropinosomes accounts forloss of their cellular function and leads to disturbed amphi-some formation. FEBS Lett. 585, 730–736.
Outeiro T. F. and Lindquist S. 2003 Yeast cells provide insightinto alpha-synuclein biology and pathobiology. Science 302,1772–1775.
Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A.,OutzenH. et al. 2007p62/SQSTM1binds directly toAtg8/LC3to facilitate degradation of ubiquitinated protein aggregates byautophagy. J. Biol. Chem. 282, 24131–24145.
Paoletti P., Vila I., Rife M., Lizcano J. M., Alberch J. andGines S. 2008 Dopaminergic and glutamatergic signalingcrosstalk in Huntington’s disease neurodegeneration: the role

Neurodegenerative diseases 699
of p25/cyclin-dependent kinase 5. J. Neurosci. 28, 10090–10101.
Palma E., Reyes-Ruiz J. M., Lopergolo D., Roseti C., BertolliniC., Ruffolo G. et al. 2016 Acetylcholine receptors from humanmuscle as pharmacological targets forALS therapy.Proc.Natl.Acad. Sci. USA 113, 3060–3065.
Pardo C. A., Xu Z., Borchelt D. R., Price D. L., Sisodia S. S. andCleveland D. W. 1995 Superoxide dismutase is an abundantcomponent in cell bodies, dendrites, and axons of motor neu-rons and in a subset of other neurons. Proc. Natl. Acad. Sci.USA 92, 954–958.
Paul P. and de Belleroche J. 2014 The role of D-serine and glycineas co-agonists of NMDA receptors inmotor neuron degenera-tion and amyotrophic lateral sclerosis (ALS). Front. Synaptic.Neurosci. 6, 10.
Paulsen J. S., Langbehn D. R., Stout J. C., Aylward E., RossC. A., Nance M. et al. 2008 Detection of Huntington’s dis-ease decades before diagnosis: the predict-HD study. J. Neurol.Neurosurg. Psychiatry 79, 874–880.
Pehar M., Vargas M. R., Cassina P., Barbeito A. G., Beckman J.S. and Barbeito L. 2005 Complexity of astrocyte-motor neu-ron interactions in amyotrophic lateral sclerosis.Neurodegener.Dis. 2, 139–146.
Peng T. I., Jou M. J., Sheu S. S. and Greenamyre J. T. 1998 Visu-alization of NMDA receptor-induced mitochondrial calciumaccumulation in striatal neurons. Exp. Neurol. 149, 1–12.
Pouladi M. A., Graham R. K., Karasinska J. M., Xie Y., San-tos R. D., Petersen A. et al. 2009 Prevention of depressivebehaviour in the YAC128 mouse model of Huntington dis-ease by mutation at residue 586 of huntingtin. Brain 132, 919–932.
Powers E. T., Morimoto R. I., Dillin A., Kelly J. W. and BalchW. E. 2009 Biological and chemical approaches to diseases ofproteostasis deficiency. Annu. Rev. Biochem. 78, 959–991.
Pozueta J., Lefort R. and ShelanskiM. L. 2013 Synaptic changesinAlzheimer’s disease and itsmodels.Neuroscience 251, 51–65.
Puzzo D., Piacentini R., Fa M., Gulisano W., Li Puma D. D.,Staniszewski A. et al. 2017 LTP and memory impairmentcaused by extracellular Abeta and Tau oligomers is APP-dependent. Elife 6, pii: e26991.
Pyo J. O., Yoo S. M., Ahn H. H., Nah J., Hong S. H., Kam T. I.et al. 2013Overexpression ofAtg5 inmice activates autophagyand extends lifespan. Nat. Commun. 4, 2300.
Radzicki D., Liu E., Deng H. X., Siddique T. and Martina M.2016 Early impairment of synaptic and intrinsic excitabilityin mice expressing ALS/Dementia-linked mutant UBQLN2.Front Cell Neurosci. 10, 216.
