Research paper Nasal administration of liquid crystal precursor mucoadhesive vehicle as an alternative antiretroviral therapy Flávia Chiva Carvalho ⇑ , Michel Leandro Campos, Rosângela Gonçalves Peccinini, Maria Palmira Daflon Gremião School of Pharmaceutical Sciences, Universidade Estadual Paulista UNESP, Araraquara, Brazil article info Article history: Received 10 September 2012 Accepted in revised form 11 November 2012 Available online 1 December 2012 Keywords: Nasal administration Liquid crystal precursor Antiretroviral Phase behavior Mucoadhesion Stimuli-sensitive drug delivery systems abstract The purpose of this study was to develop a mucoadhesive stimuli-sensitive drug delivery system for nasal administration of zidovudine (AZT). The system was prepared by formulating a low viscosity precursor of a liquid crystal phase, taking advantage of its lyotropic phase behavior. Flow rheology measurements showed that the formulation composed of PPG-5-CETETH-20, oleic acid and water (55, 30, 15% w/w), denominated P, has Newtonian flow behavior. Polarized light microscopy (PLM) revealed that formula- tion P is isotropic, whereas its 1:1 (w/w) dilution with artificial nasal mucus (ANM) changed the system to an anisotropic lamellar phase (PD). Oscillatory frequency sweep analysis showed that PD has a high storage modulus (G 0 ) at nasal temperatures. Measurement of the mucoadhesive force against excised por- cine nasal mucosa or a mucin disk proved that the transition to the lamellar phase tripled the work of mucoadhesion. Ex vivo permeation studies across porcine nasal mucosa exhibited an 18-fold rise in the permeability of AZT from the formulation. The Weibull mathematical model suggested that the AZT is released by Fickian diffusion mechanisms. Hence, the physicochemical characterization, combined with ex vivo studies, revealed that the PPG-5-CETETH-20, oleic acid, and water formulation could form a mucoadhesive matrix in contact with nasal mucus that promoted nasal absorption of the AZT. For an in vivo assessment, the plasma concentrations of AZT in rats were determined by HPLC method following intravenous and intranasal administration of AZT-loaded P formulation (PA) and AZT solution, respec- tively, at a dose of 8 mg/kg. The intranasal administration of PA resulted in a fast absorption process (T max = 6.7 min). Therefore, a liquid crystal precursor formulation administered by the nasal route might represent a promising novel tool for the systemic delivery of AZT and other antiretroviral drugs. In the present study, the uptake of AZT absorption in the nasal mucosa was demonstrated, providing new foundations for clinical trials in patients with AIDS. Ó 2012 Elsevier B.V. All rights reserved. 1. Introduction The Acquired Immunodeficiency Syndrome (AIDS) is a world- wide public health challenge. The emergence of antiretroviral ther- apy agents has significantly increased the patient’s life expectancy and quality of life [1,2]. Despite the success of antiretroviral ther- apy, they are combined in a complex therapeutic scheme, and it has several undesirable side effects that lead to a decrease in the patience compliance. Moreover, many antiretroviral drugs undergo extensive pre-systemic metabolism and instability in the gastroin- testinal environment, resulting in inadequate and erratic oral absorption. Also, some antiretroviral classes exhibit poor perme- ability and low oral absorption [3,4]. Strategies currently being investigated to overcome these limitations include the design and development of novel drug delivery systems that can improve the efficacy of existing antiretroviral drugs and increase the pa- tience compliance [3,5]. Nasal drug delivery is recognized as a very promising route for systemic effects, since the absorbed substances are transported di- rectly into the systemic circulation, thereby avoiding the first-pass metabolic effect and the side effects generally experienced follow- ing oral administration. Nasal route is less invasive than injection, and the nose allows the self-medication that improves patience compliance as well [6]. A very rapid rate of absorption can be achieved by nasal application of some drugs, because the nasal mu- cosa is highly vascularized and has a large surface area available for drug deposition and absorption [6,7]. However, there are some fac- tors that limit intranasal absorption. These barriers include physi- cal removal of the drug from the site of deposition in the nasal 0939-6411/$ - see front matter Ó 2012 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.ejpb.2012.11.021 ⇑ Corresponding author. School of Pharmaceutical Sciences, Universidade Esta- dual Paulista UNESP, Rodovia Araraquara-Jaú, Km 01, Araraquara 14801902, SP, Brazil. Tel.: +55 16 33016961; fax: +55 16 33016760. E-mail addresses: fl[email protected] (F.C. Carvalho), michelcampos7@ yahoo.com.br (M.L. Campos), [email protected] (R.G. Peccinini), pgremiao@ fcfar.unesp.br (M.P.D. Gremião). European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227 Contents lists available at SciVerse ScienceDirect European Journal of Pharmaceutics and Biopharmaceutics journal homepage: www.elsevier.com/locate/ejpb

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227
Contents lists available at SciVerse ScienceDirect
European Journal of Pharmaceutics and Biopharmaceutics
journal homepage: www.elsevier .com/locate /e jpb
Research paper
Nasal administration of liquid crystal precursor mucoadhesive vehicle as analternative antiretroviral therapy
Flávia Chiva Carvalho ⇑, Michel Leandro Campos, Rosângela Gonçalves Peccinini,Maria Palmira Daflon GremiãoSchool of Pharmaceutical Sciences, Universidade Estadual Paulista UNESP, Araraquara, Brazil
a r t i c l e i n f o
Article history:Received 10 September 2012Accepted in revised form 11 November 2012Available online 1 December 2012
Keywords:Nasal administrationLiquid crystal precursorAntiretroviralPhase behaviorMucoadhesionStimuli-sensitive drug delivery systems
0939-6411/$ - see front matter � 2012 Elsevier B.V. Ahttp://dx.doi.org/10.1016/j.ejpb.2012.11.021
⇑ Corresponding author. School of Pharmaceuticaldual Paulista UNESP, Rodovia Araraquara-Jaú, Km 0Brazil. Tel.: +55 16 33016961; fax: +55 16 33016760.
E-mail addresses: [email protected] (F.C.yahoo.com.br (M.L. Campos), [email protected] (M.P.D. Gremião).
a b s t r a c t
The purpose of this study was to develop a mucoadhesive stimuli-sensitive drug delivery system for nasaladministration of zidovudine (AZT). The system was prepared by formulating a low viscosity precursor ofa liquid crystal phase, taking advantage of its lyotropic phase behavior. Flow rheology measurementsshowed that the formulation composed of PPG-5-CETETH-20, oleic acid and water (55, 30, 15% w/w),denominated P, has Newtonian flow behavior. Polarized light microscopy (PLM) revealed that formula-tion P is isotropic, whereas its 1:1 (w/w) dilution with artificial nasal mucus (ANM) changed the systemto an anisotropic lamellar phase (PD). Oscillatory frequency sweep analysis showed that PD has a highstorage modulus (G0) at nasal temperatures. Measurement of the mucoadhesive force against excised por-cine nasal mucosa or a mucin disk proved that the transition to the lamellar phase tripled the work ofmucoadhesion. Ex vivo permeation studies across porcine nasal mucosa exhibited an 18-fold rise in thepermeability of AZT from the formulation. The Weibull mathematical model suggested that the AZT isreleased by Fickian diffusion mechanisms. Hence, the physicochemical characterization, combined withex vivo studies, revealed that the PPG-5-CETETH-20, oleic acid, and water formulation could form amucoadhesive matrix in contact with nasal mucus that promoted nasal absorption of the AZT. For anin vivo assessment, the plasma concentrations of AZT in rats were determined by HPLC method followingintravenous and intranasal administration of AZT-loaded P formulation (PA) and AZT solution, respec-tively, at a dose of 8 mg/kg. The intranasal administration of PA resulted in a fast absorption process(Tmax = 6.7 min). Therefore, a liquid crystal precursor formulation administered by the nasal route mightrepresent a promising novel tool for the systemic delivery of AZT and other antiretroviral drugs. In thepresent study, the uptake of AZT absorption in the nasal mucosa was demonstrated, providing newfoundations for clinical trials in patients with AIDS.
� 2012 Elsevier B.V. All rights reserved.
1. Introduction
The Acquired Immunodeficiency Syndrome (AIDS) is a world-wide public health challenge. The emergence of antiretroviral ther-apy agents has significantly increased the patient’s life expectancyand quality of life [1,2]. Despite the success of antiretroviral ther-apy, they are combined in a complex therapeutic scheme, and ithas several undesirable side effects that lead to a decrease in thepatience compliance. Moreover, many antiretroviral drugs undergoextensive pre-systemic metabolism and instability in the gastroin-testinal environment, resulting in inadequate and erratic oral
ll rights reserved.
Sciences, Universidade Esta-1, Araraquara 14801902, SP,
Carvalho), michelcampos7@(R.G. Peccinini), pgremiao@
absorption. Also, some antiretroviral classes exhibit poor perme-ability and low oral absorption [3,4]. Strategies currently beinginvestigated to overcome these limitations include the designand development of novel drug delivery systems that can improvethe efficacy of existing antiretroviral drugs and increase the pa-tience compliance [3,5].
Nasal drug delivery is recognized as a very promising route forsystemic effects, since the absorbed substances are transported di-rectly into the systemic circulation, thereby avoiding the first-passmetabolic effect and the side effects generally experienced follow-ing oral administration. Nasal route is less invasive than injection,and the nose allows the self-medication that improves patiencecompliance as well [6]. A very rapid rate of absorption can beachieved by nasal application of some drugs, because the nasal mu-cosa is highly vascularized and has a large surface area available fordrug deposition and absorption [6,7]. However, there are some fac-tors that limit intranasal absorption. These barriers include physi-cal removal of the drug from the site of deposition in the nasal

220 F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227
cavity by the mucociliary clearance mechanisms, enzymatic degra-dation in the mucus layer, and low permeability of the nasal epi-thelium [7,8]. One strategy that can improve nasal drugabsorption is to use mucoadhesive drug delivery systems; thesemaintain the formulation in the nasal cavity for an extended timeperiod and increase absorption of the drug [6,9].
Stimuli-sensitive systems are an interesting approach toimproving the mucoadhesion properties of drug delivery systems.Such systems undergo a phase transition in response to externalphysical or chemical stimuli, such as changes in temperature, pH,and ionic strength [10]. The liquid formulation is easy to adminis-ter, whereas post-application, it is transformed into a highlyviscous rheologically structured system [11]. This strategy hasbeen achieved in hydrogel systems, which are widely explored inthe literature [10]; however, liquid crystal phases, which are morerelated to colloid and interface science, can also act as stimuli-sensitive systems and help to optimize significantly the efficiencyof drugs.
Lyotropic liquid crystals are formed by surfactant moleculesthat have a polar and a nonpolar region. In aqueous environments,they form monolayers with the polar group facing the water and,after reaching a critical micellar concentration, aggregates calledmicelles are formed. The structure of the micelles can vary accord-ing to the geometry of the molecules and the relative amount ofeach component. It is possible to obtain disconnected micelle-forming fluid systems, such as simple micellar systems or micro-emulsions. On interconnected systems that are very organized,resulting in liquid crystal structures [12,13]. Liquid crystals are de-scribed as two or three-dimensional nanometer-scale structureswith periodic hydrophilic and hydrophobic features, organized inphases that can vary among hexagonal phase, lamellar, and cubicphases [12,13]. They are also thermodynamically stable and canbe stored for long periods of time without phase separation.Depending on the relative amount of the aqueous solution andthe surfactant characteristics, these systems can undergo variousphase transformations and structured modifications. By thismeans, their consistency and rheological properties can be chan-ged, as required. They are usually formed with low energy inputby spontaneous structural organization; their production is thusrelatively simple and energy-saving [14]. Because of their highlyuniform and porous nanoscale structures, liquid crystal phases dis-play properties that favor the solubilization of drugs in the aque-ous, oily or amphiphilic compartments; moreover, they can bedesigned to form a rigid drug release matrix that can be used tocontrol drug delivery [15].
Systems composed of polyoxypropylene (5) polyoxyethylene(20) cetyl ether (PPG-5-CETETH-20) (nonionic surfactant), oleicacid, and water were shown to solubilize the antiretroviral drugzidovudine (AZT) more efficiently than conventional liquid dosageforms [16]. Furthermore, it was found that a combination of thethree components in certain proportions formed easy-flowingoptically clear isotropic liquid systems that were transformed intoliquid crystal phases in contact with artificial mucus at nasal tem-perature [17]. In this case, the stimuli-sensitive system is a liquidcrystal precursor. Dilution in aqueous fluids promotes the phasetransition to a rigid liquid crystal matrix that can prolong the res-idence time of the formulation in contact with the mucosa. More-over, surfactant systems can change the permeability of theepithelial cell layer by modifying the phospholipid bilayer [8],while some research indicates the mucoadhesion potential ofliquid crystals such as the Boyd et al. [18], Bruschi et al. [19], andrecently the work of our research group [17,20].
There are various experimental setups currently in use for thedetermination of mucoadhesive properties of polymer-systems.In recent years, these have evolved from simple measurements ofthe force of detachment to much more sophisticated and expensive
tests [11]. Adhesive strength can be assessed by measuring theforce required to detach the drug delivery system from a mucoustissue, by applying an external force in a tension-based setups [11].
The purpose of this study was to develop and evaluate a muco-adhesive stimuli-sensitive drug delivery system for nasal adminis-tration of AZT. Preparation of the system was achieved by theformulation of low viscous precursor of a liquid crystal phase bytaking advantage its lyotropic phase behavior. Based on previousstudies [17], it was used the surfactant PPG-5-CETETH-20 com-bined with oleic acid and water to obtain a liquid crystal precursorfor nasal administration of AZT. The changes in rheology werestudied by flow and oscillatory measurements. The mucoadhesiveforce was measured using a texture analyzer, to which a porcinenasal mucosa or a mucin disk was attached. The transport and drugabsorption across the nasal mucosa was studied by an in vitro tech-nique employing a diffusion chamber and an excised mucosa tis-sue. Finally, the efficacy of the new nasal delivery system wasassessed in vivo in rats. The liquid crystal precursor formulationwas administered by the intranasal route, and the AZT plasmaticconcentration profile was compared with those obtained by theintravenous injection of an AZT solution.
2. Methods
2.1. Materials
PPG-5-CETETH-20 (Procetyl� AWS) was purchased from Croda(Campinas, Brazil) and the oleic acid from Synth (Diadema, Brazil).The high-purity water was prepared with a Millipore Milli-Q Pluspurification system. Mucin from porcine stomach was from SigmaAldrich (Steinheim, Germany). Zidovudine was a gift from FURP(Fundação para o Remédio Popular, Guarulhos, Brazil).