Rajasekhar K., Suresh S. N., Manjithaya R. and Govindaraju T.2015Rationally designed peptidomimeticmodulators of abetatoxicity in Alzheimer’s disease. Sci. Rep. 5, 8139.
Ravikumar B., Moreau K., Jahreiss L., Puri C. and RubinszteinD. C. 2010 Plasma membrane contributes to the formation ofpre-autophagosomal structures. Nat. Cell Biol. 12, 747–757.
Raychaudhuri S., Loew C., Korner R., Pinkert S., Theis M.,Hayer-Hartl M. et al. 2014 Interplay of acetyltransferaseEP300 and the proteasome system in regulating heat shocktranscription factor 1. Cell 156, 975–985.
Ritz D., Vuk M., Kirchner P., Bug M., Schutz S., Hayer A. et al.2011 Endolysosomal sorting of ubiquitylated caveolin-1 is reg-ulated by VCP and UBXD1 and impaired by VCP diseasemutations. Nat. Cell Biol. 13, 1116–1123.
Rinne U. K., Rinne J. O., Rinne J. K., Laakso K. and LonnbergP. 1984 Brain neurotransmitters and neuropeptides in Parkin-son’s disease. Acta. Physiol. Pharmacol. Latinoam. 34, 287–299.
Rizzoli S. O. 2014 Synaptic vesicle recycling: steps and principles.EMBO J. 33, 788–822.
Rosen D. R. 1993 Mutations in Cu/Zn superoxide dismutasegene are associated with familial amyotrophic lateral sclero-sis. Nature 364, 362.
Rouse S. T., Marino M. J., Bradley S. R., Awad H., WittmannM. and Conn P. J. 2000Distribution and roles of metabotropicglutamate receptors in the basal gangliamotor circuit: implica-tions for treatmentofParkinson’s disease and relateddisorders.Pharmacol. Ther. 88, 427–435.
Rowan M. J., Klyubin I., Cullen W. K. and Anwyl R. 2003Synaptic plasticity in animal models of early Alzheimer’sdiseasePhilos. Trans. R. Soc. Lond. B Biol. Sci. 358, 821–828.
Ryan S. D., Dolatabadi N., Chan S. F., Zhang X., Akhtar M.W., Parker J. et al. 2013 Isogenic human iPSC Parkinson’smodel shows nitrosative stress-induced dysfunction inMEF2-PGC1alpha transcription. Cell 155, 1351–1364.
Saitsu H., Nishimura T., Muramatsu K., Kodera H., KumadaS., Sugai K. et al. 2013 De novo mutations in the autophagygene WDR45 cause static encephalopathy of childhood withneurodegeneration in adulthood. Nat. Genet. 45, 445–449,449e1.
Sanelli T., Ge W., Leystra-Lantz C. and Strong M. J. 2007Calcium mediated excitotoxicity in neurofilament aggregate-bearing neurons in vitro is NMDA receptor dependant. J.Neurol. Sci. 256, 39–51.
Sarkar S.,Davies J. E.,HuangZ., TunnacliffeA. andRubinszteinD.C. 2007aTrehalose, a novelmTOR-independent autophagyenhancer, accelerates the clearance of mutant huntingtin andalpha-synuclein. J. Biol. Chem. 282, 5641–5652.
Sarkar S., Perlstein E. O., Imarisio S., Pineau S., CordenierA., Maglathlin R. L. et al. 2007b Small molecules enhanceautophagy and reduce toxicity in Huntington’s disease mod-els. Nat. Chem. Biol. 3, 331–338.
Sasaguri H., Nilsson P., Hashimoto S., Nagata K., Saito T., DeStrooper B. et al. 2017 APP mouse models for Alzheimer’sdisease preclinical studies. EMBO J. 36, 2473–2487.
Sasaki S. 2011 Autophagy in spinal cord motor neurons insporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp.Neurol. 70, 349–359.