2.2. Phase diagram construction and preparation of liquid crystalprecursor formulations
The phase diagram was obtained in previous studies [17].Briefly, its construction was performed weighting different combi-nations of PPG-5-CETETH-20, oleic acid, and water (w/w) and mix-ing at room temperature. The samples were maintained 24 h atroom temperature to reach equilibrium. Each phase was identifiedby polarized light microscopy (PLM), as described below. Based onthe structures identified, the ternary phase diagram of the systemwas constructed, with Sigma Plot Software (Fig. 1).
The formulation chosen as precursor (point P on Fig. 1) wasused for further characterizations. AZT was incorporated into P at10 mg/mL by mixing, giving formulation PA, which was main-tained at room temperature for 24 h, and it was named as PA. Theywere prepared 24 h before the tests.
The formulations P and PA were diluted 1:1 (w/w) with artificialnasal mucus (ANM), to assess the in situ phase behavior. The ANMwas prepared by dispersing 8% (w/v) of mucin type II from porcinestomach (Sigma, St. Louis, USA) in a solution of 7.45 mg/mL NaCl,1.29 mg/mL KCl and 0.32 mg/mL CaCl2�2H2O. The componentswere mixed by magnetic stirring, and the pH was adjusted to pH5.7 with 0.1 M NaOH a solution [22].
2.3. Polarized light microscopy (PLM)
Samples were prepared by placing a drop of formulation be-tween a cover slip and a glass slide and then examined underpolarized light. An Optical Leica Microscope equipped with a digi-tal camera was used to analyze various fields of each sample atroom temperature. The isotropic or anisotropic behavior of the

Fig. 1. Ternary phase diagram of PPG-5-CETETH-20, oleic acid, and water. P is theliquid crystal precursor system, and PD is a 1:1 (w/w) dilution of P with ANM.
F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227 221
samples was recorded. Pictures were taken at a magnification of20,000�.
2.4. Rheological measurements
The rheological measurements were taken with a controlled-stress Rheostress RS1 rheometer (Haake, Karlsruhe, Germany)and analyzed with Rheowin 3.5 software. Plate–plate geometrywas used with a gap of 200 lm between plates of 35 mm diameter.Samples were carefully applied to the lower plate, ensuring thatsample shearing was minimized, and allowed to equilibrate forat least 3 min prior to analysis.
2.4.1. Determination of flow propertiesThe experiments were performed with shear rates within the
range 0.01–500 1/s, which was chosen on the basis of the strengthof resistance to the applied stresses. The rheological measurementswere performed on both the up and down curves. The flow curveswere fitted to a power law model, using the program Origin 7.0. Allrheological determinations were carried out at 25 ± 0.25 �C.
2.4.2. Oscillatory experimentsOscillatory analysis of each sample was performed after deter-
mination of its linear viscoelastic region at 32 �C, where stresswas directly proportional to strain, and the storage modulus re-mained constant. Frequency sweep analysis was performed overthe frequency range of 0.1–10 Hz, at a constant stress of 1 Pa.The systematic error in frequency of the rheometer was around0.01 Hz.
2.5. Determination of the mucoadhesive strength
2.5.1. Preparation of the mucosa modelsPorcine nasal mucosa and mucin disks were used as mucosal
models. The nasal mucosa was taken from domestic pigs by amethod adapted from Franse and collaborators [23]. The headswere separated from the animals at a local slaughterhouse andwere made an incision along the nasal septum. They were frozenand transported to the laboratory for storage. Before the experi-ment, after defrosting, the respiratory mucosa was carefullyremoved from the nasal turbinates. During the experiments, the
pieces of mucosa were kept in saline solution in an ice bath.Mucosa from the same pig was used to investigate the perfor-mance of each formulation, to eliminate systematic errors. Themucosa was attached to a 10 mm analytical probe of the textureanalyzer by a rubber ring. The mucosa was hydrated with 100 lLof ANM at 32 �C immediately before initiation of the measurement.The excess liquid was removed with tissue paper.
The mucin disks used as a mucosal model were prepared bydirect compression of 200 mg of the mucin powder to form in11 mm diameter disks. One disk was fixed on the analytical probewith double-sided adhesive tape. It was hydrated with 100 lL ofANM at 32 �C immediately before the experiment.
2.5.2. Mucoadhesion measurement by tensile strength methodA TA-XTplus texture analyzer (Stable Micro Systems, Surrey,
UK) was used for the tensile strength measurements. The mucosamodel was fastened to the upper movable probe as describedabove, and the formulation sample was located on the lower plat-form. The measurement was triggered to begin as the upper probeencountered a force of 2 mN upon contact with the sample. Theprobe was kept in contact with no force applied for 60 s, afterwhich it was raised at the speed of 0.5 mm/s, and the force neededfor detachment was registered. The tensile work (g.s), which isproportional to the area under the force–time curve, was used todescribe the mucoadhesive characteristics.
2.6. Ex vivo permeation study
In this study, AZT was analyzed by HPLC, with the UV detector setat 266 nm (Varian Pro Star 330). A reverse-phase Luna C18 column(250 � 4.6 mm i.d., 5 lm pore size) was used (Phenomenex,Torrance, EUA). The mobile phase was a mixture of methanol/water(60:40 v/v), the flow rate was set at 0.7 mL/min, and the injectionvolume was 30 lL. A calibration curve was constructed by preparingworking solutions of AZT in 20 mM monobasic phosphate buffer (pH6.8) at concentrations ranging from 0.1 to 20 lg/mL.
The nasal mucosa was obtained from pigs, being removed asdescribed in Section 2.5.1. For the experiments, the pieces ofmucosa were thawed and kept in saline solution on ice. The loadedprecursor formulation (PA) was analyzed, with an aqueous solutionof AZT (AS) for comparison. The concentration of the drug in eachsample was 10 mg/mL.
The porcine nasal mucosa (1.77 cm2) was placed between thedonor and the receptor chambers of a Franz diffusion cell (Micro-ette Plus, Hanson Research, Chatsworth, EUA). The receptor cham-ber of the cell (7 mL) was filled with 20 mM monobasic buffer (pH6.8) stirred at 300 rpm. The samples (200 lL) were dropped on themucosa in contact with the receptor chamber. The total test timewas 12 h, and 2 mL of the receptor fluid was collected and replacedby an equal volume of fresh receptor solution. Samples collectedwere filtered and injected into the HPLC system. The assay was car-ried out on six replicates for each sample.
The drug release profile was calculated from Eq. (1) [24]:
Q ¼ Ct � Vr þX
Vc � Cc ð1Þ
where Q (lg/cm2) is the total amount permeated up to time t; Ct
(lg/mL cm2) is the concentration measured at time t; Vr (mL) isthe volume of receptor solution (7 mL); Cc (lg/mL cm2) is the con-centration at the previous sampling, and Vc (mL) is the volumesampled.
The profiles obtained by the ex vivo drug release study wereanalyzed by mathematical models to identify the mechanisms in-volved in the AZT release. Besides that, the permeation parametersstudied were the pseudo-steady-state flux (J, lg/min cm) and thepermeability coefficient (Kp, cm/min) derived from the Fick’s law,

222 F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227
represented by Eq. (2) [25]. The pseudo-steady-state flux isobtained as the gradient of the linear portion of the permeationprofile. If the concentration of the permeation in the appliedvehicle is known, then the permeability coefficient Kp can be deter-mined [25].
J ¼ dQdt¼ Cv � Kp ð2Þ
where dQ (lg/cm2) is the cumulative mass of the drug that passesthrough unit area of the membrane in time t, and Cv is the concen-tration of the drug in the vehicle (donor solution) bathing themembrane.
2.7. Pharmacokinetic studies
2.7.1. Study designThe use of animals in this study was approved by the State Uni-
versity of São Paulo Animal Use and Care Committee.Two separate in vivo studies were conducted in which the nasal
and intravenous administrations were investigated. Data were ob-tained for a period of 6 h after the administration of eachformulation.
2.7.2. Animal proceduresThe male Wistar rats (weight 250–300 g) were housed one ani-
mal per cage in a controlled environment (20–22 �C, 14 h of lightper day), with free access to daily feedings of standard chow pelletsand water.
On the day prior to dosing, a cannula was surgically inserted un-der anesthetic into the femoral artery and vein of the rats (male,Wistar, 250–300 g) to enable serial blood sampling and intrave-nous (IV) administration. The animals were anesthetized with ket-amine/xylazine (8:3), dose of 100 mg/kg, by intraperitonealinjection before such surgery. Animals were then allowed to re-cover overnight prior to the administration. Rats were fasted for8 h before and after dosing and water were freely available.
2.7.3. Formulation preparation and administrationThe formulations tested were the loaded precursor PA, contain-
ing 30 mg/mL of AZT, administrated by the nasal route and AZTsolution at 10 mg/mL, by IV bolus injection. The dose of AZTadministered to all groups was 8 mg/kg. This dose was based onthe solubility of the AZT in the formulation and its maximum stip-ulated volume to be administrated by the nasal route (approxi-mately 60 lL).
Before the administration, the rats were anesthetized with ket-amine/xylazine (8:3), at a dose of 100 mg/kg, by intraperitonealinjection. Nasal administration was performed with a 20 lL micro-pipette to instill 20 lL of PA into each nostril at a time, until a totalof 60 lL had been instilled. This spaced instillation was intended tominimize the overflow into the gastrointestinal tract. The IV injec-tion was performed in the previously cannulated femoral vein.
2.7.4. Sample preparation and HPLC analysis of AZT in plasmaTo determine AZT in rat plasma, blood samples were collected
in tubes containing sodium heparin. The tubes were centrifuged(13,000 rpm for 10 min) to separate the plasma, and this wasstored at �20 �C until analysis. In order to ensure the effective sep-aration of AZT from plasma components by HPLC, the plasma wasdeproteinized. To 100 lL of plasma, it was added 10 lL of trichlo-roacetic acid solution 0.01% (v/v) and 90 lL of methanol containingthe internal standard (IS) 3-isobutyl-1methyl-xanthine (Sigma,Steinheim, Germany) at 5000 ng/mL. The mixture was vortexedfor 1 min and centrifuged for 10 min at 13,000 rpm. The superna-tant was directly injected into the HPLC system. The volume ofinjection was 50 lL.
The chromatographic system Varian Pro Star/Dynamax 210/215with a Pro Star 330 UV–Vis PDA spectrophotometric detector (setat 266 nm) was used for the determination of AZT in plasma sam-ples. The stationary phase was a Phenomenex Luna C18 column(250 mm, 4.6 mm, 5 lm), and the mobile phase was a mixture(20:20:60 v/v/v) of acetonitrile, methanol, and water containing0.01% formic acid, flowing at 0.8 mL/min in the isocratic mode.
2.7.5. Calculation of the pharmacokinetic parametersThe plasma concentration data were fitted using a one-com-
partment model with 1/Yhat2 weighting factor, without lag timeby the software WinNonlin (Phoenix™, WinNonlin� 6.2.1; Phar-sight Corp., Mountain View, CA, USA). The following pharmacoki-netic parameters were calculated: Cmax, Tmax, AUC0–1, t1/2,clearance (Cl), distribution volume (Vd), and the elimination rateconstant (kel). Cmax and Tmax were calculated directly from concen-tration data the plasma AZT time curves. AUC0–t was calculated bythe linear trapezoidal rule. AUC0–1 was calculated from the follow-ing formula: AUC0–1 = AUC0–t + Ct/ke, where Ct was the lastmeasured concentration, and ke was calculated by applying alog-linear regression analysis to the plasma AZT time curve.
2.7.6. Statistical analysisThe results were represented as mean and standard deviation.
Comparison between the results was performed by the programGraph Pad Prism 5.01 Inc. San Diego, CA, using a nonparametrict-test (Mann–Whitney) with significance accepted when p < 0.05.
3. Results
3.1. Phase diagram construction and preparation of the liquid crystalprecursor formulation
The composition of the formulation was based in previous stud-ies, which was constructed the ternary phase diagram of PPG-5-CETETH-20, oleic acid, and water represented in Fig. 1 [17]. Theconstruction of the ternary phase diagram is an excellent tool thatgives the researcher an overview of where phase transitions mayoccur. Varying the composition of the three components alloweda wide variety of formulations that differed in their mechanicalbehavior. The optically clear liquid and semi-solid systems ob-tained were spontaneously formed, showing thermodynamicstability.
The optically clear liquid systems investigated showed a darkbackground under PLM. The cubic phases are a kind of liquid crys-tal that also show a dark background when visualized by PLM, butit is well known that the apparent viscosity of cubic liquid crystalsis very high [15]. Since the optically clear liquid systems flowedeasily, they were not a cubic phase. The optically clear semisolidsystems have a gel-like appearance and are anisotropic underpolarized light, characteristic of liquid crystals, which are birefrin-gent as a result of their molecular ordering.
Since the liquid isotropic phases have low viscosity and flowfreely, they can be injected through a syringe and thus facilitate na-sal administration. For this reason, a formulation from the opticallyclear liquid systems region of the phase diagram was selected forcharacterization. The point P in Fig. 1 was chosen as a potential li-quid crystal precursor formulation, since it is liquid and changes toa semisolid gel-like phase on dilution with water, as can be seen onthe dilution line of this formulation in the ternary phase diagram.
3.2. Physicochemical characterization of the liquid crystal precursor
The formulations P and AZT-loaded P (PA) were diluted withANM (1:1 w/w), giving PD and PAD, respectively, which were

F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227 223
analyzed by PLM. Both PD and PAD showed Maltese crosses at32 �C, characteristic of lamellar phases (Fig. 2). Therefore, thedilution of P and PA with ANM promoted the phase transition ofa liquid system to a gel-like lamellar phase. The contact of P andPA with ANM increases the organization of the systems, whichthen act as liquid crystal precursors.
The relationship between the shear rate and shear stress isshown in the rheograms in Fig. 3a. For P and PA, it can be observedthat the two parameters are proportional, typical of Newtonianbehavior. Therefore, the liquid crystal precursors P and PA have aflow property that renders them easy to apply to the required site.
The oscillatory frequency sweep was carried out on formula-tions P, PD and the loaded PA and PAD. The storage modulus G0
and the loss modulus G00 were plotted against frequency, and rep-resentative rheograms are presented in Fig. 3b. P and PA werefound to be more viscous than elastic (G00 > G0). The low level of
Fig. 2. Image of sample of PD taken by PLM (20,000�) at 32 �C, showing Maltesecrosses. (For interpretation of the references to color in this figure legend, the readeris referred to the web version of this article.)
Fig. 3. (a) Flow rheogram of P and PA at 25 �C. (b) Storage and loss moduli versus frequensamples at 32 �C. (c) Work of mucoadhesion of P and PD samples measured by the muc
interaction between the system components can explain the highloss moduli. The liquid crystal samples PD and PAD showed gel-like behavior and G0 was higher than G00, both independent of fre-quency. The increase in the elastic modulus on dilution evidencedthe higher organization of the PD and PAD systems.