Sasaki S. and Maruyama S. 1994 Decreased synaptophysinimmunoreactivity of the anterior horns in motor neuron dis-ease. Acta Neuropathol. 87, 125–128.
Schaeffer V., Lavenir I., Ozcelik S., TolnayM.,Winkler D. T. andGoedert M. 2012 Stimulation of autophagy reduces neurode-generation in a mouse model of human tauopathy. Brain 135,2169–2177.
Scheff S. W. and Price D. A. 1993 Synapse loss in the temporallobe in Alzheimer’s disease. Ann. Neurol. 33, 190–199.
Schwarcz R., Whetsell Jr W. O. and Mangano R. M. 1983Quinolinic acid: an endogenous metabolite that producesaxon-sparing lesions in rat brain. Science 219, 316–318.
Selkoe D. J. 2002 Alzheimer’s disease is a synaptic failure. Sci-ence 298, 789–791.
Sepers M. D. and Raymond L. A. 2014 Mechanisms of synapticdysfunction and excitotoxicity in Huntington’s disease. DrugDiscov. Today 19, 990–996.
Sian J., Dexter D. T., Lees A. J., Daniel S., Agid Y., Javoy-AgidF. et al. 1994 Alterations in glutathione levels in Parkinson’sdisease and other neurodegenerative disorders affecting basalganglia. Ann. Neurol. 36, 348–355.
Shehata M., Matsumura H., Okubo-Suzuki R., Ohkawa N. andInokuchi K. 2012 Neuronal stimulation induces autophagyin hippocampal neurons that is involved in AMPA receptordegradation after chemical long-term depression. J. Neu-rosci. 32, 10413–10422.
Sheng M. and Sala C. 2001 PDZ domains and the organizationof supramolecular complexes. Annu. Rev. Neurosci. 24, 1–29.

700 S. N. Suresh et al.
SkibinskiG., ParkinsonN. J., Brown J.M.,Chakrabarti L., LloydS. L., Hummerich H. et al. 2005 Mutations in the endoso-mal ESCRTIII-complex subunit CHMP2B in frontotemporaldementia. Nat. Genet. 37, 806–808.
SlowE. J., vanRaamsdonk J.,RogersD.,ColemanS.H.,GrahamR. K., Deng Y. et al. 2003 Selective striatal neuronal loss ina YAC128 mouse model of Huntington disease. Hum. Mol.Genet. 12, 1555–1567.
Smith M. G. and Snyder M. 2006 Yeast as a model for humandisease. Curr. Protoc. Hum. Genet. (https://doi.org/10.1002/0471142905.hg1506s48).
Sontag E. M., Samant R. S. and Frydman J. 2017 Mechanismsand functions of spatial protein quality control. Annu. Rev.Biochem. 86, 97–122.
Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q.,Jakes R. and Goedert M. 1997 Alpha-synuclein in Lewy bod-ies. Nature 388, 839–840.
Spires-Jones T. and Knafo S. 2012 Spines, plasticity, and cogni-tion in Alzheimer’s model mice. Neural. Plast. 2012, 319836.
Steiner H. 2004 Uncovering gamma-secretase. Curr. AlzheimerRes. 1, 175–181.
Stolz A., Ernst A. and Dikic I. 2014 Cargo recognition and traf-ficking in selective autophagy. Nat. Cell Biol. 16, 495–501.
Subramaniam S., K. M. Sixt, R. Barrow and S. H. Snyder 2009Rhes, a striatal specific protein, mediates mutant-huntingtincytotoxicity. Science 324, 1327–1330.
Sun G. Z., He Y. C., Ma X. K., Li S. T., Chen D. J., Gao M.et al. 2017 Hippocampal synaptic and neural network deficitsin young mice carrying the human APOE4 gene. CNS Neu-rosci. Ther. 23, 748–758.
Tabata K., Matsunaga K., Sakane A., Sasaki T., Noda T.and Yoshimori T. 2010 Rubicon and PLEKHM1 negativelyregulate the endocytic/autophagic pathway via a novel Rab7-binding domain.Mol. Biol. Cell 21, 4162–7412.