3.3. Determination of the mucoadhesive strength
The mucoadhesive strength of the formulations was evaluatedby measuring the force required to detach the formulation fromthe mucosal model, using a TA-XTplus texture analyzer. Fig. 3cillustrates the mucoadhesive strength of formulations P and PDtested on the mucin disk and porcine nasal mucosa. The resultsfor the two mucosa models did not differ significantly when com-pared by one-way ANOVA. However, the work of mucoadhesion ofthe lamellar phase PD was around three times higher than thatof the optically clear isotropic system P. So, for systems composedof PPG-5-CETETH-20, oleic acid, and water, the results suggest thatthe adhesive force is strongly influenced by the type of mesophase.
When the P and PA formulations incorporated an equal mass ofANM, the systems passed from the fluid phase to the viscoelasticlamellar phase. For a liquid precursor formulation capable of form-ing such a strong liquid crystal matrix as that formed by the systemunder study here, it is very likely that the rheological performanceof the formulation is as important to achieving a long contact timeas are its interactions with the mucin or mucus layer at the sys-tem–mucus interface. Hence, by combining the formation of thestrong liquid crystal matrix with the mucoadhesive force demon-strated by texture analyzer measurements, PPG-5-CETETH-20/oleic acid/water systems can be designed for prolonged retentionof the formulations in the nasal cavity.
3.4. Ex vivo drug release study
The analytical method used to determine the AZT in the recep-tor fluid by HPLC was validated. The calibration curve obtained for
cy obtained by oscillatory frequency sweep of unloaded (P, PD) and loaded (PA, PAD)oadhesive strength test.
224 F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227
the concentration range 0.1–20 lg/mL was linear (y = 0.00174x �0.00013, r2 = 0.9998). The relative standard deviations (RSD) ofthe slope and y-intercept were 2.5% and 7.7%, respectively. Theanalytical method was specific to AZT, since no compound inthe formulation or the receptor fluid and solvents interfered inthe retention time of the AZT (7 min). Three working solutions,representing low, intermediate, and high concentrations withinthe range of the calibration curve (0.5, 4.0 and 10 lg/mL, respec-tively), were analyzed in triplicate to verify the precision and accu-racy of the analytical method. The RSD values obtained for theseconcentrations were 3.62%, 2.18%, and 4.51%, while the recoveryvalues were 94.64%, 99.47%, and 98.88%, respectively, proving thatthe method is precise and accurate. The limits of detection andquantitation were 0.019 lg/mL and 0.057 lg/mL, respectively.
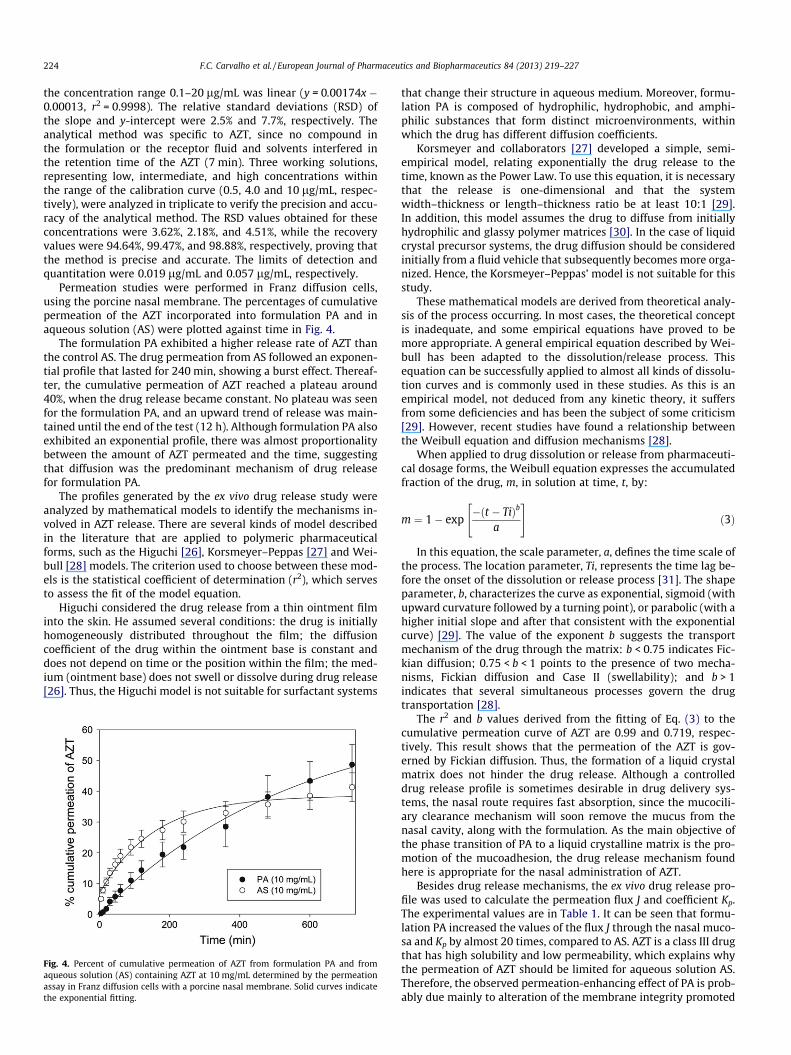
Permeation studies were performed in Franz diffusion cells,using the porcine nasal membrane. The percentages of cumulativepermeation of the AZT incorporated into formulation PA and inaqueous solution (AS) were plotted against time in Fig. 4.
The formulation PA exhibited a higher release rate of AZT thanthe control AS. The drug permeation from AS followed an exponen-tial profile that lasted for 240 min, showing a burst effect. Thereaf-ter, the cumulative permeation of AZT reached a plateau around40%, when the drug release became constant. No plateau was seenfor the formulation PA, and an upward trend of release was main-tained until the end of the test (12 h). Although formulation PA alsoexhibited an exponential profile, there was almost proportionalitybetween the amount of AZT permeated and the time, suggestingthat diffusion was the predominant mechanism of drug releasefor formulation PA.
The profiles generated by the ex vivo drug release study wereanalyzed by mathematical models to identify the mechanisms in-volved in AZT release. There are several kinds of model describedin the literature that are applied to polymeric pharmaceuticalforms, such as the Higuchi [26], Korsmeyer–Peppas [27] and Wei-bull [28] models. The criterion used to choose between these mod-els is the statistical coefficient of determination (r2), which servesto assess the fit of the model equation.
Higuchi considered the drug release from a thin ointment filminto the skin. He assumed several conditions: the drug is initiallyhomogeneously distributed throughout the film; the diffusioncoefficient of the drug within the ointment base is constant anddoes not depend on time or the position within the film; the med-ium (ointment base) does not swell or dissolve during drug release[26]. Thus, the Higuchi model is not suitable for surfactant systems
Fig. 4. Percent of cumulative permeation of AZT from formulation PA and fromaqueous solution (AS) containing AZT at 10 mg/mL determined by the permeationassay in Franz diffusion cells with a porcine nasal membrane. Solid curves indicatethe exponential fitting.
that change their structure in aqueous medium. Moreover, formu-lation PA is composed of hydrophilic, hydrophobic, and amphi-philic substances that form distinct microenvironments, withinwhich the drug has different diffusion coefficients.
Korsmeyer and collaborators [27] developed a simple, semi-empirical model, relating exponentially the drug release to thetime, known as the Power Law. To use this equation, it is necessarythat the release is one-dimensional and that the systemwidth–thickness or length–thickness ratio be at least 10:1 [29].In addition, this model assumes the drug to diffuse from initiallyhydrophilic and glassy polymer matrices [30]. In the case of liquidcrystal precursor systems, the drug diffusion should be consideredinitially from a fluid vehicle that subsequently becomes more orga-nized. Hence, the Korsmeyer–Peppas’ model is not suitable for thisstudy.
These mathematical models are derived from theoretical analy-sis of the process occurring. In most cases, the theoretical conceptis inadequate, and some empirical equations have proved to bemore appropriate. A general empirical equation described by Wei-bull has been adapted to the dissolution/release process. Thisequation can be successfully applied to almost all kinds of dissolu-tion curves and is commonly used in these studies. As this is anempirical model, not deduced from any kinetic theory, it suffersfrom some deficiencies and has been the subject of some criticism[29]. However, recent studies have found a relationship betweenthe Weibull equation and diffusion mechanisms [28].
When applied to drug dissolution or release from pharmaceuti-cal dosage forms, the Weibull equation expresses the accumulatedfraction of the drug, m, in solution at time, t, by:
m ¼ 1� exp�ðt � TiÞb
a
" #ð3Þ
In this equation, the scale parameter, a, defines the time scale ofthe process. The location parameter, Ti, represents the time lag be-fore the onset of the dissolution or release process [31]. The shapeparameter, b, characterizes the curve as exponential, sigmoid (withupward curvature followed by a turning point), or parabolic (with ahigher initial slope and after that consistent with the exponentialcurve) [29]. The value of the exponent b suggests the transportmechanism of the drug through the matrix: b < 0.75 indicates Fic-kian diffusion; 0.75 < b < 1 points to the presence of two mecha-nisms, Fickian diffusion and Case II (swellability); and b > 1indicates that several simultaneous processes govern the drugtransportation [28].
The r2 and b values derived from the fitting of Eq. (3) to thecumulative permeation curve of AZT are 0.99 and 0.719, respec-tively. This result shows that the permeation of the AZT is gov-erned by Fickian diffusion. Thus, the formation of a liquid crystalmatrix does not hinder the drug release. Although a controlleddrug release profile is sometimes desirable in drug delivery sys-tems, the nasal route requires fast absorption, since the mucocili-ary clearance mechanism will soon remove the mucus from thenasal cavity, along with the formulation. As the main objective ofthe phase transition of PA to a liquid crystalline matrix is the pro-motion of the mucoadhesion, the drug release mechanism foundhere is appropriate for the nasal administration of AZT.
Besides drug release mechanisms, the ex vivo drug release pro-file was used to calculate the permeation flux J and coefficient Kp.The experimental values are in Table 1. It can be seen that formu-lation PA increased the values of the flux J through the nasal muco-sa and Kp by almost 20 times, compared to AS. AZT is a class III drugthat has high solubility and low permeability, which explains whythe permeation of AZT should be limited for aqueous solution AS.Therefore, the observed permeation-enhancing effect of PA is prob-ably due mainly to alteration of the membrane integrity promoted

Table 1Permeation flux (J) and permeability coefficient (Kp) obtained for PA formulation andAZT aqueous solution AS, both containing the AZT at 10 mg/mL.
Sample J (lg/min cm) Kp (cm/min)
PA 0.871 ± 0.19 8710.3 ± 1994.4AS 0.046 ± 0.011 460.0 ± 112.4
F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227 225
by components in the formulation, such as the surfactant PPG-5-CETETH-20 and oleic acid. Increasing the lipophilicity normallyincreases the permeation of the compound through nasal mucosa[32]. Besides, the higher viscosity of the formulation increasesthe contact time between the drug and the nasal mucosa, therebyincreasing the time for permeation.
3.5. Pharmacokinetic studies
The bioavailability of AZT was determined from intravenousadministration for the precursor formulation PA administrated bythe nasal route and compared to the drug solution IV route. Bothgroups received the same dose of AZT (8 mg/kg).
The mean plasma concentrations of AZT for the test formulation(PA) and reference formulation (intravenous injection of AZT solu-tion) over time are shown in Fig. 5. The calculated pharmacokineticparameters of AZT are shown in Table 2.
The Fig. 5 shows that the absorption from formulation PA wasimmediate, as from the IV injection. Following intravenous admin-istration, the Cmax in 5 min was 5446 ± 1533 ng/mL and declined
Fig. 5. Mean and standard deviation of plasma concentrations of AZT following asingle-dose administration of 8 mg/kg of the formulation PA administered by thenasal route and the AZT solution by IV injection in rats (N = 5).
Table 2Pharmacokinetic parameter means and standard deviations (± SD) after administra-tion of 8 mg/kg of AZT by intravenous and intranasal routes of the AZT solution andPA formulation, respectively, in rats (N = 5).
Route of administration Intravenous IntranasalSamples AZT solution PA
AUC0-1 (ng min/mL) 296,247 ± 165,752 136,155 ± 38,354*
Cmax (ng/mL) 5446 ± 1533 2445 ± 596*
Tmax (min) 5.00 6.67 ± 4.08Vd (mL/kg) 1573 ± 467 1572 ± 344Kel (1/min) 0.021 ± 0.008 0.019 ± 0.005t1/2 (min) 37.89 ± 15.40 40.02 ± 14.85Cl (mL/min/kg) 31.89 ± 11.36 29.13 ± 9.61
* Differs from Intravenous AZT solution (P < 0.05, Mann–Whitney nonparametric t-test).
rapidly. Following intranasal administration of PA, the Cmax value2445 ± 596 ng/mL was achieved in 6.67 min and the curve declinerapidly such as the intravenous administration. The values ofAUC0–1 and Cmax of intravenous AZT solution and intranasal PA for-mulation are significantly different (P < 0.05); however, bothgroups exhibited curves with similar profile, as it can be seen inFig. 5.
The distribution of the drug was not found to be significantlydifferent (P > 0.05), since the Vd values were similar for bothgroups. The elimination was also similar for intravenous and intra-nasal groups, since the means of t1/2, kel, and Cl for the two groupswere statistically similar.
Administered nasally, this study showed that the AZT will beabsorbed as soon as it is released. The enhanced permeation effectand the mucoadhesive properties may have been responsible forthe nasal absorption of AZT.
4. Discussion
The objective of this study was to evaluate an in situ gellingstimuli-sensitive drug delivery system with mucoadhesive proper-ties to promote the retention of the formulation inside the nasalcavity, as an alternative vehicle for antiretroviral drugs for AIDStreatment. To this end, a surfactant was developed from the com-ponents PPG-5-CETETH-20, oleic acid, and water, which can formliquid crystal phases on contact with an aqueous medium.
The phase diagram is an excellent tool to visualize the behaviorof surfactant systems in response to alterations in their composi-tion. In this diagram, it is possible to see the types of systemformed on mixing components (surfactant, aqueous, and oilyphases). Consequently, the most appropriate range for the develop-ment of a drug delivery system can be chosen [20].
The phase behavior and physicochemical characterizationshowed that it is possible to obtain a readily flowing system, com-bining PPG-5-CETETH-20, oleic acid, and water, that its degree oforganization when coming into contact with artificial nasal mucus,resulting in a rigid liquid crystal matrix. The phase transition froma liquid to a semisolid lamellar phase was accompanied by a stiff-ness change that may result in increased drug residence time andpossibly enhances drug absorption via the nasal mucosa.