TanakaM.,MachidaY.,Niu S., IkedaT., JanaN.R.,DoiH. et al.2004 Trehalose alleviates polyglutamine-mediated pathologyin a mouse model of Huntington disease. Nat. Med. 10, 148–154.
Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresaR., Hill R. et al. 1991 Physical basis of cognitive alterationsin Alzheimer’s disease: synapse loss is the major correlate ofcognitive impairment. Ann. Neurol. 30, 572–580.
Thinakaran G. and Koo E. H. 2008 Amyloid precursor pro-tein trafficking, processing, and function. J. Biol. Chem. 283,29615–29619.
TrancikovaA., RamonetD. andMooreD. J. 2011Geneticmousemodels of neurodegenerative diseases. Prog. Mol. Biol. Transl.Sci. 100, 419–482.
Van Damme P., Dewil M., Robberecht W. and Van Den BoschL. 2005 Excitotoxicity and amyotrophic lateral sclerosis. Neu-rodegener. Dis. 2, 147–159.
van der Staay F. J., Arndt S. S. and Nordquist R. E. 2009 Eval-uation of animal models of neurobehavioral disorders. Behav.Brain Funct. 5, 11.
Vanhauwaert R., Kuenen S., Masius R., Bademosi A., Manets-berger J., Schoovaerts N. et al. 2017 The SAC1 domain insynaptojanin is required for autophagosome maturation atpresynaptic terminals. EMBO J. 36, 1392–1411.
Vergara R., Rick C., Hernandez-Lopez S., Laville J. A., GuzmanJ.N.,Galarraga E. et al. 2003 Spontaneous voltage oscillationsin striatal projection neurons in a rat corticostriatal slice. J.Physiol. 553, 169–182.
VermeirenC.,Hemptinne I.,VanhoutteN.,TilleuxS.,MaloteauxJ. M. and Hermans E. 2006 Loss of metabotropic glutamatereceptor-mediated regulation of glutamate transport in chemi-cally activated astrocytes in a rat model of amyotrophic lateralsclerosis. J. Neurochem. 96, 719–731.
Vijayan V. and Verstreken P. 2017 Autophagy in the presynapticcompartment in health and disease. J. Cell Biol. 216, 1895–1906.
Vogels O. J., Oyen W. J., van Engelen B. G., Padberg G. W. andHorstink M. W. 1999 Decreased striatal dopamine-receptorbinding in sporadic ALS: glutamate hyperactivity? Neurol-ogy 52, 1275–1277.
Vogels O. J., Veltman J., Oyen W. J. and Horstink M. W. 2000Decreased striatal dopamine D2 receptor binding in amy-otrophic lateral sclerosis (ALS) and multiple system atrophy(MSA):D2receptordown-regulationversus striatal cell degen-eration. J. Neurol. Sci. 180, 62–65.
von Lewinski F. and Keller B. U. 2005 Ca2+, mitochondriaand selective motoneuron vulnerability: implications for ALS.Trends Neurosci. 28, 494–500.
Vonsattel J. P.,MyersR.H., StevensT. J.,FerranteR. J.,BirdE.D.andRichardson Jr. E. P. 1985Neuropathological classificationof Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577.
Wainger B. J., Kiskinis E., Mellin C., Wiskow O., Han S. S.,Sandoe J. et al. 2014 Intrinsic membrane hyperexcitability ofamyotrophic lateral sclerosis patient-derived motor neurons.Cell Rep. 7, 1–11.
Walker F. O. 2007 Huntington’s disease. Lancet 369, 218–228.
Walther D. M., P. Kasturi, M. Zheng, S. Pinkert, G. Vecchi,P. Ciryam. et al. 2017 Widespread proteome remodeling andaggregation in aging C. elegans. Cell 168, 944.
Wang F., Durfee L. A., Huibregtse J. M. 2013 A cotranslationalubiquitination pathway for quality control of misfolded pro-teins.Mol. Cell 50, 368–378.