For an in situ gelling formulation capable of forming such astrong liquid crystal matrix as the PPG-5-CETETH-20/oleic acid/water system, it is very likely that the rheological performance ofthe formulation is as crucial to achieving a long contact time asare the possible interactions with the mucin or mucus layer atthe system–mucus interface [17]. Hence, by combining the forma-tion of the liquid crystal matrix with the mucoadhesive forceshown by texture analyzer measurements, PPG-5-CETETH-20/oleicacid/water systems can be designed for prolonged retention of theformulations in the nasal cavity.
The transport and drug absorption across the nasal mucosa wasstudied by an ex vivo technique employing a diffusion chamber andan excised mucosa tissue. In vitro approaches are efficient andinexpensive and can be used at an early stage of drug developmentto choose among different formulations. An ethical advantage isthat the material was obtained from a slaughterhouse, and the sac-rifice of animals for only a small piece of an animal for a small pieceof tissue was thus avoided [21]. The ex vivo drug release results re-ported here underline the potential of the PPG-5-CETETH-20, oleicacid, and water systems as a promising permeation enhancer in na-sal delivery systems for hydrophilic antiretroviral drugs. It can besuggested that the amphiphilic and lipophilic compounds, namelythe surfactant PPG-5-CETETH-20 and oleic acid, may cause a dis-turbance in the cell lipid bilayers, forming pores at the surface ofthe epidermis. Oleic acid is known in literature for its enhancing

226 F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227
effect on the in vitro permeability of drugs due to the reducing ofthe lipid packing order in epithelial cell membranes [33]. This ef-fect could increase the fluidity of the lipid layer and decrease thediffusion resistance, assisting the permeation of compoundsthrough the mucosa.
The in vivo results revealed that the liquid crystal precursor for-mulation achieved a fast absorption of the drug. The time taken toreach the maximum concentration for nasal administration of PAwas 6.67 min, and the literature reports that conventional com-mercial oral formulations achieve the Cmax approximately in 0.5–1.5 h [34]. Therefore, mucoadhesive drug delivery systems seemto give a better absorption of the delivered drugs than the oralroute, mainly through their ability to adhere to mucus and residelonger in the nasal cavity before being cleared by the mucociliaryclearance system [7,8,32].
During the in vivo experiment, the plasma concentration curveobserved for the nasal route had a profile comparable to that forthe IV injection, indicating that the nasal route imposes few barri-ers to the absorption and the drug is directly absorbed into theblood circulation. Despite of the same behavior, the AUC0-1 ofthe nasal route was smaller than the IV route. So, it is necessaryto find out what is the pathway of the drug that did not contributeto the AZT in the blood. Considerable evidence exists from animaland human studies that, following nasal administration, drugs canbe transported directly from the nasal cavity to the central nervoussystem (CNS), via the olfactory epithelium. The olfactory epithe-lium is the only part of the CNS that is exposed directly to the envi-ronment and has could serve as a portal for drug delivery to thebrain [7]. Therefore, it may be suggested that part of the AZT wastransported to the CNS. The work of Ved and Kim [35], who studiedin vivo absorption and brain distribution in rabbits, showed thatAZT could pass preferentially into the cerebrospinal fluid and braintissues from the nasal cavity, possibly via the olfactory pathway. Inthat study, the intranasal drug delivery system for AZT wasformulated with a thermo-reversible gelling system prepared withpoloxamer 407 in aqueous buffer solution containing n-tridecyl-b-D-maltoside as permeation enhancer [35]. The ability of thehuman immunodeficiency virus (HIV) to enter and be harboredin the brain tissues results in numerous early and late stage abnor-malities of the CNS. The CNS symptoms may start as acute enceph-alitis and headache and progress to the AIDS dementia complex[35]. Hence, intranasal stimuli-sensitive drug delivery systemscould provide a promising therapeutic option for the treatmentfor CNS disorders caused by HIV.
The distribution and the elimination of the drug were not chan-ged by the nasal administration of PA. This result indicates that theliquid crystal precursor did not prolong the drug release, and theAZT is absorbed as soon as it is released from the formulation. Thisis an advantage in case of nasal administration, since the control-ling of the drug release must not be governed before the absorptionprocess, because the formulation cannot stay so long inside the na-sal cavity, otherwise it can alter the physiology of the nose. For na-sal administration, the formulation has to promote the absorptionas soon as possible, and then, it has to be cleared by the mucocil-iary clearance. The liquid crystal precursor formulation showedto attend this characteristic. However, in order to prolong the drugrelease by nasal route via a stimuli-sensitive drug delivery system,it is necessary to associate complementary strategies. For example,the maintenance of the drug release can be achieved by the encap-sulation of the drug into nano-carriers. Due to the nano-size, thecarriers can across the mucosa and achieve the blood circulation.Therefore, they can control the drug release, increasing the half-lifeand bioavailability of the drug.
Summarizing, the results obtained in this work evidenced thatthe nasal route has valuable advantages for systemic drug delivery,since it is characterized by a rapid clinical response. In this study,
the peak serum concentration occurred in 6.67 min, while for thecommercially available oral formulations, the peak serum concen-trations occur within 0.5–1.5 h [34].
Besides that, the nasal administration can avoid the side effectsdue to the first-pass metabolism caused when the drug is adminis-tered by the oral route.
For hydrophilic antiretroviral drugs such as AZT, the pharmaco-kinetic properties observed after intranasal administration of PAare particularly suitable for the rapid systemic absorption of thedrug. Despite of the low permeability of the nasal mucosa, a drugpermeation-enhancing effect of the PPG-5-CETETH-20/oleic acid/water system was identified by diffusion cell method. The rheolog-ical and mucoadhesive properties combined with the permeation-enhancing effect may be responsible for the rapid onset of actionachieved after intranasal administration of the liquid crystal pre-cursor formulation. The nasal administration of this new drugdelivery system can be an alternative for minimizing the risk ofsystemic adverse events of antiretroviral drugs and improve thecompliance of the patience to the therapy. Further studies are nec-essary to investigate the safety of this new drug delivery systemand quantify the AZT in the anatomical reservoirs of HIV such asthe CNS. These results open the way to consider future local appli-cations of antiretroviral drugs for AIDS treatment.
5. Conclusions
According to this study, the nasal absorption of AZT could besignificantly improved by administration an intranasal liquid crys-tal precursor formulation. The rheological and mucoadhesive prop-erties of this carrier in combination with the permeationenhancement observed in animal tests suggest that the PPG-5-CE-TETH-20, oleic acid and water formulation administered by the na-sal route might represent a promising novel tool for the systemicdelivery of AZT and other antiretroviral drugs.
Acknowledgement
This work was supported by FAPESP (Fundação de Amparo à Pes-quisa do Estado de São Paulo).
References
[1] L. Geocze, S. Mucci, M.A. D Marco, L.A. Nogueira-Martins, V.A. Citero, Qualidadede vida e adesão ao tratamento anti-retroviral de pacientes portadores de HIV,Rev. Saúde Pública 44 (2010) 743–749. http://dx.doi.org/10.1590/S0034-89102010000400019.
[2] D.D. Richman, D.M. Margolis, M. Delaney, W.C. Greene, D. Hazuda, R.J.Pomerantz, The challenge of finding a cure for HIV infection, Science 323(2009) 1304–1307, http://dx.doi.org/10.1126/science.1165706.
[3] E. Ojewole, I. Mackraj, P. Naidooa, T. Govender, Exploring the use of novel drugdelivery systems for antiretroviral drugs, Eur. J. Pharm. Biopharm. 70 (3)(2008) 697–710, http://dx.doi.org/10.1016/j.ejpb.2008.06.020.
[4] N.K. Saksena, D.N. Haddad, Viral reservoirs an impediment to HAART: newstrategies to eliminate HIV-1, Curr. Drug Targets Infect. Disord. 3 (2003) 179–206, http://dx.doi.org/10.2174/1568005033481187.
[5] F.C. Carvalho, R.M.M. Mainardes, M.P.D. Gremião, Exploring thenanotechnology-based drug delivery systems for AIDS treatment, in: F.H.Kasenga (Ed.), Understanding HIV/AIDS Management and Care – PandemicApproaches in the 21st Century, InTech, Rijeka, 2011, pp. 367–384.
[6] M.I. Ugwoke, R.U. Agu, N. Verbeke, R. Kinget, Nasal mucoadhesive drugdelivery: background, applications, trends and future perspectives, Adv. DrugDeliv. Rev. 57 (2005) 1640–1665, http://dx.doi.org/10.1016/j.addr.2005.07.009.
[7] R.M. Mainardes, M.C.C. Urban, P.O. Cinto, M.V. Chaud, R.C. Evangelista, M.P.D.Gremião, Lipossomes and micro/nanoparticles as colloidal carriers for nasaldrug delivery, Curr. Drug Deliv. 3 (2006) 275–285.
[8] L. Illum, Nasal drug delivery – possibilities, problems and solutions, J. Control.Rel. 87 (2003) 187–198.
[9] F.C. Carvalho, M.L. Bruschi, R.C. Evangelista, M.P.D. Gremião, Mucoadhesivedrug delivery systems, Braz. J. Pharm. Sci. 46 (2010) 1–17.
[10] C. He, S.W. Kim, D.S. Lee, In situ gelling stimuli-sensitive block copolymerhydrogels for drug delivery, J. Control. Rel. 127 (2008) 189–207, http://dx.doi.org/10.1016/j.jconrel.2008.01.005.

F.C. Carvalho et al. / European Journal of Pharmaceutics and Biopharmaceutics 84 (2013) 219–227 227
[11] G.P. Andrews, T.P. Laverty, D.S. Jones, Mucoadhesive polymeric platforms forcontrolled drug delivery, Eur. J. Pharm. Biopharm. 71 (2009) 505–518, http://dx.doi.org/10.1016/j.ejpb.2008.09.028.
[12] M. Malmsten, Surfactants and Polymers in Drug Delivery, Marcel Dekker, NewYork, 2002.
[13] S. Singh, Phase transitions in liquid crystals, Phys. Rep. 324 (2000) 107–269.[14] M. Makai, E. Csányi, Z. Némenth, J. Pálinkás, Structure and drug release of
lamellar liquid crystals containing glycerol, Int. J. Pharm. 256 (2003) 95–107.[15] J.C. Shah, Y. Sadhale, D.M. Chilukuri, Cubic phase gels as drug delivery systems,
Adv. Drug Deliv. Rev. 47 (2001) 229–250.[16] F.C. Carvalho, V.H.V. Sarmento, L.A. Chiavacci, M.S. Barbi, M.P.D. Gremião,
Development and in vitro evaluation of surfactant systems for controlledrelease of zidovudine, J. Pharm. Sci. 99 (2010) 2367–2374.
[17] F.C. Carvalho, M.S. Barbi, V.H.V. Sarmento, L.A. Chiavacci, F.M. Netto, M.P.D.Gremião, Surfactant systems for nasal AZT delivery: structural, rheological andmucoadhesive properties, J. Pharm. Pharmacol. 62 (2010) 430–439, http://dx.doi.org/10.1211/jpp/62.04,0004.
[18] B.J. Boyd, D.V. Whittaker, S.M. Khoo, G. Davey, Lyotropic liquid crystallinephases formed from glycerate surfactants as sustained release drug deliverysystems, Int. J. Pharm. 309 (2006) 218–226.
[19] M.L. Bruschi, D.S. Jones, H. Panzeri, O. Freitas, M.P.D. Gremião, E.H.G. Lara,Semisolid systems containing propolis for the treatment of periodontaldisease: in vitro release kinetics, syringeability, rheological, textural, andmucoadhesive properties, J. Pharm. Sci. 96 (2007) 2074–2089.
[20] F.C. Carvalho, H.R. Silva, G.M. Luz, M.S. Barbi, D.S. Landgraf, L.A. Chiavacci,V.H.V. Sarmento, M.P.D. Gremião, Rheological, mechanical and adhesiveproperties of surfactant-containing systems designed as a potential platformfor topical drug delivery, J. Biomed. Nanotecnol. 8 (2012) 1–10.
[21] K. Osth, J. Grasjo, E.A. Bjork, A new method for drug transport studies on pignasal mucosa using a horizontal using chamber, J. Pharm. Sci. 91 (2002) 1259–1273.
[22] C. Callens, J. Ceulemans, A. Ludwig, P. Foreman, J.P. Remon, Rheological studyon mucoadhesivity of some nasal powder formulations, Eur. J. Pharm.Biopharm. 55 (2003) 323–328.
[23] N. Franse, E. Bjork, K. Edsman, Changes in the mucoadhesion of powderformulations after drug application investigated with a simplified method, J.Pharm. Sci. 97 (2008) 3855–3864, http://dx.doi.org/10.1002/jps.
[24] The United States Pharmacopoeial Convention 35th, Pharmaceutical, Forum,vol. 35, 2009, pp. 1–28.
[25] A.C. Williams, Transdermal and Topical Drug Delivery, Pharmaceutical Press,London, 2003.
[26] J. Siepmann, N.A. Peppas, Higuchi equation: derivation, applications, use andmisuse, Int. J. Pharm. 418 (2011) 6–12, http://dx.doi.org/10.1016/j.ijpharm.2011.03.051.
[27] R.W. Korsmeyer, R. Gurny, E. Doelker, P. Buri, N.A. Peppas, Mechanisms ofsolute release from porous hydrophilic polymers, Int. J. Pharm. 15 (1983) 25–35.
[28] V. Papadopoulou, K. Kosmidis, M. Vlachou, P. Macheras, On the use of theWeibull function for the discernment of drug release mechanisms, Int. J.Pharm. 309 (2006) (2006) 44–50, http://dx.doi.org/10.1016/j.ijpharm.2005.10.044.
[29] P. Costa, J.M.S. Lobo, Modeling and comparison of dissolution profiles, Eur. J.Pharm. Sci. 3 (2001) 123–133.
[30] C.S. Brazel, N.A. Peppas, Modeling of drug release from swellable polymers,Eur. J. Pharm. Biopharm. 49 (2000) 47–58.
[31] U.V. Banakar, Pharmaceutical Dissolution Testing, Marcel Dekker, New York,1992.
[32] P. Arora, S. Sharma, S. Garg, Permeability issues in nasal drug delivery, DrugDiscov. Today 7 (2002) 967–975.
[33] J.A. Nicolazzo, B.L. Reed, B.C. Finnin, Buccal penetration enhancers—how dothey really work?, J Control. Rel. 105 (2005) 1–15, http://dx.doi.org/10.1016/j.jconrel.2005.01.024.
[34] GlaxoSmithKline, Retrovir� (zidovudine), Tablets, Capsules, and Syrup. InitialU.S. Approval: 1987, revised: September 2008.
[35] P.M. Ved, K. Kim, Poly(ethylene oxide/propylene oxide) copolymer thermo-reversible gelling system for the enhancement of intranasal zidovudinedelivery to the brain, Int. J. Pharm. 411 (2011) 1–9, http://dx.doi.org/10.1016/j.ijpharm.2011.02.040.
Related Documents