WangG., Liu X., GaertigM. A., Li S. and Li X. J. 2016 Ablationof huntingtin in adult neurons is nondeleterious but its deple-tion in young mice causes acute pancreatitis. Proc. Natl. Acad.Sci. USA 113, 3359–3364.
Webster C. P., Smith E. F., Bauer C. S., Moller A., HautbergueG. M., Ferraiuolo L. et al. 2016 The C9orf72 protein interactswith Rab1a and the ULK1 complex to regulate initiation ofautophagy. EMBO J. 35, 1656–1676.
Williams A., Sarkar S., Cuddon P., Ttofi E. K., Saiki S., SiddiqiF. H. et al. 2008 Novel targets for Huntington’s disease in anmTOR-independent autophagy pathway. Nat. Chem. Biol. 4,295–305.
Winslow A. R., Chen C. W., Corrochano S., Acevedo-ArozenaA., Gordon D. E., Peden A. A. et al. 2010 alpha-Synucleinimpairsmacroautophagy: implications forParkinson’s disease.J. Cell Biol. 190, 1023–1037.
Wong Y. C. and Holzbaur E. L. 2014 The regulation ofautophagosome dynamics by huntingtin and HAP1 is dis-ruptedby expressionofmutant huntingtin, leading todefectivecargo degradation. J. Neurosci. 34, 1293–1305.
Yang Y., Hentati A., DengH. X., Dabbagh O., Sasaki T., HiranoM. et al. 2001 The gene encoding alsin, a protein with threeguanine-nucleotide exchange factor domains, is mutated in aform of recessive amyotrophic lateral sclerosis.Nat. Genet. 29,160–165.
Yao C., Johnson W. M., Gao Y., Wang W., Zhang J., DeakM. et al. 2013 Kinase inhibitors arrest neurodegeneration incell and C. elegans models of LRRK2 toxicity. Hum. Mol.Genet. 22, 328–344.
Yoshikai S., Sasaki H., Doh-ura K., Furuya H. and SakakiY. 1990 Genomic organization of the human amyloid beta-protein precursor gene. Gene 87, 257–263.
Yu W., Polepalli J., Wagh D., Rajadas J., Malenka R. and Lu B.2012 A critical role for the PAR-1/MARK-tau axis in mediat-ing the toxic effects of Abeta on synapses and dendritic spines.Hum. Mol. Genet. 21, 1384–1390.

Neurodegenerative diseases 701
Zaffagnini G. and Martens S. 2016 Mechanisms of SelectiveAutophagy. J. Mol. Biol. 428, 1714–1724.
Zavodszky E., SeamanM. N., Moreau K., Jimenez-Sanchez M.,BreusegemS.Y.,HarbourM.E. et al. 2014Mutation inVPS35associated with Parkinson’s disease impairs WASH complexassociation and inhibits autophagy. Nat. Commun. 5, 3828.
Zhao Y. G., Sun L., Miao G., Ji C., Zhao H., Sun H. et al.2015 The autophagy gene Wdr45/Wipi4 regulates learningand memory function and axonal homeostasis. Autophagy 11,881–890.
Zhou H., Huang C., Chen H., Wang D., Landel C. P., Xia P. Y.et al. 2010 Transgenic rat model of neurodegeneration causedby mutation in the TDP gene. PLoS Genet. 6, e1000887.
ZhouL.,WangW.,HoppelC.,Liu J. andZhuX.2017Parkinson’sdisease-associated pathogenic VPS35 mutation causes com-plex I deficits. Biochim. Biophys. Acta 1863, 2791–2795.
Zona C., Ferri A., Gabbianelli R., Mercuri N. B., Bernardi G.,Rotilio G. et al. 1998 Voltage-activated sodium currents in acell line expressing a Cu, Zn superoxide dismutase typical offamilial ALS. Neuroreport 9, 3515–3518.
Related Documents











